
Various methods used in biochemistry - Diplomovka
39
COMENIUS UNIVERSITY IN BRATISLAVA Faculty of Natural Sciences Department of Biochemistry Application of genomics in biochemistry Bachelor thesis Hana ŠIŠKOVÁ Study programme - Biochemistry Supervisor of thesis: Mgr. Silvia Poláková, PhD. BRATISLAVA 2010
Transcript of Various methods used in biochemistry - Diplomovka

COMENIUS UNIVERSITY IN BRATISLAVA
Faculty of Natural Sciences
Department of Biochemistry
Application of genomics in biochemistry
Bachelor thesis
Hana ŠIŠKOVÁ
Study programme - Biochemistry
Supervisor of thesis: Mgr. Silvia Poláková, PhD.
BRATISLAVA 2010

1

2
Declaration
I hereby declare that this thesis is the result of my own original work and I have done
it with the use of literature listed in the references.
In Bratislava, April 15th 2010 ..........................................
Author signature

3
Acknowledgement
I am very thankful to my supervisor, Mgr. Silvia Poláková, PhD., whose
encouragement, guidance and support from the initial to the final level enabled me to
develop an understanding of the subject.

4
ABSTRAKT
Hana Šišková: Využitie genomiky v biochémii
Univerzita Komenského v Bratislave, Prírodovedecká fakulta, Katedra biochémie
Bakalárska práca, 38 strán, 13 grafických príloh, 2010
Genomika je mladá vedná disciplína, ktorej rozvoj značne napomáha štúdiu
metabolických dráh v organizmoch. Táto práca mala za cieľ predstaviť základné oblasti
genomiky a postgenomiky, vysvetliť ich princípy a prínos pre biochémiu za účelom
pochopenia metabolizmu organického celku. Práca sa štruktúrou snažila chronologicky
objasniť vývoj metód pre štúdium bunkového metabolizmu; od pre-genomických metód,
využívaných pred objavom metód sekvenovania genómu, cez metódy genomiky, ktoré
pomáhajú odhaľovať vzťah medzi genómovou štruktúrou a funkciou, až po postgenomické
metódy zamerané na objasnenie fyziológie eukaryotickej a prokaryotickej bunky.
Kľúčové slová: genomika, funkčná genomika, komparatívna genomika, transkriptomika,
proteomika, metabolomika, metagenomika
ABSTRACT
Hana Šišková: Application of genomics in biochemistry
Comenius University in Bratislava, Faculty of Natural Sciences, Department of Biochemistry
Bachelor thesis, 38 pages, 13 supplements, 2010
Genomics is a young scientific discipline, the development of which has considerably
facilitated the study of metabolic pathways in living organisms. The aim of this work was to
present the fields of genomics and post-genomics, and to explain their principles and use in
biochemistry in order to understand the metabolism of an organic entity. This work attempted
to present chronologically the development of methods used to study the cell metabolism:
from pre-genomic methods, used before the discovery of genome sequencing methods,
through the methods of genomics that help to reveal the relationship between genome
structure and function, and to post-genomics methods aimed at the clarification of the
physiology of the eukaryotic and prokaryotic cell.
Key words: genomics, functional genomics, comparative genomics, transcriptomics,
proteomics, metabolomics, metagenomics

5
TABLE OF CONTENTS
Prologue ..................................................................................................................................... 6
1. Methods to reveal metabolic pathways in the pre-genomic world .................................. 7
2. How can genomics help in the study of metabolic pathways.......................................... 10
2.1 Definition of genomics ................................................................................................... 10
2.2 History of genomics ....................................................................................................... 11
2.3 Comparative genomics ................................................................................................... 13
3. Postgenomics ....................................................................................................................... 19
3.1 Use of functional genomics to determinate gene and protein function .......................... 19
3.1.1 ORF clones .............................................................................................................. 21
3.1.2 Knock-out mutants ................................................................................................... 22
3.1.3 Transcriptome analysis ........................................................................................... 23
3.1.4 Proteome analysis ................................................................................................... 26
3.1.5 Metabolome analysis ............................................................................................... 27
3.2 Metagenomics - approach for studying complex communities in natural environment 29
Summary ................................................................................................................................. 31
Zhrnutie ................................................................................................................................... 32
Used abbreviations ................................................................................................................. 33
References ............................................................................................................................... 34

6
Prologue
The discovery of the DNA structure by Watson and Crick in 1953 and the use of
genome sequencing methods by Fred Sanger‘s group in the 1970s have given rise to the new
field of genomics. Many scientific groups have started to sequence the genomes of some
simpler organisms and with the use of bioinformatics the genome databases accessible
worldwide have started to appear. These databases enabled further development of
comparative genomics, as an approach to determine unknown genes based on the comparison
with the sequences of known genes. Once the genes were aligned, scientists found a new
challenge for themselves: to determine the molecular, cellular and physiological functions for
the numerous proteins encoded by the sequenced genomes. This task has required the
integrated application of chemical and biological techniques and has promoted the further
development of the fields such as functional genomics, transcriptomics, proteomics and
metabolomics. Some scientists went even further by switching from the analysis of an isolated
organism to the research of complex communities in their natural environment, as presented
by the field of metagenomics.

7
1. Methods to reveal metabolic pathways in the pre-genomic world
Today in biochemistry we have many specialised methods to help us analyse the
metabolic pathways in various organisms. But before these methods appeared, scientists often
had to improvise in their research in order to get some notable success. The work of Hans
Krebs and Melvin Calvin is a typical case, and worth recounting.
When Hans Krebs wanted to measure the amounts of ammonia taken up and urea
formed in the liver, he had first to design a salt solution that would be similar enough to the
saline composition of blood. In his experiments, he showed that the liver made some urea
from added ammonium salts and that the energy for this synthesis was supplied by the
breakdown of stored carbohydrates. The production of urea was stimulated to various degrees
by the addition of different amino acids, but one amino acid — ornithine — stimulated it to a
much greater extent than any other tested.
Since 1904 it had been known that the amino acid arginine could be hydrolysed to
ornithine and urea by the enzyme arginase. In addition, it was known that the liver is rich in
arginase (Kossel and Dakin, 1904). Therefore, the question was whether the ornithine could
not be contaminated with arginine. Krebs, in his further research, not only gave a negative
answer to this question but also revealed that in the presence of ammonia, the addition of one
molecule of ornithine brought the extra formation of more than 20 molecules of urea. Krebs
assumed that ornithine acted as a catalyst in the reaction. Although arginine seemed to be the
intermediate regenerating the ornithine, it looked unlikely that arginine would be formed from
ornithine and ammonia in one step. Krebs postulated that citrulline might be the missing
intermediate of the cycle. He also proved his hypothesis when he obtained a few milligrams
of citrulline from two scientists who by a happy coincidence had just isolated this substance
(Wada, 1930; Ackermann, 1931). And so the ornithine cycle was discovered (Krebs and
Henseleit, 1932) (Fig.1).
Figure 1. a) The ornithine cycle formulated by Krebs and his assistant Henseleit in 1932. b) Modern
version of the ornithine cycle (Kornberg, 2000).

8
Hans Krebs has also helped to discover two other very important cycles in living
organisms. First, he participated in the discovery of the citric acid cycle. Together with
William A. Johnson studied how small organic acids are oxidized to CO2 and H2O to yield
energy for the body. He based his research on the previous studies of the Hungarian
biochemist Albert Szent–Györgyi, who used the breast muscles of pigeons to study this
process. Albert Szent–Györgyi chose this tissue as it was well adapted to extract the energy
from the food in order to power the bird flight. He showed that the rate of oxygen uptake was
greatly increased when any of three four-carbon dicarboxylic acids - fumarate, succinate or
malate - were added. He concluded that these substances were limiting in the cell and
stimulated oxidation of endogenous glucose, but the catalytic effect of these compounds
remained a puzzle for him. Krebs and Johnson argued that this could not proceed in one step.
They showed that succinate could be synthesised if pyruvate was present and that citrate was
formed from oxaloacetate if pyruvate was also added (Krebs and Johnson, 1937a). But the
most important observation was that citrate was not only oxidised, but that it was
continuously re-formed. They saw that they had a cycle, not a simple pathway, and that
addition of any of the intermediates could generate all of the others (Krebs and Johnson,
1937b). The existence of a cycle, together with the entry of pyruvate into the cycle in the
synthesis of citrate, gave a clear explanation for the catalytic effect of succinate, fumarate, and
malate. The citric acid cycle, formulated later by Kornberg and Krebs, is shown in Figure 2a.
Additionally, Krebs participated in the discovery of the glyoxylate cycle – the ‗bypass‘
of the citric acid cycle, when he wanted to explain how some organisms could grow on
acetate as the only carbon source (Kornberg and Krebs, 1957) (Fig.2b).
Figure 2. Main stages of the citric acid cycle (a) and the glyoxylate cycle (b), formulated by Kornberg and
Krebs,1957 (adapted from Kornberg, 2000).

