
UN MODÈLE EN PATHOLOGIE COMPARÉE - vetagro … a- Définition de l’Organisation Mondiale de la...
126
VETAGRO SUP CAMPUS VÉTÉRINAIRE DE LYON Année 2014 - Thèse n° 43 LE DIABÈTE SUCRÉ DU CHAT : UN MODÈLE EN PATHOLOGIE COMPARÉE ? THÈSE Présentée à l’UNIVERSITÉ CLAUDE-BERNARD - LYON I (Médecine - Pharmacie) et soutenue publiquement le 26 septembre 2014 pour obtenir le grade de Docteur Vétérinaire par Célia CHENIVESSE Née le 29 juillet 1991 à Nice (06)
Transcript of UN MODÈLE EN PATHOLOGIE COMPARÉE - vetagro … a- Définition de l’Organisation Mondiale de la...

VETAGRO SUP CAMPUS VÉTÉRINAIRE DE LYON
Année 2014 - Thèse n° 43
LE DIABÈTE SUCRÉ DU CHAT : UN MODÈLE EN PATHOLOGIE COMPARÉE ?
THÈSE
Présentée à l’UNIVERSITÉ CLAUDE-BERNARD - LYON I (Médecine - Pharmacie)
et soutenue publiquement le 26 septembre 2014 pour obtenir le grade de Docteur Vétérinaire
par
Célia CHENIVESSE Née le 29 juillet 1991
à Nice (06)

2

3

4

5
REMERCIEMENTS
A Monsieur le Professeur Charles THIVOLET,
De la faculté de Médecine de Lyon,
Qui nous a fait l’honneur d’accepter la présidence de ce jury de thèse,
Pour l’intérêt porté à notre travail,
Hommages respectueux.
A Monsieur le Professeur Jean-Luc CADORE,
De Vetagro Sup, Campus Vétérinaire de Lyon,
Pour avoir accepté d’encadrer notre travail,
Pour sa disponibilité sans faille et ses conseils avisés,
Pour sa gentillesse et sa compréhension dans les moments difficiles,
Qu’il trouve ici l’expression de notre respect le plus profond.
A Madame le Docteur Marine HUGONNARD,
De Vetagro Sup, Campus Vétérinaire de Lyon,
Pour avoir accepté de juger ce travail et de faire partie de ce jury de thèse,
Pour sa gentillesse et sa disponibilité,
Sincères remerciements.

6
A mes parents,
A ma famille,
A mes amis,
Merci pour tout.

7
TABLE DES MATIERES
REMERCIEMENTS ............................................................................................. 5
TABLE DES MATIERES .................................................................................... 7
TABLE DES TABLEAUX ................................................................................. 13
TABLE DES ABREVIATIONS ......................................................................... 15
INTRODUCTION ............................................................................................... 17
I) Le diabète sucré : définition, présentation et pathogénie ....................... 19
1) Définitions et rappels .................................................................................................... 19
a- Définition de l’Organisation Mondiale de la Santé (OMS) .................................. 19
b- Rappels anatomiques : le pancréas ........................................................................ 19
c- La glycémie et sa régulation ................................................................................... 21
i. Définitions et mesures .............................................................................................. 21
ii. Mécanismes et acteurs de la régulation .................................................................. 22
2) Epidémiologie et étiologie du diabète sucré ................................................................ 25
a- Des facteurs de risque : l’âge, le genre et la stérilisation ..................................... 28
i. Influence de l’âge ..................................................................................................... 28
ii. Influence du genre ................................................................................................... 29
iii. Rôle de la stérilisation ............................................................................................ 30
b- Des prédispositions génétiques ............................................................................... 30
c- Des facteurs environnementaux : rôle de l’obésité ............................................... 33
d- Maladies et facteurs iatrogéniques ......................................................................... 37
3) Pathogénie du diabète sucré ........................................................................................ 38
a- La résistance à l’insuline ......................................................................................... 38

8
b- Les dépôts amyloïdes ............................................................................................... 40
c- L’inflammation et la résistance à l’insuline .......................................................... 41
i. Les adipokines .......................................................................................................... 41
ii. Les adipocytokines .................................................................................................. 42
iii. Les médiateurs lipidiques de l’inflammation .......................................................... 43
d- L’inflammation et la fonction des cellules bêta ..................................................... 44
e- Cas particulier de la glucotoxicité réversible chez le chat ................................... 46
II) Le diabète sucré : démarche diagnostique et tableau clinique ................ 48
1) Une évolution à bas bruit ............................................................................................. 48
a. Le syndrome métabolique ....................................................................................... 48
b. Les signes cliniques .................................................................................................. 52
c. Méthodes diagnostiques .......................................................................................... 54
i. Démarche diagnostique ........................................................................................... 54
ii. Méthodes complémentaires ..................................................................................... 56
iii. Diagnostic étiologique ............................................................................................ 57
2) Les complications du diabète sucré ............................................................................. 58
a. Les complications microvasculaires ....................................................................... 60
i. Les rétinopathies diabétiques .................................................................................. 60
ii. Les néphropathies diabétiques ................................................................................ 60
iii. Les neuropathies diabétiques .................................................................................. 60
iv. Les affections des pieds liés au diabète ................................................................... 61
b. Les complications macrovasculaires ...................................................................... 61
i. Les maladies cardiovasculaires ............................................................................... 61
ii. Les maladies cérébrovasculaires ............................................................................ 64
iii. Les maladies vasculaires périphériques ................................................................. 64
3) La crise acidocétosique ................................................................................................ 65
a. Pathogénie ................................................................................................................ 65
b. Présentation clinique ............................................................................................... 66

