
Vpliv mikrovalov na stabilnost dvojne vijačnice DNK ... · časovnem okvirju niso imeli istega...
70
Magistrsko delo Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop Avgust, 2017 Uroš Veselič
Transcript of Vpliv mikrovalov na stabilnost dvojne vijačnice DNK ... · časovnem okvirju niso imeli istega...

Magistrsko delo
Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
Avgust, 2017 Uroš Veselič

Uroš Veselič
Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
Magistrsko delo
Maribor, 2017

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK:
Računalniški pristop
Magistrsko delo študijskega programa II. stopnje
Študent: Uroš Veselič
Študijski program: magistrski študijski program II. stopnje Kemija
Predvideni strokovni naslov: magister kemije
Mentor: izr. prof. dr. Urban Bren
Somentor: asist. Martin Gladović, mag. kem.
Maribor, 2017


Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
I
Kazalo
Kazalo ........................................................................................................................................ I Izjava........................................................................................................................................ II
Zahvala ................................................................................................................................... III Povzetek .................................................................................................................................. IV Abstract .................................................................................................................................... V Seznam tabel ........................................................................................................................... VI Seznam slik ........................................................................................................................... VII
Uporabljeni simboli in kratice ................................................................................................ IX
1 Uvod .................................................................................................................................. 1
1.1 Opredelitev problema ................................................................................................. 1 1.2 Pregled literature ........................................................................................................ 1 1.3 Namen, hipoteze in cilji ............................................................................................. 2
2 Teoretični del ..................................................................................................................... 3 2.1 Molekula DNK ........................................................................................................... 3
2.2 Mikrovalovi ................................................................................................................ 6
2.3 Metode računalniške kemije ...................................................................................... 8 2.4 Kvantno-kemijski modeli ........................................................................................... 9
2.4.1 Ab-initio metode ................................................................................................. 9
2.4.2 Semiempirične metode ..................................................................................... 11 2.5 Molekulska mehanika .............................................................................................. 12
2.5.1 Polje sil ............................................................................................................. 12
2.5.2 Monte Carlo simulacije..................................................................................... 18
2.5.3 Molekulska dinamika........................................................................................ 19 2.6 Uporabljeni računalniški programi .......................................................................... 21 2.7 Analiza rezultatov MD simulacij ............................................................................. 23
2.7.1 RMSD ............................................................................................................... 23 2.7.2 RMSF ................................................................................................................ 23
2.7.3 Analiza klastrov ................................................................................................ 24 3 Eksperimentalni del ......................................................................................................... 25
3.1 Računalniška oprema ............................................................................................... 25 3.2 Topologija odseka molekule DNK .......................................................................... 26
3.3 Ustvarjanje sistema map in podmap simulacij ......................................................... 27 3.4 Začetne minimizacije DNK molekule ..................................................................... 28
3.5 Zagon simulacij molekulske dinamike .................................................................... 29
3.6 Programi za analizo trajektorij simulacij ................................................................. 31 4 Rezultati in diskusija ....................................................................................................... 34
4.1 Vizualna predstavitev rezultatov .............................................................................. 34
4.2 Analiza RMSD in RMSF ......................................................................................... 37 4.3 Analiza razdalje med verigama DNK ter števila prisotnih vodikovih vezi ............. 45 4.4 Analiza energijskih trajektorij .................................................................................. 46 4.5 Analiza klastrov ....................................................................................................... 51
5 Zaključek ......................................................................................................................... 52
6 Literatura ......................................................................................................................... 53 7 Življenjepis ...................................................................................................................... 55

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
II
Izjava
Izjavljam, da sem magistrsko delo izdelal/a sam/a, prispevki drugih so posebej označeni.
Pregledal/a sem literaturo s področja magistrskega dela po naslednjih geslih:
Vir: Science direct (http://www.sciencedirect.com/)
Gesla: Število referenc
Shielding effect AND DNA solutions 4
Molecular dynamics simulation 8
Microwaves OR Microwave energy 5
GROMOS 5
Skupno število pregledanih člankov: 22
Skupno število pregledanih knjig: 6
Maribor, avgust 2017 Uroš Veselič
podpis

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
III
Zahvala
Zahvaljujem se mentorju, izr. prof. dr. Urbanu Brenu, za
približanje področja kvantne kemije in biomolekularnih simulacij
na predavanjih in za možnost opravljanja magistrskega dela prav
iz tega področja ter za vso pomoč pri izvedbi in pisanju zaključne
naoge. Velika zahvala tudi mag. Martinu Gladoviću za vso pomoč
in podporo pri izvajanju magistrskega dela.
Zahvalil bi se tudi svoji družini za izjemno podporo in motivacijo
skozi študijska leta.

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
IV
Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
Povzetek
Vplivi sodobne tehnologije na vsakdanjik človeka so že dalj časa tema številnih znanstvenih
raziskav z različnih področij. Ne moremo zanikati, da sodobne tehnologije izredno
pripomorejo k razumevanju sveta okoli nas in nam v veliko primerih olajšajo in rešujejo
življenje. Poleg koristi, ki jih prinaša sodobna tehnologija, pa je potrebno ovrednotiti tudi
njene morebitne stranske učinke oziroma neželjene posledice uporabe sodobne tehnologije v
vsakdanjem življenju. Mikrovalovno valovanje je nedvomno pomemben del sodobne tehnike
v obliki mobilne telefonije, brezžičnih omrežij, radarjev, satelitov in GPS naprav. V zadnjih
letih je bilo opravljenih veliko študij na temo vpliva mikrovalov na človeka in okolje. V sklopu
magistrskega dela smo z računalniškim pristopom ugotavljali, ali elektromagnetno valovanje
v območju mikrovalov vpliva na stabilnost dvojne vijačnice DNK. S simulacijami molekulske
dinamike izbranega segmenta DNK v vodni razopini z razklopom temperature sistema na
translacijsko in rotacijsko temperaturo smo ugotavljali, ali imajo mikrovalovi sposobnost
povečati (ne)stabilnost v dvojni vijačnici DNK oziroma celo povzročiti popoln razpad dvojne
vijačnice. Po izsledkih dosedanjih študij na temo mikrovalov razlog za destabilizacijo dvojne
vijačnice DNK tiči v tem, da mikrovalovi vzbudijo rotacijsko gibanje polarnih molekul vode,
kar pa zmanjša senčenje elektrostatskega odboja med dvema negativno nabitima
komplementarnima verigama DNK. S tem se po poročanju dosedanjih raziskav tako poveča
možnost razpada verige in s tem tudi potencial za morebitne spremembe dednega materiala v
celicah, kar zaradi sprememb v sintezi in strukturi molekule DNK lahko privede celo do
nastanka rakavih obolenj.
Analiza rezultatov simulacij molekulske dinamike je pokazala, da mikrovalovi v 10 ns
časovnem okvirju niso imeli istega vpliva na stabilnost fragmenta DNK kot konvencionalno
termično gretje. Analiza rezultatov je pokazala, da je fragment pri simulacijah mikrovalovnega
gretja ostal v bolj stabilnem stanju kot pri termičnem gretju.
Ključne besede: Mikrovalovi, DNK, molekulska dinamika, ločeni termostati
UDK: 615.849:54.022(043.2)

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
V
The influence of microwave irradiation on the stability of double-stranded DNA: A computational approach
Abstract
The influence of modern technology on our everyday lives has been a topic of a number of
scientific papers. We cannot deny that modern techologies contribute a great deal to our
understanding of the world and make our lifes easier in so many ways. However, despite all
the benefits that modern techonolgy brings we have to consider possible unwanted side effects
and consicuences of this techonolgy on our everyday live. Microwaves are undoubtely applied
in the most important achievements of modern techonolgy like radars, satellites, mobile
technology, wireless networks and GPS devices. In recent years, there has been quite a lot of
studies reporting the effects of microwaves on people and environment in recent years. In our
study we have been investigating the influence of microwave irradiation on the stability of
double-stranded DNA using a computational approach. We have used molecular dynamics
simulations with separate thermostates for rotational and translational degrees of freedom to
examine whether microwaves decrease the stability of double stranded DNA or even induce a
total destruction of the double helix. So far, studies have showed that the reason behind the
instability of the DNA when exposed to microwave irradiation lies in the increased rotational
temperature of the water molecules which in turn decreases the shielding of electrostatic
charges of the negatively charged chains of double-stranded DNA and hence increases the
potential of possible deffects in the genetic material of the cells and can due to changes in
synthesis and structure of the DNA molecule lead even to carcinogenesis.
The analysis of our results of 10 ns molecular dynamics simulations showed that microwave
irradiation did not have the same influence on the stability of the DNA fragment as did
conventional thermal heating. Analysis showed that the fragment remained in a more stable
state when exposed to microwave irradiation than in simulations of conventional thermal
heating.
Key words: Microwaves, DNA, molecular dynamics, seperate thermostats
UDK: 615.849:54.022(043.2)

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
VI
Seznam tabel
Tabela 1: RMSD in RMSF analize 10 ns dolgih simulacij .................................................... 37
Tabela 2: TSER analiza 10 ns dolgih simulacij ...................................................................... 45
Tabela 3: Dejanske povprečne temperature posameznih komponent sistema ........................ 46
Tabela 4: Analiza trajektorij neveznih potencialnih energij ................................................... 47
Tabela 5: Analiza klastrov ...................................................................................................... 51

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
VII
Seznam slik
Slika 1: Osnovni gradniki molekule DNK ............................................................................... 3
Slika 2: 2'-deoksiriboza in riboza ............................................................................................. 4
Slika 3: Zvita oblika dvojne vijačnice DNK pri 300K ............................................................. 5
Slika 4: Dipolna polarizacija pod vplivom oscilirajočega mikrovalovnega sevanja ................ 6
Slika 5: Ionska prevodnost ....................................................................................................... 6
Slika 6: Ponazoritev dihedralnih kotov med atomom klora in vodika (modro), ter dvema
atomoma klora (rdeče) [8] ...................................................................................................... 14
Slika 7: Nepravilni torzijski kot v molekuli benzena. Vodik je izrinjen iz planarne ravnine. 15
Slika 8: Krivulja Lennard-Jones potenciala ............................................................................ 16
Slika 9: Vizualni prikaz Monte Carlo metode. Slikice označene z rdečo piko predstavljajo
zavrnjene konformacije sistema ............................................................................................. 18
Slika 10: Ponazoritev Verletovega algoritma ......................................................................... 20
Slika 11: Koraki simulacije molekulske dinamike ................................................................. 21
Slika 12: VMD program ......................................................................................................... 22
Slika 13: Topologija DNK fragmenta .................................................................................... 26
Slika 14: Datotečna struktura simulacij .................................................................................. 27
Slika 15: Začetna minimizirana in termalizirana konformacija izbranega fragmenta DNK pri
300K ....................................................................................................................................... 28
Slika 16: .arg datoteka ............................................................................................................ 29
Slika 17: .imd datoteka ........................................................................................................... 29
Slika 18: .imd datoteka za pripravo simulacije s povišano rotacijsko temperaturo vode ....... 30
Slika 19: Vhodna datoteka za analizo s programom ene_ana ................................................ 31
Slika 20: Vhodna datoteka za izračun celotnega RMSD ........................................................ 32
Slika 21: Vhodna datoteka za izračun notranjega RMSD ...................................................... 32
Slika 22: Vhodna datoteka za analizo RMSF ......................................................................... 32
Slika 23: Vhodna datoteka za zagon programa Frameout ...................................................... 33
Slika 24: Vhodna datoteka za zagon programa TSER............................................................ 33
Slika 25: Vhodne datoteke za analizo klastrov ....................................................................... 33
Slika 26: Začetna (levo) in končna (desno) konformacija molekule DNK pri 300 K ............ 34
Slika 27: Začetna (levo) in končna (desno) konformacija molekule DNK pri povišani zgolj
rotacijski temperaturi na 400 K .............................................................................................. 34
Slika 28:Začetna (levo) in končna (desno) konformacija molekule DNK pri 400 K ............. 35
Slika 29:Začetna (levo) in končna (desno) konformacija molekule DNK pri povišani zgolj
rotacijski temperaturi na 500 K .............................................................................................. 35
Slika 30: Začetna (levo) in končna (desno) konformacija molekule DNK pri 500 K ............ 35
Slika 31: Začetna (levo) in končna (desno) konformacija molekule DNK pri povišani zgolj
rotacijski temperaturi na 600 K .............................................................................................. 36
Slika 32: Začetna (levo) in končna (desno) konformacija molekule DNK pri 600 K ............ 36
Slika 33: RMSD analiza za simulacijo 300K ......................................................................... 38

