
What happens to proteins after translocation into ER? -Protein glycosylation!!! Carbohydrate (sugar)...
80
happens to proteins after translocation to ER? -Protein glycosylation!!! • Carbohydrate (sugar) molecules are linked to amino acids in protein • carried out by enzymes that re side in ER • sugar molecules are further mo dified in Golgi oteins that get translocated to ER get lated, few cytosolic proteins are glycosylated f glycosylation (?) : s of glycosylation have little effect on protein fu resistance to proteases in-protein interactions depend on glycosylation
-
Upload
amice-newman -
Category
Documents
-
view
217 -
download
0
Transcript of What happens to proteins after translocation into ER? -Protein glycosylation!!! Carbohydrate (sugar)...

What happens to proteins after translocation into ER?-Protein glycosylation!!!
• Carbohydrate (sugar) molecules are linked to amino acids in protein• carried out by enzymes that reside in ER• sugar molecules are further modified in Golgi
Most proteins that get translocated to ER getGlycosylated, few cytosolic proteins are glycosylated
Functions of glycosylation (?) :• inhibitors of glycosylation have little effect on protein functions• increases resistance to proteases•some protein-protein interactions depend on glycosylation

Carbohydrate (sugar) molecules :
glucose (Glc)
galactose (Gal)
mannose (Man)
fucose (Fuc)
N-acetyl-glucosamine (GlcNAc)
N-acetyl-galactosamine (GalNAc)
sialic acid (NANA)

Glycoproteins
N-glycosidic linkage
CH2-CO-NH-GlcNAc-GlcNAc-….
O-glycosidic linkage
CH2-O-GalNAc-….
peptidebackbone
peptidebackbone

Typical N-linked oligosaccharide:
CH2-CO-NH-GlcNAc-GlcNAc-ManMan-GlcNAc-Gal-NANA
Man-GlcNAc-Gal-NANAFuc
Typical O-linked oligosaccharide:
CH2-O-GalNAc-GalNAc-NANA
NANA

After translocation into the ER all proteins move on to the Golgi(both soluble and integral membrane proteins)• Some proteins function in the ER and reside there permanently signal peptidase, glycosylation enzymes, etc.• ER resident proteins contain an ER retention signal, 4 amino acids at C-terminus of protein• These proteins move onto Golgi, then retrieved, brought back to ER,
• How do proteins move between ER and Golgi? VESICULAR TRANSPORT

Movement between Golgi and ER:• form two distinct, membrane-bounded compartments• components are transferred via transport vesicles that bud off from ER and fuse to the Golgi. • vesicles contain both lumenal (soluble) and integral membrane proteins

Protein Maturation in Golgi: I. Glycosylation
lysosomalenzymesonly


Summary of protein trafficking:
FREE RER Signal peptide
ReceptorMediated endocytosis

● Protein phosphorylationPhosphoBase v. 2.0 (http://www.cbs.dtu.dk/databases/PhosphoBase/)Enzymes:1. Basophilic protein kinases
(e.g. protein kinase C (PKC))2. Acidophilic protein kinases
(e.g. protein kinase CK2)3. Proline-directed protein kinases
(e.g. protein kinase cdc2)4. Protein tyrosine kinases
(e.g. epidermal growth factor receptor, EGFR)
5. Protein serine/threonine kinases
Source: Lehninger pg 1053

● Protein methylations
Source: Lehninger pg 1053
Protein methylation:
1. Increases hydrophobicity
2. May alter the charge of the protein ( e.g. if a carboxyl group of Glu is methylated)

Proteomics and posttranslational modifications

Proteomics and posttranslational modifications
Patterson and Aebersold, Nature Genetics (supp.), 33, 311 (2003)
protein-ligandprotein-ligandinteractionsinteractions
protein-ligandprotein-ligandinteractionsinteractions
proteinproteincomplexescomplexes(machines)(machines)
proteinproteincomplexescomplexes(machines)(machines)
protein familiesprotein families(activity or structural)(activity or structural)
protein familiesprotein families(activity or structural)(activity or structural)
post-translationalpost-translationalmodified proteinsmodified proteins
post-translationalpost-translationalmodified proteinsmodified proteins
Eukaryotic cell.Examples of protein properties are shown, including the interaction of proteins and protein modifications.