9
c. Prise en charge ......................................................................................................... 66
III) Le diabète sucré : prise en charge et prévention ..................................... 69
1) L’insuline ...................................................................................................................... 73
a. Les analogues de l’insuline à action rapide ........................................................... 76
b. Les analogues de l’insuline à action intermédiaire à lente ................................... 77
i. L’insuline glargine ................................................................................................... 77
ii. L’insuline detemir ................................................................................................... 80
iii. Autres formulations ................................................................................................. 81
2) Les hypoglycémiants oraux .......................................................................................... 82
a. Hypoglycémiants à action dépendante du tractus gastro-intestinal ................... 83
b. Les inhibiteurs du co-transporteur 2 sodium-glucose .......................................... 84
c. Hypoglycémiants à action sur le système nerveux central ................................... 84
d. Nouvelles options thérapeutiques ........................................................................... 85
i. Chez l’homme .......................................................................................................... 85
ii. Chez le chat ............................................................................................................. 87
3) Intérêt des mesures diététiques .................................................................................... 89
a. Perte de poids et activité physique ......................................................................... 89
b. Intérêt de certaines complémentations alimentaires ............................................ 93
CONCLUSION ................................................................................................... 96
BIBLIOGRAPHIE .............................................................................................. 97

10

11
TABLE DES FIGURES
Figure 1 : Le pancréas chez le chat (HUDSON, et al., 1993) .................................................. 20
Figure 2 : Devenir du glucose sanguin .................................................................................... 23
Figure 3 : Homéostasie normale du glucose, à jeun et en post-prandial (NOLAN, et al., 2011) .................................................................................................................................................. 25
Figure 4 : Rôle des gènes et de l'environnement dans le développement de l'obésité et du diabète de type 2 (KAHN, et al., 2014) .................................................................................... 28
Figure 5 : Holter glycémique chez un chat (TRIBOULIN, 2010) ........................................... 57
Figure 6 : Estimations de Kaplan-Meier de la probabilité de décès suite à une maladie cardiovasculaire, chez 1059 sujets ayant un diabète de type 2 et 1378 sujets non diabétiques, avec et sans antécédent d'infarctus du myocarde (HAFFNER, et al., 1998) ............................ 63
Figure 7 : Molécules thérapeutiques pour le diabète de type 2 chez l'homme, évolution chronologique (KAHN, et al., 2014) ........................................................................................ 69
Figure 8 : Molécules thérapeutiques pour le diabète de type 2 chez l'homme, classement par organe-cible(KAHN, et al., 2014) ............................................................................................ 70
Figure 9 : Bypass gastrique Roux en-Y par laparoscopie : technique de l'université de Pittsburgh (SCHAUER, et al., 2003) ....................................................................................... 90

12

13
TABLE DES TABLEAUX
Tableau I : Effet de la race et de l'âge sur la prévalence du diabète mellitus dans les populations occidentales (CAMPBELL, 2000) ....................................................................... 26
Tableau II : Estimations du nombre d'adultes âgés de 20 à 79 ans atteints de diabète mellitus, et prévalence, par régions du monde, entre 2010 et 2030 (NOLAN, et al., 2011) ................... 27
Tableau III : Critères pour le diagnostic clinique du syndrome métabolique (ALBERTI, et al., 2009) ......................................................................................................................................... 49
Tableau IV : Recommandations actuelles, par organisme, des seuils de tour de taille pour l'obésité abdominale (ALBERTI, et al., 2009) ......................................................................... 51
Tableau V : Caractéristiques du diabète de type 2 chez l’homme (CAMPBELL, 2000) ........ 53
Tableau VI : Complications chroniques du diabète de type 2 chez l'homme (CAMPBELL, 2000) ......................................................................................................................................... 59
Tableau VII : Facteurs de risque pour les maladies cardiovasculaires relativement aux antécédents d'infarctus du myocarde, chez des sujets avec diabète de type 2 et non diabétiques (HAFFNER, et al., 1998) ......................................................................................................... 62
Tableau VIII : Taux d’affections cardiovasculaires lors d'un suivi sur 7 ans, relativement aux antécédents d'infarctus du myocarde, chez des sujets avec diabète de type 2 et non diabétiques (HAFFNER, et al., 1998) ......................................................................................................... 63
Tableau IX : Molécules injectables pour le traitement de l'hyperglycémie dans le diabète de type 2 chez l'homme (KAHN, et al., 2014) .............................................................................. 71
Tableau X : Molécules per os pour le traitement de l'hyperglycémie dans le diabète de type 2 chez l'homme (KAHN, et al., 2014) ......................................................................................... 72
Tableau XI : Exemples d'insulines pour les chats diabétiques (MARTIN, et al., 2000) .......... 75
Tableau XII: Paramètres de modifications du dosage d'insuline pour la glargine ou la zinc protamine chez les chats diabétiques (RAND, et al., 2005) ..................................................... 79
Tableau XIII : Cibles thérapeutiques et mécanismes non encore testés pour le diabète de type 2 (KAHN, et al., 2014) ............................................................................................................. 87

14

15
TABLE DES ABREVIATIONS
ADA : American Diabetes Association
AHA : American Heart Association
AMM : Autorisation de Mise sur le Marché
ATPIII : National Cholesterol Education Program Adult Treatment Panel III
BID : Bis In Die
DPP4: Dipeptidyl Peptidase 4
GH: Growth Hormone
GIP: Gastric Inhibitory Polypeptide
GLP-1: Glucagon-Like Peptide 1
GLUT4 : Glucose Transporter type 4
HbA1C: Hémoglobine glyquée A1C
HDL : High Density Lipoprotein
IDF : International Diabetes Federation
IKKß : I Kappa Kinase Bêta
IL-1ß : Interleukin 1 Bêta
IL-6 : Interleukin 6
IRS-1 : Insulin Receptor Substrate 1
IRS-2 : Insulin Receptor Substrate 2
JNK : c-Jun N terminal Kinase
IMC : Indice de Masse Corporelle

16
IRS: Insulin Receptor Substrate
LDL : Low Density Lipoprotein
LPS: LipoPolySaccharid
MCP-1: Monocyte Chemotactic Protein 1
NEC: Note d’Etat Corporel
NHLBI : National Heart, Lung and Blood Institute
NPH: Neutral Protamine Hagedorn
OMS : Organisation Mondiale de la Santé (WHO)
PZI : Protamine Zinc Insulin
SGLT2: Sodium Glucose co-Transporter 2
SID: Semel In Die
TLR4 : Toll Like Receptor 4
TNF-α: Tumor Necrosis Factor Alpha
UKPDS: United Kingdom Prospective Diabetes Study
WHO : World Health Organization (OMS)