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
VIII
Slika 34: RMSD analiza za simulacijo R400K ....................................................................... 39
Slika 35: RMSD analiza za simulacijo 400K.......................................................................... 39
Slika 36: RMSD analiza za simulacijo R500K ....................................................................... 40
Slika 37: RMSD analiza za simulacijo 500K.......................................................................... 40
Slika 38: RMSD analiza za simulacijo R600K ....................................................................... 41
Slika 39: RMSD analiza za simulacijo 600K.......................................................................... 41
Slika 40: RMSF analiza simulacije 300K ............................................................................... 42
Slika 41: RMSF analiza simulacije R400K ............................................................................ 42
Slika 42: RMSF analiza simulacije 400K ............................................................................... 42
Slika 43: RMSF analiza simualcije R500K ............................................................................ 43
Slika 44: RMSF analiza simulacije 500K ............................................................................... 43
Slika 45: RMSF analiza simulacije R600K ............................................................................ 44
Slika 46: RMSF analiza simulacije 600K ............................................................................... 44
Slika 47: Spreminjanje nevezne potencialne energije s časom pri simulaciji 300K. Zelena
barva ponazarja interakcije DNK-DNK. Modra predstavlja energijo DNK-voda. Rdeča
predstavlja povprečje obeh interakcij, svetlo modra barva pa predstavlja vsoto obeh interakcij
................................................................................................................................................. 47
Slika 48: Spreminjanje nevezne potencialne energije s časom pri simulaciji R400K. Zelena
barva ponazarja interakcije DNK-DNK. Modra ponazarja energijo DNK-voda. Rdeča
predstavlja povprečje obeh interakcij, svetlo modra barva pa predstavlja vsoto obeh interakcij
................................................................................................................................................. 48
Slika 49: Spreminjanje nevezne potencialne energije s časom pri simulaciji 400K. Zelena
barva ponazarja interakcije DNK-DNK. Modra ponazarja energijo DNK-voda. Rdeča
predstavlja povprečje obeh interakcij, svetlo modra barva pa predstavlja vsoto obeh interakcij
................................................................................................................................................. 48
Slika 50: Spreminjanje nevezne potencialne energije s časom pri simulaciji R500K. Zelena
barva ponazarja interakcije DNK-DNK. Modra ponazarja energijo DNK-voda. Rdeča
predstavlja povprečje obeh interakcij, svetlo modra barva pa predstavlja vsoto obeh interakcij
................................................................................................................................................. 49
Slika 51: Spreminjanje nevezne potencialne energije s časom pri simulaciji 500K. Zelena
barva ponazarja interakcije DNK-DNK. Modra ponazarja energijo DNK-voda. Rdeča
predstavlja povprečje obeh interakcij, svetlo modra barva pa predstavlja vsoto obeh interakcij
................................................................................................................................................. 49
Slika 52: Spreminjanje nevezne potencialne energije s časom pri simulaciji R600K. Zelena
barva ponazarja interakcije DNK-DNK. Modra ponazarja energijo DNK-voda. Rdeča
predstavlja povprečje obeh interakcij, svetlo modra barva pa predstavlja vsoto obeh interakcij
................................................................................................................................................. 50
Slika 53: Spreminjanje nevezne potencialne energije s časom pri simulaciji 600K. Zelena
barva ponazarja interakcije DNK-DNK. Modra ponazarja energijo DNK-voda. Rdeča
predstavlja povprečje obeh interakcij, svetlo modra barva pa predstavlja vsoto obeh interakcij
................................................................................................................................................. 50

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
IX
Uporabljeni simboli in kratice
Simboli
a Pospešek (𝑚
𝑠2)
ci Koeficient linearne kombinacije (/)
E Energija sistema (𝑘𝑐𝑎𝑙
𝑚𝑜𝑙)
F Sila (N)
H Hamiltonov operator (/)
i Imaginarna komponenta (/)
Kl, Kθ, Kφ Harmonske konstante (različne enote)
l razdalja (m)
m masa (kg)
n multipliciteta (/)
r Krajevni vektor (Ǻ)
t Čas (s)
u dihedralni kot (rad)
V Potencial (𝑘𝑐𝑎𝑙
𝑚𝑜𝑙)
Grški simboli
ℏ Reducirana Planckova konstanta (𝑒𝑉𝑠
𝑟𝑎𝑑)
Δ Razlika/sprememba (/)
θ Valenčni kot (rad)
σ Razdalja trka (m)
ε Globina potencialnega vodnjaka (𝑘𝑐𝑎𝑙
𝑚𝑜𝑙)
ψ Valovna funkcija (/)
𝜕 Parcialni odvod (/)
φ Torzijski kot (rad)
γ Fazni kot (rad)

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
X
Kratice
AM1, PM3 Semi-empirični metodi
DNK Deoksiribonukleinska kislina (ang. DNA)
HF Hartee-Fock metoda
IR Infrardeče valovanje
MAE Ekstrakcija z mikrovalovi (ang. Microwave Assisted Extraction)
MC Monte Carlo metoda
MD Molekulska dinamika
NMR Nuklearna magnetna resonanca (ang. Nuclear Magnetic Resonance)
RMSD Koren povprečnega kvadrata razlike (ang. Root Mean Square Deviation)
RMSF Koren povprečnega kvadrata fluktuacije (ang. Root Mean Square Fluctuation)
RNK Ribonukleinska kislina (ang. RNA)
SCF Samouglašeno polje (ang. Self-Consistent Field)
XRD Difrakcija z rentgenskimi žarki (ang. X-ray Diffraction)

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
1
1 Uvod
1.1 Opredelitev problema
Mikrovalovi so del spektra elektromagnetnega valovanja z valovnimi dolžinami od 1 mm do
1 m oziroma frekvencami valovanja od 300 MHz do 300 GHz. Tehnologija nas spremlja že
kar nekaj časa tudi v vsakdanjem življenju. Uporabljamo jo v gospodinjstvu v obliki
mikrovalovnih pečic, v napravah za mobilno telefoniranje, satelitih, radarjih brezžičnih
omrežjih.
Kljub številnim uporabnim in trgovsko zanimivim lastnostim imajo mikrovalovi tudi svojo
temno plat. Raziskave namreč kažejo, da mikrovalovno gretje v primerjavi s konvencionalnim
gretjem spodbuja taljenje DNK, kar lahko vodi do poškodb dednega materiala ter posledično
do nastanka raka [1]. Obsevanje z mikrovalovi vzbudi rotacijsko gibanje polarnih molekul
vode oziroma viša njihovo rotacijsko temperaturo, kar pa lahko povzroči razpad dvojne
vijačnice DNK [1]. To bomo preverili s simulacijo molekulske dinamike z ločenima
termostatoma za rotacijske in translacijske prostostne stopnje.
1.2 Pregled literature
Po pregledu obstoječe literature na temo vpliva mikrovalov na stabilnost DNK smo zasledili,
da je bila dokazana povezava med povišano rotacijsko temperaturo in zmanjšano hidratacijo
polarnih molekul ter povišano hidratacijo nepolarnih zvrsti [2]. Dosedanja literatura prav tako
potrjuje vpliv mikrovalovnega sevanja na razpad dvojne vijačnice deoksioligonukleotidov
krepko pod njihovo termalno talilno temperaturo, in to neodvisno od dolžine
deoksioligonukleotidov [1]. Ugotovitve dosedanjih raziskav [3] kažejo tudi, da rotacijsko
gibanje molekul vode poviša stopnjo združevanja hidrofobnih molekul v bolj kompaktne
strukture zaradi destabilizacije vodnih molekul v njihovi prvi hidracijski lupini. V literaturi
[4] smo prav tako zasledili, da se je uporaba mikrovalov in njihov vpliv na stabilnost DNK že
uveljavila kot tehnika za fragmentacijo genomske DNK. To nam daje dobro iztočnico za
preučitev vpliva mikrovalov na stabilnost dvojne vijačnice DNK in s tem možnost vpogleda v
morebitno povezavo med mikrovalovnim sevanjem in mutagenezo ter posledičnim pojavom
genetskih obolenj.

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
2
1.3 Namen, hipoteze in cilji
Cilj magistrskega dela je izvesti simulacijo molekulske dinamike z ločenima termostatoma za
rotacijsko in translacijsko temperaturo pri razilčnih temperaturah sistema in na podlagi analize
vrednosti RMSF (root mean square fluctuation), RMSD (root mean square deviation),
vodikovih vezi in radialne porazdelitvene funkcije, klastrov in medsebojnih razdalj med
posameznimi atomi preveriti, ali rotacijsko vroča voda vpliva na stabilnost dvojne vijačnice
DNK.
Osnovna hipoteza magistrskega dela se glasi, da polarne molekule vode z višjo rotacijsko
temperaturo povzročajo taljenje dvojne vijačnice DNK, saj zmanjšajo senčenje
elektrostatskega odboja med negativno nabitima komplementarnima verigama DNK.

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
3
2 Teoretični del
2.1 Molekula DNK
Molekula DNK oziroma molekula deoksiribonukleinske kisline je poleg RNK ali
ribonukleinske kisline ena izmed glavnih predstavnikov nukleinskih kislin. Te dolge biološke
makromolekule so sestavljene iz dveh verig deoksiribonukleotidov, verigi pa sta stabilizirani
z vodikovimi vezmi med nukleotidi. Na tem mestu je treba razložiti nekaj osnovnih pojmov,
ki se pogosto uporabljajo pri opisovanju funkcije in strukture DNK. To so pojmi nukleobaze,
nukleozida, nukleotida in nukleinske kisline.
Strukturo molekule DNK lahko opišemo kot zaporedje nukleotidov, natančneje
deoksiribonukleotidov. Nadalje pa nukleotide opišemo kot fosfatne estre tako imenovanih
nukleozidov, ki so sestavljeni iz molekule sladkorja in molekule nukleobaze. Skupino
nukleobaz sestavlja pet molekul. To so adenin, gvanin, citozin in timin. Nukleobaze glede na
vrsto molekule delimo na purine in pirimidine. Med purinske molekule spadata gvanin in
adenin, med pirimidinske pa citozin in timin. Na Sliki 1 so ponazorjeni vsi omenjeni gradniki
molekule DNK.
Slika 1: Osnovni gradniki molekule DNK

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
4
Posamezni gradniki DNK (nukleobaze) so topni v vodi v dokaj širokem območju pH (od 6 do
8) [5]. Pri nižjih pH vrednostih pa nukleozidi postanejo nabiti. Zaradi prisotnosti sladkorjev
so deoksiribonukleozidi bolj topni kot same nukleobaze, vendar je pri pogojih vzdrževanih v
celicah deoksiribonukleozid nevtralna kemijska zvrst. Med posameznimi gradniki DNK imajo
še večjo topnost fosforilirani nukleozidi zaradi prisotnih fosfodiesterskih vezi. Fosfatne
skupine v DNK nosijo negativni naboj, kar omogoči vezavo številnih proteinov in pozitivnih
kovinskih ionov na ogrodje molekule DNK preko elektrostatskih interakcij. Zaradi fleksibilne
strukture pentoznega sladkorja in glikozidne vezi, lahko molekula DNK zavzame več možnih
konformacij, ki vplivajo na obliko oziroma zvitje molekule. V molekuli pentoznega sladkorja
2-deoksi-D-riboze sta zaradi napetosti v obroču eden ali dva ogljikova atoma zamaknjena
izven planarne lege. Na podlagi tega ločimo tako imenovano južno oziroma »south«
konformacijo ter severno oziroma »north« konformacijo [5]. Po dogovoru južna oziroma C2'-
endo konformacija pomeni lego C2' atoma nad planarno ravnino, severna oziroma C3'-endo
konformacija pa pomeni lego C3' atoma nad planarno ravnino sladkorja. Atom leži nad
planarno ravnino, ko je sladkor orientiran tako, da kažeta C5' atom ter nukleobaza proč od
obroča sladkorja. Različne konformacije so možne tudi pri bazah nukleozidov oziroma
nukleotidov. Enojna glikozidna vez namreč omogoča prosto rotacijo, kar privede do tako
imenovane »anti« in »syn« konformacije. Do »anti« konformacije pride, kadar atom H8 pri
purinih oziroma atom H6 pri pirimidinih leži nad molekulo sladkorja, do »syn« konformacije
pa kadar ista dva atoma ležita proč od sladkorja. Vse konformacije skupaj vplivajo na strukturo
dvojne vijačnice DNK.
Slika 2: 2'-deoksiriboza in riboza
Posamezne deoksiribonukleotide v celoto do deoksiribonukleinske kisline povežejo
fosfodiesterske vezi. Te povežejo 5'-hidroksilno skupino enega nukleotida s 3'-hidroksilno
skupino naslednjega nukleotida. Tako se zaporedje verige oligonukleotidov na enem koncu
zaključi s 3'-OH skupino, na drugem pa 5'-OH skupino: 3'-terminus in 5'-terminus. Po
dogovoru privzamemo, da se zaporedje oligonukleotida začne in nadaljuje v smeri od 5'-
terminusa do 3'-terminusa. Polimerizacija se nadaljuje do deoksiribonukleinske kisline
oziroma DNK, ki hrani celoten genetski zapis organizma in predstavlja eno izmed
najpomembnejših makromolekul v živih bitjih. Medtem, ko je hrbtenica (ang. backbone)
DNK dokaj podobna v vseh vrstah DNK (rastline, živali, ljudje), pa se zaporedje in število
posameznih baz v verigi močno razlikuje od organizma do organizma. Zaporedje oziroma
sekvenca baz je namreč razlog za takšno raznolikost in kompleksnost organizmov v naravi.

