
VOLTAMMETRY AT GLASSY CARBON ELECTRODESpublications.iupac.org/pac-2007/1984/pdf/5608x1095.pdf ·...
36
Pure & Appi. Chem., Vol. 56, No. 8, pp. 1095—1129, 1984. 0033—4545/84 $3.00+0.00 Printed in Great Britain. Pergamon Press Ltd. ©1984 IUPAC INTERNATIONAL UNION OF PURE AND APPLIED CHEMISTRY ANALYTICAL CHEMISTRY DIVISION COMMISSION ON ELECTROANALYTICAL CHEMISTRY* VOLTAMMETRY AT GLASSY CARBON ELECTRODES Prepared for publication by M. GROSS' and J. JORDAN2 1Laboratoire d'Electrochimie, Universite Louis Pasteur, Strasbourg, France 2Davey Laboratory, Pennsylvania State University, University Park, USA *Membership of the Commission during the preparation of this report (1981—83) was as follows: Chairman: J. Jordan (USA); Secretary: K. Izutsu (Japan); Titular Members: A. K. Covington (UK); J. Juillard (France); R. C. Kapoor (India); E. Pungor (Hungary); Associate Members: J. F. Coetzee (USA); W. Davison (UK); R. A. Durst (USA); M. Gross (France); H. Kao (China); K. M. Kadish (USA); R. Kalvoda (Czechoslovakia); Y. Marcus (Israel); T. Mussini (Italy); H. W. Nurnberg (FRG); M. Senda (Japan); D. E. Smith (USA); N. Tanaka (Japan); National Representatives: D. D. Perrin (Australia); B. Gilbert (Belgium); W. C. Purdy (Canada); R. Neeb (FRG); K. TOth (Hungary); S. K. Rangarajan (India); W. F. Smyth (Ireland); E. Grushka (Israel); Z. Galus (Poland); G. Johansson (Sweden); J. Buffle (Switzerland); B. Birch (UK); J. Osteryoung (USA); M. Branica (Yugoslavia).
Transcript of VOLTAMMETRY AT GLASSY CARBON ELECTRODESpublications.iupac.org/pac-2007/1984/pdf/5608x1095.pdf ·...

Pure & Appi. Chem., Vol. 56, No. 8, pp. 1095—1129, 1984. 0033—4545/84 $3.00+0.00
Printed in Great Britain. Pergamon Press Ltd.©1984 IUPAC
INTERNATIONAL UNION OF PUREAND APPLIED CHEMISTRY
ANALYTICAL CHEMISTRY DIVISION
COMMISSION ON ELECTROANALYTICAL CHEMISTRY*
VOLTAMMETRY AT GLASSYCARBON ELECTRODES
Prepared for publication byM. GROSS' and J. JORDAN2
1Laboratoire d'Electrochimie, Universite Louis Pasteur,Strasbourg, France
2Davey Laboratory, Pennsylvania State University,University Park, USA
*Membership of the Commission during the preparation of this report (1981—83) was asfollows:
Chairman: J. Jordan (USA); Secretary: K. Izutsu (Japan); Titular Members: A. K.Covington (UK); J. Juillard (France); R. C. Kapoor (India); E. Pungor (Hungary);Associate Members: J. F. Coetzee (USA); W. Davison (UK); R. A. Durst (USA); M. Gross(France); H. Kao (China); K. M. Kadish (USA); R. Kalvoda (Czechoslovakia); Y. Marcus(Israel); T. Mussini (Italy); H. W. Nurnberg (FRG); M. Senda (Japan); D. E. Smith (USA);N. Tanaka (Japan); National Representatives: D. D. Perrin (Australia); B. Gilbert(Belgium); W. C. Purdy (Canada); R. Neeb (FRG); K. TOth (Hungary); S. K. Rangarajan(India); W. F. Smyth (Ireland); E. Grushka (Israel); Z. Galus (Poland); G. Johansson(Sweden); J. Buffle (Switzerland); B. Birch (UK); J. Osteryoung (USA); M. Branica(Yugoslavia).

VOL TAMMETRY
at
GLASSY CARBON ELECTRODES
a) b)M. Gross and 1. lordana) Laboratoire dElectrochimie, Institut Le Bel
Université Louis Pasteur4 rue Blaise Pascal
F - 67000 Strasbourg (France)
b) Department of Chemistry152 Davey Laboratory
Pennsylvania State UniversityUniversity Park, Pennsylvania 16802 (USA)
Abstract
Recommended pretreatment procedures are described for obtaining reproducible andaccurate results. Based on a worldwide survey, information is tabulated on the workingrange of potentials of glassy carbon in water, organic solvents, and molten salts.Voltammetric characteristics of numerous redox couples are documented.
Contents
I. THE STARTING MATERIALS (Appendix A)
II. PREPARING THE GLASSY CARBON ELECTRODE SURFACE FOR ELECTRO-CHEMICAL MEASUREMENTS
1. Physical treatment2. Chemical pretreatment3. Electrochemical pretreatment
III. GEOMETRIC CHARACTERISTICS OF THE GLASSY CARBON ELECTRODES
IV. ELECTROACTIVITY RANGE OF THE GLASSY CARBON ELECTRODE (Appendix B)
V. REDOX SYSTEMS STUDIED ON GLASSY CARBON ELECTRODES (Appendix CTables (1) and (2))
VI. IDENTIFICATION OF THE SOURCES QUOTED IN THIS REPORT (Appendix D)
VII. TABLE OF ABBREVIATIONS AND SYMBOLS
APPENDIXES A, B , C and D.
1096

Voltammetry at glassy carbon electrodes 1097
The present report is intended to describe and to recommend specific treatments in the preparationof glassy caron.(G.C.) as an electrode material.
The information summarized in this report illustrates the unique capabilities of glassy carbonelectrodes with respect to other electrode materials. G.C. electrodes may be employed to obtainaccurate and reproducible results on a large variety of redox systems and in a wide range of solvents.However, this goal can be achieved only if the G.C. electrode receives the proper pretreatment and isthen handled appropriately.
Chemically modified glassy carbon electrodes, especially Hg-plated glassy carbon electrodes (thin filmmercury electrodes, MFE), while important in ultra-trace analytical chemistry, are not reviewed inthe present report. Specific papers have been published on analytical applications of such electrodes.(L. Mart, H.W. Nürnberg, P. Valenta, Fresenius Z. Anal. Chem., 300, 350-362 (1980)).
1. THE STARTING MATERIALS
Nine different commercial sources of G.C. were identified and are listed in Appendix A.
The first paper published on G.C. was by Sh. Yamada and H. Sato (Nature, 193 (4812), p.26 I (1962)).
II. PREPARING THE G.C. ELECTRODE SURFACE FOR ELECTROCHEMICALMEASUREMENTS
Careful checking of answers to an ad hoc questionnaire (see VI below) and of the published literatureleads to the conclusion that many different procedures have been used by various investigators.
However the following general trends emerge from intercomparison of these procedures, as necessarysteps to warrant good reproducibility of the electrochemical results.
1. Physical treatment
Two successive steps are usually involved in the polishing of the G.C. surface
Abrasion with emery paper of increasing fineness (typically 240, 320, 400 and 600 meshper sq.i. or 32, 23, 15, 8, 5, 3, 1 pm SiC , 2 mm. each).
Polishing with water (alumina or chromium III oxide) or oil (diamond) suspensions withdecreasing particle size (typically from 5 - 10 to 0.5 pm or 0.25 pm).
It is critical to rinse carefully the surface of the electrode (with water or another solvent)between two polishing steps.
Some authors (5) (20) (28) recommend to use ultrasonic vibrations to clean the surface (dislodgingimpurities held in pores at the surface). Typical solvents are : hexane, acetone, water. Fewminutes each.Also it was observed (39) (42) that vacuum desorption under 1 torr or less, at normal (42) or high(500°C) temperatures (39), increases the reproducibility of the results (39) (42) and the reversibility(42) of the electrode reactions.
Specially relevant to this question is the recent study (44) of the relationship between the heattreatment temperature of G.C. and the kinetic parameters of various redox reactions. This questionwas also discussed in previous papers (39) (41).
2. Chemical pretreatment
Depending on the intended use of the specific chemical pretreatments may be added to thephysical treatment.
- to obtain an electrode surface free of oxygen containing groupssurface treatments with non oxidizing acids (MCI for instance) (48) in solution free ofoxidizing species ; polarization of the electrode at a potential slightly negative to SCE.
- to obtain an electrode surface free of adsorbed specieswash the electrode with ethanol or chloroform, then wipe the surface with a wet filterpaper.

1098 COMMISSION ON ELECTROANALYTICAL CHEMISTRY
3. Electrochemical pretreatment
The purpose of this pretreatment is to increase the rate of the electron transfer steps at theelectrode surface and also to warrant reasonable reproducibility of the experimental results. Severalauthors made extensive use of electrochemical pretreatments of G.C. electrodes (4) (14) (15) (19) (21)(23) (25) (32) (33) (37) (40) (43 (45). The practice has also been reviewed critically (41) (44).
This pretreatment involves applications of several "polarization cycles" to the electrode, in order toobtain a specified reproducible "surface condition".
However, the potential limits of the cycles have to be set far enough from zero volt (versus SCE), inorder for the activation to occur.
As a general rule, the activation of the G.C. electrode surface occurs when the potential is cycled atintermediate scan rates (typically 0.1 VIs) between moderately negative potentials (i.e. about - 0.5 V)and more positive potentials, viz. + 0.9 to + 1.50 V/SCE (37) (40) (43) (44) (45). It should be noted thatsuch "activated electrodes" exhibit improved reproducibility and reversibility, at the expense of theanalytical sensitivity. The latter is handicapped by enhancement of residual currents (14) (44) (50).
It should also be kept in mind that the G.C. electrode is irreversibly damaged at too positivepotentials (>2 Volts/SCE) and by high anodic currents (>100 mA) (12).
III. GEOMETRIC CHARACTERISTICS OF THE G.C. ELECTRODES
When used for analytical purposes, the preferred geometry is a circular cross-section of a cylindricalrod of small area, whereas other geometries are involved in preparative electrolysis, which requireslarge effective areas (e.g. plates (52), reticulated vitreous carbon (32)).
"Rod cross section" electrodes are usually prepared by inserting rod-shaped electrodes (commercialdiameters range typically from 3 to 8 mm), of about 10 mm in length, into tubes of teflon, orplexiglass, or pyrex, and by sealing the rod to the tube with epoxy (for instance Epo-Tek 349, EpoxyTechnology, Watertown, Mass).
However, it is preferable to use rod electrodes inserted in tight-fitting teflon tubes, in order to avoidcontamination of ambient electrolytes by epoxy.
Typically, fitting is achieved by "forcing" the carbon rod into a teflon sheath whose inner (bore)diameter is about 15 % smaller than the rod.
Electrical contact with the electrode is made via the top of the carbon rod with the aid of silverepoxy or mercury.
IV. ELECTROACTIVITY RANGE OF THE G.C.ELECTRODE
The electroactivity range of G.C. is remarkably large in a wide variety of solvents, as documented inAppendix B. A working range of 3 Volts is common.
However, it should be noted that the electroactivity range of G.C. electrodes is dependent on theprevailing "surface condition" (4 1,44).
V. REDOX SYSTEMS STUDIED ON G.C. ELECTRODES
are listed in Appendix C and include the following couples.
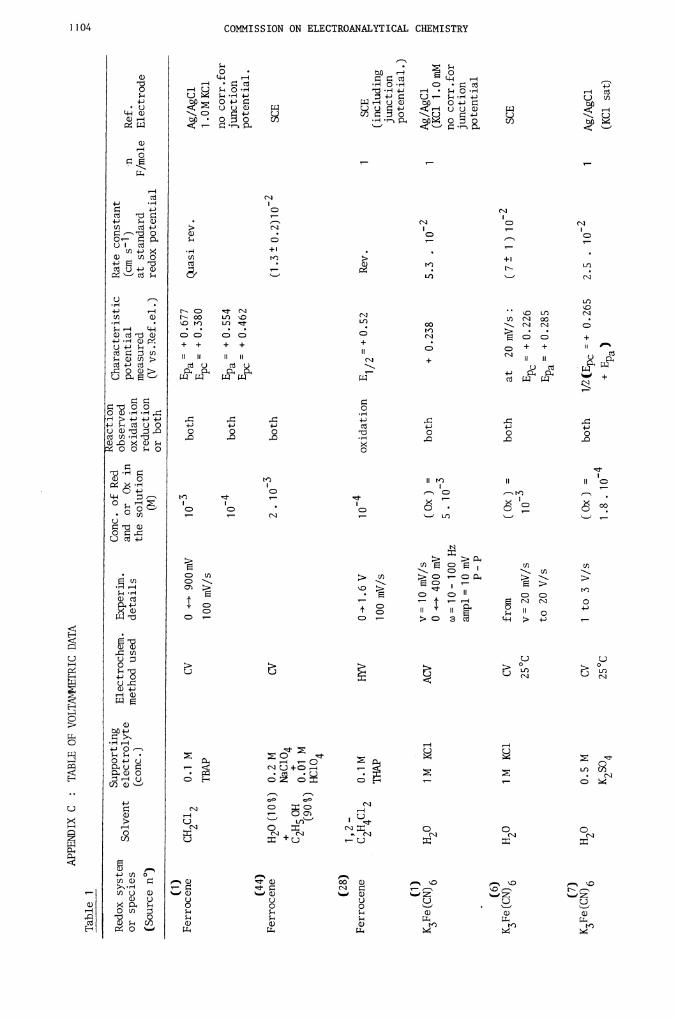
(1) Ferrocene/Ferricinium1
Table (1)
(2) Fe(CN) / Fe(CN) J
(store solutions of Fe(CN) in the dark, to avoid photodegradation).
(3) Other redox systems : Table (2)