17
INTRODUCTION
À la fin du XIXème siècle, la médecine comparée apparaît aux yeux de plusieurs
scientifiques comme une branche futuriste et probablement essentielle de la discipline. En
effet, pour Rush et Virchow, comprendre et comparer les mécanismes à l’origine des
fonctionnements et dysfonctionnements des hommes et des animaux représente l’avenir de la
science. C’est la notion d’ « une seule médecine », profitable à l’espèce humaine autant
qu’aux espèces animales qui lui seraient confrontées.
Si cette piste reste finalement très peu exploitée au XXème siècle, et se cantonne aux
modèles que représentent les animaux de laboratoire, les progrès de la génétique permettent à
la science clinique comparative de prendre véritablement son essor dès le XXIème siècle.
L’héritage génétique que partage l’Homme avec l’animal, qu’il s’agisse du chien, du chat, ou
du singe, permet une approche nouvelle et approfondie de pathologies aussi diverses que
l’hypertension artérielle, les diarrhées, ou encore le diabète de type 2 (MICHELL, 2005).
En trouvant le modèle animal qui, dans sa pathophysiologie, se rapproche le plus de
l’homme pour une affection en particulier, il est d’abord possible de s’affranchir des limites
éthiques liées aux expérimentations sur les animaux de laboratoire. D’autre part, la
compréhension des mécanismes sous-jacents à cette pathologie, depuis l’étiologie jusqu’au
traitement en passant par la pathogénie, sera facilitée, plus fiable, et par la suite bénéfique
pour les deux espèces souffrant la comparaison.
Dans notre étude, nous nous intéresserons au diabète sucré de type 2, et plus
particulièrement à la comparaison en tous points de cette grande affection endocrinienne chez
le chat et chez l’homme. En effet, si l’espèce féline a été décrite par plusieurs auteurs comme
étant particulièrement proche de l’espèce humaine dans le développement de cette maladie,
nous chercheront à détailler en quoi ses caractéristiques permettent ou non de la définir
comme un modèle en pathologie comparée.
Le diabète sucré de type 2 a pour origine l’incapacité relative du pancréas à répondre à
des demandes métaboliques augmentées et à compenser une résistance à l’insuline. La gravité
de l’intolérance au glucose dépendra alors de ces deux capacités pancréatiques. En effet, si la
sécrétion d’insuline est insuffisante, une hyperglycémie s’installe, corrélée à une hyper
insulinémie. Dès lors, la résistance à l’insuline s’aggrave, avec une sécrétion d’insuline ne

18
pouvant la compenser. L’incapacité fonctionnelle conséquente des cellules pancréatiques
conduit au développement de ce diabète, aussi appelé non insulinodépendant (SPELLMAN,
2010).
C’est une affection dont la prévalence ne cesse d’augmenter au cours de notre siècle,
aussi bien dans la population humaine que dans la population féline, atteignant aujourd’hui
une proportion très importante d’individus.
C’est pourquoi il semble cohérent, à notre époque, de se pencher sur la pertinence de
l’étude du diabète sucré du chat en médecine comparée.
Dans un premier temps nous nous intéresserons à la présentation et à la pathogénie du
diabète de type 2 au sein de ces deux espèces. Puis, nous comparerons les aspects
diagnostiques et cliniques de cette maladie chez le chat et chez l’homme.
La dernière partie de ce travail portera sur la prévention et le traitement du diabète sucré, tant
au niveau de sa prise en charge actuelle, que des perspectives d’avenir que pourrait justement
nous offrir la médecine comparée, pour une espèce comme pour l’autre.

19
I) Le diabète sucré : définition, présentation et pathogénie 1) Définitions et rappels
a- Définition de l’Organisation Mondiale de la Santé (OMS)
D’après l’Organisation Mondiale de la Santé,
« Le diabète sucré est un groupe de maladies métaboliques caractérisées par une
hyperglycémie chronique résultant d’un défaut de sécrétion ou d’action de l’insuline ou de ces
deux anomalies associées Le diabète sucré provoque chez l’homme de graves lésions
affectant de nombreuses parties du corps, en particulier les nerfs et les vaisseaux sanguins. »
(WORLD HEALTH ORGANIZATION, 2006).
Dans le cadre de notre étude, nous considérerons le diabète sucré de type 2, tel qu’il
est décrit chez l’homme, ou diabète non insulinodépendant, ou encore diabète « gras ». Il
s’agit d’un diabète acquis, conséquence d’une anomalie de sécrétion de l’insuline et d’une
résistance à l’insuline ; en opposition au diabète de type 1, d’origine génétique, et
correspondant à une véritable carence insulinique.
En effet, dans l’espèce féline, le diabète sucré apparenté au diabète de type 1, donc
insulinodépendant stricto sensu, est rarissime. C’est pourquoi nous mènerons la comparaison
entre cette endocrinopathie chez l’homme et chez le chat en se basant sur ce qui est décrit
comme diabète sucré de type 2 dans l’espèce humaine.
Il faut noter que l’on peut rencontrer un dernier type de diabète sucré, soit le diabète de
type 3, aussi appelé diabète gestationnel. Ce dernier est décrit chez le chien, plus rarement
chez le chat, et en moindre proportion chez la femme.
b- Rappels anatomiques : le pancréas
Le pancréas est une glande lobulaire, située dans le cadrant crânial droit de la cavité
abdominale, en rapport étroit avec le duodénum. Sa forme varie d’une espèce à l’autre car il
se moule sur les organes voisins. Cependant, chez l’homme comme chez le chat, il est
constitué de trois parties : le corps, situé en partie moyenne, et les deux extrémités - ou lobes-
gauche et droit (fig. 1). Le lobe droit correspond à la tête du pancréas, tandis que le lobe

20
gauche représente la queue de cet organe. C’est au niveau de la tête que s’individualisent les
canaux excréteurs du pancréas.
Figure 1 : Le pancréas chez le chat, d'après HUDSON, et al., 1993
Le pancréas est constitué de deux parties sécrétoires : une partie exocrine à l’origine
de la sécrétion d’enzymes pancréatiques, et une partie endocrine.
La glande exocrine représente le volume le plus important du pancréas, s’organisant
en lobules pancréatiques ou acini. Ces derniers produisent le suc pancréatique, déversé par les
canaux sécréteurs dans le duodénum.
Au sein des acini, on retrouve la partie exocrine, soient des cellules organisées en
groupes appelés îlots pancréatiques ou îlots de Langerhans, représentant 2 à 3% de la masse
cellulaire pancréatique totale. C’est cette partie qui est responsable de la sécrétion des
hormones régulant la glycémie, donc qui nous intéresse particulièrement dans cette étude. Ces
hormones se retrouvent dans le milieu interstitiel et diffusent ensuite grâce au large réseau de
capillaires traversant le pancréas. Puis, elles sont transportées jusqu’au foie via la veine cave,
et enfin libérées dans le reste de l’organisme.

