
Welcome message10enqo.eventos.chemistry.pt/images/eBook_of_Abstracts.pdf · Organic Chemist Award;...
333
Welcome message The Organic Chemical Division of the Portuguese Chemical Society (SPQ) cordially invites you to attend the 10º Portuguese National Meeting of Organic Chemistry and the 1st Portuguese-Brazilian Organic Chemistry Symposium. The meeting will be held in the beautiful city of Lisbon at the Pharmacy Faculty of the Lisbon University from 4-6 of September 2013. This is a unique event that will bring together the Portuguese and Brazilian organic chemistry communities in a scientific program of high quality that spans over the many aspects of modern organic chemistry, though with a particular focus on the interface of organic chemistry with biological sciences. This event will count with 10 plenary lectures, 6 of which delivered by eminent international speakers, over 28 oral communications, 16 flash presentations and a poster session. The Organic Chemical Division will award 3 young scientists with the Portuguese Young Organic Chemist Award; the Portuguese Award for Best PhD and Master Thesis. Three prizes will be awarded to poster presentations in the following categories chromatographic, mass and NMR techniques. Lisbon is a historic capital, a potpourri of unusual character and charm, where 800 years of cultural influences mingle with modern trends and life styles. It is therefore, the perfect scenario to receive you all and promote fruitful scientific discussion. Welcome to Lisbon, Prof. Carlos A. M. Afonso (Conference Chairman)
Transcript of Welcome message10enqo.eventos.chemistry.pt/images/eBook_of_Abstracts.pdf · Organic Chemist Award;...

Welcome message
The Organic Chemical Division of the Portuguese Chemical Society (SPQ) cordially
invites you to attend the 10º Portuguese National Meeting of Organic Chemistry and the 1st
Portuguese-Brazilian Organic Chemistry Symposium. The meeting will be held in the
beautiful city of Lisbon at the Pharmacy Faculty of the Lisbon University from 4-6 of
September 2013. This is a unique event that will bring together the Portuguese and
Brazilian organic chemistry communities in a scientific program of high quality that spans
over the many aspects of modern organic chemistry, though with a particular focus on the
interface of organic chemistry with biological sciences.
This event will count with 10 plenary lectures, 6 of which delivered by eminent international
speakers, over 28 oral communications, 16 flash presentations and a poster session.
The Organic Chemical Division will award 3 young scientists with the Portuguese Young
Organic Chemist Award; the Portuguese Award for Best PhD and Master Thesis. Three
prizes will be awarded to poster presentations in the following categories chromatographic,
mass and NMR techniques.
Lisbon is a historic capital, a potpourri of unusual character and charm, where 800 years of
cultural influences mingle with modern trends and life styles. It is therefore, the perfect
scenario to receive you all and promote fruitful scientific discussion.
Welcome to Lisbon,
Prof. Carlos A. M. Afonso
(Conference Chairman)

Scientific committee
Chairman: Carlos A. M. Afonso, Fac. Pharmacy, Uni. Lisbon
Ana Maria Félix Trindade Lobo, FCT, New University of Lisbon
António Manuel d’Albuquerque Rocha Gonsalves, FCT, Uni. Coimbra
Artur Manuel Soares da Silva, Uni. Aveiro
José Cavaleiro, Uni. Aveiro
Luiz Fernando da Silva Junior, Uni. São Paulo
Madalena Maria de Magalhães Pinto, Fac. Pharmacy, Uni. Porto
Maria Fernanda de Jesus R. P. Proença, Uni. Minho
Rui Moreira, Fac. Pharmacy, Uni. Lisbon
Victor Armando Pereira Freitas, Uni. Porto
Vitor Ferreira, Uni. Federal Fluminense
Organizing committee
Chairman: Carlos A. M. Afonso, Fac. Pharmacy, Uni. Lisbon
Alexandre Trindade, Fac. Pharmacy, Uni. Lisbon
Ana Ressurreição, Fac. Pharmacy, Uni. Lisbon
Ana Margarida Madureira, Fac. Pharmacy, Uni. Lisbon
André Martins, Fac. Pharmacy, Uni. Lisbon
Andreia Rosa, Fac. Pharmacy, Uni. Lisbon
Andreia Rosatella, Fac. Pharmacy, Uni. Lisbon
Ângelo Rocha, IST, Uni. Lisbon
Anthony J. Burke, Uni. Évora
Catarina Rodrigues, Fac. Pharmacy, Uni. Lisbon
Carlos Monteiro, Fac. Pharmacy, Uni. Lisbon
Fábio Santos, Fac. Pharmacy, Uni. Lisbon
Filipa Siopa, Fac. Pharmacy, Uni. Lisbon
Francesco Montalbano, Fac. Pharmacy, Uni. Lisbon
Gonçalo Farias, Fac. Pharmacy, Uni. Lisbon
Jaime Coelho, Fac. Pharmacy, Uni. Lisbon
João António, Fac. Pharmacy, Uni. Lisbon
João Rosa, Fac. Pharmacy, Uni. Lisbon
Joice Lana, Fac. Pharmacy, Uni. Lisbon
Krassimira Guerra, Fac. Pharmacy, Uni. Lisbon
Leonardo Mendes, SPQ
Luís Frija, Fac. Pharmacy, Uni. Lisbon
Luís Gomes, Fac. Pharmacy, Uni. Lisbon
Maria M. M. Santos, Fac. Pharmacy, Uni. Lisbon
Nuno Maulide, Max-Planck-Institut
Noélia Duarte, Fac. Pharmacy, Uni. Lisbon
Pedro M. P. Góis, Fac. Pharmacy, Uni. Lisbon
Pedro M. S. D. Cal, Fac. Pharmacy, Uni. Lisbon
Roberta Paterna, Fac. Pharmacy, Uni. Lisbon
Susana Lucas, Fac. Pharmacy, Uni. Lisbon
Svilen P. Simeonov, Fac. Pharmacy, Uni. Lisbon

Sponsors and acknowledgements
Organization
Institutional support
Golden Sponsors
Atral Cipan
Bial
Hovione

Sponsors
Dias de Sousa, S.A.
Enzymatic
European Journal of Organic Chemistry
Fundação Jacqueline Dias de Sousa
Izasa
LaborSpirit, Lda
Macrisan, Lda
Qlabo
Rotoquímica
Solchemar
Tecnocroma
Thieme
Unicam

Delta Cafés
Super Bock
Godal
Oficial carrier
Hotels and Hostels supporting the congress:
VIP Grand Hotel and SPA
Sana Metropolitan
Villarica
Turim Ibéria
Hotel 3K Europa
Vip inn Berna
VIP executive Zurique
Turim Europa
SANA Reno
The Independente
Shiado Hostel
Restaurants supporting the congress:
Museu da Cerveja
Faculty of Pharmacy Bar
Faculty of Dental Medicine Bar
Club i
Puro Acaso

General information
Meeting venue
The conference venue will be at the Pharmacy Faculty of the Lisbon University (FFUL),
from 4th
till 6
th of September, 2013.
Faculdade de Farmácia da UL
Av. Prof. Gama Pinto
1649-003 Lisboa – Portugal
Telefone: +351 217946400
Fax: +351 217946470
http://www.ff.ul.pt/
How to arrive to venue:
By metro: The nearest metro station is Cidade Universitária (in yellow line), about 3
minutes walk from the venue.
By train: The nearest train station is Entrecampos, about 15 minutes walk from the
venue.
By bus: 731, 735, 738, 755, 764, 768 are the main bus services going by Cidade
Universitária.
By car: The GPS coordinates of the venue are 38.749599, -9.157169. During the
congress is possible to park your car for free inside the faculty parking lot.

Venue map

Where to eat
Restaurant Average Prices
Including
Faculty of Pharmacy Bar
3.75 - 5 € Soup, main dish (meat or fish; vegetarian under order)
and fruit – 3.75 € Plus drink and coffee – 5 €
Faculty of Dental Medicine
5 € Soup, main dish (meat or fish; vegetarian under
order), drink and dessert
Club i (ISCTE)
9-12 € Soup (or dessert), main dish, drink and coffee
Puro Acaso (Complexo)
5-7 € Main dish (Buffet), drink and dessert
1 - Faculty of Pharmacy (Venue entrance) 2 - Faculty of Pharmacy Bar 3 - Faculty of Dental Medicine 4 - Puro Acaso (Complexo) 5 - Club i (ISCTE)

Access to internet
In the faculty library (level 0 floor under the auditorium) the participants can access a
computer room. In addition in the building it is possible to access the wireless network using
the following credentials:
Wireless credentials Guest User Name - fful Password - fful01 Profile - guest-UL
Language
English is the official language of the congress. Voltage
In Portugal the line voltage is 220V. Insurance
Participants are responsible for arranging their own health and accident insurance. Banking
Several banks and ATMs are located within 5 minutes walk distance from the FFUL. Most of
the restaurants announced herein will only accept money.

Social program
Welcome reception will be held in the venue in the congress day 1 (4th September) at
18.30. The registration fee includes the access to the welcome ceremony.
Conference dinner will take place in the congress day 2 (5th September) in Museu da
Cerveja at Terreiro do Paço, 20h. In this day we will walk from the Laboratório Chimico in
the direction of the restaurant, passing by several sightseeing places of Lisbon (Miradouro
de São Pedro de Alcântara, Largo do Camões/Chiado, Rossio, Baixa and Terreiro do
Paço) giving the opportunity for those how have never been in Lisbon to experience a
small piece of this lovely city. The admission price for the dinner is 26 euros (if you desire
to attend please inform the organization members at the reception).
Museu da Cerveja
Terreiro do Paço – Ala Nascente – Nº 62 a 65
1100-148 Lisboa
Telefone: 00 351 210 987 656
Email: [email protected]
Metro station: Terreiro do Paço (Blue line)

Scientific information
Oral communications
The congress has a large number of oral presentations covering several topics of
research in the field of catalysis, organic synthesis, natural products and medicinal
chemistry. The oral communications are divided in:
Plenary sessions (40 minutes),
Oral communications in the 1st Portuguese-Brazilian Organic Chemistry
Symposium (20 minutes),
Invited national and selected oral communications (15 minutes)
Flash communications (10 minutes).
These timings include time for scientific discussion.
Speakers on the 4th September are kindly ask to contact the reception desk to
provide their presentation data and verify the presentation preview. The remaining speakers
are asked, if possible, to leave their presentations 24 hours in advance.
Poster presentations
Two poster sessions will be held in the congress, giving the opportunity for
exchange of ideas and networking between all the congress participants. The first poster
session will take place in the day 1, while the second poster session will be divided between
the two mornings of day 2 and 3:
Poster session 1 – PC1 to PC96 (10.30-11.00 and 16.00-16.30, 4th Sep.)
Poster session 2 – PC97 to PC195 (10.30-11.00, 5th
and 6th Sep.)
The maximum size for the posters is 120x90 cm.

Special event of 5th
September
During the 10º Portuguese National Meeting of Organic Chemistry and the 1st
Portuguese-Brazilian Organic Chemistry Symposium will take place in the Laboratório
Chimico a special session named “Portuguese Organic Chemistry: A future full of history…”.
The Laboratório Chimico was established in the Escola Politécnica de Lisboa in 1844. At
the time, the laboratory was one of the centres of scientific progress in Europe. Generations
of future chemists, pharmacists,
doctors and even engineers studied
there. Currently displays its vast
collection of scientific instruments
and equipment. It is well worth
visiting one of the last examples of
a 19th century European teaching
and research laboratory.
Hence, no better place to hold the
honorary plenary lecture of
Professor José Cavaleiro entitled “Porphyrins and Related Macrocycles: Synthetic Studies
and Potential Applications”, along with flash presentation of five young Portuguese
researchers which applied for the Portuguese Award for Best Young Organic Chemist 2013.
How to arrive to Laboratório Chimico (Rua da Escola Politécnica, 58):
By metro: The nearest metro station (5 min walk) is Rato (in yellow line).

Awards
During the 10º Portuguese National Meeting of Organic Chemistry and the 1st
Portuguese-Brazilian Organic Chemistry Symposium will be awarded three young scientists
with the Portuguese Young Organic Chemist Award; the Portuguese Award for Best PhD
and Master Thesis. Three prizes will be also awarded to the best poster presentations in the
following categories: chromatographic, mass and NMR techniques.

Timetable
4 September 5 September 6 September
9:00-9:40 Opening Ceremony
(9:30 – 9:50)
PL2 Darren Dixon (U. Oxford)
PL5 Karl Gademann (U. Basel)
9:40-10:00 SLB-IOC3
Amélia Pilar (FC-UL)
SLB-IOC6
Maria J. U. Ferreira (FF-UL)
10:00-10:15 PL1
C.-J. Li (McGill Uni.) 9:50-10:30
OC3
M. Manuel Marques (FCT-UNL)
IOC11
Ana Costa (U. Algarve)
10:15-10:30 IOC8
Rui Pinto (FF-UC) IOC12
Lázló E. Kiss (Bial)
10:30-11:00 Coffee break
And poster session 1
Coffee break
And poster session 2
Coffee break
And poster session 2
11:00-11:40 SLB-PL1
Carlos Correia (UNICAM)
PL3
Jeffrey W. Bode (ETH Zurich)
SLB-PL4
Rui Moreira (FF-UL)
11:40-12:00 SLB-IOC1
Teresa Melo (FCT-UC)
SLB-IOC4
Nuno Maulide (MPI)
SLB-IOC7
Paula Gomes (FC-UP)
12:00-12:15 IOC1
Paula Branco (FCT-UNL)
IOC9
M. Rita Ventura (ITQB-UNL)
OC5
José Soares (FF-UP)
12:15-12:30 IOC2
Mariette M. Pereira (FCT-UC) IOC10
Ricardo Mendonça (Hovione) IOC13
A. Antunes (IST-UTL)
12:30-14:00 Lunch Lunch Lunch
14:00-14:40 SLB-PL2
Antonio Luiz Braga (UFC)
PL4
Benjamin List (MPI)
PL6
Dean Toste (Uni. California)
14:40-15:00 SLB-IOC2
M. M. M. Raposo (U. Minho)
SLB-IOC5
Antonio C. B. Burtoloso (USP)
SLB-IOC8
Pierre M. Esteves (UFRJ)
15:00-15:15 IOC3
M. C. Barreto (U. Azores)
OC4
João Tomé (U. Aveiro)
IOC14
Anthony Burke (U. Évora)
15:20-15:30
FCA1
A. Fernandes
(FC-UP)
FCB5
C. Dias
(FC-UL)
FCA9
L. Klein-Junior
(FF-UFRGS)
FCB13
A. Phillips
(FCT-UNL)
PhD-Award-OC6
(15:15-15:30)
15:30-15:40
FCA2
N. Lourenço
(IST-UTL)
FCB6
E. Silva
(U. Aveiro)
FCA10
L. Lourenço
(U. Aveiro)
FCB14
C. Kanta
(MPI)
Awards
15:40-15:50
FCA3
T. Dias (U. Minho)
FCB7
H. Faustino (USC)
FCA11
P. Cal (FF-UL)
FCB15
N. Pereira (FCT-UC)
Closing Ceremony
15:50-16:00
FCA4
S. Lucas
(FF-UL)
FCB8
S. Niyomchon
(MPI)
FCA12
J. Nunes
(UCL-UK)
FCB16
N. Candeias
(TUT-FI)
16:00-16:30 Coffee break
And poster session 1 Coffee break
Organic Chemistry A world of opportunities
within the CPLP
community
16:30-16:45 IOC4
Diana Pinto (U. Aveiro)
16:45-17:00 IOC5
Paulo Coelho (UTAD)
17:00-17:15 IOC6
João P. Telo (IST-UTL) FC17-FC21 Portuguese Young Organic
Chemistry Award
SLB-PL3 José Cavaleiro
Uni. Aveiro
Venue at
Laboratorio Chimico
17:15-17:30 OC1
M. João Queiroz (U. Minho)
17:30-17:45 IOC7
Dália Barbosa (Atral-Cipan)
17:45-18:00 OC2
Ana C. Fernandes (IST-UTL)
18:00-18:30 SPQ - Organic Chemistry
Division Meeting
Reception Conference Dinner Museu da Cerveja

Scientific program
Wednesday, September 4, 2013
Session 1 | Chairman: Rui Moreira, Carlos Correia | Room A
9.30-9.50 Opening Ceremony
9.50-10.30 PL1 | Chao-Jun Li (McGill University, Canada) Exploration of New Chemical Reactivities for Synthetic Efficiency
10.30-11.00 Coffee break and poster session 1
Session 2 | Chairman: António Luiz Braga, Ana M. Lobo | Room A
11.00-11.40 SLB-PL1 | Carlos Roque Duarte Correia (University of Campinas, Brazil) Enantioselective Heck Reactions with Aryldiazonium Salts. Challenges and Synthetic Opportunities
11.40-12.00 SLB-IOC1 | Teresa M. V. D. Pinho e Melo (FCT, University of Coimbra, Portugal) Heterocycles via Pericyclic Reactions of Aza- and Diazafulvenium Methides
12.00-12.15 IOC1 | Paula S. Branco (FCT, New University of Lisbon, Portugal) A Recyclable Ferrite–Pd Magnetic Nanocatalyst for the Buchwald-Hartwig reaction
12.15-12.30 IOC2 | Mariette M. Pereira (University of Coimbra, Portugal) Developments in Enantioselective Immobilized BINOL-based Tandem Reactions
12.30-14.00 Lunch
Session 3 | Chairman: M. Fernanda Proença, Paulo Coelho | Room A
14.00-14.40 SLB-PL2 | Antonio Luiz Braga (Federal University of Santa Catarina, Brazil) Synthesis of [Se,N]-Small Molecules: Chiral Ligands and Potentially Bioactive Compounds
14.40-15.00 SLB-IOC2 | M. Manuela M. Raposo (University of Minho, Portugal) 2,4,5-Tri(hetero)arylimidazoles: Design, Synthesis and Characterization as Novel TPA Chromophores and Optical Chemosensors
15.00-15.15 IOC3 | Maria do Carmo Barreto (University of Azores, Portugal) Biological activities of oxygen and nitrogen heterocyclic compounds
Session 4 | Chairman: Teresa Melo, M. M. Raposo (Room A) | Mariette M. Pereira, M. João Queiroz (Room B)
15.20-15.30 Room A Room B FCA1 | A. Fernandes (FC, University of Porto, Portugal) Chemical structure and stability of microencapsulated anthocyanins
FCB5 | C. Dias (FC, University of Lisbon, Portugal) New antimicrobial structures with anti-ageing potential: an efficient synthesis towards 2-deoxy glycosides and their thio analogues
15.30-15.40 FCA2 | Nuno M. T. Lourenço (Instituto Superior Técnico, Portugal) Enzymatic Resolution of Secondary Alcohols in Miniemulsion Media
FCB6 | Eduarda M. P. Silva (University of Aveiro, Portugal) Synthesis of N-substituted 1,2-dihydropyridines by 6π-electrocyclisation of (E,E)-cinnamylidene acetophenones

15.40-15.50 FCA3 | Tatiana A. Dias (University of Minho, Portugal) The acid-catalysed reaction of 2-hydroxychalcones with carbon acids
FCB7 | Hélio Faustino (University of Santiago de Compostela, Spain) Gold (I) catalyzed intermolecular cycloadditions of allenamides: a simple route to small and medium sized carbocycles
15.50-16.00 FCA4 | Susana D. Lucas (FF, University of Lisbon) Hit-to-Lead Optimization of kojic Acid Derivatives toward COPD Drug Discovery
FCB8 | Supaporn Niyomchon (Max-Planck-Institut für Kohlenforschung, Germany) Regio- and Enantioselective Cyclobutene Allylations
16.00-16.30 Coffee break and poster session 1
Session 5 | Chairman: Vitor Freitas, Paula Branco | Room A
16.30-16.45 IOC4 | Diana C. G. A. Pinto (University of Aveiro, Portugal) New syntheses of potential biologically active xanthones and benzoxanthones
16.45-17.00 IOC5 | Paulo J. Coelho (University of Trás-os-Montes e Alto Douro, Portugal) Synthesis of 1-vinylidene-naphthofurans: A thermally reversible photochromic system that colours only when adsorbed on silica gel
17.00-17.15 IOC6 | João P. Telo (Instituto Superior Técnico, Portugal) Electronic Communication in Linear Oligo(azobenzene) Radical Anions
17.15-17.30 OC1 | Maria João R. P. Queiroz (University of Minho, Portugal) New strategies for the synthesis of 2-(hetero)arylthieno[2,3-b] or [3,2-b]pyridine scaffolds from 2,3-dihalopyridines
17.30-17.45 IOC7 | Dália Barbosa (AtralCipan, Portugal) Isolation and Identification of Impurities in Tetracycline Derivatives
17.45-18.00 OC2 | Ana C. Fernandes (Instituto Superior Técnico, Portugal) Synthesis, Characterization and Citotoxic Activity of Cyclopentadienyl Ruthenium(II) Complexes with Carbohydrate Derived Ligands
18.00-18.30 SPQ Organic Chemistry Division Meeting
Thursday, September 5, 2013
Session 6 | Chairman: José Cavaleiro, João Tomé | Room A
9.00-9.40 PL2 | Darren J. Dixon (University of Oxford, UK) Enantioselective Cooperative Catalysis and Complexity Building Reaction Cascades in Library and Natural Product Synthesis
9.40-10.00 SLB-IOC3 | Amélia P. Rauter (FC, University of Lisbon, Portugal) Sugar-based surfactants as selective antimicrobial agents: a multidisciplinary approach
10.00-10.15 OC3 | M. Manuel B. Marques (FCT, New University of Lisbon, Portugal) Pd-catalysed amination on a soluble polymer support: a sustainable version of homogeneous C-N cross-coupling reaction
10.15-10.30 IOC8 | Rui M. A. Pinto (FCT, University of Coimbra, Portugal) New Chemical Processes with Bismuth(III) Salts: Applications of Bismuth(III) Reagents and Catalysts to Steroid and Terpenoid Chemistry

10.30-11.00 Coffee break and poster session 2
Session 7 | Chairman: Antonio C. B. Burtoloso, Amélia Pilar | Room A
11.00-11.40 PL3 | Jeffrey W. Bode (ETH-Zürich, Switzerland) Chemical Protein Synthesis with the KAHA Ligation
11.40-12.00 SLB-IOC4 | Nuno Maulide (Max-Planck-Institut für Kohlenforschung, Germany) Sulfur reloaded: New S(IV)-mediated transformations
12.00-12.15 IOC9 | M. Rita Ventura (ITQB, New University of Lisbon, Portugal) Study and Modulation of Inter-species Quorum Sensing by AI-2 Analogues
12.15-12.30 IOC10 | Ricardo Mendonça (Hovione FarmaCiencia SA, Portugal) Development of a practical and efficient synthesis of an Active Pharmaceutical Ingredient
12.30-14.00 Lunch
Session 8 | Chairman: André L. Meleiro Porto, Nuno Maulide | Room A
14.00-14.40 PL4 | Benjamin List (Max-Planck-Institut für Kohlenforschung, Germany) Asymmetric Counteranion Directed Catalysis (ACDC): A General Approach to Enantioselective Synthesis
14.40-15.00 SLB-IOC5 | Antonio C. B. Burtoloso (University of São Paulo, Brazil) α,β-Unsaturated Diazoketones as Useful Platforms in the Synthesis of Pyrrolidine, Piperidine and Indolizidine Alkaloids
15.00-15.15 OC4 | João P. C. Tomé (University of Aveiro, Portugal) Photoactive molecules by design
Session 9 | Chairman: M. Rita Ventura, João P. Telo (Room A) | M. Manuel Marques, Ricardo Mendonça (Room B)
15.20-15.30 Room A Room B FCA9 | Luiz C. Klein-Júnior (FF, Federal University of Rio Grande do Sul, Brazil) Two new monoterpene indole alkaloids from Psychotria umbellata Vell.
FCB13 | Ana M. F. Phillips (FCT, New University of Lisbon, Portugal) Organocatalytic Asymmetric Synthesis of Cyclopropylphosphonates
15.30-15.40 FCA10 | Leandro M. O. Lourenço (University of Aveiro, Portugal) Synthesis, photophysical and photodynamic activities of amphiphilic phthalocyanine-cyclodextrin conjugates
FCB14 | Chandra Kanta De (Max-Planck-Institut für Kohlenforschung, Germany) Catalytic Asymmetric Benzidine Rearrangement
15.40-15.50 FCA11 | Pedro M. S. D. Cal (FF, University of Lisbon, Portugal) Iminoboronates: A New Strategy for Reversible Protein Modification
FCB15 | Nelson A. M. Pereira (University of Coimbra, Portugal) Novel Synthesis of dipyrromethanesvia hetero-Diels-Alder reactionof azo- and nitrosoalkenes with pyrrole
15.50-16.00 FCA12 | João P. M. Nunes (Department of Chemistry, University College London, UK) New 2-amino-4-functionalized Cyclopentenones from 2-furaldehyde via a One-pot Method
FCB16 | Nuno R. Candeias (Tampere University of Technology, Finland) Dirhodium(II) Complexes Derived from Natural Amino Acids as Catalysts in Aqueous Asymmetric Intramolecular C-H insertion of α-Diazo Acetamides

16.00-16.30 Coffee break
Session 10 | Chairman: Artur Silva | Laboratorio Chimico
17.00-19.00 Portuguese Young Organic Chemistry Award
FC17 | Luís C. Branco (FCT, New University of Lisbon, Portugal) Carbon Dioxide Approaches for Organic Synthetic Processes
FC18 | Mário J. F. Calvete (FCT, University of Coimbra, Portugal) Synthesis and Applications of Tetrapyrrolic Macrocyclic Systems
FC19 | Pedro M. P. Góis (FF, University of Lisbon, Portugal) Shaping new Biologically Active Compounds with a Boron Tether
FC20 | Maria M. M. Santos (FF, University of Lisbon, Portugal) Synthesis of novel spirooxindoles with potential application as anticancer agents and probes
FC21 | Alexandre F. Trindade (FF, University of Lisbon, Portugal) Fast and simple synthesis of folates based in copper-free click chemistry
and
SLB-PL3 | José A. S. Cavaleiro (University of Aveiro, Portugal) Porphyrins and Related Macrocycles: Synthetic Studies and Potential Applications
Friday, September 6, 2013
Session 11 | Chairman: António Rocha Gonsalves, Anthony Burke | Room A
9.00-9.40 PL5 | Karl Gademann (University of Basel, Switzerland) Addressing the Real Brain Drain: Nerve Regeneration by Synthetic Natural Product
9.40-10.00 SLB-IOC6 | Maria J. U. Ferreira (FF, University of Lisbon, Portugal) Dual-acting Antimalarial Triterpenoids from an African Medicinal Plant
10.00-10.15 IOC11 | Ana M. Rosa da Costa (University of Algarve, Portugal) Artificial and natural polymers: from synthesis and chemical modification to biomedical applications
10.15-10.30 IOC12 | László E. Kiss (BIAL, Portugal) Synthesis and pharmacological evaluation of novel COMT inhibitors
10.30-11.00 Coffee break and poster session 2
Session 12 | Chairman: Pierre Mothé Esteves, Ana Costa | Room A
11.00-11.40 SLB-PL4 | Rui Moreira (FF, University of Lisbon, Portugal) New Chemical Tools to Study the Biology of Malaria
11.40-12.00 SLB-IOC7 | Paula Gomes (FC, University of Porto, Portugal) Straightforward organic chemistry against an intricate infectious disease: new chloroquine and quinacrine analogues as dual-stage antimalarial leads
12.00-12.15 OC5 | José Soares (FF, University of Porto, Portugal) Multidimensional optimization of pyranoxanthones with potential antitumor activity

12.15-12.30 IOC13 | Alexandra M. M. Antunes (Instituto Superior Técnico, Portugal) Bioactivation of the anti-HIV drug abacavir to an electrophilic aldehyde: in vitro and in vivo approaches
12.30-14.00 Lunch
Session 13 | Chairman: M. J. U. Ferreira, Paula Gomes | Room A
14.00-14.40 PL6 | F. Dean Toste (University of California, USA) Enantioselective Catalysis With Cations and Anions
14.40-15.00 SLB-IOC8 | Pierre Mothé Esteves (Federal University of Rio de Janeiro, Brazil) A Brief Saga into the Electrophilic Aromatic Substitution Mechanisms
15.00-15.15 IOC14 | Anthony J. Burke (University of Évora, Portugal) Ten Years of Catalytic "Asymmetric" Activity at CQE-UE: The First Decade
15.15-15.30 PhD-Award-OC6
15.30-15.40 Awards
15.40-16.00 Closing Ceremony
16.00-17.30 Organic Chemistry A world of opportunities within the CPLP community

Plenary Lectures
PL1 | Exploration of New Chemical Reactivities for Synthetic Efficiency Chao-Jun Li
PL2 | Enantioselective Cooperative Catalysis and Complexity Building Reaction Cascades in Library and Natural Product Synthesis
Darren J. Dixon
PL3 | Chemical Protein Synthesis with the KAHA Ligation Jeffrey Bode
PL4 | Asymmetric Counteranion Directed Catalysis (ACDC): A General Approach to Enantioselective Synthesis
Benjamin List
PL5 | Addressing the Real Brain Drain: Nerve Regeneration by Synthetic Natural Products Karl Gademann
PL6 | Enantioselective Catalysis with Cations and Anions F. Dean Toste
SLB-PL1 | Enantioselective Heck Reactions with Aryldiazonium Salts. Challenges and Synthetic Opportunities Carlos Roque Duarte Correia
SLB-PL2 | Synthesis of [Se,N]-Small Molecules: Chiral Ligands and Potentially Bioactive Compounds
Antonio Luiz Braga
SLB-PL3 | Porphyrins and Related Macrocycles: Synthetic Studies and Potential Applications
José A. S. Cavaleiro
SLB-PL4 | New Chemical Tools to Study the Biology of Malaria Rui Moreira
Invited Oral Communications
SLB-IOC1 | Heterocycles via Pericyclic Reactions of Aza- and Diazafulvenium Methides Teresa M. V. D. Pinho e Melo
SLB-IOC2 | 2,4,5-Tri(hetero)arylimidazoles: Design, Synthesis and Characterization as Novel TPA Chromophores and Optical Chemosensors
M. Manuela M. Raposo
SLB-IOC3 | Sugar-based surfactants as selective antimicrobial agents: a multidisciplinary approach Amélia P. Rauter
SLB-IOC4 | Sulfur reloaded: New S(IV)-mediated transformations Nuno Maulide
SLB-IOC5 | α,β-Unsaturated Diazoketones as Useful Platforms in the Synthesis of Pyrrolidine, Piperidine and Indolizidine Alkaloids
Antonio C. B. Burtoloso
SLB-IOC6 | Dual-acting Antimalarial Triterpenoids from an African Medicinal Plant M. J. U. Ferreira

SLB-IOC7 | Straightforward organic chemistry against an intricate infectious disease: new chloroquine and quinacrine analogues as dual-stage antimalarial leads P. Gomes
SLB-IOC8 | A Brief Saga into the Electrophilic Aromatic Substitution Mechanisms Pierre Mothé Esteves
IOC1 | A Recyclable Ferrite–Pd Magnetic Nanocatalyst for the Buchwald-Hartwig reaction Paula S. Branco
IOC2 | Developments in Enantioselective Immobilized BINOL-based Tandem Reactions Mariette M. Pereira
IOC3 | Biological activities of oxygen and nitrogen heterocyclic compounds Maria do Carmo Barreto
IOC4 | New syntheses of potential biologically active xanthones and benzoxanthones Diana C. G. A. Pinto
IOC5 | Synthesis of 1-vinylidene-naphthofurans: A thermally reversible photochromic system that colours only when adsorbed on silica gel Paulo J. Coelho
IOC6 | Electronic Communication in Linear Oligo(azobenzene) Radical Anions João P. Telo
IOC7 | Isolation and Identification of Impurities in Tetracycline Derivatives Dália Barbosa
IOC8 | New Chemical Processes with Bismuth(III) Salts: Applications of Bismuth(III) Reagents and Catalysts to Steroid and Terpenoid Chemistry
Rui M. A. Pinto
IOC9 | Study and Modulation of Inter-species Quorum Sensing by AI-2 Analogues M. Rita Ventura
IOC10 | Development of a practical and efficient synthesis of an Active Pharmaceutical Ingredient
Ricardo Mendonça
IOC11 | Artificial and natural polymers: from synthesis and chemical modification to biomedical applications
Ana M. Rosa da Costa
IOC12 | Synthesis and pharmacological evaluation of novel COMT inhibitors László E. Kiss
IOC13 | Bioactivation of the anti-HIV drug abacavir to an electrophilic aldehyde: in vitro and in vivo approaches
Alexandra M. M. Antunes
IOC14 | Ten Years of Catalytic "Asymmetric" Activity at CQE-UE: The First Decade Anthony J. Burke
Oral Communications
OC1 | New strategies for the synthesis of 2-(hetero)arylthieno[2,3-b] or [3,2-b]pyridine scaffolds from 2,3-dihalopyridines
Maria João R. P. Queiroz

OC2 | Synthesis, Characterization and Citotoxic Activity of Cyclopentadienyl Ruthenium(II) Complexes with Carbohydrate Derived Ligands
Ana C. Fernandes
OC3 | Pd-catalysed amination on a soluble polymer support: a sustainable version of homogeneous C-N cross-coupling reaction M. Manuel B. Marques
OC4 | Photoactive molecules by design João P. C. Tomé
OC5 | Multidimensional optimization of pyranoxanthones with potential antitumor activity J. Soares
Flash Communications
FC1 | Chemical structure and stability of microencapsulated anthocyanins A. Fernandes
FC2 | Enzymatic Resolution of Secondary Alcohols in Miniemulsion Media N. M. T. Lourenço
FC3 | The acid-catalysed reaction of 2-hydroxychalcones with carbon acids Tatiana A. Dias
FC4 | Hit-to-Lead Optimization of kojic Acid Derivatives toward COPD Drug Discovery Susana D. Lucas
FC5 | New antimicrobial structures with anti-ageing potential: an efficient synthesis towards 2-deoxy glycosides and their thio analogues
C. Dias
FC6 | Synthesis of N-substituted 1,2-dihydropyridines by 6π-electrocyclisation of (E,E)-cinnamylidene acetophenones Eduarda M. P. Silva
FC7 | Gold (I) catalyzed intermolecular cycloadditions of allenamides: a simple route to small and medium sized carbocycles
Hélio Faustino
FC8 | Regio- and Enantioselective Cyclobutene Allylations Supaporn Niyomchon
FC9 | Two new monoterpene indole alkaloids from Psychotria umbellata Vell. Luiz C. Klein-Júnior
FC10 | Synthesis, photophysical and photodynamic activities of amphiphilic phthalocyanine-cyclodextrin conjugates
Leandro M. O. Lourenço
FC11 | Iminoboronates: A New Strategy for Reversible Protein Modification Pedro M. S. D. Cal
FC12 | New 2-amino-4-functionalized Cyclopentenones from 2-furaldehyde via a One-pot Method
J. P. M. Nunes
FC13 | Organocatalytic Asymmetric Synthesis of Cyclopropylphosphonates A. M. Faísca Phillips
FC14 | Catalytic Asymmetric Benzidine Rearrangement Chandra Kanta De

FC15 | Novel synthesis of dipyrromethanes via hetero-Diels-Alder reaction of azo- and nitrosoalkenes with pyrrole
Nelson A. M. Pereira
FC16 | Dirhodium(II) Complexes Derived from Natural Amino Acids as Catalysts in Aqueous Asymmetric Intramolecular C-H insertion of α-Diazo Acetamides
Nuno R. Candeias
FC17 | Carbon Dioxide Approaches for Organic Synthetic Processes Luís C. Branco
FC18 | Synthesis and Applications of Tetrapyrrolic Macrocyclic Systems Mário J. F. Calvete
FC19 | Shaping new Biologically Active Compounds with a Boron Tether Pedro M. P. Góis
FC20 | Synthesis of novel spirooxindoles with potential application as anticancer agents and probes Maria M. M. Santos
FC21 | Fast and simple synthesis of folates based in copper-free click chemistry Alexandre F. Trindade
Poster Communications
PC1 | Novel room-temperature choline carboxylate zwitterionic ionic liquids as potential electrolytes
A. Rocha
PC2 | New synthetic routes for 3-styrylflavones using Wittig and Heck reactions Djenisa H. A. Rocha
PC3 | Synthesis and Structural Characterization of Two Dioxo-thia-triaza Macrocyclic Compounds
M. F. Cabral
PC4 | Synthesis of 2-{2-[5(4)-aryl-2H-[1,2,3]-triazol-4(5)-yl]vinyl}chromen-4-ones Hélio Albuquerque
PC5 | Synthesis of the major building blocks towards a PGN fragment Marina J. Dias Pires
PC6 | β-Amino alcohol-catalyzed direct asymmetric aldol reactions in aqueous micelles Afroditi Pinaka
PC7 | Detection of nitroaromatic explosive compounds by fluorescent oxacyclophane-tethered calix[4]arenes A. I. Costa
PC8 | Conversion of non-expensive camphor and environmentally non-desired CO2 into fine chemicals
Alexandra P. S. Roseiro
PC9 | A Novel and Facile Synthetic Approach to N9-Substituted Guanines
Alice M. Dias
PC10 | Synthesis by MAOS of alkylated derivatives of a bioactive natural flavone A. Pereira

PC11 | Microwave Assisted vs Conventional Synthesis of Chalcones, Dihydrochalcones and Flavanones as New Potential SGLTs Inhibitors for the Treatment of Diabetes
Ana Rita Jesus
PC12 | 3-aminocoumarins: synthesis and reaction with active carbonyl compounds A. Rodrigues
PC13 | Syntheses and equilibria of sugar-based hydrazone bolaamphiphiles A. M. Sánchez
PC14 | Directly Linked Porphyrin-Phthalocyanine Conjugates: Synthesis and Pyridylfullerene Supramolecular Assemblies
Ana M. V. M. Pereira
PC15 | Microwave-assisted synthesis of α-aminoacylamides and α,α’-diacylimides by Ugi four-component reaction Ana Paula Paiva
PC16 | Synthesis of new dehydropeptides N-conjugated with an oxazole moiety A. Martins
PC17 | Synthesis of new sugar nucleoside precursors of potential application for Alzheimer’s disease
Andreia Almeida
PC18 | Synthesis of calix[4]pyrroles bearing sulfonamide groups Andreia S. F. Farinha
PC19 | Studies toward chemoenzymatic synthesis of nonracemic 2-phenylpropionic acid Anna Zadlo
PC20 | Synthesis of new benzaldehyde derivatives and their transformations into Corroles Bernardo A. Iglesias
PC21 | Synthesis of chiral spiro-β-lactams from 6-alkylidenepenicillanates Bruna S. Santos
PC22 | Synthesis of Tetra-Phosphonated Porphyrins as Organic Ligands for the Preparation of Metal-Organic Frameworks
Carla F. Pereira
PC23 | Intrinsically asymmetric 1,3-dibenzyl-oxacyclophane-tethered calix[4]arenes: synthesis and characterization
C. Teixeira
PC24 | Study of Oxazol-5-(4H)-ones fragmentation using Electrospray tandem mass spectrometry
Catarina A. B. Rodrigues
PC25 | Amino acid based hydrazones: synthesis and evaluation as new chemosensors for ion recognition Susana P. G. Costa
PC26 | Synthesis and Nonlinear Optical Properties of Heterocyclic Cationic Chromophores Containing Piridinium, Quinolinium and Benzothiazolium Acceptor Groups
M. Cidália R. Castro
PC27 | Anionic Surfactants Derived From Threonine and 4-Hydroxyproline Cidália Silva Pereira
PC28 | Synthesis of glyconjugates as precursors of targeted Mn(II)-based MRI contrast agents
C. Barroso

PC29 | Synthesis of new metalloporphyrins with potential catalytic action for development of conducting polymers
Cláudia M. B. Neves
PC30 | Synthesis and Characterization of Novel Thiazolo[5,4-d]thiazoles as Two Photon Absorbers (TPA)
R. Cristina M. Ferreira
PC31 | A theoretical study of stacking interactions between buckybowls and fullerene C60 D. Josa
PC32 | Schiff Base Tridentate Ligands Derived from Camphoric Acid for Enantioselective Alkylation of Aldehydes
Dina Murtinho
PC33 | Regiospecific Synthesis of N-Substituted 2-Oxopurine-6-carboxamidines Diogo Sampaio
PC34 | A cascade condensation-cyclization reaction leading to novel triazachrysene derivatives
Elina Marinho
PC35 | Schiff bases derived from salicylaldehydes and anilines. A quantitative analysis of substituent electronic effects
E. Matamoros
PC36 | Stereoselective Synthesis of N-Acylhydrazones from Diazo Compounds and Aldehydes via NHC catalysis
Fábio M. F. Santos
PC37 | Synthesis of new chromene derivatives and pharmacological evaluation for adenosine receptors
M. Fernanda Proença
PC38 | Thiopheno[3’,4’-a]chromeno[3,4-d]pyrroles from Thiazolidine-4-carboxilic Acid Fernanda M. Ribeiro Laia
PC39 | New Photochromic Intermediates for 3D-Data Storage Filipa Siopa
PC40 | Novel pentacationic N-Fused Pentaphyrin Flávio Figueira
PC41 | Study on the microwave–assisted Diels-Alder reactions of 5-styryl-1H-pyrazoles with different dienophiles
Inês C. S. Cardoso
PC42 | The Catalytic Asymmetric Acetalization Ji Hye Kim
PC43 | Hydroxylation of flavon-3-ol derivatives Joana L. C. Sousa
PC44 | XPS studies of tetrapyrrolic macrocycles Joana F. B. Barata
PC45 | A new anion receptor based on a nanomagnet-porphyrin hybrid João M. M. Rodrigues
PC46 | Synthesis of cyclitols and biological evaluation J. N. Lana
PC47 | Synthesis and antioxidant activity of ferrocenyl derivatives J. Albertino Figueiredo

PC48 | Bioconversion of ketones by mycelia of marine-derived fungi and organic composting
Lenilson C. Rocha
PC49 | Chemical synthesis of a bioavailable anthocyanin metabolite: cyanidin-4’-O-methylglucoside
L. Cruz
PC50 | Spectroscopic study of the rearrangement of 2,6-diisopropyl-1,4-quinone Luísa M. Ferreira
PC51 | Ionic liquids derived from sugars as chiral selectors Manuela Pereira
PC52 | Benzo[a]phenoxazinium chlorides possessing chlorinated terminals: synthesis, photophysics and photostability studies
Marcello M. T. Carvalho
PC53 | A new route to the synthesis of well-defined molecularly imprinted polymers (MIPs) by ATRP: application as adsorbents for solid phase extraction
M. Simões
PC54 | An Improved Approach to 1H,3H-Thiazolo[3,4-a]benzimidazoles Maria I. L. Soares
PC55 | Brønsted Acid Catalyzed Ring Opening of Aziridines: Taming and Directing the Nucleophilicity of Carboxylic Acids
Mattia R. Monaco
PC56 | Metal-Ligand Systems and Structural Diversity Based on Cyclic Peptides Michele Panciera
PC57 | The Synthesis of 2-Aryl-hypoxanthines: A Novel and Efficient Synthetic Approach Nádia Senhorães
PC58 | Synthesis of Prodelphinidins – work in progress Natércia Teixeira
PC59 | Synthesis and Characterized of New Open Chain 2,2’-Bipyridyl-Linked Pyridine-, Bipyridine-, or Phenanthroline-Derivatived Schiff Base Ligands
Nesrin Beynek
PC60 | Synthesis of Functional Organic Bipyridinium Salts Noémi Jordão
PC61 | New 2-(2,6-diarylpyridin-4-yl)porphyrin derivatives as fluorescent probes for metal cations
Nuno M. M. Moura
PC62 | Synthesis of Different Thioamide Derivatives using Lawesson’s Reagent O. Ortet
PC63 | Organobase catalyzed conjugate addition of 4-hydroxypyran-2-ones on chalcones: Synthesis of novel warfarin analogues and hemiketal tautomers - a diastereoselective access to warfarin-bicyclic ketal structures
Oualid Talhi
PC64 | New Hydrophilic Calix[4]arene-Carbazole Conjugates Patrícia D. Barata
PC65 | Synthesis and NMR conformational studies of new dihomooxacalix[4]arene tetraurea derivatives. Cone versus partial cone conformation
Paula M. Marcos

PC66 | Luminol turns green: New luminol analogues with increased aromaticity and green chemiluminescence
Periyasami Govindasami
PC67 | Asymmetric phthalocyanines bearing phenylacetylene units Raquel Nunes da Silva
PC68 | Synthesis of Novel Task-Specific Ionic Liquids Bearing an Anhydride Moiety R. Teixeira
PC69 | Synthesis and Photophysical Characterization of Novel Triphenylamine-benzimidazole Derivatives
Rosa M. F. Batista
PC70 | Reduction of 2,2,2-trifluoroacetophenoneby marine-derived fungus Mucorracemosus CBMAI 847
Sandra S. Ribeiro
PC71 | Novel Donor-acceptor Heterocyclic Systems Bearing Phthalazine, Thiophene and Furan groups for DSSC: Synthesis and Characterization
Sara S. M. Fernandes
PC72 | Preparation of aC-glycosylcinnamoyloxyacetophenone: valuable intermediate for the synthesis of a C-glycosyl-2-styrylchromone
Sara M. Tomé
PC73 | The Aza Wharton reaction and their applicability in the stereoselective synthesis of hydroxy-cyclopentenamines and cyclohexenamines
S. A. G. Silva
PC74 | Sugar-based precursors of new potential inhibitors of xanthine oxidase: synthesis and characterization
S. Cunha
PC75 | Supramolecular architectures based on transition metal bis-1,2-dithiolene complexes with N-coordinating groups
D. Simão
PC76 | Synthesis of an Indole-based Antimalarial Library Sofia A. Santos
PC77 | Synthesis of -Amino Acid Esters Carrying Bicyclo[3,3,0]octane and Bicyclo[4,3,0]nonane Skeletons
Sonata Krikštolaitytė
PC78 | Hetero-Diels-Alder of 3-Tetrazolyl-1,2-diaza-1,3-butadiene with Dipyrromethanes Susana M. M. Lopes
PC79 | Synthesis of New Ortho Substituted Anilide Atropisomers – A Second Look at the Hydrolysis of Quaternary N-Alkylbenzazol-3-ium Salts
S. S. Ramos
PC80 | New scalable synthetic protocol for the production of 5-(hydroxymethyl)furfural (HMF)
Svilen P. Simeonov
PC81 | Chromeno-imidazo[1,2-a]pyridines: synthesis and anticancer activity Marta Costa
PC82 | Solid phase microwave assisted synthesis of Peptaibolin mimetics bearing α,α-dialkylglycines
V. I. B. Castro

PC83 | Pyrido[2,3-b]indolizines: a one-pot synthesis in green media A. Brito
PC84 | Synthesis of tetraoxane-pyridonimine antimalarials J. Magalhães
PC85 | Biosynthesis of phenazine and phenoxazinone derivatives catalysed by CotA laccase
Ana Catarina Sousa
PC86 | Smart Magnetic Liquids as novel magnetic materials A. A. Rosatella
PC87 | Simple and more sustainable approaches for one pot enzymatic resolution of sec-alcohols
C. M. Monteiro
PC88 | Application of L-proline salts for asymmetric organocatalysis Karolina Zalewska
PC89 | Iodine(III)-Mediated Beta-Lactam Formation via C-H Insertion/C-C Bond Formation: A Diazo- And Metal-Free Approach
Luis F. R. Gomes
PC90 | Self-assembly and hydrogelation behavior of new dehydropeptides P. M. T. Ferreira
PC91 | Exploratory chemistry for the synthesis of antimicrobial agents starting from sugars Vasco Cachatra
PC92 | Synthesis of fused quinolone-benzazepines Vera L. M. Silva
PC93 | Synthesis of novel aryl-fused 1,4-oxazin-3-ones Małgorzata Śmist
PC94 | Development of catalytic hydrogenation of CO2 promoted by ionic liquids Ewa Bogel-Lukasik
PC95 | Synthesis of Enantiomerically Pure Chiral N-tert-Butylsulfinamide-alkene ligands via a novel Catalytic Method
Albertino Goth
PC96 | Rhodium Catalysed Tandem Hydroformylation/Arylation Reactions Ana R. Almeida
PC97 | Oxazolidinone Polycyclitol. Synthesis of Novel Aminocarbasugars with Oxazolidinone Ring
Latif Kelebekli
PC98 | Rhodium Catalyzed Enantioselective Arylation of Glyoxylate Derivatives using Organoboron Reagents
Carolina S. Marques
PC99 | BoImAr: Borylation(Catalytic)-Imination-Arylation(Catalytic) - A New Synthetic Approach to Promising Alzheimer and Parkinson Drugs
Daniela Peixoto
PC100 | Highly enantioselective synthesis of substituted Δ1-pyrrolines
D. I. S. P. Resende
PC101 | Thiazolidines for the Enantioselective Alkylation of Aromatic Aldehydes M. Elisa da Silva Serra

PC102 | Studies on the Lewis acid catalysed cycloaddition reactions of (E)-N-(2-acetylphenyl)-3-arylacrylamides with ortho-benzoquinodimethane
Gustavo da Silva
PC103 | BorArAm - Catalytic Asymmetric Arylating Cyclizations: A New Route to Chiral Bicyclic Amines
H. Viana
PC104 | Valorization of biomass derived intermediates based on metal organo catalysis Jaime A. S. Coelho
PC105 | Synthesis of 3-arylquinolin-4(1H)-ones by Suzuki cross-coupling reactions under PTC conditions using ohmic heating
Joana Pinto
PC106 | Application of Click Chemistry Reaction in Iminosugars Derivatives M. Domingues
PC107 | Efficient EDA Addition and Ring-Expansion Reaction of Isatins Catalyzed by a DBU/Rh(II) Metal-Organo System: On Route to the Synthesis of Viridicatin Alkaloids
R. Paterna
PC108 | Oxo-rhenium(V) Complexes containing heterocyclic ligands as highly efficient catalysts for the reduction of sulfoxides
Sara C. A. Sousa
PC109 | Microwave-assisted CuI catalysis to improve the synthesis of aminated thioxanthones
PC110 | Biomimetic oxidation of benzofuran derivatives with H2O2 using metalloporphyrin catalysts
S. M. G. Pires
PC111 | Aryl C-N bond formation by ligand catalyzed electrophilic amination Tahir Daşkapan
PC112 | Synthesis of new Biginelli Compounds E. Akbas
PC113 | Reactivity of 2-(Tetrazol-5-yl)-2H-Azirines: Synthesis of 4-(1H-Tetrazol-5-yl)-1H-Imidazoles
Ana L. Cardoso
PC114 | Synthesis of new di(hetero)aryl amide or triazole thienopyridine thioethers through Cu-catalyzed reactions as potential inhibitors of VEGFR2
Agathe Begouin
PC115 | Thiol-ene reactions: a “click” to α-Peptide Nucleic Acid Building Blocks A. S. Ressurreição
PC116 | Development of a New Anti-Cancer Photosensitizer. From Basic Research to the Clinical Trials
Artur R. Abreu
PC117 | New Benzimidazole-based COX-2 Inhibitors – a drug design approach Luísa C. R. Carvalho
PC118 | Galactodendritic photosensitizers for target carbohydrate receptors and trigger phototoxicity in bladder cancer cells
Patrícia M. R. Pereira
PC119 | Synthesis and Carbonic Anhyrase Inhibitory Properties of Bromophenol Derivatives Ertan Şahin

PC120 | Synthesis and anti-microbial activity studies of some novel epicinchonina-1,2,3-triazole derivatives
Joana Magalhães
PC121 | Synthesis of polysulfated diosmin conjugates as potential orally-active antithrombotic agents M. Patrão
PC122 | Synthesis of new 18F-labelled Porphyrins and their potential application for in vivo Molecular Imaging with PET
Ana Simões
PC123 | Design and Synthesis of New Probes for AI-2 Quorum-Sensing Receptor Studies Ana Sofia Miguel
PC124 | Porphyrin Derivatives and Potential Applications against Cutaneous Leishmaniasis Ana T. P. C. Gomes
PC125 | Triazene Prodrugs for the Treatment of Malignant Melanoma A. M. C. Granada
PC126 | Secondary Metabolites from a Marine Streptomycessp Ana M. Lobo
PC127 | Synthesis and Antimicrobial Activity of 5-Aminoimidazoles Incorporating a Substituted N-phenyl Amidrazonoyl Moiety
Ana Isabel F. Ribeiro
PC128 | Organic compounds isolated from Juniperus brevifolia bark Ana M. L. Seca
PC129 | Cycloaddition Reaction of Spiro[2.4]hepta-4,6-dien-1-ylmethanol and PTAD: A New Rearrangement
Abdullah Menzek
PC130 | Synthesis and photolysis studies of 5-aminolevulinic acid conjugates based on 2-oxo-naphtho[1,2-b]pyran
Ana M. S. Soares
PC131 | Biological Activities of Vernoniacondensata: Acetylcholinesterase Inhibition and Caco-2 Cells Toxicity A. Arantes
PC132 | Deoxyvitisins: a new set of pyrano-3-deoxyanthocyanidins André Sousa
PC133 | Development of chemical tools to study the mechanism of action of potent quinazolines targeting the liver stage of malaria infection
André Dias
PC134 | -Gauch Effect in Benzobicyclo3.2.1octenes Cavit Kazaz
PC135 | Isolation, quantification and seasonal variation of labdanolic acid from Cistus ladaniferus
André N. C. Martins
PC136 | Indole Alkaloids from Tabernaemontanaelegans: Isolation and Molecular Derivatization
A. Paterna
PC137 | Hit Optimization of a new class of p53-MDM2 interaction inhibitors Ângelo Monteiro

PC138 | New Phosphonic Acids and Esters Derived from Indazole: Synthesis and Biological Activity Evaluation António P. S. Teixeira
PC139 | NMR and IR conformational studies of the influence of bulky C-tetrasubstituted amino acids on Peptaibolinmimetics
C. M. Carvalho
PC140 | Synthesis and characterization of psoralen analogues based on dibenzothiophene C. Francisco
PC141 | Synthesis of novel antiplasmodial agents containing squaramide and 4-amino-7-chloroquinoline moieties
Carlos J. A. Ribeiro
PC142 | Design, synthesis and evaluation of tacrine-cinnamic acid derivatives as potencial bi-functional anti-Alzheimer drug candidates
Catarina Quintanova
PC143 | Remaking of Dietary Antioxidants: Targeting MitoBEN’s to Mitochondria as a New Therapeutic Strategy
Catarina Oliveira
PC144 | Anti-inflammatory Activity of Genista tenera n-Butanol Extract and Evaluation of its Stability under Gastrointestinal Conditions
Daniela Batista
PC145 | Synthesis of hybrids compounds with multistage antimalarial activity Daniela Miranda
PC146 | Synthesis of 2’,4’-dihydroxy-3,4,5-trimethoxy-3’-propylchalcone analogues with potential antitumor activity
D. Pereira
PC147 | De novo Design and Synthesis of New Potent Human Neutrophil Elastase Inhibitors
E. F. P. Ruivo
PC148 | Synthesis of Derivatives of 3-Hydroxypyrrolidine and (3,4)-Dihydroxypyrrolidine:
Inhibitors of Rat Intestinal Glucosidases Elisabete P. Carreiro
PC149 | Anthracene-Derived Bis-Aminophosphonates: Synthesis, NMR Characterization and Biological Activity
PC150 | Chemoselective biohydrogenation of α,β-unsaturated ketones by mycelia of marine-derived fungal strain Penicillium citrinum CBMAI 1186
André L. M. Porto
PC151 | Synthesis and biological data of new 7-carboranylmethyl-benzo[b]acridin-12(7H)-one derivatives as potential BNCT Agents
A. Filipa F. Silva
PC152 | Discovery of New Heterocycles with Activity against Human Neutrophile Elastase Based On A Boron Promoted One-Pot Assembly Reaction
Francesco Montalbano
PC153 | Push-Pull-Push Fluorophores for Cellular Imaging Samuel Guieu
PC154 | Carbohydrate-derived compounds as dual inhibitors for Acetylcholinesterase and Aβ aggregation M. Isabel Ismael

PC155 | α-Aminophosphonic Acid Diesters, Synthesis, NMR Characterization, In Vitro Antitumor Evaluation And Safety Testing I. Tsacheva
PC156 | New synthetic approach to prolinemimetics of PLG derived from 2-azabicyclo[2.2.1]heptane system
Ivo E.Sampaio-Dias
PC157 | Quantification of phenolic compounds of corkable to migrate to hydroalcoholic solution - Intestinal absorption and biological properties
J. Azevedo
PC158 | Antioxidant and anti-inflammatory activity of Franciscan Friars Aloe syrup Liliana M. L. Silva
PC159 | Benzothiazolium Salt as a New Ligand in Affinity Chromatography for Protein Purification
L. P. Alves
PC160 | Synthesis and Singlet Oxygen Evaluation of New Unsymmetrical Squarylium Cyanine Dyes L. V. Reis
PC161 | Novel epi-Cinchonine-Triazole Derivatives: Synthesis and Evaluation of Biological Activity
Anthony J. Burke
PC162 | Valorization of Diterpenes Isolated from Cistus ladaniferus L. M. T. Frija
PC163 | Anti-tumor activity of diterpenoids against multidrug resistant phenotypes M. Reis
PC164 | Isobacteriochlorin derivative of 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin on the photodynamic inactivation of bioluminescent E. coli
Mariana Mesquita
PC165 | Probing the Chemical Space Around Aurone Scaffold – Improving Antimalarial Activity
M. P. Carrasco
PC166 | Nitroimidazolyl hydrazones are better amoebicides than their cyclized 1,3,4-oxadiazoline analogues: In vitro studies and Lipophilic efficiency analysis
Mohmmad Younus Wani
PC167 | Synthesis of Purine Nucleosides from Glucurono-6,3-lactone and their Potential as Anti-Alzheimer Agents
Nuno M. Xavier
PC168 | Chiral Induced Cyclocondensation Reactions: A Versatile Approach to Obtain a New Class of NMDAR Antagonists
Nuno A. L. Pereira
PC169 | Synthesis of α-mannopyranoside of 7α-acetoxy-6β-hydroxyroyleanone Patrícia Rijo
PC170 | Resveratrol methoxyl dimer with antibacterial activity Patrícia Máximo
PC171 | Stereoselective Synthesis of N-, S-, and C-Glycoside 8-epi-Castanospermine analogues as Glycosidase Inhibitors
Rita Gonçalves-Pereira

PC172 | Tetraoxane – pyrimidine nitrile hybrids as dual-acting antimalarials R. Oliveira
PC173 | New Unsymmetrical Squarylium Cyanine Dyes as Potential Photosensitizers for Cancer Photodynamic Therapy
S. G. Fagundes
PC174 | A Small Library of Lathyrane Diterpenes Through Molecular Derivatization S. Neto
PC175 | Targeted Delivery of MitoCIN’S: a New Therapeutic Approach for Oxidative Stress Related Diseases
Sofia Benfeito
PC176 | Synthesis of Chalcones with Potential Activity in the p53-MDM2 Interaction S. Carvalho
PC177 | Ethylenediamine-derived affinity ligands immobilized on Sepharose to isolate BSA, lysozime and RNase A
V. C. Graça
PC178 | Synthesis of Naphtalene Derivatives as New Class of Putative G-Quadruplex Ligands
A. R. P. Duarte
PC179 | Designing and syntheses of new cyclopentenones as key precursors of antiviral nucleoside (N)-MCT and novel hydrophobic compounds with promising anti-proliferative activity
Krassimira P. Guerra
PC180 | Pyrazinoic esters – human plasma stability and mycobacterial activation in free and liposomal form
Marta Oliveira
PC181 | Indole: A “Privileged Structure” in Medicinal Chemistry Mónica S. Estevão
PC182 | Mass spectrometry as a tool to provide mechanistic insights into metabolic reactions: CID of quinoloimines derivatives
Paulo J. Amorim Madeira
PC183 | Towards more potent Jolkinol D derivatives: how can docking studies guide chemical derivatization?
R. J. Ferreira
PC184 | Synthesis and surface activity of alkyl 2-deoxyglycosides as original structures for utilization as antimicrobial agents
Patrícia Serra
PC185 | Peptide synthesis: different approaches for specific drug delivering of chemotherapeutical agents
João D. Pereira
PC186 | Synthesis and the Asymmetric Resolution of Dopamine and Rotigotine Analogues 2-Amino-6,7-dimethoxyindane and 2-Amino-7,8-dimethoxy-1,2,3,4-tetrahydronaphthalene
Süleyman Göksu
PC187 | Further studies on a promising strategy for the synthesis of iminosugars, polyhydroxylated prolines and polyhydroxylated pipecolic acids
PC188 | Enzymatic synthesis of fatty acid sugar esters. Optimization by response surface methodology
O. Selaïmia-Ferdjani

PC189 | Heteropolyacids accelerated multi-component synthesis of N-phenylquinazolin-4-amines by using Silica-Supported Preyssler Nanoparticles in Green Solvent
Ali Gharib
PC190 | Synthesis of calix[4]arene nanotubes Ali Osman Karatavuk
PC191 | On the use of ionic liquids as green solvents in reactions catalyzed with glycosidases
Salim Ferdjani
PC192 | Click reaction: synthesis and characterization of novel Triazol-Quinazoline A. Ouahrouch
PC193 | Synthesis, structure, spectroscopic and thermal properties of some macrocyclic complexes
Nagihan Ersoy
PC194 | Synthesis of New Heterocyclic-Linked Bis-Indole Systems I. Fazil Sengul
PC195 | Synthesis of Halo-Indenones: Gold-Catalysed Oxidative Diyne Cyclisations Laura Nunes dos Santos Comprido

Plenary lectures

PL1
Exploration of New Chemical Reactivities for Synthetic Efficiency
Chao-Jun Li
Department of Chemistry and FQRNT Center for Green Chemistry and Catalysis, McGill University, 801
Sherbrooke Street West, Montreal, QC H3A 2K6 Canada
The efficient making of new molecules is central to any new product in the pharmaceutical
and fine chemical industries. It is equally useful to modify existing biomolecules directly in
their natural states. However, state-of-the-art synthetic methods rely extensively on
protection-deprotection, halogenation-dehalogenation, oxidation-reduction, and
functionalization-defunctionalization. To address these challenges, our laboratory is
exploring novel chemical reactivities for synthesizing chemical products in three aspects: (1)
explore highly efficient chemistry to utilize the current chemical feedstock more efficiently: to
avoid protection-deprotection, halogenation-dehalogenation, and allow direct CH/CH
coupling; and (2) explore new chemistry that can utilize renewable resources readily. In
many cases, the use of water plays the key role for the success of such reactions. This talk
will focus our laboratory’s efforts on developing C-C bond formations, with a special
attention on the catalytic nucleophilic additions of alkynes in water and their applications in
the direct modification of biomolecules such as carbohydrates and peptides under
physiological ambient conditions in water (Figure 1).
Figure 1
Acknowledgements: We thank NSF, NSERC, CFI, CRC, FQRNT, CCVC, CREATE for support of our research over the years. References: 1. Li, C.-J.; Trost, B. M. Proc. Natl. Acad. Sci (USA), 2008, 105, 13197. 2. Li, C.-J. Acc. Chem. Res. 2002, 35, 533. 3. Li, C.-J. Chem. Rev. 2005, 105, 3095. 4. Li, C.-J. Acc. Chem. Res. 2010, 43, 581. 5. Li, C.-J. Acc. Chem. Res. 2009, 42, 335.

PL2
Enantioselective Cooperative Catalysis and Complexity Building
Reaction Cascades in Library and Natural Product Synthesis
Darren J. Dixon
Department of Chemistry, University of Oxford, Oxford, OX1 3TA, UK
Enantiomerically pure compounds with the capacity to activate simultaneously electrophilic
substrates and pro-nucleophilic reagents towards one another, offer numerous opportunities
for the discovery of powerful new catalytic asymmetric carbon-carbon and carbon-
heteroatom bond forming reactions. In this presentation, new families of bifunctional
catalysts and their use in highly enantioselective Michael addition reactions, Mannich
reactions, aldol reactions and alkylation reaction as well as other synthetically relevant
transformations, will be described.1-3
The application of these and other catalysts,
separately and in concert, to the discovery of new one-pot reaction cascade processes to
generate novel, multifunctional stereochemically defined scaffolds and architectures useful
for library and target synthesis will also be discussed.
Figure 1: Manzamine alkaloid natural products.
Further application of selected methodologies as pivotal carbon-carbon bond forming steps
in the total synthesis of a range of manzamine and daphniphyllum alkaloids4-6
will then be
discussed (Figure 1). These syntheses serve to illustrate how complex natural product
targets can be rapidly accessed when combinations of catalyst-controlled reactions, one-pot
multistep procedures and powerful route-shortening cascades are designed into the overall
synthetic sequence.
Acknowledgements: We thank the EPSRC for a generous Leadership Fellowship to D.J.D. References: 1. J. Ye, D. J. Dixon, P. S. Hynes, Chem. Comm. 2005, 4481. 2. A. L. Tillman, J. Ye, D. J. Dixon, Chem. Comm. 2006, 1191. 3. F. Sladojevich, A. Trabocchi, A. Guarna, D. J. Dixon, J. Am. Chem. Soc. 2011,133,1710. 4. P. Jakubec, D. Cockfield and D. J. Dixon J. Am. Chem. Soc. 2009, 131, 16632. 5. M. Yu, C. Wang, A. F. Kyle, P. Jakubec, D. J. Dixon, R. R. Schrock, A. H. Hoveyda, Nature 2011, 479, 88. 6. P. Jakubec, A. Hawkins, W. Felzmann and D. J. Dixon, J. Am. Chem. Soc. 2012, 134, 17482.

PL3
Chemical Protein Synthesis with the KAHA Ligation
Jeffrey Bode
Laboratorium für Organische Chemie, Department of Chemistry and Applied Biosciences, ETH-Zürich,
Zürich, Switzerland 8093
The chemical synthesis of proteins is critical to the understanding of biological function of
proteins, particularly those containing posttranslational modifications or involved in covalent
protein–protein conjugates. In order to expand the scope of chemical protein synthesis and
to improve the ease with which proteins can be chemically synthesis, our group has sought
to identify new ligation reactions that give native peptide bonds under mild, chemoselective
conditions. This work has led to the discovery of the α-ketoacid–hydroxylamine amide-
forming (KAHA) ligation, which operates in the presence of unprotected functional groups,
requires no reagents or catalysts, and proceeds under aqueous conditions.
This talk will describe the development of the KAHA ligation, the reaction mechanism,
methods to prepare peptide segments containing the key functional groups, and the
application of this reaction to the chemical synthesis of proteins, including challenging
targets such as membrane associated proteins. Ongoing efforts at multiple segment
ligations to prepare larger peptides will also be discussed. Finally, we will discuss possible
solutions to the still unsolved problem of ligating large, unprotected molecules at
submillimolar concentrations with equimolar stoichiometry.
References: 1. Pattabiraman, V. R.; Ogunkoya, A. O.; Bode, J. W. "Chemical Protein Synthesis by Chemoselective α-Ketoacid–Hydroxylamine (KAHA) Ligations with 5-Oxaproline" Angew. Chem. Int. Ed. 2012, 51, 5114-5118. 2. Pusterla, I; Bode, J. W. “The mechanism of the α-ketoacid–hydroxylamine (KAHA) amide forming ligation”, Angew. Chem. Int. Ed. 2012, 50, 513–516. 3. Bode, J. W.; Fox, R. M.; Baucom, K. D. “Chemoselective Amide Ligations by Decarboxylative
Condensations of N-Alkylhydroxylaminesand α-Ketoacids”, Angew. Chem. Int. Ed. 2006, 45, 1248–1252.
O
NH
DMSO/H2O
HN
OO
NH
Me
Me
OH
OCOOHCOOH
COOH COOH
COOH COOH
CONH2
NH
H2N NH2
CONH2 NH3+
O
OH
Peptide Segment 2
O
NH
Me
Me
O
NH
HN
OH
COOHCOOH
COOH COOH
COOH COOH
CONH2
NH
HN NH2
CONH2 NH3+
O
OH
KAHA ligation
H2N
NH
H2N NH2
NH
H2N NH2
NH3+
CONH2
COOH
COOHCOOH NH3+
OH
CONH2
HOOC
Peptide Segment 1
HOOC
H2N
NH
HN NH2
NH
H2N NH2
NH3+
CONH2
COOH
COOHCOOH NH3+
OH
CONH2
HOOC HOOC
Peptide Segment 1 Peptide Segment 2

PL4
Asymmetric Counteranion Directed Catalysis (ACDC):
A General Approach to Enantioselective Synthesis
Benjamin List
Max-Planck-Institut für Kohlenforschung, Mülheim an der Ruhr, Germany
Most chemical reactions proceed via charged intermediates or transition states. Such “polar
reactions” can be influenced by the counterion, especially if conducted in organic solvents,
where ion pairs are inefficiently separated by the solvent. Although asymmetric catalytic
transformations involving anionic intermediates with chiral, cationic catalysts have been
realized, analogous versions of inverted polarity with reasonable enantioselectivity, despite
attempts, only recently became a reality. In my lecture I will present the development of this
concept, which is termed asymmetric counteranion-directed catalysis (ACDC) and illustrate
its generality with examples from organocatalysis, transition metal catalysis, and Lewis acid
catalysis.
References: Mahlau M.; List B. Angew. Chem. Int. Ed. 2013, 52, 518-533.

PL5
Addressing the Real Brain Drain:
Nerve Regeneration by Synthetic Natural Products
Karl Gademann
Department of Chemistry and National Competence Center of Research (NCCR) 'Chemical Biology', St.
Johanns-Ring 19, CH-4056 Basel, Switzerland
The reconstitution of neuronal networks by neurotrophins has been demonstrated as a
viable strategy for addressing neuritic atrophy with regard to neurodegenerative diseases or
spinal cord lesions.1
However, limited efficacy was found for these proteins, due to poor
bioavailability and the difficulty of reaching the central nervous system ('the blood/brain
barrier'). Small molecule neurotrophins could overcome many of these problems and
present thus a promising chemical alternative.2 In addition, with regard to applications such
as autologous nerve grafting for the treatment of spinal cord lesions, immobilized small
molecules could offer unique advantages when compared to their protein counterparts.
In this communication, we will report (1) on the development of a versatile molecular
platform for the generation of biologically active surfaces3 and (2) on the total synthesis and
biological evaluation of a series of small molecule neurotrophin mimics.4 Both lines of
research have converged,5 and the design and biological evaluation of natural product
functionalized surfaces for nerve regeneration is presented. Potential applications with
regard to nerve grafting and guiding or the man/machine interface are discussed.
References: 1. Review. Aebischer, P.; Ridet, J. L. Trends Neurosci. 2001, 24, 533 2. Review: Qi, J.; Luo, Y.; Gao, L. Mini-Rev Med Chem 2011, 11, 658. 3.Gomes, J.; Grunau, A.; Lawrence, A. K.; Eberl, L.; Gademann, K. Chem Commun 2013, 49, 155–157; Malisova, B.; Tosatti, S.; Textor, M.; Gademann, K.; Zuercher, S. Langmuir 2010, 26, 4018; Saxer, S.; Portmann, C.; Tosatti, S.; Gademann, K.; Zuercher, S.; Textor, M. Macromolecules 2010, 43, 1050; Wach, J.-Y.; Bonazzi, S.; Gademann, K. Angew Chem Int Edit 2008, 47, 7123; Zürcher, S.; Wäckerlin, D.; Bethuel, Y.; Malisova, B.; Textor, M.; Tosatti, S.; Gademann, K. J Am Chem Soc 2006, 128, 1064. 4. Liffert, R.; Hoecker, J.; Jana, C. K.; Woods, T. M.; Burch, P.; Jessen, H. J.; Neuburger, M.; Gademann, K. Chem. Sci. 2013, 4, 2851–2857; Burch, P.; Binaghi, M.; Scherer, M.; Wentzel, C.; Bossert, D.; Eberhardt, L.; Neuburger, M.; Scheiffele, P.; Gademann, K. Chem. Eur. J. 2013, 19, 2589; Elamparuthi, E.; Fellay, C.; Neuburger, M.; Gademann, K. Angew Chem Int Edit 2012, 51, 4071; Jessen, H. J.; Schumacher, A.; Shaw, T.; Pfaltz, A.; Gademann, K. Angew Chem Int Edit 2011, 50, 4222; Jana, C. K.; Hoecker, J.; Woods, T. M.; Jessen, H. J.; Neuburger, M.; Gademann, K. Angew Chem Int Edit 2011, 50,
8407. 5. Hoecker, J.; Liffert, R.; Burch, P.; Wehlauch, R.; Gademann, K. Org. Biomol. Chem. 2013, 11, 3314.

PL6
Enantioselective Catalysis with Cations and Anions
F. Dean Toste
Department of Chemistry, University of California, Berkeley CA, USA 94720-1480
The past decade has witnessed remarkable development in the use of cationic gold(I)
complexes as homogenous catalysts for the transformation of carbon-carbon π-bonds.1
Several years ago, we demonstrated that the reactivity of these complexes could be
controlled by modification of the counter anion to these cationic transition metal complexes.2
This discovery provided a general platform for inducing enantioselectivity in reaction not
only using cationic transition metal complexes, but also with reactive cationic reagents and
intermediates. For example, we have applied this concept towards the development of
enantioselective electrophilic fluorination under chiral anion phase transfer conditions
(Figure 1).3 The use of these ionic interactions to control selectivity of cationic species has
generally relied on small molecular anions.4 As an extension of this concept, we have been
exploring the use of supramolecular assemblies as the anionic component in reactions
catalyzed by cationic transition metal complexes. For example, cationic phosphinegold(I)
complexes encapsulated by an anionic Ga4L6 tetrahedral demonstrated higher turnover
numbers, rate acceleration, produced different products and are well-tolerated by the
enzymes compared to the unencapsulated catalysts.5
Figure 1: Enantioselective Fluorination of Phenols by Chiral Anion Phase Transfer Catalysis.
Acknowledgements: With thank the National Institutes of Health – Institute of General Medical Science (NIHGMS) and the Department of Energy (DOE) at Lawrence Berkeley National Laboratory for financial support. References: 1. D. J. Gorin, F. D. Toste, Nature 2007, 446, 395-403. 2. G. A. Hamilton, E. J. Kang, M. M. Blázquez, F. D. Toste, Science 2007, 317, 496-499. 4. V. Rauniyar, A. D. Lackner, G. L. Hamilton, F. D. Toste, Science 2011, 334, 1681-1684. 3. R. J. Phipps, G. L. Hamilton, F. D. Toste, Nature Chem. 2012, 4, 603-614. 5. Z. J. Wang, K. N. Clary, R. G. Bergman, K. N. Raymond, F. D. Toste, Nature Chem. 2013, 5, 100-103.

SLB-PL1
Enantioselective Heck Reactions with Aryldiazonium Salts.
Challenges and Synthetic Opportunities
Caio Costa Oliveira, Ricardo Almir Angnes, Cristiane Storck Schwalm, Carlos Roque Duarte
Correia
Chemistry Institute – State University of Campinas, São Paulo – Brazil
Enantioselective catalysis has revolutionized the field of organic synthesis and has brought
significant scientific and economic benefits for our society. The enantioselective arylation of
olefins in particular (Heck reaction) has been a subject of intense academic and industrial
interest due to its potential for providing enantiomeric enriched medicines, fragrances and
new materials, which are in general more selective and less toxic than the racemic
counterpart. In this context, the Pd-catalyzed coupling of arenediazonium salts to olefins
(Heck-Matsuda reaction) stands as a more practical and reliable method to access
structurally complex organic molecules than the conventional Heck protocols. The Heck-
Matsuda arylations can be easily performed in the lab under aerobic conditions without
requiring expensive and/or toxic phosphine ligands. The first examples of these reactions
were described by Tsutomu Matsuda in 1977. However, in spite of the many advantages
and the long-term existence of this reaction, its enantioselective version has, until recently,
constituted a considerable challenge due to the intrinsic incompatibility between the
ordinary phosphine ligands and the arenediazonium salts. In this lecture, the first examples
of effective enantioselective Heck-Matsuda reactions will be presented using chiral
bisoxazoline ligands.1 Some recent developments from our lab will also be highlighted.
O
N
O
NBn Bn
15-100 minutesup to 95% yield
up to > 95:5 er
R
OH
+Pd(TFA)2, ZnCO3
MeOH
O
O
N2BF4
R
(E) or (Z)
R
R
O
H
R
up to 92.5:7.5 er
Scheme: Intermolecular Enantioselective Heck-Matsuda Arylations. Synthesis of β–Aryl Lactones and β–
Aryl Aldehydes. Acknowledgements: We thank the Brazilian funding agencies FAPESP, CNPq and CAPES for financial support. References: 1. a) Correia, C. R. D.; Oliveira, C. C.; Salles Jr., A. G.; and Santos, E. A. F. “The first examples of the enantioselective Heck–Matsuda reaction: arylation of unactivated cyclic olefins using chiral bisoxazolines” Tetrahedron Lett. 2012, 53, 3325-3328. b) Oliveira,
C. C.; Angnes, R. A. and Correia, C.
R. D. “Intermolecular Enantioselective Heck-Matsuda Arylations of Acyclic Olefins. Application to the
Synthesis of -Aryl--Lactones and -Aryl Aldehydes” J. Org. Chem. 2013, 78, 4373-4385.

SLB-PL2
Synthesis of [Se,N]-Small Molecules: Chiral Ligands and Potentially
Bioactive Compounds
Antonio Luiz Braga
Departamento de Química – CFM, Universidade Federal de Santa Catarina, 88040-900, Florianópolis –
SC, Brasil
Chiral [Se,N]-Small Molecules have found growing application as ligands or catalysts in
asymmetric catalysis over the past few years.1,2
The large majority of these catalysts or
ligands is derived from either readily available chiral amino alcohols or other natural sources
in a few high-yielding synthetic steps.2,3
Additionally, the relevance of the biological and medicinal properties of organoselenium
compounds is growing in a rapid pace, mainly due to their antioxidant, antitumor,
antimicrobial, and antiviral properties.4 In this context, we have developed short and efficient
routes to access these type of molecules from amino acid or other natural sources, aiming
to evaluate their bioactivities and/or catalytic properties. In our talk we will show our
contribution in these subjects, such as the preparation of ephedrine-based diselenide
(Figure 1): A promiscuous catalyst suitable to mimic the seleno-enzyme glutathione
peroxidase (GPx) and to promote enantioselective C-C coupling reactions.
Figure 1: Ephedrine-based diselenide.
Acknowledgements: We are grateful to CNPq, INCT-Catálise, CAPES and FAPESC-Pronex for financial support. References: 1. Wirth, T. Angew. Chem. Int. Ed. 2000, 39, 3741. 2. a) Braga, A. L.; Lüdtke, D. S.; Vargas, F.; Braga, R. C. Synlett 2006, 1453. b) Braga, A. L.; Lüdtke, D. S.; Vargas, F. Curr. Org. Chem. 2006, 10, 1921. 3. Godoi, M.; Paixão, M. W.; Braga, A. L. Dalton Trans. 2011, 40, 11347;
4. a) Nascimento, V.; Alberto, E. E.; Tondo, W. D.; Dambrowski, D.; Detty, M. R.; Nome, F.; Braga, A. L. J. Am. Chem. Soc. 2012, 134, 138. b) Nogueira, C. W.; Zeni, G.; Rocha, J. B. T. Chem. Rev. 2004, 104, 6255.

SLB-PL3
Porphyrins and Related Macrocycles: Synthetic Studies and
Potential Applications
José A. S. Cavaleiro
Department of Chemistry and QOPNA Research Unit, University of Aveiro, 3810-193 Aveiro, Portugal
Porphyrin derivatives play in Nature vital functions (e. g., respiration, photosynthesis, drug
detoxification). Synthetic studies leading to the structural elucidation of such compounds
have been carried out during the last century. For example, the syntheses of protoporphyrin
IX (which forms the iron complex ruling respiration and detoxification processes) and of the
photosynthetic pigment chlorophyll a have been reported, respectively, by Fisher in 1929
and Woodward in 1960.1a,b
Subsequently, studies related with biosynthesis, mode of action
and catabolism of such compounds and also with processes mimicking Naturehave been
performed. As a result, “new chemical avenues” for such type of compounds were brought
up, in relation with the needs to have better synthetic procedures and knowledge about their
potential applications in several fields, mainly in Medicine.
In recent decades the Aveiro group has been involved in developing synthetic
methodologies leading to new porphyrinmacrocycles and related derivatives (chlorins,
bacteriochlorins, corroles). It has been shown that porphyrinmacrocycles can react under
cycloaddition conditions as dienes, dienophiles and dipolarophiles. A wide range of new
derivatives can be obtained in such way. Other derivatives can also be obtained by direct
functionalization of the macrocycle or by substituent transformations. Potential applications
for the new synthesized products have been considered. Studies have been carried out by
looking at the assessment to generate reactive oxygen species and at the involvement of
such species in photodynamic therapy (PDT) of cancer cells and in the photoinactivation of
microorganisms; the action of metalloporphyrins as oxidative catalysts in the oxidation of
organic substrates at CH(sp3) and C(sp
2) centers, using hydrogen peroxide as the oxygen
donor, has also been evaluated.2a-d
This lecture will consider the main features of such work
performed at the University of Aveiro.
Acknowledgements: Thanks are due to all students and colleagues involved in the work. Thanks are also due to the University of Aveiro and to all portuguese funding institutions (INIC, JNICT, FCT) for funding and PhD/Postdoc awarded grants. Nowadays thanks are due to Fundação para a Ciência e a Tecnologia (FCT), European Union, QREN, FEDER and COMPETE for funding the QOPNA research unit (project PEst-C/QUI/UI0062/2011) and the National NMR Network. References: 1. a) Fisher H.; Zeile K. Liebigs Ann. Chem., 1929, 468, 98. b) Woodward R. B. J. Am. Chem. Soc., 1960, 82, 3800. 2. a) Silva A. M. G.; Cavaleiro J. A. S.; Porphyrins in Diels-Alder and 1,3-dipolar cycloaddition reactions, Progress in Heterocyclic Chemistry, 2008, 19, 44, Gribble G. W.; Joule J. A.,(Eds.), Elsevier, Amsterdam. b) Cavaleiro J. A. S.; Tomé J. P. C.; Faustino M. A. F.; Synthesis of Glycoporphyrins, Top. Heterocycl.Chem., 2007, 7, 179, E. S. H. El-Ashry, (Ed.), Heterocycles from Carbohydrate Precursors, Springer. c) Cavaleiro J. A. S.; Faustino M. A. F.; Tomé J. P. C.; Porphyrinyl-type sugar derivatives: synthesis and biological evaluations,Specialist Periodical Reports, CarbohydateChemistry--Chemical and Biological Approaches, 2009, 35, 199; Rauter A. P.; Lindhorst T. K., (Eds.), Royal Society of Chemistry, London. d) Cavaleiro J. A. S.; Tomé A. C.; Neves M. G. P. M. S.; meso-Tetraarylporphyrin Derivatives: New Synthetic Methodologies, Handbook of Porphyrin Science (With Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine), 2010, 2, 193; Kadish K. M, Smith K. M., Guilard R., (Eds.), World Scientific Publishing Co., Singapore.

SLB-PL4
New Chemical Tools to Study the Biology of Malaria
Rui Moreira
iMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal
Antimalarial drugs currently in use engage a reduced number of validated targets, and their
efficacy is being undermined by the spread of parasite resistance. In addition, chemical
diversity among these drugs is limited, which also contributes to the emergence of cross-
resistance. Antimalarial drug discovery has traditionally focused on the optimization of
known lead compounds to achieve efficacious drug exposures with the lowest possible
dose. Recently, ligand- and structure-based design approaches complemented by cell-
based screening have been developed to identify innovative and readily synthesizable hit
and lead compounds. Here, we review how chimeric compounds (e.g. 1-3) have been
designed and synthesized to engage different molecular targets in malaria parasites,
enabling efficient elimination of parasites both in vitro and in vivo.1-4
In addition, we will
report how structure-based design and target agnostic cell-based screening led to the
discovery of novel small molecules that will help to overcome our limited understanding of
Plasmodium biology.5-7
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support (PEst-OE/SAU/UI4013/2011 and REDE/1501/REM/2005) References: 1. Capela R., Oliveira R., Moreira R., Gonçalves L., Domingos A., Gut J., Rosenthal P. J., Lopes F. Bioorg. Med. Chem. Lett., 2009, 19, 3229. 2. Verissimo E., Gibbons P., Araujo N.,
Cristiano M. L. S.,
Rosenthal P. J., Gut J., Moreira R., Guedes R. C., O’Neill P. M., J. Med. Chem., 2010, 53, 8202. 3. Capela R., Cabal G. G., Rosenthal P. J., Gut J., Mota M. M., Moreira R., Lopes F., Prudêncio M. Antimicrob. Agents Chemother., 2011, 55, 4698. 4. Oliveira R., Newton A. S., Guedes R. C., Miranda D., Amewu R. K., Srivastava A., Gut J., Rosenthal P. J., O’Neill P. M., Ward S. A., Lopes F., Moreira R., ChemMedChem, 2013, in press. 5. Lavrado J., Cabal G., Prudêncio M., Mota M. M., Gut J., Rosenthal P. J., Diaz C., Guedes R., Santos D. dos, Bichenkova E., Douglas K. T., Moreira R., Paulo A. J. Med. Chem., 2011, 54, 734. 6. Rodrigues T., Moreira R., Gut J., Rosenthal P. J., O’Neill P. M., Biagini G. A., Lopes F., dos Santos D. J. V. A., Guedes R. C. Bioorg. Med. Chem., 2011, 19, 6302-6308. 7. Rodrigues T., da Cruz F. P., Prudêncio M., Lafuente-Monasterio M., Gonçalves D., Sitoe A. R., Bronze M. R., Gut J., Rosenthal P. J., Javier Gamo F.-, Mota M. M., Lopes F., Moreira R., J. Med. Chem., 2013, in press.


Oral Communications

SLB-IOC1
Heterocycles via Pericyclic Reactions of Aza- and Diazafulvenium
Methides
Teresa M. V. D. Pinho e Melo
Department of Chemistry, University of Coimbra, 3004-535 Coimbra, Portugal
Aza- and diazafulvenium methide systems 1-3 are versatile building blocks for the synthesis
of pyrroles and pyrazoles.1 These extended dipoles participate in sigmatropic [1,8]H shifts
and 1,7-electrocyclizations giving vinyl pyrroles and pyrazoles. Under flash vacuum
pyrolysis conditions these heterocycles undergo interesting rearrangements. Aza- and
diazafulvenium methides can be intercepted by dipolarophiles. The 4,5-dimethoxycarbonyl
derivatives 1 and 2 act exclusively as 1,7-dipoles affording products resulting from the
addition across the 1,7-positions. These 1,7-cycloadducts include chlorin and
bacteriochlorin type macrocycles (e.g. 5) as well as steroidal analogues (e.g. 6), compounds
with relevance in medicinal chemistry. In contrast with this chemical behavior, 5-
trifluoromethylazafulvenium methides 3 can participate in both 1,7- and 1,3-dipolar
cycloadditions. Recently, the generation and reactivity of benzodiazafulveniummethides4
has also been described (Scheme 1). In this lecture, details of our contribution to the
chemistry of these “higher-order” azomethine ylides and azomethine imines will be
discussed.
Scheme 1: Generation and reactivity of aza- and diazafulvenium methides.
Acknowledgements: Thanks are due to FCT (PEst-C/QUI/UI0313/2011), FEDER, COMPETE and QREN for financial support. References:
1. a) Pinho e Melo, T. M. V. D.; Soares, M. I. L.; Gonsalves, A. M. d'A. R.; Paixão, J. A., Beja, A. M.; Silva, M. R. J. Org. Chem., 2005, 70, 6629-6638. b) Pinho e Melo, T. M. V. D.; Nunes, C. M.; Soares, M. I. L.; Paixão, J. A., Beja, A. M.; Silva, M. R. J. Org. Chem., 2007, 72, 4406-4415. c) Pereira, N. A. M.; Fonseca, S. M.; Serra, A. C.; Pinho e Melo, T. M. V. D.; Burrows, H. D. Eur. J. Org. Chem. 2011, 3970-3979. d) Peláez, W. J.; Pinho e Melo, T.M. V. D. Tetrahedron 2013, 69, 3646-3655. e) Soares, M. I. L.; Nunes, C. M.; Gomes, C. S. B.; Pinho e Melo, T. M. V. D. J. Org. Chem. 2013,78, 628-637.
N X
1
2
3
4
675- SO2
1 X = CR Azafulvenium Methides2 X = N Diazafulvenium Methides
Sigmatropic[1,8]H
N XO2S
N
NH N
HN
Ar
Ar
Ar
Ar
NN
MeO2C CO2Me
N NO2S
N NS N N
O
O
2. MCPBA
1.
Vinylpyrroles orVinylpyrazoles
[8p+2p]Cycloaddition
N X
a
ba b
N
3 5-Trifluoromethyl-azafulvenium methides
F3C
4 Benzodiazafulvenium Methides
5
1-Me or 7-Mederivatives
CO2Me
CO2Me
CO2Me
CO2Me
AcO
H
HH
N N
MeO2CCO2Me
MeO
6

SLB-IOC2
2,4,5-Tri(hetero)arylimidazoles: Design, Synthesis and
Characterization as Novel TPA Chromophores and Optical
Chemosensors
M. Manuela M. Raposo
Centre of Chemistry, University of Minho, Campus de Gualtar, 4710-057, Braga, Portugal
2,4,5-Tri(hetero)aryl-imidazoles are a versatile class of compounds with a wide range of applications in diverse areas such as medicinal or materials chemistry due to their biological activity, as well their optoelectronic properties.
Our earlier studies showed that the optical
and thermal properties of these derivatives could be tuned by substitution of aryl groups at positions 2, 4 and 5 by 5-membered heterocycles such as thiophene and furan. This raises
the potential for several innovative applications of these -conjugated systems in nonlinear optics (e.g. second harmonic generators (SHG)), chemosensors, OLEDs and DNA intercalators.
1
Recent results from our research group concerning the design, synthesis and characterization of novel 2,4,5-tri(hetero)aryl-imidazoles 1 (Figure 1), as two-photon absorption (TPA) chromophores and/or as optical chemosensors will be presented and discussed.
N
NH
R2
R2
R1 = H, MeO, N,N-dialkylamino, CN, NO2
R2 =
BridgeR1
S
BridgeX
S
X = N-alkyl, O, SX
= ,
, ,1
Figure 1: Structure of novel 2,4,5-tri(hetero)aryl-imidazoles.
Acknowledgements: Thank are due to Fundação para a Ciência e Tecnologia (Portugal) and FEDER-COMPETE for financial support through Centro de Química (PEst-C/QUI/UI0686/2011 (F-COMP-01-0124-FEDER-022716)) and Centro de Física - Universidade do Minho (Project PTDC/CTM/105597/2008 (FCOMP-01-0124-FEDER-009457)), a PhD grant to R.C.M. Ferreira (SFRH/BD/86408/2012) and a a post-doctoral grant to R.M.F. Batista (SFRH/BPD/79333/2011). The NMR spectrometer Bruker Avance III 400 is part of the National NMR Network and was purchased with funds from FCT and FEDER. References: 1. a) Batista R. M. F.; Oliveira E.; Costa S. P. G.; Lodeiro C.; Raposo M. M. M. Org. Lett. 2007, 9, 3210. b) Costa S. P. G.; Belsley M.; Lodeiro C.; Raposo M. M. M. Tetrahedron 2008, 64, 9230. c) Batista R. M. F.; Costa S. P. G.; Belsley M.; Raposo M. M. M. Dyes Pigments 2009, 80, 329. d) Pina J.; Seixas de Melo J.; Batista R. M. F.; Costa S. P. G.; Raposo M. M. M. J. Phys. Chem B 2010, 114, 4964. e) Oliveira E.; Batista R. M. F.; Costa S. P. G.; Raposo M. M. M.; Lodeiro C. Inorg. Chem. 2010, 49, 10847. f)
Pedras B.; Batista R. M. F.; Tormo L.; Costa S. P. G.; Raposo M. M. M.; Orellana G.; Capelo J. L.; Lodeiro C. Inorg. Chim. Acta 2012, 381, 95. g) Batista R. M. F.; Costa S. P. G.; Raposo M. M. M. J. Photochem. Photobiol. Chem. 2013, 259, 33.

SLB-IOC3
Sugar-based surfactants as selective antimicrobial agents: a
multidisciplinary approach
Alice Martins,a Vasco Cachatra,
a C. Dias,
a J. Pais,
a Patrícia Serra,
a M. S. Santos,
b
Amélia P. Rautera
aCarbohydrate Chemistry Group, Center of Chemistry and Biochemistry, Faculdade de Ciências da
Universidade de Lisboa, Campo Grande, 1749-016 Lisboa, Portugal; bStructure and Reactivity Group, Center of Chemistry and Biochemistry, Faculdade de Ciências da
Universidade de Lisboa, Campo Grande, 1749-016 Lisboa, Portugal
Antibiotics resistance is a global threat that encourages research on new antimicrobial
molecular entities with new mechanisms of action. In this work we present the synthesis of
new antimicrobial glycosides which structure differs in both glycon and aglycon
components. Our previous studies on alkyl deoxy hexopyranosides of the D and L series in
their α- and β-anomeric configuration have shown that high surface activity is a pre-requisite
for their antimicrobial properties, and their selectivity appeared to be linked to the anomeric
configuration of the sugar and to its deoxygenation pattern.1 Hence, chemical approaches to
glycon deoxygenation and structurally diverse aglycons will be presented, based on a
simple but efficient methodology comprising the reaction of glycals with alcohols or their
heteroanalogues, catalysed by triphenylphosphane hydrobromide. D- and L-glycosides with
aglycons exhibiting alkyl chains of different size, their fluorinated or branched chain
analogues, and those chains with an internal or terminal amide functionality as well as
thioglycosides were synthesized. The surface activity of the aqueous solutions of several
glycosides was evaluated in terms of adsorption and aggregation parameters. Compounds’
bioactivity towards Bacillus anthracis and their acute cytotoxicity will be disclosed, revealing
promising structures in view of efficacy and also of low toxicity, when compared to that of
chloramphenicol. An overview of the key structural features regarding glycon and aglycon
chemical composition and glycon configuration for this new family of antibiotics will be
presented, highlighting the correlation of their aggregation and adsorption physical data with
the antibacterial activity.
Acknowledgements: We thank QREN for financial support of the Project QREN-SI I&DT co-promotion FACIB – Project nr. 21547, and Fundação para a Ciência e a Tecnologia for financial support of PEST-OE/QUI/UI0612/2013 and of Alice Martins post-Doc grant SFRH/BPD/42567/2007. References:
1. Martins A.; Santos M. S.; Dias C; Serra P.; Cachatra V.; Pais J.; Caio, J.; Teixeira, V. H.; Machuqueiro, M.; Silva, M. S.; Pelerito A.; Justino J.; Goulart M.; Silva, F. V.; Rauter A. P. Eur. J. Org. Chem. 2013, 8, 1448 and references cited therein.

SLB-IOC4
Sulfur reloaded: New S(IV)-mediated transformations
Nuno Maulide
Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany
Sulfur ylides occupy a prominent place among so-called “textbook reagents” in organic
chemistry.1 Recently, they have also garnered interest as potential carbene donors for metal
complexes.2 Nevertheless, the standard syntheses of sulfur ylides are still multi(≥ 2)step
procedures and applications in transition metal catalysis remain limited.1
We have developed a new concept of “ylide transfer” for the direct, one-step synthesis of
sulfur ylides 2 from carbonyl and heteroaromatic compounds 1 (Scheme 1).3
Scheme 1
In this communication, results from those studies will be presented, as well as interesting applications of the ylide products in transition metal catalysis. Furthermore, an intriguing alternative pathway that results in a powerful direct arylation of carbonyl compounds (Scheme 2), as well as other recent developments, shall be discussed.
4,5
Scheme 2
Acknowledgements: We thank the Max-Planck-Society, the Deutsche Forschungsgemeinschaft (MA 4861/4-1 and 4-2) and the Max-Planck-Institut für Kohlenforschung for support.
References: 1. a) M. B. Smith, J. March, Advanced Organic Chemistry; Wiley: New York, 2001 and references therein. 2. See, e.g.: a) P. Müller, D. Fernandez, P. Nury, J.-C. Rossier, Helv. Chim. Acta 1999, 82, 935. b) M. Gandelman, B. Rybtchinski, N. Ashkenazi, R. M. Gauvin, D. Milstein, J. Am. Chem. Soc. 2001, 123, 5372. c) I. K. Mangion, I. K. Nwamba, M. Shevlin, M. A. Huffan, Org. Lett. 2009, 11, 3566. 3. X. Huang, R. Goddard, N. Maulide, Angew. Chem. Int. Ed. 2010, 49, 8979. 4. a) X. Huang, N. Maulide, J. Am. Chem. Soc. 2011, 133, 8510; b) X. Huang, M. Patil. C. Farès, W. Thiel, N. Maulide, J. Am. Chem. Soc. 2013, 135, 7312. 5. a) X. Huang, S. Klimczyk, N. Maulide, Chem. Sci. 2013, 4, 1105; b) X. Huang, B. Peng, M. Luparia, L. Gomes, L. F. Veiros, N. Maulide, Angew. Chem. Int. Ed. 2012, 51, 8886. 6. a) González S. D.-; Marion N.; Nolan S. P. Chem. Rev. 2009, 109, 3621. b) Hahn F. E. Angew. Chem. Int. Ed. 2006, 45, 1348. 7. Nair V.; Menon R. S.; Biju A. T.; Sinu C.R.; Paul R. R.; Jose A.; Sreekumar V. Chem. Soc. Rev. 2011, 40, 5336.

