
UNIVERSIDADE DE SÃO PAULO - Biblioteca Digital de Teses e … · 2016. 2. 17. · 1 UNIVERSIDADE...
159
UNIVERSIDADE DE SÃO PAULO Faculdade de Ciências Farmacêuticas Programa de Pós-Graduação em Fármacos e Medicamentos Área de Insumos Farmacêuticos Potenciais candidatos a novos antineoplásicos: síntese e avaliação da atividade antitumoral de análogos da queleritrina planejados por simplificação molecular Rosania Yang Dissertação para obtenção do Título de Mestre Orientador: Prof. Dr. Roberto Parise Filho São Paulo 2015
Transcript of UNIVERSIDADE DE SÃO PAULO - Biblioteca Digital de Teses e … · 2016. 2. 17. · 1 UNIVERSIDADE...

0
UNIVERSIDADE DE SÃO PAULO
Faculdade de Ciências Farmacêuticas
Programa de Pós-Graduação em Fármacos e Medicamentos
Área de Insumos Farmacêuticos
Potenciais candidatos a novos antineoplásicos: síntese e
avaliação da atividade antitumoral de análogos da
queleritrina planejados por simplificação molecular
Rosania Yang
Dissertação para obtenção do
Título de Mestre
Orientador:
Prof. Dr. Roberto Parise Filho
São Paulo
2015

1
UNIVERSIDADE DE SÃO PAULO
Faculdade de Ciências Farmacêuticas
Programa de Pós-Graduação em Fármacos e Medicamentos
Área de Insumos Farmacêuticos
Potenciais candidatos a novos antineoplásicos: síntese e
avaliação da atividade antitumoral de análogos da
queleritrina planejados por simplificação molecular
Rosania Yang
Versão Original
Dissertação para obtenção do
Título de Mestre
Orientador:
Prof. Dr. Roberto Parise Filho
São Paulo
2015

2

3
Rosania Yang
Potenciais candidatos a novos antineoplásicos: síntese e
avaliação da atividade antitumoral de análogos da queleritrina
planejados por simplificação molecular
Comissão Julgadora
da
Dissertação para obtenção de Título de Mestre
Prof. Dr. Roberto Parise Filho
orientador/presidente
________________________________________________
Prof. Dr. Roberto Parise Filho
_________________________________________________
2º examinador
__________________________________________________
3º examinador
São Paulo, ____ de ___________ de 2015.

4
DEDICATÓRIA
Dedico este trabalho aos meus pais, meus exemplos de vida. Mesmo nas
piores situações, souberam aproveitar o que a vida lhes ofereceu, mostrando que o
aprendizado e o esforço no trabalho sempre compensam.
À minha melhor amiga, Natália Miyuki Cazorla, a irmã que escolhi, pois sua
força e apoio foram indispensáveis para a minha trajetória.

5
AGRADECIMENTOS
Ao departamento de Farmácia da Faculdade de Ciências Farmacêuticas da
Universidade de São Paulo, pela oportunidade de desenvolver este projeto. Às
agências CAPES, CNPq e FAPESP, pelo auxílio financeiro concedido.
Ao meu orientador, Prof. Dr. Roberto Parise Filho, pela oportunidade
oferecida e por ter acreditado em mim. Não é apenas um exemplo de profissional,
cuja ética e competência são inquestionáveis, mas também trata-se de uma pessoa
de bom coração. Sempre se preocupou com a formação profissional e pessoal de
seus alunos. Além disso, me proporcionou momentos de conforto, me deu bons
conselhos e boas conversas. Não teria escolhido melhor orientador para essa
jornada.
Aos meus pais, por sempre me incentivarem a estudar e sempre se
esforçarem para que isso fosse possível. Sem sua ajuda, eu não teria conseguido
realizar esse mestrado. Ao meu irmão, Ronaldo Yang, a pessoa da minha família
que mais me compreende e cujos conselhos sempre foram valiosos para mim.
À minha querida amiga Sarah Fernandes Teixeira, que, em pouquíssimo
tempo, se mostrou uma pessoa valiosa tanto no âmbito profissional, quanto no
pessoal. Representa um exemplo de profissional para mim. Sempre me deu bons
conselhos, sabendo quando eu precisava levar bronca. Junto ao Ricardo Alexandre
de Azevedo, me deu grande suporte nos ensaios biológicos, além de se
preocuparem com o meu aprendizado como um todo.
Ao meu colega de grupo e amigo Maurício Temotheo Tavares, a pessoa que
mais merece minha gratidão. Sempre se mostrou solícito a me ajudar e, em diversos
momentos que precisei de um conselho ou conforto, eu sabia que podia contar com
ele. Não posso deixar de comentar os momentos de extroversão, os quais renderam
boas risadas e momentos de alívio em nossa rotina.
À Elys Lima, por sempre ser o meu ombro amigo no laboratório, por enxugar
minhas lágrimas nos momentos de tristeza e tensão, e por sempre estar ao meu
lado. Seus conselhos sempre me trouxeram boas reflexões e mudaram meu ponto
de vista em diversos assuntos.

6
Ao Fernando de Moura Gatti, pois, sem sua ajuda, eu não teria concluído
metade do meu trabalho. Sempre disposto a trocar informações sobre pesquisa, sua
criatividade e sede por conhecimento foram determinantes para o desenvolvimento
do meu trabalho. Além disso, partilhou de diversos momentos de descontração
comigo, dentro e fora do laboratório.
Aos amigos Ricardo Massarico Serafim, Natanael Segretti, Mariana Frojuello
Segretti e Fredson Torres, por toda a ajuda que me deram no mestrado. Seus
conselhos me ajudaram no desenvolvimento do trabalho e a refletir sobre minha
postura profissional. Vale mencionar os momentos de descontração também, que
renderam boas risadas e que ajudaram a deixar a rotina da pesquisa mais
prazerosa.
Às amigas e colegas de departamento Marina Cândido Primi, Drielli Gomes
Vital e Juliana Magalhães pelos diversos conselhos, momentos de desabafo e
compreensão, pelas risadas e pela companhia. Obrigada por acompanhar minha
jornada e sempre me manter positiva em relação ao meu trabalho.
À Thais Batista Fernandes que, não só me ofereceu sua amizade, como
também partilhou comigo muitos momentos do mestrado. Sua companhia foi muito
importante para a troca de aprendizado, de momentos de frustrações e alegrias, o
que tornou essa jornada mais agradável.
À Lorena Cristine Paes, por toda a disposição em ensinar a parte de química
analítica. Não só me ensinou as técnicas, mas também a ser bem criteriosa no
desenvolvimento e análise experimental. Obrigada pela paciência e pela amizade.
Aos colegas de grupo Gustavo Vasco, Micael Cunha, Nuno Tavares (e a
agregada Rafaela Landucci), Eugen Nesadal e o recém-integrante Alfredo Souza,
por proporcionarem momentos de alegria nos últimos seis meses. Obrigada por
todas as risadas, brincadeiras, conselhos, trocas de informações científicas e por
sempre me mostrarem que eu devo acreditar mais na minha capacidade.
Aos demais colegas de departamento Luciana de Moura Bueno, Silvestre
Modestia, Gláucio Monteiro, Soraya Santos, Profa. Dra. Jeanine Giarolla Vargas,
Marco Arribas e Juliana Pachioni, pelas conversas, conselhos e pelo apoio durante o
mestrado.

7
À Profa. MSc Bárbara Vaz, por ter me orientado na graduação e cujos
conselhos me ajudaram na decisão por fazer mestrado na área de química
farmacêutica.
Aos pesquisadores Dr. Ricardo Alexandre de Azevedo, Dr. Kleber Adilson
Ferreira e Prof. Dr. José Alexandre Marzagão Barbuto, do Laboratório de Imunologia
dos tumores, pelo suporte nos ensaios biológicos, pelos ensinamentos e pelo
espaço concedido para a realização dos experimentos. Também não posso deixar
de agradecer os demais pesquisadores do laboratório Aline Arruda, Thiago Patente,
Mariana Pereira e Cecília Pessoa, os quais me acolheram como se eu fosse
integrante do grupo.
Ao Prof. Dr. Gustavo Trossini, pelo apoio e orientação nos estudos de
modelagem molecular. Muito obrigada por sempre se mostrar disposto a ajudar e
discutir sobre o meu projeto.
À Prof. Tit. Elizabeth Igne Ferreira e ao Prof. Dr. Jean Leandro dos Santos, os
quais fizeram parte da minha banca de qualificação. Muito obrigada pelas ideias e
correções do meu trabalho. Suas contribuições foram muito relevantes para mim.
Aos funcionários Célia (ICB), Charles, Inês, David e Irineu, pela ajuda que me
deram no decorrer do mestrado.
À minha amiga da Unisa, Renata Sousa, por todas as palavras de incentivo e
carinho que me deu ao longo dessa jornada. Sua amizade significa muito para mim.
Também não posso deixar de agradecer as outras amigas da Unisa, Karla Cristina,
Telma Cristina e Liliane Cabral, por todo o incentivo e apoio que me deram para
realizar o mestrado.
Aos meus amigos do peito, Natália Miyuki Cazorla, Caroline Bidinotto
Tincani, Talita Rodrigues, Thuane Araújo, Leandro Hayashi, Thiago Contine, Gabriel
Buss, Richard Salvazzini, Shirley Takiuti, Felipe Hiroshi, Marcella Ortega, Felipe
Iqueuti, Jackeline Aguiar, Natanne Socreppa e Leandro Lemos, os quais nunca
entenderam o que eu faço no mestrado, mas sempre se mostraram interessados no
assunto e me apoiaram em diversos momentos.
E vale um agradecimento a todas as outras pessoas que me ajudaram de
maneira direta ou indireta.

8
“A vida é um dom – uma interconexão quase impossível de biologia, química, eletricidade e engenharia que se soma a um todo milagroso, muito maior do que a soma de suas partes. O universo inteiro está equipado para a desordem [...] Toda a vida na Terra, por menor que seja, é uma oposição à natureza entrópica do universo. Considerando-se todas as forças a favor da desordem, o mero fato de vivermos – e por tanto tempo, e tão bem – já é um milagre. Por isso, em vez de tomarmos nossa saúde como certa, devemos valorizá-la com toda a reverência que ela merece. [...]”
A Sobrevivência dos Mais Doentes – Dr. Sharon Moalem

9
YANG, R. Potenciais candidatos a novos antineoplásicos: síntese e avaliação da
atividade antitumoral de análogos da queleritrina planejados por simplificação
molecular. 2015. 158 p. Dissertação (Mestrado) – Faculdade de Ciências Farmacêuticas,
Universidade de São Paulo, São Paulo, 2015.
RESUMO
Câncer é uma das principais causas de mortes no mundo. Foi responsável por aproximadamente 8,2 milhões de óbitos em 2012 e estima-se 13,2 milhões de mortes em 2030, o que o posiciona como um problema de saúde pública. A quimioterapia torna-se necessária para o tratamento, pois regride o crescimento tumoral e diminui as chances de metástases, as quais são responsáveis por 90% dos óbitos decorrentes do câncer. Linhagens tumorais que apresentam alta expressão de proteínas antiapoptóticas da família Bcl-2 mostram-se resistentes à ação de quimioterápicos antineoplásicos disponíveis na terapêutica. Portanto, o desenvolvimento de compostos que inibem essas proteínas, torna-se interessante para o tratamento do câncer. A queleritrina, um alcaloide presente em diversas espécies da família Papaveraceae, representa um atraente protótipo para o desenvolvimento de inibidores de Bcl-2, visto que esta molécula apresenta atividade citotóxica por meio da inibição da Bcl-XL, proteína supressora da apoptose. Face ao exposto, o objetivo deste trabalho foi sintetizar análogos da queleritrina planejados por simplificação molecular e testar sua atividade citotóxica. A síntese dos compostos foi realizada em duas etapas, com metodologias baseadas em reações de adição nucleofílica com posterior redução. Os análogos foram caracterizados por espectroscopia de ressonância magnética nuclear (RMN) 1H e 13C, cromatografia líquida de alta eficiência (CLAE) e análise elementar (CHN). A citotoxicidade dos compostos sintetizados e da queleritrina foi avaliada por meio do método de determinação da viabilidade celular por biorredução do sal de tetrazólio (MTT) nas linhagens não-tumorigênicas HUVEC e LL24, e nas linhagens tumorais Jurkat, SK-Mel-28 e A549. Os resultados obtidos no ensaio de citotoxicidade demonstraram que os compostos sintetizados não possuem citotoxicidade significante nas concentrações testadas. Ao avaliar o potencial citostático dos compostos frente à linhagem mutirresistente A549, o composto 2f (3-OH e 4-OH) apresentou atividade antiproliferativa interessante na concentração de 75 µM. Por não apresentar toxicidade em células não tumorais, o composto 2f mostra-se mais seletivo do que seu protótipo. Estudos de modelagem molecular sugerem que a perda da planaridade da queleritrina seja o principal responsável pela diminuição da atividade biológica. Dessa forma, embora os compostos tenham apresentado propriedades biológicas diferentes da esperada, os resultados obtidos podem auxiliar no planejamento de novas moléculas com atividade e seletividade potencialmente superiores à queleritrina, representando assim, potenciais candidatos a fármacos que podem ampliar o arsenal terapêutico para o tratamento de neoplasias.
Palavras-chave: câncer, queleritrina, simplificação molecular, compostos amínicos,
atividade citostática.

10
YANG, R. Potenciais candidatos a novos antineoplásicos: síntese e avaliação da
atividade antitumoral de análogos da queleritrina planejados por simplificação
molecular. 2015. 158 p. Dissertação (Mestrado) – Faculdade de Ciências Farmacêuticas,
Universidade de São Paulo, São Paulo, 2015.
ABSTRACT
Cancer is a leading cause of death and was responsible for approximately 8.2 million of deaths in 2012 and 13.2 million of demises are estimated for 2030, which made cancer a public health problem. Chemotherapy is required for cancer treatment, because it decreases the chances of metastasis and regresses tumor growth. However, tumor cell lines which overexpress anti-apoptotic Bcl-2 proteins present resistance to most of anticancer drugs. In this context, the development of molecules with inhibitory activity against these proteins is a promising therapeutic strategy for cancer treatment. Chelerythrine, an alkaloid distributed in several species of Papaveraceae family, has shown considerable antiproliferative activity due to inhibition of Bcl-XL anti-apoptotic protein, which represents an attractive prototype to anticancer drug design. Then, the aim of this study was to synthesize chelerythrine analogues designed by molecular simplification and evaluate their cytotoxic activity. The synthetic strategy was performed into two steps, with methodologies based on nucleophilic addition reaction followed by reduction. The compounds were characterized by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy, high performance liquid chromatography (HPLC) and elemental analysis (CHN). The cytotoxic activity of the analogues were evaluated through MTT assay, a colorimetric assay for assessing cell viability, against human umbilical vein endothelial cells (HUVEC) and lung fibroblasts (LL24) as non-tumorigenic cell lines, and also against tumor cell lines, such as human lymphoblastoid (Jurkat), human melanoma cells (SK-Mel-28) and carcinomic human alveolar basal epithelial cell line (A549). The compounds have not shown significant citotoxicity at the tested concentrations. However, the evaluation of cytostatic activity on multiresistant A549 cell line has demonstrated an antiproliferative potential of compound 2f (3-OH and 4-OH) at 75 µM. Since compound 2f (3-OH and 4-OH) did not show citotoxicity on non-tumorigenic cell lines, it presented higher selectivity than chelerythrine. Molecular modeling results indicate that the lack of planarity of the chelerythrine analogues might be responsible for their lower citotoxicity. Thus, it is expected that the results might help directing the design of new compounds with superior activity and more selective than chelerythrine, thereby representing potential drug candidates, which may enhance the therapeutic arsenal for cancer treatment.
Key words: cancer, chelerythrine, molecular simplification, amine compounds, cytostatic activity.

11
LISTA DE FIGURAS
Figura 1. Gráfico dos principais tipos de câncer que afetaram a população mundial
em 2012 .................................................................................................................... 21
Figura 2. Ciclo celular e suas fases .......................................................................... 23
Figura 3. Vias bioquímicas indutoras da apoptose ................................................... 27
Figura 4. Interações entre as proteínas da família Bcl-2 na apoptose ...................... 29
Figura 5. A sequência de nucleotídeos do Oblimersen® (1) e as estruturas químicas
dos inibidores de Bcl-2 ABT-737 (2), ABT-263 (navitoclax) (3), ABT-199 (4), GX 15-
070 (obatoclax) (5) e AT 101 (6) ............................................................................... 34
Figura 6. Estruturas químicas dos alcaloides da vinca: vincristina (7), vimblastina (8),
vinorelbina (9) e vindesina (10) ................................................................................. 37
Figura 7. Estruturas químicas da camptotecina (11), topotecana (12) e irinotecano
(13) ............................................................................................................................ 38
Figura 8. Estruturas químicas dos alcaloides benzofenantridínicos sanguinarina (14)
e queleritrina (15) ...................................................................................................... 39
Figura 9. Estruturas químicas da cocaína (16) e procaína (17) ................................ 42
Figura 10. Estruturas químicas da quinina (18) e mefloquina (19) ........................... 43
Figura 11. Representação das modificações propostas na molécula de queleritrina
.................................................................................................................................. 45
Figura 12. Representação esquemática das sínteses dos análogos. Reagentes e
condições: (i) piperonilamina, NaBH(OAc)3, DCM, t.a., N2, 3-5 h; (ii) HCl etérico; (iii)
formaldeído, ácido fórmico, 110 ºC, 2-4 h ................................................................. 48
Figura 13. Representação esquemática da formação de aminas secundárias a partir
de iminas de (a) forma indireta e (b) forma direta ou aminação redutiva .................. 56
Figura 14. Representação das estruturas de ressonância de iminas com um grupo
eletronegativo ligado ao nitrogênio: (a) oxima e (b) sulfonil-hidrazona ..................... 57
Figura 15. Mecanismo de formação de amina secundária por aminação redutiva ...
.................................................................................................................................. 58
Figura 16. Espectro de RMN 1H do produto 2d ........................................................ 60
Figura 17. Espectro de RMN 13C do produto 2d ....................................................... 61
Figura 18. Mecanismo de formação de amina terciária por SN2 ............................... 62
Figura 19. Mecanismo de formação de amina terciária por metilação redutiva ........ 64

12
Figura 20. Espectro de RMN 1H do produto 3d ........................................................ 65
Figura 21. Espectro de RMN 13C do produto 3d ....................................................... 66
Figura 22. Estruturas químicas do gefitinibe (20) e temozolomida (21) .................... 70
Figura 23. (a) Efeito do composto 2f na Formação de colônias em A549, de maneira
dose-dependente. (b) O gráfico representa a media de três experimentos diferentes.
* p≤0.05; ** p≤0.01 comparado ao grupo controle (paired Student’s t test) ............... 71
Figura 24. Efeitos do composto 2f sobre o ciclo celular na linhagem A549, o qual o
histograma (a) representa o grupo controle, (b) o composto 2f a 100 µM, (c)
composto 2f a 75 µM e (d) 50 µM; a fase G0/G1 está em azul, S em verde e G2/M
em vermelho. (e) ilustra a % da distribuição de células em cada fase do ciclo. Os
resultados representam a média de dois experimentos diferentes ........................... 73
Figura 25. Estruturas químicas do composto 2f (22), da catequina (23), da
quercetina (24) e do ácido 3,4-di-idroxibenzóico (25), com o grupo catecólico em
destaque ................................................................................................................... 74
Figura 26. Representação de propriedades eletrônicas da queleritrina e dos
compostos 2a, 2c e 2f, por meio do mapa de potencial eletrostático, vetor do
momento dipolo (representados por setas amarelas), e mapas de orbitais
moleculares HOMO e LUMO ..................................................................................... 77
Figura 27. (a) Representação do alinhamento entre as estruturas da queleritrina e
do análogo 2f, nos quais os átomos de carbono estão representados em verde e
amarelo, respectivamente, os de oxigênio em vermelho e o nitrogênio em azul em
ambas. Os átomos de hidrogênio foram suprimidos para a melhor visualização. Em
(b), representação das distâncias interatômicas (em preto) mensuradas para a
queleritrina e em (c), representação das distâncias interatômicas (em preto) obtidas
para o análogo 2f. Ainda em (b) e (c), os ângulos diedros das estruturas da
queleritrina e do composto 2f, respectivamente, foram calculadas a partir dos
átomos circulados em vermelho ................................................................................ 78

13
LISTA DE TABELAS
Tabela 1. Distribuição proporcional dos dez tipos de câncer mais incidentes
estimados para 2015 por sexo, exceto pele não-melanoma ..................................... 22
Tabela 2. Fármacos inibidores de Bcl-2 em desenvolvimento .................................. 33
Tabela 3. Valores de IC50 da queleritrina em diversas linhagens celulares .............. 40
Tabela 4. Resultados obtidos na síntese dos análogos da queleritrina das séries das
aminas secundárias e terciárias ................................................................................ 55
Tabela 5. Análise elementar das aminas secundárias .............................................. 61
Tabela 6. Análise elementar das aminas terciárias ................................................... 66
Tabela 7. Valores de IC50 (µM) da queleritrina e do composto mais ativo das séries
.................................................................................................................................. 68
Tabela 8. Parâmetros físico-químicos calculados para a queleritrina e os compostos
2a, 2c e 2f ................................................................................................................. 79

14
LISTA DE SIGLAS E ABREVIATURAS
3D – tridimensional
ɛHOMO – energia de orbital HOMO
ɛLUMO – energia de orbital LUMO
A1 – Bcl-2 related gene A1 (gene A1 relacionado ao Bcl-2)
𝐴𝑎 – absorbância das amostras
𝐴𝑏 – absorbância do branco
𝐴𝑐 – absorbância do controle positivo
ACS – American Cancer Society (Sociedade Americana do Câncer)
AIF – Apoptosis-Inducing Factor (fator indutor de apoptose)
Apaf-1 – Apoptotic Protease Activing Factor (fator apoptótico 1 de ativação de
protease)
Apo2L – Apoptosis antigen 2 (antígeno de apoptose 2)
Apo3L – Apoptosis antigen 3 (antígeno de apoptose 3)
ABC – ATP binding cassete
Bad – Bcl-2-Associated Death promoter (promotora de morte associada à Bcl-2)
Bak – Bcl-2 Antagonist/Killer protein (antagonista/assassino de Bcl-2)
Bax – Bcl-2-Associated X protein (Bcl-2 associada à proteína X)
Bcl-2 – B-Cell Lymphoma 2 protein (proteína de linfoma 2 de células B)
Bcl-w – B-Cell Lymphoma w protein (proteína de linfoma w de células B)
Bcl-XL – B-Cell Lymphoma-Extra Large protein (proteína de linfoma de células B
extra grandes)
BH – Bcl-2 Homology (homólogos de Bcl-2)
BH3 – Bcl-2 homology region 3 (homólogo da região 3 de Bcl-2)
Bid – BH3-interacting domain Death agonist (agonista de morte que interage com o
domínio BH3)
Bik – Bcl-2-Interacting Killer (assassino que interage com Bcl-2)
Bim – Bcl-2-Interacting Mediator of cell death (mediador de morte celular que
interage com Bcl-2)
Bmf – Bcl-2-Modifying Factor (fator modificador de Bcl-2)
Bok - Bcl-2-Related Ovarian Killer (assassino ovariano que interage com Bcl-2)
BSA – bovine serum albumin (albumina sérica bovina)
CAD – Caspase Activated DNase (DNase ativada por caspase)

15
CCD – cromatografia em camada delgada
Cel – célula
CLAE – cromatografia líquida de alta eficiência
CLog P – logaritmo do coeficiente de partição calculado
COX-2 – ciclooxigenase-2
CRU – Cancer Research UK (Instituto do Câncer do Reino Unido)
Cyt c – citocromo C
DCM – diclorometano
DMEM – Dulbecco’s Modified Eagle’s medium
DIABLO – Direct IAP-Binding Protein with Low pI (proteína de ligação-IAP direta
com baixo pI)
DMSO – dimetilsulfóxido
DNA – Deoxyribonucleic acid (ácido desoxirribonucleico)
DNase – desoxirribonuclease
DR3 – Decoy Receptor 3 (Receptor Decoy 3)
DR4 – Decoy Receptor 4 (Receptor Decoy 4)
DR5 - Decoy Receptor 5 (Receptor Decoy 5)
EGFR – epidermal growth factor receptor (receptor de fator de crescimento
epidermal)
ELISA – Enzyme Linked Immuno Sorbent Assay (ensaio de imunoabsorção ligado à
enzima)
EROS – espécies reativas de oxigênio
FADD – Fas Associated Death Domain (domínio de morte associada a Fas)
FasL – Ligante Fas
FasR – Receptor Fas
G0 – Gap 0 (intervalo celular 0)
G1 – Gap 1 (intervalo celular 1)
G2 – Gap 2 (intervalo celular 2)
GST-π – glutationa S-transferase Pi
HBA – Hydrogen Bond Acceptor (sítio aceptor de ligação de hidrogênio)
HBD - Hydrogen Bond Donor (sítio doador de ligação de hidrogênio)
HOMO – Highest Occupied Molecular Orbital (orbital molecular ocupado de maior
energia)
HPV – Human Papiloma Virus (vírus do papiloma humano)