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
5
Tako kot pri vseh molekulah, tudi molekula DNK teži k čim nižji potencialni energiji. To
pomeni, da molekula DNK ne predstavlja popolnoma linearno zaporedje
deoksiribonukleotidov, temveč se vsi atomi oziroma gradniki molekule v prostoru razporedijo
tako, da je v molekuli čim manj napetosti oziroma čim manj elektrostatskih odbojev in
steričnih motenj. Izkaže se, da je najugodnejša oblika molekule DNK oblika dvojnega heliksa
ali dvojne vijačnice (Slika 3). Razlog za vijačno obliko je ta, da so v primeru vijačnice
interakcije hidrofobnih nukleobaz z vodo minimalne. V primeru dvojne vijačnice so
interakcije hidrofobnih baz z molekulami vode skoraj popolnoma onemogočene, obenem pa
omogočijo zelo dobro hidratacijo hidrofilnih skupin sladkorja in fosfata. K dodatni stabilizaciji
oblike dvojne vijačnice pa pripomorejo še vodikove vezi, ki se vzpostavijo med
komplementarnimi bazami v vijačnici. Komplenetarne baze so pari na nasprotnih straneh
dvojne vijačnice, ki se preko vodikovih vezi povežejo med seboj. Tako sta komplementarni
bazi s tremi vodikovimi vezmi citozin in gvanin ter komplementarni bazi z dvema vodikovima
vezema adenin in timin.
Slika 3: Zvita oblika dvojne vijačnice DNK pri 300K

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
6
2.2 Mikrovalovi
Mikrovalovi so elektromagnetno valovanje in poleg vidne svetlobe, ultravijoličnih,
rentgenskih, infrardečih, radijskih in gama žarkov sestavljajo elektromagnetni spekter. So
nepogrešljivi del elektronskih naprav na vseh področjih življenja. V vsakodnevnem življenju
mikrovalove najpogosteje asociiramo z mikrovalovno pečico. Vendar pa poleg rabe v
gospodinjstvu mikrovalove uporabljamo za delovanje navigacijskih sistemov, mobilnih
telefonov, satelitov, sistemov za nadzor prometa – radarjev ter v raziskovalne namene, kot je
ekstrakcija s pomočjo mikrovalov oziroma tako imenovana MAE (microwave assisted
extraction) in mikrovalovna kataliza.
Gledano iz molekularnega vidika nam mikrovalovi v kemiji omogočajo raznovrstno uporabo
preko dveh osnovnih mehanizmov: dipolne polarizacije in ionske prevodnosti [2]. Dipolna
polarizacija pomeni ureditev molekul vode in ostalih polarnih molekul oziroma njihovo
orientacijo pod vplivom zunanjega oscilirajočega elektromagnetnega polja, ki deluje na
polarne molekule, pri katerih so prisotni dipoli neodvisni drug od drugega in se lahko
neodvisno vrtijo okoli svoje osi, za razliko od ionske polarizacije (Slika 4).
Slika 4: Dipolna polarizacija pod vplivom oscilirajočega mikrovalovnega sevanja
Do pojava ionske prevodnosti pa pride, kadar se hidratizirana nabita molekula ali ion ob
prisotnosti oscilirajočega mikrovalovnega sevanja orientira tako, da se predhodno simetrična
hidratizirana struktura v prisotnem polju deformira pod vplivom oscilirajočega valovanja
(Slika 5)
Slika 5: Ionska prevodnost

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
7
Oba opisana mehanizma z različnima deležema prispevata k rotacijski vzbujenosti vodnih
molekul v sistemu [2]. Kadar pride do kontinuirnega obsevanja z mikrovalovi, presežna
rotacijska energija ne more povsem disipirati na vibracijsko in translacijsko prostostno
stopnjo, kar privede do povišane rotacijske temperature v primerjavi z vibracijsko in
translacijsko temperaturo. Ravno takšen pojav smo želeli doseči pri preučevanju našega
sistema. To nam je omogočilo, da smo v simulaciji ustvarili približno takšno okolje, kot bi ga
srečali v realnem sistemu gretja raztopine makromolekule z mikrovalovi.

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
8
2.3 Metode računalniške kemije
Ljude so že od nekdaj želeli avtomatizirati reševanje računskih problemov. Bodisi izvajati
preprostejše izračune v krajšem času kot sicer, preverjati točnost lastnih izračunov ali pa jih
uporabljati kot pomoč pri opravilih na najrazličnejših področjih življenja. Eno izmed teh
področij je nedvomno tudi kemija. Kemiki so že vse od začetka znanosti imeli opravka s
številnimi izračuni, obdelavo podatkov, kot so na primer lastnosti elementov, molekul, reakcij
in informacijami o vedno naraščajočem številu rezultatov eksperimentov, ki so osnova za
nadaljnji razvoj kemijske znanosti (določanje konstante ravnotežja, hitrosti reakcij idr.). Zato
so tudi kemiki imeli željo po uporabi računalnika pri svojem delu, kjer bi hranili vse
pomembne informacije in kar bi jim omogočilo hitrejše reševanje problemov v prihodnosti.
Tako se je začela razvijati veja kemije, ki se imenuje računalniška kemija. Čeprav so
računalniki v obliki, kot jih poznamo danes prisotni že vse od začetka 20. stoletja, je
računalniška kemija kot ločeno področje kemije obveljala šele leta 1998, ko sta Walter Kohn
in John Pople prejela Nobelovo nagrado za uporabo računalniških metod v kemiji.
Pod pojmom računalniške kemije razumemo združeno uporabo fizikalno-kemijskih zakonov
in parametrov, matematičnih modelov ter računalniških programov za reševanje kemijskih
problemov. Primarni namen računalniške kemije je sestaviti tako dovršen matematični model,
ki kar se da podrobno opiše nek pojav v realnem svetu, da ga lahko avtomatiziramo in
uporabimo pri reševanju vseh nadaljnjih podobnih pojavov. Takšni pojavi so lahko
določevanje prehodnih stanj molekul, ravnotežnih konstant, hitrosti kemijskih reakcij, veznih
in neveznih energij sistemov, porazdelitev in gostot elektronov ter nabojev, zvijanja proteinov
v različnih okoljih in določevanje termodinamskih parametrov.
Velike prednosti računalniškega pristopa k reševanju kemijskih problemov so:
• Nizki stroški izvajanja: Računalnik nam omogočam da določen izračun oz. simulacijo
ponovimo večkrat in vsakič po želji spremenimo pogoje in model reševanja, kar bi v
eksperimentalnem laboratoriju vzelo neprimerno več časa in občutno zvišalo stroške
zaradi porabe reaktantov in opreme.
• Spreminjanje pogojev: Uporaba simulacij nam omogoči nastavitev točno takšnih
pogojev, kot jih sami želimo, bodisi izjemno veliki tlaki ali ekstremno visoke
temperature, brez uporabe drage opreme.
• Varnost: V računalniških simulacijah lahko iz varne »razdalje« preko računalnika
izvajamo reakcije ali preučujemo nevarne sisteme, kot so na primer kancerogene,
eksplozivne in strupene spojine.

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
9
Računalniška kemija zajema veliko število metod za opisovanje vseh vrst sistemov, ki se
nenehno izboljšujejo in dopolnjujejo. Metode lahko razdelimo v dve večji skupini. To so
kvantno-kemijski modeli ter klasični modeli. Metode, ki zajemajo reševanje po kvantno-
kemijskih modelih, uporabljajo osnove kvantne mehanike, to je reševanje Schrödingerjeve
enačbe, za izračun fizikalno-kemijskih lastnosti snovi. Metode, ki uporabljajo klasične
modele, pa se opirajo na izračune na osnovah klasične oziroma Newtonove mehanike, to je
reševanje Newtonovih enačb gibanja.
2.4 Kvantno-kemijski modeli
V to skupino sodijo metode, ki temeljijo na osnovah kvantne mehanike in reševanju
Schrödingerjeve enačbe. Nadalje lahko to skupino razdelimo na dve podskupini. To so ab-
initio metode in semi-empirične metode. Ab-initio metode, kot že ime pove (od začetka), so
metode, pri katerih uporabljamo izključno fizikalne konstante in fizikalne lastnosti atomov v
sistemu ter pridobimo rešitve izključno z reševanjem Schrödingerjeve enačbe. Pri semi-
empiričnih metodah nekoliko popustimo stroge zahteve in v izračunih uporabljamo še
podatke, ki jih pridobimo izkustveno iz eksperimentov, ter približke, ki nam pohitrijo in
poenostavijo reševanje problemov.
2.4.1 Ab-initio metode
Ab-initio metode, osnovane na principih kvantne mehanike, pri izračunih zajemajo izključno
fizikalno-kemijske konstante in reševanje Schrödingerjeve enačbe s čim manj
predpostavkami. Osnova kvantne mehanike je Schrödingerjeva enačba (Enačba 1).
{−ℏ2
2𝑚(
𝜕2
𝜕𝑥2+
𝜕2
𝜕𝑦2+
𝜕2
𝜕𝑧2) + 𝑉} 𝜓(𝑟, 𝑡) = 𝑖ℏ
𝜕𝜓(𝑟, 𝑡)
𝜕𝑡 (1)
Zgornja enačba opisuje valovno funkcijo enega delca ψ (npr. elektrona) v prostoru z zunanjim
poljem V, s koordinatami x, y, z in maso m. ℏ predstavlja Planckovo konstanto deljeno z 2π, i
pa predstavlja kvadratni koren od -1 oziroma imaginarno komponento. ψ(r,t) torej predstavlja
valovno funkcijo delca, to je funkcijo ki opiše in okarakterizira njegovo gibanje. Kadar se
zunanje polje oziroma potencial ne spreminja s časom, lahko enačbo poenostavimo:
{−ℏ2
2𝑚(
𝜕2
𝜕𝑥2+
𝜕2
𝜕𝑦2+
𝜕2
𝜕𝑧2) + 𝑉} 𝜓(𝑟) = 𝐸𝜓(𝑟) (2)
Tu E predstavlja lastno vrednost valovne funkcije, oziroma energijo našega sistema. Obstaja
še alternativni zapis:
{−ℏ2
2𝑚∇2 + 𝑉} 𝜓(𝑟) = 𝐸𝜓(𝑟) (3)
Kjer velja ∇2= (𝜕2
𝜕𝑥2 +𝜕2
𝜕𝑦2 +𝜕2
𝜕𝑧2)

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
10
Nadalje lahko enačbo 3 še poenostavimo s vpeljavo operatorja – matematične operacije, ki
deluje na našo funkcijo. Takšen operator se imenuje Hamiltonov operator H.
𝐻 = −ℏ2
2𝑚∇2 + 𝑉 (4)
Vpeljava Hamiltonskega operatorja nam Schrödingerjevo enačbo poenostavi v sledeč zapis,
kjer z rešitvijo enačbe dobimo energijo sistema:
𝐻𝜓(𝑟) = 𝐸𝜓(𝑟) (5)
Schrödingerjeva enačba tako spada med parcialne diferencialne enačbe, pri katerih imamo
spremenljivke v odvodih oziroma diferencialih funkcije. Za rešitev takšne enačbe je potrebno
najti takšno funkcijo, imenovano lastna funkcija, pri kateri po delovanju Hamiltonskega
operatorja na to funkcijo kot rezultat dobimo to isto funkcijo in še neko numerično vrednost
E, ki jo imenujemo lastna vrednost funkcije 𝜓(𝑟) in predstavlja skalar, ki ustreza energiji delca
v izbranem sistemu.
Ab-initio metode so zaradi kompleksnosti reševanja Schrodingerjeve enačbe analitično
rešljive samo za majhne sisteme enega elektrona. Kvantno-mehanske metode zato zahtevajo
zelo zmogljive računalniške sisteme, ki so sposobni izvajati operacije z veliko količino
podatkov. So pa rezultati takšnih izračunov izredno točni.
Ena od prvih metod za reševanje Schrödingerjeve enačbe je tako imenovana Hartree-Fock
metoda (krajše HF). Ta metoda upošteva dva približka za lažje računanje. V metodi je
upoštevana neločljivost elektronov, torej možnost njihove zamenjave. Izračunana je torej
povprečna vrednost odbojnih sil med elektroni in ne interakcije vsakega elektrona s vsakim
posebej. S tem poenostavi nalogo izračuna ene Schrodingerjeve enačbe za več elektronov
skupaj v več enostavnejših enačb za en elektron. Rešitev vsake takšne enačbe predstavlja
valovno funkcijo (orbitalo) in njeno energijo, v kateri se elektron nahaja v povprečnem
potencialu oziroma »oblaku« ostalih elektronov. Te eno-elektronske funkcije so imenovane
bazne funkcije. Pri reševanju Schrödingerjeve enačbe za neko molekulo si računanje
poenostavimo s vpeljavo teorije molekulskih orbital (teorija MO). Teorija molekulskih orbital
obravnava molekulo podobno kot atom. Tako kot ima atom svoje orbitale in valovne funkcije,
teorija molekulskih orbital predpostavi da ima molekula molekulske orbitale, ki so
kombinacija posameznih atomskih orbital atomov v molekuli. To pomeni, da lahko valovno
funkcijo molekulske orbitale zapišemo kot linearno kombinacijo N baznih funkcij oziroma
valovnih funkcij N elektronov v sistemu.
𝜓𝑖 = ∑ 𝑐𝑣𝑖𝜙𝑣𝑖 (6)
𝑁
𝑣=1
cvi – koeficient linearnega razvoja
𝜙vi – valovna funkcija posameznega elektrona

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
11
Koraki izračuna po Hartree-Fock metodi so naslednji:
• Izmislimo si začetni sistem funkcij 𝜓𝑖
• Izračunamo nove koeficiente linearne kombinacije in njihove energije
• Postopek ponavljamo in primerjamo izračunane energije in koeficiente, dokler ne
konvergirajo k izbrani vrednosti.
Temu iterativnemu postopku pravimo SCF (ang. Self-consistent field method) oziroma
metoda samouglašenega polja.
2.4.2 Semiempirične metode
Semiempirične metode ponujajo poenostavitev reševanja zapletenih in dolgotrajnih abinitio
metod. Gre sicer še vedno za metode osnovane na osnovah kvantne mehanike, vendar z
dodatki empiričnih ali izkustvenih približkov in poenostavitev. Pri semiempiričnih metodah
uporabljamo podoben sistem reševanja kot pri Hartree-Fock metodi, vendar kvantne izračune
kombiniramo z eksperimentalnimi podatki. To so lahko na primer eksperimentalno določeni
atomski radiji, ionski potenciali, NMR premiki idr. To privede do pohitritve naših izračunov,
kar je velika prednost semiempiričnih metod pred abinitio metodami. Prav tako nam te metode
omogočajo obravnavanje večjih sistemov do nekaj 100 atomov. Za razliko od abinitio metod
so semiempirične manj natančne, saj moramo upoštevati morebitne sistematične napake, ki
izvirajo iz eksperimentalnih podatkov in uporabljenih približkov. Kljub manjši natančnosti gre
za zanesljive metode, ki lahko zelo dobro napovejo obnašanje sistema in nam dajo pomembne
informacije.
Ena izmed bolj enostavnih semiempiričnih metod je Hückelova metoda. Ta metoda se
osredotoči izključno na obravnavo π-elektronov v aromatskih sistemih. To se zdi kot zelo
obsežna poenostavitev, vendar s to metodo lahko uspešno in preprosto obravnavamo
aromatične sisteme in nam služi za obravnavo aromatičnosti sistemov, teorije barv in tudi
prevodnosti polimerov. Omejitev te metode je, da ne upošteva interakcij z ostalimi atomi.
Kljub temu pa lahko pridobimo uporabne informacije o aromatskih sistemih. Poleg Hückelove
teorije sta med bolj znanimi semiempiričnimi metodami še AM1 in PM3, ki služita za
obravnavo molekularne elektronske strukture; razlika med njima pa je v uporabi različnih
števil Gaussovih baznih funkcij za opis valovnih funkcij v atomih.