SLB-IOC5
α,β-Unsaturated Diazoketones as Useful Platforms in the Synthesis
of Pyrrolidine, Piperidine and Indolizidine Alkaloids
Antonio C. B. Burtoloso
Instituto de Química de São Carlos, Universidade de São Paulo, 13560-970, São Carlos, SP, Brazil
Diazocompounds are a very interesting class of compounds that can promote a wide range
of reactions, such as cyclopropanations, insertion reactions, ylide formation, dimerization,
and elimination and formation of ketenes by the Wolff rearrangement, among others. An
interesting class of these diazocompounds is the α,β-unsaturated diazoketones,1 which has
received little attention when compared to the saturated ones due to the difficulty of its
preparation by the usual existing methods. Herein, we would like to describe two new
methodologies for the preparation of α,β-unsaturated diazoketones with E and Z geometry
and their use as efficient platforms in the synthesis of pyrrolidines,1 indolizidines
2,3 and
piperidines (Scheme 1).
Scheme 1: α,β-unsaturated diazoketones as platforms in the synthesis of alkaloids.
Acknowledgements: We thank FAPESP and CNPq for financial support. References: 1) Pinho, V. D.; Burtoloso, A. C. B. J. Org. Chem. 2011, 76, 289. 2) Pinho, V. D.; Burtoloso, A. C. B. Tetrahedron Lett. 2012, 53, 876; 3) Bernardim, B.; Pinho, V. D.; Burtoloso, A. C. B. J. Org. Chem. 2012, 77, 9926.

SLB-IOC6
Dual-acting Antimalarial Triterpenoids from an African Medicinal
Plant
M. J. U. Ferreira
iMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal
Despite the efforts to eradicate malaria during the last decade, it remains a major global
health problem, particularly in many of the poorest countries in the world. The increasing
prevalence of drug-resistant Plasmodium falciparum strains is one of the greatest
challenges in malaria control. In order to overcome drug-resistance, new antimalarial drugs
are urgently needed.
Natural product-derived compounds have played a major role in drug discovery and
development. In case of malaria drug discovery, the great significance of plant-derived
drugs for the treatment of the disease is highlighted by quinine, artemisinin and their
derivatives, which are currently the mainstay of the antimalarial therapy.
As part of our search for bioactive compounds from medicinal African plants, we have
carried out a preliminary screening of different plants species for their antimalarial activity.
Momordica balsamina L. (Cucurbitaceae) was found to be the most active plant. M.
balsamina, also referred to as the balsam apple, or African pumpkin, is an extensively
cultivated vegetable consumed in many tropical and subtropical regions of the world. It has
also been widely used in traditional medicine in Africa to treat various diseases, mostly
diabetes, and malaria symptoms.
Bioassay-guided fractionation of the methanol extract of the aerial parts of Momordica
balsamina led to the isolation of several cucurbitane-type triterpenoids. These compounds
and acylated derivatives were evaluated for their antimalarial activity against the erythrocytic
stages of the Plasmodium falciparum chloroquine-sensitive strain 3D7 and the chloroquine-
resistant clone Dd2.1
Evaluation of the activity of some compounds against the liver stage of P. berghei was also
carried out2, measuring the luminescence intensity in Huh-7 cells infected with a firefly
luciferase-expressing P. berghei line, PbGFP-Luccon. Toxicity of compounds was assessed
on the same cell line through the fluorescence measurement of cell confluency. Moreover,
toxicity towards human cells of compounds was also investigated in the MCF-7 breast
cancer cell line, showing that most of them were not toxic or exhibited weak toxicity. In
blood stages of P. falciparum, several compounds displayed antimalarial activity, revealing
some alkanoyl ester derivatives the highest antiplasmodial effects, with IC50 values in the
nanomolar range. The highest antiplasmodial activity against the liver stages of P. berghei
was also displayed by ester derivatives, with high inhibitory activity and no toxicity.
References: 1. a) Ramalhete C.; Lopes D., Mulhovo S.; Molnár J., Rosário V.E.; Ferreira M.J.U. Bioorg. Med. Chem. 2010, 18, 5254. b) Ramalhete C.; Lopes D., Mulhovo S.; Molnár J., Rosário V.E.; Ferreira M.J.U. Bioorg. Med. Chem. 2011, 19, 330. 2. Ramalhete C.; da Cruz F.P., Lopes D.; Mulhovo S.; Rosário V. E.; Prudêncio M.; Ferreira M.J.U. Bioorg. Med. Chem. 2011, 19, 7474.

SLB-IOC7
Straightforward organic chemistry against an intricate infectious
disease: new chloroquine and quinacrine analogues as dual-stage
antimalarial leads
B. Pérez,a C. Teixeira,
b A. Gomes,
a J. R. B. Gomes,
b P. Gomes
a
aCIQ-UP, Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade do Porto, 4169-
007 Porto, Portugal; bCICECO, Departamento de Química, Universidade de Aveiro, 3810-193 Aveiro,
Portugal.
A child dies every minute from malaria. Despite this intricate infectious disease is known for
millennia, and associated deaths have decreased by about 30% in Africa since 2006,
eradication is far from being achieved in the near future.1 There are well identified obstacles
to malaria eradication, namely, the complexity of the malaria parasite’s life cycle,
widespread resistance to cheaper and most popular antimalarial drugs, and lack of efficient
vaccines or multi-stage antimalarials, able to efficiently deplete liver- and blood-stage forms
of Plasmodium parasites from the human body. Another drawback in malaria therapy is the
high cost of first-line drugs, which instigates traffic of fake antimalarials.2
For over a decade, we have been working on the chemical modification of known drugs by
means of simple and inexpensive synthetic organic chemistry, aiming at the low-cost
improvement of their therapeutic properties.3 In this connection, we have recently focused
our research towards development of potential dual-action antimalarials, obtained by
conjugation of cinnamic acids to aminoquinoline or acridine cores from classical antimalarial
drugs (Scheme 1).This led to discovery of novel chloroquine and quinacrine analogues as
dual-stage antimalarial leads.4
Scheme 1: Synthetic route towards N-cinnamoylated analogues of the classic antimalarials chloroquine
and quinacrine.
Acknowledgements:This work is supported by FEDER through the COMPETE program (ref. FCOMP-01-0124-FEDER-020963) andby Portuguese National Funds through FCT –Fundação para a Ciência e a Tecnologia (ref. PTDC/QUI-QUI/116864/2010). BP and CT thank FCT for their doctoral (SRFH/BD/86166/2012) and post-doctoral (SFRH/BPD/62967/2009) grants, respectively. References: 1. World Malaria Report, World Health Organization 2012, ISBN 978 92 4 156453 3. 2. a) Reddy, D.; Bannerji, J.Lancet Infect. Dis. 2012, 12, 829; b) http://www.fakedrugskill.org/ (accessed on May 28
th, 2013).
3. a) Vale, N.et al. J. Med. Chem. 2009, 52, 7800; b) Matos, J. et al. Antimicrob. Agents Chem other.
2012, 56, 1564; c) Pérez, B.et al. Eur. J. Med. Chem. 2012, 54, 887. 4. a) Pérez, B. et al. ChemMedChem 2012, 7, 1537; b) Pérez, B. et al. Med. Chem. Commun. 2012, 3, 1170; c) Pérez, B. et al. J. Med. Chem. 2013, 56, 556; d) Pérez, B. et al. Bioorg. Med. Chem. Lett. 2013, 23, 610; e) Gomes, A. et al. submitted (2013).

SLB-IOC8
A Brief Saga into the Electrophilic Aromatic Substitution
Mechanisms
Pierre Mothé Esteves
Instituto de Química, Universidade Federal do Rio de Janeiro, Av. Athos da Silveira Ramos, 149, CT,
Bl.A-622, Cid. Univ., Rio de Janeiro, 21941-909
A discussion about the aromatic substitution mechanism, based on internal single electron
transfer between the reagents and its consequences for developing new electrophilic
reactions will be presented. An mechanistic continuum is proposed for explaining different
reactivity and observations. This investigation lead to the development new methods for
halogenation and nitration of aromatic compounds.
Acknowledgements: We thank the CNPq and FAPERJ for financial support. References: 1. Esteves, P. M.; Barboza, A. G. H., Carneiro, J. W. M., Laali, K. K., Prakash, G. K. S., Olah, G. A., Rasul, Golam, Cardoso, S. P. J. Am. Chem. Soc. 2003, 125, 4836. 2. Esteves, P. M.; Queiroz, J.F.; Carneiro, J.W.M; Sabino, A. A.; Sparapan, R.; M. N. J. Org. Chem. 2006, 71, 6192.

IOC1
A Recyclable Ferrite–Pd Magnetic Nanocatalyst for the
Buchwald-Hartwig reaction
Sofia Sá, Manoj B. Gawande, Paula S. Branco
Requimte, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de
Lisboa, Campus FCT, 2829-516 Caparica, Portugal
Functionalized magnetic nanoparticles (MNPs) are heterogeneous catalyst supports which
have emerged as viable alternatives to conventional materials because they are robust,
inert, inexpensive, reusable, and recyclable using a simple magnet.1
Magnetite is a well-
known material, also known as ferrite (Fe3O4), which has been used as a versatile support
for functionalization of metals, organocatalyst, and chiral catalysts.2
Arylamines are
compounds of increasing importance as evidenced by its application on the synthesis of
artificial dyes, or the synthesis of biologically active compounds such as pharmaceuticals
and agrochemicals. The homogeneous palladium-catalyzed aromatic aminations
independently developed by Buchwald3a
and Hartwig3b
were a great success because of
their wide range of applicability under relatively mild conditions. Here we report the
immobilization of palladium on the surface of magnetite, and the catalyst thus prepared
Fe3O4-Pd was successively applied in the arylation of amines and amides (Scheme 1).
Scheme 1: Buchwald-Hartwig reaction catalyzed by functionalized magnetic nanoparticles,
Fe3O4-Pd. Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support through grant PEst-C/EQB/LA0006/2011. M. B. Gawande also thanks PRAXIS program for the award of research fellowship (SFRH/BPD/64934/2009). References: 1. a) Polshettiwar V.; Luque R.; FihriA.; ZhuH.; Bouhrara M.; Basset J.-M.; Chem. Rev., 2011, 111 3036. b) Polshettiwar V.; Varma, R. S.; Green Chem., 2010, 12, 743. 2. a) P. D. Stevens P. D.; Li G. F.; Fan J. D.; Yen M.; Gao Y.;Chem. Commun., 2005, 4435. b) Liu, J.; Peng X.; Sun W.; Zhao Y.; Xia C.; Org. Lett., 2008, 10, 3933. c) Shi F.; Tse M. K.; Zhou S.; Pohl M.-M.; Radnik J. R.; H bnerS.; J hnisch K.; Br ckner A.; Beller M.; J. Am. Chem. Soc., 2009, 131, 1775. 3. a) Guram A. S.;Rennels R. A.; Buchwald S.; Angew. Chem. Int. Ed. Engl. 1995, 34, 1348. b) Louie J.;Hartwig J. F.; Tetrahedron Lett. 1995, 36, 3609.

IOC2
Developments in Enantioselective Immobilized BINOL-based
Tandem Reactions
Mariette M. Pereira,a Ângela C. B. Neves,
a Carlos J. P. Monteiro,
a Rui. M. B. Carrilho,
a César
A. Henriques,a Artur R. Abreu,
b Gonçalo N. Costa,
b Marta Pineiro,
a Mário J. F. Calvete,
a
Sónia A.C. Carabineiroc, José L. Figueiredo,
c Auguste Fernandes,
d M. Filipa Ribeiro
d
aChemistry Department, University of Coimbra, 3004-535 Coimbra, Portugal.
bLuzitin, SA, Edificio Bluepharma, São Martinho do Bispo, 3045-016 Coimbra, Portugal.
cLCM – Laboratory of Catalysis and Materials – Associate Laboratory LSRE/ LCM, Faculty of
Engineering, University of Porto, Porto, Portugal. dInstituto de Biotecnologia e Bioengenharia, Centro de Engenharia Biológica e Química, Instituto
Superior Técnico, Avenida Rovisco Pais, 1049-001, Lisboa, Portugal.
The synthesis of chiral compounds is a challenging area of contemporary synthetic organic
chemistry due to the broad applications of synthetic chiral molecules in medicine and
materials. This is the main reason for the increased interest in developing new asymmetric
catalytic process. Based on amplified environmental concerns, asymmetric tandem
reactions emerged as a powerful strategy to improve the synthetic efficiency and reduce the
amounts of solvents. On this context, hydroformylation1 is considered a key tool for the one-
step transformation of olefins into valuables aldehydes, which can be transformed into high
value compounds in a sequential process. Moreover, immobilization of chiral ligands into
solid supports allows the conjugation of the selectivity of homogeneous catalytic processes
with the easy recovery of the catalyst characteristic of heterogeneous catalysts.2
In this communication we present the recent developments of immobilized BINOL-based
ligands and its application in Tandem hydrofromylation/hydrocyanation or alkylation
reactions. A comparative study between homogeneous and heterogenized catalyst will be
presented and discussed.
References: 1. Carrilho, R.M.B., Neves, Â.C.B., Lourenço, M.A.O., Abreu, A.R., Rosado, M.T.S., Abreu, P. E., Eusébio, M. E., Kollár, L., Bayón, J. C., Pereira, M. M., J. Organomet. Chem. 2012, 698, 28. 2. Neves, Â. C. B., Calvete, M. J. F., Pinho e Melo, T. M. V. D., Pereira, M. M., Eur. J. Org. Chem., 2012, 6309.

IOC3
Biological activities of oxygen and nitrogen heterocyclic
compounds
Maria do Carmo Barretoa, Diana C. G. A. Pinto
b, Djenisa H. A. Rocha
b, Miguel X.
Fernandesc, Inês J. Sousa
c, Artur M. S. Silva
b
aCIRN / DCTD, Universidade dos Açores, 9501-801 Ponta Delgada, Portugal;
bDepartment of Chemistry
&QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal; cCentro de Química da Madeira,
Universidade da Madeira, Campus Universitário da Penteada 9000-390 Portugal
Oxygen and nitrogen heterocyclic compounds often display a wide array of biological
activities, being for example very active antioxidants, antifungal, antitumor, antiviral,
cannabinoid agonists and anti-inflammatory1. In this context, we review some of the more
relevant structures, such as chromone and pyrazole derivatives (Fig. 1), which have
biological activities that render these molecules excellent scaffolds for extremely active
drugs. After this introduction, we report the biological activities of oxygen heterocyclic
compounds synthesized by our group. In vitro cytotoxicity was assessed by the MTT
method and anticholinesterasic activity by a modification of the Ellman method2. Most of the
compounds tested were extremely active against HeLa tumor cell line, both in lag and in log
phases of growth. However, they were equally toxic to Vero non tumor reference cell line,
which means that further structural modifications need to be carried out if these molecules
are meant to be used as chemotherapeutic agents. Concerning anticholinesterasic activity,
results were extremely favourable, since several of the compounds tested were very strong
inhibitors of the enzyme. Computational docking using the FlexScreen program suggested
that interaction with the enzyme is mainly at the peripheral anionic site of the
acetylcholinesterase active site gorge. The excellent results obtained for anticholinesterasic
activity indicate that some of these compounds have potential as drugs in the treatment of
Alzheimer’s disease. Future structural modifications, directed by indications provided by
computer-guided approaches, will be tested by in vitro bioassays in order to enhance both
activity and selectivity.
Figure 1: (1) Chromone and (2) pyrazole.
Acknowledgements: Thanks are due to CIRN (University of the Azores) and to DRCTC for funding the unit. Thanks are also due to the University of Aveiro, Fundação para a Ciência e a Tecnologia (FCT), European Union, QREN, FEDER and COMPETE for funding the Organic Chemistry Research Unit (project PEst-C/QUI/UI0062/2011), Centro de Química da Madeira (Project PEst-OE/QUI/UI0674/2011) and the Portuguese National NMR Network (RNRMN). References: 1. a) Sharma, S.K.; Kumar, S.; Chand, K.; Kathuria, A.; Gupta, A.; Jain, R., Curr. Med. Chem. 2011, 18, 3825. b) Gomes, A.; Neuwirth, O.; Freitas, M.; Couto, D.; Ribeiro, D.; Figueiredo, A.G.P.R.; Silva, A.M.S.; Seixas, R.S.G.R.; Pinto, D.C.G.A.; Tomé, A.C.; Cavaleiro, J.A.S.; Fernandes, E.; Lima, J.L.F.C. Bioorg. Med. Chem. 2009, 17, 7218. c) Cumella J.; Hernández-Folgado L., Girón R., Sánchez E., Morales P., Hurst DP, Gómez-Cañas M., Gómez-Ruiz M., Pinto D.C., Goya P., Reggio P.H., Martin M.I.; Fernández-Ruiz J.; Silva A.M.; Jagerovic N. ChemMedChem. 2012, 5, 452. 2. a) Moujir, L.M.; Seca, A.M.L.; Silva, A.M.S.; Barreto, M.C. Planta Med. 2008, 74,751. b) Arruda, M.; Viana, H.; Rainha, N.; Neng, N.R.; Rosa, J.S.; Nogueira, J.M.F.; Barreto, M.C. Molecules 2012, 3082.

IOC4
New syntheses of potential biologically active xanthones and
benzoxanthones
Djenisa H. A. Rocha,a Stéphanie B. Leal,
a Ana M. L. Seca,
a,b Diana C. G. A. Pinto,
a Artur M.
S. Silvaa
aDepartment of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal;
bDCTD, University
of Azores, 9501-801 Ponta Delgada, Portugal
Xanthones have a rather restricted occurrence among higher plants, being found almost
exclusively in Guttiferae and Gentianaceae.1
Natural and synthetic xanthone derivatives
have been described as exhibiting several important biological properties, such as anti-
tumor,2a
anti-inflammatory2b
and antioxidant2c
activities which make them attractive to the
pharmaceutical industry.
The synthesis of xanthones, adequately functionalized for a specific application, is a
challenging task. In this presentation, will be disclosed our recent studies with this unique
family of compounds, namely the one-pot photoinduced electrocyclisation of (E)-3-
styrylflavones 1 and in situ oxidation of cycloadducts to give 5-phenyl-7H-benzo[c]xanthen-
7-one derivatives 2,3a
and aromatization studies of (E)-3-aryl-4-benzylidene-8-hydroxy-3,4-
dihydro-1H-xanthene-1,9(2H)-diones 33b
into 4-benzyl-1,8-dihydroxy-3-phenyl-9H-xanthen-
9-one derivatives 4 (Figure 1).
Figure 1: Xanthone derivatives structure.
Acknowledgements: We would like to thank Fundação para a Ciência e a Tecnologia (FCT, Portugal), the European Union, QREN, FEDER, COMPETE, for funding the Organic Chemistry Research Unit (QOPNA) (project PEst-C/QUI/UI0062/2011) and the Portuguese National NMR Network (RNRMN). D. H. A. Rocha thanks FCT for her PhD grant (SFRH/BD/68991/2010). References: 1. Gales, L.; Damas, A.M. Curr. Med. Chem. 2005, 12, 2499. 2. a) Pedro M.; Cerqueira, F.; Sousa, M.E.;Nascimento, M.S.J. Pinto, M. Bioorg. Med. Chem. 2002, 10, 3725. b) Park, H.H.; Park, Y.-D.; Han, J.-M.; Im, K.-R.; Lee, B.W.; Jeong, I.Y.; Jeong, T.-S.; Lee, W.S. Bioorg. Med. Chem. Lett. 2006, 16, 5580. c) Suvarnakuta, P.; Chaweerungrat, C.; Devahastin, S. Food Chem. 2011, 125, 240. 3. a) Rocha, D.H.A.; Pinto, D.C.G.A.; Silva, A.M.S.; Patonay, T.; Cavaleiro, J.A.S. Synlett, 2012, 559. b) Pinto, D.C.G.A.; Seca, A.M.L.; Leal, S.B.; Silva, A.M.S.; Cavaleiro, J.A.S. Synlett, 2011, 2005.

IOC5
Synthesis of 1-vinylidene-naphthofurans: A thermally reversible
photochromic system that colours only when adsorbed on silica gel
Céu M. Sousaa, Jerome Berthet
b, Stephanie Delbaere
b, Paulo J. Coelho
a
aUniversidade de Trás-os-Montes e Alto Douro, 5001-801 Vila Real (Portugal)
bUniversité Lille Nord de France, CNRS UMR 8516, UDSL, F-59006, Lille (France)
A set of new 1-vinylidene-1,2-dihydro-naphtho[2,1-b]furans were unexpectedly obtained in
the reaction of 2-naphthol with easily obtained 1,1,4,4-tetraarylbut-2-yne-1,4-diols at room
temperature in the presence of a catalytic amount of p-toluenesulfonic acid (Scheme 1). A
mechanism for the formation of this allenic compound was proposed involving ether
formation, Claisen rearrangement, enolization and dehydration. The reaction is compatible
with different substituents in the aromatic ketone and afforded naphthofurans 1a-c in 19-
47% yield.
Scheme 1: Synthesis of 1-vinylidenenaphtho[2,1-b]furans 1 from aromatic ketones.
Surprisingly, when adsorbed in silica gel, these new compounds exhibit photochromism at
room temperature while not in solution and in the solid state. UV or sunlight irradiation
leads, in few seconds, to the formation of intense pink/violet to green colours that bleach
completely in few minutes in the dark (Scheme 2). This phenomenon is reproducible and
can be repeated several times without any sign of degradation. These new compounds also
exhibit reversible acidochromic properties in solution: addition of trifluoroacetic acid leads to
the formation of an intense violet colour that return immediately to the uncoloured initial
state upon addition of NEt3.
Scheme 2: Photochromic equilibrium for new 1-vinylidene-naphthofuran 1a (Ar=Ph) and photographs of
silica gel doped with this compound before and after UV irradiation (365 nm) Acknowledgements: We thank the Fundação para a Ciência e Tecnologia (FCT) for financial support through the research unit Centro de Química-Vila Real (POCTI-SFA-3-616)

IOC6
Electronic Communication in Linear Oligo(azobenzene) Radical
Anions
João P. Telo
Centro de Química Estrutural, Instituto Superior Técnico, Technical University of Lisbon, Av. Rovisco
Pais, 1049-001 Lisboa, Portugal.
The azobenzene molecular unit has been extensively studied in the recent past due to the
potential use of its cis-trans photoisomerization reaction in optical switches, information
storage or mechanical devices. Although the photochemistry of the azo group has received
enormous attention, the potential use of azobenzene system as charge-carrier in organic
devices has been seldom addressed in literature. The ability to transport charge depends
on the efficiency with which the charge moves within the -conjugated system. With this
purpose, the use of mixed-valence chemistry offers a unique tool to study how the
localization of charge in organic radical ions depends on the structural and chemical
features.1
Although the photochemistry of the azo group has received enormous attention, the
potential use of azobenzene systems as charge-carriers in organic devices has been
seldom addressed in the literature. In this context, it is of significant interest to understand
how the chemical structure affects the electronic coupling in negatively charged
azobenzene oligomers. We studied in this work the radical anions of seven azobenzene
oligomers, where the charge is mainly centered in the diazo groups, by optical and EPR
spectroscopy.
Acknowledgements: Support by Fundação Para a Ciência e Tecnologia through its Centro de Química Estrutural and Projects PEst-OE/QUI/UI0100/2011 and PTDC/QUI-QUI/101433/2008 is gratefully acknowledged. References: 1. a) Á. Moneo, M.F.N.N. Carvalho, J.P. Telo, J. Phys. Org. Chem 2012, 25, 559. b) J.P. Telo, Á. Moneo, M.F.N.N. Carvalho, S.F. Nelsen, J. Phys. Chem A. 2011, 115, 10738. c) Hoekstra, R.M; Telo, J.P; Wu, Q.; Stephenson, R.M.; Nelsen, S.F.; Zink, J.I., J. Am. Chem. Soc. 2010, 132, 8825. d) S.F. Nelsen, M.N. Weaver, J.P. Telo, J. Am. Chem. Soc. 2007, 129, 7036.

IOC7
Isolation and Identification of Impurities in Tetracycline Derivatives
Dália Barbosa, Carlos Anjo
AtralCipan, Rua da estação, 1649-003 Lisboa, Portugal
Identification, qualification and quantification of impurities are critical tools for assessing the
safety and quality of pharmaceutical drug substances. In order to market a drug, active
pharmaceutical ingredients (API) the manufacturer should ensure that the different classes
of impurities due of synthesis and degradation are addressed and adequately controlled in
the drug substance. Cipan produces API’s for more than fifty years and has been always
committed to respond to the increasingly demanding guidances1 on API´s production and
qualification. The identification and characterization of impurities is of utmost importance in
today´s regulatory environment and a sine-qua-on capability if one wants to sell in the most
regulated markets.
The focus of this work is to develop an efficient process of synthesis of possible impurities
of tetracycline’s derivatives produced by Cipan (Figure 1).
Synthesis of some impurities and/or degradation products of tetracycline derivatives
produced will be described. These include epimers and some other potential impurities. The
synthesized compounds as well as the corresponding intermediates were characterized.
R1 R2 R3 R4
Tetracycline H CH3 OH H
Sancycline H H H H
Minocycline N(CH3)2 H H H
Figure 1: Chemical structures of Tetracycline derivatives References: 1. Snodin J. D.; McCrossen S.; Regulatory Toxicology and Pharmacology 2012, 63, 298 2. Nair V.; Menon R. S.; Biju A. T.; Sinu C.R.; Paul R. R.; Jose A.; Sreekumar V. Chem. Soc. Rev. 2011, 40, 5336.

IOC8
New Chemical Processes with Bismuth(III) Salts: Applications of
Bismuth(III) Reagents and Catalysts to Steroid and Terpenoid
Chemistry
Jorge A. R. Salvador,a,b
Rui M. A. Pinto,a,b,§
aLaboratório de Química Farmacêutica, Faculdade de Farmácia, Universidade de Coimbra, 3000-548
Coimbra, Portugal; bCentro de Neurociências e Biologia Celular, Universidade de Coimbra, 3004-517
Coimbra, Portugal §Current affiliation at Department of Veterinary Medicine, Escola Universitária Vasco
da Gama, Mosteiro S. Jorge de Milréu, Estrada da Conraria, 3040-714 Castelo Viegas, Coimbra, Portugal
Steroids and terpenes constitute a large and structurally diverse family of natural products
and are considered important scaffolds for the synthesis of molecules of pharmaceutical
interest.1
The growing relevance of green and sustainable chemistry and the application of its guiding
principles to the development of new reactions and chemical processes is changing the
face of chemistry.2 Bismuth(III) salts are known for their low toxicity, making them potential
valuable reagents for large-scale synthesis, which becomes more obvious when dealing
with products such as active pharmaceutical ingredients or synthetic intermediates.3
In this communication, the work developed in our lab in the application of bismuth(III) salts
as reagents and/or catalyst to the chemistry of steroids and terpenoids is presented. The
reactivity of Bi(III) salts towards epoxysteroids has been studied, leading to the novel
reaction conditions for the Ritter reaction.4 Modulating these reaction conditions, new
bismuth-based processes for the synthesis of either β-substituted alcohols5 or olefinic 18-
nor and 18,19-dinorsteroids have been developed.6 Bismuth(III) salts were found suitable
catalysts for rearrangement reactions and a new Bi(III)-based process for the Wagner-
Meerwein rearrangement of lupanes have been reported.7 On the other hand, a novel
reaction for direct synthesis of 12-oxo-oleanolic acid derivatives have been developed
starting from the corresponding -hydroxylactones.8 A high value catalytic process for the
selective cleavage of the C17-dihydroxyacetone side chain of corticosteroids was performed
using bismuth(III) triflate, and highly functionalized 17-ketosteroids were obtained in good
yields.9
Figure 1: Examples of compounds obtained using the bismuth(III)-based new chemical processes
developed in our research group. References: 1. a) Hanson, J. R. Nat. Prod. Rep. 2010, 27, 887; b ) Gershenzon, J.; Dudareva, N. Nat. Chem. Biol. 2007, 3, 408. 2. a) R. A. Sheldon Chem. Soc. Rev., 2012, 41, 1437; b) J. L. Tucker Org. Proc. Res. Dev, 2006, 10, 315. 3. Salvador, J.A.R; Figueiredo, S., Pinto, R.M.A., Silvestre, S.M. Future Med. Chem. 2012, 4, 1495. 4. R. M. A. Pinto, J. A. R. Salvador, C. Le Roux Synlett, 2006, 2047. 5. R. M. A. Pinto, J. A. R. Salvador and C. Le Roux Tetrahedron, 2007, 63, 9221. 6. a) R. M. A. Pinto, J. A. R. Salvador, C. Le Roux, et al. Steroids, 2008, 73, 549; b) R. M. A. Pinto, J. A. R. Salvador, C. Le Roux, R. et al., Tetrahedron, 2009, 65, 6169. 7. J. A. R. Salvador, R. M. A. Pinto, R. C. Santos, et al., Org. Biomol. Chem., 2009, 7, 508. 8. a) J. A. R. Salvador, V. M. Moreira, R. M. A. Pinto, et al., Adv. Synth. Catal., 2011, 353, 2637; b) J. A. R. Salvador, V. M. Moreira, R. M. A. Pinto, et al., Belstein J. Org Chem., 2012, 8, 164. 9. R. M. A. Pinto, J. A. R. Salvador, C. Le Roux and J. A. Paixão, J. Org. Chem., 2009, 74, 8488.

IOC9
Study and Modulation of Inter-species Quorum Sensing by AI-2
Analogues
Osvaldo S. Ascenso,a Ana Sofia Miguel,
a Fábio Rui,
a,b João C. Marques,
b Christopher D.
Maycock,a,c
Karina B. Xavier,a,b
M. Rita Venturaa
aInstituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa, 2780-157 Oeiras, Portugal; bInstituto Gulbenkian de Ciência, 2780-156 Oeiras, Portugal;
cFaculdade de Ciências, Universidade de
Lisboa, 1749-016 Lisboa, Portugal
Autoinducer-2 (AI-2) is a signalling molecule for bacterial inter-species communication.
Examples of quorum sensing regulated behaviours are biofilm formation, virulence-factor
expression, antibiotic production and bioluminescence. Ultimately, the understanding of the
molecular mechanisms that bacteria use to regulate their behaviours can lead to the
development of new therapies to control bacterial infections, and also to develop
biotechnological applications for the control of industrial scale production of beneficial
bacterial products, such as antibiotics or recombinant proteins.
A synthesis of (S)-4,5-dihydroxypentane-2,3-dione (DPD, 1, Fig. 1), the precursor of AI-2,
has been developed starting from methyl glycolate.1 Using the same synthetic strategy and
starting from methyl (S)- and (R)-lactates, four new analogues have been prepared and
tested (Fig. 1). The new analogues had one more asymmetric center and the configuration
of the new substituent exerted an important influence in its biological activity. Other
analogues have been synthesised and tested for their quorum sensing activity, leading to
useful structure/activity conclusions.
Studies towards the preparation of new DPD fluorescent markers, using different linkers, will
also be presented. These new DPD probes are important tools to determine novel AI-2
receptors in important human pathogens such as Pseudomonas aeruginosa, and Bacillus
anthracis. The discovery of new receptors involved in AI-2 signalling can lead to strategies
to manipulate virulence in these pathogens and other closely related bacteria.
Figure 1: DPD and analogues synthesis.
Acknowledgements: We thank the Fundação para a Ciência e Tecnolgia for financial support -PTDC/QUI-BIQ/113880/2009. References: 1. Ascenso, O. S.; Marques, J. C.; Santos, A. R.; Xavier, K. X.; Ventura, M. R.; Maycock, C. D. Bioorg. Med. Chem. 2011, 19, 1236. 2. Rui, F.; Marques, J. C.; Miller, S. T.; Maycock, C. D.; Xavier, K. B.; Ventura, M. R. Bioorg. Med. Chem. 2012, 20, 249.

IOC10
Development of a practical and efficient synthesis of an Active
Pharmaceutical Ingredient
Ricardo Mendonça
Hovione FarmaCiencia SA, Sete Casas, 2674-506 Loures, Portugal;
Progression towards a scalable synthesis of an API, culminating in the first GMP
manufacturing campaign, is described. Through process development, the discovery route
was improved into an efficient industrial process. Hazardous reagents and solvents were
substituted by more eco-friendly alternatives whilst improving the quality of the final API.
Telescoping of the process reduced the total number of isolated intermediates. Also,
crystallization development was of crucial importance to ensure product quality without the
use of chromatographic steps.

IOC11
Artificial and natural polymers: from synthesis and chemical
modification to biomedical applications
Ana M. Rosa da Costa
Departamento de Química e Farmácia, Faculdade de Ciências e Tecnologia & CIQA – Centro de
Investigação em Química do Algarve, Universidade do Algarve, Campus de Gambelas, 8005-139 Faro,
Portugal
Living polymerization processes, in particular the Reversible Addition-Fragmentation chain
Transfer (RAFT) polymerization, offer many benefits, which include the ability to control
molecular weight and polydispersity, as well as to prepare block copolymers and other
complex architecture polymers, difficult to obtain otherwise. By virtue of the RAFT
mechanism, which relies in the introduction of a specific chain transfer agent (CTA) into the
polymerization medium, polymers prepared by this technique are α,ω-functionalized.
Moreover, RAFT polymerization is compatible with a great variety of monomers and
reaction media.1
Polysaccharides are promising materials for drug delivery systems due to their
biocompatibility, degradation behavior, and nontoxic profile on administration. Recently,
there has been a growing interest in the chemical modification of these polymers in order to
create derivatives with tailored properties for specific purposes. In particular, regarding the
development of appropriate vehicles for drug delivery, such modifications include the
introduction of small functional groups.2
A few examples of synthesis of artificial polymers by RAFT and of chemical modification of
polysaccharides will be presented, as well as their application in the development of
systems for biosensing, imaging, and drug and gene delivery.3
Acknowledgements: We thank Fundação para a Ciência e Tecnologia (FCT, Portugal) for financial support under projects PEst-OE/QUI/UI4023/2011, PEst-OE/EQB/LA0023/2011, PTDC/SAU-FCF/100291/2008, and PTDC/SAU-BEB/098475/2008. References: 1. a) Rizzardo E. et al. Macromolecules 1998, 31, 5559. b) McCormick C.L.; Lowe A.B. Acc. Chem. Res. 2004, 37, 312. c) Charleux B. et al. Macromolecules 2004, 37, 6329. d) van Zyl A.J.P. et al. Polymer 2005, 46, 3607. 2. a) Liu Z.; Jiao Y.; Wang Y.; Zhou C.; Zhang Z. Adv. Drug. Deliv. Rev. 2008, 60, 1650. b) Mizrahy S.; Peer D. Chem. Soc. Rev. 2012, 41, 2623. c) Baldwin A.D.; Kiick K.L. Biopolymers 2010, 94, 128. 3. a) Mouffouk F.; Rosa da Costa A.M.; Martins J.; Zourob M.; Abu-Salah K.M.; Alrokayan S.A. Biosens. Bioelectron. 2011, 26, 3517. b) Alrokayan S.A.H.; Mouffouk F.; Rodrigues dos Santos N.; Rosa da Costa A.M. WO2011113616. c) Braz L.; Grenha A.; Ferreira D.; Rosa da Costa A.M.; Sarmento B. Pharm Anal Acta 2012, 3, 115. d) Dionísio M.; Cordeiro C.; Remuñán-López C.; Seijo B.; Rosa da Costa A.M.; Grenha A., Eur. J. Pharm. Sci. 2013 (in press). e) Oliveira A.V.; Silva A.P.; Bitoque D.B.; Silva G.A.; Rosa da Costa A.M. J Pharm Bioall Sci, 2013, 5, 111.

IOC12
Synthesis and pharmacological evaluation of novel COMT inhibitors
László E. Kiss,a Patrício Soares-da-Silva
a,b
aBIAL - Portela & Cª., S.A. À Av. da Siderurgia Nacional, 4745-457 S. Mamede do Coronado, Trofa,
Portugal; bDepartamento de Farmacologia e Terapêutica, Faculdade de Medicina, Universidade do
Porto, 4200-319 Porto, Portugal
Catechol-O-methytransferase (COMT) is a magnesium-dependent enzyme found in both
the CNS and the periphery, which plays a key role in the inactivation of endogenous
catechol neurotransmitters and xenobiotics. Inhibition of COMT provides therapeutic
benefits in patients afflicted with Parkinson’s disease (PD) undergoing treatment with the
gold standard, levodopa. PD is a chronic neurological disorder associated with a reduction
in striatal levels of the endogenous neurotransmitter dopamine. Levodopa is a biological
precursor of dopamine, which is able to modulate cerebral levels of dopamine by
penetrating into the brain. Clinical efficacy of the therapy can be dramatically improved by
inhibiting the metabolic deactivation of levodopa in peripheral tissues. COMT inhibitors help
to sustain the continuous delivery of dopamine to the striatum and thereby motor-related
symptoms of PD are diminished.
A novel series of aryl- and heteroaryl-oxadiazolyl nitro-catechol derivatives of general
structure 1 were prepared and evaluated for their COMT inhibitory ability in different animal
species.1 One compound from this series, namely opicapone (BIA 9-1067) exhibited potent,
long-acting and peripheral inhibition of COMT.2 Opicapone is currently under clinical phase
III evaluation for the treatment of PD.3,4
Figure 1:Novel aryl- and heteroaryl-oxadiazolyl nitro-catechol derivatives.
Chemical development and pharmacological evaluation of opicapone and its related
analogues will be presented.
References: 1. László E. Kiss, Ferreira H.S., Torrão L, Bonifácio M.J, Palma P.N, Soares-da-Silva P, Learmonth D.A. J. Med. Chem., 2010, 53, 3396. 2. Almeida L, Rocha J.F, Falcão A, Palma P.N, Loureiro A.I, Pinto R, Bonifácio M.J, Wright L.C, Nunes T, Soares-da-Silva P.Clin Pharmacokinet 2013, 52,139. 3. Lees A, Costa R, Oliveira C, Lopes N, Nunes T, Soares-da-Silva P, Movement Disord., 2012, 27, S127. 4. Ferreira JJ, Rocha J.F, Santos A, Nunes T, Soares-da-Silva P, Movement Disord., 2012, 27, S118.

IOC13
Bioactivation of the anti-HIV drug abacavir to an electrophilic
aldehyde: in vitro and in vivo approaches.
Alexandra M. M. Antunes
Centro de Química Estrutural, Instituto Superior Técnico (CQE-IST), Universidade Técnica de Lisboa,
Av. Rovisco Pais 1, 1049-001 Lisboa, Portugal
The nucleoside reverse transcriptase inhibitor abacavir is associated with acute
hypersensitivity reactions, often leading to drug discontinuation. Moreover, an association
between long-term abacavir use and increased risk of myocardial infarction, though still
controversial, has been reported. Bioactivation to a reactive aldehyde, capable of modifying
self-proteins, is thought to be involved at the onset of these adverse reactions. We have
proposed that a conjugated aldehyde is the electrophilic intermediate primarily responsible
for reaction with the N-terminal valine of hemoglobin via Schiff base formation in vitro1 and
subsequently obtained evidence for this pathway in rats administered abacavir2. More
recently, we investigated abacavir bioactivation to aldehydes in humans by assessing the
presence of abacavir adducts with the N-terminal valine of hemoglobin in HIV-infected
patients on a standard anti-HIV regimen containing abacavir3. Following N-alkyl Edman
degradation and HPLC-ESI-MS/MS analysis, the ABC-Valine Edman adduct was identified
by comparison with a synthetic standard in 3/10 patients (50±16 years old; 560±280 CD4).
These results represent the first report of abacavir metabolism to a conjugated aldehyde in
humans. Moreover, by demonstrating that abacavir can be bioactivated to a metabolite
subsisting long enough in vivo to undergo protein haptenation we are providing important
clues to the possible role of this metabolic pathway at the onset of abacavir-induced toxic
events. Therefore, the search for causal relationships between the formation of abacavir-
derived protein adducts and the occurrence of abacavir-induced toxic events in human
patients is worth pursuing in further toxicological studies with larger cohorts.
Figure 1: Alcohol dehydrogenase (ADH) mediated bioactivation pathway of the anti HIV drug abacavir to
the electrophilic conjugated aldehyde and its haptenation mechanism. Acknowledgements: We thank the Fundação para a Ciência e Tecnolgia for financial support (PTDC/SAU-TOX/111663/2009, PTDC/QUI-QUI/113910/2009, and PEstOE/QUI/UI0100/2013). References: 1. Charneira C, Godinho ALA, Oliveira MC, Pereira SA, Monteiro EC, Marques MM, Antunes AMM Chem. Res. Toxicol. 2011, 24, 2129-2141. 2. Charneira C, Grilo NM, Pereira SA, Godinho ALA, Monteiro EC, Marques MM, Antunes AMM Br. J. Pharmacol. 2012 167, 1353-1361. 3. Grilo NM, Antunes AMM, Caixas C, Marinho AT, Charneira C, Gouveia S, Oliveira MC, Marques MM, Pereira SA Tox. Lett. 2013 219, 59-64

IOC14
Ten Years of Catalytic "Asymmetric" Activity at CQE-UE: The First
Decade
Anthony J. Burke
Department of Chemistry and Chemistry Center of Évora, University of Évora, Rua Romão Ramalho, 59,
7000 Évora, Portugal.
Catalytic asymmetric synthesis stands out as the most efficeint and elgant means of
accesing enantiomerically pure compounds. Considering the fact that about 80% of all the
pharmaceuticals on the market are chiral, this strategy is therefore a highly useful
technology for the pharmaceutical industry. Our mission over the last 10 years has been the
development of novel catalytic systems of use to the pharmaceutical industry. The
development of these catalytic systems (Figure 1), their application, including their
immobilization to solid supports will be discussed in this communication.1-3
Figure 1: Some of the chiral ligands and organocatalysts developed to date in our lab.
Acknowledgements: We are grateful for a series of projects financed from FCT during the last 10 years, including the project - Molecular Innovation and Drug Discovery (ALENT-57-2011-20) financed from the FEDER-INALENTEJO program ALENT-07-0224-FEDER-001743, as well as PEst-OE/QUI/UI0619/2011. Chiratecnics Lda (www.chiratecnics.com) is acknowledged for supporting later projects. References: 1. (a) Chercheja, S.; Carreiro, E.P.;
Burke, A.J.; Ramalho, J.P.; Rodrigues, A.I. J. Mol. Catalysis A:
Chemical, 2005, 236, 38. (b) Burke, A.J.; Carreiro, E.P.; Chercheja, S.; Moura, N.M.M.; Prates Ramalho,
J. P.; Rodrigues, A.I.; Carla I. M. Santos, J. Organomet. Chem. 2007, 692, 4863. (c) E. P. Carreiro, AJ. Burke, J.P. Prates Ramalho and A.I. Rodrigues, Tetrahedron: Asymmetry, 2009, 20, 1272. (d) Carreiro, E.P.; Moura, N.M.M.; Burke, A.J.; Eur. J. Organic Chem. 2012, 518-28. 2. a) Marques, C. S.; Burke, A. J. Eur. J. Org. Chem. 2010, 1639. b) Marques, C. S.; Burke, A. J. ChemCatChem 2011, 3, 635. c) Marques, C. S.; Burke, A. J. Eur. J. Org. Chem. 2012, 4232–4239. d) Marques, C. S.; Burke, A. J. Tetrahedron: Asymmetry 2013, in press. e) Marques, C. S.; Burke, A. J. Tetrahedron 2012, 68, 7211–7216. 3. a) Burke, A.J.; Marinho, V.I.; Prates Ramalho, J.P. Chirality, 2011, 23, 383-8. b) Burke, A.J.; Rodrigues, AI. Marinho, V.I. Tetrahedron: Asymmetry, 2008, 19, 454.

OC1
New strategies for the synthesis of 2-(hetero)arylthieno[2,3-b] or
[3,2-b]pyridine scaffolds from 2,3-dihalopyridines
Maria João R. P. Queiroz, Agathe Begouin, Daniela Peixoto
Centro de Química, Escola de Ciências, Universidade do Minho Campus de Gualtar 4710-057 Braga
In the past few years, our research group has synthesised new thieno[3,2-b]pyridines as
antitumor agents.1 Recently, we have been interested in the synthesis of new 2-
(hetero)arylthienopyridine scaffolds starting from 2,3-dihalopyridines (Scheme 1). Two
methodologies were used: either a one-pot Sonogashira coupling followed by a reaction
with Na2S, giving 2-(hetero)arylthienopyridines (Method A),2
or a reaction with NaSMe
followed by a Sonogashira coupling and a halocyclization, affording the corresponding 3-
halo-2-(hetero)arylthienopyridines (Method B).3 The key step of the latter method is the
formation of the required bromo(methylthio)pyridines by a regiocontrolled SNAr with NaSMe,
that has to be performed before the Sonogashira coupling. If not, an addition of the SMe to
the triple bond of the Sonogashira product occurs instead of the substitution of the chlorine
or fluorine atom of the pyridine. In the light of this observation, the reaction mechanism of
Method A using Na2S will also be discussed.
Z
WBr
MeSZ
W
S(Het)Ar
Y
Z
W
X
Br
W = N, Z = CH, X = F or ClW = CH, Z = N, X = Cl
1) NaSMe2) Sonogashira
coupling
3) I2 or Br2
1) Sonogashira coupling
2) Na2S
Y = I or Br
Method AMethod B
Z
W
SAr(Het)
26 compounds15 compounds
Scheme 1: Synthesis of 2-(hetero)arylthienopyridines from 2,3-dihalopyridines
Further functionalizations were also performed on the compounds obtained, allowing the
synthesis of new thienopyridine derivatives that will be studied as antitumor and/or
antiangiogenic agents having tyrosine kinase membrane growth factor receptors of tumor or
endothelial cells as targets.
Acknowledgements: We thank FCT–Portugal for financial support through the NMR Portuguese network (Bruker 400 Avance III-Univ Minho). FCT and FEDER-COMPETE-QREN-EU for financial support through the research unity PEst-C/QUI/UI686/2011, the research project PTDC/QUI-QUI/111060/2009 and the post-Doctoral grant attributed to A.B.(SFRH/BPD/36753/2007) also financed by POPH and FSE. References: 1. a) Queiroz M.-J. R. P et al. Eur. J. Med. Chem. 2010, 45, 5628. b) Queiroz M.-J. R. P et al. Eur. J. Med. Chem. 2010, 45, 5732. c) Queiroz M.-J. R. P et al. Eur. J. Med. Chem. 2011, 46, 236. d) Queiroz M.-J. R. P et al. Eur. J. Med. Chem. 2011, 46, 5800. e) Queiroz M.-J. R. P et al. Molecules, 2012, 17,
3834. 2. Peixoto D.; Begouin A.; Queiroz M.-J. R. P. Tetrahedron 2012, 68, 7082. 3. Begouin A.; Peixoto D.; Queiroz M.-J. R. P. Synthesis 2013, 45, 1489.

OC2
Synthesis, Characterization and Citotoxic Activity of
Cyclopentadienyl Ruthenium(II) Complexes with Carbohydrate
Derived Ligands
Ana C. Fernandes,a Pedro Florindo,
a Inês J. Marques,
b Carla D. Nunes
b
aCentro de Química Estrutural, Instituto Superior Técnico, Universidade Técnica de Lisboa,1049-001
Lisboa, Portugal; bCentro de Química e Bioquímica, Faculdade de Ciências da Universidade de Lisboa,
Campo Grande 1749-016 Lisboa, Portugal
Organometallic complexes containing monosaccharide ligands represent a small but
challenging field in modern chemistry. Carbohydrates are the largest class of natural
compounds and thereby readily available and renewable. They provide a large number of
functional groups and several stereogenic centres per molecule, and each of the hydroxyl
groups offers the opportunity of selective modification and coordination [1, 2]. Particularly,
the synthesis of ruthenium compounds bearing carbohydrate derived ligands is an almost
unexplored area.
As part of our endeavour to produce a library of carbohydrate-containing organometallic
compounds, we here report the synthesis and cytotoxic evaluation against human HeLa
cancer cells (cervical carcinoma) of ten new η5-cyclopentadienylruthenium cationic
complexes of general formula [Ru(η5-C5H5)(PP)Ln][PF6], in which Ln are galactose and
fructose carbohydrate derivative ligands, functionalized with nitrile, tetrazole and 1,3,4-
oxadiazole N-coordinating moieties (Scheme 1). The electronic density and the
stereochemichal environment of the metal centre are played using two different phosphanes
as coligands, PPh3 and Dppe. All new compounds were characterized by IR, 1H,
13C,
31P-
NMR spectroscopies.
Scheme 1: Synthesis of the Ru(II) organometallic complexes.
Acknowledgements: The authors thank Fundação para a Ciência e Tecnologia for financial support through projects PTDC/QUI-QUI/102114/2008, PEst-OE/QUI/UI0100/2013 and PEst-OE/QUI/UI0612/2013. References: 1. H.-U. Blaser, Chem. Rev. 92, 1992, 935-952. 2. P., Anastas, T. Williamson, Green Chemistry: Frontiers in Benign Chemical Syntheses and Processes; Oxford University Press, New York, 1998.

OC3
Pd-catalysed amination on a soluble polymer support: a sustainable
version of homogeneous C-N cross-coupling reaction
Luísa C. R. Carvalho,a Marina J. Dias Pires,
a Eduarda Fernandes,
b M. Manuel B. Marques
a
aREQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de
Lisboa, Campus de Caparica, 2829-516 Caparica, Portugal; bREQUIMTE, Departamento de Ciências
Químicas, Rua Jorge Viterbo Ferreira nº 228, 4050-313 Porto, Portugal
Heterocyclic systems represent an important class of compounds in biologically active
pharmaceuticals, natural products, and materials, and therefore the selective
functionalization of these molecules is of great interest.1a
The aryl amine moiety can be
found in a wide variety of heterocyclic compounds, and metal-catalyzed cross-coupling
reactions of anilines with aryl halides constitute the main methods for assembling this type
of substructure.1b
During the last year we have been focused on the development of novel
benzimidazole based compounds as COX inhibitors, and Pd-catalysed aryl amination
reaction has been used to produce key intermediates.2
Recently, polyethylene glycol (PEG) appears as an environmental friendly alternative to the
use of volatile, toxic and hazardous organic solvents.3 Thus, we decided to investigate the
possibility of using PEG 2000 as a soluble polymeric support for the preparation of arylated
heteroarenes. Herein, we will present an innovative palladium-catalysed amination protocol
on a solid support, solvent-free and homogeneous medium that can be considered as a
sustainable version of this key reaction (Scheme 1). This work opens the possibility to build
aromatic scaffolds while avoiding both organic solvents and purification steps with the
advantage of an easy and fast monitoring.
Scheme 1: Metal-catalysed cross-coupling reaction on PEG.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support SFRH/BD/63407/2009 and PEst-C/EQB/LA0006/2011. References: 1. a) Cho S. H.; Kim J. Y.; Kwak J.; Chang S. Chem. Soc. Rev. 2011, 40, 5068; b) Zheng N.; Anderson K. W.; Huang X.; Nguyen H. N.; Buchwald S. L. Angew. Chem. Int. Ed. Engl. 2007, 46, 7509. 2. Carvalho L. C. R.; Fernandes E.; Marques M. M. B. Chem. – Eur. J. 2011, 45, 12544. 3. Colacino E.; Martinez J.; Lamaty F.; Patrikeeva L. S.; Khemchyan L. L.; Ananikov V. P.; Beletskay I. P. Coord. Chem. Rev. 2012, 256, 2893.

OC4
Photoactive molecules by design
João P. C. Tomé
QOPNA and Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal
Porphyrinsand Chlorins constitute a group of natural compounds which play key roles in
several vital functions of life, such as: respiration, photosynthesis and many enzymatic
processes. The possibility of mimic those functions and explore several others, especially
when combined with light, have been highly explored by the scientific community.1
The
possibility of decorate the periphery of their cores, with different motifs and select their
central metals opens the possibility to fine-tune the physico-chemical
properties/functionalities of novel molecules/materials to be used in many scientific and
technological areas, especially when combined with nanostructures.2
In Aveiro, in
collaboration with other national and international groups, we have successfully been
designing and synthesizing several of those photoactive molecules/materials (Figure 1) to
be used in: i) photomedicine, mainly as photosensitizers for antibiotic resistant pathogenic
microorganisms photodynamic inactivation (PDI) and for cancer photodynamic therapy
(PDT); ii) optical (chemo)sensors, to detect and trap anionic pollutants from contaminated
environments; iii) (photo)catalysts, to be used in industrial and environmental applications.3
In this short communication, it will be highlighted some of our recent works, presenting the
usedsynthetic strategies and some of the obtained photo-physical, -chemical and –
biological results in the indicated applications.
Figure 1: Some photoactive compounds from Aveiro
Acknowledgements: Thanks are due to the Universities of Aveiro, FCT (Portugal) and FEDER for funding the projectsPTDC/QUI/65228/2006 and PTDC/CTM/101538/2008; CRUP (Portugal) for funding the integrated actions Luso–Espanholas/2012 (E-110/12); and to the European Commission for the Marie Curie Initial Training NetworksFP7-PEOPLE-2012-ITN/316975. References:
1. a) J.A.S. Cavaleiro, M.A.F. Faustino, J.P.C. Tome, “Porphyrinyl-type sugar derivatives: synthesis and biological applications”, in Carbohydrate Chemistry, The Royal Society of Chemistry, 2009, 35, 199. b) A.M.V.M. Pereira, A. Hausmann, J.P.C. Tomé, O. Trukhina, M. Urbani, M.G.P.M.S. Neves, J.A.S. Cavaleiro, D.M. Guldi, T. Torres, Chem. Eur. J., 2012, 18, 3210. c) A. Varotto, C.-Y. Nam, I. Radivojevic, J.P.C. Tomé, J.A.S. Cavaleiro, C.T. Black, C.M. Drain, J. Am. Chem. Soc., 2010, 132, 2552. 2. a) F. Figueira, J.A.S. Cavaleiro, J.P.C. Tome, J. PorphyrinsPhthalocyanines, 2011, 15, 517. b) C.M.B. Carvalho, E. Alves, L. Costa, J.P.C. Tomé, M.A.F. Faustino, M.G.P.M.S. Neves, A.C. Tomé, J.A.S. Cavaleiro, M.A. Almeida, M.A. Cunha, Z. Lin, J. Rocha, ACS Nano, 2010, 4, 7133. 3. a) C.F.A.C. Gomes, S. Silva, M.A.F. Faustino, M.G.P.M.S. Neves, J.A.S. Cavaleiro, A. Almeida, Â. Cunha, J.P.C. Tome, Photochem. Photobiol. Sci., 2013, 12, 262-271. b) S. Silva, P.M.R. Pereira, P. Silva, F.A. Almeida Paz, M.A.F. Faustino, J.A.S. Cavaleiro, J.P.C. Tomé, Chem. Commun., 2012, 48, 3608.

OC5
Multidimensional optimization of pyranoxanthones with potential
antitumor activity
J. Soares,a C. Azevedo,
a,b A. Palmeira,
a D. Sousa,
a,c R. Lima,
a,c M. Pedro,
a
M. Vasconcelos,c C. Afonso,
a S. Reis,
d M. Pinto
a
aCEQUIMED-UP, Department of Chemistry, Laboratory of Organic and Pharmaceutical Chemistry,
Faculty of Pharmacy, University of Porto, Rua Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal; bDepartment of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 25,
5230 Odense, Denmark; cCancer Drug Resistance Group, Instituto de Patologia e Imunologia Molecular
da Universidade do Porto (IPATIMUP), Rua Dr. Roberto Frias s/n, 4200-465 Porto, Portugal; dREQUIMTE, Laboratory of Applied Chemistry, Faculty of Pharmacy, University of Porto, Rua Jorge
Viterbo Ferreira, 228, 4050-313 Porto, Portugal
Pyranoxanthones are a family of O-heterocycles that have a wide variety of biological
activities, particularly antitumor activity.1,2
Our group has been focusing on the synthesis of
a library of pyranoxanthone derivatives and two compounds (XC10 and XP13) emerged as
potential antitumoragents.2 Considering that on the pipeline that drives “hit-to-lead”
compounds to drug candidates, multidimensional optimization, which involves the control of
both pharmacodynamic and pharmacokinetic behaviors, is essential, it has been envisioned
for these “hit compounds (scheme 1).
In this work, we report the synthesis of XC10 and XP13 and seven analogues using
classical and non-classical methodologies. Moreover, their growth inhibitory activity in four
human tumor cell lines was evaluated. Among the several factors that affect
pharmacokinetics, the binding to plasma proteins is one of the most prominent. Therefore,
the interaction of XP13, XC10 and the synthesized analogues with human serum albumin
(HSA) was studied. The binding to HSA was evaluated by fluorescence quenching
technique and UV–Vis absorption derivative spectroscopy. The crucial distance between
the bound compound and an albumin tryptophan residue was investigated by Förster
resonance energy transfer technique.3 In order to shed some light on the binding of XC10
and analogues to HSA, an in silico study was also performed. Using AutoDockVina
program, molecular docking to a HSA crystal structure (pdb code: 2VUE) was performed,
and the ligand conformations and docking scores were analyzed.
The obtained results allowed us to guide the design of new drug candidates with better
pharmacodynamic and pharmacokinetic profiles.
Scheme 1: Multidimensional optimization of XC10 and XP13
Acknowledgements: FCT for financial support under the project CEQUIMED – PEst-OE/SAU/UI4040/2011 and for the grants of PTDC/SAU-FCT/100930/2008, SFRH/BD/41165/2007 and SRH/BPD/68787/2010. To FEDER funds and COMPETE program under the project FCOMP-01-0124-FEDER-011057 and also to U.Porto/Banco Santander Totta (PP_IJUP2011-58).
References: 1. Pinto, M.M.M., Castanheiro R.A.P., Curr. Org. Chem, 2009, 13, 1215. 2. a) Azevedo C.; Afonso C.; Sousa D.; Lima R.; asconcelos M.; Pedro M.; Barbosa J.; Corr a A.; Reis S.; Pinto M. Bioorg. Med. Chem. 2013, 21, 2941. b) Palmeira, A.; Paiva, A.; Sousa, E.; Seca, H.; Almeida, M.; Lima, R.; Fernandes, M.; Pinto, M.; Vasconcelos, M.Chem. Biol. & DrugDes. 2010, 76 (1), 43. 3. Bogdan M.; Pirnau A.; Floare C.; Bugeac C. J. Pharmaceut. Biomed. 2008, 47 (4-5), 981.

Flash Communications

FC1
Chemical structure and stability of microencapsulated anthocyanins
A. Fernandes, Nuno Mateus, Victor de Freitas
Centro de Investigação em Química (CIQ), Departamento de Química e Bioquímica Faculdade de
Ciências, Universidade do Porto, Rua do Campo Alegre, 687, 4169-007 Porto, Portugal.
The development of food colorants from natural sources has been globally increased.1 In
this context, anthocyanins which are natural, water soluble pigments are considered
suitable food pigments. Although it’s potential as colorant, their industrial application is
limited and a correct knowledge of the factors that govern anthocyanin’s color stability
seems to be decisive in terms of a putative application in food matrices.2 The fact that
anthocyanins co-exist in aqueous solution in a complex network of equilibrium species
depending on pH is an important factor related to the color displayed by these compounds.
In acidic media anthocyanins are stable while with the increase in the pH result in an
unstable blue quinoidal base. Some of the color stabilization strategies rely on the formation
of molecular complexes, for instance by encapsulation techniques in a carbohydrate matrix.
Maltodextrins with various average molecular weights belong to the most popular capsule
materials used in the food field and both hydrophobic and hydrophilic compounds have
been encapsulated in maltodextrins. The encapsulated anthocyanins with maltodextrin
result in a more stable product than anthocyanins alone that can be used readily as a food
ingredient.3; 4
The knowledge of the nature of intermolecular interactions between the species presented
in the carbohydrate-anthocyanin complexes is of fundamental importance in the
understanding of the factors that determine their stability. It is therefore essential to
elucidate the structure of anthocyanin-loaded particles and to clarify the mechanisms of
anthocyanin immobilization in the carbohydrate matrix. The encapsulated complexes
between cyanidin-3-O-glucoside (cy3glc) and maltodextrin (MDE) at different pHs are going
to be investigated using a NMR spectroscopy approach. Diffusion ordered NMR
spectroscopy (DOSY) and study of nuclear Overhauser effects (NOE) are going to be used
to determine the selective intermolecular interactions and structure of these complexes in
aqueous solution. Moreover the differences in thermal stability at three pH’s (pH 2, 3 and 5)
between free and encapsulated anthocyanins are going to be investigated.
Acknowledgements: This work received financial support from FEDER funds through COMPETE, POPH/FSE, QREN and FCT (Fundação para a Ciência e Tecnologia) from Portugal by one PhD scholarship (SFRH/BD/65350/2009) and through project PEst-C/QUI/UI0081/2011. To all financing sources the authors are greatly indebted. References: 1. Burin, V. M., Rossa, P. N., Ferreira-Lima, N. E., Hillmann, M. C. R., & Boirdignon-Luiz, M. T. (2011). Anthocyanins: optimisation of extraction from Cabernet Sauvignon grapes, microcapsulation and stability in soft drink. International Journal of Food Science & Technology, 46(1), 186-193. 2. Brouillard, R. (1982). In P. Markakis (Ed.). Anthocyanins as Food Colors. New York: Academic Press. 3. Robert, P., Gorena, T., Romero, N., Sepulveda, E., Chavez, J., & Saenz, C. (2010). Encapsulation of polyphenols and anthocyanins from pomegranate (Punica granatum) by spray drying. International Journal of Food Science & Technology, 45(7), 1386-1394. 4. Tonon, R. V., Brabet, C., & Hubinger, M. D. (2010). Anthocyanin stability and antioxidant activity of spray-dried açai (Euterpe oleracea Mart.) juice produced with different carrier agents. Food Research International, 43(3), 907-914.

FC2
Enzymatic Resolution of Secondary Alcohols in Miniemulsion Media
C. M. Altas,a L. P. Fonseca,
a,b N. M. T. Lourenço
a
aIBB-Institute for Biotechnology and Bioengineering, Centre for Biological and Chemical Engineering,
b
Department of Bioengineering, Instituto Superior Técnico, Av. Rovisco Pais, 1, 1049-001 Lisboa,
Portugal
Over the last years we have been pursuing the development of attractive, competitive and
more environmentally friendly processes for the enzymatic resolution of secondary alcohols.
In this line, our effort has been made on the development of new strategies for resolution of
free secondary alcohols, namely by the use of more sustainable acylating agents.1 More
recently, we become interested on the use of miniemulsions as sustainable media for the
enzymatic resolution of secondary alcohols.
Miniemulsions are characterized by a two phase system in which stable nanodroplets,
between 20 and 500nm, of an organic phase are dispersed in a second continuous
aqueous phase.2,3
These nanodroplets are stabilized against coalescence by the addition of
appropriate surfactants, which provide either electrostatic or steric stabilization. These
features make miniemulsions a very appealing media for different chemical reactions.
Herein, a low temperature miniemulsion media for the preparative enzymatic resolution of 1-
phenylethanol is described (Scheme 1). The media is characterized by the use of a
miniemulsion that allows the enantioselective enzymatic hydrolysis of 1-phenylethyl
alkanoates at low temperatures. The central feature of this methodology is the low
temperature miniemulsion system that drives the reaction equilibrium by the precipitation of
one of the products. The preparative miniemulsion enzymatic reaction of 1-phenylethyl
alkanoates at 4ºC allowed the preparation of both free enantiomers in good yields and
enantiomeric excess.
Scheme 1: Miniemulsion methodology for the enzymatic resolution of 1-phenylethanol.
Acknowledgements: We thank Fundação para a Ciência e Tecnologia (POCI 2010) and FEDER (SFRH/BD/41175/2007 and PTDC/QUI-QUI/119210/2010) for financial support. We thank Novozymes for the generous enzyme gift. References: 1. Lourenço N.M.T., Afonso C.A.M. Angew. Chem. Int. Ed., 2007, 46, 8178; Monteiro, C.A.; Lourenço N. M. T.; Afonso C.A.M., Tetrahedron: Asymmetry, 2010, 952-956; Lourenço N.M.T, Monteiro C.M., Afonso C.A.M, Eur. J. Org. Chem., 2010, 6938–6943. 2. Landfester K., Annu Rev Mater Res, 2006, 36, 231-279. 3. Solans C., Izquierdo P., Nolla J., Azemar N., Garcia-Celma M.J., Curr. Opin. Colloid Interface Sci., 2005, 10, 102-110.
NaO
O
8
NaOO
8
ONaO
8
ONa
O8
NaOO
8
NaO
O 8
ONaO
8
ONa
O
8
E
E
E
E
E
E
E
E
ONaO
8 E
H2O
H2O
H2O
H2O
H2O
H2O
H2O
H2O
H2O
Ph O
O
n
Ph O
O
n Ph OH(R)(S)
rac
HO
O
n

FC3
The acid-catalysed reaction of 2-hydroxychalcones with carbon
acids
Tatiana A. Dias, Marta Costa, M. Fernanda Proença
University of Minho, Centre of Chemistry, School of Sciences, Campus of Gualtar, Braga, 4710-057,
Portugal
Chalcones have been used as important precursors of the chromene scaffold, present in a
wide variety of compounds including natural products. Important pharmacological properties
have been identified and this nucleus has been an inspiration for the synthesis of
analogues, since some derivatives exhibit remarkable biological activities namely
anticancer, anti-inflammatory, antioxidant, antitubercular, anti-microbial and anti-HIV.1, 2
Chalcones can be prepared by the Aldol condensation of a ketone and an aldehyde, a
reaction that is usually performed in aqueous basic media. The condensation of an active
methylene compound with salicylaldehydes in the presence of base catalysis is also a well-
known reaction that directly leads to the formation of 2-imino or 2-oxo-2H-chromenes.3, 4
Previous experimental results in our research group, on the reactivity of α,β-unsaturated
carbonyl compounds, revealed that they react with carbon acid derivatives, leading to
distinct chromene-based structures. The careful control of experimental conditions and the
nature of the carbon acid proved to be crucial for the product isolated.
In this work, a study on the reactivity of the 2-hydroxychalcones 1 with different carbon acid
derivatives 2, in the presence of acid catalysis (Scheme 1), will be presented. A proposal
for the mechanistic pathways leading to the formation of the corresponding products 3 and
4 will also be discussed.
O
OH
OCH3R1
O
R2 H
1 24
O
X
Y
CN
3
X and Y, from COR1
R2=H R2=CN
H
O
O
O
Scheme 1: Reaction of chalcone 1 with carbon acids 2.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support through the Portuguese NMR network (RNRMN), the Project F-COMP-01-0124-FEDER-022716 (ref. FCT PEst-C/QUI/UI0686/2011) FEDER-COMPETE, a PhD grant assigned to Tatiana Dias (SFRH/BD/88245/2012), a BPD grant awarded to Marta Costa (SFRH/BPD/79609/2011) and the University of Minho. References: 1. Sandhya B., Giles D., Mathew V., Basavarajaswamy G., Abraham R., Eur. J. Med. Chem. 2011, 46, 4696–4701. 2. Kempen I., Hemmer M., Counerotte S., Pochet L., Tullio P., Foidart J., Blacher S., Noel A., Frankenne F., Pirotte B., Eur. J. Med. Chem. 2008, 43, 2735–2750. 3. Dias T., Proença F., Tetrahedron Lett. 2012, 53, 5235-5237. 4. Proença F., Costa M., Tetrahedron 2010, 66, 4542–4550.

FC4
Hit-to-Lead Optimization of kojic Acid Derivatives toward COPD
Drug Discovery
Susana D. Lucas, L. A. R. Carvalho, H. F. Correia, Lídia M. Gonçalves, Rita C. Guedes,
Rui Moreira
iMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal.
Combination of emphysema and chronic asthmatic bronchitis — a deadly duo known as
chronic obstructive pulmonary disease (COPD) affects millions of people worldwide that find
distressingly difficult to breathe. The primary cause of COPD is tobacco smoke but other
risk factors include air pollution or occupational dusts and chemicals. COPD is not curable
and the available treatment only helps to control its symptoms. Therefore, COPD-related
deaths are projected to increase dramatically, being COPD predicted by WHO to become
the third leading cause of death by 2030.1 Human Neutrophil Elastase (HNE) is a serine
protease which plays a major role through COPD inflammatory process wherein due to an
imbalance between protease and anti-protease, an excess of HNE is produced hydrolyzing
elastin, the structural protein which gives the lungs their elasticity.2
Recently we developed a virtual screening protocol toward HNE hit generation that led us to
the kojic derivative 1 (Figure 1),3 which is a 18 µM acyl-enzyme inhibitor that showed to be
selective for HNE when compared with parent proteases. Hence we envisaged a lead
optimization campaign toward an activity and drugability gain along the kojic acid scaffold,
which was the aim of the present work (Figure 1). Kojic acid derivatives were synthesized
with different small and hydrophobic ester moieties as it is preferred for HNE S1 pocket
recognition, while we introduced several thioether-linked building blocks as recognition
pattern on the opposite counterpart of the acylating function. On the other hand we studied
the effect of having a pyrone versus an aryl--pyridone scaffold. Lead optimization protocol
allowed the synthesis of very promising compounds with activities in the nM range with
good selectivity profiles.
Figure 1
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support, Pest-OE/SAL/UI4013/2011, SFRH/BPD/64265/2009 (SDL). References: 1. a) source: http://www.who.int/respiratory/copd/en. b) Brody H. (Supp. Editor) Nature 2012, 489, S1. 2. Lucas S.D.; Costa E., Guedes R. C.; Moreira, R. Med. Res. Rev. 2013, 33, S1, E73. 3. Lucas, S.D.; Gonçalves, L.M.; Cardote, T.A.F.; Correia, H.F.; Moreira, R.; Guedes, R.C.; Med. Chem. Commun., 2012, 3, 1299.

FC5
New antimicrobial structures with anti-ageing potential: an efficient
synthesis towards 2-deoxy glycosides and their thio analogues
C. Dias,a Alice Martins,
a R. A. Staniford,
b Amélia P. Rauter
a
aGrupo da Química dos Glúcidos, Centro de Química e Bioquímica, Departamento de Química e
Bioquímica, Faculdade de Ciências da Universidade de Lisboa, Ed C8, Piso 5, Campo Grande, 1749-
016 Lisboa, Portugal; bKrebs Institute, Department of Molecular Biology and Biotechnology, University of
Sheffield, S10 2TN, Sheffield, UK
Sugar-based surfactants are a very appealing class of compounds due to their low toxicity
and their synthesis from renewable resources. Alkyl deoxy-arabino-hexopyranosides with a
potent antimicrobial activity in several Bacillus species have been previously described by
our research group.1 Aiming at a better insight to the relationship between these lead
structures and their bioactivity, new alkyl 2-deoxyglycosides as well as their thio analogues
were synthesized. Their preparation was accomplished by reaction of 1,5-anhydro-3,4-di-O-
acetyl-2,6-dideoxy-L-arabino-hex-1-enitol (3,4-di-O-acetyl-6-deoxy-L-glucal) or its
benzylated analogue with the corresponding alcohol or thiol, using triphenylphosphane
hydrobromide (TPHB) as catalyst, followed by deprotection (Scheme 1). The previously
reported reaction conditions2 were optimized and microwave assisted reactions were run for
the first time to synthesize the deoxy thio glycosides. 3,4-Di-O-benzyl-6-deoxy-L-glucal
revealed to be the most efficient starting material, as it diminished the formation of the
Ferrier secondary product, and its synthesis will also be presented.
The antimicrobial activity of these newly synthesized compounds was studied on Bacillus
species, namely B. cereus and B. anthracis, and some of them showed a remarkable and
reproducible activity against the latter.
Moreover, infections in elderly populations are known to be not only more frequent but also
more severe, being this susceptibility often related to neurodegenerative diseases such as
dementia and Alzheimer’s.3 A preliminary study assessing the anti-amyloidogenic potential
of these 2-deoxy glycosides has previously demonstrated that these compounds interact
with soluble cystatin B, opening a new window into a new line of investigation, pertaining to
antibiotic compounds showing neuroprotective activity. An investigation to check the ability
of these new structures to protect cells against Aβ induced toxicity is currently on going and
will also be presented.
Scheme 1: Synthesis of alkyl deoxy-arabino-hexopyranosides using TPHB as catalyst.
Acknowledgements: The authors thank Fundação para a Ciência e Tecnologia for financial support and for the research grant of Catarina Dias (FRH/BDE/51998/2012). Thanks are also due to QREN – COMPETE program for the support of FACIB project (QREN – SI I&DT Co-Promoção Projecto nº 21547) and to Fundação para a Ciência e Tecnologia for the PhD grant (SFRH/BD/78236/2011) and for financial support (PEst-OE/QUI/UI0612/2013).
References: 1. a) Rauter A. P.; Lucas S.; Almeida T.; Sacoto D.; Ribeiro V.; Justino J.; Neves A.; Silva F. V. M.; Oliveira M. C.; Ferreira M. J.; Santos M. S.; Barbosa E.; Carbohydr. Res. 2005, 340, 191.; b) Silva F.; Goulart M.; Justino J.; Neves A.; Santos F.; Caio J.; Lucas S.; Newton A.; Sacoto D.; Barbosa E.; Santos M. S.; Rauter A.P.; Bioorg. Med. Chem. 2008, 16, 4083.
2. Rauter A. P.; Almeida T.; Vicente A. I.; Ribeiro V.; Bordado J. C.; Marques J. P.; Ribeiro F. R.; Ferreira M. J.; Oliveira C.; Guisnet M. Eur. J. Org. Chem. 2006, 2429. 3. Gavazzia G.; Krause K.-H. Lancet Infec. Dis. 2002, 2 (11), 659.

FC6
Synthesis of N-substituted 1,2-dihydropyridines by 6π-
electrocyclisation of (E,E)-cinnamylidene acetophenones
Pedro A. M. M. Varandas, Eduarda M. P. Silva, Artur M. S. Silva
Department of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal
From the five theoretically possible isomeric dihydropyridines, the 1,2- or the 1,4-dihydro
structure are the most commonly found in known dihydropyridines.1 The 1,4-
dihydropyridines are known to possess a wide range of biological and pharmacological
actions namely as calcium-channel modulating agents in the treatment of cardiovascular
disease; as multidrug-resistance-reversing agents in cancer chemotherapy and as
antimycobacterial and anticonvulsant agents.2 The less studied 1,2-dihydropyridines consist
of an important scaffold for the preparation of 2-azabicyclo[2.2.2]octanes (isoquinuclidines).3
The isoquinuclidine ring system is widely found in natural products such as ibogaine and
dioscorine alkaloids, which have a large spectrum of interesting biological properties.3
Because of the lack of general methods for the regioselective synthesis of highly
functionalised 1,2-dihydropyridines, their potential remains largely unexplored.4 Our group
has recently started a project considering the synthesis of this templates using (E,E)-
cinnamylidene acetophenones 1 (Scheme 1) as versatile starting materials. (E,E)-
cinnamylidene acetophenones 1 are a major group of α,β,γ,δ-diunsaturated ketones, widely
used in a variety of synthetic transformations namely by our research group. In the present
communication we describe a novel one-pot synthetic route to prepare N-substituted 1,2-
dihydropyridines 3 using compound 1 as starting material and several primary amines. This
reaction proceeds by forming a ketimine intermediate 2 that through a 6π-electrocyclisation
afforded the desired product 3 in moderate yields.
Scheme 1: Synthesis of N-substituted 1,2-dihydropyridines 3 via a 6π-electrocyclisation of ketimine2.
Acknowledgements: Thanks are due to the University of Aveiro, Portuguese Foundation for Science and Technology (FCT), European Union, QREN, FEDER and COMPETE for funding the QOPNA research unit (project PEst-C/QUI/UI0062/2011). E. M. P. Silva is grateful to FCT for a Post-Doc grant (SFRH/BPD/66961/2009). References: 1. (a) Kuthan J.; Kurfürst A. Ind. Eng. Chem. Prod. Res. Dev. 1882, 21, 191. (b) Eisner U.; Kuthan J. Chem. Rev. 1972, 72, 1. 2. Edraki N.; Mehdipour A. R.; Khoshneviszadeh M.; Miri R. Drug Discov. Today 2009, 14, 1058. 3. Faruk-Khan M. O.; Levi M. S.; Clark C. R.; Ablor-Deppey S. Y.; Law S.-L.; Wilson N. H.; Borne R. F. Isoquinuclidines: A Review of Chemical and Pharmacological Properties. In Stud. Nat. Prod. Chem., 1st ed., Vol. 34; Atta-ur-Rahman, Ed.; Academic Press-Elsevier, 2008, 753. 4. Silva E. M. P.; Varandas P. A. M. M.; Silva A. M. S. Synthesis 2013, submitted.

FC7
Gold (I) catalyzed intermolecular cycloadditions of allenamides: a
simple route to small and medium sized carbocycles
Hélio Faustino,a José L. Mascareñas,
a Fernando López
a,b
aCentro Singular de Investigación en Química Biológica y Materiales Moleculares (CIQUS) and
Departamento de Química Orgánica, Universidad de Santiago de Compostela. 15782, Santiago de
Compostela, Spain; bInstituto de Química Orgánica General, CSIC, 28006, Madrid, Spain.
In recent years there have been extraordinary advances in the development of new Au-
catalyzed processes. In this context, our group demonstrated in 2009 the possibility of using
allenes in intramolecular (4 + 2) and (4 + 3) cycloadditions of allenedienes, including their
enantioselective versions.1 Herein, we describe our efforts in the development of
intermolecular Au-catalyzed cycloadditions with allenic scaffolds. In particular, we will
demonstrate that the use of allenamides, a particularly accessible and versatile type of
allenic scaffold, allowed the discovery of highly selective and even enantioselective gold-
catalyzed (4 + 2) cycloadditions to 1,3-dienes.2 The detection of minor side products
resulting from a (2 + 2) cycloaddition between the allenamide and one of the double bonds
of the diene, prompted us to specifically pursued the development of a gold-catalyzed
intermolecular (2 + 2) cycloaddition, which could be achieved using appropriate alkenes and
a phosphite-gold catalyst.3 Finally, very recent examples of a simple and highly versatile
cascade cycloaddition between allenamides and carbonyl-tethered alkenes, including
several enantioselective examples, will be also described. This method enables a
straightforward and highly efficient entry to oxa-bridged seven-, eight- and even nine-
membered rings.4
Acknowledgements: HF thank the Fundação para a Ciência e Tecnologia for a doctoral fellowship SFRH/BD/60214/2009. References: 1. (a) López, F.; Mascareñas, J. L. Beilstein J. Org. Chem. 2011, 7, 1075; (b) Alonso, I.; Trillo, B.; López, F.; Montserrat, S.; Ujaque, G.; Castedo, L.; Lledós, A.; Mascareñas, J. L. J. Am. Chem. Soc. 2009, 131, 13020-13030. (c) Alonso, I.; Faustino, H.; López, F.; Mascareñas, J. L. Angew. Chem. Int. Ed. 2011, 11496-11500 2. (a) Faustino, H.; López, F.; Castedo, L.; Mascareñas, J. L. Chem. Sci. 2011, 2, 633. (b) Francos, J.; Grande-Carmona, F.; Faustino, H.; Iglesias-Sigüenza, J.; Díez, E. J. Am. Chem. Soc. 2012, 134, 14322. 3. Faustino, H.; Bernal, P.; Castedo, L.; López, F.; Mascareñas, J. L. Adv. Synth. Catal. 2012, 354, 1658. 4. Faustino, H.; Alonso, I.; Mascareñas, J. L.; López, F. Angew. Chem. Int. Ed. 2013, DOI: 10.1002/anie.201302713 – Early view.

FC8
Regio- and Enantioselective Cyclobutene Allylations
Supaporn Niyomchon, Davide Audisio, Marco Luparia, Nuno Maulide
Max-Planck-Institut für Kohlenforschung – Kaiser-Wilhelm-Platz , D-45470 Mülheim an der Ruhr,
Germany
Palladium catalysed asymmetric allylic alkylation (AAA) is a powerful synthetic method for
the preparation of optically active compounds.1 Recently, our laboratory has focused on the
palladium-catalysed reactions of bicyclic lactone 1 with stabilized (“soft”) nucleophiles to
generate highly functionalised cyclobutenes with impressive diastero- and
enantioselectivities.2,3
We have now investigated the behaviour of this system in the
presence of nonstabilized (“hard”) nucleophiles. In this presentation, we report our
preliminary results on the catalytic, asymmetric regioselective allylation of lactone 1 with
allyl boronates 2 (Scheme 1) as well as exploratory mechanistic studies.4
Scheme 1: Pd-catalyzed allylation of lactones 1 with allyl pinacol boranes 2
Acknowledgements: We are grateful to the Max-Planck-Society and the Max-Planck-Institut für Kohlenforschung for generous support of our research programs. This work was funded by the Deutsche Forschungsgemeinschaft (Grant MA-4861/3-1) and the European Research Council. References: 1. F. Frébault, M. Luparia, M. T. Oliveira, R. Goddard, N. Maulide, Angew. Chem. Int. Ed., 2010, 49, 5672. 2. M. Luparia, M. T. Oliveira, D. Audisio, F. Frébault, R. Goddard, N. Maulide, Angew. Chem. Int. Ed., 2011, 50, 12631. 3. D. Audisio, M. Luparia, M. T. Oliveira, D. Klütt, N. Maulide, Angew. Chem. Int. Ed., 2012, 51, 7314. 4. S. Niyomchon, D. Audisio, M. Luparia, N. Maulide, Org. Lett., 2013, 15, 2318.

FC9
Two new monoterpene indole alkaloids
from Psychotria umbellata Vell.
Luiz C. Klein-Júnior,a Vitor A. Kerber,
a Carolina dos Santos Passos,
a Jean-Charles Quirion,
b
Amélia T. Henriquesa
aLaboratório de Farmacognosia, Programa de Pós-Graduação em Ciências Farmacêuticas, Faculdade de Farmácia da Universidade Federal do Rio Grande do Sul, Av. Ipiranga, 2752 – 90 610-000, Porto Alegre, Brasil;
bLaboratoire d’Hétérochimie Organique, IRCOF-INSA de Rouen, Pl. E. Blondel, BP 08,
76131 Mont-Saint-Aignan cedex, France.
Psychotria genus (Rubiaceae) is divided in three subgenera: Psychotria (pantoprical),
Tetramerae (species of Africa and Madagascar) and Heteropsychotria (neotropical). The
latest subgenus is chemically characterized by monoterpene indole alkaloids (MIAs). In a
previous study, it was reported the isolation of an unusual MIA from P. umbellata, named
psychollatine (1), formed by the condensation of a geniposide derivative with triptamine.1
Considering it, in the present study it is reported the isolation and characterization of two
new psychollatine derivatives from P. umbellata. The ethanol extract of the leaves of the
plant was dissolved in HCl 2% and partitioned with CH2Cl2. Later, the acid phase was
basified and re-partitioned with CH2Cl2, resulting in the alkaloid fraction (766 mg). This
fraction was submitted to VCC using CHCl3/MeOH as eluent. One of the subfraction was
purified by PTLC using CHCl3/MeOH (85:15) in the presence of NH3 vapor as eluent,
resulting in compounds 2 (43 mg) and 3 (29 mg). Both 2 and 3 displayed pseudo-molecular
ions at m/z [M + H]+ 587 in the CIMS spectra, matching with C30H38N2O10.
13C NMR (100
MHz) confirmed the presence of 30 carbon atoms and the proton-bearing carbons were
assigned from HMQC spectra. Both alkaloids displayed a 1H NMR (400 MHz) pattern of
tetrahydro-β-carboline glucosidic MIA, similarly to 1. Comparing 2 and 3 to 1, is verified that
their mass spectra are compatible with an additional –C3H6O substituent. For 2 and 3, the 1H-
1H COSY spectrum showed correlations of H-25 with H-24a, H-24b, and 3H-26. Taking
together these 1H-
1H COSY correlations with the
13C NMR chemical shifts displayed by C-
25, and the multiplicity of the 1H NMR signals attributed to H-25, it is possible to assign that
C-25 is bound to an oxygen, a methyl, and a methylene groups, consisting of an
isopropanol moiety. Moreover, based on the analyses of the 1H and
13C NMR spectra, it is
possible to suggest that the C-24 is bound to the N-4 of the 1 nucleus in both compounds.
In order to confirm the structures attributed for alkaloids 2 and 3, these two compounds
were synthesized with racemic propylenoxide and psychollatine. The two resulting
compounds displayed the same Rf on TLC that products 2 and 3 and similar 1H NMR
spectra. In addition, to find the relative configuration at C-25, the synthesis was performed
using the S-isomer of propylenoxide, demonstrating that compound 2 is N4-[1-(2-α-
hydroxypropyl)]-psychollatine and compound 3 is N4-[1-(2-β-hydroxypropyl)]-psychollatine.
NH
NH
O
COOMe
GlcOH
H
HNH
N
O
COOMe
GlcOH
H
HCH3
R
1 2 R = -OH3 R = -OH
3
2425
26
Acknowledgements: We thank to CNPq, CAPES and FAPERGS for their financial support. References: 1. Kerber, V. A.; Passos, C. S.; Verli, H.; Fett-Neto, A. G.; Quirion, J. P.; Henriques, A. T. J. Nat. Prod. 2008, 71, 697.

FC10
Synthesis, photophysical and photodynamic activities of
amphiphilic phthalocyanine-cyclodextrin conjugates
Leandro M. O. Lourenço,a Patrícia M. R. Pereira,
a Elisabete Maciel,
a
Maria R. M. Domingues,a Rosa Fernandes,
b Maria G. P. M. S. Neves,
a
José A. S. Cavaleiro,a João P. C. Tomé
a
aQOPNA and Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal;
bLaboratory of
Pharmacology and Experimental Therapeutics, IBILI, Faculty of Medicine, University of Coimbra, 3000-548 Coimbra, Portugal
Phthalocyanines (Pcs) and cyclodextrins (CDs) have been intensively studied due to their
applications in many scientific fields, being their use as photosensitizers (PSs) in
photodynamic therapy (PDT) one of the most promising.1 PDT uses a blend of visible light,
oxygen and a PSto cause an efficient and selective methodology for the treatment of
several diseases.2 Three novel Pcs conjugated with α-, β- and γ-CDs have been prepared
and assessed for application as new PS agents (Figure 1) by photophysical, photochemical
and in vitro photobiological studies. The simple nucleophilic substitution of two β-fluorine
atoms on the hexadecafluorophthalocyaninato zinc(II) (PcF16) enabled the preparation of
these hybrids. The conjugates Pc-α-CD and Pc-γ-CD demonstrated high efficiency to
interact with human serum albumin, to generate singlet oxygen and were highly phototoxic
against UM-UC-3 human bladder cancer cells. The lower photodynamic activity of the Pc-β-
CD can be attributed to its higher aggregation tendency, leading to a lower efficiency to
generate reactive oxygen species inside the cells. The promising photoactivity of Pc-α-CD
and Pc-γ-CD ensure the potential candidacy as PDT drugs.
Figure 1: Representation of the Pc-CD dyads 1-3.
Acknowledgements: Thanks are due to the University of Aveiro, IBILI, FCT (Portugal), European Union, QREN, FEDER and COMPETE for funding QOPNA Research Unit (project PEst-C/QUI/UI0062/2011), the Portuguese National NMR Network, and the projects PTDC/CTM/101538/2008 and PTDC/QUI/65228/2006. Leandro M.O. Lourenço (SFRH/BD/64526/2009) and Patrícia M. R. Pereira (SFRH/BD/85941/2012) thanks FCT for their PhD grants. References:
1. a) Silva S.; Pereira P. M. R.; Silva P.; Paz F. A. A.; Faustino M. A. F.; Cavaleiro J. A. S.; Tomé J. P. C., Chem. Commun., 2012, 48 (30), 3608. b) Pereira J. B.; Carvalho E. F. A; Faustino M. A.; Neves M. G. P. M. S.; Cavaleiro J. A. S.; Gomes N. C. M.; Cunha A.; Almeida A.; Tomé J. P. C. Photochem. Photobiol., 2012, 88(3), 537. 2. Senge M. O.; Radomski M. W. Photodiagn. Photodyn., 2013, 10 (1), 1.

FC11
Iminoboronates: A New Strategy for Reversible Protein Modification
Pedro M. S. D. Cal,a J. Vicente,
a Luís F. Veiros,
b C. Cordeiro,
c Pedro M. P. Góis
a
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal; bCQE, Departamento de Engenharia Química e Biológica Complexo I, Instituto Superior
Técnico, 1049-001 Lisboa, Portugal; cCentro de Química e Bioquímica, Departamento de Química e
Bioquímica, Faculdade de Ciências da Universidade de Lisboa, 1749-016 Lisboa Portugal
Proteins are biomolecules that carry out the majority of cell’s functions, acting as their
structural blocks, as catalysts in all processes concerning cellular metabolism, and as
regulators of cell cycle, differentiation, growth, division, motility, death, among others.1
Furthermore, therapeutic peptides or proteins show crucial importance in some pathologies,
namely, insulin, GNRH/LHRH agonists, sandostatin, calcitonin or platelet aggregation
inhibitors. However, since these molecules are unstable to some conditions, peptide and
protein enhancement technologies are being developed in order to have more suitable
delivery systems.2
Selective modification of native proteins is one of the approaches to
change the protein’s properties without shifting its function or natural structure.
We present a new strategy to modify the lysine’s ε-amino group and the protein’s
N-terminal, based on the formation of stable iminoboronates in aqueous media. The
modification of these amine groups can be reverted upon the addition of fructose,
dopamine, or glutathione.3
Moreover, derivatives of these modifying agents were
synthesized in order to confer some biological properties that the biomolecules didn’t
possess naturally, namely, enhanced pharmokinetics (PEGylation), fluorescence or even
the ability of conjugation with drugs (Figure 1).
Figure 1: Selective modification of native proteins using derivatives of 2-carbonylbenzeneboronic acid (R=H – formyl derivative, R=CH3 – acetyl derivative)
Acknowledgements: We thank the Fundação para a Ciência e a Tecnologia for financial support from projects and scholarships (PEst-OE/SAU/UI4013/2011; PEst-OE/QUI/UI0100/2011; PTDC/QUI-QUI/118315/2010, SFRH/BD/72376/2010). References: 1. Graaf, A. J.; Kooijman, M.; Hennink, W. E.; Mastrobattista, E. Bioconjugate Chemistry 2009, 20 (7), 1281. 2. Pichereau, C.; Allary, C., European Biopharmaceutical Review 2005, Winter 05’ issue 3. Cal, P. M. S. D.; Vicente, J. B.; Pires, E.; Coelho, A. V.; Veiros, L. F.; Cordeiro, C.; Góis, P. M. P. J. Am. Chem. Soc. 2012, 134 (24), 10299.
X being: Bioorthogonal species, Fluorescent probes, PEGylating agents and Drugs

FC12
New 2-amino-4-functionalized Cyclopentenones from 2-furaldehyde
via a One-pot Method
J. P. M. Nunes,a,b
Stephen Caddick,b Carlos. A. M. Afonso
a,c
aCQFM - Centro de Química-Física Molecular, IN - Institute of Nanosciences and Nanotechnology,
Instituto Superior Técnico, Av. Rovisco Pais P-1049-001 Lisboa, Portugal; bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK;
ciMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal
Functionalized cyclopentenones are amongst the most valuable precursors available for
synthesis of active biomolecules and derivates. In particular, diamino cyclopentenones are
useful for the synthesis of Agelastatin A1 whereas 2-aza-cyclopentenones can be useful for
the synthesis of (–)-Cephalotaxine,2 Palau’amine
3 and (+)-Nakadomarin.
4 However, most
syntheses towards functionalized 2-aza-cyclopentenones remain multi-step strategies.
Thus, we were interested in reports of a one step transformation in the presence of Lewis
acids that has afforded trans-diamino cyclopentenones from 2-furaldehyde in high yields.1b
This method has been expanded to obtain 4-aminocyclopentenones via the aza-Piancatelli
rearrangement.5
We have reported6 the conversion of 2-furaldehyde into bifunctionalized cyclopentenones
via 1,4-addition of nucleophiles to intermediate trans-diamino cyclopentenones, coupled
with β-elimination. Our studies have established a new one-pot method that afforded a
variety of functionalized 2-amino-4-functionalized cyclopentenones with a variety of
nucleophiles comprising mostly thiols as well as amines and alkoxides with yields in the 60-
80% range (Scheme 1). Further enamine modification was also achieved under mild
conditions.
Scheme 1: Synthesis of new 2,4-bifunctionalized cyclopentenones from 2-furaldehyde. Acknowledgements: We thank the Fundação para a Ciência e a Tecnologia and FEDER (Ref. SFRH/BD/31678/2006 and POCI/QUI/56582/2004) References: 1. a) Davis F. A.; Deng J. Org Lett. 2005, 7 (4), 621-623. b) Li S. W.; Batey R. A. Chem. Commun. 2007, 3759-3761. 2. Berhal F.; Pérard-Viret J.; Royer J. Tetrahedron: Asymmetry 2010, 21, 325–332. 3. Namba K.; Kaihara Y.; Yamamoto H.; Imagawa H.; Tanino K.; Williams R. M.; Nishizawa M. Chem. Eur. J. 2009, 15, 6560–6563. 4. Inagaki F.; Kinebuchi M.; Miyakoshi N.; Mukai C. Org. Lett. 2010, 12 (8), 1800-1803. 5. Veits G. K.; Wenz D. R.; deAlaniz J. R. Angew. Chem. Int. Ed. 2010, 49, 9484 –9487. 6. Nunes J. P. M.; Afonso, C. A. M.; Caddick, S., RSC Adv. 2013, accepted.

FC13
Organocatalytic Asymmetric Synthesis of
Cyclopropylphosphonates
A. M. Faísca Phillips, M. T. Barros, A. I. R. Reis
Departamento de Química, Faculty of Sciences and Technology, Universidade Nova de Lisboa, Campus
de Caparica, Quinta da Torre, 2829-516 Caparica, Portugal
Phosphonates have a wide range of applications in medicine, agriculture and materials
science. Their bioactivity derives from their close structural similarity to the phosphates that
occur widely in living organisms and to carboxylic acids of which they are isosters. If there is
a chiral centre present in the molecule, its configuration is often critical for activity. There are
a few cyclopropylphosphonates with proven biological activity: insecticidal, anti-malarial,
anti-viral, enzyme inhibitors and glutamate metabotropic receptor agonists.
Aminocyclopropylphosphonates are constrained analogues of amino acids, and have been
used for the synthesis of interesting peptidomimetics. Some aminocyclopropylphosphonates
are also known to be potent inhibitors of HCV NS3 protease, a promising target for therapy
against the hepatitis C virus, which presently is estimated to infect 170 million people
worldwide.1 As a follow-up on our interest in developing chiral methodology for the synthesis
of biologically active phosphonates,2 we have now developed a novel organocatalytic
method to synthesize chiral -cyclopropylphosphonates based on a domino process. In this
communication we present the results obtained and preliminary results on synthetic
applications of the new methodology.
Acknowledgements: A. M. Faísca Phillips thanks Fundação para a Ciência e Tecnologia for financial support. References:
1. a) Barros M. T.; Faísca Phillips A. M. Eur. J. Org. Chem., IYC Special Issue: Women in Chemistry 2011, 4028; b) Faísca Phillips A. M.; Barros M. T. Org. Biomol. Chem., 2012, 10, 404. 2. Pyun H. J.; Chaudhary K; Somoza J. R.; Sheng X. C.; Kim C. U. Tetrahedron Lett. 2009, 50, 3833.

FC14
Catalytic Asymmetric Benzidine Rearrangement
Chandra Kanta De, Fabio Pesciaioli, Benjamin List*
Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr (Germany)
The [3,3]-diaza Cope rearrangement is a powerful synthetic motif that can be utilized to
generate a C–C-bond at the expense of a N-N-bond. It forms the basis of important and
fundamental acid catalyzed transformations such as the Fischer indolization and the
benzidine rearrangement (Figure 1).1 Recently, our group has established the first catalytic
asymmetric variant of the Fischer indolization. In light of the mechanistic similarities
between the two reactions, we set up a program towards developing an asymmetric
benzidine rearrangement. Such a transformation may find utility in the synthesis of highly
useful chiral biaryls such as 1,1´-binaphthyl-2,2´-diamine (BINAM).
A Brønsted acid catalyzed asymmetric benzidine rearrangement was developed, providing
different electronically and structurally diverse axially chiral 2,2´-binaphthyl diamine (BINAM)
derivatives with high enantioselectivity.2
Figure 1: The diaza Cope rearrangement and its synthetic utilization.
Acknowledgements: We gratefully acknowledge generous financial support from the Max-Planck-Society and the Alexander von Humboldt Foundation (fellowship for C.K.D). References:
1. S. Müller, M. J. Webber, B. List, J. Am. Chem. Soc. 2011, 133, 18534–18537; b) A. Martínez, M. J. Webber, S. Müller, B. List, Angew. Chem. Int. Ed. 2013, 52, 1–5. 2. C. K. De, F. Pesciaioli, B. List, Angew. Chem. Int. Ed. 2013, in press.

