
VIII VIII JORNADAS CHILENAS JORNADAS CHILENAS JORNADAS ... · Palabras claves: Enantioselectividad,...
25
VIII VIII VIII VIII JORNADAS CHILENAS JORNADAS CHILENAS JORNADAS CHILENAS JORNADAS CHILENAS DE CATÁLISIS Y ADSOR DE CATÁLISIS Y ADSOR DE CATÁLISIS Y ADSOR DE CATÁLISIS Y ADSORCIÓN CIÓN CIÓN CIÓN LIBRO DE RESÚMENES Organiza DIVISION DE CATÁLISIS Y ADSORCIÓN, SOCIEDAD CHILENA DE QUÍMICA 19 al 21 noviembre 2014, Olmué, Chile
Transcript of VIII VIII JORNADAS CHILENAS JORNADAS CHILENAS JORNADAS ... · Palabras claves: Enantioselectividad,...

VIII VIII VIII VIII JORNADAS CHILENAS JORNADAS CHILENAS JORNADAS CHILENAS JORNADAS CHILENAS
DE CATÁLISIS Y ADSORDE CATÁLISIS Y ADSORDE CATÁLISIS Y ADSORDE CATÁLISIS Y ADSORCIÓNCIÓNCIÓNCIÓN
LIBRO DE RESÚMENES
Organiza
DIVISION DE CATÁLISIS Y ADSORCIÓN, SOCIEDAD CHILENA DE QUÍMICA
19 al 21 noviembre 2014,
Olmué, Chile

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile II
EDITORIAL
Nos hemos reunido en Olmué, Región de Valparaíso, para realizar las VIII Jornadas
Chilenas de Catálisis y Adsorción, evento que se viene realizando en forma ininterrumpida
cada dos años en el mes de noviembre. El año 2000 en Concepción, el 2002 en Santiago, el
2004 en Talca, el 2006 en Santiago, el 2008 en Talca, el 2010 en Concón y el 2012 en
Termas de Catillo. El objetivo de este encuentro es fomentar el intercambio de ideas,
crear nuevos vínculos, fortalecer los existentes así como fomentar lazos de amistad.
Agradecemos profundamente a nuestros conferencistas Profesores Carmen Claver,
Andrea Beltramone y Pedro Ávila por haber aceptado nuestra invitación, por vuestra
colaboración académica y amistad. Hacemos extensivos los agradecimientos a los
integrantes del Comité Científico por la revisión y sugerencias para mejorar los trabajos
presentados.
En esta oportunidad hemos aceptado 31 trabajos de investigadores chilenos y españoles,
un aumento respecto a los 26 presentados en el evento anterior, lo que representa un
mayor interés y compromiso de los investigadores chilenos en desarrollar la Catálisis en
nuestro país.
Esperamos que disfruten estas jornadas y agradecemos vuestra activa participación.
Dr. Nestor Escalona B
Presidente Comité Organizador Dra. Gina Pecchi S
Presidenta Comité Científico

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile III
COMITÉ ORGANIZADOR
Dr. Néstor Escalona (Presidente)
Dr. Patricio Baeza (Vice-presidente)
Dr. Romel Jiménez
(Tesorero)
Dr. Gonzalo Aguila (Secretario)
COMITÉ CIENTÍFICO
Dra. Gina Pecchi (Presidente)
Dr. Pedro Aguirre
Dr. Rafael García
Dr. Juan Ojeda
Dr. Patricio Reyes
Dr. René Rojas
Dra. Mirza Villarroel
Dra. Lorena Wilson
EDICIÓN
Mag Sc Robinson Dinamarca
ORGANIZA
División de Catálisis y Adsorción, Sociedad Chilena de Química

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile IV
AUSPICIADORES

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile V
CONFERENCISTAS INVITADOS
Plenaria 1.
Dra. Carmen Claver. Universitat Rovira i Virgili, Terragona, España.
Estrategias para la Química Sustentable. Catalizadores inmovilizados y nanocatalizadores.
Plenaria 2.
Dra. Andrea Beltramone. Universidad Tecnológica Nacional Córdoba, Córdoba, Argentina.
Mejoramiento de Diesel utilizando catalizadores mesoporosos.
Plenaria 3.
Dr. Pedro Ávila. Instituto de Catálisis y Petroleoquímica, Madrid, España.
La migración electroforética como herramienta en la preparación y caracterización de
catalizadores.

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile VI
PROGRAMA
Horario Miércoles 19 Jueves 20 Viernes 21 08:00 - 08:20 R Morales B Aranda
08:20 - 08:40 E Camú C Rivera
08:40 - 09:00 K Letelier R Rojas
09:00 - 09:20 P Araya R Bassi
09:20 - 09:40 R Portela D Salinas
09:40 - 10:00 M Romero Saez S Guerrero
10:00 - 10:15 CAFÉ CAFÉ
10:15 - 10:35 O Trofymchuck H Oliva
10:35 - 10:55 R Villanueva C Matus
10:55 - 11:15 Y Beltrán K Leiva
11:15 - 11:35 L Delgado R Dinamarca
11:35 - 11:55 A Sánchez A Dongil
11:55 - 12:55 PLENARIA 2 PLENARIA 3
13.00 – 14:30 ALMUERZO CLAUSURA/ALMUERZO
14:30 - 14:50 D Osorio
14:50 - 15:10 T Celis
15:10 - 15:30 R Barton
15:30 - 15:50 A Mera
15:50 - 16:00 CAFÉ
16:00 - 16:20 S Alejandro
16:20 - 16:40 C Herrera
16:40 - 17:00 C Valdés
17:00 - 17:20 Inscripciones Presentación
INAUGURACION Empresas
18:00 - 19:00 PLENARIA 1
19:00 -19:20 M Ávila
19:20 – 19:40 C Álvarez
21:00 CENA CENA

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile VII
PRESENTADORES EN SALA
Horario Miércoles 19 Jueves 20 Viernes 21
08:00 - 10:00 Rafael García Francisco Javier Gil
10:15 - 11:55 Pedro Aguirre Doris Ruiz
11:55 - 12:55 Gina Pecchi Sichem Guerrero
14:30 - 15:50
René Rojas
16:05 - 17:45
Daniela Salinas
18:00- 19:00 Doris Ruiz

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile VIII
Miércoles 19 noviembre 2014
17:00 - 17:45 Inscripciones y entrega de material
17:45 - 18:00 Inauguración
18:00 - 19:00 ESTRATEGIAS PARA LA QUÍMICA SUSTENTABLE. CATALIZADORES INMOVILIZADOS Y NANOCATALIZADORES Carmen Claver
19:00 -19:20 HIDROGENACÍON DE α-CETOÉSTERES SOBRE 1%Pt(DIOP)/SiO2. EFECTO DE LAS CONDICIONES DE REACCIÓN Mauricio Ávila, Claudio Mella, Patricio Reyes, Doris Ruiz
19:20 – 19:40 CONVERSIÓN DE GUAIACOL SOBRE CATALIZADORES DE Re SOPORTADOS EN Al2O3 MODIFICADO CON CeO2 Claudia Alvarez, C. Sepúlveda, R. Garcia, JLG. Fierro, N. Escalona
20:30 CENA