Voltammetry at glassy carbon electrodes 1099
VI. IDENTIFICATION OF THE SOURCES QUOTED IN THIS REPORT
Two types of source materials were used
a) answers to a questionnaire distributed to 332 prominent electrochemists worldwide.Forty seven (47) responses were received.
b) published papers.
Each source has been assigned a code number and is identified accordingly, both in the foregoing textand in the following tabulations. The code numbers are listed in Appendix D.
VII. TABLE OF ABBREVIATIONS AND SYMBOLS
ACV Alternating Current VoltammetryAN AcetonitrileASV Anodic Stripping VoltammetryBN BenzonitrileCA ChronoamperometryCV Cyclic VoltammetryDCP Direct Current PolarographyDMF DimethylformamideDPP Differential Pulse PolarographyDPV Differential Pulse VoltammetryEl Electrolysis
Halfwave potentialE Potential of anodic peak in C V
paE Potential of cathodic peak in C Vpc +Fc/Fc Ferrocene/FerriciniumGC Glassy CarbonGCE Glassy Carbon ElectrodeHYV Hydrodynamic VoltammetryIRREV IrreversibleLSV Linear scanning voltammetryM Mole . litre -1Me2SO Dimethylsulfoxiden-PrCN n-propionitrileNHE Normal Hydrogen ElectrodeNPP Normal Pulse PolarographyPC Propylene CarbonatePot PotentiometryPY Direct current polarographyQRev Quasi reversibleRev ReversibleRDE Rotating disk electrodeRDEV Voltammetry on Rotating disk electrodeSCE Calomel Electrode in KCI Saturated solutionSHE Standard Hydrogen ElectrodeSR Scan RateSSCE Calomel Electrode in NaCI satured solutionTBAP Tetra-n-butyl-ammonium perchlorateTBAPF6 Tetrabutyl ammonium hexafluorophosphateTEAP Tetraethyl am monium perchiorateTEAPTS Tetraethyl ammonium para-toluene sulfonylTHAP Tetrahexyl ammonium perchiorateTHF TetrahydrofuranTMAP Tetramethyl ammonium perchlorate
1100 COMMISSION ON ELECTROANALYTICAL CHEMISTRY
APPENDIX A : STARTING MATERIALS
(I) Le Carbone Lorraine, Département Produits Spéciaux, 37 a 41, rue Jean Jaurès, F-92230Gennevilliers (France), Telex F-620847 LCLGV, Tel. (1) 799.98.41.
(2) China Scientific Instruments and Materials Corporation, 75, W. Dengshi Street, Beijing (China),Cable adress : "CSIMC" Beijing.
(3) Fluorocarbon Company, Process Systems Division, 1432 So. Allec St., Anaheim, California,92803 (USA), Tel. 714/956-7330.
(4) Tokai Carbon Co/IMC Industry Group & Associates, Ming-Yu International Building, 8-8,4-chome Ginza, Chuo-Ku, Tokyo 104 (Japon), Telex 3-22 316, 3-22 923, Tel. (535) 4381.
*(5) Atomergic Chemetals Corp., 100 Fairchild Avenue, Plainview, New York, N.Y. 11803 (USA).
(6) Sign Elektrographit, Postfach 1160, D-8901 Meitingen (RFA) (quality "Sigradur"), Telex 053823 Sign d, Tel. (08271) 83 3147.
*(7) Deutsche Carbon, D-6000 Frankfurt, Postfach 56 0209 (RFA).
(8) Metrohm Ltd, CH-9100 Herisau (Switzerland).
* Sellers only : (5), (7) : Le Carbone Lorraine.
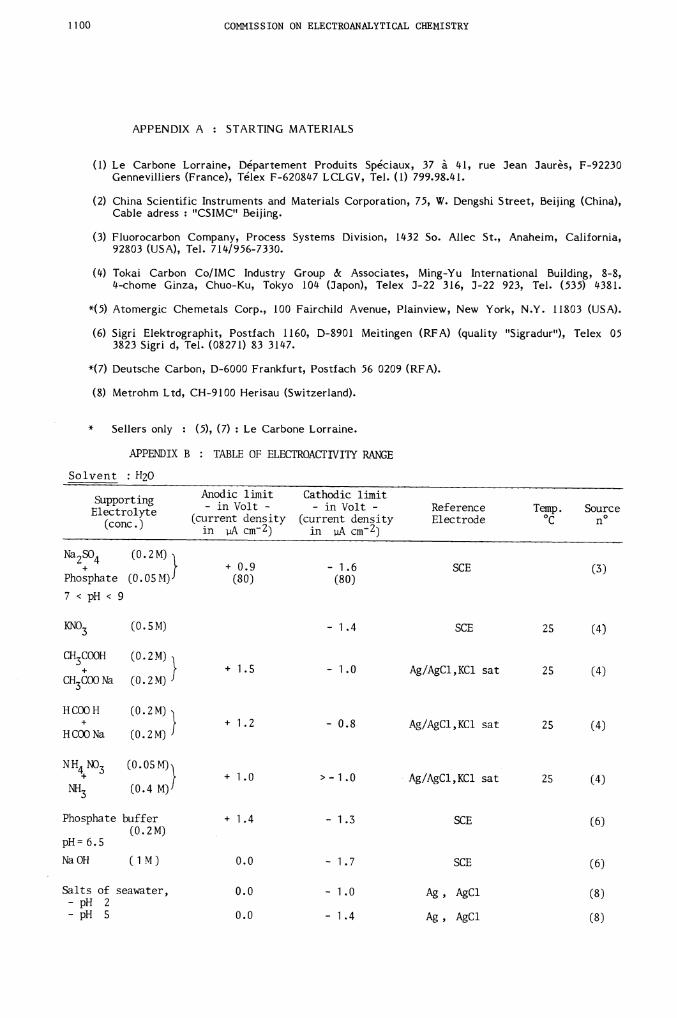
APPENDIX B : TABLE OF ELECTROACTIVITY RANGE
Solvent : H20
SupportingElectrolyte
(conc.)
Na SO (0.2M)2 4 + 0.9 - 1.6 SCE (3)Phosphate (O.O5M) (80) (80)
7 < pH < 9
KNO3 (0.5M) — 1.4 SCE 25 (4)
CH3COOH (0.2M)+ + 1.5 - 1.0 Ag/AgCl,KC1 sat 25 (4)
CH3COONa (0.2M)
HCOOH (O.2M)+ + 1.2 - 0.8 Ag/AgC1,KC1 sat 25 (4)
HCOONa (0.2M)
NH4NO3 (O.O5M)+ + 1 .0 > - 1 .0 Ag/AgCl,KC1 sat 25 (4)
NH3 (0.4 M)
Phosphate buffer + 1 .4 - 1 .3 SCE (6)(0.2M)
pH = 6.5NaOH (1M) 0.0 - 1.7 SCE (6)
Salts of seawater, 0.0 - 1.0 Ag, AgCl (8)- pH 2- pH 5 0.0 - 1.4 Ag, AgCl (8)
Anodic limit Cathodic limit- in Volt - - in Volt - Reference Temp. Source(current densityin crn2)
(currentin iiA
densitycm2)
Electrode °C n°

Voltammetry at glassy carbon electrodes 1101
SupportingElectrolyte
(conc.)
Anodic limit- in Volt -
(current densityin iA cm—2)
(;'pp)
Cathodic- in V
(currentin pA
( *
limit -
cit -densitycm2)3A)
ReferenceElectrode
Temp.°C
Sourcen°
K01-1 (0.1 M) + 0.5 - 1.4 SHE (14)
Acetate buffer + 1.4 - 0.7 SHE (14)pH = 4.7
5.102M(NH4CH300
CH3COOH)buffer—after polishing on + 1.5 — 1.25 SCli (15)emery paper 400 (56) (56)
-after polishing + 1.6 - 1.4 SCE (15)
with O.Sp paste (28) (28)
H2S04 (1 N) + 1.2 — 0.30 SCE 20 (17)
(45) (45)
NaClO4 (1 M) + 1.0 + 1.5 SCE (19)
(1); (1)*
CH3COOH (0.1H)1 + 1.1 — 0.5 SCE (20)
CH3COONH4 (0.2 M)' (7.9) (7.9)pH = 5.0
Phosphate andacetate buffers of ' + 1 -0.8 Ag/AgC1,KCI (1M) (22)
pH=5 to 8, in CH3OHand CH3CN
(1M) + 1.5 — 1.0 SCE (23)
I (1DM) + 1.2 — 1.4 SCE
(25) (25)
HBr (1M) + 0.7 - 1.5 SCE (23)
None (electron + 0.5 - 1.2 SCE (25)
photoeiniss ion)
Na2SO4 (0.2M)— 1.2 SSCE 130)
Me4NC1O4 (0.1M)> + 1.4 - 1.6 SSCE (30)
KC1 (5 . io2 M) + 1.1 — 0.8 SCE (35)
(10) (10)
HC1 (1M) + 0.7 -1.0 SCE (6)
HC1 (0.O1M) > + 0.075 — 1.20 SCE (21)
HC1O4 (0.1M) + 1.2 — 0.8 SCE (19)
(1)* (1)
HC1O4 (0.O1M) > + 0.075 - 1.20 SCE (21)
1-1C104(0.O1M)
(0.1M) + 0.45 — 2.3 SCE (23)
J (10) (10)
NaSCN 1 (2M) + 0.35 - 2.5 SCE
1102 CONMISSION ON ELECTROANALYTICAL CHEMISTRY
SupportingElectrolyte
(conc.)
Anodic- in V
(currentin iA
limitolt -
densitycm-2)
Cathodic- in V
(currentin pA
limitolt -
density-2)
ReferenceElectrode
Temp.°C
Sourcen°
HC1O4+KC1O4-2
+ 1.7 - 1.0 SCE (25)
(8.10 M)pH = 1 .5
HC1O4 + KC1O4÷
N20 + 1 .7 - 1 .5 SCE (25)pH = 7
HC1O4 (0.5M) + 0 — 0.4 NI-IE (27)(1) (1)
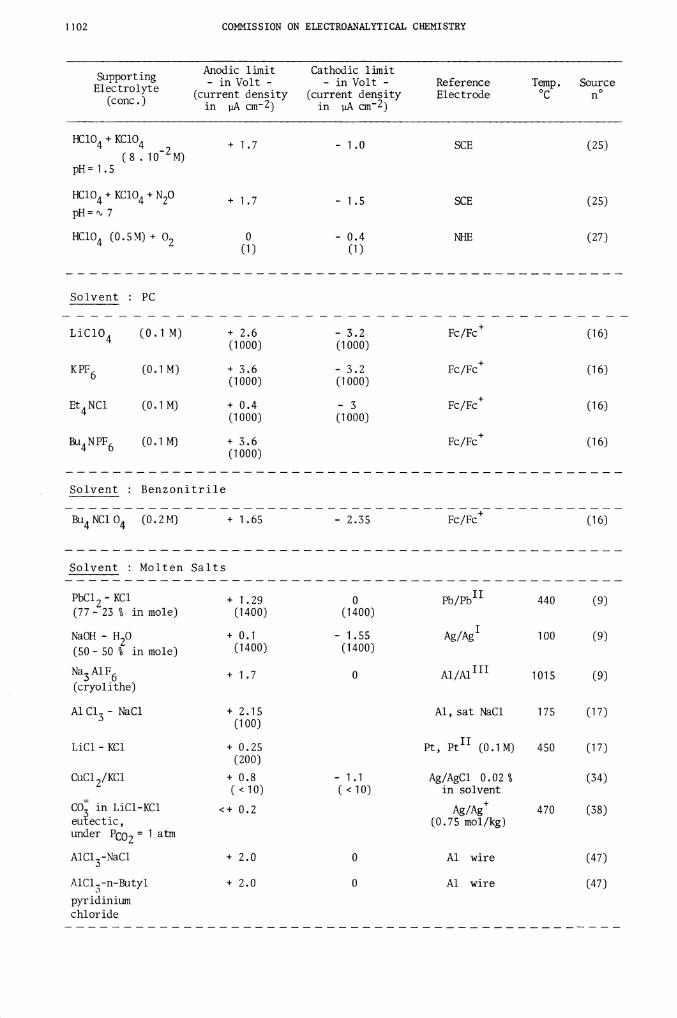
Solvent : PC
LiClO (0.1 M) + 2.6 - 3.2 Fc/Fc (16)(1000) (1000)
KPF (0.1 M) ÷ 3.6 - 3.2 Fc/Fc (16)6(1000) (1000)
Et NC1 (0.1 M) + 0.4 - 3 Fc/Fc (16)
(1000) (1000)
Bu NPF6 (0.1M) + 3.6 Fc/Fc (16)
(1000)
Solvent : Benzonitrile
Bu4NC1O4 (0.2M) + 1.65 - 2.35 Fc/Fc (16)
Solvent : Molten Salts
PbCl2-KC1 + 1.29 0 Pb/Pb11 440 (9)(77— 23 % in mole) (1400) (1400)
NaOH -H20
+ 0.1 - 1.55 Ag/Ag1 100 (9)
(50- 50 % in mole) (1400) (1400)
Na3A1F6 + 1.7 0 Al/Al11' 1015 (9)(cryolithe)
Al Cl3 — NaCl + 2.15 Al, sat NaC1 175 (17)(100)
LiCl—KC1 + 0.25 Pt, Pt1' (0.1M) 450 (17)
(200)
CuCl2/KC1+ 0.8 - 1 .1 Ag/AgC1 0.02 % (34)
<10) (<10) in solvent
CO in LiC1KC1 <+ 0.2 Ag/Ag 470 (38)
eutectic, (0.75 mol/kg)under ft02 = 1 atm
A1C13-NaCl+ 2.0 0 Al wire (47)
AlCl3-n-Butyl+ 2.0 0 Al wire (47)
pyridiniumchloride

