
Use of Multicolor Spectral Karyotyping in Genetic … of Multicolor Spectral...Use of Multicolor...
8
Use of Multicolor Spectral Karyotyping in Genetic Analysis of Pleuropulmonary Blastoma MAJA BARNARD, 1 JANE BAYANI, 2,3 RONALD GRANT, 4 IKUKO TESHIMA, 1,3 PAUL THORNER, 1,3 AND JEREMY SQUIRE 1,2,3 * 1 Department of Pediatric Laboratory Medicine, The Hospital for Sick Children, 555 University Avenue, Toronto, Ontario, Canada M5G 1X8 2 Ontario Cancer Institute, Princess Margaret Hospital, 610 University Avenue, University Health Network, Toronto, Ontario, Canada M5G 2M9 3 Department of Laboratory Medicine and Pathobiology, Banting Institute Faculty of Medicine, University of Toronto, 100 College Street, Toronto, Ontario, Canada M5G 1L5 4 Department of Pediatrics, The Hospital for Sick Children, 555 University Avenue, Toronto, Ontario, Canada M5G 1X8 Received April 8, 1999; accepted August 20, 1999. ABSTRACT Pleuropulmonary blastoma (PPB) is a rare, malignant intrathoracic pediatric tumor. It arises from the lung, pleura, or mediastinum and its pathogenesis and rela- tionship to other pediatric solid tumors is not well un- derstood. In this study, a case of PPB in a 3-year-old girl was studied using a combination of molecular genetic methods and cytogenetics. Molecular analysis of the commonly encountered fusion translocation gene prod- ucts of pediatric solid tumors failed to detect a rear- rangement. Cytogenetic analysis, supplemented by mul- ticolor spectral karyotyping (SKY), identified an unbalanced translocation between chromosomes 1 and X, resulting in additional copies of 1q, an extra copy of Xq, and loss of part of Xp. In addition, trisomy 8 was detected. The identification of new chromosomal alter- ations and confirmation of previously reported ones in this rare neoplasm helps to improve our understanding of its pathogenesis and association with other pediatric tumors. Key words: pleuropulmonary blastoma, spectral karyo- typing, chromosome translocation INTRODUCTION Pleuropulmonary blastoma (PPB) is a rare and highly malignant intrathoracic childhood neo- plasm. It arises from the lung, pleura, or medias- tinum with up to 25% of cases being extrapulmo- nary [1]. The neoplastic component is purely mesenchymal in phenotype. Unlike classic adult- type pulmonary blastoma, there is no malignant epithelial component. Three types are recognized, with type 1 being multicystic, type 2 containing both cystic and solid areas, and type 3 being exclu- sively solid. Some of the recurrent chromosomal alterations in eight reported cases include trisomy 8 in seven cases [1– 4], partial trisomy 2 in two cases [4,5], and partial deletion of chromosome 17 in three cases [1,3]. Most common pediatric tu- mors are associated with consistent chromosomal abnormalities that provide insight into the patho- genesis of these neoplasms (reviewed in [6]). PPB is an uncommon tumor and its pathogenesis and relationship to other pediatric tumors is not well understood. To help characterize this neoplasm at *Corresponding author, at Princess Margaret Hospital/Ontario Cancer Institute, 610 University Avenue, Room 9-721, Toronto, Ontario, Canada M5G 2M9. Pediatric and Developmental Pathology 3, 479 – 486, 2000 DOI: 10.1007/s100240010094 Pediatric and Developmental Pathology © 2000 Society for Pediatric Pathology
Transcript of Use of Multicolor Spectral Karyotyping in Genetic … of Multicolor Spectral...Use of Multicolor...

Use of Multicolor Spectral Karyotyping inGenetic Analysis of PleuropulmonaryBlastoma
MAJA BARNARD,1 JANE BAYANI,2,3 RONALD GRANT,4 IKUKO TESHIMA,1,3
PAUL THORNER,1,3 AND JEREMY SQUIRE1,2,3*1Department of Pediatric Laboratory Medicine, The Hospital for Sick Children, 555 University Avenue, Toronto,Ontario, Canada M5G 1X82Ontario Cancer Institute, Princess Margaret Hospital, 610 University Avenue, University Health Network, Toronto,Ontario, Canada M5G 2M93Department of Laboratory Medicine and Pathobiology, Banting Institute Faculty of Medicine, University ofToronto, 100 College Street, Toronto, Ontario, Canada M5G 1L54Department of Pediatrics, The Hospital for Sick Children, 555 University Avenue, Toronto, Ontario,Canada M5G 1X8
Received April 8, 1999; accepted August 20, 1999.
ABSTRACTPleuropulmonary blastoma (PPB) is a rare, malignantintrathoracic pediatric tumor. It arises from the lung,pleura, or mediastinum and its pathogenesis and rela-tionship to other pediatric solid tumors is not well un-derstood. In this study, a case of PPB in a 3-year-old girlwas studied using a combination of molecular geneticmethods and cytogenetics. Molecular analysis of thecommonly encountered fusion translocation gene prod-ucts of pediatric solid tumors failed to detect a rear-rangement. Cytogenetic analysis, supplemented by mul-ticolor spectral karyotyping (SKY), identified anunbalanced translocation between chromosomes 1 andX, resulting in additional copies of 1q, an extra copy ofXq, and loss of part of Xp. In addition, trisomy 8 wasdetected. The identification of new chromosomal alter-ations and confirmation of previously reported ones inthis rare neoplasm helps to improve our understandingof its pathogenesis and association with other pediatrictumors.