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
12
2.5 Molekulska mehanika
Druga skupina metod, uporabljenih v računalniški kemiji, predstavlja pristop reševanja
lastnosti sistema s strani klasične fizike in z reševanjem Newtonovih enačb gibanja, za razliko
od kvantno-kemijskih modelov, kjer imamo opravka s Schrödingerjevo enačbo. Te metode,
imenovane metode molekulske mehanike, se izognejo reševanju Schrödingerjeve enačbe in
namesto tega uporabijo enačbe klasične fizike za opis večjih molekul na ravni vzmeti in
kroglic, ki posnemajo atome in vezi, namesto določevanja valovne funkcije za vsak posamezni
elektron v molekuli. Te metode lahko obravnavajo velike sisteme, do nekaj 1000 atomov, kar
je praktično neizvedljivo s kvantno-kemijskimi metodami, saj bi zahtevale nepredstavljivo
veliko računalniške moči. Dve najpogostejši metodi molekulske mehanike, ki jih srečamo pri
obravnavi večjih biomolekularnih sistemov, sta molekulska dinamika (MD) in Monte Carlo
(MC). Glavna razlika med tema metodama je, da pri molekulski dinamiki računamo enačbe
gibanja delcev v sistemu v časovnem zaporedju korak za korakom, pri Monte Carlo
simulacijah pa v vsakem koraku spremenimo koordinate naključno izbranega delca v sistemu
in nato preračunamo potencialno energijo sistema.
2.5.1 Polje sil
Molekulska mehanika se izogne reševanju zapletenih kvantno-kemijskih enačb tako, da uvede
tako imenovano molekularno polje sil. Molekularno polje sil je sestavljeno iz empirične
potencialne funkcije in empiričnih parametrov. Empirična potencialna funkcija vsakemu
delcu v sistemu pripiše ustrezno potencialno energijo in nam pove, na kakšen način se
potencialna energija spreminja glede na spremembo položaja v sistemu.
𝑉 = 𝑓(𝑟𝑁) (7)
V – potencialna energija (𝑘𝑐𝑎𝑙
𝑚𝑜𝑙)
r – koordinata (položajni vektor)
N – število atomov v molekuli
Empirična potencialna funkcija se sestoji iz preprostejših enačb klasične Newtonove fizike, ki
dobro opišejo obnašanje molekule z analogijo vzmeti in kroglic. Tako na primer s klasičnimi
enačbami za harmonski oscilator lahko opišemo potencialno energijo vezi, valenčnih,
dihedralnih in nepravih torzijskih kotov. Osnovno obliko empirične potencialne funkcije opiše
enačba 8.
𝑉(𝑟𝑁) = 𝑉𝑣𝑒𝑧𝑛𝑖(𝑟𝑁) + 𝑉𝑛𝑒𝑣𝑒𝑧𝑛𝑖(𝑟𝑁) (8)

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
13
Vezni del potenciala sestavlja potencialna energija kovalentnih vezi 𝑉𝑘𝑜𝑣𝑎𝑙.𝑣𝑒𝑧𝑖(𝑟𝑁), energija
valenčnih kotov 𝑉𝑣𝑎𝑙.𝑘𝑜𝑡(𝑟𝑁), energija dihedralnih kotov 𝑉𝑑𝑖ℎ.𝑘𝑜𝑡(𝑟𝑁) ter energija nepravih
torzijskih kotov 𝑉𝑛𝑒𝑝.𝑡𝑜𝑟.𝑘𝑜𝑡(𝑟𝑁). K neveznemu delu empirične potencialne funkcije pa
spadata van der Waalsova 𝑉𝑣𝑑𝑊(𝑟𝑁) in elektrostatska interakcija 𝑉𝑒𝑙.𝑠𝑡𝑎𝑡(𝑟𝑁).
𝑉𝑣𝑒𝑧𝑛𝑖(𝑟𝑁) = 𝑉𝑘𝑜𝑣𝑎𝑙.𝑣𝑒𝑧𝑖(𝑟𝑁) + 𝑉𝑣𝑎𝑙.𝑘𝑜𝑡(𝑟𝑁) + 𝑉𝑑𝑖ℎ.𝑘𝑜𝑡𝑖(𝑟𝑁) + 𝑉𝑛𝑒𝑝.𝑡𝑜𝑟.𝑘𝑜𝑡(𝑟𝑁) (9)
𝑉𝑛𝑒𝑣𝑒𝑧𝑛𝑖(𝑟𝑁) = 𝑉𝑣𝑑𝑊(𝑟𝑁) + 𝑉𝑒𝑙.𝑠𝑡𝑎𝑡(𝑟𝑁) (10)
Potencial 𝑉𝑘𝑜𝑣𝑎𝑙.𝑣𝑒𝑧𝑖(𝑟𝑁) opiše Hookov zakon.
𝑉𝑘𝑜𝑣𝑎𝑙.𝑣𝑒𝑧𝑖(𝑟𝑁) = 1
2𝐾𝑙(𝑙 − 𝑙0)2 (11)
𝐾𝑙 – Harmonska konstanta
𝑙 – dolžina skrčene ali raztegnjene vzmeti (vezi)
𝑙0 – ravnovesna razdalja vzmeti (vezi)
Kadar je harmonska konstanta velika, pomeni, da imamo opravka s tršo vzmetjo oziroma
močnejšo vezjo med atomoma. Harmonsko konstanto in ravnovesno razdaljo vzmeti pa
dobimo eksperimentalno, torej sta empirična parametra. Vrednost harmonske konstante
dobimo z IR analizo (infrardeča spektroskopija), vrednost ravnovesne razdalje vezi pa
pridobimo po Braggovem zakonu z meritvami rentgenske difrakcije [6].

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
14
Podobno zvezo uporabimo za opis potencialne funkcije valenčnih kotov, le da namesto
ravnovesne razdalje vpeljemo pojem ravnovesnega kota med atomi v molekuli, namesto
harmonske konstante vezi pa uporabimo harmonsko konstanto valenčnih kotov
𝑉𝑣𝑎𝑙.𝑘𝑜𝑡(𝑟𝑁) = 1
2𝐾𝜃(𝜃 − 𝜃0)2 (12)
𝐾𝜃 – Harmonska konstanta
𝜃 – trenutni valenčni kot
𝜃0 – ravnovesni valenčni kot
Obnašanje energije dihedralnih kotov opišemo z enačbo 13. Dihedralni kot je kot definiran z
dvema ravninama, ki ju definirajo po trije atomi [7]. Povedano drugače je to kot med dvema
vezema (Slika 6)
Slika 6: Ponazoritev dihedralnih kotov med atomom klora in vodika (modro), ter dvema atomoma klora (rdeče) [8]
𝑉𝑑𝑖ℎ.𝑘𝑜𝑡(𝑟𝑁) = 1
2𝐾(1 + cos(𝑛𝑢 − 𝛾)) (13)
K – amplituda
n – multipliciteta
u – velikost dihedralnega kota
𝛾 – fazni kot

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
15
Splošna zveza za opis nepravilnih torzijskih kotov je podobna zvezi, ki opisuje valenčne kote.
Nepravilni torzijski kot je kot med dvema ravninama, vsako s tremi atomi, v nelinearni verigi
[7] (Slika 7). Podatek o ravnovesnem kotu pridobimo s kristalografskimi meritvami, vrednost
harmonske konstante za kote pa določimo s kvantno mehanskimi izračuni.
𝑉𝑛𝑒𝑝.𝑡𝑜𝑟.𝑘𝑜𝑡(𝑟𝑁) = 1
2𝐾𝜑(𝜑 − 𝜑0)2 (15)
𝐾𝜑 – Harmonska konstanta nepravilnih torzijskih kotov
𝜑 – trenutni nepravilni torzijski kot
𝜑0 – ravnovesni nepravilni torzijski kot
Slika 7: Nepravilni torzijski kot v molekuli benzena. Vodik je izrinjen iz planarne ravnine.

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
16
Podobno zvezo, kot smo jo uporabili za opis kovalente vezi in valenčnih kotov, uporabimo
tudi za opis nepravih torzijskih kotov. Pri kotih, ki so prisotni pri π-konjugiranih sistemih,
izpustimo člen ravnovesnega kota, saj je njegova velikost 0°, ker se nahaja v planarni ravnini
molekule. Zato se enačba za opis nepravih torzijskih kotov nekoliko poenostavi:
𝑉𝑛𝑒𝑝.𝑡𝑜𝑟.𝑘𝑜𝑡(𝑟𝑁) = 1
2𝐾𝜑𝜑2 (14)
Potencialno energijo prispevkov neveznih interakcij sestavljajo van der Waalsove in
elektrostatske sile. Van der Waalsove sile so kratkosežne in šibke medmolekulske sile, ki jih
imenujemo tudi Londonove disperzijske sile. van der Waals in London sta namreč ugotovila,
da tudi atomi brez elektrostatskih interakcij, kot so na primer žlahtni plini, ne sledijo
idealnemu obnašanju, ampak se lahko utekočinijo, kar zahteva prisotnost nekakšnih privlačnih
interakcij. To sta opisala kot privlačne disperzijske interakcije, ki so šibke in kratkosežne;
potencial je sorazmeren z 1
𝑟6 , kjer je 𝑟 razdalja med atomoma. Lennard-Jones pa je
disperzijsko interakcijo združil s Paulijevo odbojno silo v enoten Lennard-Jonesov potencial
[6].
Slika 8: Krivulja Lennard-Jones potenciala

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
17
Pri približevanju dveh neskončno oddaljenih atomov potencialna energija doseže minimum
na določeni negativni vrednosti imenovani globina potencialne jame 𝜖, ki predstavlja
ravnovesno razdaljo med tema dvema atomoma r*AB, σ pa predstavlja razdaljo, kjer
potencialna energija spremeni predznak oziroma kjer so privlačne disperzijske sile enake
odbojnim zaradi Paulijevega izključitvenega načela.
Potencialno funkcijo, ki opisujejo van der Waalsove interakcije, zapišemo z enačbo 16.
𝑉𝑣𝑑𝑊(𝑟𝑁) = 4𝜖 [(𝜎
𝑟)
12
− (𝜎
𝑟)
6
] (16)
oziroma
𝑉𝑣𝑑𝑊(𝑟𝑁) = 𝐴
𝑟12−
𝐵
𝑟6 (17)
kjer sta 𝐴 = 4𝜖𝜎12 in 𝐵 = 4𝜖𝜎6
𝜖 – globina potencialnega vodnjaka
𝑟 – razdalje med atomoma
𝜎 – razdalja trka
Potencialno funkcijo elektrostatskega privlaka pa opišemo s Coloumbovo zvezo.
𝑉𝑒𝑙.𝑠𝑡𝑎𝑡(𝑟𝑁) =𝑒1𝑒2
4𝜋𝜀0𝑟12 (18)
𝑒1 𝑖𝑛 𝑒2 – delna naboja ionov
𝜀0 – dielektrična konstanta
𝑟12 – razdalja med delcema