Complexity of the ProteomeComplexity of the Proteome• Protein processing and modification comprise an importa
nt third dimension of information, beyond those of DNA sequence and protein sequence.
• Complexity of the human proteome is far beyond the more than 30,000 human genes(20,000-25,000).
• The thousands of component proteins of a cell and their post-translational modifications may change with the cell cycle, environmental conditions, developmental stage, and metabolic state.
• Proteomic approaches that advance beyond identifying pProteomic approaches that advance beyond identifying proteins to elucidating their post-translational modificationroteins to elucidating their post-translational modifications are needed.s are needed.

Proteomic Analysis of Post-translational Modifications
– Mass spectrometry and other biophysical methods can be used to determine and localize potential PTMs. However, PTMs are still challenging aspects of proteomics with current methodologies

Isolation of modified proteins:
Modification analysis is usually done by comparison of experimental data to a known amino acid sequence.
A central consideration in the characterization of Modifications is the need for as large an amount of the protein as possible. Why?
-typically not homogeneous, a very small fraction of total…
● Recombinantly expressed proteins or recombinantly expressed proteins that are modified in vitro. ( baculovirus and Mammalian expression system are often significant differences.)
● Chromatographic purifications +antibody precipitations ----the modifications of a single protein.
● 2D +different staining.

The ‘pearls-on-string’pattern is a telltale indication of protein phosphorylation, although the introduction of charge heterogeneity by deamidation of Asn or Gln residues to form carboxylic acids can also generate such a pattern.

PTM mapping of a purified protein:A variety of techniques can be used to determine the modified amino acids.
Don’t forget the traditional method: Amino acid sequencing for amino- or carboxy-terminal processing!

Proteomic analysis of PTMs
Mann and Jensen, Nature Biotech. 21, 255 (2003)

● A Proteomics Approach to Understanding Protein Ubiquitination
1)Nature Biotechnology,21,921-926(2003)

S.cerevisiae Strain SUB592(in which all ubiquitin genes were removed and a 6xHis-myc-ubiquitin-coding plasmid was introduced) and the control strain SUB280(similar to SUB592 except for the introduction of a wild-type ubiquitin plasmid)


Summary:

2) Genome Research,13:1389-1394(2003)
---The Comparative Proteomics of Ubiquitination in Mouse
It was a bioinformatics work.
Mus musculus proteome (based upon proteins from SWISS-PROT and TrEMBL and additional peptides predicted by Ensembl) was obtained from the EBI Proteome Analysis Database(http://www.ebi.ac.uk/proteome/)
The Representative Transcript and Protein Set(RTPS)
UA InterPro domains----UA proteins

● Proteomic Analysis of glycoprotein




Isotope-coded glycosylation-site-specific tagging(IGOT)

What ahout O-linked glycoproteins?
● There is no enzyme comparable to PNGase F for removing intact O-linked sugars.
● Sequentially remove monosaccharides by using a panel of exoglycosidase until only the Galbeta1,3 GalNAc core remains attached to the serine or threonine residue.
● The core can then be released by O-glycosidase.
●The chemical method, such as beta-elimination, may be more generally useful and effective.


Glycoprotein Gel StainGlycoprotein Gel Stain
CandyCane glycoprotein molecular weight standards containing alternating glycosylated and nonglycosylated proteins were electrophoresed through a 13% polyacrylamide gel. After separation, the gel was stained with SYPRO Ruby protein gel stain to detect all eight marker proteins (left). Subsequently, the gel was stained by the standard periodic acid–Schiff base (PAS) method in the Pro-Q Fuchsia Glycoprotein Gel Stain Kit to detect the glycoproteins alpha2-macroglobulin, glucose ox
idase, alpha1-glycoprotein and avidin.
Pro-Q™ Glycoprotein Stain (DDAO phosphate)Molecular Formula: C15H18Cl2N3O5P (MW 422.20)
Detection of glycoproteins and total protein on an SDS-polyacrylamide gel using the Pro-Q Fuchsia Glycoprotein Gel Stain Kit.