9
Another well-known scientist, Melvin Calvin, used the method of C14
labelling in his
experimental observation of the carbon cycle in photosynthesis. The C14
isotope was cheaply
available in large amounts after 1945 thanks to the number of nuclear reactors that had been
constructed. Calvin studied the carbon reduction sequence in photosynthesis on the unicellular
green alga Chlorella, which turned out to be a very good biological material. In his
experiment, Calvin injected some C14
labelled CO2 into the stream of nonradioactive CO2,
both of which entered the plant materials. At the end of the different time periods, the plants
were killed and the extraction of material for analysis was initiated. The isolation and
identification procedures were then carried out in ion-exchange columns. Later, when Martin
and Synge had developed the method of partition chromatography, Calvin used this method as
his principal analytical tool. He spread the plant material on a sheet of filter paper by two-
dimensional chromatography and he placed the paper in contact with photographic film, thus
exposing the film at those points of the paper where the compounds he was interested in were
located. Thanks to this method, together with his co-workers A.A. Benson and J.A. Bassham,
he was able to identify the first product of photosynthesis – the phosphoglycerate, as well as
other intermediates of the photosynthetic carbon cycle, known also as Calvin-Benson-
Bassham cycle (Calvin, 1962) (Fig.3).
Figure 3. The photosynthetic carbon cycle (Calvin, 1962).

10
2. How can genomics help in the study of metabolic pathways
2.1 Definition of genomics
A new age of study of metabolic pathways came with the emergence of genomics as
a new field in the omics studies. ‗Omics‘ is a general term for a broad discipline of science
and engineering for analysing the interactions of biological information objects in
various omes (ome is the Greek suffix for ‗whole‘, so it means the organic totality of
something). Since the mid-'90s researchers are rapidly taking up omes and omics, as shown
by the explosion of the use of these terms.1 These include studies such as genomics,
proteomics, metabolomics, expressomics, materiomics and interactomics. When we talk about
genes that are differentially expressed and proteins produced by the cell, the terms like
transcriptome and proteome are used; and since the living cell is a complex structure, there is
a complexome; all of the parts are connected ultimately with the metabolome, and together
these omes house the phenome, which interacts with an environome (Fig.4).
Figure 4. The diagram suggests other omes that constitute a living multicellular organism (Scriver, 2001).
The main focus in the omics study is on:
1) mapping information objects such as genes and proteins,
2) finding interaction relationships among the objects,
3) engineering the networks and objects to understand and manipulate the regulatory
mechanisms.
So genomics, as the omics study, is a field studying the complexity of genes in
individual organisms, populations, and species.
1 The internet site omics.org provides a list of more than 400 categories of different omes which gives an
evidence of rising tendency in use of this term, however not necessarily just in the field of molecular biology.

11
2.2 History of genomics
Much in the way that the biochemical studies based on radioactively-labelled
biological molecules paved the way for the discovery of the basic mechanisms involved in the
regulation of metabolic pathways, genomic DNA sequences are today paving the way for the
elucidation of global mechanisms for genetic regulation.
The work, in which radioisotopes were used for the elucidation of metabolic pathways,
resulted in a monograph titled Studies of Biosynthesis in Escherichia coli that guided research
in biochemistry for the next 20 years and helped to establish this bacterium as a model
organism for biological research (Roberts et al., 1955). In the following years, most of the
metabolic pathways required for the biosynthesis of intermediary metabolites were revealed
and new methods were developed to identify and characterise the enzymes involved in these
pathways. These studies defined the biosynthetic pathways for the building blocks of
macromolecules such as proteins and nucleic acids, and enabled the discovery of the
mechanisms for metabolic regulation such as end product inhibition, allostery, and
modulation of enzyme activity by protein modification. A bigger breakthrough in the
biosynthesis of macromolecules presented a discovery of the structure of the DNA helix
(Watson and Crick, 1953), which provided a basis for the new field of molecular biology.
This field gained even more importance with further discoveries of restriction endonucleases
and DNA ligases. These new enzymes, together with a discovery of a method of polymerase
chain reaction by Kary Mullis in 1983, enabled construction of recombinant DNA molecules
composed of DNA sequences from different organisms.
As for genomics itself, it was established by Fred Sanger group in 1970s when they
developed a gene sequencing technique and completed the first genomes. In 1972, Walter
Fiers and his team at the Laboratory of Molecular Biology of the University of Ghent were
the first to determine the sequence of a gene – it was the gene for Bacteriophage MS2 coat
protein (Fiers et al., 1972). In 1976, the team determined the complete nucleotide-sequence of
bacteriophage MS2-RNA (Fiers et al., 1976). In 1977, Frederick Sanger sequenced the first
DNA-based genome in its entirety - the bacteriophage Φ-X174 (5,368 bp) (Sanger et al.,
1977).
The new era of genomics was initiated in 1986 at an international conference in Santa
Fe, New Mexico, sponsored by the US Department of Energy. At this meeting, the leading
scientists from around the world endorsed unanimously the desirability and feasibility of
implementing a human genome program. This meeting resulted in a 1988 study by the

12
National Research Council titled ―Mapping and Sequencing the Human Genome‖ that
recommended that the USA support a human genome program and presented an outline for a
multiphase plan. In 1990, an international scientific research project was launched at the US
National Institutes of Health (NIH) in order to determine the complete human genome – The
Human Genome Project.
In 1991 Craig Venter at the NIH developed a way of finding human genes without
having to sequence the entire human genome. He estimated that only about 3% of the genome
is composed of genes that express mRNA and suggested that the most efficient way to find
genes would be to use the processing machinery of the cell. At any time, only a part of cell‘s
DNA is transcriptionally active. These ―expressed‖ segments of DNA are transcribed to
mRNA and then with the help of the enzyme reverse transcriptase can be transcribed into
complementary DNA (cDNA). These stable cDNA fragments are called expressed sequence
tags (EST). These cDNA sequences are then assembled into longer sequences representing
large parts of many human genes. This is done with computer programs that match
overlapping ends of EST‘s. In 1992 Venter left NIH and later became one of the founders of
Celera Genomics Corporation that in 1999 announced its parallel research project of The
Human Genome project.
Meanwhile, the sequencing proceeded very fast. In 1995, the first free-living organism
was sequenced in the Institute for Genomic Research – the Haemophilus influenzae (1.8 Mb)
(Fleischmann et al, 1995). Then, the complete genomes of many bacteria, viruses and
eukaryote organisms (mostly fungi) were determined. For scientific research, it was an
important milestone when the genomes of the most studied organisms, Escherichia coli and
budding yeast (Saccharomyces cerevisiae) were sequenced (Goffeau et al., 1996). Other
important sequences of model organisms were: Caenorhabditis elegans (The C. elegans
Sequencing Consortium, 1998), Arabidopsis – the first sequenced plant (The Arabidopsis
Genome Initiative, 2000), Drosophila melanogaster (Adams et al., 2000) and the mouse Mus
musculus as the first sequenced mammal (Mouse Genome Sequencing Consortium, 2002).
In 1998 the Human Genome Program announced a plan to complete the human
genome sequence by 2003, the 50th
anniversary of the discovery of the DNA structure. The
goals of this plan were to:
Achieve coverage of at least 90% of the genome in a working draft based on mapped
clones by the end of 2001.
Finish one-third of the human DNA sequence by the end of 2001.
Finish the complete human genome sequence by the end of 2003.

13
Make the sequence totally and freely accessible.
In 2000, both the director of the Human Genome project and Celera Genomics
announced that they completed the draft versions of the human genome two years ahead of
schedule. These drafts were published in special issues of Science and Nature in 2001 and the
sequence is now online at the National Center for Biotechnology Information (NCBI).
Genome sequencing enabled the identification of many genes that had been previously
unknown. Many projects, such as The Human Genome Project, are still focusing on the
genome sequencing, but the knowledge of full genomes has created the possibility for the
other field such as comparative and functional genomics, for example.
2.3 Comparative genomics
Comparative genomics is a young field of genomics that studies the relationship of
genome structure and function across different biological species or strains. It uses the
similarities and differences of genes in different organisms in order to analyse their function
and to gain a better understanding of how species have evolved. A very effective approach to
find the most likely function of an unidentified gene is based on projection of experimentally
established functions of proteins from one species to another on the basis of homology,
determined by sequence similarity. This projection is supported by a set of powerful tools and
public archives (such as GenBank and Swiss-Prot), as well as by a significant body of
literature. Researchers have learned a lot about the function of human genes by examining
their counterparts in simpler model organisms. Many different features can be considered by
genome comparison: sequence similarity, gene location, the length and number of coding
regions within genes (exons), the amount of noncoding DNA in each genome, and highly
conserved regions maintained in organisms as simple as bacteria and as complex as humans.
The comparative approach is an ideal way to solve the missing genes problem. A
metabolic reconstruction attempts to develop a detailed overview of an organism‘s
metabolism from an analysis of its genomic sequence. As the reconstruction technology is
focused on identifying present components, it also gives a notion of components that should
be present but cannot be identified. In this case, there is an attempt to connect functional roles
to genes that have not yet been characterized. Two categories of missing genes can be
distinguished: globally missing (for functions without any representative sequenced genes
from any organism) and locally missing (for functions previously connected to one sequenced