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
18
2.5.2 Monte Carlo simulacije
Druga pogosta metoda, ki prav tako uporablja enačbe klasične fizike pri simulacijah fizikalno-
kemijskih sistemov, je metoda Monte Carlo. Gre za stohastično metodo, kar pomeni, da nas
ne zanima stanje oziroma obnašanje sistema po časovnih korakih, kjer iz enega koraka
preidemo v naslednjega po nekem časovnem intervalu, temveč ansambel reprezentativnih
stanj sistema pridobimo z naključnimi spremembami pozicij delcev v sistemu. Koraki metode
Monte Carlo potekajo po naslednjem zaporedju [9]:
• Izberemo si začetne koordinate delcev v sistemu
• Delec naključno premaknemo v drugo lego - generiramo nove naključne koordinate
• Izračunamo razliko potencialov med staro in novo konfiguracijo ΔV
• Naključno generiramo vrednost med 0 in 1
• Če je generirana vrednost manjša ali enaka energijsko pogojeni verjetnosti 𝑒−𝛽∆𝑉,
naključno tvorjeno konfiguracijo sprejmemo v ansambel reprezentetivnih struktur.
• Simulacijo ponavljamo od prvega koraka, le da namesto začetne konformacije
uporabimo zadnjo sprejeto konformacijo
Slabost te metode je, da nam ne nudi spremljanja dinamičnih lastnosti, saj ne pozna časovnega
koncepta, ker nove konformacije izbiramo naključno in ne v časovnem sosledju. Prednost
metode pa je, da lahko hitro določimo ansambel reprezentativnih konfiguracij in na podlagi
tako imenovane ergodijske hipoteze, ki pravi da je časovno povprečje enako ansambelskemu
povprečju, z Monte Carlo simulacijo dobimo povprečje neke količine enako, kot če bi izvajali
simulacijo molekulske dinamike.
Slika 9: Vizualni prikaz Monte Carlo metode. Slikice označene z rdečo piko predstavljajo zavrnjene konformacije
sistema

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
19
2.5.3 Molekulska dinamika
Pri simulacijah molekulske dinamike privzamemo, da imajo jedra atomov v sistemu dovolj
veliko maso, da jih lahko obravnavamo s klasičnimi enačbami gibanja Newtonove mehanike.
Na podlagi tega približka lahko izvedemo simulacijo molekulske dinamike z reševanjem
diferencialne enačbe 2. Newtonovega zakona. Ta pravi, da če na telo z maso m deluje neka
sila, bo telo začelo enakomerno pospeševati. Z drugimi besedami, sila je enaka produktu mase
in pospeška nekega telesa. Z rešitvijo diferencialne enačbe 2. Newtonovega zakona lahko
dovolj dobro napovemo razvoj našega izbranega sistema pri simulacijah molekulske
dinamike.
−𝑑𝑉
𝑑𝑟= 𝑚
𝑑2𝑟
𝑑𝑡2 (19)
V – potencialna energija sistema
r – vektor, ki vsebuje koordinate delcev v sistemu (vektor z dolžino 3N)
t – čas
Če privzamemo neko skupino delcev s položaji ri, pozicije teh delcev na naslednjem časovnem
koraku t + Δt, lahko opišemo z razvojem v Taylorjevo vrsto.
𝑟𝑖+1 = 𝑟𝑖 +𝜕𝑟
𝜕𝑡(∆𝑡) +
1
2
𝜕2𝑟
𝜕𝑡2(∆𝑡)2 + ⋯ (20)
oziroma
𝑟𝑖+1 = 𝑟𝑖 + 𝑣𝑖(∆𝑡) +1
2𝑎𝑖(∆𝑡)2 + ⋯ (21)
𝑣𝑖 – hitrosti ob času 𝑡
𝑎𝑖 - pospeški ob času 𝑡
Podobno so pozicije istih atomov v prejšnjem koraku podane z zvezo
𝑟𝑖−1 = 𝑟𝑖 − 𝑣𝑖(∆𝑡) +1
2𝑎𝑖(∆𝑡)2 + ⋯ (22)
Če enačbi za predhodni korak (𝑟𝑖−1) in prihodnji korak (𝑟𝑖+1) seštejemo, dobimo enačbo za
izračun položaja atomov v naslednjem koraku iz trenutnih in predhodnih položajev ter
trenutnega pospeška, ki ga izračunamo iz sile ali potenciala. Ta postopek imenujemo Verletov
algoritem.
𝑟𝑖+1 = (2𝑟𝑖 − 𝑟𝑖−1) + 𝑎𝑖(∆𝑡)2 + ⋯ (23)
𝑎𝑖 = 𝐹𝑖
𝑚𝑖= −
1
𝑚𝑖
𝑑𝑉
𝑑𝑟𝑖 (24)

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
20
Vidimo, da moramo ob vsakem časovnem koraku posebej izračunati sile med delci v sistemu
s katerimi nato izračunamo pozicije delcev v novem časovnem koraku. S krajšanjem
časovnega koraka dobimo boljši približek dejanskemu gibanju delcev v sistemu, vendar je
posledično potrebno opraviti več izračunov v istem časovnem okvirju.
Verletov algoritem si lahko za lažjo predstavo ponazorimo tudi grafično.
Slika 10: Ponazoritev Verletovega algoritma
Pridobitev konfiguracije ri
Pridobitev konfiguracije ri-1
Izračun sil na vse atome Fi
preko ri
Izračun položajev atomov z naslednji korak ri+1
Če je potrebno korake ponovimo

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
21
2.6 Uporabljeni računalniški programi
Pri izvajanju simulacij in analizi rezultatov tekom raziskovanja smo uporabljali naslednje
računalniške programe:
• GROMOS
• VMD
• PYMOL
GROMOS je kratica za Groningen Molecular Simulation paket. Je računalniški program za
simulacije molekulske dinamike (MD) in modeliranje večjih sistemov biomolekul (nekaj 1000
atomov). Prvotno razvit na Nizozemskem leta 1978, od leta 1990 pa se razvoj programskega
paketa nadaljuje v Švici [10]. Program zajema dve osnovni zmogljivosti: simulacije
molekulske dinamike in simulacije biomolekul z uporabo stohastične dinamike (SD) ter
analizo molekularnih trajektorij in energij pridobljenih z računalniškimi simulacijami z
uporabo polj sil, osnovanih na eksperimentalnih podatkih [11]. Skozi leta se je programski
jezik spremenil iz prvotnega Fortrana v C++. Zato se tudi programski paketi v novejših
verzijah GROMOS-a imenujejo MD++, spremljajoči programi pa GROMOS++. GROMOS
sestavlja 2x105 vrstic programske kode, ki ponuja široko paleto uporabnosti simulacij
molekulske dinamike in možnost analize širokega spektra parametrov ter izdelave vizualne
podpore in grafov za lažjo interpretacijo rezultatov [10]. Shematski prikaz izvedbe simulacije
molekulske dinamike s programom GROMOS, je prikazan na Sliki 9.
Slika 11: Koraki simulacije molekulske dinamike

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
22
VMD je program, namenjen vizualizaciji rezultatov simulacije, in nam omogoča vizualno
analizo obnašanja sistema tekom simulacije v izbranem časovnem okviru ter omogoči zajem
slik stanja sistema ob izbranem času simulacije [12].
Slika 12: VMD program
PYMOL je podobno kot VMD program, ki nam omogoča vizualno analizo, izvoz grafik in
manipulacijo s strukturami v sistemu ter upravljanje z oznakami posameznih atomov in
dodajanje informacij k delcem v sistemu [13].

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
23
2.7 Analiza rezultatov MD simulacij
2.7.1 RMSD
Kadar želimo med seboj primerjati vse konformacije, ki smo jih dobili iz simulacije
molekulske dinamike, skoraj vedno uporabimo kriterij RMSD (ang. Root Mean Square
Deviation). Ta kriterij nam pove, ali so si izbrane konformacije nekega sistema med seboj
podobne in koliko se med seboj razlikujejo. RMSD nam poda informacijo o podobnosti
oziroma različnosti dveh konformacij iste molekule.
𝑅𝑀𝑆𝐷 = √∑ ((𝑥𝑖
1 − 𝑥𝑖0)2 + (𝑦𝑖
1 − 𝑦𝑖0)2 + (𝑧𝑖
1 − 𝑧𝑖0)2)𝑁
𝑖=1
𝑁 (25)
Iz enačbe za RMSD, kjer x, y, z predstavljajo koordinate v 3D sistemu atomov, lahko vidimo,
da bi v idealnem primeru, kjer bi se dve konformaciji popolnoma ujemali, RMSD funkcija
imela vrednost 0. To bi pomenilo da se neka konformacija (1) popolnoma ujema s referenčno
strukturo (0), katero pa dobimo na podlagi informacij NMR ali XRD analiz.
Kadar želimo med seboj primerjati dve konformaciji, moramo poiskati minimum RMSD
funkcije. To dosežemo z računanjem najboljšega prekrivanja dveh konformacij z minimizacijo
RMSD. Z drugimi besedami to pomeni poiskati lego, pri kateri sta si dve konformaciji najbolj
podobni (najnižja vrednost RMSD). To dosežemo z odvajanjem funkcije po 𝑥𝑖1, 𝑦𝑖
1 in 𝑧𝑖1 po
algoritmih minimiziranja RMSD Ferro-Hermans ali Kabash.
𝜕𝑅𝑀𝑆𝐷
𝜕𝑥𝑖1 = 0 ,
𝜕𝑅𝑀𝑆𝐷
𝜕𝑦𝑖1 = 0 𝑖𝑛
𝜕𝑅𝑀𝑆𝐷
𝜕𝑧𝑖1 = 0 (26)
2.7.2 RMSF
Funkcija RMSF (ang. Root Mean Square Fluctuation) nam pove, kako se posamezen atom v
molekuli tekom simulacije giba okoli svoje povprečne lege. Nizke RMSF vrednosti pomenijo,
da se položaj delcu manj spreminja skozi simulacijo. RMSF vrednost nam tako lahko da
informacijo o fleksibilnih ali »mehkih« regijah v biomolekuli, saj se fluktuacije različnih regij
v strukturi razlikujejo.
𝑅𝑀𝑆𝐹 = √1
𝑁𝑡∑(𝑟𝑖 − ⟨𝑟⟩)2
𝑁𝑡
𝑖=1
(27)
𝑁𝑡 – število časovnih korakov
𝑟𝑖 – trenutna lega atoma
⟨𝑟⟩ – povprečna lega atoma

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
24
2.7.3 Analiza klastrov
Analiza klastrov oziroma »grozdov« (ang. Cluster) nam pove, katera konformacija našega
sistema prevladuje tekom naše simulacije in koliko konformacij doseže to najpogostejšo
strukturo v našem časovnem okvirju simulacije [11]. Analizo klastrov tako uporabimo za
identificiranje podobnih ali najpogostejših skupin struktur v našem sistemu. S tem dobimo
dobro predstavo o najpogostejših strukturah, ki so obstojne v sistemu pod izbranimi pogoji.
Analiza in določevanje klastrov je osnovana na računanju RMSD matrike med vsemi pari
struktur, ki smo jih dobili tekom simulacije. Strukture združi v klastre glede na odrezno
vrednost RMSD, ki jo empirično določimo. Za referenčno strukturo najpogosteje vzamemo
strukturo eksperimentalno pridobljeno z NMR ali XRD analizo. Nato konformacije znotraj
ustrezne razdalje od referenčne strukture izločimo iz ponovne analize v naslednjem koraku.
Sledeči korak poteka enako kot prejšnji, z iskanjem naslednje najpogostejše konformacije
glede na vrednosti RMSD.

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
25
3 Eksperimentalni del
V sklopu eksperimentalnega dela magistrske naloge smo izvedli simulacije molekulske
dinamike konvencionalnega ter mikrovalovnega gretja vodne raztopine DNK z uporabo dveh
ločenih termostatov za rotacijsko in translacijsko temperaturo vode. Simulacije
konvencionalnega gretja smo izvajali pri temperaturah 300 K, 400 K, 500 K in 600 K. Sistem
pri 300 K je služil za referenco. Simulacije mikrovalovnega gretja smo izvedli tako, da smo
temperaturo DNA in translacijsko temperaturo vode obdržali na 300K, rotacijsko temperaturo
vode pa zvišali na 400 K, 500 K in 600 K. Te simulacije smo poimenovali R400K, R500K ter
R600K. Simulacije smo izvajali 20 ns, razen 300K in R400K, kjer je DNK obstojna dalj časa.
Tu so simulacije potekale 50 ns.
3.1 Računalniška oprema
Ker smo se pri magistrski nalogi lotili reševanja problema z računalniškim pristopom, smo
raziskovalni del naloge opravljali izven laboratorija v računalniškem okolju. Vse simulacije
in analizo smo izvajali na dveh lokalnih računalnikih, vsak s po dvema Intel Xeon E5 2630-
v4 procesorjema, kar skupaj nanese 20 jeder oz. 40 niti, 32 GB delovnega pomnilnika in
grafično kartico GeForce GTX 1050Ti. Vsi programi se izvajajo znotraj operacijskega sistema
Linux X Ubuntu, saj ponuja odprtokodni sistem boljše prilagajanje funkcij uporabniku in ker
vsi uporabljeni programi zaradi učinkovitejšega dela delujejo v sistemu Linux, saj pri
aplikaciji ne uporabljajo uporabniku prijaznega grafičnega vmesnika in s tem pridobijo na
zmogljivosti, ki je potrebna za izvajanje zahtevnih računov.
Računalniški programi v katerih smo izvajali simulacije in analize so bili nameščeni na
lokalnih računalnikih imenovanih Nautilus in Castor. Dostop do teh dveh računalnikov in s
tem do programov pa je bil možen preko domačega računalnika z operacijskim sistemom
Linux preko ukaza SSH. Tako smo se do obeh delovnih računalnikov lahko povezovali od
doma kot tudi iz šolskega računalnika in sproti preverjali potek ter stanje simulacij.
Pri našem delu smo se srečali z naslednjimi najpogostejšimi vrstami datotek:
• .top – vsebuje vse informacije o topologiji naše biomolekule
• .pdb – vsebuje podatke o koordinatah in lastnostih biomolekule in podatke, ki
omogočajo vizualizacijo biomolekule v programih VMD in PYMOL.
• .dat – vsebuje podatke o rezultatih analiz trajektorij naših simulacij
• .out – vsebuje zbrane rezultate energijske analize simulacij molekulske dinamike
• .pdf – vsebuje grafične ponazoritve dobljenih rezultatov
• .arg – vsebuje informacije in navodila za zagon simulacij in analiznih programov

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
26
3.2 Topologija odseka molekule DNK
Preden smo začeli s simulacijami molekulske dinamike, smo morali izdelati topologijo naše
biomolekule. Topologija vsebuje vse informacije o topološki strukturi, tipu atomov,
konstantah vezi, kotih, elektrostatskih interakcijah in prostorski orientiranosti izbrane
biomolekule.
Slika 13: Topologija DNK fragmenta
Slika 10 prikazuje del podatkov, ki jih vsebuje datoteka s topologijo naše biomolekule. ATNM
predstavlja številko atoma v molekuli, MRES številko nukleotida, PANM ime atoma v
topljencu, IAC kodo tipa atoma, MASS predstavlja maso atoma v topljencu, CG naboj atomov,
CGC kodo skupine naboja (0 ali 1), INE številko izštetih atomov ter INE14 število interakcij.