Key words: pleuropulmonary blastoma, spectral karyo-typing, chromosome translocation
INTRODUCTIONPleuropulmonary blastoma (PPB) is a rare andhighly malignant intrathoracic childhood neo-plasm. It arises from the lung, pleura, or medias-tinum with up to 25% of cases being extrapulmo-nary [1]. The neoplastic component is purelymesenchymal in phenotype. Unlike classic adult-type pulmonary blastoma, there is no malignantepithelial component. Three types are recognized,with type 1 being multicystic, type 2 containingboth cystic and solid areas, and type 3 being exclu-sively solid. Some of the recurrent chromosomalalterations in eight reported cases include trisomy8 in seven cases [1–4], partial trisomy 2 in twocases [4,5], and partial deletion of chromosome 17in three cases [1,3]. Most common pediatric tu-mors are associated with consistent chromosomalabnormalities that provide insight into the patho-genesis of these neoplasms (reviewed in [6]). PPBis an uncommon tumor and its pathogenesis andrelationship to other pediatric tumors is not wellunderstood. To help characterize this neoplasm at
*Corresponding author, at Princess Margaret Hospital/Ontario CancerInstitute, 610 University Avenue, Room 9-721, Toronto, Ontario,Canada M5G 2M9.
Pediatric and Developmental Pathology 3, 479–486, 2000
DOI: 10.1007/s100240010094
Pediatric and Developmental Pathology
© 2000 Society for Pediatric Pathology

the genetic level, we examined a case of PPB in a3-year-old girl using molecular methods and cyto-genetic analysis including the use of the novel tech-nique of multicolor spectral karyotyping (SKY).
CASE REPORTA 3-year-old girl presented with a long-standinghistory of failure to thrive and a brief history ofprogressive shortness of breath. Chest X-ray dem-onstrated a left-sided cystic mass with a pleuraleffusion and compression of the airways. She un-derwent a left thoracotomy and a complete resec-tion of a pulmonary mass. Following the diagnosisof type 2 PPB on histologic examination, she wastreated with chemotherapy consisting of four cy-cles of VAdriaC and four cycles of Ifosfamide andVP-16. She tolerated chemotherapy reasonablywell, with several hospital admissions for fever andneutropenia. A CT scan performed 7 months latershowed no evidence of tumor recurrence.
METHODSSamples of the tumor were frozen and then sub-mitted for electron microscopy, cytogenetic, andmolecular analysis, and the remainder was fixed informalin. Immunohistochemical studies were per-formed on representative formalin-fixed, paraffin-embedded sections using an avidin-biotin-peroxi-dase complex method.
CytogeneticsCytogenetic studies were carried out on a short-term culture prepared according to standard pro-tocols using colcemid and hypotonic KCl solution.Cells in suspension were harvested after 1 day andthe in situ cultures after 5 days. Five metaphasesfrom one in situ culture slide were available foranalysis. Karyotype analysis was performed usingstandard techniques [7]. Three G-banded meta-phase cells were analyzed to identify consistentchromosomal abnormalities and to characterizemarker chromosomes.
Spectral karyotypingThe commercially available SKY™ kit from Ap-plied Spectral Imaging (ASI, Carlsbad, CA) wasused for spectral karyotyping. The slide treatment,posthybridization detection, and washes weredone according to standard protocols and manu-
facturer’s instructions [8]. SKY analysis was per-formed on five metaphases in which chromosomeswere also identified by RGB display image andinverted DAPI banding methods. Images werestored in a computer for color classification andfurther analysis using the SKYVIEW software(ASI), and for comparison with the findings fromG-banding.
RESULTSPathologyA segment of lung tissue weighing 20 g and mea-suring 5.5 3 5 3 3 cm was received. It contained apleural-based 5 3 3.5 3 3 cm nodule. On bisectingthe nodule, a 3.5-cm cyst was identified, containingmultiple soft, tan and blue-black nodules measur-ing up to 1.1 cm in diameter. The cyst was partiallylined by trabeculated and papillary tan tissue.