Voltammetry at glassy carbon electrodes 1103
Solvent : CH3CN
Anodic limit Cathodic limitSupportingElectrol te - in Volt - - in Volt - Reference Temp. Source
conc (current density (current density Electrode °C n°'- -' in pA cm2) in pA cm2)
TEAP (0.1 M) + 1.1 - 2.25 Ag,AgNO3 (0.O1M) 25 (4)
LiClO4÷ 2.8 - 2.7 SCE+ junction (6)
TEAl' (0.1 M) + 2.2 Ag/Ag (0.01 M) (12)
TEAP - 2.1 SCE+ junction (18)
50 % C2H5OH÷ - 1.75 SCE+junction (18)
NaOH (pH ca.13.3)
C4H9NPF (0.5 M) - 2.2 Ag/Ag (0.1 M) (24)6(5000)
Et NBF (0.1OM) + 3.5 (26)(5000)
Solvent : THF
LiClO4 (0.3 M) — 2.2 (12)
Solvent : n-CH3 - (CH2) 2 - CN
(n-Bu)4NC1O4 (0.1OM) - 2.5 Ag/Ag (0.01 M)- 60 (26)
Solvent DMF
THAP 0.1 M) + 1.4 — 2.0 SCE 25 (28)
(325) (325)
Solvent : 1,2 - C2H4C12
THAP (0.1 M) + 1.6 — 1.9 SCE 25 (28)
(325 (325)
APP
FM)I
X C
TABLE OF V
OL
TA
MM
F1R
IC D
AT
A
(28)
Ferrocene
AC
V
v=lO
mV
/s
0 ÷÷ 4
00 mV
w=10—100 Hz
ampl= 1
0 m
V
p-p
(Ox)
=
bo
th
l/2(E
pc+
0.2
65
2.5
. io
2 1.8.
+ E
P)
SCE
(including
junction
potential.)
Ag/AgCl
(KC1 1.0mM
no corr.for
junction
potential
SCE
Table 1
CH2C12
0.1 M
(1)
Ferrocene
(44)
Ferrocene
Supporting
Redox system
Solvent electrolyte
or species
(conc.)
(Source n°)
Electrochem.
method used
Exp
erim
. de
tails
Conc. of Red
and or Ox in
the solution
(M)
Reaction
observed
oxidation
reduction
or both
- Cha
ract
eris
tic
potential
measured
(V vs.Ref.el.)
Rate constant
(cm s1)
at standard
redox potential
n
F/mole Ref.
Electrode
CV
0
900mV
100 mV/s
CV
HY
V
0-'-1.6V
100 mV/s
both
Epa=
E
+ 0
.677
+ 0
.380
çiasi rev.
both
Epa=
+ 0
.554
÷ 0
.462
2. 10
both
(1.3±0.2)10_2
TB
AP
0.2 H
NaC
1O4
+
0.01
M
HC1O4
0.1 M
THAP
1M KC1
1M KC1
0.5 H
Ag/AgC1
1 .OMKC1
no corr.for
junction
potential.
SCE
H20 (10%)
+
C2H
5 OH
(90%)
1,2—
C2H4C12
(1)
K3Fe(CN)6
H20
(6)
K3Fe(CN)6
H20
(7)
K3Fe(CN)6
H20
0
CV
25°C
(Ox)
=
5.
(Ox)
=
1o
oxidation
E1/2=+0.52
Rev.
both
+ 0.
238
5.3 . io
2
both
at 20 mV/s:
(7 ± 1) io2
Ep
+ 0
.226
Epa
+ 0
.285
from
v=
20 mV/s
to 20 V/s
K2S04
25°C
CV
lto3V/s
1
Ag/AgC1
(KC1 sat)

Supporting
Redox system
Solvent electrolyte
or species
(conc.)
(Source n°)
Electrochem.
method used
lixperim.
details
Conc. of Red
and or Ox in
the solution
()
Reaction
observed
oxidation
reduction
or both
Characteristic
potential
measured
(V vs.Ref.el.)
Rate constant
(cm s1)
at standard
redox potential
n
F/mole
Ref.
Electrode
HO
0.5M
2
KC1
from -0.40
CV
to +0.75V
at
20 mV/s
(Red) =
1.0
. i0
both
Ep = +
0.2
4 = +
0.1
4 Quasi rev.
1
SCE
H20
0.1 M
H2504
H20
Phosphate
buffer 1
M
pH =
7
H20
0.1 M
phosphate
buffer
+
1M KC1
pH = 3
7.4
H 0
0.1 M
2
phosphate
buffer
pH =
7.5
H20
0.5 M
K2S04
RDE V
CV
10
0 mV/s
CV
50 m
V/s
Turbulent
tubular
electrode
(convective
voltam.)
CV
22°C
polished +
heat
ed
(520 — 540°C)
2.69 ml.miiT
v= 0.2V.miri
1
. 10
1
. 10
3M
5 . io
2
.
1 . io
6 of
each
5.0
. io
of
each
both
E1/2=+ 0.46
both
reduction
both
E
=
p,c
(Ep,a)
+ 0
.235
(+
0.29
2)
+0.
231
(+0.
281)
Rev.
-4
"P10
(2
1)
1.5±
0.2
io2
on anodically
'activated" GCE
(10)
K4Fe(CN)6
(14)
K Fe(CN)
K4Fe (C
N):
(39)
K3Fe(CN)6
(40)
K
3Fe(
CN
)6
(44)
K
3Fe(
CN
)6
K4F
e(C
N)6
1
SHE
Ag/
AgC
l (4
M KC1)
Ag/AgCl
(0.1
MK
C1)
SCE
C
Ui

Table 2
System
studied
(Red/Ox)
(Source
Supporting
Solvent
electrolyte
(conc.)
n°)
Electrochem.
method used
Experim.details
(potential range
explored,
mi-
tial potential,
scan rate, etc
..
Conc. of
Red
and/or
Ox
in the
solution
in
Mole/l
Reaction
observed
Oxidation
Reduction
or Both
Characteristic
Potential
measured
(V/ref.)
(E1/2, Epeak
other ...)
Rate
constant
(cm s1)
at
standard
redox
potential
n
F/mole E112 or
Epeak of
pij
ferro -
ferr
icya
nide
or
ferrocene/
ferricinium
(1)
bis-hydroxy-
methyl ferro-
H20
KF -O.1M
CV
0 ÷
- 90
0 m
V
50 mV/s
10
Both
Ep,a0.280
Ep,cO.2lS
Quasi-rev
cene
H20
KNO3 -0.1M
CV
0 ÷
- 90
0 m
V
50 mV/s
10
Both
Ep,
a027
7 £p
,c°2
04
Quasi-rev
Quasi-rev
H20
KC1
-0.1 M
CV
0 -
-÷ 9
00 m
V
50 mV/s
10
Both
Ep,a 0.268
Ep,cO.2lO
ReV
CH
2C12
T
BA
P -0
.1M
C
V
0 ÷
-* 9
00
mV
50 mV/s
10
Both
Ep,a0.502
'p,c 0.422
Quasi-rev
CH3CN
CH3CN
CH3CN
TBAP -0.1M
TEAPTS
—0.1M
TBAPF6
- 0
.1 M
CV
CV
CV
0
0
0 ÷-* 900 mV
--* 90
0 m
V
5OmV/s
÷- 9
00 m
V
50 m
V/s
Bot
h
Bot
h
Bot
h
Ep,a0.424
Epc 0.355
Ep,a 0.409
Ep,cO.33O
13p,a
0.401
Ep,c = 0
.332
Quasi-rev
Quasi-rev
Quasi-rev
All potentials are
vs .
A
g/A
gCl (1 .OmKCl)
no correction for junc-
tion potential.
Ref
eren
e:
HIE
Fc
(2)
CH
3CN
0.
1 M
N
aClO
4 C
V
0.1
-0. 7
V, 0. 1V/s
1 . 1
Bot
h (E
P)a
0.41
0.
35
Rev
Ag/AgC1
/ 0.
1M N
aC1
BH
EF
c C
H3C
N
0.1M NaC1O4
CV
0.1 -0.7V, 0.1V/s
1 . 10
Both
0.41
0.35
Rev
/ AFc
CH3CN
0.1 M NaC1O4
CV
0.1 -0. 7V, 0. 1V/s
1 . 1 0
Both
0.42
0.38
Rev
/ BAFc
CH3CN
0.1 M N
aC1O4
CV
0.1 - 0
. 7V, 0. 1V/s
1 . 1 0
Both
0.42
0.38
Rev
/
tt

c-hydroxyferrocene-
--HEFc
acetoferrocene --- A
Fc
1 , 1 '-bis a-hydroxyl ferrocene---BHEFc
1,1'-bis-acetoferrocene
BAFc
U)
Cl)
NA
DH
=
1 ,
4 - D
ihyd
roni
cotin
amid
e adenine dinucleotide.
—3
ca. 5
. 10 -3
ca. 5.10
(2)
HEFc
BHEFc
AF
BAFc
System
studied
(Red/Ox)
(Source
Supporting
Solvent
electrolrte
(conc.)
n°)
Electrochem.
method used
Experim.details
(potential range
explored,
mi-
tial potential,
scan rate, etc
...
Conc. of
Red
and/or
Ox
in the
solution
in
Mole/l
Reaction
observed
Oxidation
Reduction
or Both
Characteristic
Rate
Potential
constant
measured
(cm s-i)
(V/ef.)
at
Ei/2
Epeak
standard
other ...)
redo
x potential
fl F
/mol
e E112 or
Epeak of
ferro-
ferricyanide
or
ferrocene/
ferricinium
CEp)a
(EPJc
0.1 M
+0.1M NaC1
aqueous
aqueous
aqueous
aqueous I
CV
1
0.1
-0.7V, 0.1V/s
1
. 10
(3)
NADH
+
NA
D
+
+ 2
e
H20
0.2MNa2SO4
0.05M phos-
phate
Both
0.29
0.22
Rev
/ Reference:
Both
0.32
0.25
Rev
/ Ag/AgCl,
Both
0.27
0.20
Rev
/ 0.1 M
Cl
/ /
/ /
/
Ox
+ 0.
4 to
+ 0.
5 V/
SCE
lrrev
2
+ 0.15V
Strong
adsorp-
tion of
NAD
PY and
N.P.P.
at the
RDE
from 0.OV to
0. 9V
-6
from 10
to iü3
7 <
pH
< 9
+ CV on
scan rate up
RDE
to 50Vs1
(4)
CV
OV—-1.5V
Tl(I)/Tl
H20
KNO3 (0.SM)
CA
CD
CD
frI 0 C
D
Cl)
Red
Ep0.7
Red
-1.OV
1
Reference:
SCE