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile IX
Jueves 20 noviembre 2014
08:00 - 08:20 NANOPARTÍCULAS DE Ni-MODIFICADAS COMO CATALIZADORES EN LA REACCION DE HIDROGENACIÓN DE XILOSA Ruddy Morales, Gina Pecchi, Marco Fraga
08:20 - 08:40 ESTUDIO DE LA DESNITROGENACIÓN DE PIRIDINA POR ADSORCIÓN SOBRE Ni(x)/Al2O3 Esteban Camú Cecilia Peralta, Mirza Villarroel, Juan Ojeda, Patricio Baeza
08:40 - 09:00 ESTUDIO CATALÍTICO DE NUEVOS COMPLEJOS DE Ni(II) CONTENIENDO LIGANDOS PNP Y PN Karina Letelier, Catalina Pérez-Zúñiga, Pedro Aguirre , Sergio A Moya
09:00 - 09:20 ACTIVIDAD DE CATALIZADORES Cu-Ce SOPORTADOS EN LA REACCIÓN WATER-GAS SHIFT.EFECTO DE LA ESTRUCTURA CRISTALINA DEL SOPORTE ZrO2 Paulo Araya, Gonzalo Aguila, Sichem Guerrero
09:20 - 09:40 HÍBRIDOS DE ARCILLA NATURAL Y TIO2 PARA FOTOCATÁLISIS Raquel Portela, Ingrid Jansson, Silvia Suárez, Mirza Villarroel, Benigno Sánchez, Pedro Ávila
09:40 - 10:00 ELIMINACIÓN DE COMPUESTOS ORGÁNICOS VOLÁTILES CLORADOS MEDIANTE OXIDACIÓN CATALÍTICA SOBRE Fe-ZEOLITAS Manuel Romero-Sáez, D Divakar, A Aranzabal, JR González-Velasco, JA González-Marcos
10:00 - 10:15 CAFÉ
10:15 - 10:35 EL DESEMPEÑO DE COMPLEJOS METILALIL NIQUEL Y SUS ADUCTOS DE BORO EN LA ACTIVACIÓN DE ETILENO: UNA PERSPECTIVA DE TFD CONCEPTUAL Oleksandra Trofymchuk, Daniela E Ortega, Soledad Gutiérrez-Oliva, René S Rojas, Alejandro Toro-Labbé
10:35 - 10:55 ESTUDIO DEL SISTEMA Co Mo SOPORTADO EN γ-Al2O3, CONFORMADO EN EXTRUIDOS Y MONOLITOS DE SEPIOLITA, PARA LA HDS DE GASOIL Regina Villanueva, Patricio Baeza, Mirza Villarroel, Francisco Gil-Lambías, Pedro Ávila
10:55 - 11:15 INFLUENCIA DE LA SUSTITUCIÓN DE LANTANO POR CERIO EN LA ACTIVIDAD PARA EL REFORMADO SECO DE METANO Yayne Beltrán, Ruddy Morales, Gina Pecchi, Romel Jiménez
11:15 - 11:35 HIDROGENÓLISIS DE GLICEROL SOBRE CATALIZADORES DE Cu/SiO2. Luna Delgado, Karen Cruces, Catherine Sepúlveda, Néstor Escalona, Rafael García
11:35 - 11:55 EFECTO DE LA ADICIÓN DE P(C6H5)3 Y Na3(C6H5O7) EN LA SÍNTESIS DE NANO PARTÍCULAS DE Pd COMO CATALIZADORES DE HIDROGENACIÓN Ariel Sánchez, J Luis García Fierro, Patricio Reyes, Doris Ruiz
11:55 - 12:55 MEJORAMIENTO DE DIESEL UTILIZANDO CATALIZADORES MESOPOROSOS Andrea Beltramone
13.00 – 14:30 ALMUERZO
14:30 - 14:50 SÍNTESIS Y CARACTERIZACIÓN DE ADUCTOS BISAMIDÍNICOS CON ÁCIDOS DE LEWIS Y SU UTILIZACIÓN EN LA ACTIVACIÓN CATALÍTICA DE CO

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile X
Danay Osorio Meléndez, Alexandra Becerra Agudelo, René Rojas Guerrero
14:50 - 15:10 CATALIZADORES HETEROGÉNEOS SOPORTADOS PARA LA PRODUCCIÓN DE BIODIESEL. EFECTO DEL TIPO DE METAL ALCALINO, TEMPERATURA DE CALCINACIÓN, Y DEL TIPO DE SOPORTE Tania Celis, Giuseppe Baeza, Gonzalo Aguila, Paulo Araya, Sichem Guerrero
15:10 - 15:30 THE EFFECT OF NICKEL LOADING ON THE HYDRODEOXYGENATION OF GUAIACOL, CATALYZED BY NI/HZSM-5 SYNTHESIZED BY DEPOSITION-PRECIPITATION Ryan Barton, Nestor Escalona, Rafael García, Marion Carrier, Cristina Segura, Steven Peretti
15:30 - 15:50 MICROESFERAS DE BiOI PARA LA DEGRADACIÓN FOTOCATALÍTICA DE ÁCIDOS HIDROXIBENZOICOS Adriana Mera, David Contreras, Hector D Mansilla
15:50 - 16:05 CAFÉ
16:00- 16:20 MODELACIÓN DE LA ADSORCIÓN DE TOLUENO SOBRE ZEOLITA: EFECTO DE LA COMPOSICIÓN DE LOS CATIONES DE COMPENSACIÓN Serguei Alejandro, Héctor Valdés, Marie-Helene Manero, Claudio A Zaror
16:20 - 16:40 ADSORCIÓN DE CO2(g) EN CARBONES ACTIVADOS: EFECTO DE LOS GRUPOS FUNCIONALES SUPERFICIALES Carla Herrera, Rafael García, Catherine Sepúlveda, Néstor Escalona
16:40 - 17:00 EFECTO DE LOS RADICALES EN LA DESCOMPOSICIÓN DE FENOL EN PRESENCIA DE H2O2 Y ÓXIDO DE MANGANESO Cristian Valdés, Carlos Navarro, Claudia Campos, Jorge Villaseñor
20:30 CENA

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile XI
Viernes 21 noviembre 2014
08:00 - 08:20 NUEVOS COMPUESTOS DE Pd(II) CONTENIENDO LIGANDOS P^N Y SU USO EN AMINOCARBONILACIÓN DE ARILHALUROS Braulio Aranda, Pedro Aguirre
08:20 - 08:40 HIDROGENACIÓN SELECTIVA DE p-CLORONITROBENCENO CON CATALIZADORES DE Pd SOPORTADO SOBRE MATERIALES DE CARBONO Camila Rivera, AB Dongil, D Ruiz, JLG Fierro, P Reyes
08:40 - 09:00 ACTIVACIÓN REMOTA DE COMPLEJOS DE NÍQUEL CON ÁCIDOS DE LEWIS, USO EN POLIMERIZACIÓN DE ETILENO René Rojas, OleksandraTrofymchu, Manuel Escobar
09:00 - 09:20 EFECTO DEL SOPORTE EN CATALIZADORES DE RENIO EN LA HIDRODENITROGENA CIÓN DE QUINOLINA Romina Bassi, Mirza Villarroel, Néstor Escalona, José Luis García Fierro, Catalina Pérez, Patricio Baeza
09:20 - 09:40 EFECTO DEL CONTENIDO DE POTASIO Y NATURALEZA DEL SOPORTE EN LA REACCION DE COMBUSTIÓN DE NEGRO DE HUMO Daniela Salinas, Gina Pecchi, Ximena García
09:40 - 10:00 ADSORCION DE OXIDO NITRICO EN CATALIZADORES DE M/Cu/(Y, Gd, Li)CeO2
(M=Na, K, Rb, Li)
Sichem Guerrero, Gonzalo Águila, Paulo Araya
10:00 - 10:15 CAFE
10:15 - 10:35 EFECTO DEL METAL EN LA HIDROGENACIÓN DE CETOPANTOLACTONA SOBRE NANOPARTÍCULAS EN SiO2 Héctor Oliva, Ariel Sánchez, Mauricio Ávila, Claudio Mella, Doris Ruiz
10:35 - 10:55 ESTUDIO DE LA ELIMINACIÓN DE 4 – NITROFENOL EN MATRIZ ACUOSA, MEDIANTE ADSORCIÓN SOBRE CARBON ACTIVO Camila Matus, Esteban Camú, Mirza Villarroel, Juan Ojeda, Patricio Baeza
10:55 - 11:15 EFECTO INHIBITORIO DE MOLECUAS REPRESENTATIVAS DEL BIO-OIL EN LA CONVERSION DEL 2-METOXIFENOL SOBRE EL CATALIZADOR ReOx/SiO2 Katherine Leiva, Catherine Sepulveda, Rafael García, Nestor Escalona
11:15 - 11:35 MANGANITAS DE LANTANO SUSTITUIDAS EN EL SITIO B PARA SER UTILIZADAS COMO CATALIZADORES EN LA REACCION DE COMBUSTIÓN DEL DIMETIL ÉTER Robinson Dinamarca, Eduardo J Delgado, Octavio Peña, Gina Pecchi
11:35 - 11:55 SÍNTESIS DE NANOCOMPOSITES Ni/CeO2/grafeno: ESTUDIO COMPORTAMIENTO CATALÍTICO EN LA REACCIÓN DESPLAZAMIENTO DE GAS DE AGUA Ana Belen Dongil, L Pastor-Pérez, A Sepúlveda-Escribano, P Reyes
11:55 - 12:55 LA MIGRACIÓN ELECTROFORÉTICA COMO HERRAMIENTA EN LA PREPARACIÓN Y CARACTERIZACIÓN DE CATALIZADORES. Pedro Avila
13:00 – 13:15 Clausura
13:15 – 14:30 ALMUERZO