16
Hrk - Harakiri
HtrA2 – High Temperature Required protein A2
IAP – Inhibitors of Apoptosis Proteins (proteínas inibidoras de apoptose)
IARC – International Agency for Research on Cancer (Agência Internacional de
Pesquisa do Câncer)
IC50 – concentração inibitória 50 %
INCA – Instituto Nacional do Câncer “José Alencar Gomes da Silva”
LBDD – Ligand-Based Drug Design (planejamento de fármaco a partir do ligante)
LUMO - Lowest Unoccupied Molecular Orbital (orbital molecular não ocupado de
menor energia)
M – mitose
Mcl-1 – Myeloid Cell Leukemia-1 protein (proteína de célula mieloide de leucemia 1)
MMFF – Merck Molecular Force Field (campo de força molecular Merck)
MPE – mapa do potencial eletrostático
MTT – brometo de 3-(4,5-dimetiltiazol-2-1)2,5-difenil tetrazólio
NCI – National Cancer Institute (Instituto Nacional do Câncer)
Noxa – (Latin for Damage) Phorbol-12-myristate-13-acetate-induced protein 1
p21 – proteína de 21 kDa
p53 – proteína de 53 kDa
PBS – Phosphate Buffered Saline (tampão salina fosfato)
PM – peso molecular
PM6 – Parametric Method 6 (método paramétrico 6)
Puma – p53 Upregulated Modulator of Apoptosis (proteína moduladora de apoptose
modulada positivamente por p53)
R – ponto de restrição
Rf – Retenction fator (fator de retenção)
RMN – ressonância magnética nuclear
RNA – Ribonucleic acid (ácido ribonucleico)
RNase – ribonuclease
Ro5 – Regra dos Cinco de Lipinski
RPM – rotações por minuto
RPMI – Roswell Park Memorial Institute
S – fase celular de síntese de DNA

17
SBDD – Structure-Based Drug Design (planejamento de fármaco a partir da
estrutura do alvo)
SFB – soro fetal bovino
Smac – Second Mitochondria-derived Activator of Caspases (segundo ativador
mitocôndrial de caspase)
TNF – Tumor Necrosis Factor (fator de necrose tumoral)
TNFR1 - Tumor-Necrosis Factor Receptor 1 (receptor de fator de necrose tumoral 1)
TRADD – Tumor Necrosis Factor Receptor type 1 – Associated Death Domain
(domínio de morte associada ao TNF)
TRAIL – TNF-related apoptosis-inducing ligand (ligante indutor de apoptose
relacionado ao TNF)
UV – ultravioleta
WHO – World Health Organization (Organização Mundial da Saúde)

18
SUMÁRIO
1 INTRODUÇÃO ....................................................................................................... 20
1.1 Câncer ............................................................................................................. 20
1.1.1 Aspectos epidemiológicos ......................................................................... 20
1.1.2 Fisiopatologia............................................................................................. 23
1.1.3 Apoptose ................................................................................................... 25
1.1.4 Família Bcl-2 .............................................................................................. 28
1.2 Tratamento ...................................................................................................... 31
1.2.1 Inibidores de Bcl-2 ..................................................................................... 33
1.2.2 Produtos naturais ....................................................................................... 36
1.3 Planejamento racional ....................................................................................... 41
2 OBJETIVOS E JUSTIFICATIVA ............................................................................ 44
3 PLANEJAMENTO DOS ANÁLOGOS ................................................................... 45
4 MATERIAL E MÉTODOS ...................................................................................... 47
4.1 Material ............................................................................................................ 47
4.2 Métodos sintéticos ......................................................................................... 48
4.2.1 Síntese das aminas secundárias ............................................................... 48
4.2.2 Síntese das aminas terciárias .................................................................... 49
4.3 Métodos analíticos ......................................................................................... 50
4.3.1 Cromatografia em camada delgada (CCD) e cromatografia em coluna .... 50
4.3.2 Espectrometria por ressonância magnética nuclear (RMN) ...................... 50
4.3.3 Cromatografia líquida de alta eficiência (CLAE) acoplado ao UV .............. 50
4.3.4 Análise elementar (CHN) ........................................................................... 51
4.4 Métodos biológicos ........................................................................................ 51
4.4.1 Cultura celular ............................................................................................ 51
4.4.2 Avaliação da citotoxicidade ....................................................................... 52

19
4.4.3 Ensaio clonogênico .................................................................................... 53
4.4.4 Análise do ciclo celular .............................................................................. 53
4.5 Métodos computacionais .............................................................................. 54
5 RESULTADOS E DISCUSSÃO ............................................................................. 55
5.1 Síntese e caracterização das aminas secundárias ..................................... 56
5.2 Síntese e caracterização de aminas terciárias ............................................ 62
5.3 Ensaios biológicos ......................................................................................... 67
5.3.1 Ensaio de citotoxicidade ............................................................................ 67
5.3.2 Ensaio clonogênico e ciclo celular ............................................................. 70
5.4 Modelagem molecular .................................................................................... 75
6 CONCLUSÕES E PERSPECTIVAS ...................................................................... 81
7 REFERÊNCIAS BIBLIOGRÁFICAS ...................................................................... 84
8 ANEXOS ................................................................................................................ 99

20
1 INTRODUÇÃO
1.1 Câncer
1.1.1 Aspectos epidemiológicos
Câncer é o termo genérico designado a um grupo de doenças que pode afetar
qualquer parte do organismo. Esse grupo de doenças também pode ser denominado
como neoplasia maligna, na qual a palavra neoplasia significa “novo crescimento”,
ou tumor maligno (WHO, 2015).
O câncer foi considerado por muito tempo uma doença de países
desenvolvidos. Porém, nas últimas décadas, tornou-se um problema de saúde
pública mundial (INCA, 2012). Isso se fundamenta no fato de que os indivíduos
vivenciam um prolongamento da expectativa de vida e envelhecimento populacional.
Dessa forma, aumenta-se a incidência de doenças crônico-degenerativas, tais como
as doenças cardiovasculares e o câncer (INCA, 2014).
Embora as taxas de incidência sejam mais elevadas em países mais
desenvolvidos, (CRU, 2011), os países em desenvolvimento e com poucos recursos
são os mais afetados. Cerca de 70% das mortes por neoplasias são atribuídos a
esses locais (WHO, 2015), principalmente porque países em desenvolvimento
concentram mais pessoas de baixa renda, e estas, por sua vez, têm prognóstico
menos favorável após o diagnóstico da doença (ACS, 2014).
Este padrão está associado a barreiras financeiras, estruturais e pessoais aos
auxílios médico-hospitalares, dentre eles o reduzido acesso a cuidados preventivos,
ao tratamento adequado, bem como o diminuto acesso a informações. Desta forma,
frequentemente o diagnóstico é feito quando a doença está em um estado
avançado, no qual o tratamento não é capaz de atribuir qualidade e sobrevida ao
paciente (ACS, 2014).
Existem mais de 200 tipos de câncer, cada um com uma diferente causa,
quadro de sintomas e tratamento (CRU, 2014). Em 2012, os principais tipos de
tumores malignos que acometeram a população mundial foram os de pulmão,

21
1590000
745000 723000 694000521000
400000
0200000400000600000800000
10000001200000140000016000001800000
Pulmão Fígado Estômago Colorretal Mama Esôfago
Principais tipos de Câncer em 2012
fígado, estômago, colorretal, de mama e de esôfago, conforme a Figura 1 (WHO,
2015).
Figura 1. Gráfico dos principais tipos de câncer que afetaram a população mundial em 2012 (WHO, 2015).
Ademais, a incidência destas neoplasias varia de acordo com a distribuição
de recursos dos países. Naqueles com maior renda per capita, os cânceres de
pulmão, cólon e reto, mama e próstata predominam. Já em países de menor produto
interno bruto, a predominância é dos cânceres de pulmão, estômago, fígado, mama
e colo de útero (ACS, 2014; INCA, 2014; WHO, 2015).
Alguns fatores podem contribuir para a instalação do câncer. Maus hábitos de
vida, como o tabagismo, são responsáveis pelo aumento da incidência de cânceres,
como o de pulmão. Além disso, o estilo de vida adotado pela população no que se
refere à má alimentação e ao sedentarismo, aumenta a incidência de tumores
malignos como os de cólon e reto, estômago, fígado, mama e próstata. O menor
acesso a programas preventivos como o exame Papanicolau e a vacinação contra o
Papilomavírus humano (HPV) é responsável pela predominância de câncer de colo
do útero em países em desenvolvimento (ACS, 2014; INCA, 2014; WHO, 2015).
Ainda assim, os padrões podem mudar, já que historicamente cânceres de
pulmão, mama, cólon e reto não apresentavam a mesma importância e magnitude
que apresentam atualmente (INCA, 2014; WHO, 2015). Como o aumento na
prevalência de câncer no mundo está diretamente relacionado ao tabagismo,
sedentarismo, má alimentação, uso de álcool, exposição à poluição, suscetibilidade

22
ao Papilomavírus humano (HPV) e ao vírus da hepatite, o câncer se configura como
uma doença que pode ser prevenida ao modificar ou evitar esses fatores de risco
(ACS, 2014; WHO, 2015).
No Brasil, segundo dados do Instituto Nacional do Câncer “José Alencar
Gomes da Silva” (INCA), estima-se que para o ano de 2015 ocorram
aproximadamente 576 mil novos casos de câncer, incluindo os casos de pele não-
melanoma e, excluindo este último, estima-se um total de 394 mil novos casos. A
distribuição dos dez tipos de câncer mais incidentes por sexo está representada na
Tabela 1, na qual pode-se observar que os tumores mais incidentes em homens e
mulheres no Brasil são, respectivamente, próstata e mama (INCA, 2014).
Tabela 1. Distribuição proporcional dos dez tipos de câncer mais incidentes estimados para 2015 por sexo, exceto pele não-melanoma (Adaptado de INCA, 2014).
Homens Mulheres
Localização Primária
Casos Novos
Percentual Localização
Primária Casos Novos
Percentual
Próstata 68800 22,8% Mama Feminina 57120 20,8% Traqueia,
Brônquio e Pulmão
16400 5,4% Cólon e Reto 17530 6,4%
Cólon e Reto 15070 5,0% Colo do Útero 15590 5,7%
Estômago 12870 4,3% Traqueia,
Brônquio e Pulmão
10930 4,0%
Cavidade Oral 11280 3,7% Glândula Tireóide
8050 2,9%
Esôfago 8010 2,6% Estômago 7520 2,7% Laringe 6870 2,3% Corpo do Útero 5900 2,2% Bexiga 6750 2,2% Ovário 5680 2,1%
Leucemias 5050 1,7% Linfoma não
Hodgkin 4850 1,8%
Sistema Nervoso Central
4960 1,6% Leucemias 4320 1,6%
Atualmente, o câncer é uma das principais causas de mortes no mundo, o
qual foi responsável por aproximadamente 8,2 milhões de óbitos e 14,1 milhões de
casos novos em 2012 (IARC, 2012; WHO, 2015). De acordo com o INCA, em 2030,
a projeção do número de óbitos relacionados ao câncer pode alcançar 13,2 milhões
de indivíduos e a incidência será de 21,4 milhões de pessoas (INCA, 2014).
Diante da magnitude e do impacto da doença, medidas preventivas e a
descoberta de novas alternativas para o combate ao câncer tornam-se necessárias.

23
Os tratamentos utilizados atualmente são muitas vezes ineficazes e muitas
linhagens tumorais já não respondem à farmacoterapia disponível, ou até mesmo
desenvolvem resistência a esses agentes. Desta forma, a introdução de novas
opções terapêuticas torna-se um dos focos para o controle do câncer e suas
consequências (AVENDAÑO; MENÉNDEZ, 2008).
1.1.2 Fisiopatologia
Os cânceres surgem a partir de alterações na homeostase celular, com um
componente genético fortemente associado. Estas alterações culminam no
crescimento desordenado de células anômalas, incapazes de formar uma estrutura
funcional e que têm a capacidade de invasão a tecidos adjacentes e órgãos
distantes (FLOOR et al., 2012; PATRICK, 2009).
Em uma célula normal, o ciclo celular é regulado por inúmeros mecanismos
que visam a correta divisão celular, a qual é dividida em dois estágios: a mitose, que
é subdividida em prófase, metáfase, anáfase e telófase; e a intérfase, que é o
intervalo entre duas mitoses e também é subdivida em G0, G1, S e G2, conforme
Figura 2 (FISCHER, 2008).
Figura 2. Ciclo celular e suas fases (VERMEULEN; VAN BOCKSTAELE; BERNEMAN, 2003).

24
A preparação para a síntese de DNA ocorre na fase G1. Nesta fase, a célula
encontra-se suscetível às condições ambientais e caso elas não sejam favoráveis, o
ciclo é interrompido. Entretanto, se o ciclo celular já tiver ultrapassado o ponto de
restrição (R) (Figura 2), o ciclo celular progredirá independente dos estímulos
ambientais (GUEMBAROVSKI; CÓLUS, 2008).
O processo de síntese replicação do DNA ocorre na fase S, enquanto na fase
G2 ocorre a preparação para a mitose, estado no qual ocorre a divisão nuclear.
Quando a célula se caracteriza em um estado de quiescência, ou repouso,
permanecendo na intérfase, ela encontra-se na fase G0, a qual sob estímulo, pode
retornar à fase G1 e dar continuidade ao ciclo celular (VERMEULEN; VAN
BOCKSTAELE; BERNEMAN, 2003).
O controle do ciclo celular é mediado pelo ponto de restrição e pelos pontos
de checagem, que podem pausar o ciclo celular em qualquer etapa do processo. Isto
ocorre quando é identificado algum dano ou alteração no DNA, para que haja os
reparos ou, na sua impossibilidade, indução da célula à apoptose (GABRIELLI;
BROOKS; PAVEY, 2012).
A desregulação do ciclo celular e crescimento descontrolado das células
podem surgir por meio de mutações que ocorrem em duas classes de genes: os
proto-oncogenes e os genes supressores de tumor. Os proto-oncogenes possuem a
função de estimular a proliferação celular e, quando sofrem mutação, passam a ser
denominados de oncogenes, os quais permitirão ganho de função à célula mutada
(LUO; SOLIMINI; ELLEDGE, 2009).
Em contrapartida, os genes supressores de tumor codificam proteínas
envolvidas no controle negativo do ciclo celular e, quando mutados, perdem sua
função. A perda desse mecanismo, em conjunto com as alterações em proto-
oncogenes, levam a uma característica marcante da maioria das células tumorais,
que é a proliferação desordenada das células (VOGELSTEIN; KINZLER, 2004).
A tumorigênese, ou seja, o processo de formação tumoral, é descrita como
um processo de várias etapas. Diversas mutações genéticas estão envolvidas e
permitem que as células atingidas adquiram novas capacidades (HANAHAN;
WEINBERG, 2011). Hanahan e Weinberg (2011) sugerem oito alterações essenciais

25
na fisiologia de uma célula normal que permitem o crescimento tumoral maligno.
Dentre elas estão a autossuficiência de sinais de crescimento, a insensibilidade a
sinais de supressão tumoral, a evasão a apoptose, o potencial replicativo ilimitado, a
indução de angiogênese, a invasão tecidual e metástase, reprogramação metabólica
e evasão ao sistema imune.
A apoptose tem importância na homeostase tecidual, pois pode evitar a
perpetuação de potenciais mutações oncogênicas ao induzir a célula mutada à
morte. Como a evasão à apoptose é um mecanismo presente em muitas células
cancerígenas, este processo é um dos alvos para o desenvolvimento de agentes
anticâncer. Além disso, a atenuação da apoptose é observada em tumores que
progridem a estágios malignos e são resistentes à terapia (BROWN; ATTARDI,
2005; HANAHAN; WEINBERG, 2011).
1.1.3 Apoptose
A apoptose é o fenômeno de indução à morte celular programada que ocorre
em diversos processos fisiológicos e patológicos (FULDA; DEBATIN, 2006). Pode
ocorrer durante o desenvolvimento e o envelhecimento tecidual, sendo um
mecanismo homeostático necessário para a manutenção da população celular
(ELMORE, 2007). A desregulação desse processo pode causar um desequilíbrio
entre proliferação e morte celular, favorecendo o aparecimento de doenças como o
câncer (IGNEY; KRAMMER, 2002).
Esse fenômeno se caracteriza por uma série de mudanças morfológicas
celulares, dentre as quais há a condensação nuclear (picnose) e fragmentação
(cariorréxis). Nota-se também a formação de uma protuberância irregular da
membrana plasmática que culmina na produção de corpos apoptóticos (MARIÑO et
al., 2014).
Os corpos apoptóticos, por sua vez, são constituídos de citoplasma com as
organelas compactadas com ou sem um fragmento nuclear. Estes são
subsequentemente fagocitados e degradados por macrófagos, células parenquimais
ou células neoplásicas. A apoptose não envolve um processo inflamatório, o que a
diferencia de outras vias de morte celular, como a necrose (ELMORE, 2007).

26
A apoptose apresenta um mecanismo altamente complexo e sofisticado que
envolve uma cascata de eventos moleculares dependentes de ativação energética.
Basicamente, engloba a ativação de cisteína proteases, denominadas de caspases,
as quais possuem atividade proteolítica e têm como função a amplificação da via de
sinalização apoptótica (FULDA; DEBATIN, 2006).
As caspases são sintetizadas como proformas inativas (procaspases) e
quando ativadas, clivam outras procaspases nas proximidades dos resíduos de
aspartato presentes nessas enzimas. Além disso, as caspases podem se autoativar
ou ativar umas às outras em sequência (ELMORE, 2007).
Na apoptose, essas proteases podem ser divididas em ativadoras e
executoras. As ativadoras (caspases-8 e 9) clivam proformas inativas de caspases
executoras (caspases-3, 6 e 7), e estas, por sua vez, clivam outros substratos
proteicos da célula resultando no processo apoptótico (FAN et al., 2005).
Em princípio, a apoptose pode ser ativada por duas vias alternativas, uma via
extrínseca mediada por receptores de morte celular, e uma via intrínseca, mediada
pela mitocôndria. Entretanto, há evidências de que essas vias estão interligadas,
pois alguns fatores de uma via podem influenciar na outra. Ademais, ambas as vias
convergem na ativação das caspases executoras, conforme Figura 3 (FULDA;
DEBATIN, 2006; IGNEY; KRAMMER, 2002).
A via extrínseca da apoptose ocorre em resposta a estímulos aos receptores
de morte. Estes pertencem à superfamília de receptores de fator de necrose tumoral
(TNF – Tumor Necrosis Factor) que são ativados por determinados ligantes e
transmitem o sinal de morte da superfície celular para vias sinalizadoras
intracelulares (Figura 3). Os FasL/FasR, TNF-α/TNFR1, Apo3L/DR3, Apo2L/DR4 e
Apo2L/DR5 são alguns exemplos de ligantes e seus respectivos receptores
(ELMORE, 2007; FULDA; DEBATIN, 2006; IGNEY; KRAMMER, 2002).
Após a ativação dos receptores de morte, ocorre o recrutamento de diversas
proteínas citoplasmáticas. Dentre elas, há a proteína adaptadora de domínio de
morte associada a Fas (FADD – Fas Associated Death Domain), resultante da
interação FasL/FasR; e a proteína adaptadora de domínio de morte associada ao
TNF (TRADD – Tumor Necrosis Factor Receptor type 1 – Associated Death
Domain), resultante da interação TNF-α/TNFR1, a qual recruta diretamente o FADD

27
(Figura 3). Esta última forma um complexo com a procaspase-8 que resulta em sua
ativação para caspase-8. Uma vez ativada, a caspase-8 inicia o processo de
apoptose por meio da clivagem das caspases executoras 3, 6 e 7, que culmina em
uma cascata proteolítica e posterior morte celular (Figura 3) (ASHKENAZI, 2008).
Figura 3. Vias bioquímicas indutoras da apoptose (FERNANDES, 2015).
A via intrínseca ou mitocondrial, por sua vez, é iniciada por estímulos
produtores de sinais intracelulares que podem agir de maneira positiva ou negativa
sobre os eventos celulares (Figura 3). Sinais negativos envolvem a ausência de
fatores de crescimento, hormônios e citocinas que conduzem a falhas na supressão
de morte celular, induzindo à apoptose. Os estímulos que podem agir de maneira
positiva são a radiação, toxinas, hipóxia, hipertermia, infecções virais e radicais livres
(ELMORE, 2007).

28
Todos esses estímulos provocam modificações na membrana mitocondrial,
que resulta em formação de poros, perda do potencial transmembrânico e liberação
de dois grupos principais de proteínas proapotóticas do espaço intermembrana para
o citosol (Figura 3). O primeiro grupo consiste de citocromo c, Smac/DIABLO e a
serina protease HtrA2/Omi. Estas proteínas ativam a via mitocondrial caspase-
dependente (FULDA; DEBATIN, 2006).
O citocromo c se liga à Apaf-1 e à procaspase-9, formando o apoptossomo, o
qual é responsável pela ativação da caspase-9. Já a Smac/DIABLO e a HtrA2/Omi
são relacionadas à promoção de apoptose por meio da inibição de proteínas
inibidoras de apoptose (IAP – Inhibitors of Apoptosis Proteins) (ELMORE, 2007;
RENAULT; CHIPUK, 2014).
O segundo grupo de proteínas proapoptóticas, formado pelo fator indutor de
apoptose (AIF – Apoptosis-Inducing Factor), endonuclease G e DNase ativada por
caspase (CAD – Caspase Activated DNase), é liberado da mitocôndria tardiamente,
após a célula já ter iniciado o processo de morte (FULDA; DEBATIN, 2006).
O controle e regulação da apoptose associada à via mitocondrial são
realizados por membros da família Bcl-2 de proteínas, as quais tem papel
fundamental na permeabilidade da membrana desta organela. A proteína supressora
de apoptose p53 tem papel crucial na regulação desta família de proteínas,
induzindo-as ao efeito pró-apoptótico ou antiapoptótico, dependendo dos membros
envolvidos no processo (Figura 3) (MARTINOU; YOULE, 2011).
1.1.4 Família Bcl-2
A família Bcl-2 de proteínas representa o centro regulador da apoptose por
meio da via mitocondrial. Os membros desta família podem ser divididos de acordo
com a sua estrutura e função. Considerando-se a sua estrutura, as proteínas podem
conter quatro sequências homólogas, denominadas de domínios homólogos de Bcl-
2 (BH – Bcl-2 Homology). De acordo com a sua função, as proteínas podem ser
antiapoptóticas ou proapotóticas (PLACZEK et al., 2010).