21
Des différences morphologiques et fonctionnelles permettent de distinguer les populations
cellulaires qui les constituent.
Les cellules α représentent 20 à 30% des îlots, se situent à leur périphérie et
synthétisent le glucagon, hormone hyperglycémiante.
Les cellules ß sont majoritaires, environ 60 à 80% du pancréas exocrine, localisées au
centre des îlots, et sécrétant l’insuline, hormone hypoglycémiante.
Les cellules δ, soit 10% de la glande exocrine, sont situées aléatoirement entre les
deux types cellulaires et sécrètent la somatostatine, à l’origine de l’inhibition de la sécrétion
de glucagon et d’insuline.
On distingue également des cellules γ, subdivisées en cellules PP et D. Le sous-type
PP, que l’on retrouve dans le lobe droit et le corps du pancréas, produit le peptide
pancréatique, dont le rôle est encore peu connu, mais a priori en rapport avec le contrôle de la
satiété. Les cellules D sécrètent le peptide vasoactif intestinal.
Enfin, on retrouve, en faible nombre, des cellules EE ou entérochromaffines, à
l’origine de la sécrétion de sérotonine, motiline et substance P (VENEL, 2009 ; TRIBOULIN,
2010).
Dans le cadre de l’étude du diabète sucré, les trois premiers types cellulaires décrits
seront ceux à considérer en priorité.
c- La glycémie et sa régulation
i. Définitions et mesures
La glycémie correspond à la concentration de glucose sanguin.
Son dosage peut s’effectuer grâce à différentes techniques, chimiques ou enzymatiques,
sur plasma. Certains appareils miniaturisés portatifs permettent des mesures rapides de la
glycémie à l’aide d’une seule goutte de sang. Bien qu’ils soient très intéressants dans la
pratique, il faudra veiller à vérifier la compatibilité de ces machines, issues généralement
de la médecine humaine, avec l’usage vétérinaire.
Dans de nombreuses espèces, dont l’espèce humaine et l’espèce féline, la glycémie
normale correspond à une valeur d’environ 1 g/L, soit 5,5 mmol/L.
Cependant, cette valeur s’échelonne sur une fourchette plus importante chez le chat que
chez l’homme. De plus, une particularité féline est l’influence majeure et instantanée du

22
stress sur la concentration en glucose, qui va augmenter très rapidement dans de telles
conditions. C’est pourquoi les objectifs de la thérapie dans le cadre du diabète sucré de
type 2 ne seront pas les mêmes pour les deux espèces étudiées en ce qui concerne la
normalisation de la glycémie.
On définit la glucosurie (ou glycosurie) comme la présence de glucose dans les urines.
Cette dernière est anormale et on l’observe lors de l’élévation de la glycémie. Cette valeur
dépasse le seuil rénal et le glucose est alors éliminé dans les urines.
On peut également la détecter grâce à un test rapide, une bandelette urinaire, cependant
peu informative d’un point de vue quantitatif.
A nouveau, sa présence chez le chat n’est pas diagnostique d’une hyperglycémie
chronique, ni forcément pathologique, puisque l’on décrit chez cette espèce une
hyperglycémie de stress, qui est transitoire.
ii. Mécanismes et acteurs de la régulation
La glycémie est un paramètre très contrôlé par l’organisme (fig. 2).
Tout d’abord, le glucose est apporté par l’alimentation, puis il est distribué dans l’organisme
suite à son passage dans le sang : le foie, les muscles et le tissu adipeux notamment. Le
stockage se fait sous deux formes : le glycogène et les triglycérides.
D’autres tissus, au contraire, ne peuvent stocker le glucose, et leur besoin en celui-ci est élevé,
ils sont donc très sensibles aux variations de la glycémie. Il s’agit du système nerveux central,
de la médulla rénale et des globules rouges.
Lors de chute de la glycémie, trois mécanismes se mettent en place afin de libérer du
glucose dans la circulation : la glycolyse, la néoglucogenèse et la lipolyse.

23
Figure 2: Devenir du glucose sanguin
Physiologiquement, la glycémie doit correspondre à des valeurs comprises entre 3 et 8
mmol/L.
En-dessous de 3 mmol/L, on parle d’hypoglycémie, et au-delà de 8 mmol/L, l’individu est en
hyperglycémie.
Parmi les facteurs de régulation physiologique de la glycémie, on distingue deux
catégories de molécules : l’insuline, et les hormones hyperglycémiantes.
L’insuline représente la seule hormone hypoglycémiante de l’organisme. Elle est
synthétisée par le pancréas et permet d’empêcher la persistance de l’hyperglycémie
secondaire à la prise alimentaire. Le passage du glucose dans les tissus périphériques est ainsi
favorisé, pour une utilisation immédiate, ou pour le stockage. De plus, l’insuline inhibe la
néoglucogenèse, la libération d’acides aminés par les muscles ainsi que la formation de corps
cétoniques. L’hyperglycémie survient donc lorsqu’il y a un déficit total ou fonctionnel
d’insuline. Ceci se produit lorsque le glucose issu de l’alimentation ou de la néoglucogenèse
s’accumule dans le sang, ou lors de chute de la sécrétion d’insuline, ou encore si son
utilisation par les tissus est moindre.
D’autre part, on retrouve les hormones hyperglycémiantes, qui sont au contraire
beaucoup plus diversifiées.