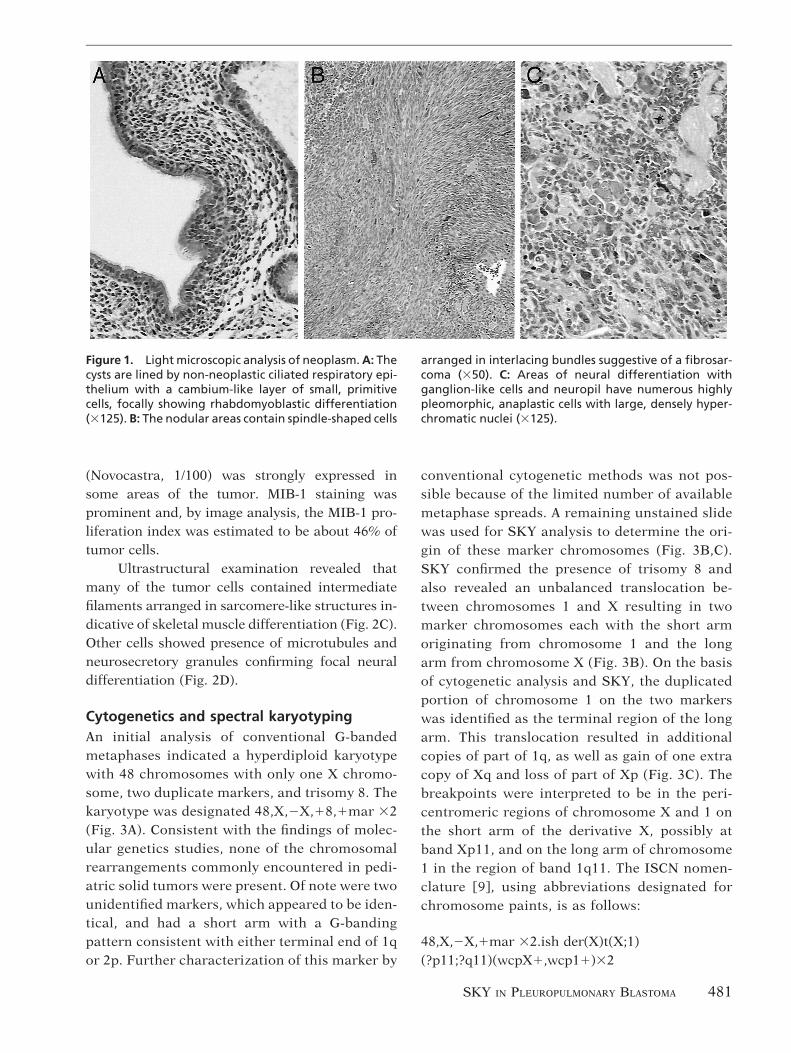
Microscopic examination showed a neoplasmcomposed of both epithelium-lined cysts and solidnodules. The cysts were lined by non-neoplasticciliated respiratory epithelium. Beneath the epi-thelium, small, primitive cells with ovoid to spin-dle-shaped nuclei and minimal cytoplasm were ar-ranged in a cambium-like layer (Fig. 1A). Furtheraway, these primitive cells were intermixed withmore differentiated cells showing rhabdomyoblas-tic features. The nodular areas were composed ofcells with variable differentiation. In addition toprimitive cells, there were spindle-shaped cells ar-ranged in interlacing bundles suggestive of a fibro-sarcoma (Fig. 1B), and areas of neural differentia-tion with ganglion-like cells and neuropil (Fig. 1C).Numerous highly pleomorphic, anaplastic cellswith large, densely hyperchromatic nuclei wereseen in the solid component. No obvious foci ofcartilaginous differentiation were identified. Smallentrapped airways were present in the cystic areas,but there were no malignant glandular structures.
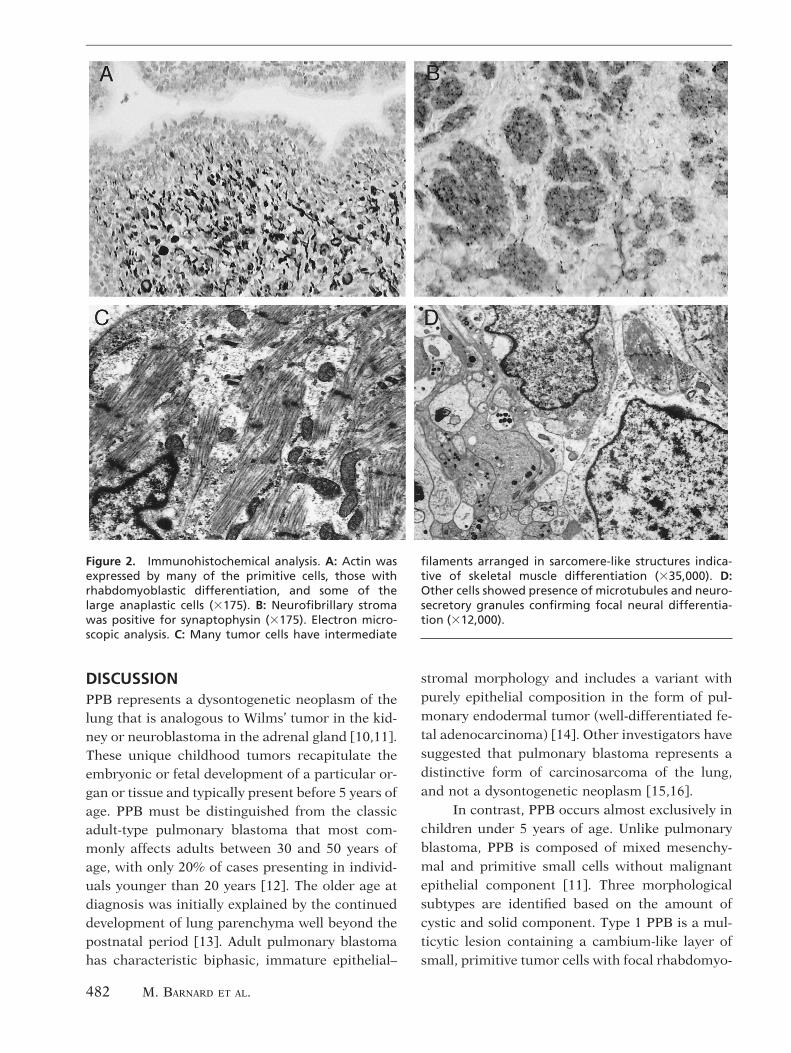
Immunohistochemical studies showed thatmost of the tumor cells expressed vimentin. Boththe entrapped and lining epithelia were positive forkeratin. Actin and desmin were expressed by manyof the primitive cells, those with rhabdomyoblasticdifferentiation, and some of the large anaplasticcells (Fig. 2A). S-100 did not highlight any areas ofchondroblastic differentiation. The neurofibrillarystroma was positive for neuron-specific enolase,chromogranin, and synaptophysin (Fig. 2B). p53