29
As proteínas que apresentam os quatro domínios BH, podem atuar como
supressores de apoptose (por exemplo, Bcl-2, Bcl-XL, Bcl-w, Mcl-1 e A1) (GARCÍA-
SÁEZ, 2012) ou promotores de apoptose (por exemplo, Bax, Bak e Bok) (LONDON
et al., 2012). O mecanismo pelo qual essas proteínas regulam a apoptose está
representado na Figura 4 (DEWSON; KLUCK, 2010).
Figura 4. Interações entre as proteínas da família Bcl-2 na apoptose (PLACZEK et al., 2010).
As proteínas denominadas de BH3-only (Bid, Bim, Puma, Noxa, Bad, Bmf,
Hrk e Bik), as quais possuem apenas o domínio homólogo BH3, desempenham a
função de promover a apoptose pela sensibilização ou ativação das proteínas
proapoptóticas efetoras (Figura 4) (DEWSON; KLUCK, 2010).
A proteína Bid, uma proteína proapoptótica do tipo BH3-only ativadora, atua
na indução da apoptose de maneira direta, ao se ligar à Bak, Bax e/ou Bok ativando-
as (1). Esta ativação resulta na dimerização entre Bak, Bax e/ou Bok (2) e
consequente oligomerização destas proteínas (3). O oligodímero formado se liga na
membrana mitocondrial e, consequentemente, promove a liberação de citocromo c
(4). Este, em conjunto a outras substâncias, forma o apoptossomo, um complexo

30
que tem como função a ativação da caspase 9, a qual dará continuidade ao
processo de apoptose (PLACZEK et al., 2010).
As proteínas antiapoptóticas, tais como Bcl-2, Bcl-XL, Bcl-w, Mcl-1 e A1, tem
como função a conjugação com proteínas proapoptóticas efetoras, tais como Bak,
Bax e Bok, representadas pelo evento (5), na Figura 4. A interação de uma proteína
antiapoptótica com uma proapoptótica efetora interfere na atividade da última, visto
que as proteínas supressoras de apoptose vão ocupar sítios de ligação nos quais as
proteínas BH3-only interagem, resultando na ausência de ativação de Bak, Bax e
Bok (CZABOTAR et al., 2014).
A promoção da apoptose pode ocorrer de maneira indireta, mediada pela
proteína Bid, a qual pode se ligar às proteínas da família antiapoptótica, promovendo
a inibição das mesmas (6). Em outras palavras, as proteínas responsáveis pela
supressão da apoptose sofrem inibição pelo Bid e perdem a capacidade de se ligar a
Bak, Bax e Bok (SCHROEDER et al., 2012).
A inibição dos membros antiapoptóticos da família Bcl-2 também pode ocorrer
por meio da ligação das proteínas proapoptóticas BH3-only do tipo sensibilizadoras,
como a Noxa e a Bad (6). Este mecanismo é denominado de sensibilização à
apoptose porque Noxa e Bad não se ligam a Bak, Bax e/ou Bok e, portanto, não
desempenham função na ativação direta das proteínas efetoras (SCHROEDER et
al., 2012).
Altos níveis de concentração dos agentes supressores de apoptose da família
de proteínas Bcl-2 estão associados a tumores resistentes à quimioterapia
(VOGLER et al., 2009). O tipo de proteína superexpressa pode variar entre os
diversos tipos de câncer existentes. A própria Bcl-2, por exemplo, tem alta
expressão em linfoma folicular de células B, no qual essa proteína foi descoberta
primeiramente, dando origem ao seu nome (IGNEY; KRAMMER, 2002).
A Mcl-1, por sua vez, apresenta-se superexpressa em tumores
hematopoéticos e linfóides, incluindo o mieloma múltiplo e leucemia linfoide crônica
(LESSENE; CZABOTAR; COLMAN, 2008), enquanto a proteína Bcl-w está
relacionada aos tumores de origem epitelial, tais como o adenocarcinoma colorretal
e o câncer gástrico (LEE et al., 2003). A alta expressão de Bcl-XL é observada em
tumores como melanoma, câncer de mama, de próstata, de pulmão, linfoma de

31
células B, câncer colorretal e adenocarcinomas em geral (MENA et al., 2012;
RAGHAV; VERMA; GANGENAHALLI, 2012).
Amudson e colaboradores (2000) coordenaram um estudo utilizando o painel
NCI (National Cancer Institute) de 60 linhagens de células tumorais humanas de
diferentes origens. Este estudo demonstrou forte correlação entre altos níveis de
Bcl-XL e sensibilidade das células tumorais à maioria dos fármacos utilizados na
terapêutica. Desta forma, a Bcl-XL representa um alvo interessante para o
desenvolvimento de novos fármacos que podem apresentar atividade biológica em
linhagens tumorais resistentes aos quimioterápicos da terapia atual (ADAMS; CORY,
2007; KAZI et al., 2011; VOGLER et al., 2009).
1.2 Tratamento
O tratamento do câncer pode variar de acordo com o tempo de detecção, pois
quanto mais cedo for o diagnóstico, menor será o tamanho do neoplasma e mais
eficiente será o tratamento. Além disso, o tipo de tratamento estabelecido varia de
acordo com tecido afetado, com a velocidade de crescimento tumoral, com o grau de
diferenciação das células que compõem o tumor maligno e se este sofreu
metástase. Desta forma, mais de uma estratégia para o correto tratamento do
câncer pode ser necessária, e dentre elas há a cirurgia, a radioterapia e a
quimioterapia (INCA, 2012; WHO, 2012).
O que muitas vezes agrava o quadro do paciente e torna o tratamento do
câncer exponencialmente mais difícil é a metástase. Responsável por 90% das
mortes oriundas de quadros neoplásicos, a metástase é um processo extremamente
complexo, que ocorre por meio de uma série de eventos. Esses podem ser
representados pela invasão de tecidos adjacentes ao de origem do tumor, transporte
das células pela circulação sanguínea ou linfática, seguida pela fixação e
crescimento de novos focos tumorais em órgãos ou tecidos secundários (CHAFFER;
WEINBERG, 2011; MEHLEN; PUISIEUX, 2006; WAN; PANTEL; KANG, 2013).
Em busca de um tratamento que diminua as chances de metástases dos
tumores malignos e que seja capaz de regredir o seu crescimento, torna-se
necessária uma abordagem sistêmica. Esta é obtida por meio do tratamento
quimioterápico, pois as células tumorais, por estarem em constante divisão, são

32
mais suscetíveis aos fármacos antineoplásicos, dado o aumento da sua atividade
metabólica (DE ALMEIDA et al., 2005).
O tratamento quimioterápico também se mostra importante em casos nos
quais os tumores têm a característica de não serem sólidos, como os que afetam o
sistema hematopoiético. Como as células encontram-se disseminadas, esses tipos
tumorais têm uma dependência maior da quimioterapia antineoplásica, pois esta tem
uma capacidade maior de difundir-se (INCA, 2008).
Os fármacos disponíveis na terapia anticâncer têm diversos mecanismos de
ação e agem em diferentes etapas do ciclo celular. Dentre eles, podem-se destacar
os que atuam diretamente no DNA, por meio de alquilação; os antimetabólitos, que
atuam diretamente ou indiretamente na síntese de DNA; os intercalantes de DNA; os
inibidores de mitose e os inibidores de tirosina-quinase (DE ALMEIDA et al., 2005).
Entretanto, a maioria dos fármacos é altamente tóxica, principalmente sobre
células de rápida divisão celular o que causa os efeitos adversos como queda de
cabelo, náuseas e destruição da medula óssea (ROCHE, 2013). Além disso, muitos
pacientes exibem ou desenvolvem resistência aos agentes quimioterápicos
antineoplásicos existentes, ou seja, uma resistência intrínseca ou adquirida
(GARRAWAY; JANNE, 2012).
A resistência intrínseca é observada desde o início do tratamento e pode
estar relacionada ao crescimento lento das células, diminuição da captação dos
fármacos pelas células ou por propriedades bioquímicas/genéticas das células
(PATRICK, 2009). Entre estas propriedades, pode-se citar a alta expressão de
proteínas antiapoptóticas da família Bcl-2, que, como citado anteriormente, está
diretamente relacionada à resistência de diversas linhagens tumorais aos agentes
antineoplásicos utilizados na terapêutica (AMUNDSON et al., 2000; NÉMATI et al.,
2014; SAVRY et al., 2013).
Na resistência adquirida, o tumor inicialmente apresenta sensibilidade ao
fármaco e torna-se resistente ao longo do tratamento. Isto pode ocorrer devido a
uma mistura de células tumorais sensíveis e células resistentes ao fármaco. Com a
exposição do tumor ao agente antineoplásico, somente as células resistentes
sobrevivem e o neoplasma torna-se menos sensível ao quimioterápico (PATRICK,
2009).

33
Adicionalmente, células tumorais podem adquirir resistência a partir de
alterações em genes regulatórios, decorrentes da exposição ao próprio
quimioterápico (WU; HSIEH; WU, 2011). Portanto, torna-se evidente a importância
da introdução de novos fármacos para a terapia antineoplásica (GARRAWAY;
JANNE, 2012).
1.2.1 Inibidores de Bcl-2
Tendo em vista a necessidade de se ampliar as opções terapêuticas contra o
câncer, o desenvolvimento de compostos capazes de promover a inibição dos
membros antiapoptóticos da família Bcl-2 torna-se relevante, uma vez que
favoreceriam a apoptose em células cancerígenas, além de sensibilizá-las frente a
outros agentes quimioterápicos (KANG; REYNOLDS, 2009; MUKHERJEE et al.,
2010). Neste sentido, alguns agentes que possuem estas proteínas como alvo foram
desenvolvidos, conforme mostra a Tabela 2, e suas respectivas estruturas químicas
estão agrupadas na Figura 5.
Tabela 2. Fármacos inibidores de Bcl-2 em desenvolvimento.
Composto Alvo Estágio
Oblimersen® Bcl-2 Fase III
ABT-737 Bcl-2, Bcl-XL, Bcl-w Fase I/II
ABT-263 (navitoclax) Bcl-2, Bcl-XL, Bcl-w Fase I/II
ABT-199 (venetoclax) Bcl-2 Fase I/II
GX 15-070 (obatoclax) Bcl-2, Bcl-XL, Bcl-w, Mcl-1 Fase I/II
AT 101-(R-(-)-gossypol) Bcl-2, Bcl-XL, Bcl-w, Mcl-1 Fase I/II
(Fonte: KANG; REYNOLDS, 2009; SOUERS et al., 2013; YAP; WORKMAN, 2012).
O Oblimersen® (1) é um fármaco sintético oligonuclotídeo antisenso de DNA
de 18 bases e fita única, que foi desenvolvido para diminuir a expressão de RNA
mensageiro da proteína Bcl-2 (CHESON, 2007). Este fármaco está envolvido em
estudos clínicos para o tratamento de câncer de mama, melanoma, linfoma, entre
outros tumores, tanto em monoterapia ou associado a outro fármaco
(CLINICALTRIALS, 2015).

34
Figura 5. A sequência de nucleotídeos do Oblimersen® (1) e as estruturas químicas dos inibidores de
Bcl-2 ABT-737 (2), ABT-263 (navitoclax) (3), ABT-199 (4), GX 15-070 (obatoclax) (5) e AT 101 (6).
O Oblimersen® se liga seletivamente aos primeiros seis códons do RNAm,
formando um complexo que recruta a enzima RNase. Esta, consequentemente,
degrada a fita de RNA, liberando o fármaco para atuar em outras fitas. Como
resultado, ocorre a diminuição da expressão de proteínas Bcl-2, o que leva ao

35
aumento da sensibilidade das células neoplásicas aos agentes quimioterápicos
antineoplásicos (CHESON, 2007).
O ABT-737 (2) (Figura 5) foi um dos primeiros inibidores a serem
desenvolvidos pelo planejamento baseado no fragmento (Fragment-Based Drug
Design – FBDD), e possui alta afinidade com as proteínas Bcl-2, Bcl-XL, Bcl-w
(ZHOU et al., 2012). Este fármaco está sendo testado em ensaios ex vivo de câncer
ovariano, sozinho e em associação (CLINICALTRIALS, 2015). Porém, o progresso
dos estudos foi prejudicado devido a sua baixa solubilidade e biodisponibilidade por
via oral (TSE et al., 2008).
O ABT-263 (3) (Figura 5), ou navitoclax, foi planejado a partir da otimização
do ABT-737 e tem afinidade pelas mesmas proteínas que seu precursor apresenta.
Ao contrário do ABT-737, o navitoclax pode ser administrado por via oral (ZHOU et
al., 2012), e já está sendo testado em tumores sólidos, linfomas e leucemias, em
monoterapia e em associação (CLINICALTRIALS, 2015).
Já o ABT-199 (4) (Figura 5), foi planejado para exibir seletividade apenas para
a proteína Bcl-2, pois os inibidores de Bcl-XL estão relacionados à toxicidade sobre
plaquetas, provocando trombocitopenia (SOUERS et al., 2013). O ABT-199 está em
teste para leucemia mieloide aguda e linfoma não-Hodgkin (CLINICALTRIALS,
2015).
O GX 15-070 (5) (Figura 5), ou obatoclax, é um análogo da cicloprodigiosina
com capacidade de inibição na Bcl-2, Bcl-XL, Bcl-w e Mcl-1 (GOARD; SCHIMMER,
2013). Este composto está em estudo clínico para a terapia de diversos tipos de
tumor, dentre eles linfomas e câncer de pulmão, tanto em monoterapia quanto em
associação (CLINICALTRIALS, 2015).
Por fim, o AT 101 (6), um atropoisômero do gossipol, é um composto
polifenólico isolado de sementes e raízes da planta do algodão e também exibe
capacidade de inibição na Bcl-2, Bcl-XL, Bcl-w e Mcl-1. Evidências sugerem que a
citotoxicidade do AT 101 também pode ser resultante de mecanismos adicionais,
como clivagem do DNA ou geração de espécies reativas de oxigênio (NI et al.,
2013).
O AT 101 está envolvido em estudos clínicos frente a diversos tumores como
os de próstata, pulmão e linfoma (CLINICALTRIALS, 2015). Este composto, bem

36
como o GX 15-070, se originou por meio da descoberta de produtos naturais com
potencial de inibição às proteínas da família Bcl-2, ressaltando a importância dessa
abordagem na descoberta de candidatos a fármaco.
1.2.2 Produtos naturais
O uso de produtos naturais para o tratamento do câncer reporta-se a tempos
remotos. Cerca de 60% dos agentes utilizados atualmente na terapia antineoplásica
advém de fontes naturais. O reino vegetal é o que mais contribui na utilização de
substâncias naturais para o tratamento de diversas doenças (BHANOT; SHARMA;
NOOLVI, 2011; COSTA-LOTUFO et al., 2010).
Este fato é devido à grande variedade e complexidade de metabólitos
secundários que as plantas podem fornecer. Esses compostos caracterizam-se pela
disposição de diversos heterociclos e centros quirais em suas estruturas, o que
justifica a sua variedade e complexidade. Muitos metabólitos secundários originam-
se a partir da adaptação e regulação das plantas ao ambiente, bem como em sua
proteção contra predadores (CHIN et al., 2006; MONTANARI; BOLZANI, 2001;
NEWMAN; CRAGG, 2012).
Os alcaloides merecem destaque dentre os diversos tipos de metabólitos
secundários existentes. Caracterizados por conter átomos de nitrogênio e anéis
heterocíclicos, essas substâncias estão presentes em muitas terapias para diversas
doenças, servindo tanto como fármaco, quanto protótipo para o planejamento de
novos compostos bioativos (KITTAKOOP; MAHIDOL; RUCHIRAWAT, 2014).
A vincristina (7) e a vimblastina (8) (Figura 6), de origem natural, junto à
vinorelbina (9) e à vindesina (10) (Figura 6), de origem semissintética, representam
um sucesso terapêutico no emprego de alcaloides para o tratamento do câncer
(FUMOLEAU; GUIU, 2013). Conhecidos como alcaloides da vinca, os compostos de
origem natural podem ser encontrados nas folhas da espécie Catharanthus roseus,
conhecida popularmente como vinca (BRANDÃO et al., 2010).
Os alcaloides da vinca atuam como desestabilizadores de microtúbulos,
atividade esta que resulta na lentidão ou bloqueio da mitose, ou seja, da divisão
celular (GIGANT et al., 2005). Devido a isso, são altamente empregados na terapia

37
contra o câncer, visto que as células tumorais estão em constante divisão.
Entretanto, a neuropatia apresenta-se como um importante efeito colateral
decorrente destes fármacos. Acredita-se que os microtúbulos sejam componentes
chave para os neurônios, o que justificaria tal neurotoxicidade (BRANDÃO et al.,
2010).
Figura 6. Estruturas químicas dos alcaloides da vinca: vincristina (7), vimblastina (8), vinorelbina (9) e
vindesina (10).
A camptotecina (11) (Figura 7), que pode ser encontrada na espécie vegetal
Camptotheca acuminata, é outro exemplo de alcaloide empregado para a terapia
anticâncer. Entretanto, devido à toxicidade promovida pelo metabólito carboxilato da
camptotecina, esta foi descontinuada durante os ensaios clínicos (CAO et al., 2005).
Contudo, a camptotecina apresenta um mecanismo de ação interessante
devido à sua atuação na inibição da síntese de DNA e RNA. Em estudos

38
mecanísticos mais aprofundados, verificou-se que essa atividade era devido à
interação da camptotecina com a topoisomerase I, que desempenha papel
importante nos processos de replicação e empacotamento de DNA (ULUKAN;
SWAAN, 2002).
Figura 7. Estruturas químicas da camptotecina (11), topotecana (12) e irinotecano (13).
Devido a isso, houve uma busca por derivados que apresentassem o mesmo
mecanismo de ação, porém com uma toxicidade menor. Dessa forma, a
camptotecina originou a topotecana (12) (Figura 7) e o irinotecano (13) (Figura 7),
ambos alcaloides de origem sintética. Embora menos tóxicos que seu protótipo,
esses compostos são empregados na terapia antineoplásica com precaução, pois
podem causar toxicidade em doses altas (XU; BARCHET; MAGER, 2010).
Um grupo de compostos que vem sendo bem estudado é o dos alcaloides
quaternários benzofenantridínicos. Essas substâncias podem ser encontradas em
diversas espécies da família Papaveraceae, tais como Sanguinaria canadensis,
Dicranostigma lactucoides, Chelidonium majus, Macleaya cordata and M. microcarpa
(SUCHOMELOV et al., 2007).

39
A sanguinarina (14) (Figura 8) e a queleritrina (15) (Figura 8) são consideradas
os alcaloides mais importantes deste grupo por demonstrarem propriedades
biológicas de maior interesse farmacológico (LIU et al., 2006).
Figura 8. Estruturas químicas dos alcaloides benzofenantridínicos sanguinarina (14) e queleritrina
(15).
A queleritrina está associada a diversos efeitos biológicos, apresentando ação
antimicrobiana (ZUO et al., 2008), anti-inflamatória (PENCIKOVA et al., 2012), na
doença de Alzheimer (BRUNHOFER et al., 2012) e efeito antitumoral (KAH et al.,
2008).
Hebert e colaboradores (1990) relacionaram a queleritrina à inibição de
proteína quinase C (PKC) no sítio de ligação do substrato dessa enzima, que é
muito relacionada à promoção de tumores (KOIVUNEN; AALTONEN; PELTONEN,
2006).
Além disso, Yu e colaboradores (2000) identificaram outro mecanismo pelo
qual a queleritrina exibe atividade antitumoral. Esse mecanismo consiste na ativação
das proteínas quinases c-Jun N-terminal (JNK) e a p38, relacionadas à regulação da
diferenciação celular e apoptose.
A hipótese mais discutida e aceita é que a queleritrina interage com proteínas
antiapoptóticas da família Bcl-2 (CHAN et al., 2003; KAH et al., 2008; LIU; WANG,
2012). Alguns estudos demonstraram que o alcaloide se liga à Bcl-XL em uma região
hidrofóbica da proteína, denominada sulco BH, que é próxima ao domínio BH3, no
qual as proteínas proapoptóticas tendem a se ligar.
A ligação da queleritrina no sulco BH da Bcl-XL promove sua inibição e o
deslocamento e liberação da proteína proapotótica (Bid, Bak ou Bax), anteriormente

40
ligada na região BH3 da Bcl-XL, induzindo, assim, a promoção da apoptose
(BERNARDO et al., 2008; ZHANG et al., 2006).
Ainda em relação à atividade antitumoral, a queleritrina exibiu atividade
citotóxica em diversas linhagens celulares humanas. Dentre elas a de carcinoma de
próstata humano (LNCaP e DU-145) (MALÍKOVÁ et al., 2006), de adenocarcinoma
de mama humano (MDA-MB-231) (MATKAR; WRISCHNIK; HELLMANN-
BLUMBERG, 2008) e de câncer gástrico humano (BGC-823) (ZHANG et al., 2012).
Esses dados demonstram a abrangência de linhagens nas quais a queleritrina pode
atuar.
Entretanto, observa-se uma baixa seletividade desse alcaloide às linhagens
tumorais em comparação às linhagens não tumorigênicas, como por exemplo a de
cultivo primário de fibroblastos gengivais humano (FGH) (MALÍKOVÁ et al., 2006) e
fibroblastos de pele humana (KF-II) (SLANINOVÁ et al., 2007). Isso pode ser
observado pelos valores de IC50 nas diversas linhagens relatadas, demonstrados na
Tabela 3.
Tabela 3. Valores de IC50 da queleritrina em diversas linhagens celulares.
Linhagens celulares IC50 (µM) da queleritrina Referências
LNCaP 22,0 (MALÍKOVÁ et al., 2006)
DU-145 6,0 (MALÍKOVÁ et al., 2006)
MDA-MB-231 6,7 (MATKAR; WRISCHNIK; HELLMANN-BLUMBERG,
2008)
BCG-823 6,31 (ZHANG et al., 2012)
FGH 4,7 (MALÍKOVÁ et al., 2006)
KF-II 1,5 (SLANINOVÁ et al., 2007)
LNCaP e DU-145 – carcinoma de próstata humano; MDA-MB-231 – adenocarcinoma de mama humano; BGC-
823 – câncer gástrico humano; FGH – cultivo primário de fibroblastos gengivais humano; KF-II – fibroblastos de
pele humana.
Sendo assim, o desenvolvimento de análogos mais seletivos e menos tóxicos
da queleritrina torna-se relevante, tendo em vista que este composto apresenta
diversos mecanismos de ação relatados e citotoxicidade em diversas linhagens
tumorais (BERNARDO et al., 2008; ZHANG et al., 2006).

41
1.3 Planejamento de fármacos
Com a existência de diversas moléculas com grande potencial para tornarem-
se fármacos, a descoberta de novas entidades químicas sempre foi um grande
desafio. Com a modernização do processo de planejamento de novos compostos
bioativos, essa descoberta pôde ser conduzida de maneira mais racional, baseado
no mecanismo farmacológico envolvido no processo fisiopatológico. A esta forma de
descoberta, é atribuído o termo planejamento racional de fármacos (BARREIRO;
FRAGA, 2005; CHAST, 2008; NEUMEYER, 2013; WEINER; WILLIAMS, 1994).
O advento do projeto genoma humano revolucionou a área da proteômica
(ANDERSON, 2003), que é o estudo de todas as interações, modificações,
isoformas e características estruturais das proteínas existentes em uma célula
(TYERS et al., 2003). A proteômica auxiliou no conhecimento de diversas
biomacromoléculas envolvidas nos processos fisiopatológicos, que foram
identificadas, então, como alvos terapêuticos para o planejamento de compostos
bioativos (SCHAFFHAUSEN, 2012). O planejamento racional a partir dessas
biomacromoléculas denomina-se de planejamento de fármaco a partir da estrutura
do alvo (SBDD – Structure-Based Drug Design) (AMARAL et al., 2003; MCKAY et
al., 2012).
Com o conhecimento da estrutura dos alvos terapêuticos surge a possibilidade
de elaborar substâncias com perfis farmacológicos melhor estabelecidos. Com a
informação acerca dos ligantes e de como eles interagem na biomacromolécula,
pode-se prever com maior precisão a resposta celular consequente (ANDERSON,
2003). A ressonância magnética nuclear (RMN) e a cristalografia de raio-x, permitiu
o acesso à estruturas tridimensionais (3D) dessas biomacromoléculas, possibilitando
a obtenção de maiores informações sobre a interação de ligantes nos alvos
(SCHAFFHAUSEN, 2012).
Quando não há informações sobre o alvo envolvido no processo fisiopatológico
ou o conhecimento da estrutura da biomacromolécula é incipiente, o planejamento
também pode se dar a partir de ligantes, denominado, então, de planejamento de
fármaco a partir do ligante (LBDD – Ligand-Based Drug Design). Leva-se em
consideração as propriedades estruturais, conformacionais e físico-químicas de uma

42
dada molécula, denominada de protótipo, que pode ser um ligante endógeno, um
composto natural ou outras substâncias com propriedades biológicas interessantes
para uma determinada doença (SHIM; MACKERELL, JR., 2011).
A abordagem por LBDD tem como objetivo a otimização dos ligantes
selecionados e pode ser aplicada com o intuito de melhorar parâmetros como
potência, seletividade, toxicidade e perfil farmacocinético. Dessa forma, o
planejamento de um fármaco pode se dar racionalmente, e a modificação molecular
torna-se base para que isso ocorra (SILVERMAN, 2004).
De maneira geral, as modificações moleculares podem alterar a
estereoquímica, as dimensões, a flexibilidade das estruturas moleculares, bem como
propriedades físico-químicas, como basicidade, lipofilicidade e distribuição
eletrônica. Portanto, as modificações moleculares podem alterar a resposta biológica
que um ligante pode desencadear, visto que as propriedades estruturais,
conformacionais e físico-químicas determinam a forma como o ligante interage com
o receptor (GALDINO; PITTA; BOUCHERLE, 2011).
Uma das modalidades de modificação molecular é a simplificação molecular,
uma estratégia que, ao ser empregada, pode reduzir rotas sintéticas e gerar
compostos com atividade biológica semelhante ou superior ao seu protótipo. Além
disso, é possível obter informações sobre a mínima estrutura química necessária
para a atividade biológica, ou seja, o grupo farmacofórico (PINACHO CRISÓSTOMO
et al., 2009).
Os anestésicos locais derivados da cocaína ilustram esse tipo de modificação,
conforme Figura 9. A simplificação empregada na estrutura do alcaloide cocaína (16)
gerou o composto procaína (17), que manteve o efeito anestésico local característico
de seu protótipo, sem apresentar as propriedades narcóticas (MORICE; WERMUTH,
2008).
Figura 9. Estruturas químicas da cocaína (16) e procaína (17).