H20
0.2M Phosphate
DPV
buff
er, p
H=6.5
H20
0.2M Phosphate
DPV
buff
er, p
H=6.5
RD
E 40
0 RI4
Range -0.3V to
- 0
.6V
RDE 4
00 R
RVI
Ran
ge +
0.5V
to
+ 1
.5V
RDE 4
00 P24
+0.
5 —
+
1.5V
Irrev
Irrev
2 e
Reference:
SCE
System
studied
(Red/Ox)
(Source
Supporting
Solvent
electrolyte
(conc.)
n°)
Electrochem.
method used
Experim.details
(potential range
explored,
mi-
tial potential,
scan rate, etc
..
Conc. of
Red
and/or
Ox
in the
solution
in
Mole/l
Reaction
observed
Oxidation
Reduction
or Both
Characteristic
Potential
measured
(V/red.)
(E1/2
Epeak
other ...)
Rate
constant
( 1
) at
standard
redox
potential
n
F/mole E112 or
Epeak
IRKS
ferro-
ferricyanide
or
ferrocene/
ferricinium
(4)
03
Ru(acac)3
CH3CN
TEAP (0.1 M)
CV
1 .1 V — - 2.
0 V
CA
- 1.4V
H20
CH3COOH
HYV
(Ru(H-edta)
- (H
20))
(6)
H20
S4O
S20
(0.2
M)
+
CH
3CO
ON
a
(0.2M)
1M HC1
DPV
Reference:
Ag/AgI'K3
0.01
M
Ref
eren
ce:
Ag/AgCl,
KC1 sat
1
1
0.5
1
(III)/(II)
E
Red
-
1.12
Ox
—1.12
(III) / (V
I)
Red
0.
70
Ox
0.62
(III)/(II)
Red
2Ru(
III)
/ E
1/2
0.8
Ru(
IV)-
R
u (I
II)
Ru (
III)
/ 0
05
Ru(
IV)
-
Ru
(III
)/
0 3
Ru(JJ)
Red
F -0.475V
Ox
E+1.3OV
id5 -
10
Ox
F. +
0.9
5V
Irrev
2 e
System
studied
(Red/Ox)
Supporting
Solvent
electrolrte
(conc.)
Electrochem.
method used
Experim.details
(potential range
explored,
mi-
tial potential,
Conc. of
Red
and/or
Ox
Reaction
observed
Oxidation
Reduction Characteristic
Potential
measured
(V/ref.)
1ate
constant
(cms)
at
n
F/mole E112 or
Epeak of
pijj
ferro-
ferricyanide
(Source n°)
scan rate, etc
...
in the
solution
in
Mole/l
or Both
(E1/2, Epeak
other ...)
standard
redox
potential
or
ferrocene/
ferricinium
(6)
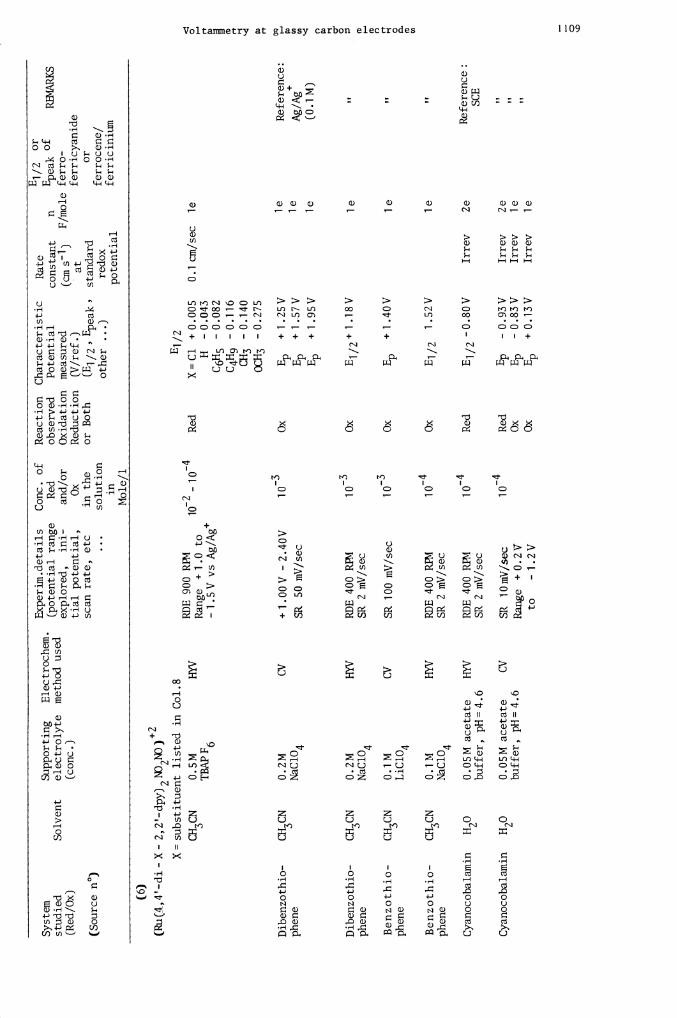
(Ru(4,4'-di-X- 2,2'-dpy)2fl2NO)2
X=substituent listed in Col.8
E1/2
RDE 9
00 R
BVI
io_2io4
Red
XC1 +
0.0
05
0.1cm/sec
le
CH3CN
0.5 M
Range + 1
.0
to
H - 0
.043
TBAPF6
-1 .5V vs Ag/Ag
C61-15
- 0.
082
C4H9 -0.116
CH3 —0.140
OCH3
—0.
275
Dibenzothio-
CH3CN
0.2
M
CV
+ 1
.00 V - 2
. 40V
1
Ox
E
+ 1
.25 V
1 e
Reference:
U)
U)
phen
e N
aC1O
4 S
R
50 m
V/s
ec
E
+ 1
.57 V
1 e
Ag/Ag
U)
F
÷1.95V
le
(0.1M)
U) 0
Dib
enzo
thio
- CH3CN
0.2M
HYV
BIJE 40
0 RFM
10
Ox
E1/2+1.18V
le
CD
phen
e N
aC1O
4 S
R 2
mV
/sec
CD
C)
Ben
zoth
io-
CH3CN
0.1 M
CV
SR 100 mV/sec
Ox
E
+ 1
.40V
1 e
0
phen
e LiClO4
CD
U)
Ben
zoth
io-
CH3CN
0.1M
HYV
RDE
400 RFM
Ox
E1/2 1.52V
le
phene
NaClO4
SR 2 mV/sec
Cyanocobalamin
H20
0.05 M acetate
HYV
RDE 400 REV1
Red
E1 /2 -0.80 V
Irrev
2e
Reference:
buffer, pH= 4.6
SR 2 mV/sec
SCE
Cyanocobalamin
H20
0.05M acetate
CV
SR lOniV/sec
Red
E
p - 0
.93 V
Irrev
2e
buffer, pH=4.6
Range +0.2V
Ox
E
-0.83V
Irrev
le
to
-1.2V
Ox
F
±0.13V
Irrev
le
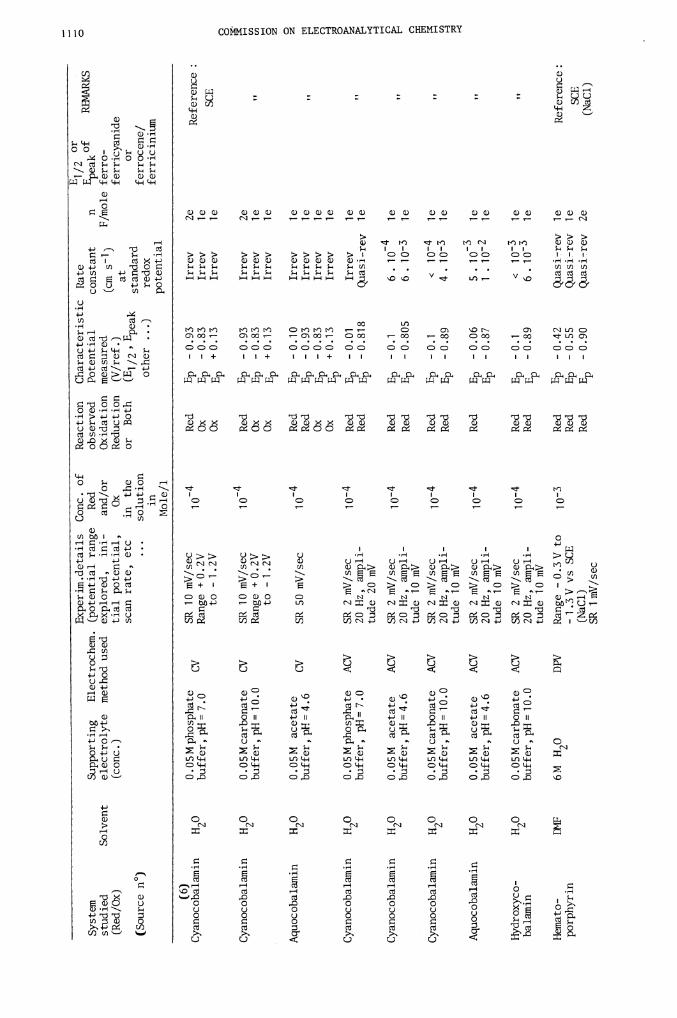
System
Supporting
Electrochem. Experiin.details
(potential range Conc. of
Red
Reaction
observed
Characteristic
Potential
Rate
constant
n
E112 or
Epeak0f
RRvIARKS
studied
(Red/Ox)
(Source n°)
Solvent
electrolyte
(conc.)
method used
explored,
mi-
tial potential,
scan rate, etc
...
and/or
Ox
in the
solution
in
Mole/l
Oxidation
Reduction
or Both
measured
(V/ref.)
(E1/2 Epeak
other ...)
(cm s1)
at
standard
redox
potential
F/mole ferro-
ferricyanide
or
ferrocene/
ferricinium
(6)
0.05M phosphate
SR 10 mV/sec
Red
E -
0.9
3 Irrev
2e
Reference
Cyanocobalamin
H20
buffer, pH = 7
.0
Range + 0
. 2V
Ox
E
- 0
.83
Irrev
le
SCE
to -1.2V
Ox
E +
0.13
Irrev
le
Cyanocobalamin
H20
0.05M carbonate
CV
SR 10 mV/sec
Red
Ep -0.93
Irrev
2e
buffer, pH = 1
0.0
Range + 0
. 2V
Ox
Ep
- 0
.83
Irrev
1 e
to -1.2V
Ox
E +0.13
Irrev
le
Aquocobalamin
H20
0.05M acetate
CV
SR 50 mV/sec
io4
Red
Ep -0.10
Irrev
le
buffer, pH = 4
.6
Red
E -
0.9
3 Irrev
1 e
Ox
E -0.83
Irrev
le
Ox
+0.13
Irrev
le
Cyanocobalamin
H20
0.05M phosphate
ACV
SR 2 mV/sec
i0
Red
E
- 0
.01
Irre
v le
buffer, pH=7.0
20Hz, ampli-
Red
Ep -0.818
Jasi-rev le
tude 20 mV
Cyanocobalamin
H20
0.05M acetate
ACV
SR 2 mV/sec
ici-4
Red
E
- 0
.1
6
10
le
buffer, pH =4.6
20 Hz, ainpli-
Red
E
- 0
.805
6: 1
ü
1 e
tude 10 mV
Cyanocobalamin
H20
0.05 M carbonate
ACV
SR 2 mV/sec
1
Red
E
p - 0
.1
<
1 0
1 e
buffer,pH=10.0
20Hz, ampli-
Red
E -0.89
4. 103
le
tude 10 mV
Aquocobalamin
H20
0.05M acetate
ACV
SR 2 mV/sec
Red
Ep
- 0
.06
5. 1
0
le
buffer, pH=4.6
20Hz, ampli-
E -0.87
1 .10-2
le
tude 10 mV
Hydroxyco-
H20
0.O5Mcarbonate
ACV
SR 2 mV/sec
j-4
Red
E -0.1
< i0
le
balamin
buffer, pH= 10.0
20 Hz, ampli-
Red
-0.89
6. io
le
tude 10 mV
Hemato-
IX'IF
6M H20
DPV
Ran
ge - 0
.3 V to
1
Red
E
p - 0
.42
Qiasi-rev le
Reference:
porphyrin
- 1
.3 V vs SCE
Red
Ep
- 0
.55
iasi
-rev
1 e
SCE
(NaC1)
Red
-0.90
Qiasi-rev 2e
(NaCl)
SR 1 mV/sec
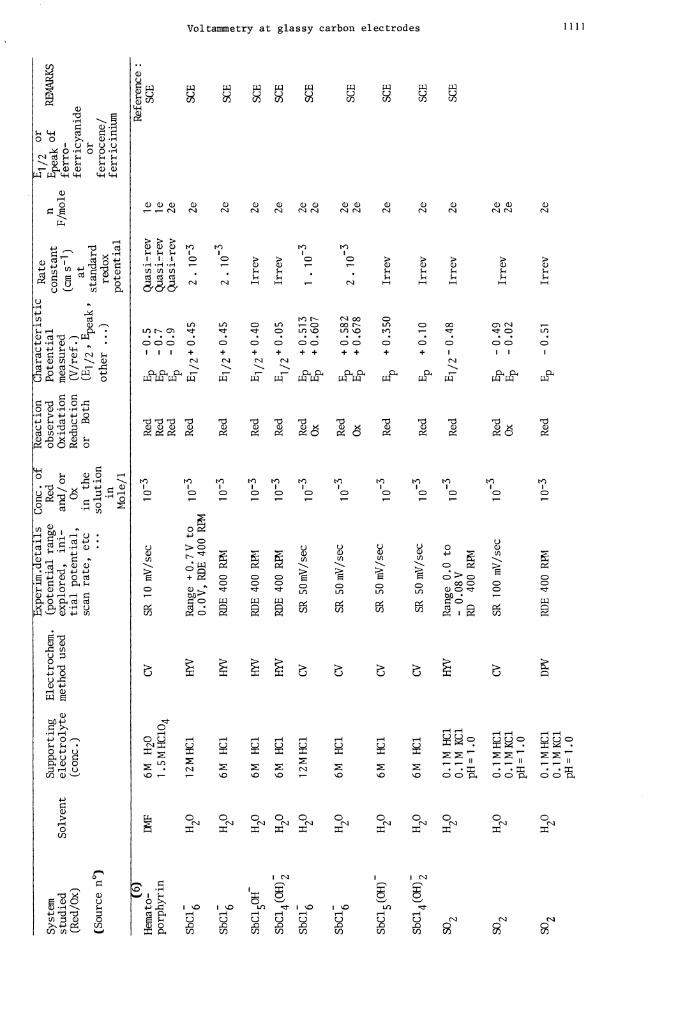
IX'4F
6M H20
1 .5MHC1O4
Ran
ge +
0.7V
to
io
0.O
V, R
DE
40
0 R
1vI
RD
E 40
0 R
FM
i0
Red
E
- 0
.5
Quasi-rev
Red
Ep
- 0
.7
Quasi-rev
Red
E
-0.9
Quasi-rev
Red
E112+0.45
2.10
Red
E112+ 0.45
2 .
Reference:
SCE
System
studied
(Red/Ox)
(Source
Supporting
Solvent
electrolyte
(conc.)
n°)
Electrochem.
method used
Experim.details
(potential range
explored, mi-
tial potential,
scan rate, etc
...
Conc. of
Red
and! or
Ox
in the
solution
in
Mole/l
Reaction
observed
Oxidation
Reduction
or Both
Characteristic
Potential
measured
(V/ref.)
(E1/2, Epeak
other ...)
Rate
constant
( s1
) at
standard
redox
potential
n
F/mole
E1/2 or
Epeak of
REMARKS
ferro-
ferricyanide
or
ferrocene/
ferricinium
le
le
2e
2e
(b)
Hemato-
porphyrin
SbC
l6
H20
12MHC1
SbCl6
H2O
6M HC1
SbC15OH
H20
6M HC1
SbC14 (OH)2
H20
6M HC1
SbCl
H20
12MHC1
SbCl6
H20
6M HC1
SbCl5(OH)
H20
6M HC1
SbCl4(OH)2
H20
6M HC1
SO
HO
0.
1MH
C1
2 2
0.1M KC1
pH = 1
.0
SO
HO
0.
1MH
C1
2 2
0.1MKC1
pH = 1
.0
SO
HO
0.1MHC1
2
2
0.1MKC1
pH = 1
.0
CV
SR
10
mV
/sec
HY
v
HJ
HYV
RIJ
E 40
0 RFM
HYV
RD
E 40
0 RfM
CV
SR 50 mV/sec
CV
SR 50 mV/sec
CV
SR 50 mV/sec
CV
SR 50 mV/sec
HYV
Range 0.0 to
- 0.
08V
RD
400R
fM
CV
SR 100 mV/sec
DPV
RUE
400 P24
io—3
io—3
1o
1o
1o
1o
SCE
2e
SCE
Red
E1/2+ 0.40
Irrev
2e
SCE
Red
E1/2 + 0
.05
Irrev
2e
SCE
Red
E
+ 0
.513
1
.
2e
SCE
Ox
E
+ 0
.607
2e
Red
+ 0
.582
2.
2e
SCE
Ox
E
+ 0
.678
2e
Red
E
+ 0
.350
Irrev
2e
SCE
Red
E
+ 0
.10
Irrev
2e
SCE
Red
E1/2 0.48
Irrev
2e
SCE
Red
Ep
-0.49
Irrev
2e
Ox
-0.02
2e
Red
E
-0.51
Irrev
2e
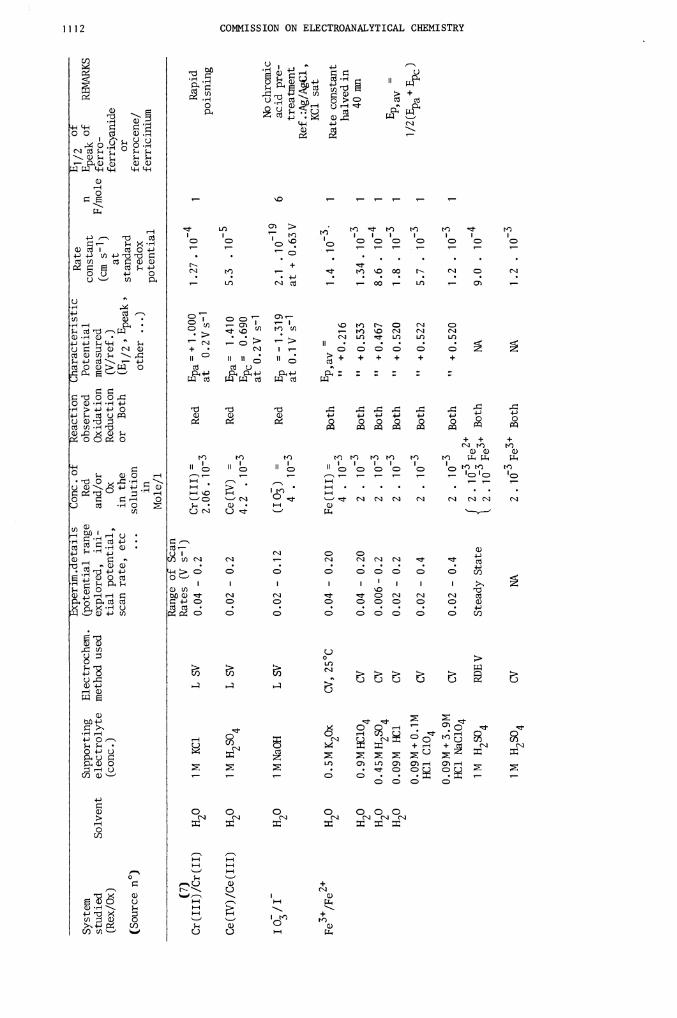
0.09M+0.1M
CV
HC1 dO4
0.09M + 3.
9M
HC
1 NaClO4
1M H
2S04
RDEV
2
.
Red
Epa= 1.410
Ep= 0.690
at 0.2V si
2
. i0
3 Both
"
+ 0
.467
2
.
Bot
h "
+ 0
.520
2
. 10
Both
2
. 10
Both
"
+ 0
.520
{
Bot
h
Rap
id
pois
ning
System
studied
(Rex/Ox)
(Source
Supporting
Solvent
electrolyte
(conc.)
n°)
Electrochem.
method used
Experim.details
(potential range
explored, mi—
tial potential,
scan rate, etc
...
Conc. of
Red
and/or
Ox
in the
solution
in
Mole/l
Reaction
observed
Oxidation
Reduction
or Both
Characteristic
Potential
measured
(V/ref.)
(E1/2, Epeak
other ...)
Rate
constant
(cm s1)
at
standard
redox
potential
n
F/mole F112 of
Epeak of
REMARKS
ferro-
ferricynide
or
ferrocene/
ferricinium
(7)
Cr (
111)/Cr (II)
Ce(IV)/Ce(III)
I Os/I
Fe3/Fe2
Range 0± Scan
Rates (V l)
H20
1M KC1
L SV
0.04 — 0
.2
Cr(III)
2.06 .
10
H 0
1 M H SO
L SV
0.02 -
0.2
Ce(IV) =
2 2
4
4.2 .i0-
II2O
1MNaOH
L SV
0.02 - 0
.12
(103)
=
4 . 10
H20
0.5MK2Ox
CV,25°C
0.04
- 0
.20
Fe(III)=
4
. i0
Red
Epa+l.000
at 0.2Vs1
H2O
H20
H20
0.9M
HC
1O4
0.45
MH
2S04
0.
09M
HC1
CV
CV
CV
Red E =
-1.319
at 0.1V s
Both
Ep,av =
0.21
6
1.27.10k
1
5.3
.
2.1
.
6 at
+ 0.
63V
1.4 .10
1
8.6 . 10
1
1.8 . 10
1
1.2 . 10
1
0.04 — 0
.20
0.006-0.2
0.02 — 0
.2
0.02 — 0
.4
0.02 - 0
.4
Steady State
Both
"
+0.
533
1.34.
CV
No chromic
acid pre-
treatment
Ref.:Ag/AgCl,
KC1 sat
Rate constant
halved in
40 am
Ep,av =
l/2(E
+ Ep
c)
+0.522
5.7 . 1o
1 M H2S04
CV
N
A
2 .
Fe3 Both
NA
1.2 .
NA
9.
0 .