480 M. BARNARD ET AL.
(Novocastra, 1/100) was strongly expressed insome areas of the tumor. MIB-1 staining wasprominent and, by image analysis, the MIB-1 pro-liferation index was estimated to be about 46% oftumor cells.
Ultrastructural examination revealed thatmany of the tumor cells contained intermediatefilaments arranged in sarcomere-like structures in-dicative of skeletal muscle differentiation (Fig. 2C).Other cells showed presence of microtubules andneurosecretory granules confirming focal neuraldifferentiation (Fig. 2D).
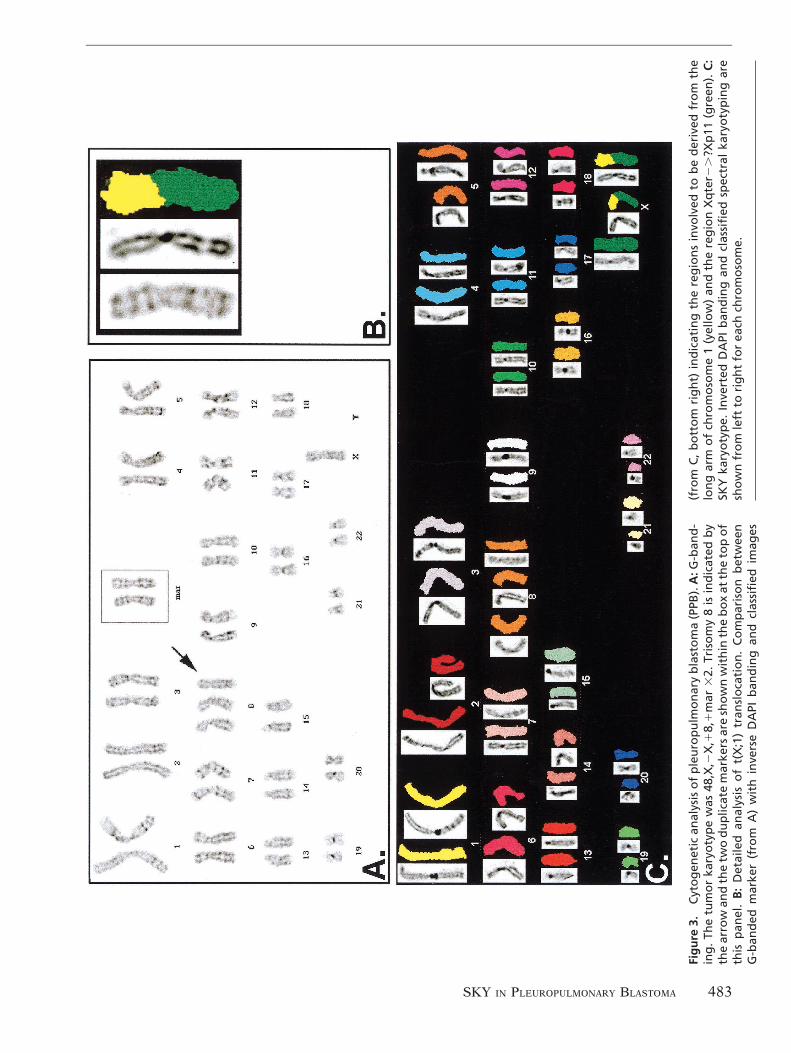
Cytogenetics and spectral karyotypingAn initial analysis of conventional G-bandedmetaphases indicated a hyperdiploid karyotypewith 48 chromosomes with only one X chromo-some, two duplicate markers, and trisomy 8. Thekaryotype was designated 48,X,2X,18,1mar 32(Fig. 3A). Consistent with the findings of molec-ular genetics studies, none of the chromosomalrearrangements commonly encountered in pedi-atric solid tumors were present. Of note were twounidentified markers, which appeared to be iden-tical, and had a short arm with a G-bandingpattern consistent with either terminal end of 1qor 2p. Further characterization of this marker by
conventional cytogenetic methods was not pos-sible because of the limited number of availablemetaphase spreads. A remaining unstained slidewas used for SKY analysis to determine the ori-gin of these marker chromosomes (Fig. 3B,C).SKY confirmed the presence of trisomy 8 andalso revealed an unbalanced translocation be-tween chromosomes 1 and X resulting in twomarker chromosomes each with the short armoriginating from chromosome 1 and the longarm from chromosome X (Fig. 3B). On the basisof cytogenetic analysis and SKY, the duplicatedportion of chromosome 1 on the two markerswas identified as the terminal region of the longarm. This translocation resulted in additionalcopies of part of 1q, as well as gain of one extracopy of Xq and loss of part of Xp (Fig. 3C). Thebreakpoints were interpreted to be in the peri-centromeric regions of chromosome X and 1 onthe short arm of the derivative X, possibly atband Xp11, and on the long arm of chromosome1 in the region of band 1q11. The ISCN nomen-clature [9], using abbreviations designated forchromosome paints, is as follows:
48,X,2X,1mar 32.ish der(X)t(X;1)(?p11;?q11)(wcpX1,wcp11)32
Figure 1. Light microscopic analysis of neoplasm. A: Thecysts are lined by non-neoplastic ciliated respiratory epi-thelium with a cambium-like layer of small, primitivecells, focally showing rhabdomyoblastic differentiation(3125). B: The nodular areas contain spindle-shaped cells
arranged in interlacing bundles suggestive of a fibrosar-coma (350). C: Areas of neural differentiation withganglion-like cells and neuropil have numerous highlypleomorphic, anaplastic cells with large, densely hyper-chromatic nuclei (3125).