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile 1
HIDROGENACÍON DE -CETOÉSTERES SOBRE 1%Pt(DIOP)/SiO2. EFECTO DE LAS CONDICIONES DE REACCIÓN.
Mauricio Ávila, Claudio Mella, Patricio Reyes, Doris Ruiz*.
Facultad de Ciencias Químicas, Universidad de Concepción, Concepción, Chile, 4070371.
Email: [email protected], [email protected].
Palabras claves: Enantioselectividad, -cetoésteres, Nanopartículas
INTRODUCCIÓN
La creciente demanda de productos quirales principalmente por la industria farmacéutica ha
hecho de la catálisis una de las disciplinas más estudiadas en los últimos años.
Considerando la importancia de catalizadores en síntesis de productos enantioméricamente
puros se ha estudiado la síntesis de catalizadores de Pt mediante la reducción in situ del
precursor [Pt2(dba)3] en presencia de (-)DIOP que permite la obtención de nanopartículas
(NPs) de superficies quirales con bajo diámetro metálico. El catalizador 1%Pt(DIOP)/SiO2
fue utilizado en la hidrogenación enantioselectiva de los sustratos en figura 1: piruvato de
etilo (PE), cetopantolactona (CP), benzoilformiato de etilo (BFE) y benzoilformiato de
metilo (BFM). Un estudio adicional permitió evaluar el efecto de las condiciones de
reacción en la actividad y selectividad de CP modificando el solvente, masa de catalizador,
concentración de sustrato y presión de H2.
Figura 1. Sustratos de hidrogenación: CP, PE, BFM y BFE.
EXPERIMENTAL Las nanopartículas quirales de Pt se sintetizaron en un reactor batch recubierto de vidrio en
40 mL de THF y atmósfera inerte desde una mezcla de 150 [mg] (0,137 [mmol]) de
[Pt2(dba)3] con (-)DIOP en relación 10Pt: 1DIOP. Posteriormente se mantuvo en agitación
durante 20 h a 3 bar de H2. Las NPs limpias y secas se impregnaron en SiO2 al 1% de Pt.
El catalizador 1%Pt(DIOP)/SiO2 fue caracterizado por Adsorción-Desorción de N2 a 77 K,
Difracción de Rayos X de Polvo (DRX), Termogravimetría (TG), Microscopía Electrónica
de Transmisión (TEM), absorción atómica y Espectroscopía Fotoelectrónica de rayos X
(XPS). Los ensayos catalíticos se realizaron a 298 K, en un reactor batch recubierto de
vidrio. Los solventes utilizados fueron 2-propanol, tolueno, metanol y THF, la relación
sustrato/ metal varió de 100 a 2000 y la presión de H2 de 10 a 40 bar. Las condiciones en
los primeros ensayos fueron: 100 mg de catalizador con relación sustrato/metal de 100:1 y
50 mL de ciclohexano como solvente. El análisis se efectuó en un equipo GCMS clarus 680
acoplado a SQ8T-headspace.

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile 2
RESULTADOS Y DISCUSIÓN
Los resultados obtenidos por TEM, DRX, XPS y TG mostraron que el método de síntesis
resulta efectivo en la obtención del catalizador a partir de NPs quirales estabilizadas. El
diámetro metálico fue de 1.7 nm con elevado contenido de metal en superficie. El
catalizador 1%Pt(DIOP)/SiO2 generó a los pocos minutos de reacción la conversión total de
los sustratos PE y CP con excesos enantioméricos (ee) promedios del 11%. Los sustratos
BFE y BFM mostraron conversiones menores al 18% en iguales condiciones. El estudio
del efecto del solvente permite observar la influencia de la naturaleza polar de cada
disolvente en la actividad del catalizador. Al aumentar la actividad en CP disminuye
drásticamente la enantioselectividad. El estudio de la masa del catalizador permitió
establecer los rangos óptimos de cantidad de catalizador en que sólo se evaluaban las etapas
químicas del proceso, encontrando una masa óptima de catalizador de 50 [mg]
de1%Pt(DIOP)/SiO2. El efecto de la concentración del sustrato, permitió evaluar el
comportamiento del catalizador en la reacción determinando que la concentración óptima
de sustrato es 0.020 [mol/L] de CP.
Mediante el estudio del efecto de la presión
de H2 se logró determinar que la velocidad
de la reacción de CP prácticamente no
depende de la presión y que a 10 [bar] los
resultados son promisorios en cuanto a
actividad y selectividad. Respecto a la
utilización de (-)DIOP como agente de
inducción quiral, no se alcanzó elevada
enantioselectividad probablemente debido a
la conformación inadecuada en superficie
del ligando. Los estudios de reciclo
muestran una leve disminución de la
actividad catalítica en comparación a una
reacción con catalizador fresco, sin embargo
es posible asumir una alta estabilidad de la
fase metálica sobre el soporte.
CONCLUSIONES
Los sistemas catalíticos ensayados presentan conversiones del 100% en los primeros
minutos de reacción de CP. El estudio de las condiciones de reacción en el sustrato
cetopantolactona permitió concluir que las condiciones óptimas de reacción para este
sistema son 50 [mg] de catalizador, 40 [bar] H2, 0.020 [mol/L] de CP y utilizando tolueno
como disolvente. El incremento de la actividad se relaciona directamente con el aumento de
la presión de H2, la polaridad del solvente y con la disminución de moléculas de sustrato en
el medio. Contrario a esto, la enantioselectividad disminuye a medida que aumenta la
actividad.
AGRADECIMIENTOS Proyecto FONDECYT INICIACION 11121631.
REFERENCIAS
[1] G. Martin, P. Maki-Arvela, D. Yu Murzin, T. Salmi, Catal. Lett. 143, (2013) 1051-1060.
[2] C. Torres, C. Campos, J.L G. Fierro, P. Reyes, D. Ruiz, J. Mol. Catal.A: 392, (2014) 321–328.
0
10
20
30
40
50
60
70
80
90
100
0 20 40 60 80 100
% C
on
versi
ón
Tiempo [min]
10 bar
20 bar
30 bar
40 bar
Figura 2. Conversión vs. Tiempo de CP a diferentes presiones de H2
con 50 [mg] de catalizador, 0.010 [mol/L] de CP a 25 [ C] y 800 [rpm].
0
10
20
30
40
50
60
70
80
90
100
0 20 40 60 80 100
% C
on
vers
ión
Tiempo [min]
10 bar
20 bar
30 bar
40 bar
Ln P
-Ln
K