14
form of a gene in one group of species, but expected to exist in an alternative form in another
group of species)2.
A typical missing gene study is split in three phases: revealing missing genes,
identification and ranking of candidate genes and experimental verification. In the first phase
of the missing gene search, there has to be defined a ‗functional context‘, which usually
includes the other enzymes that participate in the same pathway or variants of the pathway.
When the closely related functions have been determined, a table showing which of these
functions are present or absent in model organisms can be built. This table shows, which
organisms have variants of the pathway, which do not, and where the situation is ambiguous
(Table 1). Once the gene inventory has been completed, the missing genes are identified
according to the presence of pathway‗s variants in model organisms – a process of metabolic
reconstruction.
Table 1. Gene inventory showing which of closely related functions is present or absent within a diverse set of
model organisms. The table contains a row for each of the enzymatic functions and a column for each organism
(adapted from Osterman, 2003).
2 The term missing genes problem, as well as its division into these two categories, has been used by Osterman,
2003.

15
In the second phase, various techniques of genome context analysis are used to
produce an initial list of candidate genes for a sought functional role. Let‘s examine the major
techniques closely:
Clustering on the chromosome – this technique uses the fact that on prokaryotic
chromosomes the genes from the same pathway tend to cluster. They are often
organized into operons, defined as a set of adjacent genes all under the transcriptional
control of the same operator. This enables to infer ‗functional coupling‘ between
genes (Overbeek et al., 1999). Using the genome-scanning tools, one can find cases
where multiple genes orthologous to members of the gene inventory occur in close
proximity.
Protein fusion events – this technique uses searching for a pair of genes from one
genome that appear to be fused into a single gene in another genome, which provides
further evidence of potential functional coupling (Enright et al., 1999).
Occurrence profiles – this technique, also called ‗phylogenetic profiling‘, is based on
assumption that two proteins from the same cellular pathway are expected to either
both occur or both not occur in any specific organism (Pellegrini et al., 1999). The
higher version of this technique generates instances of potential functional coupling
for a pair of proteins on the basis of their occurrence profiles.
Shared regulatory sites – This technique is based on identification of so-called
regulons (ensemble of genes which have coordinated expression). Co-regulation of a
pair of genes provides evidence that these genes may be functionally coupled
(McGuire, 2000).
These techniques produce candidate genes that can be further prioritised based on
strength and consistency of evidence. For gene discovery, as well as for additional candidate
ranking, there are methods revealing and analysing putative folds (Pawlowski et al., 2001),
long-range sequence similarities (Bateman et al., 2002) and conserved motifs (Falquet, 2002).
In most cases, the number of highly ranked gene candidates is very limited and they can be
quickly subject to experimental verification by traditional experimental techniques.
Among all of the contemporary techniques of genome context analysis, gene
clustering on the chromosome presents the most critical contribution to missing gene
discovery in spite of fact that this technique is almost exclusively applicable for the
comparative analysis of prokaryotic genomes. Large-scale sequencing and comparative

16
analysis of many prokaryotic genomes provide growing evidence that for an overwhelming
majority of eukaryotic metabolic enzymes, it is possible to find functional counterparts in one
or more prokaryotes (Osterman, 2003).
The comparative approach can also help to find new genes based on the comparison of
closely related organisms coming from the same family. For instance, this method was used to
identify a large amount of new genes in one of the most favourite model organisms –
Caenorhabditis elegans. Stein et al. (2003) used this approach to compare this soil nematode
with Caenorhabditis briggsae - both species diverged from a common ancestor roughly 100
million years ago and yet are morphologically almost indistinguishable. They have the same
chromosome number and genome sizes. After the C. briggsae genome had been sequenced to
a high-quality draft stage and compared to the finished C. elegans sequence, the researchers
predicted approximately 19,500 protein-coding genes in the C. briggsae genome, roughly the
same as in C. elegans. 12,200 of these have clear C. elegans orthologs (two genes are
orthologous if they diverged after a speciation event, when a new species forms from an
existing one), other 6,500 have one or more clearly detectable C. elegans homologs, and
approximately 800 C. briggsae genes showed no equivalent in C. elegans. Based on
comparison to C. briggsae, the authors found strong evidence for 1,300 new C. elegans genes.
Apparently, comparisons of the two genomes can help us to understand the evolutionary
forces that modified nematode genomes. Surprisingly, C. briggsae and C. elegans, despite
their abundant differences at the genomic level, remain morphologically almost
indistinguishable, whereas mouse and human, for example, are more similar genetically, but
show dramatic anatomic and behavioural differences.
The comparative approach can be effectively used also for studying such a complex
organism as the human being. In order to explore the function of different human genes, the
comparative study of the laboratory mouse Mus musculus and Homo sapiens turned out to be
a very valuable approach. In the roughly 75 million years since the divergence of the human
and mouse lineages, evolution has changed their genome sequences and caused them to
diverge by nearly one substitution for every two nucleotides, as well as by deletion and
insertion (Mouse Genome Sequencing Consortium, 2002). The divergence is low enough that
orthologous sequences can still be aligned but high enough so that many functionally
important elements can be recognised by their greater degree of conservation (Fig.5). An
example of some similarities and differences revealed by the comparative analysis of mouse
and human genomes:

17
The mouse genome is about 14% smaller than the human genome, which probably
reflects a higher rate of deletion in the mouse lineage.
At the nucleotide level, approximately 40% of the human genome can be aligned to
the mouse genome. These sequences seem to represent most of the orthologous
sequences that remain in both lineages from the common ancestor.
The mouse and human genomes each seem to contain about 30,000 protein-coding
genes. The proportion of mouse genes with a single identifiable orthologue in the
human genome seems to be approximately 80%. The proportion of mouse genes
without any homologue currently detectable in the human genome (and vice versa)
seems to be less than 1% (Mouse Genome Sequencing Consortium, 2002).
Figure 5. The genetic similarity (or homology) of superficially dissimilar species – mice and humans. The full
complement of human chromosomes can be cut, schematically at least, into about 150 pieces (only about 100 are
large enough to appear in this illustration), then reassembled into a reasonable approximation of the mouse
genome. The colors of the mouse chromosomes and the numbers alongside indicate the human chromosomes
containing homologous segments. This piecewise similarity between the mouse and human genomes means that
insights into mouse genetics are likely to illuminate human genetics as well (adapted from Human Genome
Program, U.S. Department of Energy, To Know Ourselves, 1996) .
Comparing of human and mouse genomes has also allowed the identification of novel
genes. An interesting example is the discovery of a gene called APOAV that encodes a
previously unknown member of the apolipoprotein gene family. After the identification of
the gene APOAIV, which was considered to be the last catalogued from this gene family
(Boguski, 1984), it was quite surprising to find out that there‘s a gene APOAV and that the

18
field of lipid metabolism and cardiovascular disease had not been so much explored as it was
thought. The comparative analysis helped to identify a region of conserved sequence,
approximately 25 kilobases from APOAIV, which proved to contain the APOAV gene
(Pennacchio et al., 2001). As the AIV/CIII/AI gene locus influence plasma lipid levels in
humans, Pennacchio et al. studied lipid levels in knockout and transgenic mice and found that
the apoAV protein has a strong inverse correlation with plasma triglyceride levels, which
presents a risk factor for coronary artery disease. Further studies showed that genetic variation
in the APOAV locus influences plasma triglyceride levels in humans (Talmud et al., 2002).

19
3. Postgenomics
Genome sequencing projects have provided researchers with a large quantity of
molecular information, which presented a solid basis for new techniques that enabled the
exploration of new genes. However, in the postgenomic era, other scientific fields have
emerged, taking these techniques even further by studying how genes are transcribed into
mRNA (transcriptomics), how mRNA is translated into proteins (proteomics), and what
chemical fingerprints cellular processes leave behind (metabolomics). All these methods are
„postgenomic― in the sense that they require complete genome sequences for optimal
performance.
3.1 Use of functional genomics to determinate gene and protein function
Once the genomes of a large number of organisms have been sequenced, scientists
found a new challenge for themselves: to determine the molecular, cellular and physiological
functions for the numerous proteins encoded by eukaryotic and prokaryotic genomes. This
task requires the integrated application of chemical and biological techniques, as several
factors complicate the research:
The realisation that numerous protein products can be derived from a single gene as a
result of multiple mechanisms, including alternative splicing, proteolytic processing
and post-translational modifications (PTM).
The fact that proteins with highly related sequences can perform different functions in
vivo, and conversely, proteins lacking structural similarity show similar activities.
The differences in protein function in vitro (purified material) and in vivo (part of
complex metabolic and signalling networks), where they can be regulated by covalent
modification and protein-protein interactions (Fig.6).
An example of a clear case where gene or protein sequence alone cannot be used to
predict function is provided by the family of (β/α)8 - barrel enzymes. Two proteins of this
family, mandelate racemase and muconate lactonizing enzyme, have been shown to share
conserved structures and active site functional groups, even though they catalyze different
reactions (Neidhart, 1990). Since then, other members of this family have also been shown to
perform a wide range of mechanistically diverse chemical transformations.