FC15
Novel synthesis of dipyrromethanes via hetero-Diels-Alder reaction
of azo- and nitrosoalkenes with pyrrole
Nelson A. M. Pereira,a Susana M. M. Lopes,
a Américo Lemos,
b Arménio C. Serra,
a
Teresa M. V. D. Pinho e Meloa
aDepartment of Chemistry, University of Coimbra, 3004-535 Coimbra, Portugal,
bCIQA, FCT, Universityof
Algarve, Campus de Gambelas, 8005-139 Faro, Portugal
Porphyrins and 4,4-difluoro-4-bora-3a,4a-diaza-s-indacenes (BODIPY) are a class of
compounds with great potential, namely as sensitizers in multiple applications.1
Dipyrromethanes are important building blocks in the synthesis of these compounds. This
has led to an increasing interest in developing new synthetic methods to obtain novel meso
substituted dipyrromethanes.2 Generally, acids or Lewis acids are necessary to catalyze the
5-substituted dipyrromethane synthesis. However, polymerization of pyrrole and the
formation of by-products require carefully controlled reaction conditions. In this
communication, we will present a new acid free catalyzed one-pot synthetic strategy for 5-
substituted dipyrromethanes (e.g compounds 6a and 6b, Scheme 1). Hetero-Diels-Alder
reactions of azoalkenes and nitrosoalkenes are extremely important in preparing
heterocyclic compounds containing nitrogen such 1,2-oxazines, pyridazines
andisoxazolines.3
Herein, we present a novel bis-hetero-Diels-Alder reaction with azo- and
nitrosoalkenes, giving meso functionalized dipyrromethanes. Details of this synthetic
strategy will be disclosed.
Scheme 1: Synthetic route to 5-substituted dipyrromethanes from hydrazones or oximes and pyrrole.
Acknowledgements: Thanks are due to FCT (Grant: SFRH/BD/61573/2009; Project: PEst-C/QUI/UI0313/2011), FEDER, COMPETE and QREN for financial support. References: 1. (a) Li L.-L.; Diau E. W.-G. Chem. Soc. Rev. 2013, 42, 291; (b) Ethirajan M.; Chen Y.; Joshi P.; Pandey R. K. Chem. Soc. Rev. 2011, 40, 340; (c) Ulrich G.; Ziessel R.; Harriman A. Angew. Chem., Int. Ed. 2008,47, 1184. 2.Gryko D. T.; Gryko D.; Lee C.-H. Chem. Soc. Rev. 2012, 41, 3780. 3. (a) Lemos A. Molecules 2009, 14, 4098;(b) Attanasi O. A.; De Crescentini L.; Favi G.; Filippone P.; Mantellini F.; Perrulli F. R.; Santeusanio S. Eur. J. Org. Chem. 2009, 3109; (c) Pereira N. A. M.; Lemos A.; Serra A. C.; Pinho e Melo T. M. V. D. Tetrahedron Letters, 2013, 54, 1553.

FC16
Dirhodium(II) Complexes Derived from Natural Amino Acids
as Catalysts in Aqueous Asymmetric Intramolecular C-H insertion
of α-Diazo Acetamides
Nuno R. Candeias,a,b
Pedro M. P. Góis,b Carlos A. M. Afonso
b
aTampere University of Technology, Department of Chemistry and Bioengineering, Tampere, Finland
biMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal
The use of dirhodium stabilized carbenes in intramolecular C-H insertion of -diazo
substrates is a powerful methodology for the synthesis of highly valuable compounds.1 The
extrapolation of intramolecular C-H insertion in organic solvents to aqueous medium
indicates that the success of the transformation is dependent on the hydrophobic nature of
the metalocarbene formed.2
Recently, we have demonstrated the preparation of a new type of homochiral dirhodium
complexes based on natural amino acids (1a-c).
3 Such complexes can be either isolated or
formed in situ by ligand exchange of dirhodium(II) tetraacetate. These new complexes were
tested in the aqueous asymmetric intramolecular C-H insertion of α-diazo acetamides
providing the corresponding lactams in good yields and moderate enantioselectivities. The
catalytic system could be reused six times by simple extraction of the reaction product with
ethyl ether (Scheme 1). The asymmetric preparation of α-phosphono-β-lactams in water
and dichloroethane was achieved in up to 66 % ee (Scheme 2), making this the most
enantioselective method for the preparation of such compounds. The reaction scope
regarding the amide substituents and α-substituent influence was also studied.
Scheme 1
Scheme 2 Acknowledgements: This work was supported by the Fundação para a Ciência e Tecnologia and FEDER (Ref. PTDC/QUI/66695, PTDC/QUI/66695, PTDC/QUI-QUI/099389/2008 SFRH/BPD/46589/2008, and SFRH/BD/61220/2009). References: 1 a) C. A. Merlic, A. L. Zechman, Synthesis 2003, 1137; b) H. M. L. Davies, S. J. Hedley, Chem. Soc.
Rev. 2007, 36, 1109; c) N. R. Candeias, C. A. M. Afonso, P. M. P. Gois, Org. Bioorg. Chem. 2012, 10, 3357; d) P. M. P. Gois, C. A. M. Afonso, Eur. J. Org. Chem. 2004, 3773. 2. a) N. R. Candeias, P. M. P. Gois, C. A. M. Afonso, Chem. Commun. 2005, 391; b) N. R. Candeias, P. M. P. Gois, C. A. M Afonso, J. Org. Chem. 2006, 71, 5489; c) N. R. Candeias, P. M. P. Gois, L. F. Veiros, C. A. M. Afonso, J. Org. Chem. 2008, 73, 5926. 3. N.R. Candeias, C. Carias, L.F.R. Gomes, V. André, M.T. Duarte, P.M.P. Gois, C.A.M. Afonso, Adv. Synth. Catal. 2012, 354, 2921.

FC17
Carbon Dioxide Approaches for Organic Synthetic Processes
Luís C. Branco, Karolina Zalewska, Gonçalo Carrera, Manuel Nunes da Ponte
REQUIMTE, Departamento de Química, FCT-UNL, Faculdade de Ciências da Universidade Nova de Lisboa, 2829-516 Caparica, Portugal
In recent years, the use and capture of Carbon Dioxide (CO2) became a hot research topic
including their application for organic and pharmaceutical chemistry.1 The possibility to use
carbon dioxide as useful reagent for different synthetic approaches or supercritical CO2 for
efficient extraction and separation processes has been reported.1 For many synthetic
approaches the incorporation of CO2 as alternative reagent or green solvent can improve
significantly the efficiency (yields, purity, reaction conditions) for several organic processes.
The combination of ionic liquids and supercritical fluids has been reported for many organic
transformations in particular catalytic reactions.2 The possibility to use scCO2 in order to
extract the pure products without IL or catalyst contamination is one of the advantages for
these processes. Several publications proof the advantages for ILs and scCO2
combinations in order to recycle the catalytic media during many reaction cycles without
loss of efficiency.
In this communication, we described the applicability of carbon dioxide approaches in two
different organic synthetic processes:3
a) The use of CO2 as reagent for the preparation of reversible chiral and non-
chiralcarbamate salts by the reaction with different amines (e.g. primary alkyl and aryl
amines or polyamines), aminoacids and pharmaceutical compounds in the presence of an
organic superbase (e.g. DBU or tetramethylguanidine). According with the optimized
reaction conditions, it is possible to tune the chemical and thermal stability as well as
potential application of the final salts.
b) The potential use of scCO2 for extraction and separation processes in the case of
three asymmetric catalytic reactions in the presence of ionic liquids and/or chiral ionic
liquids as solvent or chiral media respectively. In particular, Sharpless asymmetric
dihydroxylation of olefins (in the presence of osmium catalyst), asymmetric Aldol and
Michael reactions (in the presence of chiral organocatalysts based on chiral ILs) will be
presented.
The peculiar properties of carbon dioxide including as supercritical fluid open excellent
perspectives for the application in novel organic synthetic transformations as well as their
use in industrial processes.
Scheme 1: Reversible carbamate salts obtained by the reaction of CO2 with primary alkyl and aryl
amines, polyamines, aminoacids and pharmaceutical compounds in the presence of organic superbases (DBU or TMG).
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support (PEst-C/EQB/LA0006/2011 and PTDC/CTM/103664/2008 projects and SFRH/BD/67174/2009 for KZ PhD grant).
References: 1. a) Goodrich, B. F.; de la Fuente, J. C.; Gurkan, B. E.; Zadigian, D. J.; Price, E. A.; Huang, Y; Ind. Eng. Chem. Res. 2011, 50, 111. b) Camper, D.; Bara, J. E.; Gin, D. L.; Noble, R. D.; Ind. Eng. Chem. Res. 2008, 47, 8496. 2. Afonso, C. A. M.; Branco, L. C.; Candeias, N. R.; Gois, P. M. P.; Lourenço, N. M. T.; Mateus, N. M. M.; Rosa, J. N.; Chem. Commun., 2007, 2669. 3. a) Carrera, G. V. M.; da Ponte, M. N.; Branco, L. C.; Tetrahedron, 2012, 68, 7408. b) Branco, L. C.; Serbanovic, A.; da Ponte, M. N.; Afonso, C. A. M.; ACS Catalysis 2011, 1, 1408.

FC18
Synthesis and Applications of Tetrapyrrolic Macrocyclic Systems
Mário J. F. Calvete, Sara M. A. Pinto, Ângela C. B. Neves, César A. Henriques,
Carlos J. P. Monteiro, Mónica Silva, Hugh D. Burrows, Mariette M. Pereira
Departmento De Química, Faculdade de Ciências e Tecnologia, Universidade de Coimbra, Rua Larga,
3004-535, Coimbra, Portugal
Tetrapyrrolic macrocycles occupy a prominent spotlight in the field of Organic Chemistry,
within materials chemistry, besides its remarkable significance in physics, biology and
medicine.1 Their versatility grants them the possibility of modification in numerous ways;
each new modification yielding derivatives demonstrative of new chemistry, with a vast
array of materials relevance.
Here we expect to provide a sight on our group’s recent contribution to this field, with
emphasis placed on the use of these materials as components in building materials where
the symmetry and photophysical properties of the molecule are the key points for the
construction of higher entities: in this communication we present here the synthetic
methodology, together with some relevant photophysical data obtained from the application
of our tetrapyrrolic macrocyclic materials (Figure 1).2
Figure 1 Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support from Programa Compete and QREN/FEDER/FCT through Program CIÊNCIA 2008. PTDC/QUI-QUI/112913/2009 and PTDC/QUI-QUI/099730/2008 are also ackonowledged. References: 1. Handbook of Porphyrin Science: With Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine (Volumes 6-10), Kadish, K. M. Smith, K. M. Guilard R. (Eds.), World Scientific Publishing Co. Pte. Ltd. 2010; 2. a) Silva M.; Azenha M. E.; Pereira, M. M.; Burrows H. D.; Sarakha M.; Forano C.; Ribeiro M. F.; Fernandes A. Appl. Catal. B: Environ. 2010,100, 1. b) Neves A. C. B., Pinto S. M. A.; Henriques C. A.; Rosado M. T. S.; Mallavia, R.; Burrows, H. D.; Pereira M. M.; Calvete M. J. F. J. Molec. Struct., 2012,
1029, 199. c) Pinto S. M. A.; Lourenco M. A. O.; Calvete M. J. F.; Abreu A. R.; Rosado M. T. S.; Burrows H. D.; Pereira M. M. Inorg. Chem. 2011, 50(17), 7916. d) Marques, A. T.; Pinto, S. M. A.; Monteiro, C. J. P.; Melo, J. S. S.; Burrows, H. D.; Scherf, U.; Calvete, M. J. F.; Pereira M. M. J. Polym. Sci. A: Polym. Chem. 2012, 50, 1408.

FC19
Shaping new Biologically Active Compounds with a Boron Tether
Francesco Montalbano, Pedro M. S. D. Cal, Pedro M. P. Góis
iMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal
The quest for functionally diverse compound collections is emerging as a powerful method
for identifying chemical probes that may more efficiently interact with biological
macromolecules.
It is now accepted that small molecules displaying more complex
architectures enriched with oxygen and hydrogen bond donors and acceptors embebedded
in cyclic systems comprising multiple sp3-hybridized carbons and stereogenic centres, are
privileged candidates for these campaigns.
To prepare such compounds, medicinal
chemists rely on labor intensive synthetic strategies that combine a plethora of complex
methodologies frequently used in a multi-step sequence, that often yield the target
compounds in low yields determining the need for extensive purification steps. Therefore,
based on the recent interest of medicinal chemistry for boron containing compounds, we
conceived that small drug-like molecules could be easily created by assemble of simple
building blocks promoted by a boron tether. This innovative strategy would enable the
straightforward generation of discrete molecular surfaces with distinct pharmacophores that
may be readily tuned for optimal interaction with the biological macromolecule. In this
communication will be presented the development of this strategy and its use in the design
of biologically active compounds.1,2
Acknowledgements: The author acknowledge Fundação para a Ciência e Tecnologia (PEst-OE/SAU/UI4013/2011; PEst-OE/QUI/UI0100/2011; PTDC/QUI-QUI/118315/2010; PTDC/CTM-NAN/115110/2009 SFRH/BD/61419/2009 and 72376/2010) are acknowledged for financial support.) References: 1. F. Montalbano, N. R. Candeias, L. F. Veiros, V. André, M. T. Duarte, M, R. Bronze, R. Moreira, P. M. P. Gois, Org. Lett. 2012, 14, 988-991.
2. P. M. P. Gois et al, PTC patent pending: “Boron heterocycles as new inhibitors of human neutrophil
elastase (HNE)” F. Montalbano, P. M. S. D. Cal, L. M. D. Gonçalves, R. Moreira, P. M. P. Gois. PCT/IB2012/057064.

FC20
Synthesis of novel spirooxindoles with potential application as
anticancer agents and probes
Carlos J. A. Ribeiro, Joana D. Amaral, Cecília M. P. Rodrigues, Rui Moreira,
Maria M. M. Santos
iMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal
Compounds possessing a spiro-oxindole core represent attractive synthetic targets due to
their interesting biological properties and consequently with potential application in
medicinal chemistry. In particular, spiropyrrolidine oxindole derivative MI-219 is already
entering phase I clinical trials as anticancer agent through inhibition of p53-MDM2
interaction.1
Taking advantage of our expertise in the synthesis of indole and oxindole
derivatives, we present here our recent results in the synthesis and biological screening of
novel spiroisoxazoline oxindoles as p53-MDM2 interaction inhibitors.
The construction of a spiroisoxazoline oxindole scaffold has been established via an
efficient 1,3-dipolar cycloaddition of 3-methylene indolin-2-ones and nitrile oxides. The nitrile
oxides were synthesized in situ from the corresponding chorooximes in the presence of
triethylamine or zinc (Scheme 1). This protocol represents the first time that zinc is used as
the dehydrochlorinating agent in a 1,3-dipolar cycloaddition reaction, providing an easy
access to spiroisoxazoline oxindoles with structural diversity in high yields.2 In addition, the
bioscreen of these new spiroisoxazoline oxindole has led to the finding of seven compounds
with antiproliferative profile superior to the p53-MDM2 interaction inhibitor nutlin-3, and that
induced cell death by apoptosis.
Scheme 1: Synthesis of spiroisoxazoline oxindoles.
Acknowledgements: We thank Fundação para a Ciência e a Tecnologia for funding projects PTDC/QUI-QUI/111664/2009, PTDC/SAU-FAR/110848/2009, PTDC/SAU-ORG/119842/2010 and Pest-OE/SAU/UI4013/2011, and fellowships SFRH/BD/69258/2010 (CJAR) and SFRH/BPD/47376/2008 (JDA). References: 1. Azmi A. S.; Philip P. A.; Beck F. W. J. et al., Oncogene 2011, 30, 117. 2. Ribeiro C. J. A.; Kumar S. P.; Moreira R.; Santos M. M. M. Tetrahedron Lett. 2012, 53, 281.

FC21
Fast and simple synthesis of folates based in copper-free click
chemistry
Alexandre F. Trindade,a,b
Raquel F. M. Frade,a Ermelinda M. S. Maçôas,
b
Catarina A. B. Rodrigues,a José M. G. Martinho,
b Carlos A. M. Afonso
a
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal; bCQFM, Centro de Química-Física Molecular, IN-Institute of Nanosciences and
Nanotechnology, Instituto Superior Técnico,1049-001 Lisboa, Portugal
In the last decades, folic acid has been employed to actively target tumor cells constituting
one very active field of research in medicinal chemistry, since their receptors were found to
be over-expressed in a considerable percentage of tumor cell lines. Nowadays, a hand-full
of small folic acid conjugates with drugs or fluorescent probes were described and
evaluated in clinical trials.1 The synthetic tools available for conjugation into folic acid are
relatively underdeveloped and remain highly challenging.2 We developed a novel approach
for folic acid conjugation based in copper-free strain-promoted alkyne/azide cycloadditions
allowing a fast and selective synthesis of new valuable folic acid conjugates. This work
deals with the challenging synthesis of folic acid conjugate 1 (scheme 1) and its efficient
conjugation into fluorescent probes, heterogeneous supports and polymers (pegylation).
Furthermore, a folate-rhodamine conjugate was studied by confocal fluorescence imaging in
malignant cells.
Scheme 1: General strategy for the synthesis of new folic acid conjugates
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support (SFRH/ BPD/73932/2010, PTDC/QUI-QUI/101187/2008, RECI/CTM-POL/0342/2012, PTDC/QUI-QUI/099389/ 2008). References: 1. Xia, W.; Low, P. S. J. Med. Chem. 2010, 53, 6811 2. (a) Luo, J.; Smith, M. D.; Lantrip, D. A.; Wang, S.; Fuchs, P. L. J. Am. Chem. Soc. 1997, 119, 10004 (b) Wang, S.; Lee, R. J.; Mathias, C. J.; Green, M. A.; Low, P. S. Bioconjugate Chem. 1996, 7, 56 (c) Nomura, M.; Shuto, S.; Matsuda, A. J. Org. Chem. 2000, 65, 5016

Poster Communications

PC1
Novel room-temperature choline carboxylate zwitterionic ionic
liquids as potential electrolytes
A. Rocha,a N. M. T. Lourenço
a
aIBB – Institute for Biotechnology and Bioengineering, Centre for Biological and Chemical Engineering,
Instituto Superior Técnico, Av. Rovisco Pais 1, 1049-001 Lisboa, Portugal
In the last decade room-temperature ionic liquids (RTIL) have attracted great interest from
the scientific community for the replacement of current carbonate-based electrolyte systems
in lithium-ion batteries, which have inherited safety problems due to their high volatility and
flammability. RTILs on the other hand are non-flammable and non-volatile. However most
electrochemical systems require target carrier-ions such as lithium cations or protons for
assembling their corresponding cells. Conventional RTILs lack these ions so their target ion
mobility is too low. They also can’t be used as liquid matrixes because their component ions
migrate along the potential gradient, inhibiting target ion transport.1 One solution is the
synthesis of zwitterions, which by having the cation and anion tethered to each other
prevents their migration.1c)
These zwitterions usually present melting points above 100ºC
but mixing them with lithium salts gives RTILs. Many studies have shown that zwitterions
promote target ion transport and can act as excellent target ion conductors.1,2
Imidazolium-
based zwitterions are by far the most common ones in the literature. However toxicological
studies have raised questions about their biodegradability.3a)
With this in mind and due to the increasing environmental awareness regarding man-made
chemicals, we envisioned novel zwitterions based on cations with less potential to be
harmful, like choline which is essential for normal functioning of all cells.3
Herein we describe in detail two synthetic routes used for synthesis of choline carboxylate
zwitterions and the RTILs obtained from their conjugation with lithium bis(trifluoromethyl
sulfonyl)imide (Scheme 1). In view of their use as potential electrolytes the physical-
chemical characterization of these attractive ionic liquids was performed. In terms of
conductivity the most promising ionic liquid showed an excellent conductivity between
8.66×10-5 S/cm at 25ºC and 1.80×10
-3 S/cm at 90ºC. These zwitterionic ionic liquids
maintained a liquid state and no dissociation was observed even after six months of storage
at room temperature.4
Scheme 1: Synthetic routes for the preparation of choline-based zwitterionic ionic liquids
Acknowledgements We thank Fundação para a Ciência e Tecnologia and FEDER (SFRH/BPD/41175/2007, PTDC/CTM/ 100244/2008, PTDC/EQU-EQU/104552/2008) for financial support. References 1. a) Narita A.; Shibayama W.; Ohno H. J Mater Chem 2006, 16, 1475. b) Yoshizawa M.; Narita A.; Ohno H. Aust J Chem 2004, 57, 139. c) Yoshizawa M.; Hirao M.; Ito-Akita K.; Ohno H. J Mater Chem 2001, 11, 1057. 2. Yoshizawa-Fujita M.; Tamura M.; Takeoka Y.; Rikukawa M. Chem Commun 2011, 47, 2345. 3. a) Petkovic M.; Seddon K.R.; Rebelo N.P.N.; Pereira C.S. Chem Soc Rev 2011, 40, 1383. b) Meck W.H.; Williams C.L. Devel Brain Res 1999, 118, 51. 4. Rocha Â.; Carvalho T.; Vidinha P.; Lourenço N.M.T. ChemPlusChem 2012, 77, 1106.

PC2
New synthetic routes for 3-styrylflavones using Wittig and Heck
reactions
Djenisa H. A. Rocha, Diana C. G. A. Pinto, Artur M. S. Silva
Department of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal
3-Methylflavones, 3-bromoflavones and 3-styrylchromones are oxygenated heterocyclic
compounds bearing different 3-substituents on the chromone nucleus. Some have drawn
considerable interest owing to their occurrence in nature, their significant importance in
organic synthesis and their biologic activity. 3-Bromoflavones belong to an important class
of organic intermediates that are used as precursors in different routes towards other
flavone derivatives. They also can be used as potent antibacterial and antiviral agents.1 3-
Styrylchromones exhibit anti-fungal and anti-bacterial activities.2 Due to their possible
significance many synthetic strategies have been employed for their synthesis.3a
Although
there is only one strategy towards the synthesis of 3-styrylflavones 5,3b
a type of compound
that joined the flavone core with a styryl group. We have been interested in developing new
synthetic routes towards these derivatives. Consequently we have developed efficient
strategies for their synthesis using 3-methylflavones 2 and 3-bromoflavones 1 as
precursors.4
We will present our current work on this issue, the Wittig reaction of commercial
triphenylphosphonium salt with 4-chloro-2-phenyl-2H-chromene-3-carbaldehyde 4.
Furthermore we will compare this method with our previous reported two synthetic routes for
3-styrylflavones via Heck reaction of 3-bromoflavones 1 with styrenes and Wittig reaction of
flavon-3-ylmethyltriphenylphosphonium salts 3 with benzaldehydes.4 The mechanistic and
structural studies of the new compounds will be discussed based on spectral data recorded
from 1D and 2D NMR experiments.
Scheme 1 Acknowledgements: We would like to thank Fundação para a Ciência e a Tecnologia (FCT, Portugal), the European Union, QREN, FEDER, COMPETE, for funding the Organic Chemistry Research Unit (QOPNA) (project PEst-C/QUI/UI0062/2011) and the Portuguese National NMR Network (RNRMN). D. H. A. Rocha thanks FCT for her PhD grant (SFRH/BD/68991/2010). References: 1. Das D. P.; Parida K. M. J. Mol. Catal. A, 2006, 253, 70. 2. Sonaware S. A.; Chavan V. P.; Karale B. K.; Shingare M. S. Indian J. Heterocycl. Chem. 2002,12, 65. 3. a) e .g. Silva, V. L. M.; Silva, A. M. S.; Pinto, D. C. G. A.; Cavaleiro, J. A.S.; Vasas, A.; Patonay, T. Monastsh. Chem., 2008, 139, 1307. b) Lokshin, V.; Heynderickx, A.; Samat, A.; Pèpe, G.; Guglielmetti, R. Tetrahedron Lett., 1999, 40, 6761. 4. a) Rocha D. H. A; Pinto D. C. G. A.; Silva A. M. S.; Patonay T.; Cavaleiro, J. A. S. Synlett, 2012, 23, 559. b) Rocha D. H. A; Pinto D. C. G. A.; Silva A. M. S. submitted.

PC3
Synthesis and Structural Characterization of Two Dioxo-thia-triaza
Macrocyclic Compounds
M. F. Cabral,a,b
J. Costa,a,b
J. Franco Machado,b S. Gonçalves,
a J. Padilla,
b
M. J. Villa Brito,c,d
N. Torresa,b
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal; bFaculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto,
1649-003 Lisboa, Portugal; cCQE, Instituto Superior Técnico, Universidade Técnica de Lisboa, Av.
Rovisco Pais, 1049-001, Lisboa, Portugal; dDQB, Faculdade de Ciências da Universidade de Lisboa,
Campo Grande, 1749-001, Lisboa, Portugal
In the last years we have been concerned on the design of ligands that can be useful for
several medical applications. Following our previous studies1
and taking into account this
proposal, we now report the synthesis and characterization of two macrocyclic compounds
containing both nitrogen and sulfur as donor atoms, 1-thia-4,7,10-triazacyclododecane-
3,11-dione (dioxo-[12]aneN3S) and 1-thia-4,8,12-triazacyclotetradecane-3,13-dione(dioxo-
[14]aneN3S), cf. Figure 1. The synthesis of dioxo-[12]aneN3S was described by Tabushi et
al.2
and, in the present work,we adopted a similar synthetic method. The cyclization was
performed via aminolysis of the dimethyl ester of an α,ω-dicarboxylic acid with a
polyethylene polyamine. The new macrocyclic compounddioxo-[14]aneN3S was prepared
using a similar methodology with some improvements. The compounds were obtained in
good yields, after purification using chromatographic and recrystallization techniques. The
characterization of these macrocyclic compounds was performed by ESI-MS/MS analysis, 1H and
13CNMR (including APT, COSY, HSQC, HMBC and NOESY experiments) and
infrared spectroscopies.
Figure 1: Macrocyclesdioxo-[12]aneN3S and dioxo-[14]aneN3S, respectively.
Acknowledgements: We thank the Fundação para a Ciência e a Tecnologia (FCT) for theproject REDE/1518/REM/2005 for ESI-MS/MS experiments at LCLEM, Faculdade de Farmácia daUniversidade de Lisboa, Portugal. References: 1. Fernandes A. S.; Cabral M. F.; Costa J.; Castro M.; Delgado R.; Drew M. G. B.; Félix V. J. Inorg. Biochem. 2011, 105, 292. 2. Tabushi I.; Okino H.; Kuroda Y. Tetrahedron Lett. 1976, 48, 4339.
NH
NH
S
HN
O
O
NH
NH
S
HN
O
O

PC4
Synthesis of
2-{2-[5(4)-aryl-2H-[1,2,3]-triazol-4(5)-yl]vinyl}chromen-4-ones
Hélio Albuquerque,a Clementina M. M. Santos,
a,b José A. S. Cavaleiro,
a Artur M. S. Silva
a
aDepartment of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal;
bDepartment of
Vegetal and Production Technology, School of Agriculture, Polytechnic Institute of Bragança, 5301-855
Bragança, Portugal
Chromones are a family of oxygen-containing heterocyclic compounds that have been
shown particular relevant biological activity.1 In what concerns to 2-methylchromones, their
reactivity is well-known and allowed to exploit many different kinds of chemical reactions.
The acidic character of the 2-methyl group, due to the low electron density at C-2 caused by
carbonyl group enable this class of compounds to undergo oxidation, photolysis,
cycloaddition and condensation reactions.2
In this communication we will highlight the condensation reaction of 2-methylchromone 23
with propargyl aldehydes 34 in order to obtain (E)-2-(4-arylbut-1-en-3-ynyl)-4H-chromen-4-
ones 4. The high reactivity of these alkynes with sodium azide provided 2-{2-[5(4)-aryl-2H-
[1,2,3]-triazol-4(5)-yl]vinyl}chromen-4-ones 5 (Scheme 1) in good yields. The required
starting material 2-methylchromone 2 was prepared in a three step sequence starting from
2’-hydroxy-acetophenone 1 while the propargyl aldehydes 3 were obtained from the
reaction of the appropriate iodobenzene with propargyl alcohol and further MnO2 oxidation.
Experimental procedures and spectroscopic characterization of compounds 4 and of all the
intermediates will be presented and discussed.
OOH
O
O
A
3 steps
1 2
3
I
2 steps
B
R
OHC
R
O
R
O
C
1 step
4
R = H, Br, OCH3, CH3, NO2
O
O
NNH
N
R
D
1 step
5
Scheme 1: Synthetic route for the preparation of 2-{2-[5-aryl-2H-[1,2,3]-triazol-4(5)-yl]vinyl}chromen-4-ones 5.
Acknowledgements: Thanks are due to the University of Aveiro, Fundação para a Ciência e Tecnologia (FCT, Portugal), European Union, QREN, FEDER and COMPETE for funding the QOPNA Research Unit (project PEst-C/QUI/UI0062/2011) and the Portuguese National NMR Network. Hélio Albuquerque also thanks FCT for his fellowship (SFRH/BI/51556/2011). References: 1. Sharma, S.K.; Kumar, S.; Chand, K.; Kathuria, A.; Gupta, A.; Jain, R. Curr. Med. Chem. 2011, 18, 3825. 2. Ibrahim, M. A.; Ali, T. E.; Alnamer, Y. A.; Gabr, Y. A. Arkivoc 2010, (i), 98. 3. Hirao, I.; Yamaguchi, M.; Hamada, M. Synthesis 1984, 1076.
4. a) Tretyakov, E. V.; Tkachev, A. V.; Rybalova, T. V.; Gatilov, Y. V.; Knight, D. W.; Vasilevsky, S. F.; Tetrahedron 2000, 56, 10075. b) Wadsworth, D. H.; Gecr, S. M.; Detty, M. R. J. Org. Chem. 1987, 52, 3662. c) Bumagin, N. A.; Ponomaryov, A. B.; Beletskaya, I. P. Synthesis 1984, 728.

PC5
Synthesis of the major building blocks towards a PGN fragment
Marina J. Dias Piresa, Sérgio R. Filipe,
b M. Manuel B. Marques
a*
aREQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de
Lisboa, 2829-516 Caparica, Portugal; bInstituto de Tecnologia Química e Biológica, Universidade Nova
de Lisboa, Av. Da República (EAN), Oeiras, Portugal
Oligosaccharides and glycoconjugates are abundant in nature and play a large role in
biological systems, making them attractive for biological and chemical research.1
Indeed,
the most relevant and naturally occurring glycoconjugates contain residues of 2-amino-2-
deoxy-β-D-glucopyranosyl (D-glucosamine) moieties connected to other residues via a 1,2-
trans-glycosidic linkage. Particularly 2-N-acetamido-2-deoxyglycosides, abundant in nature,
contain glucosamine units that can be glycosylated through O-3, O-4, and O-6 positions.2
The vertebrate and invertebrate innate immune system recognizes invading pathogens by
some associated molecular pathways, such as the PGN (peptidoglycan).3,4
PGNis the major
component of the bacterial cell wall and is constituted by glycan chains of alternating
β(1−4)-linked N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) residues
(Figure 1), cross-linked by short peptide bridges. In order to understand the role of the PGN
in bacterial infections, fragments of homogeneous PGN are required, but their availability
and limited purification still remain a critical point.
Due to the increasing interest on these systems our research is focused on the
development of efficient synthesis of glucosamine disaccharides building blocks to achieve
the construction of PGN. The challenge lies on the regioselective protection of hydroxyl
groups and the stereoselective glycosylation of the glucosamine moieties.5,6
Figure 1: Representation of a PGN unit
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support for the projects PTDC/SAU-IMU/111806/2009 PTDC/QEQ-QOR/2132/2012and for fellowship SFRH/BD/89518/2012. References: 1. Ernst B.; Magnan J. L., Nat. Rev. Drug Discovery 2009, 8, 661. 2. Bongat A. F. G.; Demchenko A. V., Carbohydr. Res. 2007, 342, 374. 3. Filipe S. R.; Tomasz A.; Ligoxygakis P. EMBO J. 2005, 6, 327. 4. Swaminathan C. P. et al. PNAS 2006, 103, 684. 5. Enugala R.; Carvalho L. C. R.; Marques M. M. B. Synlett 2010, 2711. 6. Enugala. R.; Carvalho L. C.; Pires M. J. D.; Marques M. M. B. Chem. Asian J. 2012, 7, 2482.

PC6
β-Amino alcohol-catalyzed direct asymmetric aldol reactions in
aqueous micelles
Afroditi Pinaka,a,b
Georgios C. Vougioukalakis,a,c
Dimitra Dimotikali,b Elina Yannakopoulou,
a
Bezhan Chankvetadze,d Kyriakos Papadopoulos
a
aInstitute of Physical Chemistry, NCSR Demokritos, 15310 Athens, Greece.
bDepartment of Chemical
Engineering, NTU Athens, 15780 Athens, Greece. cLaboratory of Organic Chemistry, Department of
Chemistry, University of Athens, 15771 Athens, Greece. dInstitute of Physical and Analytical Chemistry, School of Exact and Natural Sciences, Tbilisi State
University, 0170 Tbilisi, Georgia
Over the last decade, the direct asymmetric aldol reaction has undergone a renaissance
following the rebirth of organocatalysis. In 2000, List and co-workers reported that (S)-
proline efficiently catalyzes the intermolecular asymmetric aldol reaction under mild
conditions.1
Since then, there have been a great number of reports on asymmetric aldol
reactions catalyzed by proline derivatives, affording enantiopure products in high yields.
However, only a small number of non proline-derived chiral organocatalysts for aqueous
aldol reactions have been reported to date. Although chiral primary β-amino alcohols are
easily accessible from inexpensive α-amino acids, there are only a handful of reports
regarding their application in organocatalytic asymmetric aldol reactions thus far.2 Moreover,
there are no reports on the catalytic efficiency of chiral primary β-amino alcohols in aldol
reactions performed in micellar media. In this context, we herein present a detailed study on
the catalytic efficiency of chiral primary β-amino alcohols in the aldol reaction of simple
ketones with aromatic aldehydes in aqueous micelles (Scheme 1). Throughout our
experiments, we found that a family of cheap and easily accessible β-amino alcohols,
obtained in one step from naturally occurring amino acids, successfully catalyze the
asymmetric aldol reaction between a series of ketones and aromatic aldehydes. These aldol
reactions furnished the corresponding β-hydroxy ketones with up to 93% isolated yield and
89% ee. (S)-2-phenylglycinol and Triton X-100 proved to be the best organocatalyst and
surfactant, respectively.
Scheme 1: Direct asymmetric aldol reaction between ketones and aromatic aldehydes catalyzed by β-
amino alcohols in aqueous micellar media Acknowledgements: The authors thank NCSR Demokritos and the National Technical University of Athens for financial support. A.P. is grateful to NCSR Demokritos for a PhD fellowship. References: 1. List B.; Lerner R.A.; Barbas C.F. III J. Am. Chem. Soc. 2000, 122, 2395.
2. a) Mase N.; Nakai Y.; Ohara N.; Yoda H.; Takabe K.; Tanaka F.; Barbas C.F. III. J. Am. Chem. Soc. 2006, 128, 734. b) Pinaka A.; Vougioukalakis G.C.; Dimotikali D.; Psycharis V.; Papadopoulos K. Synthesis 2012, 44, 1057.

PC7
Detection of nitroaromatic explosive compounds by fluorescent
oxacyclophane-tethered calix[4]arenes
C. Teixeiraa, A. I. Costa
a, J. V. Prata
a
aLaboratório de Química Orgânica, Área Departamental de Engenharia Química and Centro de
Investigação de Engenharia Química e Biotecnologia, Instituto Superior de Engenharia de Lisboa,
Instituto Politécnico de Lisboa, R. Conselheiro Emídio Navarro, 1, 1959-007, Lisboa, Portugal.
Small-molecules-based fluorescent sensors like p-phenylene ethynylene trimers integrating
calix[4]arene receptors exhibit an extraordinary sensing ability toward the detection of
certain nitroaromatic compounds (NACs).1
New calix[4]arene-oxacyclophanes were preliminary described very recently along with their
ability to act as chemical sensing agents for the detection of nitroanilines.2
Herein we report on the chemosensing ability of these oxacyclophane-tethered
calix[4]arenes1 and 2 (Figure 1) in the detection of NACs such as nitrobenzene (NB), 2,4-
dinitrotoluene (2,4-DNT), 2,4,6-trinitrotoluene (TNT) and picric acid (PA) (Figure 2).
It was found by a Stern-Volmer analysis that both fluorophores displayed high sensitivities
toward NACs detection (eg.CALIX-OCP1 (Ksv/M-1): PA (1400), TNT (280), DNT (250) and
NB (85) under air-equilibrated conditions and right-angle illumination). The response of the
two fluorophores in the solid state upon exposure to NB, 2,4-DNT, TNT and PA vapors were
also evaluated through steady-state fluorescence quenching experiments with the materials
as neat films. Host and guest structural factors affecting the sensory efficiency will be
discussed.
Fig. 1: Molecular Structure of CALIX-
OCPs (1 and 2).
Fig. 2: Photoluminescence quenching spectra of CALIX-OCP 1 with picric acid in fluid phase. Inset: Stern-Volmer plot (λexc = 350 nm).
Acknowledgements: We thank the Fundação para a Ciência e a Tecnologia/MCTES (Portugal) for partial financial support (PEst-OE/EQB/UI0702/2012). References: 1. Costa, A. I., Prata, J. V., Sensors and Actuators B 2012, 161, 251.
2. Teixeira, C., Costa, A. I., Prata, J. V., Abstracts of XXIIIENSPQ, 12-14 July 2013, Aveiro, Portugal.

PC8
Conversion of non-expensive camphor and environmentally non-
desired CO2 into fine chemicals
Alexandra P. S. Roseiro, M. Fernanda N.N. Carvalho, Adelino M. Galvão, Ana S.O. Knittel,
Pedro F. Pinheiro
Centro de Química Estrutural, Instituto Superior Técnico, Universidade Técnica de Lisboa, Lisboa,
Portugal.
The new synthesis of 3-camphor carboxylic acid (OC11H15COOH, CCA-H) from camphor
and carbon dioxide under mild experimental conditions considerably improves the existing
procedure.1 In the process the water soluble lithium carboxylate (OC11H15COOLi, CCA-Li) is
also obtained.
CCA-H and CCA-Li were used as precursors for the synthesis of camphor N-carboxamides
(1) by condensation with the appropriate amines. By tuning the reaction of CCA with
diamines it is possible to obtain bi-camphor species (2) or N-carboxamides (Scheme 1).
Scheme 1: R = H (CCA-H) or Li (CCA-Li); i) LiBun, CO2 ii) NH2R´; iii) NH2ZNH2
The formulation of all species was based on elemental analysis and the structural
characterization on IR and NMR (1H,
13C, 2-D) spectroscopy. The structural characterization
of CCA-H by single crystal X-ray diffraction analysis shows it crystallizes in the exo form.
Acknowledgements: We thank Fundação para a Ciência e Tecnologia (FCT) for financial support (PEst-OE/QUI/UI0100/2011) and the NMR Network for facilities. References 1. W.W. Shumway, N.K. Dalley, D.M. Birney, J. Org. Chem. 2001, 66, 5832-5839.

PC9
A Novel and Facile Synthetic Approach to N9-Substituted Guanines
Nádia Senhorães, Alice M. Dias,
M. Fernanda Proença
Department/Centre of Chemistry, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal
Purine nucleobases are fundamental biochemicals in living organisms. Playing several key
rolesin the cell, they have been a valuable inspiration for drug design.1
The importance of
the guanine scaffold has been well demonstrated from the reported applications of synthetic
guanines as anti-cancer and anti-viral agents such as the guanine analogue acyclovir and
the growing interest in designer PNAs and G-quartets. Despite that, routes to synthetic
guanines are still scarsedas guanines pose several problems to synthetic chemists due to
their polifunctional nature and poor solubility.2
Here we report a novel and facile synthetic
method to N9-substituted guanines.
In our research group, a number of substituted purines were prepared by efficient and
inexpensive synthetic methods, involving a common imidazole precursor, the 5-amino-4-
cyanoformimidoyl imidazole 1. Recently, a comprehensive study on the reactivity of
imidazoles 1 with nucleophiles under acidic conditions led us to develop experimental
methods to incorporate primary amines into the 4-cyanoformimidoyl group.3 These results
prompted us to investigate the reaction of imidazoles 1 with cyanamide in order to generate
intermediates 2 as key precursors for the synthesis of guanines 4. Condensation of
imidazoles 1 with cyanamide occurred by displacement of ammonia and led to the formation
of the new N-substituted imidoyl cyanide intermediate 2. Amides 3 were obtained when this
intermediate was combined with water. Finally, intramolecular cyclization of 2 gave the
target guanines 4. The new compounds 3 and 4 were characterized on the basis of
elemental analysis, IR and NMR spectroscopy, including 13
C and 2D techniques.
Scheme 1:The synthesis of guanines 4 from hydrolysis of imidazoles 2
Acknowledgements:We thank the University of Minho and the Foundation for the Science and Technology (FCT, Portugal) for financial support to NMR Portuguese network (PTNMR, BrukerAvance III 400-Univ. Minho); FCT and FEDER (European Fund for Regional Development)-COMPETE-QREN-EU for financial support to the Research Centre, CQ/UM [PEst-C/QUI/UI0686/2011 (FCOMP-01-0124-FEDER-022716)] and a PhD grant awarded to Nádia Senhorães (SFRH/BD/73721/2010). References: 1. Legraverend, M.; Grierson,D. S. Bioorg. Med. Chem., 2006, 14, 3987. 2. Fletcher, S; Shahani, V. M.; Lough, A. J.; Gunning, P. T. Tetrahedron, 2010, 66, 4621. 3. Dias, A. M.; Vila-Chã, A. M.; Costa, A. L.; Cunha, D; Senhorães, N; Proença, M. F.; Synlett, 2011, 2675.

PC10
Synthesis by MAOS of alkylated derivatives of a bioactive natural
flavone
A. Pereiraa,b
, S. Cravoa,b
, L. Saraivac,d
, M. Pintoa,b
, H. Cidadea,b
aCentro de Química Medicinal da Universidade do Porto (CEQUIMED-UP), Rua de Jorge Viterbo
Ferreira, 228, 4050-313 Porto, Portugal; bLaboratório de Química Orgânica e Farmacêutica, Departamento de Ciências Químicas, Faculdade de
Farmácia, Universidade do Porto, Rua de Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal; cREQUIMTE, Faculdade de Farmácia, Universidade do Porto, Porto, Portugal;
dLaboratório de Microbiologia, Departamento de Ciências Biológicas, Faculdade de Farmácia,
Universidade do Porto, Rua de Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal.
One of the main interests of CEQUIME-UP is the search of new pharmacologically active
compounds from natural and synthetic origin, focusing on potential antitumor compounds. In
this respect, several small-molecules have been evaluated for their activity as inhibitors of
the growth of human tumor cell lines. From these studies several hit compounds have been
emerged, particularly prenylated flavonoids.1 Inspired by the potential of these flavonoids as
antitumor agents, we have synthesized eleven alkylated analogues using a natural flavone
as the starting material. The synthetic approach was based on the reaction with alkyl
bromides in alkaline medium under microwave irradiation. The structure elucidation of
synthesized compounds was established on the basis of NMR techniques (1H NMR,
13C
NMR, HSQC and HMBC).
Acknowledgements: This work is funded through national funds from FCT – Fundação para a Ciência e a Tecnologia under the project CEQUIMED – PEst-OE/SAU/UI4040/2011, FEDER funds and COMPETE program under the projects FCOMP-01-0124-FEDER-011057 and FCOMP-01-0124-FEDER-015752. References: 1. Neves M.P., Cidade H., Pinto M., Silva A.M.S., Gales L., Damas A.M., Lima R.T., Vasconcelos M.H., Nascimento M.S.J. Eur J Med Chem. 2011,46, 2562.

PC11
Microwave Assisted vs Conventional Synthesis of Chalcones,
Dihydrochalcones and Flavanones as New Potential SGLTs
Inhibitors for the Treatment of Diabetes
Ana Rita Jesus, Amélia P. Rauter
Grupo da Química dos Glúcidos, Centro de Química e Bioquímica/Departamento de Química e
Bioquímica, Faculdade de Ciências da Universidade de Lisboa, Campo Grande Edificio C8, 5o Piso,
1749-016 Lisboa, Portugal
Diabetes is one of the most common metabolic disorders in the world. The usual therapy for
the treatment of diabetes is based on lowering the blood glucose levels by injecting insulin
(type 1 diabetes), or using oral hypoglycaemic agents (type 2 diabetes). However a new
therapy has been investigated consisting on inhibition of glucose uptake by sodium glucose
co-transporters (SGLTs).1-3
These are membrane proteins that transport glucose into the
body. Generally glucose is filtered in kidneys but most of it (> 99%) is reabsorbed and
returns to bloodstream. Glucose up-take leads to higher blood glucose levels, which are
inadvisable in diabetic patients. Thus, inhibiting this process allows the excretion of excess
glucose in the urine, lowering plasma glucose levels.4,5
Phlorizin is a dihydrochalcone glucoside and was the first reported SGLT inhibitor. However
it is poorly absorbed in gastrointestinal tract because it is easily hydrolyzed by phlorizin
lactase.6
In this work several chalcones, dihydrochalcones and flavanones were synthesized by conventional methods and using microwave irradiation (Scheme 1) as potential SGLTs inhibitors for the treatment of diabetes.
Scheme 1. Synthetic pathway for the synthesis of the target molecules Acknowledgements: We thank the Fundação para a Ciência e Tecnolgia for the PhD grant (SFRH/BD/78236/2011) and for financial support (PEst-OE/QUI/UI0612/2013). References: 1. R. Rajesh, P. Naren, S. Vidyasagar, Unnikrishnan, S. Pandey, M. Varghese, S. Gang, Intern. J. Phar. Scienc. Res. 2010, 1(2) 139 2. R. K. Vats, V. Kumar, A. Kothari, A. Mital, U. Ramachandran, Curr. Science 2005, 88 (2), 241 3. M. Isaji, Kidney Int., 2011, 79, S14 4. S. Han, D. L. Hagan, J. R. Taylor, L. Xin, W. Meng, S.A. Biller, J. R. Wetterau, W. N. Washburn, J. M. Whaley, Diabetes, 2008, 57 (6), 1723 5. E. C. Chao, R. R. Henry, Nat. Rev. Drug Disc., 2010, 9 (7), 551 6. J. R. L. Ehrenkranz, N. G. Lewis, C. R. Kahn, J. Roth, Diabetes/Metab. Res. Rev., 2005, 21, 310

PC12
3-aminocoumarins: synthesis and reaction with active carbonyl
compounds
A. Rodrigues, Marta Costa, M. Fernanda Proença
Department of Chemistry, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal
The 3-aminocoumarin core is an important building block, present in many bioactive
structures.1 Novobiocin 1 and clorobiocin 2 are probably the most important examples of
antibiotics with this core unit (Figure 1). Despite the enormous effort of the scientific
community to understand the biosynthesis of novobiocin2 and to reproduce the synthesis in
the laboratory, in a pathway with affordable costs and environmentally friendly experimental
conditions, no satisfactory pathway has yet emerged.3 The experience in the synthesis of
chromene-based scaffolds using eco-friendly approaches acquired over the last few years4
encouraged the search for a viable synthetic pathway to isolate the 3-aminocoumarin core
unit with a variety of substituents in the aromatic moiety. In this work we explored the
synthetic pathway to isolate 3-aminocoumarin derivatives 5 in good yield, obtained from the
reaction of the starting materials 3 and 4 in a few steps (Scheme 1). The reactivity of
compounds 5 with active carbonyl species was studied and will be discussed. Isolated
chromenes 6 were fully characterized.
Figure 1: Novobiocin 1 and clorobiocin 2.
Scheme 1: Synthesis of 3-aminocoumarin derivatives 5 and reaction with active carbonyl compounds.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support from University of Minho and FCT through the Portuguese NMR network (RNRMN), the Project F-COMP-01-0124-FEDER-022716 (ref. FCT Pest-C/QUI/UI0686/2011) FEDER-COMPETE and BPD grant awarded to Marta Costa (SFRH/BPD/79609/2011). References: 1. a) Bhaskar R. K.; Adam S. D.; Brian S. J. B. Bioorg. Med. Chem. Lett. 2011, 21, 7170-7174. b) Bhaskar R. K.; Laura B. P.; Huiping Z.; George V.; Jeffrey H.; Brian S. J. B. J. Med. Chem. 2011, 54,
6234-6253. 2. Lutz H. Nat. Prod. Rep. 2009, 26, 1241-1250. 3. a) Frank W. L. J. Chem. Soc. 1912, 101, 1758-1765. b) Joseph A. B. et al. J. Am. Chem. Soc. 2006, 128, 15536. c) Amit A. K.; Jamie K.; C. Chad W.; Natasha D. W.; Graham J. B. Tetrahedron Lett. 2007, 48, 5077-5080. 4. a) Fernanda P.; Marta C. Green Chem. 2008, 10, 995-998. b) Filipe A.; Marta C.; Marián C.; José B.; Elisabet G; M. Fernanda P.; Jordi M.; María I. L. Eur. J. of Med. Chem. 2012, 54, 303-310.

PC13
Syntheses and equilibria of sugar-based hydrazone
bolaamphiphiles
A. M. Sánchez, M. Ávalos, R. Babiano, P. Cintas, J. L. Jiménez, J. C. Palacios
Departamento de Química Orgánica e Inorgánica, Grupo QUOREX, Universidad de Extremadura, Avda.
de Elvas s/n, 06006 Badajoz (España)
Both hydrazides and hydrazones derived from sugars have been extensively studied.1
However, there have seldom been antecedents on sugar bishydrazides aimed at the
chemoselective preparation of glycoconjugates.2 Herein we describe a facile protocol for the
synthesis of bolaamphiphilic hydrazones derived from unprotected sugars (D-mannose,
D-glucuronolactone and 4-formyl phenyl-β-D-allopyranoside) and linked by hydrocarbon
spacers derived from dicarboxylic acids: carbonic, oxalic, malonic, succinic, adipic, azelaic
(nonanedioic), sebacic (decanedioic), and dodecanedioic acids.
The resulting products contain four chiral centers plus two C=N bonds and two amide bonds
with restricted rotation. Overall, up to ten stereoisomers could be potentially generated. The
barrier to rotation about the amide bond, inferred from dynamic 1H-NMR spectra, is ~17
Kcal·mol-1. This experimental value is consistent with a computational estimation (~18
Kcal·mol-1) by DFT methods at the M062X/6-311G(d,p) level
3 and taking into account the
solvent effect4. The theoretical analysis reveals that stereoisomers with the E,E,E,E
configuration are always favored. In addition, the corresponding Z-E equilibria as well as
ring-chain interconversions in solution have been studied by 1H-and
13C-NMR analysis.
Acknowledgements: We thank the Ministerio de Ciencia e Innovación (MICINN) and FEDER (Project CTQ2010-18938) for financial support and the Centro de Investigación, Innovación Tecnológica y
Supercomputación (CenitS) de Extremadura for allowing us the use of supercomputer LUSITANIA. References: 1. Takeda, Y. Carbohydr. Res., 1979, 77, 9-23. 2. Flinn, N. S.; Quibell, M.; Monk, T. P.; Ramjee, M. K.; Urch, C. J. Bioconjugate Chem., 2005, 16, 722-
728. 3. Frisch, M.J. et al., Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009. 4. Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B, 2009, 113, 6378-6396.

PC14
Directly Linked Porphyrin-Phthalocyanine Conjugates: Synthesis
and Pyridylfullerene Supramolecular Assemblies
Ana M. V. M. Pereira,a Anita Hausmann,
b João P. C. Tomé,
a Olga Trukhina,
c Maxence
Urbani,c Maria G. P. M. S. Neves,
a José A. S. Cavaleiro,
a Dirk M. Guldi,
b Tomás Torres
c,d
aDepartamento de Química e QOPNA, Universidade de Aveiro, 3810-193 Aveiro, Portugal;
bInstitute for
Physical Chemistry, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstr. 3, 91058 Erlangen,
Germany; cDepartamento de Química Orgánica, Universidad Autónoma de Madrid, Cantoblanco, 28049
Madrid, Spain; dIMDEA-Nanociencia, Facultad de Ciencias, Cantoblanco, 28049 Madrid, Spain
Constructing electron donor-acceptor conjugates/hybrids, featuring long-lived charge-
separated states, constitutes a tremendous challenge for chemists.1 The emergent interest
on porphyrin-phthalocyanine (P-Pc) arrays2 comes from the complementary absorptions of
Ps and Pcs - covering a large
part of the solar spectrum - and
their mutual interactions through
photoinduced energy and/or
electron transfer. In most cases,
an efficient energy transfer drives
the excited state energy from the
Ps to Pcs, when the distance
between the cromophores is
small. Owing to their ability to
accept reversibly up to six
electrons, fullerenes (C60) have
been extensively tested in studies regarding the construction of photocurrent-generating
devices. In particular, coordination to the central metal ion of the Ps or Pcs macrocycles by
ligands containing the C60 unit is an excellent method to produce complex photoactive
arrays from simple building blocks.3
In this communication we will present synthetic approaches for the preparation of novel P-
Pc conjugates 1in which the phthalocyanine is directly linked to β-pyrrolic position of the
porphyrin. The study of photoinduced electron transfer processes in P-Pc~C60 hybrids,
assembled through metal coordination with different pyridylfullerenes (2 and 3), will be also
described.4
Acknowledgements: Thanks are due to Fundação para a Ciência e a Tecnologia (FCT), European
Union, QREN, FEDER and COMPETE for funding the QOPNA research unit (project PEst-
C/QUI/UI0062/2011) and the Portuguese National NMR Network, also supported by funds from FCT.
Ana M. V. M. Pereira is grateful to FCT for a post-doc fellowship (SFRH/BPD/64693/2009). Continuous
financial support from MEC, Spain, and the Comunidad de Madrid are also acknowledged. We are
grateful for the financial support from the Deutsche Forschungsgemeinshaft and FCI.
References: 1. Benniston A. C.; Harriman A. Mater. Today 2008,11, 26. 2. Lo P.-C.; Leng X.; Ng D. K. P. Coord. Chem. Rev.2007,251, 2334 and references therein. 3. a) D’Souza F.; Ito O.Chem. Commun. 2009, 4913. b) Bottari G.; de la Torre G.; Torres T. Chem. Rev.2010, 110, 6768. 4. a) Tomé J. P. C.; Pereira A. M. V. M.; Alonso C. M. A.; Neves, M. G. M. P. S.; Tomé A. C.; Silva A. M. S.; Cavaleiro J. A. S.; Martínez-Díaz M. V.; Torres T.; Aminur Rahman, Ramey J.; Guldi D. M. Eur. J. Org. Chem. 2006, 257. b) Pereira A. M. V. M.; Hausmann A.; Tomé J. P. C.; Trukhina O.; Urbani M.; Neves M. G. P. M. S.; Cavaleiro J. A. S.; Guldi D. M.; TorresT. Chem. Eur. J. 2012, 18, 3210.

PC15
Microwave-assisted synthesis of α-aminoacylamides and
α,α’-diacylimides by Ugi four-component reaction
Ruben Almeida,a Maria Clara Costa,
a,b Ana M. Rosa da Costa,
b,c Carlos Nogueira,
d
Ana Paula Paivae
aCC-Mar,
bDepartamento de Química e Farmácia, Faculdade de Ciências e Tecnologia,
cCIQA,
Universidade do Algarve, Campus de Gambelas, 8005-139 Faro, Portugal; dLaboratório Nacional de
Energia e Geologia, I.P., Campus do Lumiar, 1649-038 Lisboa; eCQB, Departamento de Química e
Bioquímica, Faculdade de Ciências da Universidade de Lisboa, 1749-016 Lisboa, Portugal
Since their discovery by Ugi et al., back in 1959, the Ugi four-component reactions (U-
4CRs), have given access to a myriad of structures. Typically, the one-pot reaction of an
aldehyde or ketone, a primary amine, a carboxylic acid and an isocyanide allows the rapid
preparation of peptidomimetic α-aminoacylamide derivatives. When the primary amine is
replaced by a secondary amine, a α,α’-diacylimide is formed instead.1 Here, we report the
microwave-assisted synthesis of two α,α’-diacylimides (5 a,b) and one α-aminoacylamide
(8) by U-4CR (Scheme 1). In the first case, either cyclohexanal (1a) or benzaldehyde (1b)
and cyclohexanoic (2a) or benzoic acid (2b), respectively, were reacted with dimethylamine
(3) and dodecylisocyanide (4), the latter obtained from dodecylamine by the “carbylamine
reaction”.2 In the second case, the reaction occurred between dodecanal (1c), methylamine
(6), cyclohexanoic acid (2a) and cyclohexylisocianide (7).
Scheme 1: Synthesis of α,α’-diacylimides (5 a,b) and α-aminoacylamide (8) by microwave-assisted Ugi
four-component reaction. Acknowledgements: We thank Fundação para a Ciência e Tecnologia (FCT, Portugal) for financial support under projects PTDC/QUI-QUI/109970/2009, PEst-OE/QUI/UI4023/2011, PEst-OE/QUI/UI0612/2011, and PEst-C/MAR/LA0015/2011. References: 1. Dömling A.; Ugi I. Angew. Chem. Int. Ed. 2000, 39, 3168. 2. Jungermann E.; Smith, F.W. J. Amer. Oil Chem. Soc. 1959, 36, 388.
R1CHO
1 a: R1 = cyclohexyl b: R1 = phenyl
+ R2COOH
2 a: R2 = cyclohexyl b: R2 = phenyl
+
+
3
H25C12NC
4
NN R2
R1
O O
C12H25
5 a: R1,R2 = cyclohexyl b: R1,R2 = phenyl
MeOHW, 50 W
60 ºC 10 h
R1CHO
1 c: R1 = C11H23
+ R2COOH
2 a: R2 = cyclohexyl
+
+CH3NH2
6
H11C6NC
7
MeOHW, 50 W
60 ºC 24 h
C6H11 N
HN
C6H11
O
O
C11H23
8
NH

PC16
Synthesis of new dehydropeptides N-conjugated with an oxazole
moiety
P. M. T. Ferreira, J. A. Martins, G. Pereira, A. Martins
Chemistry Centre, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal
Oxazoles are important structural motifs of a wide range of biologically active molecules.
There have been many synthetic efforts to developed efficient and mild methodologies for
the preparation of oxazoles. In our research group we have been interested in the synthesis
of dehydroamino acids and in their application as substrates in several types of reactions.
Thus we developed an efficient method to prepare oxazoles from N-acyldehydro
aminobutyric acids by treatment with I2/K2CO3 followed by DBU.1 In order to extend the
scope of this reaction to other N-acyldehydroamino acids it was decided to test the method
proposed by Du et al.2 for the synthesis of oxazoles from enamines. Several dehydroamino
acid derivatives were treated with phenyliodine diacetate (PIDA) to give the corresponding
oxazoles in moderate to good yields. Some of these compounds, after cleavage of the
methyl ester were coupled to dehydrodipeptides using conventional peptide synthesis
protocols (Scheme 1).
Scheme 1: Synthesis of new dehydrodipeptides conjugated with na oxazole moiety.
The hydrogelation capability of these compounds will be tested.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia and FEDER/COMPETE for
financial support through CQ-UM and National NMR Network (Bruker 400) and I3N Strategic Project LA 25:2011-2012 References: 1. Ferreira, P.M.T.; Monteiro, L.S.; Pereira, G. Amino Acids 2010, 39, 499–513. 2. Zheng, Y.; Li, X.; Ren, C.; Zhang-Negrerie, D.; Du, Y.; Zhao, K.; J. Org. Chem. 2012, 77 10353-10361.

PC17
Synthesis of new sugar nucleoside precursors of potential
application for Alzheimer’s disease
Andreia Almeida, Vasco Cachatra, Amélia P. Rauter
Grupo da Química dos Glúcidos, Centro de Química e Bioquímica/Departamento de Química e
Bioquímica, Faculdade de Ciências da Universidade de Lisboa, Campo Grande Edifício C8, 5o Piso,
1749-016 Lisboa, Portugal
Alzheimer’s disease (AD) is clinically characterized by a progressive memory loss and other
cognitive impairments. Although the etiology of AD is not completely identified, amyloid-β
plaques and tau neurofibrillary tangles are characteristic of this pathology.1 The current
therapeutic options are acetylcholinesterase inhibitors (AChEIs), which increase
neurotransmission at cholinergic synapses in the brain and reduce temporarily the cognitive
deficit.1 Butyrylcholinesterase (BuChE) is an enzyme also involved in cholinergic
neurotransmission, which has received increased attention in the past years. With AD
progression, the activity of AChE decreases while that of BuChE rises in an attempt to
modulate ACh levels in cholinergic neurons. Recently it was reported that BuChE is present
in AD beta-amyloid plaques but its role is still unknown.2 This discovery also encouraged the
search for new and selective inhibitors of this enzyme. We present herein a simple, efficient
and non-expensive approach to synthesize the sugar moiety of nucleosides type 3, which
are selective inhibitors of butyrylcholinesterase.3
The sugar bicycle 2 is built starting from
methyl α-D-glucopyranoside (1) through regioselective protection, oxidation, Wittig reaction,
cyclization and reduction.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support (PEst-OE/QUI/UI0612/2013). References: 1.Tijms, B. M., Wink, A. M., Haan, W., Flier, W. M., Stam, C. J., Scheltens, P., Barkhof, F., Neurobiology of Aging, 2013, 34, 2023– 2036. 2. Darvesh S., Cash M. K., Reid G. A., Martin E., Mitnitski A., Geula C., J. Neuropathol. Exp. Neurol., 2012, 71(1), 2-14.
3.F. Marcelo, F. V. M. Silva, M. Goulart, J. Justino, P. Sinay, Y. Bleriot, A. P. Rauter, Bioorg. Med.
Chem., 2009, 17, 5106-5116.

PC18
Synthesis of calix[4]pyrroles bearing sulfonamide groups
Mónica R. C. Fernandes, Andreia S. F. Farinha, Augusto C. Tomé, José A. S. Cavaleiro
Department of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro
Human activities, such as food and energy production, are responsible for major biological
and chemical pollution hazards, including high levels of antibiotics, pesticides and
anthropogenic anions.1
The development of selective sensors to monitor these pollutants is
an important task. In recent years, we have been focused in the synthesis of calix[4]pyrrole
derivatives that can acts aschromogenic anion sensors.2,3
In this communication we report
the synthesis of calix[4]pyrroles and dipyrromethanes bearing sulfonamide groups. The new
compounds were obtained from the reaction of 4-acetylbenzenesulfonamides 1 with pyrrole
under different experimental conditions. The details of these syntheses and the structural
characterization of the new compounds will be described.
Scheme 1
Acknowledgements: Thanks are due to Fundação para a Ciência e a Tecnologia (FCT), European
Union, QREN, FEDER and COMPETE for funding the QOPNA research unit (project PEst-C/QUI/UI0062/2011) and the Portuguese National NMR Network. Andreia S. F. Farinha also thanks FCT for the post-doc fellowship (SFRH/BPD/73060/2010). References: 1. Lee, C.; Miyaji, H.; Yoon, D.; Sessler, J. L. Chem. Commun. 2008, 24. 2. Farinha, A. S. F.; Tomé, A. C.; Cavaleiro, J. A. S. Tetrahedron Lett. 2010, 51, 2184. 3. Farinha, A. S. F.; Tomé, A. C.; Cavaleiro, J. A. S. Tetrahedron 2010, 66, 7595.

PC19
Studies toward chemoenzymatic synthesis of nonracemic 2-
phenylpropionic acid
Anna Zadlo, Ryszard Ostaszewski
Institute of Organic Chemistry Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland
With the increasing demand for enantiopure compounds, the preparation of single
enantiomer from racemic mixture has become a key step in the development of fine
chemicals for pharmaceuticals, agricultural chemicals, flavours and fragrances. 2-
Arylpropionic acids belong to important class of non-steroidal anti-inflammatory drugs. They
are widely used to control symptoms of arthritis and related connective tissue diseases.1
However, it is well documented that only the S-enantiomer is active. In the case of
Naproxen the S-enantiomer is 28 times more active than the corresponding R-enantiomer.
Also S-Ibuprofen is 160 times more potent than the R-enantiomer in vitro. It is already
known that the accumulation of the R-enantiomer can cause serious side effects such as
gastrointestinal pain.2 Among the existing enantioselective processes, biocatalysis based on
kinetic resolution seems to be the simplest and one of the most efficient methods. It
employs enzymes as biocatalyst to selectivly transform only one enantiomer of the racemic
substrate into a product. If the rates are sufficiently different one enantiomer may be
completly converted before the other begins to react. Reaction yield and enantioselectivity
can be modulated by the reaction conditions, such as temperature, solvent, type of enzyme
or alkoxy group donor.3
The results of studies on enzymatic kinetic resolution and dynamic kinetic resolution of 2-
phenylpropionic acid as a model compound will be provided (Scheme).
Scheme: Kinetic resolution of 2-phenylpropionic acid.
Acknowledgements: This work was supported by project ”Biotransformations for pharmaceutical and cosmetics industry” No. POIG.01.03.01-00-158/09-01 part-financed by the European Union within the European Regional Development Fund. References: 1. Guieysse D., Cortés J., Puech-Guenot S., Barbe S., Lafaquiére V., Monsan P., Siméon T., André I., Remaud-Siméon M., ChemBioChem 2008, 9, 1308-1317. 2. Adams S. S., Bresloff P., Manson G. C., J. Pharmacol. 1976, 28, 156-157. 3. Kamal A., Azhar M. A., Krishnaji T., Malik M., Azeeza S., Coord. Chem. Rev. 2008, 252, 569-592.

PC20
Synthesis of new benzaldehyde derivatives and their
transformations into Corroles
Bernardo A. Iglesias, Joana F. B. Barata, Maria G. P. M. S. Neves, José A. S. Cavaleiro
Department of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal
Certain tetrapyrrolic macrocycles like corroles are being key targets for many research
groups.1 The contracted skeleton, with a direct C-C linkage between two adjacent pyrrole
rings, provide unique physical-chemistry properties and reactivity features.2,3
The synthetic
approaches and the chemical functionalization of corroles are being studied in great
detail.4,5
Here we present our results on corrole macrocycle synthesis using two different
acid catalysis methodologies (Scheme 1). The results obtained on the synthesis and
structural characterization will be reported and discussed.
Scheme 1: Formation of pyridyl substituted corrole derivatives by condensation between
dipyrromethanes and functionalized aldehydes. Acknowledgements: Thanks are due to “Fundação para a Ciência e a Tecnologia”-FCT/Lisbon, QREN, FEDER and COMPETE programs for funding the Organic Chemistry Research Unit (Proj. Pest-C/QUI/UI0062/2011) and Project PTDC/QUI-QUI/121857/2010. Thanks are also due to the national NMR network. B.A.I. and J. F. B. B. thanks CNPq (Brazil) and FCT-MCTES (Portugal) (SFRH/BPD/63237/ 2009) for their postdoc grants, respectively. References: 1. Gross, Z.; Galili, N.; Simkhovich, L.; Saltsman, I.;Botoshansky, M.; Blaser, D.; Boese, R.; Goldberg, I.;
Org, Lett., 1999, 1, 599-602. 2. Paolesse, R.; Synlett, 2008, 15, 2215-2230. 3. Aviv-Harel, I.; Gross, Z.; Coord. Chem. Rev., 2011, 255, 717-736. 4. Flamigni, L.; Gryko, D.T.; Chem. Soc. Rev., 2009, 38, 1635-1646.
5. Barata, J.F.B.; Santos, C.I.M.; Neves, M.G.P.M.S.; Faustino, M.A.F.; Cavaleiro, J.A.S.; Functionalization of Corroles; Top. Heterocycl. Chem., Springer-Verlag, Berlin, 2013, in press.

PC21
Synthesis of chiral spiro-β-lactams from 6-alkylidenepenicillanates
Bruna S. Santos,a Clara S. B. Gomes
b, Teresa M. V. D. Pinho e Melo
a
aDepartment of Chemistry, University of Coimbra, 3004-535 Coimbra, Portugal;
bCentro de Química
Estrutural, Departamento de Engenharia Química, Instituto Superior Técnico,Technical University of
Lisbon, 1049-001 Lisboa, Portugal
The β-lactam ring is the core of the biological activity of a large class of antibiotics and also
several β-lactamase inhibitors.1
The indiscriminate and massive use of antibiotics has
resulted in an increasing antibacterial resistance, often caused by the action of enzymes
known as β-lactamases, which cleave the reactive β-lactam bond of the antibiotic, making it
ineffective. The demand for more efficient β-lactam antibiotics and β-lactamase inhibitors
led us to explore the reactivity of 6-diazopenicillanates and 6-alkylidenepenicillanates for the
functionalization at C-6, keeping the penicillanate nucleus, as an approach to new penicillin
analogues.2
Sheehan et al. have previously described the 1,3-dipolar cycloaddition reactions of 6-
alkylidenepenicillanates with diphenyldiazomethane to give spiro-1-pyrazolinepenicillanates,
which undergo thermally induced ring contraction to afford spirocyclopropylpenicillanates.3
We observed that this ring contraction reaction can also be carried out under microwave
irradiation in quantitative yield. Interestingly, we have observed a different regioselectivity in
the reaction of 6-alkylidenepenicillanates (e. g. 1) with diazomethane, which afforded
compounds 3 and 4, stereoselectively. The observed stereoselectivity can be explained
considering that the products result from the addition to the less sterically hindered α-side of
the β-lactam. The synthesis of spirocyclopropyl-β-lactam 5 via microwave-induced
denitrogenation of spiro-β-lactam 4 was also achieved. In this communication, further details
on the synthesis of this type of new chiral spiro-β-lactams, including derivatives from
different 6-alkylidenepenicillanates, will be disclosed.
Scheme 1: Synthesis of chiral spiro-β-lactams via stereoselective 1,3-dipolar cycloaddition of 6-
alkylidenepenicillanates. Acknowledgements: Thanks are due to FCT (Grants: SFRH/BD/63622/2009 and SFRH/BPD/64623/2009); Projects: PEst-C/QUI/UI0313/2011 and PEst-OE/QUI/UI0100/2013; FEDER, COMPETE and QREN for financial support. We acknowledge AtralCipan for providing the 6-aminopenicillanic acid. References:
1. Comprehensive Heterocyclic Chemistry II (Eds.: Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V.), Elsevier, Oxford, 1996. 2. a) Santos, B. S.; Nunes S. C. C.; Pais, A. A. C. C.; Pinho e Melo, T. M. V. D. Tetrahedron 2012, 68, 3720-3737. b) Santos, B. S.; Pinho e Melo, T. M. V. D. Eur. J. Org. Chem. 2013, in press. 3. Sheehan, J. C.; Chacko, E.; Lo, Y. S.; Ponzi, D. R.; Sato, E. J. Org. Chem. 1978, 43, 4856–4859.

PC22
Synthesis of Tetra-Phosphonated Porphyrins as Organic Ligands
for the Preparation of Metal-Organic Frameworks
Carla F. Pereira,a,b
Sérgio M. F. Vilela,a,b
Flávio Figueira,a Filipe A. Almeida Paz,
b
João P. C. Toméa
aDepartment of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal;
bDepartment of Chemistry & CICECO, University of Aveiro, 3810-193 Aveiro, Portugal
Porphyrins (Pors), and their respective derivatives,are valuable compounds in various
scientific fields due to their unique physico-chemical properties.1
Following our recent
scientific activity2 we are now developing novel Pors carrying phosphonate groups at the
periphery of their cores, which will then be employed as ligands for the preparation of Metal-
Organic Frameworks (MOFs) that may exhibit (photo)catalytic activity. In this
communication we report the synthesis of dietoxyphosphonated porphyrins (4a, 4b and 8a,
8b) by different methodologies.
Scheme 1: Synthesis of tetra-phosphonated porphyrins. Acknowledgements: We would like to thank FCT for their general financial support (R&D projects PTDC/CTM/101538/2008 and PTDC/QUI-QUI/098098/2008), for the PhD research grants SFRH/BD/86303/2012 (to CFP), SFRH/BD/66371/2009 (to SMFV), SFRH/BD/46788/2008 (to FF). We further wish to thank the European Union, QREN, FEDER, COMPETE, CICECO (PEst-C/CTM/LA0011/2011), QOPNA (PEst-C/QUI/UI0062/2011) and the Chemistry Department for their general funding scheme. References: 1. (a) Kadish, K.; Smith, K. M.; Guilard, R. Handbook of Porphyrin Science, Eds., vol. 1-15. Singapore: World Scientific Publisher, 2010-2011; (b) Choi, E. Y.; Barron, P. M.; Novotny, R. W.; Son, H. T.; Hu, C.;Choe, W. Inorg. Chem., 2009, 48, 426. 2. (a) Vilela, S. M. F.; Ananias, D.; Gomes, A. C.; Valente, A. A.; Carlos, L. D.; Cavaleiro, J. A. S.; Rocha, J.; Tomé, J. P. C.; Paz, F. A. A. J. Mater. Chem., 2012, 22, 18354; (b) Pereira, A.M.V.M.; Hausmann, A.;
Tomé, J.P.C.; Trukhina, O.; Urbani, M.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S.; Guldi, D.M.; Torres, T.Chem. Eur. J., 2012, 18, 3210.

PC23
Intrinsically asymmetric 1,3-dibenzyl-oxacyclophane-tethered
calix[4]arenes: synthesis and characterization
C. Teixeira, A. I. Costa, J. V. Prata
Laboratório de Química Orgânica, Área Departamental de Engenharia Química and Centro de
Investigação de Engenharia Química e Biotecnologia, Instituto Superior de Engenharia de Lisboa,
Instituto Politécnico de Lisboa, R. Conselheiro Emídio Navarro, 1, 1959-007, Lisboa, Portugal.
Calix[4]arene scaffolds have been successfully used in the design and synthesis of neutral
compounds’ molecular receptors of several origins. In particular, our most recent work has
been directed to the synthesis and application of several calix[4]arene architectures as
chemosensors, either as single-molecule or polymer-based sensors of nitroaromatic
explosives and explosive taggants.1-4
An import feature in the complexing ability of calix[4]arene host molecules is their structural
rigidity, particularly when the recognition and reporting events are to be undertaken in fluid
phases. Our latest efforts toward the synthesis of such conformationally rigid calixarene
receptors are here described along with their main photophysical properties.
Thus, the narrow rim 1,3-oxacyclophane tethered calix[4]arene derivatives (CALIX-OCP-diI;
3 and 4) were prepared from CALIX-diBz-diBr (1 and 2) which were in turn obtained from
the parent calix[4]arene-tetraol. The reporting sub-unit (p-phenyleneethynylene segments)
was then attached orthogonally to the bicyclic receptors yielding CALIX-OCPs 5 and 6. The
asymmetric nature of CALIX-OCPs will be documented by NMR data.
Scheme 1: Synthesis of 1,3-dibenzyl-oxacyclophane-tethered calix[4]arenes.
Acknowledgements: We thank the Fundação para a Ciência e a Tecnologia/MCTES (Portugal) for partial financial support (PEst-OE/EQB/UI0702/2012). References: 1. Costa, A. I., Prata, J. V., Sensors and Actuators B 2012, 161, 251. 2. Costa, A. I., Pinto, H. D., Ferreira, L.F.V., Prata, J.V., Sensors and Actuators B 2012, 161, 702. 3. Barata, P.D., Costa, A.I., Prata, J.V., React. Funct. Polym. 2012, 72, 627. 4. Barata, P.D., Prata, J.V., Supramol. Chem. 2013, in press.

PC24
Study of Oxazol-5-(4H)-ones fragmentation using Electrospray
tandem mass spectrometry
Catarina A. B. Rodrigues,a, b
Ana Dias,c M. Conceição Oliveira,
c Carlos A. M. Afonso,
a
José M. G. Martinhob
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal; bCQFM, Centro de Química-Física Molecular, IN-Institute of Nanosciences and
Nanotechnology, Instituto Superior Técnico; cCQE, Departamento de Engenharia Química e Biológica
Complexo I, Instituto Superior Técnico,1049-001 Lisboa, Portugal
Oxazol-5-(4H)-ones are small and simple molecules that have the general structure
presented in Scheme 1 (a). This class of compounds have diverse biological activity and,
due to the presence of numerous reactive sites (see Scheme 1 (a)), constitute precursors
for the synthesis of other biological active heterocyclic molecules.1 When the structure has
an exocyclic double bond in position 4 (Scheme 1 (b)) a highly conjugated system is
achieved that gives oxazol-5-(4H)-ones interesting photochemical properties.2
In the present work, electrospray tandem mass spectrometry was used to fully characterize
a series of oxazol-5-(4H)-ones. The studied compounds are similar, being only different in
the R1
substituent of oxazolone core. Despite the differences in the aryl substituents at
position 4, all the oxazolones follow a similar fragmentation that ends with fragment ions m/z
105 corresponding to a benzoyl cation (B1), as reported in oxazolones EI MS studies.3 It
can be concluded that the major fragmentation pathways of oxazolones under ESI/MS
conditions occur preferentially in the oxazolone core with losses of CO, CO2, HCN,
benzaldehyde and/or phenylacetonitriles. An example of fragmentation pathway is proposed
in Scheme 2.
Scheme 1: General structure of Oxazol-5-(4H)-ones (a) and unsaturated Oxazol-5-
(4H)-one (b).
Scheme 2: Proposed fragmentation pathways for
4-(4-dimethylamino-benzylidene)-2-phenyl-oxazol-5-(4H)-one ([M+H]
+, m/z 293).
Acknowledgements: We thank the Fundação para a Ciência e Tecnolgia for financial support. (ref. SFRH/BD/48145/2008, RECI/CTM-POL/0342/2012 and PTDC/CTM-POL/114367/2009) and the IST Node of RNEM for MS facility. References: 1. Fisk J. S., Mosey R. A., Tepe J. J., Chem. Soc. Rev., 2007, 36, 1432–1440; 2. Rodrigues C. A. B., Mariz I. F. A. , Macoas E. M. S., Afonso C. A. M., Martinho J. M. G., Dyes and Pigments 2012; 95(3), 713–722. 3. Sanchez-Viesca F., Berros M., Flores J. P., Rapid Commun. Mass Spectrom., 2003; 17, 498–502.

PC25
Amino acid based hydrazones: synthesis and evaluation as new chemosensors for ion recognition
Cátia I. C. Esteves, M. Manuela M. Raposo, Susana P. G. Costa
Centre of Chemistry, University of Minho, Campus de Gualtar, 4710-057, Braga, Portugal
Anionic and cationic species are key players in biological, environmental, and chemical
processes and there is an interest in the design of artificial receptors for molecular
recognition studies. Optical receptors are preferred due the possibility of using low cost and
widely available instruments, and consist of a reporting unit, able to change its fluorescence
or absorption properties, and a binding unit, for the selective recognition of the
anionic/cationic substrate. Colorimetric chemosensors allow a straightforward “naked-eye”
detection, whereas fluorescent chemosensors are more sensitive and versatile, offering
subnanometer spatial resolution.1
Metallic cations can be complexed through N, O and S
donor atoms as in amino acids, at the main and side chains, and in aromatic heterocycles,
which are most usually fluorophores. Therefore, the insertion of suitable heterocyclic
systems at the side chain of natural amino acids can add extra functionality to the amino
acid.2 Anion coordination is based on hydrogen bonding and electrostatic interactions and at
amino acids these processes can arise from side and main chain OH and NH groups,
whereas in heterocycles the presence of NH groups or suitable substituent’s such as urea
and thiourea groups can provide a site for coordination.3
Therefore, the combination of the above mentioned entities would result in a ditopic system
capable of detecting both anions and cations. Bearing in mind our research on the synthesis
and application of colorimetric/fluorimetric probes for anions and cations based on
heterocycles and amino acids,4 we now report the synthesis of novel hydrazones based on
a phenylalanine core, obtained from condensation of formylated phenylalanine with different
heterocyclic hydrazides (Figure 1). The evaluation of the new amino acid hydrazones as
fluorimetric chemosensors was carried out by performing spectrofluorimetric titrations in
acetonitrile and in aqueous solutions, in the presence of relevant organic and inorganic
anions, and of alkaline, alkaline-earth and transition metal cations. 1H NMR titrations were
also conducted in order to gain further insight into the site and mechanism of coordination
for anions and cations.
Figure 1: Structure of novel hydrazones based on a phenylalanine.
Acknowledgements: Thanks are due to Fundação para a Ciência e Tecnologia (FCT-Portugal) and FEDER-COMPETE for financial support through Centro de Química [PEst-C/QUI/UI0686/2011 (F-COMP-01-0124-FEDER-022716)] and a PhD grant to C.I.C. Esteves (SFRH/BD/68360/2010). The NMR spectrometer BrukerAvance III 400 is part of the National NMR Network and was purchased with funds from FCT and FEDER.
References: 1. a) Cho D.-G.; Sessler J.L. Chem. Soc. Rev. 2009, 38, 1647. b) Prodi L.; Bolletta F.; Montalti M.; Zaccheroni N. Coord. Chem. Rev. 2000, 205, 59. c) Gale P.A.; Caltagirone, C. In Chemosensors, Wang B.; Anslyn E. V. (Eds.) John Wiley & Sons, 2011, pp 395. 2. Shimazaki Y.; Takani M.; Yamauchi O.Dalton Trans. 2009, 38, 7854. 3. Santos-Figueroa L.E.; Moragues M.E.; Raposo M.M.M.; Batista R.M.F.; Ferreira R.C.M.; Costa S.P.G.; Sancenón F.; Martínez-Máñez R.; Ros-Lis J.V.; Soto J. Org. Biomol. Chem. 2012, 10, 7418. 4. a) Batista R.M.F.; Ferreira R.C.M.; Raposo M.M.M.; Costa S.P.G. Tetrahedron 2012, 68, 7322. b) Esteves C.I.C.; Raposo M.M.M.; Costa S.P.G. Amino Acids 2011, 40, 1065.

PC26
Synthesis and Nonlinear Optical Properties of Heterocyclic Cationic
Chromophores Containing Piridinium, Quinolinium and
Benzothiazolium Acceptor Groups
M. Cidália R. Castro,a M. Belsley,
b M. Manuela M. Raposo
a
aCentre of Chemistry, University of Minho, Campus de Gualtar, 4710-057, Braga, Portugal; bCentre of Physics, University of Minho, Campus de Gualtar, 4710-057, Braga, Portugal
The need for new materials that may find uses in optoelectronic and all-optical data
processing technologies has inspired a great deal of research with organic nonlinear optical
(NLO) compounds. A diverse range of materials has been studied over the past few
decades, including piridinium and benzothiazolium salts in part due to their stability and the
ease of tailoring them for specific physical properties. As a result, stilbazolium
chromophores have been widely applied in such diverse areas as frequency-up conversion,
optical power limiting, fluorescent probes, laser scanning fluorescent microscopy and
molecular electronics.1
As part of an on-going investigation to develop efficient donor-
acceptor heterocyclic systems for NLO applications2 we synthesized compounds 1-2
bearing thiophene and pyrrole rings as efficient π-electron donor moieties and
simultaneously as modulated -bridges, functionalized with N-methyl- pyridinium,
quinolinium or benzothiazolium acceptor groups substituted at thiophene or pyrrole rings
(Figure 1). Compounds 1-2 were synthesized through Knoevenagel condensation of the
precursor aldehydes with the heterocyclic salts in ethanol at reflux, in the presence of a
catalytic amount of piperidine. On the other hand, the aldehydes were prepared through two
synthetic procedures: Suzuki cross-coupling reaction or cyclisation of 1,4-dicarbonilic
compounds with Lawesson´s reagent followed by Vilsmeier formylation. In this
communication we report on the synthesis and characterization of the optical properties of
the novel cationic chomophores 1-2. These results indicate that, these compounds have
good potential for a variety of NLO applications namely as chromophores for second
harmonic generation (SHG).
N
S
R
A
n = 1-2
N
S
Rn = 1-2
AN CH3A = N CH3 N
S
H3C
, ,
1 2
R = alkyl
Figure 1: Structure of donor-acceptor (bi)thienylpyrrole cationic chromophores.
Acknowledgements: Thank are due to Fundação para a Ciência e Tecnologia (Portugal) and FEDER-COMPETE for financial support through the Centro de Química and Centro de Física - Universidade do Minho, Projects PTDC/QUI/66251/2006 (FCOMP-01-0124-FEDER-007429), PTDC/CTM/105597/2008 (FCOMP-01-0124-FEDER-009457), PEst-C/QUI/UI0686/2011 (F-COMP-01-0124-FEDER-022716) and a PhD grant to M.C.R. Castro (SFRH/BD/78037/2011). The NMR spectrometer Bruker Avance III 400 is part of the National NMR Network and was purchased within the framework of the National Program for Scientific Re-equipment, contract REDE/1517/RMN/2005 with funds from POCI 2010 (FEDER) and FCT. References: 1. a) Brown A. S.; Bernal L.-M.; Micotto T. L.; Smith, E. L.; Wilson J. N. Org. Biomol. Chem. 2011, 9, 2142. b) Zajac M.; Hrobarik P.; Magdolen P.; Zahradník P. Tetrahedron 2008, 64, 10605. 2. a) Castro M. C. R.; Belsley M.; Fonseca A. M. C.; Raposo M. M. M. Tetrahedron 2012, 68, 8147. b) Raposo M. M. M.; Castro M. C. R.; Schellenberg P.; Fonseca A. M. C.; Belsley M. Tetrahedron 2011, 67, 5189.

PC27
Anionic Surfactants Derived From Threonine and 4-Hydroxyproline
Cidália Silva Pereira, Cristiana I. C. Santos, M. Luísa C. do Vale, Eduardo F. Marques,
José E. Rodriguez-Borgesa
CIQ-UP, Centro de Investigação em Química, Faculdade de Ciências da Universidade do Porto, 4169-
007 Porto, Portugal
Surfactants are chemical compounds which present two distinct regions, a polar and an
apolar one, in the same molecule. They find widespread utility in household products, being
popularly known as soaps and detergents, in food and pharmaceutical formulations, among
others. Surfactants are characterized by their tendency to adsorb on surfaces and at
interfaces reducing surface tension. The growing demand for environmentally friendly
surfactants requires the design of new molecules that fulfill this need.1 Natural surfactants
seem to be a good way to achieve this requirement. Thus, the synthesis of surfactants
derived from amino acids brings some advantages in what concerns to biocompatibility and
toxicity.2
This work describes the synthesis of new anionic monomeric single-chained surfactants
derived from threonine and 4-hydroxyproline. The compounds contain long alkyl chains
linked to the nitrogen atom through an amide bond, as schematically illustrated in figure 1.
Attempts to introduce a second alkyl chain at the hydroxyl group are being performed,
aiming at the obtention of monomeric double-chained surfactants. Furthermore, the basic
physico-chemical properties (CMC, surface molecular area, effectiveness of surface
tension) of all the synthesized compounds will be determined.
Figure 1: Synthesis of threonine/4-hydroxyproline based surfactants.
Acknowledgements: Thanks are due to FCT (Fundação para a Ciência e Tecnologia) and FEDER-Compete for financial support through projects PTDC/QUI-QUI/115212/2009 and Pest/C-QUI/UI0081/2011. References: 1. S. Goreti Silva, J. Enrique Rodriguez-Borges, Eduardo F. Marques, M. Luísa C. do Vale, Tetrahedron, 2009, 65, 4156. 2. S. Goreti Silva, Cláudia Alves, Ana M. S. Cardoso, Amália S. Jurado, Maria C. Pedroso de Lima, M. Luísa C. do Vale, Eduardo F. Marques, Eur. J. Org. Chem., 2013, 1758.

PC28
Synthesis of glyconjugates as precursors of targeted Mn(II)-based
MRI contrast agents
C. Barroso, J. André, A. Esteves
Centro de Química, Universidade do Minho, Campus de Gualtar, 4710-057 Braga, Portugal
Magnetic resonance imaging (MRI) is a non-invasive technique for medical diagnosis. A
considerable part of its success is based on the use of paramagnetic contrast agents (CAs)
which, by far, are based on Gd(III) chelates.1 Mn(II) chelates are anyway promising CAs due
to the electronic properties and to the labile water exchange displayed by this metal ion.2 In
this context we are interested in the synthesis of triazapolycarboxylate-type macrocyclic
ligands for the complexation of Mn(II), using 1 (Figure 1) as starting compound.
Carbohydrate mediated cellular recognition plays an important role in nature, being a
possible way to address CAs to specific targets, but reports on carbohydrate-based probes
are still relatively scarce.3It is also well known that the triazole linkage at the anomeric
position of carbohydrates is stable under a large variety of acidic or basic conditions.4
In this communication we report the synthesis of compounds 3 by alkylation of NO2AtBu
(tert-butyl-1,4,7-triazacyclonane-1,4-diacetate) 1 with the glycoconjugates 2 bearing a 1,2,3-
triazole unit (Figure 1). These substrates were prepared by click reaction approach5
between the corresponding glycosylazide and a commercial acetylenic bromide. All
compounds were fully characterized by 1H-NMR,
13C-NMR and bidimensional NMR
techniques and the results will be presented.
Figure 1
Acknowledgements: The authors acknowledge to the Foundation for the Science and Technology (FCT, Portugal) for financial support to the NMR Portuguese network (PTNMR, BrukerAvance III 400-Univ. Minho). FCT and FEDER (European Fund for Regional Development)-COMPETE-QREN-EU for financial support to the Research Centre and CQ/UM [PEst-C/QUI/UI0686/2011 (FCOMP-01-0124-FEDER-022716)]. References: 1. Balogh E. et al.,Inorg. Chem., 2007, 46, 238. 2. Drahoš B., Lukeš I., TóthÉ., Eur. J. Inorg. Chem., 2012, 1975. 3. Geraldes C.F.G.C., Djanashvili K., Peters J.A., Future Med. Chem., 2010, 2, 409; André J. P.et al.,
Chemistry - a European Journal, 2004, 10, 5804 4. B.H.M. Kuijpers et. al., Org. Lett., 2004, 6, 3123 5. F. Himo et al., J. Am. Chem. Soc., 2005, 127, 210

PC29
Synthesis of new metalloporphyrins with potential catalytic action
for development of conducting polymers
Cláudia M. B. Neves,a I. M. Ornelas,
b A. S. Viana,
b Maria G. P. M. S. Neves,
a J. P. Correia,
b
José A. S. Cavaleiroa
aDepartment of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal;
bDepartment of Chemistry and Biochemistry & CQB, University of Lisbon, 1749-016 Lisbon, Portugal
The Direct Borohydride Fuel Cell (DBFC) is currently recognized as one of the most
promising chemical energy converter. The preparation of new electrocatalysts is one of the
most important subjects in that field. It is known that some conducting polymer matrices
containing metal-nitrogen active sites have revealed high activity for the electrochemical
oxygen reduction.1
This hypothesis is supported by the very good performance of
metalloporphyrins and phtalocyanines in these reactions.2 Also, the classical conducting
polymers such the polypyrrole and the polyaniline have revealed interesting activity for the
molecular oxygen electroreduction.3 Therefore, it is intended to associate the advantageous
features of both materials by preparing conducting polymer films based on polyaniline
incorporating metalloporphyrins with potential catalytic action. The route to prepare the
electrocatalysts for oxygen reduction involves the synthesis of specially designed
metalloporphyrins (with Co and Mn) bearing aniline-type moieties suitable for
electropolymerization. From these monomers, polymer films will be electrogenerated on
conducting substrates.
In this communication the synthetic routes to achieve novel metalloporphyrins specially
functionalized with an aniline-type moiety will be shown.4 The ability of such compounds to
form thin films onto solid surfaces by electropolymerization will be also demonstrated. The
electrochemical behaviour of these novel compounds will be investigated, and the anodic
oxidation of such tailored monomers will be studied in situ by ellipsometry and
electrogravimetric techniques.
Acknowledgements: Thanks are due to the Universities of Aveiro and Lisbon, to “Fundação para a Ciência e a Tecnologia” (FCT, Portugal) / Project PTDC/QUI-QUI/121857/2010. We also thank the European Union, QREN, FEDER and COMPETE for funding the QOPNA research unit (project PEst-C/QUI/UI0062/2011), and the Portuguese National NMR Network also supported by funds from FCT. Cláudia M. B. Neves thanks PTDC/QUI-QUI/121857/2010 for her grant. References: 1. a) Qin H. Y.; Liu Z. X.; Yin W. X.; Zhu J. K.; Li Z. P. J. Power Sources 2008, 185, 909. b) Bashyam, R.; Zelenay P.; Nature 2006, 443, 63. c) Ma J.; Wang J.; Liu Y. J. Power Sources 2007, 172, 220. 2. a) Tyurin,V. S.; Radyushkina K. A.; Levina O. A.; Tarasevich M. R. Russ. J. Electrochem. 2001, 37, 843. b) Ma J.; Wang J.; Liu Y. J. Power Sources 2007,172, 220. 3. Khomenko V. G.; Barzukov V. Z.; Katashinskii A. S. Electrochim. Acta 2005, 50, 1675.
4. a) Kadish K. M.; Smith K. M.; Guilard R. (Eds.),The Porphyrin Handbook (Academic Press, London, 2000). b) Lacerda P. S. S.; Silva A. M. G.; Tomé A. C.; Neves, M. G. P. M. S.; Silva A. M. S.; Cavaleiro J. A. S.; Llamas-Saiz A. L. Angew. Chem. Int. Ed. 2006, 45, 5487.

PC30
Synthesis and Characterization of Novel Thiazolo[5,4-d]thiazoles as
Two Photon Absorbers (TPA)
R. Cristina M. Ferreira,a Cátia P. R. Dias,
a Susana P. G. Costa,
a M. Belsley,
b
M. Manuela M. Raposoa
aCentre of Chemistry, University of Minho, Campus de Gualtar, 4710-057, Braga, Portugal; bCentre of Physics, University of Minho, Campus de Gualtar, 4710-057, Braga, Portugal
Recently, an increasing interest in the design of NLO chromophores for two photon
absorption (TPA) applications has been motivated by the vast applications of TPA
phenomenon in fields such as materials science (3D data storage and microfabrication,
optical power limiting), biology and medicine (fluorescence imaging, two-photon microscopy
(TPM), photodynamic therapy). For in vivo imaging, two-photon processes allow finer
resolution and in-depth tissue penetration, with reduced cell damage. High two-photon
excited fluorescence (TPEF) quantum yields, good photostability, adequate solubility, and
optimized response wavelengths are also required in fields such as fluorescence
microscopy or upconversion lasing.1 Incorporation of heterocycles into -conjugated
systems allows the fine tuning of electrooptical properties and often results in strong
fluorescence, mandatory for certain TPA-based applications. Recently, we have studied the
potential of push-pull systems bearing thiophene, pyrrole and benz-X-azole heterocycles
with high thermal and photophysical stabilities, interesting emissive properties and second
and third order optical responses.2 In this context, thiazolo[5,4-d]thiazoles 1 were
synthesized by condensation of the precursor aldehydes with dithiooxamide in DMF at
reflux. Bromination of 1a with NBS in DMF gave precursor 2a which was submitted to
Suzuki coupling with several heterocyclic boronic acids giving conjugated systems 2b-c.
The TPA cross sections of compounds 1-2 using the Z-scan technique will be presented
and discussed.
N
S N
S
X
a X = Sb X = NMe
N
NH
N
c d e
1a-e
R1 R1
R1 =
N
S N
S S
S
R2
R2 2a-c
R1-CHO
R2 = NCHO
cba
Br
Figure 1: Structure of thiazolo[5,4-d]thiazole TPA chromophores.
Acknowledgements: Thank are due to Fundação para a Ciência e Tecnologia (Portugal) and FEDER-COMPETE for financial support through Centro de Química (PEst-C/QUI/UI0686/2011 (F-COMP-01-0124-FEDER-022716)) and Centro de Física [PTDC/CTM/105597/2008 (FCOMP-01-0124-FEDER-009457)], and a PhD grant to R.C.M. Ferreira (SFRH/BD/86408/2012). The NMR spectrometer Bruker Avance III 400 is part of the National NMR Network and was purchased with funds from FCT and FEDER.
References: 1. a) Pawlicki M.; Collins H. A.; Denning R. G.; Anderson H. L. Angew. Chem. Int. Ed. 2009, 48, 3244. b) He G. S.; Tan L. -S.; Zheng Q.; Prasad P. N. Chem. Rev. 2008, 108, 1245. 2. a) Genin E.; Hugues
V.; Clermont G.; Herbivo
C.; Comel
A.; Castro M. C. R.; Raposo M. M. M.;
Blanchard-Desce M. Photochem. Photobiol. Sci. 2012, 11, 1756. b) Pina J.; Seixas de Melo, J.; Batista R. M. F.; Costa S. P. G.; Raposo M. M. M. Phys. Chem. Chem. Phys. 2010, 12, 9719. c) Batista R. M. F.; Costa S.P.G.; Malheiro E. L.; Belsley M.; Raposo M. M. M. Tetrahedron 2007, 63, 4258. d) Pina, J.;
Seixas de Melo, J.; Burrows, H. D.; Batista, R. M. F.; Costa, S.P.G.; Raposo, M. M. M. J. Phys. Chem. A 2007, 111, 8574. e) Costa, S. P. G.; Batista, R. M. F.; Cardoso, P.; Belsley, M.; Raposo, M. M. M. Eur. J. Org. Chem. 2006, 3938.

PC31
A theoretical study of stacking interactions between buckybowls
and fullerene C60
D. Josa,a J. Rodríguez-Otero,
a E. M. Cabaleiro-Lago,
b I. González-Veloso,
a L. A. Santos,
c
T.C. Ramalhoc
aCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS), Universidade
de Santiago de Compostela, Rúa Jenaro de la Fuente, s/n, Santiago de Compostela, 15782, Spain; bDepartamento de Química Física, Universidade de Santiago de Compostela, Facultad de Ciencias,
Avda. de Alfonso X o Sabio, s/n, 27002, Lugo, Spain; cDepartamento de Química, Universidade Federal de Lavras, Campus Universitário, Lavras-MG, 37200-
000, Brazil
The first strong evidence of π···π interactions between corannulene and C60was published
in 2007, when Sygula and co-workers synthesized the molecular tweezers made up two
units of corannulene that can trap one fullerene (Figure 1).1 Since then, the interest by
concave-convex π···π interactions has been revived. The modification of the tweezers by a
different functionalization of buckybowls, which constitute the main part of tweezers, could
improve their efficiency and selectivity. Therefore, the aim of this work is to carry out a
detailed theoretical study of the effects that can enhance the stacking interactions between
buckybowls and fullerene C60 to achieve our ultimate goal that is predict how to modify the
molecular tweezers to improve their efficiency and selectivity. All complexes studied were
optimized at the B97-D/TZVP level using resolution of identity approximation (RI)
implemented in TURBOMOLE 5.10 program suite.2 Counterpoise corrections were applied
to all reported interaction energies to avoid BSSE.3
Figure 1: Molecular tweezers synthesized by Sygula and co-workers.
1
Acknowledgements: The authors want to express their gratitude to the CESGA (Centro de Supercomputación de Galicia) for the use of their computers. D. Josa thanks the Spanish Ministry of Education for FPU scholarship. References: 1. Sygula A.; Fronczek F.R.; R. Sygula; Rabideau P. W.; Olmstead. M. M. J. Am. Chem. Soc. 2007, 129, 3842. 2. Ahlrichs R.; M. Bär; M. Häser; H. Horn; Kölmel C. Chem. Phys. Lett. 1989, 162, 165. 3. Boys S. F.; Bernardi F. Mol. Phys. 1970, 19, 553.