Glycome-Glycomics
Glycoproteome-Glycoprotomics
Glycan array

● Proteomics and Protein phosphorylation

Phosphorylation• Analysis of the entire complement of phosphorylated proteins in cell
s: “phosphoproteome”• Qualitative and quantitative information regarding protein phosphory
lation important• Important in many cellular processes
– signal transduction, gene regulation, cell cycle, apoptosis
• Most common sites of phosphorylation: Ser, Thr, Tyr
• MS can be used to detect and map locations for phosphorylation– MW increase from addition of phosph
ate group
– treatment with phosphatase allows determination of number of phosphate groups
– digestion and tandem MS allows for determination of phosphorylation sites

Enrichment strategies to analyze phosphoproteins/peptides
• Phosphospecific antibodiesPhosphospecific antibodies– Anti-pY quite successful– Anti-pS and anti-pT not as successful, but may be used (M. Grønb
org, T. Z. Kristiansen, A. Stensballe, J. S. Andersen, O. Ohara, M. Mann, O. N. Jensen, and A. Pandey, “Approach for Identification of Serine/Threonine-phosphorylated Proteins by Enrichment with Phospho-specific Antibodies.” Mol. Cell. Proteomics 2002, 1:517–527.
• Immobilized metal affinity chromatography (IMAC)Immobilized metal affinity chromatography (IMAC)– Negatively charged phosphate groups bind to postively charged m
etal ions (e.g., Fe3+, Ga3+) immobilized to a chromatographic support
– Limitation: non-specific binding to acidic side chains (D, E)• Derivatize all peptides by methyl esterification to reduce non-sp
ecific binding by carboxylate groups.• Ficarro et al., Nature Biotech. (2002), 20, 301.





Chemical derivatization to enriChemical derivatization to enrich for phosphoproteinsch for phosphoproteins
• Developed because other methods based on affinity/adsorption (e.g., IMAC) displayed some non-specific binding
• Chemical derivatization methods may be overly complex to be used routinely
• Sensitivity may not be sufficient for some experiments (low pmol)

Phosphoprotein StainPhosphoprotein Stain
PeppermintStick phosphoprotein molecular weight standards separated on a 13% SDS polyacrylamide gel. The markers contain (from largest to smallest) beta-galactosidase, bovine serum albumin (BSA), ovalbumin, beta-casein, avidin and lysozyme. Ovalbumin and beta-casein are phosphorylated. The gel was stained with Pro-Q Diamond phosphoprotein gel stain (blue) followed by SYPRO Ruby protein gel stain (red). The digital images were pseudocolored.
Phospho

Phosphoprotein StainPhosphoprotein Stain
Visualization of total protein and phosphoproteins in a 2-D gel
Proteins from a Jurkat T-cell lymphoma line cell lysate were separated by 2-D gel electrophoresis and stained with Pro-Q Diamond phosphoprotein gel stain (blueblue) followed by SYPRO Ruby protein gel stain (redred). After each dye staining, the gel was imaged and the resulting composite image was digitally pseudocolored and overlaid.
T.H. Steinberg et al., Global quantitative phosphoprotein analysis using Multiplexed Proteomics technology, Proteomics 2003, 3, 1128-1144

RAW 264.7 exposed to DEP
Global Analysis of Protein PhosphorylationGlobal Analysis of Protein Phosphorylation
Pro-Q DiamondPro-Q Diamond Sypro RubySypro Ruby
Xiao, Loo, and Nel - UCLA
IEF
9.53.54.5 5.1 5.5 6.0 7.0 8.4
53
4
12
6 7
20
30
37
98
55
9.53.54.5 5.1 5.5 6.0 7.0 8.4
30
37
98
55
20
89
10
1112
13
14
TNFTNF convertase convertaseMAGUK p55MAGUK p55
PDIPDIProtein phosphatase 2AProtein phosphatase 2A
JNK-1JNK-1p38 MAPK alphap38 MAPK alpha
ERK-1ERK-1ERK-2ERK-2ErbB-2ErbB-2
TNFTNFHSP 27HSP 27