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile 3
CONVERSIÓN DE GUAIACOL SOBRE CATALIZADORES DE Re
SOPORTADOS EN Al2O3 MODIFICADO CON CeO2
Claudia Alvarez, C. Sepúlvedaa, R. Garciaa, J. L. G. Fierrob, N. Escalonaa
aUniversidad de Concepción, Facultad de Ciencias Químicas, Casillas 160c, Concepción, Chile.
b Instituto de Catálisis y Petroleoquímica, CSIC, Cantoblanco, 28049 Madrid, España.
Palabras claves: guaiacol, hidrotratamiento, Renio.
INTRODUCCIÓN
Debido a la alta demanda del petróleo y a su continuo incremento en su valor, la biomasa
ha tomado fuerza en las últimas décadas como alternativa para la obtención de productos
químicos. Uno de los procesos de trasformación de la biomasa es la pirólisis rápida, en la
cual se obtiene el bio-oil como producto mayoritario. Sin embargo, el bio-oil presenta altos
contenidos de compuestos oxigenados que debe ser removido. Un proceso que puede
mejorar la propiedades del bio-oil es la hidrodesoxigenación catalítica (HDO) [1]. Diferentes
catalizadores han sido estudios en las reacciones de HDO[2]. Los catalizadores basados en
Re han presentado alta actividad en la conversión del 2-metoxifenol (guaiacol), como
molécula modelo del bio-oil. Por otro lado, la modificación de las propiedades ácido-base
del soporte, por adición de un sólido con características básicas puede proporcionar una
mejor actividad y favorecer la selectividad hacia la formación de productos de HDO [2]. Por
este motivo, en este trabajo se estudiaron dos fases activas de Re soportados sobre γ-Al2O3
modificado con CeO2 para la reacción de conversión del guaiacol.
EXPERIMENTAL
Distintas cargas metálicas de CeO2 (5- 18%) fueron impregnadas sobre la γ-Al2O3 mediante
exceso de solvente. Las muestras fueron secadas y calcinadas a 500 ºC por 4 horas. Para el
soporte γ-Al2O3 con mayor recubrimiento de CeO2 se impregnaron distintas cantidades de
Re (5 - 25%) mediante impregnación húmeda. La activación de los catalizadores en estado
oxidado se realizó a 400°C por 4 h. La actividad catalítica de los catalizadores en estado
oxidado y reducido se realizó en un reactor batch a 300°C y 5 MPa de presión de H2. Se
tomaron muestras periódicamente durante 4 h y se analizaron por GC-FID. Los
catalizadores fueron caracterizados por adsorción-desorción de N2, TPR, quimisorción de
CO, DRX, TEM, XPS, migración electroforética (ME) y TPR-S.
RESULTADOS Y DISCUSIÓN
Mediante ME se encontró que con un 14% de CeO2 se alcanza el máximo recubrimiento
del soporte. La superficie específica y el volumen de poro del soporte disminuyen
gradualmente a medida que aumenta el contenido de Re, sugiriendo que el Re se deposita
de forma homogénea. Por otro lado, mediante TPR de los catalizadores se observó un pico
de reducción muy intenso a 350°C, el cual fue asignado a la reducción del óxido de Re+7 a
Re0. Los resultados de DRX de los catalizadores de ReOx(x)/CeO2-Al2O3 muestran que
bajo el 15 % de Re no se observa planos de difracción asignadas algunas especie de Re,
sugiriendo una alta dispersión de las especies de Re. Por el contrario, sobre el 15 % de Re
se observa especies cristalinas de Re asignadas a ReO4-, que aumentan su intensidad a
medida que aumenta el contenido de Re. Los resultados de conversión en función del
tiempo sobre los catalizadores de ReOx soportados sobre CeO2-Al2O3 presentaron, a un 10

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile 4
% de conversión, a fenol como principal producto y a ciclohexano, benceno y anisol en
menor cantidad. Por el contrario, los catalizadores metálicos presentaron a fenol como
principal producto y catecol, metifenol y anisol en menor cantidad, a un 10 % de
conversión. Estos resultados muestran que la conversión del guaiacol sobre estos
catalizadores sigue un mecanismo de reacción diferente. En la Figura 1 se resumen las
velocidades iniciales de desaparición de guaiacol de los catalizadores oxidados y reducidos
en función del contenido metálico.
Fig.1: Velocidades iniciales de conversión guaiacol sobre los catalizadores de Re/CeO2-Al2O3 y ReOx/CeO2-Al2O3.
La Fig. 1 muestra que los catalizadores en fase oxidada en la reacción de conversión de
guaiacol presento una mayor actividad catalítica que los catalizadores metálicos. Esta figura
también muestra, que la velocidad inicial de los catalizadores en fase oxidada va
aumentando a medida que aumenta el contenido de Re. Por el contrario, en los
catalizadores metálicos se observa un máximo de actividad al 10% de Renio y luego
disminuye gradualmente. Esta tendencia puede ser correlacionada con la dispersión de la
fase activa sobre el soporte. Medidas de XPS, quimisorción de CO y TEM confirmaran esta
tendencia.
CONCLUSIONES
El CeO2 modifico la acidez del soporte disminuyendo la formación de catecol en
comparación a los catalizadores sin modificar. El catalizador de ReOx presento una mayor
actividad catalítica que el catalizador de Re metálico. La distribución de los productos se
modificó con la adición de CeO2, sugiriendo que el mecanismo de reacción de conversión
de guaiacol es diferente en ambos catalizadores.
AGRADECIMIENTO Proyecto FONDECYT 1100512.
REFERENCIAS [1] A.V. Bridgwater, Biomass and Bioenergy 38 (2012)68.
[2] C. Sepúlveda, R. Garcia, N. Escalona, D. Laurenti, L. Massin, M. Vrinat. Catal. Lett. 141(2011) 987.
0 5 10 15 20 25 30
0
10
20
30
Vi *1
0-6 (m
olg
-1s-1)
Vi*1
0-6 (
mo
lg-1s
-1)
Contenido metalico de Re (%)
ReOx/CeO2-Al
2O
3
Re/CeO2-Al
2O
3
0
2
4
6
8
10

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile 5
NANOPARTÍCULAS DE Ni-MODIFICADAS COMO CATALIZADORES EN LA
REACCION DE HIDROGENACIÓN DE XILOSA
Ruddy Moralesa, Gina Pecchia, Marco Fragab
aDepartamento de Fisico-Quimica, Facultad de Ciencias Químicas, Universidad de Concepción, Concepción(Chile),
bDivision de catálisis, Instituto Nacional de Tecnologia, Rio do Janeiro(Brasil).
Palabras clave: Níquel, hidrogenación, xilosa
INTRODUCCIÓN
La reducción catalítica selectiva de azucares naturales a sus respectivos alcoholes es una
manera amigable de producir endulzantes alternativos. El xilitol se destaca por sus
propiedades como endulzante y tanto su importancia como su producción a nivel mundial
ha ido en aumento[1-3]. La hidrogenación de xilosa se lleva a cabo principalmente en
reactores batch sobre catalizadores de Raney níquel finamente dispersos. Se ha determinado
previamente que los catalizadores de Ni son más activos que la mayoría de los metales
nobles en esta reacción[2, 4]. Las principales ventajas de estos catalizadores son su bajo
costo y alta actividad y selectividad, sin embargo, la mayor desventaja de los catalizadores
de Raney níquel es su desactivación y perdida de carga metálica. En este trabajo,
perovskitas parcialmente sustituidas con Ni fueron usadas como precursores para producir
nanopartículas de Ni en un ambiente químico modificado para ser usadas en la reacción de
hidrogenación de xilosa a xilitol.
EXPERIMENTAL
Se sintetizaron óxidos mixtos de fórmula La1-XCeXAl0,18Ni0,82O3 (x=0.0, 0.1, 0.5 y 0.7), con
carga de níquel correspondiente a 20% en masa. Los sólidos se sintetizaron por el método
de autocombustión[5], a partir de los nitratos metálicos y glicina y luego fueron calcinados
a 973K. Los sólidos fueron caracterizados por difracción de rayos X (DRX), reducción
térmica programada (TPR), adsorción de N2 a 77K, microscopia electrónica de transmisión
(TEM) y espectroscopia de absorción atómica (AAS). La actividad catalítica se evaluó en la
reacción de hidrogenación de xilosa a xilitol, utilizando catalizadores previamente
reducidos en flujo de H2 a 773K. La reacción se llevó a cabo en un reactor batch trifásico,
usando 1g de xilosa en un volumen de 80mL de agua y 100mg de catalizador, a 373K y
20bar de H2 por 6h. La reacción se monitoreo tomando alícuotas periódicamente. Los
catalizadores se caracterizaron luego de la reducción y luego de la reacción en fase liquida;
los sólidos reducidos fueron inmediatamente pasivados bajo un flujo de O2 diluido a 243K.
RESULTADOS Y DISCUSIÓN Los resultados de DRX caracterización muestran
comportamientos diferentes según alto (xCe=0.5, 0.7) o bajo (xCe=0.0, 0.1) contenido de Ce.
Los resultados de DRX (Fig. 1) muestran que el sólido sin Ce presenta estructura tipo
perovskita (JCPDF 33-0711), mientras con el aumento en la sustitución por Ce aparecen
fases segregadas gradualmente. Para altos grados de sustitución, la estructura perovskita no
esta presente y solo una solución solida de óxidos de Ce-La es encontrada. Los
catalizadores reducidos muestran poco, o nada, de cambio en la estructura y las fases
presentes. Los resultados de TPR muestran solo dos picos de reducción correspondientes a
la reducción del Ni. Las micrografías TEM muestran que el Ni se encuentra disperso en el
soporte oxido sin formar nanopartículas luego de la reducción. Los resultados de actividad