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
27
3.3 Ustvarjanje sistema map in podmap simulacij
Preden smo začeli z zagonom začetnih 1 ns simulacij minimizacije smo si ustvarili pregleden
sistem map in podmap, ki nam je v nadaljevanju omogočil nemoteno delo in avtomatično
poganjanje programov za analizo rezultatov simulacij molekulske dinamike. Shema
ustvarjenih map je prikazana na spodnji sliki.
Slika 14: Datotečna struktura simulacij
DNA
Topologija molekule DNK
Simulacije
t300_cv_01ns
t400_cv_01ns
t500_cv_01ns
t600_cv_01ns
t300_cv_50ns
tR400_cv_50ns
t400_cv_20ns
tR500_cv_20ns
t500_cv_20ns
tR600_cv_20ns
t600_cv_20ns
.cnf datoteke

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
28
3.4 Začetne minimizacije DNK molekule
Preden smo začeli z zagonom simulacije molekulske dinamike z dvema ločenima
termostatoma za rotacijsko in translacijsko prostostno stopnjo vode, smo morali izbrano
začetno molekulo DNK energijsko minimizirati in termalizirati na izbrano temperaturo.
Začetni podatki o molekuli namreč vsebujejo informacije o poziciji in stanju atomov v
molekuli pri izredno nizki temperaturi in v vakuumu. Zato moramo molekulo DNK najprej
postopoma sergrevati do začetne temperature, dodati topilo ter definirati konstanten volumen,
v katerem bodo tekle simulacije. Tako smo pognali 5 0.1 ns simulacij začetnih termalizacij do
temperatur pri katerih smo v nadaljevanju izvajali simulacije molekulske dinamike.
Začetne minimizacije in termalizacije smo izvedli pri naslednjih temperaturah:
• 300 K
• 400 K
• 500 K
• 600 K
Po uspešno izvedenih začetnih simulacijah smo našo molekulo imeli pripravljeno za zagon 50
ns oziroma 20 ns simulacij s konvencionalnim gretjem in ločenima termostatoma za rotacijsko
in translacijsko temperaturo vode.
Slika 15: Začetna minimizirana in termalizirana konformacija izbranega fragmenta DNK pri 300K

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
29
3.5 Zagon simulacij molekulske dinamike V vse predhodno ustvarjene mape simulacij smo pripravili datoteke .lib, .imd ter .arg, ki so
vsebovale informacije o parametrih simulacije ter ukaze za izračune, katere smo želeli izvesti
na našem sistemu skupaj z navodili o shranjevanju in izpisu rezultatov v ustrezni obliki,
primerni za kasnejšo obdelavo. Za začetno konformacijo pri vsaki simulaciji smo vzeli zadnjo
konformacijo predhodne termalizacijske simulacije in priredili .imd in .arg datoteke pri vsaki
simulaciji, kot je prikazano na spodnjih slikah.
Slika 16: .arg datoteka
.arg datoteki smo za vsako simulacijo določili ustrezno ime, datotečno pot do končne
konformacije predhodne termalizacijske simulacije in z ukazom @script definirali število 1 ns
korakov simulacije, kar bo ustvarilo simulacijo dolgo 50 ns oziroma 20 ns.
Slika 17: .imd datoteka
V .imd datoteki smo za vsako simulacijo nastavili ustrezne parametre simulacije, označene na
mestih vidnih na Sliki 14. Slika prikazuje .imd datoteko za simulacijo, pri kateri so vsi
termostati nastavljeni na 300 K. V nadaljevanju smo izvedli še 3 simulacije pri 400 K, 500 K
in 600 K, kar je pomenilo, da smo zgornjo .imd datoteko priredili tako, da smo v stolpec
TEMP0 vpisali vse tri vrednosti enake temperature. Tako smo imeli pripravljene datoteke za
zagon simulacij z enakimi temperaturami topljenca, translacijske in rotacijske temperature
topila.

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
30
Cilj magistrskega dela je bil ugotoviti, ali rotacijsko vzbujena voda s strani mikrovalov vpliva
na stabilnost dvojne vijačnice DNK, zato smo nadalje pripravili še 3 simulacije, pri katerih pa
smo povišali samo rotacijsko temperaturo topila, kar je simuliralo eksperiment, kjer bi v
laboratoriju raztopino obsevali samo z mikrovalovi. Te simulacije smo si pripravili na enak
način kot prejšnje, le da smo spremenili vhodne podatke tako, da smo povišali samo
temperaturo tretjega termostata, ki kontrolira rotacijsko temperaturo topila.
Slika 18: .imd datoteka za pripravo simulacije s povišano rotacijsko temperaturo vode
Simulacije smo zagnali na računalniku Nautilus po 5 simulacij naenkrat. Vsaki simulaciji smo
pripisali 3 jedra procesorja od 20 možnih jeder, saj se je to izkazalo za najbolj učinkovit
izkoristek procesorske moči. Vsako simulacijo smo pognali po enakem postopku. Najprej smo
ustrezno uredili in priredili .imd in .arg datoteke, kot je opisano zgoraj. Nato smo s programom
mk_script, ki služi za ustvarjanje potrebnih datotek za zagon simulacije in preverjanje vhodnih
podatkov, preverili ustreznost naših vhodnih podatkov in ustvarili datoteke potrebne za zagon
simulacije - .run, .omd, .imd, .cnf. Nato smo s programom screen odprli novo okno, v katerem
smo z ukazom ./md_dna_1.run pognali simulacijo. Vsaka simulacija je trajala približno 14
dni.

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
31
3.6 Programi za analizo trajektorij simulacij
Po uspešno zaključenih simulacijah molekulske dinamike z ločenima termostatoma za
translacijsko in rotacijsko prostostno stopnjo vode smo opravili analizo rezultatov, to je energij
in geometrij trajektorij simulacije. Z analiznimi orodji programov GROMOS, VMD in
PYMOL smo iz simulacije zbrali naslednje parametre in jih primerjali med seboj:
• Temperatura sistema (program ene_ana)
• Nevezne energije sistema (program ene_ana)
• RMSD (program RMSD)
• RMSF (program RMSF)
• Analiza klastrov (program cluster)
• Razdalja med verigami (program TSER)
• Prisotnost vodikovih vezi (program TSER)
• Vizualna interpretacija dinamike topljenca (program Frameout)
Analizo trajektorij smo začeli tako, da smo si za vsako simulacijo ustvarili sistem map, kamor
smo shranjevali vhodne podatke za zagon analiz, kakor tudi izhodne podatke analiz trajektorij.
Mape smo poimenovali enako, kot so se imenovali programi, katere smo v nadaljevanju
uporabili, ter za vsako vrsto analize napisali svojo vhodno .arg datoteko z ukazi,
predstavljenimi spodaj.
Datoteka .arg s vhodnimi podatki za zagon analiznega programa ene_ana je vsebovala
datotečno pot do topologije začetne molekule DNK, pot do knjižnice podatkov in pot do
datotek .tre.gz, ki so vsebovale trajektorije energije molekule v simulaciji ter vhodne podatke
@prop, ki definirajo vrsto analize v programu ene_ana. Kot kaže spodnja slika, smo želeli
analizirati celotno, potencialno ter kinetično energijo sistema, izvedeti podatke o temperaturah
posameznih komponent sistema, nevezne energije topljenec-topljenec in topljenec-topilo,
nevezne van der Waalsove in elektrostatske energije ter njihov seštevek.
Slika 19: Vhodna datoteka za analizo s programom ene_ana

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
32
RMSD analizo smo izvedli v dveh korakih. Najprej smo ustvarili .arg datoteko z navodili za
zagon RMSD analize po celotni DNK molekuli (od 1. do 24. baznega para), nato pa še novo
.arg datoteko, v kateri smo zahtevali RMSD analizo po notranjih baznih parih (od 2. do 23.
baznega para), saj smo predvidevali, da se lahko tekom simulacije končni bazni pari bolj
prosto gibajo kot notranji, zato smo želeli primerjati obe možnosti. Vhodna datoteka za zagon
RMSD programa je izgledala sledeče:
Slika 20: Vhodna datoteka za izračun celotnega RMSD
Slika 21: Vhodna datoteka za izračun notranjega RMSD
RMSF analizo smo pognali na podoben način kot analizo RMSD, le da smo v vhodni datoteki
za RMSF dodali vrstico @atomsfit, ki nam je definirala referenčno konformacijo, na podlagi
katere se je nato računal RMSF. Kot referenčno strukturo smo vzeli DNK fragment v raztopini
pri 300K.
Slika 22: Vhodna datoteka za analizo RMSF

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
33
Program Frameout nam kot izhodno datoteko vrne datoteko .pdb, ki je primerna za odpiranje
v programih VMD ali PYMOL in nam omogoča vizualno interpretacijo rezultatov ter možnost
predvajanja animacije naše 50 ns oziroma 20 ns simulacije, ki nam še dodatno pomaga
razumeti dinamiko topljenca tekom simulacij. Vhodna datoteka za zagon programa Frameout
je sledeča:
Slika 23: Vhodna datoteka za zagon programa Frameout
S programom TSER smo določili število in dolžino prisotnih vodikovih vezi v molekuli DNK
tekom 50 ns oziroma 20 ns simulacije, kot tudi razdaljo med verigami in čas razpada fragmenta
DNK. .arg datoteka za zagon programa je bila naslednja:
Slika 24: Vhodna datoteka za zagon programa TSER
Analiza tako imenovanih klastrov nam določi strukture ali konformacije sistema, ki so najbolj
pogoste v naših simulacijah. Poišče nam strukturne »sosede«, ki so po vrednosti RMSD zelo
podobni oziroma imajo razliko RMSD vrednosti manjšo od predhodno izbrane odrezne
razdalje. Za zagon programa CLUSTER moramo ustvariti tri ločene datoteke .arg
predstavljene spodaj.
Slika 25: Vhodne datoteke za analizo klastrov

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
34
4 Rezultati in diskusija
Po približno 4 tednih izvajanja simulacij smo začeli z analizo trajektorij in rezultatov serije 50
ns oziroma 20 ns simulacij. Pri analizah smo uporabili že prej omenjene programe. V
nadaljevanju so predstavljeni in ovrednoteni rezultati analiz.
4.1 Vizualna predstavitev rezultatov Zaradi lažje predstave nas je najprej zanimala vizualna ponazoritev rezultatov naših simulacij
molekulske dinamike. Tako smo .pdb datoteko izdelano s programom Frameout odprli v
programu za vizualizacijo rezultatov VMD. Začetne in končne konformacije vseh 9 simulacij
so predstavljene na spodnjih slikah.
Slika 26: Začetna (levo) in končna (desno) konformacija molekule DNK pri 300 K
Slika 27: Začetna (levo) in končna (desno) konformacija molekule DNK pri povišani zgolj rotacijski temperaturi na
400 K

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
35
Slika 28:Začetna (levo) in končna (desno) konformacija molekule DNK pri 400 K
Slika 29:Začetna (levo) in končna (desno) konformacija molekule DNK pri povišani zgolj rotacijski temperaturi na
500 K
Slika 30: Začetna (levo) in končna (desno) konformacija molekule DNK pri 500 K

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
36
Slika 31: Začetna (levo) in končna (desno) konformacija molekule DNK pri povišani zgolj rotacijski temperaturi na
600 K
Slika 32: Začetna (levo) in končna (desno) konformacija molekule DNK pri 600 K