Identification of Low Abundance Proteins
• The identification of low abundance proteins in the presence of high abundance proteins is problematic (e.g., “needle in a haystack”)
• Pre-fractionation of complex protein mixtures can alleviate some difficulties– gel electrophoresis, chromatography, etc
• Removal of known high abundance proteins allows less abundant species to be visualized and detected
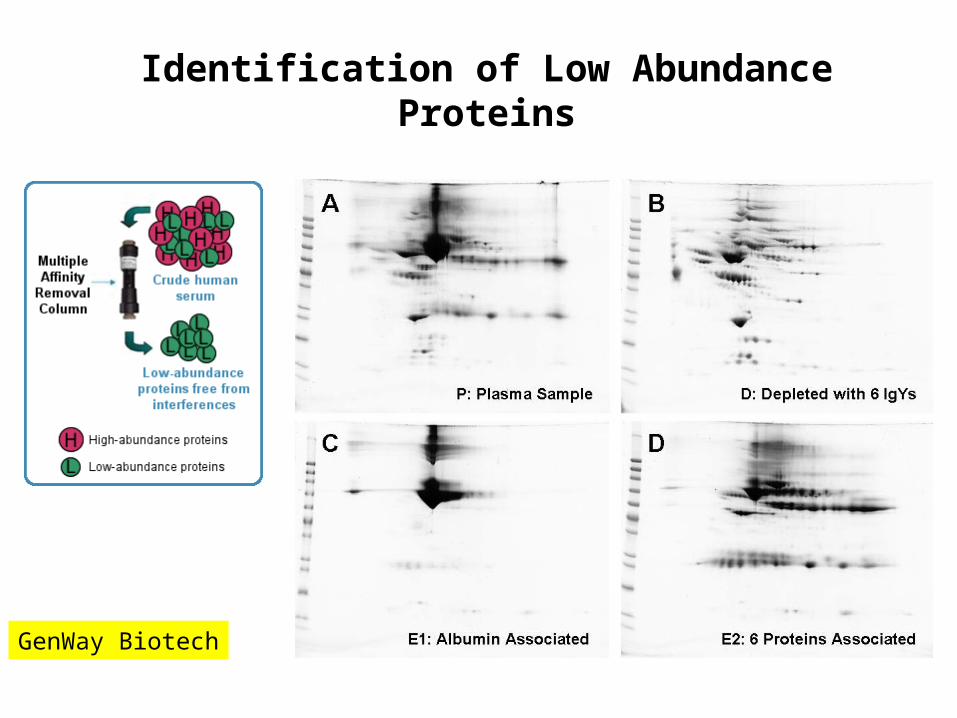
Identification of Low Abundance Proteins
GenWay Biotech

Proteinchip for analysis of protein phosphorylation

Molecular Cell, Vol. 13, 649–663, March 12, 2004
Endostatin’s Antiangiogenic Signaling Network

Methods: DNA+Antibody Chip, Real-time PCR & Immunocytochemistry
◆ Human Unigene Chip II containing 74834 elements covering 90% of all human genes. This represents one of the largest gene sets interrogated in array-based gene expression studies to date. ratio-voting criteria (more than 2.0 and less than 0.5)
◆ analyzed the alteration in gene expression after a 4 hr treatment of primary isolated human dermal microvascular of endothelial cells (HDMVEC) with 200 ng/ml of human recombinant endostatin (Pichia Pastoris). 16 characteristic genes were chosen for real-time quantitative RT-PCR analysis using Taq-man technology
◆ Differentially phosphorylated proteins were examined using antibody array technology. After 30 min incubation with 500 ng/ml human recombinant endostatin, whole cell protein lysates from HDMVECs were hybridized on an antibody array containing 400 immobilized antibodies against well- studied signaling proteins. The phosphorylation status of the tyrosine residues from the cellular proteins bound to the array were detected using a horse peroxidase-conjugated anti-phosphotyrosine antibody.
◆Immunocytochemistry

Results:
●Genome-Wide Expression Profiling: 6635 Unigene clusters to 24 different functional groups

● Protein-Level Regulation:

● Endostatin Downregulates Id1 and Id3
● Endostatin Downregulates HIF1-alpha
● Ephrins and TNF in Endostatin Signaling
● Endostatin Modulates NF-B Signaling
● AP-1 Transcription Factors Are Downregulated By Endostatin
● Coagulation Cascade and Adhesion Molecules in Endostatin Signaling
● STATs in Endostatin Signaling
● ETS-1 in Endostatin Signaling



Bioinformatics and protein modifications

This database is freely accessible on the Internet through resources provided by the European Bioinformatics Institute (http://www.ebi.ac.uk/RESID), and by the National Cancer Institute – Frederick Advanced BiomedicalComputing Center (http://www.ncifcrf.gov/RESID).