20
Figure 6. Comparison of the endogenous (in vivo) and exogenous (in vitro) environments typically experienced
by proteins, highlighting several natural modes for their post-translational regulation (adapted from Saghatelian
and Cravatt, 2005).
An opposite case is an example of sequence-unrelated enzymes that perform similar
reactions. By comparing the 4-CBA dehalogenation pathway operons from different bacteria,
Zhuang et al. identified conserved sequence homologs responsible for the 4-CBA-CoA ligase
and 4-CBA-CoA dehalogenase activities in this pathway (Zhuang, 2003). However, the 4-
hydroxybenzoate-CoA thioesterase activity was found to be catalysed by distinct sets of
sequence-unrelated enzymes (Fig.7). This discovery leads to the speculation that natural
sources of 4-CBA may exist and that the divergence in 4-CBA pathways in different bacteria
did not occur as a recent adaptation to exposure to industrially produced 4-CBA.
Figure 7. The 4-hydroxybenzoate-CoA (HBA) thioesterase enzymes in the 4-chlorobenzoate dehalogenation
pathways of Arthrobacter and Pseudomonas share no sequence homology but perform the same reaction
(adapted from Saghatelian and Cravatt, 2005).

21
The new approaches, that enable to overpass the obstacles and characterise the
activities of proteins, are such techniques as production of ORF clones, targeted gene
disruption (knockout mutants) and RNA interference (RNAi).
Most of these techniques focus more on determining the function of enzymes than of
other proteins. This is caused by simpler determination of enzyme‘s function because it
generally equates to catalytic activity. Enzymes also have well-defined active sites, which
possess conserved catalytic residues that can be exploited by many approaches in
experimental analysis. However, many of the principles established for the postgenomic
examination of enzymes could be applied to other types of proteins as well, such as receptors
and ion channels, that also share common molecular and structural features.
3.1.1 ORF clones
Although more than 400 bacterial genomes have been sequenced, the function of all
the gene products has not yet been assigned for any organism. E. coli K12, the best studied
organism, is estimated to contain about 4400 genes, of which about 2000 have not been
characterized experimentally, and of these 20% remain difficult even to predict their function.
The latest estimates reveal that about 700 to 800 of total ORFs have no attributable function.
An ORF is a portion of an organism's genome, which contains a sequence of bases that could
potentially encode a protein. Therefore, new technologies are being developed to identify
these functions. A basic genetic tool for studying gene function is provided by ORF clones,
since they present a template for PCR amplification and for preparing purified gene products.
In E. coli, the whole set of PCR amplified ORF fragments were cloned into a special
plasmid vector, and these clones represent the only comprehensive collection of E. coli ORFs
that is available to the public (Mori et al., 2004).
Many groups have started to collect ORFs in a genome-wide manner, so-called
‗ORFeome cloning‘ (Nagase et al., 2008). ORFeome collections can be used for experiments
on single ORFs (for example to study protein localisation) or to study entire ORFeomes (for
structural experiments). They can be also used in module-scale experiments, where a
particular pathway or biological function can be characterised. But the greatest value can be
extracted from ORFeome collections in large-scale experiments. Until recently, these studies
were impossible to be practised because of low numbers of cloned ORFs, and because the
ORFs that were available were not in the same vector or were not expression ORFs. With the

22
availability of ORFeome collections, three important approaches could have been further
developed: structural genomics, proteome-wide mapping of PPI (notably using the Y2H
system) and cell-based assays (Temple et al., 2006). In the emerging field of structural
genomics, there have been several large-scale initiatives3 that aim to generate protein
structures based on available ORFs. Some success with this approach has been shown with
ORFeome collection for Caenorhabditis elegans (Luan et al., 2004). High-throughput Y2H
approaches of PPI detection generally consist of testing all available combinations of proteins
as DNA-binding domain and activation domain fusion proteins (Fields and Song, 1989). In
cell-based assays, expression ORFs are transfected4 into mammalian cells. Changes in cell-
shape or protein localisation can be detected and analysed automatically thanks to the
technology allowing ‗high-content screening‘ of cells. This method was used in a live cell
assay to identify proteins that increase proliferation when over-expressed (Harada et al.,
2005).
3.1.2 Knock-out mutants
A tool that could provide basic information and insight into the function of genes is a
systematic mutational analysis. Creation of knock-out mutants is a technique used in the
reverse genetics approach, which is based on discovering the function of a gene by analysing
its phenotypic effects. However, some complications arise (such as incomplete disruption of
the targeted gene or polar effects on the downstream genes) because of the nature of
transposon mutagenesis. This problem can be avoided only by the set of in-frame deletion
mutants. Most bacteria are not transformable with linear DNA because of the presence of
intracellular exonucleases that degrade transformed linear DNA (Mori, 2004). It was shown
that many bacteriophages encode their own homologous recombination systems (Smith,
1988) and that the λ Red (g, b, exo) function promotes a greatly enhanced rate of
recombination over that exhibited by the recBC sbcB or recD mutants when using linear
DNA. So the λ Red system has been used as a basis for development of a convenient
procedure that provides an efficient way to isolate replacement mutants using PCR fragments
encoding an antibiotic resistance gene and having only 40 to 50 nucleotides of flanking
regions (Datsenko and Wanner, 2000) (Fig. 8).
3 For example the Protein Structure Initiative (http://www.structuralgenomics.org/)
4 Transfection is the process of introducing nucleic acids into cells by non-viral methods
(http://www.promega.com/paguide/chap12.htm)

23
Fig. 8. Construction of deletion mutants (Mori, 2004).
Some organisms have developed the ability to protect against gene deletions. This
ability is called genetic robustness and it has been demonstrated in some model oganisms,
such as S. cerevisiae. Its genome contains duplicates that often share similar functions, and
the loss of one paralog may be buffered by others (Hsiao and Vitkup, 2008). In spite of this
robustness, the genome of S. cerevisiae can still be used for functional characterization by the
technique of gene deletion. Winzeler et al. (1999) constructed a library of 6925 S. cerevisiae
strains, each with a precise deletion of one of 2026 ORFs (more than one-third of the ORFs in
the genome). 17 percent were essential for viability in rich medium. The phenotypes of more
than 500 deletion strains were assayed in parallel, 40 percent of them
showed quantitative
growth defects in either rich or minimal medium.
3.1.3 Transcriptome analysis
The transcriptome is the set of all RNA molecules (including mRNA, rRNA, tRNA,
and non-coding RNA) produced in the cell. Because it includes all mRNA transcripts in the
cell, the transcriptome reflects the genes that are being actively expressed at any given time.
For the analysis of global gene expression, many novel techniques have been developed,
including DNA microarray or DNA chip technology (Lockhart et al., 1996). A DNA
microarray is an orderly arrangement of tens to hundreds of thousands of unique DNA
molecules of known sequence, usually on a glass slide. DNA molecules can be individually
synthesised on a rigid silicon plate (DNA chips) or prepared from pre-synthesised DNA

24
(synthetic oligonucleotides or PCR products) that are spotted and immobilised on a slide
glass. DNA microarrays were first used to study E. coli gene regulation (Blattner et al., 1997)
but this technology has rapidly expanded and been applied to study various aspects of
transcriptional regulation (Tao et al., 1999). Microarrays present the accumulation of large
amounts of functional genomic information. The experimental design enables to show
respective internal and external contributions to the physiological state of a biological system
(Fig. 9).
Figure 9. Microarrays in systems analysis. Intrinsic and extrinsic forces affect biological processes such as gene
expression, which can be examined with microarrays (Schena, 1998).
This method also appeared to be helpful by revealing novel biochemical pathways.
When the genomes of the most studied organisms, E.coli and yeast, had been sequenced, it
was thought that the primary metabolic pathways, reactants, intermediates, products and
enzymes in these organisms were elucidated and fully understood. But with the help of
comparative genomics, Loh et al. showed that there is still an undiscovered central
biochemical pathway in E. coli K12 (Loh et al., 2006). The use of microarray technology
showed that the b1012 operon is highly expressed under the control of the transcriptional
activator nitrogen regulatory protein C (NtrC) and probably involved in catabolism of
alternative nitrogen sources (Zimmer, 2000). When Loh et al. tested strains carrying a mini
Tn5 insertion in several of the seven genes in the operon, he found that these mutants could
not grow on uracil and uridine as sole nitrogen source at room temperature, but the control
wild-type strain could grow. This experiment showed two surprising features that have been
overlooked so far: E. coli K12 can grow on uracil as the sole nitrogen source at room
temperature but not at 37.8 C and this phenotype is directly connected to the b1012 operon.