SKY IN PLEUROPULMONARY BLASTOMA 481
DISCUSSIONPPB represents a dysontogenetic neoplasm of thelung that is analogous to Wilms’ tumor in the kid-ney or neuroblastoma in the adrenal gland [10,11].These unique childhood tumors recapitulate theembryonic or fetal development of a particular or-gan or tissue and typically present before 5 years ofage. PPB must be distinguished from the classicadult-type pulmonary blastoma that most com-monly affects adults between 30 and 50 years ofage, with only 20% of cases presenting in individ-uals younger than 20 years [12]. The older age atdiagnosis was initially explained by the continueddevelopment of lung parenchyma well beyond thepostnatal period [13]. Adult pulmonary blastomahas characteristic biphasic, immature epithelial–
stromal morphology and includes a variant withpurely epithelial composition in the form of pul-monary endodermal tumor (well-differentiated fe-tal adenocarcinoma) [14]. Other investigators havesuggested that pulmonary blastoma represents adistinctive form of carcinosarcoma of the lung,and not a dysontogenetic neoplasm [15,16].
In contrast, PPB occurs almost exclusively inchildren under 5 years of age. Unlike pulmonaryblastoma, PPB is composed of mixed mesenchy-mal and primitive small cells without malignantepithelial component [11]. Three morphologicalsubtypes are identified based on the amount ofcystic and solid component. Type 1 PPB is a mul-ticytic lesion containing a cambium-like layer ofsmall, primitive tumor cells with focal rhabdomyo-
Figure 2. Immunohistochemical analysis. A: Actin wasexpressed by many of the primitive cells, those withrhabdomyoblastic differentiation, and some of thelarge anaplastic cells (3175). B: Neurofibrillary stromawas positive for synaptophysin (3175). Electron micro-scopic analysis. C: Many tumor cells have intermediate
filaments arranged in sarcomere-like structures indica-tive of skeletal muscle differentiation (335,000). D:Other cells showed presence of microtubules and neuro-secretory granules confirming focal neural differentia-tion (312,000).
482 M. BARNARD ET AL.
Fig
ure
3.C
yto
gen
etic
anal
ysis
of
ple
uro
pu
lmo
nar
yb
last
om
a(P
PB).
A:G
-ban
d-
ing
.Th
etu
mo
rka
ryo
typ
ew
as48
,X,2
X,1
8,1
mar
32.
Tris
om
y8
isin
dic
ated
by
the
arro
wan
dth
etw
od
up
licat
em
arke
rsar
esh
ow
nw
ith
inth
eb
ox
atth
eto
po
fth
isp
anel
.B
:D
etai
led
anal
ysis
of
t(X
;1)
tran
slo
cati
on
.C
om
par
iso
nb
etw
een
G-b
and
edm
arke
r(f
rom
A)
wit
hin
vers
eD
API
ban
din
gan
dcl
assi
fied
imag
es
(fro
mC
,b
ott
om
rig
ht)
ind
icat
ing
the
reg
ion
sin
volv
edto
be
der
ived
fro
mth
elo
ng
arm
of
chro
mo
som
e1
(yel
low
)an
dth
ere
gio
nX
qte
r2.
?Xp
11(g
reen
).C
:SK
Yka
ryo
typ
e.In
vert
edD
API
ban
din
gan
dcl
assi
fied
spec
tral
kary
oty
pin
gar
esh
ow
nfr
om
left
tori
gh
tfo
rea
chch
rom
oso
me.
SKY IN PLEUROPULMONARY BLASTOMA 483

blastic differentiation beneath the surface epithe-lium. The exclusively solid type 3 PPB has mixedblastematous and sarcomatous features with areasof rhabdomyoblastic, cartilaginous, and fibrosar-coma-like differentiation. Foci of giant, bizarre-appearing, pleomorphic cells with hyperchromaticnuclei can be identified. The combination of bothcystic and solid areas is present in type 2 PPB. Theprognosis appears more favorable for type 1 PPB,but there is a definite pathogenetic linkage be-tween the three types, as indicated by type 1 casesthat have progressed or recurred as a higher type[17].