24
Elles comprennent le glucagon, synthétisé par les cellules α des îlots de Langerhans ; la
somatostatine, produite par les cellules δ pancréatiques, qui inhibe la synthèse de glucagon et
d’insuline ; les catécholamines ; l’hormone de croissance GH (growth hormone) et enfin les
hormones sexuelles.
C’est la sécrétion de ces molécules qui est principalement à l’origine de l’hyperglycémie.
À jeun, la concentration sanguine en glucose est déterminée par l’équilibre entre la
production endogène de glucose, principalement via la glycogénolyse et la gluconéogenèse
hépatiques, et son utilisation par les tissus indépendant de l’insuline, comme le cerveau. Cette
endo-production prévient l’hypoglycémie, ajoutée à un faible ratio plasmatique insuline sur
glucagon.
Si le cerveau dépend énergétiquement du glucose, d’autres tissus, comme le cœur ou les
muscles squelettiques, s’approvisionnent majoritairement en nutriments autres que le glucose,
c’est-à-dire les acides gras non estérifiés provenant de la lipolyse du tissu adipeux.
Après un repas, notamment s’il contient des hydrates de carbone, la glycémie
augmente suite à l’absorption de nourriture, ce qui stimule la sécrétion d’insuline par les
cellules ß et inhibe celle du glucagon par les cellules α. La production endogène de glucose
est alors inhibée, et son exploitation par les tissus périphériques sensibles à l’insuline, tels que
le cœur, les muscles squelettiques, et le tissu adipeux, est activée.
Ces mécanismes entraînent le relargage des hormones incrétines, en particulier GLP-1
(glucagon-like peptide 1), qui amplifient la sécrétion d’insuline stimulée par le glucose et
l’inhibition glucidique de celle du glucagon. La lipolyse des tissus adipeux est arrêtée et
l’anabolisme est favorisé.
La glycémie revient dans les valeurs du jeûne au bout de 2 heures (fig. 3) (NOLAN, et al.,
2011).

25
Figure 3 : Homéostasie normale du glucose, à jeun et en postprandial, d'après NOLAN, et al., 2011
2) Epidémiologie et étiologie du diabète sucré
Le patient type présentant un diabète sucré possède des caractéristiques très similaires
chez l’homme et le chat. En effet, on retrouve en général un individu d’âge moyen, mâle et
très souvent obèse.
Le diabète mellitus de type 2 représente entre 75 et 90% des cas de diabète chez
l’homme selon les populations.

26
La prévalence dans les pays occidentaux est estimée à 2 à 6 %, dont seulement la moitié serait
diagnostiquée. Ce chiffre est cependant à revoir à la hausse chez les individus plus âgés et non
blancs. Ainsi, au-delà de 65 ans, on considère que 10 à 20 % des personnes sont affectées.
Quant aux populations adultes non blanches vivant dans des pays occidentaux, elles sont
touchées à hauteur de 10 à 40 % par le diabète de type 2 (tab I) (CAMPBELL, 2000).
La tranche d’âge la plus touchée dans les pays en voie de développement correspond à
la classe des travailleurs, soit 40 à 60 ans ; alors que le diabète de type 2 affecte
prioritairement les individus âgés de plus de 60 ans dans les pays développés (SHAW, et al.,
2010).
Tableau I : Effet de la race et de l'âge sur la prévalence du diabète mellitus dans les populations occidentales, d’après CAMPBELL, 2000
On estimait le nombre de patients souffrant de diabète mellitus de type 2 à travers le
monde, en l’an 2000, à 157 millions (AMOS, 1997). En 2009, cette estimation grimpait à 285
millions de personnes avec un chiffre devant atteindre 438 millions en 2030, soit 7,8% de la
population adulte (tab II) (IDF, 2009).
Les prédictions n’envisagent qu’une croissance très importante de cette maladie, compte tenu
du changement de mode de vie, de plus en plus sédentaire et incluant la consommation de
graisses saturées en grand nombre ; et de l’allongement de la durée de vie.
L’espérance de vie d’un patient diabétique de type 2 est réduite de 30 à 40 %, soit 10 ans de
vie en moins pour la tranche d’âge des 40-70 ans (DUNCAN, 1992).

27
Tableau II : Estimations du nombre d'adultes âgés de 20 à 79 ans atteints de diabète mellitus, et prévalence, par régions du monde, entre 2010 et 2030, d'après NOLAN, et al., 2011
Chez les chats et les chiens, le diabète mellitus est l’endocrinopathie la plus
diagnostiquée.
Le diabète de type 2 touche principalement les félins âgés de 6 à 10 ans et les animaux
obèses représentent les principales victimes de la maladie (COURTIN-DONAS, 2011).
Selon les populations étudiées, la prévalence du diabète chez les chats varie de 1 pour
50 à 1 pour 400 (BARAL, et al., 2003). On constate que cette prévalence est plus faible chez
l’espèce féline par rapport à l’espèce humaine. Cependant, les chiffres sont probablement
sous-évalués dans la mesure où les critères diagnostiques ne sont pas les mêmes chez le chat
(RAND, et al., 2001). Si chez l’homme un patient est considéré diabétique lorsque sa
glycémie est supérieure ou égale à 7 mmol/L, on diagnostique cette maladie chez le chat
lorsque des symptômes apparaissent. Or il est décrit que cet état clinique survient lorsque la
concentration sanguine en glucose dépasse le seuil rénal, soit environ 16 mmol/L chez les
chats sains (KRUTH, et al., 1982 ; RAND, et al., 2004).
Dans l’espèce féline, tout comme chez l’homme, et contrairement à l’espèce canine, c’est le
diabète de type 2 qui est le plus représenté (RAND, et al., 2004).

28
Ainsi, en considérant les découvertes histologiques et les signes cliniques, le diabète de type 2
représente 80 à 95% des diabètes félins (RAND, 1999).
Le développement du diabète est multifactoriel (fig. 4). On sait que la prédisposition
génétique existe et joue un rôle important, malgré tout, ce sont la mauvaise alimentation, le
manque d’activité physique et l’obésité qui, chez l’homme, représentent les principaux
facteurs de risque (LI, et al., 2011). Chez le chat également, l’étiologie est complexe et
plurielle, incluant des facteurs génétiques, mais aussi des interactions environnementales
(RAND, et al., 2004).
Figure 4: Rôle des gènes et de l'environnement dans le développement de l'obésité et du diabète de type 2, d’après KAHN, et al., 2014
a- Des facteurs de risque : l’âge, le genre et la stérilisation
i. Influence de l’âge
D’après (HENSON, et al., 2006), le diabète sucré de type 2 se déclare chez des chats
âgés de plus de 6 ans en moyenne, tandis que l’on retrouve une moyenne plus élevée à 8 ans
dans une étude de 2003 (BARAL, et al., 2003), avec un pic d’incidence entre 10 et 13 ans.
Chez les chats Birmans, l’incidence du diabète de type 2 passe de 1 pour 50 dans la
population totale à 1 pour 10 chez les individus âgés de plus de 8 ans.