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile 6
catalítica muestran que todos los catalizadores son activos en la reacción de hidrogenación
de xilosa a xilitol como se revela en las curvas de conversión (Fig. 2). Los catalizadores con
bajo contenido de Ce (xCe=0.0, 0.1) muestran actividad catalítica más alta, apuntando a la
importancia de la estructura perovskita. En cuanto a la selectividad hacia xilitol, esta se
encuentra entre 40 y 60% en condiciones de isoconversión (~30%). Otros productos son
principalmente productos de hidrogenólisis, como glicerol, acetol y etilén glicol. Los
resultados de AAS antes y después de las pruebas catalíticas indican para los sólidos con
bajo grado de sustitución (xCe=0.0, 0.1) una carga de Ni constante y cercana a la
estequiométrica indicando alta estabilidad a reacciones en medio acuoso. Mientras que los
sólidos con grados de sustitución más altos (xCe=0.5, 0.7) muestran perdida de Ni por
leaching luego de la prueba catalítica.
10 20 30 40 50 60 70 80 90
Ni20% xCe
=0.7 700°C
Ni20% xCe
=0.5 700°C
Ni20% xCe
=0.1 700°C
Ni20% xCe
=0.0 700°C
in
ten
sity (
a.u
.)
20 50 100 150 200 250 300 350 400
0
20
40
60
80
100
Ni20% xCe
=0.0
Ni20% xCe
=0.1
Co
nve
rsio
n (
%)
tiempo (min)
Ni20% xCe
=0.5
Ni20% xCe
=0.7
Figura 1: DRX de los sólidos calcinados a 700°C Figura 2: curvas de conversión en la reacción de
hidrogenación de xilosa
CONCLUSIONES Se obtuvieron sólidos con estructura perovskita, los cuales fueron
utilizados como precursores para la formación de nanopartículas de Ni. Los catalizadores
utilizados presentaron alta estabilidad durante la reacción de hidrogenación de xilosa en
medio acuoso y alta actividad comparado con los catalizadores que se utilizan de forma
industrial. Además presentan características que dan la posibilidad de ser utilizadas en
diferentes procesos y reacciones catalíticas. La estructura de tipo perovskita permite la
estabilización delníquel impidiendo su carácter pirofórico y por lo tanto manteniendo el
carácter metálico de las partículas, las cuales se mantienen en tamaños muy pequeños.
AGRADECIMIENTOS Proyecto FONDECYT 1130005 y beca CONICYT 21130572
REFERENCIAS [1] J.-P. Mikkola, T. Salmi, R. Sjöholm, J. Chem Techn & Biotec 74 (1999) 655-662.
[2] J. Wisniak, M. Hershkowitz, R. Leibowitz, S. Stein, Product R&D 13 (1974) 75-79.
[3] M. Yadav, D.K. Mishra, J.-S. Hwang, Applied Catalysis A: General 425–426 (2012) 110-116.
[4] J. Wisniak, M. Hershkowitz, S. Stein, Product R&D 13 (1974) 232-236.
[5] L.A. Chick, L.R. Pederson, G.D. Maupin, J.L. Bates, L.E. Thomas, G.J. Exarhos, Materials Letters
10 (1990) 6-12.

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile 7
ESTUDIO DE LA DESNITROGENACIÓN DE PIRIDINA POR ADSORCIÓN
SOBRE Ni(x)/-Al2O3.
Esteban Camúa, Cecilia Peraltaa, Mirza Villaroelb, Juan Ojedac, Patricio Baezaa.
a Pontificia Universidad Católica de Valparaíso, Valparaíso, Chile, Casilla 4059.
b Universidad de Santiago, Facultad de Química y Biología, Casilla 40, Correo 33, Santiago c Universidad de Valparaíso, Valparaíso, Casilla 92v, Chile
Palabras claves: Adsorción, piridina, desnitrogenación.
INTRODUCCIÓN Uno de los inconvenientes de los procesos de hidrodesnitrogenación (HDN), es la
incapacidad de eliminar compuestos de nitrógeno refractarios [1]. Para poder eliminar estas
moléculas se necesitan condiciones severas de tratamiento, sin embargo esto conlleva una
disminución en el índice de octanos (gasoil) y cetanos (diésel), es por esto que se hace
necesario desarrollar un método que permita reducir los costos para la obtención de un
combustible sin impurezas [2]. En el proceso de adsorción mediante π complejación los
cationes metálicos pueden captar los electrones π enlazantes de la molécula con nitrógeno
depositándolo en su orbital s vacío y, además, sus orbitales d ceden la densidad de
electrones a los π* de los orbitales p de los anillos aromáticos de la molécula con nitrógeno
[3,4]. Uno de los cationes que deberían tener considerables capacidades de adsorción de
este tipo de moléculas corresponde al Ni2+, cuya configuración es [Ar] 3d8 4s0, implicando
una adsorción planar, facilitando la retención de estas moléculas refractarias al proceso de
HDN [5]. Teniendo en cuenta lo mencionado anteriormente, el objetivo de este trabajo es
estudiar la capacidad de adsorción (C.A.) mediante π complejación de un sistema
compuesto por níquel, a distintas concentraciones, soportado en -Al2O3 para la adsorción
de piridina (Py), los que son refractarios al proceso de HDN.
EXPERIMENTAL
Las muestras de Ni(x) (x= 2, 4, 6%) soportado en -Al2O3 se prepararon por impregnación
húmeda, posteriormente fueron secadas a 105°C por 12 h. Finalmente las muestras fueron
calcinadas a 500°C por 4 h. Las muestras fueron caracterizadas mediante adsorción –
desorción BET, acidez superficial y migración electroforética (ME). La determinación de la
adsorción de compuestos nitrogenados tales como Piridina (Py), en los distintos
adsorbentes estudiados, se realizó en un reactor pyrex de lecho fijo vertical, y en
condiciones como en estudios anteriores [6, 7]. Las muestras se analizaron, luego de ser
recolectadas en intervalos regulares de tiempo de 5 minutos, mediante GC-FID en un
cromatógrafo de gases marca Shimadzu GC-2010, hasta la saturación del adsorbente. La
capacidad de adsorción (C.A.) de los materiales estudiados, se determinó mediante el
cálculo del área bajo la curva de adsorción.
RESULTADOS Y DISCUSIÓN
Los resultados BET indican que existe una leve disminución en el área específica de la
muestra al impregnar con el metal, lo cual se debe a la deposición del metal en la superficie
de la -Al2O3. En la tabla 1 se muestra los resultados de caracterización de fuerza ácida y
migración electroforética para los distintos adsorbentes utilizados. Se observa que al
incorporar el metal, la fuerza ácida aumenta con el contenido metálico alcanzando un