PC32
Schiff Base Tridentate Ligands Derived from Camphoric Acid for
Enantioselective Alkylation of Aldehydes
Dina Murtinho, Camila Ogihara, M. Elisa da Silva Serra
Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade de Coimbra, 3004-535
Coimbra, Portugal
Enantioselective C-C bond forming reactions are one of the most important organic
synthetic processes. In this area alkylation of aldehydes with dialkylzinc has received
considerable attention, allowing the preparation of chiral secondary alcohols, important
precursors in organic synthesis. Many types of ligands, especially bidentate, have been
used in these reactions, namely amino alcohols, diamines, sulfonamides, amongst others.1
Tridentate ligands have been less used and studied.2
In recent years we have prepared new ligands derived from natural compounds, namely
camphoric acid, for enantioselective alkylation and trimethylsilylcyanation of aldehydes.3
Herein, we describe the synthesis of new tridentate Schiff base ligands derived from natural
camphoric acid and their application in the enantioselective alkylation of aldehydes with
diethylzinc. Ligands 5, with different substituents in the aromatic ring, were prepared in
several steps starting from (+)-camphoric acid. (Scheme 1).
Scheme 1: Synthetic sequence for camphoric acid derived tridentate Schiff base ligands.
With these ligands the enantioselective alkylation of aldehydes with diethylzinc, gave
products with ee up to 69%.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia (Project Pest-C/QUI/UI0313/2011)), FEDER, COMPETE and QREN for financial support. References: 1. Pu L.; Yu H.-B. Chem. Rev., 2001, 101, 757. 2. a) Tanaka, T. Y. Y.; Hayashi, M. J. Org. Chem. 2006, 71, 7091-7093. b) Csillag K.; Nemeth L.; Martinek T. A.; Szakonyi, Z.; Fulop, F. Tetrahedron: Asymmetry 2012, 23, 144. c) Yang, X. F. H. T.; Zhang, G. Y. Tetrahedron: Asymmetry 2008, 19, 1670. 3. Murtinho, D.; Serra M. E. S.; Gonsalves, A. M. d. A. R. Tetrahedron: Asymmetry 2010, 21, 62. Serra, M. E. S.; Murtinho, D.; Goth, A.; Gonsalves, A. M. d. A. R.; Abreu, P. E.; Pais, A. A. C. C. Chirality 2010, 22, 425.

PC33
Regiospecific Synthesis of N-Substituted 2-Oxopurine-6-
carboxamidines
Diogo Sampaio, Alice M. Dias,
M. Fernanda Proença
Department/Centre of Chemistry, University of Minho, Campus de Gualtar, 4710-057 Braga,Portugal
Purine-based compounds display a wide range of applications as chemical-biology tools
and/or therapeutic agents.1 Their potency and selectivity depends on the position and
nature of the substituents on the ring.1
In particular, 9-benzyl-6-arylpurines, exhibited
antimycobacterium and antibacterial activity.2-4
In our research group, a number of substituted purines were prepared by efficient and
inexpensive synthetic methods, involving a common imidazole precursor, the 5-amino-4-
cyanoformimidoyl imidazole 1. The N-tosyl 2-oxo-purine-6-carboxamidines 2 were easily
obtained by a multi-step one-pot reaction from imidazoles 1 and tosyl isocyanate.5
In
previous work, these purine derivatives reacted easily with hydrazine and methylamine by
displacement of the tosylamine group, but reaction with different alkyl amines showed
experimental draw backs that prevented the isolation of the corresponding 2-oxopurines 3.
Recent studies on the reaction of 2 with a series of alkyl amines (Scheme 1) led us to the
selective synthesis of purines 3, which could be isolated in moderate-excellent yield using
simple and efficient experimental procedures. The new compounds 3 were characterized on
the basis of elemental analysis, IR and NMR spectroscopy, including 13
C and 2D
techniques.
Scheme 1: The synthesis of 2-oxopurines 3 from 2-oxopurines 2 and primary alkylamines
Acknowledgements: We thank the University of Minho and FCT for financial support to NMR
portuguese network (PTNMR) FCT and FEDER-COMPETE-QREN-EU for financial support to the Research Centre [PEst-C/QUI/UI0686/2011 (FCOMP-01-0124-FEDER-022716)]. References: 1. Legraverend, M.; Grierson, D. S. Bioorg. Med. Chem., 2006, 14, 3987. 2. Luca L. D. Curr. Med. Chem., 2006, 13, 1. 3. Helmick, R. A.; Fletcher, A. E.; Gardner, A. M. et al., Antimicrob. Agents Chem other., 2005, 49, 1837. 4. Lóránd, T.; Kocsis, B. Mini-Rev. Med. Chem., 2007, 7, 900-911. 5. Booth, B. L. ; Cabral, I. M. ; Dias, A. M.; Freitas, A. P.; Matos Beja, A. M.; Proença, M. F. ; Ramos Silva, M. J. Chem. Soc. Perkin Trans 1, 2001, 1241.

PC34
A cascade condensation-cyclization reaction leading to novel
triazachrysene derivatives
Elina Marinho, M. Fernanda Proença
Centre of Chemistry, School of Sciences, University of Minho, Campus de Gualtar, 4710-057 Braga,
Portugal
Nitrogen-containing heterocycles are often present in a broad range of biologically active
compounds.1 Azachrysenes are tetracyclic aromatic compounds containing nitrogen in one
of the ring positions. Substituted diaza- and triazachrysenes are less known and their
synthesis and biological properties are mainly reported in patented work. These compounds
proved to be potent topoisomerase-targeting agents with exceptional cytotoxic activity and
have been studied as anticancer agents.2
In this work, aryl and heteroaryl o-aminonitriles 1 were reacted with triethylorthoformate 2, in
the presence of acid catalysis and proved to be versatile reagents for the synthesis of a
number of triazachrysene derivatives 3 (Figure 1). These compounds, isolated as salts,
were generated through a cascade condensation-cyclization reaction, following a pathway
similar to that previously reported for anthranilonitrile.3
The synthetic approach will be
discussed in detail. All the compounds were characterized by elemental analysis and
spectroscopic (IR, 1H,
13C, HMQC and HMBC) techniques.
NH2
CN
N
N
N
NH
OEt
OEt
H OEt+
1 2 3
HX
. HX
chrysene
N
azachrysene
Figure 1: Synthesis of aryl/heteroaryl triazachrysene 3.
Acknowledgements: The authors gratefully acknowledge the UMinho and FCT (Portugal) for financial support to the NMR Portuguese Network (PTNMR, Bruker Avance III 400-Univ. Minho). FCT and FEDER (European Fund for Regional Development)-COMPETE-QREN-EU for financial support to the Research Centre, CQ/UM [Pest-C/QUI/UI0686/2011 (FCOMP-01-0124-FEDER-022716)] and a PhD grant awarded to Elina Marinho (SFRH/BD/73659/2010). References: 1. Shen Z.; Hong Y.; He X.; Mo W.; Hu B.; Sun N.; Hu X. Organic Letters, 2010, 12, 552.
2. Ruchelman A. L.; Singh K. S.; Wu X.; Ray A.; Yang J. M.; Li T. K.; Liu A.; Liub L. F.; LaVoie E. J. Bioorganic & Medicinal Chemistry Letters, 2002, 12, 3333. 3. Marinho E.; Araújo R.; Proença F. Tetrahedron 2010, 66, 8681.

PC35
Schiff bases derived from salicylaldehydes and anilines. A
quantitative analysis of substituent electronic effects
E. Matamoros; M. Ávalos; R. Babiano; P. Cintas; J. L. Jiménez; J. C. Palacios
Departamento de Química Orgánica e Inorgánica, Grupo QUOREX, Universidad de Extremadura, Avda.
de Elvas s/n, 06006 Badajoz (Spain)
This communication describes an in-depth study on the structure and quantitative
measurement of substituent electronic effects in Schiff bases derived from salicylaldehydes
and anilines. To this end, a set of forty two compounds (shown below) with different
substitution patterns has been obtained.
Solid-state FT-IR data show imine structures in all cases, a fact further corroborated by X-
ray diffraction structures of compounds 2a and 5a. In solution, however, the Schiff bases
exhibit rapid equilibria between imine and enamine structures, although NMR data suggest
the prevalence of imine tautomers. Moreover, DFT calculations at the M062X/6-
311++G(d,p) level reveal that imines are invariably favored over enamine tautomers by4
kcal·mol-1. The low energies for the transition structures (<7 kcal·mol
-1) account for a rapid
equilibrium at room temperature. 13
C-NMR data have been employed to evaluate the equilibrium constant (KT) between the
tautomers. Lastly, Hammett relationships could be obtained to measure the electronic
effects of substituents on such equilibria as well as their influence on proton and carbon
chemical shifts, both phenolic and iminic ones.
Acknowledgements: We thank the Ministerio de Ciencia e Innovación (MICINN) and FEDER (Project CTQ2010-18938) for financial support and the Centro de Investigación, Innovación Tecnológica y Supercomputación (CenitS) de Extremadura for allowing us the use of supercomputer LUSITANIA.

PC36
Stereoselective Synthesis of N-Acylhydrazones from Diazo
Compounds and Aldehydes via NHC catalysis
Fábio M. F. Santos,a João N. Rosa,
a Vânia André,
b M. T. Duarte,
b Luís F. Veiros,
b
Pedro M. P. Góisa
aiMed.UL, Faculty of Pharmacy, University of Lisbon, Av. Prof. Gama Pinto, 1649-003 Lisbon, Portugal
bCentro de Química Estrutural, Departamento de Engenharia Química, Instituto Superior Técnico,
Universidade Técnica de Lisboa, 1049-001 Lisboa, Portugal
An unprecedented N-heterocyclic carbine reaction is presented. This innovative reaction
promoted the stereoselective synthesis of N-acylhydrazones from the addition of aldehydes
to diazo compounds in yields up to 91%. Enals exclusively afforded N-acylhydrazones,
which are important biologically active scaffolds.1,2
The observed regioselectivity was studied based on DFT calculations, in which, the reaction
of the vinylogous Breslow intermediate via the acyl anion pathway was compared with the
competing homoenolate, enol, and acyl azolium pathways. DFT calculations also revealed
that this unusual reaction is under orbital control, rather than being ruled by charge.1
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support (PEst-OE/SAU/UI4013/2011; PEst-/QUI/UI0100/2011; PTDC/QUIQUI/099389/2008; SFRH/BPD/48219/2008). References: 1. Santos, F. M. F; Rosa, J. N.; André, V.; Duarte, M. T.; Veiros, L. F.; Góis, P. M. P. Org. Lett. 2013, 15, 1760. 2. Carvalho S. A.; Feitosa L. O.; Soares M.; Costa T. E.M.M.; Henriques M. G.; Salomão K.; Castro S. L.; Kaiser M.; Brun R.; Wardell J. L.; Wardell S. M.S.V.; Trossini G. H.G.; Andricopulo A. D.; Silva E. F.; Fraga C. A.M. Eur. J. Med. Chem. 2012, 54, 512.

PC37
Synthesis of new chromene derivatives and pharmacological
evaluation for adenosine receptors
M. Fernanda Proença,a Marta Costa,
a F. Areias,
b M. Castro,
b J. Brea,
b M. Loza
b
aDepartment of Chemistry, University of Minho, Portugal;
bDepartment of Pharmacology, University of Santiago de Compostela, Spain
Adenosine is an endogenous purinergic nucleoside, occurring in all cells of the body that
modulates many physiological and pathological conditions related to cardiovascular,
immune, metabolic and neurological functions.1 Cellular signaling by adenosine occurs
through four known adenosine receptor subtypes (A1, A2A, A2B, and A3), belonging to the G
protein-coupled receptor (GPCR) superfamily. The therapeutical interest in the development
of new drugs active on adenosine receptors is evidenced by the continuous patent claims
on new compounds modulating these receptors or new uses for selective ligands.2
Compounds active on these receptors displayed pharmacological activity namely for the
treatment of cardiovascular, inflammatory or neurodegenerative diseases and cancer.3
These active molecules usually belong to the purine family, but compounds with the
pyrazolo-triazolo-pyrimidine, dihydropyridine and quinazoline-urea core unit were also
identified as active.3 The chromene scaffold is present in a variety of biologically active
compounds and their synthesis has been widely explored in the literature.4 The interaction
of chromene derivatives with adenosine receptors was never reported, to the best of our
knowledge. This work describes a one-pot procedure for the synthesis of novel chromene
derivatives 3 and 4 from the reaction of 2-oxo-2H-chromene-3-carbonitriles 1 and
cyanoacetamides 2 (Scheme 1).
Scheme 1: Synthetic pathway for the isolation of compounds 3 and 4.
These new scaffolds proved to be active at human adenosine receptors and several hits
were identified in this study with affinities in the submicromolar range. A detailed discussion
of the synthetic method and affinities of the compounds will be presented.
Acknowledgements: We gratefully acknowledge the financial support from University of Minho and FCT through the Portuguese NMR network (RNRMN), the Project F-COMP-01-0124-FEDER-022716 (ref. FCT PEst-C/QUI/UI0686/2011) FEDER-COMPETE and BPD grant awarded to Marta Costa (SFRH/BPD/79609/2011). References: 1. a) Hilaire, C. St.; Carroll, S.; Chen H.; Ravid, K. J. Cell Physiol. 2009, 218, 35. b) Baraldi, P.; Tabrizi, M.; Gessi, S.; Borea, P. Chem. Rev. 2008, 108, 238. 2. Wilson, C. N.; Mustafa, S. J. Handb. Exp. Pharmacol. 2009; 193, v-vi. 3. a) Baraldi, P.; Tabrizi, M.; Gessi, S.; Borea, P. Chem. Rev. 2008, 108, 238. b) Müller, C.; Jacobson, K. Biochim. Biophys. Acta 2011, 1808, 1290. 4. For recent examples see: a) Chimenti, F. et al. Bioorg. Med. Chem. Lett. 2010, 20, 4922. b) Tang, J. et al. Bioorg. Med. Chem. 2010, 18, 4363. c) Robert, S. et al. J. Med. Chem. 2008, 51, 3077. d) Proença, F.; Costa, M. Green Chem. 2008, 10, 995.

PC38
Thiopheno[3’,4’-a]chromeno[3,4-d]pyrroles from Thiazolidine-4-
carboxilic Acid
Fernanda M. Ribeiro Laia,a Clara S. B. Gomes,
b Teresa M. V. D. Pinho e Melo
a
aDepartment of Chemistry, University of Coimbra, 3004-535 Coimbra, Portugal;
bCentro de Química Estrutural, Departamento de Engenharia Química, Instituto Superior Técnico,
Technical University of Lisbon, 1049-001 Lisboa, Portugal
Chromene and chromane derivatives are particularly interesting molecules, since these
heterocycles are substructures present in many biologically important compounds.1 In the
recent past, we have been interested in exploring the intermolecular cycloaddition of non-
stabilized ylides generated via the decarboxylative condensation of thiazolidine-4-carboxilic
acids.2 In this context, we decided to explore the intramolecular version of this reaction, as a
route to new chromenopyrrole derivatives.
The decarboxylative condensation of thiazolidine-4-carboxilic acid with alkynyl and allenyl
ethers of salicylaldehydes led to the synthesis of 1,3,3a,10b-tetrahydro-5H-thiopheno[3’,4’-
a]chromeno[3,4-d]pyrrole and 1,3,3a,4-tetrahydro-10bH-thiopheno[3’,4’-a]chromeno[3,4-
d]pyrrole derivatives, respectively. Exclusive or selective formation of the product, resulting
from the cycloaddition of the anti-dipole was observed (Scheme 1). Further details of this
study will be presented.
Scheme 1: Synthesis of thiopheno[3’,4’-a]chromeno[3,4-d]pyrrole and pyrrolo[1,2-c]thiazole derivatives.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia (SFRH/BD/69941/2010, SFRH/BPD/64623/2009, PEst-C/QUI/UI0313/2011 and PEst-OE/QUI/UI0100/2013), FEDER, COMPETE and QREN for financial support. References: 1. a) Nicolaou, K. C.; Pfefferkorn, J. A.; Roecker, A. J.; Cao, G.-Q.; Barluenga, S.; Mitchell, H. J. J. Am. Chem. Soc. 2000, 122, 9939-9953; b) Purushothaman, S.; Prasanna, R.; Niranjana, P.; Raghunathan, R.; Nagaraj, S.; Rengasamy, R. Bioorg. Med. Chem. Lett. 2010, 20, 7288-7291; c) Ardill, H.; Grigg, R.; Sridharan, V.; Surendrakumar, S. Tetrahedron 1988, 44, 4953-4966.
2. Cardoso, A. L.; Kaczor, A.; Silva, A. M. S.; Fausto, R.; Pinho e Melo, T. M. V. D.; Gonsalves, A. M. d’A. Tetrahedron 2006, 62, 9861-9871.

PC39
New Photochromic Intermediates for 3D-Data Storage
Filipa Siopa,a Catarina A. B. Rodrigues,
a,b Inês F. A. Mariz,
b Ermelinda M. S. Maçôas,
b
José M. G. Martinho,b Carlos A. M. Afonso
a
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal; bCQFM and IN, IST, Universidade Técnica de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa,
Portugal
Photochromism is a reversible transformation of a molecule between two different chemical
forms with different absorption spectra induced by absorption of electromagnetic radiation
(Figure 1).1 Typically, photochromic compounds (PCs) have application in photonic
devices, such as optical memories and display devices. More recently their biological
applications have been explored.2-4
Diarylethene derivatives, specially
dibenzothienylethenes containing the central 1,2-perfluorcyclopentene fragment, are
promising compounds in the field of optical data storage and optical switches. Those
compounds have notable thermally irreversible photochromic behavior, high
photoisomerization quantum yields and outstanding fatigue resistance.1-3, 5
Different PCs have been designed and synthesized, aiming the development of
photochromic molecules with high potential for application in photonic and data storage
(Scheme 1). Preliminary studies regarding the application of these compounds showed that
compound 1 is a promising system for high speed/high-density data storage.
Figure 1: Photochromic transformation.
Scheme 1: Synthesized photochromic compounds.
Acknowledgements: We thank the Fundação para a Ciência e Tecnolgia for financial support SFRH/BPD/88666/2012 and PTDC/CTM-POL/114367/2009. References: 1. Kawata, S.; Kawata, Y., Chem Rev 2000, 100, 1777-1788. 2. Irie, M., Chem Rev 2000, 100, 1685-1716. 3. Soh, N.; Yoshida, K.; Nakajima, H.; Nakano, K.; Imato, T.; Fukaminato, T.; Irie, M., Chem. Commun. 2007, 5206-5208.
4. Kim, Y.; Jung, H. Y.; Choe, Y. H.; Lee, C.; Ko, S. K.; Koun, S.; Choi, Y.; Chung, B. H.; Park, B. C.; Huh, T. L.; Shin, I.; Kim, E., Angew. Chem. Int. Ed. 2012, 51, 2878-2882. 5. Irie, M.; Miyatake, O.; Uchida, K.; Eriguchi, T., J. Am. Chem. Soc. 1994, 116, 9894-9900.

PC40
Novel pentacationic N-Fused Pentaphyrin
Flávio Figueira, José A. S. Cavaleiro, João P. C. Tomé
QOPNA and Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal
In recent years, expanded porphyrins, having a macrocycle larger than porphyrins have
emerged as a novel class of functional molecules in the light of their fascinating optical,
electrochemical, and coordination properties.1
The chemistry of such systems, often offers
highly intriguing functional dyes, making it clear that expanded porphyrins have an
important role to play in areas as diverse as X-ray
cancer therapy, photodynamic therapy, and anion
recognition.2
In this specific case, the non-linear
optical properties of these materials are of special
interest, in part for energy transfer with molecular
control, and in part for potential applications in
optical communications, data storage, electro-
optical signal processing.3
Also, lightharvesting
applications with supramolecular assemblies
based on carbon nanotubes and other pi acceptor
entities have attracted much attention due to their
unique optoelectronic properties. Nanotubes are
attractive entities for collecting electrons in
photovoltaic devices, whereas their near infrared emission capabilities together with their
versatile functionalization could be valuable for nanomedicine and biolabeling
applications.4
As part of our studies focused on porphyrinoid derivatives with potential non-
linear optical properties, and supramolecular assemblies we report here the synthesis and
structural characterization, of a [24] N-Fused pentaphyrin functionalized with 4-
mercaptopyridine in the para phenyl positions. Further cationization of the pyridyl moieties
with methyl iodide provides 1 (Figure). It is expected that this compound will open a new
range of synthesis and application opportunities for expanded porphyrins.
Acknowledgements: Thanks are due to FCT (Fundação para a Ciência e a Tecnologia) and FEDER for funding the QOPNA Unit and the project PTDC/CTM/101538/2008. F. Figueira also thanks FCT for his PhD grant (SFRH/BD/46788/2008). References: 1. a) Lash, T. D. Angew. Chem., Int. Ed., 2000, 39, 1763; b) Sessler J. L; Seidel, D.; Lynch V. J. Am. Chem. Soc., 1999, 121, 11257–11258; c) Furuta,H; Maeda, H; Osuka, A.Chem. Commun., 2002, 1795. 2. a) Jasat, A.; Dolphin. D.Chem. Rev.,1997, 97, 2267; b) Sessler,J. L.; Tvermoes, N. A.; Davis, J.; Anzenbacher P.; Jursikova, K.; Sato, W.; Seidel, D.; Lynch, V.; Black, C. B.; Try, A.; Andrioletti, B; Hemmi, G.; Mody, T. D.; Magda, D. J.; Krall, V.; Pure Appl. Chem., 1999, 71, 2009. 3. a) Pawlicki, M.; Collins, H. A.; Denning, R. G.;. Anderson, H. Angew. Chem. Int. Ed. 2009, 48, 3244; b) Tanaka, Y.; Saito, S.; Mori, S.; Aratani, N.; Shinokubo, H.; Shibata, N.; Higuchi, Y.; Yoon, Z. S.; Kim; Noh, S. B; Park, J. K.; Kim, D.; Osuka, A. Angew. Chem. 2008, 120, 693; c) Lim, J. M.; Yoon, Z. S.; Shin, J.- Y.; Kim, K. S.; Yoon, M.-C.; Kim, D. Chem. Commun., 2009, 261. 4. Guldi, D. M.; Rahman, G. M. A.; Sgobba, V.; Ehli, C. Chem. Soc. Rev., 2006, 35, 471-487.

PC41
Study on the microwave–assisted Diels-Alder reactions of
5-styryl-1H-pyrazoles with different dienophiles
Inês C. S. Cardoso, Vera L. M. Silva, Artur M. S. Silva
Department of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal
Pyrazoles have been extensively studied and several methods have been developed for
their synthesis due to their widespread application in the fields of agriculture, industry and
medicine.1
With continuing interest on the synthesis and transformation of styryl-1H-
pyrazoles through Diels-Alder cycloaddition reactions to obtain novel indazole based
heterocyclic compounds,2 we decided to study the reactivity of (E)-3(5)-(2-hydroxyphenyl)-
5(3)-styryl-1H-pyrazoles 1 as dienes in the reaction with electron rich and electron poor
dienophiles. It is well-known that vinylpyrazoles are very reluctant to participate as dienes in
cycloaddition reactions involving the pyrazole ring, owing to the loss of aromaticity involved
in these reactions. Therefore very reactive dienophiles, high temperatures and pressures
are required and these reactions are usually slow giving rise to adducts in moderate to low
yields.3,4
Although this fact indicates that (E)-3(5)-(2-hydroxyphenyl)-5(3)-styryl-1H-
pyrazoles 1 should also have low reactivity, we have already performed the Diels-Alder
reaction of pyrazoles 1 with N-methylmaleimide and maleic anhydride. In the first case,
under the tested experimental conditions, we obtained the expected tetrahydroindazoles in
moderate to low yields but in the second case, with maleic anhydride, the corresponding
tetrahydroindazoles were not obtained. More recently, we started to study the reactivity of
(E)-3(5)-(2-hydroxyphenyl)-5(3)-styryl-1H-pyrazoles 1 towards other dienophiles (Figure 1).
In this communication we will present our experimental results as well as the
stereochemistry of the obtained cycloadducts and the structural characterization of the new
1H-indazoles which were unequivocally assigned by NMR.
Figure 1: Structure of the diene (E)-3(5)-(2-hydroxyphenyl)-5-styryl-1H-pyrazole and selected
dienophiles. Acknowledgements: Thanks are due to the University of Aveiro, “Fundação para a Ciência e a Tecnologia” (FCT, Portugal), European Union, QREN, FEDER and COMPETE for funding the QOPNA research unit (project PEst-C/QUI/UI0062/2011), to the Portuguese National NMR network, also supported by funds from FCT and to the project QREN (FCOMP-01-0124-FEDER-010840) (FCT PTDC/QUI-QUI/102454/2008). References: 1. Elguero, J.; Goya, P.; Jagerovic, N.; Silva, A. M. S., In: Attanasi O. A., Spinelli D. (Eds), Targets in Heterocyclic Systems-Chemistry and Properties, Italian Society of Chemistry, Camerino, Italy, 2002, Vol. 6., p 52. 2. Silva, V. L. M.;Silva, A. M. S., Pinto, D. C. G. A.; Elguero, J.; Cavaleiro, J. A. S. Eur. J. Org. Chem., 2009, 4468. 3. a) Medio-Simón, M.; Laviada, M. J. A.; Sepúlveda-Arques, J., J. Chem. Soc., Perkin Trans 1, 1990, 2749. b) Sepúlveda-Arques, J.; Abarca-González, B.; Médio-Simón, M., Adv. Heterocycl. Chem., 1995, 63, 339. 4. Médio-Simón, M.; Sepúlveda-Arques, J., Tetrahedron 1986, 42, 6683.

PC42
The Catalytic Asymmetric Acetalization
Ji Hye Kim, Ilija Čorić, Sreekumar Vellalath, Benjamin List*
Max Planck Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, D-45470 Mülheim an der Ruhr,
Germany
An asymmetric version of the acid catalyzed acetalization of aldehydes is described.1 A
novel member of the chiral confined Brønsted acid family significantly outperformed
previously established catalysts,2 providing cyclic acetals with excellent enantioselectivity.
(Scheme 1) Our asymmetric acetalization can be conveniently applied to kinetic resolution
of diols.
Scheme 1: Asymmetric Acetalization of Aldehydes. Acknowledgements: Max Planck Society, European Research Council and Max Planck Institut für Kohlenforschung analytical department (HPLC, GC, NMR, X-Ray) are gratefully acknowledged. References: 1. Kim J. H.; Čorić I.; ellalath S.; List B. Angew. Chem. Int. Ed. 2013, 52, 4474. 2. Čorić I.; List B. Nature 2012, 483, 315.

PC43
Hydroxylation of flavon-3-ol derivatives
Joana L. C. Sousa, Artur M. S. Silva
Department of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal
Flavon-3-ols constitute a subclass of the flavonoid family which present a 3-hydroxyflavone
backbone. These oxygen heterocyclic compounds act as free radical scavengers so they
have attracted attention as potential antioxidant agents. The number and position of the
hydroxyl groups are one of the most important structural characteristics to present a strong
antioxidant activity.1 We have been studying the selective hydroxylation of flavonols in order
to prepare novel polyhydroxylated ones with different substitution patterns. This
transformation can be performed by several methods, including the reaction with the 2-
iodoxybenzoic acid (IBX)/sodium dithionite system,2 the Elbs persulfate oxidation
3 and the
Baeyer-Villiger oxidation.4
In this communication, we will present some of the obtained results for the hydroxylation of
polysubstituted 3-hydroxy-flavones, which were prepared by us. The used method
consisted in the Vilsmeier-Haack formylation of polysubstituted flavon-3-ols with POCl3
followed by a Baeyer-Villiger-type oxidation using H2O2 as the oxidant in the presence of
H2SO4 as the catalyst (Scheme 1). All the experimental details of this study and the
structural characterization of the new compounds will also be presented.
Scheme 1: Vilsmeier-Haack formylation of polysubstituted flavon-3-ols followed by a Baeyer-Villiger-type
oxidation. Acknowledgements: Thanks are due to the University of Aveiro, Fundação para a Ciência e a Tecnologia (FCT, Portugal) and European Union, QREN, FEDER and COMPETE for funding the Organic Chemistry Research Unit (project PEst-C/QUI/UI0062/2011) and the Portuguese National NMR Network. J. L. C. Sousa is also grateful to FCT for her PhD grant (SFRH/BD/76407/2011). References: 1. Heim K. E.; Tagliaferro A. R.; Bobilya D. J. J. Nutr. Biochem. 2002, 13, 572. 2. Barontini M.; Bernini R.; Crisante F.; Fabrizi G. Tetrahedron 2010, 66, 6047. 3. Park H.; Dao T. T.; Kim H. P. Eur. J. Med. Chem. 2005, 40, 943. 4. González R. R.; Gambarotti C.; Liguori L.; Bjørsvik H.-R. J. Org. Chem. 2006, 71, 1703.

PC44
XPS studies of tetrapyrrolic macrocycles
Joana F. B. Barata, Maria G. P. M. S. Neves, José A. S. Cavaleiro, Tito Trindade
Department of Chemistry, CICECO and QOPNA, University of Aveiro, Campus Santiago 3810-193,
Aveiro.
Several spectroscopic methods has been a standard modus operandi for establishing the
structure and bonding of chemical compounds. With this purpose nearly every available
technique has been applied to tetrapyrrolicmacrocyles, namely porphyrins, corroles and
their derivatives. These macrocycles are versatile functional building blocks in many
biological and biochemical processes.
XPS, X-ray Photoelectron Spectroscopy, has been extensively used to analyse the donor
ability of porphyrins and the charge potentials present throughout a porphyrin molecule,
particularly with regard to the electronic effects of substituents and changes at the central
core.1,2
XPS has been applied for the study of the adsorption and metalation of this family of
molecules on metal surfaces.3 However to the best of our knowledge, it have never been
reported an comprehensive XPS study concerning the C1s, N1s and F1s binding energies
of porphyrins and corroles, related with the symmetry of the macrocycles. In this
communication we will present and compare the C1s, N1s and F1s binding energies of two
porphyrins (5,10,15,20-tetra(pentafluorophenyl)porphyrin TPFP, 5-phenyl-10,15,20-tris
(pentafluorophenyl)porphyrin) PTPFP and one corrole (5,10,15-tris(pentafluorophenyl)
corrole TPFC) Figure 1.
Figure 1: Structures of the tetrapyrrolicmacrocycles used in this work
Acknowledgements:Thanks are due to Fundaçãopara a Ciência e a Tecnologia (FCT, Portugal), European Union, QREN, FEDER and COMPETE for funding the QOPNA and CICECO research units (project PEst-C/QUI/UI0062/2011 and Pest-C/CTM/LA0011/2011). J. F. B. Barata thanks FCT-MCTES (Portugal) for her Post-Doctoral grant SFRH/BPD/63237/2009. References: 1. Karweik, D. H.; Winograd, N. Inorg. Chem. 1976, 15, 2336. 2. Gassman, P. G.; Ghosh, A.; Almlöf, J. J. Am. Chem. Soc. 1992, 114, 9990. 3. Tatyana, E.; Shubina, T. E.; Marbach, H.; Flechtner, K.; Kretschmann, A.; Jux, N.; Buchner, F.; Steinru, H.-P.; Clark, T.; Gottfried J. M. J. Am. Chem. Soc. 2007, 129, 9476.

PC45
A new anion receptor based on a nanomagnet-porphyrin hybrid
João M. M. Rodrigues,a Andreia S. F. Farinha,
a Tito Trindade,
b Augusto C. Tomé,
a
José A. S. Cavaleiro,a João P. C. Tomé
a
aQOPNA,
bCICECO, Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal
Even though regularly ignored in terms of their importance, anions are almost present
everywhere. The design and development of new materials able to recognize and host
efficiently and selectively anionic species, have assumed great importance in several areas
in the past few years, including chemical and biological processes, environment, energy
and biology.1 Also, silica nanoparticles (SNPs) and magnetic SNPs have been intensively
studied due to their unique chemical and physical properties.2 When these SNPs are
combined with organic molecules the resulting hybrids find application in many scientific
areas, such as in catalysis, biomedical imaging, materials chemistry and sensing.3 They can
provide an increased value in environmental applications, since they can be easily
removed/recovered and reused providing new support for the development of "green"
technologies in different areas.4 In this communication, we report the synthesis and studies
of a new anion receptor based on a porphyrin-silica coated magnetic nanoparticle hybrid 1
(Figure 1). Anion binding studies were performed using typical anionic substrates, such as
acetate, bromide, fluoride, nitrate, nitrite and dihydrogen phosphate and were conducted by
UV-Vis spectroscopy.
Figure 1: New nanomagnet-porphyrinhybrid 1
Acknowledgements:Thanks are due to the Universidade de Aveiro (UA), Fundação para a Ciência e a Tecnologia (FCT), European Union, QREN, FEDER and COMPETE for funding the QOPNA (project PEst-C/QUI/UI0062/2011), CIECO (project Pest-C/CTM/LA0011/2011) and project PTDC/CTM/101538/2008. João M.M. Rodrigues and Andreia S. F. Farinha also thank FCT for their PhD (SFRH/BD/81014/2011) and post-doc grants (SFRH/BPD/73060/2010), respectively. References: 1. Beer P. D.; Gale P. A. Angew. Chem. Int. Ed. 2001, 40, 486. 2. Figueira F.; Cavaleiro J. A. S.; Tome J. P. C. J. Porphyrins Phthalocyanines 2011, 15, 517. 3. Cormode D. P.; Davis J. J.; Beer P. D. J. Inorg. Organomet. Polym. Mater. 2008, 18, 32. 4. Carvalho C. M. B.; Alves E.; Costa L.; Tome J. P. C.; Faustino M. A. F.; Neves M. G. P. M. S.; Tome A. C.; Cavaleiro J. A. S.; Almeida A.; Cunha A.; Lin Z.; Rocha J. ACS Nano 2010, 4, 7133.

PC46
Synthesis of cyclitols and biological evaluation
J. N. Lana, A. A. Rosatella, Carlos A. M. Afonso
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal
In 1966, McCasland anticipated that “pseudo-sugars may be found acceptable in place of
corresponding true sugars to some but not all enzymes or biological systems, and thus
might serve to inhibit growth of malignant or pathogenic cells”.
Carbasugars are important molecules, that
can be divided in carbafuranoses and
carbapyranoses, in the last ones are
included aminocyclitols such as
validamine, valienamine, valiolamine,
validamycin A (antibiotic) and analogues
(Figure 1), that possess enzyme inhibitory
activity toward certain glycosidases. As a
result, these molecules have been
important lead compounds for the
development of potent and specific
inhibitors of glycosidases involved in the
intestinal metabolism of sugars. Among
them, valiolamine possesses very potent activity against maltase and sucrase, voglibose is
a clinically useful medicine to control diabetes1 and cyclophellitol may promote inhibition of
the human immunodeficiency virus (HIV) and with possible antimetastatic therapeutic
activity.2
Recently, we reported a method for the direct trans-dihydroxylation of olefins by aqueous
hydrogen peroxide catalyzed by p-toluenesulfonic acid (PTSA) at 50°C. The expected
mechanism resulted of epoxidation followed by fast ring opening by water in which both
steps are catalysed by PTSA.3,4
In addition, was observed that 1,4-cyclohexadiene
originates only one product and in high yield (77%, trans, trans-cyclohexane-1,2,4,5-tetraol
diasteriomer (Scheme 1)).4 This outstanding stereoselectivity and formation of high
hydroxylated cyclohexane opened new opportunities for the development of new
competitive synthetic routes to cyclitols, including carbasugars.
Scheme 1: Direct thans-dihydroxylation
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia (POCI 2010), FEDER (Ref.: PTDC/QUI-QUI/119823/2010, SFRH/BPD/75045/2010) and Bolsa Universidade de Lisboa/Fundação Amadeu Dias 2012/2013 for financial support. References: 1. Ogawa, S.; Kanto, M. J. Nat. Prod. 2007, 70, 493 and references cited therein. 2. Arjona, O.; Gómez, A. M.; López, J. C.; Plumet, J. Chem. Rev. 2007, 107, 1919. 3. Rosatella, A.A.; Afonso, C.A. J. Chem. Education, 2011, 88, 1002. 4. Rosatella, A.A.; Afonso, C.A. Adv. Synth. Catal. 2011, 353, 2920.
Figure 1: Reported aminocyclitols with biological activity.

PC47
Synthesis and antioxidant activity of ferrocenyl derivatives
J. Albertino Figueiredo,a S. Santos,
a M. Isabel Ismael,
a Ana C. Fernandes
b
aDepartamento de Química, Unidade de Materiais Têxteis e Papeleiros, Universidade da Beira Interior,
6201-001 Covilhã, Portugal bCentro de Química Estrutural, Complexo I, Instituto Superior Técnico, 1049-
001 Lisboa, Portugal
Since ferrocene was first prepared and characterized half a century ago, its chemistry and
applications have been extensively studied. Ferrocene is a sandwich organometallic
compound with the chemical formula Fe(C5H5)2. It is a prototypical metallocene that consists
of two cyclopentadienyl rings that are bound on the opposite sides of the central iron (Fe)
atom.1,2
Ferrocene has a rich chemistry stemming from the accessibility of a large number of
derivatives and their facile redox properties; this has led to their use in industry, including
petroleum, plastics, textiles, and metallurgy. Current areas of interest in ferrocene chemistry
include use in catalysis, as sensors, and as immunoassay reagents.1 Many ferrocenyl
compounds display interesting cytotoxic, antitumor, antifungal, antioxidant and DNA-
cleaving activity.3
In this work will be presented the synthesis of several ferrocenyl derivatives (figure 1), and
the determination of antioxidant activity of the products, by the DPPH method.4
It was
possible to compare the influence of the side chains in the oxidant activity of the products 1-
7.
Fe
R 1 R = H2 R = COCH33 R = CHO4 R = CH2OH5 R = CH2OCH2CCH6 R = CHNNH[C6H3(NO2)2]7 R = COCH2(C6H4Cl)
Figure 1: Ferrocenyl derivatives.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia (FCT) for financial support through project PTDC/QUI-QUI/102114/2008. References: 1. Gyömöre Á.; Csámpai A.; J. Organomet. Chem., 2011, 696, 533-539. 2. Trivedi R.; Deepthi S. B.; Giribabu L.; Sridhar B.; Sujitha P.; Kumar C. G.; Ramakrishna K. V. S.; Eur. J. Inorg. Chem., 2012, 2267-2277. 3. Maguene G. M.; Jakhlal J.; Ladyman M.; Vallin A.; Ralambomanana D. A.; Bousquet T.; Maugein J.; Lebibi J.; Pélinski L.; Eur. J. Med. Chem., 2011, 46, 31-38. 4. Kumar, A.; Sharma, P.; Kumari, P.; Lal Kalal, B.; Bioorg. Med. Chem. Lett., 2011, 21, 4353–4357.

PC48
Bioconversion of ketones by mycelia of marine-derived fungi and
organic composting
Lenilson C. Rochaa, João V. Comasseto
a, Suzan P. Vasconcellos
b, André L. M. Porto
c
aLaboratório de Síntese de Compostos de Selênio e Telúrio, Instituto de Química de São Carlos,
Universidade de São Paulo, Avenida Prof. Lineu Prestes, 748, Cidade Universitária, 05508-900, São
Saulo, SP, Brazil. bChefe do Setor de Microbiologia, Imunologia e Parasitologia, Departamento de
Ciências Biológicas, Universidade Federal de São Paulo, Rua Artur Riedel, 275, Jardim Eldorado,09972-
270,Campus Diadema, SP, Brazil. cLaboratório de Química Orgânica e Biocatálise, Instituto de Química
de São Carlos, Universidade de São Paulo, Avenida João Dagnone, nº 1100, Ed. Química Ambiental,
Jardim Santa Angelina, 13563-120, São Carlos, SP, Brazil.
The asymmetric reduction of ketones plays an important role in synthetic chemistry since
chiral secondary alcohols are versatile synthons, especially for the preparation of
enantiomerically pure pharmaceuticals.1 New methodologies have been used in the
bioconversion of ketones. In this work, mycelia of (Penicilliumcitrinun CBMAI 1186 and
Trichoderma sp. CBMAI 932) marine-derived and (Aspergillus sp.146 and FPZSP152)
organic composting fungi were employed the bioconversion of ketones 1-2. Fungal mycelia
were incubated at 32 ºC for 12 days on an orbital shaker (150 rpm) and harvested by
Buchner filtration. The biocatalytic reactions were carried out with 5.0 g (wet weight) of
mycelia and 50 mg of ketones 1 and 2 previously dissolved in 300 μL DMSO and added to
a phosphate buffer solution (Na2HPO4/KH2PO4, 100 mL, 0.1 M), both reaction were realized
in various pH values, i.e. 4, 5, 6, 7 and 8 (Scheme 1). The reactions were performed for 12
days in an orbital shaker (130 rpm, 32 oC). The mycelia of marine-derived fungi and organic
composting were able to catalyze the bioreduction of ketone 1 and 2 with good conversion
and enantiomeric excess. The corresponding 4-phenylbutan-2-ol (1a) was observed in
ketone 1 bioconversion, however, in ketone 2 reduction 4-phenylbutan-2-one (1),
4-phenylbutan-2-ol (1a) and 4-phenylbut-3-en-2-ol (2a) were observed. In the case of α,β-
unsatured ketone 2, selectivity between the two functionalities was observed.
Scheme 1. Bioreduction of ketones 1-2 by mycelia of fungi.
Acknowledgments: The authors thank the Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP) for financial support and scholarship. References: 1.(a) Rocha, L. C, Ferreira, V. F, Luiz, R. F, Sette, L. D, Porto, A. L. M. Stereoselective bioreduction of 1-(4-methoxyphenyl)ethanone by whole cells of marine-derived fungi. Marine biotech. 2012, 14, 358–362; (b) Nakamura, K. Yamanaka, R. Matsuda, T. Harada, T. Recent developments in asymmetric reduction of ketones with Biocatalysts. Tetrahedron: Asymmetry, 2003, 14, 2659–2681; (c) K. Nakamura,T. Matsuda. Biocatalytic Reduction of Carbonyl Groups. Curr. Org. Chem.2006, 10, 1217–1246.

PC49
Chemical synthesis of a bioavailable anthocyanin metabolite: cyanidin-4’-O-methylglucoside
L. Cruz, Nuno Mateus, Victor de Freitas
Centro de Investigação em Química, Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade do Porto, Portugal
Anthocyanins are naturally occurring polyphenols with potential benefits to human health.1
In the human organism, anthocyanins are metabolized to different derivatives (glucuronides,
methylethers and sulfates), which are further found in plasma and should have different
biological effects from their precursors. This enterohepatic recycling opens a new filed of
interest that remains a challenge: the biological properties of anthocyanin metabolites. To
evaluate their activity, it is important to have sufficient quantities of these molecules which
cannot be obtained commercially or by isolation from biological fluids. In fact, some
anthocyanin metabolites (methylated, glucuronides and glutathione adducts from cyanidin
and delphinidin-3-glucosides) were recently obtained through enzymatic synthesis by our
research group, however, the yields are poor and the synthesis expensive.2 In order to
obtain higher amounts of anthocyanin metabolites, chemical synthesis as already achieved
for other flavonoid metabolites such as catechins and quercetins glucuronides, methylethers
and sulfates should allow to obtain good quantities. The achievement of anthocyanin
metabolites likely to occur in vivo, is crucial in order to have proper standards currently
lacking in several biological studies. Known synthetic routes to obtain anthocyanidin and
other flavylium pigments result from the construction of the C-ring by cyclization which
involves the coupling together of two halves, the so-called “Eastern” and “Western” portions
of the molecule. Due to the inherent instability of anthocyanins in neutral and basic
conditions, only a few syntheses have been reported to date. Among them, the acidic aldol
condensation between a phenolic aldehyde and an aryl ketone pioneered by Robert
Robinson in the early 1900s remains efficient until date and has subsequently been further
developed.3
Although the method is classical, it has never been driven towards the
synthesis of metabolites.
We recently reported the synthesis of cyanidin-4’-O-methylglucoside, a metabolized form of
cyanidin-3-glucoside previously found in vivo (Scheme 1).4
Scheme 1: Synthesis of cyanidin-4’-O-methylglucoside: (i) HCl (g), AcOEt, 0ºC to RT, overnight; (ii)
Triethylsilane, Pd/C 10%, MeOH, RT, 10 min; (iii) KOH, H2O/MeOH 1:1, RT, 15 min. Acknowledgements: Luís Cruz gratefully acknowledges the Post. Doc. Grant from Fundação para a Ciência e a Tecnologia (FCT) (SFRH/BPD/726 52/2010) and to FCT project grant (PTDC/AGR-TEC/2227/ 2012). References: 1. Erdman, J. W., Jr.; Balentine, D.; Arab, L.; Beecher, G.; Dwyer, J. T.; Folts, J.; Harnly, J.; Hollman, P.; Keen, C. L.; Mazza, G.; Messina, M.; Scalbert, A.; Vita, J.; Williamson, G.; Burrowes, J. J. Nutr. 2007,
137, 718S. 2. Fernandes, I.; J. Azevedo; A. Faria; C. Calhau; De Freitas, V.; Mateus, N. J. Agr. FoodChem. 2009, 57, 735. 3. Dangles, O.;Elhajji, H. Helv. Chim. Acta 1994, 77, 1595. 4. Cruz, L; Mateus, N.; De Freitas, V. Tetrahedron Lett. 2013, 54, 2865.

PC50
Spectroscopic study of the rearrangement of 2,6-diisopropyl-1,4-
quinone
Margarida Espadinha, Paula S. Branco,
Luísa M. Ferreira
REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de
Lisboa, 2829-516 Caparica, Portugal
The anesthetic propofol (1) phase I free-metabolites 2,6-diisopropyl-1,4-quinol (2) and 2,6-
diisopropyl-1,4-quinone (3) may be associated with some potential toxic effects of this
substance1,2
On standing compound 3 gave rise to the rearrangement product 4 in a
chloroform solution (Figure 1). This product was isolated in 46% yield which is in
accordance with a redox reaction occurring with the sacrifice of one molecule of quinone.
To elucidate the mechanism of this rearrangement NMR and EPR techniques were used to
clarify the isopropyl-allylic rearrangement.
Figure 1: Rearrangement compound 3, phase I metabolite of the anesthetic propofol (1).
Acknowledgements: We thank the NMR facilities from REQUIMTE (Portugal) for NMR spectra acquisition. References: 1. Favetta P.; Guittona J.; Degouted C.S.; Van Daele L.; Boulieu R. J. Chromatogr. B 2000, 742, 25-35. 2. Silva A.; Amorim P.; Ferreira L.; Branco P.; de Pinho P.G.; Ferreira D. A. Eur. J. of Anesth. 2012, 29, 136.

PC51
Ionic liquids derived from sugars as chiral selectors
João Domingos, Ana Maria, Nuno Costa, Sara Matos, Marco Gomes da Silva, Manuela Pereira
REQUIMTE/CQFB, Chemistry Department, Faculdade de Ciências e Tecnologia, Universidade Nova de
Lisboa, Campus da Caparica, Caparica, 2829-516 Caparica, Portugal
Ionic liquids (ILs) are very simple low melting point organic salts. The combination of
organic cations and bulky inorganic anions provides the possibility to create tailor-made ILs
derived from sugars,1
with different properties, to be used as catalysts in asymmetric
synthesis, chiral selectors in NMR studies or as stationary phases in chiral chromatographic
analysis.2
New imidazolium and pyridinium triflate ionic liquids derived from isosorbide, isomannide
and cyclodextrins were synthesized in one pot free solvent procedure. The ability to use
theseionic liquids as chiral selectors was demonstrated by 19
F-NMR spectra and also when
they are used as stationary phases in gas liquid chromatography. (Scheme 1)
Some of these ionic liquids were used as stationary phases in homemade capillary columns
for enantio GC and its chiral selectivity explored in the separation of ethers, esters and
lactones racemic mixtures.
Scheme 1: Synthesis of imidazolium and pyridinium triflate ionic liquids from cyclodextrins and
isosorbide or isomannide and an application as stationary phase in enantio-GC. Acknowledgements: This work was supported by the Fundação para a Ciência e Tecnologia (FCT,
Portugal) through PTDC/QUI-QUI/100672/2008. References: 1. Pereira, M. M. A. Mini-Rev. Org. Chem. 2012, 9, 243-260. 2. Huang, K.; Zhang, X. T.; Armstrong, D. W.; J. Chromatogr. A, 2010, 1217, 5261-5273.

PC52
Benzo[a]phenoxazinium chlorides possessing chlorinated
terminals: synthesis, photophysics and photostability studies
Marcello M. T. Carvalho,a B. Rama Raju,
a,b Paulo J. G. Coutinho,
b M. Sameiro T. Gonçalves
a
aCentre of Chemistry, Centre of Physics
b, University of Minho, Campus of Gualtar, 4710-057 Braga,
Portugal
Fluorescent probes are essential in fluorescence based bioanalytical techniques that have
been widely used in a diversity of scientific areas, including environment, biology, pharmacy
and medicine. They are involved in studies with cells, proteins, enzyme activity, DNA and
RNA, among others. The interaction of fluorophores with targets can be accomplished by
covalent or non-covalent linkages, carried out through reactive groups suitable for chemical
binding to biomolecules or by a variety of mechanisms, such as electrostatics, hydrophobics
and hydrogen-bonding, respectively.1 As a continuation of our research in the synthesis and
characterisation of fluorescence probes,2
the present work aims to describe the preparation
of a new set of benzo[a]phenoxazinium chlorides possessing isopentylamino, (2-
cyclohexylethyl)amino and phenethylamino substituents at 5-position and 3-chloropropyl
groups at the amine in 9-position of the polycyclic system (Figure 1). The later terminals
allow covalent labelling of targets by nucleophilic substitution, besides the intrinsic non-
covalent ability of benzo[a]phenoxazinium chlorides synthesized. Fundamental
photophysics as well as photochemical stability studies were carried out and will be
discussed.
Figure 1: Structure of benzo[a]phenoxazinium chlorides.
Acknowledgements: Thanks are due to Fundação para a Ciência e Tecnologia (FCT, Portugal) for financial support to the NMR portuguese network (PTNMR, BrukerAvance III 400-Univ. Minho), FCT and FEDER (European Fund for Regional Development)-COMPETE-QREN-EU for financial support to the Research Centres CQ/UM [Strategic Project PEst-C/QUI/UI0686/2011 (FCOMP-01-0124-FEDER-022716)] and CFUM [Strategic Project PEst-C/FIS/UI0607/2011 (F-COMP-01-0124-FEDER-022711)]. A post-doctoral grant to B. R. Raju (SFRH/BPD/62881/2009) is also acknowledged to FCT, POPH-QREN, FSE. We thank the Fundação para a Ciência e Tecnolgia for financial support. References: 1. a) Gonçalves, M. S. T. Chem. Rev. 2009, 109, 190. b) Pansare,V. J.; Hejazi, S.; Faenza, W. J.; Prud’homme, R. K. Chem. Mater. 2012, 24, 812. 2. a) Raju, B. R.; Firmino, A. D. G.; Costa, A. L. S.; Coutinho, P. J. G.; Gonçalves, M. S. T. Tetrahedron 2013, 69, 2451. b) Firmino, A. D. G.; Raju, B. R.; Gonçalves, M. S. T. Eur. J. Org. Chem. 2013, 1506.

PC53
Scheme 1: Schematic procedure of the ATRP process.
A new route to the synthesis of well-defined molecularly imprinted
polymers (MIPs) by ATRP: application as adsorbents for solid phase
extraction
M. Simõesa, N. Martins
b, M. J. Cabrita
b, Anthony J. Burke
a, R. Garcia
b
aCentro de Química de Évora e Departamento de Química da ECTUE,
bICAAM- Instituto de Ciências Agrárias e Ambientais Mediterrânicas - Universidade de Évora
The use of pesticides to prevent pests can cause their presence in food matrices. To date
several methodologies have been explored for the quantification of pesticides in various
food matrices, which comprise several extraction procedures and the use of
chromatographic techniques. However, the analysis of these compounds in vegetable
matrices with high fat content is still a difficult task due to the inherent complexity of the
sample. Recently, the use of molecularly imprinted polymers (MIPs) have been successfully
applied for selective trace analysis of pesticides in various food matrices. These polymeric
materials have recognition sites with high molecular specificity for the target analyte and can
be regarded as synthetic receptors with artificially made binding sites.1
Atom transfer radical polymerization (ATRP) is a catalytic process using a metal complex, in
which the transitionmetal (Mt) can exist in two
different oxidation states.2
This synthetic
technique was chosen for the preparation of
well defined MIPs with predetermined molecular
weights, narrow molecular weight distributions,
and high degrees of chain end functionalities
(Scheme 1).
In this presentation we will discuss our efforts at
the synthesis of well-defined MIPs - used as
adsorbents for solid phase extraction of
pesticides from olive oil - via the ATRP
procedure, including their physico-chemical
characterization.
Acknowledgements: This work is funded by FEDER (Regional Development European Fund), COMPETE (Operational Program Factors for Competitivity) and FCT (Foundation for Science and Technology), under the Strategic Project PEst-C/AGR/UI0115/2011 and Project PTDC/AGR-ALI/117544/2010. References: 1. Martín- Esteban A., Trends in Analytical Chemistry, 2013, 45, 169-181. 2. Matyjaszewski, K.; Tsarevsky, N. V. Nat. Chem., 2009, 1, 276.

PC54
An Improved Approach to 1H,3H-Thiazolo[3,4-a]benzimidazoles
Maria I. L. Soares, Teresa M. V. D. Pinho e Melo
Department of Chemistry, University of Coimbra, 3004-535 Coimbra, Portugal
The study of pericyclic reactions of dipolar systems, azafulvenium methides and
diazafulvenium methides, is one of our research interests.1a,b
Currently we are interested in
extending this chemistry to benzodiazafulvenium methides. In fact, we recently described
the generation and reactivity of benzo-2,3-diazafulvenium methide derivatives.1c
We are
now interested in exploring the chemistry of benzo-2,5-diazafulvenium methides generated
from 2,2-dioxo-1H,3H-thiazolo[3,4-a]benzoimidazoles as an approach to new
benzo[d]imidazole derivatives. These are an interesting class of heterocyclic compounds
since many benzimidazole2 and, particularly, thiazolo[3,4-a]benzimidazole
3a derivatives are
known to exhibit a wide variety of biological activities. In order to proceed with this research
we need to implement an efficient approach to 1H,3H-thiazolo[3,4-a]benzimidazole
derivatives. The one-pot synthesis of thiazolo[3,4-a]benzimidazoles from the appropriate
1,2-phenylendiamine, an aldehyde and 2-mercaptoacetic acid is known.3 However, when
we applied these condensation-cyclization reaction conditions for the synthesis of our
thiazolo[3,4-a]benzimidazoles, the target products were obtained in low yield and often by-
products, such as benzimidazole 2, were obtained (Scheme 1). Thus, we outlined an
alternative three-step approach which involves the mono protection of 1,2-
phenylenediamine followed by a condensation reaction to give thiazolidin-4-one derivatives
4, thereby preventing the formation of by-products such as 2. Microwave irradiation of 4
affords the target thiazolo[3,4-a]benzimidazoles 5 via an efficient and clean deprotection-
cyclization reaction (Scheme 1). In some cases compound 5 was also obtained in the
second step of this sequence. Further details of this study will be disclosed.
Scheme 1: Synthesis of 1H,3H-thiazolo[3,4-a]benzimidazoles 5.
Acknowledgements: Thanks are due to FCT (PEst-C/QUI/UI0313/2011), FEDER, COMPETE and QREN for financial support.
References: 1. a) Pinho e Melo, T. M. V. D.; Soares, M. I. L.; Gonsalves, A. M. d'A. R.; Paixão, J. A., Beja, A. M.; Silva, M. R. J. Org. Chem., 2005, 70, 6629-6638. b) Pinho e Melo, T. M. V. D.; Nunes, C. M.; Soares, M. I. L.; Paixão, J. A., Beja, A. M.; Silva, M. R. J. Org. Chem., 2007, 72, 4406-4415. c) Soares, M. I. L.; Nunes, C. M.; Gomes, C. S. B.; Pinho e Melo, T. M. V. D. J. Org. Chem. 2013, 78, 628-637. 2. a) Kim, J. S.; Sun, Q.; Gatto, B.; Yu, C.; Liu, A.; Liu, L. F.; La Voie, E. J. Bioorg. Med. Chem. 1996, 4, 621-631. b) Ram, S.; Wise, D. S.; Wotring, L. L.; McCall. J. W.; Townsend, L. B. J. Med. Chem. 1992, 35, 539-547.
3. a) Chimirri, A.; Monforte, P.; Musumeci, L.; Rao, A.; Zappalà, M.; Monforte, A.-M. Arch. Pharm. Pharm. Med. Chem. 2001, 334, 203-208. b) Chimirri, A.; Grasso, S.; Monforte, P.; Romeo, G.; Zappalà, M. Synthesis 1988, 244-246.

PC55
Brønsted Acid Catalyzed Ring Opening of Aziridines:
Taming and Directing the Nucleophilicity of Carboxylic Acids.
Mattia R. Monaco, Belén Poladura, Miriam Diaz, Markus Leutzsch, Benjamin List
Max Planck Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, D-45470 Mülheim an der Ruhr,
Germany
Chiral 1,2-aminoalcohols are valuable compounds due to their abundance in medicinal and
natural products and for this reason the scientific community has invested great efforts in
the development of synthetic routes for their preparation.1 Ring opening of aziridines, three-
membered nitrogen containing rings, with oxygen nucleophiles represents a very interesting
approach towards this scaffold, but, despite being well explored in racemic procedures, a
catalytic asymmetric variant is unknown to date.
While asymmetric Brønsted acid catalysis has flourished during the last decade, the
activation of aziridines still represents an elusive target.2
Here we report a novel catalytic
strategy for the title transformation employing a chiral acid as the organocatalyst and
carboxylic acids as the nucleophiles of choice. Cyclic and acyclic meso-aziridines could be
desymmetrized into the desired protected trans 1,2-aminoalcohols with very high yield (up
to 99%) and excellent stereoselectivity (e.r. up to >99.5:0.5), while racemic terminal
aziridines could undergo a very effective kinetic resolution (s factor up to 51). (Scheme 1)
The use carboxylic acids in such an organocatalytic reaction is strategically valuable
because they represent a masked form of the free hydroxyl moiety, which is often desired.3
Scheme 1: Organocatalytic access to 1,2-aminoalcohols
Acknowledgements: Max-Planck-Gesellschaft, European Research Council and Max-Planck-Institut für Kohlenforschung analytical department (HPLC, GC, X-Ray, NMR) are gratefully acknowledged. References: 1. Bergmeier S. C. Tetrahedron 2000, 56, 2561. 2. a) Kampen D.; Resinger C. M.; List B. Top. Curr. Chem. 2010, 291, 395. b) Larson S. E.; Baso J. C.; Li G.; Antilla J. C. Org. Lett. 2009, 11, 5186. 3. Monaco M. R.; Poladura B.; Diaz M.; Leutzsch M.; List B. 2013 manuscript in preparation

PC56
Metal-Ligand Systems and Structural Diversity Based on Cyclic
Peptides
Michele Panciera, Manuel Amorín, Luís Castedo, Juan R. Granja
Departamento de Química Orgánica y Centro Singular de Investigación en Química Biológica y
Materiales Moleculares (CIQUS). Campus Vida. Universidad de Santiago de Compostela. 15782
Santiago de Compostela, Spain
Our research group is implicated in the design and synthesis of supramolecular structures
based on self-assembling process of α,-cyclic peptides (CP).1 The self-assembling process
of α,-CPs takes place through a β-sheet-type hydrogen bonding interactions and it is, in
general, independent of the peptide sequence allowing the preparation of a large variety of
structures with a variety of applications, such as electron and energy transfer process,
molecular tweezers and so on.2 In this context, we have found that the β-sheet register
derived from no-symmetrical CPs played an important role in hierarchical supramolecular
processes, due to the formation of several non equivalent dimers of which only one, in most
of the cases, is functionally active and it can be controlled by metal coordination. Here, we
present the designed and synthesis of microparticles based on CPs that has a ligand
attached to one of its side chain that is able to coordinate metals (Figure 1). Our results
show the potential of this metal coordination in the design of new supramolecular catalytic
systems.3
Figure 1: Supramolecular nanometric design based on cyclic peptides.
Acknowledgements: This work was supported by the Spanish Ministry of Economy and Competivity (MEC) and the ERDF [(CTQ2010-15725, and Consolider Ingenio 2010 (CSD2007-00006)], by the Xunta de Galicia (GRC2010/132). M.A. thanks the Spanish MEC for his Ramón y Cajal contract and M.P. thanks to the Spanish Ministry of Foreign Affairs and Cooperation for her MAEC-AECID fellowship grand. References: 1. a) Brea, R. J.; Reiriz, C.; Granja, J. R. Chem. Soc. Rev. 2010, 39, 1448; b) Amorín, M.; Castedo, L.; Granja, J. R. J. Am. Chem. Soc. 2003, 125, 2844. 2. Brea, J. R.; Pérez-Alvite, M. J.; Panciera, M.; Mosquera, M.; Castedo, L.; Granja, J. R. Chem. Asian J. 2011, 6, 110. 3. a) Panciera, M.; Amorín, M.; Castedo, L.; Granja, J. R. Chem. Eur. J. 2013, 19, 4826; b) Panciera, M.;
Amorín, M.; Castedo, L.; Granja, J. R. In preparation.

PC57
The Synthesis of 2-Aryl-hypoxanthines: A Novel and Efficient
Synthetic Approach
Nádia Senhorães, Alice M. Dias, M. Fernanda Proença
Departamento de Química, Universidade do Minho, Campus de Gualtar, 4710-057 Braga, Portugal
[email protected]; [email protected]
The 6-oxopurine nucleus is a ubiquitous central core present in many natural important
small molecules. Acyclic nucleoside phosphonates (ANPs) constitute a group of compounds
with a wide range of biological activities including antiviral, cytostatic, antiparasitic and
immunomodulatory effects. In particular, compounds with hypoxanthine as a nucleobase,
found important application as antimalarial and antiviral agents.1,2
In our research group, a number of substituted purines have been obtained from a common
imidazole precursor, the 5-amino-4-cyanoformimidoyl imidazoles (1). In a previous work,
amidines 2 were easily prepared from 1, benzoic anhydride and ammonia by simple and
inexpensive methods.3 Recent results demonstrated that, under appropriated reaction
conditions, amidines 2 undergo a facile hydrolysis leading to imides 3. These new
intermediates 3 could be easily converted into the target 2-arylhypoxanthines 4 by
intramolecular cyclization (Scheme 1). The novel molecules 3 and 4 were isolated in good
yield and their structures were assigned on the basis of elemental analysis, IR and NMR
spectroscopy, including 13
C and 2D techniques. Synthetic results and structural data will be
presented and discussed.
Scheme 1: Synthesis of hypoxanthines 4
Acknowledgements: The authors gratefully acknowledge the University of Minho and the Foundation for the Science and Technology (FCT, Portugal) for financial support to the NMR portuguese network (PTNMR, Bruker Avance III 400-Univ. Minho). FCT and FEDER (European Fund for Regional Development)-COMPETE-QREN-EU for financial support to the Research Centre, CQ/UM [PEst-C/QUI/UI0686/2011 (FCOMP-01-0124-FEDER-022716)] and a PhD grant awarded to Nádia Senhorães (SFRH/BD/73721/2010). References: 1. M. B. Allwood, B. Cannan, D. M. F. van Aaltena, I. M. Eggleston, Tetrahedron, 2007, 12294–12302. 2. D. Hockova, D. T. Keough, Z. Janeba,T.-H. Wang, J. Jersey, L. W. Guddat. J. Med. Chem. 2012, 55, 6209−6223 3. N. Senhorães, A. M. Dias, L. M. Conde, M. F. Proença, Synlett, 2011, 181-186.

PC58
Synthesis of Prodelphinidins – work in progress
Natércia Teixeira, Nuno Mateus, Victor de Freitas
Centro de Investigação em Química, Departamento de Química e Bioquímica, Faculdade de Ciências da
Universidade do Porto, Rua do Campo Alegre, 687, 4169-007, Porto, Portugal
Prodelphinidins are polymeric tannins composed of gallocatechin or epigallocatechin units.
They were found in barley, beer,1 pomegranate peels,
2 redcurrant
3 and green tea
4.The
interest on this kind of compounds is growing due to their significant bioactivities.5a,b
They
are extremely difficult to identify and purify from nature and there are only a few standards
to compare. Synthesis allows the access to sufficient amounts of compounds to perform
chemical, biochemical and pharmacological studies and to have standards to compare and
identify these compounds on natural sources. Figure 1 shows a generic procedure of
synthesis of a prodelphinidin, involving five steps. Briefly, the synthesis starts with the
protection of (-)-epigallocatechin (1) hydroxyl groups, giving (-)-epigallocatechin4Bn (2) that
will act as upper unit in the dimer; the second step is the protection of the down unit, (+)-
catechin (3), giving (+)-catechin4Bn (4); the third step is to benzylate the upper unit at C4,
giving (-)-epigallocatechin4Bn(Bn) (5); the fourth step is the condensation of both units
giving epigallocatechin-catechin9Bn (6); and finally, the removal of the protection groups.
Several methods are been studied for the de-benzylation since this is the most sensible
step.
Figure 1: Generic synthesis of a prodelphinidin.
6
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support through
a Ph.D. grant (SFRH/BD/70053/2010).
References: 1. Dvorakova, M., Moreira, M. M., Dostalek, P. Skulilova, Z., Guido, L. F., Barros, A. A. J. Chrom. A, 2008, 1189, 398-405. 2. Plumb, G. W., de Pascual-Teresa, S., Santos-Buelga, C., Rivas-Gonzalo, J. C., Williamson, G. Redox Report 2002, 7, 41-46. 3. Pascual-Teresa de, S., Santos-Buelga, C., Rivas-Gonzalo, J. C. J. Agric. Food Chem. 2000, 48, 5331-
5337. 4. Cheng, H. Y., Lin, C. C., Lin, T. C. Antivir. Chem. Chemother., 2002, 13 (4), 223-229. 5. a) Ferreira, D., Li, X.-C-, Nat. Prod. Rep., 2000, 17, 193. b) Ferreira, D., Li, X.-C-, Nat. Prod. Rep., 2002, 19, 517. 6. Ahmed, I., PhD Thesis, Paderborn, 2007

PC59
Synthesis and Characterized of New Open Chain 2,2’-Bipyridyl-
Linked Pyridine-, Bipyridine-, or Phenanthroline-Derivatived Schiff
Base Ligands
Nesrin Beynek, Nurcihan Tan
Department of chemistry, Trakya University, Edirne 22000, Turkey
Schiff-base ligands recently have taken more attention in bioinorganic and bioorganic in
medicine because of the fact that it has antimicrobial and chemotherapy features besides
they are used in different industrial areas: Environment chemistry, agriculture, and paint
industry. In this study, we describe the synthesis and characterization of three new versatile
acyclic schiff base containing 2,2’-bipyridyl-linked pyridine, bipyridine or phenanthroline
groups in their framework. The structure of these compounds was elucidated by using
spectroscopic methods.

PC60
Synthesis of Functional Organic Bipyridinium Salts
Noémi Jordão, Fernando Pina, Luís C. Branco
REQUIMTE, Departamento de Química, FCT-UNL, Faculdade de Ciências da Universidade Nova de
Lisboa, 2829-516 Caparica, Portugal
4,4’-bipyridine is a useful organic precursor for the preparation of functional bipyridinium
salts. These ionic species are electroactive, and its toxicity arises from the ability of this
dication to interrupt biological electron transfer. In general, they can be used as herbicides,
redox indicators, functional organic electrochromic materials or coordination polymers.1
In this context, we developed more sustainable synthetic methods for the preparation of
mono and disubstituted bipyridinium cations including the incorporation of different alkyl or
ether side substituents. Novel bis(bipyridinium) salts with different linkers (n-alkyl or ether)
have been also developed. All bipyridinium and bis(bipyridinium) cations were combined
with several organic anions such as bistriflimide [NTf2] and docusate [AOT] (Figure 1).
These compounds were obtained in moderate to high yields as well as higher purity levels.
All organic bipyridinium salts were characterized by 1H and
13C NMR and FTIR spectra,
calorimetric studies (melting point, glass and decomposition temperature) and ESI-MS
spectra.
Figure 1: Structure of functional organic bipyridinium and bis(bipyridinium) salts prepared in this work.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support of this work (PTDC/CTM-NAN/120658/2010). References: 1. a) Mortimer R. J., Annu. Rev. Mater. Res. 2011, 41, 241. b) Grenier M. C. et al, Bioorg. Med. Chem. Lett. 2012, 22, 4055. c) Pepitone M. F. et al, Org. Lett. 2007, 9, 801.

PC61
New 2-(2,6-diarylpyridin-4-yl)porphyrin derivatives as fluorescent probes for metal cations
Nuno M. M. Moura,a Cristina Nuñez,
b Maria A. F. Faustino,
a Filipe A. Almeida Paz,
c
José A. S. Cavaleiro,a Maria G. P. M. S. Neves,
a José Luis Capelo,
c Carlos Lodeiro
c
aDepartment of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal;
bBIOSCOPE Group,REQUIMTE-CQFB, Chemistry Department, Faculty of Science and Technology, University NOVA of Lisbon2829-516, Monte da Caparica, Portugal;
cDepartment of Chemistry and
CICECO, University of Aveiro, 3810-193 Aveiro, Portugal
The modification of readily available meso-tetraarylporphyrins, namely those with β-formyl
groups, is recognized as an important tool to give access to new porphyrin derivatives with
adequate features to be used in medicine, catalysis, electronic devices, sensors or dyes for
solar cells.1
The development of new fluorescent sensors and chemosensors for metal ion
recognition is an area in high expansion due to the sensitivity, selectivity and response time
of the approach when compared with others analytical techniques usually used for metal ion
detection.2
Synthetic strategies based on well-established transformations of the formyl group are
being explored to achieve such modifications.3
The reaction of 2-formyl-5,10,15,20-
tetraphenylporphyrin with aryl methyl ketones and ammonium acetate, in the presence of
La(OTf)3, affords benzoporphyrins and 2-(2,6-diarylpyridin-4-yl)porphyrin derivatives. This
methodology was used to prepare, for the first time, a 2-(2,2’:6’,2’’-terpyridin-4’-
yl)porphyrin.4
In this communication we describe the synthesis, photophysical parameters and the study
of sensing ability towards several metal ions of new 2-(2,6-diarylpyridin-4-yl)porphyrins.5
Scheme 3.
Acknowledgements: Authors are grateful to the Universidade de Aveiro, Fundação para a Ciência e a Tecnologia (FCT) European Union, QREN, FEDER and COMPETE for funding the QOPNA Research Unit (project PEst-C/QUI/UI0062/2011), the Portuguese National NMR Network (RNRMN) and CICECO (PEst-C/CTM/LA0011/2011) for specific funding towards the purchase of the single-crystal X-ray diffractometer as well as to REQUIMTE-FCT-UNL for their facilities. Thanks are also due to the Xunta de Galicia (Spain) through project 09CSA043383PR and Scientific PROTEOMASS Association (Ourense-Spain) for financial support. N.M.M. Moura and C. Nuñez thanks FCT/MEC for Post-Doctoral grant SFRH/BPD/84216/2012 and Xunta de Galicia (Spain) for post-doctoral contract (I2C program), respectively.
References: 1. Handbook of Porphyrin Science, Kadish K. M.; Smith K. M.; Guilard, R. (Eds.) Vols. 10-12, World Scientific Publishing Co: Singapore, 2010. 2. a) Dujols V.; Ford F.; Czarnik A. W. J. Am. Chem. Soc. 1997, 119, 7386. b) Ishida M.; Naruta Y.; Tani F. Angew. Chem. Int. Ed. 2010, 49, 91. C) Lodeiro C.; Capelo J. L.; Mejuto J. C.; Oliveira E.; Santos H. M.; Pedras B.; Nuñez C. Chem. Soc. Rev. 2010, 39, 2948. 3. a) Silva A. M. G.; Tomé A. C.; Neves M. G. P. M. S.; Silva A. M. S.; Cavaleiro J. A. S. J. Org. Chem. 2002, 67, 726. (b) Silva A. M. G.; Lacerda P. S. S; Tomé A. C.; Neves M. G. P. M. S.; Silva A. M. S.; Cavaleiro J. A. S.; Makarova E. A.; Lukyanets E. A. J. Org. Chem. 2006,71, 8352. (c) Tomé A. C.; Neves M. G. P. M. S.; Silva A. M. S.; Cavaleiro J. A. S. J. Porphyrins Phthalocyanines, 2009,13, 408. 4. Moura N. M. M.; Faustino M. A. F.; Neves M. G. P. M. S.; Paz F. A. A.; Silva A. M. S.; Tomé A. C.; Cavaleiro J. A. S. Chem. Comm. 2012, 48, 6142.
5.Moura,N. M. M.; Nuñez,C.; Santos, S. M.; Faustino, M. A. F.; Cavaleiro, J. A. S.; Neves, M. G. P. M. S.; Capelo, J. L.; Lodeiro, C. ChemPlusChem 2013, in press.

PC62
Synthesis of Different Thioamide Derivatives using Lawesson’s
Reagent
O. Ortet,a,b,*
, Ana Paula Paivaa
aCentro de Química e Bioquímica , Faculdade de Ciências da Universidade de Lisboa (DQB-FCUL), C8,
Campo Grande, 1749-016 Lisboa, Portugal; bDepartamento de Ciência e Tecnologia, Universidade de
Cabo Verde, 379C, Praia, Ilha de Santiago, Cabo Verde
Nowadays, thioamide derivatives have a large application, namely in pharmaceutical
industry. Additionally, they can be used as metal extractants in hydrometallurgy, because
they may allow a good compromise between efficiency and selectivity and, at the same
time, they have a more “environmental-friendly” composition (with nitrogen and sulfur as
heteroatoms, and without phosphorus atoms). One of the ways to synthesize thioamide
derivatives is starting from their corresponding amides, and making their conversion using
Lawesson´s reagent.
Various new thionating reagents have been prepared and used for synthesis of
organosulfur compounds in the last few years.1-3
The well-known Lawesson´s reagent (LR =
2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane-2,4-disulfide), one of the best
thionating compounds, converts a wide range of carbonyl groups to thiocarbonyls. In this
communication, we present the synthesis and characterization of three different thioamide
derivatives using Lawesson´s reagent (1) as a thionating agent (Scheme 1). Hence, N-
cyclohexyl-N-methyloctanthioamide (2), N,N´-dicyclohexyl-N,N´-dimethylthiodiglycoldithio
amide (3) and N,N´-dimethyl-N,N´-diphenylthiodiglycoldithioamide (4) were obtained. These
compounds were characterized by adequate techniques, including FTIR, 1D and 2D NMR,
GC-MS, LC-MS, and determination of melting point for the solid compounds.
Scheme 1: General synthetic scheme
Acknowledgements: We thank “Fundação para a Ciência e a Tecnologia” (FCT- Portugal) for financial support under project PEst-OE/QUI/UI0612/2011, and Osvaldo Ortet PhD grant SFRH/BD/78289/2011. Thanks are also due to J. M. Nogueira and colleagues for the use of the GC-MS equipment, as well as to Ana M. Rosa da Costa, from the University of Algarve, for her help in LC-MS analyses. References: 1. Ortet O.; Paiva A. P.; Separation Science and Technology 2010, 45, 1130. 2. Polshettiwar V.; Kaushik M. P.; Journal of Sulfur Chemistry 2006, 27, 353. 3. Kodama Y.; Ori M.; Nishio T.; Helvetica Chimica Acta 2005, 88, 187.
N
O
P
S P
S
S
S
R´
R´
DryToluene
R´: OCH3
N
S
N2
RN
SN
S
R
S
N2
RN
SN
O
R
O
(3) R: C6H11(4) R: C6H5
(1) LR -
(2)
DryToluene

PC63
Organobase catalyzed conjugate addition of 4-hydroxypyran-2-ones
on chalcones: Synthesis of novel warfarin analogues and hemiketal
tautomers - a diastereoselective access to warfarin-bicyclic ketal
structures
Oualid Talhi,a José A. Fernandes,
b Diana C. G. A. Pinto,
a Filipe A. Almeida Paz,
b
Artur M. S. Silvaa
aQOPNA and
bCICECO, Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal
Warfarin and analogues have long been studied for their anticoagulant activity.1 The
warfarin basic skeleton is built by coupling 4-hydroxycoumarin to benzylideneacetone via
conjugate addition.2 In this study, we present an efficient organobase catalyzed conjugate
(or Michael) addition of 4-hydroxypyran-2-ones on the α,β-unsaturated ketone system of
chalcones. The synthetic strategy has led to the production of new series of warfarin
analogues. The study of the tautomeric equilibrium featuring in the resulting Michael
adducts is performed by solution 1H NMR spectroscopy which evidences the presence of an
opened acyclic form together with cyclic hemiketal tautomer in equilibrium. The use of the B
ring ortho-hydroxy-substituted chalcones in this reaction has led to a diastereoselective
synthesis of warfarin-bicyclic ketal analogues, this bicyclo[3.3.1]nonane-like structure has
been finely elucidated by X-ray diffractometry (Scheme 1).
O O
O
R1OH
R2
O O
O
R2
OHR1
O O
O
R1
OH
R2
+ organobase
O
O
O
R1
O
R2
O O
O
R2
OHR1
OH
R
R = OH
R = H
- H2O
Scheme 1: Synthesis of novel warfarin analogues and hemiketal tautomers - a diastereoselective access
to warfarin-bicyclic ketal structures.
Acknowledgements: We would like to thank Fundação para a Ciência e a Tecnologia (FCT, Portugal),
the European Union, QREN, FEDER, COMPETE, for funding the Organic Chemistry Research Unit
(QOPNA) (project PEst-C/QUI/UI0062/2011) and the Portuguese National NMR Network (RNRMN).
European Community’s Seventh Framework Programme (FP7/2007-20139 under grant agreement nº
215009) is also acknowledged for financial support.
References:
1. a) Holbrook, A. M.; Pereira, J. A.; Labiris, R.; McDonald ,H.; Douketis, J. D.; Crowther, M.; Wells, P. S.
Arch. Intern. Med. 2005, 165, 1095, b) Au, N.; Rettie, A. E. Drug Metab. Rev. 2008, 40, 355.
2. a) Dong, J.; Du, D-. M. Org. Biomol. Chem. 2012, 10, 8125. b) Blicke, F. F.; Swisher, R. D. J. Am.
Chem. Soc. 1934, 56, 902.

PC64
New Hydrophilic Calix[4]arene-Carbazole Conjugates
Patrícia D. Barata, J. V. Prata
Laboratório de Química Orgânica, Área Departamental de Engenharia Química and Centro de
Investigação de Engenharia Química e Biotecnologia, Instituto Superior de Engenharia de Lisboa,
Instituto Politécnico de Lisboa, R. Conselheiro Emídio Navarro, 1, 1959-007, Lisboa, Portugal.
Fluorescent compounds based on synthetic small molecules are powerful tools to visualize
biological events in living cells and organisms.1 Solubility in aqueous media is essential for
the sensing ability of sensors in a biological environment which can be achieve by the
incorporation of ionic functionality.
In this communication we report the synthesis, characterization and photophysical
properties of new fluorescent bis-calix[4]arene-carbazole conjugates (CALIX-CO2H-CBZs)
incorporating hydrophilic functionalities as pendant groups(-CO2H) which were designed
having in mind their potential application as sensors for the detection of biomolecules (eg.
globular proteins and carbohydrates). Thus, treatment of 1,4-diiodo-2,5-bis(bromomethyl)
benzene2 with the trimethyl ester derivative of p-tert-butyl calix[4]arene 1
3 in the presence of
base furnished the desired bis-calix[4]arene compound 2 as a white solid in good yield.
CALIX-CO2Me-CBZs were in turn obtained from 2 by double Sonogashira cross-coupling
reactions with 2-diethynyl-9-propyl-9H-carbazole and 3-diethynyl-9-propyl-9H-carbazole
using PdCl2(PPh3)2/CuI as catalytic system. The triesters CALIX-CO2Me-CBZs were further
hydrolyzed, yielding the corresponding acid derivatives (CALIX-CO2H-CBZs) (Scheme 1) in
good overall yields. The new compounds are strongly fluorescent and were structurally
characterized by FT-IR and 1H/
13C NMR.
Scheme 1: Synthesis of Hydrophilic Calix[4]arene-Carbazole Conjugates.
Acknowledgements: We thank Fundação para a Ciência e a Tecnologia/MCTES (Portugal) for partial financial support (PEst-OE/EQB/UI0702/2011). References: 1. Terai T.; Nagano T. Pflugers Arch - Eur J Physiol 2013, 465, 347 2. Gaefke, G.; Enkelmann, V.; Höger, S. Synthesis 2006, 17, 2971 3. Abidi, R.; Oueslati, I.; Amri H.; Thuéry, P.; Nierlich, M.; Asfarid, R.; Vicens, J.Tetrahedron Letters 2001, 42, 1685.

PC65
Synthesis and NMR conformational studies of new
dihomooxacalix[4]arene tetraurea derivatives. Cone versus partial
cone conformation
Paula M. Marcos,a,b
Filipa A. Teixeira,a Ana S. Augusto,
a José R. Ascenso
c
aCentro de Ciências Moleculares e Materiais, FCUL, Edifício C8, 1749-016 Lisboa, Portugal;
bFaculdade
de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa; bIST, Complexo I, Av.
Rovisco Pais, 1049-001 Lisboa
Calixarenes represent an extremely versatile class of macrocyclic receptors, able to bind
and selectively transport ions and neutral molecules. Lately, the study of anion receptors
based on calixarenes has considerably increased.1 The important role of anions in both
biological and environmental areas is one of the reason for this growth. The NH groups of
urea derivatives are strong hydrogen bond donors, and this property has been widely used
for the construction of neutral anions receptors.
For some years we have been synthesising homooxacalixarenes and studying their binding
properties towards a variety of cations.2,3
Recently, we have extended our research to the
study of anion complexation.4
In the present work we report the synthesis and the NMR conformational analysis of two p-
tert-butyl-dihomooxacalix[4]arene derivatives modified with substituted urea groups at the
lower rim via a butyl or propyl spacer, respectively. Alkylation of the parent compound with
bromoalkyl phthalimides and K2CO3 in refluxing MeCN gave the corresponding
phthalimides. After, these compounds reacted with hydrazine in refluxing EtOH furnishing
the amines, which by addition of tert-butylisocyanate yielded finally to the urea derivatives 1
and 2. 1H,
13C, COSY and NOESY NMR experiments were carried out in CDCl3 at r.t. to
establish the urea conformations. Preliminary studies concerning the anion binding
properties of these ureas will also be presented.
Acknowledgements: Authors thank Fundação para a Ciência e a Tecnologia, Project ref. PTDC/QUI/69858/2006. References: 1. Matthews, S. E.; Beer, P. D. Supramol. Chem. 2005, 17, 411. 2. Marcos, P. M.; Ascenso, J. R.; Segurado, M. A. P.; Cragg, P. J.; Michel, S.; Hubscher-Bruder, V.; Arnaud-Neu, F. Supramol. Chem. 2011, 23, 93. 3. Marcos, P. M.;.Teixeira, F. A.; Segurado, M. A. P.; Ascenso, J. R.; Bernardino, R. J.; Cragg, P. J.; Michel, S.; Hubscher-Bruder, V.; Arnaud-Neu, F. J. Phys. Org. Chem. 2013, 26, 295.
4. Marcos, P. M.; Proença, C. S.; Teixeira, F. A.; Ascenso, J. R.; Bernardino, R. J.; Cragg, P. J. Tetrahedron 2013 (in press).

PC66
Luminol turns green: New luminol analogues with increased
aromaticity and green chemiluminescence
Periyasami Govindasami, Liliana Martelo, Carlos Baleizão, Mario N. Berberan e Santos
Centro de Química-Física Molecular and Institute of Nanoscience and Nanotechnology, Instituto Superior
Tecnico, 1049-001 Lisboa, Portugal
Luminol is best known due to the strong blue light emission upon specific oxidative
conditions. The chemiluminescence of luminol has been widely applied in bio-analytical
chemistry, namely biosensors and heavy metal identification. Of all applications, probably
the most relevant is the identification of hemic iron from cleaned blood-stains.
Given that human eye sensitivity is maximal in the green part of the spectrum, a green
chemiluminescent compound with the chemical properties of luminol should surpass the
applicability range and detection limits of this compound. Herein we present the synthesis
and the chemiluminescence studies of five new naphthalenic luminol analogues, with
increased aromaticity and different N-substituents. These new compounds show a green
chemiluminescence, and in some cases have higher emission quantum yield and
chemiluminescence duration, when compared with luminol.
Acknowledgements: This work was supported by Fundação para a Ciência e a Tecnologia (FCT),
Portugal and POCI 2010 (FEDER) within project PTDC/QUI-QUI/123162/2010. P.G. and L.M. acknowledges grants from FCT (SFRH/BPD/66457/2009 and SFRH/BD/78032/2011, respectively).
N H
N H
O
O
N H 2
N H
N H
N H
N H
O
O
O
O
N P h P h
N H
N H
O
O
N H 2
N H
N H
O
O
N H
N H
N H
O
O
N H
P h
P h
L u m i n o l
N P h
P h

PC67
Asymmetric phthalocyanines bearing phenylacetylene units
Raquel Nunes da Silva,a Leandro M. O. Lourenço,
a Augusto C. Tomé,
a Ângela Cunha
b
aDepartment of Chemistry & QOPNA, Universityof Aveiro, 3810-193 Aveiro, Portugal bDepartment of Biology & CESAM, Universityof Aveiro, 3810-193 Aveiro, Portugal
Phthalocyanines (Pcs) are photoactive molecules that can absorb and emit light in a large
range of the UV-Vis spectrum.1 Due to their unusual optical and redox properties,
photophysical conductivity and chemical stability, these compounds have been applied in
several areas, namely catalysis, sensing systems, optical information storage and in the
construction of supramolecular structures with different applications in materials science.2
Besides the above applications, Pcs also have an important role in medicine, particularly in
photodynamic therapy (PDT) in which, by combination of visible light and molecular oxygen,
reactive oxygen species (ROS) are generated and cytotoxic effects at the target cells are
induced.3
In this communication the synthesis and structural characterization of the asymmetric Pcs 1-
3 (Figure 1) will be discussed. These Pcs bear phenylacetylene units that can be used for
further structural modification.
Figure 1: Zinc tert-butyl phenylacetylene Pcs 1-3.
Acknowledgements: Thanks are due to the University of Aveiro, FCT (Portugal), European Union, QREN, FEDER and COMPETE for funding the project PTDC/QEQ-QOR/1273/2012, the QOPNA Research Unit (project PEst-C/QUI/UI0062/2011),the CESAM Research Unit (PEst-C/MAR/LA0017/2011) and the Portuguese National NMR network. R. Nunes da Silva andL. M. O. Lourenço thank FCT for their PhD grants. (SFRH/BD/87598/2012 and SFRH/BD/64526/2009, respectively). References: 1. Kadish K.; Smith K. M.;Guilard R. The Porphyrin Handbook. 1 ed.; Academic Press: New York, 2003; Vol. 11-20, p 3310. 2.a) Carvalho E. F. A.; Calvete M. J. F.; Cavaleiro J. A. S.; Dini D.; Meneghetti M.;Tomé A. C. Inorg. Chim. Acta 2010, 363, 3945;b) de la Torre G.; Claessens C. G.; Torres T. Chem. Commun. 2007, 28, 2000. 3.a) Carvalho E. F. A.; Calvete M. J. F.; Tomé A. C.;Cavaleiro J. A. S. Tetrahedron Lett. 2009,50,
6882;b) Pereira J. B.; Carvalho E. F. A.; Faustino M. A. F.; Fernandes R.; Neves M. G. P. M. S.; Cavaleiro J. A. S.; Gomes N. C. M.; Cunha A.; Almeida A.; Tomé J. P. C. Photochem. Photobiol. 2012, 88, 537.

PC68
Synthesis of Novel Task-Specific Ionic Liquids Bearing an
Anhydride Moiety
R. Teixeira, N. M. T. Lourenço
IBB-Institute for Biotechnology and Bioengineering, Centre for Biological and Chemical Engineering,
Instituto Superior Técnico, 1049-001 Lisboa, Portugal
Task-specific ionic liquids (TSILs) are, by definition, ILs that incorporates a covalently linked
reactive functionality. By introducing different functional groups on the structure of the IL
cation, it is possible to improve or add new properties to the ILs. Due to their high versatility,
TSILs have found applications in diverse areas, from catalysis and organic synthesis to the
construction of nanostructured and ion conductive materials.1
Among the different reported TSILs, those containing anhydride moieties have not been
explored so far, to the best of our knowledge. Anhydrides are an important class of
compounds in organic chemistry. They can work, for instance, as reactive intermediates in
peptide synthesis, or in prodrugs design, where they show some advantages over the
commonly used ester function.2
In this work, TSILs bearing an anhydride function were prepared by condensation of two
identical IL carboxylic acids, through a carbodiimide-mediated coupling (Scheme 1). They
are composed of two imidazolium cations and two hexafluorophosphate (PF6-) anions.
Furthermore, the alkyl chain linking both imidazolium cations to the anhydride moiety in the
center of the molecule has a variable length (n = 3, 5 or 9).
These IL anhydrides have been envisaged for applications in biocatalysis. More specifically,
their potential use as acylating agents in the enzymatic resolution of secondary alcohols will
be explored taking advantage of: (i) their ionic properties to avoid complex separation
processes; and (ii) the irreversibility in acyl transfer conferred by the anhydride
functionality.3
Scheme 1: Synthesis of task-specific ionic liquid anhydrides through a carbodiimide coupling reaction.
DIC: N,N’-diisopropylcarbodiimide. Acknowledgements: We thank Fundação para a Ciência e Tecnologia (POCI 2010), FEDER (SFRH/BPD/41175/2007) and PTDC/EQU-EQU/122106/2010 for the financial support. References: 1. a) Lee, S. Chem. Commun. 2006, 1049. b) Rocha, A.; Carvalho, T.; Vidinha, P.; Lourenço, N. M. T. ChemPlusChem 2012, 77, 1106. 2. Mizrahi, B.; Domb, A. J. AAPS PharmSciTech 2009, 10, 453. 3. a) Lourenço, N. M. T.; Afonso, C.A.M. Angew. Chem. Int. Ed. 2007, 45, 8178. b) Wu, W.-H.; Akoh, C. C.; Phillips, R. S. Enzyme Microb. Technol. 1996, 18, 536.

PC69
Synthesis and Photophysical Characterization of Novel
Triphenylamine-benzimidazole Derivatives
Rosa M. F. Batista,a J. Pina,
b J. Seixas de Melo,
b Susana P. G. Costa,
a
M. Manuela M. Raposoa
aCentre of Chemistry, University of Minho, Campus de Gualtar, 4710-057, Braga, Portugal;
bDepartment of Chemistry, University of Coimbra, Rua Larga, 3004-535, Coimbra, Portugal
Research on organic luminescent materials has been intensely pursued due to their
importance in technological applications related to signaling, fluorescent
biosensory/chemosensory materials, molecular switches and organic light emitting diodes
(OLEDs). Organic fluorophores such as triphenylamine and benzimidazole derivatives have
attracted a particular attention owing to their high emission efficiency being widely used as
electron transporters and emitting layers for OLEDs.1
Recently, we have been investigating the potential of heterocyclic systems bearing
functionalized (benz)imidazole derivatives exhibiting high thermal stability, interesting
emissive and chemosensory properties.2 In this communication we report the synthesis and
photophysical characterization of triphenylamine-benzimidazoles (compounds 1a-d) which
were synthesized by a one step reaction through the Na2S2O4 reduction of several
commercially available o-nitroanilines in the presence of triphenylamine aldehyde in DMSO
at 120 °C. Compounds 1a-d bear different functionalization at position 5 of the
benzimidazole with electron-donor or acceptor groups (Figure 1). A comprehensive spectral
and photophysical investigation of these compounds including absorption, fluorescence and
triplet-triplet absorption spectra, together with quantum yields of fluorescence, internal
conversion, intersystem crossing and singlet oxygen and rate constants for the radiative
and radiationless processes has been undertaken in solution at room temperature. It is
shown that compounds 1a-d exhibit high fluorescence quantum yields (F= 0.70-0.78).
Additionally, a comparison between the optical and photophysical properties of 1a-d will be
also presented and discussed.
N
N
N
H
R
1a R = H
1b R = OH
1c R = OMe
1d R = CN
Figure 1: Structure of triphenylamine-benzimidazoles 1a-d. Acknowledgements: Thank are due to Fundação para a Ciência e Tecnologia (Portugal) and FEDER-COMPETE for financial support through Centro de Química-UM [PEst-C/QUI/UI0686/2011 (F-COMP-01-0124-FEDER-022716)] and Program 652 C2008-DRH05-11-842. A post-doctoral grant to R.M.F. Batista (SFRH/BPD/79333/2011) is also acknowledged The NMR spectrometer Bruker Avance III 400 is part of the National NMR Network and was purchased with funds from FCT and FEDER. References: 1. Zhong C.; Duan C.; Huang F.; Wu H.; Cao Y. Chem. Mater. 2011, 23, 326. 2. a) Batista R. M. F.; Costa S. P. G.; Malheiro E. L.; Belsley M.; Raposo M. M. M. Tetrahedron 2007, 63,
9842. b) Pina J.; Seixas de Melo J.; Batista R. M. F.; Costa S. P. G.; Raposo M. M. M. J. Phys. Chem. B 2010, 114, 4964. c) Batista R. M. F.; Ferreira, R. C. M.; Costa S. P. G.; Raposo M. M. M. Tetrahedron 2012, 68, 7322.

PC70
Reduction of 2,2,2-trifluoroacetophenoneby marine-derived fungus
Mucorracemosus CBMAI 847
Sandra S. Ribeiro, André L. M. Porto
Laboratório de Química Orgânica e Biocatálise, Instituto de Química de São Carlos, Universidade de
São Paulo, Avenida João Dagnone, nº 1100, Ed. Química Ambiental, J. Santa Angelina, 13563-120, São
Carlos, SP, Brazil.
Fluorinated chiral alcohols are interesting building blocks for pharmaceuticals and
agrochemicals, owing to the unique properties of the fluorine atom.1
The enantioselective
reduction of ketones is one of the most convenient and useful methods for the preparation of
chiral secondary alcohols.2
Recent investigations by our group showed the preparation of
enantiopure organofluorine compounds by lipase-catalyzed under microwave irradiation and
orbital shaking.3
In this work, two reactions were performed in 50 mL, one on Erlenmeyer
flask in an orbital shaker (130 rpm, 40°C, 6h) and the other round-bottomed flask under
microwave irradiation (40°C, 200W, 6h) in a CEM Discover reactor. The reaction mixture in
each flask was 1.0 g of fungal mycelia in 25 mL enzymatic broth in various concentrations of
the ketone 1 (pH 7, 6 h, Table 1). The mycelia of fungus M. racemosus CBMAI 847
catalyzed the selective reduction of the carbonyl group, with excellent conversion (up to
99%) and selectivityto give S-alcohol 1 under microwave irradiation and orbital shaker. It
was the first study of reduction of fluoroketone by marine fungus under microwave
irradiation.
Table 1.Reduction of 2,2,2-trifluoroacetophenone 1 by marine-derived fungus
Mucorracemosus CBMAI 847 (6h, pH).
(mmol/L) c (%) 1 c (%) (S)-1 ee (%) (S)-1
Orbital shaker (40°C, 130 rpm)
2.9 - 100 74
5.7 - 100 74
8.5 44 56 90
14.0 61 39 95
Microwave irradiation (40°C, 200W)
2.9 62 38 73
5.7 36 64 91
8.5 49 51 92
14.0 72 28 96
c: concentration of ketone 1 determined by GC-FID analysis; c: conversion to alcohol (S)-1 determined by GC-FID analysis; ee: enantiomeric excess.
Acknowledgements: The authors thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and the Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP) for financial support and scholarship. References: 1. Thomas C. Rosen, T. C.; Feldmann, R.; Dünkelmann, P.; Daubmann, T. Tetrahedron Lett., 2006, 47,
4806. 2. Nennajdenko, V. G.; Smolko, K. I.; Balenkova, E. S. Tetrahedron: asymmetry, 2001, 12, 1259. 3. Ribeiro S. S.; Raminelli C.; Porto, A. L. M. J. Fluorine. Chem. (accepted, June 2013).