29
Chez l’humain également, plus l’âge avance, plus le risque de développer cette
maladie est important. En effet, la fonction des cellules ß pancréatiques se détériore au fil des
années, ce qui rend plus probable l’apparition de l’endocrinopathie (CHIU, et al., 2000).
ii. Influence du genre
Dans l’espèce humaine, ce sont les hommes qui sont le plus touchés par le diabète
non-insulinodépendant. Ce fait épidémiologique est très fortement corrélé à la prédisposition
de ce sexe à développer de l’obésité, et plus particulièrement une obésité centrale, par rapport
à la femme chez qui les graisses se répartissent en périphérie. Cet élément est un facteur de
risque clairement mis en évidence pour le diabète sucré (cf I-2-c) (GEER, et al., 2009 ;
KISSEBAH, 1996).
Chez le chat, ce sont également les individus de sexe mâle qui présentent un plus fort
risque de développer un diabète de type 2 par rapport aux femelles (RAND, et al., 2004).
On attribue ce fait épidémiologique à deux facteurs.
Tout d’abord les chats mâles ont de manière générale des valeurs plus basses de
sensibilité à l’insuline par rapport aux femelles lorsqu’ils présentent un poids normal. Ce
chiffre est de 37% inférieur et se détériore encore lors de la prise de poids (APPLETON, et
al., 2001). De même, les chats mâles obèses ont tendance à présenter une sensibilité à
l’insuline moins importante que les femelles. De plus, seuls les mâles ont des concentrations
basales d’insuline augmentées de façon significative après la prise de poids, et ces taux en
valeur absolue sont plus élevés de manière générale que chez les chats de sexe féminin.
D’autre part, les chats mâles sont prédisposés à l’obésité (WALKER, et al., 1977 ;
ROBERTSON, 1999). Dans une étude de 2001 (cf. I-1-c), des chats avaient libre accès à la
nourriture durant environ 10 mois. Les mâles ont alors pris plus de poids que les femelles
(54% du poids corporel par rapport à 39%) et étaient plus lourds que celles-ci de manière
significative (7,26 kg contre 5.69 kg) alors que les poids initiaux étaient comparables
(respectivement 4,78 kg et 4,12 kg). La masse graisseuse des chats mâles s’est aussi avérée
significativement plus importante que celle des femelles (3,2 kg contre 2,3 kg) avec 4,1 kg
contre 3,4 kg de masse corporelle maigre. Il a alors été également montré que plus la masse
grasse était importante, moins l’insuline était efficace pour réduire le taux de glucose
plasmatique (APPLETON, et al., 2001).

30
Ces découvertes concernant une sensibilité à l’insuline moindre et des concentrations plus
élevées d’insuline chez les chats mâles expliquent pourquoi ce sexe présente un risque plus
élevé de développer de l’obésité et un diabète de type 2 par rapport au sexe féminin
(PANCIERA, et al., 1990).
iii. Rôle de la stérilisation
Ce paramètre est logiquement plus documenté chez le chat que chez l’homme.
Ainsi, il a été démontré que la stérilisation est un des facteurs de risque majeurs pour l’obésité
féline en induisant un changement dans le métabolisme basal. L’obésité étant elle-même une
cause prédisposante au diabète sucré (cf. I-2-c), la cascade logique qui en découle prédispose
les individus de l’espèce féline stérilisés à cette maladie.
Les mâles comme les femelles augmentent leur prise alimentaire et donc leur masse
corporelle suite à la stérilisation. La suppression des hormones sexuelles a une influence sur
les centres cérébraux contrôlant la prise alimentaire et le taux métabolique, d’où une
augmentation de la prise énergétique et une balance énergétique positive (KANCHUK, et al.,
2003 ; MARTIN, et al., 2006).
Une étude sur des rongeurs (GEARY, et al., 1994) en 1994 a montré que la stérilisation
lève l’inhibition œstrogénique pour la prise alimentaire chez les femelles, cela pouvant être
contrecarré par la mise en place d’un traitement à base d’œstradiol.
En 2007, une étude a prouvé qu’une administration quotidienne d’œstradiol empêche
l’augmentation de la prise alimentaire suite à la stérilisation, aussi bien chez les chats obèses
mâles que femelles (CAVE, et al., 2007).
b- Des prédispositions génétiques
Une prédisposition génétique a clairement été mise en évidence chez l’homme pour le
diabète de type 2. En effet, on a pu associer la défaillance des cellules des îlots pancréatiques
à l’expression de plusieurs variants géniques, notamment ceux codant pour des facteurs de
transcription, des enzymes impliquées dans le métabolisme du glucose, et des protéines et
molécules chaperonnes de l’insuline (SAXENA, et al., 2007).
D’autre part, l’appartenance ethnique est un facteur de risque chez l’homme. Comme
nous l’avons décrit précédemment, on retrouve une très forte prévalence du diabète non
insulinodépendant dans certaines populations indigènes, comme les Indiens d’Amérique, les