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile 8
máximo al contenido de 4%, esto se debe principalmente a que a bajos contenidos se
forman aluminatos y la modificación de la estructura de la alúmina, provoca que aumente la
acidez de los grupos OH superficiales, mientras que a mayor contenido (6%) la presencia
de óxidos ha sido reportada previamente [8]. Además, se muestran los Puntos Isoeléctricos
(PIE), los Punto de Carga Cero (PCC) y la fracción de superficie cubierta (XM) de los
adsorbentes estudiados en este trabajo. En primer lugar, se observa que el PCC de las
muestras con Ni disminuye desde el PIE de la alúmina, debido a que al incorporar Ni a la
superficie de la alúmina hasta un 4%, se forma en primer lugar el aluminato, el cual posee
un PIE menor, al contenido superior (6%), se observa la formación de óxido de Ni sobre la
superficie debido al aumento del PCC, en ese caso no se calculó la XM, pues existen 3
especies, alúmina, aluminato y óxido. Se observa además que el Ni posee una mayor
fracción de superficie cubierta en sus composiciones de 2 y 4% aumentando con el
contenido de Ni, esto es relevante ya que se ha informado que las especies con una alta
capacidad de retrodonación son bien dispersas.
Los resultados de
capacidad de adsorción
(C.A.) muestran que aún
en ausencia de un metal,
la -Al2O3 permite la
adsorción de py, la cual
depende fuertemente de
su fuerza ácida. Al
incorporar al metal, se
observa que existe una
disminución en la C.A. debido probablemente a la disminución de la fuerza ácida por la
incorporación de Ni y la formación de aluminato. Luego a un 6%, la C.A. aumenta debido
probablemente a la presencia de NiO en la superficie aumentando la adsorción por un
fenómeno de retro donación electrónica [4]
CONCLUSIONES La incorporación de Ni en -Al2O3 provoca una disminución de la
capacidad de adsorción de piridina en este tipo de adsorbentes, debido probablemente a la
disminución en la fuerza ácida del soporte, lo cual se debe principalmente a la formación de
aluminato sobre la superficie. Esto se observa al aumentar la cantidad de Ni, hasta un
contenido de Ni de un 6%, a este contenido que es el mayor estudiado en este trabajo se
observa un leve aumento de la capacidad de adsorción de Py, relacionado a un aumento de
la cantidad de NiO superficial generando este aumento por retro-donación electrónica, entre
el adsorbato y el adsorbente.
AGRADECIMIENTOS Proyecto FONDECYT 1140808 y beca CONICYT 21130871.
REFERENCIAS [1] G.C. Laredo, S. Leyva, R. Alvarez, M.T. Mares, J. Castillo, J. Cano, Fuel, 81, 1341 (2002).[2] L. Qiao, J.
Wang, J. Hazard. Mater. 176, 220 (2010).[3] A.J. Hernandez-Maldonado, R.T. Yang, Catal. Rev. 46, 111
(2004).[4] R.T. Yang, A.J. Hernandez-Maldonado, F.H. Yang, Science 301, 79 (2003).[5] A.J. Hernandez-
Maldonado, R.T. Yang, Ind. Eng. Chem. Res. 42, 123 (2003).[6] P. Baeza, G. Aguila, F. Gracia, P. Araya.,
Catal. Commun. 9, 751 (2008).[7] P. Baeza, G Aguila, G. Vargas; J. Ojeda, P. Araya Appl. Catal. B, 133, 111
(2012).[8] F.J. Gil Llambias, A.M. Escudey Castro, J. Santos Blanco, J. Catal. 83, 225 (1983).
Tabla 1: Resultados de caracterización de adsorbentes estudiados
Adsorbente
Fuerza ácida ME C.A.
(mg Py g Ads-1) F.A.
(mV)
Sitios ácidos
(mEq·g-1)
PIE o
PCC XM
-Al2O3 -127,0 1,00 8,7 -- 22,13
Ni(2)/-Al2O3 -52,6 1,45 8,11 0,10 16,04
Ni(4)/-Al2O3 -6,8 1,34 6,80 0,33 14,58
Ni(6)/-Al2O3 -49,5 0,95 7,45 --- 16,06

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile 9
ESTUDIO CATALÍTICO DE NUEVOS COMPLEJOS DE Ni(II) CONTENIENDO
LIGANDOS PNP Y PN.
Karina Letelier a(*), Catalina Pérez-Zúñiga a, P. Aguirre b, S. A. Moya a. a) Departamento de Química de los Materiales, Facultad de Química y Biología, Universidad de Santiago
de Chile, Av. Libertador Bernardo O´Higgins 3363, Santiago, Chile.
b) Departamento de Química Inorgánica y Analítica, Facultad de Ciencias Químicas y Farmacéuticas,
Universidad de Chile, Casilla 233 Santiago 1. Santiago, Chile..
(*) E-mail: [email protected]
INTRODUCCIÓN
La hidrogenación catalítica juega un rol muy importante a nivel industrial para la
generación de productos en química fina [1]. El desarrollo de la química organometálica, ha
permitido la aplicación de muchos de estos compuestos como catalizadores en química
orgánica para la síntesis de compuestos orgánicos con elevados rendimientos [2].
Las reacciones de hidrogenación convencionales llevan implícito el uso del hidrógeno
como reactivo. Sin embargo, resulta posible que en presencia de un complejo metálico, una
molécula dadora (DH2) transfiera hidrógeno a un sustrato insaturado (por ejemplo una
amina) que actúa como aceptor. Frecuentemente las moléculas donantes que se utilizan son
ácido fórmico o 2-propanol, ésta última simultáneamente cumple el rol de solvente. En esta
reacción se han utilizado diferentes metales, tales como Ru y Pd [3], sin embargo
compuestos de níquel no han sido reportados como catalizadores para esta reacción.
EXPERIMENTAL
En este estudio catalítico se utilizaron nuevos catalizadores de níquel (II) con ligandos del
tipo PNP (PPh2NHpyNHPPh2) [4] y PN (PPh2NHpy)2
[5]. Los nuevos compuestos
[NiBr(PNP)]Br y [NiPPh3(PNP)2](CF3SO3)2 (Fig. 1) fueron caracterizados y empleados
como catalizadores en la reacción de transferencia de hidrógeno hacia el sustrato N-
bencilideanilina.
Fig. 1. Estructuras de los complejos de Níquel (II).
La reacción catalítica fue estudiada variando las condiciones de reacción (bases, relación
base catalizador y dador de hidrógeno) manteniendo constante la relación sustrato
/catalizador 200/1. La actividad catalítica fue analizada por GC-FID obteniéndose
diferentes resultados dependiendo de las condiciones utilizadas (Tabla 1) en la reacción de
transferencia de hidrógeno (Fig. 2) con ambos complejos de níquel (II).

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile 10
RESULTADOS Y DISCUSIÓN
Fig. 2. Reacción de hidrogenación de N-bencilidenalinina
% Conversión
[NiBr(PNP)]Br [Ni(Ph2P-NH-Py)2](CF3SO3)3
Horas
(h)
iPrOH/ter-
BuOK
HCOOH/Et3N
HCOOH
iPrOH/ter-
BuOK HCOOH/Et3N
HCOOH
0 9 75 53 6 63 75
1 10 90 86 12 75 90
2 11 93 100 9 75 93
3 11 91 100 8 76 93
4 18 87 100 4 80 93
5 2 87 100 4 85 93
Tabla 1. Resultados de las actividades catalíticas de los complejos [NiBr(PNP)]Br y [Ni(Ph2P-NH-
Py)2](CF3SO3)3, en la reacción de transferencia de hidrógeno del sustrato N-bencilideanilina.
Los catalizadores de níquel sintetizados en este trabajo representan una alternativa a los
metales ya estudiados debido al bajo costo de este metal, considerando que se obtiene
100% de actividad catalítica con ácido fórmico en condiciones suaves de temperatura. Con
ambos complejos de Ni(II) se observó un aumento gradual en las conversiones al disminuir
la presencia de base en el proceso catalítico, obteniéndose hasta un 100 % de conversión
para [NiBr(PNP)]Br y 93% para [Ni(Ph2P-NH-Py)2](CF3SO3)3 en ausencia total de base,
utilizando como dador de hidrógeno ácido fórmico. Los resultados muestran bajas
concentraciones de productos de hidrolisis en medio ácido y conversiones variables en
medio básico.
CONCLUSIONES
Los nuevos complejos de Níquel (II) conteniendo ligandos del tipo PNP y PN muestran
excelente actividad en la reacción de hidrogenación de N-bencilideanilina. La disminución
de la conversión podría ser consecuencia de la hidrolisis del sustrato en condiciones básica,
lo cual no se observa en condiciones acidas.
AGRADECIMIENTOS Proyectos FONDECYT 1120685 y 1120149; Beca CONICYT
21110810 y Beca Gastos Operacionales 21110810.
REFERENCIAS: [1] Cheng-Chao Pai, Ching-Wen Lin, Chi-Ching Lin, Chih-Chiang Chen, Albert , S. C.
Chan; J. Am. Chem. Soc., 2000, 122, 11513. [2] B. Plietker; Angew. Chem. Int. Ed., 2006, 45, 1469.[3] Sean
E. Clapham, Alen Hadzovic, Robert H. Morris; Coordination Chemistry Reviews, 2004, 248, 2201[4] David
Benito-Garagorri, Eva Becker, Julia Wiedermann, Wolfgang Lackner, Martin Pollak, Kurt Mereiter, Joanna
Kisala, and Karl Kirchner, Organometallics 2006, 25, 1900. [5] P. Aguirre, C. Lagos, S. A. Moya, C. Zúñiga,
C. Vera-Oyarce, E. Sola, G. Peris, JC. Bayón, Dalton Transactions, 2007, 5419.