43
Outro exemplo de sucesso terapêutico alcançado por meio da simplificação
molecular é a mefloquina (19) (Figura 10). Originada a partir da quinina (18) (Figura
10), um alcaloide componente da casca de Cinchona officinalis, a mefloquina possui
atividade antimalárica e tornou-se importante ao suplantar os fármacos mais antigos
da classe dos antimaláricos (BARREIRO, 2002).
Figura 10. Estruturas químicas da quinina (18) e mefloquina (19).
Como pode ser observado, o emprego da simplificação molecular, ainda que
tenha sido aplicado nos exemplos citados de maneira empírica, é uma estratégia de
extrema relevância. Com as ferramentas disponíveis atualmente, como por exemplo,
o uso de banco de dados e modelagem molecular, a estratégia de simplificação
molecular pode ser empregada de maneira racional (BARREIRO, 2002; PINACHO
CRISÓSTOMO et al., 2009; TOROPOV et al., 2012).
A aplicação de métodos computacionais em complemento ao planejamento
de compostos bioativos pode facilitar o processo de desenvolvimento.
Adicionalmente, pode integrar todas as informações obtidas a partir das estruturas e
correlacioná-las à atividade biológica dos compostos planejados. Dessa forma,
torna-se possível traçar uma relação-estrutura atividade dos compostos a partir da
modelagem molecular, o que pode contribuir de forma impactante no planejamento
racional de fármacos (SANT’ANNA, 2009).
Face ao exposto, pode-se observar que o processo de desenvolvimento de
fármacos é desafiador, demanda tempo, recursos e necessita de uma abordagem
multidisciplinar. Por isso, o planejamento racional de fármacos pode oferecer
subsídios de modo a reduzir alguns problemas provenientes do processo de
desenvolvimento, podendo ao mesmo tempo aumentar a probabilidade de formação
de uma nova molécula bioativa (GALDINO; PITTA; BOUCHERLE, 2011).

44
2 OBJETIVOS E JUSTIFICATIVA
De acordo com os dados acerca do câncer, pode-se notar que este grupo de
doenças representa problema de grande impacto mundial. Devido à problemática da
terapia antineoplásica, composta principalmente de quimioterápicos tóxicos e pouco
seletivos, é de extrema importância que sejam realizadas pesquisas na área para o
alcance de uma terapia mais eficiente e que aumente a qualidade de vida dos
pacientes acometidos.
Dessa forma, o objetivo geral deste trabalho foi o planejamento, a síntese e a
avaliação da citotoxicidade de análogos da queleritrina planejados por simplificação
molecular, no intuito de se obter compostos mais seletivos para as linhagens
tumorais. Esse projeto ainda conta com os seguintes objetivos específicos:
Síntetizar e purificar os análogos propostos da queleritrina;
Avaliar a citotoxicidade dos compostos em culturas de células de linhagens
não-tumorigênicas de células endoteliais da veia umbilical humana (HUVEC)
e fibroblasto de pulmão (LL24), e em linhagens tumorais como a linfoblastoide
humana (Jurkat), de melanoma humano (SK-Mel-28) e em carcinoma de
pulmão humano epitelial alveolar basal (A549);
Realizar estudos mecanísticos complementares nas linhagens tumorigênicas
as quais os análogos mais promissores apresentarem atividade biológica;
Determinar, por estudos teóricos, as propriedades eletrônicas e lipofílicas, tais
como os mapas de potencial eletrostático, mapas de orbital molecular, vetor
de momento dipolo, CLog P (logaritmo do coeficiente de partição calculado),
peso molecular, HBA (sítios aceptores de ligação de hidrogênio) e HBD (sítios
doadores de ligação de hidrogênio) dos compostos sintetizados para
correlacionar com a atividade biológica.

45
3 PLANEJAMENTO DOS ANÁLOGOS
Foi utilizado como protótipo a queleritrina, um produto natural que possui 5
anéis em sua estrutura, representados pelas letras A, B, C, D e E (Figura 11). Foram
propostas modificações moleculares à estrutura do protótipo, cujo intuito foi o auxílio
nos estudos de elucidação de grupo farmacofórico, dos grupos acessórios e a
obtenção dos análogos em etapas sintéticas reduzidas.
Figura 11. Representação das modificações propostas na molécula de queleritrina.
Para todas as séries de análogos, as regiões A e B foram mantidas, pois em
estudo coordenado por Bernardo e colaboradores (2008), ao retirar a estrutura
benzodioxol da queleritrina e mantendo os anéis C, D e E na mesma composição a
desse alcaloide, verificou-se ausência de atividade sobre a Bcl-XL nas
concentrações testadas no estudo. Dessa forma, pode-se supor que o benzodioxol
pode ser necessário para a atividade da queleritrina.
Já nas regiões C e D, foi proposta a abertura dos anéis, com o intuito de
verificar se estes representam grupos acessórios, bem como obter os análogos em
etapas sintéticas reduzidas. No nitrogênio presente na região D, pode ser adicionado
uma metila ou hidrogênio, visando à verificação da contribuição destes

46
grupos/átomos para a atividade biológica. Além disso, a carga formal positiva será
retirada no intuito de aumentar a lipofilicidade do composto com possível melhoria
dos parâmetros farmacocinéticos que os análogos podem apresentar em relação à
queleritrina.
Por fim, na região E, foram feitas variações de posição e de número de
grupos metoxilas e hidroxilas ligados ao anel, pois no estudo coordenado por
Bernardo e colaboradores (2008), foi observado também que ao substituir os grupos
metoxilas por hidroxilas houve aumento de potência dos análogos. A variação de
posição dos substituintes no anel auxiliará na verificação se esse aumento é
promovido apenas nas posições R1 e R2 (Figura 11), conforme verificado nesse
mesmo estudo.

47
4 MATERIAL E MÉTODOS
4.1 Material
Para o desenvolvimento do presente trabalho utilizaram-se as vidrarias e
equipamentos usualmente empregados em laboratório, em conjunto aos reagentes e
solventes listados a seguir:
Reagentes
Piperonilamina (Sigma-Aldrich);
Benzaldeído (Vetec);
4-metoxibenzaldeído (Merck);
3,4-dimetoxibenzaldeído (Sigma-Aldrich);
3,4,5-trimetoxibenzaldeído (Sigma-Aldrich);
3-metoxi-4-hidroxibenzaldeído (Sigma-Aldrich);
3,4-di-hidroxibenzaldeído (Sigma-Aldrich);
Triacetoxiboroidreto de sódio (Sigma-Aldrich);
Ácido acético glacial (Jt. Baker);
Ácido fórmico (Merck);
Formaldeído (Quimex).
Solventes
Diclorometano (Synth);
Acetona (Synth);
Acetato de etila (Synth);
Hexano (Synth)
Metanol (Synth);
Éter etílico (Synth);
Trietilamina (Vetec);
Ácido clorídrico (Synth);
Ácido sulfúrico (Synth);

48
Água destilada;
Clorofórmio deuterado (Cambridge Isotope Laboratories);
Dimetilsulfóxido deuterado (Cambridge Isotope Laboratories).
4.2 Métodos sintéticos
Os compostos análogos à queleritrina foram divididos em duas séries, das
quais a série 2a-g é caracterizada pelo grupo funcional amina secundária e a série
3a-d, pela amina terciária, conforme Figura 12.
Figura 12. Representação esquemática das sínteses dos análogos. Reagentes e condições: (i)
piperonilamina, NaBH(OAc)3, DCM, t.a., N2, 3-5 h; (ii) HCl etérico a 6,8 M; (iii) formaldeído, ácido
fórmico, 110 ºC, 2-4 h.
4.2.1 Síntese das aminas secundárias
Para a formação das aminas secundárias, o método selecionado foi adaptado
a partir de reações realizadas por Abdel-Magid e colaboradores (1996), no qual o
agente redutor é adicionado junto à amina e ao aldeído, sem o isolamento prévio da
imina, com a redução seletiva deste grupo ao invés do aldeído. Experimentalmente,
2 mmol do aldeído e 2,3 mmol de ácido acético glacial (146 µL, 1,15 eq) foram
dissolvidos em 10 mL de diclorometano e, após 30 minutos, 2,2 mmol do nucleófilo
(281,6 µL, 1,1 eq de piperonilamina) foram adicionados ao sistema. A mistura
reacional foi mantida sob agitação e atmosfera inerte de nitrogênio por 30 minutos e,
então, 2,8 mmol (593,6 mg, 1,4 eq) do agente redutor NaBH(OAc)3 foram
adicionados. O sistema foi mantido sob as mesmas condições até o consumo total

49
do aldeído e o acompanhamento da reação foi realizado por cromatografia em
camada delgada (CCD), com fase móvel de hexano/acetato de etila 4:1 (v/v) e 5%
de trietilamina para os compostos 2a e 2b; fase móvel de hexano/acetato de etila 1:1
(v/v) e 5% de trietilamina para os compostos 2c, 2d e 2e; ou fase móvel de
clorofórmio/metanol 9:1 (v/v) para o composto 2f. Ao final da reação, acrescentou-se
10 mL de solução de NaOH 1,25 M para os compostos 2a-e e 10 mL de água
destilada para o composto 2f.
Para purificação, realizou-se extração com diclorometano (3 x 20 mL),
solução NaOH 1,25 M (1 x 20 mL), e solução saturada de NaCl (1 x 60 mL) para os
compostos 2a-e. Para o composto 2f, realizou-se extração com diclorometano (1 x
20 mL), acetato de etila (3 X 20 mL) e solução saturada de NaHCO3 (1 x 20 mL).
Posteriormente, a fase orgânica foi seca com Na2SO4 anidro e removeu-se o
solvente por rotaevaporação. Realizou-se coluna cromatográfica quando necessário.
O produto resultante foi transformado em sal de cloridrato por meio de uma solução
saturada de HCl a 6,8 M em éter e o sólido foi posteriormente lavado com éter ou
clorofórmio gelado. O armazenamento e as análises foram feitos com os compostos
na forma de cloridrato.
4.2.2 Síntese das aminas terciárias
Para a formação das aminas terciárias, o método selecionado foi adaptado a
partir de reações realizadas por Minarini e colaboradores em 2008.
Experimentalmente, 1,0 mmol da amina secundária na forma de cloridrato foi
solubilizada em 8 mL de ácido fórmico (98-100%) sob refluxo de aproximadamente
120 ºC. Logo em seguida, 8 mL de formaldeído (36-38%) foram adicionados gota a
gota. O acompanhamento da reação foi realizado por cromatografia em camada
delgada (CCD), com fase móvel de hexano/acetato de etila 4:1 (v/v) e 5% de
trietilamina para os compostos 3a e 3b e fase móvel de hexano/acetato de etila 1:1
(v/v) e 5% de trietilamina para os compostos 3c e 3d. A condições reacionais foram
mantidas até o consumo total da amina secundária.
Para purificação, a solução foi basificada com NaOH 10 M e, em seguida,
procedeu-se a uma extração com diclorometano (2 x 60 mL) e acetato de etila (2 x
60 mL). Posteriormente, a fase orgânica foi seca com Na2SO4 anidro e o solvente foi

50
removido sob pressão reduzida. A coluna cromatográfica foi utilizada quando
necessário. O produto resultante foi purificado através da formação de sal de
cloridrato por meio de uma solução saturada de HCl a 6,8 M em éter. O
armazenamento e a caracterização dos compostos foram feitos com as aminas na
forma de cloridrato.
4.3 Métodos analíticos
4.3.1 Cromatografia em camada delgada (CCD) e cromatografia em
coluna
O acompanhamento das reações foi realizado por CCD, utilizando
cromatofolhas de alumínio cobertas com sílica GF254 (Merck). A observação das
placas foi feita por luz ultravioleta (UV) e a revelação destas, com as soluções de
vanilina, 2,4-dinitrofenil-hidrazina ou ácido fosfórico molibdato, conforme
necessidade. Foram utilizadas colunas de vidro e sílica gel 60 (Merck) para a
separação e purificação dos compostos obtidos.
4.3.2 Espectrometria por ressonância magnética nuclear (RMN)
As análises espectroscópicas de RMN 1H e 13C foram realizadas em
espectrômetro Bruker 300 MHz, modelo Advanced DPX-300, na Faculdade de
Ciências Farmacêuticas da Universidade de São Paulo.
4.3.3 Cromatografia líquida de alta eficiência (CLAE) acoplado ao UV
As análises cromatográficas por CLAE foram feitas no laboratório de química
analítica do departamento de Farmácia da Faculdade de Ciências Farmacêuticas da
universidade de São Paulo. As purezas cromatográficas dos compostos sintetizados
foram determinadas no cromatógrafo Shimadzu Proeminence com coluna analítica
C-18 da marca Gemini® (5µ, 150 x 4,6 mm). A detecção foi conduzida por

51
ultravioleta (UV) em 230 nm. A separação foi realizada em gradiente de eluição com
fase móvel de metanol e solução aquosa de ácido trifluoracético (0,1%).
4.3.4 Análise elementar (CHN)
A análise elementar das moléculas foi realizada na Central Analítica do
Instituto de Química da USP em analisador elementar Perkin Elmer® 2400 CHN. A
variação dos valores experimentais não poderá exceder a ± 0,5% dos valores
teóricos calculados para cada composto.
4.4 Métodos biológicos
Os ensaios de atividade antitumoral contaram com a colaboração dos
pesquisadores Dr. Ricardo A. Azevedo, Dr. Adilson K. Ferreira e da mestranda
Sarah Fernandes Teixeira, vinculados ao Laboratório de Imunologia de Tumores do
Instituto de Ciências Biomédicas da USP (ICB/USP) do Prof. Dr. José Alexandre M.
Barbuto.
4.4.1 Cultura celular
Para a realização dos ensaios foram utilizadas as linhagens não-
tumorigênicas de células endoteliais da veia umbilical humana (HUVEC) e
fibroblasto de pulmão (LL24), e linhagens tumorais linfoblastoide humana (Jurkat),
de melanoma humano (SK-Mel-28) e em carcinoma de pulmão humano epitelial
alveolar basal (A549).
As linhagens HUVEC, Jurkat, SK-Mel-28 e A549 foram cultivadas em meio de
cultura RPMI-1640, pH 7.0, suplementado com 10% de soro fetal bovino (SFB)
inativado, penicilina (100 U/mL) e estreptomicina (100 μg/mL). A linhagem LL24 foi
cultivada em meio de cultura DMEM, pH 7.0, suplementado com 20% de soro fetal
bovino (SFB) inativado, penicilina (100 U/mL) e estreptomicina (100 μg/mL). Os

52
frascos de cultura de 75 cm2 foram mantidos em estufa contendo atmosfera úmida
com 5% de CO2 a 37°C.
4.4.2 Avaliação da citotoxicidade
Para a avaliação da viabilidade celular foi utilizado o teste colorimétrico do
MTT [brometo de 3-(4,5-dimetiltiazol-2-1)2,5-difenil tetrazólio]. Foram plaqueadas em
triplicata 104 células das linhagens utilizadas em 100 µL em placas de 96 poços e
cultivadas por 2 horas em estufa contendo 5% de CO2 a 37°C. Após o período de
incubação, as células foram tratadas durante 24 horas com os compostos
sintetizados, a queleritrina e o paclitaxel (como controle positivo) nas concentrações
de 50, 100 e, em alguns casos, 150 µM, para a triagem dos compostos. Para a
determinação da concentração inibitória (IC50), foram escolhidas 5 concentrações
diferentes que variaram de acordo com o resultado da triagem dos compostos. Após
o tratamento foram adicionados 5 μL de MTT (5 mg/mL) às células e estas foram
incubadas por 2 horas. Após este período, o meio foi removido e acrescentou-se 100
μL de Dimetilsulfóxido (DMSO) para dissolver os cristais de formazan formados e
precipitados. A quantificação da absorbância foi realizada em leitor de ELISA com
comprimento de onda de 540 nm. Os resultados de citotoxicidade, em triplicada intra
e interexperimento, foram transformados em porcentagem de células viáveis,
conforme a equação a seguir:
% células viáveis= (𝐴𝑎−𝐴𝑏)×100 𝐴𝑐−𝐴𝑏
Onde: 𝐴𝑎 = absorbância das amostras; 𝐴𝑏 = absorbância do branco; 𝐴𝑐 =
absorbância do controle positivo.
Os dados foram plotados no software Prism 5 versão 5.03 (GraphPad
Software, Inc, 2009), onde calculou-se a IC50 segundo o tutorial fornecido pelo
Prism.

53
4.4.3 Ensaio clonogênico
O composto mais promissor do ensaio de citotoxicidade em A549 foi avaliado
também quanto ao seu potencial de inibição da proliferação celular por meio do
ensaio clonogênico.
Para a avaliação do efeito inibitório do composto sobre a proliferação das
células, foram plaqueadas, em triplicata, 200 células da linhagem A549 em 500 µL
em placas de 12 poços e cultivadas por 2 horas em estufa contendo 5% de CO2 a 37
°C. Após o período de incubação, as células foram tratadas durante 24 horas com o
composto selecionado nas concentrações de 100, 75 e 50 µM. Após o tratamento, o
meio foi trocado por 1 mL de meio não metabolizado e as células foram cultivadas
por 10 dias. O meio foi, então, removido e os poços foram lavados com 500 µL de
tampão salina fosfato (PBS) não estéril por duas vezes e as colônias formadas
foram fixadas com solução de glutaraldeído 6% e coradas com cristal de violeta
0,5% diluídos em PBS. As colônias foram contadas e as placas foram fotografadas
para a comparação do crescimento das colônias nos diferentes tratamentos em
relação ao controle não tratado. Os dados foram plotados no software Prism 5
versão 5.03 (GraphPad Software, Inc, 2009), onde obtiveram-se o gráfico e os dados
estatísticos do ensaio.
4.4.4 Análise do ciclo celular
Para o estudo das fases do ciclo celular, as células da linhagem A549 foram
plaqueadas em placas de 12 poços na concentração de 1 x 105 céls/poço e
cultivadas por 2 horas em estufa contendo 5% de CO2 a 37°C. Para a sincronização,
o meio foi trocado por RPMI sem adição de soro fetal bovino. Após 12 h de
incubação, o meio foi removido e as células foram tratadas com o composto
estudado no ensaio clonogênico, nas concentrações utilizadas no mesmo, diluído
em meio RPMI suplementado com 10% de SFB e incubadas em estufa contendo 5%
de CO2 a 37°C. Após 24 h de incubação, as células foram extraídas dos poços,
lavadas três vezes com solução de PBS/BSA 0,5% azida 0,05% e centrifugadas por
10 min a 1200 RPM. O pellet foi ressuspenso em 2 mL de etanol 70% gelado e

54
mantido sob refrigeração por 1 h. Logo após, a amostra foi centrifugada e o pellet
resultante foi ressuspenso em 300 μL de solução composta de 0,1% de Triton X-
100, 0,1% de citrato de sódio, 50 μg/mL de Iodeto de Propídeo e 10 μg/mL de
RNAse (Sigma); e incubado a 37 ºC por 1h na ausência de luz. Os dados foram
adquiridos em citometria de fluxo (FACSCalibur). Para a determinação da
porcentagem de células em cada fase do ciclo, utilizou-se o algoritmo Dean Jett-Fox,
do software FlowJo (TreeStar Inc., Ashland, OR, EUA).
4.5 Métodos computacionais
Os estudos de modelagem molecular foram realizados em colaboração com o
prof. Dr. Gustavo Henrique Goulart Trossini, do Departamento de Farmácia da FCF-
USP. Foram utilizados os programas Spartan’10 versão 1.1.0 (Wavefunction Inc,
2011), HyperChem versão 8.0.7 (Hypercube Inc) e ViewerLite versão 4.2 (Accelrys
Inc, 2001) em computador PC Windows 7 professional de configuração Intel(R)
Xeon(R) CPU 31270, 3.40 GHz, RAM 12.0 GB, sistema operacional de 64 bits.
Os modelos tridimensionais das estruturas dos compostos foram construídos
considerando-se sua forma catiônica, uma vez que a queleritrina possui carga formal
positiva e as aminas sintetizadas encontram-se, em maioria, protonadas em pH
fisiológico. A otimização da geometria se deu pelos métodos de mecânica molecular
MMFF, semi-empírico PM6 e Hartree-Fock 6-31G* no programa Spartan’10.
A partir das estruturas otimizadas foram calculados os mapas de potencial
eletrostático, mapas de orbital molecular, vetor de momento dipolo, CLog P
(logaritmo do coeficiente de partição calculado), peso molecular, HBA (sítios
aceptores de ligação de hidrogênio) e HBD (sítios doadores de ligação de
hidrogênio) utilizando o método Hartree-Fock 6-31G*. O alinhamento dos ligantes foi
realizado pelo programa HyperChem versão 8.0.7 (Hypercube Inc) e a determinação
de distâncias interatômicas, bem como dos ângulos diedros das estruturas foi
realizada no programa ViewerLite versão 4.2 (Accelrys Inc, 2001) (SANT’ANNA,
2009).

55
5 RESULTADOS E DISCUSSÃO
Foram sintetizados dez compostos das séries propostas, dentre eles seis
aminas secundárias e quatro aminas terciárias. Os valores de rendimento, fator de
retenção, aspecto físico e pureza cromatográfica podem ser verificados na Tabela 4.
Os seus respectivos espectros de RMN 1H e 13C, cromatogramas e dados de análise
elementar constam no Anexo I.
Tabela 4. Resultados obtidos na síntese dos análogos da queleritrina das séries das aminas
secundárias e terciárias.
Composto Estrutura CLAE Aspecto físico Rf Rendimento
global (%)
2a
t = 11,9 min
pureza >95% Sólido branco 0,371 44
2b
t = 6,1 min
pureza >98%
Sólido branco 0,331 54
2c
t = 7,7 min
pureza >99%
Sólido branco 0,302 54
2d
t = 8,0 min
pureza >99%
Sólido branco 0,292 39
2e
t = 8,1 min
pureza >98%
Sólido branco 0,222 45
2f
t = 8,1 min
pureza >96% Sólido amarelo 0,113 4
3a
t = 12,2 min
pureza >95% Sólido branco 0,731 31
3b
t = 6,0 min
pureza >98% Sólido branco 0,571 28
3c
t = 9,3 min
pureza >99% Sólido branco 0,472 23
3d
t = 7,7 min
pureza >97% Sólido branco 0,472 22
1 Fator de retenção, obtido com o sistema eluente acetato de etila e hexano na proporção 1:4 e 2,5% de trietilamina; 2 Fator de retenção, obtido com o sistema eluente acetato de etila e hexano na proporção 1:1 e 5% de trietilamina; 3 Fator de retenção, obtido com o sistema eluente clorofórmio e metanol na proporção 9:1.

56
5.1 Síntese e caracterização das aminas secundárias
Para a formação de aminas secundárias, a melhor metodologia descrita na
literatura e geralmente a de primeira escolha é a redução de iminas
(BHATTACHARYYA, 1995; PELTER; ROSSER; MILLS, 1984; SALVATORE; YOON;
JUNG, 2001).
Essa metodologia pode ser desenvolvida de duas formas (Figura 13): (a)
forma indireta, na qual há a formação, isolamento da imina e posterior redução e se
processa em duas etapas e; (b) forma direta ou aminação redutiva, na qual há a
formação da imina e redução imediata desse grupo funcional, processando-se, desta
forma, em apenas uma etapa (ABDEL-MAGID et al., 1996).
Figura 13. Representação esquemática da formação de aminas secundárias a partir de iminas de (a)
forma indireta e (b) forma direta ou aminação redutiva (CHO; KANG, 2005).
Para este trabalho, a principal dificuldade na utilização da forma indireta
consistiu no fato de que as iminas que seriam obtidas são instáveis e acabavam se
decompondo à amina primária e aldeído. Isto se deve, provavelmente, à falta de um
grupo eletronegativo ligado diretamente ao nitrogênio da ligação imínica, pois esse
tipo de substituinte pode participar da conjugação da dupla ligação entre o carbono e
o nitrogênio, diminuindo a carga parcial positiva sobre o carbono e sua
suscetibilidade a um ataque nucleofílico (Figura 14) (CLAYDEN; GREEVES;
WARREN, 2012a).