31
Aborigènes australiens et les Insulaires du Pacifique. Cette prédisposition génétique est
exacerbée lorsqu’elle est combinée à un mode de vie occidental (RAVUSSIN, et al., 1994).
Malgré le fait que le diabète de type 2 soit fortement héréditaire, on ne peut expliquer la
majorité des cas de cette maladie exclusivement par ces facteurs génétiques.
Dans l’espèce féline, on suspecte également une prédisposition génétique. Cette notion
est supportée en grande partie par la prédisposition raciale des Birmans, chez qui 1 chat sur
50 est atteint de diabète de type 2 en Australie (alors que la fréquence est en moyenne d’1
pour 200 chez les chats européens) (HENSON, et al., 2006 ; BARAL, et al., 2003).
Dans certaines familles de chats Birmans, plus de 10% de la portée est affectée par ce type de
diabète.
On ne connaît pas encore précisément les facteurs génétiques qui prédisposent les
chats au diabète. Cependant on sait que chez les Birmans, l’hérédité n’est ni liée au sexe, ni
dominante (WADE, et al., 1999).
Une étude de 2010 a montré l’existence d’une mutation dans la séquence codant pour
le récepteur 4 de la mélanocortine (MC4R) dans le génome des chats. Or, on sait que chez
l’homme, un polymorphisme dans une zone similaire à celle du MC4R prédispose à l’obésité.
Le polymorphisme du gène MC4R pourrait de ce fait être un important facteur de
prédisposition au développement du diabète et/ou de l’obésité chez les chats (FORCADA, et
al., 2010).
En 2003, une étude a conduit à procéder à l’examen histologique d’un panel de tissus
pancréatiques issus d’individus appartenant à différentes races félines. Les analyses mettent
en évidence les mêmes pathologies touchant les îlots pancréatiques, qu’il s’agisse
d’échantillons provenant de chats diabétiques européens ou de chats diabétiques Birmans. On
peut en déduire que la mutation spécifique à la race birmane n’entraîne pas directement un
dépôt excessif de polypeptide amyloïde ou de tout autre produit métabolique à l’origine de la
destruction des cellules ß. Ces données, bien que peu nombreuses, nous orienteraient plutôt
vers des gènes ayant une action sur la sensibilité à l’insuline (RAND, et al., 2004).
A l’inverse, on constate que d’autres races pures félines sont sous-représentées dans
l’expression de cette maladie, corroborant l’hypothèse d’un support génétique au diabète de
type 2.

32
Une prédisposition génétique est également suspectée mais non démontrée chez le chat en
ce qui concerne la résistance à l’insuline, comme cela a été avéré dans l’espèce humaine
(RAND, et al., 2004). En effet, chez l’homme, comme chez le chat, il existe un seuil de
sensibilité à l’insuline, qui, lorsqu’il est bas, augmente le risque de développer une intolérance
au glucose lors de la prise de poids chez des individus minces (cf. I-2-a). Chez l’homme, on
sait que cette résistance à l’insuline est déterminée génétiquement et aggravée par des facteurs
environnementaux (PORTE, 1991 ; HARRIS, 1995).
Depuis de nombreuses années, une théorie évolutionniste a été développée pour expliquer
les fondements du diabète de type 2. La résistance à l’insuline sous-jacente chez certains
individus de l’espèce humaine -ou faible sensibilité à l’insuline-, serait associée à un gène,
appelé « thrifty gene » ou littéralement « gène économe ». Ce gène se serait exprimé
originellement afin de permettre aux chasseurs-cueilleurs de rentabiliser la nourriture qu’ils
parvenaient à se procurer. Cette théorie repose sur le fait que la résistance sélective à
l’abaissement du taux de glucose, et non pas à la formation de graisse par l’insuline, aurait
facilité la conversion efficace de l’énergie en gras lorsque la nourriture était abondante. A
l’inverse, quand les nutriments se faisaient rares, la réserve graisseuse était utilisée et la
résistance à l’insuline permettait de maintenir une glycémie normale (NEEL, 1962).
La théorie de la « carnivore connection » ou « connexion carnivore » développe l’idée
selon laquelle la résistance aux effets de l’insuline pour diminuer le taux de glucose s’est
développée au cours de l’Âge de Glace, pour maintenir l’euglycémie avec un régime
alimentaire riche en protéines et pauvre en glucides (BRAND MILLER, et al., 1994).
Cependant, une telle capacité s’avère au contraire délétère lorsqu’elle est associée à un
mode de vie aisé et abondant, tel que la plupart des hommes le connaissent à l’époque
actuelle. En effet, cet héritage de résistance à l’insuline associée à des facteurs de risque
environnementaux, comme le manque d’activité physique et la consommation en trop grande
quantité de glucides raffinés et hautement digestibles (cf. I-1-c), exige une sécrétion
importante et prolongée d’insuline supplémentaire par les cellules ß. Ce mécanisme aboutit à
l’épuisement de ces cellules, puis finalement au diabète de type 2 (RAND, et al., 2004).
Il est intéressant de noter que les chats ont subi au fil des années les mêmes changements
de style de vie que les groupes ethniques d’hommes prédisposés au diabète, dans la mesure où
ils sont passés d’animaux prédateurs à des chats d’intérieur, qui ne dépendent à présent plus
du résultat de leur chasse pour se nourrir correctement (NEEL, 1962). L’activité des chats est

33
donc en moyenne nettement diminuée à notre époque, passant d’un mode de vie à l’extérieur
à un confinement à l’intérieur.
Parallèlement à cela, les félins sont passés pour la plupart d’une alimentation strictement
carnivore tout au cours de l’évolution, à une alimentation commerciale, constituée, de manière
modérée à importante, de glucides, soit plus de 50% de calories supplémentaires. Cette
transition depuis un régime alimentaire pauvre en glucides et riche en protéines,
caractéristique du chat sauvage, jusqu’à une alimentation riche en glucides peut ainsi être, en
association avec la chute globale de l’activité physique, en partie responsable de l’expansion
récente du diabète de type 2 dans la population féline (RAND, et al., 2004).
c- Des facteurs environnementaux : rôle de l’obésité
Chez l’homme, on note l’existence d’une véritable « épidémie d’obésité », sa prévalence
ne cessant d’augmenter fortement dans le monde entier ces 30 dernières années, surtout dans
les pays les plus développés. On s’attend à ce qu’elle croisse encore d’une façon marquée
dans les deux prochaines décennies (KELLY, et al., 2008 ; WANG, et al., 2008).
De nombreux facteurs contribuent à l’établissement et à la perduration de cette pandémie
mondiale, qu’il s’agisse d’éléments environnementaux (l’alimentation et le comportement
alimentaire, ou encore le statut socio-économique), génétiques, ou de caractéristiques
physiologiques.
Une étude en 2005, intéressant 106 pays, soit 88% de la population mondiale, a mis en
évidence que 23,2% des adultes étaient en surpoids (24% chez les hommes et 22,4% chez les
femmes), et 9,8% étaient obèses (7,7% chez les hommes et 11,9% chez les femmes). Le
nombre d’individus en surpoids devrait encore augmenter de 937 millions à 1.35 milliard en
2030, celui des personnes obèses de 396 millions à 573 millions (KELLY, et al., 2008).
La définition basique de l’obésité est l’accumulation anormale ou excessive de graisse
corporelle. C’est une maladie complexe et multifactorielle résultant de l’interaction de
facteurs génétiques et environnementaux.
La mesure la plus utilisée pour rendre compte du tissu adipeux corporel est l’indice de
masse corporelle (IMC), défini par le poids en kilogrammes divisé par le carré de la taille en
mètre (kg/m²). D’après l’Organisation Mondiale de la Santé (OMS), le surpoids et l’obésité
sont caractérisés par un IMC respectivement supérieur ou égal à 25 kg/m² et 30 kg/m²
(SHAMSEDDEEN, et al., 2011).