An association between PPB and congenitalcystic lung lesions in the same lung, opposite lung,or in a sibling of a patient with PPB has beenreported and provides support for a hypothesisthat PPB arises from a precursor developmentalanomaly. Extrapulmonary, intrathoracic PPBsthat are attached to pleura, mediastinum, or dia-phragm share this anatomical feature with somecongenital malformations, such as extralobar se-questrations. However, the absence of epithelial-lined spaces in metastatic deposits favors the sug-gestion that PPB can induce formation ofepithelial-lined cysts in the lung, and that cystictransformation represents a secondary changerather than a precursor lesion [17].
While there is essentially no evidence to linkPPB with adult pulmonary blastoma, its relation-ship with other pediatric tumors is less clear [18].In 25% of cases of PPB, a heritable predispositionto other dysplastic and neoplastic diseases hasbeen observed in patients and their young rela-tives. Associated conditions include cystic nephro-mas, sarcomas, medulloblastomas, germ cell tu-mors, thyroid neoplasias, and hematologicalmalignancies. Therefore, PPB is a strong markerfor familial disease but it does not appear to be acomponent of other well-characterized childhoodor adult cancer syndromes [3].
The most consistent genetic finding in PPB istrisomy 8, reported in seven out of eight publishedkaryotypes [1–4]. This mutation was also identifiedin our case of PPB. Trisomy 8 has been reported inother solid tumors, including Ewing sarcoma [19],primary esthesioneuroblastoma [20], leiomyosar-coma [21], embryonal rhabdomyosarcoma [22], al-veolar soft part sarcoma [23], and breast carci-
noma [24]. It is one of the most commonchromosomal abnormalities in myeloid disorders,particularly in acute nonlymphocytic leukemiaand myelodysplastic syndrome [25], and accompa-nies progression of chronic myelogenous leukemiato blast crisis [25]. While this mutation appears tobe associated with tumor progression in some ma-lignancies [25], its occurrence in benign tumors[26–28] suggests that an extra copy of chromo-some 8 may be a primary event in tumor develop-ment. This may have occurred in one reported caseof PPB, where trisomy 8 was identified in the initialbiopsy but not in the resected specimen [4]. Pres-ence of trisomy 8 in various other neoplasms indi-cates that this mutation is a nonspecific, commonfinding in tumors. When duplication of chromo-somal material is associated with a neoplasm, itsdevelopment can usually be attributed to amplifi-cation of oncogenes. Gains of chromosome 8 inadult lung neoplasms are thought to be associatedwith increased expression of the MYC oncogene at8q24 [29], but its role in PPB has not been eluci-dated.
The identification of partial trisomy 2 in twocases of PPB [4,5], also observed in cases of hepa-toblastoma and embryonal rhabdomyosarcoma,have led to an interesting speculation about a pos-sible relationship between these tumors [30–32].In particular, PPB and embryonal rhabdomyosar-coma share clinical and pathological features andsome of the first reported PPB cases were initiallyinterpreted as “primary pulmonary rhabdomyosar-comas” [33]. Both tumors have areas of blastema-tous proliferation and cells showing rhabdomyo-blastic differentiation. The main difference is thatcells of PPB tend to have polyphenotypical expres-sion, rather than predominantly skeletal muscledifferentiation. In addition, rhabdomyoblastic dif-ferentiation can be seen in other dyontogeneticneoplasms, such as teratoma, Wilms’ tumor, andmedulloblastoma [17]. Absence of trisomy 2 in theadult-type pulmonary blastoma was interpreted asadditional proof of its distinctiveness from PPB[5].
The loss of 17p chromosomal material, re-ported in three cases of PPB [1,3], raises the pos-sibility of the involvement of p53, a tumor suppres-sor gene located on 17p13.1. However, initialinvestigations did not show immunopositivity for
484 M. BARNARD ET AL.

p53 in cases of PPB [34]. This is in contrast to ourcase, which showed strong expression of p53 insome areas of the tumor (data not shown). Anothertumor suppressor gene implicated in the etiologyof PPB is the distal Wilms’ tumor gene (WT2) onchromosome 11p. While PPB and Wilms’ tumorare considered distinct entities, some investigatorshave demonstrated in PPB a loss of heterozygosityon chromosome 11p15.5 in the region of WT2 [17],a finding also reported in rhabdomyosarcoma [35].These chromosomal regions remain to be fully in-vestigated in patients with PPB and their families.
Our finding of an unbalanced translocationbetween chromosomes 1 and X, resulting in addi-tional copies of 1q, one extra copy of Xq, and lossof part of Xp, has not been previously reported andwarrants further investigation. The presence of du-plicated translocation t(X;1) in our case suggeststhat gain of the marker may have conferred a se-lective advantage. Amplifications of 1q have beenrepeatedly demonstrated in various soft tissue andbone sarcomas [36], including several pediatricsarcomas, such as the Ewing family of tumors.Gain of 1q represents the most common chromo-somal change in invasive breast carcinoma; and inphyllodes tumor of the breast, has been associatedwith stromal overgrowth, recurrence, and aggres-sive behavior [37]. Recent analysis of 46 cases ofWilms’ tumor using comparative genomic hybrid-ization (CGH) identified gain of 1q in 9 tumors(20%) and, in 3 cases, in areas of nephroblastoma-tosis associated with these tumors, indicating thatgain of 1q may be an early event in tumor [38]. Ithas been suggested that several genes localized tothis region may play a role in development and/orprogression of both mesenchymal and epithelialtumors.
