
UREA CYCLE - jsmu.edu.pk Lecture 3. Urea cycle.pdf · •Hyperammonemia type I CPS I . Carbamoyl...
32
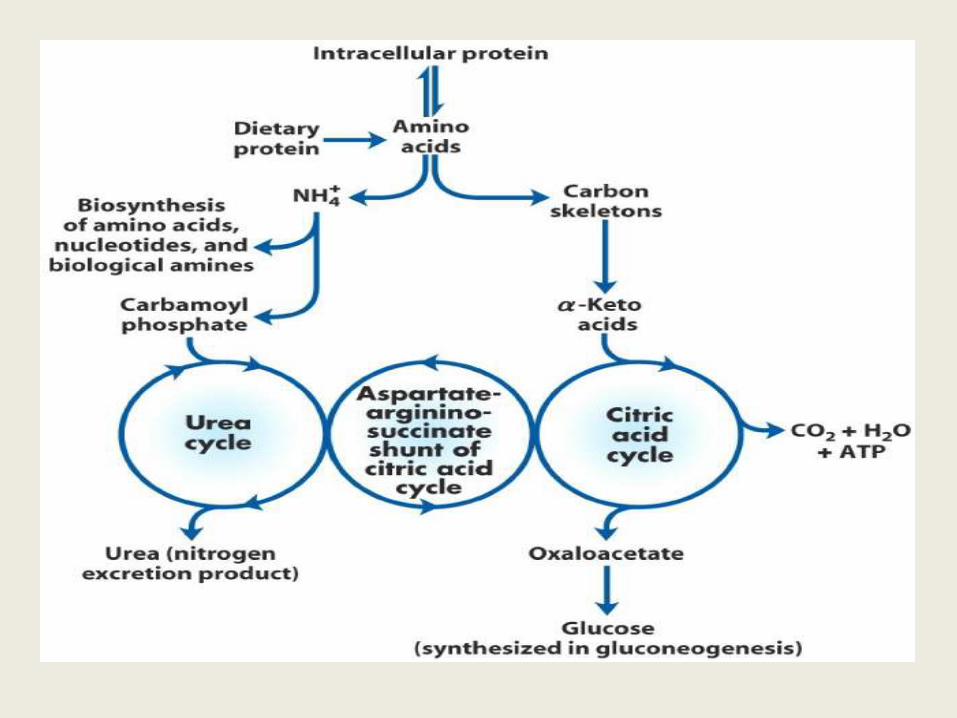
UREA CYCLE
Transcript of UREA CYCLE - jsmu.edu.pk Lecture 3. Urea cycle.pdf · •Hyperammonemia type I CPS I . Carbamoyl...

UREA CYCLE

Urea cycle with Clinical Significance
H2N C
O
NH2
urea
ornithine NH3 + CO2
H2O
NH3H2O
H2O
urea
arginase
Arg citrulline

Urea formation (Urea cycle)
Characteristics of urea cycle
• Urea is the major disposal form of amino groups
• It accounts for 90% of the nitrogen containing components of urine
• The urea cycle is the sole source of endogenous production of arginine
• Urea formation takes place in liver,
• Urea excretion occurs through kidney

Urea cycle- An overview
• Urea synthesis is a cyclic process. • The first two reactions of urea synthesis
occur in the matrix of the mitochondrion, the remaining reactions occur in the cytosol
• Since the Ornithine consumed in 2nd reaction is regenerated in last reaction, so there is no net loss or gain of Ornithine, Citrulline, argininosuccinate, or arginine.
• Ammonium ion, CO2, ATP, and aspartate are, however, consumed.
• Aspartate can however be resynthesized from the released fumarate by a series of reactions

Urea formation (Urea cycle)
• 6 amino acids participate in urea formation, which are-
• Ornithine • Citrulline • Aspartic acid • Argino succinic acid • Arginine and • N-Acetyl Glutamate

Step-1- Formation of Carbamoyl-Phosphate
• CPSI is strongly activated by N-acetyl glutamate, which controls the overall rate of urea production.
• Hyperammonemia type I
CPS I