25
This operon has been then renamed to rut (pyrimidine utilization). Loh et al. then used the
databases to compare the seven deduced protein sequences with other sequences in the
database. This approach revealed that these seven genes are present in several proteobacteria,
such as Shigella, Yersinia and Agrobacterium, and that the Rut proteins are homologous to
enzymes characterised in other organisms. It was then possible to clearly deduce a probable
function for the final protein. The chemical analysis then completed the bioinformatics
approach. When E. coli K12 was grown on radioactively labelled substrates, a three-carbon
product was detected. It was obvious that the carbon atoms corresponding to the uracil
positions 4, 5 and 6, were predominantly secreted as 3-hydroxypropionic acid but the precise
enzymatic activities and intermediates in the reaction remained unknown (Loh et al., 2006).
However, the Rut homologous enzymes in other organisms enable the use of comparative
genomics to make speculations about the reaction mechanism that is alternative to the
reductive and oxidative pathway of degradation of pyrimidines (Fig.10).
Figure 10. Pyrimidine catabolic pathways. Known reductive (1) and oxidative (2) pathways for catabolism of
pyrimidine rings (A, upper and lower, respectively) and the novel pathway discovered by Loh et al. (2006),
adapted from Loh et al., 2006.
26
3.1.4 Proteome analysis
In prokaryotic organisms, in which related genes are often linked within an operon, the
use of the comparative genomics method for the assignment of protein function is a very
powerful tool. However, for the global characterisation of proteins encoded by an eukaryotic
genome, some additional technologies are needed. These are described by the field of
proteomics (Patterson et al., 2003). Proteomic methods quantify the abundance levels of
proteins. Nonetheless, many proteins (especially enzymes) are regulated by PTM mechanisms
in vivo (Kobe and Kemp, 1999), so it can happen that their translation levels do not correlate
with their activities. To deal with this problem, a chemical proteomic strategy has been
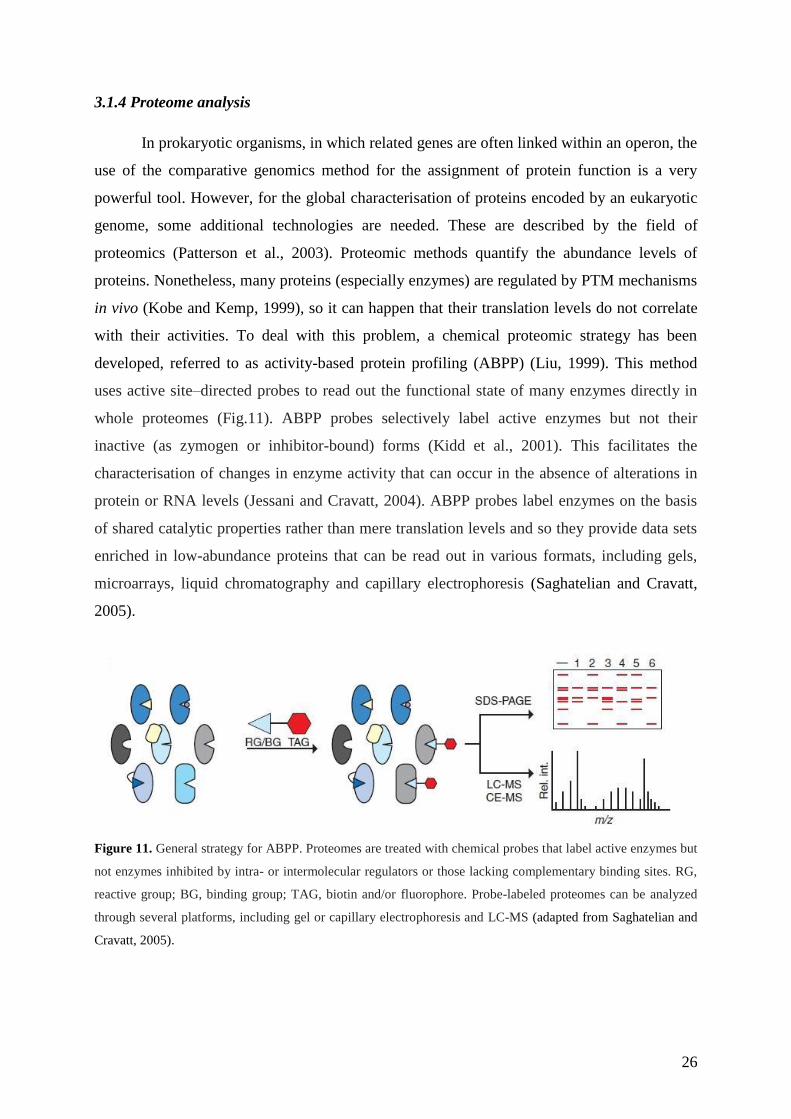
developed, referred to as activity-based protein profiling (ABPP) (Liu, 1999). This method
uses active site–directed probes to read out the functional state of many enzymes directly in
whole proteomes (Fig.11). ABPP probes selectively label active enzymes but not their
inactive (as zymogen or inhibitor-bound) forms (Kidd et al., 2001). This facilitates the
characterisation of changes in enzyme activity that can occur in the absence of alterations in
protein or RNA levels (Jessani and Cravatt, 2004). ABPP probes label enzymes on the basis
of shared catalytic properties rather than mere translation levels and so they provide data sets
enriched in low-abundance proteins that can be read out in various formats, including gels,
microarrays, liquid chromatography and capillary electrophoresis (Saghatelian and Cravatt,
2005).
Figure 11. General strategy for ABPP. Proteomes are treated with chemical probes that label active enzymes but
not enzymes inhibited by intra- or intermolecular regulators or those lacking complementary binding sites. RG,
reactive group; BG, binding group; TAG, biotin and/or fluorophore. Probe-labeled proteomes can be analyzed
through several platforms, including gel or capillary electrophoresis and LC-MS (adapted from Saghatelian and
Cravatt, 2005).