The role of the X chromosome in develop-ment of PPB is of less certain significance. Severalgenes that may be involved in tumor developmentor progression have been localized to the long armof chromosome X. The Xp11-q13 amplicon con-taining androgen receptor gene is present in re-lapsed prostate cancer [39]. Certain inhibitor ofapoptosis protein (IAP) genes have been assignedto chromosome Xq25 [40]. The role of these genesin PPB has not been investigated. The consequenceof partial loss of Xp is unclear, since no well-defined tumor suppressor genes have been local-
ized to this region. The t(X;1) translocation mayalso result in the production of a fusion proteinwhose function is yet unknown.
At present, this is the first karyotype of PPBanalyzed using SKY. This technique provides forsimultaneous hybridization of 24 chromosome-specific painting probes combinatorial labeledwith five different fluorochromes or their combi-nations. Each chromosome pair is assigned aunique color, and changes in color pattern are usedto detect subtle, as well as highly complex, numer-ical and structural karyotype rearrangements.Therefore, SKY allows for rapid screening of theentire genome for chromosomal alterations thatmay not be detected by G-banding [8]. In our case,SKY enabled us to identify the composition of thetwo marker chromosomes that could not be iden-tified by G-banding. However, this technique re-quires viable dividing tumor cells to produce meta-phase spreads.
Many pediatric tumors have characteristicgenetic changes used in diagnosis and prognosis(reviewed in [6]). Similar genetic findings in histo-logically unrelated embryonal tumors might indi-cate a common genetic pathway in their develop-ment. Additional studies, using SKY andcomplementary techniques, will be required to ob-tain such information in PPBs and improve ourunderstanding of its pathogenesis and relationshipwith other pediatric tumors.
A C K N O W L E D G M E N T S
This work was supported by the National CancerInstitute of Canada with funds from the CanadianCancer Society.
R E F E R E N C E S1. Kelsey AM, McNally K, Birch J, Mitchell EL. Case of extra
pulmonary, pleuropulmonary blastoma in a child: patho-logical and cytogenetic findings. Med Pediatr Oncol 1997;29:61–64.
2. Novak R, Dasu S, Agamanolis D, Herold W, Malone J,Waterson J. Trisomy 8 is a characteristic finding in pleu-ropulmonary blastoma. Pediatr Pathol Lab Med 1997;17:99–103.
3. Priest JR, Watterson J, Strong L, et al. Pleuropulmonaryblastoma: a marker for familial disease. J Pediatr 1996;128:220–224.
4. Sciot R, Dal Cin P, Brock P, et al. Pleuropulmonary blas-toma (pulmonary blastoma of childhood): genetic linkwith other embryonal malignancies? Histopathology 1994;24:559–563.
5. Yang P, Hasegawa T, Hirose T, et al. Pleuropulmonary
SKY IN PLEUROPULMONARY BLASTOMA 485

blastoma: fluorescence in situ hybridization analysis indi-cating trisomy 2. Am J Surg Pathol 1997;21:854–859.
6. Thorner PS, Squire JA. Molecular genetics in the diagnosisand prognosis of solid pediatric tumors. Pediatr DevPathol 1998;1:337–365.
7. Verma RS, Babu A. Human Chromosomes. New York:McGraw-Hill, 1995.
8. Schrock E, du Manoir S, Veldman T, et al. Multicolorspectral karyotyping of human chromosomes. Science1996;273:494–497.
9. Mitelman F, ed. ISCN (1995): International System forHuman Cytogenetic Nomenclature. New York: S. Karger,1995.
10. Manivel JC, Priest JR, Watterson J, et al. Pleuropulmonaryblastoma. The so-called pulmonary blastoma of child-hood. Cancer 1988;62:1516–1526.
11. Dehner LP, Watterson J, Priest J. Pleuropulmonary blas-toma. A unique intrathoracic-pulmonary neoplasm ofchildhood. Perspect Pediatr Pathol 1995;18:214–226.
12. Dehner LP. Pleuropulmonary blastoma is the pulmonaryblastoma of childhood. Semin Diagn Pathol 1994;11:144–151.
13. Spencer H. Pulmonary blastomas. J Pathol Bacteriol 1961;82:161–165.
14. Koss MN, Hochholzer L, O’Leary T. Pulmonary blastomas.Cancer 1991;67:2368–2381.
15. Souza RC, Peasley ED, Takaro T. Pulmonary blastomas. Adistinctive group of carcinosarcomas of the lung. AnnThorac Surg 1965;1:259–268.
16. Bauermeister DE, Jennings ER, Beland AH, Judson HA.Pulmonary blastoma, a form of carcinosarcoma. Report ofa case of 24 years’ duration without treatment. Am J ClinPathol 1966;46:322–329.
17. Priest JR, McDermott MB, Bhatia S, Watterson J, ManivelJC, Dehner LP. Pleuropulmonary blastoma. A clinicopath-ological study of 50 cases. Cancer 1997;80:147–161.