57
Figura 14. Representação das estruturas de ressonância de iminas com um grupo eletronegativo
ligado ao nitrogênio: (a) oxima e (b) sulfonil-hidrazona (CLAYDEN; GREEVES; WARREN, 2012a).
A forma direta é vantajosa nos casos em que o equilíbrio de formação da
imina não é favorável o suficiente para permitir o seu isolamento (SCHELLENBERG,
1963), como no caso deste trabalho. Porém demanda a utilização de um agente
redutor seletivo para a ligação imínica e que não reduza o aldeído (CLAYDEN;
GREEVES; WARREN, 2012a).
O uso do cianoboroidreto de sódio (NaBH3CN) foi introduzido por Borch,
Bernestein e Durst em 1971. Este agente redutor foi relatado como um composto
versátil que, em condições adequadas, teria a capacidade de reduzir seletivamente
as iminas. Ao testar a metodologia descrita por Borch (1972), foi observado um
baixo rendimento e a formação de muitos subprodutos, o que dificultou a purificação
do composto. Além disso, o NaBH3CN é descrito na literatura como um composto
que pode gerar subprodutos altamente tóxicos como o HCN e o NaCN e, portanto,
está sendo pouco utilizado atualmente (CHO; KANG, 2005; GRENGA et al., 2009;
SAXENA; BORAH; SARMA, 2000).
A metodologia de Abdel-Magid e colaboradores (1996), baseada nas
descobertas de Gribble e colaboradores (1974), descreve também a formação de
aminas secundárias por meio da aminação redutiva. Esta metodologia contempla o
uso do triacetoxiboroidreto de sódio (NaBH(OAc)3) como agente redutor (Figura 15),
visto que há relatos de que este reagente pode reduzir seletivamente o grupo imina,
e em menores proporções, o aldeído (ABDEL-MAGID; MARYANOFF; CARSON,
1990). Devido a vantagem de produzir menos produtos tóxicos e gerar maiores

58
rendimentos dos compostos desejados, essa foi a metodologia utilizada neste
trabalho.
Figura 15. Mecanismo de formação de amina secundária por aminação redutiva (ABDEL-MAGID;
MEHRMAN, 2006; EVANS; CHAPMAN; CARREIRA, 1988).
A redução seletiva do NaBH(OAc)3 é atribuída à sua característica de agente
redutor moderado, que é devido à presença dos três grupos acetoxi promotores de
efeito estérico e elétron-retirador sobre o boro. Estes efeitos estabilizam a ligação do
boro com o hidrogênio, responsável pela propriedade redutora do composto
(ABDEL-MAGID; MEHRMAN, 2006; EVANS; CHAPMAN; CARREIRA, 1988).
Foi empregado o uso de ácido acético glacial na reação, pois na ausência
deste, a reação se processava mais lentamente, favorecendo a formação de
subprodutos que não foram caracterizados. O excesso de ácido acético
(quantidades superiores a 2 eq) também pode favorecer a formação de subprodutos,
porém estes podem estar relacionados à redução do aldeído (ABDEL-MAGID;
MEHRMAN, 2006).
Os produtos foram obtidos na forma de sal de cloridrato que variaram a
coloração de branco (2a-e) a amarelo claro (2f). Cada composto se diferencia pela
presença e quantidade de substituintes metoxila e hidroxila. O produto 2a é

59
caracterizado pela ausência de substituintes e os demais compostos são
caracterizados pela presença de uma metoxila na posição 4 (2b); duas metoxilas em
3 e 4 (2c); três em 3, 4 e 5 (2d), uma metoxila na posição 3 e uma hidroxila na
posição 4 (2e) e duas hidroxilas nas posições 3 e 4 (2f).
A grande vantagem da obtenção dos produtos na forma salina consiste na
melhoria de certas propriedades, tais como o aumento da estabilidade das
substâncias e o aumento de sua solubilidade em meio aquoso. Esta última pode ser
favorável para o uso dos compostos em ensaios biológicos in vitro, visto que o meio
utilizado é formado principalmente de água, assim como os fluidos biológicos
(PAULEKUHN; DRESSMAN; SAAL, 2007; SERAJUDDIN, 2007).
Os rendimentos das reações variaram de 4 a 54 %, valores os quais são
considerados de moderados a pouco apreciáveis. Estes podem estar relacionados à
formação de subprodutos, como, por exemplo, as aminas terciárias formadas pela
condensação da piperonilamina com duas moléculas de aldeído, ou aos métodos de
purificação utilizados (ABDEL-MAGID; MEHRMAN, 2006).
O análogo 2f possui característica anfortérica, ou seja, a molécula pode atuar
como base ou ácido (IUPAC, 2014). Esta característica é atribuída à presença do
grupo amino básico e das duas hidroxilas fenólicas ácidas e, dependendo do pH do
meio, ficam protonados ou desprotonados (CLAYDEN; GREEVES; WARREN,
2012b; MCMURRY, 2011a). Dessa forma, ao realizar uma extração básica, ácida ou
neutra, detectou-se baixa concentração da forma molecular do composto na fase
orgânica, o que gerou grande perda do composto, uma vez que a fase aquosa
concentrou não apenas o produto, como diversas impurezas provenientes da
reação, o que justifica o baixo rendimento de 4%.
A Figura 16 ilustra o espectro de RMN 1H do composto 2d, um representante
da série das aminas secundárias, na qual observa-se a formação da amina
secundária pela ausência dos deslocamentos em 9,88 ppm e em 3,77 ppm que são
característicos da carbonila do aldeído e do metileno ligado ao nitrogênio da
piperonilamina respectivamente. Na amina secundária, estes sinais foram
substituídos pelos deslocamentos em 4,03-4,01 ppm (H7 e H9) dos metilenos
ligados ao nitrogênio.

60
Figura 16. Espectro de RMN 1H do produto 2d.
Na figura 17, o espectro de RMN 13C do produto 2d exemplifica o perfil dos
espectros dessa série, no qual os deslocamentos em 49,6 e 49,4 ppm dos carbonos
C9 e C7, respectivamente, indicam a presença da amina secundária.
Concomitantemente, pode-se observar a ausência dos deslocamentos químicos em
191,0 e 46,2 ppm característicos da carbonila do aldeído e do metileno ligado ao
nitrogênio da piperonilamina, respectivamente, indicando que houve a formação da
amina secundária.
H7,9
H7,9

61
Figura 17. Espectro de RMN 13C do produto 2d.
Para complementar a caracterização, os compostos foram submetidos à
análise elementar (Tabela 5), e os resultados obtidos encontram-se dentro dos
limites de variação aceitáveis (±0,50% de variação).
Tabela 5. Análise elementar das aminas secundárias.
Composto Análise elementar (%)
Teórico Amostra Desvio
2a
C= 64,87
H= 5,81
N= 5,04
C= 64,43
H= 5,61
N= 5,09
C= 0,44
H= 0,20
N= 0,05
2b
C= 62,44
H= 5,89
N= 4,55
C= 62,44
H= 5,80
N= 4,69
C= 0,00
H= 0,09
N= 0,14
2c
C= 60,44
H= 5,97
N= 4,15
C= 60,36
H= 6,05
N= 4,36
C= 0,08
H= 0,08
N= 0,21
C7,9

62
2d
C= 58,78
H= 6,03
N= 3,81
C= 58,28
H= 5,91
N= 3,74
C= 0,50
H= 0,12
N= 0,07
2e
C= 59,35
H= 5,60
N= 4,33
C= 59,05
H= 5,58
N= 4,43
C= 0,30
H= 0,02
N= 0,10
2f
C= 58,17
H= 5,21
N= 4,52
C= 58,06
H= 5,86
N= 4,26
C= 0,11
H= 0,65
N= 0,26
Determinaram-se também as purezas cromatográficas dos compostos por
cromatografia líquida de alta eficiência (CLAE) e todas as aminas secundárias
sintetizadas nesse trabalho mostraram-se com pureza de 95% (Tabela 4),
considerada aceitável para dar prosseguimento aos ensaios biológicos.
5.2 Síntese e caracterização de aminas terciárias
A formação de aminas terciárias N-metiladas pode ocorrer por meio da reação
de substituição nucleofílica do iodeto de metila com as aminas secundárias
formadas (Figura 18).
Figura 18. Mecanismo de formação de amina terciária por SN2 (REGAN; CRAIG; BRAUMAN, 2002).
Trata-se de uma substituição nucleofílica de segunda ordem (SN2), pois o
grupo alquílico do iodeto de metila é primário e formaria carbocátions instáveis.
Dessa forma, a velocidade de reação depende do ataque do nucleófilo (amina
secundária) ao substrato (iodeto de metila) e não da formação de um intermediário
(carbocátion), ou seja, o aumento da velocidade é dependente da concentração de
ambos os reagentes (SMITH; MARCH, 2007).

63
Ao testar a metodologia de formação de aminas terciárias N-metiladas por
SN2, o iodeto de metila foi escolhido como agente alquilante devido ao fato do iodo
constituir um bom grupo de saída, pois o iodeto resultante é um ânion estável
(CAREY, 2000). As aminas constituem bons nucleófilos, uma vez que possuem um
par eletrônico disponível (SALVATORE; YOON; JUNG, 2001).
O emprego de bases em reações que aconteçam via SN2 é desejável, visto
que essas podem aumentar a nucleofilicidade das aminas secundárias, por meio da
abstração do hidrogênio ligado ao nitrogênio (MCMURRY, 2011b).
A acetona, solvente utilizado na metodologia, é uma escolha comum para a
realização desse tipo de reação, pois é polar o suficiente para dissolver o nucleófilo,
porém não tão polar a ponto de solvatá-lo, tornando o nucleófilo mais reativo perante
o substrato (CLAYDEN; GREEVES; WARREN, 2012c; SOLOMONS, T. W. G.
FRYHLE, 2011).
Entretanto, as aminas secundárias são passíveis de dialquilação (DA SILVA;
ESTEVAM; BIEBER, 2007), ou seja, além de gerar as aminas terciárias, formavam-
se também os sais de amônio quaternário, mesmo controlando a estequiometria
com o uso em excesso da amina.
Em busca de metodologia mais seletiva para a formação de aminas terciárias
N-metiladas, a metilação redutiva mostrou-se mais adequada para a formação das
aminas desejadas. Também denominada de reação de Eschweiler-Clark, foi
nomeada em homenagem ao químico alemão Wilhelm Eschweiler, quem
inicialmente reportou essa reação em 1905 e ao químico britânico Hans Thacher
Clarke, que em colaboração com Gillespie e Weisshaus em 1933, reportou o uso da
reação para a metilação de aminas e aminoácidos (CLARKE; GILLESPIE;
WEISSHAUS, 1933; ESCHWEILER, 1905).
A reação de Eschweiler-Clark tem o mecanismo semelhante à aminação
redutiva, porém ao invés de possuir a versatilidade de escolha de diversos aldeídos
ou cetonas, a metilação redutiva ocorre apenas com o formaldeído e utiliza somente
o ácido fórmico como agente redutor e também como solvente (Figura 19) (PINE;
SANCHEZ, 1971).

64
Figura 19. Mecanismo de formação de amina terciária por metilação redutiva (PINE, 1968).
A reação de Eschweiler-Clarke tem como principal vantagem, a formação
seletiva de aminas terciárias a partir de aminas secundárias, se comparado à reação
de alquilação. Isto se deve ao fato das aminas terciárias não formarem iminas com o
formaldeído, portanto não sendo possível a formação de sal de amônio quaternário,
que é o principal subproduto da alquilação de aminas secundárias (DA SILVA;
ESTEVAM; BIEBER, 2007; SALVATORE; YOON; JUNG, 2001).
Dessa forma, os compostos foram obtidos a partir da metilação redutiva e
transformados em sal de cloridrato. E, assim como seus precursores, os sais
possuem coloração branca e cada produto se diferencia pela presença e quantidade
de substuintes metoxila. Os rendimentos globais das reações foram de 31% para o
3a; 13% para o 3b; 23% para o 3c e 22% para o 3d.
As aminas terciárias correspondentes aos compostos 2e e 2f não foram
obtidas com sucesso devido à presença de hidroxila fenólica em sua estrutura. Os
sais das aminas terciárias já demonstravam ser mais higroscópicos que os sais das
aminas secundárias e a presença destas hidroxilas só aumentou essa característica,
além de ser observado sinais de degradação dos compostos mesmo armazenados
protegidos de luz e umidade. Dessa forma, a purificação e o armazenamento desses
compostos não foi possível e estratégias para melhorar estes processos devem ser
feitas para a obtenção desses análogos.

65
Para as aminas terciárias obtidas com sucesso, confirmou-se a sua formação
por meio de RMN 1H, principalmente pelo surgimento do deslocamento químico em
2,59 ppm (H25) referente à metila ligada ao nitrogênio (Figura 20), exemplificado
pelo composto 3d. A mudança dos deslocamentos químicos de 4,03-4,01 ppm (H7 e
H9, Figura 12) da amina secundária, para 4,24-4,01 ppm (H9 e H7) no espectro de
RMN 1H da amina terciária formada, também confirma a formação do produto
desejado.
Figura 20. Espectro de RMN 1H do produto 3d.
Na figura 21, no RMN 13C, nota-se a presença do deslocamento químico em
38,6 ppm, correspondente à metila ligada ao nitrogênio (C25), exemplificado pelo
espectro de RMN 13C do produto 3d. O aparecimento deste sinal indica a formação
da amina terciária, corroborando com o que fora indicado no espectro RMN 1H.
H7,9
H25
H7,9

66
Figura 21. Espectro de RMN 13C do produto 3d.
Além de RMN 1H e 13C, realizou-se também a análise elementar dos
compostos (Tabela 6), e os resultados obtidos encontram-se dentro dos limites de
variação aceitáveis (±0,50% de variação).
Tabela 6. Análise elementar das aminas terciárias.
Composto Análise elementar (%)
Teórico Amostra Desvio
3a
C= 65,86
H= 6,22
N= 4,80
C= 65,40
H= 5,78
N= 4,74
C= 0,46
H= 0,44
N= 0,06
3b
C= 63,45
H= 6,26
N= 4,35
C= 63,03
H= 6,22
N= 4,31
C= 0,42
H= 0,04
N= 0,04
3c
C= 61,45
H= 6,30
N= 3,98
C= 61,44
H= 6,34
N= 4,12
C= 0,01
H= 0,04
N= 0,14
C25

67
Foram determinadas também as purezas cromatográficas das aminas
terciárias (Tabela 4) por cromatografia líquida de alta eficiência (CLAE) e a pureza
de todos os compostos sintetizados encontraram acima de 95%, um valor aceitável
para dar prosseguimento aos ensaios in vitro.
5.3 Ensaios biológicos
5.3.1 Ensaio de citotoxicidade
Para determinar o potencial citotóxico dos compostos sintetizados, bem como
da queleritrina, foi utilizado o ensaio de biorredução do MTT (brometo de 3-(4,5-
dimetiltiazol-2-il)-2,5-difeniltetrazólio) ou sal de tetrazólio. Trata-se de um método
colorimétrico que avalia a atividade metabólica celular (BILLACK; RADKAR;
ADIABOUAH, 2008).
O produto resultante do metabolismo mitocondrial do MTT é o formazan, uma
substância de cor azul escura que é insolúvel em meio aquoso. Este composto pode
ser quantificado por espectrofotometria após lise celular induzida e solubilização em
solvente adequado, neste caso, o dimetilsulfóxido (DMSO) (SCHERLIES, 2011).
Essa técnica tem como fundamento traçar uma correlação entre a quantidade
de formazan produzido e a concentração de células viáveis no meio. Desta forma,
quanto maior for a concentração de células viáveis no meio, maior será a taxa de
biotranformação do MTT e consequentemente, a densidade óptica dos poços será
mais elevada (AZIZ, 2006).
Portanto, é possível estimar a taxa de viabilidade das células expostas aos
compostos sintetizados neste trabalho. O principal problema que é observado nos
ensaios biológicos em geral é a dificuldade de solubilização dos compostos em meio
3d
C= 59,76
H= 6,34
N= 3,67
C= 59,50
H= 6,27
N= 3,79
C= 0,26
H= 0,07
N= 0,12

68
aquoso. Porém, isso não foi observado com os análogos da queleritrina, uma vez
que se trata de compostos na forma de cloridrato.
Em princípio, foram preparadas soluções dos compostos em DMSO a 20 mM,
e a partir dessas, foram feitas diluições sucessivas de 100 e 50 μM para a triagem
dos compostos e de 150 a 12,5 μM para a determinação de IC50. É importante para
a correta condução dos ensaios que a concentração final de DMSO seja de no
máximo 1%, pois é amplamente conhecido o potencial citotóxico deste solvente,
quando em concentrações superiores a 2% (BRAYTON, 1986).
Dos compostos testados, apenas a queleritrina apresentou citotoxidade nas
linhagens não-tumorigênicas Huvec e LL24 e em todas as linhagens tumorais
testadas, tais como a Jurkat, SK-Mel-28 e A549. Esse resultado indica a falta de
seletividade da queleritrina em relação às diversas linhagens, o que corrobora com
dados de outros trabalhos (MALÍKOVÁ et al., 2006; MATKAR; WRISCHNIK;
HELLMANN-BLUMBERG, 2008; SLANINOVÁ et al., 2007; ZHANG et al., 2012). Os
valores de IC50 podem ser observados na Tabela 7.
Tabela 7. Valores de IC50 (µM) da queleritrina e do composto mais ativo das séries.
Linhagens celulares
IC50 (µM) ± DP
queleritrina 2f
SKMel-28 9,76 ± 2,15 > 100
Jurkat 3,13 ± 1,80 > 100
A549 7,46 ± 1,67 118,31 ± 17,9
Huvec 4,93 ± 1,73 > 100
LL24 3,73 ± 0,69 > 100
Dos compostos sintetizados, nenhum apresentou atividade citotóxica nas
concentrações testadas nas linhagens não-tumorigênicas Huvec e LL24, bem como
nas linhagens tumorais SK-Mel-28 e Jurkat. Na linhagem A549, o único composto
que apresentou citotoxicidade foi o 2f, conforme Tabela 7.
A determinação da curva dose-resposta e cálculo de IC50 para o 2f em A549
foi realizado com cinco pontos e concentração máxima de 150 µM. Essa

69
concentração ultrapassa os valores escolhidos para a triagem dos compostos nas
outras linhagens celulares. Entretanto, a escolha de uma concentração mais alta é
justificável uma vez que a A549 é uma linhagem multirresistente (JARAMILLO et al.,
2008; LAMORAL-THEYS et al., 2010; MATHIEU et al., 2004).
Dentre os diversos mecanismos de resistência da linhagem A549 relatados na
literatura, a alta expressão de ciclooxigenase-2 (COX-2) (DOHADWALA et al., 2001)
e a resistência à TRAIL (AZIJLI et al., 2012; VOORTMAN et al., 2007) conferem
fenótipo resistente à apoptose para a linhagem A549. Já a alta expressão de
transportadores ABC (ATP binding cassete) e de glutationa S-transferase Pi (GST-
π), também presentes na A549, aumenta o efluxo de fármacos antineoplásicos e a
destoxificação das células tumorais (DONG et al., 2014; MATHIEU et al., 2004; VAN
DER DEEN et al., 2005), respectivamente. Estas características conferem
resistência da A549 a compostos antitumorais, tais como a cisplatina (BARR et al.,
2013; WU et al., 2010), a gencitabina (IKEDA et al., 2011; WANG et al., 2008) e o
paclitaxel (GONÇALVES et al., 2001; SUN et al., 2011).
Como pode ser observado, os compostos planejados não apresentaram
atividade citotóxica interessante no painel de linhagens determinado para este
trabalho. Contudo, isso não descarta uma possível atividade anticâncer, uma vez
que a atividade citostática pode ser considerada na análise de uma atividade
antitumoral (BASSO et al., 2002; DRAPPATZ; WEN, 2004; RIXE; FOJO, 2007).
Além disso, fármacos citotóxicos são relatados como compostos mais passíveis de
resistência tumoral, assim como são responsabilizados pelos efeitos adversos mais
severos da terapia antineoplásica (BOCK; LENGAUER, 2012).
A capacidade citostática, combinada com o potencial citotóxico de compostos
antineoplásicos, representam uma modalidade terapêutica mais eficiente do que o
uso desses diferentes tipos de agentes sozinhos (RIXE; FOJO, 2007). Isto se deve
ao fato de que os compostos citostáticos podem retardar o surgimento de resistência
aos agentes citotóxicos ao diminuir a taxa de proliferação celular e surgimento de
células resistentes. Além disso, substâncias citostáticas podem desacelerar o
crescimento dos tumores entre as doses dos antineoplásicos citotóxicos
(GARDNER; FERNANDES, 2004; KIM; CHEN; LIAO, 2007).
O uso do gefitinibe (20) (Figura 22) para o tratamento do glioma maligno, um
tipo de câncer de pobre prognóstico, ilustra a aplicabilidade de compostos

70
citostáticos para uma melhor resposta terapêutica (CHANG et al., 2014; DRAPPATZ;
WEN, 2004). Este efeito citostático é observado em concentrações menores que 25
µM e, ultrapassando esse valor, o efeito citotóxico do gefitinibe é predominante
(CHANG et al., 2014).
Figura 22. Estruturas químicas do gefitinibe (20) e temozolomida (21).
O mecanismo de ação do gefitinibe consiste na inibição do receptor de fator
de crescimento epidermal (EGFR – epidermal growth factor receptor), cuja
expressão está exarcebada neste tipo de tumor (JUN et al., 2012; PÉDEBOSCQ et
al., 2008). O efeito do gefitinibe sobre o EGFR foi capaz de aumentar a sensibilidade
dos gliomas resistentes à radioterapia (ANDERSSON et al., 2007), e, quando
utilizado no início da doença, melhorou a resposta tumoral à temozolomida (21)
(Figura 22), um composto antitumoral citotóxico (PRADOS et al., 2008).
As evidências demonstram que o potencial citostático dos compostos pode
desempenhar um papel importante na quimioterapia antineoplásica. Devido a isso,
realizou-se o ensaio clonogênico do composto 2f com o intuito de verificar se,
apesar da baixa citotoxicidade apresentada pelo composto, este possui atividade
citostática.
5.3.2 Ensaio clonogênico e ciclo celular
O ensaio clonogênico, ou ensaio de formação de colônia é um método que
mensura a sobrevivência e capacidade de proliferação celular a partir de uma única

71
célula. É considerada uma colônia se o aglomerado for formado por pelo menos 50
células (CELIS et al., 2005).
Um pequeno número de células é plaqueado em placas com poços grandes
para que uma célula não fique próxima à outra. As células passam, então, a ter
contato com a substância a ser testada por um período curto de tempo, e logo após,
são incubadas por um período maior sem o contato com os compostos (MUNSHI;
HOBBS; MEYN, 2005). Para a visualização das colônias, utiliza-se o glutaraldeído
para a fixação das células e o cristal de violeta para a marcação das colônias. A
contagem é feita pelo microscópio (FRANKEN et al., 2006).
Após o tratamento da A549 com o composto 2f, observou-se uma atividade
inibitória do crescimento de colônias em todas as concentrações testadas (50 µM, 75
µM e 100 µM), conforme Figura 23.
Figura 23. (a) Efeito do composto 2f na Formação de colônias em A549, de maneira dose-
dependente. (b) O gráfico representa a média de três experimentos diferentes. * p≤0.05; ** p≤0.01
comparado ao grupo controle (paired Student’s t test).
No ensaio, o grupo controle apresentou uma média de 60 colônias, enquanto
os grupos tratados apresentaram médias de 9, 19 e 34 colônias nas concentrações

72
100 µM, 75 µM e 50 µM do 2f, respectivamente. A formação de colônias foi reduzida
de maneira dose-dependente, conforme ilustra a Figura 23.
O ensaio clonogênico mimetiza o desenvolvimento de um tumor a partir de
uma célula, o que o aproxima ao comportamento tumoral in vivo (ZHOU et al., 2015).
Diferente do ensaio de citotoxicidade por MTT, o ensaio clonogênico é analisado
após um período maior depois do tratamento e pode-se então, observar os efeitos
do composto testado de modo mais aproximado à resposta terapêutica, ou seja,
analisa-se os efeitos do composto a longo prazo (BROWN; ATTARDI, 2005).
O fato do composto 2f inibir o crescimento de colônias, indica seu potencial
como substância anticâncer. Ademais, como o composto 2f não demonstrou
toxicidade na linhagem de fibroblasto de pulmão humano (LL24), mostra-se ser mais
seletivo do que a queleritrina.
Para complementar os resultados obtidos no ensaio clonogênico, procedeu-se
à análise do ciclo celular a fim de verificar alterações induzidas pelo composto 2f.
Esta análise mostrou que o composto 2f provocou aumento na porcentagem de
células na fase G0/G1. Enquanto o grupo controle apresentou 36,55% da população
celular em G0/G1, os grupos tratamento apresentaram 54,55%, 50,6 % e 49,2 %
para as concentrações 100 µM, 75 µM e 50 µM do 2f, respectivamente (Figura 24).
Em detrimento, houve redução da população celular nas fases S e G2/M,
ilustrado pela diminuição das áreas verdes (fase S) e vermelhas (fase G2/M) dos
histogramas do grupo tratado (Figura 24). Isto caracteriza interrupção do ciclo celular
na fase G0/G1, o que pode indicar que as células estão em quiescência, ou seja,
em repouso, fenótipo típico de células que não se proliferam (JANUMYAN et al.,
2008). Este resultado corrobora com o achado no ensaio clonogênico, que indica
que o composto 2f tem o potencial de inibir a proliferação celular.
A capacidade de interromper o ciclo celular na fase G0/G1 também foi
observado em estudos relacionados ao gefitinibe (20) (Figura 22) nas linhagens
tumorais de adenocarcinoma de pulmão (A549) (HUANG et al., 2013), câncer
pancreático humano (PANC-1) (ZHOU et al., 2009) e câncer de mama humano
(HCC1937) (CORKERY et al., 2009). Como o gefitinibe é descrito como composto
citostático (MILLAR; LYNCH, 2003), a interrupção do ciclo celular na fase G0/G1
indica relação entre este fenômeno ao potencial citostático de um composto.