27
3.1.5 Metabolome analysis
The metabolome can be described as the total set of metabolites in a cell (Tweeddale
et al., 1998). The field of metabolomics has emerged with the objective to advance new
technologies that can profile the molecular composition of cells and tissues in order to
characterise the endogenous substrates of enzymes. The challenges that arise in this field are
caused by the fact that metabolites are not directly linked to the genetic code, but are instead
the products of complex enzymatic networks. Also, they are not linear polymers composed of
a defined set of monomers, but rather a diverse collection of structures with different chemical
and physical properties. These features have inspired the development of several approaches
for metabolome analysis, such as flux analysis using isotopic tracer, pathway reconstruction
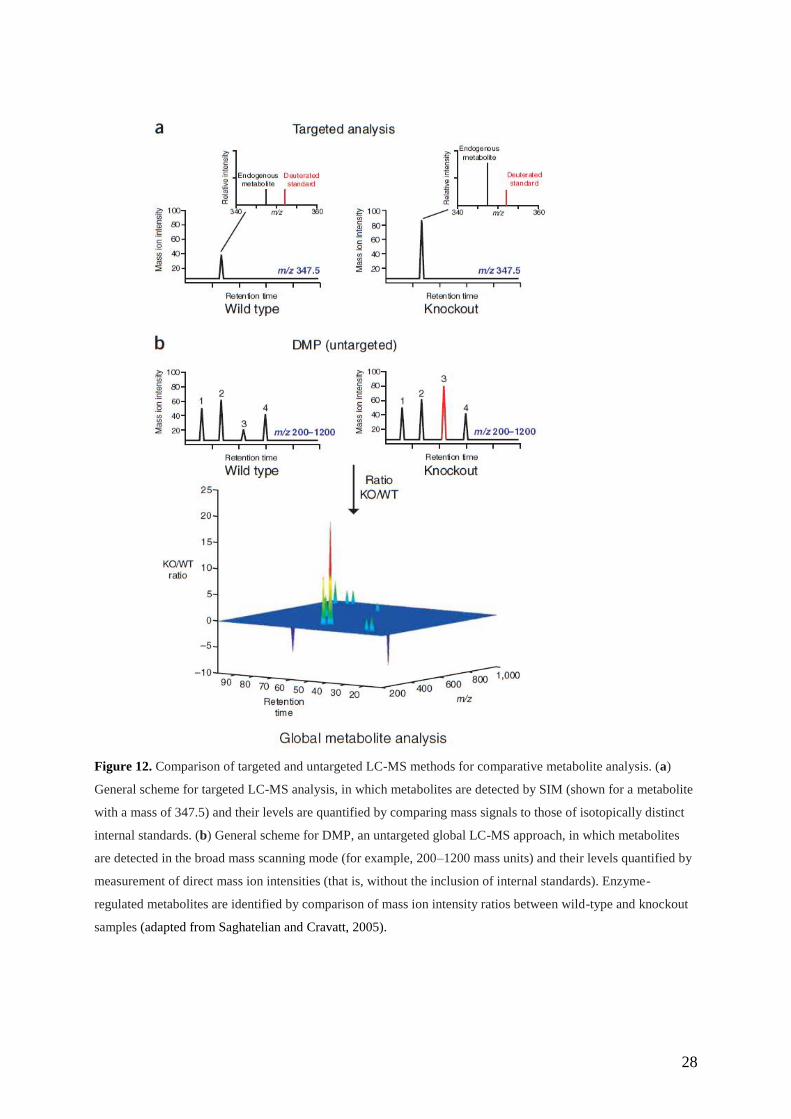
and in particular, metabolite profiling. The method of metabolite profiling (Fig.12) can be
either targeted (to quantify known metabolites in complex biological samples) or untargeted
(to report on the full spectrum of substrates used by a given enzyme in vivo, potentially
including the discovery of new metabolites) (Saghatelian and Cravatt, 2005).
The approach of global metabolite profiling has been used for functional genomics
studies in plants (Fiehn et al., 2000) and yeast (Raamsdonk et al., 2001). In E. coli,
metabolites were labeled with C-14 glucose and identified by 2D thin-layer chromatography
after extraction by cold methanol (Maharjan and Ferenci, 2003). Recently, a powerful
analytical method using capillary electrophoresis-electrospray ionization mass spectrometry
(CE-ESI-MS) has been developed. This dramatically increases the number of metabolites that
can be measured simultaneously (Soga et al., 2003).
28
Figure 12. Comparison of targeted and untargeted LC-MS methods for comparative metabolite analysis. (a)
General scheme for targeted LC-MS analysis, in which metabolites are detected by SIM (shown for a metabolite
with a mass of 347.5) and their levels are quantified by comparing mass signals to those of isotopically distinct
internal standards. (b) General scheme for DMP, an untargeted global LC-MS approach, in which metabolites
are detected in the broad mass scanning mode (for example, 200–1200 mass units) and their levels quantified by
measurement of direct mass ion intensities (that is, without the inclusion of internal standards). Enzyme-
regulated metabolites are identified by comparison of mass ion intensity ratios between wild-type and knockout
samples (adapted from Saghatelian and Cravatt, 2005).

29
3.2 Metagenomics - approach for studying complex communities in natural environment
Historically, the study of organisms (notably microbes) has focused on single species
in pure culture. Therefore, understanding of complex communities of these organisms has
been perceived only through understanding of their individual members. However, their
functions are often conducted within complex communities - intricate, balanced, and
integrated entities that adapt swiftly and flexibly to environmental change. Metagenomics,
still a very new science, has emerged to overcome these obstacles of unculturability and
genomic diversity of most microbes, the understanding of wich is essential for advancement
in clinical and environmental microbiology. In Greek, meta means ‗transcendent‘. This shows
that metagenomics focuses on transcending the individual organism to pinpoint the genes in
the community and the influence of each other‘s activities in serving collective functions.
In a metagenomics study, DNA is extracted directly from the natural environment. The
mixed sample can be then analysed directly or cloned into a form maintainable in laboratory
bacteria. This enables the creation of a library that contains the genomes of all the microbes
found in the environment (Fig. 13). This library is not organised into neat volumes, each
containing the genome of one community member. Instead, it is composed of millions of
clones, each holding a random fragment of DNA. These DNA fragments are translated into
proteins by bacteria growing in the laboratory. Clones producing ―foreign‖ proteins are then
tested for various capabilities, such as vitamin production or antibiotic resistance. This
enables researchers to discover new antibiotics and resistance mechanisms without knowing
anything about the underlying gene sequence, the structure of the desired protein, or the
microbe of origin (National Academy of Sciences, 2007).
An example of an application of metagenomic approach is a study of human
microbiome. The term ‗microbiome‗ refers to the fact that the human body provides an
environment for our 100 trillion microbial partners. Considering that we contain perhaps 10
times more microbial than human cells and at least 100 times more microbial than human
genes, it is obvious that human metabolome is actually a mixture of human and microbial
parts (National Academy of Sciences, 2007). The first truly metagenomic study of the human
microbiome consisted of sequencing the microbial communities taken from the colons of two
healthy adults. Further analysis of 78 million base pairs of unique DNA sequence showed
that, in comparison with previously sequenced human and microbial genomes, the gut
metagenome contains genes responsible for the breakdown of otherwise indigestible plant-
derived polysaccharides that form a substantial part of modern diets, as well as for the

30
detoxification of consumed xenobiotics and the synthesis of essential amino acids and
vitamins (Gill et al., 2006).
Figure 13. Construction of a metagenomic library: 1 - prokaryotic cells are extracted from the environmental
sample (e.g. soil, sediment, seawater), 2 - the total DNA content (the metagenome) is extracted and purified, 3 -
DNA fragments are cloned into a cloning vector and transformed into a host cell, 4 - the result is thousands of
cells, each carrying a DNA fragment from the metagenome (adapted from
http://wiki.biomine.skelleftea.se/wiki/index.php/Metagenomics).
The metagenomic approach gives an idea of the future, in which it may be possible to
optimise the nutritional requirements of overfed or underfed people on the basis of their gut
microbial ecology, or to forecast the risk of particular types of cancer for individuals or
populations. Researchers in this field are led by programs, such as Human Gut Microbiome
Initiative (HGMI) or Human Microbiome Project (HMP), that are trying to develop
a reference set of microbial genome sequences and preliminary characterisation of human
microbiome (National Institutes of Health, USA, 2008). Greater knowledge of the microbial
communities in other parts of human body, such as the oral cavity, skin or female
reproductive tract will also improve our ability to prevent, diagnose, and treat diseases at
those sites.

31
Summary
It has been only a few decades since the field of genomics appeared, yet it has become
a very popular scientific study among molecular biologists as well as biochemists. In these
few years, the field of genomics has been subjected to turbulent evolution and has given rise
also to many other fields, which are presented in this work.
At the beginning of the genomic era, the main goal of scientists was to sequence the
genomes of numerous organisms. But once the sequencing has been done, further exploration
of the sequenced genomes has become a challenge. The field of comparative genomics has
evolved in order to identify the missing genes and to make the complete image of the
genomes. The field of transcriptomics has started to examine the levels of gene expression in
a given cell population. And the field of proteomics has focused on the structures and
functions of the protein complement of a cell.
However, the biggest challenge in biology has remained: to determine gene function.
To achieve this goal today, scientists can take advantage of the vast data produced by
genomic and proteomic projects. In addition, the complete set of ORF clones as well as the
libraries of knock-out mutants showed to be very helpful. The resulting organisms (either with
an extra gene or with a deleted/disrupted gene) can be screened for phenotypes that provide
clues to the function of the studied gene.
Metabolomics presents the next step of the systematic study as it aims to analyse the
complete set of metabolites. Thus, while transcriptomic and proteomic analyses do not tell the
whole story of what might be happening in a cell, metabolic profiling can give an
instantaneous snapshot of the physiology of the cell.
The metagenomic approach could be placed on the top of this pyramid as it goals to
study all the aforementioned points within complex communities and in their natural
environment.
In biochemical research, we have witnessed the progressive transition from the
application of pre-genomic methods, such as radioisotope labeling, to genome sequencing and
advanced methods used for further genome, proteome and metabolome analysis. Using these
methods, we can get a better understanding of the physiology in any organism. In addition,
the obtained complexe knowledge of the cell physiology will improve our ability to prevent,
diagnose, and treat various diseases.