18. Baraniya J, Desai S, Kane S, et al. Pleuropulmonary blas-toma. Med Pediatr Oncol 1999;32:52–56.
19. Maurici D, Perez-Atayde A, Grier HE, Baldini N, Serra M,Fletcher JA. Frequency and implications of chromosome 8and 12 gains in Ewing sarcoma. Cancer Genet Cytogenet1998;100:106–110.
20. Van Devanter DR, George D, McNutt MA, Vogel A,Luthardt F. Trisomy 8 in primary esthesioneuroblastoma.Cancer Genet Cytogenet 1991;57:133–136.
21. Lessick M, Israel J, Szego K, Wong P. Leiomyosarcoma ina patient with trisomy 8 mosaicism. J Med Genet 1990;27:643–644.
22. Dietrich CU, Jacobsen BB, Starklint H, Heim S. Clonalkaryotypic evolution in an embryonal rhabdomyosarcomawith trisomy 8 as the primary chromosomal abnormality.Genes Chromosom Cancer 1993;7:240–244.
23. Craver RD, Heinrich SD, Correa H, Kao YS. Trisomy 8 inalveolar soft part sarcoma. Cancer Genet Cytogenet 1995;81:94–96.
24. Rohen C, Meyer-Bolte K, Bonk U, et al. Trisomy 8 and 18 asfrequent clonal and single-cell aberrations in 185 primarybreast carcinomas. Cancer Genet Cytogenet 1995;80:33–39.
25. Heim S, Mitelman F. Cancer Cytogenetics, 2nd ed. NewYork: Wiley-Liss, 1995.
26. Kouho H, Aoki T, Hisaoka M, Hashimoto H. Clinicopath-ological and interphase cytogenetic analysis of desmoidtumours. Histopathology 1997;31:336–341.
27. Dal Cin P, Sciot R, Van Poppel H, Baert L, Van Damme B,Van den Berghe H. Chromosome analysis in angiomyoli-poma. Cancer Genet Cytogenet 1997;99:132–134.
28. Mark J, Dahlenfors R. Cytogenetical observations in 100human benign pleomorphic adenomas: specificity of thechromosomal aberrations and their relationship to sites oflocalized oncogenes. Anticancer Res 1986;6:299–308.
29. Johnson BE. The role of MYC, JUN, and FOS oncogenes inhuman lung cancer. In: Pass HI, Mitchell JB, Johnson DH,Turrisi AT, eds. Lung Cancer: Principles and Practice.Philadelphia: Lippincott-Raven, 1996;83–98.
30. Bardi G, Johansson B, Pandis N, et al. Trisomy 2 as thesole chromosomal abnormality in a hepatoblastoma.Genes Chromosom Cancer 1992;4:78–80.
31. Fletcher JA, Kozakewich HP, Pavelka K, et al. Consistentcytogenetic aberrations in hepatoblastoma: a commonpathway of genetic alterations in embryonal liver and skel-etal muscle malignancies? Genes Chromosom Cancer1991;3:37–43.
32. Rodriguez E, Reuter VE, Mies C, Bosl GJ, Chaganti RS.Abnormalities of 2q: a common genetic link between rhab-domyosarcoma and hepatoblastoma? Genes ChromosomCancer 1991;3:122–127.
33. Allan BT, Day DL, Dehner LP. Primary pulmonary rhab-domyosarcoma of the lung in children. Report of two casespresenting with spontaneous pneumothorax. Cancer 1987;59:1005–1011.
34. Pacinda SJ, Ledet SC, Gondo MM, et al. p53 and MDM2immunostaining in pulmonary blastomas and broncho-genic carcinomas. Hum Pathol 1996;27:542–546.
35. Besnard-Guerin C, Newsham I, Winquist R, Cavanee WK.A common region of loss of heterozygosity in Wilms’ tu-mor and embryonal rhabdomyosarcoma distal toD11S988 locus on chromosome 11p15.5. Hum Genet1996;97:163–170.
36. Knuutila S, Bjorkqvist A-M, Autio K, et al. DNA copynumber amplifications in human neoplasms. Review ofcomparative genomic hybridization studies. Am J Pathol1998;152:1107–1123.
37. Lu Y-J, Birdsall S, Osin P, Gusterson B, Shipley J. Phyl-lodes tumors of the breast analyzed by comparativegenomic hybridization and association of increased 1qcopy number with stromal overgrowth and recurrence.Genes Chromosom Cancer 1997;20:275–281.
38. Steenman M, Redeker B, de Meulemeester M, et al. Com-parative genomic hybridization analysis of Wilms tumors.Cytogenet Cell Genet 1997;77:296–303.
39. Visakorpi T, Hyytinen E, Koivisto P, et al. In vivo ampli-fication of the androgen receptor gene and progression ofhuman prostate cancer. Nat Genet 1995;9:401–406.
40. Rajcan-Separovic E, Liston P, Lefebvre C, Korneluk RG.Assignment of human inhibitor of apoptosis protein (IAP)genes xiap, hiap-1 and hiap-2 to chromosomes Xq25 and11q22-q23 by fluorescence in situ hybridization. Genomics1996;37:404–406.
486 M. BARNARD ET AL.


