Carbamoyl phosphate synthetase Ⅰ (CPSⅠ) is an
allosteric enzyme and is absolutely dependent up
on N-acetylglutamic acid (AGA) for its activity.
Carbamoyl phosphate synthetaseⅠ:
Occurs in mitochondria of liver cells. It is involved in urea synthesis.
Carbamoyl phosphate synthetaseⅡ:
Present in cytosol of liver cells which is involved in pyrimidine synthesis.
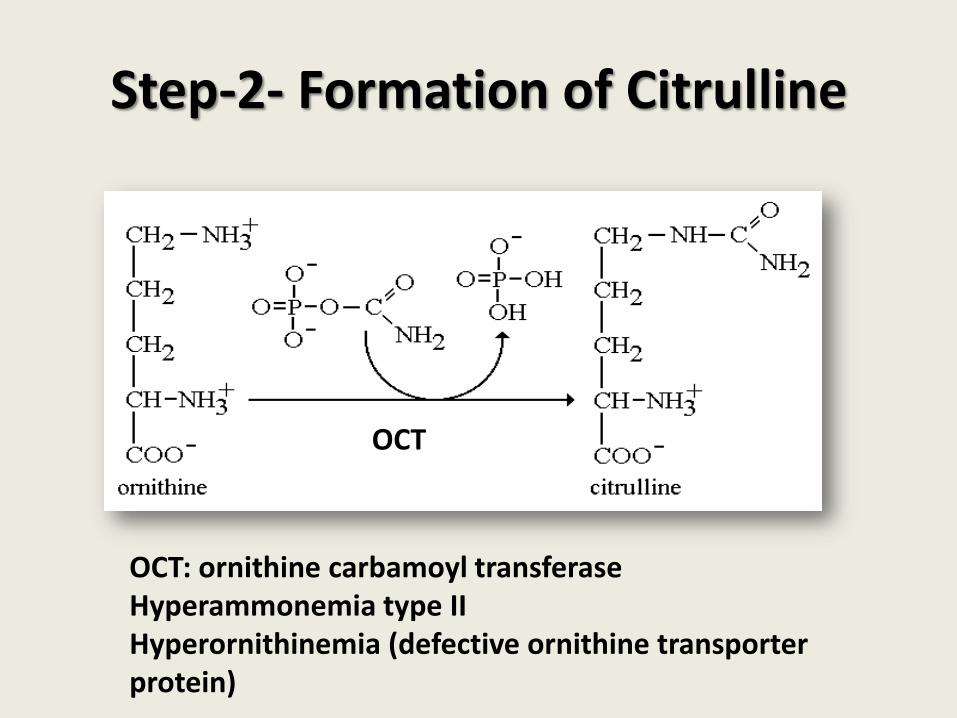
Step-2- Formation of Citrulline
OCT: ornithine carbamoyl transferase Hyperammonemia type II Hyperornithinemia (defective ornithine transporter protein)
OCT

Step 3- Formation of arginine (in cytosol)
NH
£¨CH £©2 3
CHNH2
COOH
NH2
C O
citrulline
+
COOH
H2-N-C-H
CH2
COOH
ATP AMP+PPi
NH
£¨CH £©2 3
CHNH2
COOH
NH2
C
COOH
N-C-H
CH2
COOH
arginino succinate
Asp
ASS
ASS: argininosuccinate synthetase
Citrullinemia

Step-4- Cleavage of Argino succinate
(ASL)
ASL: argininosuccinate lyase
Argininosuccinic aciduria
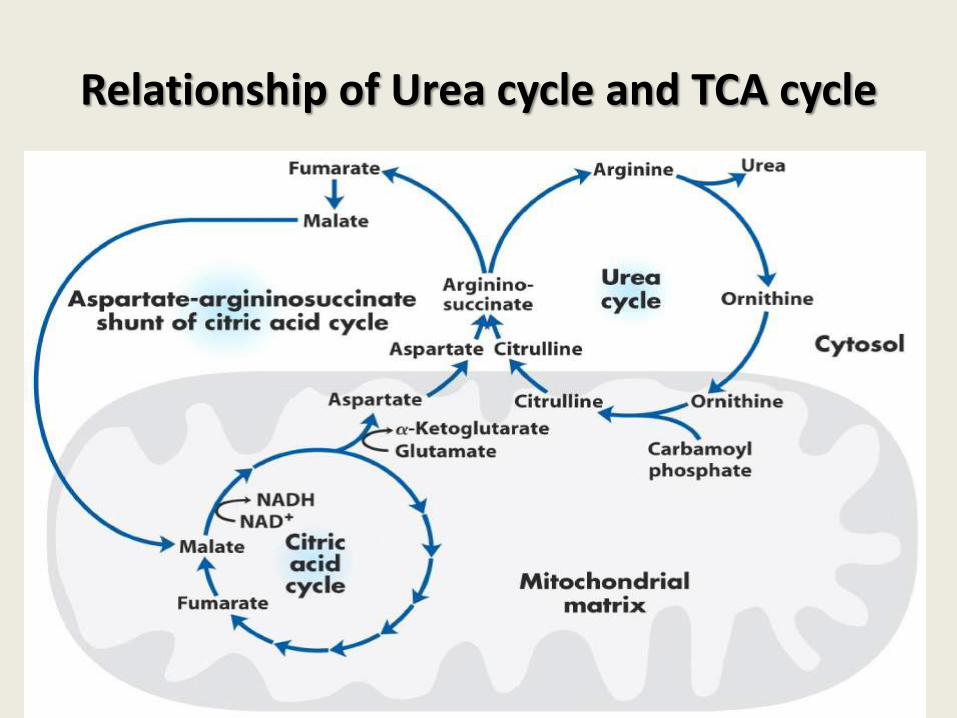
Relationship of Urea cycle and TCA cycle