Several qui-
none
/ h
ydro
- quinones
e.g.
Ag/Ag (16)
Ag(bipy) /
Ag(
bipy
) 2+
(02-glow discharge
treated CC)
E'°= 0.50
Oxid.of
E'°
= 0.59
Ag(I) to
Ag(II)
io
Co(III)-*
0.1
; -1.3
Co(II) -
Co
(I)
Rev
1;1
2
Reversibility of the
system increases dras-
tically after exposing
the polished GC sur-
face to an 02 glow-
discharge. Reference
Fc/Fc
System
studied
(Red/Ox)
(Source
Supporting
Solvent
electrolyte
(conc.)
n°)
Electrochem.
method used
Experim.details
(potential range
explored, mi-
tial potential,
scan rate, etc
...
Conc. of
Red
and/or
Ox
in the
solution
inMole/l
(*in p.p.b.)
Reaction
observed
Oxidation
Reduction
or Both
Characteristic
Potential
measured
(V/ref.)
(E1/2, Epeak,
other ...)
pate
constant
(s)
at
standard
redox
potential
r
F/mole B112 or
1peak of
R]JvlApJ(5
ferro-
ferricyanide
or
ferrocene/
ferricinium
(8)
Trace metals
Cd/Cd
Pb/Pb
Cu/Cu4
Bi/B
i"
Zn/Zn
(11)
W(CN)
H2O
H2O
H20
H2O
H20
NaCl/HC1
Differential
from + 1 .0 V
From
5
Firs
t
0.5M
pulse
anodic
to
SR 10
OV
mV/s
to
0.001 ppb
Red,
then
(Sea water)
stripping
voltanetry
Ox
0.2M KC1
2.5 io
K4W(CN)8
Both
Epa + 0.37
Epc - 0
.21
Rev
CV
SR 100 mV/s
0.2M KC1
CV
SR 100 mV/s
Sat
(Ox)
1
0
NaO3S 4,S03Na
PC
BU
4NPF
6 PO
T
PC
BU
4NPF
6 PO
T,C
V,R
DE
CA
,EL
BN
BU4NPF6
CV
rP
Ref
eren
ce :
SCE
= -
0.50
5
= -
0.59
Co TPP
0
CD
CD
C)
0 0.
CD
Cl)
Rev
1
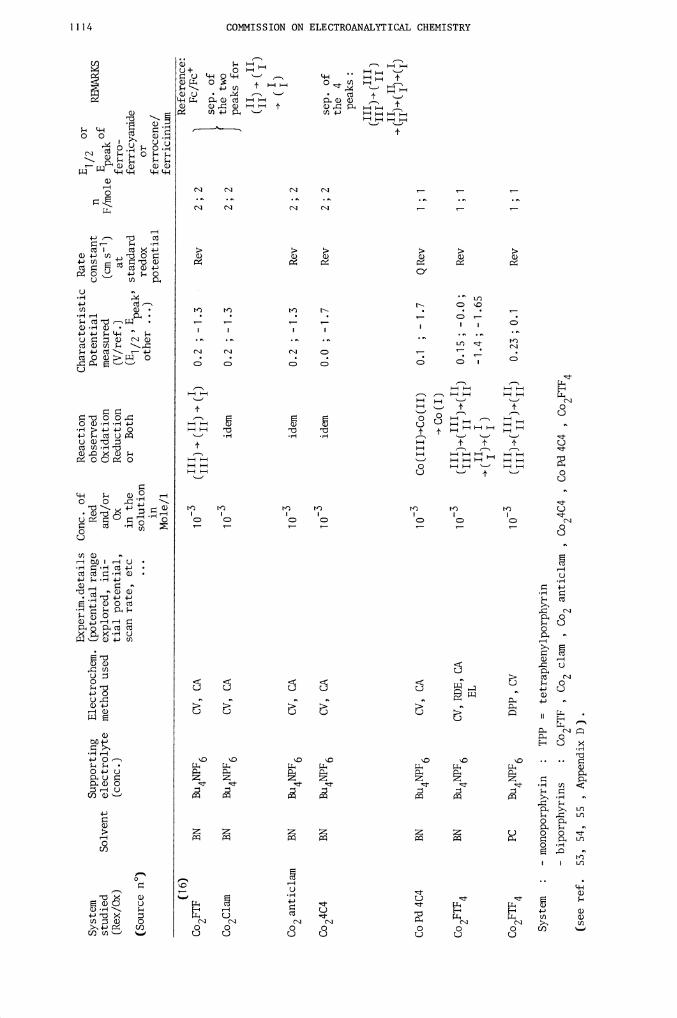
Experim.details Conc. of
Reaction
Characteristic Rate
E
or
System
Supporting
Electrochem. (potentialrange
Red
observed
Potential
constant
n
REMARKS
studied
Solvent electrolyte method used
explored, mi-
and/or
Oxidation
measured
(cms1)
F/mole peak°
(Rex/Ox)
(conc.)
tial potential,
Ox
Reduction
(V/ref.)
at
ferro-
scan rate, etc
in the
or Both
(E1/2 E e
ak' standard
ferricyanide
(Source n )
...
solu
tion
other
)
redo
x or
in
potential
ferrocene/
Mole/l
ferricinium
16
Reference:
Co2FTF
BN
4F6
CV, CA
10
0.2
; -1.3
Rev
2; 2
Fc/Fc
sep. of
Co2Clam
BN
Bu4NPF6
CV, C
A
idem
0.2
;
- 1
.3
2; 2
kWOr
(II)÷(I)
I
7
-'()
Co2
antic
lam
B
N
BU
4NPF
6 C
V,
CA
10
idem
0.2 ; - 1
.3
Rev
2; 2
Co24C4
BN
Bu4NPF6
CV, C
A
idem
0.0 ; -1.7
Rev
2; 2
sep. of
the 4
peaks:
III
II II
()÷( )*
()
CoPd4C4
BN
BU4NPF6
CV, CA
Co(III}÷Co(II)
0.1
; -1.7
QRev
1; 1
- C
o (I)
Co2FTF4
BN
BU4NPF6
CV, RDE, CA
0.15
; -0.0 ;
Rev
1; 1
EL
II
I
-1.4; -1.65
Co2FTF4
PC
Bu4NPF6
DPP, CV
10
()-(')÷()
0.23
; 0.1
Rev
1; 1
System
:
- m
onop
orph
yrin
:
TPP =
tetr
aphe
nylp
orph
yrin
- bi
porp
hyrin
s :
Co2FTF ,
Co2
clam
,
Co2
antic
lam
,
Co2
4C4
,
Co Pd 4C4 ,
Co2
FT
F4
(see
ref.
53, 54, 55
,
App
endi
x D
).