73
Figura 24. Efeitos do composto 2f sobre o ciclo celular na linhagem A549, o qual o histograma (a)
representa o grupo controle, (b) o composto 2f a 100 µM, (c) composto 2f a 75 µM e (d) 50 µM; a fase
G0/G1 está em azul, S em verde e G2/M em vermelho. (e) ilustra a % da distribuição de células em
cada fase do ciclo. Os resultados representam a média de dois experimentos diferentes.
O análogo 2f foi o único composto que apresentou atividade biológica frente à
linhagem A549. Uma particularidade deste composto é a presença do grupo
catecólico em sua estrutura, como observado na Figura 25 (22). Este tipo de grupo é
encontrado em diversas substâncias naturais com potencial antitumoral, tais como a
catequina (23) (SUGANUMA; SAHA; FUJIKI, 2011; YU et al., 2014), a quercetina

74
(24) (KUMAR; PANDEY, 2013) e o éster fenetil do ácido caféico (25) (CHUU et al.,
2012).
Figura 25. Estruturas químicas do composto 2f (22), da catequina (23), da quercetina (24) e do éster
fenetil do ácido caféico (25), com o grupo catecólico em destaque.
Os principais mecanismos relatados para compostos catecólicos são a
geração de espécies reativas de oxigênio (EROS), danos ao DNA por oxidação ou
formação de adutos e danos à proteínas por arilação ou oxidação sulfidrílica (Figura
26) (SCHWEIGERT; ZEHNDER; EGGEN, 2001).
Os mecanismos de geração de EROS e de danos ao DNA em geral induzem
à morte celular (KANG et al., 2012). Como este efeito não foi observado como
predominante após o tratamento pelo composto 2f, o mecanismo por danos a
proteínas pode ser uma hipótese a ser considerada.
Além disso, estudos mecanísticos acerca da catequina, quercetina e do éster
fenetil do ácido caféico, indicam que estes, em doses sub-citotóxicas, induzem
interrupção do ciclo celular na fase G0/G1 em diversas linhagens, assim como o
composto 2f sobre a linhagem A549. Nesses estudos, o acúmulo de células na fase
G0/G1 aponta para um provável mecanismo de modulação de proteínas-alvo
específicas, as quais estão inseridas em vias de sinalização necessárias para o
desenvolvimento tumoral. Observou-se também que, possivelmente, os compostos

75
catecólicos exercem alguma influência na atividade de algumas enzimas (CHUU et
al., 2012; KUMAR; PANDEY, 2013; SCHWEIGERT; ZEHNDER; EGGEN, 2001;
SUGANUMA; SAHA; FUJIKI, 2011; YU et al., 2014). Entretanto, estudos
mecanísticos mais aprofundados teriam que ser conduzidos para a compreensão do
exato mecanismo de ação do 2f.
5.4 Modelagem molecular
A queleritrina, o composto 2f (análogo ativo) e os compostos 2a e 2c
(compostos inativos), foram submetidos aos estudos de modelagem molecular.
Desse modo, foi possível determinar teoricamente propriedades físico-químicas e
estereoeletrônicas que podem ser correlacionadas com a atividade biológica.
Ao empregar modificações moleculares em uma dada estrutura, podem-se
alterar propriedades físico-químicas e, consequentemente, a sua atividade biológica.
Nesse contexto, os estudos de modelagem molecular tem grande valia na discussão
sobre relação estrutura-atividade, uma vez que por meio do cálculo de propriedades
pode-se correlacionar os efeitos biológicos com as características físico-químicas de
uma molécula e, desse modo, determinar os perfis farmacocinéticos e
farmacodinâmicos de um composto (ARROIO; HONÓRIO; DA SILVA, 2010).
Iniciaram-se os estudos pela construção dos modelos tridimensionais dos
quatro compostos. Após a construção, a geometria molecular das estruturas foi
otimizada e, por meio do cálculo de carga de ponto único, foram calculadas
propriedades estereoeletrônicas.
Entre as propriedades, foram determinados: mapa de potencial eletrostático,
mapas dos orbitais moleculares HOMO (orbital molecular ocupado de maior energia)
e LUMO (orbital molecular não ocupado de menor energia) e de momento dipolo.
Propriedades hidrofóbicas também foram levadas em consideração, tais como o
logaritmo do coeficiente de partição calculado – CLog P, uma vez que este
parâmetro reflete a propriedade lipofílica dos compostos e, consequentemente,
possibilita a extrapolação da capacidade dos compostos de serem solubilizados e
absorvidos nos sistemas biológicos (TAVARES, 2004).

76
Em relação ao mapa de potencial eletrostático (MPE) (Figura 26), a coloração
apresentada indica que regiões de tom vermelho intenso (60,000 kJ/mol) possuem
maior densidade eletrônica, enquanto que regiões de colocação azul escuro
(580,000 kJ/mol) possuem menor distribuição de densidade eletrônica. Nota-se uma
diferença da distribuição eletrônica das moléculas estudadas, principalmente
influenciada pela rigidez ou flexibilidade dos compostos e pela presença ou ausência
de metoxilas e hidroxilas nas estruturas (Figura 26).
No MPE é possível observar que a estrutura planar da queleritrina, a qual é
composta por anéis aromáticos fundidos, favorece a distribuição mais uniforme da
densidade eletrônica em diversas regiões da molécula, padrão ilustrado pela
coloração verde (Figura 26). Além disso, o sistema conjugado dos anéis aromáticos
diminui a carga positiva sobre o nitrogênio da queleritrina (WHEELER; HOUK, 2009).
Esses fenômenos não foram observados nos análogos 2a, 2c e 2f, uma vez
que, com a simplificação molecular, os compostos perderam o sistema de
conjugação entre os anéis aromáticos e o nitrogênio das estruturas. Como os
compostos 2a, 2c e 2f não apresentaram atividade citotóxica significante, a perda da
planaridade da queleritrina e a falta de distribuição uniforme da densidade eletrônica
sobre a molécula podem estar relacionadas à diminuição da atividade citotóxica dos
análogos.
Assim como no MPE, o valor encontrado de vetor de momento dipolo (μ) da
queleritrina apresentou-se distinto, se comparado aos compostos sintetizados. O
vetor de momento dipolo representa o produto da soma total de cargas positivas ou
negativas e a distância entre seus centroides (DE-SÁ-JÚNIOR et al., 2013). A
queleritrina apresenta valor μ = 4,82 Debye, enquanto 2a, 2c e 2f apresentam
valores mais próximos entre si, de 6,45, 7,10 e 6,16 Debye, respectivamente. Como
o momento dipolo pode variar de acordo com geometria da molécula, a queleritrina
possui um valor μ menor em comparação aos análogos, devido à sua estrutura
planar e conjugada (SINEGOVSKAYA; KEIKO; TROFIMOV, 1987). Além disso, com
os resultados teóricos obtidos, pode-se inferir que quanto menor o valor de μ, maior
a atividade biológica, visto que a queleritrina e o composto 2f (composto ativo) foram
os que apresentaram os valores mais inferiores de μ.

77
Figura 26. Representação de propriedades eletrônicas da queleritrina e dos compostos 2a, 2c e 2f, por meio do mapa de potencial eletrostático, vetor do
momento dipolo (representados por setas amarelas), e mapas de orbitais moleculares HOMO e LUMO.
77

78
Em relação aos valores de energia do HOMO (ɛHOMO) e LUMO (ɛLUMO), nota-
se que a queleritrina apresenta menor diferença (Gap) entre ɛHOMO e ɛLUMO, cujo
∆ɛ(LUMO-HOMO) = 8,98 eV, se comparado aos análogos 2a, 2c e 2f, os quais possuem
os valores semelhantes de Gap de 11,64, 11,71 e 11,69 eV, respectivamente. O
valor inferior de Gap encontrado para a queleritrina sugere que ela seja menos
estável e mais reativa (ZHANG; MUSGRAVE, 2007).
A menor diferença entre os valores de ɛHOMO e ɛLUMO resulta em uma
transferência acentuada de energia entre os orbitais, permitindo ressaltar as
características elétron-aceptoras ou doadoras dos grupos funcionais da queleritrina,
logo acentuando a sua reatividade e diminuindo a sua estabilidade
(GOPALAKRISHNAN; KALAIARASI, 2014).
As estruturas da queleritrina e do composto 2f foram alinhados no programa
HyperChem versão 8.0.7 (Hypercube Inc) e a imagem foi gerada pelo programa
ViewerLite versão 4.2 (Accelrys Inc, 2001) (Figura 27). Esse alinhamento foi
realizado a fim de observar se as distâncias interatômicas de determinados grupos
funcionais se mantiveram mesmo realizando a simplificação molecular sobre a
queleritrina.
Figura 27. (a) Representação do alinhamento entre as estruturas da queleritrina e do análogo 2f, nos
quais os átomos de carbono estão representados em verde e amarelo, respectivamente, os de
oxigênio em vermelho e o nitrogênio em azul em ambas. Os átomos de hidrogênio foram suprimidos
para a melhor visualização. Em (b), representação das distâncias interatômicas (em preto)
mensuradas para a queleritrina e em (c), representação das distâncias interatômicas (em preto)

79
obtidas para o análogo 2f. Ainda em (b) e (c), os ângulos diedros das estruturas da queleritrina e do
composto 2f, respectivamente, foram calculadas a partir dos átomos circulados em vermelho.
Ao calcular as distâncias interatômicas entre o grupo metilenodioxi e o
nitrogênio de ambas as estruturas, obtiveram-se os valores 6,61 Å e 6,66 Å para a
queleritrina e o 2f, respectivamente (Figura 27). Além disso, a distância entre o
nitrogênio e o carbono do anel aromático ligado a um oxigênio é de 4,77 Å e 4,97 Å
para a queleritrina e o 2f, respectivamente.
Entretanto, os ângulos diedros obtidos entre o benzodioxol, o nitrogênio e o
carbono do anel aromático ligado a um oxigênio de ambas as estruturas,
apresentaram grande diferença. Para a queleritrina, o valor obtido foi de 96,5 º e
para o 2f, o valor foi de 164,1 º, e este valor superior reflete em uma flexibilidade
estrutural acentuada apresentada pelo composto 2f (KEEPERS et al., 1982; MAO,
1992).
Esses dados sugerem que a simplificação molecular pode não ter alterado a
distância interatômica entre grupos característicos das moléculas de maneira
significativa. Entretanto, a perda da planaridade da queleritrina pode ter sido
responsável pela diminuição da atividade biológica, fato também observado ao
analisar o mapa de potencial eletrostático.
Ademais, foram calculados outros parâmetros físico-químicos adicionais como
o peso molecular, CLogP e os sítios de ligação de hidrogênio, conforme Tabela 8.
Estes parâmetros permitem estimar a biodisponibilidade oral dos compostos com
base na Regra dos Cinco de Lipinski (Ro5). A “Regra dos cinco de Lipinski” – Ro5 -,
é definida como um conjunto de parâmetros capazes de identificar compostos com
problemas de absorção e permeabilidade (LIPINSKI et al., 1997).
Tabela 8. Parâmetros físico-químicos calculados para a queleritrina e os compostos 2a, 2c e 2f.
Composto PM CLogP HBA HBD
queleritrina 348,37 4,05 5 0
2a 277,75 2,62 3 0
2c 337,80 2,36 5 0
2f 309,74 1,84 5 2

80
Esta regra abrange características físico-químicas como massa molecular,
lipofilicidade e sítios doadores e aceptores de ligação hidrogênio. Dessa forma,
determina que uma boa absorção e permeação sejam mais comuns quando:
O peso molecular é menor ou igual a 500 Daltons (Da);
O CLog P é menor ou igual a 5;
O número de grupos aceptores de ligação hidrogênio é menor ou igual a 10;
O número de grupos doadores de ligação hidrogênio é menor ou igual a 5.
Sendo assim, verifica-se, portanto, que o composto 2f apresenta valores
compatíveis com o determinado por Lipinski, podendo ser considerado uma
molécula com características mais favoráveis de apresentar atividade quando
administrado por via oral.

81
6 CONCLUSÕES E PERSPECTIVAS
O presente trabalho consistiu na síntese de dez compostos, dentre os quais,
seis da série de aminas secundárias e quatro da série de aminas terciárias. Cada
série foi composta por um análogo não substituído (2a/3a), um análogo 4-
monometoxilado (2b/3b), um análogo 3,4-dimetoxilado (2c/3c), um 3,4,5-
trimetoxilado (2d/3d) e os análogos 3-metoxi-4-hidroxilado (2e) e 3,4-di-idroxilado
(2f) pertencem somente à série de aminas secundárias.
A síntese proposta para a formação da amina secundária pela metodologia
com o agente redutor NaBH(OAc)3 proporcionou a formação dos compostos de
modo satisfatório. Entretanto, os rendimentos obtidos nessa série foram de
moderados a pouco satisfatórios e podem ser devido aos métodos de purificação
utilizados e à possível formação de subprodutos. As estruturas das aminas
secundárias foram confirmadas por meio de análise espectrométrica de RMN 1H e
13C, cromatografia líquida de alta eficiência e análise elementar.
A partir das aminas secundárias, testaram-se metodologias de obtenção das
aminas terciárias e a reação de metilação redutiva demonstrou ser a mais
adequada, uma vez que não havia a formação de subprodutos comuns de reações
como a substituição nucleofílica. As estruturas dos compostos, assim como as
aminas secundárias, foram confirmadas por meio de análise espectrométrica de
RMN 1H e 13C, cromatografia líquida de alta eficiência e análise elementar.
Os compostos com grau de pureza cromatográfica acima de 95% foram
testados quanto ao seu potencial citotóxico em comparação à queleritrina. O
alcaloide não demonstrou seletividade ao comparar a citotoxicidade em linhagens
tumorigênicas e não-tumorigênicas, o que ressalta a necessidade do planejamento
de análogos mais seletivos.
Em contrapartida, os compostos sintetizados não apresentaram atividade
citotóxica significativa em nenhuma linhagem testada. Apenas o composto 2f
apresentou potencial citostático frente à linhagem A549 no ensaio clonogênico. O
seu mecanismo foi corroborado por outro estudo, a análise do ciclo celular, no qual
observou-se a interrupção do ciclo celular na fase G0/G1, o que pode indicar que as
células estão em quiescência, fenótipo típico de células que não se proliferam.

82
Essa atividade é promissora, visto que o composto 2f apresentou-se ativo em
uma linhagem tumoral sem ser citotóxico à uma linhagem não-tumorigênica, o que
indica maior seletividade do análogo se comparado à queleritrina.
Os estudos de modelagem molecular inferem que a perda da planaridade da
queleritrina pode ser a principal responsável pela diminuição da atividade biológica.
Entretanto, o composto mais ativo apresentou propriedades distintas que o difere
dos outros análogos inativos e da queleritrina, fato que pode ser relacionado ao seu
potencial citostático. Além disso, o composto 2f apresenta os parâmetros físico-
químicos avaliados pela Ro5 dentro dos valores preconizados, apresentando uma
característica drug-like, passível de ser administrado por via oral.
Dessa forma, novas modificações moleculares devem ser planejadas a fim de
otimizar a atividade da queleritrina para fornecer compostos mais ativos e mais
seletivos. Vale ressaltar também, a otimização do composto 2f, pois o seu potencial
citostático também é interessante para a atividade antitumoral.
Tendo em vista os resultados obtidos, sugere-se:
Realizar a reação de proteção das hidroxilas fenólicas do composto 2f,
a fim de obter maiores rendimentos;
Obter a amina terciária a partir do análogo 2f para verificar se a metila
ligada ao nitrogênio pode influenciar na atividade, uma vez que a
influência dessa modificação não foi observada nos análogos
metoxilados;
Realizar estudos mecanísticos complementares do composto 2f, como,
por exemplo, o western blot, a fim de verificar se o mesmo influencia na
expressão de proteínas relacionadas ao ciclo celular, como as ciclinas,
as enzimas ciclina-dependentes, a p21 e p53, ou não relacionadas ao
ciclo celular;
Realizar estudos de atividade biológica do análogo 2f para verificar o
seu potencial em terapia combinada com um fármaco citotóxico, como
a oxaliplatina, que é utilizada no tratamento de câncer de pulmão
(KNIGHT et al., 2004);
Obter outros análogos da queleritrina planejados por simplificação
molecular, com maior rigidez estrutural;

83
Obter outros compostos que contenham padrão de mono-, di- e
trissubstituição por hidroxilas fenólicas e em diferentes posições.

84
7 REFERÊNCIAS BIBLIOGRÁFICAS
ABDEL-MAGID, A. F. et al. Reductive Amination of Aldehydes and Ketones with
Sodium Triacetoxyborohydride. Studies on Direct and Indirect Reductive Amination
Procedures. The Journal of Organic Chemistry, v. 61, n. 11, p. 3849–3862, 1996.
ABDEL-MAGID, A. F.; MEHRMAN, S. J. A Review on the Use of Sodium
Triacetoxyborohydride in the Reductive Amination of Ketones and Aldehydes.
Organic Process Research & Development, v. 10, n. 5, p. 971–1031, 2006.
ABDEL-MAGID, A.; MARYANOFF, C.; CARSON, K. Reductive amination of
aldehydes and ketones by using sodium triacetoxyborohydride. Tetrahedron
Letters, v. 31, n. 39, p. 5595–5598, 1990.
ACS. Cancer Facts & Figures 2014. [s.l: s.n.]. Disponível em:
<http://www.cancer.org/acs/groups/content/@research/documents/webcontent/acspc
-042151.pdf>.
ADAMS, J. M.; CORY, S. The Bcl-2 apoptotic switch in cancer development and
therapy. Oncogene, v. 26, n. 9, p. 1324–1337, 2007.
AMARAL, P. D. A. et al. Química combinatória: moderna ferramenta para a obtenção
de candidatos a protótipos de novos fármacos. Revista Brasileira de Ciências
Farmacêuticas, v. 39, n. 4, p. 351–363, 2003.
AMUNDSON, S. A et al. An Informatics Approach Identifying Markers of
Chemosensitivity in Human Cancer Cell Lines An Informatics Approach Identifying
Markers of Chemosensitivity in Human. Cancer Research, p. 6101–6110, 2000.
ANDERSON, A. C. The process of structure-based drug design. Chemistry &
Biology, v. 10, p. 787–797, 2003.
ANDERSSON, U. et al. Treatment schedule is of importance when gefitinib is
combined with irradiation of glioma and endothelial cells in vitro. Acta oncologica
(Stockholm, Sweden), v. 46, n. 7, p. 951–60, 2007.
ARROIO, A.; HONÓRIO, K. M.; DA SILVA, A. B. F. Propriedades químico-quânticas
empregadas em estudos das relações estrutura-atividade. Quimica Nova, v. 33, n.
3, p. 694–699, 2010.
ASHKENAZI, A. Targeting the extrinsic apoptosis pathway in cancer. Cytokine &
Growth Factor Reviews, v. 19, n. 3-4, p. 325–331, 2008.
AVENDAÑO, C.; MENÉNDEZ, J. C. Cancer chemoprevention. In: Medicinal
Chemistry of Anticancer Drugs. [s.l.] Elsevier, 2008. p. 417–429.
AZIJLI, K. et al. Kinome profiling of non-canonical TRAIL signaling reveals RIP1-Src-
STAT3 dependent invasion in resistant non-small cell lung cancer cells. Journal of
Cell Science, p. 4651–4661, 2012.

85
AZIZ, D. M. Assessment of bovine sperm viability by MTT reduction assay. Animal
Reproduction Science, v. 92, n. 1-2, p. 1–8, 2006.
BARR, M. P. et al. Generation and Characterisation of Cisplatin-Resistant Non-Small
Cell Lung Cancer Cell Lines Displaying a Stem-Like Signature. PLoS ONE, v. 8, n. 1,
p. e54193, 2013.
BARREIRO, E. J. Estratégia de simplificação molecular no planejamento racional de
fármacos: a descoberta de novo agente cardioativo. Química Nova, v. 25, n. 6b, p.
1172–1180, 2002.
BARREIRO, E. J.; FRAGA, C. A. M. A questão da inovação em fármacos no Brasil:
Proposta de criação do programa nacional de fármacos (PRONFAR). Quimica
Nova, v. 28, n. SUPPL., p. 56–63, 2005.
BASSO, U. et al. Non-cytotoxic therapies for malignant gliomas. Journal of neuro-
oncology, v. 58, n. 1, p. 57–69, 2002.
BERNARDO, P. H. et al. Structure # Activity Relationship Studies of Phenanthridine-
Based Bcl-X Inhibitors Structure-Activity Relationship Studies of Phenanthridine-
Based Bcl-X L Inhibitors. Journal of Medicinal Chemistry, p. 6699–6710, 2008.
BHANOT, A.; SHARMA, R.; NOOLVI, M. N. Natural sources as potential anti-cancer
agents: A review. International Journal of Phytomedicine, v. 3, n. 1, p. 09–26,
2011.
BHATTACHARYYA, S. Reductive Alkylations of Dimethylamine Using Titanium(IV)
Isopropoxide and Sodium Borohydride: The Journal of Organic Chemistry, v. 60, n.
d, p. 4928–4929, 1995.
BILLACK, B.; RADKAR, V.; ADIABOUAH, C. In Vitro evaluation of the cytotoxic and
anti-proliferative properties of resveratrol and several of its analogs. Cellular and
Molecular Biology Letters, v. 13, n. 4, 2008.
BOCK, C.; LENGAUER, T. Managing drug resistance in cancer: lessons from HIV
therapy. Nat Rev Cancer, v. 12, n. 7, p. 494–501, 2012.
BORCH, R. F. Reductive Amination With Sodium Cyanoborohydride: N,N-
Dimethylcyclohexylamine. Organic Syntheses, v. 52, p. 124, 1972.
BORCH, R. F.; BERNSTEIN, M. D.; DUSRT, D. The cyanoborohydridoborate anion
as a selective reducing agent. Journal of the American Chemical Society, v. 93, n.
12, p. 2897–2904, 1971.
BRANDÃO, H. N. et al. Química e farmacologia de quimioterápicos antineoplásicos
derivados de plantas. Quimica Nova, v. 33, n. 6, p. 1359–1369, 2010.
BRAYTON, C. F. Dimethyl sulfoxide (DMSO): a review. The Cornell Veterinarian, v.
76, p. 61–90, 1986.

86
BROWN, J. M.; ATTARDI, L. D. The role of apoptosis in cancer development and
treatment response. Nature reviews. Cancer, v. 5, n. 3, p. 231–237, 2005.
BRUNHOFER, G. et al. Exploration of natural compounds as sources of new
bifunctional scaffolds targeting cholinesterases and beta amyloid aggregation: The
case of chelerythrine. Bioorganic and Medicinal Chemistry, v. 20, n. 22, p. 6669–
6679, 2012.
CAO, Z. S. et al. Synthesis and anti-tumor activity of alkenyl camptothecin esters.
Acta Pharmacologica Sinica, v. 26, n. 2, p. 235–241, 2005.
CAREY, F. A. Nucleophilic substitution. In: Organic chemistry. 4. ed. [s.l.]
McGrawHill Companies Inc, 2000. p. 302–338.
CELIS, J. E. et al. Cytotoxicity and cell growth assays. In: Cell Biology: A
laboratory handbook. [s.l.] Academic Press, 2005. p. 2328.
CHAFFER, C. L.; WEINBERG, R. A. A Perspective on Cancer Cell Metastasis.
Science, v. 331, n. 6024, p. 1559–1564, 2011.
CHAN, S. L. et al. Identification of chelerythrine as an inhibitor of BclXL function.
Journal of Biological Chemistry, v. 278, n. 23, p. 20453–20456, 2003.
CHANG, C.-Y. et al. Autophagy contributes to gefitinib-induced glioma cell growth
inhibition. Experimental cell research, v. 327, n. 1, p. 1–11, 2014.
CHAST, F. A History of Drug Discovery. In: KUBINYI, H. (Ed.). . The Practice of
Medicinal Chemistry. 3. ed. [s.l.] Academic Press, 2008. p. 3–62.
CHESON, B. D. Oblimersen for the treatment of patients with chronic lymphocytic
leukemia. Therapeutics and clinical risk management, v. 3, n. 5, p. 855–70, 2007.
CHIN, Y.-W. et al. Drug discovery from natural sources. AAPS Journal, v. 8, n. 2, p.
E239–E253, 2006.
CHO, B. T.; KANG, S. K. Direct and indirect reductive amination of aldehydes and
ketones with solid acid-activated sodium borohydride under solvent-free conditions.
Tetrahedron, v. 61, n. 24, p. 5725–5734, 2005.
CHUU, C.-P. et al. Caffeic acid phenethyl ester suppresses the proliferation of
human prostate cancer cells through inhibition of p70S6K and Akt signaling
networks. Cancer prevention research (Philadelphia, Pa.), v. 5, n. 5, p. 788–97,
2012.
CLARKE, H. T.; GILLESPIE, H. B.; WEISSHAUS, S. Z. The Action of Formaldehyde
on Amines and Amino Acids. Journal of The American Chemical Society, v. 55, n.
11, p. 4471–4587, 1933.
CLAYDEN, J.; GREEVES, N.; WARREN, S. Nucleophilic substitution at C=O with
loss of carbonyl oxygen. In: Organic chemistry. 2. ed. New York: Oxford, 2012a. p.