PC71
Novel Donor-acceptor Heterocyclic Systems Bearing Phthalazine,
Thiophene and Furan groups for DSSC: Synthesis and
Characterization
Sara S. M. Fernandes, M. Manuela M. Raposo
Centre of Chemistry, University of Minho, Campus de Gualtar, 4710-057, Braga, Portugal
The ever increasing consumption of fossil fuels, causing global warming and environmental
pollution, has led to a greater focus on renewable energy sources and sustainable
development. Among several new energy technologies, solar cells utilizing the sun as an
energy source are the most promising. The commercially available solar cells are currently
based on expensive inorganic silicon semiconductors. Consequently, organic solar cells,
appear to be a highly promising and cost-effective option for the photovoltaic energy sector.
Therefore, dye-sensitized solar cells (DSSCs) based on dye sensitizers adsorbed on
nanocrystalline TiO2 electrodes have received significant attention because of their high
incident solar light-to-electricity conversion efficiency, colorful and decorative natures and
low cost of production. As a result, in the last two decades, a wide range of structural
modifications to the donor, acceptor and -bridge have been carried out having in mind the
preparation of organic chromophores with high performance for DSSCs.1
As part of an on-going research to develop efficient donor-acceptor substituted heterocyclic
systems for several optoelectronic applications2 we synthesized phthalazines 3-4
functionalized at the furan or aryl rings with cyanoacetic or rhodanine anchoring groups
(Figure 1). Donor-acceptor compounds 3-4 were synthesized through Suzuki coupling of
bromo-thienylphthalazine 2 with commercially available (hetero)aryl-boronic acids. On the
other hand, precursor 2 was prepared by reaction of thienylphthalazinone 1 with POBr3.3 In
this communication we report on the synthesis and characterization of novel heterocyclic
dyes 3-4. The experimental results indicate that, these compounds could have potential
application as sensitizers for DSSCs.
N NS
O R
N NS
R
4a-c3a-c
a R= CHO
b R= CH=C(CN)(CO2H)
N
S
O
S
CO2HcN N
BrS
2
N NH
OS
1
Figure 1: Structure of donor-acceptor thienylphthalazine dyes 3-4.
Acknowledgements: Thank are due to Fundação para a Ciência e Tecnologia (Portugal) and FEDER-COMPETE for financial support through the Centro de Química PEst-C/QUI/UI0686/2011 (F-COMP-01-0124-FEDER-022716) and a PhD grant to S.S.M. Fernandes (SFRH/BD/87786/2012). The NMR spectrometer Bruker Avance III 400 is part of the National NMR Network and was purchased within the framework of the National Program for Scientific Re-equipment, contract REDE/1517/RMN/2005 with funds from POCI 2010 (FEDER) and FCT. References: 1. Misha A.; Fischer M. K. R.; Bäuerle P. Angew Chem. Int. Ed. 2009, 48, 2479. 2. a) Castro M. C. R.; Schellenberg P.; Belsley M.; Fonseca A. M. C.; Fernandes S. S. M.; Raposo M. M. M. Dyes Pigments 2012, 95, 392. b) Coelho P.; Castro M. C. R.; Fonseca A. M. C.; Fernandes S. S. M.; Raposo M. M. M. Tetrahedron Lett. 2012, 53, 4502. 3. Raposo M. M. M.; Sampaio A. M. B. A.; Kirsch G. J. Heterocyclic Chem. 2005, 42, 1245.

PC72
Preparation of aC-glycosylcinnamoyloxyacetophenone: valuable
intermediate for the synthesis of a C-glycosyl-2-styrylchromone
Sara M. Tomé, Artur M. S. Silva
Department of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal
Remarkable progress has recently been achieved on the chemistry of C-glycosyl flavonoids,
a natural widespread family of polyphenolic heterocycles.1
The prominent interest on these
compounds lies on the combination of the distinguishing hydrolytic stability of their C-
glycosidic bond and the high hydrophilic character of the carbohydrate unit together with the
renowned biological activities of flavonoids.2
In this communication we present our study concerning the synthesis of a polymethoxylated
C-glycosyl-2-styrylchromone. As an alternative route to the unproductive direct C-
glycosylation of 2-styrylchromones we explored the Kumazawa’s3
C-glycosylation of 2’-
hydroxyacetophenone 1 via a O->C glycoside Fries-type rearrangement by using 2,3,4,6-
tetra-O-benzyl-α-D-glucopyranosyl fluoride and trifluoride etherate as an activator leading to
C-glycosylacetophenone 2. Applying the Baker-Venkataraman method, the optimized
treatment of compound 2 with the appropriated cinnamic acid in the presence of N,N-
dicyclohexylcarbodiimide (DCC)4 and 4-pyrrolidinopyridineresulted on the synthesis of C-
glycosylcinnamoyloxyacetophenone 3. The cinamoyl group transposition of the latter
compound and subsequent cyclodehydration is a conceivable route towards the synthesis
of C-glycosyl-2-styrylchromone 4 (Scheme 1).
Scheme 1: Studied route towards the synthesis of polymethoxylatedC-glycosyl-2-styrylchromone
Acknowledgements: Thanks are due to the University of Aveiro, Fundação para a Ciência e Tecnologia (FCT, Portugal), European Union, QREN, FEDER and COMPETE for funding the QOPNA Research Unit (project PEst-C/QUI/UI0062/2011) and the Portuguese National NMR Network. Sara Tomé also thanks University of Aveiro and the Organic Chemistry Research Unit for her fellowship (BIIC/QUI/5173/2011). References: 1. a) Talhi O.; Silva A. M. S. Curr. Org. Chem. 2012, 16, 859. b) Veitch N. C.; Grayer R. J. Nat. Prod. Rep. 2008, 25, 555. 2. Rice-Evans C. A.; Packer, L. Flavonoids in Health and Disease, 2
nd ed., Marcel Dekker, New York,
2003. 3. a) Kumazawa T.; Kimura T.; Matsuba S.; Sato S.; Onodera J. Carbohydr. Res. 2001, 334, 183. b) Kumazawa T.; Ohki K., Ishida M., Sato S., Onodera J.-i.; Matsuba S., Bull. Chem. Soc. Jpn. 1995, 68, 1379. 4. Pinto D. C. G. A.; Silva A. M. S.; Cavaleiro J. A. S. New J. Chem. 2000, 24, 85.

PC73
The Aza Wharton reaction and their applicability in the
stereoselective synthesis of hydroxy-cyclopentenamines and
cyclohexenamines
S. A. G. Silva,a P. Rodrigues,
a Christopher D. Maycock
a,b
aInstituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa, 2780-157 Oeiras, Portugal;
bDepartamento de Química e Bioquímica, Faculdade de Ciências, Universidade de Lisboa, Campo
Grande, 1749-016 Lisboa, Portugal.
The Wharton reaction is a reductive rearrangement of acyl epoxides, discovered some time
ago.1 It promotes the transformation of α,β-epoxiketones into allylic alcohols, using
hydrazine (Scheme 1). It has been widely used in different complex syntheses. Recently
the Wharton reaction was used for the preparation of allylic amines from tosyl aziridines.2
Azabicycloketones can also be prepared from α-iodoenones with a variety of N-
substutions.3 This method constitutes a very versatile and simple way to prepare and
resolve aziridines. Taking advantage of the ready availability of the respective aziridines, a
range of cyclic allylic amines was prepared in some cases enantiomerically pure by
application of the Wharton reaction. Thus, an alternative route to cyclic allylic amines has
been found, which constitutes a simple strategy for the production of important building
blocks and ligands for organic synthesis.
Scheme 1: Wharton Reaction.
The selective aziridination of 4-hydroxy substituted 2-iodocycloenones is a key step in this
method. Applying the Wharton ring opening vicinal aminoalcohols of cyclopentene and
cyclohexene were produced in the same manner. It was possible to selectively prepare cis
or trans cyclic aminoalcohols by tuning the amine used (Scheme 2).
Scheme 2: Stereoselective synthesis of cyclic aminoalcohols
Acknowledgements: S. A. G. Silva and P. Rodrigues thank the Fundação para a Ciência e Tecnologia
for the grants SFRH/BD/84309/2012 and SFRH/BD/27423/2006. This work has been supported by Fundação para a Ciência e a Tecnologia through projects POCI/QUI/62794/2004 and PTDC/QUI-QUI/104056/2008. References: 1. P. Wharton, D. Bohlen J. Org. Chem.1961, 26, 3615-3616; 2. H. Jiang, N. Holub, K. A. Jørgensen P. Natl. Acad. Sci. USA 2010, 107 (48), 20630-20635; 3. M. T. Barros, C. D. Maycock; M. R. Ventura Tetrahedron Lett. 2002, 43, 4329-4331.

PC74
Sugar-based precursors of new potential inhibitors of xanthine
oxidase: synthesis and characterization
S. Cunha, A. Esteves
Centro de Química, Universidade do Minho, Campus de Gualtar, 4710-057 Braga, Portugal
Xanthine oxidase (XO) catalyses the sequential oxidation of hypoxanthine and of xanthine
to uric acid and acts as an important biological source of free radicals.1 Allopurinol, a purine
analogue, is the only commercially available xanthine oxidase inhibitor and it is used as a
potent drug against gout.2 However, some adverse effects of allopurinol led to the
development of other xanthine oxidase inhibitors,3 namely pyrazolopyrimidines that showed
to be generally more active than allopurinol.4 Otherwise, it is well known that
glycoconjugates have an enormous potential in drug design. Glycoconjugates containing
the 1,2,3-triazole unit find application in medicinal chemistry, particularly in those cases
where this unit acts as a bridge between an amino acid/peptide and the sugar moiety.5
In this communication we report the synthesis of several glycoconjugates of type 3
containing a 1,2,3-triazole unit built by click chemistry strategy between the acetylenic
compounds of type 2 and glycosylazide (Scheme 1). All compounds were fully
characterized by 1H-NMR,
13C-NMR and elemental analysis and/or mass spectra and the
results obtained will be presented.
Scheme 1
Acknowledgements: The authors acknowledge to the Foundation for the Science and Technology (FCT, Portugal) for financial support to the NMR Portuguese network (PTNMR, Bruker Avance III 400-Univ. Minho). FCT and FEDER (European Fund for Regional Development)-COMPETE-QREN-EU for financial support to the Research Centre and CQ/UM [PEst-C/QUI/UI0686/2011 (FCOMP-01-0124-FEDER-022716)] References: 1. Orth P.; Fedida D.; Spector T. Biochem. Pharmacology, 2006, 71, 1747. 2. Pacher P.; Nivorozhkin A.; Szabó C. Pharmacol. Rev., 2006, 58, 87. 3. Khanum S. A. Bioorg. Med. Chem. 2011, 19, 211. 4. Gupta S. et al. Eur. J. Med. Chem., 2008, 48, 771 5. Kuijpers B. H. M.et. al. Org Lett. 2004, 6, 3123.

PC75
Supramolecular architectures based on transition metal
bis-1,2-dithiolene complexes with N-coordinating groups
A. C. Cerdeira,a D. Belo,
a S. Rabaça,
a L. C. J. Pereira,
a J. T. Coutinho,
a I. C. Santos,
a
R. T. Henriques,b O. Jeannin,
c M. Fourmigué,
c M. Almeida,
a D. Simão
d
aCampus Tecnológico e Nuclear Instituto Superior Técnico, Universidade Técnica de Lisboa Estrada
Nacional 10 (km 139,7), 2695-066 Bobadela LRS - Portugal; bInstituto de Telecomunicações, Polo de
Lisboa, Av.Rovisco Pais,1049-001 Lisboa, Portugal; cInstitut des Sciences Chimiques de Rennes,
Université Rennes 1 & CNRS, Campus de Beaulieu, F-35042 Rennes, France; dCentro de Química
Estrutural, Departamento de Engenharia Química, Instituto Superior Técnico da Universidade Técnica de
Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal
Transition metal complexes with dithiolene ligands have been extensively studied since the
sixties due to their unusual redox behaviour, their remarkable electric and magnetic
properties, making these complexes interesting as key building blocks for preparing
conducting and magnetic molecular materials. Dithiolene ligands containing N-coordinating
groups are particularly interesting, since they present an additional pole to selectively
coordinate different transition metals, which can lead to a variety of coordination structures,
from the simple discrete bimetallic complexes to linear chains and two- or three-dimensional
networks.1 In this work, we describe the results of heterobimetallic coordination structures
based on the combination of metal cations with transition metal bisdithiolene complexes
with extended dithio-azo ligands2 (Figure 1).
Figure 1: Crystal structure of [Na(18C6)][Ni(4-pedt)2].2H2O and extended dithio-azo ligands.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support, SFRH/BD/46543/2008. References: 1. Rabaça S., Almeida M., Coord. Chem. Rev., 2010, 254, 1493-1508. 2. Cerdeira A. C., Belo D., Rabaça S., Pereira L. C. J., Coutinho J. T., Simão D., Henriques R. T., Jeannin O., Fourmigué M. and Almeida M., Eur. J. Inorg. Chem. 2013, accepted for publication.

PC76
Synthesis of an Indole-based Antimalarial Library
Sofia A. Santos,a,b
Ralph Mazitschek,a Rui Moreira,
b Alexandra Paulo
b
aCenter for Systems Biology, Massachusetts General Hospital, Boston, MA 02114, USA
bResearch Institute for Medicines and Pharmaceutical Sciences (iMed.UL), Faculty of Pharmacy,
University of Lisbon. Av. Prof. Gama Pinto, 1649-003 Lisbon, Portugal
[email protected] / [email protected]
Malaria, the most devastating of the parasitic diseases, is a major global health problem
due to the rapid emergence and spread of multidrug-resistant strains of Plasmodium
falciparum, the most lethal of the four malaria parasite species that infect humans.1
Although the continued attempts to develop a vaccine for malaria, small molecule drugs
remain the sole treatment option. As a result, there is an urgent need for novel drugs,
preferably new chemotypes acting on new parasite targets in order to delay or overcome
the selection of clinical resistance.2 We report here the development and optimization of a
robust synthetic route towards obtaining novel 3-(piperidin-4-yl)-1H-indole analogues
starting from commercially available synthetic building blocks (Scheme 1). The derivatives
were prepared by condensation of the appropriate indole with N-benzyl-4-piperidone in the
presence of a base and subsequent reduction under hydrogenation conditions. The
additional chemical blocks can be linked to the free amine group by reductive amination.
Scheme 1: Synthetic route for novel 3-(piperidin-4-yl)-1H-indole analogues.
Acknowledgements: to FCT (Portugal) for financial support (SFRH/BD/80162/2011and Pest-OE/SAU/UI4013/2011). References: 1. Wells et al. Nat. Rev. Drug Discov. 2009, 8, 879. 2. Ekland E. H, Fidock D. A. Int. J. Parasitol. 2008, 38, 743-747.

PC77
Synthesis of -Amino Acid Esters Carrying Bicyclo[3,3,0]octane and
Bicyclo[4,3,0]nonane Skeletons
Sonata Krikštolaitytė,a Wolfgang Holzer,
b Gerald Giester,
c Algirdas Šačkus
a
aDepartment of Organic Chemistry, Kaunas University of Technology, Kaunas, Lithuania;
bDepartment of
Drug and Natural Product Synthesis, Faculty of Life Sciences, University of Vienna, Vienna, Austria; cInstitute of Mineralogy and Crystallography, Faculty of Geosciences, Geography and Astronomy,
University of Vienna, Vienna, Austria
Quaternary α-amino acids constitute a powerful approach for generating structurally defined
peptides as conformational probes and bioactive agents.1 In the present work we report the
stereoselective synthesis of quaternary α-amino acid esters possessing the functionalized
bicyclo[3,3,0]octane and bicyclo[4,3,0]nonane skeletons. The stepwise alkenylation and
alkynylation procedure2 of the Schöllkopf’s bislactim auxiliary 1 and subsequent
intramolecular Pauson-Khand cyclization of the obtained substrates 2a,b yielded the
bicyclic unsaturated ketones 3a,b (Scheme 1).
N
N
Me
Me OMe
MeO
N
N
Me
Me OMe
MeO
1 2a,b( )n
N
N
Me
Me OMe
MeO( )
n
OH
N
N
Me
Me OMe
MeO( )
n
OH
3a,b
4a,b
5
O
H2N
MeO2C
H
H
6
O
H
H
H2N
MeO2C
Hhydrolysis
2-4a n = 1; b n = 2
Scheme 1: Stereoselective synthesis of quaternary amino acid esters carrying bicyclic skeletons
Hydrogenation of compounds 3a,b gave the chiral substrates 4a,b. The subsequent mild
acid hydrolysis provided rigid bicyclic amino acid esters 5 and 6. The structure of the
synthesized compounds was established by methods of 1H-,
13C-,
15N-NMR-spectroscopy
and single crystal X-ray analysis.
References: 1. a) Cativela C.; Ordónez M. Tetrahedron: Assym. 2009, 20, 1.b) Trabocchi A.; Scarpi D.; GuarnaA.
Amino Acids 2008, 34, 1. 2. a) Krikštolaitytė S.; Šačkus A.; Rømming C.; Undheim K. Tetrahedron: Assym. 2001, 12, 393. b) Wang J.; Falck-Pedersen M. L.; Rømming C.; Undheim K. Synth. Commun. 2001, 31, 1141.

PC78
Hetero-Diels-Alder of 3-Tetrazolyl-1,2-diaza-1,3-butadiene with Dipyrromethanes
Susana M. M. Lopes,a Nelson A. M. Pereira,
a Américo Lemos,
b
Teresa M. V. D. Pinho e Meloa
aDepartment of Chemistry, University of Coimbra, 3004-535 Coimbra, Portugal;
bCIQA, FCT, University of Algarve, Campus de Gambelas, 8005-139 Faro, Portugal.
Dipyrromethanes are of wide interest as building blocks in organic synthesis, namely in the
synthesis of porphyrins and porphyrin analogues. On the other hand, dipyrromethanes are
the precursors of BODIPY dyes (4,4-difluoro-4-bora-3a,4a-diaza-s-indacenes) whose
photophysical properties make them the ideal fluorescent scaffold for the development of
high performance imaging probes.1 Recently, we have described a methodology to
functionalized dipyrromethanes via hetero-Diels-Alder of azo- and nitrosoalkenes.2 On the
other hand, we have been interested in obtaining new compounds that contain a tetrazole
moiety, exploring the carboxylic acid/tetrazole bioisosterism as strategy to find compounds
with potential biological activity.3 In this context, we describe the generation and Diels-Alder
reaction of 3-tetrazolyl-1,2-diaza-1,3-butadiene (3) with dipyrromethanes (e.g. 1) giving
open chain hydrazones (e.g. 4) and tetrahydro-pyridazines (e.g. 5). The synthesis of
compound 5 proves that compound 4 is formed via Diels-Alder reaction, followed by ring
opening. In fact, conversion of 5 into 4 is quantitative. The deprotection of the tetrazole
group of 4 or 5 leads to new functionalized dipyrromethanes (e.g. 6). Details of this study
will be disclosed.
Scheme 1: Synthesis of functionalized dipyrromethanes via hetero-Diels-Alder reaction.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia (Grant
SFRH/BPD/84423/2012 and Project Pest-C/QUI/UI0313/2011), FEDER, COMPETE and QREN for
financial support.
References:
1. Ulrich, G.; Ziessel, R.; Harriman, A. Angew. Chem. Int. Ed. 2008, 47, 1184.
2. Pereira, N. A. M.; Lemos, A.; Serra, A. C.; Pinho e Melo, T. M. V. D. Tetrahedron Lett. 2013, 54, 1553.
3. a) Lopes, S. M. M.; Lemos, A.; Pinho e Melo, T. M. V. D. Tetrahedron Lett. 2010, 51, 6756. b) Lopes,
S. M. M.; Lemos, A.; Pinho e Melo, T. M. V. D.; Palacios, F. Tetrahedron 2011, 67, 8902. c) Lopes, S. M.
M..; Brigas, A. F.; Palacios, F.; Lemos, A.; Pinho e Melo, T. M. V. D. Eur. J. Org. Chem. 2012, 2152.

PC79
Synthesis of New Ortho Substituted Anilide Atropisomers – A
Second Look at the Hydrolysis of Quaternary N-Alkylbenzazol-3-ium
Salts
S. S. Ramos,a,b
L. V. Reis,c R. E. F. Boto,
a,d P. F. Santos,
c P. Almeida
a,d
aDepartment of Chemistry, University of Beira Interior, 6201-001 Covilhã, Portugal;
bUMTP-UBI-Unit of
Textile and Paper Materials, University of Beira Interior, 6200-001 Covilhã, Portugal; cDepartment of
Chemistry and Chemistry Center, University of Trás-os-Montes e Alto Douro, 5001-801 Vila Real,
Portugal; dCICS-UBI-Health Sciences Research Centre, University of Beira Interior, Av. Infante D.
Henrique, 6200-506 Covilhã, Portugal
The hydrolysis of 3-alkyl-2-methylbenzazol-3-ium iodides 1 was reevaluated performing the
reaction in boiling ethanol with triethylamine as catalytic base and isolating the unstable
intermediate 2 by trapping it with different alkyl iodides, in the presence of sodium
hydroxide, in moderate to good yields. Also, the anilides 3 so obtained were converted by
reduction with lithium aluminium hydride in the presence of aluminium chloride in diethyl
ether to the corresponding anilines 4 with good yields (Figure 1).
Surprisingly, we observed that compounds 3, anilide derivatives with a single ortho group
not so bulky, are enantiomeric atropisomers (so they resist to racemization) that result from
the twisting on the Ar-N bond, and this feature is central to the achiral auxiliary strategy.
First, this feature was observed in anilides bearing two ortho groups (which have usually
very high barriers to rotation, very often from <20 to 30 kcal/mol)1 and then in anilides
bearing a single but large ortho group.2 Atropisomerism involved in the above reaction has
been studied using dynamic NMR spectroscopy and the corresponding rotation barrier was
determined. The measured free energy of activation to interconversion of the rotamers
ranged from 17.1 to 20.5 kcal/mol.
Figure 1: Synthesis of anilides 3 and anilines 4; reagents and conditions: a) NEt3, 96% ethanol,
reflux; b) ethanol, NaOH, R2I, reflux; c) LiAlH4/AlCl3 (1:1), diethyl ether, from 0 ºC to r.t. Acknowledgements: This work was financed by FCT (Project PTDC/QUI-QUI/100896/2008) and COMPETE (Project PEst-C/SAU/UI0709/2011). References: 1. Oki M. In Applications of Dynamic NMR Spectroscopy to Organic Chemistry, VCH: Deerfield Beach. FL, 1985, pp 160-193.
2. As representative examples: a) Curran D. P.; Qi H.; Geib S. J.; DeMello N. C. J. Am. Chem. Soc. 1994, 116, 3131-3132; b) Clayden J. Angew. Chem. Int. Edit. 1997, 36, 949-951.
X = S, O or Se; R1 = Et, Pr or Hex; R2 = Et, Pr or Hex
4321
c)X
N
R1
R2
X
N
R1
R2
Ob)X
N
R1
OH
I
X
N
R1
a)

PC80
New scalable synthetic protocol for the production
of 5-(hydroxymethyl)furfural (HMF)
Svilen P. Simeonov, Carlos A. M. Afonso
iMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal.
5-hydroxymethylfurfural (HMF) can be obtained from carbohydrates like fructose, glucose,
sucrose, cellulose which are a main class of biomass compounds and is considered a
promising biorenewable intermediate for the industry. Several methods for the synthesis of
HMF have been already reported in the literature using diferent solvents and homogeneous
and heterogeneous catalysts.1 Ionic liquids have been also found to be good reaction media
for the conversion of sugars to HMF.2 The main problem that industry is faced to regarding
with the big scale HMF production is its water solubility that results in non-favorable
extraction distribution coefficient. In order to overcome the main synthetic problems an
integrated, simple, efficient reusable and scalable methodology for fructose, glucose,
sucrose or inulin dehydration into HMF in outstanding yield (>90%) and purity (>99%) is
described using wet tetraethyl-ammonium bromide as reaction medium.3
The procedure
allows the HMF isolation just by reaction medium crystallization that can be efficiently
reused or, alternatively the transformation can be performed under continuous operation.
Furthermore the method has been combined with the well industrially established enzymatic
glucose isomerization into fructose. High overall yield of 87% HMF with 99% purity over 8
cycles has been achieved using nitric acid as catalyst (Scheme 1).4
Scheme 1: Integrated approach for the production and isolation of HMF from glucose.
Additionally fructose dehydration into HMF has been developed as student’s laboratory
protocol either in batch and flow conditions.5
Acknowledgements: We would like to acknowledge Fundação para a Ciência e Tecnologia (POCI 2010) and PTDC/QUI-QUI/119823/2010, SFRH/BD/67025/2009 for financial support. References: 1.Rosatella, A. A.; Simeonov, S. P.; Frade, R. F. M.; Afonso, C. A. M. GreenChem, 2011, 13, 754. 2. Zakrzewska, M. E.; Bogel-Łukasik, E.; Bogel-Łukasik, R. Chem. Rev., 2010, 111, 397. 3. Simeonov, S.P., Coelho, J.A.S., Afonso, C. A. M., ChemSusChem,2012, 5, 1388. 4. Simeonov, S.P., Coelho, J.A.S., Afonso, C. A. M., ChemSusChem, DOI: 10.1002/cssc.201300176. 5. Simeonov, S.P. and Afonso, C. A. M., submitted.

PC81
Chromeno-imidazo[1,2-a]pyridines: synthesis and anticancer
activity
Marta Costa,a M. Fernanda Proença,
a O. Pereira,
b C. F. Lima,
c C. Pereira-Wilson
b
aDepartment of Chemistry, University of Minho, Braga, Portugal;
bCBMA/
cCITAB - Department of
Biology, University of Minho, Braga, Portugal
Cancer is a devastating disease and despite the huge efforts of the scientific community to
come up with a cure, no real improvements were attained with the newer drugs marketed
during the last decade. Limited effectiveness, toxicity and eventual resistance remain a
serious problem. The biological complexity of the disease usually requires the use of a
combination of drugs acting on different receptors.1 Another plausible strategy involves the
use of a single molecule capable of interfering with multiple altered pathways.
As part of a drug discovery collaboration program, a qualitative virtual target profiling3 was
performed on a library of 5500 novel compounds designed by our organic chemistry group.
This in silico screening identified a number of molecules bearing the chromene unit as
potentially active on different receptor families known to play an important role in some of
the pathways associated with cell proliferation and apoptosis in cancer cells.
In this work a detailed discussion of the scope of the synthetic method for chromenes 1-3
(Scheme 1) will be presented. The anticancer potential of these compounds was evaluated
in a colorectal carcinoma cell line HCT116 by the MTT assay. IC50 concentration of the
compounds was calculated, and the most active compounds tested for their ability to induce
cell cycle arrest and cell death by apoptosis.
Scheme 1: Synthetic chromene based compounds 1, 2 and 3.
Acknowledgements: we gratefully acknowledge the financial support from University of Minho and FCT through the Portuguese NMR network (RNRMN), the Project F-COMP-01-0124-FEDER-022716 (ref. FCT PEst-C/QUI/UI0686/2011) FEDER-COMPETE and BPD grant awarded to Marta Costa (SFRH/BPD/79609/2011). References: 1. Keith C.; Borisy A.; Stockwell B. Nat. Rev. Drug Discovery 2005, 4, 71. 2. Antonello A. et al. J. Med. Chem. 2006, 49, 6642.
3. Gregori-Puigjané E.; Mestres J. J. Chem. Inf. Model 2006, 46, 1615.

PC82
Solid phase microwave assisted synthesis of Peptaibolin mimetics
bearing α,α-dialkylglycines
V. I. B. Castro, C. M. Carvalho, R.D.V. Fernandes, Susana P. G. Costa,
S. M. M. A. Pereira-Lima
Centrode Química, Universidade do Minho, Campus de Gualtar, 4710-057 Braga, Portugal
Peptides and proteins mediate virtually all biological aspects of living organisms, from the
regulation of gene expression, to muscle movement or signal transmission between cells. It
is therefore expected that most diseases are, to a certain extent, related to peptide or
protein malfunctioning, lack or over expression. Despite this fact, the number of therapeutic
peptides is still very small due to the fact that they are promptly hydrolysed by acids present
in our stomach or hydrolytic enzymes present in the blood stream.
In order to prevent enzyme recognition and increase peptide half-lives most
pharmacological peptides have unnatural amino acids introduced into their sequence.This is
usually achieved by taking advantage from current peptide synthetic strategies, but there
are some examples of naturally occurring peptides that bear this interesting feature. One
example are Peptaibols, a class of peptides with antimicrobial activity extracted from several
strains of fungi, that are characterized by the presence of a C-terminal amino alcohol and
α,α-dialkylglycines, such as Aib, Deg and Iva, in their composition. These amino acids are
tetrasubstituted at the central carbon atom and restrict the rotation around peptide bonds,
yielding peptides with more defined conformations and more resistant to biodegradation.1,2
Despite the above interesting structural features, the steric hindrance and lower reactivity of
α,α-dialkylglycines results in an inevitable synthetic challenge whenever conventional
methods are used for their incorporation into peptide chains. With the development and
current availability of microwave-assisted synthetic equipment, such methodologies have
enabled to overcome poor coupling reactions and have been used for the synthesis of large
and difficult peptide sequences.3–5
Bearing these facts in mind, we report our research on the synthesis of the shortest
Peptaibol, Peptaibolin (Ac-Leu-Aib-Leu-Aib-Phol) and several mimetics incorporating
unnatural α,α-dialkylglycineson the Aib positions, using microwave-assisted solid phase
peptide synthesis performed on a CEM discover SPS device.
Acknowledgements: The authors acknowledge Fundação para a Ciência e Tecnologia (Portugal) for financial support through project PTDC/QUI-BIQ/118389/2010 (FCOMP-01-0124-FEDER-020906) and PEst-C/QUI/UI0686/2011 (F-COMP-01-0124-FEDER-022716), FEDER-COMPETE. The NMR spectrometer Bruker Avance III 400 is part of the National NMR Network and was purchased with funds from FCT and FEDER. References: 1. Aravinda, S.; Shamala, N.; Balaram, P. Chem. Biodivers. 2008, 5, 1238–1262. 2. Pinto, F. C. S. C.; Pereira-Lima, S. M. M. A.; Maia, H. L. S. Tetrahedron 2009, 65, 9165–9179. 3. Pedersen, S. L.; Tofteng, a P.; Malik, L.; Jensen, K. J. Chem. Soc. Rev. 2012, 41, 1826–1844. 4. Rizzi, L.; Cendic, K.; Vaiana, N.; Romeo, S. Tetrahedron Lett. 2011, 52, 2808–2811. 5. Bacsa, B.; Horváti, K.; Bõsze, S.; Andreae, F.; Kappe, C. O. J. Org. Chem. 2008, 73, 7532–7542.

PC83
Pyrido[2,3-b]indolizines: a one-pot synthesis in green media
A. Brito, Marta Costa, M. Fernanda Proença
Department of Chemistry, University of Minho, Braga, Portugal.
Natural occurring indolizine derivatives were identified as anticancer, antiviral, anti-
inflammatory, anti-tuberculosis, analgesic and antioxidant agents.1 As a result the indolizine
nucleus is considered an important scaffold for the synthesis of new compounds with
potential biological activity. Various synthetic approaches have been reported in the
literature2 however fused tricyclic structures combining the indolizine and pyridine moieties
are rather uncommon. To our knowledge, the only synthesis of pyrido[2,3-b]indolizines
reported in the literature uses the reaction of 2-amino-3-(arylcarbonyl)indolizine-1-
carbonitrile with substituted acetophenones.3
The work that will be presented describes a novel eco-friendly regioselective approach for
the synthesis of novel substituted pyrido[2,3-b]indolizine-10-carbonitriles 3. The cascade
transformation involves the reaction of α,β-unsaturated carbonyl compounds 2 with
dipyridinium dichloride 1. The reaction was carried in ethanol and water, using sodium
acetate as catalyst (Scheme 1). Experimental details and mechanistic studies will be
presented. The products were fully characterized including X-ray spectroscopic analysis.
Scheme 1: Synthesis of pyrido[2,3-b]indolizine-10-carbonitriles 3.
Acknowledgements: We gratefully acknowledge the financial support from University of Minho and FCT through the Portuguese NMR network (RNRMN), the Project F-COMP-01-0124-FEDER-022716 (ref. FCT PEst-C/QUI/UI0686/2011) FEDER-COMPETE and BPD grant awarded to Marta Costa (SFRH/BPD/79609/2011). References: 1. For some examples see: a) Shen, Y.; Lv, P.; Chen, W.; Liu, P.; Zhang, M.; Zhu, H. Eur. J. Med. Chem. 2010, 45, 3184-3190; b) Bolle, L.; Andrei, G.; Snoeck, R.; Zhang, Y.; Lommel, A.; Otto, M.; Bousseau, A.; Roy, C.; Clercq, E.; Naesens, L. Biochem. Pharmacol. 2004, 67, 325-336; c) Gundersen, L.; Charnock, C.; Negussie, A.; Risea, F.; Teklu, S. European Journal of Pharmaceutical Sciences 2007, 30, 26-35; d) Teklu, S.; Gundersen, L.; Larsen, T.; Malterud, K.; Rise, F. Bioorg. Med. Chem. 2005, 13, 3127-3139.
2. Katritzky, A.; Rees, C.; Scriven, E. In Comprehensive Heterocyclic Chemistry II, Pergamon, Oxford, 1996, Vol. 8, 237-248. 3. Kakehi, A.; Suga, H.; Sato, S. Heterocycles 2009, 77, 471-481.

PC84
Synthesis of tetraoxane-pyridonimine antimalarials
J. Magalhães, M. J. Perry, A. Bavetta, A. P. Francisco, Rui Moreira, Francisca Lopes
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal
Elimination of malaria requires compounds that can act both in blood and liver stages.1 In a
synthetic program of hybrid antimalarials, we are synthesizing compounds with a 1,2,4,5-
tetraoxane moiety linked to 4-aminopyridines (Figure 1). Tetraoxane are cyclic peroxides
with potent in vitro and in vivo antimalarial activity,2 while 4-pyridonimimes have been
recently reported as novel chemotypes very effective in inhibiting liver stage P. berghei
parasites.3 Tetraoxanes were prepared in a one pot reaction by reacting 4-substituted
cyclohexanones with hydrogen peroxide and formic acid to give the corresponding gem-
dihydroperoxide. Adamantan-2-one and Re2O7 were then added to complete conversion to
the 1,2,4,5-tetraoxane esters. The synthesis of the hybrid molecules posed unique synthetic
challenges and we now report the synthetic methodologies developed to obtain the target
molecules.
Figure 1: General structure of the synthesized compounds
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for the research grant PTDC/SAU-FAR/118459/2010 and PEst-OE/SAU/UI4013/2011. References: 1. Wells, T.N.C., Burrows, J. N., Baird, J. K. Trends in Parasitology, 2010, 26, 145.
2. J. L. Vennerstrom, H.-N. Fu, W. Y. Ellis, A. L. Ager, J. K. Wood, S. L. Andersen, L. Gerena, W. K. Milhous, J. Med. Chem. 1992, 35, 3023-3027. 3. Rodrigues T. Novel mitochondrial electron transport-chain inhibitors as potential antimalarial agents. PhD thesis, University of Lisbon, 2010.
N
N R 2
R 1
O O
O O

PC85
Biosynthesis of phenazine and phenoxazinone derivatives catalysed
by CotA laccase
Ana Catarina Sousa,a,b
M. Conceição Oliveira,b Lígia O. Martins,
c M. Paula Robalo
a,b
aÁrea Departamental de Engenharia Química, Instituto Superior de Engenharia de Lisboa Rua Cons.
Emídio Navarro, 1, 1959-007 Lisboa, Portugal. bCentro de Química Estrutural, Complexo I, Instituto
Superior Técnico, Universidade Técnica de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal. cInstituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa, Av. da República, 2780-15
Oeiras, Portugal.
Heterocyclic compounds with phenazine and phenoxazinone frameworks displaying
biological activity have been used in medicinal applications, in the pharmaceutical industry
and due to their chromophoric structure, have also been applied as dyes or pigments in
many other industries. Most of the known procedures for the synthesis of these compounds
involve chemical condensations in aggressive conditions of temperature and oxidants.1,2
The use of cleaner alternative methods to organic synthesis in milder reaction conditions,
such as biocatalysis, represent an attractive route to the increasing demandof eco-friendly
processes in the chemical industry. Therefore, biocatalytic processes and in particular the
utilization of laccases (EC 1.10.3.2, benzenediol: oxygen oxidoreductase) are increasingly,
being used for the oxidation of a wide range of phenols and aniline derivatives followed by
homo and heterocoupling reactions.3 In this work we report the biosynthesis of coloured
phenazine and phenoxazine derivatives by CotA laccase (Scheme 1).The redox properties
of the substituted aromatic amines, used as substrates, were studied by cyclic voltammetry
and the kinetic parameters for the oxidation reactions were also determined. Compounds 1,
2 and 3 were obtained in good yields and identified by FTIR, 1D and 2D-NMR and ESI/MS
techniques.
CotA-Laccasephosphate buffer
24h, 37ºC
O2 H2O
R
N
SO3H
R
N
SO3HR = NH2 or NHPh
R
NH2
SO3H
O2 H2O
H2N
NH2
OH
O
H2N
O
N
NH2
(1) R = NH2 (2) R = NHPh
(3)
CotA-Laccasephosphate buffer
24h, 37ºC
Scheme 1: Biocatalytic synthesis of phenazine and phenoxazinone derivatives Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support (PTDC/BIO/72108/2006), to Portuguese NMR and MS Networks (IST-UTL Center) for providing access to facilities. References: 1. Polak, J., Jarosz-Wilkolazka, A.; Process Biochem. 2012, 47, 1295. 2. Witayakran, S., Ragauskas, A.J.; Adv.Synth. Catal. 2009, 351, 1187.
3. Morozova, O. V., Shumakovich, G. P., Gorbacheva M. A., Shleev, S. V., and Yaropolov A. I., Biochemistry, 2007, 72, 1136.

PC86
Smart Magnetic Liquids as novel magnetic materials
A. A. Rosatella, Carlos A. M. Afonso
iMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal
Magnetic fluids are liquids that have a strong response to an applied magnetic field, due to
magnetic particles (usually iron based particles, such as magnetite or hematite) suspended
in a carrier fluid (generally organic solvents, or water), Figure 1. Depending on the magnetic
particle size, these fluids can be magnetic fluids (MF) or magneto rheological fluids (MRF).
Some of these fluids are called smart fluids, due to their capability to change drastically a
physical property upon application of a magnetic field. These and other important
applications have gather scientists interested in magnetic materials although, the use of
magnetic fluids has some drawbacks: a) The synthesis of the magnetic particle, which
usually requires the use of toxic and expensive precursors, in organic solvents at high
temperatures.1 Additionally, the size of the particle is difficult to control. b) The stability of
the magnetic particle is difficult to achieve due to the possibility of agglomeration.2 c) The
carrier fluid, usually organic solvents or water, can affect magnetic and physical properties
of the material.2
Figure 1: Schematic example of a magnetic fluid.
Figure 2: Different MILs synthetized in our
laboratory, based on iron (III), gadolinium (III),
cobalt (II) and manganese (II) chloride
Recently, it was described a new type of magnetic materials based on paramagnetic metal
salts - Magnetic Ionic Liquids (MILs).3 These salts are the combinations of different organic
cations, with anionic metal complexes possessing magnetic properties (Figure 2). Usually
the metal anions are metal halides that are not air stable, and some can react with moister,
losing their magnetic properties. Another drawback of these materials is the unclear metal
structure, and sometimes different anions can be formed.
In this work we present the synthesis of stable magnetic fluids in a way to overcome the
disadvantages of magnetic fluids and MILs referred before.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia (POCI 2010) and FEDER (Ref.:SFRH/BPD/75045/2010) for financial support. References: 1. Kciuk, M.; Turczyn, R. Journal of Achievements in Materials and Manufacturing Engineering 2006, 18,
127. 2. Lu, A. H.; Salabas, E. L.; Schuth, F. Angew. Chem. Int. Ed. 2007, 46, 1222. 3. Hayashi, S.; Hamaguchi, H. O. Chem. Lett. 2004, 33, 1590.

PC87
Simple and more sustainable approaches for one pot enzymatic
resolution of sec-alcohols
C. M. Monteiro,a,b
N. M. T. Lourenço,c Carlos A. M. Afonso
a
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal; bCQFM, Centro de Química-Física Molecular, IN-Institute of Nanosciences and
Nanotechnology, Instituto Superior Técnico,1049-001 Lisboa, Portugal; cDepartment of Bioengineering,
IBB- Institute for Biotechnology and Bioengineering, Centre for Biological and Chemical Engineering I. S.
T., 1049-001 Lisboa, Portugal
Enantiomerically pure sec-alcohols are valuable molecules for several applications, namely
due to their biological relevance and versatile functional group transformation. In several
cases, where both enantiomers are important, the resolution of racemic alcohols is an
attractive approach. Over the last decade, our quest has been the development of
appealing, competitive and more sustainable processes for the enzymatic-resolution of
secondary alcohols. Therefore, our effort have been made on the development of new
strategies for the one-pot resolution-separation of free sec-alcohols by the use of new
acylating agents.1 The resolution-separation is based on the selective reaction of one
alcohol enantiomer with acylating agent, where one enantiomer stays in medium, leaving
the other enantiomer free to be removed. The anchored enantiomer can be isolated by a
second enzymatic reversible reaction. With this approach is possible to obtain both free
enantiomers using only the biocatalyst and a sustainable acylating agent. The main
advantage of this approach is the possibility to circumvent the limitations of the common
existing technology, specifically the use of chromatography separations, the use of organic
solvents and post-chemical transformations for the isolation of free enantiomers. This
methodology is quite simple, robust and reliable allowing the reuse of the medium and
enzyme. Herein, is present the progresses achieved on the different strategies for the
enzymatic one-pot resolution-separation of several sec-alcohols.
Scheme: Methodology for separation-resolution of secondary alcohols.
Acknowledgements: We thank Fundação para a Ciência e Tecnologia (POCI 2010) and FEDER (SFRH/BPD/41175/2007 and SFRH/BD/48395/2008), PTDC/QUI-QUI/119210/2010 and ACS Green Chemistry Institute (Ref. GCI-PRF#49150-GCI) for the financial support and also Novozymes and Amano enzymes for their generous enzyme supply. References: 1. Lourenço, N. M. T., Afonso, C. A. M. Angew. Chem. Int. Ed., 2007, 46, 8178; Monteiro, C. M.; Lourenço N. M. T.; Afonso C. A. M., Tetrahedron: Asymmetry, 2010, 952-956; Monteiro, C. M.; Lourenço N. M. T.; Afonso C.A.M., J. Chem. Educ., 2010, 87, 423-425, Lourenço N. M. T, Monteiro C. M., Afonso
C. A. M, Eur. J. Org. Chem., 2010, 6938–6943; Monteiro, C. M.; Lourenço N. M. T.; Ferreira F. C., Afonso C. A. M., submitted.

PC88
Application of L-proline salts for asymmetric organocatalysis
Karolina Zalewska, Luís C. Branco
REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de
Lisboa, 2829-516 Caparica, Portugal
Carbon-Carbon bond forming reactions based on asymmetric organocatalysis have been
gaining extensive interest over the past decade, and excellent results have been reported in
recent years.1,2
The current research has been concentrated on the development of more
efficient and selective catalytic systems using salts derived from natural acids for this
functional transformation. Expensive and often toxic transition metals as organocatalysts
have been replaced by amino acids, in particular L-proline based systems can catalyze
these reactions under mild conditions.3a,b
In this context, we focused on the Michael Addition and Aldol Reaction with the goal of
developing novel L-proline based salts as efficient and alternative active catalysts. Using
sustainable synthetic approaches, it was possible to obtain L-proline based salts using the
L-proline moiety as chiral organic cation or anion combined with appropriate counter-ions
(selected cations based on ammonium-type, [choline]; phosphonium-type, [P6,6,6,14] and
imidazolium-type, [emim], [C3Omim], [C2OHmim], [C2OHDmim]) or anions based on
bistriflimide, [NTf2] and docusate [AOT]). These L-proline based salts were employed as
catalysts for asymmetric Michael Addition of ketones to nitrostyrene (Scheme 1). The
reactions were carried out using water, ethanol or ionic liquids as reaction media. The
process affords synthetically valuable chiral products in good yields when proline scaffold
was used as anion. In the case of asymmetric aldol reaction between cyclohexanone or
acetone and 2-, or 4-nitrobenzaldehyde it was also possible to obtain the desired pure chiral
products in moderate to high yields and enantiomeric excesses comparable with the
conventional systems (using neutral L-proline as organocatalyst) (Scheme 2). Further
studies on the possibility to recovery the catalytic media using supercritical CO2 (scCO2) will
be evaluated.
Scheme 1: Asymmetric Michael addition of ketone to nitrostyrene using L-proline based salts as chiral
catalysts.
Scheme 2: Asymmetric Aldol Reaction using L-proline based salts as chiral catalysts.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support (PEst-C/EQB/LA0006/2011, PTDC/CTM/103664/2008 projects and doctoral fellowship KZ- SFRH/BD/67174/2009).
References: 1 Lu A.; Liu T.; Wu R.; Wang Y.; Wu G.; Zhou Z.; Fang J.; Tang C. J. Org. Chem. 2011, 76, 3872. 2 Vasiloiu M.; Rainer D.; Gaertner P.; Reichel C.; Schröder C.; Bica K. Catalysis Today 2013, 200, 80. 3 a) Bica K; Gaertner P. Eur. J. Org. Chem. 2008, 19, 3235. b) Branco L.; Zalewska K. Minireviews in Organic Chemistry, 2013, in press.

PC89
Iodine(III)-Mediated Beta-Lactam Formation via C-H Insertion/C-C
Bond Formation: A Diazo- And Metal-Free Approach
Luis F. R. Gomesa,b
, Nuno Maulideb, Carlos A. M. Afonso
a
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal; bMax-Planck-Institut f r ohlenforschung, aiser- ilhelm-Platz , 4 47 M lheim an der Ruhr,
Germany
Diazo compounds are known for their explosive, carcinogenic and toxic behaviour. Their
propensity towards facile dinitrogen liberation being generally responsible for those
hazards. They are nonetheless widely used in laboratory scale mainly due to the easy and
smooth metallocarbene formation. The dirhodium (II) tetrapaddlewheel catalysts can react
with diazo compounds in a clean reaction affording a stabilized yet very reactive
metallocarbene that can undergo several reactions such as cyclopropanation, ylide
formation or C-H insertion. Among these reactions C-H insertion typically provides excellent
yields and enantioselectivities, besides intermolecular versions are also available. The high
structure complexity assembly in two steps – diazo formation and its decomposition – has
also been used in total synthesis where a C-H insertion step can offer new and innovative
synthetic disconnections. Despite these advantages, on industrial scale processes the
explosive behaviour of diazos is a stringent limitation.
To circumvent the diazo functional group we have studied the already known
phenyliodonium ylides. These ylides are surrogates of the diazo moiety but higher catalyst
loadings are usually required, given the oxidizing properties of I(III) derivatives. Additionally,
the difficult purification and low stability of phenyliodonium ylides renders them less
appealing from a synthetic perspective (in spite of their reduced hazards). Herein, we
present a novel approach for the employment of iodonium ylides that bypasses the need for
isolation of those compounds, but rather engages them in a transition-metal free in situ
insertion step. The scope and limitations of this intriguing reaction shall be presented.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia (SFRH / BD / 61220 / 2009) and Acções Integradas Luso-Alemãs 2011 (A-14/11) for financial support. References: 1. a) Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L.; Chem. Rev., 2010, 110, 704. b) Davies, H. M. L.; Manning, J. R.; Nature, 2008, 451, 417 c) Archambeau, A.; Miege, F.; Meyer, C.; Cossy, J.; Angew. Chem. Int. Ed., 2012, 51, 11540 2. a) Müller, P.; Acc. Chem. Res., 2004, 37, 243 b) Moriarty, R. M.; Tyagi, S.; Kinch, M.; Tetrahedron, 2010, 66, 5801.

PC90
Self-assembly and hydrogelation behavior of new dehydropeptides
H. Vilaça,a O. Oliveira,
a G. Pereira,
a L. Hillioub,
b C. F. Lima,
c J. A. Martins,
a
P. M. T. Ferreiraa
aChemistry Centre, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal;
bInstitute for
Polymers and Composites/I3N, Department of Polymer Engineering, University of Minho, Campus de
Azurém, 4800-058, Guimarães, Portugal; cCITAB, Department of Biology, University of Minho, Campus
de Gualtar, 4710 - 057 Braga, Portugal
Recently, a group of low molecular weight peptides modified with bulky aromatic motifs has
been identified as a new class of hydrogelators.1 The biocompatibility and biodegradability
of this type of hydrogels make them ideal for biomedical applications.2 The enzymatic
hydrolysis of peptide based hydrogelators can be regarded as a serious disadvantage of
this type of materials. One way to circumvent this problem and thus increase biostability
consists in using non-proteinogenic amino acids. In our laboratories we have developed an
efficient method for the synthesis of dehydroamino acid derivatives using
butylpyrocarbonate, dimethylaminopyridine as catalyst and N,N,N’,N’-tetramethylguanidine.
Due to the high reaction yields and to the simple work-up procedures we were able to
prepare these compounds in large amounts and to use them as substrates in other types of
reactions to obtain new amino acids.3 Herein we describe the synthesis and hydrogelation
behavior of small dehydrodipeptides (Scheme 1).
Scheme 1: Structures of the new hydrogelators.
The new hydrogels were characterized using TEM, CD and fluorescence spectroscopy and
rheology. The enzymatic resistance of some of these hydrogelators was also evaluated.
These ultra short peptide hydrogelators are attractive candidates for biomedical
applications.
Acknowledgements: We thank the Fundação para a Ciência e Tecnolgia FEDER/COMPETE for financial support through CQ-UM and National NMR Network (Bruker 400) and I3N Strategic Project LA 25:2011-2012. H. Vilaça also thanks FCT for the PhD grant (SFRH/BD/72651/2010), co-funded by the European Social Fund. References: 1. Ryan, D. M; Nilsson, B. L. Polym. Chem. 2012, 3, 18-33. 2. a) Li, X.; Du, X.; Li, J.; Gao, Y.; Pan, Y.; Shi, J.; Zhou, N.; Xu, B. Langmuir 2012, 28, 13512-13517. b) Liang, G. L.; Yang, Z. M.; Zhang, R. J.; Li, L. H.; Fan, Y. J.; Kuang, Y.; Gao, Y.; Wang, T.; Lu, W. W.; Xu, B. Langmuir 2009, 25, 8419-8422. 3. a) Ferreira, P.M.T.; Monteiro, L. S.; Pereira, G.; Ribeiro, L.; Sacramento, J.; Silva, L. Eur. J. Org. Chem. 2007, 5934-5949. b) Ferreira, P. M. T.; Monteiro, L. S.; Pereira, G.; Castanheira, E. M. S.; Frost, C. G. Eur. J. Org. Chem. 2013, 3, 550-556. c) Pereira, G.; Vilaça, H.; Ferreira, P. M. T. Amino Acids, 2013, 44, 2, 335-334.

PC91
Exploratory chemistry for the synthesis of antimicrobial agents
starting from sugars
Vasco Cachatra, Andreia Almeida, Amélia P. Rauter
Grupo da Química dos Glúcidos, Centro de Química e Bioquímica/Departamento de Química e
Bioquímica, Faculdade de Ciências da Universidade de Lisboa, Campo Grande Edificio C8, 5o Piso,
1749-016 Lisboa, Portugal
Pyricularia oryzae is a fungus responsible for the disease known as rice blast that is the
most devastating disease affecting rice worldwide, both in terms of distribution and damage
caused. It affects about 85 countries, where rice is usually cultivated, and its remarkable
ability to overcome plant defenses is responsible for the destruction of an amount of rice
crops that would feed 60 million people annually. Since rice is an important food source
around the world it is imperative that a potent antibiotic is developed.1
Miharamycins (3) are a group of natural products that have been isolated form
Streptomyces miharaensis that exhibit a potent antimicrobial activity against various
microbes, particularly Pyricularia oryzae. It shows low toxicity and it does not inhibit
cholinesterases, which is an important feature for agrochemicals.2 Although various
synthesis have been proposed for the sugar core, they usually give mixtures of isomers that
are difficult to separate and use toxic reagents. In this work we present a synthesis for the
myharamycins sugar moiety (2) with simple and steroselective reactions starting from (1).
Scheme 1: Synthetic route towards the miharamycins. Acknowledgements: The authors thank FCT for Vasco Cachatra’s PhD grant (SFRH/BD/90359/2012) and for financial support of CQB Strategic Project PEst-OE/QUI/UI0612/2011. References: 1. a) K. Manibhusshan, Rice Blast Disease, 1994, 1 - 2; b) H. Kato, Pesticide Outlook, 2001, 23-25. 2. a) T. Tsuruoka, H. Yumoto, N. Ezaki, T. Niida, Sci. Reports of Meiji Kasha, 1967, 9, 1-4; b) T.
Shomura, K. Hamamoto, T. Ohashi, S. Amano, J. Yoshida, C. Moriyama, T. Niida, Sci. Reports Seika Kasha, 1967, 9, 5-10; c) T. Noguchi, Y. Yasuda, T. Niida, T. Shomura, Ann. Phytopath. Soc. Japan, 1968, 34, 323-327.

PC92
Synthesis of fused quinolone-benzazepines
Vera L. M. Silva, Inês M. M. Ribau, Artur M. S. Silva
Department of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal.
Quinolones and azepines are found in a wide range of natural products and are very well-
known due to their wide application in medicine, mainly as antibacterial, antiviral,
antidepressant and anticancer drugs.1,2
In addition the interest in seven-membered
azaheterocycles, such as azepines, by synthetic chemists, has persistently been increased
due to the high reactivity and some unique properties of these compounds.3 Here we
present our strategy for the synthesis of fused quinolone-benzazepine-type compounds.
This strategy comprises the synthesis of acrylamide 3, through the condensation of 2’-
aminoacetophenone 1 with 2-nitrocinnamic acid, methylation and in situ cyclization of 3 to
(E)-1-methyl-2-(2-nitrostyryl)quinolin-4(1H)-one 4 followed by C-3 halogenation and
reduction of the nitro group to give the corresponding amine 5. Finally the intramolecular
palladium (Buchwald-Hartwig)4 or copper (Ullmann)
4 mediated N-arylation of 5, leads to
fused quinolone-benzazepine 6 that can be further derivatized in the nitrogen atom of the
azepine nucleus. The developed methodology is technically simple, proceeds under
relatively mild conditions and forms potential biologically valuable products that are difficult
to synthesise by other methods. The experimental results and the structural characterization
of these new compounds will be also reported.
Scheme 1: Synthetic strategy for the synthesis of fused quinolone-benzazepine-type compounds
Acknowledgements: Thanks are due to the University of Aveiro, “Fundação para a Ciência e a Tecnologia” (FCT, Portugal), European Union, QREN, FEDER and COMPETE for funding the QOPNA research unit (project PEst-C/QUI/UI0062/2011) and to the Portuguese National NMR network, also supported by funds from FCT. References: 1. a) Mitscher L. A. Chem. Rev. 2005, 105, 559. b) Hahn F. E. Angew. Chem. Int. Ed. 2006, 45, 1348. 2. Kawase M., Saito S, Motohashi N. Int. J. Antimicrob. Agents, 2000, 14, 193.
3. Bremner J. B., Samosorn S. in Katritzky A. R. et al. (Eds) Compr. Heterocyclic Chem. Vol 13, Seven Membered Heterocyclic Rings, Elsevier, 2008, p. 560. 4. Fischer C., Koenig B. Beilstein J. Org. Chem. 2011, 7, 59.

PC93
Synthesis of novel aryl-fused 1,4-oxazin-3-ones
Małgorzata Śmist, Halina Kwiecień
Department of Organic Synthesis and Drug Technology, West Pomeranian University of Technology,
71-065 Szczecin, Poland
Aryl-fused 1,4-oxazine derivatives are well known for interesting biological and
pharmacological properties. Both natural and synthetic bezno- and naphth[1,4]oxazin-3-
ones were reported as compounds exhibiting various biological activity. For example,
substituted benzo[1,4]oxazines have been reported as hypoxia target compounds for
cancer therapeutics1
and as potassium channel modulators.2 Some of benzo[1,4]oxazepin-
5-ones are also known for showing anti-ischemic effects.3 On the other hand naphth[1,2-
b][1,4]oxazines can be useful in the treatment of parkinsonism, depression or hypertension4
whereas a series of spiroindolinonaphth[1,2-b][1,4]oxazines were reported as an excellent
group of photochromic dyes.5
The aim of our research was to obtain novel derivatives of aryl-fused 1,4-oxazin-3-ones.
Therefore we synthesised several derivatives having halogens, methyl, nitro or amino group
on the aromatic ring. Generally, the synthesis starts from 2-bromoalkanoates (2) and
commercially available 2-nitrophenols (1a) or 2-nitronaphthol (1b). The process is based on
catalytic reduction of proper nitroesters (3a, 3b), which undergo simultaneous
intramolecular cyclization to the target compounds (4a, 4b) (Scheme 1).
NO2
OH
R1
R3
R2
R
O
O
Br
K2CO3, DMF
NO2
O
R1
R3
R2
R
O
O
H2, Pd/C
HN
O
R1
R4
R2
O
R
1a
2
3 4
R = C2H5, C3H7, C4H9, R1 =H, Cl, R2 =H, F, CH3, R3=H, Cl, NO2, R4=H, Cl, NH2
NO2
OH O
NO2
R
OO
NH
O
R
OR
O
O
Br
K2CO3, DMF
2 H2, Pd/C
3a1b 4a
Scheme 1: Synthesis of aryl-fused 1,4-oxazin-3-one derivatives. Acknowledgements: This work is co-financed by the European Union from the European Agricultural Social and State Budget, the Operational Programme Human Resources Priority VIII, Action 8.2. Transfer of knowledge, 8.2.2. Regional Innovation Strategies, the project realized by Wojewódzki Urząd Pracy in Szczecin "An investment in knowledge is a motor of innovation in the region – 2
nd edition”.
References: 1. B. C. Das, A.V. Madhukumar, J. Anguiano, S. Mani, Bioorg. Med. Chem. Lett., 2009, 19, 4204. 2. S. Sebille, P. de Tullioa, S. Boveriea, M. H. Antoineb, P. Lebrunb, B. Pirotte, Curr. Med. Chem., 2004, 11, 1213. 3. K. Kamei et al., Bioorg. Med. Chem., 2006, 14, 1978. 4. J.H. Jones, U.S. Patent 4 420 480, 1983. 5. D.A. Clarke, B.M. Heron, C.D. Gabbutt, J.D. Hepworth, S.M. Partington, S.N. Corns, WO Patent 9920630, 1999.

PC94
Development of catalytic hydrogenation of CO2 promoted by ionic
liquids
Joana Afonso, Catarina I. Melo, Manuel Nunes da Ponte,* Ewa Bogel-Lukasik
#
REQUIMTE, Universidade Nova de Lisboa, Faculdade de Ciências e Tecnologia, Quinta da Torre, 2829-
516 Caparica, Portugal;
*[email protected]; #[email protected]
In recent years, CO2 sequestration and storage, whereby captured, purified and pressurized
CO2 is sent by pipeline into geological structures,1 has appeared as an practical means of
climate change mitigation. An attractive alternative to storage is to use it as feedstock to
produce fuels.2a,b
For reduction of CO2 with H2 homogeneous catalysis was provided in
supercritical carbon dioxide (scCO2) to produce formic acid,3 and in biphasic ionic liquid +
supercritical CO2 mixtures.4 Ru3(CO)12 homogeneous catalyst was promoted by iodide salts
in order to produce one-carbon products.5 [Ru(acac)3] with Triphos, 1,1,1-
tris(diphenylphosphinomethyl)ethane and the ruthenium(II)-complex [(Triphos)Ru-(TMM)]
(TMM, trimethylenemethane)6 in methanesulfonic acid and EtOH, or MeOH was applied at
lower temperature of 413K and a decreased time of reaction. Among heterogeneous
catalysts, low-cost Ni-based catalysts at 773-1123K were the most active for methanol
production.7 Reduction of CO2 with H2 to higher hydrocarbons was favoured by the
heterogeneous Fe(17%)-Mn(12%)-K(8%)/Al2O38 at 563K at a H2:CO2 ratio of 3:1.
Our work is focused on reduction of CO2 with hydrogen in biphasic ionic liquid /(CO2+H2)
mixtures in high-pressure CO2 at 413K, 14MPa, H2:CO2=1:3. We are exploring the effect of
the ionic liquid as solvent in order to obtain selectivity for different products of the reduction
- formic acid, methanol or methane. The reaction takes place in the IL phase where the
catalyst is immobilized (in case of homogeneous catalyst used) or is suspended (in case of
heterogeneous catalyst used). Pd, Ru and Rh heteregeneous and Ru homogeneous
catalysts are used for this propose. The products obtained are collected and analysed using
a GC method.
Acknowledgements: This work was supported by Fundação para a Ciência e a Tecnologia (FCT, Portugal), through the project PTDC/EQU/EPR/103505/2008 and by the European Commission within the Seventh Framework Programme (Project CEOPS: CO2 - Loop for Energy storage and conversion to Organic chemistry Processes through advanced catalytic Systems, NMP3-SL-2012-309984).
References:
1. Zhang, X. P.; Zhang, X. C.; Dong, H. F.; Zhao, Z. J.; Zhang, S. J.; Huang, Y. Energ. Environ. Sci. 2012, 5 (5), 6668 2. a) Yang, J.; Xian, M.; Su, S.; Zhao, G.; Nie, Q.; Jiang, X.; Zheng, Y.; Liu, W. PLoS ONE 2012, 7 (4), e33509. b) Federsel, C.; Jackstell, R.; Beller, M. Angew. Chem. Int. Ed. 2010, 49 (36), 6254. 3. Jessop, P. G.; Ikariya, T.; Noyori, R. Nature 1994, 368 (6468), 231. 4. Liu, F. C.; Abrams, M. B.; Baker, R. T.; Tumas, W. Chem. Commun. 2001, (5), 433. 5. Tominaga, K.; Sasaki, Y.; Kawai, M.; Watanabe, T.; Saito, M. Chem. Commun. 1993, (7), 629. 6. Wesselbaum, S.; vom Stein, T.; Klankermayer, J.; Leitner, W. Angew. Chem. Int. Ed. 2012, 51, 1. 7. Ma, J.; Sun, N. N.; Zhang, X. L.; Zhao, N.; Mao, F. K.; Wei, W.; Sun, Y. H. Catal. Today 2009, 148 (3-4), 221. 8. Dorner, R. W.; Hardy, D. R.; Williams, F. W.; Willauer, H. D. Catal. Commun. 2011, 15 (1), 88.

PC95
Synthesis of Enantiomerically Pure Chiral N-tert-Butylsulfinamide-
alkene ligands via a novel Catalytic Method
Albertino Goth, Anthony J. Burke
Departamento Química, Centro de Química de Évora e LADECA, Universidade de Évora,
Rua Romão Ramalho 59, 7000 Évora, Portugal
[email protected], [email protected]
N-Sulfinyl imines are versatile intermediates in the asymmetric synthesis of chiral amines,
such as α-branched amines, α,α-dibranched amines, α- and β-amino acids, aziridines and
α- and β-aminophosphonic acids.1
In this communication we will discuss our efforts at the asymmetric catalytic synthesis of N-
tert-butylsulfinamide-alkenes (Figure), which we wish to use as valuable chiral ligands in
the asymmetric catalytic synthesis of bioactive compounds for treating neurodegrenative
diseases such as Alzheimer's and Parkinson's.2
O
S
NH2
+
O
S
N
R
H
O
R H
Chiral catalyst
n = 0,1
B
O
O
O
SNH
RH n = 0,1
Figure: Enantioselective catalytic allylation/vinylation of sulfinimines
Acknowledgements: This work is supported by the project: INMOLFARM - Molecular Innovation and Drug Discovery (ALENT-57-2011-20) financed from the FEDER-INALENTEJO program ALENT-07-0224-FEDER-001743, as well as PEst-OE/QUI/UI0619/2011. References: 1. a) Guangcheng Liu, Derek A. Cogan, Timothy D. Owens, Tony P. Tang, and Jonathan A. Ellman, J. Org. Chem. 1999, 64, 1278-1284; b) Xiangqing Feng, Yazhou Wang, Beibei Wei, Jing Yang, Haifeng Du, Org. Lett., 2011, 13, 13; c) Elisa. Serra, Dina Murtnho, Albertino Goth, Antonio Rocha Gonsalves, Chirality, 2010, 22, 425–431
2. a) Darryl W. Low, Graham Pattison, Martin D. Wieczysty, Gwydion H. Churchill, Hon Wai Lam, Org. Lett., 2012, 14, 10; b) Paivi Tolstoy, Samantha X. Y. Lee, Christof Sparr, and Steven V. Ley, Org. Lett., 2012, 14, 18; c) Rodney A. Fernandes, Jothi L. Nallasivam, Org. Biomol. Chem. 2012, 10, 7789.

PC96
Rhodium Catalysed Tandem Hydroformylation/Arylation Reactions
Ana R. Almeida,a Artur R. Abreu,
a Pedro M. P. Góis,
b Juan C. Bayón,
c Mariette M. Pereira
a
aDepartamento de Química, Universidade de Coimbra, Coimbra, Portugal;
biMed.UL, Faculdade de
Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa, Portugal; cDepartament
de Química, Universitat Autònoma de Barcelona, Bellaterra, 08193 Barcelona, España
Aryl alcohols are key structural elements in a large number of pharmacologically active
compounds.1 In this context, tandem processes, starting from simple olefins, that allow the
preparation of high value compounds are attractive synthetic routes in organic chemistry. In
order to achieve the highest possible efficiency of the processes the combination of tandem
reaction sequences with enantioselective chemical transformations is crucial to attend the
modern synthetic demands.2
From the best of our knowledge there are no examples in the literature describing
sequential hydroformylation/arylation reaction using the catalyst in both steps.
In this communication we will present the results for sequential rhodium catalysed
hydroformylation of olefins,3 followed by in situ aldehyde arylation with different boronic
acids,4 using the same catalyst (Scheme 1). Some preliminary results of asymmetric
tandem hydroformylation/arylation reactions will be also presented and discussed.
Scheme 1: Tandem Hydroformylation/Arylation reactions. Acknowledgements: The authors thank for financial support to Programa Compete and QREN/FEDER/FCT (PTDC/QUI-QUI/112913/2009). A.R. Almeida also thanks to FCT for PhD grant SFRH/BD/73190/2010. References: 1. Bolshan Y.; Chen C.-Y.; Chilenski J. R.; Gosselin F.; Mathre D. J.; O’Shea P. D.; Roy, A.; Tillyer R. D. Org.Lett. 2004, 6, 111. 2. Eilbracht P.; Schmidt A. M. Top. Organomet. Chem. 2006, 18, 65.
3. Peixoto A. F.; Melo D. S.; Fernandes T. F.; Fonseca Y.; Gusevskaya E. V.; Silva A. M. S.; Contreras R. R.; Reyes M.; Usubillaga A.; Santos E. N. ; Pereira, M. M.; Bayón J. C. Appl. Catal., A 2008, 340, 212. 4. Trindade A. F.; Góis P. M. P.; Veiros L. F.; André V.; Duarte M. T.; Afonso C. A. M.; Caddick S.; Cloke, F. G. N. J. Org. Chem. 2008, 73, 4076.

PC97
Oxazolidinone Polycyclitol. Synthesis of Novel Aminocarbasugars
with Oxazolidinone Ring
Latif Kelebekli,a Neslihan Balci
b
aDepartment of Chemistry, Ordu University, 52200 Ordu, Turkey
bDepartment of Chemistry, Ataturk University, 25240 Erzurum, Turkey
Oxazolidinones are a new class of synthetic antimicrobial agents which are now clinically
useful.1 Aminocarbasugars which are carbasugars with amino cores such as validamine 1
and voglibose 2 have been used for the treatment of type 2 diabetes.
2 Aminocarbasugars
and their derivatives are an important subclass of cyclitols and are thought to be more
potent drug candidates than natural sugars, since they are much more stable towards
hydrolysis.3 Considering the fundamental importance of polyhydroxylated and oxazolidinone
ring compounds, we aimed at the first synthesis and family of oxazolidinone polycyclitols.
Compounds 3 which are a new aminocarbasugars were synthesized starting from p-
benzoquinone. Reduction of related to the diketones with NaBH4 followed by acetylation
gave diol-acetate. The treatment of diol-acetate with p-toluenesulfonyl isocyanate in the
presence of Pd(0)-catalyst gave oxazolidones.4 Oxidation of two double bonds reaction in
oxazolidone compounds with OsO4 followed by acetylation afforded oxazolidone
hexaacetates. Hydrolysis of the acetate groups furnished the desired oxazolidinone
polycyclitols.5
Acknowledgements: We are most indebted to The Scientific and Technical Research Council of Turkey (TUBITAK, Grant No: TBAG-108T112) for financial support of this work. References: 1. Zappia G.; Gacs-Baitz E.; Monache G. D.; Misiti D.; Nevola L.; Botta B. Current Organic Synthesis, 2007, 4, 81. 2. Berecibar A.; Grandjean C.; Siriwardena A. Chem. Rev. 1999, 99, 779. 3. Horii S.; Iwasa T.; Mizuta E.; Kameda Y. J. Antibiot. 1971, 24, 59. 4. a) Trost B. M.; Van Vranken D. L. J. Am. Chem. Soc. 1993, 115, 444. b) Kelebekli L.; Çelik M.; Şahin E.; Kara Y.; Balci M., Tetrahedron Lett., 2006, 47, 7031, 5. Kelebekli L.; Balci N.; Şahin E. Tetrahedron 2012, 68,1886.

PC98
Rhodium Catalyzed Enantioselective Arylation of Glyoxylate
Derivatives using Organoboron Reagents
Carolina S. Marques,a Anthony J. Burke,
a Mehmet Dindaroğlu,
b Hans-Günther Schmalz
b
aDepartment of Chemistry and Chemistry Center of Évora, University of Évora, Rua Romão Ramalho, 59,
7000 Évora, Portugal. bDepartment ofChemistry,University of Cologne,Greinstrasse 4,50939
Köln,Germany.
The α-hydroxyester structural function is widespread in natural products and is a convenient
building block in organic synthesis.1
For example, a group of glycosphingolipids, known as
cerebrosides found in animal muscle and nervecell membranes along with (+)-Wikstromol,
an antitumor compound found in plants, contain in their backbone an α-hydroxyester unit
(Figure 1). Although a number of synthetic methods exist in the literature, the development
of new efficient methods for the stereoselective synthesis of chiral α-hydroxyesters remains
a challenge. As an extension of our work on the development of new transition-metal
catalysts to perform the catalytic arylation of activated imine substrates with boronic acids
and derivatives,2 we decided to investigate the arylation of glyoxylate esters as a route to
such products. Herein, we report our results on the asymmetric catalytic version using Rh(I)
catalysts and chiral phosphine-phosphite ligands as well as new N-heterocyclic carbene
(NHC) ligands (Scheme 1).3
Scheme 1: Enantioselective synthesis of mandelate derivatives with Rh(I) catalysts.
Figure 1.
Acknowledgements: We are grateful for the award of a PhD Grant to C.S.M. (SFRH/BD/45132/2008) from the Fundação para a Ciência e a Tecnologia (FCT) 2010. Chiratecnics Lda is acknowledged for donating some precursors and its interest in this project. We acknowledge Lab-RMN at FCT-UNL for the acquisition of the NMR spectra. The University of Vigo (Spain) is gratefully acknowledged for MS analysis. References: 1. Coppola, G. M.; Schuster, H. F. α-Hydroxy Acids in Enantioselective Synthesis; VCH: Weinheim, 1997, pp 1-511; 2. a) Marques, C. S.; Burke, A. J. Eur. J. Org. Chem. 2010, 1639–1643; b) Marques, C. S.; Burke, A. J. ChemCatChem 2011, 3, 635–645; c) Marques, C. S.; Burke, A. J. Eur. J. Org. Chem. 2012, 4232–4239. d) Marques, C. S.; Burke, A. J. Tetrahedron: Asymmetry 2013, http://dx.doi.org/10.1016/j.tetasy.2013.04. 011. 3. a) Marques, C. S.; Burke, A. J. Tetrahedron 2012, 68, 7211–7216; b) Marques, C. S.; Burke, A. J. Tetrahedron: Asymmetry 2013, http://dx.doi.org/10.1016/j.tetasy.2013.04.011; c) Dindaroğlu, M.; Falk, A. ; Schmalz, H.-G. Synthesis 2013, 45, 527-535 ; d) Dindaroğlu, M.;et al. Tetrahedron: Asymmetry 2013, http://dx.doi.org/10.1016/j.tetasy.2013.04.008.

PC99
BoImAr: Borylation(Catalytic)-Imination-Arylation(Catalytic) - A New
Synthetic Approach to Promising Alzheimer and Parkinson Drugs
Daniela Peixoto, Carolina S. Marques, Paulo J. Mendes, Anthony J. Burke
Departamento e Centro de Química, Universidade de Évora, Rua Romão Ramalho 59, 7000 Évora.
Although dramatic progress has been made in understanding the pathogenesis of
neurodegenerative conditions of the aged population such as Alzheimer's disease,
Parkinson's disease and Fronto-Temporal dementia, to date most of these diseases are
incurable. Because of the aging population, these disorders pose a serious challenge to the
health care system. Loss of synapses is probably the common neuropathological feature
leading to dementia in these neurodegenerative disorders.1
Parkinson's disease is a progressive neurodegenerative condition caused by loss of
dopamine producing cells in the substantia located in the basal ganglia causing motor,
autonomic and cognitive impairments.2 Rasagiline is a potent, selective, irreversible inhibitor
of monoamine oxidase (MAO) which is an anti-Parkinson drug.3
Herein, we present our innovative approach to the synthesis of several chiral amine4
-benzolactam analogues which involves a one-pot borylation-Imination-Arylation (BoImAr)
sequence - the last step being a key intramolecular catalytic arylation reaction (Scheme 1).
NH
Rasagiline
CO2H
Br(I)
R1
R1 = OR or alkane
H2NOR
OR
R1 NH
O
OR
ORBr(I)
NH
O
HNR2
R1
Borylation Imination Arylation
n
n
n*
Scheme 1
Acknowledgements: This work is supported by the project: INMOLFARM - Molecular Innovation and Drug Discovery (ALENT-57-2011-20) financed from the FEDER-INALENTEJO program ALENT-07-0224-FEDER-001743, as well as PEst-OE/QUI/UI0619/2011 (CQE-UE). References: 1. Rockenstein, E.; Crews, Leslie.; Masliah, E. Advanced Drug Delivery Reviews, 2007, 59, 1093. 2. Chenoweth, L.; Sheriff, J.; McAnally, L.; Tait; F. Nurse Education Today, 2013, 33, 458. 3. Weinreb O.; Amit T.; Bar-Am O.; Youdim, M. B. H. Progress in Neurobiology, 2010, 92, 330. 4. Marques, C. S.; Burke, A. J. Chem. Cat. Chem., 2011, 3, 635.

PC100
Highly enantioselective synthesis of substituted Δ1-pyrrolines
D. I. S. P. Resende, Cristina G. Oliva, Artur M. S. Silva
QOPNA, Departamento de Química, Universidade de Aveiro, Campus Universitário de Santiago, 3810-
193 Aveiro, Portugal
Nitrogen-containing heterocycles are important core structures in natural and synthetic
biologically active componds.1 In particular 3,4-dihydro-2H-pyrroles, also called
Δ1-pyrrolines, can be found in a wide variety of biologically active compounds such as
hemes, chlorophylls andbacteriochlorins.2
Recent studies in our laboratory have led to the
development of a new methodology for the highly asymmetric organocatalytic 1,4-Michael
addition of nitromethane to different 1,5-diarylpenta-2,4-dien-1-ones, leading to (R,E)- and
(S,E)-1,5-diaryl-3-(nitromethyl)-5-pent-4-en-1-ones 1 with excellent levels of
enantioselectivity (up to 99%) and isolated yields (up to 97%).3
These compounds,
containing a nitro group, are attractive starting materials for the synthesis of various
synthetic compounds and can undergo a series of transformations. They can be easily
converted in ketones, reduced to amines or transformed into carboxyl groups, imines or
hydroxylamines.
Here we report the highly enantioselective one pot synthesis of Δ1-pyrrolines 2 through a
reduction of the nitro group and subsequent nucleophilic ring closure. We have tested four
different reductive systems widely used for this type of reductive cyclization and we have
observed that the reducing system employed plays an important role on these
transformations. Depending on the reduction method that is applied (Zn, Sn, Fe, Pd/C), we
can obtain either the Δ1-pyrroline 2 or the corresponding pyrroline N-oxide 3, both with high
levels of enantioselectivity (Scheme 1).
Scheme 1: Synthesis of Δ
1-pyrrolines2 and pyrrolineN-oxide 3.
Acknowledgements: Thanks are due to the University of Aveiro, Portuguese Foundation for Science and Technology (FCT), European Union, QREN, FEDER and COMPETE for funding the QOPNA research unit (project PEst-C/QUI/UI0062/2011), and the Portuguese NMR Network. D. I. S. P. Resende thanks also FCT for her PhD grant (SFRH/BD/62696/2009). References: 1. a) Royer, J “Asymmetric Synthesis of Nitrogen Heterocycles” Wiley-VCH: Weinheim, 2009; b) Wijdeven, M. A.; Willemsen, J.; Rutjes F. P. J. T. Eur. J. Org. Chem. 2010, 15, 2831–2844. 2. a) Kadish, K. M., Smith, K. M., Guilard, R., Eds.; "The Porphyrin Handbook", Academic Press: San Diego, 2000; Vol. 1.; b) Reddy, K. R.; Lubian, E.; Pavan, M. P.; Kim, H.-J.; Yang, E.; Holten, D.; Lindsey, J. S. New J.Chem. 2013, 37, 1157; 3. a) Oliva, C. G..; Silva, A. M. S.; Paz, F. A. A.; Cavaleiro, J. A. S. Synlett 2010, 1123. b) Oliva, C. G.; Silva, A. M. S.; Resende, D. I. S. P.; Paz, F. A. A.; Cavaleiro, J. A. S. Eur. J. Org. Chem. 2010, 18, 3449.

PC101
Thiazolidines for the Enantioselective Alkylation of Aromatic
Aldehydes
M. Elisa da Silva Serra, Dina Murtinho, Teresa M. V. D. Pinho e Melo
Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade de Coimbra, 3004-535
Coimbra, Portugal
The enantioselective alkylation of aldehydes using diethylzinc in the presence of chiral
ligands constitutes a convenient process for the synthesis of optically active secondary
alcohols with high optical. Numerous types of ligands have been used to induce chirality in
these reactions, among which are some mentions to 1,3-thiazolidine-4-carboxylic esters.1
Very few references, however, have been made to the use of hydroxymethylthiazolidines.2
Continuing our studies on enantioselective catalysis, we synthesized
hydroxymethylthiazolidines 3 and 4 and tested their ability to induce chirality in the
enantioselective alkylation of aldehydes with ZnEt2 (Scheme 1). These results were
compared to those obtained using the corresponding methyl-1,3-thiazolidine-4-carboxylate
esters 1 and 2.
All reactions gave excellent conversions to 1-phenylpropan-2-ol and enantioselectivities of
up to 89%.
In this communication we will describe the synthesis of the chiral ligands and present the
results of the enantioselective alkylations catalyzed by these ligands. Results concerning
the synthesis and use of analogous compounds derived from D-penicillamine will also be
presented. The results of the alkylations using all ligands will be discussed, namely, effects
of steric bulk and the sense of chirality of the product.
Scheme 1: Synthesis of chiral thiazolidines.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia (Project Pest-C/QUI/UI0313/2011), FEDER, COMPETE and QREN for financial support. References: 1. a) Jin, M-J; Kim, S-H. Bull. Korean Chem. Soc., 2002, 23, 509. b) Meng, Q L; LI, Y. L; HE, Y; Guan, Y. Tetrahedron: Asymmetry 2000, 11, 4225. 2. Kim, S-H.; Chung, S. T.; Jin, M-J. J. Ind. Eng. Chem., 1998, 4, 345.

PC102
Studies on the Lewis acid catalysed cycloaddition reactions of (E)-
N-(2-acetylphenyl)-3-arylacrylamides with ortho-
benzoquinodimethane
Gustavo da Silva, Vera L. M. Silva, Artur M. S. Silva
Department of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal
The development of new methodologies for the synthesis of quinolin-4(1H)-ones and their
transformation into other heterocycles has been stimulated by the wide spectrum of
pharmacological activity attributed to these molecules, due to their potential applications as
antibiotics and as chemotherapeutic and fluorescent labeling agents.1
Following our research interest in the synthesis of novel quinolin-4(1H)-ones, this work aims
at the reactivity study of (E)-N-(2-acetylphenyl)-3-arylacrylamides 1 as dienophiles in Lewis
acid catalysed cycloaddition reactions with the very reactive ortho-benzoquinodimetane
used as diene, affording new N-(2-acetylphenyl)-3-aryl-1,2,3,4-tetrahydronaftalene-2-
carboxamides 2. In the present communication, the intramolecular cyclization of these
cycloadducts to 2-(3-aryl-1,2,3,4-tetrahydronaphthalen-2-yl)quinolin-4(1H)-ones 3, followed
by dehydrogenation to 2-(3-arylnaphthalen-2-yl)quinolin-4(1H)-ones 4 will be reported. The
influence on the reactivity of electron-withdrawing and electron-donating groups on the aryl
ring of the acrylamides (Scheme 1) will be also discussed. Besides our experimental results
we will present the structural characterization of the new synthesised compounds.
Scheme 1 Reagents and conditions: (i) AlCl3, 1,2,4-TCB reflux; (ii) NaH, THF, 80 ºC;
(iii) DDQ, 1,4-dioxane, 110 ºC. Acknowledgements: Thanks are due to the University of Aveiro, “Fundação para a Ciência e a Tecnologia” (FCT, Portugal), European Union, QREN, FEDER and COMPETE for funding the QOPNA research unit (project PEst-C/QUI/UI0062/2011) and to the Portuguese National NMR network, also supported by funds from FCT. Gustavo da Silva also thanks project QREN (FCOMP-01-0124-FEDER-010840) (PTDC/ QUI-QUI/ 102454/ 2008) for his research grant. References: 1. (a) Michael, J. P. Nat. Prod. Rep. 2008, 25, 166. (b) Hachiya, I., Mizota, I., Shimizu, M. Heterocycles. 2012, 85, 993. (c) Eggleston, M. & Park, S.-Y. Infection Control. 1987, 8,119.

PC103
BorArAm - Catalytic Asymmetric Arylating Cyclizations: A New
Route to Chiral Bicyclic Amines
H. Viana, C. S. Marques, Paulo J. Mendes, Anthony J. Burke
Departamento de Química, Centro de Química de Évora e LADECA, Universidade de Évora
Rua Romão Ramalho 59, 7000 Évora, PORTUGAL.
[email protected], [email protected]
Nowadays neurodegenerative diseases such as Alzheimer’s disease and Parkinson
disease represent a worldwide health threat. Rasagiline is one well-known medication for
the treatment of Parkinson's disease, but more and cheaper alternatives are required.1
For
this reason, our group is currently investigating a new catalytic asymmetric arylating2
cyclization route - borylation-arylation-amination (BorArAm) (Scheme 1) giving useful
potential lead compounds based on the rasagiline core structure for treating these diseases.
Our results will be discussed in this communication.
X = NR, O, S
X
I(Br)
n
O
X
OH
X
B
n
O
OR
OR
1
2
3X
NHR
1 Borylation2 Arylation3 Amination
n
BorArAm
N
Rasagline
Scheme 1: Reaction sequence for the synthesis of chiral bicyclic amines.
Acknowledgements: This work is supported by the project: INMOLFARM - Molecular Innovation and Drug Discovery (ALENT-57-2011-20) financed from the FEDER-INALENTEJO program ALENT-07-0224-FEDER-001743, as well as PEst-OE/QUI/UI0619/2011 (CQE-UE). References: 1. Orly Weinreb, Tamar Amit, Orit Bar-Am, Moussa B.H. Youdim, Progress in Neurobiology, 2010, 92, 330–344; 2. C. S. Marques, A. J. Burke, ChemCatChem, 2011, 3, 635-645.

PC104
Valorization of biomass derived intermediates based on metal
organo catalysis
Jaime A. S. Coelho,a Alexandre F. Trindade,
b Carlos A. M. Afonso
a
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal; bCentro de Química-Física Molecular, IN-Institute of Nanosciences and Nanotechnology,
Instituto Superior Técnico,1049-001 Lisboa, Portugal
The efficient use of biomass has recently attracted considerable attention as a potential
alternative to limited petroleum resources for fuel and chemicals productions.1
Furanics,
such as 5-hydroxymethylfurfural (HMF), are one of the major class of compounds obtained
from carbohydrates. HMF is already a feedstock for common polyester building blocks,2
including 2,5-furandicarboxylic acid, 2,5-bis(hydroxymethyl)furan, and a precursor to 2,5-
dimethylfuran, which is a promising alternative liquid transportation fuel.3 Recent examples
towards the HMF valorization are the synthesis of caprolactam and ranitidine (Zantac).4,5
Increasing interest in this molecule led to rapid progress in the HMF synthesis from simple
sugars and therefore multiple conditions are now known.6 Very recently our group was also
significantly contributed for this area by the development of the straightforward production
and isolation of HMF from fructose7 and the more challenging integrated chemo-enzymatic
production of HMF from glucose.8
In the course of our studies through the exploration of a novel approach for the
transformation of HMF derivatives into valuable molecules a homo-Mannich type reaction
was discovered. Herein we would like to present the results obtained.
Scheme 1: Synthesis and valorization of HMF.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia(PEst-OE/SAU/UI4013/2011 and SFRH/BD/73971/2010) for financial support. References: 1. Christensen, C. H. et. al., ChemSusChem 2008, 1, 283-289; 2. See for example: Corma, A., Chem. Rev. 2007, 107, 2411-2502; 3. Recent example: Leshkov, Y. R. et. al., Nature 2007, 447, 982-986; 4. Buntara, T. et al., Angew.Chem. Int. Ed.2011, 50, 7083 7087; 5. Mascal, M., Green Chem.2011, 13, 3101-3102. 6. Rosatella, A. A. et al., Green Chem. 2011, 13, 754-793; 7. Simeonov, S. P., Coelho, J. A. S. and Afonso, C. A. M., ChemSusChem 2012, 5, 1388; 8. Simeonov, S. P., Coelho, J. A. S. and Afonso, C. A. M., ChemSusChem 2013, 6, 997-1000.

PC105
Synthesis of 3-arylquinolin-4(1H)-ones by Suzuki cross-coupling
reactions under PTC conditions using ohmic heating
Joana Pinto,a Vera L. M. Silva,
a Ana M. G. Silva,
b Luís M. N. B. F. Santos,
c Artur M. S. Silva
a
aDepartment of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal.
bREQUIMTE,Department of Chemistry and Biochemistry Faculty of Sciences, University of Porto, 4169-
007 Porto, Portugal. cCentro de Investigação em Química, Departamento de Química e Bioquímica,
Faculdade de Ciências da Universidade do Porto, Rua Campo Alegre 687, 4169-007 Porto, Portugal.
[email protected]; [email protected]; [email protected]
Quinolin-4(1H)-ones are commercially available for clinical use and represent one of the
major classes of antibiotics inthe world.Their clinical applications are diverse and include the
treatment of urinary, respiratory, gastrointestinal and gynaecological infections,
osteomyelitis, chronic prostatitis, sexually transmitted diseases and some skin, bone and
soft tissues infections.1 Considering the referred important applications and in continuation
of our previous work on the synthesis and transformations of quinolin-4(1H)-ones,2
now we
will present our recent results on the synthesis of 3-arylquinolin-4(1H)-ones 3. These
compounds were obtained through Suzuki cross-coupling reaction of3-iodo-1-
methylquinolin-4(1H)-one 1 with the appropriate arylboronic acids 2 under phase transfer
conditions (PTC) using ohmic heating (H). This is an advanced thermal processing
method where the reaction mixture, usually an aqueous medium, which serves as an
electrical resistor, is heated by passing electricity through it.3 Electrical energy is dissipated
into heat with high efficiency, which results in a high speed heating rate, allowing a rapid
and uniform heating (temperature homogeneity) and an increased directional dynamics of
charged species and dipole orientation in solution. Thus it is expected to achieve a
significant reduction of reaction time and an increase of energetic efficiency of the chemical
transformations by using ohmic heating. In addition, we will explore the influence of the
electrodes material, in contact with the reaction medium, in the Suzuki coupling reaction.
The experimental results and structural characterization of the obtained 3-arylquinolin-
4(1H)-ones will be also reported.
Figure 1: Synthesis of 3-arylquinolin-4(1H)-ones by Suzuki cross-coupling reactions under phase
transfer conditions.
Acknowledgements: Thanks are due to the University of Aveiro, “Fundação para a Ciência e a Tecnologia” (FCT, Portugal), European Union, QREN, FEDER and COMPETE for funding the QOPNA research unit (project PEst-C/QUI/UI0062/2011), to the Portuguese National NMR network, also supported by funds from FCT and to the project QREN (FCOMP-01-0124-FEDER-010840) (PTDC/QUI-QUI/102454/2008). Joana Pinto also thanks FCT for her PhD grant (SFRH/BD/77807/2011).
References: 1. a) J.-I. Alós, Enferm. Infecc. Microbiol. Clin 2003, 21, 261; b) G. L. A. S. M. Mella, M. Q. Muñoz, C. C. Perez, J. L. Labarca, G. R. Gonzalez, H. T. Bello, M. Y. Dominguez e R. Z. Zemelman, Rev. Chil. Infect. 2000, 17, 53. 2. A. I. S. Almeida; A. M. S. Silva; J. A. S. Cavaleiro. Synlett. 2010, 3, 462; b) R. S. G. R. Seixas; A. M. S. Silva; J. A. S. Cavaleiro. Synlett. 2010, 15, 2257; c) R. S. G. R. Seixas; A. M. S. Silva; I. Alkorta; J. Elguero. Monatsh Chem, 2011, 142, 731. 3. Pinto, J.; Silva, V. L. M.; Silva, A. M. G.; Silva, A. M. S.; Costa, J. C. S.; Santos, L. M. N. B. F.; Enes, R.; Cavaleiro, J. A. S.; Vicente, A. A. M. O. S.; Teixeira, J. A. C. Green Chem, 2013, 15, 970.

PC106
Application of Click Chemistry Reaction in Iminosugars Derivatives
M. Isabel Ismael,a J. Albertino Figueiredo,
a M. Domingues,
a Marie Schuler,
b
Arnaud Tatibouëtb
aDpto. de Química, Unidade I&D Materiais Têxteis e Papeleiros, Universidade da Beira Interior, 6201-
001 Covilhã, Portugal, bUniversity of Orléans, Institut de Chimie Organique et Analytique, Rue de
Chartres, Orléans, 45067, France
Iminosugars are small organic compounds that mimic carbohydrates by replacing with a
nitrogen atom, the oxygen in the sugar ring.1 With the goal of improving the biological
activity of the synthesized molecules, click ligation has been used to prepare iminosugars
linked with sugars. Compounds synthesized have potential biological activities in cancer,
infectious diseases, hepatitis B and C, neurodegenerative diseases, lysosomal storage
disorders, cystic fibrosis and type II diabetes.2,3
Nowadays, "Click-Chemistry" is an efficient approach for the synthesis of numerous
compounds making reactions of carbon-heteroatom bond formation. The Copper catalysed
azide-alkyne cycloaddition (CuAAC) is a mild and selective reaction that gives 1,2,3-
triazoles as products.4
Iminosugar
PG= CH3 or isopropylidene
Iminosugar
Scheme 1: Synthesis of iminosugars derivatives.
In this work were made the coupling CuAAC reactions to prepare triazoles from an alkyne
derived iminosugar in the presence of an azido sugar, using copper(I) as a reaction
promoter (Scheme 1). The resulting compounds were identified by NMR and IR
spectroscopy.
References: 1. Kotland, A.; Accadbled, F.; Robeyns, K.; Behr, J-B J. Org. Chem. 2011, 76, 4094. 2. Winchester, B. G. Tetrahedron: Asymmetry 2009, 20, 645. 3. Compain, P.; Martin, O. R. Iminosugars: From Synthesis Therapeutic Applications; John Wiley & Sons Ltd., 2007. 4. (a) Colombo, M.; Bianchi, A. Molecules 2010, 15, 178. (b) Pengju, J.; Atherton, J.; Page, M.Org. Biomol. Chem. 2012, 10, 7965.

PC107
Efficient EDA Addition and Ring-Expansion Reaction of Isatins
Catalyzed by a DBU/Rh(II) Metal-Organo System: On Route to the
Synthesis of Viridicatin Alkaloids
R. Paterna,a Nuno R. Candeias,
b Pedro M. P. Góis
a
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal; bDepartment of Chemistry and Bioengineering, Tampere University of Technology, Finland.
An efficient and novel 4-steps route for the synthesis of the viridicatin alkaloids via Suzuki-
Miyaura coupling reaction of aryl-boronic acids with 3-hydroxy-4-bromoquinolin-2(1H)-ones
prepared from 3-hydroxy-4-ethylesterquinolin-2(1H)-ones will be presented. The 3-hydroxy-
4-arylquinolin-2(1H)-ones core, include several natural products like viridicatin,1 viridicatol
2
and 3-O-methyl viridicatin,3 which have been reported as very promising inhibitors against
the human immunodeficiency virus replication induced by tumour necrosis4 and as
promising lead compounds for the development of new anti-inflammatory agents.5 We have
developed a new one-pot NHC-dirhodium(II)/DBU catalyzed Eistert ring expansion reaction
of isatins with ethyl diazoacetate to afford the 3-hydroxy-4-ethylesterquinolin-2(1H)-ones
core, regioselectively and in good to excellent yields. The DFT calculations performed on
this system support a mechanism in which the key step is the metallocarbene formation
between the 3-hidroxyindole-diazo intermediate and the dirhodium(II) complex. Finally,
vidicatin alkaloids were synthesised in yields up to 80 % via a Suzuki-Miyaura cross
coupling of the 3-hydroxy-4-bromoquinolin-2(1H)-one core with aryl-boronic acid.
Scheme 1: Ring – Expansion Reaction of Isatins
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support (PEst-OE/SAU/UI4013/2011; PTDC/QUIQUI/099389/2008; SFRH/BD/78301/2011). References: 1. Cunningham, K. G.; Freeman, G. G. Biochem. J. 1953, 53, 328; (b) Bracken, A.; Pocker, A.; Raistrick, H. Biochem. J. 1954, 57, 587; (c) Luckner, M.; Mothes, K. Tetrahedron Lett. 1962, 3, 1035. 2. Luckner, M.; Mothes, K. Arch. Pharm. 1963, 296, 18; (b) Mohammed, Y. S.; Luckner, M. Tetrahedron Lett. 1963, 4, 1953; (c) Wei, M. -Y.; Yang, R. -Y.; Shao, C. -L.; Wang, C. -Y.; Deng, D. -S.; She, Z. -G.; Lin, Y. -C. Chem. Nat. Compd. 2011, 47, 322. 3. Austin, D. J.; Meyers, M. B. J. Chem. Soc. 1964, 1197 4. Ribeiro, N.; Tabaka, H.; Peluso, J.; Fetzer, L.; Nebigil, C.; Dumont, S.; Muller, C. D.; Désaubry, L. Bioorg. Med. Chem. Lett. 2007, 17, 5523.
5. Heguy, A.; Cai, P.; Meyn, P.; Houck, D.; Russo, S.; Michitsch, R.; Pearce, C.; Katz, B.; Bringmann, G.; Feineis, D.; Taylor, D. L.; Tyms, A. S. Antiviral Chem. Chemother.1998, 9, 149.

PC108
Oxo-rhenium(V) Complexes containing heterocyclic ligands as
highly efficient catalysts for the reduction of sulfoxides
Sara C. A. Sousa,a Joana R. Bernando,
a Mariusz Wolff,
b Barbara Machura,
b
Ana C. Fernandesa
aCentro de Química Estrutural, Instituto Superior Técnico, Universidade Técnica de Lisboa, Av. Rovisco
Pais, 1049-001 Lisboa, Portugal; bDepartment of Crystallography, Institute of Chemistry, University of
Silesia, 9th Szkolna St., 40-006 Katowice, Poland.
In continuation of our studies about the use of oxo-complexes in organic reductions,1 in this
communication we describe the catalytic activity of several oxo-rhenium complexes
containing heterocyclic ligands2-5
in the reduction of sulfoxides using silanes and boranes as
reductants (Scheme 1).
In general, all the complexes are excellent catalysts for the deoxygenation of sulfoxides,
although the catalytic systems PhSiH3/[ReOBr2(hmpbta)(PPh3)] and
HBpin/[ReOBr2(hmpbta)(PPh3)] proved to be highly efficient for the reduction of aromatic
and aliphatic sulfoxides, tolerating different functional groups.
Scheme 1: Reduction of sulfoxides catalyzed by oxo-rhenium complexes.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support of this work through projects PTDC/QUI-QUI/110080/2009 and PEst-OE/QUI/UI0100/2013. S. C. A. Sousa (SFRH/BD/63471/2009) and J. R. Bernardo (SFRH/BD/90659/2012) thank FCT for grants and M. Wolff (UMO-2011/03/N/ST5/04522) thanks the Polish National Science Centre for grant. References: 1. S. C. A. Sousa, I. Cabrita, A. C. Fernandes, Chem. Soc. Rev. 2012, 41, 5641. 2. B. Machura, M. Wolff, R. Kruszynski, J. Kusz, Polyhedron, 2009, 28, 1211. 3. B. Machura, R. Kruszynski, J. Kusz, Polyhedron, 2008, 27, 1679. 4. B. Machura, R. Kruszynski, J. Kusz, Polyhedron, 2007, 26, 3455. 5. B. Machura, M. Wolff, J. Kusz, R. Kruszynski, Polyhedron, 2009, 28, 2949.