32
Zhrnutie
Je to len pár desaťročí, čo vznikol nový študijný odbor genomika, avšak za ten čas sa
tento odbor stihol stať veľmi obľúbeným medzi molekulárnymi biológmi ako aj medzi
biochemikmi. Za tých pár rokov prešiel biochemický výskum turbulentnou evolúciou
a zapríčinil vznik mnohých ďalších odborov, ktoré sú prezentované v tejto práci.
Na počiatku genomickej éry bolo hlavným cieľom vedcov sekvenovať genómy
mnohých organizmov. Ale potom ako bolo sekvenovanie spravené, novou výzvou sa stal
hlbší výskum týchto genómov. Vznikol nový odbor, komparatívna genomika, za účelom
identifikovania chýbajúcich génov a vytvorenia kompletného obrazu genómu. Odbor
transkriptomiky začal skúmať úrovne génovej expresie v daných bunkových populáciách a
odbor proteomiky sa zase sústredil na štruktúry a funkcie proteínových súčastí bunky.
Avšak najväčšia výzva v molekulárnej biológii ostala: určiť funkciu génu.
V súčastnosti na dosiahnutie tohto cieľa vedci môžu využívať rozsiahle databázy, ktoré sú
výsledkom mnohých genomických a proteomických projektov. Taktiež ako veľmi nápomocné
sa ukázali kompletné sety ORF klonov ako aj genomické knižnice knock-out mutantov.
Výsledné organizmy (buď s génom navyše alebo s odstráneným génom) môžu byť testované
na fenotypy, ktoré nám poskytujú kľúč k funkcii študovaného génu.
Metabolomika predstavuje ďalší krok v tomto systematickom výskume, nakoľko sa
zameriava na analýzu kompletného setu metabolitov. Vďaka tomu, zatiaľ čo transkriptomické
a proteomické analýzy nám nepovedia všetko, čo sa deje v bunke, tak metabolické
profilovanie nám poskytuje okamžitú ukážku fyziológie bunky.
Na vrchol tejto genomickej pyramídy by mohol byť umiestnený metagenomický
prístup, nakoľko jeho cieľom je výskum všetkých vyššie uvedených bodov v rámci
komplexných komunít v ich prirodzenom prostredí.
Boli sme svedkom postupného vývoja biochemického výskumu, od využívania pre-
genomických metód, ako je napr. rádioizotopické značenie, až po sekvenovanie genómu
a používanie pokročilejších metód pre hlbšiu analýzu genómu, proteómu a metabolómu.
S použitím týchto metód budeme môcť získať lepšie porozumenie o fyziológii v každom
študovanom organizme. Vďaka tomu, tieto získané komplexné vedomosti o fyziológii bunky
tiež zlepšia naše schopnosti predchádzať rôznym chorobám, ako aj rozšíria možnosti ich
včasnej diagnostiky a liečenia.

33
Used abbreviations
ABPP - activity-based protein profiling
CBA - chlorobenzoate
DMP - discovery metabolite profiling
EST – expressed sequence tags
LC-MS - liquid chromatography – mass spectrometry
NIH - National Institutes of Health
ORF – open reading frames
PCR – polymerase chain reaction
PPI – protein-protein interactions
PTM – post-translational modifications
SIM - selected ion monitoring
Y2H – yeast-2-hybrid

34
References
Ackermann, D. (1931). Über den biologischen Abbau des Arginins zu Citrullin. Biochem.
Z., 203, 66–69
Adams, M. D., Celniker, S. E., Holt, R. A., Evans, C. A., Gocayne, J. D., Amanatides, P. G.,
Scherer, S. E., Li, P. W., Hoskins, R. A., & Galle, R. F. (2000). The genome sequence of
Drosophila melanogaster. Science, 287(5461), 2185-2195.
Arabidopsis Genome Initiative. (2000). Analysis of the genome sequence of the flowering
plant Arabidopsis thaliana. Nature, 408(6814), 796-815.
Bateman, A., & Haft, D. H. (2002). HMM-based databases in InterPro. Brief Bioinform, 3,
236-245.
Blattner, F. R., Plunkett, G., 3rd, Bloch, C. A., Perna, N. T., Burland, V., Riley, M.,
Collado-Vides, J., Glasner, J. D., Rode, C. K., Mayhew, G. F. et al. (1997). The complete
genome sequence of Escherichia coli K-12. Science, 277, 1453-1474.
Boguski, M. S., Elshourbagy, N., Taylor, J. M., & Gordon, J. I. (1984). Rat apolipoprotein
A-IV contains 13 tandem repetitions of a 22-amino acid segment with amphipathic helical
potential. Proc Natl Acad Sci USA, 81(16), 5021-5025.
C. elegans Sequencing Consortium. (1998). Genome sequence of the nematode C. elegans:
a platform for investigating biology. Science, 282(5396), 2012-2018.
Calvin, M. (1962). The Path of Carbon in Photosynthesis. Science, 135(3507), 879-889
Datsenko, K. A. & Wanner, B. L. (2000). One-step inactivation of chromosomal genes in
Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA, 97, 6640-6645.
Enright, A. J., Iliopoulos, I., Kyrpides, N. C., & Ouzounis, C. A. (1999). Protein interaction
maps for complete genomes based on gene fusion events. Nature, 402, 86-90.
Falquet, L., Pagni, M., Bucher, P., Hulo, N., Sigrist, C. J., Hofmann, K., & Bairoch, A.
(2002). The PROSITE database, its status in 2002. Nucleic Acids Res, 30, 235-238.
Fiehn, O., Kopka, J., Dormann, P., Altmann, T., Trethewey, R. N. & Willmitzer, L. (2000).
Metabolite profiling for plant functional genomics. Nat. Biotechnol., 18, 1157-1161.
Fields, S., & Song, O. (1989). A novel genetic system to detect protein-protein interactions.
Nature, 340(6230), 245-246.
Fiers, W., Contreras, R., Duerinck, F., Haegeman, G., Iserentant, D., Merregaert, J., Min
Jou, W., Molemans, F., Raeymaekers, A., Van den Berghe, A., Volckaert, G., & Ysebaert,
M. (1976). Complete nucleotide sequence of bacteriophage MS2 RNA: primary and
secondary structure of the replicase gene. Nature, 260 (5551), 500–507

35
Fleischmann, R., Adams, M., White, O., Clayton, R., Kirkness, E., Kerlavage, A., Bult, C.,
Tomb, J. F., Dougherty, B. A., Merrick, J. M., et al. (1995). Whole-genome random
sequencing and assembly of Haemophilus influenzae Rd. Science, 269, 496-512.
Gill, S. R., Pop, M., Deboy, R. T., Eckburg, P. B., Turnbaugh, P. J., Samuel, B. S., Gordon,
J. I., Relman, D. A., Fraser-Liggett, C. M., & Nelson, K. E. (2006). Metagenomic analysis
of the human distal gut microbiome. Science, 312 (5778), 1355-1369.
Goffeau, A., Barrell, B. G., Bussey, H., Davis, R. W., Dujon, B., Feldmann, H., Galibert, F.,
Hoheisel, J. D., Jacq, C., Johnston, M., Louis, E. J., Mewes, H. W., Murakami, Y.,
Philippsen, P., Tettelin, H., & Oliver, S. G. (1996). Life with 6000 genes. Science,
274(5287), 563-567.
Harada, J.N., Bower, K.E., Orth, A.P., Callaway, S., Nelson, C.G., Laris, C., Hogenesch,
J.B., Vogt, P.K., & Chanda, S.K. (2005). Identification of novel mammalian growth
regulatory factors by genome-scale quantitative image analysis. Genome Res, 15, 1136-
1144.
Hsiao, T-L., & Vitkup, D. (2008). Role of Duplicate Genes in Robustness against
Deleterious Human Mutations. PLoS Genet, 4(3), e1000014
Jessani, N. & Cravatt, B.F. (2004). The development and application of methods for activity
based protein profiling. Curr. Opin. Chem., Biol. 8, 54–59.
Kidd, D., Liu, Y. & Cravatt, B.F. (2001). Profiling serine hydrolase activities in complex
proteomes. Biochemistry, 40, 4005–4015.
Kobe, B. & Kemp, B.E. (1999). Active site-directed protein regulation. Nature, 402, 373–
376.
Kornberg, H. (2000). Krebs and his trinity of cycles. Nat Rev Mol Cell Biol., 1(3), 225-228.
Kornberg, H. L., & Krebs, H. A. (1957). Synthesis of cell constituents from C2-units by a
modified tricarboxylic acid cycle. Nature, 179, 988–991
Kossel, A., & Dakin, H. D. (1904). Über die Arginase. Z. Physiol. Chem., 41, 321–331
Krebs, H. A., & Henseleit, K. (1932). Untersuchungen über die Harnstoffbildung im
Tierkorper. Z. Physiol. Chem., 210, 33–66
Krebs, H. A., & Johnson, W. A. (1937a). Metabolism of ketonic acids in animal tissues.
Biochem. J., 31, 645–660
Krebs, H. A., & Johnson, W. A. (1937b). The role of citric acid in intermediate metabolism
in animal tissues. Enzymologia 4, 148–156
Liu, Y., Patricelli, M.P. & Cravatt, B.F. (1999). Activity-based protein profiling: the serine
hydrolases. Proc. Natl. Acad. Sci. USA, 96, 14694–14699.