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
37
Na zgornjih slikah začetnih in končnih konformacij molekule DNK že vidimo nek trend, ki se
pojavlja pri končnih konformacijah vsake simulacije. Pri 300 K vidimo da bistvene razlike,
vsaj vizualno, ne vidimo, kar je tudi v skladu s pričakovanji, saj je DNK pri temperaturi 300
K oziroma 27 °C še stabilna.
Pri simulaciji 400K smo začeli opažati razpad dvojne vijačnice DNK fragmenta. Pri simulaciji
R400K pa nam vizualna analiza pove, da je naša molekula DNK obdržala strukturo približno
enako prvotni. Enako obnašanje opazimo pri vse nadaljnjih simualcijah. Simulacije, ki
simulirajo konvencionalno, termično gretje (500K in 600K) rezultirajo v razpadu dvojne
vijačnice DNK fragmenta. Simualcije, ki simulirajo mikrovalovno gretje, pa se ne končajo z
razpadom dvojne vijačnice.
Na podlagi vizualne analize lahko sklepamo, da pri simulacijah konvencionalnega termičnega
gretja prej pride do razpada dvojne vijačnice DNK, ker je temperatura sistema nad talilno
temperaturo DNK, zato je razpad fragmenta pri simulacijah 400K, 500K in 600K pričakovan.
4.2 Analiza RMSD in RMSF
Tabela 1 prikazuje povprečja in standardne odmike vrednosti RMSD in RMSF za vsako
simulacijo. V tabeli so zbrane RMSD vrednosti dveh različnih sistemov. Prvi stolpec podaja
vrednosti RMSD za notranji del DNK fragmenta, kar pomeni da pri analizi nismo upoštevali
zunanjih dveh baz (na obeh koncih verige), saj nas je zanimalo, ali nestabilni končni deli
fragmenta vplivajo na RMSD analizo. Z primerjavo drugi stolpec RMSD v Tabeli 1 podaja
rezultate analize za celoten DNK fragment.
Simulacija RMSD (res. 2-23)
[nm] RMSD (res. 1-24)
[nm] RMSF (backbone)
[nm]
〈 〉 σ 〈 〉 σ 〈 〉 σ
300K 0,287 0,061 0,299 0,062 0,183 0,090
R400K 0,421 0,124 0,470 0,151 0,273 0,226
400K 1,028 0,287 1,041 0,288 0,721 0,213
R500K 0,277 0,035 0,287 0,036 0,142 0,061
500K 1,295 0,286 1,328 0,292 1,046 0,294
R600K 0,370 0,046 0,379 0,045 0,205 0,128
600K 1,616 0,381 1,687 0,391 1,321 0,257
Tabela 1: RMSD in RMSF analize 10 ns dolgih simulacij

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
38
Iz analize RMSD in RMSF vrednosti lahko vidimo, da je ponovno prisoten ponavljajoč vzorec,
ki loči simulacije 400K, 500K in 600K od simulacij R400K, R500K in R600K. Opazimo, da
mikrovalovno gretje drugače vpliva na gibanje fragmenta DNK, kot termično gretje. Opaziti
je namreč, da so RMSD vrednosti termičnega gretja višje kot pri simulacijah mikrovalovnega
gretja. Višje vrednosti RMSD pomenijo večje odstopanje od referenčne strukture, kar nakazuje
na bolj fleksibilen fragment DNK pri simulacijah termičnega gretja.
Enako obnašanje posledično ugotovimo tudi pri analizi RMSF funkcije. Simulacije 400K,
500K in 600K imajo večjo fluktuacijo atomov v strukturi kot pa simulacije mikrovalovnega
gretja R400K, R500K in R600K. Ponavljajoč trend lahko ponovno pojasnimo s tem, da v
primeru konvencionalnega termičnega gretja, ko sta poleg rotacijske zvišani še translacijska
temperatura topila in temperatura topljenca, naša molekula DNK razpada hitreje in s tem
posledično v večji meri spremeni strukturo. Na Slikah 33-39 so grafično predstavljeni rezultati
RMSD analiz. Rdeči graf predstavlja RMSD analizo po celotnem fragmentu 24 baznih parov,
zelena barva pa predstavlja analizo po notranjih baznih parih (od 2. do 23. baznega para) brez
baznih parov na vsakem koncu fragmenta.
Slika 33: RMSD analiza za simulacijo 300K

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
39
Slika 34: RMSD analiza za simulacijo R400K
Slika 35: RMSD analiza za simulacijo 400K

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
40
Slika 36: RMSD analiza za simulacijo R500K
Slika 37: RMSD analiza za simulacijo 500K

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
41
Slika 38: RMSD analiza za simulacijo R600K
Slika 39: RMSD analiza za simulacijo 600K
Na Slikah 40-46 pa so grafično predstavljeni rezultati RMSF analiz.

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
42
Slika 40: RMSF analiza simulacije 300K
Slika 41: RMSF analiza simulacije R400K
Slika 42: RMSF analiza simulacije 400K

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
43
Slika 43: RMSF analiza simualcije R500K
Slika 44: RMSF analiza simulacije 500K

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
44
Slika 45: RMSF analiza simulacije R600K
Slika 46: RMSF analiza simulacije 600K
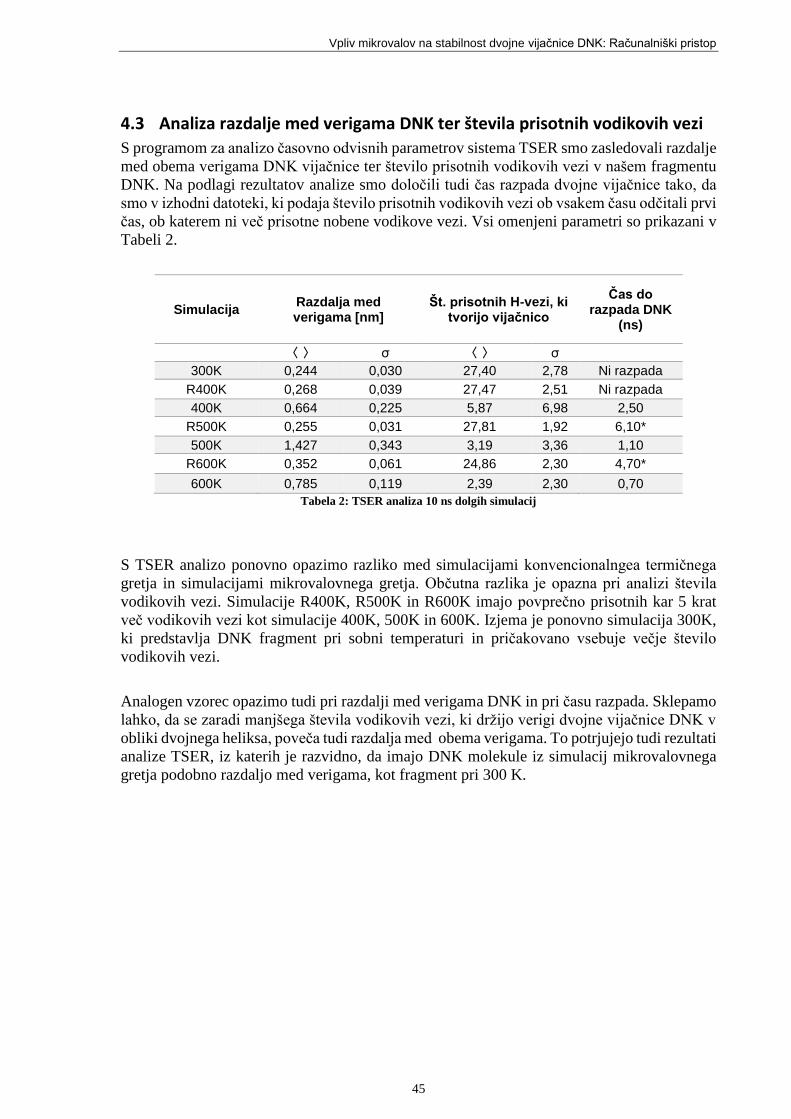
Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
45
4.3 Analiza razdalje med verigama DNK ter števila prisotnih vodikovih vezi
S programom za analizo časovno odvisnih parametrov sistema TSER smo zasledovali razdalje
med obema verigama DNK vijačnice ter število prisotnih vodikovih vezi v našem fragmentu
DNK. Na podlagi rezultatov analize smo določili tudi čas razpada dvojne vijačnice tako, da
smo v izhodni datoteki, ki podaja število prisotnih vodikovih vezi ob vsakem času odčitali prvi
čas, ob katerem ni več prisotne nobene vodikove vezi. Vsi omenjeni parametri so prikazani v
Tabeli 2.
Simulacija Razdalja med verigama [nm]
Št. prisotnih H-vezi, ki tvorijo vijačnico
Čas do razpada DNK
(ns)
〈 〉 σ 〈 〉 σ
300K 0,244 0,030 27,40 2,78 Ni razpada
R400K 0,268 0,039 27,47 2,51 Ni razpada
400K 0,664 0,225 5,87 6,98 2,50
R500K 0,255 0,031 27,81 1,92 6,10*
500K 1,427 0,343 3,19 3,36 1,10
R600K 0,352 0,061 24,86 2,30 4,70*
600K 0,785 0,119 2,39 2,30 0,70
Tabela 2: TSER analiza 10 ns dolgih simulacij
S TSER analizo ponovno opazimo razliko med simulacijami konvencionalngea termičnega
gretja in simulacijami mikrovalovnega gretja. Občutna razlika je opazna pri analizi števila
vodikovih vezi. Simulacije R400K, R500K in R600K imajo povprečno prisotnih kar 5 krat
več vodikovih vezi kot simulacije 400K, 500K in 600K. Izjema je ponovno simulacija 300K,
ki predstavlja DNK fragment pri sobni temperaturi in pričakovano vsebuje večje število
vodikovih vezi.
Analogen vzorec opazimo tudi pri razdalji med verigama DNK in pri času razpada. Sklepamo
lahko, da se zaradi manjšega števila vodikovih vezi, ki držijo verigi dvojne vijačnice DNK v
obliki dvojnega heliksa, poveča tudi razdalja med obema verigama. To potrjujejo tudi rezultati
analize TSER, iz katerih je razvidno, da imajo DNK molekule iz simulacij mikrovalovnega
gretja podobno razdaljo med verigama, kot fragment pri 300 K.

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
46
Trenda razdalje med verigama in števila vodikovih vezi kažeta, da je v primeru termičnega
gretja fragment DNK vedno manj stabilen. To pomeni, da se vodikove vezi hitreje pretrgajo
in čas do razpada fragmenta se krajša.
V časovne okviru simulacij (10 ns) smo dejanski razpad dvojne vijačnice opazili samo pri
simulacijah 400K, 500K in 600K. Pri simulacijah R500K in R600K smo pri časih, ki so v
tabeli označeni z zvezdico opazili zgolj znaten preskok v vrednosti RMSD, vendar ne
popolnega razpada, kot bi pričakovali.
4.4 Analiza energijskih trajektorij
S programom ene_ana za analizo energij in z njimi povezanih lastnosti smo izvedli analizo
energijskih trajektorij, kjer smo izračunali skupno nevezno potencialno energijo in posamezne
prispevke nevezne potencialne energije – elektrostatsko potencialno energijo ter van der
Waalsovo potencialno energijo. Dobili in analizirali smo tudi temperature komponent sistema
pri vsaki simulaciji, ki so prikazane v Tabeli 3. Do odstopanja dejanskih povprečnih
temperatur posameznih komponent sistema od teoretičnih (300 K, 400 K, 500 K in 600 K)
pride, ker so v kondenziranih sistemih prostostne stopnje sklopljene in se na primer del
energije rotacijskega gibanja preko medmolekulskih trkov (disipacije) pretvori v translacijsko
ali vibracijsko gibanje.
Simulacija T DNK [K] T vode, trans. [K] T vode, rot. [K]
〈 〉 σ 〈 〉 σ 〈 〉 σ
300K 299,799 0,090 299,655 0,008 301,306 0,014
R400K 300,219 0,062 303,312 0,014 397,693 0,020
400K 399,878 0,100 399,579 0,016 401,595 0,030
R500K 300,898 0,057 308,389 0,016 492,619 0,027
500K 499,623 0,101 499,442 0,014 501,779 0,032
R600K 301,636 0,068 314,101 0,022 586,800 0,033
600K 599,336 0,104 599,357 0,020 602,052 0,029
Tabela 3: Dejanske povprečne temperature posameznih komponent sistema

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
47
Simulacija Nevezna energija (DNK-
topilo + DNK - DNK) [kcal/mol]
Nevezna energija VDW (DNK - topilo + DNK-
DNK) [kcal/mol]
Nevezna energija EL.STAT (DNK - topilo +
DNK -DNK) [kcal/mol]
〈 〉 σ 〈 〉 σ 〈 〉 σ
300K -5812,35 349,17 -1735,12 47,42 -4077,23 347,69
R400K -6281,07 546,74 -1793,71 59,72 -4487,36 512,71
400K -6593,51 719,65 -1633,90 75,99 -4959,61 712,91
R500K -6042,06 319,55 -1773,15 36,83 -4268,91 317,83
500K -6571,07 613,54 -1537,23 83,55 -5033,84 598,71
R600K -6316,57 333,84 -1765,54 43,00 -4551,02 330,76
600K -6428,59 720,78 -1353,74 113,17 -5074,84 679,97
Tabela 4: Analiza trajektorij neveznih potencialnih energij
Analiza trajektorij skupne nevezne potencialne energije in potencialne energije njenih
komponent iz Tabele 4 nam poda zanimive rezultate. Če pogledamo vrednost nevezne van der
Waalsove energije opazimo, da pri simulacijah konvencionalnega termičnega jakost van der
Waalsovih interakcij z višanjem temperature pada. To je v skladu s pričakovanji, saj se z
zmanjšanjem števila prisotnih vodikovih vezi povečajo razdalje med verigama, kar vpliva na
znižanje potencialne van der Waalsove energije, ki je kratkosežna in hitro pada z večanjem
razdalje. Jakost nevezne elektrostatske energije pa pri istih simulacijah narašča s temperaturo,
prav tako tudi jakost celokupne nevezne potencialne energije. To bi lahko razložili s tem, da
sistem z višanjem temperature dobiva bolj neurejeno strukturo, ki odstopa od začetne, in s tem
se negativno nabite fosfatne skupine bolj izpostavijo hidrataciji vode. Večanje jakosti
elektrostatskih interakcij pri simulacijah mikrovalovnega gretja pa si lahko razlagamo kot
posledico povečanega in ne zmanjšanega senčenja nabojev fosfatne skupine s strani vode
Slika 47: Spreminjanje nevezne potencialne energije s časom pri simulaciji 300K. Zelena barva ponazarja
interakcije DNK-DNK. Modra predstavlja energijo DNK-voda. Rdeča predstavlja povprečje obeh interakcij, svetlo
modra barva pa predstavlja vsoto obeh interakcij
[kcal/m
ol]