PC109
Microwave-assisted CuI catalysis to improve the synthesis of
aminated thioxanthones
A. S. Gomes, S. Cravo, E. Sousa, M. Pinto
Centro de Química Medicinal – Universidade do Porto (CEQUIMED-UP), Laboratório de Química
Orgânica e Farmacêutica, Departamento de Ciências Químicas, Faculdade de Farmácia, Universidade
do Porto, Rua Jorge Viterbo Ferreira nº 228, 4050-313 Porto, Portugal
Thioxanthones have been described as interesting antitumor derivatives.1
A library of
aminated thioxanthones with potent effects as inhibitors of leukemia tumour cells growth
that also circumvent Pgp-mediated multidrug resistance was obtained in our group
CEQUIMED-UP.2 The aminated thioxanthones were obtained by copper catalysed aromatic
nucleophilic substitution reaction. Nonetheless, the the reaction yields were low, being a
constrain to achieve these compounds in quantity to pursue in vivo studies.
In order to improve these yields, several parameters concerning reaction conditions were
investigated in the synthesis of the hit compound, 1-{[2-(diethylamino)ethyl]amino}-4-
propoxy-9H-thioxanthen-9-one (TXA1, Scheme 1). Through several reactions with
thioxanthone (1-chloro-4-propoxy-9H-thioxanthen-9-one, TX), 2 equivalents of amine (N,N-
diethylethylenediamine, A1), 2 equivalents of base (K2CO3), 0.4 mL of solvent and catalytic
amounts of a metallic catalyst, the reactions were assisted by microwave irradiation in
closed vessel with magnetic stirring (Scheme 1). Different reaction conditions were
modified, such as solvent (protic polar and aprotic polar), catalyst (CuI, CuO, Pd(0), Pd(II),
montemorillonite K10) and presence/absence of ligands. After work-up, each reaction
product was analysed by HPLC.
Scheme 1:Synthesis of 1-{[2-(diethylamino)ethyl]amino}-4-propoxy-9H-thioxanthen-9-one (TXA1).
The overall results showed that higher yields were achieved by a copper-catalysed reaction
in the synthesis of TXA1. The use of CuI, methanol as solvent and a ligand furnished TXA1
in 40% yield with the lowest amount of undesirable substitution products. Future work will
consist on applying these conditions to synthesize other aminated thioxanthones and to
evaluate the versatility of these conditions.
Acknowledgements: FCT Pest-OE/SAU/UI4040/2011, FEDER COMPETE, FCOMP-01-0124-FEDER-011057, U. Porto/Santander Totta. References:
1. Paiva A. M.; Sousa M. E. and Pinto M.M. Curr. Med. Chem. 2013, 20, 2438-2457. 2. Palmeira A.; et al. Biochem. Pharmacol. 2012. 83(1): 57-68.

PC110
Biomimetic oxidation of benzofuran derivatives with H2O2 using
metalloporphyrin catalysts
S. M. G. Pires,a S. L. H. Rebelo,
b M. M. Q. Simões,
a I. C. M. S. Santos,
a Artur M. S. Silva,
a
Maria G. P. M. S. Neves,a José A. S. Cavaleiro
a
aQOPNA, Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal
bREQUIMTE, Department of Chemistry, Faculty of Sciences, University of Porto, 4169-007 Porto,
Portugal
Benzofuran is a unique scaffold, being various natural and synthetic benzofuran derivatives
associated with a wide range of biological activities and hence displaying a diverse array of
applications in the field of medicine.1
Antibacterial, antifungal, anticonvulsant, anti-
inflammatory, antitumor, imaging, anti-HIV, antidiabetic, and antioxidant activities are
amongst the most widely referred therapeutic uses. So, the development of new synthetic
procedures to this type of compounds is a relevant area of research, contributing to the
discovery of new benzofuran derivatives with potentially improved properties and also
allowing, throw the isolation and identification of metabolites from drugs and drug
candidates presenting this moiety, a better understanding of the implied biological
processes.1
Metalloporphyrin complexes (MPorph), widely known biomimetic catalysts of the
cytochrome P450 enzymes, are able to catalyze numerous oxidation processes observed in
vivo under benign and environmentally clean conditions,2
contrastingly with the commonly
used more aggressive approaches.3
In this communication the results concerning the
oxidation of benzofurans (1-3) by hydrogen peroxide at room temperature, using distinct
porphyrin catalysts (I-III) will be presented. Furthermore, the catalytic activity of either
manganese or iron complexes, allowing the achievement of different oxidation products, will
be also discussed (Figure 1).
Figure 1: Metalloporphyrin complexes and substrates tested in the catalytic experiments
Acknowledgements: Thanks are due to Fundação para a Ciência e a Tecnologia (FCT, Portugal), European Union, QREN, FEDER and COMPETE for funding the QOPNA research unit (project PEst-C/QUI/UI0062/2011) and the Portuguese National NMR Network, supported by FCT funds. S. M. G. Pires thanks FCT for her PhD Grant (SFRH/BD/64354/2009) and S. L. H. Rebelo thanks program ON.2 SAESCTN-PIIDT/1/2011 for her grant. References: 1. a) Kumar D. R. H., Karvekar M. D., E. -J. Chem., 2010, 7, 636-640. b) Kamal M., Shakya A. K., Jawaid T., Int. J. Pharm. Sci., 2011, 1, 1-15. 2. a) Mansuy D., C. R. Chimie, 2007, 10, 392-413. b) Pires S. M. G., Simões M. M. Q, Santos I. C. M. S., Rebelo S. L. H., Pereira M. M., Neves M. G. P. M. S., Cavaleiro J. A. S., Appl. Catal. A: Gen., 2012, 439-440, 51-56. 3. Gingerich S. B., Jennings P. W., J. Org. Chem, 1983, 48, 2606-2608.
N
N N
N
Ar
Ar
Ar
ArM
Cl
Ar=
Cl
Cl
F F
F
FF
Ar=
I
I I
M= Mn;
M= Mn;
I I I
F F
N
FF
Ar=M= Fe;
O
R
R'
1- R= H; R'=H
2- R= H; R'= CH3
3- R= CH3; R'= H

PC111
Aryl C-N bond formation by ligand catalyzed electrophilic amination
Tahir Daşkapan, Semra Çiçek, Adem Korkmaz
Ankara University, Science Faculty, Department of Chemistry, 6 Beşevler, Ankara-Turkey
Arylamines are important building blocks in the synthesis of various important organic
compounds such as pharmaceuticals, agrochemicals, polymers, dyes, xerographic and
photographic materials. Therefore, development of new synthetic methods for arylamines
has attracted the attention of many researchers. Electrophilic amination of an easily
available organometallic reagent is an important preparation method for amines and it
continues to be an active area for research in synthetic organic chemistry.1,2
Organocopper reagents are indispensible in the field of synthetic organic chemistry. They
have been used not only for the synthesis of simple organic molecules, but also for the
synthesis of various types of natural products with high chemo-, regio- and
stereoselectivity.2
To date, most aminating reagents, for the electrophilic amination of organocopper reagents
were based on sp3-hybridized nitrogen species.
1 In this work, we described a practical
ligand catalyzed process for electrophilic amination of diarylcuprates with ketoximes, which
produced arylamines in high yields (Scheme 1).
Scheme 1: Ligand catalyzed electrophilic amination of diarylcuprates.
All reactions involving organometallic reagents were performed in flame-dried glassware
with standard syringe / cannula techniques under an atmosphere of dry, oxygen-free argon.
Diarylcuprates were prepared by addition of 2 mol equiv. of corresponding arylmagnesium
bromide in THF to a suspantion of 1 mol equiv. of copper iodide in THF at -3 ºC. The final
product, arylamine was isolated as its N-benzoyl derivative that was identified by its melting
point and FT-IR spectrum.
Acknowledgements: Financial support from Ankara University Research Foundation (grant no. 09B4240005) is greatly acknowledged by the authors. References 1. Daşkapan, T. Arkivoc 2011, v, 230. 2. Ciganek, E. Organic Reactions, (Editor: Denmark, S. E.) Vol. 72, John Wiley & Sons, Inc., 2009. 3. Krause, N. Modern Organocopper Chemistry, Wiley-VCH, Weinheim, 2002.

PC112
Synthesis of new Biginelli Compounds
E. Akbas, A. Erdogan
Yuzuncu Yil University, Faculty of Sciences, Department of Chemistry, Zeve Campus, 65100-Van,
Turkey.
The new pyrimidine compounds have been prepared via an application of the one-pot three
component Biginelli condensation procedure1 (Scheme 1).
Scheme 1: Synthesis of new pyrimidene compounds
The compounds which are included S atom, reacted with ethyl 2-bromoacetate and ethyl 2-
bromopropionate obtained new pyrimidine compounds (Scheme 2). This reaction likes the
Eschenmoser coupling reaction.2 The structures of the compounds have been confirmed by
spectroscopic data analysis. Our investigations will continue on this subject, and the results
will be published when our studies are complete.
Scheme 2: Synthesis of new pyrimidene compounds Acknowledgements: This work was supported by the Yuzuncu Yil University of Turkey (2011-FED-B084and2012-YNL-MRK003). References: 1. Biginelli P. Gazz. Chim. Ital., 1893, 23, 360. 2. Eschenmoser, A.;Wintner, C. E. Science. 1977, 196, 1410–1426. 3. Aslanoğlu, F.; Akbaş, E.; Sönmez, M.; and Anil, B. Phosphorus, Sulfur, and Silicon and the Related Elements, 2007, 182, 1589-1597. 4. Akbaş, E.; Aslanoğlu, F.; Anıl, B.; Şener, A. J. Heterocyclic Chem.2008, 45, 1457-1460. 5. Akbas, E.; Berber, I.; Akyazi, I.; Anil, B.; and Yildiz, E. Letters in Organic Chemistry, 2011, 8(9), 663-667.
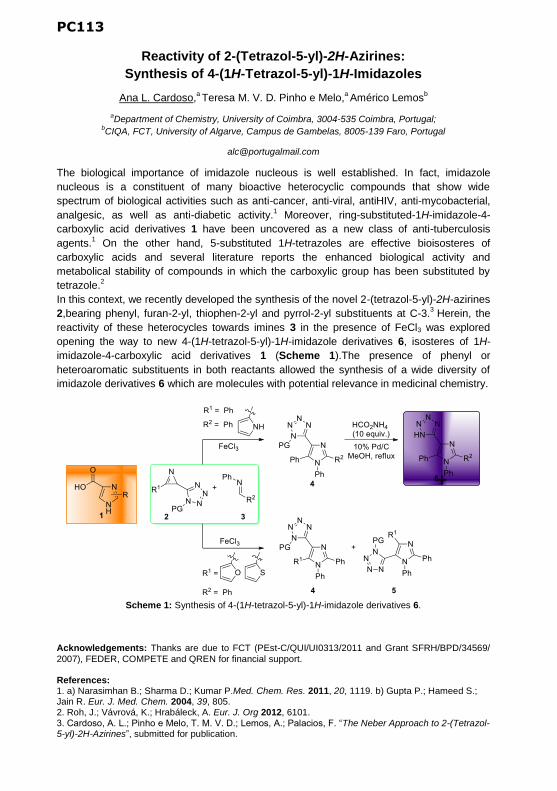
PC113
Reactivity of 2-(Tetrazol-5-yl)-2H-Azirines:
Synthesis of 4-(1H-Tetrazol-5-yl)-1H-Imidazoles
Ana L. Cardoso,a Teresa M. V. D. Pinho e Melo,
a Américo Lemos
b
aDepartment of Chemistry, University of Coimbra, 3004-535 Coimbra, Portugal;
bCIQA, FCT, University of Algarve, Campus de Gambelas, 8005-139 Faro, Portugal
The biological importance of imidazole nucleous is well established. In fact, imidazole
nucleous is a constituent of many bioactive heterocyclic compounds that show wide
spectrum of biological activities such as anti-cancer, anti-viral, antiHIV, anti-mycobacterial,
analgesic, as well as anti-diabetic activity.1 Moreover, ring-substituted-1H-imidazole-4-
carboxylic acid derivatives 1 have been uncovered as a new class of anti-tuberculosis
agents.1 On the other hand, 5-substituted 1H-tetrazoles are effective bioisosteres of
carboxylic acids and several literature reports the enhanced biological activity and
metabolical stability of compounds in which the carboxylic group has been substituted by
tetrazole.2
In this context, we recently developed the synthesis of the novel 2-(tetrazol-5-yl)-2H-azirines
2,bearing phenyl, furan-2-yl, thiophen-2-yl and pyrrol-2-yl substituents at C-3.3
Herein, the
reactivity of these heterocycles towards imines 3 in the presence of FeCl3 was explored
opening the way to new 4-(1H-tetrazol-5-yl)-1H-imidazole derivatives 6, isosteres of 1H-
imidazole-4-carboxylic acid derivatives 1 (Scheme 1).The presence of phenyl or
heteroaromatic substituents in both reactants allowed the synthesis of a wide diversity of
imidazole derivatives 6 which are molecules with potential relevance in medicinal chemistry.
Scheme 1: Synthesis of 4-(1H-tetrazol-5-yl)-1H-imidazole derivatives 6.
Acknowledgements: Thanks are due to FCT (PEst-C/QUI/UI0313/2011 and Grant SFRH/BPD/34569/
2007), FEDER, COMPETE and QREN for financial support. References: 1. a) Narasimhan B.; Sharma D.; Kumar P.Med. Chem. Res. 2011, 20, 1119. b) Gupta P.; Hameed S.; Jain R. Eur. J. Med. Chem. 2004, 39, 805. 2. Roh, J.; Vávrová, K.; Hrabáleck, A. Eur. J. Org 2012, 6101. 3. Cardoso, A. L.; Pinho e Melo, T. M. . D.; Lemos, A.; Palacios, F. “The Neber Approach to 2-(Tetrazol-5-yl)-2H-Azirines”, submitted for publication.

PC114
Synthesis of new di(hetero)aryl amide or triazole thienopyridine
thioethers through Cu-catalyzed reactions as potential inhibitors of
VEGFR2
Agathe Begouin, Joana F. Campos, Maria João R. P. Queiroz
Centro de Química, Escola de Ciências, Universidade do Minho, Campus de Gualtar 4710-057 Braga
VEGFR2 (Vascular Endothelial Growth Factor Receptor 2) is a transmembrane receptor
primarily expressed in endothelial cells that specifically binds to VEGF released by the
tumor, thus triggering the autophosphorylation of the receptor, which activates the signalling
pathways of proliferation and migration of endothelial cells and leads to the formation and
the expansion of new blood vessels (vasculogenesis and angiogenesis) towards the tumor.1
Recently, several thienopyridine derivatives have been shown to be promising inhibitors of
the intracellular tyrosine kinase domain of VEGFR2.2 In this work, we describe the synthesis
of various diarylamides through a Cu-catalyzed C-N coupling of 7-(3-bromophenylthio)
thieno[3,2-b]pyridine with aryl amides and, di(hetero)aryltriazoles from 3-(thieno
[3,2-b]pyridin-7-ylthio)aniline via the corresponding azide through a one-pot Cu(I)-catalyzed
azide-alkyne 1,3-dipolar cycloaddition ("click reaction"). The synthesis of the azide was
performed under mild conditions with tBuONO and TMSN3. The 1,4-disubstituted 1,2,3-
triazoles were obtained in good yields from a variety of (hetero)aryl alkynes with no need for
isolation of the azide intermediate.
Scheme 1: Synthesis of new di(hetero)aryl amide or triazole thienopyridine thioethers
The compounds prepared will be submitted to docking studies using the tyrosine kinase
domain of the VEGFR2 and the best predicted inhibitors will be studied either in enzymatic
or in cellular assays using VEGF-stimulated Human Umbilical Vein Endothelial Cells
(HUVECs).
Acknowledgements: We thank FCT–Portugal for financial support through the NMR Portuguese network (Bruker 400 Avance III-Univ Minho). FCT and FEDER-COMPETE-QREN-EU for financial support through the research unity PEst-C/QUI/UI686/2011, the research project PTDC/QUI-QUI/111060/2009 and the post-Doctoral grant attributed to A.B.(SFRH/BPD/36753/2007) also financed by POPH and FSE. References: 1. Hicklin D. J.; Ellis L. M. J. Clin. Oncol. 2005, 23, 1011. 2. a) Munchhof M. J. et al. Bioorg. Med. Chem. Lett. 2004, 14, 21. b) Claridge S. et al. Bioorg. Med.
Chem. Lett. 2008, 18, 2793.

PC115
Thiol-ene reactions: a “click” to α-Peptide Nucleic Acid Building
Blocks
A. S. Ressurreição,a A. R. P. Duarte,
a J. Iley,
b Rui Moreira
a
aResearch Institute for Medicines and Pharmaceutical Sciences (iMed.UL), Faculty of Pharmacy,
University of Lisbon, Av. Prof. Gama Pinto 1649-019 Lisbon, Portugal; bChemistry Department, The
Open University, Walton Hall, Milton Keynes MK7 6AA, UK.
The introduction of the ‘‘click’’ chemistry concept, focusing on constructing carbon–
heteroatom linkages in a modular and highly efficient manner, revolutionized the way that
research is being conducted in numerous fields. In particular, the reaction of thiols with
reactive carbon–carbon double bonds (thiol-ene coupling, TEC), has become an
outstanding tool for functionalization of polymers and molecules of biological interest.1
With the aim of developing new alpha-Peptide Nucleic Acids2 (α-PNAs), structural mimics of
DNA where the ribose-phosphate backbone has been replaced by a peptide polymer to
which the nucleobases are linked, we have investigated the applicability of the TEC reaction
for the incorporation of nucleobases into cysteine amino acids or cysteine containing
dipeptides. In this report, we will describe an efficient synthetic route to cysteine-based α-
PNA building blocks, containing all four nucleobases (5, Figure 1). Futhermore, the strategy
designed results in building blocks with orthogonal protecting groups and compatible with
Fmoc-based solid-phase peptide synthesis (SPPS) protocols.
Figure 1: Synthesis of cysteine-based α-PNA building blocks via thiol-ene reaction (B
PG: Boc-protected
nucleobase). Acknowledgements: We thank the Fundação para a Ciência e Tecnolgia for financial support: Pest-
OE/SAU/UI4013/2011, BPD/64859/2009 (A.S.R.) and SFRH/BD/70491/2010 (A.R.D.). References: 1. a) Lowe, A. B.; Polym. Chem. 2010, 1, 17-362; b) Hoyle, C. E.; Bowman, C. N. Angew. Chem. Int. Ed. 2010, 49, 1540-1573. 2. a) Egholm M., Buchardt, O. et al., Nature 1993, 365, 566-568; b) Roviello, G. N.; Benedetti, E.; Pedone, C.; Bucci, E. M. Amino Acids 2010, 39, 45-57.

PC116
Development of a New Anti-Cancer Photosensitizer.
From Basic Research to the Clinical Trials
Artur R. Abreu,a Nuno P. F. Gonçalves,
a Carlos J. P. Monteiro,
a,c Gonçalo N. Costa,
a
Luís B. Rocha,a,b
Janusz M. Dąbrowski,d Elsa Silva,
c Fábio A. Schaberle,
a
Mariette M. Pereira,c Luis G. Arnaut,
a,c Sérgio Simões,
a,b Sebastião J. Formosinho
c
aLuzitin, SA, Edificio Bluepharma, São Martinho do Bispo, Coimbra, Portugal;
bBluepharma, SA, Rua
Bayer, São Martinho do Bispo, Coimbra, Portugal; cChemistry Department, University of Coimbra,
Coimbra, Portugal; dFaculty of Chemistry, Jagiellonian University, Krakow, Poland
Over the past few years, near infrared (NIR) absorbing tetrapyrrolicmacrocycle compounds,
like chlorins and bacteriochlorins, have been used for several applications.1 Our particular
interest in photomedicine, namely photodynamic therapy of cancer (PDT), led us to design
molecules with strong absorption in the infrared, but nearly transparent in the visible region
(380-720 nm).2
The development of a new drug is a long and laborious task that involves a
multidisciplinary scientific team, from pure organic chemists to biochemists, including,
physicians, statisticians, physicists, and analysts, among others. The new drug
development process is well characterized and comprises different stages from pure R&D to
human clinical trials.
Luzitin SA is a spin-off company which main goal is the development of a family of patented
photosensitizer compounds, with reckoned antitumor activity in the Photodynamic Therapy
field and clear advantages over the competitor products already on the market.
In this communication we present some key aspects of the strategy employed in the
synthesis/discovery, characterization and selection of the lead compound (LUZ11), based
on the photophysical studies, pharmacokinetics, biodistribution and mechanism of action.3
References: 1. a) Fabian, J.; Nakazumi, H.; Matsuoka, M. Chem. Rev. 1992, 92, 1197; b) Qian, G.; Wang, Z. Y. Chem. Asian J. 2010, 5, 1006 2. a) Pereira, M. M.; Monteiro, C. J.P.; Simões, A. V.C., Pinto, S. M. A. Pinto , Abreu, A. R., Sá, G. F. F., Silva, E F. F.; Rocha, L. B. Rocha, Dabrowski, J. M., Formosinho, S. J., Simões, S.; Arnaut, L. G., Tetrahedron 2010, 66, 9545 ; b) Pereira, M. M.; Abreu, A. R.; Goncalves , N. P. F.; Calvete , M. J. F.; Simões, A. V. C.; Monteiro, C. J. P.; Arnaut , L. G.; Eusébio, M. E.; Canotilho J.; Green Chem., 2012, 14, 1666. 3. a) Silva, E. F. F., Serpa, C., Dabrowski, J. M., Monteiro, C. J. P.; Formosinho S. J.; Stochel, G.; Urbanska,K.; Simões, S.; Pereiras, M. M.; Arnaut L. G. Chem. Eur. J. 2010, 16, 9273; b) Pereira, M. M.;
Monteiro C. J. P.; Simões, A. V. C. Pinto, S. M. A.; Arnaut, L. G. Sá, G. F. F.; Silva, E. F. F.; Rocha, L. B.; Simões, S.; Formosinho, S. J. J. of porphyrins Phthalocyanines, 2009, 13, 567.

PC117
New Benzimidazole-based COX-2 Inhibitors – a drug design
approach
Luísa C. R. Carvalho,a Eduarda Fernandes,
b M. Manuel B. Marques
a
aREQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de
Lisboa, Campus de Caparica, 2829-516 Caparica, Portugal; bREQUIMTE, Departamento de Ciências
Químicas, Rua Jorge Viterbo Ferreira nº 228, 4050-313 Porto, Portugal
In the last decades, several nonsteroidal anti-inflammatory drugs (NSAIDs) were developed
and commercialized, such as ibuprofen, nimesulide, naproxen or diclofenac. These NSAIDs
were found to inhibit cycloxygenase (COX), an enzyme involved in the inflammatory
process. However following repeated administration, these drugs usually present adverse
side effects, since most of them act as nonselective inhibitors of both COX-1 and COX-2
isoforms. Whereas COX-1 is involved in several body functions, such as gastric protection,
COX-2 is responsible for the inflammation.1 This fact encouraged the development of potent
and selective COX-2 inhibitors devoid of side effects.
Benzimidazoles represent an important class of heterocyclic compounds exhibiting a wide
range of biological properties,2 particularly 1,2-disubstituted benzimidazoles.
3 Benzimidazole
based compounds were already reported as COX inhibitors,4 and our aim was to find novel
anti-inflammatory drugs based on 1,2-disubstituted benzimidazole scaffold and understand
their mode of action. Moreover, we expected to attain a more straightforward route to the
benzimidazole synthesis. Presently, the available synthetic strategies to prepare these
compounds are limited and present significant drawbacks, such as the restriction to the
commercially available starting materials and the poor regioselectivity achieved.5
Consequently, a rational design strategy was adopted, by performing docking studies,
library synthesis, STD-NMR experiments and biological activity evaluation in an iterative
approach. Herein we present our results on the synthesis of 1,2-disubstituted
benzimidazoles with potential in COX inhibitory activity (Figure 1).
Figure 1
Acknowledgements: The author acknowledges Fundação para a Ciência e Tecnologia for fellowship SFRH/BD/63407/2009. References: 1. a) Laneuville, O.; Breuer, D.; Dewitt, T.; Funk, C.; Smith, W. J. Pharmcol. Exp. Ther. 1994, 271, 927; b). Seibert, K.; Zhang, Y.; Leahy, K.; Hauser, S.; Masferrer, J.; Perkins, W.; Lee, L.; Isakson, P. C. Proc. Natl. Acad. Sci. 1994, 91, 12013. 2. Mayer, J.; Lewis, G.; McGee, C.; Bankaitis-Davis, D. Tetrahedron Lett. 1998, 39, 6655. 3. a) Ishida, T.; Suzuki, T.; Hirashima, S.; Mizutani, K.; Yoshida, A.; Ando, I.; Ikeda, S.; Adachi, T.; Hashimoto, H. Bioorg. Med. Chem. Lett. 2006, 16, 1859; b) Falco, J.; Pique, M.: Gonza, Buira, I.; Mendez, E.; Terencio, J.; Perez, C.; Princep, M.; Palomer, A.; Guglietta, A. Eur. J. Med. Chem. 2006, 41,
985. 4. Franke, L.; Byvatov, E.; Werz, O.; Steinhilber, D.; Schneider, P.; Schneider, G. J. Med. Chem. 2005,
48, 6997. 5. Carvalho, L. C. R.; Fernandes E.; Marques, M. M. B., Chem. – Eur. J. 2011, 45, 12544.

PC118
Galactodendritic photosensitizers for target carbohydrate receptors
and trigger phototoxicity in bladder cancer cells
Patrícia M. R. Pereira,a,b
Sandrina Silva,a José A. S. Cavaleiro,
a Carlos A. F. Ribeiro,
b
João P. C. Tomé,a Rosa Fernandes,
b,c
aQOPNA and Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal;
bLPET & IBILI,
Faculty of Medicine, University of Coimbra,3000-548 Coimbra, Portugal; cCenter of Investigation in
Environment, Genetics and Oncobiology, Coimbra, Portugal
Conventional photodynamic therapy (PDT) combines a non-toxic photosensitizer (PS), light
irradiation at a specific wavelength and tissue molecular oxygen to produce cytotoxic
reactive oxygen species (ROS) with the final purpose to kill cancer cells.1 Although clinical
successes and promising pre-clinical studies with PDT, the synthesis of new PSs with
outstanding photo-chemical, -physical and -biological properties for cancer treatment is still
a challenge for many chemists. In order to enhance the specific target of PSs in cancer
cells, they have been conjugated with galactose units which have specific recognition by
lectin proteins overexpressed in cancer cells and play an important role in several
biochemical signalling pathways implicated in cancer metastasis, cell growth and
inflammation.2 We have reported the synthesis
3 of a porphyrin (PorGal8) and
phthalocyanine (PcGal16) conjugated with dendrimers of galactose and more recently, we
have synthesized the related chlorin derivative (ChlGal8). The idea is that galactodendritic
PS structures should be more specialized than the well-studied galacto-PS structures, since
they have multivalent interactions with lectins promoting a synergistic increase in binding
affinity and improving PDT efficacy.
In this communication, we will report the photophysical properties of these new
galactodendritic conjugates (water solubility, singlet oxygen generation, ability to interact
with human serum albumin and galectin-1) as well as their photobiological properties from
the standpoint of its uptake by cancer cells and interaction with galactose-binding proteins;
induction of phototoxicity, apoptosis, ROS production and activity of antioxidant enzymes
after PTD.
Acknowledgements: Thanks are due to the Universities of Aveiro and Coimbra, FCT and FEDER for
funding the QOPNA and IBILI Units and the projects PTDC/CTM/101538/2008, PEst-C/SAU/UI3282/2011. Thanks to ACIMAGO (Ref. 12/12). P. Pereira and S. Silva thank to FCT for their PhD (SFRH/BD/85941/2012) and post-doctoral (SFRH/BPD/64812/2009) grants, respectively. References: 1. Allison, R. R.; Sibata, C. H. Photodyn. Ther. 2010, 7, 61. 2. Liu, F. T.; Rabinovich, G. A. Nature Reviews Cancer, 2005, 5, 29. 3. Silva, S.; Pereira, P. M.; Silva, P.; Paz, F. A.; Faustino, M. A.; Cavaleiro, J. A.; Tome, J. P. Chem Commun, 2012, 48, 3608.

PC119
Synthesis and Carbonic Anhyrase Inhibitory Properties of
Bromophenol Derivatives
Yusuf Akbaba,a Halis T. Balaydin,
b Abdullah Menzek,
a Süleyman Göksu,
a Ertan Şahin,
a
Deniz Ekincic
aAtatürk University, Faculty of Science, Department of Chemistry, 25240, Erzurum Turkey;
bArtvin Çoruh
University, Faculty of Education Science Education Department, 08000, Artvin, Turkey; cOndokuz Mayıs
University, Faculty of Agriculture, Department of Biotehnology, 55139, Samsun, Turkey
Natural bromophenols, frequently isolated from red algae of the family Rhodomelaceae,
have important biological activities.1
Bromophenols 1-3 are natural bromophenols and
compounds of 4 with Br also are bromophenol derivatives. They exhibit biological activities
such cytotoxicity and anticancer.2
Acylation of 3,4-dimethoxytoluene with 3,4-dimethoxybenzoic acidin PPA at 80oC gave
compound with a yield. By its bromination in different conditions, compounds 6-8 were
synthesized. Reaction of 5–8 with BBr3 in CH2Cl2 provided the novel phenolic compound 9
and synthetic bromophenols 10-12 (Scheme 2). The compounds were characterized and
tested against the two most studied members of the pH regulatory enzyme family, carbonic
anhydrase (CA).3
References: 1. Gribble G. W J Nat. Prod. 1992, 55, 1353. b) Gribble G. W Chem. Soc. Rev. 1999, 28, 335. 2. a) Oh K. B.; Lee J. H.; Chung S. C.; Shin J.; Shin H. J.; Kim H. K.; Lee H. S. Bioorg. Med. Chem. Lett. 2008, 18, 104. b)Xu X.; Song F.; Wang S.; Li S., Xiao F.; Zhao J.; Yang X.; Shang V.; Yang L.; Shi J. J. Nat. Prod. 2004, 67, 1661. c) Shi D.; Li J.;Guo S.; Su H.; Fan X. Chin. J. Ocean Lim. 2009, 27, 277. d) Balaydin H. T.; Gulcin I.; Menzek A.; Goksu S.; Sahin E. J. Enzym. Inhib. Med. Ch. 2010, 25, 685. e) Balaydin H. T.; Soyut H.; Ekinci D.; Goksu, S.; Beydemir S.; Menzek, A.; Sahin, E. J. Enzym. Inhib. Med. Ch. 2012, 27, 43. 3. Akbaba Y.; Balaydin H. T.; Menzek A.; Goksu S.; Sahin, E.; Ekinci D. Arch. Pharm. Chem. Life Sci. 2013, 346, 447.

PC120
Synthesis and anti-microbial activity studies of some novel
epicinchonina-1,2,3-triazole derivatives
Joana Magalhães,a Pedro Barrulas,
a Jiri Gut,
b Philip J. Rosenthal,
b Ana T. Caldeira,
a
Anthony J. Burkea
aDepartamento de Química e Centro de Química de Évora, Escola de Ciências e Tecnologia da
Universidade de Évora, Rua Romão Ramalho 59, 7000-671 Évora, Portugal. bFaculty of Medicine, San Francisco General Hospital, University of California, San Francisco, CA 94143-
0811, USA.
Cinchona alkaloids are an important class of compounds isolated from the cinchona tree.
The main cinchona alkaloids are quinine, quinidine, cinchonidine and cinchonine. They are
used in the treatment of cardiac arrhythmias and also used in the treatment of malaria.1
Although quinine is the most efficient and most used in antimalarial therapy,2 the synthesis
of new derivatives of Cinchona can provide more effective agents. Many triazole molecules
show a broad spectrum of antimicrobial activity, for example, fluconazole, a triazole, which
is used in the treatment of fungal infections contracted by AIDS patients and thus the
synthesis of triazole derivatives of cinchonine may enhance the medicinal effects of both
these sets of compounds.3 Moreover, the 1,2,3-triazole unit serves as a good "mimic" of
amide bonds, thus behaving as a "peptidomimetic". In this communication, we describe the
synthesis of a small library of new hybrid epicinchonina-1,2,3-triazoles (Scheme 1) some of
which contain an iodine in the 5- position, that were synthesized by a novel "one-pot"
catalytic process, involving the Huisgen 1,3-dipolar cycloaddition followed by iodination.4
We also discuss our recent anti-fungal and anti-malaria screening study results.
N
N
N
N
N
R
N
OH
N
1 2
X
NN
N
N
N
N
F
F
OH
FLUCONAZOLX = H, I
Scheme 1: Synthetic route to new epicinchonina-1,2,3-triazole hybrids.
Acknowledgements: We thank Miss Mara Silva, from the biotechnology laboratory in the Chemistry department (UE) for the help with the fungal inhibition studies. References: 1. L. Mink, Z. Ma, R. A. Olsen, J. N. James, D. S. Sholl, L. J. Mueller, F. Zaera , The Physico-chemical Properties of Cinchona Alkaloids Responsible for their Unique Performance in Chiral Catalysis, Top. Catal. 2008, 48 (1-4), 120-127; 2. P. Dewick, in Medicinal Natural Products: A Biosynthetic Approach, John Wiley and Sons Ltd, 2002, pp. 362-364; 3. P. Wu, V. V. Fokin, Aldrichimica Acta, 2007, 40, 7-17 4. L. Li, G. Zhang, A. Zhu, L. Zhang, J. Org. Chem. 2008, 73, 3630-3. V. Fiandanese, S. Maurantonio, A. Punzi, G. Rafaschieri, Org. Biomol. Chem. 2012, 10, 1186-95.

PC121
Synthesis of polysulfated diosmin conjugates as potential orally-
active antithrombotic agents
M. Correia-da-Silva, M. Patrão, E. Sousa, M. Pinto
Centro de Química Medicinal – Universidade do Porto (CEQUIMED-UP), Laboratório de Química
Orgânica e Farmacêutica, Departamento de Ciências Químicas, Faculdade de Farmácia, Universidade
do Porto, Rua Jorge Viterbo Ferreira nº 228, 4050-313 Porto, Portugal
Although currently available anticoagulants are effective for the prevention and treatment of
thromboembolic disorders, they have several drawbacks.1
Therefore, the search for new
orally-active anticoagulant agents is a major challenge to medicinal chemists.
In CEQUIMED-UP a new class of antithrombotic agents, namely polysulfated diosmin DS
(Figure 1), with dual anticoagulant/antiplatelet activity, was identified.2 However, by oral
administration in mice, these derivatives were not active.2 Important strategies such as the
use of oral absorption enhancers, the development of lipid conjugates of heparin, and the
incorporation of heparin in polymeric matrix systems have been carried out for the
development of heparin as oral anticoagulant.3 These strategies can facilitate its enteric
absorption reaching adequate levels for prevention and/or treatment of venous
thromboembolic disease in humans. Thus, the aim of this work was to obtain conjugates of
the active sulfated compound DS with several carriers to achieve potentially orally-active
derivatives.
Figure 1: Dual anticoagulante/antiplatelet agent diosmin 2’’,2’’’,3’’,3’’’,4’’,4’’’-O-hexasulfate (DS).
In this study, sulfation of diosmin was achieved using SO3:TEA in DMA combined with
microwave irradiation to afford DS in 60% yield. The conjugates of DS were obtained by
esterification using several coupling reagents. The structure elucidation of two new
synthesized conjugates of DS was established by spectroscopic methods, such as IR, 1H
and 13
C NMR. Future work will consist in the investigation of the anticoagulant activity and
permeability of the conjugates derivatives across mouse small intestine.
Acknowledgements: FCT, PEst-OE/SAU/UI4040/2011, FEDER, POCI, POPH/FSE/QREN for financial support and for the post-doctoral grant to M C Silva (SFRH/BPD/81878/2011). References: 1. a) Hirsh J., Warkentin T. E., Shaughnessy S. G., Anand S. S., Halperin J. L., Raschke R., Granger C., Ohman E. M., Dalen J. E. Chest 2001, 119, 64S. b) Ansell J., Hirsh J., Hylek E., Jacobson A., Crowther M., Palareti G. Chest 2008,133, 160S. 2. a) Correia-da-Silva M., Sousa E., Duarte B., Marques F., Carvalho F., Cunha-Ribeiro L. M., Pinto M. M. M. J. Med. Chem. 2011, 54, 95. b) Correia-da-Silva M., Sousa E., Duarte B., Marques F., Carvalho F., Cunha-Ribeiro L. M., Pinto, M. M. M. J. Med. Chem. 2011, 54, 5373. 3. Paliwal R., Paliwal S. R., Agrawal G. P., Vyas S. P. Med. Res. Rev. 2011, 1.

PC122
Synthesis of new 18F-labelled Porphyrins and their potential
application for in vivo Molecular Imaging with PET
Ana Simõesa, Mário J. F. Calvete, Mário
a, Mariette M. Pereira
a, Jordi Llop
b,
Antero Abrunhosac
aChemistry Department, University of Coimbra, Rua Larga 3004-535, Coimbra, Portugal;
bCICbiomaGUNE, Paseo de Miramón 20009 Donostia-San Sebastián, Espanha;
cInstituto de Ciências
Nucleares Aplicadas à Saúde, Universidade de Coimbra, Azinhaga de Santa Comba, 3000-548
Coimbra, Portugal
Molecular imaging holds the promise of non-invasive assessment for biological and
biochemical processes in living subjects using specific imaging tracers. Positron Emission
Tomography (PET) is a highly specific and sensitive molecular imaging technique with
widespread use for research and clinical application. A large number of early stage cancer
detection for application in PET studies today are performed with molecules labelled with
fluorine-18, a radionuclide possessing important characteristics including am favorable half-
life (110 min) and the ability to replace H in organic molecules.1 It’s widely recognized that
porphyrins are one of the most important prosthetic groups in biological systems and
porphyrin derivatives have recently found promising biomedical applications in detection
and treatment of a variety of tumours due to their affinities for these tissues in relation with
the nature of the side chain and the mechanism of their physico-chemical action.2-5
In this
communication we describe our recent studies on the synthesis of novel sulfonamide
substituted meso-tetraphenylporphyrins via chlorossulfonation reaction and amine
substitution, followed by mono-tosylation and automated and non-automated synthesis of
new 18F-labelled porphyrin derivatives, by fluorination via nucleophilic substitution with
K2CO3/K2.2.2/ACN (Figure 1). These results may open new directions for the development of
new theragnostic tools.
Figure 1: Nucleophilic
18F-fluorinated porphyrin synthesis
Acknowledgements: The authors thank FCT/QREN/FEDER-COMPETE Portugal; FCT for AVC Simoes PhD grant (SFRH/BD/65699/2009); Maria Puigivila, Carlos Perez, Mikel González and Aitor Lekuona from CICbiomaGUNE and Vítor Alves, Sérgio Carmo from ICNAS. References: 1. a) William K. Hagmann, Journal of Medicinal Chemistry, 15, 2008, 4359; b) Didier Le Bars, Journal of Fluorine Chemistry, 127, 2006, 1488. 2. Dabrowski J. M., Krzykawska M., Arnaut L. G., Pereira M. M., Monteiro C. J. et al., ChemMedChem, 2011, 6, 1715 3. M. M. M Pereira, C. J. P Monteiro, A. V. C. Simões; et al., J. Porphyrins Phathlocyanines 13, 2009,
567. 4. Dabrowski J. M, Luis G. Arnaut, Mariette M. Pereira et al., Med. Chem. Commun., 3, 2012, 502. 5. Pereira, M. M.; Monteiro, C. J. P.; Simoes, A. V. C.; et al., Tetrahedron, 66, 2010, 9545.

PC123
Design and Synthesis of New Probes for AI-2 Quorum-Sensing
Receptor Studies
Ana Sofia Miguel,a Karina B. Xavier,
a,b M. Rita Ventura
a
aITQB-UNL, Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa, Av. da
República, 2780-157 Oeiras, Portugal; bIGC, Instituto Gulbenkian de Ciência, 2781-901 Oeiras, Portugal
Autoinducer-2 (AI-2)1,2
(isomer mixture of (S)-4,5-Dihydroxypentane-2,3-dione, DPD) is a
signalling molecule for bacterial inter-species communication. Understanding the molecular
mechanisms that bacteria use to regulate their behaviours can lead to the development of
new therapies to control bacterial infections. The synthesis of analogues of AI-2 is an on-
going project targeting molecules, which modify the behaviour of bacteria. An important
objective of this work is trying to understand how these molecules function and also
visualize them within the biological system.
During the past few decades, technology has made great improvements to enable
visualization, identification and quantitation in biological systems. The fluorescent optical
reporters, called quantum dots (QDs) have been used for the labelling of biomolecules for a
wide range of applications. CdSe/ZnS core-shell QDs are the most used because they
possess unique optical and electronic properties, compared to standard organic dyes. By
changing the ligands attached to the surface of the dots their solubility can be tuned and the
terminal groups can be used for conjugation with a variety of biomolecules via simple
covalent and non-covalent strategies.
The synthesis, functionalization and bio-conjugation of QDs for different applications,3,4
is
one of the research lines in the group. This communication describes the synthesis of new
DPD analogues as well as the design of strategies for the attachment of DPD and
derivatives to QDs (Scheme 1) in order to better understand these molecular mechanisms
inside bacteria and to identify AI-2 quorum-sensing receptors not yet known.
Scheme 1: Representative scheme of QD attachment to DPD and DPD analogues.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support - PTDC/QUI-BIQ/113880/2009. References: 1. Ascenso, O.; Marques, J.; Santos, R.; Xavier, K.; Ventura, R.; Maycock, C. Bio. Med. Chem. 2011, 19, 1236; 2. Rui, F.; Marques, J.; Miller, S.; Maycock, C.; Xavier, K.; Ventura R. Bio. Med. Chem. 2012, 20, 249; 3. Santos, A.; Miguel, A.; Tomaz, L.; Malhó, R.; Maycock, C.; Patto, C.; Fevereiro, P.; Oliva, A. J. Nanobiotechnology 2010, 8:24; 4. Miguel, A.; Maycock, C.; Oliva, A. Nanoparticles in Biology and Medicine: Methods and Protocols, Methods in Molecular Biology, 906, Part 2, 143-155, 2012, Springer.
CdSe
ZnS
O
O
R
HO
HN
O
R= OH (DPD)R= OMe (DPD Analogue)
Hydrophilic spacerand/or Linker
QD attachment
Bioconjugated QD

PC124
Porphyrin Derivatives and Potential Applications against Cutaneous
Leishmaniasis
Ana T. P. C. Gomes,a Mônica M. Bastos,
a,b Maria G. P. M. S. Neves,
a Artur M. S. Silva,
a
Osvaldo A. Santos-Filho,b Núbia Boechat,
b José A. S. Cavaleiro
a
aUniversity of Aveiro, Department of Chemistry and QOPNA, University Campus, 3810-193 Aveiro,
Portugal; bDepartamento de Pesquisa e desenvolvimento de Fármacos, Instituto de Tecnologia em
Fármacos, FIOCRUZ, 100, Manguinhos, Rio de Janeiro, Brazil
Certain porphyrin derivatives are heterocycles currently having significant medicinal
applications, mainly in cancer photodynamic therapy, against the age-related macular
degeneration and as anti-microorganisms (viruses, bacteria) and as water disinfection
agents. Usually such heterocycles are of the porphyrin, chlorin (dihydroporphyrin) and
bacteriochlorin (tetrahydroporphyrin) types. During the last decades several publications
concerned with the synthesis, reactivity and photodynamic action of new derivatives with
potential biological applications have appeared.1 A disease considered to be a negleted one
by the World Health Organization is Leishmaniasis. This is caused by a protozoon of the
gender Leishmania. Cutaneous leishmaniasis is the most common one; it disfigures the
face and other parts of the body. Considering the heterogeneity of the disease it can be
stated that the present drugs against it are inadequate and cause severe side effects.2 It is
known that new biologically active molecules can be obtained by linking two or more
compounds with well established biological action. Such is the case with porphyrins and
triazole and thiadiazole derivatives. This communication will describe our synthetic work
carried out on the coupling of β-formylporphyrins with amino heterocycles (such as amino-
triazoles and amino-thiadiazoles and pyridine-4-carbohydrazide), which was followed by
imine reduction. Molecular docking simulations and singlet oxygen generation studies will
also be reported.3
Acknowledgements: Thanks are due to Fundação para a Ciência e a Tecnologia (FCT, Portugal), European Union, QREN, FEDER and COMPETE for funding the QOPNA research unit (project PEstC/QUI/UI0062/2011) and the Portuguese National NMR Network. Thanks are also due to Farmanguinhos /FIOCRUZ and the FCT-CAPES collaborative program for funding. ATP Gomes also thanks FCT for her post-doctoral grant (SFRH/BPD/79521/2011). References: 1. R. Bonnett, Chemical Aspects of Photodynamic Therapy, Gordon and Breach Science Publishers, 2000, Amsterdam. 2. M. M. Bastos, N. Boechat, A. T. P. C. Gomes, M. G. P. M. S. Neves, J. A. S. Cavaleiro, Rev. Virtual Quim., 2012, 4, 257-267. 3. M. M. Bastos, A. T. P. C. Gomes, M. G. P. M. S. Neves, A. M. S. Silva, O. A. Santos-Filho, N. Boechat, J. A. S. Cavaleiro, Eur. J. Org. Chem., 2013, 1485-1493.

PC125
Triazene Prodrugs for the Treatment of Malignant Melanoma
A. M. C. Granada, M. E. Mendes, M. J. Perry, A. P. Francisco
iMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal
The malignant melanoma is considered the most dangerous skin cancer, and one of the
most aggressive to humans. Its incidence in western countries is high and has been
increasing continuously, being considered itself as a concern for the medical and scientific
communities. Thus it becomes important to develop new drugs for the treatment of
malignant melanoma. We developed a new series of prodrugs of anti-tumor triazenes for
use in targeted therapeutic strategies, such MDEPT (Melanocyte-Directed Enzyme/Prodrug
Therapy). The MDEPT strategy is based on the occurrence of over expression of the
tyrosinase enzyme in malignant melanocytes, increasing the selectivity of drug distribution,
and minimizing side effects.
The compounds were synthesized as mutual prodrug resulting from binding of an alkylating
agent (triazene) to a carrier (4-S-CAP), which simultaneously is a substrate for tyrosinase
enzyme and a melanocytotoxic agent. The 4-S-CAP was synthesized via Wehrmeister
reaction (Scheme 1),1 and is joined via urea linkage to the alkylating triazene by coupling
the 4-S-CAP with a carbamate triazene intermediate 1 (Scheme 2).2
Scheme 1: 4-S-CAP Synthesis
Scheme 2: Prodrugs Synthesis (R = Br, CH3, CN, EtCO2, MeCO)
The synthesized compounds were studied concerning their chemical stability, enzymatic
activation by tyrosinase, and stability in human plasma. Results of these preliminary studies
showed that these compounds are very promising as prodrugs to apply in the melanoma
chemotherapy.
Acknowledgements: This work was supported by grant Pest-OE/SAU/UI4013/2011 from Fundação
para a Ciência e a Tecnologia, Lisbon, Portugal References: 1. Padgette, S.; Herman, H. J Med Chem, 1984, 27, 1354–1357. 2. Perry, M. J.; et al. Eur Med Chem, 2009, 44, 3228–34.

PC126
Secondary Metabolites from a Marine Streptomycessp
Marta M. Andrade,a Vasco D. Bonifácio,
a Daniel B. Silva,
a Viriato M´Bana,
a
Susana P. Gaudêncio,a Ilda S. Sanches,
b Ana C. Costa,
c Paula B. Aguiar,
c
Ana M. Lourenço,a Ana M. Lobo
a
aREQUIMTE, Dep. Química, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa,
Caparica, Portugal; bCREM, Dep. Ciências da Vida, Faculdade de Ciências e Tecnologia, Universidade
Nova de Lisboa, Caparica, Portugal; cDep. Biologia, Universidade dos Açores, S. Miguel, Açores,
Portugal
Prodigiosins, a family of naturally occurring tripyrrolic red pigments were found to have very
interesting pharmacological properties, such as antibacterial, anticancer, antimalarial and
immunosuppressive activity.1
Recently a Streptomyces sp was isolated from the Azores (S.
Miguel) ocean bed and found to contain prodigiosins. When cultured in the laboratory it
yielded two major prodigiosins namely undecylprodiginine (1) and streptorubin B (2) (Figure
1). Analogues with different carbon chains as in 4 and 5 were also detected by mass
spectral analysis.
Figure 1: Prodigiosins from a marine Streptomycessp.
Acknowledgements: We thank Fundação para a Ciência e Tecnologia for financial support through project PTDC/QUI-QUI/101813/2008 – MariNatProd-XXI, as well as project PEst-C/EQB/LA0006/2011. Thanks are also due to Professor W. Fenical (Scripps Institution of Oceanography at La Jolla, University of California, S. Diego, USA) for the biological analysis of the microorganism, Drs P. Jensen and C. Kaufmann for helping with the ocean sediments collection, and Prof Mª Rosário Bronze and Dr João Ferreira (Faculty of Farmacy/Univ. Lisbon) for the mass spectra. References: 1. a) Regourd, J.; Al-Sheikh Ali, A.; Thompson, A. J. Med. Chem. 2007, 50, 1528. b) D’Alessio, R.;
Bargiotti, A.; Carlini, O.; Colotta, F.; Ferrari, M.; Gnocchi, P.; Isetta, A.; Mongelli, N.; Motta, P.; Rossi, A.; Rossi, M.; Tibolla, M.; Vanotti, E. J. Med. Chem. 2000, 43, 2557. c) Furstner, A. Angew. Chem. Int. Ed. 2003, 42, 3582.

PC127
Synthesis and Antimicrobial Activity of 5-Aminoimidazoles
Incorporating a Substituted N-phenyl Amidrazonoyl Moiety
Ana Isabel F. Ribeiro,a Carla Gabriel,
b Alice M. Dias,
a M. Fernanda Proença,
a
Fátima Cerqueirab,c
aCentro de Química, Universidade do Minho, Campus de Gualtar, 4710-057 Braga, Portugal;
bCEBIMED,
Centro de Estudos em Biomedicina/ Faculdade de Ciências da Saúde, Universidade Fernando Pessoa,
Rua Carlos da Maia, 296, 4200-150 Porto, Portugal; cCEQUIMED-UP, Centro de Química Medicinal da
Universidade do Porto, Faculdade de Farmácia da Universidade do Porto, Rua Jorge Viterbo Ferreira,
nº 228 (E2), 4050-313 Porto, Portugal
5-Aminoimidazoles are attractive building blocks in chemical synthesis because they are
key components in many bioactive compounds. Few reports on the chemistry of these
compounds appear in the literature due to their known instability.1
In our research group, imidazoles 1 are easily accessible from an efficient method
previously developed. Studies on the reactivity of the 4-cyanoformimidoyl group with
primary amines showed that nucleophilic displacement of NH3 or HCN were easy reactions
in acidic medium.2 In a recent work, those reactions could be extended to hydrazines and
the experimental procedures were optimized for phenyl hydrazine leading to the selective
synthesis of imidazoles 2 or 3. Imidazoles 2 were screened for the antibacterial and
antifungal activities and three hit compounds were identified. These results prompted us to
prepare a series of imidazoles 3 in order to study their antibacterial and antifungal activities.
The new compounds were isolated in moderate-very good yield and were well characterized
by spectroscopic techniques, which include 2D NMR.
The in vitro screening studies of analogs 3 involved P. aeruginosa ATCC27853, E. coli
ATCC25922, S. aureus ATCC29213, C. albicans ATCC10231, C. krusei ATCC6258 and C.
parapsilosis ATCC22019. From the screened imidazoles 3, six hit compounds were
identified as active in C. krusei (MIC 6-50 µg/mL). The synthetic approach (Scheme 1) will
be discussed together with the results on the biological activity.
Scheme 1: The synthesis of imidazoles 2 and 3 from imidazoles 1 and aromatic hydrazines
Acknowledgements: We thank University of Minho and Fundação Ensino e Cultura Fernando Pessoa; FCT- Fundação para a Ciência e Tecnologia through the Portuguese NMR network (RNRMN), FCT- project PEst-C/QUI/UI0686/2011; FCT- project CEQUIMED - PESt.OE/SAU/UI4040/2011 and also QREN and COMPETE program. References: 1. Soh, C. H. et al., J. Comb. Chem. 2006, 8, 464-468 and references there in. 2. Dias, A. M.; Vila-Chã, A. M.; Costa, A. L.; Cunha, D; Senhorães, N; Proença, M. F.; Synlett, 2011, 2675.

PC128
Organic compounds isolated from Juniperus brevifolia bark
Amélia Vieira,a Marcele Martins,
a Ana M. L. Seca,
a,b Diana C. G. A. Pinto,
a Artur M. S. Silva
a
aDepartment of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal;
bDCTD, University
of Azores, 9501-801 Ponta Delgada, Portugal
The genus Juniperus is the unique genus from the Cupressaceae family that grows
spontaneously in Europe.1
Although some Juniperus species have toxic effects, they have
also many uses in folk medicine in several parts of the world, such as anT.
sive and haemostatic activities, with diuretic, antiseptic, stomachic and carminative effects,
as hypoglycaemic, antifertility agents and also as remedies of cold, urinary infection,
urticaria dysentery, hemorrhage, leukorrhea and rheumatic arthritis.1 It is also used as
remedy for tuberculosis and jaundice in Saudi Arabia (Topçu et al., 1999), as insect
repellent and for treatment of fever and dysurea in Bhutan.1 Belonging to this genus, the
Juniperus brevifolia, an endemic species from Azores Islands whose wood was used in the
past to build caravels, in works of art and furniture.2 Previous work showed that
dichloromethane and chloroform-soluble portions of the leaves acetone extract were the
most active against HeLa and Hep-2 tumour cell lines.3
Chemical investigation of these
extracts afforded more than thirty compounds, mainly abietane and pimarane derivatives,
eight of them were new natural compounds and other exhibit interesting antitumor activity.4
Recent investigation showed that the bark acetone extract has antioxidant activity similar to
that of quercetin; and also showed activity against Bacillus cereus, B. subtilis and
Micrococusluteus, while the wood acetone extract showed activity only against B. cereus.
Furthermore the bark acetone extract showed higher anti-AChE activity.5
Now we report on the purification and structural elucidation of the isolated compounds from
the most active extracts. The spectroscopic characterization details by NMR (1D and 2D)
will be presented and discussed.
Acknowledgements: We would like to thank Fundação para a Ciência e a Tecnologia (FCT, Portugal), the European Union, QREN, FEDER, COMPETE, for funding the Organic Chemistry Research Unit (QOPNA) (project PEst-C/QUI/UI0062/2013) and the Portuguese National NMR Network (RNRMN). References: 1. Seca A. M. L.; Silva A. M. S. The chemical composition of the Juniperus genus (1970–2004). In: Govil JN, Singh VK, editors. Recent progress in medicinal plants, vol 16-phytomedicines.Houston: Studium Press LLC; 2006. p. 401–522. 2. Elias R. B.; Dias E. Cad. Bot. 2008, 5, Edição Herbário da Universidade dos Açores, Angra do Heroísmo. ISBN: 978-989-630-978-7. 3. Moujir L. M.; Seca A. M. L.; Silva A. M. S.; Barreto M. C. Planta Med. 2008, 74, 751-753. 4. a) Seca A. M. L.; Silva A. M. S.; Bazzocchi I. L.; Jimenez A. I. Phytochemistry, 2008, 69, 498-505; b) Moujir L. M.; Seca A. M. L.; Araujo L.; Silva A. M. S.; Barreto M. C. Fitoterapia 2011, 82, 225-229.
5. Oliveira N.; Medeiros S.; Barreto M. C.; Seca A. M. L.; Rosa J. S. XXIII ENSPQ, Aveiro, Junho, 2013.

PC129
Cycloaddition Reaction of Spiro[2.4]hepta-4,6-dien-1-ylmethanol and
PTAD: A New Rearrangement
Abdullah Menzek, Halil Şenol, Çetin Bayrak, Ertan Şahin
Atatürk University, Faculty of Science, Department of Chemistry, 25240, Erzurum Turkey
[email protected] or [email protected]
Molecular rearrangement in organic chemistry is important subject and rearrangements
products can be starting, intermediate or result products for any aim. Furthermore, the
mechanism of rearrangement also is important for occurring products. PTAD (1)
(phenyltriazolinedione) is a good dienofile and cyclopentadiene and its derivatives are good
dienes in cycloaddition reactions.1
PTAD with cyclopentadiene and its derivative gives
adducts.
Spiro[2.4]hepta-4,6-dien-1-ylmethanol (2) which is a cyclopentadiene derivative includes
cyclopropyl methanol structure. Cyclopropyl methanol groups in adducts of compound 2 can
give skeleton rearrangement.3 A new rearrangement product 3 was obtained from reaction
of PTAD with compound 2 (Scheme 1).This rearrangement product should be formed via
intermediate A. In this study, structure of intermediate A and mechanism of formation of
new rearrangement product was discussed according to some reactions and their
spectroscopic data.
Acknowledgements: We are grateful to Ataturk University and TÜBİTAK (109T403) for their financial supports. References: 1. Fleming I. Frontier orbitals and organic chemical reactions, 1976, John Wiley & Sons, London. 2. a) Adam W.; De Lucchi O.; Erden İ. J. Am. Chem. Soc., 1980, 102, 4806. b) Adam W.; Dörr M.; Kron J.; Rosenthal J. R. J Am. Chem. Soc., 1987, 109, 7074. c) Warrener R. N.; Harrison P. A. Molecules,
2001, 6, 353. 3. a) Menzek A. Tetrahedron, 2000, 56, 8505. b) Menzek A.; Gökmen M. Helv. Chim. Acta, 2003, 86, 324.

PC130
Synthesis and photolysis studies of 5-aminolevulinic acid
conjugates based on 2-oxo-naphtho[1,2-b]pyran
Ana M. S. Soares, Susana P. G. Costa, M. Sameiro T. Gonçalves
Centro de Química, Universidade do Minho, Campus de Gualtar, 4710-057 Braga, Portugal
5-Aminolevulinic acid (5-ALA) is the first precursor in heme biosynthesis, leading to the
formation of hematin and hemin. The latter was reported to inhibit the proteasome and
induce apoptosis of malignant cells. Proteasome inhibitors as single agents or as therapy
adjuvants have entered phase I and phase II trials in solid tumours and hematologic
malignancies. Nevertheless, the hydrophilic nature of 5-ALA limits its ability to penetrate
through the skin or cell membranes and therefore restricts its use for topical treatment only,
with emphasis in photodinamic therapy (PDT).1,2
Strategies for the development of
pharmacologically inert chemical derivatives, prodrugs, which can be converted into active
drug molecules, have been explored to overcome common drawbacks such as low oral
drug absorption, lack of site specificity, chemical instability, toxicity and poor patient
acceptance. Photoactive prodrugs with a suitable photolabile group, whose reactivity can be
controlled by selecting the wavelength of the excitation light, could be an alternative to the
molecular design of prodrugs.3
Considering our research interests, that include the synthesis of fluorescent bioconjugates
based on heterocyclic compounds and photorelease of the corresponding active molecule,4
in this communication we report the synthesis of 6-amino-, 6-methoxy-, and 7-amino-2-oxo-
naphtho[1,2-b]pyrans 1a-c, their conjugation with 5-(N-butyloxycarbonyl)aminolevulinic acid
2 via an ester bond and a study of the behaviour of the resulting conjugates 3a-c in different
photocleavage conditions, namely the wavelength of irradiation and the solvent, in a
photochemical reactor with lamps of 254, 300, 350 and 419 nm (Scheme 1).
Scheme 1: Synthesis of 5-aminolevulinic naphthopyran conjugates 3a-c.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia (FCT) for financial support to the NMR portuguese network (PTNMR, BrukerAvance III 400-Univ. Minho), FCT and FEDER-COMPETE-QREN-EU for financial support to the Research Centre CQ/UM [PEst-C/QUI/UI0686/2011 (FCOMP-01-0124-FEDER-022716)], and a PhD grant to A. M. S. S. (SFRH/BD/80813/2011). References: 1.Chung C. -W.; Kim C. H.; Choi K. H.; Yoo J. -J.; Kim D. H.; Chung K. -D.; Jeong Y. -I.; Kang D. H. Eur. J. Pharm. Bio. 2012,80, 453. 2. Kaufman J. L.; Lonial S. Onkologie 2006, 29, 162. 3. Stella V. J.; Borchardt R. T.; Hageman M. J.; Oliyai R.; Maag H.; Tilley J. W. in Prodrugs: Challenges and Rewards, part1, vol. V, Springer, New York, 2007, pp. 703.
4.a) Soares A. M. S.; Piloto A. M.; Hungerford G.; Costa S. P. G.; Gonçalves M. S. T. Eur. J. Org. Chem. 2012, 922. b) Piloto A. M.; Hungerford G.; Costa S.P.G.; Gonçalves M. S. T. Photochem. Photobiol. Sci. 2013, 12, 339. c) Fernandes M. J. G.; Costa S. P. G.; Gonçalves M. S. T. New J. Chem. 2013, doi: 10.1039/C3NJ00247K.

PC131
Biological Activities of Vernoniacondensata: Acetylcholinesterase
Inhibition and Caco-2 Cells Toxicity
A. Arantes,a,b
Pedro L. Falé,a L. C. B. Costa,
c, L. Ascensão,
d M. L. Serralheiro
a,b
aCQB, Faculdade de Ciências da Universidade de Lisboa, Campo Grande, 1749-016 Lisboa, Portugal; bDQB, Departamento de Química e Bioquímica, Faculdade de Ciências da Universidade de Lisboa.
c
Universidade Estadual de Santa Cruz, Departamento de Ciências Biológicas, Baia, Brasil. dFaculdade de
Ciências. Centro de Biotecnologia Vegetal, IBB, Campo Grande 1749-016 Lisboa
Vermoniacondensata or “boldobaiano” is a plant used in Brasil to treat gastrointestinal
disorders.1 In order to explain some of these biological effects, acetylcholinesterase (AChE)
inhibition2 and antioxidant activity can be referred. Aqueous extracts of theleaves of this
plant were analysed on what concerns the AChE inhibition capacity. IC50 value of 411
µg/mL was obtained. The antioxidant activity, measured as DPPH scavenging capacity
indicated an EC50 of 20 µg/mL, a very low value similar to the antioxidant standard BHT (14
µg/mL). These activities can be ascribed to the polyphenol compounds. HPLC-DAD and
LC-MS indicated the presence of chlorogenic acid (the main component) and cynarin,
Figure 1. Both chlorogenic acid and cynarin have high AChE inhibition activity, IC50 of 12.9
µg/mL and 9.2 µg/mL, respectively. Previous results3 indicated that the gastrointestinal
digestion may decrease the biological activities found. The gastrointestinal digestion of this
aqueous extract of V. condensate was studied using pepsin at pH 1 (simulating the gastric
juice) and pH 8, pancreatin (simulating the pancreatic juices). During this digestion the
biological activities were not changed and the compounds were kept the same. As the
infusion is going to be in contact with the intestine cells, Caco-2 cell lines were used to test
the toxicity of the aqueous extract, using MTT test. IC50 value 0.56 mg/mL was obtained,
meaning that the extract is not toxic to these cells.
Figure 1: Main phenol compounds detected in V. condensata aqueous extract
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support PTDC/QUI_BIQ/113477/2009 and PEst-OE/QUI/UI0612/2013. References: 1. Grandi T.; Trindade J.; Pinto M.; Ferreira L.; Catella A., Acta Bot. Bras., 1988, 3, 185. Chem. Rev., 2009, 109, 3621. 2. Jarvie E. M.; Cellek S.; Sanger G. J., Pharmacol Res 2008, 58, 297. 3. Porfirio S.; Falé P.; Madeira P. J. A.; Florêncio M. H.; Ascensão L.; Serralheiro M. L. M., Food Chem. 2010, 122, 179.

PC132
Deoxyvitisins: a new set of pyrano-3-deoxyanthocyanidins
André Sousa, Paula Araújo,
Nuno Mateus,
Victor de Freitas
Chemistry Investigation Centre (CIQ), Department of Chemistry, Faculty of Sciences, University of Porto,
4169-007 Porto, Portugal
The chemical synthesis of deoxyvitisins (pyrano-3-deoxyanthocyanidins) is reported herein.
Three different types of compounds were synthesized from the reaction between two
deoxyanthocyanidins (deoxypeonidin and deoxymalvidin) and pyruvic acid, vinyloxy-
trimethylsilane and acetone-1,3-dicarboxylic acid (Scheme 1). Due to their pyranic ring,
pyranoanthocyanins are much more stable towards pH variations and bleaching by SO2 in
comparison to the genuine anthocyanins. Furthermore, oppositely to anthocyanins,
deoxyanthocyanidins are yellowish pigments that also have a significant increased stability
in slightly acidic solutions compared to anthocyanins and several studies have
demonstrated the potential applications of these compounds for viable commercial food
colorants, hair dyes, laser dyes, sensitizers for solar cells, molecular-level memory systems
and health-promoting phytochemicals.1
Recently, a new pigment was isolated from red Sorghum bicolor with a structure of a
pyrano-3-deoxyanthocyanidin, containing apigeninidin as a base unit.2 This was the first
report of a deoxyvitisin-type compound, which displayed a higher stability comparatively to
the corresponding anthocyanin, justifying the research developed in the chemistry of
3-deoxyanthocyanins and, in particular, the search for new colorants with significant
stability. Bearing this, the aim of this work was to synthesize deoxyvitisins of type A, B and
methylpyranodeoxyanthocyanidins and to propose a formation mechanism.
Scheme 1: Formation of pyrano-3-deoxyanthocyanidins.
The structure of the new compounds has been characterized by means of visible, MS and
NMR spectroscopy. These results show for the first time a new class of
deoxyanthocyanidins that have interesting visible spectroscopic properties, representing a
step forward towards the research for new pigments with significant stability.
References: 1. Roque, A.; Lodeiro, C.; Pina, F.; Maestri, M.; Ballardini, R.; Balzani, V. Photochromic Properties of 3-Methyl-Substituted Flavylium Salts. Eur. J. Org. Chem. 2002, 2699–2709. 2. Khalil, A.; Baltenweck-Guyot, R.; Ocampo-Torres, R.; Albrecht, P. A novel symmetrical pyrano-3-deoxyanthocyanidin from a Sorghum species. Phytochem. Lett. 2010, 3, 93–95.

PC133
Development of chemical tools to study the mechanism of action of
potent quinazolines targeting the liver stage of malaria infection
André Dias,a Inês Albuquerque,
b Miguel Prudêncio,
b Maria M. Mota,
b Lídia M. Gonçalves,
a
Rui Moreira,a A. S. Ressurreição
a
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa, Portugal;
bIMM, Faculdade de Medicina da Universidade de Lisboa, Av. Prof. Egas Moniz,1649-028
Lisboa, Portugal
Malaria is one of the most lethal infection diseases of our days and is caused by infection
with eukaryotic parasites of genus Plasmodium. According to the latest estimates of World
Health Organization, there were about 219 million cases of malaria in 2010 and an
estimated 660 000 deaths around the world. However, the emergence of resistance to
currently available antimalarials is a major obstacle in the control of the disease.1
The liver stage (LS) of malaria infection offers important advantages for prophylactic and
disease eradication strategies. In an attempt to address the lack of chemotypes, a high-
throughput phenotypic LS Plasmodium parasite screen have been developed to
systematically identify molecules with LS efficacy, disclosing a quinazoline-based hit (a
known NF-kB activation inhibitor) as a potent LS inhibitor with an IC50 value in the low nM
range.2 However, despite being a promising derivative for the LS, it is not yet known the
mechanism of action (MoA) of quinazolines as LS inhibitors.
In this report, we will describe the synthesis and structure-activity relationship (SAR) for
optimization of this hit. In the SAR study, we analyzed the variation of the methylene chain
length and the introduction of electron donors and acceptors groups in the 6- and 7-
positions of the quinazoline scaffold. The compounds synthesized were tested for in vitro
antimalarial activity and selectivity.
Based on the structural information obtained from the SAR study we will also describe our
efforts on the development of bioorthogonal turn-on probes3 for imaging malaria parasites
inside living cells, applying tetrazine/trans-cyclooctene (TCO) cycloadditions (Scheme 1).
The design and synthesis of the tetrazine-BODIPY fluorophore and TCO-quinazoline
molecules will be described.
Scheme 1: Rational development of bioorthogonal “turn-on” probes targeting the liver stage of malaria
infection. Acknowledgements: This work was supported by Fundação para a Ciência e Tecnologia (Portugal): PEst-OE/SAU/UI4013/2011 and postdoctoral fellowship to A. S. R. (BPD/64859/2009). References: 1. Rodrigues, T.; Prudêncio, M.; Moreira, R.; Mota, M. M.; Lopes, F. Targeting the Liver Stage of Malaria Parasites: A Yet Unmet Goal. J. Med. Chem. 2012, 55, 995-1012; 2. Derbyshire, E. R.; Prudêncio, M.; Mota, M. M.; Clardy, J. Liver stage malaria parasites vulnerable to diverse chemical scaffolds. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 8511-8516; 3. Devaraj, N. K.; Hilderbrand, S. A.; Upadhyay, R.; Mazitschek, R.; Weissleder, R. Bioorthogonal Turn-On Probes for Imaging Small Molecules inside Living Cells. Angew. Chem., Int. Ed. 2010, 49, 2869-2872.

PC134
-Gauch Effect in Benzobicyclo3.2.1octenes
Cavit Kazaz*a
, Arif Daştana, Metin Balci
b
aDepartment of Chemistry, Atatürk University, 25240 Erzurum, Turkey
bDepartment of Chemistry, Middle East Technical University, 06531 Ankara, Turkey
It is well known that interactions related to the van der Waals effect cause a paramagnetic
contribution to the shielding constants, which results in shifts to lower field. Tori et al.1
discovered the remarkable -syn (shielding) and -anti (deshielding) effect exerted by a
cyclopropane ring annulated to the bicyclo[2.2.1heptane skeleton upon its bridging carbon
atoms.
In this study, we aimed the synthesis of benzobicyclo3.2.1octene derivatives for
discussing -gauch effect in the benzobicyclo3.2.1octene derivatives (Figure 1).
Figure 1
Acknowledgements: We thank the Research Foundation of Ataturk University for financial support.
References: 1. Tori, K.; Ueyama, M.; Tsuji, T.; Matsumura, H.; Tanida, H.; Iwamura, H.;Kushida, K.; Nishida, T. Tetrahedron Lett. 15, 327-330 (1974). 2. Kazaz, C. Daştan, A. Balci, M. Magn. Res. Chem., 43, 75-81 (2005).

PC135
Isolation, quantification and seasonal variation of labdanolic acid
from Cistus ladaniferus
André N. C. Martins,a,b
L. M. T. Frija,a Svilen P. Simeonov,
a Carlos A. M. Afonso
a
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal; bCQFM, Centro de Química-Física Molecular, IN-Institute of Nanosciences and
Nanotechnology, Instituto Superior Técnico,1049-001 Lisboa, Portugal
Natural products continue to provide unique source of inspiration for advances in organic
chemistry and disease treatment.1 Particularly, labdane-type diterpenes
2 are an excellent
example of natural products with important applications in medicine. Several of these
derivatives possess a wide range of relevant biological properties, such as anti-fungal and
anti-bacterial, anti-mutagenic, cytotoxic, anti-inflammatory or analgesic activities. Our
interest in the study of labdane-type diterpenes emerged recently due to the possibility of
isolation of appreciable quantities of a specific diterpene, the labdanolic acid (LA, Figure 1),
from a Portuguese natural resource, i.e., the plant Cistus ladaniferus. From the extract of
this plant has been identified near 300 compounds, including fragrances such as Ambrox
and diterpenes like labdane derivatives.3 However, LA is one major compound which has
been isolated in high quantities (1.1 g per 100 g of air-dried twigs) by extraction with diethyl
ether followed by aqueous basic extraction and normal column chromatography.4
Through this investigation we have focused our efforts on the development of an analytical
method for the quantification of LA. The process depends on the derivatization of LA to form
the benzylic ester that can be analysed by HPLC. This investigation aims for the study of
the variation of LA present in Cistus ladaniferus during the year and the results obtaind will
be present and discussed in this communication.
Figure 1: Labdanolic acid (LA). Acknowledgements: Fundação para a Ciência e a Tecnologia (FCT, Portugal) and POCI 2010
(FEDER), (Ref. PTDC/QUI-QUI/119823/2010). References: 1. (a) Wilson R. M.; Danishefsky, S.; J. Org. Chem. 2006, 71, 8329; (b) Newman, D. J.; Cragg, G. M.; J. Nat. Prod. 2007, 70, 461; (c) Kingston, D. G.; J. Org. Chem. 2008, 73, 3975; Butler, M. S.; Nat. Prod. Rep. 2008¸ 25, 475; (e) Banwell, M. Tetrahedron 2008, 64, 4669; (f) Baker, D. D.; Chu, M.; Oza, U.; Rajgarhia, V.; Nat. Prod. Rep. 2007, 24, 1225. 2. Frija, L. M. T.; Frade, R. F. M.; Afonso, C. A. M.; Chem Rev, 2011, 111, 4418. 3. (a) Weyesrstahl, P.; Marschall, H.; Weirauch M.; Thefeld, K. Surburg, H.; Flavour and Fragr. J. 1998, 13, 295; (b) Gomes, P. B.; Mata, V. G.; Rodrigues, A. E.; J. Essential Oil Research 2005, 17, 160. 4. Bolster, M. G.; Jansen, B. J. M.; Groot, A. Tetrahedron 2001, 57, 5657.

PC136
Indole Alkaloids from Tabernaemontanaelegans: Isolation and
Molecular Derivatization
A. Paterna,a S. Mulhovo,
b M. J. U. Ferreira
a
aResearch Institute for Medicines and Pharmaceutical Sciences (iMed.UL), Faculty of Pharmacy,
University of Lisbon, Av. Prof. Gama Pinto, 1649-003, Lisbon, Portugal; bCentro de Estudos
Moçambicanos e de Etnociências, Universidade Pedagógica Campus de Lhanguene, Av. de
Moçambique, 21402161 Maputo, Mozambique
Tabernaemontana species (Apocynaceae) have been used in traditional medicine to treat
cancer. They are able to synthesize a high content and a wide variety of indole alkaloids,
including iboga-type alkaloids. Previously, a significant apoptosis inducing activity was
found for indole alkaloids from these species.1 Thus, the main goal of this project is to find
out new effective anticancer compounds, namely apoptosis inducers, from plants of
Tabernaemontana genus. Therefore, the roots of T. elegans, collected in Mozambique,
have been investigated. Several indole alkaloids, including one bis-indole alkaloid, were
isolated from the methanol extract of roots of this plant. In order to obtain homologous
series of compounds, two alkaloids found in larger amount, were derivatized according to
their functional groups. The structures of the compounds were characterized by
spectroscopic methods mainly 1D NMR (1H,
13C, DEPT) and 2D NMR (COSY, HMBC,
HMQC, NOESY).
Acknowledgements: We thank the Fundação para a Ciência e Tecnolgia for financial support, Portugal (projects PTDC/QUI-QUI/099815/ 2008; Pest-OE/SAU/UI4013/2011; PTDC/QEQ-MED/0905/2012; PhD grant SFRH/BD/77612/2011) References: 1. Tayyab A. Monsoor, et al. Bioorg. Med. Chem. Lett. 2009 19, 4255-4258.

PC137
Hit Optimization of a new class of p53-MDM2 interaction inhibitors
Ângelo Monteiro,a Nuno A. L. Pereira,
a J. Soares,
b L. Saraiva,
b Maria M. M. Santos
a
aResearch Institute for Medicines and Pharmaceutical Sciences (iMed.UL), Faculty of Pharmacy,
University of Lisbon, Av Prof. Gama Pinto, 1649-003 Lisbon, Portugal; bREQUIMTE, Department of
Biological Sciences, Laboratory of Microbiology, Faculty of Pharmacy, University of Porto, Rua Jorge
Viterbo Ferreira nº 228, 4050-313 Porto, Portugal
The tumor suppressor p53 plays a central role in the regulation of cell cycle, apoptosis, DNA
repair and senescence. In fact, its gene (TP53) is frequently mutated or deleted in almost
half of all human cancers, representing one of the most frequently mutated proteins found in
human tumors. In the other half of human cancers, where a wild-type (wt) p53 form is
retained, its activity is normally inhibited by overexpressed endogenous negative regulators,
such as MDM2. As a consequence, inhibiting the p53-MDM2 protein-protein interaction to
reactivate p53 function represents a promising approach to discover new drugs for cancer
therapy.1
In the last years our research group has been involved in the synthesis of novel p53
modulators. In particular, a chemical library of oxazoloisoindolinones was evaluated as p53-
MDM2 interaction inhibitors using a yeast cell-based screening approach and one
compound emerged as p53–MDM2 interaction inhibitor. Furthermore, this compound,
exhibited promising antiproliferative activities in two human tumor cell lines derived from
breast cancer (MCF7) and colon carcinoma (HCT116 p53+/+).
Here our recent results on the synthesis of novel p53-MDM2 inhibitors bearing
oxazoloisoindolinone scaffolds, in order to optimize the activity of the hit compound will be
disclosed. The compounds were synthesized by cyclocondensation of (S)-phenylalaninol
with 2-acylbenzoic acid derivatives via reflux in toluene under Dean–Stark conditions. In all
cases, only one diastereoisomer product was observed.2
To study the effect of the
corresponding enantiomers asp53-MDM2 interaction inhibitors, compounds were also
synthesized starting from (R)-phenylalaninol with 56–85 % (Scheme 1).
Scheme 1: Synthesis of novel p53-MDM2 inhibitors bearing oxazoloisoindolinone scaffolds.
Acknowledgements:This work was supported by FCT (Fundação para a Ciência e a Tecnologia) through projects PTDC/QUI-QUI/111664/2009, PEst-OE/SAU/UI4013/2011 and FEDER funds through the COMPETE program under the project FCOMP-01-0124-FEDER-015752 (ref. PTDC/SAU-FAR/110848/2009). References: 1. Cheok C. F.; Verma C. S.; Baselga J.; Lane D. P. Nat. Rev. Clin. Oncol. 2011, 8, 25.
2. Pereira N. A. L.; Sureda F. X.; Turch M.; Amat M.; Bosch J.; Santos M. M. M. Monatsh Chem 2013,
144, 473.

PC138
New Phosphonic Acids and Esters Derived from Indazole: Synthesis
and Biological Activity Evaluation
Silvânia S. Afonso,a António P. S. Teixeira,
a,b M. Rosário Martins,
a,c Fátima C. Teixeira
d
aDepartamento de Química, Escola de Ciências e Tecnologia, Universidade de Évora, R. Romão
Ramalho, 59, 7000-671 Évora, Portugal; bCentro de Química de Évora, IIFA, Universidade de Évora, R.
Romão Ramalho, 59, 7000-671 Évora, Portugal; cICAAM, IIFA, Universidade de Évora - Pólo da Mitra,
Apartado 94, 7002-554 Évora, Portugal; dLNEG,Estrada do Paço do Lumiar, 22, 1649-038 Lisboa,
Portugal
Bisphosphonates (BPs) are a group of compounds derived from bisphosphonic acid and
their salts, with a P-C-P structure which confers higher metabolic and chemical stability.
They are an important class of drugs with therapeutic applications in the treatment of
diseases of bone mineral metabolism such as osteoporosis and Paget's disease. These
compounds have also shown activity in other areas, such as antitumor and antiparasitic
activities.1 The use of these compounds in therapy is affected by their low oral bioavailability
due to low lipophilicity and the presence of charges at physiological pH. The improvement
of their pharmacokinetic properties can be obtained by the use of ester derivatives as
prodrugs.2
In this work, we present the synthesis and characterization of various phosphonic acids and
esters derivatives of indazole (Figure 1). The biological activity of these compounds were
evaluated including their toxicity, antioxidant and antimicrobial activities.3
Bisphosphonic
acid derivatives showed broad spectra with high activity against Gram – and Gram +
pathogenic and commensal bacteria. These compounds also present antioxidant activity by
the β-carotene/linoleic acid method.
Figure 1: Structure of studied phosphonates derived from indazole.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support (POCI/QUI/55508/2004, PPCDT/QUI/55508/2004 and pluriannual CQE funding) and to the Portuguese NMR Network (IST-UTL). References: 1. a) Fleisch H. Bisphoshponates in Bone Disease: from the Lab to Patient, 4th ed., Academic Press, San Diego, 2000. b) Bartl R., Frisch B., von Tresckow E., Bartl C.Bisphosphonates in Medical Practice, Springer-Verlag, Heidelberg, 2007. c) Zhang S., Gangal G., Uludağ H. Chem. Soc. Rev., 2007, 36, 507. 2. Ezra A., Golomb G. Adv. Drug Deliv. Rev., 2000, 42, 175. 3. NCCLS (2005), Performance Standards for Antimicrobial Susceptibility Testing: Seventh Informational Supplement M100-S15, National Committee for Clinical Laboratory Standards, Wayne, PA, USA.

PC139
NMR and IR conformational studies of the influence of bulky
C-tetrasubstituted amino acids on Peptaibolinmimetics
C. M. Carvalho, V. I. B. Castro, Susana P. G. Costa, S. M. M. A. Pereira-Lima
Centro de Química, Universidade do Minho, Campus de Gualtar, 4710-057 Braga, Portugal
Amino acid chain constraining techniques can result in more rigid peptide structures and
improvement of the molecular stability, which may also influence the bioactivity of the
resulting peptide. In the case of peptaibols, a family of natural antimicrobial peptides, the
presence of α,α-dialkylglycines such as Aib, Iva and Deg in their composition yields highly
helical structures, which directly correlates to their mechanism of action. Therefore, these
peptides have been the focus of structure-activity relationship studies in view of the
development of new peptidomimetic therapeutic agents.
Bearing these facts in mind, and our research interests in the synthesis and application of
α,α-dialkylglycines,2
we now report our conformational studies on a model peptide,
Peptaibolin (Ac-Leu-Aib-Leu-Aib-Phol), which is the shortest member of the peptaibol family
(Figure 1). Mimetics incorporating unnatural α,α-dialkylglycines bearing longer and bulkier
side chains (α,α-diethylglycine, Deg; α,α-dipropylglycine, Dpg; α,α-di-isobutylglycine, Dibg)
were studied in order to gain insight about the conformational preferences resulting from
substitution of the native Aib residues. An Ala analogue was studied as a control in the
conformational study. The influence imposed by the bulkier and more hydrophobic amino
acids in the peptide chain was evaluated through IR spectroscopy and 1D and 2D NMR
techniques, revealing certain conformational preferences which will be discussed.
Figure 1: Structure of Peptaibolin and its mimetics.
Acknowledgements: The authors acknowledge Fundação para a Ciência e Tecnologia (Portugal) for financial support through project PTDC/QUI-BIQ/118389/2010 (FCOMP-01-0124-FEDER-020906) and PEst-C/QUI/UI0686/2011 (F-COMP-01-0124-FEDER-022716), FEDER-COMPETE. The NMR spectrometer Bruker Avance III 400 is part of the National NMR Network and was purchased with funds from FCT and FEDER. References: 1. a) Duclohier H. Chem. Biodivers. 2007, 4, 1023. b) Brogden K.A. Nat. Rev. Microbiol. 2005, 3 238. c) Bocchinfuso G.; Palleschi A.; Orioni B.; Grande G.; Formaggio F.; Toniolo C.; Park Y.; Hahm K.-S.; Stella L. J. Pept. Sci. 2009, 15, 550. 2.a) Costa S. P. G.; Maia H. L. S.; Pereira-Lima S. M. M. A. Org. Biomol. Chem. 2003, 1, 1475. b) Pinto F. C. S. C.; Pereira-Lima S. M. M. A.; Maia H. L. S. Tetrahedron 2009, 65, 9165.

PC140
Synthesis and characterization of psoralen analogues based on
dibenzothiophenes
C. Francisco,a L. Rodrigues,
b A. Esteves
a
aChemistry Centre, School of Sciences, University of Minho, 4710-057 Braga, Portugal;
bInstitute for
Biotechnology and Bioengineering, University of Minho, 4710-057 Braga, Portugal
Psoralens are natural products (linear furocoumarins) present in several plant families,
which have been shown to possess a wide spectrum of biological activities including
cytotoxic, phytotoxic, photosensitizing, insecticidal, and antibacterial to antifungal effects.1
Furthermore, psoralens have been suggested as potential therapeutics for the treatment of
skin disorders (e.g. vitiligo, leukoderma and psoriasis),1
cutaneous T-cell lymphoma,
autoimmune diseasesand several types of cancer.2 Previously, in our group, some psoralen
type compounds were prepared and found to inhibit the in vitro growth of different human
tumor cell lines.3
The novel psoralen analogues of type 2 (Figure 1) were prepared by Pechmann reaction.
These compounds were obtained from the precursor dibenzothiophen-4-ol 1 by
condensation with ethyl acetoacetate or ethyl-2-chloroacetoacetate, in the presence of
sulphuric acid, at room temperature. In its turn, dibenzothiophen-4-ol 1 was obtained from
commercial dibenzothiophene by reaction with TMEDA and B(BuO)3 in the presence of n-
butyllithium. All these compounds were fully characterized by 1H NMR,
13C NMR, elemental
analysis and/or mass spectra. Moreover, anti-proliferative effect of compounds of type 2 on
human cancer cell lines (MDA-MB231, HeLa and TCC-SUP) was evaluated using a
commercial MTS assay. All these results will be presented in this communication.
Figure 1: Structures of the compound 1 and compound type 2.
Acknowledgements: We thank the Foundation for the Science and Technology (FCT, Portugal) for financial support to the NMR Portuguese network (PTNMR, BrukerAvance III 400-Univ. Minho). FCT and FEDER (European Fund for Regional Development)-COMPETE-QREN-EU for financial support to the Research Centre, CQ/UM [PEst-C/QUI/UI0686/2011 (FCOMP-01-0124-FEDER-022716)], (Pest-C/EQB/LA0006/2011) and the PhD grant to C.S. Francisco (SFRH/BD/48636/2008). References: 1. Gambari R.; Lampronti I.; Bianchi N.; Zuccato C.; Viola G.; Vedaldi D.; Dall´Acqua F. Top Heterocycl. Chem. 2007, 9, 265. 2. Chilin A.; Marzano C.; Guiotto A.; Manzini P.; Baccichetti F.; Carlassare F.; Bordin F. J. Med. Chem. 1999, 42, 2936. 3. Francisco C. S.; Rodrigues L. R.; Cerqueira N.; Oliveira-Campos A.; Rodrigues L. M. Eur. J. Med. Chem. 2012, 47, 370.

PC141
Synthesis of novel antiplasmodial agents containing squaramide
and 4-amino-7-chloroquinoline moieties
Carlos J. A. Ribeiro,a S. Praveen Kumar,
a Jiri Gut,
b Lídia M. Gonçalves,
a
Philip J. Rosenthal,b Rui Moreira,
a Maria M. M. Santos
a
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal, bDepartment of Medicine, San Francisco General Hospital, University of California, San
Francisco, CA 94143, USA
Based on the hypothesis that linking two active molecules to form a hybrid compound can
enhance efficacy and/or reduce propensity to resistance relative to the parent drugs,1
we
synthesized compounds containing a squaric acid core and one or two 7-chloroquinolines
moieties, linked together by different linear or heterocyclic diamines (R2) (1 and 2, Scheme
1).
In a previous work we scoped the potentialities of squaric acid scaffold as antiplasmodial
agent by synthesizing derivatives containing different aromatic and aliphatic amines, aza
vinyl sulfones and aza-dipeptide falcipain recognition moieties. The best compound showed
a IC50= 0,99 µM against chloroquine resistant Plasmodium falciparum W2 strain.2
This novel series of compounds now described presents improved antiplasmodial activity.
Three compounds, 2a (IC50 = 99 nM), 2b (IC50 = 95 nM), and 2c (IC50 = 105 nM) had greater
in vitro potency than chloroquine (IC50 = 140 nM) against P. falciparum W2 strain. In
addition, they were noncytotoxic against NIH 3T3 and Hek 293T cells.3
Scheme 1: Synthesis of squaricaminoquinoline derivatives.
Acknowledgements: This work was supported by Fundação para a Ciência e a Tecnologia (Portugal) through postdoctoral fellowship to S. P. K. (SFRH/BPD/44481/2008), doctoral fellowship to C. J. A. R. (SFRH/BD/69258/2010) and grant PEst-OE/SAU/UI4013/2011. References: 1. Muregi F. W.; Ishih A.Drug Dev. Res. 2010, 71, 20. 2. Kumar S. P.; Gloria P. M. C.; Goncalves L. M.; Gut J.; Rosenthal P. J.; Moreira R.; Santos M. M. M. Med. Chem. Comm. 2012, 3, 489. 3. Ribeiro C. J. A.; Kumar S. P.; Gut J; Gonçalves L. M.; Rosenthal P. J., Moreira R.; Santos M. M. M. Eur. J. Med. Chem. (submitted).

PC142
Design, synthesis and evaluation of tacrine-cinnamic acid
derivatives as potencial bi-functional anti-Alzheimer drug
candidates
Catarina Quintanova,a,b
Rangappa Keri,a Sérgio M. Marques,
a M. L. Serralheiro,
b M. Amélia
Santosa
aCentro de u mica Estrutural, Instituto Superior Te nico, Av. Rovisco Pais , 4 -001 Lisboa, Portugal bCentro de Química e Bioquímica, Faculdade de Ciências da Universidade de Lisboa, 1749-016 Lisboa,
Portugal.
Alzheimer’s disease (AD) is one of the most complexes human degenerative disorders. Its
multifactorial nature has limited the understanding of the mechanisms involved and, as a
consequence, no disease-modifying treatment is still available. Although many different
theories have arisen in the past decades to explain the true origin of AD, their discovery
lead to define the common hallmarks of the disease, such as β-amyloid (Aβ) deposits in
senile plaques and neurofibrillary tangles (phosphorylated tau protein).With the progression
of the disease, neuronal cells are dead and cholinergic neurons are selectively lost, which
leads to cognitive impairment. Other features have been found in patient brains, namely
related with high levels of oxidative stress. The current treatments for AD involve the use of
acetylcholinesterase (AChE) inhibitors, such as tacrine and donepezil, however their effect
does not stop the progression of the disease, only providing symptomatic benefits.1,2
Although the exact role of these molecular markers in the pathophysiology of the disease is
still unclear, they have been the targets in the recent development of multi-targeted agents
against AD.
Following our recent developments on multifunctional anti-neurodegeneratives,3
in the
present study a novel series of tacrine hybrid compounds were designed and studied,
involving the conjugation of tacrine withone moiety of cinnamic acid derivatives or the
corresponding 2,4-pentadienoic analogues (Scheme 1). These molecular units were
chosen for their capacity of binding the active site of AChE and to potentially inhibit Aβ
aggregation, besides their anti-oxidant activity. Based on molecular modelling studies the
secinnamic derivatives are capable of binding to the peripheral anionic site (PAS) of AChE,
thus suggesting the potential of these compounds to block the AChE-induced Aβ
aggregation.2 The compounds were prepared and fully characterized, and then assessed
for their inhibitory activity and their radical scavenging ability. Although their antioxidant
activity is not very significant, their ability to inhibit AChE activity is promising, since their
IC50 is in the low micromolar range.
Scheme 1: Simplified scheme of synthesis of tacrine-cinnamic acid derivatives
Acknowledgments: We acknowledge the Portuguese Fundação para Ciência e Tecnologia (F.C.T.) for
financial support with project PEst-OE/QUI/UI0100/2013 and the postdoc grant SFRH/BPD/75490/2010 (R.S.K).
References: 1. Hennessy, E.J., Buchwald, L., J. Org. Chem., 2005, 70, 7371-7375; 2. Cavalli, A, Bolognesi, M.L., Minarini, A., Rosini, M., Tumiatti, V., Recanatini, M., Melchiorre, C., J. Med. Chem., 2008, 51, 347-372; 3. Keri, R. S. Keri, Quintanova, C., Marques, S. M., Esteves, A. R., Cardoso, S. M., Santos, M. A., Bioorg. Med. Chem., 2013 (in press).

PC143
Remaking of Dietary Antioxidants: Targeting MitoBEN’s to
Mitochondria as a New Therapeutic Strategy
Catarina Oliveira,a Sofia Benfeito,
a José Teixeira,
a Ricardo Amorim,
a Alexandra Gaspar,
a
Jorge Garrido,a,b
Eugenio Uriarte,c Paulo J. Oliveira,
d Fernanda Borges
a
aCIQ/Department of Chemistry and Biochemistry, Faculty of Sciences, University of Porto, Portugal;
bDepartment of Chemical Engineering, Superior Institute of Engineering of Porto (ISEP), IPP, Portugal; cDepartment of Organic Chemistry, Faculty of Pharmacy, University of Santiago Compostela, Spain;
dCNC/Center for Neuroscience and Cell Biology, University of Coimbra, Portugal
[email protected]; [email protected]
Mitochondria are cellular organelles fundamental for the survival of the cells, being their
dysfunction associated with some diseases, named mitochondrial diseases that encloses,
for example, mitochondrial myopathies, diabetes, cancer, neurodegenerative diseases,
such as Parkinson and Alzheimer diseases, and amyotrophic lateral sclerosis.
Besides providing cellular energy (ATP), mitochondria are involved in other biological
processes, such as signaling, differentiation, cell growth and death, also in the production of
reactive oxygen species (ROS). In fact, it was estimated a daily production of 1011
ROS in a
typical aerobic cell.
The neurodegenerative diseases constitute a public health problem affecting millions of
people worldwide. The overall process in the genesis of neurodegenerative diseases may
be considered antagonistic to the one that mediates cancer since in cancer occurs
uncontrolled proliferation of the cells and in neurodegeneration the final result is cell death.
In both cases, mechanisms of necrosis and/or apoptosis as well as mitochondrial
dysfunction are intrinsically involved.
Therefore, it is urgent to find an approach to reduce or delay the progression of
neurodegenerative processes and, consequently, the associated ageing processes. In this
context, mitochondria are currently considered an important target and the selective
inhibition/minimization of the mitochondria oxidative damage is considered a promising
therapeutic solution for this type of disorders.
Accordingly, the present project encompasses the rational design and synthesis of new
hydroxybenzoic acid derivatives, presenting antioxidant activity, with positive charges at
physiological pH, thus displaying the ability of accumulation inside mitochondria.
In order to achieve this goal, structural changes were performed in natural phenolic
antioxidants present in human diet (protocatechuic and gallic acids) by inserting an aliphatic
carbon chain spacer linked to a triphenylphosphonium cation (TPP+). The synthesized
MitoBEN’s present antioxidant activity, measured by using ABTS, DPPH, and GO methods
and also redox potential determinations, and their performance in mitochondrial and
neuronal systems is being attained. There is the hope that, in a near future, this new
therapeutic approach can improve the lifestyle of people who suffer from diseases related to
oxidative stress, namely of neurodegenerative nature.
The synthesized compounds, methods and applications are in patenting process.
Acknowledgements: This work was supported by the Fundação para a Ciência e Tecnologia (FCT),
Portugal (PTDC/QUI-QUI/113687/2009 and Pest/C-QUI/UI0081/2011). A. Gaspar (SFRH/BD/43531/ 2008) and J.Teixeira (SFRH/BD/79658/2011) thank FCT grants.

PC144
Anti-inflammatory Activity of Genista tenera n-Butanol Extract and
Evaluation of its Stability under Gastrointestinal Conditions
Daniela Batista, Alice Martins, Pedro L. Falé, M. L. Serralheiro, M. Eduarda M. Araújo, Paulo A. Madeira, Carlos Borges, Amélia P. Rauter
Centro de Química e Bioquímica/Departamento de Química e Bioquímica, Faculdade de Ciências da
Universidade de Lisboa, Campo Grande, Edifício C8, 5o Piso, 1749-016 Lisboa, Portugal
The butanol extract obtained from the aerial parts of Genista tenera, an endemic plant to
the island of Madeira, Portugal, has proven to be quite efficient as an antioxidant,
antidiabetic and acetylcholinerase inhibitor.1 Therefore, its stability under gastrointestinal
conditions was tested and analysed by HPLC-DAD and monitored by the DPPH method.
The analysis of the extract, after being submitted to in vitro digestion with artificial gastric
and pancreatic juices, showed that none of its constituents suffered hydrolysis. These
results suggest that this extract may pass through the gastrointestinal tract keeping its
composition, and therefore its activity. Extract chemical composition was analyzed by
electrospray tandem mass spectrometry (ESI-MS/MS) in the negative ion mode, allowing
the identification of the flavonoid glycosides 5,8-diglucosylgenistein, 6,8-diglucosylapigenin
and 5-O-methylgenistein-7-O-glucoside as major constituents.
Evaluation of the anti-inflammatory activity of this antidiabetic extract is now assessed by an
in vitro assay2 involving the inhibition of cyclooxygenase (COX, or Prostaglandin-H
synthase, PGHS), a key enzyme in the synthesis of prostaglandin H2, which is a precursor
for the biosynthesis of prostaglandins, thromboxanes, and prostacyclins. PGs support the
release of further mediators of inflammation and cause the typical symptoms at sites of
inflammation. Therefore, PGHS has been regarded for a long time as an important target of
most non-steroidal anti-inflammatory drugs (NSAIDs) like indomethacin or diclofenac. We
found that this extract inhibited COX-1 enzyme in a dose dependent manner: 20.3 ± 3.3%
(0.100 mg.mL-1), 42.9 ± 9.1% (0.200 mg.mL
-1) and 47.8 ± 5.8% (0.300 mg.mL
-1). These
preliminary results encourage the further study of this promising bioactive extract for
nutraceutical purposes.
Acknowledgements: The authors thank the Fundação para a Ciência e a Tecnologia for financial support (Projects PTDC/QUI/67165/2006, PEst-OE/QUI/UI0612/2013 and REDE/1501/REM/2005). References: 1. Rauter A. P.; Ferreira J.; Martins A.; Santos R. G.; Serralheiro M. L.; Borges C.; Araújo M. E.; Silva F.; Goulart M.; Justino J.; Rodrigues J.; Edwards E.; Thomas-Oates J.; Noronha J. P.; Pinto R.; Mota-Filipe H. J. Ethnopharmacol. 2009, 122, 384. 2. Gierse J. K.; Koboldt C. M.; Walker M. C.; Seibert K.; Isakson P. C. Biochem. J. 1999, 339, 607.