36
Lockhart, D. J., Dong, H., Byrne, M. C., Follettie, M. T., Gallo, M. V., Chee, M. S.,
Mittmann, M., Wang, C., Kobayashi, M., Horton, H. & Brown, E. L. (1996). Expression
monitoring by hybridization to high-density oligonucleotide arrays. Nat. Biotechnol., 14,
1675-1680.
Loh, K. D., Gyaneshwar, P., Markenscoff Papadimitriou, E., Fong, R., Kim, K., Parales, R.,
Zhou, Z., Inwood, W., & Kustu, S. (2006). A previously undescribed pathway for
pyrimidine catabolism. PNAS, 103(13), 5114-5119.
Luan, C.H., Qiu, S., Finley, J.B., Carson, M., Gray, R.J., Huang, W., Johnson, D., Tsao, J.,
Reboul, J., Vaglio, P., et al. (2004). High-throughput expression of C. elegans proteins.
Genome Res., 14(10B), 2102-2110.
Maharjan, R. P. & Ferenci, T. (2003). Global metabolite analysis: the influence of
extraction methodology on metabolome profiles of Escherichia coli. Anal. Biochem., 313,
145-154.
McGuire, A. M., Hughes, J. D., & Church, G. M. (2000). Conservation of DNA regulatory
motifs and discovery of new motifs in microbial genomes. Genome Res, 10, 744-757.
Min Jou, W., Haegeman, G., Ysebaert, M., & Fiers, W. (1972). Nucleotide sequence of the
gene coding for the bacteriophage MS2 coat protein. Nature, 237(5350), 82–88.
Mori, H. (2004). From the sequence to cell modeling: comprehensive functional genomics
in Escherichia coli. J Biochem Mol Biol., 37(1), 83-92.
Mouse Genome Sequencing Consortium. (2002). Initial sequencing and comparative
analysis of the mouse genome. Nature, 420(6915), 520-562.
Nagase, T., Yamakawa, H., Tadokoro, S., Nakajima, D., Inoue, S., Yamaguchi, K., Itokawa,
Y., Kikuno, R.F., Koga, H., & Ohara, O. (2008). Exploration of human ORFeome: high-
throughput preparation of ORF clones and efficient characterization of their protein
products. DNA Res., 15(3), 137-49.
Neidhart, D.J., Kenyon, G.L., Gerlt, J.A. & Petsko, G.A. (1990). Mandelate racemase and
muconate lactonizing enzyme are mechanistically distinct and structurally homologous.
Nature, 347, 692–694.
Osterman, A., & Overbeek, R. (2003). Missing genes in metabolic pathways: a comparative
genomics approach. Curr. Opin. Chem, Biol. 7, 238–251.
Overbeek, R., Fonstein, M., D‘Souza, M., Pusch, G. D., & Maltsev, N. (1999). The use of
gene clusters to infer functional coupling. Proc Natl Acad Sci USA, 96, 2896-2901.
Patterson, S.D. & Aebersold, R. (2003). Proteomics: the first decade and beyond. Nat.
Genet., 33, 311–323.
Pawlowski, K., Rychlewski, L., Zhang, B., & Godzik, A. (2001). Fold predictions for
bacterial genomes. J Struct Biol, 134, 219-231.

37
Pellegrini, M., Marcotte, E. M., Thompson, M. J., Eisenberg, D., & Yeates, T. O. (1999).
Assigning protein functions by comparative genome analysis: protein phylogenetic profiles.
Proc Natl Acad Sci USA, 96, 4285-4288.
Pennacchio, L. A., Olivier, M., Hubacek, J. A., Cohen, J. C., Cox, D. R., Fruchart, J. C.,
Krauss, R. M., & Rubin, E. M. (2001). An apolipoprotein influencing triglycerides in
humans and mice revealed by comparative sequencing. Science, 294(5540), 169-173.
Raamsdonk, L. M., Teusink, B., Broadhurst, D., Zhang, N., Hayes, A., Walsh, M. C.,
Berden, J. A., Brindle, K. M., Kell, D. B., Rowland, J. J., et al. (2001). A functional
genomics strategy that uses metabolome data to reveal the phenotype of silent mutations.
Nat. Biotechnol., 19, 45-50.
Roberts, R. B., Abelson, P. H., Cowie, D. B., Bolton, E. B., & Britten, J. R. (1955). Studies
of Biosynthesis in Escherichia coli. Carnegie Institution of Washington, Publication 607
Saghatelian, A., & Cravatt, B. F. (2005). Assignment of protein function in the postgenomic
era. Nat Chem Biol., 1(3), 130-142.
Sanger, F., Air, G. M., Barrell, B. G., Brown, N. L., Coulson, A. R., Fiddes, C. A.,
Hutchison, C. A., Slocombe, P. M., & Smith, M. (1977). Nucleotide sequence of
bacteriophage phi X174 DNA. Nature, 265(5596), 687–695
Scriver, C.R. (2001). Garrod's foresight; our hindsight. J. Inherit. Metab. Dis., 24, 93-116
Schena, M., Heller, R.A., Theriault, T.P., Konrad, K., Lachenmeier, E., & Davis, R.W.
(1998). Microarrays: biotechnology's discovery platform for functional genomics. Trends
Biotechnol., 16(7), 301-306.
Smith, G. R. (1988). Homologous recombination in procaryotes. Microbiol. Rev., 52, 1-28.
Soga, T., Ohashi, Y., Ueno, Y., Naraoka, H., Tomita, M. & Nishioka, T. (2003).
Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J.
Proteome Res., 2, 488-494.
Stein, L. D., Bao, Z., Blasiar, D., Blumenthal, T., Brent, M. R., Chen, N., Chinwalla, A.,
Clarke, L., Clee, C., Coghlan, A., et al. (2003). The genome sequence of Caenorhabditis
briggsae: a platform for comparative genomics. PLoS Biol, 1(2), e45.
Talmud, P. J., Hawe, E., Martin, S., Olivier, M., Miller, G. J., Rubin, E. M., Pennacchio,
L.A., & Humphries, S. E. (2002). Relative contribution of variation within the
APOC3/A4/A5 gene cluster in determining plasma triglycerides. Hum Mol Genet., 11(24),
3039-3046.
Tao, H., Bausch, C., Richmond, C., Blattner, F. R. & Conway, T. (1999). Functional
genomics: expression analysis of Escherichia coli growing on minimal and rich media. J.
Bacteriol., 181, 6425-6440.

38
Temple, G., Lamesch, P., Milstein, S., Hill, D.E., Wagner, L., Moore, T., & Vidal, M.
(2006). From genome to proteome: developing expression clone resources for the human
genome, Hum. Mol. Genet.,15, R31–R43.
The National Academy of Sciences. (2007). The New Science of Metagenomics: Revealing
the Secrets of Our Microbial Planet. The National Academies Press, ISBN: 0-309-10677-X
Tweeddale, H., Notley-McRobb, L. & Ferenci, T. (1998). Effect of slow growth on
metabolism of Escherichia coli, as revealed by global metabolite pool (―metabolome‖)
analysis. J. Bacteriol., 180, 5109-5116.
Wada, M. (1930). Über Citrullin, eine neue Aminosaure im presssaft der Wassermelone,
Citrullis vulgaris schrad. Biochem. Z., 224, 420–429
Watson, J. D., & Crick, F. H. C. (1953). A structure for deoxyribose nucleic acid. Nature,
171-173.
Winzeler, E.A., Shoemaker, D.D., Astromoff, A., Liang, H., Anderson, K., Andre, B.,
Bangham, R., Benito, R., Boeke, J.D., & Bussey, H. et al., (1999). Functional
characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science,
285(5429), 901-906.
Zhuang, Z., Gartemann, K.H., Eichenlaub, R. & Dunaway-Mariano, D. (2003).
Characterization of the 4-hydroxybenzoyl-coenzyme A thioesterase from Arthrobacter sp.
strain SU. Appl. Environ. Microbiol., 69, 2707–2711
Zimmer, D. P., Soupene, E., Lee, H. L., Wendisch, V. F., Khodursky, A. B., Peter, B. J.,
Bender, R. A. & Kustu, S. (2000). Nitrogen regulatory protein C-controlled genes of
Escherichia coli: scavenging as a defense against nitrogen limitation. Proc. Natl. Acad. Sci.
USA, 97, 14674–14679.
Internet sources:
Human Genome Program, U.S. Department of Energy, To Know Ourselves. [online] 1996.
[cit.18.12.2009]. Available from
<http://www.ornl.gov/sci/techresources/Human_Genome/publicat/tko/>
Metagenomic library. [online] [cit.10.2.2010]. Available from
<http://wiki.biomine.skelleftea.se/wiki/index.php/Image:Metagenomic_library.jpg>
National Institutes of Health, USA. [online] Dec. 2008. [cit. 28.2.2010]. Available from
<http://nihroadmap.nih.gov/hmp/initiatives.asp>
Omes and omics in Biotechnology and Bioscience. [online] [cit.29.1.2010]. Available from
< http://omics.org>
Protein Structure Initiative. [online] [cit.12.2.2010]. Available from
<http://www.structuralgenomics.org/>


