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
48
Slika 48: Spreminjanje nevezne potencialne energije s časom pri simulaciji R400K. Zelena barva ponazarja
interakcije DNK-DNK. Modra ponazarja energijo DNK-voda. Rdeča predstavlja povprečje obeh interakcij, svetlo
modra barva pa predstavlja vsoto obeh interakcij
Slika 49: Spreminjanje nevezne potencialne energije s časom pri simulaciji 400K. Zelena barva ponazarja
interakcije DNK-DNK. Modra ponazarja energijo DNK-voda. Rdeča predstavlja povprečje obeh interakcij, svetlo
modra barva pa predstavlja vsoto obeh interakcij
[kcal/m
ol]
[kcal/m
ol]

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
49
Slika 50: Spreminjanje nevezne potencialne energije s časom pri simulaciji R500K. Zelena barva ponazarja
interakcije DNK-DNK. Modra ponazarja energijo DNK-voda. Rdeča predstavlja povprečje obeh interakcij, svetlo
modra barva pa predstavlja vsoto obeh interakcij
Slika 51: Spreminjanje nevezne potencialne energije s časom pri simulaciji 500K. Zelena barva ponazarja
interakcije DNK-DNK. Modra ponazarja energijo DNK-voda. Rdeča predstavlja povprečje obeh interakcij, svetlo
modra barva pa predstavlja vsoto obeh interakcij
[kcal/m
ol]
[kcal/m
ol]

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
50
Slika 52: Spreminjanje nevezne potencialne energije s časom pri simulaciji R600K. Zelena barva ponazarja
interakcije DNK-DNK. Modra ponazarja energijo DNK-voda. Rdeča predstavlja povprečje obeh interakcij, svetlo
modra barva pa predstavlja vsoto obeh interakcij
Slika 53: Spreminjanje nevezne potencialne energije s časom pri simulaciji 600K. Zelena barva ponazarja
interakcije DNK-DNK. Modra ponazarja energijo DNK-voda. Rdeča predstavlja povprečje obeh interakcij, svetlo
modra barva pa predstavlja vsoto obeh interakcij
[kcal/m
ol]
[kcal/m
ol]

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
51
4.5 Analiza klastrov
Analiza klastrov nam je pomagala določiti skupine najpogostejših konformacij tekom naših
simulacij. Klaster oziroma »grozd« predstavlja skupino podobnih konformacij molekule
oziroma konformacijskih sosedov. Več različnih klastrov kot zasledimo v simulaciji, bolj
nestabilna je naša preiskovana struktura. V primeru, ko bi se ves čas simulacije molekula zgolj
malo spreminjala, bi posledično zasledili več podobnih struktur, katerih vrednost RMSD ne bi
presegala odrezne vrednosti, skupno število klastrov pa bi bilo majhno.
Rezultati analize klastrov posameznih simulacij so prikazani v Tabeli 5.
Temperatura Št. clustrov, ki so
prisotni vsaj 5% časa
Št. clustrov, ki skupaj predstavljajo več kor
95% vseh struktur
Št. vseh clustrov v
10ns
300K 2 2 4
R400K 2 1 5
400K 5 42 59
R500K 2 1 2
500K 0 153 178
R600K 5 4 6
600K 0 425 450
Tabela 5: Analiza klastrov
Vidimo, da lahko potegnemo vzporednice z rezultati ostalih analiz. Kot smo predvidevali, je
struktura DNK pri simulaciji 300K ostala stabilna, kar nam potrjuje tudi analiza klastrov, ki
nam pove, da so bili v času 10 ns simulacije prisotni zgolj 4 klastri. To pomeni, da so si bile
strukture DNK tekom simulacije 300K tako podobne, da smo jih lahko razvrstili v samo 4
različne skupine klastrov, glede na izbran odrezni kriterij RMSD (vrednost 0,3 nm). Naša
pričakovanja, da se bo tudi pri analizi klastrov pokazal enak trend kot pri prejšnjih analizah,
smo lahko potrdili. Pri vseh simulacijah termičnega gretja 400K, 500K in 600K namreč
opazimo izjemno veliko število prisotnih klastrov napram simulacijam mikrovalovnega gretja
R400K, R500K in R600K.

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
52
5 Zaključek
Rezultati magistrske naloge so pokazali, da mikrovalovi v naših simulacijah molekulske
dinamike v 10 ns časovnem okvirju niso imeli istega vpliva na stabilnost fragmenta DNK kot
konvencionalno termično gretje. Analiza rezultatov je pokazala, da je fragment pri simulacijah
mikrovalovnega gretja ostal v bolj stabilnem stanju kot pri termičnem gretju.
Ker je analiza neveznih interakcij nakazala večanje senčenja nabojev fosfatne skupine s strani
vode poseldično jačanje elektrostatskih interakcij, v našem časovnem okvirju ni prišlo do
razpada ali vidne destabilizacije fragmenta DNK. Ta ugotovitev lahko pomaga pri bodočih
raziskavah stabilnosti DNK pod vplivom mikrovalov pri določevanju časa razpada in njene
stabilnosti v različnih topilih. Iz dobljenih rezultatov magistrske naloge lahko sklepamo, da
mikrovalovi celo pripomorejo k stabilizaciji molekule DNK, podobno kot pripomorejo k
stabilizaciji hidrofobnih beljakovin, kajti podobno kot beljakovine bi se lahko obnašale tudi
nukleobaze, ki predstavljajo hidrofobni del DNK. Pridobljeni rezultati so nam dali dobro
izhodišče za nadaljevanje raziskovanja na področju vpliva mikrovalov na stabilnost DNK, saj
je to področje še dokaj neraziskano in ponuja možnost številnih novih spoznanj, ki lahko
pripomorejo k zagotavljanju večje varnosti potrošniških izdelkov, kot so na primer mobilni
telefoni, mikrovalovne pečice navigacijske naprave idr. Poleg tega ima lahko preučevanje
stabilnosti DNK pomembne posledice v genetiki in genomiki, predvsem pri preučevanju
hibridizacije molekul DNK in mehanizmov transkripcije, ki so pomembni pri odkrivanju in
zdravljenju genetskih bolezni. Rezultati, ki smo jih pridobili pri raziskovanju nenazadnje
potrjujejo uspešno izvedbo magistrskega dela oziroma simulacij, saj različne analize podatkov
nakazujejo isti vzorec obnašanje in se med seboj dopolnjujejo, kar kaže na kvalitetno izvedene
simulacije in uspešno opravljeno analizo.

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
53
6 Literatura
[1] W. F. Edwards, D. D. Young,A. Deiters The effect of microwave irradiation on DNA
hybridization, Org Biomol Chem, 7 (12), 2009
[2] U. Bren, D. Janezic Individual degrees of freedom and the solvation properties of
water, J Chem Phys, 137 (2), 2012
[3] T. Mohoric, U. Bren, V. Vlachy Fast rotational motion of water molecules increases
ordering of hydrophobes in solutions and may cause hydrophobic chains to collapse, J Chem
Phys, 143 (24), 2015
[4] Y. Yang, J. Hang Fragmentation of genomic DNA using microwave irradiation. J
Biomol Tech, 24 (2), 2013
[5] T. M. Devlin Textbook of biochemistry: with clinical correlations. Hoboken: Wiley-
Liss, 2006
[6] C. J. Cramer Essentials of Computational Chemistry, Theories and Models. Wiley:
Chichester, England, 2002
[7] R. Richard Oxford Dictionary of Chemistry. Oxford University Press, 2016
[8] UCLA, Dihedral angle,
http://www.chem.ucla.edu/~harding/IGOC/D/dihedral_angle.html (14.8.2017)
[9] A. R. Leach Molecular Modeling: Principles and Applications 2nd Edition. Pearson
Education Limited, 2001
[10] U. o. Groningen, About the GROMOS software for biomolecular simulation,
http://www.gromos.net/ (12.8.2017)
[11] H. J. C. B. W. F. van Gunsteren The GROMOS Software for (Bio)Molecular
Simulation. Groningen: Biomos, 2016
[12] T. a. C. B. Group, VMD - Visual Molecular Dynamics,
http://www.ks.uiuc.edu/Research/vmd/ (12.8.2017)
[13] Schrodinger PyMOL, https://pymol.org/ (12.8.2017)
[14] C. R. Calladine, H. R. Drew, B. F. Luisi,A. A. Travers Understanding DNA, Third
Edition. Oxford: Academic Press, 2004
[15] R. Cammack Oxford Dictionary of Biochemistry and Molecular Biology. New York:
Oxford University Press, 2000
[16] G. F. Clifford E. Dykstra, Kwang S. Kim and Gustavo E. Scuseria Theory and
Applications of Computational Chemistry: The First Forty Years. Elsevier, 2005
[17] J. A. Glasel Introduction to Biophysical Methods for Protein and Nucleic Acid
Research. San Diego: Academic Press, 1995
[18] D. J. Griffiths Introduction to quantum mechanics. New Jersey: Prentice Hall, 1995
[19] A. Hinchliffe Molecular modeling for begginers. West Sussex: John Wiley & Sons
Ltd, 2003
[20] F. Jensen Introduction to computational chemistry 2nd edition. West Sussex: John
Wiley & Sons Ltd, 2007
[21]
[22] G. R. Clark, C. J. Squire, L. J. Baker, R. F. Martin,J. White Intermolecular interactions
and water structure in a condensed phase B-DNA crystal. Nucleic Acids Research, 2000
[23] V. Garaj-Vrhovac, D. Horvat,Z. Koren The effect of microwave radiation on the cell
genome. Mutat Res, 1990
[24] E. D. Kadtsyn, A. V. Anikeenko,N. N. Medvedev Molecular dynamics simulation of
a DNA duplex labeled with triarylmethyl spin radicals. Journal of Molecular Liquids, 2016

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
54
[25] C. Leal, E. Moniri, L. Pegado,H. Wennerström Electrostatic Attraction between DNA
and a Cationic Surfactant Aggregate. The Screening Effect of Salt. The Journal of Physical
Chemistry B, 2007
[26] T. Mohoric, U. Bren,V. Vlachy Effects of Translational and Rotational Degrees of
Freedom on the Hydration of Ionic Solutes as Seen by the Popular Water Models. Acta Chim
Slov, 2015
[27] K. M. Rahman,D. E. Thurston Effect of microwave irradiation on covalent ligand-
DNA interactions. Chem Commun (Camb), 2009
[28] N. Schmid, C. D. Christ, M. Christen, A. P. Eichenberger,W. F. van Gunsteren
Architecture, implementation and parallelisation of the GROMOS software for biomolecular
simulation. Computer Physics Communications, 2012
[29] J. Šponer, G. Bussi, P. Stadlbauer, P. Kührová, P. Banáš, B. Islam, S. Haider, S.
Neidle,M. Otyepka Folding of guanine quadruplex molecules–funnel-like mechanism or
kinetic partitioning? An overview from MD simulation studies. Biochimica et Biophysica
Acta (BBA) - General Subjects, 2017
[30] T. Špringer, H. Šípová, H. Vaisocherová, J. Štěpánek,J. Homola Shielding effect of
monovalent and divalent cations on solid-phase DNA hybridization: surface plasmon
resonance biosensor study. Nucleic Acids Research, 2010
[31] Z.-J. Tan,S.-J. Chen Nucleic Acid Helix Stability: Effects of Salt Concentration,
Cation Valence and Size, and Chain Length. Biophysical Journal, 2006
[32] S. Yuan, H. C. S. Chan, S. Filipek,H. Vogel PyMOL and Inkscape Bridge the Data and
the Data Visualization. Structure, 2016
[33] H. Zhao,K. Shen Microwave-induced inactivation of DNA-based hybrid catalyst in
asymmetric catalysis. International Journal of Biological Macromolecules, 2016
[34] Zhi-Jie Tan,S.-J. Chen Nucleic Acid Helix Stability: Effects of Salt Concentration,
Cation Valence and Size, and Chain Length. Biophysical Journal, 2006
[35] L. B. Jassie, Microwave heating: Theory and Practice,
http://allchemy.iq.usp.br/agregando/wpa/Palestra2.pdf (25.5.2017)

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
55
7 Življenjepis

Vpliv mikrovalov na stabilnost dvojne vijačnice DNK: Računalniški pristop
56


