87
222–239.
CLAYDEN, J.; GREEVES, N.; WARREN, S. Organic structures. In: Organic
chemistry. 2. ed. New York: Oxford, 2012b. p. 15–42.
CLAYDEN, J.; GREEVES, N.; WARREN, S. Nucleophilic substitution at saturated
carbon. In: Organic chemistry. 2. ed. New York: Oxford, 2012c. p. 328–359.
CLINICALTRIALS, C. T. U. S. Clinical Trials. Disponível em:
<http://www.clinicaltrials.gov/>. Acesso em: 22 ago. 2015.
CORKERY, B. et al. Epidermal growth factor receptor as a potential therapeutic
target in triple-negative breast cancer. Annals of oncology : official journal of the
European Society for Medical Oncology / ESMO, v. 20, n. 5, p. 862–867, 2009.
COSTA-LOTUFO, L. V et al. A Contribuição dos produtos naturais como fonte de
novos fármacos anticâncer : estudos no laboratório nacional de oncologia
experimental da Universidade Federal do Ceará. Revista Virtual de Química, v. 2,
n. 1, p. 47–58, 2010.
CRU, C. R. U. Cancer cases and deaths across the world and in the UK:
October 2011. Disponível em: <http://publications.cancerresearchuk.org/
cancerstats/statsworldwide/cancer-global-ppt.html>. Acesso em: 29 jun. 2014.
CRU, C. R. U. All cancers combined key facts. Disponível em:
<http://publications.cancerresearchuk.org/publicationformat/formatfactsheet/keyfacts
all.html>. Acesso em: 29 jun. 2014.
CZABOTAR, P. E. et al. Control of apoptosis by the BCL-2 protein family:
implications for physiology and therapy. Nature Reviews Molecular Cell Biology, v.
15, n. 1, p. 49–63, 2014.
DA SILVA, R. A.; ESTEVAM, I. H. S.; BIEBER, L. W. Reductive methylation of
primary and secondary amines and amino acids by aqueous formaldehyde and zinc.
Tetrahedron Letters, v. 48, n. 43, p. 7680–7682, 2007.
DE ALMEIDA, V. L. et al. Câncer e agentes antineoplásicos ciclo-celular específicos
e ciclo-celular não específicos que interagem com o DNA: Uma introdução. Quimica
Nova, v. 28, n. 1, p. 118–129, 2005.
DE-SÁ-JÚNIOR, P. L. et al. RPF101, A new capsaicin-like analogue, Disrupts the
microtubule network accompanied by arrest in the G2/M phase, Inducing apoptosis
and mitotic catastrophe in the MCF-7 breast cancer cells. Toxicology and Applied
Pharmacology, v. 266, n. 3, p. 385–398, 2013.
DEWSON, G.; KLUCK, R. M. Bcl-2 family-regulated apoptosis in health and disease.
Cell Health and Cytoskeleton, v. 2, p. 9–22, 2010.
DOHADWALA, M. et al. Non-small cell lung cancer cyclooxygenase-2-dependent

88
invasion is mediated by CD44. The Journal of biological chemistry, v. 276, n. 24,
p. 20809–12, 2001.
DONG, Z. et al. MicroRNA-31 inhibits cisplatin-induced apoptosis in non-small cell
lung cancer cells by regulating the drug transporter ABCB9. Cancer Letters, v. 343,
n. 2, p. 249–257, 2014.
DRAPPATZ, J.; WEN, P. Y. Non-cytotoxic drugs as potential treatments for gliomas.
Current opinion in neurology, v. 17, p. 663–673, 2004.
ELMORE, S. Apoptosis: A Review of Programmed Cell Death. Toxicologic
Pathology, v. 35, n. 4, p. 495–516, 2007.
ESCHWEILER, W. Ersatz von an Stickstoff gebundenen Wasserstoffatomen durch
die Methylgruppe mit Hülfe von Formaldehyd. Berichte der deutschen chemischen
Gesellschaft, v. 38, n. 1, p. 880–882, 1905.
EVANS, D. A.; CHAPMAN, K. T.; CARREIRA, E. M. Directed Reduction of beta-
Hydroxy Ketones Employing Tetramethylammonium Triacetoxyborohydride. Journal
of the American Chemical Society, v. 110, p. 3560–3578, 1988.
FAN, T.-J. et al. Caspase Family Proteases and Apoptosis. Acta Biochimica et
Biophysica Sinica, v. 37, n. 11, p. 719–727, 2005.
FERNANDES, T. B. Planejamento , síntese e avaliação do potencial antitumoral
de compostos arilsulfonil-hidrazônicos Planejamento , síntese e avaliação do
potencial antitumoral de compostos arilsulfonil-hidrazônicos. [s.l: s.n.].
FISCHER, P. M. Cell cycle inhibitors: current status and future directions. In: Cancer
Drug Design and Discovery. Londres: Elsevier, 2008. p. 253–XVIII.
FLOOR, S. L. et al. Hallmarks of cancer: of all cancer cells, all the time? Trends in
Molecular Medicine, v. 18, n. 9, p. 509–515, 2012.
FRANKEN, N. A P. et al. Clonogenic assay of cells in vitro. Nature protocols, v. 1,
n. 5, p. 2315–2319, 2006.
FULDA, S.; DEBATIN, K.-M. Extrinsic versus intrinsic apoptosis pathways in
anticancer chemotherapy. Oncogene, v. 25, n. 34, p. 4798–4811, 2006.
FUMOLEAU, P.; GUIU, S. New Vinca Alkaloids in Clinical Development. Current
Breast Cancer Reports, v. 5, n. 1, p. 69–72, 2013.
GABRIELLI, B.; BROOKS, K.; PAVEY, S. Defective Cell Cycle Checkpoints as
Targets for Anti-Cancer Therapies. Frontiers in Pharmacology, v. 3, n. February, p.
1–6, 2012.
GALDINO, S. L.; PITTA, I. R.; BOUCHERLE, A. Descoberta de fármacos. In:
Química medicinal: métodos e fundamentos em planejamento de fármacos.
São Paulo: EDUSP, 2011. p. 7–46.

89
GARCÍA-SÁEZ, A J. The secrets of the Bcl-2 family. Cell Death and
Differentiation, v. 19, n. 11, p. 1733–1740, 2012.
GARDNER, S. N.; FERNANDES, M. Cytostatic anticancer drug development.
Journal of experimental therapeutics & oncology, v. 4, n. 1, p. 9–18, 2004.
GARRAWAY, L. A.; JANNE, P. A. Circumventing Cancer Drug Resistance in the Era
of Personalized Medicine. Cancer Discovery, v. 2, n. 3, p. 214–226, 2012.
GIGANT, B. et al. Structural basis for the regulation of tubulin by vinblastine. Nature,
v. 435, n. 7041, p. 519–522, 2005.
GOARD, C. A; SCHIMMER, A. D. An evidence-based review of obatoclax mesylate
in the treatment of hematological malignancies. Core evidence, v. 8, p. 15–26, 2013.
GONÇALVES, A. et al. Resistance to Taxol in lung cancer cells associated with
increased microtubule dynamics. Proceedings of the National Academy of
Sciences of the United States of America, v. 98, n. 20, p. 11737–11742, 2001.
GOPALAKRISHNAN, S. B.; KALAIARASI, T. Comparative Phytochemical Screening
of the Fruits of Cucumis Trigonus Roxb . and Cucumis Sativus Linn . v. 3, n. 4, p.
1455–1468, 2014.
GRENGA, P. N. et al. Reductive amination agents: comparison of Na(CN)BH3 and
Si-CBH. Tetrahedron Letters, v. 50, n. 48, p. 6658–6660, 2009.
GRIBBLE, G. W. et al. Reaction of sodium borohydride in acidic media. I. reduction of
indoles and alkylation of aromatic amines with carboxylic acids. Journal of the
American Chemical Society, v. 7812, n. 1964, p. 7812–7814, 1974.
GUEMBAROVSKI, R. L.; CÓLUS, I. M. S. Câncer: uma doença genética. Revista
Genética na Escola, v. 3, p. 4–7, 2008.
HANAHAN, D.; WEINBERG, R. A. Hallmarks of cancer: the next generation. Cell, v.
144, n. 5, p. 646–74, 2011.
HERBERT, J. M. et al. Chelerythrine is a potent and specific inhibitor of protein
kinase C. Biochemical and Biophysical Research Communications, v. 172, n. 3,
p. 993–999, 1990.
HUANG, Y. et al. Combined inhibition of the EGFR and mTOR pathways in EGFR
wild-type non-small cell lung cancer cell lines with different genetic backgrounds.
Oncology Reports, v. 29, n. 6, p. 2486–92, 2013.
IARC, I. A. FOR R. ON C. Globocan 2012: Estimated cancer incidence, mortality
and prevalence worldwide in 2012. Disponível em:
<http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx>. Acesso em: 21 fev. 2014.
IGNEY, F. H.; KRAMMER, P. H. Death and anti-death: tumor resistance to apoptosis.
Nature Reviews Cancer, v. 2, p. 277–288, 2002.

90
IKEDA, R. et al. Isolation and characterization of gemcitabine-resistant human non-
small cell lung cancer A549 cells. International Journal of Oncology, v. 38, p. 513–
519, 2011.
INCA, I. N. DO C. J. DE A. G. DA S. A situação do câncer no Brasil. [s.l: s.n.].
INCA, I. N. DO C. J. DE A. G. DA S. Estimativa 2012: incidência de câncer no
Brasil. Rio de Janeiro. Rio de Janeiro: Instituto Nacional do Cancer José Alencar
Gomes da Silva, 2012.
INCA, I. N. DO C. J. DE A. G. DA S. Estimativa 2014: incidência de câncer no
Brasil. Rio de Janeiro. Rio de Janeiro: [s.n.].
IUPAC, I. U. OF P. AND A. C. Amphoteric. Disponível em:
<http://goldbook.iupac.org/A00306.html>. Acesso em: 21 out. 2015.
JANUMYAN, Y. et al. G0 function of BCL2 and BCL-xL requires BAX, BAK, and p27
phosphorylation by mirk, revealing a novel role of BAX and BAK in quiescence
regulation. Journal of Biological Chemistry, v. 283, n. 49, p. 34108–34120, 2008.
JARAMILLO, M. L. et al. Differential sensitivity of A549 non small lung carcinoma cell
responses to epidermal growth factor receptor pathway inhibitors. Cancer Biology &
Therapy, v. 7, n. 4, p. 557–568, 2008.
JUN, H. J. et al. Acquired MET Expression Confers Resistance to EGFR Inhibition In
a Mouse Model of Glioblastoma Multiforme. Oncogene, v. 31, n. 25, p. 3039–3050,
2012.
KAH, F. W. et al. Chelerythrine induces apoptosis through a Bax/Bak-independent
mitochondrial mechanism. Journal of Biological Chemistry, v. 283, n. 13, p. 8423–
8433, 2008.
KANG, M. A. et al. DNA damage induces reactive oxygen species generation
through the H2AX-Nox1/Rac1 pathway. Cell Death and Disease, v. 3, n. 1, p. e249,
2012.
KANG, M. H.; REYNOLDS, C. P. Bcl-2 Inhibitors: Targeting Mitochondrial Apoptotic
Pathways in Cancer Therapy. Clinical Cancer Research, v. 15, n. 4, p. 1126–1132,
2009.
KAZI, A. et al. The BH3 α-helical mimic BH3-M6 disrupts Bcl-XL, Bcl-2, and MCL-1
protein-protein interactions with Bax, Bak, Bad, or Bim and induces apoptosis in a
Bax- and Bim-dependent manner. Journal of Biological Chemistry, v. 286, n. 11, p.
9382–9392, 2011.
KEEPERS, J. W. et al. Molecular mechanical studies of DNA flexibility: coupled
backbone torsion angles and base-pair openings. Proceedings of the National
Academy of Sciences of the United States of America, v. 79, n. 18, p. 5537–
5541, 1982.

91
KIM, S. L.; CHEN, H. C.; LIAO, S. K. Non-cytotoxic and sublethal paclitaxel treatment
potentiates the sensitivity of cultured ovarian tumor SKOV-3 cells to lysis by
lymphokine-activated killer cells. Anticancer Research, v. 27, n. 2, p. 841–850,
2007.
KITTAKOOP, P.; MAHIDOL, C.; RUCHIRAWAT, S. Alkaloids as important scaffolds
in therapeutic drugs for the treatments of cancer, tuberculosis, and smoking
cessation. Current topics in medicinal chemistry, v. 14, n. 2, p. 239–52, 2014.
KNIGHT, L. A. et al. The in vitro effect of gefitinib (’Iressa') alone and in combination
with cytotoxic chemotherapy on human solid tumours. BMC cancer, v. 4, p. 83,
2004.
KOIVUNEN, J.; AALTONEN, V.; PELTONEN, J. Protein kinase C (PKC) family in
cancer progression. Cancer Letters, v. 235, n. 1, p. 1–10, 2006.
KUMAR, S.; PANDEY, A. K. Chemistry and biological activities of flavonoids: an
overview. The Scientific World Journal, v. 2013, p. 1–16, 2013.
LAMORAL-THEYS, D. et al. Simple di- and trivanillates exhibit cytostatic properties
toward cancer cells resistant to pro-apoptotic stimuli. Bioorganic and Medicinal
Chemistry, v. 18, n. 11, p. 3823–3833, 2010.
LEE, H. W. et al. Bcl-w is expressed in a majority of infiltrative gastric
adenocarcinomas and suppresses the cancer cell death by blocking stress-activated
protein kinase/c-Jun NH2-terminal kinase activation. Cancer Research, v. 63, n. 5,
p. 1093–1100, 2003.
LESSENE, G.; CZABOTAR, P. E.; COLMAN, P. M. BCL-2 family antagonists for
cancer therapy. Nature Reviews Drug Discovery, v. 7, n. 12, p. 989–1000, 2008.
LIPINSKI, A. C. et al. Experimental and computational approaches to estimate
solubility and permeability in drug discovery and development settings. Advanced
Drug Delivery Reviews, v. 23, p. 3–25, 1997.
LIU, Q. et al. A simple and sensitive method of nonaqueous capillary electrophoresis
with laser-induced native fluorescence detection for the analysis of chelerythrine and
sanguinarine in Chinese herbal medicines. Talanta, v. 70, n. 1, p. 202–207, 2006.
LIU, Q.; WANG, H.-G. Anti-cancer drug discovery and development: Bcl-2 family
small molecule inhibitors. Communicative & integrative biology, v. 5, n. 6, p. 557–
65, 2012.
LONDON, N. et al. In silico and in vitro elucidation of BH3 binding specificity toward
Bcl-2. Biochemistry, v. 51, n. 29, p. 5841–5850, 2012.
LUO, J.; SOLIMINI, N. L.; ELLEDGE, S. J. Principles of Cancer Therapy: Oncogene
and Non-oncogene Addiction. Cell, v. 136, n. 5, p. 823–837, 2009.

92
MALÍKOVÁ, J. et al. The effect of chelerythrine on cell growth, apoptosis, and cell
cycle in human normal and cancer cells in comparison with sanguinarine. Cell
Biology and Toxicology, v. 22, n. 6, p. 439–453, 2006.
MAO, B. Molecular-dynamics investigation of molecular flexibility in ligand binding.
The Biochemical journal, v. 288, p. 109–116, 1992.
MARIÑO, G. et al. Self-consumption: the interplay of autophagy and apoptosis.
Nature Reviews Molecular Cell Biology, v. 15, n. 2, p. 81–94, 2014.
MARTINOU, J.-C.; YOULE, R. J. Mitochondria in Apoptosis: Bcl-2 Family Members
and Mitochondrial Dynamics. Developmental Cell, v. 21, n. 1, p. 92–101, 2011.
MATHIEU, A. et al. Development of a chemoresistant orthotopic human nonsmall cell
lung carcinoma model in nude mice: analyses of tumor heterogenity in relation to the
immunohistochemical levels of expression of cyclooxygenase-2, ornithine
decarboxylase, lung-related resista. Cancer, v. 101, n. 8, p. 1908–18, 2004.
MATKAR, S. S.; WRISCHNIK, L. A.; HELLMANN-BLUMBERG, U. Production of
hydrogen peroxide and redox cycling can explain how sanguinarine and chelerythrine
induce rapid apoptosis. Archives of Biochemistry and Biophysics, v. 477, n. 1, p.
43–52, 2008.
MCKAY, P. B. et al. Consensus Computational Ligand-Based Design for the
Identification of Novel Modulators of Human Estrogen Receptor Alpha. Molecular
Informatics, v. 31, n. 3-4, p. 246–258, 2012.
MCMURRY, J. Alcohols and phenols. In: Organic chemistry. 8. ed. Belmont:
Brooks/Cole, 2011a. p. 620–675.
MCMURRY, J. Reactions of alkyl halides: nucleophilic substitutions and eliminations.
In: Organic Chemistry. 8. ed. Belmont: Brooks/Cole, 2011b. p. 372–423.
MEHLEN, P.; PUISIEUX, A. Metastasis: a question of life or death. Nature Reviews
Cancer, v. 6, n. 6, p. 449–458, 2006.
MENA, S. et al. Glutathione and Bcl-2 targeting facilitates elimination by
chemoradiotherapy of human A375 melanoma xenografts overexpressing bcl-xl, bcl-
2, and mcl-1. Journal of Translational Medicine, v. 10, n. 1, p. 8, 2012.
MILLAR, A. W.; LYNCH, K. P. Rethinking clinical trials for cytostatic drugs. Nature
reviews. Cancer, v. 3, n. July, p. 540–545, 2003.
MINARINI, A. et al. Design, synthesis, and biological evaluation of pirenzepine
analogs bearing a 1,2-cyclohexanediamine and perhydroquinoxaline units in
exchange for the piperazine ring as antimuscarinics. Bioorganic & Medicinal
Chemistry, v. 16, n. 15, p. 7311–7320, 2008.
MONTANARI, C. A.; BOLZANI, V. DA S. Planejamento racional de fármacos

93
baseado em produtos naturais. Química Nova, v. 24, n. 1, p. 105–111, 2001.
MORICE, C.; WERMUTH, C. G. Ring Transformations. In Vivo, p. 343–362, 2008.
MUKHERJEE, P. et al. Targeting the BH3 domain mediated protein-protein
interaction of Bcl-Xl through virtual screening. Journal of Chemical Information and
Modeling, v. 50, p. 906–923, 2010.
MUNSHI, A.; HOBBS, M.; MEYN, R. E. Clonogenic cell survival assay. Methods in
molecular medicine, v. 110, p. 21–28, 2005.
NÉMATI, F. et al. Targeting Bcl-2/Bcl-XL Induces Antitumor Activity in Uveal
Melanoma Patient-Derived Xenografts. PLoS ONE, v. 9, n. 1, p. e80836, 2014.
NEUMEYER, J. L. History and Evolution of Medicinal Chemistry. In: Foye’s
Principles of Medicinal Chemistry. 7. ed. Baltimore: Lippincott Williams & Wilkins,
2013. p. 1–10.
NEWMAN, D. J.; CRAGG, G. M. Natural products as sources of new drugs over the
30 years from 1981 to 2010. Journal of Natural Products, v. 75, n. 3, p. 311–335,
2012.
NI, Z. et al. Natural Bcl-2 inhibitor (−)– gossypol induces protective autophagy via
reactive oxygen species–high mobility group box 1 pathway in Burkitt lymphoma.
Leukemia & Lymphoma, v. 54, n. 10, p. 2263–2268, 2013.
PATRICK, G. L. Anticancer agents. In: An introduction to medicinal chemistry. 4.
ed. New York: Oxford, 2009. p. 518–578.
PAULEKUHN, G. S.; DRESSMAN, J. B.; SAAL, C. Trends in active pharmaceutical
ingredient salt selection based on analysis of the Orange Book database. Journal of
medicinal chemistry, v. 50, n. 26, p. 6665–72, 2007.
PÉDEBOSCQ, S. et al. Cytotoxic and apoptotic effects of bortezomib and gefitinib
compared to alkylating agents on human glioblastoma cells. Journal of
experimental therapeutics & oncology, v. 7, n. 2, p. 99–111, 2008.
PELTER, A.; ROSSER, R. M.; MILLS, S. Reductive aminations of ketones and
aldehydes using borane–pyridine. J. Chem. Soc., Perkin Trans. 1, p. 717–720,
1984.
PENCIKOVA, K. et al. Investigation of sanguinarine and chelerythrine effects on
LPS-induced inflammatory gene expression in THP-1 cell line. Phytomedicine, v.
19, n. 10, p. 890–895, 2012.
PINACHO CRISÓSTOMO, F. R. et al. Design and synthesis of a simplified inhibitor
for XIAP-BIR3 domain. Bioorganic & Medicinal Chemistry Letters, v. 19, n. 22, p.
6413–6418, 2009.
PINE, S. H. The Eschweiler-Clark methylation of amines: An organic chemistry

94
experiment. Journal of Chemical Education, v. 45, n. 2, p. 118, 1968.
PINE, S. H.; SANCHEZ, B. L. Formic acid-formaldehyde methylation of amines. The
Journal of Organic Chemistry, v. 36, n. 6, p. 829–832, 1971.
PLACZEK, W. J. et al. A survey of the anti-apoptotic Bcl-2 subfamily expression in
cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer
therapy. Cell death & disease, v. 1, n. 5, p. e40, 2010.
PRADOS, M. D. et al. Phase-1 trial of gefitinib and temozolomide in patients with
malignant glioma: a North American brain tumor consortium study. Cancer
Chemother Pharmacol, v. 61, n. 6, p. 1059–1067, 2008.
RAGHAV, P. K.; VERMA, Y. K.; GANGENAHALLI, G. U. Peptide screening to
knockdown Bcl-2’s anti-apoptotic activity: implications in cancer treatment.
International journal of biological macromolecules, v. 50, n. 3, p. 796–814, 2012.
REGAN, C. K.; CRAIG, S. L.; BRAUMAN, J. I. Steric effects and solvent effects in
ionic reactions. Science, v. 295, n. March, p. 2245–2247, 2002.
RENAULT, T. T.; CHIPUK, J. E. Death upon a Kiss: Mitochondrial Outer Membrane
Composition and Organelle Communication Govern Sensitivity to BAK/BAX-
Dependent Apoptosis. Chemistry & Biology, v. 21, n. 1, p. 114–123, 2014.
RIXE, O.; FOJO, T. Is cell death a critical end point for anticancer therapies or is
cytostasis sufficient? Clinical Cancer Research, v. 13, n. 24, p. 7280–7287, 2007.
ROCHE, V. F. Cancer and chemotherapy. In: Foye’s Principles of Medicinal
Chemistry. 7. ed. Baltimore: Lippincott Williams & Wilkins, 2013. p. 1199–1266.
SALVATORE, R. N.; YOON, C. H.; JUNG, K. W. Synthesis of secondary amines.
Tetrahedron, v. 57, n. 37, p. 7785–7811, 2001.
SANT’ANNA, C. M. R. Molecular modeling methods in the study and design of
bioactive compounds: An introduction. Revista Virtual de Química, v. 1, n. 1, 2009.
SAVRY, A. et al. Bcl-2—Enhanced Efficacy of Microtubule-Targeting Chemotherapy
through Bim Overexpression: Implications for Cancer Treatment. Neoplasia, v. 15, n.
1, p. 49–IN17, 2013.
SAXENA, I.; BORAH, R.; SARMA, J. C. Reductive amination of aromatic aldehydes
and ketones with nickel boride. Journal of the Chemical Society, Perkin
Transactions 1, n. 4, p. 503–504, 2000.
SCHAFFHAUSEN, J. Advances in structure-based drug design. Trends in
Pharmacological Sciences, v. 33, n. 5, p. 223, 2012.
SCHELLENBERG, K. The synthesis of secondary and tertiary amines by borohydride
reduction. The Journal of Organic Chemistry, v. 28, n. 1961, p. 3259–3261, 1963.
SCHERLIES, R. The MTT assay as tool to evaluate and compare excipient toxicity in

95
vitro on respiratory epithelial cells. International Journal of Pharmaceutics, v. 411,
n. 1-2, p. 98–105, 2011.
SCHROEDER, G. M. et al. Pyrazole and pyrimidine phenylacylsulfonamides as dual
Bcl-2/Bcl-xL antagonists. Bioorganic and Medicinal Chemistry Letters, v. 22, n.
12, p. 3951–3956, 2012.
SCHWEIGERT, N.; ZEHNDER, A. J. B.; EGGEN, R. I. L. Chemical properties of
catechols and their molecular modes of toxic action in cells, from microorganisms to
mammals. Environmental Microbiology, v. 3, n. 2, p. 81–91, 2001.
SERAJUDDIN, A. T. M. Salt formation to improve drug solubility. Advanced Drug
Delivery Reviews, v. 59, n. 7, p. 603–616, 2007.
SHIM, J.; MACKERELL, JR., A. D. Computational ligand-based rational design: role
of conformational sampling and force fields in model development. MedChemComm,
v. 2, n. 5, p. 356, 2011.
SILVERMAN, R. B. Drug discovery, design and development. In: The organic
chemistry of drug design and drug action. Amsterdam: Elsevier Academic Press,
2004. p. 8–105.
SINEGOVSKAYA, L. M.; KEIKO, V. V; TROFIMOV, B. A. Rotational Isomerism of
Vinyl Ethers and Sulfides. Sulfur reports, v. 7, n. 5, p. 337–375, 1 dez. 1987.
SLANINOVÁ, I. et al. Screening of minor benzo(c)phenanthridine alkaloids for
antiproliferative and apoptotic activities. Pharmaceutical Biology, v. 45, n. 2, p.
131–139, 2007.
SMITH, M. B.; MARCH, J. Aliphatic substitution: nucleophilic and organometallic. In:
March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure.
6. ed. Nova Jersey: John Wiley & Sons, Inc, 2007. p. 425–656.
SOLOMONS, T. W. G. FRYHLE, C. B. Ionic reactions: nucleophilic substitution and
elimination reactions of alkyl halides. In: Organic Chemistry. 10. ed. Nova Jersey:
John Wiley & Sons, Inc, 2011. p. 230–284.
SOUERS, A. J. et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves
antitumor activity while sparing platelets. Nature Medicine, v. 19, n. 2, p. 202–208,
2013.
SUCHOMELOV, J. et al. Short communication HPLC quantification of seven
quaternary benzo [ c ] phenanthridine alkaloids in six species of the family
Papaveraceae r a. v. 44, p. 283–287, 2007.
SUGANUMA, M.; SAHA, A.; FUJIKI, H. New cancer treatment strategy using
combination of green tea catechins and anticancer drugs. Cancer science, v. 102, n.
2, p. 317–23, 2011.