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile 11
ACTIVIDAD DE CATALIZADORES CU-CE SOPORTADOS EN LA REACCIÓN
WATER-GAS SHIFT. EFECTO DE LA ESTRUCTURA CRISTALINA DEL
SOPORTE ZrO2
Paulo Arayaa, Gonzalo Aguilaa, Sichem Guerrerob a Departamento de Ingeniería Química y Biotecnología, Facultad de Ciencias Físicas y Matemáticas, Universidad de
Chile, Santiago, Chile b Facultad de Ingeniería y Ciencias Aplicadas, Universidad de los Andes, Santiago, Chile
Palabras claves: Cobre, Ceria, Circonia, Water-Gas Shift
INTRODUCCIÓN
Las celdas de combustible se han convertido en una real alternativa para obtener energía
limpia a partir de H2 [1]. Uno de los tipos de celdas más estudiadas es la de “Membrana de
Intercambio de Protones” (PEM-FC). Sin embargo, este tipo de celda de combustible no
puede admitir una corriente de alimentación con un contenido de CO mayor a 10-20 ppm,
ya que el CO se adsorbe sobre los electrodos de platino, produciendo una disminución en su
rendimiento [2]. El CO puede eliminarse a través de la conocida reacción “Water-Gas
Shift” (CO + H2O ↔ CO2 + H2), donde se disminuye el contenido CO y al mismo tiempo se
aumenta la cantidad de H2 en la corriente. Esta reacción se realiza en etapas sucesivas, a
alta y baja temperaturas. Para la última etapa de baja temperatura, el contenido de CO es
reducido a alrededor de 1%, utilizándose catalizadores de Cu/ZnO o Cu/ZnO/Al2O3 [3]. Sin
embargo, ambos catalizadores presentan problemas de estabilidad y carácter pirofórico.
Dentro de los sistemas alternativos más estudiados en WGS, se encuentran los catalizadores
de CuO-CeO2, principalmente debido a su alta actividad [4,5], que se explica por la
existencia de sitios de interface “Cu-CeO2”, que aumentan la reducibilidad de Ce, y por lo
tanto, aumenta la actividad [4]. Otro sistema que muestra buenas actividades catalíticas es
Cu-ZrO2 [6], que ya fue estudiado por nuestro grupo en un trabajo previo, y se mostró que
la actividad en la reacción WGS depende del tipo de estructura cristalina de la ZrO2 usada y
del tamaño de partícula de Cu [7]. Considerando las buenas propiedades de los anteriores
sistemas, en este trabajo se estudió el efecto de la estructura cristalina de ZrO2 en la
actividad en la reacción WGS de catalizadores Cu-Ce/ZrO2.
EXPERIMENTAL
Los catalizadores fueron preparados por el método de impregnación, con distintos
contenidos de Cu y Ce sobre ZrO2, utilizando dos tipos de estructura cristalina para ZrO2: i)
monoclínica, y ii) tetragonal. Los catalizadores impregnados fueron calcinados en mufla a
500°C por 3h. También se prepararon catalizadores de Cu soportado en CeO2, utilizando el
método de impregnación con distintos contenidos de Cu, y calcinando a 500°C por 3 h. Los
catalizadores fueron caracterizados por adsorción de N2, difracción de rayos X, reducción
por temperatura programada con H2, y espectroscopia infrarroja DRIFT. La actividad
catalítica en la reacción WGS se midió usando un reactor tubular de lecho fijo, cargando
0,2 g de catalizador. Previo a la reacción, el catalizador fue reducido in situ a 350°C por 1
h. La alimentación utilizada consistió en un flujo de 70 mL/min de 2%CO y 6%H2O. La
temperatura del reactor se aumentó hasta 300°C y se mantuvo constante por 2 horas. Se
tomaron muestras gaseosas en la entrada y la salida del reactor, que fueron analizadas en un
equipo GC equipado con un detector TCD y una columna CTR-1 (Alltech), que permite
determinar la concentración de H2, CO y CO2.

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile 12
RESULTADOS Y DISCUSIÓN
Los resultados de actividad en la reacción WGS demuestran que la fase cristalina posee una
fuerte influencia en catalizadores Cu/ZrO2, y no afecta la actividad de los catalizadores Cu-
Ce/ZrO2. Junto con los resultados de caracterización, lo anterior se puede explicar al
proponer que existe una diferencia en el mecanismo de reacción entre estos dos sistemas.
Es decir, en los catalizadores Cu/ZrO2 la reacción WGS ocurriría a través de un mecanismo
“asociativo”, donde la estabilidad de la especie intermediaria formiato depende de la
estructura cristalina del soporte, lo cual influye fuertemente en la actividad de estos
catalizadores. Mientras que en los catalizadores Cu-Ce/ZrO2 la reacción WGS ocurriría a
través de un mecanismo “redox”, que no depende de la estructura cristalina de ZrO2, sino
de la fuerza del sitio redox, que en este caso corresponde a la interface “Cu-CeO2”,
responsables de la alta actividad obtenida (ver Figura 1).
0
10
20
30
40
50
60
70
0 50 100 150
CO
Co
nve
rsió
n/%
Time/minutes
(A)
2%Cu (B) 6%Cu
Figura 1: Conversión de CO en la reacción WGS en el tiempo a 300°C. (A) Catalizadores con 2%Cu:
(2Cu/Zr-t () 2Cu/Zr-m () 2Cu-8Ce/Zr-t () 2Cu-8Ce/Zr-m. (B) Catalizadores con 6%Cu: (6Cu/Zr-t
() 6Cu/Zr-m () 6Cu-8Ce/Zr-t () 6Cu-8Ce/Zr-m. (Zr-m: ZrO2 monoclínica; Zr-t: ZrO2 tetragonal)
CONCLUSIONES
La estructura cristalina del soporte ZrO2 sólo afecta la actividad en la reacción WGS a los
catalizadores Cu/ZrO2, y no influye en los catalizadores Cu-Ce/ZrO2. La razón de este
comportamiento se debería a la diferencia en los mecanismo de reacción en cada sistema
catalítico, siendo tipo “asociativo” para Cu/ZrO2, y tipo “redox” para Cu-Ce/ZrO2.
Finalmente, las actividades de los catalizadores Cu-Ce/ZrO2 son levemente inferiores a las
actividades de Cu/CeO2, que es uno de los más activos en la reacción WGS.
AGRADECIMIENTOS Proyecto FONDECYT 1110184, Empresa MEL Chemicals.
REFERENCIAS [1] F. Joensen, J. Rostrup-Niesen, J. Power Sources 105 (2002) 195-201.
[2] D. Trimm, Applied Catalysis A 296 (2005) 1-11.
[3] C. Satterfield, Heterogeneous Catalysis in Industrial Practice, McGraw-Hill, New York (1980)
[4] Y. Li, Q. Fu, M. Flytzani-Stephanopoulos, Applied Catalysis B 27 (2000) 179-191.
[5] R. Si, J. Raitano, N. Yi, L. Zhang, S. Chan, M. Flytzani-Stephanopoulos, Catal Today 180 (2012) 68-80.
[6] J. Ko, C. Bae, Y. Jung, D. Kim, Catalysis Letters 105 (2005) 157-161.
[7] G. Aguila, S. Guerrero, P. Araya, Catalysis Communications 9 (2008) 2550-2554.

VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile 13
HÍBRIDOS DE ARCILLA NATURAL Y TiO2 PARA FOTOCATÁLISIS
Raquel Portelaa, Ingrid Janssonb, Silvia Suárezb, Mirza Villarroelc, Benigno Sánchezb y
PedroÁvilaa a Instituto de Catálisis y Petroleoquímica, Consejo Superior de Investigaciones Científicas, Madrid, España
bCentro de Investigaciones Energéticas y Medioambientales, Madrid, España. c Universidad de Santiago de Chile, Facultad de Química y Biología, Santiago, Chile, Casilla 40.,
Palabras clave: silicatos, adsorción, VOCs
INTRODUCCIÓN La oxidación fotocatalítica permite eliminar compuestos orgánicos con la ventaja competitiva de
que opera a temperatura ambiente, como los sistemas de adsorción, y transforma los contaminantes
en CO2 y H2O, como en la combustión catalítica, evitando los inconvenientes de la saturación de los
adsorbentes o las elevadas temperaturas de la combustión. Materiales híbridos con un componente
poroso y otro semiconductor constituyen fotocatalizadores soportados que se benefician de la
sinergia entre sus componentes para mejorar sus propiedades[1, 2].En esta comunicación se
estudian híbridos basados enTiO2 y silicatos naturales, y se evalúan sus propiedades para el
tratamiento de compuestos orgánicos utilizando el formaldehído como molécula representativa de la
contaminación en el aire interior.
EXPERIMENTAL Los fotocatalizadores fueron preparados por extrusión mezclando al 50% en peso TiO2 con un
silicato 2:1fibroso, sepiolita, y uno laminar 2:1, bentonita (ambos con Mg), y dos aluminosilicatos,
uno laminar 1:1, caolinita, y una zeolita, mordenita. Los materiales conformados se calcinaron a
300, 500 y 800 °C y se caracterizaron por porosimetría de intrusión de Hg, isotermas de adsorción
de N2,DRX, SEM y espectroscopía UV-VIS. También se determinó la resistencia a la rotura y el
TiO2expuesto en la superficie externa a partir del punto de carga cero (migración electroforética)
mediante la ecuación de Parks modificada [3]. La evaluación en régimen dinámico de la capacidad
de adsorción y de oxidación fotocatalítica se realizó con caudales entre 100 y 900 mL min-1y una
concentración de formaldehído en aire de 15 ppm.
RESULTADOS Y DISCUSIÓN En la Tabla 1y la Figura 1 se muestran algunos resultados de los materiales tratados a 500ºC.
Tabla 1. Propiedades de las muestras silicato/TiO2tratadas a 500°C
Muestra Área BET V poro PCC P ruptura Ads. dinámica Cristalito
(m² g-1) (cm3 g-1) (pH) (kg cm-2) (micromol g-1) (nm)
STi 96 0,66 2,5 590 371 13
BTi 67 0,42 2,6 540 319 20
KTi 60 0,54 3,3 148 422 20
MTi 50 0,45 2,4 179 1444 17
Todos los compuestos presentan mesoporosidad similar, que se colapsa progresivamente con el
incremento de la temperatura de tratamiento, y una macroporosidad relacionada con la distancia
interparticular, que es ligeramente superior en los compuestos de mordenita (Figura 1, Izq). La
muestra con mayor porosidad y área específica es la preparada con sepiolita, la cual presenta una
resistencia mecánica muy buena, al igual que la bentonita, que sin embargo tiene una porosidad
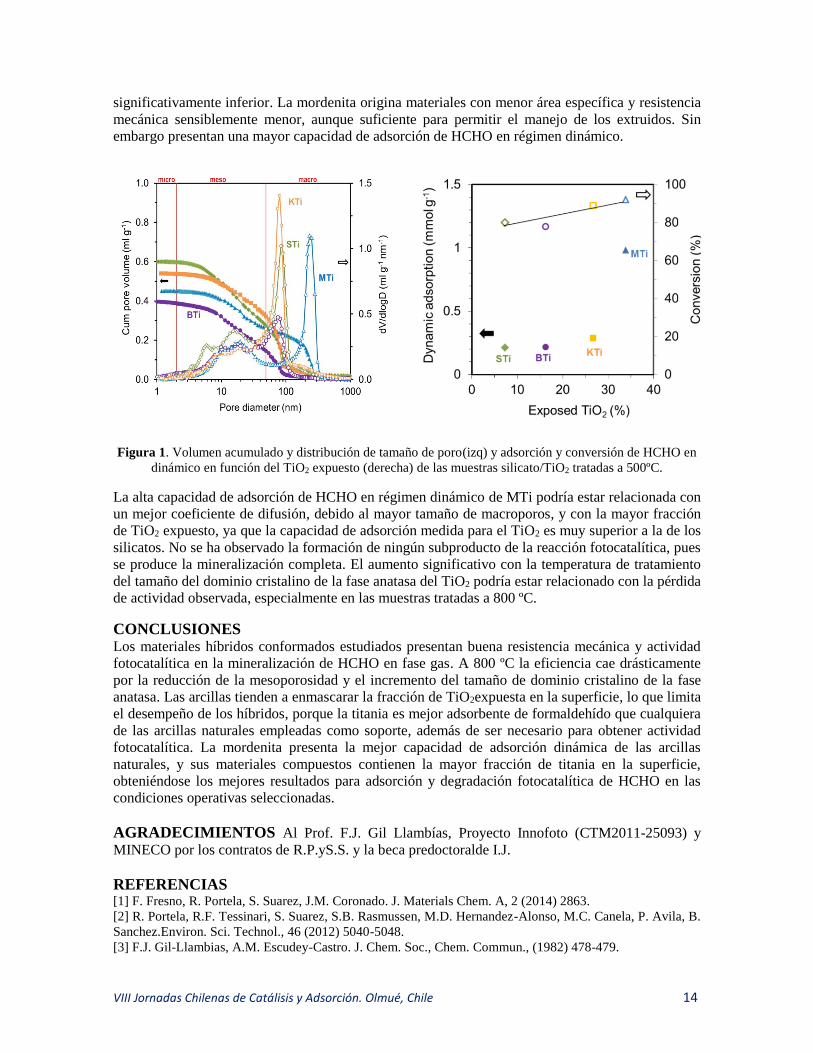
VIII Jornadas Chilenas de Catálisis y Adsorción. Olmué, Chile 14
significativamente inferior. La mordenita origina materiales con menor área específica y resistencia
mecánica sensiblemente menor, aunque suficiente para permitir el manejo de los extruidos. Sin
embargo presentan una mayor capacidad de adsorción de HCHO en régimen dinámico.
Figura 1. Volumen acumulado y distribución de tamaño de poro(izq) y adsorción y conversión de HCHO en
dinámico en función del TiO2 expuesto (derecha) de las muestras silicato/TiO2 tratadas a 500ºC.
La alta capacidad de adsorción de HCHO en régimen dinámico de MTi podría estar relacionada con
un mejor coeficiente de difusión, debido al mayor tamaño de macroporos, y con la mayor fracción
de TiO2 expuesto, ya que la capacidad de adsorción medida para el TiO2 es muy superior a la de los
silicatos. No se ha observado la formación de ningún subproducto de la reacción fotocatalítica, pues
se produce la mineralización completa. El aumento significativo con la temperatura de tratamiento
del tamaño del dominio cristalino de la fase anatasa del TiO2 podría estar relacionado con la pérdida
de actividad observada, especialmente en las muestras tratadas a 800 ºC.
CONCLUSIONES Los materiales híbridos conformados estudiados presentan buena resistencia mecánica y actividad
fotocatalítica en la mineralización de HCHO en fase gas. A 800 ºC la eficiencia cae drásticamente
por la reducción de la mesoporosidad y el incremento del tamaño de dominio cristalino de la fase
anatasa. Las arcillas tienden a enmascarar la fracción de TiO2expuesta en la superficie, lo que limita
el desempeño de los híbridos, porque la titania es mejor adsorbente de formaldehído que cualquiera
de las arcillas naturales empleadas como soporte, además de ser necesario para obtener actividad
fotocatalítica. La mordenita presenta la mejor capacidad de adsorción dinámica de las arcillas
naturales, y sus materiales compuestos contienen la mayor fracción de titania en la superficie,
obteniéndose los mejores resultados para adsorción y degradación fotocatalítica de HCHO en las
condiciones operativas seleccionadas.
AGRADECIMIENTOS Al Prof. F.J. Gil Llambías, Proyecto Innofoto (CTM2011-25093) y
MINECO por los contratos de R.P.yS.S. y la beca predoctoralde I.J.
REFERENCIAS
[1] F. Fresno, R. Portela, S. Suarez, J.M. Coronado. J. Materials Chem. A, 2 (2014) 2863.
[2] R. Portela, R.F. Tessinari, S. Suarez, S.B. Rasmussen, M.D. Hernandez-Alonso, M.C. Canela, P. Avila, B.
Sanchez.Environ. Sci. Technol., 46 (2012) 5040-5048.
[3] F.J. Gil-Llambias, A.M. Escudey-Castro. J. Chem. Soc., Chem. Commun., (1982) 478-479.


