SR 0.1 V s_i
3 10
for CV
io2
for macro-
electrolyses
CV
SR 0.1 V s
3 10
System
studied
(Red/Ox)
(Source n°)
Solvent
Supporting
electrolyte
(conc.
Electrochem.
method used
Experim.details
(potential range
explored,
mi-
tial potential,
scan ra
te,
etc ...
Conc. of
Red
and/or
Ox
in t
he
solu
tion
in Mole/l
( in pg/l)
('':
1)02
)
Rea
ctio
n ob
serv
ed
Oxi
datio
n R
educ
tion
or
Bot
h
Characteristic
Potential
measured
(V/ref.)
(E1/2, E
peak
. ot
her
...)
•
Rat
e constant
(cm _1)
at
stan
dard
redox
potential
n
F/mole E1,2 or
eak of
ferro-
ferricyanide
or
ferrocene/
ferricinium
CH2CN
TEAP
0.1M
C
V
and
CH
2CN
macroelec-
trolyses
CH
2CN
B
u4N
BF4
=
C
(12)
/
OR
\
/ C
=C
/ N C
=C
/
NS
R
(15)
C
e3/C
e
Cu
Pb
Cd
(17)
02
/H20
Cl/C
l2
Cr(
III)
/Cr(
II)
Ace
tate
bu
ffer
5.
1 M
HC
1/K
C1
pH = 3
Sea water
pH =
2
H20
RD
E
2V/m
in
100
a 150 t
/mn
Pre-
elec
trol
ys is
at 1 V/ECS
-0.8V
OV
3 in
n pr
e-el
ec-
trol
ysis
DPP SR2m\1s1
Prussian Blue
(PB)
mod
ifie
d CC
LSV
Ano
dic
stri
ppin
g Pulse Pola-
rography.
CV
CV
CV
Ox
0.95<E<1.59
Irrev
2
0.95<E<1.32
Irrev
2
Ox
Ox
0.87
<zE
<1.
60
Irre
v 2
8.10
R
ed
1e
a at
8 . i0
-0.
2V/E
CS
"z1
pg/l
as o
n
Hg
elec
trod
e
1 at
m O
x 02
Red
. 13
1/2=
+
0.2
Irre
v 4
I3=
+O
.2
02 Red is
for ferro-
accelerated
ferricyanide
on PB film
0.052
(450
°C)
Rep
rod.
Hg
film
onGCE
Fused
LiCl - K
C1
(450
°C)
LiC
1 - K
C1
(450
°C)
Cl0/ClO2
CV
System
studied
(Red/Ox)
(Source
Supporting
Solvent
electrolyte
(conc.)
n°)
Electrochem.
method used
Experien.details
(potential range
explored,
mi-
tial potential,
scan rate, etc
••
Conc. of
Red
and/or
Ox
in the
solution
in
Mole/ 1
Reaction
observed
Oxidation
Reduction
or Both
Characteristic
Potential
measured
(V/ref.)
(E1/2, Epeak
other ...)
nate
constant
( -1
at
standard
redox
potential
n
F/mole E112 or
EspeakOf
MARKS
ferro-
fenicyanide
or
ferrocene/
ferricinium
(17)
Fe(I
II)/
Fe(I
I)
LiC
l-K
Cl
CV
(450
°C)
Cu(
II)/
Cu(
I)
LiCl-KC1
CV
(450°C)
Cl/Cl2
Fused
NaCl-A1C13
CV
i0=8.6
(175 C)
uA/cm
(175
°C)
t,
(18)
SO2/
SO
tMF
CV
SR 10-500 mV/s
iO-1
E -
0.9V
Qiasi
0.65
AN
0.1
TEAP
El.
(19)
water
var.acids
HYV
+ 0
.7 ,
-0
.1 V
0-6. 10
Red
+ 0
.44
÷ +
0.2
5 ÷io-2
1
-
Fe
water
var.compl.
HYV
+ 0
.06
+ -
0.32
5 10
1
recalc
agents
> io
to SHE
CuO/Cu2+
water
var.inorg.
HYV
Red
various
1 + 1
Reference:
salts, HOAc,
or
SCE
ethylene-
2
diamine
(For details see
:
J. Electroanal.Chem.
38, 349 (1972)
; 39, 229 (1972)
; 44, 117 (1973)).
HYV
1.5 ÷--1.OV
i-5_
iü-2
Red
E.
=1.OV
mit
Hg(II)/Hg(I)
H2S04
1M
"
i05_
i02
Red
0.42
i0
na0.55
System
studied
(Red/Ox)
(Source
Supporting
Solvent
electrolyte
(conc.)
n°)
Electrochem.
Method used
Experiin.details
(potential range
explored,
mi-
tial potential,
scan rate, etc
••
Conc. of
Red
and/or
Ox
in the
solution
in
Mole/l
Reaction
observed
Oxidation
Reduction
or Both
Characteristic
Potential
measured
(V/ref.)
(E1/2, eak'
other ...)
P.ate
constant
( s-
i) at
standard
redox
potential
n
F/mole
or
Epeak of
pipj
ferro-
ferricyanide
or
ferrocene/
ferricinium
(20)
3, 4-Dihydro-
xybenzylamine
(23)
Sb (V) /Sb(III)
Ce(IV) /Ce(III)
Sn(IV)/Sn(II)
Cu(II)/Cu(I)
Ti (IV) /Ti (III)
Hg(II) /Hg(I)
Epa 0.275
Epc 0.125
at
SR 50 mVs
HO
0.1M
CV
+0.7V -0.OV
2
CflzCOOH
E-
.
=
± 0.OV
mit
0.2M
CH3COO H4
SR 5 - 200 mV 51
pH = 5
.0
9.5M
HW
1.2V÷--1.0V
HC1
SR1OmV.s1
E.
=
0.6V
mit
H2S04
1M
HYV
1.5 -÷-1.0V
E.
=
1.2
mit
4M HC1
+1M-ffir
1.2 ÷--0.5V
9.5M LiBr
HYV
0.0
-0.7V
KC1
0.5M
I-WV
1.2 +-÷-1.OV
(+HC1)
(0.1M)
II2SO4
1- 1
0
HYV
1.2
+-÷ —
0.8V
H2S04
1 M
1
Both
Epa = 0
.49
Epc =
0.2
0
at
SR 50 mVs1
1o_3_102
Red
E1/2 =
0.47
(3000 rpm
and
1 mM)
io3_io2
Red
0.9 (3000 rpm)
102M
Ox
0.4
(300
0 rp
m)
i03÷
÷iO
2 7 mM
Red
-0.3
io3-
io2
1oth
1o-4
O
x 0.
28 (
3000
rpm
) 1 , 5
mM
0.43
(30
00rp
m)
10-3
M
3.2 .
Qiasi-rev 2
4.10
2 1
(4.6
-9.5
) 1
io
1
<<10
<<iO-5
Reference:
SCE
Ref
eren
ce
SCE
na =
0.
2 C
C is stable
in 9.5 M
a= 0
.25
n 0.35
na= 0.32
a=0.49-0.53
f30.
32—
0
a =
0.5
5
2 .
5.6.
2
0 0 CD
CD
N
0 CD
C
l)
2
+0.8 to —2.2 Volt
CV
0.02V/sto 200V/s
400 to 10000rpm
11W
Rate
constant
(cm s-i)
at
standard
redox
potential
Supporting
Solvent
electrolyte
(conc.)
Electrochem.
method used
txper:im .details
(potential range
Conc. of
explored,
mi-
Red
tial potential,
and/or
scan rate, etc
Ox
in the
solution
in
___________
Mol
e/l
React ion Characteristic
observed
Potential
Oxidation measured
Reduction (V/ref.)
or Both
(E1/2, Epeak,
other ...)
n
F/mole
System
studied
(Red/Ox)
(Source n°)
(24)
I/TI
CH3CN
TBAPF6
0.5 M
ITT/TV
U
V/V
T
VIl/VITI
IX/X
XI/XII
Note : (Ru(di-X-dpy)2
I/TI , X
= C
l ITT/TV , X
=
H
(25)
H20
HC1O4
R1RKS
or
Epeak of
ferro-
ferricyanide
or
ferrocene/
ferriciniuni Reference:
Ag/Ag
(0.1 M)
(NO))/2
V/VT, X = C6H5 (phenyl)
VIT/VTTT, X = C4H9 (But)
to
E1/2 0.005
10—i
Red
E1/2 - 0
.043
Red
E1/2 —0.082
Red
E1/2 -0.116
Red
E1/
2 -0.140
Red
E1/2 — 0
.275
IX/X ,
X = C
H3
XI/X
II',
X = O
CH
3
unknown
Red
in this
very
method
obvious
Perhaps
Ox
Interactions
Electron
C-Hydrogen
+
photo -
C-H20
KC1O4
emission
C-Oxygen
0.08M
in electro-
lytes
0.06
1
0.19
1
0.09
1
0.07
1
0.15
1
0.12
1
very
fast
Reactivity of
photogenerated
atomic hydrogen
H - H
3O
H — H2
+
CV
Activation of
the electrode
Study of the
interactions
H
c1
H
MeNNt
CH CN
Me'
\/4
3
U)
Cl)
C
l)
C)
U)
Ref
.: SCE
CD
CD
C
) 0 0 (D
Cl)
Ref
.: SCE
/s#Br(red)
Br
n-Pr
CN
System
studied
(Red/Ox)
(Source
Supporting
Solvent
electrolyte
(conc.)
n°)
Electrochem.
method used
Experim. details
{potential range
explored,
mi-
tial potential,
scan rate, etc
...
Conc. of
Red
and/or
Ox
in the
solution
in
Mole/l
Reaction
observed
Oxidation
Reduction
or Both
- C
hara
cter
istic
Potential
measured
(V/ref.)
(E1/2, Epeak.
other ...)
iate
constant
( s1
at
standard
redox
potential
fl F
/mol
e
E112 or
Epç0f
REMARKS
ferro-
ferricyanide
or
ferrocene/
ferricinium
(26)
TBAP
0.1OM
TMP
CV
0.10M
TEAP
0.1O
M
TEAP
0.1O
M
CH
O
CH
3CN
(CH3)3CCHO
/
CH
3CN
rela
t sp
ecie
s
From - 1.
5V
LSV
to -2.7V
SR 0.5- 2 V/s
- 60
to
-
80°C
Fro
m -0.2 V
to +0.5V
SR 0.02— 0.1V/s, 21°C
CV
From +1.0 V
to +3.5V
SR 0.5V/s
CV
From
+2V
to
- 2.
2V
CV
From
ca. - 0
.3V
to
+0.1 V
SR
0.05V/s,
25°C
Referece:
Ag/Ag
(0.01 M)
I,
+
othe
rs
1.5 io
Red
E=-2.0,-2.3
Irrev
2
of each
conformer
io
Ox/Red
E1/
2 =+0.20
1.8.
1O
3 1
2. io-2
Ox
E =
+ 3
.0,
Irre
v +
2.9
1
- 5
. 1
Ox/Red
Many
-
2. 1
0
Ox/Red
Ep,c ,a
Rev
-0.04 -.-—0.15
as a function
of pH
57 % EtOH/
NaOH +
43 % H20
NaNO3
(Vol/Vol)
(OH-) = i0
2+
SM
System
studied
(Red/Ox)
(Source
Supporting
Solvent
electrolyte
(conc.)
n°)
Electrochem.
method used
Thçperim.details
(potential range
explored,
mi-
tial potential,
scan rate, etc
...
Conc. oT
Red
and/or
Ox
in the
olution
in
Mole /1
Reaction
observed
Oxidation
Reduction
or Both
Characteristic
Potential
measured
(V/ref.)
(E1/2 Epeak
other ...)
Rate
constant
(cm s-i)
at
standard
redox
potential
n
F/mole
E172 or
Epeak of
ferro-
ferricyanide
or
ferrocene/
ferricinium
(2 7)
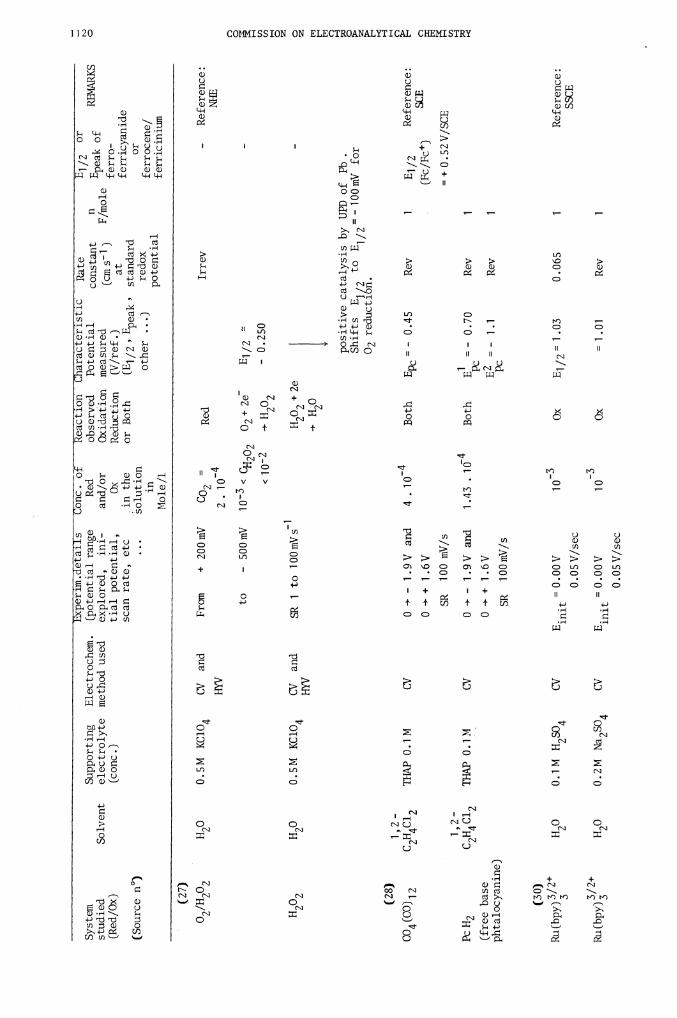
02202
H20
0.5M KC1O4
CV and
From
+ 200mV
CO2 =
Red
Irrev
-
Ref
eren
ce:
HYV
2
.
to
-
500m
V i0< CH202
O2+2e
E1/2
<10-2
-- H
202
— 0.
250
H2O2
H20
0.5M KC1O4
CV and
SR 1 to 100mVs
H2O2+2e
HYV
÷H20
positive catalysis by UPD of Pb.
Shifts E1/2 to E1/2-lOOmV f
or
02 reduction.
(28)
1,2—
CO4 (CO)12
C2H4C12
THAP 0.1 M
CV
0 - -
1 .9 V and
4 .
1 0
Both
Epc = -
0.45
Rev
1
B1 /2
Reference:
SCE
0 -
+ 1
.6V
(Fc/Fc)
SR 100 mV/s
+0.52V/SCE
1,2—
Pc H2
C2H4 Cl2
THAP 0.1 M
CV
0 -' -
1 .9 V and
1 .43
.
1 (i
Both
E1 = -
0.70
Rev
1
Pc
(free base
0
+
1 .6V
B2 = -
1 .1
Rev
1
phtalocyanine)
Pc
SR
100 mV/s
(30)
Ru(bpy)/2
H2O
0.1M H SO
CV
E.
.
=0.
OO
V
10's
Ox
E1/2= 1.03
0.065
1
Reference:
2 4
mit
SSCE
0.05 V/sec
-3
Ru(bpy)2
H2O
0.2M Na2SO4
CV
E.
.
=0.
OO
V
10
Ox
=1.01
Rev
1
mit
0.05 V/sec
System
studied
(Red/Ox)
(Source
Supporting
Solvent
electrolyte
(conc.)
n°)
Electrochem.
method used
Exp
erim
. det
ails
(p
oten
tial r
ange
explored, mi-
tial potential,
scan rate, etc
...
Con
c.
of
Red
and/or
Ox
in the
solution
in
Mole/l
Rea
ctio
n ob
serv
ed
Oxidation
Reduction
or Both
Cha
ract
eris
tic
Pote
ntia
l measured
(V/ref.)
(Ei/2, Epeak
other ...)
Pate
co
nsta
nt
(cm _1)
at
standard
redox
potential
n
F/mole
B1 /2
or
E
peak
of
REMARKS
ferro-
ferricyanide
or
ferrocene/
ferricinium
(30)
Reference:
Ru(bpy)'2
H20/CH3CN 0.1 M NAP
CV
Et =
0.00
V
1 0
Ox
E1 /2= 1 .15
Rev
1
SSCE
50/50
SR
0.OSV/s
Os(bpy)'2
H20
0.2M 24
CV
"
Ox
= 0
.60
Rev
1
Cp2FeTMA2'
H2O
0.2M Na2504
CV
i0
Ox
= 0
.38
Rev
1
bpy = 2,
2' bipyridine.
Cp2Fe TMA =
(Tet
ram
ethy
lani
mon
io)f
erro
cene
. No correction for junction potential.
(33)
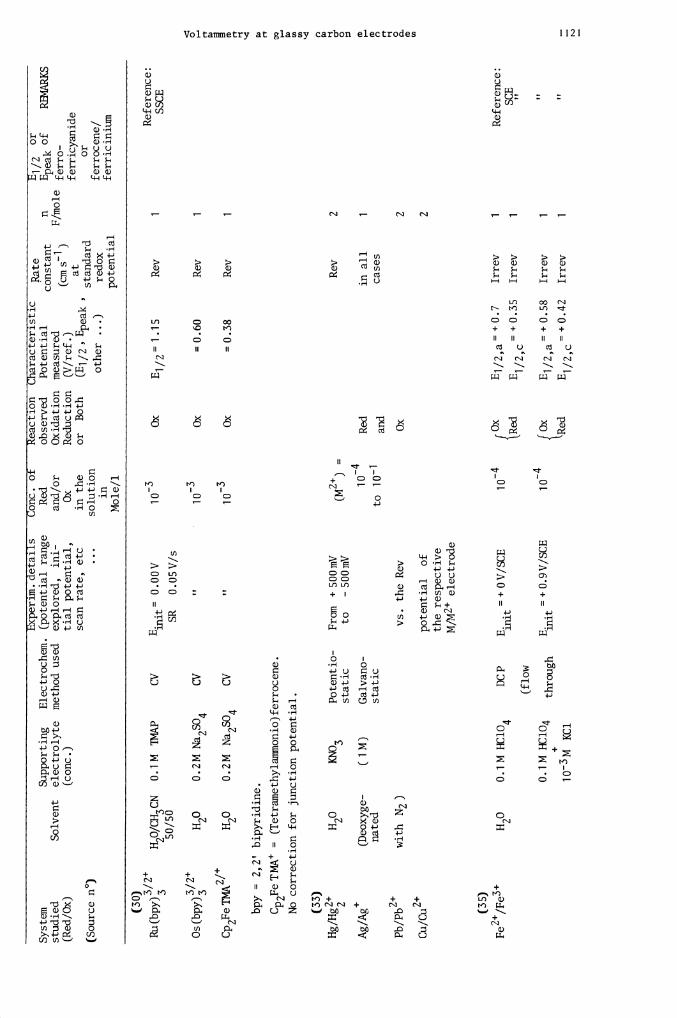
Hg/
Hg
H2O
KNO3
Potentio-
From + 500mV
(M2) =
Rev
2
static
to
- 500mV
Ag/Ag
(Deoxyge-
(1 M)
Galvano-
1
Red
in all
1
CD
CD
nate
d static
to 10-i
and
cases
0
Pb/P
b2
with N2)
vs. the Rev
Ox
2
CD
Cu/
Cu2
potential of
2
the respective
M/M2 electrode
(35)
Fe2/
Fe
HO
0.1M HC1O
DCP
E.
=+OV/SCE
Ox
E1/2,a+O.7
Irrev
1
Reference:
2
4
mit
{Red
E1 /2 ,c = +
0.35
Irrev
i
SE
(flow
0.1MHC1O4
throug
E
=+0.9V/5CE
fOx
E1/2,a=+O.S8
Irrev
h
mit
i03M
KC1
Red
Ei/2c=+O42
Irrev