96
SUN, Q.-L. et al. Comparative proteomic analysis of paclitaxel sensitive A549 lung
adenocarcinoma cell line and its resistant counterpart A549-Taxol. Journal of
cancer research and clinical oncology, v. 137, n. 3, p. 521–32, 2011.
TAVARES, L. C. QSAR: A abordagem de Hansch. Quimica Nova, v. 27, n. 4, p.
631–639, 2004.
TOROPOV, A. A et al. Calculation of molecular features with apparent impact on
both activity of mutagens and activity of anticancer agents. Anti-Cancer Agents
Med. Chem., v. 12, p. 807–817, 2012.
TSE, C. et al. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor.
Cancer Research, v. 68, n. 9, p. 3421–3428, 2008.
TYERS, M. et al. From genomics to proteomics. Nature, v. 422, n. 6928, p. 193–7,
2003.
ULUKAN, H.; SWAAN, P. W. Camptothecins: a review of their chemotherapeutic
potential. Drugs, v. 62, n. 14, p. 2039–2057, 2002.
VAN DER DEEN, M. et al. ATP-binding cassette (ABC) transporters in normal and
pathological lung. Respiratory research, v. 6, p. 59, 2005.
VERMEULEN, K.; VAN BOCKSTAELE, D. R.; BERNEMAN, Z. N. The cell cycle:a
review of regulation,deregulation and therapeutic targets in cancer. Cell
Proliferation, v. 36, n. 3, p. 131–149, 2003.
VOGELSTEIN, B.; KINZLER, K. W. Cancer genes and the pathways they control.
Nat Med, v. 10, n. 8, p. 789–799, 2004.
VOGLER, M. et al. Bcl-2 inhibitors: small molecules with a big impact on cancer
therapy. Cell Death and Differentiation, v. 16, n. 3, p. 360–367, 2009.
VOORTMAN, J. et al. TRAIL therapy in non-small cell lung cancer cells: sensitization
to death receptor-mediated apoptosis by proteasome inhibitor bortezomib. Molecular
Cancer Therapeutics, v. 6, n. 7, p. 2103–2112, 2007.
WAN, L.; PANTEL, K.; KANG, Y. Tumor metastasis: moving new biological insights
into the clinic. Nature Medicine, v. 19, n. 11, p. 1450–1464, 2013.
WANG, W. et al. The difference between multi-drug resistant cell line A549/Gem and
its parental cell A549. The Chinese-German Journal of Clinical Oncology, v. 7, n.
11, p. 615–619, 2008.
WEINER, D. B.; WILLIAMS, W. B. (EDS.). Chemical and structural approaches to
rational drug design. [s.l.] CRC Press, Inc, 1994.
WHEELER, S. E.; HOUK, K. N. Through-space effects of substituents dominate
molecular electrostatic potentials of substituted arenes. Journal of Chemical Theory
and Computation, v. 5, p. 2301–2312, 2009.

97
WHO, W. H. O. Treatment of cancer. Disponível em:
<http://www.who.int/cancer/treatment/en/>. Acesso em: 18 set. 2012.
WHO, W. H. O. Cancer. Disponível em:
<http://www.who.int/mediacentre/factsheets/fs297/en/>. Acesso em: 4 out. 2015.
WU, C. P.; HSIEH, C. H.; WU, Y. S. The emergence of drug transporter-mediated
multidrug resistance to cancer chemotherapy. Molecular Pharmaceutics, v. 8, n. 6,
p. 1996–2011, 2011.
WU, J. et al. Trichostatin A sensitizes cisplatin-resistant A549 cells to apoptosis by
up-regulating death-associated protein kinase. Acta pharmacologica Sinica, v. 31,
n. 1, p. 93–101, 2010.
XU, C.; BARCHET, T. M.; MAGER, D. E. Quantitative structure–property
relationships of camptothecins in humans. Cancer Chemotherapy and
Pharmacology, v. 65, n. 2, p. 325–333, 2010.
YAP, T. A.; WORKMAN, P. Exploiting the Cancer Genome: Strategies for the
Discovery and Clinical Development of Targeted Molecular Therapeutics. Annual
Review of Pharmacology and Toxicology, v. 52, n. 1, p. 549–573, 2012.
YU, R. K13 Activation of p38 and c-Jun N-terminal Kinase Pathways and Induction of
Apoptosis by Chelerythrine Do Not Require Inhibition of Protein Kinase C. Journal of
Biological Chemistry, v. 275, n. 13, p. 9612–9619, 2000.
YU, Y. et al. Green tea catechins: a fresh flavor to anticancer therapy. Apoptosis, v.
19, n. 1, p. 1–18, 2014.
ZHANG, G.; MUSGRAVE, C. B. Comparison of DFT methods for molecular orbital
eigenvalue calculations. Journal of Physical Chemistry A, v. 111, n. 8, p. 1554–
1561, 2007.
ZHANG, Y. H. et al. Chelerythrine and Sanguinarine Dock at Distinct Sites on BclXL
that are Not the Classic BH3 Binding Cleft. Journal of Molecular Biology, v. 364, n.
3, p. 536–549, 2006.
ZHANG, Z. et al. Chelerythrine chloride from Macleaya cordata induces growth
inhibition and apoptosis in human gastric cancer BGC-823 cells. Acta
Pharmaceutica Sinica B, v. 2, n. 5, p. 464–471, 2012.
ZHOU, H. et al. Structure-based design of potent Bcl-2/Bcl-xL inhibitors with strong in
vivo antitumor activity. Journal of Medicinal Chemistry, v. 55, p. 6149–6161, 2012.
ZHOU, W. et al. Design and optimization of hybrid of 2,4-diaminopyrimidine and
arylthiazole scaffold as anticancer cell proliferation and migration agents. European
Journal of Medicinal Chemistry, v. 96, p. 269–280, 2015.
ZHOU, X. et al. Gefitinib inhibits the proliferation of pancreatic cancer cells via cell

98
cycle arrest. Anatomical record (Hoboken, N.J. : 2007), v. 292, n. 8, p. 1122–7,
2009.
ZUO, G. Y. et al. Antibacterial alkaloids from Chelidonium majus Linn
(Papaveraceae) against clinical isolates of methicillin-resistant Staphylococcus
aureus. Journal of Pharmacy and Pharmaceutical Sciences, v. 11, n. 4, p. 90–94,
2008.

99
8 ANEXOS

100
ANEXO I
Espectros de RMN 1H e 13C
Cromatograma de HPLC
Análise Elementar

101
Cloridrato de (2H-1,3-benzodioxol-5-ilmetil)(benzil)amina
Espectro de RMN 1H do composto 2a (RPF800)
RMN 1H (300 MHz, DMSO-d6, δ = ppm): 9,60
(s, 2H, H-19,20), 7,56-7,53 (m, 2H, H-12,14),
7,46-7,40 (m, 3H, H-11,13,15), 7,18 (s, 1H, H-
6), 7,00 (dd, J1 = 8,0 Hz, J2 = 1,2 Hz, 1H, H-4),
6,95 (d, J = 7,9 Hz, 1H, H-3), 6,05 (s, 2H, H-
17), 4,09 (s, 2H, H-9), 4,05 (s, 2H, H-7)
DMSO
H2O-DMSO
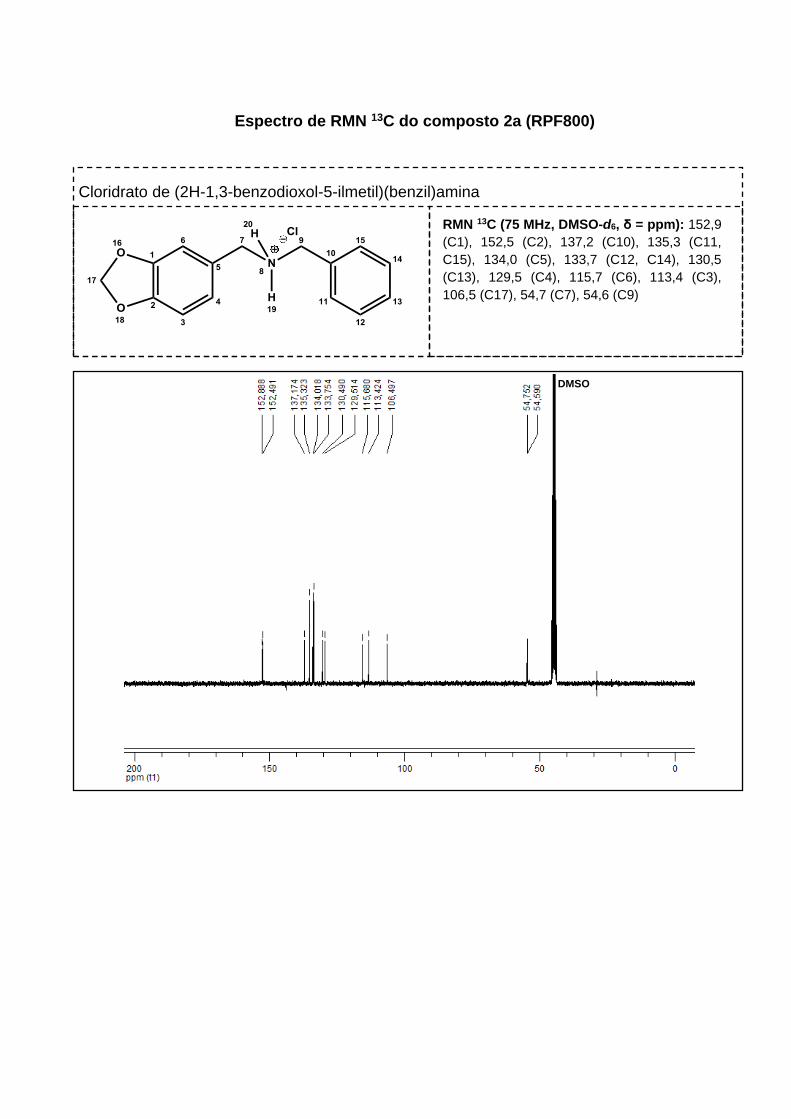
102
Cloridrato de (2H-1,3-benzodioxol-5-ilmetil)(benzil)amina
Espectro de RMN 13C do composto 2a (RPF800)
RMN 13C (75 MHz, DMSO-d6, δ = ppm): 152,9
(C1), 152,5 (C2), 137,2 (C10), 135,3 (C11,
C15), 134,0 (C5), 133,7 (C12, C14), 130,5
(C13), 129,5 (C4), 115,7 (C6), 113,4 (C3),
106,5 (C17), 54,7 (C7), 54,6 (C9)
DMSO

103

104

105

106
Cloridrato de (2H-1,3-benzodioxol-5-ilmetil)[(4-metoxifenil)metil]amina
Espectro de RMN 1H do composto 2b (RPF801)
RMN 1H (300 MHz, DMSO-d6, δ = ppm): 9,68
(s, 2H, H-21,22), 7,48 (d, J = 8,6 Hz, 2H, H-
11,15), 7,21 (s, 1H, H-6), 7,00 (dd, J1 = 8,0 Hz,
J2 = 1,3 Hz, 1H, H-3), 6,97 (s, 1H, H-4), 6,93
(d, J = 7,8, 2H, H-12,14), 6,04 (s, 2H, H-19),
4,00 (t, J = 5,7, 4H, H-7,9), 3,77 (s, 3H, H-17)
DMSO
H2O-DMSO

107
Cloridrato de (2H-1,3-benzodioxol-5-ilmetil)[(3,4-dimetoxifenil)metil]amina
Espectro de RMN 13C do composto 2b (RPF801)
RMN 13C (75 MHz, DMSO-d6, δ = ppm): 159,6
(C13), 147,6 (C1), 147,2 (C2), 131,7 (C11, C15),
125,3 (C5), 124,2 (C10), 123,7 (C4), 113,9 (C12,
C14), 110,4 (C6), 108,2 (C3), 101,2 (C19), 55,2
(C17), 49,2 (C7), 48,9 (C9)
DMSO

108

109

110

111
Cloridrato de (2H-1,3-benzodioxol-5-ilmetil)[(3,4-dimetoxifenil)metil]amina
Espectro de RMN 1H do composto 2c (RPF802)
RMN 1H (300 MHz, DMSO-d6, δ = ppm): 9,60
(s, 2H, H-22,23), 7,28 (s, 1H, H-18), 7,20 (s, 1H,
H-6), 7,04-6,93 (m, 4H, H-3,4,14,15), 6,04 (s,
2H, H-8), 4,00 (s, 4H, H-10,12), 3,77 (s, 3H, H-
21), 3,76 (s, 3H, H-22)
DMSO
H2O-DMSO

112
Cloridrato de (2H-1,3-benzodioxol-5-ilmetil)[(3,4-dimetoxifenil)metil]amina
Espectro de RMN 13C do composto 2c (RPF802)
RMN 13C (75 MHz, DMSO-d6, δ = ppm): 149,1
(C17), 148,6 (C1), 147,6 (C16), 147,2 (C2),
125,3 (C13), 124,2 (C5), 124,0 (C14), 122,7
(C4), 113,9 (C18), 111,5 (C6), 110,4 (C15),
108,1 (C3), 101,2 (C8), 55,6 (C21), 55,5 (C22),
49,2 (C10), 49,2 (C12)
DMSO

113

114

115

116
Cloridrato de (2H-1,3-benzodioxol-5-ilmetil)[(3,4,5-trimetoxifenil)metil]amina
Espectro de RMN 1H do composto 2d (RPF803)
RMN 1H (300 MHz, DMSO-d6, δ = ppm): 9,66
(s, 2H, H-24,25), 7,20 (s, 1H, H-6), 7,01 (d, J =
8,0 Hz, 1H, H-4), 6,96 (s, 1H, H-3), 6,94 (s, 2H,
H-11,15), 6,04 (s, 2H, H-23), 4,03-4,01 (m, 4H,
H-7,9), 3,79 (s, 6H, H-19,21), 3,66 (s, 3H, H-
20)
DMSO
H2O-DMSO

117
Cloridrato de (2H-1,3-benzodioxol-5-ilmetil)[(3,4,5-trimetoxifenil)metil]amina
Espectro de RMN 13C do composto 2d (RPF803)
RMN 13C (75 MHz, DMSO-d6, δ = ppm): 152,7
(C12, C14), 147,6 (C1), 147,3 (C2), 137,6
(C13), 127,3 (C10), 125,2 (C5), 124,3 (C4),
110,4 (C6), 108,2 (C3), 107,6 (C11, C15),
101,2 (C23), 60,0 (C20), 56,0 (C19, C21), 49,6
(C9), 49,4 (C7)
DMSO

118

119

120

121
Cloridrato de 4-{[(2H-1,3-benzodioxol-5-ilmetil)amino]metil}-2-metoxifenol
Espectro de RMN 1H do composto 2e (RPF804)
RMN 1H (300 MHz, DMSO-d6, δ = ppm): 9,50
(s, 2H, H-22,23), 9,22 (s, 1H, H-20), 7,21 (d, J
= 1,4 Hz, 1H, H-6), 7,18 (d, J = 1,4 Hz, 1H, H-
18), 7,00 (dd, J1 = 8,0 Hz, J2 = 1,3 Hz, 1H, H-
3), 6,94 (d, J = 7,9 Hz, 1H, H-4), 6,88 (dd, J1 =
8,0 Hz, J2 = 1,3 Hz, 1H, H-15), 6,79 (d, J = 8,0
Hz, 1H, H-14), 6,04 (s, 2H, H-8), 3,98 (t, J =
5,5 Hz, 4H, H-10,12), 3,78 (s, 3H, H-21)
DMSO
H2O-DMSO

122
Cloridrato de 4-{[(2H-1,3-benzodioxol-5-ilmetil)amino]metil}-2-metoxifenol
Espectro de RMN 13C do composto RPF804 (2e)
RMN 13C (75 MHz, DMSO-d6, δ = ppm): 147,6
(C1), 147,4 (C17), 147,2 (C2), 147,1 (C16),
125,4 (C13), 124,2 (C5), 122,9 (C14), 122,3
(C4), 115,2 (C15), 114,4 (C18), 110,4 (C6),
108,2 (C3), 101,2 (C8), 55,7 (C21), 49,4 (C12),
49,1 (C10)
DMSO

123

124

125

126
Cloridrato de 4-{[(2H-1,3-benzodioxol-5-ilmetil)amino]metil}benzeno-1,2-diol
Espectro de RMN 1H do composto 2f (RPF806)
RMN 1H (300 MHz, DMSO-d6, δ = ppm): 9,28
(s, 2H, H-11), 9,12 (s, 2H, H-19,20), 7,13 (s,
1H, H-6), 6,98 (d, J = 8,0 Hz, 1H, H-4), 6,93 (d,
J = 8,0 Hz, 1H, H-3), 6,89 (s, 1H, H-18), 6,76
(s, 2H, H-14,15), 6,03 (s, 2H, H-11), 3,99 (t, J =
6 Hz, 2H, H-10), 3,89 (t, J = 6 Hz, 2H, H-12)
DMSO
H2O-DMSO

127
Cloridrato de 4-{[(2H-1,3-benzodioxol-5-ilmetil)amino]metil}benzeno-1,2-diol
Espectro de RMN 13C do composto 2f (RPF806)
RMN 13C (75 MHz, DMSO-d6, δ = ppm): 147,6
(C1), 147,3 (C2), 146,0 (C17), 145,2 (C16),
125,2 (C13), 124,1 (C5), 122,2 (C14), 121,3
(C4), 117,5 (C18), 115,5 (C15), 110,2 (C6),
108,2 (C3), 101,3 (C8), 49,4 (C10), 49,3 (C12)
DMSO

128

129

130

131
Cloridrato de (2H-1,3-benzodioxol-5-ilmetil)(benzil)metilamina
Espectro de RMN 1H do composto 3a (RPF850)
RMN 1H (300 MHz, CDCl3, δ = ppm): 12,55 (s,
1H, H-20), 7,69-7,64 (m, 2H, H-12,14), 7,45-
7,43 (m, 3H, H-11,13,15), 7,23 (s, 1H, H-6),
7,09 (d, J = 7,8 Hz, 1H, H-4), 6,83 (d, J = 7,9
Hz, 1H, H-3), 5,99 (s, 2H, H-17), 4,30-4,08 (m,
4H, H-7,9), 2,59 (s, 3H, H-19)
TMS
CHCl3

132
Cloridrato de (2H-1,3-benzodioxol-5-ilmetil)(benzil)amina
Espectro de RMN 13C do composto 3a (RPF850)
RMN 13C (75 MHz, CDCl3, δ = ppm): 149,1
(C1), 148,4 (C2), 131,3 (C11, C15), 130,0
(C10), 129,4 (C12, C14), 128,8 (C5), 125,6
(C13), 122,0 (C4), 111,4 (C6), 108,8 (C3),
101,7 (C17), 59,0 (C7), 58,9 (C9), 38,5 (C19)
CHCl3

133

134

135

136
Cloridrato de (2H-1,3-benzodioxol-5-ilmetil)[(4-metoxifenil)metil]metilamina
Espectro de RMN 1H do composto 3b (RPF851)
RMN 1H (300 MHz, DMSO-d6, δ = ppm): 10,95
(s, 1H, H-22), 7,55 (s, 1H, H-15), 7,55 (s, 1H, H-
11), 7,28 (d, J = 1,1 Hz, 1H, H-6), 7,06 (dd, J1 =
8,0 Hz, J2 = 1,3 Hz, 1H, H-4), 7,01 (s, 1H, H-3),
6,96 (d, J = 7,8 Hz, 2H, H-12,14), 6,06 (s, 2H, H-
19), 4,32-4,25 (m, 2H, H-7), 4,12-4,04 (m, 2H,
H-9), 3,78 (s, 3H, H-17), 2,44 (d, J = 4,2 Hz, H-
21)
DMSO
H2O-DMSO
DMSO

137
Cloridrato de (2H-1,3-benzodioxol-5-ilmetil)[(4-metoxifenil)metil]metilamina
Espectro de RMN 13C do composto 3b (RPF851)
RMN 13C (75 MHz, DMSO-d6, δ = ppm): 159,9
(C13), 148,0 (C1), 147,4 (C2), 132,9 (C11,
C15), 125,5 (C5), 123,5 (C10), 121,8 (C4),
114,0 (C12, C14), 111,3 (C6), 108,3 (C3),
101,4 (C19), 57,7 (C7), 57,4 (C9), 55,2 (C17),
37,3 (C21)
DMSO

138

139

140

141
Cloridrato de (2H-1,3-benzodioxol-5-ilmetil)[(3,4-dimetoxifenil)metil]metilamina
Espectro de RMN 1H do composto 3c (RPF852)
RMN 1H (300 MHz, CDCl3, δ = ppm): 12,63 (s,
1H, H-24), 7,61 (s, 1H, H-18), 7,15 (s, 1H, H-
6), 7,04 (d, J = 7,9 Hz, 1H, H-14), 6,96 (d, J =
8,1 Hz, 1H, H-4), 6,85 (d, J = 7,8 Hz, 2H, H-
3,15), 6,01 (s, 2H, H-8), 4,25-4,17 (m, 2H, H-
10), 4,02-3,89 (m, 8H, H-12,21,22), 2,54 (d, J =
4,8 Hz, 3H, H-23)
TMS
CHCl3

142
Cloridrato de (2H-1,3-benzodioxol-5-ilmetil)[(3,4-dimetoxifenil)metil]metilamina
Espectro de RMN 13C do composto 3c (RPF852)
RMN 13C (75 MHz, CDCl3, δ = ppm): 150,3
(C17), 149,7 (C1), 149,1 (C16), 148,4 (C2),
125,6 (C13), 123,5 (C5), 121,9 (C14), 121,2
(C4), 114,1 (C18), 111,4 (C6), 111,0 (C15),
108,8 (C3), 101,6 (C8), 59,1 (C10), 58,9 (C12),
56,5 (C21), 55,9 (C22), 38,4 (C23)
CHCl3

143

144

145

146
Cloridrato de (2H-1,3-benzodioxol-5-ilmetil)[(3,4,5-trimetoxifenil)metil]metilamina
Espectro de RMN 1H do composto 3d (RPF853)
RMN 1H (300 MHz, CDCl3, δ = ppm): 12,63 (s,
1H, H-26), 7,16 (s, 1H, H-6), 7,05 (d, J = 8,2
Hz, 1H, H-4), 7,01 (s, 2H, H-11,15), 6,85 (d, J
= 7,9 Hz, 1H, H-3), 6,01 (s, 2H, H-23), 4,24-
4,16 (m, 2H, H-7), 4,10-4,01 (m, 2H, H-9), 3,92
(s, 6H, H-19,21), 3,86 (s, 3H, H-20), 2,59 (s,
3H, H-25)
TMS
CHCl3

147
Cloridrato de (2H-1,3-benzodioxol-5-ilmetil)[(3,4,5-trimetoxifenil)metil]metilamina
Espectro de RMN 13C do composto 3d (RPF853)
RMN 13C (75 MHz, CDCl3, δ = ppm): 153,7
(C12, C14), 149,2 (C1), 148,4 (C2), 139,1
(C13), 125,7 (C10), 124,4 (C5), 124,3 (C4),
121,7 (C6), 111,4 (C3), 108,8 (C15), 108,2
(C11), 101,7 (C23), 60,8 (C20), 59,4 (C7), 59,2
(C9), 56,7 (C19, C21), 38,6 (C25)
CHCl3

148

149

150

151
ANEXO II
Ficha da aluna

152

153

154

155
ANEXO III
Currículo Lattes

156

157

158


