Step-5-Cleavage of Arginine
• Ornithine and lysine are potent inhibitors of arginase, competitive with arginine.
• Arginine also serves as the precursor of the potent muscle relaxant nitric oxide (NO) in a Ca2+-dependent reaction catalyzed by NO synthase.
• Hyperargininemia
arginase

2ADP+Pi
CO2 + NH3 + H2O
Carbamoyl phosphate
2ATP N-acetylglutamic acid
Pi
ornithine citrulline
Amino
acids
α-ketoglutaric acid
Glutamic Acid
α-keto
acid
citrulline
Arginino succinate
Asp ATP
AMP + PPi
Arg
ornithine
urea
mitochondria
in cytosol fumarate
malic acid
oxaloacetic acid
Urea cycle

Summary of urea synthesis
• One nitrogen of urea molecule comes from ammonia, another nitrogen comes from Asp.
• HCO3- ion provides the carbon atom of urea.
• Found primarily in liver and lesser extent in kidney
• Synthesis of a urea will consume 3ATP and 4 ~P.
2NH3 + CO2 + 3ATP + 2H2O urea + 2ADP + AMP + 2Pi + PPi
Total formula:
H2N C
O
NH2
urea

Regulation factors:
1. Ratio of protein in dietary foods:
2. Carbamoyl phosphate synthetase is allosterically
activated by N-acetylglutamate
(acetyl CoA + glutamate N-acetylglutamate)
3. Rate limiting enzyme: argininosuccinate
synthetase(ASS)

Clinical significance of urea
A moderately active man consuming about 300gm
carbohydrates ,100gm of fats and 100gm of proteins
daily must excrete about 16.5gm of N daily.
95% is eliminated by the kidneys and the remaining
5%, for the most part as N, in the faeces.
in man ,normal blood level of NH3 varies from 40 to
70µg/100ml.free NH+4 concentration of fresh plasma
is less than 20µg per 100ml.
Normal blood ammonia level:

Fate of Urea

HYPERAMMONEMIAS
Ammonia has a direct neurotoxic effect on the CNS .for example ,elevated concentrations of ammonia in the blood cause the symptoms of ammonia intoxication, which include:
tremors, slurring of speech, Somnolence ,vomiting ,cerebraledema, and blurring of vision.
Hyperammonemia is a metabolic disturbance characterised by an excess of ammonia in the blood. It is a dangerous condition that may lead to encephalopathy and death. It may be primary or secondary.

2. Hereditary
hyperammonemia:
is caused by several inborn
errors of metabolism that are
characterised by reduced
activity of any of the enzymes
in the urea cycle.
1. Acquired hyperammonemia : dysfunction of liver is common cause of hyperammonemia(eg hepatic disease). porto-systemic encephalopathy: communications between portal and systemic veins. The portal blood may bypass the liver.
The two major types of hyperammonemia:

The major reasons of hyperammonemias:
3. Liver desfunction or porto-systemic encephalopathy,haemorrhage into GI tract.
2. Kidney secretion : kidney desfunction
Degradation of urea in the intestine
1. Excessive putrefaction in the intestine,
example:hemorrhage of digestive tract.
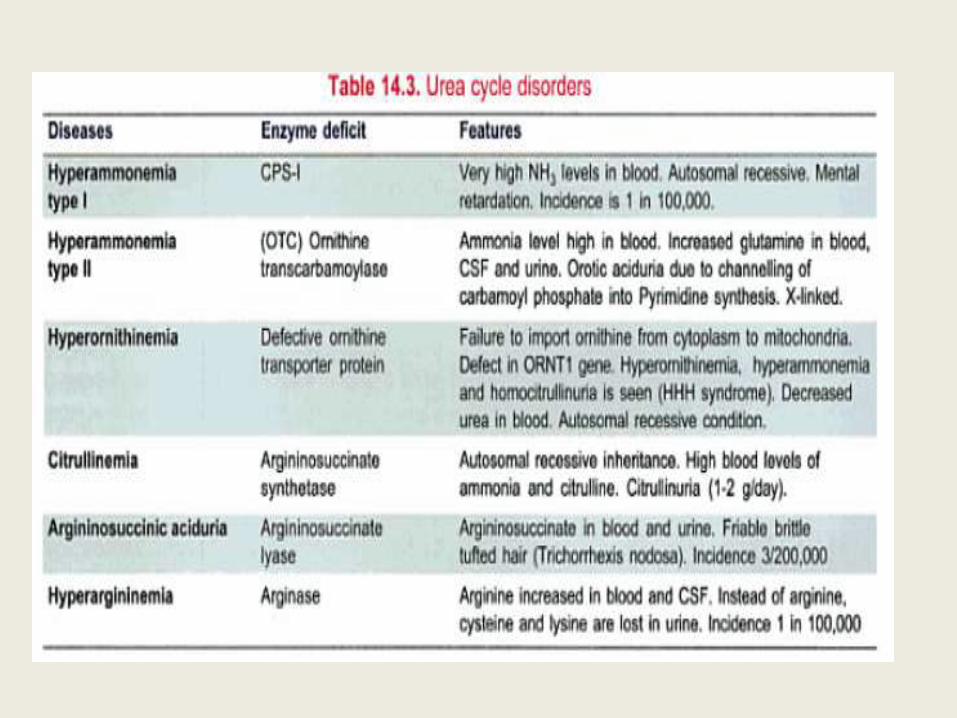