PTMs are ubiquitous and dynamic. Their presence must be: ● Predicted by computational sequence analysis ● Determined by: efficient and sensitive experimental proteomic techniques genetic, biochemical and analytical techniques

Hepatitis C Virus and Proteomics

The Hepatitis C Virus (HCV)
Genus of Hepacivirus, member of Flaviviridae, a family comprising enveloped (+)strand RNA viruses.
Complex secondary structure at the 5’NTR and part of the core
sequences serves as IRES.
Hepatitis C virus infection represents a major problem of public health with around 350 millions of chronically infected indi
viduals worldwide. The frequent evolution towards severe liver disease and cancer are the main features of HCV chronic infection.



Brechot C Cell Death and Differentiation(2003)10,S27-S38

Brechot C Cell Death and Differentiation(2003)10,S27-S38

Brechot C Cell Death and Differentiation(2003)10,S27-S38

Dubuisson J . Biochimie 85(2003)295-301

Human vesicle-associated membrane protein-associated protein A(hVAP-A)
Matthew J.Evans PNAS 2004 101:13038-43






DNA/RNA Affinity Chromatography
Protein Loading
Column Wash
Elution
Highly Purifed Transcription Factor
Contaminant proteins





Additional Readings
• R. Aebersold and M. Mann, Mass spectrometry-based proteomics, Nature (2003), 422, 198-207.
• M. B. Goshe and R. D. Smith, “Stable isotope-coded proteomic mass spectrometry.” Curr. Opin. Biotechnol. 2003; 14: 101-109.
• W. A. Tao and R. Aebersold, “Advances in quantitative proteomics via stable isotope tagging and mass spectrometry.” Curr. Opin. Biotechnol. 2003; 14: 110-118.
• S. D. Patterson and R. Aebersold, “Proteomics: the first decade and beyond.” Nature Genetics 2003; 33 (suppl.): 311-323.
• M. Mann and O. N. Jensen, “Proteomic analysis of post-translational modification.” Nature Biotech. 2003; 21: 255-261.
• D. T. McLachlin and B. T. Chait, “Analysis of phosphorylated proteins and peptides by MS.” Curr. Opin. Chem. Biol. 2001; 5: 591-602.
• S. Gygi et al., “Quantitative analysis of complex protein mixtures using isotope-coded affinity tags.” Nature Biotech. 1999; 17: 994-999.

Proteomics in Practice: A Laboratory Manual of Proteome AnalysisReiner Westermeier, Tom NavenWiley-VCH, 2002
PART II: COURSE MANUAL Step 1: Sample Preparation Step 2: Isoelectric Focusing Step 3: SDS Polyacrylamide Gel Electrophoresis Step 4: Staining of the Gels Step 5: Scanning of Gels and Image Analysis Step 6: 2D DIGE Step 7: Spot Excision Step 8: Sample Destaining Step 9: In-gel Digestion Step 10: Microscale Purification Step 11: Chemical Derivatisation of the Peptide Digest Step 12: MS Analysis Step 13: Calibration of the MALDI-ToF MS Step 14: Preparing for a Database Search Step 15: PMF Database Search Unsuccessful
PART I: PROTEOMICS TECHNOLOGY Introduction Expression Proteomics Two-dimensional Electrophoresis Spot Handling Mass Spectrometry Protein Identification by Database Searching Methods of Proteomics

Proteins and Proteomics: A Laboratory ManualRichard J. SimpsonCold Spring Harbor Laboratory (2002)
Chapter 1. Introduction to Proteomics Chapter 2. One–dimensional Polyacrylamide Gel Electrophoresis Chapter 3. Preparing Cellular and Subcellular Extracts Chapter 4. Preparative Two–dimensional Gel Electrophoresis with
Immobilized pH Gradients Chapter 5. Reversed–phase High–performance Liquid Chromatography Chapter 6. Amino– and Carboxy– terminal Sequence Analysis Chapter 7. Peptide Mapping and Sequence Analysis of Gel–resolved Proteins Chapter 8. The Use of Mass Spectrometry in Proteomics Chapter 9. Proteomic Methods for Phosphorylation Site Mapping Chapter 10. Characterization of Protein Complexes Chapter 11. Making Sense of Proteomics: Using Bioinformatics to Discover a
Protein’s Structure, Functions, and Interactions
















![Human plasma protein N-glycosylation - Springer · 2017. 8. 26. · As the previous review on plasma protein N-glycosylation originatesfrom2008[40],weherestrivetoconveythecurrent](https://static.fdocuments.net/doc/165x107/6148e5b178bdf203dd34e7ec/human-plasma-protein-n-glycosylation-springer-2017-8-26-as-the-previous-review.jpg)