prepol. at + 0
.25
3 .
10
an
d at -1.25V
(1 mm)
Red
of
dissolved
species
E
(± 0.01)
1' —
0.08
-
1 .45
1-0.11
-0.17
L -
1 .68
I +
0.0
6 1-
0.85
1-1.
20
- 1
.68
[- 1
.31
- 1
.72
1-1.18
1- 1
.75
Reference
SCE
Ep f cou-
nled to
gLassy carbon
1— 1.
08
1.50
- 0
.13
J-
1.17
- 1
.65
1÷ 0
.10
1- 0.
86
1- 1.
19
1- 1.
65
1- 1.
39
- 1
.75
1- 1.
21
- 1
.78
H20
0.1M KOH
HYV
Anodic prepol.
20 pA for 2 mm.
3. 10
Red
- 0
.022
Reference
SHE
System
studied
(Rex/Ox)
(Source
Supporting
Solvent
electrolyte
(conc.)
n°)
Electrochern.
method used
Experim. dietails
(potential range
explored,
mi-
tial potential,
scan rate, etc
...
Gonc. of
Red
and/or
Ox
in the
solution
in
Mole/l
Reaction
observed
Oxidation
Reduction
or Both
Characteristic
Potential
measured
(V/ref.)
(E1/2, Epeak,
other ...)
Rate
constant
(cm
1)
at
standard
redox
potential
n
F/mole Ei/2 or
EpeakOf
REMARKS
ferr
o-
feiricyanide
or
ferrocene/
ferricinium
(5)
Me2SO
TEAP
CV
T
Pc /
'Pa
CV
1
CV
1
CV
1
C(T- (rn-NH2) P
P)
C(F
e"T
(p-N
H2)
PP)
C(Co"T (m
-NH
2)PP
)
C (C
u'
(p-N
H2)
PP)
C (Z
n'
(p-N
H2)
PP)
C(N
i"T
(p-N
H2)
PP)
(14)
H20
0.1M KOH
Red
-1.143
ca
2
Reference
SHE
System
studied
(Red/Ox)
(Source n°)
Solvent
Supporting
electrolyte
(conc.)
Electrochem.
method used
Experim. details
(potential range
explored,
mi-
tial potential,
scan rate, etc
...
Conc. of
Red
and/or
Ox
in the
solution
in Mole/l
(p.p.b.)
Reaction
observed
Oxidation
Reduction
or Both
Characteristic
Potential
measured
(V/ref.)
(E1/2, Epeak
other ...)
Rate
constant
(cm s-i)
at
standard
redox
potential
Ti
F/l E
112 or
eeof
RAS
ferro-
ferricyanide
or
ferrocene/
ferricinium
(21)
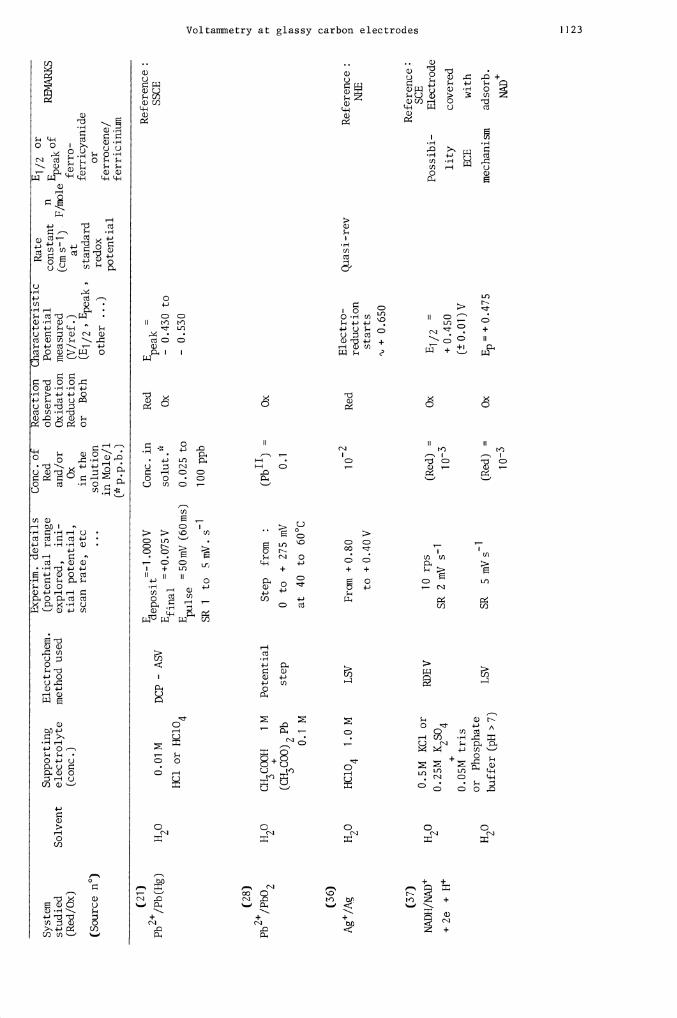
1 000V
Conc in
Red
E
=
Ref
eren
ce:
Pb2/Pb(Hg)
H20
0.01 M
DCP - A
eposit
peak
SSCE
E
.
=+
0.07
5V
solu
t. Ox
- 0.
430 to
final
HC1 or HC1O4
E1
=50mV (6Oms)
0.025 to
- 0.
530
SR 1 to 5 mV. S1
100 ppb
(28)
Pb2/PbO2
H20
C
H3C
OO
H
1 M
Potential
Step from
:
(Pb") =
Ox
00
(CH
3CO
O)2
Pb
step
0
to
+
275 mV
0.1
U)
0.1 M
at 40 to 60°C
(36)
Ag/Ag
H20
HC1O4
1 .0 M
LSV
From + 0
.80
1 o2
Red
Electro-
Qiasi -rev
Reference:
0
0
reduction
NHE
CD
to +0.40V
starts
CD
o+O
.65O
0
(37)
Reference:
0.5M KC1 or
SCE
RDE V
10 rps
(Red) =
Ox
E1 /2 =
Pos
sibi
- Electrode
H2O
0.25M K2SO4
SR 2 mV 51
+ 0
.450
lity
covered
+2e +H+
+
0.05
M tris
(±0.01)V
ECE
with
or Phosphate
H20
buffer (H 7)
LSV
SR 5 mV s
(Red) =
Ox
Ep = +
0.4
75
mechanism
adsorb.
NAD

DoPAC
(3,4-dihydroxy-
phenyl ethyl-
amine-HC1)
Ox
(+0.590
Irrev
Ox
+0.430
System
studied
(Red/Ox)
(Source
Supporting
Solvent electrolyte
(conc.)
n°)
Electrochem.
method used
Experiim. details
(potential range
explored,
mi-
tial p
otential,
scan rate, etc
Conc. of
Red
and/or
Ox
in the
solution
in
Mole/l
'Reaction
observed
Oxidation
Reduction
or Both
Characteristic
Potential
measured
(V/ref.)
(E1/2, Epeak
other ...)
Rate
cstant
(cm s)
at
standard
redox
potential
n
F/mole E1 /2 or
Epeak0f
RB'4ARKS
ferro-
ferricyardce
or
ferrocene/
ferricinium
(39)
Ascorbate
(Ascorbic
acid)
4 Me Cat
(Methyl-
catechol)
HO
2
pH
7.4
(0.1M buffer)
CV
SR5O mV. S
1 5.
Ox
E +0.390V
Ionic strengh
to
1 with
KC1
pH = 3.
0 CV
SR 50 mV. s1
5. 10
Ox
0.480
H20
pH = 3.
0
7.4
CV
CV
SR 50
SR 50 mV.
mV.
s s
5. 10
5. 10
Ox
Ox
0.415
0.140
pH=3.0
CV
SR5OmV.s1
5.10
7.4
CV
SR 50 mV. s
5.
pH= 3.0
—3
10
Irrev
DHBA
(3,4-dihydroxy-
benzylamine-
HBr)
(46)
Reterence
Ag/AgCl.
KC1 4M
Syst. more
reversible on
CC heated be-
fore use at
520°C/i torr.
+ 0
.35
+ 0
.253
+ 0
.428
+ 0
.132
+ 0
.437
+ 0
.155
+ 0
.460
+ 0
.217
Reference:
SCE
2-Ethyl-
anthraquinone
CH3OH
C2H5OH
Buffer :
(0.iM NH Cl
+
0.1M NH3)
LSV and CV
between 0
and -1.OV
20°C
Propan-2-ol
" R
ev
+ 0
.710
+ 0
.360
=
— 0
.68
± 0.01 V
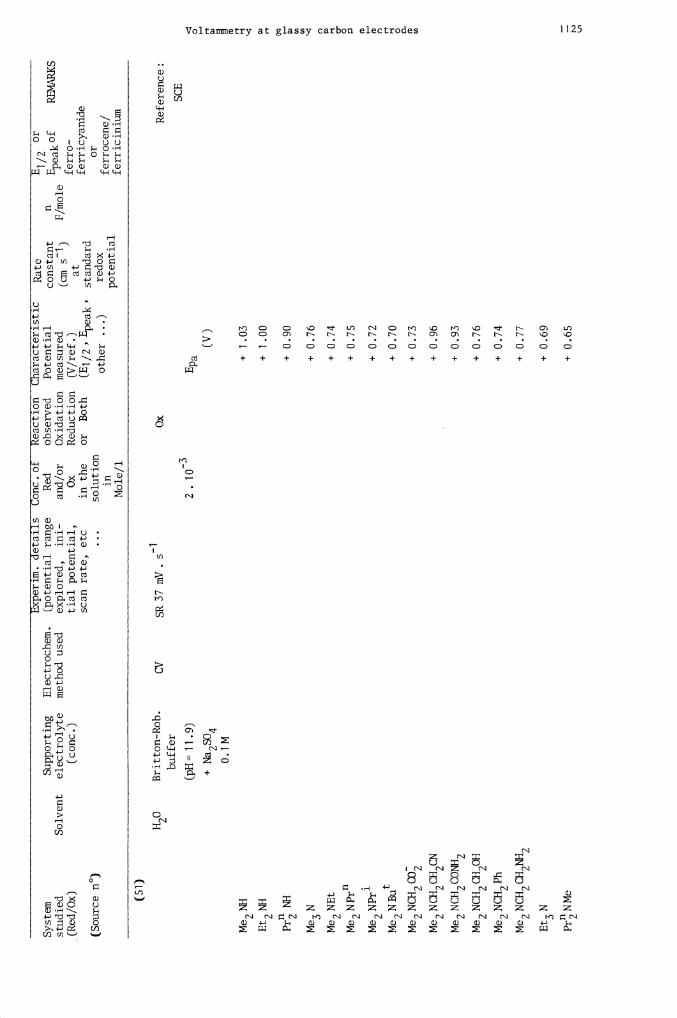
System
studied
(Red/Ox)
(Source
Supporting
Solvent electrolyte
(conc.)
n°)
Electrochem.
method used
Exp
erim
. det
ails
(potential range
explored,
mi-
tial potential,
scan rate, etc
...
Conc. of
Red
and/or
Ox
in the
solution
in
Mole/l
Reaction
observed
Oxidation
Reduction
or Both
Characteristic
Potential
measured
(V/ref.)
(Ei/2, Epeak
other ••,)
P,ate
constant
(m s1)
at
standard
redox
potential
n
F/mole
E112 or
EpeakOf
RB'1ARKS
ferr
o-
ferricyanide
or
ferrocene/
ferricinium
(51)
I12O
Britton-Rob.
CV
SR 37 mV. s
Ox
Reference:
buffer
-3
SCE
(pH = 1
1 .9)
2
. 10
E
pa
+ N
a2S
O4
(V)
0.1
M
Me2
NH
+
1.
03
Et2NH
+
1.00
Pr N
H
+
0.90
Me3
N
+
0.76
Me2
NE
t + 0.
74
Me2 NPrITI
÷ 0.75
Me2 NPr'
+
0.72
Me2 NBut
+
0.70
Me2
NC
H2C
O2
+ 0.
73
Me2 NCH2CH2CN
+ 0.
96
Me2 NCH2CONH2
+ 0.
93
Me2 NCH2CH2OH
+ 0.
76
Me2 NCH2Ph
+ 0.
74
Me2 NCH2CH2NH2
+
0.77
Et3
N
+
0.69
PrNMe
+
0.65
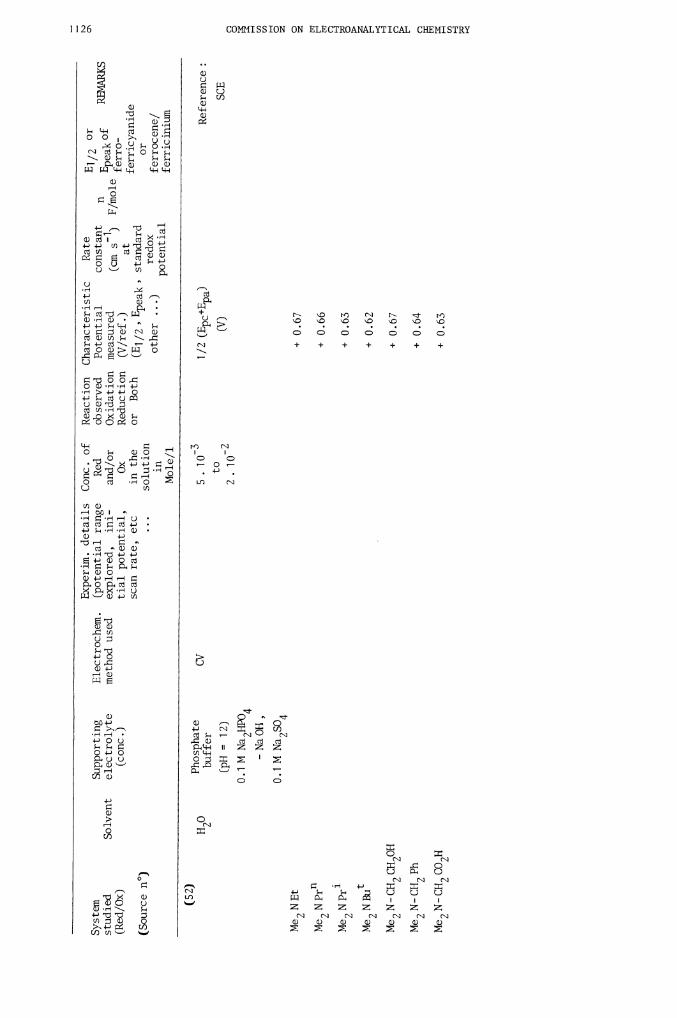
H o
Phosphate
2
buffer
(pH = 12)
0.1 M N
a21-1P04
- N
a O
H,
Me2NEt
Me2 N Pr1
Me2 N P
r1
Me2
NBUt
Me2
N-CH2 CH2OH
Me2N-CH2Ph
+
0.64
Me2 N-CH2 CO2H
+
0.63
System
studied
(Red/Ox)
(Source n°)
Supporting
Solvent
electrolyte
(conc.)
Electrochem.
method used
Experim. details
(potential range
explored,
mi-
tial potential,
scan rate, etc
...
Conc. of
Red
and/or
Ox
in the
solution
in
Mole/l
Reaction
observed
Oxidation
Reduction
or Both
Characteristic
Potential
measured
(V/ref.)
(Ei/2, Epeak,
other ...)
Rate
constant
( 1
) at
standard
redox
potential
n
F/mole El12 or
EpeakOf RKS
ferro-
ferricyanide
or
ferrocene/
ferricinium
(52)
5.10
0.1 M Na2504
to
2. io2
Reference:
SCE
1/2 (Epc+Epa)
(V)
+ 0.
67
+ 0.
66
+ 0.
63
+ 0.
62
+
0.67
0