PC145
Synthesis of hybrids compounds with multistage antimalarial
activity
Daniela Miranda, Rita Capela, Rui Moreira, Francisca Lopes
iMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal
Malaria is a potentially life-threatening and one of the world’s most prevalent tropical
diseases. The emergence of resistant parasite strains to antimalarial drugs remains a real
and ever-present danger.1 Hybrid molecules capable of acting on different targets and
different stages of the parasite’s life cycle are a promising approach to overcome the
resistance problem.2,3
Here we report the synthesis of a series of hybrid compounds that combine a 1,2,4-trioxane,
1, or a 1,2,4,5-tetraoxane, 2, with 8-aminoquinoline moieties (Figure 1). The linker used
between the two pharmacophoric moieties was an amide or amine. Compounds containing
an amide linker were synthesized by reacting a carboxilic acid intermediate of 1,2,4-trioxane
or 1,2,4,5-tetraoxane, with Primaquine (PQ), using TBTU or methyl chloroformate as
coupling agents. The amine counterpart was obtained by reductive amination of 1,2,4-
trioxane or 1,2,4,5-tetraoxanes aldehyde intermediate, with PQ and NaBH(AcO)3. Hybrid
drugs containing the artemisinin pharmacophore, 1, were also linked to PQ through a 10-
carba bond, involving Zn-mediated C-C bond formation. The structural assignment of these
compounds was made by 1H,
13C NMR (1D, 2D).
Figure 1: General structure of hybrid compounds.
Acknowledgements: FCT is acknowledged for support through the projects PTDC/SAU-FAR/118459/2010, PEst-OE/SAU/UI4013/2011 and PhD grant SFRH/BD/30418/2006 (RC). References: 1. W.H.O., World Malaria Report 2011, Geneva, Switzerland, 2011. 2. The malERA consultative group on drugs. A research agenda for Malaria eradication: Drugs. PloS Med. 2011, 8, e1000402. 3. Capela, R.; Cabal, G. G.; Rosenthal, P. J.; Gut, J.; Mota, M. M.; Moreira, R.; Lopes, F.; Prudêncio, M.; Antimicrob. Agents Chemother. 2011, 55, 4698.

PC146
Synthesis of 2’,4’-dihydroxy-3,4,5-trimethoxy-3’-propylchalcone
analogues with potential antitumor activity
D. Pereira,a,b
S. Cravo,a,b
L. Saraiva,c,d
M. Pinto,a,b
H. Cidadea,b
aCentro de Química Medicinal da Universidade do Porto (CEQUIMED-UP), Rua de Jorge Viterbo
Ferreira, 228, 4050-313 Porto, Portugal; bLaboratório de Química Orgânica e Farmacêutica, Departamento de Ciências Químicas, Faculdade de
Farmácia, Universidade do Porto, Rua de Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal; cREQUIMTE, Faculdade de Farmácia, Universidade do Porto, Porto, Portugal;
dLaboratório de Microbiologia, Departamento de Ciências Biológicas, Faculdade de Farmácia,
Universidade do Porto, Rua de Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal
During our research work on the search for potential antitumor compounds, several
chalcones were synthesized and evaluated for their inhibitory activity against the in vitro
growth of several human tumor cell lines.1,2
Particularly, a chalcone derivative, 2’,4’-
dihydroxy-3,4,5-trimethoxy-3’-propylchalcone, revealed to have a potent growth inhibitory
effect.1 Additionally this chalcone interfered with the cell cycle distribution of MCF-7 cell
line.1 Taking this into account, seven analogues of this chalcone were synthesized by a
base-catalyzed aldol reaction of appropriated substituted acetophenones with
benzaldehydes under microwave irradiation. The synthesized compounds were
characterized by spectroscopic techniques (1H NMR,
13C NMR, HSQC, HMBC). In the
future all synthesized compounds will be evaluated for their ability to inhibit the in vitro
growth of human tumor cell lines. For the most promising compounds further studies will
also be carried out in order to clarify if the growth inhibitory effect is associated with cell
cycle arrest and/or induction of apoptosis.
Acknowledgements: This work is funded through national funds from FCT – Fundação para a Ciência e a Tecnologia under the project CEQUIMED – PEst-OE/SAU/UI4040/2011, FEDER funds and COMPETE program under the projects FCOMP-01-0124-FEDER-011057 and FCOMP-01-0124-FEDER-015752. References: 1. Neves, M. P., Cravo, S., Lima, R. T., Vasconcelos, M. H., Nascimento, M. S. J., Silva, A. M. S., Pinto, M., Cidade, H., Corrêa, A. G., Bioorganic Med. Chem., 2012, 20, 25-33. 2. Neves, M. P., Lima, R. T., Choosang, K., Pakkong, P., Nascimento, M. S. J., Vasconcelos, M. H., Pinto, M., Silva, A. M. S., Cidade, H., Chem Biodivers, 2012, 9, 1133-1143.

PC147
De novo Design and Synthesis of New Potent Human Neutrophil
Elastase Inhibitors
E. F. P. Ruivo,a L. R. P. Areias,
a Lídia M. Gonçalves,
a M. T. Duarte,
b Vânia André,
b
Susana D. Lucas,a Rita C. Guedes,
a Rui Moreira
a
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal; bCentro de Química Estrutural, Instituto Superior Técnico, Universidade Técnica de
Lisboa,1049-001 Lisboa, Portugal
[email protected], [email protected]
Human neutrophil elastase (HNE) is a serine protease that plays an important role in
Chronic Obstructive Pulmonary Disease (COPD) inflammatory process wherein an excess
of HNE is produced hydrolyzing elastin, the structural protein which gives the lungs their
elasticity. COPD is a life-threatening disease being the fifth leading cause of death
worldwide. The available COPD therapeutic is limited to palliative drugs and, so far, no HNE
inhibitor got FDA approval for the treatment of COPD. Hence, there is an urgent need of an
effective HNE inhibitor, so that it will halt the progression of the disease.1
Our group has been engaged with SAR studies of a novel class of powerful inhibitors, the 4-
oxo-β-lactams (Figure 1),2 that shown to be a promising scaffold for lead development and
structure-based drug design. Hence, de novo design approach was applied to decorate the
4-oxo-β- lactam scaffold in a systematic way by a simple "Add group to ligand" tool in
Molecular Operating Environment (MOE) software,3 defining the bond from an R-group to
the scaffold and using a fragment database for virtual generation of an oxo-β-lactam library.
Resulting molecules were docked against HNE active site using previously developed
docking methodology toward HNE-based molecular docking.4 Herein we present the top-
ranked structures and the synthetic methodologies toward their development as well as the
activity against HNE of the synthesized compounds that led, so far, to very promising
activities in the 0.1 µM range.
Figure 1
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support, Pest-OE/SAL/UI4013/2011, PEst-OE/QUI/UI0100/2013, RECI/QEQ-QIN/0189/2012, SFRH/BPD/64265/2009 (SDL), SFRH/BPD/78854/2011 (VA). References: 1. Lucas S. D.; Costa E., Guedes R. C.; Moreira R. Med. Res. Rev. 2013, 33,S1, E73. 2. Mulchande J.; Oliveira R.; Carrasco M.; Gouveia L.; Guedes R. C.; Iley J.; Moreira R. J. Med. Chem. 2010, 53 (1), 241. 3. Molecular Operating Environment (MOE), 2011.10; Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2011. www.chemcomp.com. 4. Lucas, S. D.; Gonçalves, L. M.; Cardote, T. A. F.; Correia, H. F.; Moreira, R.; Guedes, R. C.; Med. Chem. Commun., 2012, 3, 1299.

PC148
Synthesis of Derivatives of 3-Hydroxypyrrolidine and (3,4)-
Dihydroxypyrrolidine: Inhibitors of Rat Intestinal Glucosidases
Elisabete P. Carreiro,a,b
Gizé Adriano,a,b
Patrícia Louro,a,b
Célia Antunes,c,d
R. A. Guedes,e
Rita C. Guedes,e Anthony J. Burke
a,b
aChemistry Department, University of Évora, Rua Romão Ramalho, 59, 7000 Évora, Portugal,
bCentro de Química de Évora, University of Évora, Rua Romão Ramalho, 59, 7000 Évora, ,Portugal.
cCentro de Neurociências e Biologia Celular de Coimbra, University of Coimbra.
dInstituto de Ciências Agrárias e Ambientais Mediterrânicas (ICAAM), University of Évora.
eResearch Institute for Medicines and Pharmaceutical Sciences – iMedUL, Faculty of Pharmacy,
Universidade de Lisboa,Av. Prof. Gama Pinto, 1649-003, Lisboa
The iminocyclitols- polyhydroxylated pyrrolidines and piperidines - are a family of important
pharmacologically active compounds in the treatment of diseases, like cancer, viral
infections and diabetes, etc. These type of molecules are both potent glycosidase and
glycosyltransferase inhibitors.1
The design and synthesis of a library of pyrrolidine
iminocyclitol inhibitors derivatives from 3-hydroxypyrrolidine and (3,4)-dihydroxypyrrolidine
(Figure 1) and with a structural similarity to 1,4-dideoxy-1,4-imino-D-arabitol (DAB-1) is
reported.1
This library was specifically designed to gain a better insight into the mechanism
of inhibition of glucosidases by these inhibitors (Figure 1).2 The inhibition studies were
carried out with the α-glucosidases from rat intestine enterocytes using DNJ as a
references. Docking studies performed on this library with a homology model of α-
glucosidase from murine intestine and also with the new crystallographic structure of human
glucosidase showed that results are consistent with the experimental. Structure- activity
relationships, ligand efficiency, and most important enzyme-ligand interactions have been
analysed. All these results will be discussed in this communication.
Figure 1
Acknowledgements: EPC thanks the Fundação para a Ciência e a Tecnologia (FCT) for a post-doctoral research fellowship (SFRH/BPD/72182/2010). References: 1. a) V. H. Lillelund, H. H. Jensen, X. Liang, M. Bols, Chem. Rev. 2002, 102, 515. b) M. Sugiyama, Z. Hong, P-H. Liang, S. M. Dean, L. J. Whalen, W. A. Greenberg, C-H. Wong, J. Am. Chem. 2007, 129,
14813, and references cited therein. 2. L. R. Guerreiro, E. P. Carreiro, L. Fernandes, T. A. F. Cardote, R. Moreira, A. T. Caldeira, R. C. Guedes, A. J. Burke, Bioorganic & Medicinal Chemistry, 2013, 21, 1911.

PC149
Anthracene-Derived Bis-Aminophosphonates: Synthesis, NMR
Characterization and Biological Activity
I. Kraicheva,a E. Vodenicharova,
a A. Kril,
b M. Topashka-Ancheva,
c I. Iliev,
b A. Georgieva,
b
T. Gerasimova,c I. Tsacheva,
a K. Troev
a
aInstitute of Polymers,BAS, Acad. G. Bonchev Street, Block 103 A, 1113 Sofia, Bulgaria
bInstitute of Experimental Morphology, Pathology and Anthropology with Museum, BAS, Acad. G.
Bonchev Street, Block 25, 1113 Sofia, Bulgaria cInstitute of Biodiversity and Ecosystems Research, BAS, 2 Gagarin Street, 1113 Sofia, Bulgaria
Aminophosphonic acid derivatives possess versatile biological activity and find a wide range
of useful applications in medicine and pharmacology, as enzyme inhibitors, metabolic
regulators, inhibitors of bone resorbtion, radiopharmaceuticals, antibacterial, antiviral and
antitumor agents. Due to their low toxicity in mammals and the resistance to chemical and
enzymatic hydrolysis, they are quite promising in the development of new drugs against
several disorders, including antiviral and anticancer agents.
Novel bis(aminophosphonate)s bearing anthracene rings, bis[N-methyl(diethoxyphos
phonyl)-1-(9-anthryl)]benzidine and 4,4’-bis[N-methyl(diethoxyphosphonyl)-1-(9-anthryl)]
diaminodiphenylmethane were synthesized. The bis-aminophosphonates were prepared via
the Kabachnik-Fields reaction from diethyl phosphite, 9-anthracenecarboxaldehyde and
diamines (4,4’-diaminodiphenylmethane and benzidine, respectively) Scheme 1. The
compounds have been characterized by elemental analysis, TLC, IR, NMR (1H,
13C,
31P)
and fluorescent spectra. The structures were also confirmed by X-ray crystallography.
The cytotoxic potential, genotoxicity and antiproliferative activity of bis-aminophosphonates,
as well as their subcellular distribution in a tumor cell culture system, are also reported. The
compounds showed optimal antiproliferative activity to human tumor cells from colon
carcinoma line HT-29. In vitro and in vivo safety testing revealed that the compounds exert
lower toxicity to normal cells as compared to well known anticancer and cytotoxic agents.
Scheme 1: Synthesis of bis(aminophosphonate)s
Acknowledgements: Thanks are due to Bulgarian National Science Fund of Ministry of Education and Science for the financial support: contract DTK-02/34 (2009).
H
PC2H5O
C2H5O
OH2N
RNH2+ +
O
2
PC2H5O
NH
R
O
NH
PO OC2H5
OC2H5 OC2H5
1, 2
3, 4
R =
R =
1, 3
2, 4
Scheme 1
2

PC150
Chemoselective biohydrogenation of α,β-unsaturated ketones by
mycelia of marine-derived fungal strain Penicillium citrinum CBMAI
1186
Irlon M. Ferreira, André L. M. Porto
Laboratório de Química Orgânica e Biocatálise, Instituto de Química de São Carlos, Universidade de São Paulo, Avenida João Dagnone, nº 1100, Ed. Química Ambiental, Jardim Santa Angelina, 13563-120,
São Carlos, SP, Brazil.
Dihydrochalcones are common in many natural products. These compounds have attracted attention because some studies have shown that they possess medicinal properties such as cytotoxic, antileishmanial
1 and anti-Trypanosoma cruzi
2 activities. Thus, several strategies
have been developed for the synthesis of dihydrochalcones. In this study, we employed biocatalysis by mycelia of marine-derived fungus Penicillium citrinum CBMAI 1186 to obtain derivatives of dihydrochalcones. The reaction proceeded in a 250 mL Erlenmeyer flask, in which 50 mg of compound 1 (reaction A), previously dissolved in DMSO, was added to 5.0 g of fungal mycelia in 100 mL of 0.1 mol L
-1 phosphate buffer, pH 7. The reaction continued
for 6 days in an orbital shaker (130 rpm, 32 ºC). The same procedure was followed for the other α,β-unsaturated ketones (Reactions B-D, Scheme 1). The mycelia of the fungus P. citrinum 1186 CBMAI catalyzed the chemoselective reduction of the carbon-carbon double bond of the chalcones, with good conversions to the corresponding dihydrochalcones. In none of the substrates employed was the carbonyl group reduced by alcohol dehydrogenases. Therefore, the reduction of α,β-unsaturated ketones by P. citrinum CBMAI 1186 showed excellent selectivity in the biohydrogenation of the double bond, catalyzed by enoate reductases.
Scheme 1: Biohydrogenation of α,β-unsaturated ketones by mycelia of marine-derived fungus P.
citrinum CBMAI 1186
Reaction R Conversiona (%)
A
98
B
49
C
72
D
97 aConversion determined by GC-MS
Acknowledgments: The authors thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and the Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP) for financial support and scholarship. References: 1. (a) Anto, R., Sukumaran, K., Kuttan, G., Rao, M., Subbaraju, V., Kuttan, R. 1995. Anticancer and antioxidant activity of synthetic chalcones and related compounds. Cancer Letters, 97, 33-37. (b) Ducki, S., Forrest, R., Hadfield, J., Kendall, A., Lawrence, N., McGown, A., Rennison, D. 1998. Potent
antimitotic and cell growth inhibitory properties of substituted chalcones. Bioorganic & Medicinal Chemistry Letters, 8, 1051-1056. 2. (a) Boeck, P., Falcao, C., Leal, P., Yunes, R., Cechinel, V., Torres-Santos, E., Rossi-Bergmann, B. 2006. Synthesis of chalcone analogues with increased antileishmanial activity. Bioorganic & Medicinal
Chemistry, 14, 1538-1545. (b) Aponte, J., Verastegui, M., Malaga, E., Zimic, M., Quiliano, M., Vaisberg, A., Gilman, R., Hammond, G. 2008. Synthesis, cytotoxicity, and anti-Trypanosoma cruzi activity of new chalcones. Journal of Medicinal Chemistry, 51, 6230-6234.

PC151
Synthesis and biological data of new 7-carboranylmethyl-
benzo[b]acridin-12(7H)-one derivatives as potential BNCT Agents
A. Filipa F. Silva,a Raquel S. G. R. Seixas,
a Artur M. S. Silva,
a Fernanda Marques
b
aDepartment of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal;
bUnidade de
Ciências Químicas e Radiofarmacêuticas, Instituto Superior Técnico, Campus Tecnológico e Nuclear, Estrada Nacional 10, 2695-066 Bobadela LRS Sacavém, Portugal
Boron neutron capture therapy (BNCT) have been received considerable attention as a
highly selective treatment modality for cancer.1
This approach is a two-step chemo-
radiotherapeutic technique that involves the selective delivery of boron rich compounds to
tumours and their subsequent irradiation with thermal neutrons, inducing a nuclear reaction
that causes the selective destruction of the targeted cells. Therefore, if boron (i.e., B-10) can
be selectively delivered to the tumour cell nucleus, the result is a targeted irradiation of
tumour cells whilst sparing normal tissues. Attach boron fragment with different tumour-
specific targeting molecules could lead to good and effective boron delivery agents for
BNCT.2
DNA intercalators such as acridine and acridone are excellent candidates since
they target the DNA and, simultaneously, act as fluorescent probes to follow them inside the
cells.3
Due to our interested in the search of novel antitumour agents, we present the synthesis
and evaluation of benzo[b]acridin-12(7H)-ones bearing carboranyl moieties as BNCT
agents. The synthesis of benzo[b]acridin-12(7H)-ones (1a-c) was already described by our
group and involves the Diels-Alder reaction of 7-ethoxycarbonyl-4-quinonole-3-
carbaldehyde with ortho-benzoquinodimethanes followed by oxidation.4
Now, we
functionalized the benzo[b]acridin-12(7H)-ones amino group with propargyl bromide
[Scheme 1, i)], followed by the cycloaddition reaction of the obtained compound with
decaborane [Scheme 1, ii)]. All benzo[b]acridin-12(7H)-one derivatives (3a-c)
wereevaluated for their in vitro cytotoxic activity in U87 human glioblastoma cells before and
after neutron irradiation treatment. Experimental procedures and biological data of the
synthesized benzo[b]acridin-12(7H)-ones derivatives will be presented and discussed.
Scheme 1: i) HC≡CCH2Br, NaH, dry THF, 40ºC; ii) B10H14, CH3CN, toluene, 80ºC.
Acknowledgements: Thanks are due to the University of Aveiro, Portuguese Foundation for Science and Technology (FCT), European Union, QREN, FEDER and COMPETE for funding the QOPNA research unit (project PEst-C/QUI/UI0062/2011), and the Portuguese NMR Network.A.F.F. Silva and R. S. G. R. Seixas thank project PEst-C/QUI/UI0062/2011, QOPNA and FCT for their grants. References: 1. Hawthorne, M. F. Angew. Chem. 1993, 105, 997. 2. Scholz, M.; Hey-Hawkins, E. Chem. Rev. 2011, 111, 7035. 3. Howell, L. A.; Gulam, R.; Mueller, A.; O’Connell, M. A.; Searcey, M. Bioorg. Med. Chem. Lett. 2010, 20, 6956. 4. Seixas, R. S. G. R.; Silva, A. M. S.; Pinto, D. C. G. A.; Cavaleiro, J. A. S. Synlett 2008, 3193.

PC152
Discovery of New Heterocycles with Activity against Human
Neutrophile Elastase Based On A Boron Promoted One-Pot
Assembly Reaction
Francesco Montalbano,a Pedro M. S. D. Cal,
a Marta Carvalho,
a Lídia M. Gonçalves,
a
Susana D. Lucas,a Rita C. Guedes,
a Luís F. Veiros,
b Rui Moreira,
a Pedro M. P. Góis
a
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal; bCQE, Departamento de Engenharia Química e Biológica Complexo I, Instituto Superior
Técnico.
Medicinal Chemists incessantly face the daunting task of discovering new drugs with
potency, specificity, and low toxicity for human diseases.1 To unravel new therapeutically
useful molecules, medicinal chemists frequently use natural products’ architecture and
biological properties as inspiration.2-4
Herein we demonstrate for the first time that a boron
promoted one-pot assembly reaction may be used to discover novel enzyme inhibitors.
Inhibitors for HNE were simply assembled in excellent yields, high diastereoselectivities and
IC50 up to 1.10 μM, based on components like salicylaldehyde, arylboronic acids and
amino acids. The combination of synthetic, biochemical, analytic and theoretical studies,
allowed the identification of the 4-methoxy or the 4-diethyl amino substituent of the
salicylaldehyde as the most important recognition moiety and the imine alkylation, lactone
ring opening as key events in the mechanism of inhibition.
Figure 1: Proposed mechanism of inhibition against HNE for 4-diethyl amino heterocycle.
Acknowledgements: Fundação para a Ciência e Tecnologia (PEst-OE/SAU/UI4013/2011; 55 PEst-OE/QUI/UI0100/2011; PTDC/QUI-QUI/118315/2010; PTDC/CTM-NAN/115110/2009; SFRH/BD/61419/2009 and 72376/2010) is acknowledged for financial support. References: 1. W. L. Jorgensen, Angew. Chem., 2012, 51, 11680-11684. 2. D. J. Newman, G. M. Cragg, J. Nat. Prod., 2012, 75, 311-335. 3. K. Grabowski, K.-H. Baringhausb, G. Schneider, Nat. Prod. Rep. 2008, 25, 892–904. 4. K. Hübel, T. Leßmanna, H. Waldmann, Chem. Soc. Rev., 2008, 37, 1316-1374.

PC153
Push-Pull-Push Fluorophores for Cellular Imaging
Sandra I. Vieira,a Odete A. B. da Cruz e Silva,
a João Rocha,
b Artur M. S. Silva,
c
Samuel Guieub,c
aUniversity of Aveiro, Neuroscience Laboratory, Center for Cell Biology, Health Sciences Department and
Biology Department, 3810-193 Aveiro, Portugal; bUniversity of Aveiro, CICECO, Department of
Chemistry, 3810-193 Aveiro, Portugal; cUniversity of Aveiro, QOPNA, Department of Chemistry,
3810-193 Aveiro, Portugal
Fluorescence labeling is an important process employed in several biology and medicine-
related techniques in order to identify cellular organelles and the subcellular localization of
molecules.1 Push-Pull fluorophores have been used successfully for this purpose,
2
especially because they present a characteristic large Stroke shift. Here we report six Push-
Pull-Push fluorophores, including some containing halogen atoms (Figure 1).3
All these
compounds exhibit good molar extinction coefficients and reasonable quantum yields. More
interestingly, their Stroke shift is large, and their emission maximum is greatly influenced by
the solvent.
300 400 500 600 700 800 9000.0
0.1
0.2
0.3
0.4
0.5
Ab
so
rba
nce
Wavelenght (nm)
Linear H
Linear Cl
Linear Br
Cyclic H
Cyclic Cl
Cyclic Br
400 500 600 700 8000
2
4
6
8
10
12
14
Em
issio
n (
a.u
.)
Wavelenght (nm)
Linear H
Linear Cl
Linear Br
Cyclic H
Cyclic Cl
Cyclic Br
Figure 1: Push-Pull-Push fluorophores and their absorption and emission spectra in dichloromethane.
The structures of these fluorophores have been studied by NMR and X-ray diffraction, and
their absorption and emission properties characterized. Further, these fluorophores have
been used for in-vitro cell imaging, and preliminary results will be reported.
Acknowledgements: Thanks are due to the University of Aveiro and the Portuguese Fundação para a Ciência e a Tecnologia (FCT), European Union, QREN, FEDER and COMPETE for funding the Organic Chemistry Research Unit (project PEst-C/QUI/UI0062/2011), the CICECO Associate Laboratory (PEst-C/CTM/LA0011/2011), the Portuguese National NMR Network (RNRMN) and the Centre for Cell Biology (PEst-OE/SAU/UI0482/2011, REEQ/1023/BIO/2005). SG also thanks the FCT for a postdoctoral grant (SFRH/BPD/70702/2010). References: 1. a) Vendrell, M.; Lee, J. S.; Chang, Y. T. Curr. Opin. Chem. Biol. 2010, 14, 383; b) Kang, N. Y.; Ha, H. H.; Yun, S. W.; Yu, Y. H.; Chang, Y. T. Chem. Soc. Rev. 2011, 40, 3613. 2. Lee, S. -C.; Kang, N.- Y.; Park, S. -J.; Yun, S. -W.; Chandran, Y.; Chang, Y.-T. Chem. Commun. 2012, 48, 6681. 3. Guieu, S.; Rocha, J.; Silva, A. M. S. J. Mol. Struct. 2013, 1035, 1.

PC154
Carbohydrate-derived compounds as dual inhibitors for
Acetylcholinesterase and Aβ aggregation
Rita Gonçalves-Pereira,a,b
Carlos Anjo,a Juan M. Benito,
b M. Isabel García-Moreno,
b
José M. García Fernández,c J. Albertino Figueiredo,
d M. Isabel Ismael,
d
Carmen Ortiz Mellet,b Amélia P. Rauter
a
aDpto. de Química e Bioquímica, Centro de Química e Bioquímica, Faculdade de Ciências, Universidade
de Lisboa, Ed. C8, 5ºPiso, Campo Grande, 1749-016 Lisboa, Portugal; bDpto. de Química Orgánica,
Facultad de Química, Universidad de Sevilla, C/Prof. García González 1, 41012 Sevilla, Spain; cInstituto
de Investigaciones Químicas, CSIC – Universidad de Sevilla, Avda. Américo Vespucio 49, 41092 Sevilla,
Spain; dDpto. de Química, Unidade I&D Materiais Têxteis e Papeleiros, Universidade da Beira Interior,
6201-001 Covilhã, Portugal
Alzheimer’s disease (AD) is the most common neurodegenerative disease in ageing. Thus,
there is great interest in developing new therapeutic strategies that target the molecular
mechanisms that cause the disease in order to delay or even stop disease progression.
Acetylcholinesterase inhibitors were the first group of compounds that showed some
promise in the treatment of AD and have been effective for the control of mild to moderate
AD.1
However, actuality, the dominant hypothesis in research of AD is the amyloid cascade
hypothesis, that suggests the accumulation of high levels of β-amyloid peptide in the brain
is responsible for triggering a sequence of events that ultimately lead to neuronal death and
consequently dementia. The literature describes that the prevention of Aβ aggregation can
be accomplished using a variety of inhibitors such as small organic molecules like
carbohydrates scaffolds.2 On the other hand, recently our research group has synthesized
carbohydrate derived compounds that are potent cholinesterase inhibitors. For this reason,
we have applied the concept to the design of dual inhibitors, and we have now synthesized
compounds (1-4) (Figure 1) starting from D-glucose and evaluated their inhibitory activity
against acetylcholinesterase and computational analysis (docking) with β-Secretase.
Preliminary results reveals that this type of compounds behaved as moderate inhibitors of
acetylcholinesterase (ranged 35% to 58%), showing the docking studies an interaction with
aspartic acid 32 that can relate to biological activity.
Figure 1: Structures of carbohydrate–based dual inhibitors.
References:
1. Figueiredo, J. A.; Ismael, M. I.; Pinheiro, J. M.; Silva, A. M. S.; Justino, J.; Silva, F. V. M.; Goulart, M.; Mira, D.; Araújo, M. E. M.; Campoy, R.; Rauter, A. P. Carbohydr. Res. 2012, 347. 2. (a). Lee, S.; Lim, H.; Masliah, E.; Lee, H. Neuroscince Res. 2011, 70, 339.(b) Liu, R.; Barkhordarian, H; Emadi, S.; Park, C. B.; Sierks, M. R. Neurobiology of Disease, 2005, 20, 74.

PC155
α-Aminophosphonic Acid Diesters, Synthesis, NMR
Characterization, In Vitro Antitumor Evaluation And Safety Testing
I. Kraicheva,a I. Tsacheva,
a E. Vodenicharova,
a A. Bogomilova,
a G. Momekov,
b A. Kril,
c
I. Iliev,c M. Topashka-Ancheva,
d T. Gerasimova,
d I. Ivanov,
c K. Troev
a
aInstitute of Polymers, Bulgarian Academy of Sciences, 1113 Sofia, Bulgaria;
bDepartment of
Pharmacology, Pharmacotherapy and Toxicology, Faculty of Pharmacy, Medical University-Sofia, 1000
Sofia, Bulgaria; cInstitute of Experimental Morphology, Pathology and Anthropology with Museum,
Bulgarian Academy of Sciences, 1113 Sofia, Bulgaria; dInstitute of Biodiversity and Ecosystems
Research, Bulgarian Academy of Sciences, 2 Gagarin Street, 1113 Sofia, Bulgaria
Aminophosphonates, structural analogues of natural α- aminocarboxylic acids, constitute a
valuable class of compounds with a wide spectrum of biological activity, including treatment
of metabolic disorders and cancer.
The pharmacological importance and utility of aminophosphonate derivatives have
stimulated extensive studies on various aspects of their chemistry and biochemistry:
synthetic routes, structural and spectral characterization and evaluation of their biological
properties. Among the numerous synthetic approaches to aminophosphonates, the addition
of dialkyl phosphites to Schiff bases in the presence of sodium alkoxide and Lewis acids is
the most convenient procedure. The synthesis of three novel α-aminophosphonic acid
diesters N,N-dimethyl-[N'-methyl(diethoxyphosphonyl)-(2-furyl)]-1,3-diaminopropane, p-[N-
ethyl(diethoxyphosphonyl) -(2-furyl)]toluidine and p-[N-ethyl(diethoxyphosphonyl)-(4-
dimethylaminophenyl)]toluidine through an addition of diethyl phosphite to N,N-dimethyl-N'-
furfurylidene-1,3-diaminopropane, N-furfurylidene p-toluidine and N-(4-
dimethylaminobenzylidene)-p-toluidine, respectively, is reported.
The synthesized α- aminophosphonates were characterized by elemental analysis, IR and
NMR (1H,
13C and
31P) spectra. The compounds were tested for antiproliferative effects
against 4 human leukemic cell lines, namely LAMA-84, K-562 (chronic myeloid leukemias),
HL-60 (acute promyelocyte leukemia) and HL-60/Dox (multi-drug resistant sub-line,
characterized by overexpression of MRP-1 (ABC-C1)) and were found to exert
concentration dependent cytotoxic effects. A representative aminophosphonate compound
was shown to induce oligonucleosomal DNA fragmentation. That implies that the induction
of cell death through apoptosis plays important role for its cytotoxicity mode of action.
In vitro antitumor activity of N,N-dimethyl-[N'-methyl(diethoxyphosphonyl)-(2-furyl)]-1,3-
diaminopropane towards a panel of epitel human cancer cell lines is reported. Safety testing
of the compound was performed both in vitro (3T3 NRU test) and in vivo on ICR mice for
genotoxicity and antiproliferative activity.
Acknowledgements: We thank the Bulgarian National Science Fund: Contract DTK 02/34 for providing financial support.

PC156
New synthetic approach to prolinemimetics of PLG
derived from 2-azabicyclo[2.2.1]heptane system
Ivo E.Sampaio-Dias,a José E. Rodriguez-Borges,
a Xerardo García-Mera
b
aCIQ - Departamento de Química e Bioquímica, Faculdade de Ciências da Universidade do Porto, Rua
do Campo Alegre, 4169-007 Porto, Portugal; bDepartamento de Química Orgánica,
Facultad de
Farmacia, Praza Seminario de Estudos Galegos (Campus Vida), 15782 Santiago de Compostela, España.
The tripeptide L-prolyl-L-leucylglycinamide (PLG) it's the endogenous tripeptide derivated
from the clivage of the hormone oxytocin.1,2
PLG takes important roles in the central
nervous system reveling to provide neuroprotection both in vitro and in vivo in pathologies
such as Parkinson's syndrome, tardive dyskinesia induced by antipsychotic drugs and
depression.3,4
Previous research points that the neuroprotective effects of this natural
tripeptide are predominantly associated to the proline amino acid residue.5 Therefore, it
becomes attractive and imperative to develop prolinemimetics analogs of this neuropeptide
in order to study the way that subtle modifications in this residue can contribute to the
development of new pharmaceuticals with greater neuroprotective activity.
In this work we synthesized three sub-classes of tripeptides analogs of the one described
before. Here we proposed convergent synthetic routes leading to the preparation of the
tripeptides P*LG, P*GL and P*VA (Figure 1) by introduction of a proline mimetic unit
resultant from methyl 2-azabicyclo[2.2.1]hept-5-ene-3-carboxilate by aza-Diels-Alder
reaction, between the imine of methyl glyoxylate and CPD through a synthesis
methodologies well known in our research group at CIQ-UP.6 The synthesis of
corresponding prolinemimetics tripeptides may come to grant an enormous contribution in to
the discovery of new potential therapeutic agents for the treatment of neurodegenerative
diseases opening ways for the development of more prevailing pharmaceuticals through a
quick and efficient route. Biological appraisal of the prolinemimetics coumponds analogs of
PLG will be held in USC.
Figure 1: Synthesized prolinemimetics coumpounds analogs of natural PLG. References: 1. Celis, M. E.; Taleisnik, S.; Walter, R., Proceedings of the National Academy of Sciences of the United States of America, 1971, 68 (7), 1428–33. 2. Petersson, M.; Uvnäs-Moberg, Physiology & Behavior, 2004, 83 (3), 475–81. 3. Verma, V.; Mann, A.; Costain, W.; Pontoriero, G.; Castellano, J., M.; Skoblenick, K.; Gupta, S. K.; Pristupa, Z.; Niznik, H. B.; Johnson, R. L.; Nair, V. D.; Mishra, R. K., J. Pharmacol. Exp.Ther., 2005, 315, 1228-1236. 4. Mann, Amandeep; Verma, V.; Basu, D.; Skoblenick, K, J.; Beyaert, M. G. R.; Fischer, A.; Thomas, N.; Johnson, R. L.; Mishra, R. K., European Jornal of Pharmacology, 2010, 641, 96-101. 5. Diego, S. A. A.; Muñoz, P.; González-Muñiz, R.; Herranz, R.; Martín-Martínez, M.; Cenarruzabeitia, E.; Frechilla, D.; Río, J.; Jimeno, M. L.; García-López, M. Teresa, Bioorganic & Medicinal Chemistry Letters, 2005, 15, 2279-2283. 6. Rodriguez-Borges, J. E.; Vale, M. L. C.; Aguiar, F. R.; Alves, M. J.; García-Mera, X., Synthesis, 2008, 6, 971-977.

PC157
Quantification of phenolic compounds of corkable to migrate to
hydroalcoholic solution - Intestinal absorption and biological
properties.
J. Azevedo,a I. Fernandes,
a P. Lopes,
b I. Roseira,
b M. Cabral,
b Nuno Mateus,
a
Victor de Freitasa
aCIQ- Centro de Investigação em Química, Faculdade de Ciências da Universidade do Porto, Rua do
Campo Alegre, 687, 4169-007 Porto, Portugal; bAmorim e Irmãos S.A. Mozelos VFR, Portugal
Cork is a suberized cellular tissue that is continuously produced by the phellogen of the cork
oak tree (Quercus suber L.) native species of the Mediterranean region. The unique
properties of this material, allowed its use in a wide range of applications, wine cork
stoppers is still its most common and valued use.1,2
Considering the enological interest of cork, this study aimed to identify and quantify, over
time (27 months), the different phenolic compounds which are able tomigrate from different
cork stoppers into bottled wine model solutions.
The compounds found in larger quantities were the acids and aldehyde phenols such as
gallic acid, protocatechuic acid, protocatechuic aldehyde, cafeic acid, vanillin, sinapic acid,
ferrulic acid and elagic acid (Figure 1). Trace amounts of more complex polyphenols such
as mongolicain A/B, valoneic acid, valoneic acid dilactone, ellagic acid-pentose,
castalagin/vascalagin, HHDP-galloyl-glucose, di-HHDP-galloyl-glucose were also
presented.
The intestinal absorption of the compounds previously identified in each wine model
solutions after 27 months of bottled was evaluated using caco-2 cell line model. The simpler
phenolic compounds were able to cross this intestinal epithelium cell model in percentages
ranging from 20 to 30%.
The antiproliferative activity of the same wine model solutions was also evaluated against
gastric and breast cancer cell lines. All samples were active against the two cell lines, which
increases the possible health benefits of cork sealed wine.
Gallic Acid Vanilin Protocatechuic
acid Cafeic acid Sinapic acid
Figure 1: Structure of some of the identified compounds. Acknowledgements: This work was supported by FCT (Fundação para a Ciência e Tecnologia) (POCI, FEDER, POPH, QREN) by the studentship grant (SFRH/BPD/86173/2012). The authors also want to thank BIOCORK Project nº 11430 for the financial support. References: 1. Casey J. Aust. Grapegrow. Winemak. 1994, 372, 39-41. 2. Jung R.; Hamatscheck J. Wein-Wiss 1992, 47, 226-234.

PC158
Antioxidant and anti-inflammatory activity of Franciscan Friars Aloe
syrup
Liliana M. L. Silva, M. Eduarda M. Araújo
Center of Chemistry and Biochemistry, Chemistry and Biochemistry Department, Faculty of Sciences,
University of Lisbon, Campo Grande Ed. C8, 1749-016 Lisbon, Portugal
The syrup of Aloe barbadensis Miller, usually known as Aloe vera (A.v.), is widely used as a
home remedy in Portugal and Brazil. It is produced from a Franciscan Friars old recipe and
there is some evidence of improvement of inflammatory pathologies. The syrup consists of
A.v. leaves, honey and brandy. The plant has anti-inflammatory and anticancer activity as
well as antioxidant activity like honey and brandy.
In this work we present the evaluation of the antioxidant activity of the acetone extract of
syrup as it is marketed by the Franciscan friars and another acetone extract syrup prepared
in the laboratory according to the recipe also provided with the former. Antioxidant activity
was evaluated by the inhibition of iron (III) to iron (II) reduction, metal chelating activity of
the syrup and DPPH assay and also of their components, honey, Aloe vera and brandy,
using a methodology described in literature.1,2
We present the results of the acetone extract
of this syrup, once it’s the most active extract of this remedy.
We found that there was some differences between the syrup marketed and the syrup
prepared in the laboratory:
inhibition of iron (III) to iron (II)reduction – friars syrup = 0.233 ± 0.009 mg AAE/mg dry
extract; laboratory prepared syrup = 0.170 ± 0.005 mg AAE/mg dry extract
metal chelating activity –friars syrup IC50 = 20.15 ± 1.90 mg/mL; laboratory prepared syrup
IC50 = 1.732 ± 0.615 mg/mL
DPPH assay – friars syrup = 0.0005 ± 0.0001 mg Trolox/mg dry extract; laboratory
prepared syrup =0.0006 ± 0,0001 mg Trolox/mg dry extract
Anti-inflammatory activity of the acetone extract of friars syrup was also investigated.
Evaluation was assessed by an in vitro assay3 involving the inhibition of cyclooxygenase
(COX, or Prostaglandin-H synthase, PGHS), a key enzyme in the synthesis of prostaglandin
H2, which is a precursor for the biosynthesis of prostaglandins, thromboxanes, and
prostacyclins. We found that this extract inhibited COX-1 enzyme in a dose dependent
manner: 26.7 ± 6.7% (0.500 mg.mL-1), 71.1 ± 3.8% (0.800 mg.mL
-1) and 98.0 ± 3.5% (1000
mg.mL-1).
These preliminary results encourage the further study of this promising active folk remedy.
References: 1. Dastmalchi, K., Dorman, D., Oinonen, P., Darwis, Y., Laakso, I., Hiltunen, R., LWT Food Science and Technology, 2008,41,391-400; 2. A. T. Mata, C. Proença, A. R. Ferreira, M. L. M. Serralheiro, J. M. F. Nogueira, M. E. M. Araújo. Food Chemistry, 2007, 103, 778–786. 3. James K. Gierse, Carol M. Koboldt, Mark C. Walker, Karen Seibert, Peter C. Isakson. Biochem.J., 1999, 339, 607.

PC159
Benzothiazolium Salt as a New Ligand in Affinity Chromatography
for Protein Purification
L. P. Alves,a,b
S. S. Ramos,a,c
R. E. F. Boto,a,b
F. Sousa,a P. Almeida
a,b
aCICS-UBI - Health Sciences Research Centre, University of Beira Interior, 6200-506 Covilhã, Portugal;
bDepartment of Chemistry, University of Beira Interior, 6201-001 Covilhã, Portugal;
cUMTP-UBI - Unit of
Textile and Paper Materials, University of Beira Interior, 6200-001 Covilhã, Portugal
A new support for Affinity Chromatography (AC) was synthesized from Sepharose CL-6B
containing a benzothiazolium salt as a ligand. These salts are common precursors in
cyanine dyes synthesis.1
Nevertheless, cyanine dyes have already been evaluated as ligand for AC as a whole, the
study of the contribution of each part of these dyes has never been done. Therefore, the
study of benzothiazolium salt as ligand for AC is herein presented, as part of our efforts to
clarify its influence on proteins purification.
The immobilization of this salt was performed by an esterification reaction, using
DCC/DMAP in DMF (Figure 1) in alternative of the use of a curing method as already
described,2 which is not effective for compounds that decompose at the used temperatures.
The AC support so obtained revealed the ability to promote selective interactions allowing
the separation of three standard proteins from an artificial mixture, namely lysozyme, α-
chymotrypsin and bovine serum albumin (BSA), using an increasing sodium chloride
stepwise gradient. Also, the application of a decreasing ammonium sulfate gradient has
been studied. In conclusion, this study allows estimating and better understanding the role
of the benzothiazolium moiety in the biomolecules purification achieved by AC based on
cyanine dyes, as well as the potential use of these salts as ligands for AC.
Figure 1: Preparation of AC Sepharose CL-6B with benzothiazolium salt as ligand.
Acknowledgements: This work was financed by FCT (Project PTDC/QUI-QUI/100896/2008) and COMPETE (Project PEst-C/SAU/UI0709/2011). L. P. Alves acknowledge the scholarship awarded (BI-1-PTDC/QUI-QUI/100896/2008). References:
1. Boto, R. E. F.; El-Shishtawy, M.; Santos, P. F.; Reis, L. V.; Almeida P. Dyes Pigments, 2007, 73, 195-205. 2. Boto, R. E. F.; Almeida, P.; Queiroz, J. A. Biomed. Chromatogr. 2008, 22, 278-288.

PC160
Synthesis and Singlet Oxygen Evaluation of New Unsymmetrical
Squarylium Cyanine Dyes
S. G. Fagundes,a D. P. Ferreira,
b D. S. Conceição,
b R. E. F. Boto,
c P. Almeida,
c
L. F. V. Ferreira,b L. V. Reis
a
aCQ-VR, Universidade de Trás-os-Montes e Alto Douro, Quinta dos Prados, Apartado 1013, 5001-801,
Vila Real, Portugal; bCQFM, Centro de Química-Física Molecular, Complexo Interdisciplinar, Instituto
Superior Técnico,1049-001 Lisboa, Portugal cCICS-UBI, Universidade da Beira Interior, 6201-001
Covilhã, Portugal.
Photodynamic therapy (PDT) is a treatment that, to be effective, requires three elements:
visible or near infrared radiation, the administration of a photosensitizer and the presence of
molecular oxygen. The basic principle of PDT involves the generation of highly toxic and
reactive oxygen (mainly singlet oxygen) species upon excitation of the sensitizer by light
absorption. The phototoxic specie called singlet oxygen is responsible for cell dead. The
high selectivity to the destruction of tumor cells over normal cells has made PDT an
attractive alternative to the traditional cancer therapies.1
A desirable sensitizer for PDT should fulfill some parameters such as: be inactive in the
dark and active only when irradiated with light of an appropriate wavelength, it should have
strong absorption at longer wavelength region, ideally in the 600-900 nm, region also called
“phototherapeutic window”, wherein the tissue penetration by light is higher; It should also
possess high triplet quantum yields with long lifetimes; it should be able to generate reactive
species such as singlet oxygen in quantitative yields and should be rapidly excreted from
the body.2
In this work we present the synthesis of new unsymmetrical squarylium cyanine dyes, that
really absorb strongly within the “phototherapeutic window”, and their singlet oxygen
generation ability (Figure 1). Dyes were obtained with good yields by methods adapted
from literature.3 Based on the values obtained for the quantum yield of singlet oxygen
production, we can say that some of these dyes are promising candidates to be used as
photosensitizers in PDT.
Yield
1 (71%)
2 (89%)
3 (75%)
4 (90%)
5 (84%)
6 (97%)
7 (78%)
X
-
-
I
I
I
I
I
Y
H
I
I
I
I
I
I
W
O-
O-
NH2
NHMe
NHPh
3-I-NHPh
NHCH2CH2OSO3-
λmax
683.6
689,3
680,2
690,5
679,9
688,8
689,3
log ε
5.34
5.32
5.28
5.32
5.35
5.27
5.32
φΔ
0.02
0.14
0.22
0.17
0.18
0.26
0.16
Figure 1: New unsymmetrical squarylium cyanine dyes. Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for partial financial support (project PTDC/QUI-QUI/100896/2008) and COMPETE (Project Pest-C/SAU/UI0709/2011). References: 1. Bonnet R., in Chemical Aspects of Photodynamic Therapy, Gordon and Breach Science Publishers, The Netherlands, 2000. 2. Avirah R. R.; Jayaram D. T.; Adarsh N.; Ramaiah D. Org. Biomol. Chem. 2012, 10, 911.
3. a) Santos P. F.; Reis L. V.; Duarte I.; Serrano J. P.; Almeida P.; Oliveira A. S.; Ferreira L. F. V. Helvetica Chimica Acta. 2005, 88, 1135. b) Kim S.; Mor G. K., Paulose M.; Varghese O. K.; Baik C.; Grimes C. A. Langmuir 2010, 26, 1348.

PC161
Novel epi-Cinchonine-Triazole Derivatives: Synthesis and
Evaluation of Biological Activity
Anthony J. Burke, Ana T. Caldeira, Pedro Barrulas, Luís Alves
Departamento de Química e Centro de Química de Évora, Escola de Ciências e Tecnologia da
Universidade de Évora, Rua Romão Ramalho 59, 7000-671 Évora, Portugal
Cinchona alkaloids are secondary metabolites found in the bark of the trees of the
Cinchona species, along with quinine, notorious for its antimalarial properties. Although the
latter is the most effective and most used in antimalarial treatment,1 the synthesis of new
cinchona derivatives could possibly bring about more effective agents. Triazole (red,
Scheme 2) formation could then potentiate the natural effects of these compounds, as well
as add its own biological activity and HIV-1 protease inhibition.2 A small library of epi-
cinchonine-triazoles (2, Scheme 1) were formed through the Huisgen 1,3-dipolar
cycloaddition,3 the final step in a series of three reactions starting with the selected raw
natural product, cinchonine (1, Scheme 1). Biological assays were conducted to determine
the antifungal and antimalarial activities of the newly formed compounds.
Scheme 1: Target synthesis of cinchonine-triazole hybrid compounds.
Acknowledgements: We thank Philip Rosenthal and Jiri Gut of the UCSF for their contribution on the antimalarial tests and the Fundação para a Ciência e Tecnologia for financial support. References: 1. Dewick P. in Medicinal Natural Products: A Biosynthetic Approach, John Wiley and Sons Ltd, 2002,
pp. 362-364 2. Wu P.; Fokin V.V. Aldrichimica Acta 2007, 40, 7-17 3. Rostovtsev V.V.; Green L.G.; Fokin V.V.; Sharpless K.B.; Angewandte Chemie 2002, 41, 2596-2599.

PC162
Valorization of Diterpenes Isolated from Cistus ladaniferus
L. M. T. Frija,a,b
H. Garcia,c C. Rodrigues,
c I. Martins,
c Nuno R. Candeias,
d Vânia André,
e
M. T. Duarte,e C. S. Pereira,
c,f Carlos A. M. Afonso,
b Raquel F. M. Frade,
b P. Fernandes
g
aCQFM, Centro de Química-Física Molecular, IN-Institute of Nanosciences and Nanotechnology, Instituto
Superior Técnico,1049-001 Lisboa, Portugal; biMed.UL, Faculdade de Farmácia da Universidade de
Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa, Portugal; cITQB, Universidade Nova de Lisboa, Av. da
República 2780-157, Oeiras, Portugal; dTampere University of Technology, Department of Chemistry and
Bioengineering, Tampere, Finland eCQE, Departamento de Engenharia Química, Instituto Superior
Técnico, 1049-001 Lisboa, Portugal; fIBET, Apartado 12, 2781-901, Oeiras, Portugal;
gIBB Institute for
Biotechnology and Bioengineering, Centre for Biological and Chemical Engineering, Instituto Superior
Técnico, 1049-001 Lisboa, Portugal
Small molecule natural products continue to hold the interest and attention of a wide
chemical community.1 This is mainly due to their biological properties and potential
applicability in health care and plant protection, as well as their frequently complex
molecular architectures and arrays of functionality, which make them a challenge for total
synthesis and a testing ground for novel methodology. As a huge class of natural products,
labdane-type diterpenes are an excellent example in terms of important applications in
medicine.2
Our interest in the study of labdane-type diterpenes emerged due to the possibility of
isolation of appreciable quantities of a specific diterpene, the labdanolic acid (1, LA), from a
readily available natural resource, i.e., the plant Cistus ladaniferus (rock rose). Throughout
this investigation, a novel approach for the extraction of LA from Cistus ladaniferus plant
was primarily developed. Subsequently, from LA, several analogues were prepared via
chemical transformation or biotransformation, including other natural products.3 Biological
assays of these analogues were done by using different tumor cell lines. Besides, apart
from the labdanolic acid, another diterpene, namely 6-oxocativic acid (2), was isolated from
Cistus ladaniferus evidencing activity towards Mycobacterium XP. Some of the molecules
presented in this communication were synthesized for the first time.
Figure 1: (left) Selected analogues of labdanolic acid (1); (right) 6-oxocativic acid (2).
Acknowledgements: Generous financial support from the Fundação para a Ciência e Tecnologia (FCT) and FEDER (PTDC/QUI/73061/2006, SFRH/BPD/43853/2008, SFRH/BD/38378/2007 and SFRH/BD/40474/2007) and Fundação Calouste Gulbenkian (21-95587-B) is gratefully acknowledged. References: 1. Newman, D. J.; Cragg, G. M. J. Nat. Prod. 2007,70, 461-477. 2. Frija, L. M. T.; Frade, R. F. M., Afonso, C. A. M. Chem. Rev. 2011,111, 4418-4452. 3. Frija, L. M. T.; Garcia, H.; Rodrigues, C.; Martins, I.; Candeias, N. R.; André, V.; Duarte, M. T.; Pereira, C. S.; Afonso, C. A. M. Phytochem. Lett. 2013, 6, 165-169.

PC163
Anti-tumor activity of diterpenoids against multidrug resistant
phenotypes
M. Reis,a A. Paterna,
a H. Lage,
b M. J. U. Ferreira
a
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal; bCharité Campus Mitte, Institute of Pathology, Berlin, Germany
The natural occurring diterpenic compounds with the jatrophane and lathyrane macrocyclic
scaffolds have been regarded as anti-tumor agents by multidrug resistance reversion and
apoptosis induction. Moreover, other class of diterpenes, namely ent-abietane lactones also
showed to have a selective anti-proliferative activity against cancer cells presenting a
multidrug resistant phenotype. These natural products are commonly isolated from plants
from Euphorbia genus. As many of this type of compounds have shown interesting
biological activities, the main goal of this work was to conduct a phytochemical study on the
plant Euphorbia piscatoria, in order to discover new diterpenes and select the most potent
compounds against multidrug resistant phenotypes for further chemical derivatization. This
strategy will allow the study of possible structure-activity relationships towards the
understanding of the underlying mechanisms of such selective multidrug resistant anti-
proliferative activity.
Euphorbia piscatoria, an endemic species from Madeira archipelago, has never been
studied for its diterpenes. Therefore, the aerial parts of this plant were exhaustively
extracted with methanol resulting on the isolation of four new diterpenes: two ent-abietane
and two lathyrane-type diterpenes. Furthermore, several known compounds were isolated,
namely, ditepenoids, a triterpene and phenolic compounds. The structures of the
compounds were characterized by spectroscopic methods mainly 1D NMR (1H,
13C, DEPT),
2D NMR (COSY, HMBC, HMQC, NOESY) and by comparison with literature data. One of
the new ent-abietane diterpenes seems to be an intermediate compound on the biogenesis
of helioscopinolide E, therefore a possible biogenetic pathway will be proposed.
From the isolated compounds, a macrocyclic lathryane showed the highest activity against
the multidrug resistant variants of the human cancer cell lines: EPP85-181 (pancreatic),
EPG85-257 (gastric) and HT-29 (colon). Consequently, was chosen for chemical
derivatizationin order to generate a small library of bioactive compounds.
Acknowledgements: This study was supported by FCT, Portugal (project PTDC/QUI-QUI/099815/
2008; PEst-OE/SAU/UI4013/2011; PTDC/QEQ-MED/0905/2012; PhD grant SFRH/BD/72915/2010).

PC164
Isobacteriochlorin derivative of 5,10,15,20-
tetrakis(pentafluorophenyl)porphyrin on the photodynamic
inactivation of bioluminescent E. coli
Mariana Mesquita,a José C. J. M. D. S. Menezes,
a Maria G. P. M. S. Neves,
a
Augusto C. Tomé,a José A. S. Cavaleiro,
a Ângela Cunha,
b Adelaide Almeida,
b
Maria A. F. Faustinoa
aDepartment of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal;
bDepartment of Biology & CESAM, University of Aveiro, 3810-193 Aveiro, Portugal
Microbial photodynamic inactivation (PDI) represents a potential alternative methodology to
inactivate microbial cells and has already shown to be effective in vitro against bacteria,
fungi, viruses and protozoa.1 The PDI approach is based on the photodynamic therapy
(PDT) concept that comprises the action of three components: a photosensitizing agent,
light of an appropriate wavelength and dioxygen.1,2
Porphyrins and their analogues such as
chlorins, bacteriochlorins, or isobacteriochlorins, have unique photophysical properties
which make them good candidates as photosensitizing agents.3 Chlorins, bacteriochlorins
and isobacteriochlorins are distinguished from the parent porphyrins by the presence of
reduced peripheral double bonds and this change in symmetry leads to strong changes in
the corresponding visible spectrum.3,4
Herein we report the results obtained in the photodynamic inactivation of bioluminescent E.
coli in the presence of neutral (1) and cationic isobacteriochlorin (2) derivatives obtained
from 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin. The experimental procedure and their
photophysical characterizations will also be presented and discussed.
Acknowledgements: Thanks are due to the University of Aveiro, Fundação para a Ciência e a Tecnologia (FCT, Portugal), European Union, QREN, FEDER and COMPETE for funding the QOPNA research unit (project PEst-C/QUI/UI0062/2011), the Portuguese National NMR network and CESAM. References: 1. M Tavares, A.; Carvalho, C. M. B.; Faustino, M. A.; Neves, M. G. P. M. S.; Tomé, J. P. C.; Tomé, A. C.; Cavaleiro, J. A. S.; Cunha, A.; Gomes, N. C. M.; Alves, E.; Almeida, A. Mar. Drugs. 2010, 8, 91-105. 2. Almeida, A.; Cunha, Â.; Faustino, M. A. F.; Tomé, A. C.; Neves, M. G. P. M. S. in Photodynamic inactivation of microbial pathogens: medical and environmental applications, Royal Society of Chemistry, 2011, p. 83. 3. Kadish, K.M.; Smith, K. M.; Guilard, R., in The Porphyrin Handbook, Academic Press, 2000, vol.6.
4. Silva, A. M. G.; Tomé, A. C.; Neves, M. G. P. M. S.; Silva, A. M. S.; Cavaleiro J. A. S. Chem. Commun., 1999, 1767.

PC165
Probing the Chemical Space Around Aurone Scaffold – Improving
Antimalarial Activity
M. P. Carrasco,a Jiri Gut,
b Lídia M. Gonçalves,
a D. J. V. A. dos Santos,
a,c
Philip J. Rosenthal,b Rui Moreira
a
aResearch Institute for Medicines and Pharmaceutical Sciences (iMed.UL), Faculty of Pharmacy,
University of Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa, Portugal, bDepartment of Medicine, San
Francisco General Hospital, University of California, San Francisco, CA 94143-0811, USA, cPresently at
REQUIMTE, Department of Chemistry & Biochemistry, Faculty of Sciences, University of Porto, R. do
Campo Alegre, 4169-007 Porto, Portugal
Malaria is responsible for causing an estimated 225 million clinical cases and one million
deaths annually1 being drug resistance to currently established antimalarial drugs such as
chloroquine (CQ) a major problem of concern. Therefore, novel and innovative inhibitors
active against Plasmodium falciparum, are urgently required to develop new treatments able
to fight malaria.2
Aurones are secondary metabolites belonging to the flavonoids family and their antimalarial
activity was already recognized.3 More recently, it was shown that the mechanism of action
of this family is most likely a CQ-like action, i.e., by inhibiting the hemozoin formation inside
the acidic digestive vacuole of the parasite.4
In an attempt to obtain new potent antimalarial agents and explore the chemical space
around this scaffold, a library of novel aurone derivatives was synthesized by introducing an
additional aromatic moiety through Suzuki–Miyaura and Buchwald–Hartwig cross-coupling
reactions. Further studies were also made to evaluate the substitution of the intracyclic
oxygen in this scaffold by a nitrogen atom (Scheme 1). The synthetic procedures applied
and some preliminary results will be presented and discussed.
Scheme 1: General structure of the synthesized compounds.
Acknowledgements: This work was financially supported by Fundação para a Ciência e Tecnologia (FCT, Portugal) through projects PTDC/SAU-FCF/098734/2008 and PEst-OE/SAU/UI4013/2011. FCT is also acknowledged for the PhD grant SFRH/BD/61611/2009. References: 1. WHO, World Malaria Report (2009), www.who.int; 2. Wu, T. et al, Curr. Med. Chem., 2011, 18, 6, 853-871; 3. Kayser, O. et al, Planta Med. 2001,67, 718-721; 4. Guiguemde, W. A. et al, Nature 2010, 465, 311-315.

PC166
Nitroimidazolyl hydrazones are better amoebicides than their
cyclized 1,3,4-oxadiazoline analogues: In vitro studies and
Lipophilic efficiency analysis
Mohmmad Younus Wani,a*
Abdul R. Bhat,b Amir Azam,
c Fareeda Athar
a
aCentre for Interdisciplinary Research in Basic Sciences, Jamia Millia Islamia, New Delhi-110025, India.
bSchool of Biotechnology,Yeungnum University, Gyeongshan, South Korea.
cDepartment of Chemistry, Jamia Millia Islamia (Central University), New Delhi-110025, India.
*Present address: Departamento de uımica, Universidade de Coimbra, Rua Larga, 3004-535 Coimbra, Portugal.
Design of a potential drug to treat infections caused by the intestinal protozoan parasite, Entamoeba histolytica, has remained an active area of research since decades.1 E. histolytica primarily lives in the colon as a harmless commensal, but is capable of causing devastating dysentery, colitis and liver abscess. More than 50 million people are estimated to suffer from the symptoms of amoebiasis such as hemorrhagic colitis and amoebic liver abscess resulting in 100,000 deaths annually. The 5-nitroimidazole drug metronidazole has long been used for the treatment of this disease, but there are concerns regarding its safety.2 Although metronidazole and its derivatives are currently employed in therapy, the paucity of effective drugs and potential clinical resistance necessitate the development of a novel drug with a good therapeutic activity and without any side effects. In this pursuit ample compounds have been synthesized and evaluated for their antiamoebic potency, but until now no compound reached clinical trials or achieved approval which is due to poor physicochemical properties associated with the compounds.1 In recent years the concepts of physicochemical properties and Lipophilic efficiency (LipE), which combines both “potency and lipophilicity,” have been shown to be useful tools in the lead optimization process. Overall physicochemical properties of drugs have been associated with bioavailability, however, to our knowledge, little has been done to examine possible correlations between compound physicochemical properties and in vitro outcomes. Within this study, we designed series of acylhydrazones (HZ1-HZl2, series 1) and their cyclized analogues (1,3,4-oxadiazolines) (OZ1-OZ12, series 2), of the 2-methyl-5-nitro-1H-imidazole scaffold; which is the core ring of many antiprotozoal drugs (Figure 1), for the purpose of finding better antiamoebic leads (Scheme 1). Physicochemical properties and lipophilic efficiency (LipE) analysis predicted higher intrinsic quality of the acylhydrazone derivatives (series 1) than their corresponding oxadiazoline analogues (series 2). In vitro antiamoebic results supported the above findings and validated that the acylhydrazone derivatives (HZ1-HZl2) show better activity than the oxadiazoline derivatives (OZ1-OZ12). MTT assay, using HepG2 cell line, revealed noncytotoxic nature of the compounds. The most promising results were observed for compound HZ5 (IC50 = 0.96 µM) and HZ9 (IC50 = 0.81 µM) both in silico and in vitro. Analysis of the Lipophilic Efficiency (LipE) of the compounds provided new insight for the design of potent and selective amoebicides.
Figure 1: Antiprotozoal drugs and the designed target compounds with a common 2-methyl- 5-nitro-1H-imidazole scaffold.
Scheme 1: Schematic representation of synthesis of acylhydrazones (Series 1) and their cyclized 1,3,4-oxadiazoline analogues (Series 2).
Acknowledgements: We thank Department of Science and Technology India (DST-India) for their financial support (Grant no. SR/FT/LS-069/2007).
References: 1. Singh S.; Bharti N.; Mohapatra P.P. Chem Rev. 2009,109,1900. 2. Wani M. Y.; Bhat A. R.; Azam A.; Choi I.; Athar F. Eur. J. Med. Chem. 2012, 48, 313.

PC167
Synthesis of Purine Nucleosides from Glucurono-6,3-lactone and
their Potential as Anti-Alzheimer Agents
Nuno M. Xavier,a Stefan Schwarz,
a,b René Csuk,
b Amélia P. Rauter
a
aGrupo da Química dos Glúcidos, Centro de Química e Bioquímica, Faculdade de Ciências da
Universidade de Lisboa, Campo Grande Edificio C8, 5º Piso, 1749-016 Lisboa, Portugal;
bBereich
Organische Chemie, Martin-Luther-Universität Halle-Wittenberg, Kurt-Mothes-Str. 2, D-06120 Halle
(Saale), Germany
The reduced concentration of the neurotransmitter acetylcholine (ACh) is a major feature of
Alzheimer's disease (AD). In this regard, acetylcholinesterase (AChE), which hydrolyses
ACh, has been a main drug target for AD. Butyrylcholinesterase (BChE), another major
cholinesterase (ChE), also hydrolyses ACh and its activity increases progressively in AD
patients.1
Hence, inhibitors of both ChEs may offer more sustained efficacy than AChE-
selective agents towards AD.
Our group previously showed the selective BChE nanomolar inhibition and low acute
toxicity of novel 2-acetamidopurine nucleosides comprising a bicyclic sugar moiety.2
Purine
derivatives have also been reported as dual AChE/BChEinhibitors.3
These aspects
prompted us to focus on the synthesis of new purine nucleosides for further evaluation of
their cholinesterase inhibitory activities.
In this communication we present the synthesis of compounds of types I-III, in which a
pyranose/furanose moiety or a bicyclic sugar lactone is linked to a purine via a C-N9- or a C-
N7-nucleosidic bond (Figure 1). Glucofuranurono-6,3-lactone was used as starting material
and converted into suitable 1-O-acetyl glycosyl donors, including furanosyl and
pyranosylglucuronoamide derivatives, for the subsequent Lewis acid-promoted coupling to
the silylated nucleobase. The results of the enzyme inhibition assays will also be disclosed.
Figure1 Acknowledgements: Fundação para a Ciência e Tecnologia (FCT) is gratefully acknowledged for the postdoctoral grants of N. M. Xavier and S. Schwarz and for financial support through the project PEst-OE/QUI/UI0612/2013. References: 1. Greig, N. H. et al. Int. Psychogeriatr. 2002, 14, 77-91 2. Marcelo, F. et al. Bioorg. Med. Chem. 2009, 17, 5106-5116. 3. Rodríguez-Franco, M. I. et al. Bioorg. Med.Chem. 2005, 13, 6795–6802.

PC168
Chiral Induced Cyclocondensation Reactions: A Versatile Approach
to Obtain a New Class of NMDAR Antagonists
Nuno A. L. Pereira,a Francesc X. Sureda,
b Mercedes Amat,
c Joan Bosch,
c
Maria M. M. Santosa
aiMed.UL, Faculty of Pharmacy, University of Lisbon, Av. Prof. Gama Pinto, 1649-003 Lisbon, Portugal;
bPharmacology Unit, Faculty of Medicine and Health Sciences, University of Rovirai Virgili, c./St Llorenç
21, t.43201 Reus, Tarragona, Spain; cLaboratory of Organic Chemistry, Faculty of Pharmacy and
Institute of Biomedicine (IBUB), University of Barcelona, Av. Joan XXIII, s/n, 08028 Barcelona, Spain
Chiral induced cyclocondensation reactions have been long described as a powerful tool to
allow chemists to access enantiopure scaffolds. These compounds are many times the
precursors of natural products with important biological activity profiles.1 Encouraged by our
latest results in the discovery of a new class of N-Methyl-D-Aspartate Receptor (NMDAR)
antagonists2 – a well know biological target for the treatment of neurodegenerative
pathologies such as Alzheirmer’s Disease – we describe here the enantiopure synthesis of
a series of (S) and (R)-phenylalaninol derived oxazolopyrrolidone lactams with moderate to
good yields (Scheme 1), bearing a wide range of substitution patterns and well defined
chiral centers. The stereospecific outcome of this reaction is controlled by a dynamic kinetic
resolution mechanism that generates enantiopure products which were evaluated as
NMDAR antagonists. These scaffolds present distinct chemical functions that are currently
being modulated to produce structure-activity relationships.
Scheme 1: Cyclocondensation reactions between racemic oxo-esters and enantiopure phenylalaninol. Acknowledgements: We thank Fundação para a Ciência e Tecnologia (Portugal) for project grants PTDC/QUI-QUI/111664/2009 and PEst-OE/SAU/UI4013/2011 and the Portuguese-Spanish-Integrated Action E-07/11 for financial support. References: 1. Newman D. J.; Craig G. M. J. Nat. Prod. 2012, 75, 311. 2. Pereira N. A. L.; Sureda F. X.; Turch M.; Amat M.; Bosch J.; Santos M. M. M. Monatsh. Chem. 2013, 144, 473.

PC169
Synthesis of α-mannopyranoside of 7α-acetoxy-6β-
hydroxyroyleanone
Patrícia Rijo,a,b
Clara Uriel,c Ana Gomez
c, Fátima Simões
b
aCBios, Universidade Lusófona de Humanidades e Tecnologias, Campo Grande 376, 1749-024 Lisboa,
Portugal; bFaculdade de Farmácia da Universidade de Lisboa, iMed.UL, Av. Prof. Gama Pinto 1649-003
Lisboa; cInstituto de Química Orgánica General, IQOG-CSIC, Juan de la Cierva 3, E-28006 Madrid,
Spain.
Cancer still remains one of the most devastating diseases with deep impact in the actual
world society. The influence of natural products leading drug cancer discovery has been
remarkable.1
Diverse therapeutic approaches, including the use of glycosides as anticancer
agents, have been developed given that a number of glycosylated natural products exhibit
high activity against a variety of human tumors. In fact, it has been described that linking a
glycosyde moiety to a given drug improves its efficacy and selectivity for cancer cells. Due
to their higher metabolic rate, the uptake of glycosylated drugs, probably mediated by
glucose transport proteins, would be much higher in cancer cells than in normal ones.2
Simões M. F. et al.3
discovered that 7-acetoxy-6-hydroxy-royleanone (1) (Figure 1)
isolated from Plectranthusgrandidentatus exhibited anticancer properties against MCF7
cells.4
In order to access to more potent antitumoral agents, royleanone derivative (2) was
obtained by esterification of the C-6 hydroxyl group of the lead royleanone (1) with
protected mannose (2R,3R,4S,5S,6R)-2-(acetoxymethyl)-6-(2,2,2-trichloro-1-iminoethoxy)
tetrahydro-2H-pyran-3,4,5-triyl triacetate (Figure 1). The novel compound was obtained and
fully characterized through spectroscopic and physicochemical methods. The glycosilated
derivative was obtained with very low yields and that is being improved. Further derivatives
are under study.4
Adjustment of the carbohydrate structures will, expectantly, have profound effect on the
molecular targeting and on the organism specificity of anticancer agents.2 Comparison of
the biological activities between the glycoside and its aglycon may reveal some structure-
effect correlations and express the advantage (or uselessness) of introducing glycosyl
moieties into this bioactive natural product (1).
Figure 1: α-mannopyranoside of 7α-acetoxy-6β-hydroxyroyleanone.
Acknowledgements: We thank Professor Benjamín Rodríguez for scientific expert advice. References: 1. Newman D. J. and Cragg G. M., Natural Products As Sources of New Drugs over the 30 Years from 1981 to 2010 Journal of Natural Products 2012 75 (3), 311-335. 2. La Ferla B., Airoldi C., Zona C., Orsato A., Cardona F., Merlo S., Sironi E., D'Orazio G., Nicotra F., Natural Glycoconjugates with Antitumor Activity, Nat. Prod. Rep., 2011, 28, 630–648. 3. Cerqueira F, Cordeiro-da-Silva A, Gaspar-Marques C, Simões M F, Pinto M. M. M., Nascimento M. S. J. Effect of Abietane Diterpenes from Plectranthusgrandidentatus on T- and B- Lymphocyte Proliferation. Bioorg Med Chem 2004; 12:217-23.
4. Uriel C., Ventura J., Gómez A. M., López J. C., Fraser-Reid B., Eur. J. Org. Chem., 2012, 3122-3131.

PC170
Resveratrol methoxyl dimer with antibacterial activity
Patrícia Máximo,a Alexandre Borges,
b Sara Monteiro,
b Ricardo Ferreira,
b Ana Lourenço,
a
Luísa M. Ferreiraa
aREQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de
Lisboa, 2829-516 Caparica, Portugal; bDisease & Stress Biology Laboratory, Instituto Superior de
Agronomia, Universidade Técnica de Lisboa, 1349-017 Lisboa, Portugal.
Plant secondary metabolism can provide a vast research source of natural/semi synthetic
bioactive compound. Stilbenoids among other phenolics represent a particular group of
interest.
Resveratrol (1), a stilbene, is biosynthesized in Vitis vinifera leafs as a response to infection,
being a phytoalexin. Prone to radical polimerization, several of its oligomers are known,
many of them possessing biological activity.1 One of these, trans--viniferin (2), was shown
to act as an antifungal metabolite in infected grapevine leafs.2
Reinvestigation of the previously reported method3
for the synthesis of trans--viniferin (2)
from resveratrol (1) allowed the isolation of the new compound resveratrol methoxyldimer
(3) as a diastereoisomeric mixture (18.9%, 3:1). Racemic trans--viniferin (2) was also
isolated (38.0%) together with an unidentified compound (14.0%) (Figure 1).
Antibacterial activity testing by the microdilution method assay4 of resveratrol (1), racemic
trans--viniferin (2) and resveratrol methoxyldimer (3) allowed the determination of MIC
values against Listeria monocytogenes, Staphylococcus aureus, Bacillus subtilis and
Enterococcus faecalis. Racemic trans--viniferin (2) is the most active, followed by
resveratrol methoxyldimer (3).
Figure 1: Structures of resveratrol (1), trans--viniferin (2) and resveratrol methoxyldimer (3).
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support (PTDC/AGR-PRO/112340/2009) and for the PhD grant SFRH/BD/61903/2009/J536313CQ5T3 (Borges, A). We thank the NMR facilities from REQUIMTE (Portugal) for NMR spectra acquisition. References: 1. Shen T.; Wang X. -N.; Lou H. -X. Nat. Prod. Rep. 2009, 26, 916. 2. Pezet R.; Perret C.; Jean-Denis J. B.; Tabacchi R.; Gindro K.; Viret O. J. Agric. Food Chem. 2003, 51,
5488. 3. Sako M.; Hosokawa H.; Ito T.; Iinuma M. J. Org. Chem.2004, 69, 2598. 4. Wiegand I.; Hilpert K.; Hancock R. E. W.; Nat. Protoc. 2008, 3, 163.

PC171
Stereoselective Synthesis of N-, S-, and C-Glycoside 8-epi-
Castanospermine analogues as Glycosidase Inhibitors
Rita Gonçalves-Pereira,a Elena M. Sánchez-Fernández,
a José M. García Fernández,
b
Carmen Ortiz Melleta
aDpto. de Química Orgánica, Facultad de Química, Universidad de Sevilla, C/Prof. García González 1, 41012 Sevilla, Spain;
bInstituto de Investigaciones Químicas, CSIC – Universidad de Sevilla, Avda.
Américo Vespucio 49, 41092 Sevilla, Spain.
Glycosidases are involved in a wide variety of pathological and biological processes.
Consequently, inhibitors of these enzymes exhibit a high therapeutic potential for the
treatment of diseases such as diabetes, bacterial and viral infections, cancer or lysosomal
storage disorders.1 For instance, (+)-castanospermine (1), a bicyclic iminosugar, is a potent
inhibitor of α- and β-glucosidases. Nonetheless, its lack of anomeric selectivity is largely
responsible for the failure of this iminosugar-type inhibitor in clinical trials. In order to
overcome this drawback, our research group has reported that ring-modified reducing
castanospermine analogues incorporating pseudoamide functionalities (sp2-iminosugars)
and α-oriented pseudoanomeric groups exhibit high α-anomeric selectivity in the inhibition
of glycosidases. Thus, by exploiting the ability of the nitrogen atom of oxazolidine-2-one to
act as a nucleophile in intramolecular addition to the masked aldehyde in D-galacto-
configured precursors, the 8-epi-castanospermine analogue (2) was synthesized in our
laboratories in the last few years.2 The inhibitory results obtained with this D-
galactopyranoside analogue underlined the critical influence of the hydroxylation profile
behaving as moderate competitive inhibitor of coffee beans α-galactosidase (Ki = 259 μM)
and exhibiting total α-anomeric selectivity (no inhibiton towards bovine liver β-
galactosidase). On the other hand, preliminary data from different configuration sp2-
iminosugars (D-gluco)3 showed that lipophilic aglycons resulted in amphiphilic compounds
which improved cell permeability and, at the same time, generated new interactions with
aminoacids at the active site of the enzyme. Bringing all these findings together, we present
in this communication the incorporation of axially-oriented substituents bearing an α-
configured N-, S-, or C-linked group at the pseudoanomeric position of the 8-epi-
castanospermine analogue (2). These new compounds (3-6) (Figure 1) have been assayed
against a panel of glycosidases allowing to evaluate structure/glycosidase inhibition
selectivity and potency relationships.
Figure 1: Structures of castanospermine, 8-epi-2-oxa-castanospermine derivative and N-, S- and C-
glycoside sp2-iminosugars.
Acknowledgements: This study was supported by the Spanish MINECO (contract numbers SAF2010-
15670 and CTQ2010-15848; co-financed by FEDER), and the Fundación Ramón Areces. References: 1. Nishimura, Y. In Iminosugars: From Synthesis to Therapeutic Applications, ed. Compain, P.; Martin, O. R. Wiley-VCH, Weinheim, Germany, 2007, 269. 2. Pérez, P. D.; García Moreno, M. I.; Mellet, C. O.; García Fernández, J. M. Eur. J. Org. Chem. 2005, 2903. 3. Sánchez Fernández, E. M.; Rísquez Cuadro, R.; Chasseraud, M.; Ahidouch, A.; Mellet, C. O.; Ouadid-Ahidouch, H.; García Fernández, J. M. Chem. Commun. 2010, 46, 5328.

PC172
Tetraoxane – pyrimidine nitrile hybrids as dual-acting antimalarials
R. Oliveira,a P. M. O’Neill,
b Rui Moreira,
a Francisca Lopes
a
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal; bDepartment of Chemistry, University of Liverpool, Liverpool, L69 3BX, United Kingdom
The urgent need of novel chemotherapeutic strategies to treat and control malaria requires
a continuous effort to discover new chemical entities endowed with potent activity against
multi-drug resistant Plasmodium falciparum strains.1,2
Herein we present the synthesis of
hybrid compounds containing tetraoxanes and pyrimidine nitrile scaffolds (1, Scheme 1).
These hybrids were designedto provide two distinct mechanisms of action, an essential
feature to avoid resistance in malaria treatment.3
Preparation of hybrid compounds 1
required a two-step synthesis of the tetraoxane moiety starting with adamantan-2-one and
the appropriate cyclohexanone, using hydrogen peroxide and rhenium oxide (VII) as oxidant
species. The different pyrimidine nitriles were obtained by a series of nucleophilic aromatic
substitutions performed on the appropriate dichloropyrimidines. Finally, amide coupling of
the two moieties gave the target compounds 1. In this communication we will discuss the
challenges faced through the synthetic pathway towards hybrids 1 and the in vitro activity
profiling against several P. falciparum strains.
Scheme 1:Retrosynthetic analysis for tetraoxane – pyrimidine nitrile hybrids1.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support (grants PEst-OE/SAU/UI4013/2011 and REDE/1501/REM/2005; fellowship SFRH/BD/63200/2009 to RO) and Fundação Luso-Americana (award to RM). References: 1) WHO, World Malaria Report, 2011 2) a) Vennerstrom J. L.; Fu H. -N.; Ellis W. Y.; Ager A. L.; Wood J. K.; Andersen S. L.;Gerena L.; Milhous W. K. J. Med. Chem. 1992, 35, 3023. b) Opsenica I.;Opsenica D.; Smith K. S.; Milhous W. K.; Šolaja B. A. J. Med. Chem. 2008, 51, 2261
3) Coterón, J. M.; Catterick, D.; Castro, J.; Chaparro, M. J.; Díaz, B.; Fernández, E.; Ferrer, S.; Gamo, F. J.; Gordo, M.; Gut, J.; de Las Heras, L.; Legac, J.; Marco, M.; Miguel, J.; Mu oz, .; Porras, E.; de La Rosa, J. C.; Ruiz, J. R.; Sandoval, E.; Ventosa, P.; Rosenthal, P. J.; Fiandor, J. M. J. Med. Chem. 2010, 53, 6129.

PC173
New Unsymmetrical Squarylium Cyanine Dyes as Potential
Photosensitizers for Cancer Photodynamic Therapy
S. G. Fagundes,a L. V. Reis,
a J. R. Fernandes,
b R. E. F. Boto,
c P. Almeida,
c A. M. Silva
d
aCQ-VR, Universidade de Trás-os-Montes e Alto Douro, Quinta dos Prados, Apartado 1013, 5001-801,
Vila Real, Portugal; bINESC-TEC and Departamento de Física, UTAD, Portugal;
cCICS-UBI,
Universidade da Beira Interior, 6201-001 Covilhã, Portugal dCITAB, Universidade de Trás-os-Montes e
Alto Douro, Vila Real, Portugal
Photodynamic Therapy is a medical treatment used as a very attractive alternative to
traditional cancer therapies. This technique involves the administration of a photosensitizer
(drug) that accumulates preferentially in malignant cells, which, by itself, is harmless and
have no effect on any healthy tissue or abnormal. Photosensitizer candidate to be used in
this technique must have some requirements, such as strong absorption (> 105 M
-1cm
-1)
ideally in the 600-850 nm region also so called "phototherapeutic window", should be able
of generating reactive species such as singlet oxygen with quantitative yields and have
minimal toxicity in the dark and be cytotoxic only when irradiated with light of an appropriate
wavelength.1
In this study, were tested, the in vitro effect of seven new unsymmetrical squarylium cyanine
dyes (Figure 1) using HepG2 and Caco-2 cells, previously seeded into 96- wells plates, to
evaluate their ability to reduce cell viability. Cells were incubated with compounds 1-7 at 0.1,
1.0, 5.0 and 10 μM, for 24 hours. For each compound, three conditions were tested: non-
irradiated, 7 min. and 14 min. of irradiation, with LED's centered on 660 nm.
We observed no significant dark toxicity for concentrations of 0.1, 1.0 and 5.0 μM, in all
tested compounds with the exception of compound 3. With irradiation we observed, none
effect of compounds 5 and 6, and a high increase in cytotoxicity for compounds 1, 2, 3, 4
and 7. It was also observed differences in sensibility to the dyes between the two cell lines.
We may conclude that the compounds 1, 2, 4 and 7 have potential to be used as efficient
photosensitizers for the cancer photodynamic therapy, in the future, being the order of
potency 1 > 2 > 7 > 4.
Figure 1: New unsymmetrical cyanine squarylium dyes.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for partial financial support (project PTDC/QUI-QUI/100896/2008) and COMPETE (Project Pest-C/SAU/UI0709/2011). References:
1. a) Bonnet R., in Chemical Aspects of Photodynamic Therapy, Gordon and Breach Science Publishers, The Netherlands, 2000. b) Avirah R. R.; Jayaram D. T.; Adarsh N.; Ramaiah D. Org. Biomol. Chem. 2012, 10, 911.

PC174
A Small Library of Lathyrane Diterpenes Through Molecular
Derivatization
S. Neto,* A. M. Matos,* N. Duarte, M. J. U. Ferreira
iMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal. *These authors contributed equally to this work.
In our search for MDR reversal agents, we have isolated from Euphorbia species, several
macrocyclic diterpenes that have shown to be very strong Pgp modulators in multidrug
resistance cancer cells.1
Multidrug resistance (MDR), a complex phenomenon involving
several mechanisms, is believed to be the major problem for the success of modern cancer
treatment. The best studied form of MDR is that resulting from the overexpression of P-
glycoprotein, an ATP-dependent efflux-pump that extrudes the drugs outside the cell,
thereby reducing its intracellular concentration and leading to loss of therapeutic efficacy.
The co-administration of compounds that are able to restore the cytotoxicity of the available
anticancer drugs is among the most promising strategies to overcome Pgp mediated MDR.2
In order to obtain a large number of analogues that allow the construction of a small library
of compounds indispensable to carry out the QSAR studies, several derivatives were
prepared from two lathyrane diterpenes previously isolated in larger amounts. The
molecular derivatization of these macrocyclic diterpenes included the preparation of new
alkanoyl and aroyl esters and Michael addition reactions to α,β-unsaturated systems.
Several derivatives were also obtained through manipulation of an epoxide ring and
reduction of carbonyl groups. The chemical structures of the compounds were deduced
from their physical and spectroscopic data, namely low and high resolution Mass
spectrometry, and extensive Nuclear Magnetic Ressonance studies, including two-
dimensional homo and heteronuclear correlation experiments.
Acknowledgements: This study was supported by FCT, Portugal (project PTDC/QUI-QUI/099815/ 2008; PEst-OE/SAU/UI4013/2011; PTDC/QEQ-MED/0905/2012) References: 1. Reis, M.; Ferreira, R.; Serly, J.; Duarte, N.; Madureira, A.; Santos, D.; Ferreira, M.J. Anti-Cancer Agents Med. Chem. 2012 , 12, 1015. 2. a) Perez- Tomas, R. Curr. Med. Chem. 2006, 13, 1859. b) Baguley, B.C. Mol. Biotechnol. 2010, 46, 308.

PC175
Targeted Delivery of MitoCIN’S: a New Therapeutic Approach for
Oxidative Stress Related Diseases
Sofia Benfeito,a Catarina Oliveira,
a José Teixeira,
a Pedro Soares,
a Alexandra Gaspar,
a
Jorge Garrido,a,b
Eugenio Uriarte,c Paulo J. Oliveira,
d Fernanda Borges
a
a CIQ/Department of Chemistry and Biochemistry, Faculty of Sciences, University of Porto, Portugal;
bDepartment of Chemical Engineering, Superior Institute of Engineering of Porto (ISEP), IPP, Portugal; cDepartment of Organic Chemistry, Faculty of Pharmacy, University of Santiago Compostela, Spain;
dCNC/Center for Neuroscience and Cell Biology, University of Coimbra, Portugal
[email protected]; [email protected]
The mitochondrion is an important organelle for the genesis of ATP in which occurs the
intracellular formation of the reactive oxygen species (ROS) and for that reason is
particularly vulnerable to oxidative damage. Thus, one way to prevent or retard the
progression of oxidative damage is the regulation of ROS production in mitochondria. In this
context, it is believed that the modulation of mitochondrial dysfunctions throughout the
antioxidant therapy can correspond to an appropriate strategy to prevent or retard the
deleterious oxidative effects present in aging and degenerative diseases, such as
Alzheimer's disease. Mitocondriotropic antioxidants can be a successful approach as they
are able to penetrate in the phospholipid bilayer of mitochondria´s membrane and have the
capacity of accumulation in the negatively charged compartments of the mitochondrial
matrix.
Hydroxycinnamic acids, and in particular caffeic acid, are phenolic compounds present in
the diet that have been used as a model for the design and development of new
antioxidants. However, despite exhibiting an interesting in vitro antioxidant activity their
application in therapy was not successful. Failure in the antioxidant therapy of compounds
of natural origin is often associated with setbacks related with their physicochemical
characteristics, particularly its low lipophilicity, which are not suitable for their biodistribution
and penetration into the target site.
In this context the aim of the present project is the rational design of new mitocondriotropic
antioxidants based on caffeic acid, namely ([(E)-2-(3-(3,4-dihydroxyphenyl)prop-2-enamido)
ethyltriphenylphosphonium methanesulfonate (Di_D1), (E)-6-(3-(3,4-dihydroxyphenyl)prop-2-
enamido)hexyltriphenylphosphoniummethanesulfonate (Di_D2) and (E)-6-(3-(3,4,5-
trihydroxyphenyl)prop-2-enamido)hexyltriphenylphosphoniummethanesulfonate (Tri_D2).
The structural modifications performed, such as modification of the length of the aliphatic
spacer and the number of hydroxyl groups will allow establishing the structure-activity
relationship and the optimization of the lead compound. The compounds were characterized
by nuclear magnetic resonance spectroscopy (1H,
13C and DEPT) and electronic impact
mass spectroscopy (MS/IE) and the antioxidant activity was evaluated by DPPH• and
ABTS• methods. In addition, the redox potentials of the synthesized compounds were
evaluated by the techniques of differential pulse and cyclic voltammetry. According to the
results it was possible to assign a hierarchy on the antioxidant efficacy and redox data of
the lipophilic antioxidants according to the order: Tri_D2 > Di_D2 > Di_D1. The study of their
performance in mitochondrial and neuronal systems is on progress.
The compounds, processes and application have been patented – patent 20121000003813.
The synthesized compounds, methods and applications are in patenting process. Acknowledgements: This work was supported by the Fundação para a Ciência e Tecnologia (FCT),
Portugal (PTDC/QUI-QUI/113687/2009 and Pest/C-QUI/UI0081/2011). A. Gaspar (SFRH/BD/43531/2008) and J.Teixeira (SFRH/BD/79658/2011) thank FCT grants.

PC176
Synthesis of Chalcones with Potential Activity in the p53-MDM2
Interaction
S. Carvalho,a,b
, J. Soares,c,d
S. Cravo,a,b
L. Saraiva,c,d
M. Pinto,a,b
H. Cidadea,b
aCentro de Química Medicinal da Universidade do Porto (CEQUIMED-UP), Rua de Jorge Viterbo
Ferreira, 228, 4050-313 Porto, Portugal bLaboratório de Química Orgânica e Farmacêutica, Departamento de Ciências Químicas, Faculdade de
Farmácia, Universidade do Porto, Rua de Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal cREQUIMTE, Faculdade de Farmácia, Universidade do Porto, Porto, Portugal
dLaboratório de Microbiologia, Departamento de Ciências Biológicas, Faculdade de Farmácia,
Universidade do Porto, Rua de Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal
The p53 tumor suppressor protein is one of the major regulators of cell proliferation and
death, and it has a crucial role in the regulation of cell cycle and apoptosis. Therefore, p53
is considered a key protein in cancer development. The activity and stability of p53 are
regulated by the endogenous negative regulator, MDM2. Hence the discovery of activators
of p53 and inhibitors of p53-MDM2 interaction is considered a promising strategy for cancer
treatment.1
Chalcones represent an outstanding class of naturally occurring compounds with interesting
biological activities, being the antitumor effect one of the most reported in the literature.2
Among chalcones, prenylated derivatives have been attracting the attention of the scientific
community because of their myriad biological activities.3,4
In fact, it has been demonstrated
that isoprenylation of chalcones significantly increased their growth inhibitory effect on
human tumor cell lines.4 Considering this, we decided to synthesize prenylated chalcones.
Chalcones were synthesized by a base-catalyzed aldol reaction of 2’-hydroxy-4’,6’-
dimethoxyacetophenone with the respective substituted benzaldehydes by MAOS. O-prenyl
derivatives were synthesized by the nucleophilic substitution of the chalcone building block
with prenyl bromide in presence of tetrabutylammonium hydroxide at room temperature.
The structure elucidation of synthesized compounds was established on the basis of NMR
techniques. Given the central role of p53 in cancer development, the modulatory effect of
these chalcones on p53-MDM2 interaction was investigated. For this purpose, phenotypic
screening assays using yeast cells co-expressing human p53 and the negative regulator
MDM2 were performed.
Acknowledgements: This work is funded through national funds from FCT – Fundação para a Ciência e a Tecnologia under the project CEQUIMED – PEst-OE/SAU/UI4040/2011, FEDER funds and COMPETE program under the projects FCOMP-01-0124-FEDER-011057 and FCOMP-01-0124-FEDER-015752. References: 1. Dickens, M. P., Fitzgeral, R., Fischer P. M., Seminars in cancer Biology 2010, 20, 1.
2. Boumendje, A.,Ronot X., Boutonnat J., Curr. Drug Targets, 2009, 10, 363-371.
3. Szliszka E., Czuba Z. P., Mazur B., Sedek L., Paradysz A., Krol W., Int. J. Mol. Sci., 2010, 11, 1-13.
4. Neves M. P., Lima R. T., Choosang K., Pakkong P., Nascimento M. S. J., Vasconcelos M. H., Pinto
M., Silva A. M. S., Cidade H., Chem Biodivers, 2012, 9, 1133-1143.

PC177
Ethylenediamine-derived affinity ligands immobilized on Sepharose
to isolate BSA, lysozime and RNase A
V. C. Graça,a M. S. M. R. Silva,
b L. V. Reis,
a F. Sousa,
b P. Almeida,
b J. A. Queiroz,
b
P. F. Santosa
aDept. Química and Centro de Química - Vila Real, Universidade de Trás-os-Montes e Alto Douro, 5001-
911 Vila Real, Portugal bCICS-UBI – Centro de Investigação em Ciências da Saúde, Universidade da
Beira Interior, 6201-001 Covilhã, Portugal
Dye-based affinity chromatography has becoming the preferred method for the purification
of proteins1 since it considerably overcomes the limitations of biological ligands, namely
their high cost and their poor resistance to biological and chemical degradation.2 In general,
the more widely used dye-ligands in protein purification by affinity chromatography have
been based on triazine dyes.3
Recently, by using affinity matrices prepared from symmetrical aminosquarylium cyanine
dyes, immobilized on cyanuric-chloride activated Sepharose via the central amino residue
acting as a spacer arm, we were able to isolate lysozyme, α-chymotrypsin and trypsin from
a mixture.4 Following this, we envisioning the immobilization of a squarylium dye on
Sepharose through one of its heterocyclic ending nuclei as a way of enhancing the mobility
of the dye ligand and, potentially, improving its interactions with proteins. The assymetric
dye chosen bears one pendent N-carboxyethyl group in one of the benzothiazole nuclei and
was linked to ethylenediamine-activated Sepharose trough 1-ethyl-3-[3-
dimethylaminopropyl]carbodiimide hydrochloride (EDC)/N-hydroxysuccinimide (NHS)
mediated amidation coupling. The prepared support was found to be able to efficiently
separate a mixture of BSA, lysozime and RNase A.
The chromatographic support prepared in the absence of the dye-ligand, for comparison
purposes, turned out to exhibit a separation performance superior to that of the dyed
support, what prompted us to try to determine the structural molecular constitution of the
support’s surface. A synthetic route leading to the final support could be devised, the
outcome of which it is believed to be the cyclization of two nearby ethylenediamine units,
involving the inclusion of a succinimide-derived residue between them and the EDC
mediated Lossen rearrangement of an intermediary hydroxamic acid.
Acknowledgements: This work was supported by FCT, the Portuguese Foundation for Science and Technology, through the project PTDC/QUI-QUI/100896/2008. V. C. Graça
and M. S. M. R. Silva also
acknowledge fellowships in the ambit of this project and COMPETE (Project PEst-C/SAU/UI0709/2011). References: 1. Labrou, N. E. J. Chromatogr. B, 2003, 790, 67. 2. Clonis, Y. D. J. Chromatogr. A, 2004, 1, 1101. 3. Labrou, N. E., Mazitsos, K., Clonis, Y. D. in: Hage, D. S. (Ed), Handbook of Affinity Chromatography, CRC Press, Boca Raton, 2006, p. 231.
4. Silva, M. S. Graça, V. C. Reis, L. R. Santos, P. F. Almeida, P. Queiroz
, J. A. Sousa, F. Biomed.
Chromatogr. 2013 (DOI 10.1002/bmc.2978).

PC178
Planar aromatic core
Synthesis of Naphtalene Derivatives as New Class of Putative
G-Quadruplex Ligands
A. R. P. Duarte, Rui Moreira, A. S. Ressurreição
iMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal
Guanine-rich nucleic acid sequences have a high propensity to self-associate into planar
guanine quartets (G-quartets) to give unusual structures called G-quadruplexes. These
motifs are particularly found at key regulatory regions of the human genome, such as
promoters, telomeric regions and untranslated regions. Therefore, stabilization of
quadruplex structures within their genomic environments by the binding of small molecules
can lead to a range of biological effects: inhibition of transcription, inhibition of translation or
telomere maintenance, depending on their context within a genome.1
Here in we describe the synthesis of small molecules (Figure 1) that incorporated some key
features that a G-quadruplex ligand should have: (i) a planar aromatic core to maximize the
– stacking and electrostatic interactions with the G-quartet but conceived to be sufficiently
small to overcome some of the druggability problems associated with large polycyclic
groups, and (ii) substituents with terminal basic groups to enhance electrostatic interactions
with quadruplex grooves.2
In order to synthesise these molecules, we used different reactions such as Suzuki-Miyaura
cross-coupling reactions and thiol-ene radical reactions (Figure 1).
Figure 1: Retrosynthetic scheme of the synthesis of the new class of putative G-quadruplex ligands. Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support through the projects Pest-OE/SAU/UI14013/2011 and grants SFRH/BPD/64859/2009 (ASR) and SFRH/70491/2010 (ARPD). References:
1. Collie G. W.; Parkinson G. N. Chem. Soc. Rev. 2011, 40, 5867. 2. Lombardo C. M.; Welsha S. J.; Strauss S. J.; Dale A. G.; Todda A. K.; Nanjunda R.; Wilson W. D.; Neidle S. Bioorg. Med. Chem. Lett. 2012, 22, 5984.
Terminal Basic
Groups
Terminal Basic
Groups
Suzuki-Miyaura Cross-coupling Reaction
Thiol-ene Reaction

PC179
Designing and syntheses of new cyclopentenones as key
precursors of antiviral nucleoside (N)-MCT and novel hydrophobic
compounds with promising anti-proliferative activity
Krassimira P. Guerra, Raquel F. M. Frade, Carlos A. M. Afonso iMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal
A (North)-2'-deoxy-methanocarba thymidine nucleoside, (N)-MCT, is an active specific
agent against herpes simplex viruses (HSV1 and 2), cowpox, orthopoxviruses and vaccinia
viruses. (N)-MCT reveals in vitro activity against Kaposi’s sarcoma-associated herpesvirus
(KSHV), and also antitumour activity in vivo against HSV-tk transduced and nontransduced
murine colon cancer (MC38).1
Based of reported total syntheses of (N)-MCT in the recent
past,1 we built up a new synthetic strategy to reach the chiral precursor 4. The principal
substrate is new adduct 2, prepared in our Morita-Baylis-Hillman (MBH) conditions2
(Scheme 1, A). The chiral compound 4 was achieved via CBS reduction and (or) Mitsunobu
inversion. (N)-MCT could be produced from 4 following highly stereoselective directed
Simmons-Smith cyclopropanation reaction and introduction of pyrimidine base. Instead, the
α, β,-unsaturated unit, which mediates the cytotoxicity, as many other biological activities of
the cyclopentenones, reacts covalently in a Michael type addition with the cysteine
sulfhydryl group of proteins. Consequently a variety of cellular functions are imbibed, which
directs the cells into apoptosis.3,4
The differences in activity within the series of
cyclopentenone adducts are attributed to the variation in the lipophilicity, the molecular
geometry and the chemical environment or the protein targets. Therefore the library of
about 70 new hydrophobic MBH cyclopentenone derivatives were designed and
synthesized in good and excellent yields. Their biological activities were scanned to a range
of tumoral cell lines and some of them were revealed auspicious anti-proliferative activity
and high selectivity (Scheme 1, B).
O
HO
O
RO
OH
O
RO
OR1
R = H, OH, R1
ref.2
R1O
R1OH
OH
cyclopropanation
NH
N
O
O
HO
HO
N-MCT
H
Mitsunobu
conditions
OR1
OR2
A B
1 2 3
(reported)(reported)
IC50 (NCI-H460) =3.9±1.1 IC50 (HT-29, MCF-7)>20
R1 = TBDPS
R1O
OHR1O 4
Scheme 1.
Acknowledgements: The authors are grateful to Fundação para a Ciência e a Tecnologia: Programa
Operacional Ciência e Inovação (POCI) 2010 and Fundo Europeu de Desenvolvimento Regional (FEDER) for financial support (SFRH/BPD/28038/2006 and PTDC/QUI-QUI/119823/2010). References: 1. Krassimira P. Guerra and Carlos A. M. Afonso, Current Org. Synthesis 2013,10, 210. 2. Krassimira P. Guerra and Carlos A. M. Afonso, Tetrahedron 2011, 67, 2562. 3. H. M. Sampath Kumar et al, Eur. J. Med. Chem., 2011, 46, 3210. 4. Carlos A. M. Afonso, V. Kurteva, S. Simeonov, J. Nunes, Krassimira P. Guerra, Chem Rev., June, 2013, submitted.