Urea cycle disorders
Carbamoyl Phosphate synthetase (CPS-1) deficiency
• Along with OTC deficiency, deficiency of CPS-I is the most severe of the urea cycle disorders.
• Defects in the enzyme carbamoyl phosphate synthase I are responsible for the relatively rare (estimated frequency 1:62,000) metabolic disease termed "hyperammonemia type 1."
• Individuals with complete CPS-I deficiency rapidly develop hyperammonemia in the newborn period.

Ornithine Transcarbamoylase deficiency
(OTC deficiency)
• The disease is characterized as X linked dominant
• A significant number of carrier females have hyperammonemia and neurologic compromise.
• The risk for hyperammonemia is particularly high in pregnancy and the postpartum period.
• The disease is much more severe in males than in females.
• The enzyme activity can range from 0% to 30% of the normal.

Citrullinemia (ASS deficiency)
• The hyperammonemia in this disorder is quite
severe.
• Affected individuals are able to incorporate some waste nitrogen into urea cycle intermediates,
• which makes treatment slightly easier.

Argininosuccinic aciduria (ASL deficiency)
• This disorder also presents with rapid-onset hyperammonemia in the newborn period.
• This enzyme defect is past the point in the metabolic pathway at which all the waste nitrogen has been incorporated into the cycle.
• Treatment of affected individuals often requires only supplementation of arginine.
• Affected individuals can also develop trichorrhexis nodosa, a node-like appearance of fragile hair, which usually responds to arginine supplementation.
• ASL deficiency is marked by chronic hepatic enlargement and elevation of transaminases.

Arginase deficiency (hyperargininemia;
ARG deficiency)
• This disorder is not typically characterized by rapid-onset hyperammonemia.
• Affected individuals develop progressive spasticity and can also develop tremor, ataxia, and choreoathetosis.
• Growth is affected

NAG Synthase deficiency
• Deficiency of this enzyme has been described in a number of affected individuals.
• Symptoms mimic those of CPSI deficiency; since CPSI is rendered inactive in the absence of NAG
• N-Acetyl glutamate is essential for Carbamoyl phosphate synthase I activity
• The NAGS gene encodes N-acetyl glutamate synthase, which catalyzes the condensation of acetyl-CoA with glutamate.
• Defects in the NAGS gene result in severe hyperammonemia, which in this specific instance may respond to administered N-acetyl glutamate.

Ornithine Transporter deficiency
• Hyperornithinemia, hyperammonemia, and homocitrullinuria syndrome (HHH syndrome) results from mutation of the ORNT1 gene that encodes the mitochondrial membrane ornithine transporter.
• The failure to import cytosolic ornithine into the mitochondrial matrix renders the urea cycle inoperable, with consequent hyperammonemia, and the accompanying accumulation of cytosolic ornithine results in Hyperornithinemia.
• In the absence of its normal acceptor ornithine, mitochondrial carbamoyl phosphate carbamoylates lysine to homocitrulline with a resulting homocitrullinuria.

Clinical manifestations in urea cycle
disorders • Infants with a urea cycle disorder often appear normal initially
but rapidly develop-
o cerebral edema
o lethargy
o anorexia
o hyperventilation or hypoventilation,
o hypothermia
o slurring of the speech,
o blurring of vision
o seizures
o neurologic posturing and
o coma.

limiting protein intake to the amount barely adequate to supply amino acids for growth, while adding to the diet the a-keto acid analogs of essential amino acids. Phenylbutyrate Prenatal testing using molecular genetic testing is available for five of the six urea cycle disorders Liver transplantation has also been used, since liver is the organ that carries out Urea Cycle.
Treatment of deficiency of Urea Cycle enzymes (some treatments depend on which enzyme is deficient)



