Voltammetry at glassy carbon electrodes 1127
APPENDIX D SOURCE LIST
a) Answers to the questionnaire
(1) Dr. John F. Evans, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota55414 (USA).
(2) Pr. Wang Erkang, Changchun Institute of Applied Chemistry, Academia Sinica, Changchun,3ilin 130021 (China).
(3) Dr. 1. Moiroux, Laboratoire de Chimie, E.N.S.E.T., 61, Avenue du Président Wilson, 94230Cachan (France).
(4) Dr. Gen P. Satô, Department of Chemistry, Sophia University, Faculty of Science andTechnology, 1, Kioi-cho 7-chome, Chiyoda-Ku, Tokyo 102 (3apan).
(5) Pr. Royce W. Murray, Kenan Laboratories of Chemistry, University of North Carolina, ChapelHill, North Carolina 27514 (USA).
(6) Pr. 3oseph Jordan, Department of Chemistry, Pennsylvania State University, 152, DaveyLaboratory, University Park, Pennsylvania 16802 (USA).
(7) Dr. Russell 3. Taylor, Department of Defence, Materials Research Laboratories, CorditeAvenue, Maribyrnong, P.O.B. 50, Ascot Vale, Victoria 3032 (Australia).
(8) Dr. Leon Mart /ICH 4, Nuclear Research Center 3uelich, P.O.B. 1913, D-5l70 3Ulich 1 (FRG).
(9) Dr. Plichon, Laboratoire de Chimie Analytique des Milieux Réactionnels, E.S.P.C.I., 10, rueVauquelin, 75231 Paris Cédex 05 (France).
(10) Pr. G.3. Patriarche - Dr. 3.M. Kauffmann, Université Libre de Bruxelles, Institut dePharmacie, Campus Plaine, C.P. 205/6, Boulevard du Triomphe, B-1050 Bruxelles (Belgium).
(11) Dr. Karl Doblhofer, Fritz-Haber Institut der Max-Planck-Gesellschaft, Faradayweg 4-4, D-l000Berlin 33 (FRG).
(12) Dr. G. Le Guillanton, Laboratoire de Synthèse et Electrochimie Organique, UniversitéCatholique de l'Ouest, B.P. 808, 49005 Angers Cédex (France).
(13) Dr. P. Zuman, Department of Chemistry, Clarkson College of Technology, Potsdam, New York13676 (USA).
(14) Dr. M. Brzina and Dr. 3. Weber, 3. Heyrovsk Institute of Physical Chemistry andElectrochemistry, Czechoslovak Academy of Sciences, U továren 254, 10200 Prague 10(Czechoslovakia).
(15) Dr. M. Molina, Laboratoire dElectrochimie Analytique, SCAA - SEA - DCAEA, CentredEtudes Nucléaires, B.P. 6, 92260 Fontenay aux Roses (France).
(16) Dr. Maurice L'Her, Departement de Chimie, Université de Bretagne Occidentale, 6, Avenue LeGorgeu, 29283 Brest Cédex (France).
(17) Dr. Isamu Uchida, Department of Applied Chemistry, Faculty of Engineering, TohokuUniversity, Sendai (3apan).
(18) Dr. Dierk Knittel, Institut fUr Physikalische Chemie, Universität Hamburg, Laufgraben 24,D-2000 Hamburg 13 (FRG).
(19) Dr. Karel tulIk, Charles University, Department of Analytical Chemistry, Albertov 2030,12840 Prague 2 (Czechoslovakia).
(20) Pr. Mitsugi Senda and Dr. Tokuji Ikeda, Department of Agricultural Chemistry, College of Agri-culture, Kyoto University, Sakyo-ku, Kyoto 606 (3apan).

1128 COISS ION ON ELECTROANALYTICAL CHEMISTRY
(21) Dr. 3uerg B. Reust, Inst. für Anorganische, Analytische und Physikalische Chemie, UniversitätBern, Freiestrasse 3, CH-3012 Bern (Switzerland),Div. of Chemical and Physical Sciences, Deakin University, Victoria 3217 (Australia).
(22) Dr. lames Anderson, Department of Chemistry, University of Georgia, Athens, Georgia 30602(USA).
(23) Dr. Paul Kiekens, Rijksuniversiteit Gent, Laboratorium voor Analytische Scheikunde (Pr.Verbeek), Krijgslaan 271-S 12, 9000 Gent (Belgium).
(24) Dr. Kevin 1. Corby, Dupont Experimental Station, E 269/304, Wilmington, Delaware 19898(USA).
(25) Dr. Pierre Vennereau, Laboratoire d'Electrochimie Interfaciale du CNRS, I, Place AristideBriand, 92195 Meudon Cédex principal (France).
(26) Dr. Dennis H. Evans, Department of Chemistry, University of Wisconsin, 1101 UniversityAvenue, Madison, Wisconsin 53706 (USA).
(27) Pr. W. Lorenz, Institut für Physikalische Chemie und Elektrochemie, Lehrstuhl III, UniversitätKarlsruhe, Kaiserstrasse 12, 7500 Karlsruhe I (FRG).
(28) Pr. M. Gross, Laboratoire dElectrochimie, Université Louis Pasteur, 4, rue Blaise Pascal,67000 Strasbourg (France).
(29) Dr. Daniel S. Polcyn, Pope Scientific, Inc., P.O.B. 495, Menomonee Falls, Wisconsin 53051(USA).
(30) Pr. Allen J. Bard, Department of Chemistry, University of Texas, Austin, Texas 78712 (USA).
(31) Pr. Maja, Istituto Di Elettrochimica e Di Chimica Fisica, Politecnico, Corso Duca degliAbruzzi, 24 Torino (Italy).
(33) Dr. B.R. Schariffer, Departamento de Quimica, Universidad Simon Bolivar, Apartado 80659,Caracas 1080 (Venezuela).
(34) Dr. H.C. Brookes, Chemistry Department, University Natal, Durban (South Africa).
(35) Pr. V.D. Linden, Chemical Analytical, Department CT. Technische Hogeschool Twente Postbus217, 7500 AE Enschede (Netherlands).
(36) Pr. R.G. Barradas, Department of Chemistry, Carleton University, Ottawa, Ontario KIS 5B6(Canada).
(37) Pr. Philip J. Elving, Department of Chemistry, University of Michigan, Ann Arbor, Michigan48109 (USA).
(39) Pr. R. Mark Wightman, Department of Chemistry, Chem. Building, Indiana University,Bloomington, Indiana 47405 (USA).
(47) Dr. 1. Robinson, Department of Chemistry, The University, Southampton S09 5NH (G.B.).
b) Published papers
(32) loseph Wang, Department of Chemistry, New Mexico State University, Las Cruces NM 88003(USA).- Electrochimica Acta, 26, n° 12, p. 1721-1726 (1981).
(38) F. Seon, G. Picard and B. Tremillon, Laboratoire dElectrochimie Analytique et Appliquée, LAau CNRS n° 216, Ecole Nationale Supérieure de Chimie de Paris, Université P. et M. Curie,11, rue P. et M. Curie, 75231 Paris Cédex 05 (France).- Electrochimica Acta, 27, n° 10, p. 1357-1368 (1982).

Voltammetry at glassy carbon electrodes 1129
(40) W.3. Blaedel and G.W. Schieffer, Department of Chemistry, University of Wisconsin, Madison,Wisconsin 53706 (USA).- 3. Electroanal. Chem., 80, p. 259-271 (1977).
(41) W.E. Van der Linden and 3.W. Dieker, Laboratory for Analytical Chemistry, University ofAmsterdam, Nieuwe Achtergracht 166, 1018 WV Amsterdam (Netherlands).- Analytica chimica Acta, 119, p. 1-24 (1980).
(42) D. Laser and M. Ariel, Department of Chemistry, Technion, Israel Institute of Technology,Haifa (Israel).- Electroanalytical Chemistry and Interfacial Electrochemistry, 52, p. 291-303 (1974).
(43) Royce C. Engstrom, Department of Chemistry, University of South Dakota, Vermillion, SouthDakota 57069 (USA).- Anal. Chem., 54, p. 2310-23 14 (1982).
(44) Luke Bjelica, Roger Parsons and M. Reeves, Department of Physical Chemistry, TheUniversity, Bristol BS8 ITS (G.B.).- Proc. Symp. Electrode Processes Electrochemical Society Monograph, p. 190 (1979).- Croatica Chemica Acta, CCACAA , 53 (2), p.211-231 (1980).
(45) W.3. Blaedel and Roger A. 3enkins, Department of Chemistry, University of Wisconsin,Madison, Wisconsin 53706 (USA).- Analytical Chemistry, 46, n° 13 (Nov. 1974).
(46) V.3. 3ennings, T.E. Forster and 3. Williams, Lanchester Polytechnic, Coventry, Warwickshire(G.B.).- Analyst, 95, p. 718-721 (August 1970).
(48) L.S. Chulkina and V.A. Zarinskii, V.1. Vernadskii Institute of Geochemistry and AnalyticalChemistry, Academy of Sciences of the USSR, Moscow (USSR).- 3. Anal. Chem. USSR, 27, p. 2044 (1972).
(49) Ruedi Eggli, Institute of Inorganic Chemistry, University of ZCirich, CH-8001 Zurich(Switzerland).- Analytica Chimica Acta, 97, p. 195-198 (1978).
(50) F. Vydra and P. Petak, 3. Heyrovsky Polarographic Institute, Czechoslovak Academy ofSciences and Ore Research Institute, Prague (Czechoslovakia).- 3. Electroanal. Chem., 24, p. 379-386 (1970).- 3. Electroanal. Chem., 33, p. 161-168, (1971).
(51) Masaichiro Masui, Hiroteru Sayo and Yoshiaki Tsuda, Faculty of Pharmaceutical Sciences,Osaka University, Toneyama, Toyonaka, Osaka-f u (3apan).- 3. Chem. Soc. (B), p. 973-976 (1968).
(52) Masaichiro Masui and Hiroteru Sayo, Faculty of Pharmaceutical Sciences, Osaka University,Toneyama, Toyonaka, Osaka-f u (3apan).- 3. Chem. Soc. (B), p. 1593-1596 (1971).
(53) 3.P. Coliman, M. Marrocco, C.M. Elliott and M. L'Her- 3. Electroanal. Chem., 124, p. 113 (1981).
(54) 3.P. Coilman, C.M. Elliott- Proc. Nat. Acad. Sci. USA, 7, (1977).
(55) 3.P. Coliman, P. Denisevich, Y. Konai, M. Marocco, C. Koval and F. Anson- 3. Amer. Chem. Soc., 102, p. 6027 (1980).

1954 SUBJECT INDEX
Transport properties of electrolytes forapplied research and technologyfrom infinite dilution to saturationin mixed-solvent solutions
Transport property measurements, insightsinto solute—solute—solvent interactionsfrom
Triglycerides in fats and oils, standardisedmethod for determining
Tungsten, Mo, and Re enolates, synthesisand C—C bond-forming reactions of
U.v. microscopy, oxidation and stabilizationstudies on polymers 1727
Watersfresh, pH determination in low ionic
strengthnatural, physical chemistry of
Wavegu ides, preparation of opticalWhite blood cells, morfometry of
X-ray scattering measurements of order innon-crystalline polymers 1627
Volume, Issue Page no. and Correctionand Year location
56, 8 (1984) 1102Solvent: Molten SaltsColumn 1, item 6
2i CuCl2/KC1CaCl2/KC].
for cholesterol oxygen oxidoreductaseread cholesterol oxidase suspension
903Section 6.5line I
903Section 7.2line 3
for (catalase—suspension 1)read (catalase—suspension)
for catalase—suspension 1 (6.5)read cholesterol oxidase suspension (6.4)
1071355369
1083
151 5R
1789
1641993
Ultra high modulus polyethylenecomposites
uPVC pipe, understanding brittle failure of
877R10151299599
Errata
57, 6 (1985) 903Section 6.4line I


