PC180
Pyrazinoic esters – human plasma stability and mycobacterial
activation in free and liposomal form
Marta Oliveira, Teresa Almeida, Susana Calado, Emília Valente, Manuela M. Gaspar, Luís Constantino
iMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto
Tuberculosis is a major infectious disease caused by Mycobacterium tuberculosis. Roughly
one third of the world population is infected with M. tuberculosis, and 1.4 million people died
in 2011 as a result of this disease. The emergence of strains resistant to first-line
antitubercular drugs has led to a need to find alternatives for tuberculosis treatment.
Pyrazinamide is a first-line drug with remarkable sterilizing activity that plays a unique role in
shortening the therapy from 9-12 months to 6 months. Pyrazinamide is a prodrug of
pyrazinoic acid (POA), unfortunately resistant strains due to mutations in the pncA gene
encoding the enzyme responsible for conversion of PZA are observed. In order to overcome
this problem, pyrazinoic acid esters were synthesized. Since the plasma is rich in esterase
activity, these prodrugs must be resistant to plasma hydrolysis in order to enter the
mycobacterial cells in the prodrug form as pyrazinoic acid is not efficiently taken up by
mycobacteria. In order to raise the stability of these compounds in plasma they have been
incorporated in liposomes. Liposomal formulations can protect further the prodrugs from
plasma degradation during the transport phase and liposomes have the additional
advantage of targeting incorporated compounds to macrophages, the cells that are infected
by M. tuberculosis. Once inside the mycobacteria these prodrugs must be activated by
mycobacteria enzymes in order to release the drug. 1
In this work we studied the stability in human plasma of prodrugs incorporated in
DMPC:DMPG (7:3) liposomes with a lipid/drug ratio 10/1, and also their activation by a
Mycobacterium smegmatis homogenate. The prodrugs studied were 1-methylundecyl
pyrazinoate, dodecyl pyrazinoate and nonyl pyrazinoate. These compounds were found to
be more active than pyrazinamide (in vitro MIC). The stability tests were performed in 80%
of human plasma and the activation tests were performed in 2% of a Mycobacterium
smegmatis homogenate (total protein concentration 13.65 mg/mL). Comparing the results
obtained for the prodrugs in the liposomal form with those obtained previously for the free
prodrugs in the same conditions we concluded that the prodrugs encapsulation in liposomes
raised their stability in human plasma. The activation in mycobacterial homogenate led to a
decrease in kobs for 1-methylundecyl pyrazinoate and dodecyl pyrazinoate whereas for nonyl
pyrazinoate this constant was about the same.
Figure 1: General structure of pyrazinoic esters
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support - project
PTDC/SAU-FCF/101950/2008.
References: 1. a) WHO. Global tuberculosis report 2012. Geneva, Switzerland, 2012. b) Caminero, José A., et al Lancet Infect Dis. 2010 10 (9), 621-29. c) WHO. WHO Model List of Essential Medicines. Seventeenth edition. Geneva, Switzerland, 2011. d) Zhang, Y. and Mitchison, D. Int J Tuberc Lung Dis. 2003. 7(1), 6-21. e) Zhang, Y. and Yew, W. W. Int J Tuberc Lung Dis. 2009. 13(11), 1320-1330. f) Pinheiro, M., Lúcio, M., Lima, JLFC and Reis, S. Nanomedicine. 2011. 6(8), 1413-28.

PC181
Indole: A “Privileged Structure” in Medicinal Chemistry
Mónica S. Estevão,a Eduarda Fernandes,
b M. Manuel B. Marques
a
aREQUIMTE – Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de
Lisboa, 2829-516 Caparica, Portugal; bDepartamento de Ciências Químicas, Faculdade de Farmácia,
Universidade do Porto, Rua de Jorge Viterbo Ferreira n.º 228, 4050-313 Porto, Portugal
The indole scaffold represents one of the most important structural subunits in drug
discovery. The demonstration that several alkaloids contain the indole nucleus and the
recognition of the importance of the essential amino acid tryptophan in human nutrition lead
to an extensive research on indole chemistry. Indeed a vast number of biologically active
natural and synthetic derivatives have been reported, with a wide range of therapeutic
targets, such as antioxidant (melatonin), anti-inflammatory (indomethacin), antimicrobial,
analgesic, anticonvulsant, antimalarial.1
Regarding the unique properties of this scaffold, three indole-based libraries were
synthesized and its biological activity investigated, searching for novel agents, including
antioxidants,2 selective COX-2 inhibitors
3 and tuberculostatic agents.
In the present work, the synthesis of several indole derivatives will be presented as well as
the biological tests results.
Figure 1: Chemical structures of the antioxidant melatonin and the NSAID indomethacin.
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia for financial support (PTDC/QUI/65187/2006) (PEst-C/EQB/LA0006/2011) and for the PhD fellowship to M. E. (SFRH/BD/46234/2008) References: 1. a) Alves F. R. S.; Barreiro E. J.; Fraga C. A. M. Mini-Rev. Med. Chem. 2009, 9, 782. b) Baumann M.; Baxendale I. R.; Ley V. S.; Nikbin N.Beilstein J. Org. Chem. 2011, 7, 442. 2. Estevão M. S.; Carvalho L. C.; Ribeiro D.; Couto D.; Freitas M.; Gomes A.; Ferreira L. M.; Fernandes E.; Marques M. M. B. Eur. J. Med. Chem. 2010, 45, 4869.
3. Estevão M. S.; Carvalho L. C. R.; Freitas M.; Gomes A.; Viegas A.; Manso J.; Erhardt S.; Fernandes E.; Cabrita E. J.; Marques M. M. B. Eur. J. Med. Chem. 2012, 54, 823.

PC182
Mass spectrometry as a tool to provide mechanistic insights into
metabolic reactions: CID of quinoloimines derivatives
Paulo J. Amorim Madeira, Ana Raquel Fernandes Sitoe,
Daniel Gonçalves,
Tiago Rodrigues, Rita C. Guedes,
Rui Moreira,
Francisca Lopes,
M. Rosário Bronze
iMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa,
Portugal
From the identification of metabolites to the study of the interactions between new chemical
entities and potential targets (proteins or even DNA),1
mass spectrometry is extensively
used in drug development. Furthermore, there are literature reports in which mass
spectrometry data were used to predict stability in aqueous medium at physiological pH and
temperature.2
Here we report the use of electrospray ionisation tandem mass spectrometry
(ESI-MS/MS) to study the gas-phase behaviour of several quinoloimines derivatives
(Scheme 1A) with potent antimalarial activity. Besides the fragmentation pathways
(Scheme 1B), we will present results from energy-dependent collision induced dissociation
(CID) experiments that will allow us to determine the appearance energies of the product
ions,3 through a breakdown diagram (example presented in Scheme 1C). These
appearance energies are related with the dissociation energy of the bond that breaks during
the CID process (N1-Csubstituent of the quinoline ringin our case) and they will be compared
with energy values calculated at the DFT level of theory.
Scheme 1: (A) General structure of the compounds under study; (B) Fragmentation pathways for the protonated quinoloimines derivatives;(C) Example of a breakdown diagram used to estimate the bond
strength.
Acknowledgements: We thank the Fundação para a Ciência e a Tecnologia (FCT) for financial support: REDE/1518/REM/2005 for the LC-MS/MS equipment; PEst-OE/SAU/UI4013/2011; PTDC/SAU-FCT/098734/2008. PJAM (SFRH/BPD/86948/2012), ARFS (SFRH/BD/51459/2011), TR (SFRH/BD/30689/2006) acknowledge FCT for their post-doctoral and PhD grants. References: 1. Ma L.; Song F.; Liu Z.; Liu S. Rapid Commun. Mass Spectrom. 2013, 27, 51 2. Vale N.; Matos J.; Moreira R.; Gomes P. Eur. J. Mass Spectrom. 2009, 15, 627 3. a) Madeira P. J. A.; Morais T. S.; Silva T. J. L.; Florindo P.; Garcia M. H. Rapid Commun. Mass Spectrom. 2012, 26, 1675; b) Zins E.-L.; Pepe C.; Schröder D. J. Mass Spectrom. 2010, 45, 1253; c) Schröder D.; Engeser M.; Brönstrup M.; Daniel C.; Spandl J.; Hartl H. Int. J. Mass Spectrom. 2003, 228, 743.
(A) (B)
X: Cl, CF3
R:
17
(C)

PC183
Towards more potent Jolkinol D derivatives: how can docking
studies guide chemical derivatization?
R. J. Ferreira,a M. J. U. Ferreira,
a D. J. V. A. dos Santos
a,b
aiMed.UL, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa, Portugal
bREQUIMTE, Department of Chemistry and Biochemistry, Faculty of Sciences, University of
Porto, Rua do Campo Alegre, 4169-007 Porto, Portugal
Multidrug resistance is a major drawback in current cancer chemotherapy, mainly due to
overexpression of the ABC transporter P-glycoprotein (P-gp) at the surface of cancer cells.
Using a macrocyclic lathyrane diterpene (Jolkinol D) isolated from Euphorbia piscatoria, a
small library of novel bioactive derivatives was synthesized in order to obtain more selective
P-gp reversal agents.1
We used molecular dynamics to refine the murine P-gp crystallographic structure published
in 2009 (PDB ID: 3G5U).2,3
The insertion of the transporter in a lipid bilayer and the addition
of a missing linker structure (short amino acid sequence between both functional halves)
greatly improved structural stability and allowed a clear definition of the drug-binding pocket
lower boundary.3 By applying molecular docking techniques to the P-gp refined structure,
our group was able to assign and characterize three drug-binding sites.4 The first two are
located next to the inner leaflet interface and were identified as substrate-binding H and R-
sites described in literature, whereas the third one, located next to the outer leaflet, is
identified as the M-site already characterized in a previous study.2 Herein, we propose a
new classification model, based on the binding free energy (ΔG) and cross interaction
capability (CIC) between the two functional halves, in order to distinguish substrates from
modulators.4
How can this new model guide chemical derivatization for more potent MDR reversers
based on the macrocyclic scaffold? In this study, a suitable approach could be made using
ΔG, CIC and ligand efficiency (LE).5 As π–π or CH–π interactions seem to prevail at M-site,
a different approach can be made for LE, being calculated as the ratio between ΔG and the
number of contacts at the M-site. In this line, a prediction of the impact of C-3 chiral center
inversion upon Jolkinol D activity (Figure 1) was assessed using a small set of diterpene
derivatives,1 showing that C-3 chiral inversion may be a valid chemical strategy in order to
increase the potency of MDR-reveral macrocyclic diterpenes.
Figure 1: Jolkinol D
Acknowledgements: We thank the Fundação para a Ciência e Tecnologia (FCT, Portugal) for financial support through projects PTDC/QUI-QUI/099815/2008, Pest-OE/SAU/UI4013/2011 and PTDC/QEQ-MED/0905/2012. Ricardo Ferreira acknowledges FCT for the PhD scholarship SFRH/BD/84285/2012.
References: 1. Reis M. A., Ferreira R.J., Santos M. M. M., dos Santos D. J. V. A., Molnár J., Ferreira M. J. U. J. Med. Chem. 2013, 56, 748–760. 2. Aller S. G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P. M., et al. Science 2009, 323, 1718–1722. 3. Ferreira R. J., Ferreira M. J. U., dos Santos, D. J. V. A. J. Chem. Theory Comput. 2012, 8, 1853–1864. 4. Ferreira R. J., Ferreira M. J. U., dos Santos, D. J. V. A. J. Chem. Inf. Model. 2013 (submitted) 5. a) Kuntz I. D., Chen K., Sharp K. A., Kollman P. A. Proc Natl Acad Sci USA 1999, 96, 9997–10002. b) Hopkins A. L., Groom C. R., Alex A. Drug Discov Today 2004, 9, 430–431.

PC184
Synthesis and surface activity of alkyl 2-deoxyglycosides as original
structures for utilization as antimicrobial agents
Patrícia Serra, Vasco Cachatra, Alice Martins, Maria Soledade Santos, Amélia P. Rauter
Centro de Química e Bioquímica/Departamento de Química e Bioquímica, Faculdade de Ciências da
Universidade de Lisboa, Campo Grande Edificio C8, 5o Piso, 1749-016 Lisboa, Portugal
The search for new drugs for pathogenic infections is currently a major topic of research as
a result of the ongoing spread of multidrug-resistance. Other important issue relates to
biohazard security matters and the lack of treatment. These facts demand an incessant
investigation of new antibacterial agents with new mechanisms of action. We have
introduced a new family of compounds structurally related to alkyl 2-deoxyglycosides, which
exhibited a potent activity against Bacillus species.1-3
These structural features may give
insights onto the relationship between structure, surface activity and bioactivity of this family
of compounds regarding Bacillus cereus, Bacillus subtilis and Bacillus anthracis and
contribute to the study of their mechanism of action. A preliminary evaluation of the
antibacterial activity on B. species showed that the most promising compound is dodecyl 2-
deoxy-α-D-threo-pentopyranoside (compound type 6). Hence, its surface activity and its
scale up were also investigated.
An underlying goal was the development of an easier and economical synthesis of the
glycal used as glycosyl donor of the glycosylation reaction. Regarding the molecular
diversity associated to derivatives synthesized from glycals, new strategies for their
synthesis are of key importance. Glycosylation with 4 of a variety of alcohols led to
compounds type 5 (Scheme 1), which were submitted to the Zémplen deacetylation to give
6 in good yields.4 The structure of the isolated compounds was confirmed by spectroscopic
analysis using NMR as a prime tool. The 2-deoxyglycosides were subjected to surface
activity studies and the results will be presented and discussed.
Scheme 1: Reagents and conditions: a) Ac2O, pyridine; b) CH3COOH/CH3COBr in MeOH; c) CH3COOH/CH3COBr, MeOH, Ac2O; d) Zn/NaH2PO4/acetone; e) TPHB, CnXmOH; f) NaOMe/MeOH
Acknowledgements: This work was supported by FEDER-QREN-SI I&DT co-promotion. The authors would like to thank the FCT for financial support ((PEst-OE/QUI/UI0612/2013). References: 1. Silva F., Goulart M., Justino J., Neves A., Santos F., Caio J., Lucas S., Newton A., Sacoto D., Barbosa E., Santos M. S., Rauter A. P., Bioorg. Med. Chem., 16, 4083-4092 (2008) 2. Rauter A. P., Lucas S., Almeida T., Sacoto D., Ribeiro V., Justino J., Neves A., Silva F. V., Oliveira M. C., Ferreira M. J., Santos M. S., Barbosa E., Carbohydr. Res, 340, 191-201 (2005) 3. Martins, A., Santos, M. S., Dias, C., Serra, P., Cachatra, V., Pais, J. P., Caio, J., Teixeira, V. H., Machuqueiro, M., Silva, M. S., Pelerito, A., Justino, J., Goulart, M., Silva, S. V., Rauter, A. P., Eur. J. Org. Chem., 2013, 1448 –1459 (2013)
4. A. P. Rauter, A. Martins, J. Caio, J. P. Pais, P. Serra, M. S. Santos, A. Pelerito, J. P. Gomes, J. Justino, R. Dias, R. Tenreiro, Sugar derivatives as inhibitors of Bacillus species, process for their preparation and utilization, PCT/IB2012/050123, submitted 2012.

PC185
Peptide synthesis: different approaches for specific drug delivering
of chemotherapeutical agents
João D. Pereira, M. J. Perry, A. P. Francisco, M. E. Mendes, Manuela M. Gaspar
Research Institute for Medicines and Pharmaceutical Sciences (iMed.UL), Faculdade de Farmácia da
Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa, Portugal
Malignant melanoma is characterized by an abnormal growth of melanocytes and it is
presented in four major stages. The fourth stage is the most aggressive form corresponding
to a distant metastatic phase.
In cancer disease, most of biochemical pathways are modified. In case of melanoma it is
detected an increasing production of a specific enzyme called tyrosinase which is over
expressed only in malignant melanocytes. Designing a prodrug that is activated only in
these cells appears to be a good therapeutic strategy.
Currently, chemotherapy for melanoma treatment uses a variety of alkylating agents, such
as Dacarbazine. Based on its structure with a triazene function and exploiting the tyrosinase
over expression in melanoma cells, our group synthesized a range of triazene prodrugs
(TPD) applying several synthetic approaches of amide coupling using different catalyzers.
Here, we used three distinct methods to synthesize the triazene prodrug (Figure 1). Parallel
to this study, it was developed an alternative carrier system, using liposomes, to solubilize
TPD without using toxic solvents, enhance the internalization of these cytotoxic agents into
tumor cells and protect molecules from premature degradation.
In vitro chemical and enzymatic stability was evaluated for free and encapsulated TPD and
the affinity of the prodrug to act as a tyrosinase substrate was also determined.
Cytotoxic studies of both formulations are currently in course using MNT-1, a human
melanoma cell line.
Figure 1: 1-(4-bromophenyl)-3-(5-(4-hydroxyphenyl)-1-pentanoyl)-3-methyltriazene

PC186
Synthesis and the Asymmetric Resolution of Dopamine and
Rotigotine Analogues 2-Amino-6,7-dimethoxyindane and
2-Amino-7,8-dimethoxy-1,2,3,4-tetrahydronaphthalene
Süleyman Göksu, Akın Akincioğlu, Leyla Polat
Department of Chemistry, Faculty of Science, Atatürk University, 25240-Erzurum, TURKEY
[email protected], [email protected]
Dopamine (1), a monoamine hormone, is a neurotransmitter and have important roles in
central nervous system (CNS)–related disorders such as schizophrenia and Parkinson’s
disease.1 Dopamine-like actions of many chemical compounds have been reported.
2 A drug
rotigotine (2), commercially known as neupro, is used in the treatment of Parkinson’s
disease as transdermal patch.3 Dopaminergic activities of 6,7-ADTN (3) and 5,6-ADTN (4)
have also been reported.4
In the present study, dopamine (1) and rotigotine (2) analogues 2-aminoindane 7 and 2-
aminotetralin 8 were synthesized starting from 3-(3,4-dimethoxyphenyl) propanoic acid (5)
and 3-(3,4-dimethoxyphenyl) butanoic acid (6) as racemic mixtures. The synthesized
compounds were converted to their corresponding diastereomers with (R)-2-
acetoxymandelyl chloride. Asymmetric resolution of diastereomeric amide mixtures was
achieved by crystallization. Hydrolysis of the corresponding diastereomers gave (+)-7, (+)-8
and (-)-8 enantiomers with high enantiomeric excess.
Acknowledgments
The authors are indebted to the Scientific and Technological Research Council of Turkey (TÜBİTAK, Grant No. 109T/241) and Atatürk University for their financial support of this work. References 1. Haadsma-Svensson, S. R., Svensson, K. A. CNS Drug Rev. 4, 42, 1998. 2. Cannon, J. G. Prog. Drug Res. 29, 303, 1985. 3. Giladi, N.; Boroojerdi, B.; Korczyn, A. D.; Burn, D. J.; Clarke, C. E.; Schapira, A. H. Mov Disord. 2007, 2398-2404 4. Göksu, S., Seçen, H., Sütbeyaz, Y. Helv. Chim. Acta 2007, 270-273.

PC187
Further studies on a promising strategy for the synthesis of
iminosugars, polyhydroxylated prolines and polyhydroxylated
pipecolic acids
Rosalino Balo, María Campos, Juan C. Estévez*, Ramon J. Estévez*
Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS), Laboratorio de
Glicoquímica, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, tfno: +34 881
815 762
Carbohydrates constitute versatile synthons of great usefulness for the stereoselective
synthesis of rich functionalized carbo- and/or heterocyclic based natural products.1 Previous
synthetic strategies developed for this purpose are usually longer and tedious, because
preservation of the hydroxy groups and the stereogenic centers is required. Accordingly,
there is an urgent need for the development of novel synthetic approaches that allow
shorter, more efficient access to these targets.1
Here we present our preliminary results on a novel strategy designed for the synthesis of
nitrogen heterocyclic compounds from sugars. They consist of the transformation of hexose
derivatives 2 into iminosugars 3, 3,4-dihidroxyprolines 3 (n=1) and 3,4,5-trihidroxypipecolic
acids 3 (n=2), proceeding as stated in the Scheme.
Scheme 1
Our ongoing work in this field also includes the synthesis and biological evaluation of a
novel morphiceptin peptidomimetic incorporating proline and pipecolic acids 4.2
Acknowledgements: We thank the Xunta de Galicia (CN2011/037) and the Spanish Ministry of Education (CTQ2009-08490) for financial support, and the former for providing postdoctoral funding to L. B. References: 1. For a book on this topic, see: S. Hanessian. Total Synthesis of Natural Products; The Chiron Approach; Pergamon; Oxford, 1993. 2. Janecka, A.; Fichna, J.; Mirowski, M.; Janecki, T. Mini-Reviews in Medicinal Chemistry 2002, 2, 565-
572.

PC188
Enzymatic synthesis of fatty acid sugar esters. Optimization by
response surface methodology
O. Selaïmia-Ferdjani, N. Bouzaouit, C. Bidjou-Haiour
Laboratoire de Synthèse Organique Modélisation et Optimisation des Procédés Chimiques, Université
Badji-Mokhtar Annaba, BP12, 23000 Annaba, Algérie
Fatty acid Sugar esters are nonionic biosurfactants, they have found widespread
applications as emulsifiers in food, detergents, cosmetics and pharmaceutical industries.1
Fatty acid sugar esters are synthesized by both chemical and enzymatic methods however,
enzymatic mediated synthesis of this compounds class has several advantages over
conventional chemical synthesis.2 Today, lipases stand amongst the most important
biocatalysts, used for novel reactions in both aqueous and non-aqueous media.3 Our study
concerns the xylose fatty acid esters synthesis catalyzed by CCL immobilized lipase,
performed in THF where unprotected xylose and fatty acids were directly used as starting
materials. Various reaction parameters affecting this synthesis, including reaction time,
temperature, quantity of molecular sieves, the molar ratio of sugar/acyl donor and the
amount of enzyme were investigated.
Statistical analysis made it possible to release the three most influential factors among the
five initial ones. The process was then optimized by response surface methodology (RSM)
based on these three major reaction parameters. The optimal conditions for the enzymatic
reaction were obtained at 60°C for 72h and using 30 mg of molecular sieves. Under these
conditions the esterification conversion C was 81%.
References: (1) Habulin, M.; Sabeder, S.; Knez, Z. Journal of Supercritical Fluids 2008, 45, 338.
(2) Plou, F. J.; Cruces, M. A.; Ferrer, M.; Fuentes, G.; Pastor, E.; Bernabe, M.; al., e. Journal of Biotechnology 2002, 96, 55. (3) Shah, S.; Solanki, K.; Gupta, M. N. Chemistry Central Journal 2007, 19, 1.

PC189
Heteropolyacids accelerated multi-component synthesis of
N-phenylquinazolin-4-amines by using Silica-Supported Preyssler
Nanoparticles in Green Solvent
Ali Gharib,a,b,*
Manouchehr Jahangir,a Mina Roshani,
a Lida Bakhtiari,
b Sara Mohadeszadeh,
b
Shirin Lagzian,b Sina Ahmadi
b
aDepartment of Chemistry, Islamic Azad University, Mashhad, IRAN,
bAgricultural Researches and
Services Center, Mashhad, IRAN
In recant years, multicomponent reactions (MCRs) have become important tools in modern
preparative synthetic chemistry because these reactions increase the efficiency by
combining several operational steps without any isolation of intermediates or change of the
conditions1 and MCRs have recently emerged as valuable tools in the preparation of
structurally diverse chemical libraries of drug-like heterocyclic compounds.2 Natural and
synthetic compounds possessing the quinazoline structural motif display a wide range of
biological activities. Recently, quinazolin-4(3H)-ones were prepared via cyclocondensation
of 2-aminobenzamides with orthoesters catalyzed by H2SO4/SiO2 under anhydrous and
microwave conditions.3 N-phenylquinazolin-4-amines derivatives were obtained in high
yields with excellent purity from the reaction of 2-aminobenzamide, orthoesters, and
substituted anilines in the presence of Silica-Supported Preyssler Nanoparticles and variuos
heteropolyacids (HPAs) in Scheme 1. In recent years, considerable effort has been devoted
to the design and controlled fabrication of nanostructured POMs for using in green
reactions. This interest has resulted in the development of numerous protocols for the
synthesis of nanostructured materials over a range of sizes. Therefore the field of nano
POMs and their applications continue to attract significant attention, so the number of
publications and patents continue to grow, and new researchers are entering the field.
However, in spite of extensive investigations on synthesis and characterization of Keggin-
type nanocatalysts,4 the synthesis of sodium 30-tungstopentaphosphate nanocatalysts has
been largely overlooked.
Scheme 1: synthesis of N-phenylquinazolin-4-amines.
References: 1. Zhu J.; Bienayme H. Multicomponent Reactions; Wiely-VCH: Weinheim: 2005. 2. Ugi I.; Dömling A.; Werner B. J. Heterocycl. Chem. 2000, 37, 647. 3. Montazeri N.; Rad-Moghadam K. Phosphorus, Sulfur, Silicon. 2004, 179, 2533. 4. Sawant D. P.; Vinu A.; Jacob N. E.; Lefebvre F.; Halligudi S. B. Journal of Catalysis. 2005, 235, 2, 341.

PC190
Synthesis of calix[4]arene nanotubes
Hayrettin Beynek, Ali Osman Karatavuk
Department of chemistry, Trakya University, Edirne 22000, Turkey
The calixarene nanotubes have been synthesized by binding various intermediate groups
and calixarene nanotubes have been used in extraction studies, conversion of gas and as
gas trap. It was seen that the calixarene nanotubes have high-efficiency in encapsulation
of different molecule and ions. It can be proposed that the calixarene nanotubes can be
developed by doing different studies.1,2
In this study, calixarene nanotubes have been prepared by using different pyridine
intermediate groups and also these calixarene nanotubes have atoms of oxygen and sulfur
which make coordination.
Scheme 1: Synthesis of calix[4]arene nanotubes.
References:
1. Zyryanov G. V.; Rudkevich D. M.; J. Am. Chem. Soc. 2004, 126, 4264.
2. Kim S. K.; Sim W.; Vicens J.; Kim J. S.; Tetrahedron Letters 2003, 44, 805.

PC191
On the use of ionic liquids as green solvents in reactions catalyzed with glycosidases
Salim Ferdjani,a,b
Zeinnedine Djeghaba,a Claude Rabiller,
c Charles Tellier
c
aLaboratoire de Chimie Organique Appliquée, Université Badji Mokhtar, BP 12, Annaba, Algérie bDépartement de Pharmacie, Faculté de Médecine Université Badji Mokhtar, Annaba, Algérie
cBiotechnologie, Biocatalyse et Biorégulation, UMR 6204 CNRS, Université de Nantes, 2, rue de la
Houssinière, 44322 Nantes cedex 03, France.
Due to the well-recognized biological role of oligosaccharides, there is still a great need for
green and low cost methods for their preparation, despite the considerable development of
efficient synthetic methods. In the last decade, enzyme-catalyzed reactions have proved to
be very efficient in the building of the glycosidic bond due to their stereoselectivity. For this
purpose, retaining glycoside hydrolases were particularly efficient, but their use was limited
by the competition between hydrolysis and transglycosylation, reactions both catalyzed by
these enzymes. One alternative to improve transglycosylation might be the use of ionic
liquids in order to reduce water activity and, hence hydrolysis.
Ionic liquids (ILs) have received attention as a promising new class of solvent for chemo-
and bio-catalytic organic synthesis.1
Due to their ability to dissolve polar substrates such as
amino-acids or carbohydrates in a low-water environment, several enzymes, mainly
esterases, lipases and proteases, have been assayed in ILs and shown to retain
biocatalytic activity even at low water activity.2 However, a limited number of studies has
been devoted to the use of ILs as solvents for reactions catalyzed by glycosidases,3-5
even
though ILs present interesting properties such as their capacity to solubilize both
carbohydrates and enzymes.
The activity and stability of a β-glycosidase (Thermus thermophilus) and two α-
galactosidases (Thermotoga maritima and Bacillus stearothermophilus) were studied in
different hydrophilic ionic liquid (IL)/water ratios. For the ILs used, the glycosidases showed
the best stability and activity in 1,3-dimethylimidazolium methyl sulfate [MMIM][MeSO4] and
1,2,3-trimethylimidazolium methyl sulfate [TMIM] [MeSO4]. A close correlation was observed
between the thermostability of the enzymes and their stability in IL media. At high IL
concentration (80%), a time dependent irreversible denaturing effect was observed on
glycosidases while, at lower concentration (30%), a reversible inactivation affecting mainly
the kcat was obtained. The results demonstrate that highly thermostable glycosidases are
more suitable for biocatalytic reactions in water-miscible ILs.6
Scheme 1: Reactions catalyzed by glycosidases and use of ionic liquids to reduce hydrolysis.
References: 1. Sheldon R. A., Lau R. M., Sorgedrager M. J., van Rantwijk F, Seddon K. R. Green Chem, 2002, 4, 147. 2. Van Rantwijk F., Sheldon R. A. Chem Rev, 2007, 107, 2757. 3. Kaftzik N., Wasserscheid P., Kragl U.. Org Process Res Dev, 2002, 6, 553. 4. Kamiya N., Matsushita Y., Hanaki M., Nakashima K., Narita M., Goto M., Takahashi H. Biotechnol. Lett., 2008, 30, 1037. 5. Lang M., Kamrat T., Nidetzky B. Biotechnol. Bioeng., 2006, 95, 1093. 6. Ferdjani S., Ionita M., Roy B., Dion M., Djeghaba Z., Rabiller C., Tellier C. Biotechnol Lett, 2011, 33, 1215.

PC192
Click reaction: synthesis and characterization of novel Triazol-
Quinazoline
A. Ouahrouch,a M. Taourirte,
a H. B. Lazrek,
b J. W. Engels
c
aLaboratoire de Chimie Bio-organique et Macromoléculaire, Faculté des Sciences et Techniques Guéliz,
Marrakech, bUnité de Chimie Biomoléculaire et Médicinale, Faculté des Sciences Semlalia, Marrakech ,
cInstitut für Organische Chemie und Chemische Biologie, J.W. Goethe Universität, Max-von- Laue Str. 7,
60438 Frankfurt am Main, Germany
The aim of this work is the combination of the two “privileged pharmacophore” heterocycles
(quinazolinone and 1,2,3-triazole) using click reaction.1 The desired products have been
prepared starting from anthranilic acid in four steps via a modified Niementowski reaction.
Heterocyclic structures are basic elements of many pharmaceuticals, agrochemicals and
veterinary products. The quinazolinone derivatives are an important class of compounds, as
they are present in a large family of products with broad biological activities. For example:
anticancers and diuretics.2-3
Also, triazoles associated with various heterocycles are one of
the research areas of interesting pharmacological activities, some analogues are used for
the treatment of hepatitis C and HIV-1.4-5
The desired products have been prepared starting
from anthranilic acid in five steps (Scheme).
Scheme: Synthesis of triazol-4-yl substituted withquinazoline derivatives.
One of these products was crystallized [R1= Phenyl, R2= -(CH2)4OH] by slow evaporation of a methanol/methylene chloride solution. X-ray analysis confirms the structure found (Figure).
6
Figure: Three dimensional view of the crystal as hemihydrate
References: 1. Krim J.; Sillahi B.; Taourirte M.; Rakib E. M.; Engels J. W. ARKIVOC, 2009, xiii, 142. 2. Chan J. H.; Hong J. S.; Kuyper L. F.; Jones M. L.; Baccanari D. P.; Tansik R. L.; Boytos C. M.; Rudolph S. K.; Brown A. D. J. Heterocycl. Chem. 1997, 34, 145. 3. Gackenheimer S. L.; Schaus J. M.; Gehlert D. R. J. Pharmacol. Exp. Ther. 1996, 732, 113. 4. De Clercq, E. Nat. Rev. Drug Discovery, 2002, 1, 13. 5. Alvarez R.; Velazquez S.; San-Felix A.; Aquaro S.; De Clercq E.; Perno C. F.; Karlsson A.; Balzarini J.; Camarasa M. J. J. Med. Chem. 1994, 37, 4185. 6. Ouahrouch A.; Lazrek H. B.; Taourirte M.; El Azhari M.; Saadid M.; El Ammari L. Acta Cryst. 2011, E67, o2029.

PC193
Synthesis, structure, spectroscopic and thermal properties of some
macrocyclic complexes
Nagihan Ersoy, Murat Turkyilmaz
Department of chemistry, Trakya University, Edirne 22000, Turkey
Azomethine complexes are studied for their antitumor, antimicrobial, antiviral, catalytic,
enzymatic and mesogenic characteristics. Azomethine metal complexes are also a focus for
scientific interest, due to their important role in biological systems, and represent an
interesting model for metalloenzymes which efficiently catalyze the reduction of oxygen.
Salicylaldehyde derivatives of azomethine compounds show a variety of biological activities,
such as antibacterial activity. In this study, new macrocyclic coordination compound will be
obtained from different diamino and dialdehayde compounds with transition metal ion
controlled Schiff base condensations. Macrocyclic ligands will be synthesized from the
pyridine based compounds.1-4
New coordination compounds will be obtained with the reaction of these ligands and
different some metal salts. The structures of synthesised ligands and their complexes will be
enlightened by using data obtained from LT MALDI-TOF MS, IR, 1H,
13C spectrums, DTA
and elementel analysis.
NOO
O O
NCH3CH2CH2CH2OH
SOCl2
HO
O
OH
O NHO OH
NaBH4
CH3CH2OH
NS
O
O
S
O
O
CH 2Cl 2
, KOH
p-tolu
ene sulfo
nyl
c
hloride
O O
O
NO O
O O
N
S S
SH
NH2
NH2 H2N
+
OH
N
S
SN
NO
N
O
References: 1. H. Masuda, Eur. J. Inorg. Chem, 2006, 3753. 2. A. S. Gaballa, M. S Asker, A. S. Bakarat, S. B. Teleb, Spectrochim. Acta, Part A 67 (2007) 114. 3. E. Keskinoglu, A.B. Gunduzalp, S. Cete, F. Hamurcu, B. Erk, Spectrochim. Acta, Part A 70 (2008) 634. 4. A. Pui, J. Pieere, Polyhedron 26 (2007) 3143.

PC194
Synthesis of New Heterocyclic-Linked Bis-Indole Systems
I. Fazil Sengul,a N. Kumar,
b D. StC. Black
b
aDepartment of chemistry, Gebze Institute of Technology, P. O. Box 141. 41400, Gebze, Kocaeli-Turkey
bDepartment of chemistry, The University of New South Wales, NSW 2052, Sydney, Australia
The indole or 2,3-benzopyrrole nucleus has been subjected to intense study over the last
fourteen decades. The indoles are a class of heterocyclic compounds, widely found in
nature, whose derivatives therefore have a strong link to natural products. These natural
indolic compounds and their synthetic analogues show a wide range of biological activities.1
More complex indoles, such as bis-indoles are very important biologically active scaffolds as
they are found in many pharmacologically active alkaloids. Bis-indole alkaloids are
heterocyclic compounds, which consist of two indoles connected to each other via linking
units.2
In addition to this, macrocyclic bis-indole systems are an important and prolific
structural class and possess interesting biological properties. The general aim of the work
described in this project was to develop dibenzofuran and carbazole linked bis-indole
compounds and explore their potential as building blocks to larger macrocyclic structures.
Specifically, 3,6-bis-(2-indolyl)heteroarenes 13 and carbazoles and 3,6-bis-(3-indoly)dibenzo
furans 24 which could undergo electrophilic substitutions and additions at the vacant C3 or
C2 position respectively were targeted. 3,6-bis-(3-indoly)dibenzofurans were also exploited
in order to provide additional reactivity at the C7 position.
.
References
1. Pindur, U.; Lemster, T. Curr. Med. Chem. 2001, 8, 1681-1698.
2. Gibbs, T. J. K.; Tomkinson, N. C. O. Org. Biomol. Chem. 2005, 3, 4043-4045.
3. Sengul, I., F.; Wood, K.; Kumar. N.; Black, D., StC.; Tetrahedron, 2012, 68, 9050-9055.
4. Sengul, I., F.; Wood, K.; Bowyer, P., K.; Bhadbhade, Rui Chen M.; Kumar, N.; Black, D., StC.;
Tetrahedron, 2012, 68, 7429-7434.

PC195
Synthesis of Halo-Indenones:
Gold-Catalysed Oxidative Diyne Cyclisations
Laura Nunes dos Santos Comprido, A. Stephen K. Hashmi
University of Heidelberg, Institute of Organic Chemistry, 69120 Heidelberg, Germany
[email protected], [email protected]
The gold-catalysed reaction of diynes is a research topic of current interest.1 We have
developed a method for the cyclisation of halodiynes to halo-indenones using simple
Ph3PAuNTf2 as a catalyst when combined with an N-oxide. Yields for this reaction are
excellent (up to 97%).
Scheme 1: Gold-catalysed halodiyne oxidative cyclisation.
An unprecedented2 halo-α-oxo carbenoid is proposed to be a key intermediate. The
obtained compounds are coloured and show strong UV-Vis absorptions. From TD-DFT
analysis of the involved orbitals a strong influence of the pendant alkene, which is not in
conjugation with the rest of the aromatic system, can be found.
Acknowledgements: L. N. d. S. C. gratefully acknowledges financial support from the ERASMUS programme. References: 1. For examples of Gold-Catalysed Diyne Cyclisations see: W. Rao, M. J. Koh, D. Li, H. Hirao and P. W. H. Chan, J. Am. Chem. Soc., 2013, 135, 7926-7932, A. S. K. Hashmi, M. Wieteck, I. Braun, P. Nösel, L. Jongbloed, M. Rudolph and F. Rominger, Adv. Synth. Catal., 2012, 354, 555-562, A. S. K. Hashmi, I. Braun, P. Nösel, J. Schädlich, M. Wieteck, M. Rudolph and F. Rominger, Angew. Chem. Int. Ed., 2012, 51, 4456-4460, A. S. K. Hashmi, I. Braun, M. Rudolph and F. Rominger, Organometallics, 2012, 31, 644-
661. 2. Laura Nunes dos Santos Comprido, MChem Advanced Project Report, The University of York (June 2013) York, U.K.

PC196
List of Participants
A Abdalilah, M Chemistry Department, McGill University Apt. 28, 3063 Durocher Street, H2X 2E8, Montreal Quebec, Canada H2X 2E8, Montréal, Canada [email protected] Abdelaaziz, Q Chemistry, Faculty of Sciences and Technics Guéliz Laboratoire de Chimie Bio-organique et Macromoléculaire, Faculté des Sciences et Techniques Guéliz, Marrakech 7000-671, Marrakech, Morocco [email protected] Abreu, ACR Chemistry Department, Luzitin, SA Edificio Bluepharma, São Martinho do Bispo 3045-016, Coimbra, Portugal [email protected] Afonso, CAM iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Akbas, E Chemistry Department, Yuzuncu Yil Universty Science Faculty 65100, VAN, Turkey [email protected] Albuquerque, HMT Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Almeida, AF Chemistry and Biochemistry Department, Faculty of Science, University of Lisbon Faculdade de Ciências da Universidade de Lisboa, Campo Grande, Edificio C8, 5º piso 1749-016, Lisboa, Portugal [email protected] Almeida, ARM Chemistry Department, University of Coimbra Rua Larga 3004-535, Coimbra, Portugal [email protected] Alves, LFG Chemistry Department, University of Évora Rua Romão Ramalho 59 7000-671, Évora, Portugal [email protected]

Alves, LPM Chemistry Department, University of Beira Interior Rua Marquês d'Ávila e Bolama 6201-001, Covilhã, Portugal [email protected] Antonio, JPM iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Antunes, AMM Centro de Química Estrutural, Instituto Superior Técnico Avenida Rovisco Pais, nº1 1049-001 , Lisboa, Portugal [email protected] Azevedo, JFC Chemistry Department, Faculty of Science, University of Porto Rua do Campo Alegre, 687 4169-007, Porto, Portugal [email protected]
B Baptista, RMF Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Barata, JFB Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Barata, PAMD Department of Chemical Engineering, Lisbon Superior Engineering Institute Rua Conselheiro Emídio Navarro, 1 1959-007, Lisboa, Portugal [email protected] Barbosa, D AtralCipan,Portugal [email protected] Barreto, MC DCTD, University of Azores Rua da Mãe de Deus 58 9501-801, Ponta Delgada, Portugal [email protected]

Barroso, CRM Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Batista, DAD Chemistry and Biochemistry Department, Faculty of Science, University of Lisbon Faculdade de Ciências da Universidade de Lisboa, Campo Grande, Edificio C8, 5º piso 1749-016, Lisboa, Portugal [email protected] Begouin, AAB Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Benfeito, EST Chemistry and Biochemistry Department, Faculty of Science, University of Porto Rua do Campo Alegre, s/n 4169-007, Porto, Portugal [email protected] Benohoud, M Chemistry Department, ICSN - CNRS Institut de Chimie des Substances Naturelles ICSN - CNRS1 Avenue de la Terrasse, bat 27 91198, Gif-sur-Yvette, France [email protected] Beynek, H Chemistry Department, Trakya University Balkan Yerleskesi 22030, Edirne, Turkey [email protected] Beynek, N Chemistry Department, Faculty of Science, Trakya Univeristy 22030, EDIRNE, Turkey [email protected] Bode, JW Departements Chemie und Angewandte Biowissens, ETH Zürich HCI F315, Lab. für Organische Chemie, Wolfgang-Pauli-Str. 10 8093, Zürich, Swiss [email protected] Bogel-Lukasik, E Chemistry Department, FCT, New University of Lisbon Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Quinta da Torre, Monte da Caparica 2829-516, Caparica, Portugal [email protected]

Borges, RS Pharmacy Department, Federal University of Pará Av Perimetral SN, Laboratório de Química Farmacêutica, Bairro do Guamá 66075-110, Belém, Brazil [email protected] Braga, AL Chemistry Department, Federal University of Santa Catarina Campus Trindade - UFSC, Carvoeira 88040-900, Florianopolis, Brazil [email protected] Branco, LAAFC REQUIMTE, Chemistry Department, FCT, New University of Lisbon Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Quinta da Torre, Monte da Caparica 2829-516, Caparina, Portugal [email protected] Branco, PCS Chemistry Department, FCT, New University of Lisbon Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Quinta da Torre, Monte da Caparica 2829-516, Caparica, Portugal [email protected] Brito, AMF Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Burke, AJ Chemistry Department, University of Évora Rua Romão Ramalho, 59 7000-671, Evora, Portugal [email protected] Burtoloso, ACB University of São Paulo Av. Trab. São-carlense SP CEP 13560-970, São Paulo, Brazil [email protected]
C Cachatra, VMC Chemistry and Biochemistry Department, Faculty of Science, University of Lisbon Faculdade de Ciências da Universidade de Lisboa, Campo Grande, Edificio C8, 5º piso 1749-016, Lisboa, Portugal [email protected]

Cal, PMSD iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Calvete, MJF Chemistry Department, University of Coimbra Rua Larga 3004-535, Coimbra, Portugal [email protected] Candeias, NFR Department of Chemistry and Bioengineering, Tampere University of Technology Korkeakoulunkatu 8 FI-33101, Tampere, Finland [email protected] Cardoso, ICS Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Cardoso, ZS Chemistry Department, University of Coimbra Rua Larga 3004-535, Coimbra, Portugal [email protected] Carrasco, MAP iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Carrega, JAM Chemistry Department, University of Évora Rua Romão Ramalho, 59 7000-671, Évora, Portugal [email protected] Carreiro, EP Chemistry Department, University of Évora Colégio Luís António Verney, Rua Romão Ramalho, 59 7000-671, Évora, Portugal [email protected] Carvalho, AMAV Chemistry and Biochemistry Department, Faculty of Science, University of Lisbon Faculdade de Ciências da Universidade de Lisboa, Campo Grande, Edificio C8, 5º piso 1749-016, Lisboa, Portugal [email protected]

Carvalho, CM Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Carvalho, LCCR REQUIMTE, Chemistry Department, FCT, New University of Lisbon Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Quinta da Torre, Monte da Caparica 2829-516, Caparica, Portugal [email protected] Carvalho, MMT Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Carvalho, SL Chemical Science, Faculty of Pharmacy, University of Porto Rua de Jorge Viterbo Ferreira, nº 228 4050-313, Porto, Portugal [email protected] Castellano, EM Organic and Inorganic Chemistry, University of Extremadura Avda de Elvas s/n 6006, Badajoz, Spain [email protected] Castro, MCR Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Castro, VIB Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Cavaleiro, JAS Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Coelho, JAS iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected]

Coelho, PJS Chemistry Department, University of Trás-os-Montes and Alto Douro Quinta dos Prados, Apartado 1013 5001-801, Vila Real, Portugal [email protected] Comprido, LNS Institute of Organic Chemistry, University of Heidelberg 69120 Heidelberg, Germany [email protected] Correira, CRD Chemistry Department, Inst. Chemistry of the State University of Campinas Caixa Postal 6154, Sala D-314 13083-862, Campinas, Brazil [email protected] Costa, AIMP Department of Chemical Engineering, Lisbon Superior Engineering Institute Rua Conselheiro Emídio Navarro, 11959-007 Lisboa 1959-007, Lisboa, Portugal [email protected] Costa, AMSR Department of Chemistry and Pharmacy, University of Algarve Faculty of Science and TechnologyCampus de Gambelas 8005-139, Faro, Portugal [email protected] Costa, MSF Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Cravo, SMMS Chemistry Department, Faculty of Pharmacy, University of Porto Rua de Jorge Viterbo Ferreira, nº 228 4050-313, Porto, Portugal [email protected] Cunha, SDA Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected]
D Daskapan, T Chemistry Department, Ankara University Science Faculty Ankara University Science Faculty, 06100 Besevler 6100, Ankara, Turkey [email protected]

De, CK Homogeneous Catalysis, Max-Planck-Institut für Kohlenforschung Kaiser-Wilhelm-Platz 1 45470, Mülheim an der Ruhr, Germany [email protected] Dias, AFRM iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Dias, AME Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Dias, CAS Chemistry and Biochemistry Department, Faculty of Science, University of Lisbon Faculdade de Ciências da Universidade de Lisboa, Campo Grande, Edificio C8, 5º piso 1749-016, Lisbon, Portugal [email protected] Dias, IES Chemistry and Biochemistry Department, Faculty of Science, University of Porto Rua do Campo Alegre, s/n 4169-007, Porto, Portugal [email protected] Dias, TAFP Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Dixon, DJ Chemistry Department, University of Oxford Chemistry Research Laboratory, 12 Mansfield Road OX1 3TA, Oxford, United Kingdom [email protected] Djilali, B Chemistry Department, University of Sidi Bel Abbes Cite S6 N38, Benhamouda; Sidi Djillali 22000, Sidi Bel Abbes, Algeria [email protected] Domingues, MAG Chemistry Department, University of Beira Interior Rua Marquês d'Ávila e Bolama 6201-001, Covilha, Portugal [email protected]

Duarte, ARP iMed.UL - Medicinal Chemistry, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Duarte, N iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected]
E Ersoy, N Chemistry Department, Trakya University Balkan Yerleskesi 22030, Edirne, Portugal [email protected] Estevão, MAS Chemistry Department, FCT, New University of Lisbon Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Quinta da Torre, Monte da Caparica 2829-516, Caparica, Portugal [email protected] Esteves, CIC Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Esteves, PM Chemistry Department, Universidade Federal do Rio de Janeiro Centro de Tecnologia, Bl. A, sala 613 21941-901, Rio Janeiro, Brazil [email protected] Estevez-Cabanas, RJ Center for Research in Biological Chemistry A, University of Santiago de Compostela Calle Jenaro de la Fuente s/n 15782, Santiago de Compostela, Spain [email protected]
F Fagundes, SIG Chemistry Department, University of Trás-os-Montes and Alto Douro Quinta dos Prados, Apartado 1013 5001-801, Vila Real, Portugal [email protected]

Farias, GDVF iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Farinha, ASF Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Faustino, HMF Centro Singular de Investigación en Química B, University of Santiago de Compostela CIQUS, C/ Jenaro de la Fuente s/n (esquina Avda. Mestre Mateo), Campus Vida 15782, Santiago de Compostela, Spain [email protected] Ferdjani, S Pharmacy, University of Annaba Université de Annaba, Faculté de médecine, département de pharmacie 23000, Annaba, Algeria [email protected] Fernandes, ACS Centro de Química Estrutural, Instituto Superior Técnico Avenida Rovisco Pais, nº1 1049-001 , Lisboa, Portugal [email protected] Fernandes, ALMAP Chemistry and Biochemistry Department, Faculty of Science, University of Porto Rua do Campo Alegre, s/n 4169-007, Porto, Portugal [email protected] Fernandes, SSM Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Ferreira, LP Chemistry Department, FCT, New University of Lisbon Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Quinta da Torre, Monte da Caparica 2829-516, Caparica, Portugal [email protected] Ferreira, MJU iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected]

Ferreira, PMVST Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Ferreira, RCM Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Ferreira, RJDG iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Ferreira, VF Chemistry Department, Federal University of Fluminense Rua Outeiro de São João Batista, s/n, Prédio da Física Velha, sala 100, Campus do Valonguinho - Centro Niterói, Brazil [email protected] Figueira, FAS Department of Chemistry and QOPNA, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Figueiredo, JAA Chemistry Department, University of Beira Interior Rua Marquês d'Ávila e Bolama 6201-001, Covilhã, Portugal [email protected] Francisco, APG iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Francisco, CS Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Freitas, VAP Chemistry Department, Faculty of Science, University of Porto Rua do Campo Alegre, s/n 4169-007, Porto, Portugal [email protected]

Frija, LMT iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected]
G Gademann, K Chemistry Department, University of Basel St. Johanns-Ring 19 CH-4056, Basel, Swiss [email protected] Gharib, A Organic chemistry, Agricultural Research & Service and Islamic Azad University 1st floor,No. 40, 16 Ghaem St, Ahmad Abad St, Mashhad, Iran 98511, Mashhad, Islamic Republic of Iran [email protected] Góis, PMP iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Goksu, S Chemistry Department, Ataturk University Atatürk University, Faculty of Sciences 25240, Erzurum, Turkey [email protected] Goksu, S Chemistry Department, Ataturk University Atatürk University Faculty of Science Department of Chemistry 25240, Erzurum, Turkey [email protected] Gomes, ATPC Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Gomes, LFR iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Gomes, PAC Chemistry and Biochemistry Department, Faculty of Science, University of Porto Rua do Campo Alegre, 687 4169-007, Porto, Portugal [email protected]

Gonçalves, AMAR Chemistry Department, University of Coimbra Rua Larga 3004-535, Coimbra, Portugal [email protected] Goth, AJB Chemistry Department, University of Évora Rua Romão Ramalho, 59 7000-671, Évora, Portugal [email protected] Govindasami, P Centro de Quimica-Fisica Molecular, Instituto Superior Técnico Avenida Rovisco Pais, nº1 1049-001, Lisboa, Portugal [email protected] Graça, VCSS Chemistry Department, University of Trás-os-Montes and Alto Douro Quinta dos Prados, Apartado 1013 5001-801 , Vila Real, Portugal [email protected] Granada, AMC iMed.UL - Química Farmacêutica e Terapêutica, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Grilo, JHF iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Guerra, KAP iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Guieu, S Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected]
I Iglesias, BA Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected]

Isca, VMS Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Ismael, MIGC Química Unidade I&D MTP, University of Beira Interior Rua Marquês d'Ávila e Bolama 6201-001, Covilhã, Portugal [email protected]
J Jesus, ARX Chemistry and Biochemistry Department, Faculty of Science, University of Lisbon Faculdade de Ciências da Universidade de Lisboa, Campo Grande, Edificio C8, 5º piso 1749-016, Lisboa, Portugal [email protected] Jordão, N Chemistry Department, FCT, New University of Lisbon Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Quinta da Torre, Monte da Caparica 2829-516, Caparica, Portugal [email protected] Josa, D Physical chemistry, University of Santiago de Compostela CIQUS, Avda. Jenaro de la Fuente s/n 15782, Santiago de Compostela, Spain [email protected] Júnior, LCK Laboratório de Farmacognosia, Federal University of Rio Grande do Sul Avenida Ipiranga, 2752 90610000, Porto Alegre, Brazil [email protected]
Junior, LFS Chemistry Department, University of São Paulo Av. Prof. Lineu Prestes, 748 - Butantã 05508-000, São Paulo, Brazil [email protected]
K Karatavuk, AO Chemistry Department, Faculty of Science, Trakya Univeristy 22030, Edirne, Turkey [email protected] Kazaz, C Chemistry Department, Ataturk University Atatürk University, Faculty of Sciences 25240, Erzurum, Turkey [email protected]

Kelebekli, L Chemistry Department, Ordu University 52200, Ordu, Turkey [email protected] Kim, JH Homogeneous Catalysis, Max-Planck-Institut für Kohlenforschung Kaiser-Wilhelm-Platz 1 45470, Mülheim an der Ruhr, Germany [email protected] Kiss, LE Bial, Portugal [email protected] Korkmaz, A Chemistry Department, Ankara University Science Faculty Ankara University Science Faculty, 06100 Besevler 6100, Ankara, Turkey [email protected] Krikstolaityte, S Department of Organic Chemistry, Kaunas University of Technology Radvilenu pl. 19 LT-50254, Kaunas, Lithuania [email protected]
L Laia, FMR Chemistry Department, University of Coimbra Rua Larga 3004-535, Coimbra, Portugal [email protected]
Lana, JNA iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] León, MAS Organic and Inorganic Chemistry, University of Extremadura Avda. Elvas s/n 6006, Badajoz, Spain [email protected] Li, C-J Chemistry Department, McGill University Otto Maass Chemistry Building, 801 Sherbrooke Street West H3A0B8 , Montreal, Canada [email protected]

Lima, JMCS Chemistry Department, Faculty of Science, Agostinho Neto University Rua Cerveira Pereira, 31 4 Esquerdo 2860, Luanda, Angola [email protected] Lima, MRMBVC Chemistry Department, ITQB, New University of Lisbon ITQB-UNL,Av. Republica, EAN 2780-157, Oeiras, Portugal [email protected] List, B Homogeneous Catalysis, Max-Planck-Institut für Kohlenforschung Kaiser-Wilhelm-Platz 1 45470, Mülheim an der Ruhr, Germany [email protected] Lobo, AMFT Chemistry Department, FCT, New University of Lisbon Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Quinta da Torre, Monte da Caparica 2829-516, Caparica, Portugal [email protected] Lopes, A Chemistry Department, University of Coimbra Universidade de Coimbra, Palácio dos Grilos, Rua da Ilha 3004-531, Coimbra, Portugal [email protected] Lopes, F. iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Lopes, SMM Chemistry Department, University of Coimbra Rua Larga 3004-535, Coimbra, Portugal [email protected]
Lourenço, LMO Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Lourenço, NMT BioEngenharia, Instituto Superior Técnico Avenida Rovisco Pais, nº1 1049-001, Lisboa, Portugal [email protected]

Lucas, SD iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected]
M Madeira, PJA iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Madureira, AM iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Magalhães, JRP iMed.UL - Medicinal Chemistry, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Marcos, PMJ CCMM Química Farmacêutica e Terapêutica, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Marinho, EMR Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Marques, CS Chemistry Department, University of Évora Colégio Luís António Verney, Rua Romão Ramalho, 59 7000-671, Évora, Portugal [email protected] Marques, MMMSB Chemistry Department, FCT, New University of Lisbon Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Quinta da Torre, Monte da Caparica 2829-516, Caparica, Portugal [email protected] Marques, MSM iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Martins, AIM

Chemistry and Biochemistry Department, Faculty of Science, University of Lisbon Faculdade de Ciências da Universidade de Lisboa, Campo Grande, Edificio C8, 5º piso 1749-016, Lisboa, Portugal [email protected] Martins, ANC iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Martins, APR Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Maulide, N Homogeneous Catalysis, Max-Planck-Institut für Kohlenforschung Kaiser-Wilhelm-Platz 1 45470, Mülheim an der Ruhr, Germany [email protected] Máximo, PIS Chemistry Department, FCT, New University of Lisbon Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Quinta da Torre, Monte da Caparica 2829-516, Caparica, Portugal [email protected] Melo, TMVDP Chemistry Department, University of Coimbra Rua Larga 3004-535, Coimbra, Portugal [email protected] Mendonça, R Hovione, Portugal [email protected] Menzek, A Chemistry Department, Ataturk University Atatürk University, Faculty of Sciences 25240, Erzurum, Turkey [email protected] Mesquita, MQ Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Miguel, ASC

Bioorganic Chemistry, ITQB, New University of Lisbon Av. da República 2780-157, Oeiras, Portugal [email protected] Miranda, DFP iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Monaco, MR Homogeneous Catalysis, Max-Planck-Institut für Kohlenforschung Kaiser-Wilhelm-Platz 1 45470, Mülheim an der Ruhr, Germany [email protected] Montalbano, F iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Monteiro, AFA iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Monteiro, CMFT iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Moreira, AMD University of Beira Interior Rua Marquês d'Ávila e Bolama 6201-001, Covilha, Portugal [email protected] Moreira, R iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Moura, NMM Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Murtinho, DMB

Chemistry Department, University of Coimbra Rua Larga 3004-535, Coimbra, Portugal [email protected]
N Neto, SCS iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Neves, CMB Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193 , Aveiro, Portugal [email protected] Niyomchon, S Homogeneous Catalysis, Max-Planck-Institut für Kohlenforschung Kaiser-Wilhelm-Platz 1 45470, Mülheim an der Ruhr, Germany [email protected] Nunes, J Chemistry Department, University College London 20 Gordon St., WC1H 0AJ, London, UK WC1H 0AJ, London, United Kingdom [email protected] Nunes, RS Chemistry and Biochemistry Department, Faculty of Science, University of Lisbon Faculdade de Ciências da Universidade de Lisboa, Campo Grande, Edificio C8, 5º piso 1749-016, Lisboa, Portugal [email protected]
O Oliveira, ACG Chemistry and Biochemistry Department, Faculty of Science, University of Porto Rua do Campo Alegre s/n 4169-007, Porto, Portugal [email protected] Oliveira, MS iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Oliveira, RMS iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Ortet, OAL

Chemistry and Biochemistry Department, Faculty of Science, University of Lisbon Faculdade de Ciências da Universidade de Lisboa, Campo Grande, Edificio C8, 5º piso 1500-175, Lisboa, Portugal [email protected]
P Paiva, APP Chemistry and Biochemistry Department, Faculty of Science, University of Lisbon Faculdade de Ciências da Universidade de Lisboa, Campo Grande, Edificio C8, 5º piso 1749-016, Lisboa, Portugal [email protected] Panciera, P Chemistry Department, University of Santiago de Compostela CIQUS, C/ Jenaro de la Fuente s/n, Campus Vida, CP 15782 , Santiago de Compostela, Spain [email protected] Paterna, A iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Paterna, R iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Patrão, MA Chemical Science, Faculty of Pharmacy, University of Porto Rua de Jorge Viterbo Ferreira, nº 228 4050-313, Porto, Portugal [email protected] Peixoto, DAS Chemistry Department, University of Évora Rua Romão Ramalho 59 7000-671, Évora, Portugal [email protected] Pereira, ACAA Chemical Science, Faculty of Pharmacy, University of Porto Rua de Jorge Viterbo Ferreira, nº 228 4050-313, Porto, Portugal [email protected] Pereira, AMVM Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Pereira, CPF

Chemistry Department, QOPNA and CICECO Universidade de Aveiro, Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Pereira, DMV Chemical Science, Faculty of Pharmacy, University of Porto Rua de Jorge Viterbo Ferreira, nº 228 4050-313, Porto, Portugal [email protected] Pereira, ECS Chemistry and Biochemistry Department, Faculty of Science, University of Porto Rua do Campo Alegre, s/n 4169-007, Porto, Portugal [email protected] Pereira, JDFM iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Pereira, MM Chemistry Department, University of Coimbra Rua Larga 3004-535, Coimbra, Portugal [email protected] Pereira, MMMA REQUIMTE, Chemistry Department, FCT, New University of Lisbon Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Quinta da Torre, Monte da Caparica 2829-516, Caparica, Portugal [email protected] Pereira, NAL iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Pereira, NAM Chemistry Department, University of Coimbra Rua Larga 3004-535, Coimbra, Portugal [email protected] Pereira, PMR Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Pereira, RAG

Organic Chemistry, University of Seville C/ Profesor García González 1 41012, Sevilla, Spain [email protected] Phillips, AMMMF Chemistry Department, FCT, New University of Lisbon Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Quinta da Torre, Monte da Caparica 2829-516, Caparica, Portugal [email protected] Pinaka, A Institute of Physical Chemistry, NCSR Demokritos Patriarchou Gregoriou and Neapoleos Str. 15310, Athens, Greece [email protected] Pinto, DCGA Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Pinto, JFG Department of Chemistry and QOPNA, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Pinto, MMM Chemistry Department, Faculty of Pharmacy, University of Porto Rua de Jorge Viterbo Ferreira, nº 228 4050-313, Porto, Portugal [email protected] Pinto, RMA Medicina Veterínária, Escola Universitária Vasco da Gama Mosteiro S. Jorge de Milréu, Estrada da Conraria 3040-714, Coimbra, Portugal [email protected] Pires, MJD Chemistry Department, FCT, New University of Lisbon Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Quinta da Torre, Monte da Caparica 2829-516, Caparica, Portugal [email protected] Pires, SMG Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Porto, ALM

Physical chemistry, Inst. of Chemistry of Sao Carlos, University of São Paulo Avenida João Dagnone, 1100, Ed. Química Ambiental, Jardim Santa Angelina 13563-120, São Carlos, Brazil [email protected] Proença, MFJRP Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected]
Q Queiroz, MJRP Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Quintanova, CIR Centro Química Estrutural, Instituto Superior Técnico Avenida Rovisco Pais, nº1 1049-001, Lisboa, Portugal [email protected] Quntar, AA Material of Engineering, Al Quds University Cremisan St., Cremisan, Jerusalem, Po box: 73319, Pelestinian authority via Israel 73319, Jerusalem, Israel [email protected]
R Ramos, SS Chemistry Department, University of Beira Interior Rua Marquês d'Ávila e Bolama 6201-001, Covilhã, Portugal [email protected] Raposo, MMM Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Rauter, AP Chemistry and Biochemistry Department, Faculty of Science, University of Lisbon Faculdade de Ciências da Universidade de Lisboa, Campo Grande, Edificio C8, 5º piso 1749-016, Lisboa, Portugal [email protected] Rebelo, JSC iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Reino, AME

Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Reis, LV Chemistry Department, University of Trás-os-Montes and Alto Douro Quinta dos Prados, Apartado 1013 5001-801, Vila Real, Portugal [email protected] Reis, MA iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Resende, DISP Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Ressurreição, ASM iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Ribeiro, AIF Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Ribeiro, CJAC iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Ribeiro, SS IQSC-USP, Inst. of Chemistry of Sao Carlos, University of São Paulo Avenida João Dagnone, 1100, Ed. Química Ambiental, Jardim Santa Angelina 13563-120, São Carlos, Brazil [email protected]

Rijo, PDM Escola de Ciências e Tecnologias da Saúde, Universidade Lusófona de Humanidades e Tecnologias Campo Grande 376 1749-024, Lisbon, Portugal [email protected] Riss, B NOVARTIS Pharma AG Novartis Campus CH-4002, Basel, Switzerland [email protected] Rocha, AMR IBB, Instituto Superior Tecnico Avenida Rovisco Pais, nº1 1049-001, Lisboa, Portugal [email protected] Rocha, DHA Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Rocha, LC Fundamental Chemistry, University of São Paulo Instituto de Química de São Carlos, Universidade de São Paulo, Avenida Prof. Lineu Prestes, 748, Cidade Universitária 05508-900, São Saulo, Brazil [email protected] Rodrigues, AIGPP Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Rodrigues, CAB iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Rodrigues, JMM Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Rosa, APC iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected]

Rosa, JMRAN iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Rosatella, AA iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Roseiro, AIPS CQE - Centro de Química Estrutural, Instituto Superior Técnico Avenida Rovisco Pais, nº1 1049-001 , Lisboa, Portugal [email protected] Ruivo, EFP iMed.UL - Medicinal Chemistry, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected]
S Sackus, A Department of Organic Chemistry, Kaunas University of Technology Radvilenu pl. 19 LT-50254, Kaunas, Lithuania [email protected] Sahin, E Chemistry Department, Ataturk University Atatürk University, Faculty of Sciences 25240, Erzurum, Turkey [email protected] Saleh, AO Medicinal and Pharmaceutical Chemistry, National Research Center Tahreer Street-Dokki- Cairo- Egypt 12311, Cairo, Egypt [email protected] Sampaio, DMF Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Santos, BCS Chemistry Department, University of Coimbra Universidade de Coimbra, Palácio dos Grilos, Rua da Ilha 3004-531, Coimbra, Portugal [email protected]

Santos, EA Chemistry Department, Federal Technological University of Paraná Campus Apucarana, Rua Marcílio Dias, 635 86812-460, Apucarana-PR, Brazil [email protected] Santos, FMF iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Santos, MMM iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Santos, PPLO Department of Chemical Engineering, Instituto Superior Técnico Avenida Rovisco Pais, nº1 1049-001 , Lisboa, Portugal [email protected] Santos, SA iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Sebastião, JSV Chemistry Department, Faculty of Science, Agostinho Neto University Rua Comandante Kuenha, número 223, 1 Andar 2860, Luanda, Angola [email protected] Seca, AML DCTD, University of Azores Rua Mãe de Deus 9501-801, Ponta Delgada, Portugal [email protected] Selaimia-Ferdjani, Q Chemistry Department, University of Annaba Université de Annaba 23000, Annaba, Algeria [email protected] Sengul, IF Chemistry Department, Gebze Institute of Technology Gebze Institute of Technology, Deparment of Chemistry, 41400, Gebze, 41400, Kocaeli, Turkey [email protected]

Senhorães, NR Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Serra, MES Chemistry Department, University of Coimbra Rua Larga 3004-535, Coimbra, Portugal [email protected] Serra, PFA Chemistry and Biochemistry Department, Faculty of Science, University of Lisbon Faculdade de Ciências da Universidade de Lisboa, Campo Grande, Edificio C8, 5º piso 1749-016, Lisboa, Portugal [email protected] Shaker, NO Chemistry Department, Faculty of Science, Al Azhar University 4, Ali Amin Street Nasr City 11371, Cairo, Egypt [email protected] Silva, AFF Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Silva, EMP Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Silva, GALF Department of Chemistry and QOPNA, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Silva, MAS Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Silva, RSON Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected]

Silva, SAG Organic Synthesis Laboratory, ITQB, New University of Lisbon Av. da RepúblicaEstação Agronómica Nacional 2780-157, Oeiras, Portugal [email protected] Silva, VLM Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Simão, DEBTP Department of Chemical Engineering, Instituto Superior Técnico Avenida Rovisco Pais, nº1 1049-001, Lisboa, Portugal [email protected] Simeonov, SP iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Simões, AVC Chemistry Department, University of Coimbra Rua Larga 3004-535, Coimbra, Portugal [email protected] Simões, MAC Chemistry Department, University of Évora Rua Romão Ramalho 59 7000-671, Évora, Portugal [email protected] Siopa, FAD iMed.UL, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Smist, M West Pomeranian University of Technology Piastów 42 71-065, Szczecin, Poland [email protected] Soares, JFX Chemistry Department, Faculty of Pharmacy, University of Porto Rua de Jorge Viterbo Ferreira, nº 228 4050-313, Porto, Portugal [email protected]

Soares, MAS Chemistry Department, University of Minho Campus de Gualtar 4710-057, Braga, Portugal [email protected] Soares, MIL Chemistry Department, University of Coimbra Rua Larga 3004-535, Coimbra, Portugal [email protected] Sousa, ACC Área Departamental de Engenharia Química, Lisbon Superior Engineering Institute Rua Conselheiro Emídio Navarro, 1 1959-007, Lisboa, Portugal [email protected] Sousa, JLC Department of Chemistry and QOPNA, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Sousa, SCA Centro de Química Estrutural, Instituto Superior Técnico Avenida Rovisco Pais, nº1 1049-001, Lisboa, Portugal [email protected]
T Talhi, O Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Teixeira, APS Chemistry Department, University of Évora Rua Romão Ramalho, 59 7000-671, Évora, Portugal [email protected] Teixeira, CMB Department of Chemical Engineering, Lisbon Superior Engineering Institute Rua Conselheiro Emídio Navarro, 1 1959-007, Lisboa, Portugal [email protected] Teixeira, RAM Departamento de Bioengenharia, Instituto Superior Técnico Avenida Rovisco Pais, nº1 1049-001, Lisboa, Portugal [email protected]

Telo, JPNC DEQB, Instituto Superior Técnico Avenida Rovisco Pais, nº1 1049-001 , Lisboa, Portugal [email protected] Tomé JPC Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Tomé, SM Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected] Torres, NAL iMed.UL - Chemical Biology and Toxicology, Faculty of Pharmacy, University of Lisbon Av. Prof. Gama Pinto 1649-003, Lisboa, Portugal [email protected] Toste, FD Department of Chemistry , University of California MC 1460 CA 94720-1460, Berkeley, United States [email protected] Trindade, AF Centro de Quimica-Fisica Molecular, Instituto Superior Técnico Avenida Rovisco Pais, nº1 1049-001, Lisboa, Portugal [email protected] Tsacheva, IT Institute of Polymers, Bulgarian Academy of Sciences Acad. G. Bonchev, Str., 103-A 1113, Sofia, Bulgaria [email protected] Turkyilmaz, M Chemistry Department, Faculty of Science, Trakya Univeristy 22030, Edirne, Turkey [email protected] Tutar, A Chemistry Department, Sakarya University Sakarya University, Science Faculty, Department of Chemistry 54187, Serdivan, Turkey [email protected]

V Viana, HRM Chemistry Department, University of Évora Colégio Luís António Verney, Rua Romão Ramalho, 59 7000-671, Évora, Portugal [email protected] Victoria, AM School of Chemistry & Physics, University of KwaZulu-Natal H Block, 3rd Floor, PMB X54001, Westville Campus 4000, Durban, South Africa [email protected] Vieira, LMLS Chemistry and Biochemistry Department, Faculty of Science, University of Lisbon Faculdade de Ciências da Universidade de Lisboa, Campo Grande, Edificio C8, 5º piso 1749-016, Lisboa, Portugal [email protected] Vinagreiro, CS Chemistry Department, University of Coimbra Rua Larga 3004-535, Coimbra, Portugal [email protected] Vodenicharova, EP Institute of Polymers, Bulgarian Academy of Sciences Acad. G. Bonchev, Str., 103-A 1113, Sofia, Bulgaria [email protected]
W Wani, MY Chemistry Department, University of Coimbra Rua Larga 3004-535, Coimbra, Portugal [email protected] Wessel, HP Chemistry Department, University of Aveiro Campus Universitário de Santiago 3810-193, Aveiro, Portugal [email protected]
X Xavier, NMRM Chemistry and Biochemistry Department, Faculty of Science, University of Lisbon Faculdade de Ciências da Universidade de Lisboa, Campo Grande, Edificio C8, 5º piso 1749-016 , Lisboa, Portugal [email protected]

Z Zadlo, A Institute of Organic Chemistry, Polish Academy of Sciences Kasprzaka 44/52 01-224, Warsaw, Poland [email protected] Zalewska, K REQUIMTE, Chemistry Department, FCT, New University of Lisbon Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Quinta da Torre, Monte da Caparica 2829-516, Caparica, Portugal [email protected]

List of Authors
A Abreu, A. R., IOC2, PC96, PC116 Abrunhosa, A., PC122 Adriano, G., PC148 Afonso, C., OC5 Afonso, C. A. M., FC12, FC16, FC21, PC24, PC39, PC46, PC80, PC86, PC87, PC89, PC104, PC135, PC162, PC179 Afonso, J., PC94 Afonso, S. S., PC138 Aguiar, P. B., PC126 Ahmadi, S., PC189 Akbaba, Y., PC119 Akbas, E., PC112 Akincioğlu, A., PC186 Albuquerque, H., PC4 Albuquerque, I., PC133 Almeida, A. R., PC96 Almeida, Adelaide, PC164 Almeida, Andreia, PC17, PC91 Almeida, M., PC75 Almeida, P., PC79, PC159, PC160, PC173, PC177 Almeida, R., PC15 Almeida, T., PC180 Altas, C. M., FC2 Alves, L., PC161 Alves, L. P., PC159 Amaral, J. D., FC20 Amat, M., PC168 Amorim, R., PC143 Amorín, M., PC56 Andrade, M. M., PC126 André, J., PC28 André, V., PC36, PC147, PC162 Angnes, R. A., SLB-PL1 Anjo, C., IOC7, PC154 Antunes, A. M. M., IOC13 Antunes, C., PC148 Arantes, A., PC131 Araújo, M. E. M., PC144, PC158 Araújo, P., PC132 Areias, F., PC37 Areias, L. R. P., PC147 Arnaut, L. G., PC116 Ascensão, L., PC131 Ascenso, J. R., PC65 Ascenso, O. S., IOC9 Athar, F., PC166 Audisio, D., FC8 Augusto, A. S., PC65 Ávalos, M., PC13, PC35 Azam, A., PC166,
Azevedo, C., OC5 Azevedo, J., PC157
B Babiano, R., PC13, PC35 Bakhtiari, L., PC189, Balaydin, H. T., PC119 Balci, M., PC134 Balci, N., PC97 Baleizão, C., PC66, Balo, R., PC187, Barata, J. F. B., PC20, PC44 Barata, P. D., PC64, Barbosa, D., IOC7 Barreto, M. C., IOC3 Barros, M. T., FC13 Barroso, C., PC28 Barrulas, P., PC120, PC161 Bastos, M. M., PC124 Batista, D., PC144 Batista, R. M. F., PC69 Bavetta, A., PC84 Bayón, J. C., PC96 Bayrak, Ç., PC129 Begouin, A., OC1, PC114 Belo, D., PC75 Belsley, M., PC26, PC30 Benfeito, S., PC143, PC175 Benito, J. M., PC154 Bernando, J. R., PC108 Berthet, J., IOC5 Beynek, H., PC190 Beynek, N., PC59 Bhat, A. R., PC166 Bidjou-Haiour, C., PC188 Black, D. S., PC194 Bode, J., PL3 Boechat, N., PC124 Bogel-Lukasik, E., PC94 Bogomilova, A., PC155 Bonifácio, V. D., PC126 Borges, A., PC170 Borges, C., PC144 Borges, F., PC143, PC175 Bosch, J., PC168 Boto, R. E. F., PC79, PC159, PC160, PC173 Bouzaouit, N., PC188 Braga, A. L., SLB-PL2 Branco, L. C., FC17, PC60, PC88 Branco, P. S., IOC1, PC50 Brea, J., PC37, Brito, A., PC83, Brito, M. J. V., PC3,

Bronze, M. R., PC182, Burke, A. J., IOC14, PC53, PC95, PC98, PC99, PC103, PC120, PC148, PC161, Burrows, H. D., FC18, Burtoloso, A. C. B., SLB-IOC5
C Cabaleiro-Lago, E. M., PC31, Cabral, M., PC157, Cabral, M. F., PC3, Cabrita, M. J., PC53, Cachatra, V., SLB-IOC3, PC17, PC91, PC184 Caddick, S., FC12 Cal, P. M. S. D., FC11, FC19, PC152 Calado, S., PC180, Caldeira, A. T., PC120, PC161 Calvete, M. J. F., IOC2, FC18, PC122 Campos, J. F., PC114 Campos, M., PC187 Candeias, N. R., FC16, PC107, PC162 Capela, R., PC145 Capelo, J. L., PC61 Carabineiro, S. A. C., IOC2 Cardoso, A. L., PC113 Cardoso, I. C. S., PC41 Carrasco, M. P., PC165 Carreiro, E. P., PC148 Carrera, G, FC17 Carrilho, R. M. B., IOC2 Carvalho, C. M., PC82, PC139 Carvalho, L. A. R., FC4 Carvalho, L. C. R., OC3, PC117 Carvalho, M., PC152, Carvalho, M. F. N. N., PC8, Carvalho, M. M. T., PC52, Carvalho, S., PC176, Castedo, L., PC56, Castro, M., PC37, Castro, M. C. R., PC26, Castro, V. I. B., PC82, PC139 Cavaleiro, J. A. S., SLB-PL3, FC10, PC4, PC14, PC18, PC20, PC29, PC40, PC44, PC45, PC61, PC110, PC118, PC124, PC164 Cerdeira, A. C., PC75, Cerqueira, F., PC127, Chankvetadze, B., PC6, Çiçek, S., PC111, Cidade, H., PC10, PC146, PC176 Cintas, P., PC13, PC35 Coelho, J. A. S., PC104 Coelho, P. J., IOC5 Comasseto, J. V., PC48 Comprido, L. N. S., PC195
Conceição, D. S., PC160 Constantino, L., PC180 Cordeiro, C., FC11 Čorić, I., PC42 Correia, C. R. D., SLB-PL1 Correia, H. F., FC4 Correia, J. P., PC29 Correia-da-Silva, M., PC121 Costa, A. C., PC126 Costa, A. I., PC7, PC23 Costa, A. M. R., IOC11, PC15 Costa, G. N., IOC2, PC116 Costa, J., PC3, Costa, L. C. B., PC131 Costa, M., FC3, PC12, PC37, PC81, PC83 Costa, M. C., PC15 Costa, N., PC51 Costa, S. P. G., PC25, PC30, PC69, PC82, PC130, PC139, Coutinho, J. T., PC75, Coutinho, P. J. G., PC52, Cravo, S., PC10, PC109, PC146, PC176 Cruz, L., PC49, Csuk, R., PC167, Cunha, Â., PC67, PC164 Cunha, S., PC74
D Dąbrowsk, J. M., PC116 Daşkapan, T., PC111 Daştan, A., PC134 De, C. K., FC14 Delbaere, S., IOC5 Dias, A. M., PC9, PC33, PC57, PC127 Dias, Ana, PC24 Dias, André, PC133 Dias, C., SLB-IOC3, FC5 Dias, C. P. R., PC30 Dias, T. A., PC81 Diaz, M., PC55 Dimotikali, D., PC6 Dindaroğlu, M., PC98 Dixon, D. J., PL2 Djeghaba, Z., PC191 Domingos, J., PC51 Domingues, M., PC106 Domingues, M. R. M., FC10 Duarte, A. R. P. , PC115, PC178 Duarte, M. T., PC36, PC147, PC162 Duarte, N., PC174
E Ekinci, D., PC119, Engels, J. W., PC192 Erdogan, A., PC112

Ersoy, N., PC193 Espadinha, M., PC50 Estevão, M. S., PC181 Esteves, A., PC28, PC74, PC140 Esteves, C. I. C., PC25 Esteves, P. M., SLB-IOC8 Estévez, J. C., PC187 Estévez, R. J., PC187
F Fagundes, S. G., PC160, PC173 Falé, P. L., PC131, PC144 Farinha, A. S. F., PC18, PC45 Faustino, H., FC7 Faustino, M. A. F., PC61, PC164 Ferdjani, S., PC191 Fernandes, A., FC1 Fernandes, A. C., OC2, PC47, PC108 Fernandes, Auguste, IOC2 Fernandes, E., OC3, PC117, PC181 Fernandes, I., PC157 Fernandes, J. A., PC63 Fernandes, J. R., PC173 Fernandes, M. R. C., PC18 Fernandes, M. X., IOC3 Fernandes, P., PC162 Fernandes, R., FC10, PC118 Fernandes, R. D. V., PC82 Fernandes, S. S. M., PC71 Fernández, J. M. G., PC154, PC171 Ferreira, D. P., PC160 Ferreira, I. M., PC150 Ferreira, L. F. V., PC160 Ferreira, L. M., PC50, PC170 Ferreira, M. J. U., SLB-IOC6, PC136, PC163, PC174, PC183 Ferreira, P. M. T., PC16, PC90 Ferreira, R., PC170 Ferreira, R. C. M., PC30, Ferreira, R. J., PC183, Figueira, F., PC22, PC40 Figueiredo, J. A., PC47, PC106, PC154 Figueiredo, J. L., IOC2 Filipe, S. R., PC5 Florindo, P., OC2 Fonseca, L. P., FC2 Formosinho, S. J., PC116 Fourmigué, M., PC75 Frade, R. F. M., FC21, PC162, PC179 Francisco, A. P., PC84, PC125, PC185 Francisco, C., PC140 Freitas, V., FC1, PC49, PC58, PC132, PC157 Frija, L. M. T., PC135, PC162
G Gabriel, C., PC127, Gademann, K., PL5 Galvão, A. M., PC8, Garcia, H., PC162, Garcia, R., PC53, García-Mera, X., PC156, García-Moreno, M. I., PC154, Garrido, J., PC143, PC175 Gaspar, A., PC143, PC175 Gaspar, M. M., PC180, PC185 Gaudêncio, S. P., PC126, Gawande, M. B., IOC1 Georgieva, A., PC149, Gerasimova, T., PC149, PC155 Gharib, A., PC189, Giester, G., PC77, Góis, P. M. P., FC11, FC16, FC19, PC36, PC96, PC107, PC152 Göksu, S., PC119, PC186 Gomes, A., SLB-IOC7 Gomes, A. S., PC109 Gomes, A. T. P. C., PC124, Gomes, C. S. B., PC21, PC38 Gomes, J. R. B., SLB-IOC7 Gomes, L. F. R., PC89, Gomes, P., SLB-IOC7, Gomez, A., PC169, Gonçalves, D., PC182, Gonçalves, L. M., FC4, PC133, PC141, PC147, PC152, PC165, Gonçalves, M. S. T., PC52, PC130 Gonçalves, N. P. F., PC116 Gonçalves, S., PC3 Gonçalves-Pereira, R., PC154, PC171 González-Veloso, I., PC31 Goth, A., PC95 Govindasami, P., PC66 Graça, V. C., PC177 Granada, A. M. C., PC125 Granja, J. R., PC56 Guedes, R. A., PC148 Guedes, R. C., FC4, PC147, PC148, PC152, PC182 Guerra, K. P., PC179 Guieu, S., PC153 Guldi, D. M., PC14 Gut, J., PC120, PC141, PC165
H Hashmi, A. S. K., PC195 Hausmann, A., PC14 Henriques, A. T., FC9 Henriques, C. A., IOC2, FC18 Henriques, R. T., PC75 Hillioub, L., PC90

Holzer, W., PC77
I Iglesias, B. A., PC20 Iley, J., PC115 Iliev, I., PC149, PC155 Ismael, M. I., PC47, PC106, PC154 Ivanov, I., PC155
J Jahangir, M., PC189 Jeannin, O., PC75 Jesus, A. R., PC11 Jiménez, J. L., PC13, PC35 Jordão, N., PC60 Josa, D., PC31
K Karatavuk, A. O., PC190 Kazaz, C., PC134 Kelebekli, L., PC97 Kerber, V. A., FC9 Keri, R., PC142, Kim, J. H., PC42, Kiss, L. E., IOC12 Klein-Júnior, L. C., FC9 Knittel, A. S. O., PC8 Korkmaz, A., PC111 Kraicheva, I., PC149, PC155 Krikštolaitytė, S., PC77 Kril, A., PC149, PC155 Kumar, N., PC194 Kumar, S. P., PC141 Kwiecień, H., PC93
L Lage, H., PC163 Lagzian, S., PC189 Laia, F. M. R., PC38 Lana, J. N., PC46 Lazrek, H. B., PC192 Leal, S. B., IOC4 Lemos, A., FC15, PC78, PC113 Leutzsch, M., PC55 Li, C.-J., PL1 Lima, C. F., PC81, PC90 Lima, R., OC5 List, B., PL4, FC14, PC42, PC55 Llop, J., PC122 Lobo, A. M., PC126 Lodeiro, C., PC61 Lopes, F., PC84, PC145, PC172, PC182 Lopes, P., PC157 Lopes, S. M. M., FC15, PC78 López, F., FC7 Lourenço, A., PC170
Lourenço, A. M., PC126 Lourenço, L. M. O., FC10, PC67 Lourenço, N. M. T., FC2, PC1, PC68, PC87 Louro, P., PC148 Loza, M., PC37 Lucas, S. D., FC4, PC147, PC152 Luparia, M., FC8
M M´Bana, V., PC126 Machado, J. F., PC3 Machura, B., PC108 Maciel, E., FC10 Maçôas, E. M. S., FC21, PC39 Madeira, P. A., PC144 Madeira, P. J. A., PC182 Magalhães, J., PC84 Magalhães, Joana, PC120 Marcos, P. M., PC65 Maria Soledade Santos, PC184 Maria, A., PC51 Marinho, E., PC34 Mariz, I. F. A., PC39 Marques, C. S., PC103 Marques, C. S., PC98, PC99 Marques, E. F., PC27 Marques, F., PC151 Marques, I. J., OC2 Marques, J. C., IOC9 Marques, M. M. B., OC3, PC5, PC117, PC181 Marques, S. M., PC142 Martelo, L., PC66 Martinho, J. M. G., FC21, PC24, PC39 Martins, A., PC16 Martins, A. N. C., PC135 Martins, Alice, SLB-IOC3, FC5, PC144, PC184 Martins, I., PC162 Martins, J. A., PC16, PC90 Martins, L. O., PC85 Martins, M., PC128 Martins, M. R., PC138 Martins, N., PC53 Mascareñas, J. L., FC7 Matamoros, E., PC35 Mateus, N., FC1, PC49, PC58, PC132, PC157 Matos, A. M., PC174 Matos, S., PC51 Maulide, N., SLB-IOC4, FC8, PC89 Máximo, P., PC170 Maycock, C. D., IOC9, PC73 Mazitschek, R., PC76 Mellet, C. O., PC154, PC171

Melo, C. I., PC94 Melo, J. S., PC69 Melo, T. M. V. D. P., SLB-IOC1, FC15, PC21, PC38, PC54, PC78, PC101, PC113 Mendes, M. E., PC125, PC185 Mendes, P. J., PC99, PC103 Mendonça, R., IOC10 Menezes, J. C. J. M. D. S., PC164 Menzek, A., PC119, PC129 Mesquita, M., PC164 Miguel, A. S., IOC9, PC123 Miranda, D., PC145 Mohadeszadeh, S., PC189 Momekov, G., PC155 Monaco, M. R., PC55 Montalbano, F., FC19, PC152 Monteiro, Â., PC137 Monteiro, C. J. P., IOC2, FC18, PC116 Monteiro, C. M., PC87 Monteiro, S., PC170 Moreira, R., SLB-PL4, FC4, FC20, PC76, PC84, PC115, PC133, PC141, PC145, PC147, PC152, PC165, PC172, PC178, PC182 Mota, M. M., PC133 Moura, N. M. M., PC61 Mulhovo, S., PC136 Murtinho, D., PC32, PC101
N Neto, S., PC174 Neves, A. C. B., IOC2, FC18 Neves, C. M. B., PC29, Neves, M. G. P. M. S., FC10, PC14, PC20, PC29, PC44, PC61, PC110, PC124, PC164 Niyomchon, S., FC8 Nogueira, C., PC15 Nunes, C. D., OC2 Nunes, J. P. M., FC12 Nuñez, C., PC61
O O’Neill, P. M., PC172 Ogihara, C., PC32 Oliva, C. G., PC100 Oliveira, C., PC143, PC175 Oliveira, C. C., SLB-PL1 Oliveira, M., PC180 Oliveira, M. C., PC24, PC85 Oliveira, O., PC90 Oliveira, P. J., PC143, PC175 Oliveira, R., PC172 Ornelas, I. M., PC29 Ortet, O., PC62
Ostaszewski, R., PC19 Ouahrouch, A., PC192
P Padilla, J., PC3 Pais, J., SLB-IOC3 Paiva, A. P., PC15, PC62 Palacios, J. C., PC13, PC35 Palmeira, A., OC5 Panciera, M., PC56 Papadopoulos, K., PC6 Passos, C. S., FC9 Paterna, A., PC136, PC163 Paterna, R., PC107 Patrão, M., PC121 Paulo, A., PC76 Paz, F. A. A., PC22, PC61, PC63 Pedro, M., OC5 Peixoto, D., OC1, PC99 Pereira, A., PC10 Pereira, A. M. V. M., PC14 Pereira, C. F., PC22 Pereira, C. S., PC162 Pereira, Cidália S., PC27 Pereira, D., PC146 Pereira, G., PC16, PC90 Pereira, J. D., PC185 Pereira, L. C. J., PC75 Pereira, M, PC51 Pereira, M. M., IOC2, FC18, PC96, PC116, PC122 Pereira, N. A. L., PC137, PC168 Pereira, N. A. M., FC15, PC78 Pereira, O., PC81 Pereira, P. M. R., FC10, PC118 Pereira-Lima, S. M. M. A., PC82, PC139 Pereira-Wilson, C., PC81 Pérez, B., SLB-IOC7 Perry, M. J., PC84, PC125, PC185 Pesciaioli, F., FC14 Phillips, A. M. F., FC13 Pina, F., PC60 Pina, J., PC69 Pinaka, A., PC6 Pineiro, M., IOC2 Pinheiro, P. F., PC8 Pinto, D. C. G. A., IOC3, IOC4, PC2, PC63, PC128 Pinto, J., PC105 Pinto, M., OC5, PC10, PC109, PC121, PC146, PC176 Pinto, R. M. A., IOC8 Pinto, S. M. A., FC18 Pires, M. J. D., OC3, PC5 Pires, S. M. G., PC110 Poladura, B., PC55 Polat, L., PC186

Ponte, M. N., FC17, PC94 Porto, A. L. M., PC48, PC70, PC150 Prata, J. V., PC7, PC23, PC64 Proença, M. F., FC3, PC9, PC12, PC33, PC34, PC37, PC57, PC81, PC83, PC127 Prudêncio, M., PC133
Q Queiroz, J. A., PC177 Queiroz, M. J. R. P., OC1, PC114 Quintanova, C., PC142 Quirion, J.-C., FC9
R Rabaça, S., PC75 Rabiller, C., PC191 Raju, B. R., PC52 Ramalho, T. C., PC31 Ramos, S. S., PC79, PC159 Raposo, M. M. M., SLB-IOC2, PC25, PC26, PC30, PC69, PC71, Rauter, A. P., SLB-IOC3, FC5, PC11, PC17, PC91, PC144, PC154, PC167, PC184 Rebelo, S. L. H., PC110, Reis, A. I. R., FC13 Reis, L. V., PC79, PC160, PC173, PC177 Reis, M., PC163 Reis, S., OC5 Resende, D. I. S. P., PC100 Ressurreição, A. S., PC115, PC133, PC178 Ribau, I. M. M., PC92 Ribeiro, A. I. F., PC127 Ribeiro, C. A. F., PC118 Ribeiro, C. J. A., FC20, PC141 Ribeiro, M. F., IOC2 Ribeiro, S. S., PC70 Rijo, P., PC169 Robalo, M. P., PC85 Rocha, A., PC1 Rocha, D. H. A., IOC3, IOC4, PC2 Rocha, J., PC153, Rocha, L. B., PC116 Rocha, L. C., PC48 Rodrigues, A., PC12 Rodrigues, C., PC162 Rodrigues, C. A. B., FC21, PC24, PC39 Rodrigues, C. M. P., FC20 Rodrigues, J. M. M., PC45 Rodrigues, L., PC140 Rodrigues, P., PC73 Rodrigues, T., PC182 Rodríguez-Borges, J. E., PC27, PC156 Rodríguez-Otero, J., PC31 Rosa, J. N., PC36
Rosatella, A. A., PC46, PC86 Roseira, I., PC157 Roseiro, A. P. S., PC8 Rosenthal, P. J., PC120, PC141, PC165 Roshani, M., PC189 Rui, F., IOC9 Ruivo, E. F. P., PC147
S Sá, S., IOC1 Šačkus, A., PC77 Şahin, E., PC119, PC129 Salvador, J. A. R., IOC8 Sampaio, D., PC33 Sampaio-Dias, I. E., PC156 Sanches, I. S., PC126 Sánchez, A. M., PC13 Sánchez-Fernández, E. M., PC171 Santos, B. S., PC21 Santos, C. I. C., PC27 Santos, C. M. M., PC4 Santos, D. J. V. A., PC165, PC183 Santos, F. M. F., PC36 Santos, I. C., PC75 Santos, I. C. M. S., PC110 Santos, L. A., PC31 Santos, L. M. N. B. F., PC105 Santos, M. A. l., PC142 Santos, M. M. M., FC20, PC137, PC141, PC168 Santos, M. N. B., PC66 Santos, M. S., SLB-IOC3 Santos, P. F., PC79, PC177 Santos, S., PC47 Santos, S. A., PC76 Santos-Filho, O. A., PC124 Saraiva, L., PC10, PC137, PC146, PC176 Schaberle, F. A., PC116 Schmalz, H.-G., PC98 Schuler, M., PC106 Schwalm, C. S., SLB-PL1 Schwarz, S., PC167 Seca, A. M. L., IOC4, PC128 Seixas, R. S. G. R., PC151 Selaïmia-Ferdjani, O., PC188 Sengul, I. F., PC194 Senhorães, N., PC9, PC57 Şenol, H., PC129 Serra, A. C., FC15 Serra, M. E. S., PC32, PC101 Serra, P., SLB-IOC3, PC184 Serralheiro, M. L., PC131, PC142, PC144 Silva, A. F. F., PC151 Silva, A. M., PC173 Silva, A. M. G., PC105

Silva, A. M. S., IOC3, IOC4, FC6, PC2, PC4, PC41, PC43, PC63, PC72, PC92, PC100, PC102, PC105, PC110, PC124, PC128, PC151, PC153 Silva, D. B., PC126 Silva, E., PC116 Silva, E. M. P., FC6 Silva, G., PC102 Silva, L. M. L., PC158 Silva, M., FC18 Silva, M. G., PC51 Silva, M. S. M. R., PC177 Silva, O. A. B. C., PC153 Silva, R. N., PC67 Silva, S., PC118 Silva, S. A. G., PC73 Silva, V. L. M., PC41, PC92, PC102, PC105 Simão, D., PC75 Simeonov, S. P., PC80, PC135 Simões, A., PC122 Simões, F., PC169 Simões, M., PC53 Simões, M. M. Q., PC110 Simões, S., PC116 Siopa, F., PC39 Sitoe, A. R. F., PC182 Śmist, M., PC93 Soares, A. M. S., PC130 Soares, J, OC5, PC137, PC176 Soares, M. I. L., PC54 Soares, P., PC175 Soares-da-Silva, P., IOC12 Sousa, A., PC132 Sousa, A. C., PC85 Sousa, C. M., IOC5 Sousa, D., OC5 Sousa, E., PC109, PC121 Sousa, F., PC159, PC177 Sousa, I. J., IOC3, Sousa, J. L. C., PC43 Sousa, S. C. A., PC108 Staniford, R. A., FC5 Sureda, F. X., PC168
T Talhi, O., PC63 Tan, N., PC59 Taourirte, M., PC192 Tatibouët, A., PC106 Teixeira, A. P. S., PC138 Teixeira, C., SLB-IOC7, PC7, PC23 Teixeira, F. A., PC65 Teixeira, F. C., PC138 Teixeira, J., PC143, PC175
Teixeira, N., PC58 Teixeira, R., PC68 Tellier, C., PC191 Telo, J. P., IOC6 Tomé, A. C., PC18, PC45, PC67, PC164 Tomé, J. P. C., OC4, FC10, PC14, PC22, PC40, PC45, PC118 Tomé, S. M., PC72 Topashka-Ancheva, M., PC149, PC155 Torres, N., PC3 Torres, T., PC14 Toste, F. D., PL6 Trindade, A. F., FC21, PC104 Trindade, T., PC44, PC45 Troev, K., PC149, PC155 Trukhina, O., PC14 Tsacheva, I., PC149, PC155 Turkyilmaz, M., PC193
U Urbani, M., PC14 Uriarte, E., PC143, PC175 Uriel, C., PC169
V Vale, M. L. C., PC27 Valente, E., PC180 Varandas, P. A. M. M., FC6 Vasconcellos, S. P., PC48 Vasconcelos, M., OC5 Veiros, L. F., FC11, PC36, PC152 Vellalath, S., PC42 Ventura, M. R., IOC9, PC123 Viana, A. S., PC29 Viana, H., PC103 Vicente, J., FC11 Vieira, A., PC128 Vieira, S. I., PC153 Vilaça, H., PC90 Vilela, S. M. F., PC22 Vodenicharova, E., PC149, PC155 Vougioukalakis, G. C., PC6
W Wani, M. Y., PC166 Wolff, M., PC108
X Xavier, K. B., IOC9, PC123 Xavier, N. M., PC167
Y Yannakopoulou, E., PC6
Z Zadlo, A., PC19 Zalewska, K., FC17, PC88


