34
Cette classification est cependant remise en cause par les différences morphologiques
interraciales. On a ainsi mis en évidence dans des populations asiatiques une prévalence
importante de diabète de type 2 et de maladies cardiaques parmi des sujets ne correspondant
pas à la définition décrite précédemment de surpoids ou d’obésité (ANONYMOUS, 2004).
D’autre part, l’IMC peut surestimer le taux de tissu adipeux chez des individus musclés. On
pourra alors utiliser, pour évaluer plus correctement les risques pour la santé, des mesures
anthropométriques, telles que le tour de taille, le tour de cou, et le rapport taille/hanches. Elles
reflètent en effet mieux l’adiposité centrale, et sont de plus en plus utilisées pour appréhender
ces risques (NIH-NHLBI, 2000).
On définit l’obésité chez le chat comme un excès de plus de 30% de poids corporel par
rapport à la normale, alors que l’on parle de surpoids au-delà de 10 à 15% au-dessus de la
normale. Il est cependant préférable de classer ces différentes notions grâce à une note d’état
corporel (NEC). Grâce à ce score, on estime que la prévalence des chats en surpoids varie de
6 à 52% (COLLIARD, et al., 2009 ; SCARLETT, et al., 1994).
Chez l’homme comme chez le chat, l’obésité a des effets néfastes sur la santé et la
longévité. Les patients humains obèses présentent une morbidité et une mortalité accrues,
notamment via un risque augmenté de diabète mellitus de type 2, maladies cardiovasculaires,
dyslipidémie, ostéoarthrite, syndrome d’apnée du sommeil et certains cancers (DANIELS,
2006). Parmi les individus obèses, les risques pour la santé sont proportionnellement
augmentés avec l’élévation de l’IMC (WHO/IASO/IOTF, 2000).
On décrit une forte corrélation entre l’obésité et le diabète de type 2 dans la mesure où le
risque de développer cette endocrinopathie est trois fois plus élevé chez des personnes en
surpoids que chez des sujets présentant un poids normal. Cette probabilité est accrue 20 fois
chez les individus obèses (FIELD, et al., 2001).
L’obésité prédispose aussi très clairement les chats à de nombreuses maladies,
notamment à la résistance à l’insuline et au diabète. Indirectement, elle favorise le diabète en
augmentant les taux de lipidose hépatique, ainsi que les troubles cardiorespiratoires par
exemple (ZORAN, 2010 ; PRAHL, et al., 2007).
L’obésité est donc un facteur prédisposant nettement à l’apparition du diabète sucré de type 2.
Chez l’homme, 70% des patients diabétiques de type 2 sont obèses (CAMPBELL, 2000).

35
D’autre part, on considère que les individus présentant un IMC supérieur à 40 kg/m²
présentent un risque 7 fois plus élevé de développer un diabète que ceux présentant un IMC
dans la norme (MOKDAD, et al., 2003).
Il a été prouvé que même de faibles augmentations de l’IMC et de la taille des cellules
adipeuses sont corrélées à un risque significatif de développer du diabète (COLDITZ, et al.,
1995). Tout comme chez le chat, l’obésité est un facteur de risque très important pour cette
maladie dans l’espèce humaine car elle induit une résistance à l’insuline et une hyper
insulinémie (PORTE, 1991 ; APPLETON, et al., 2001).
Dans une étude menée en 2001, un accès libre à de la nourriture très appétante et
hautement calorique a été donné à des chats pendant environ 10 mois. S’en est suivie une
prise de poids de 1.9 kg en moyenne, soit 44,2% de la masse corporelle. La sensibilité à
l’insuline de ces sujets était alors réduite de plus de moitié (APPLETON, et al., 2001). En
réalité, chez deux tiers des chats, la sensibilité à l’insuline chute en-dessous de la fourchette
décrite pour les chats normaux. De plus, après la prise de poids, 25% des chats de l’étude
avaient une sensibilité à l’insuline dont les valeurs correspondaient à celles décrites chez des
chats diabétiques. La mise en place rapide de l’hyper insulinémie est le facteur de risque le
plus important dans le développement de l’intolérance au glucose liée à l’obésité
(FELDHAHN, et al., 1999 ; RAND, et al., 2004).
On retrouve des résultats similaires dans l’espèce humaine. On constate ainsi dans
différentes études un abaissement de la sensibilité à l’insuline allant de 44 à 72% chez des
individus obèses par rapport à ceux présentant un poids normal (BERGMAN, et al., 1987 ;
TANIGUCHI, et al., 1995).
Il faut noter que la manière dont se dépose la graisse est également à prendre en
compte. On distingue l’obésité centrale -ou obésité abdominale-, de l’obésité périphérique.
Dans l’espèce humaine, on constate que la répartition des graisses est fortement liée au sexe.
Pour un même IMC, les hommes ont tendance à stocker moins de graisse, avec une
distribution centrale du tissu adipeux ; alors que les femmes présentent globalement une
proportion supérieure d’adiposité, distribuée en priorité en périphérie (GEER, et al., 2009).
La distribution centrale correspond à un dépôt des graisses prioritairement au niveau du tronc
et reflète une prépondérance d’adiposité viscérale. L’obésité périphérique, quant à elle, est
corrélée à une distribution du gras dans les membres et les hanches, en particulier dans la
partie inférieure du corps (GEER, et al., 2009).

36
Il a été démontré que ces différentes silhouettes ont une influence sur la sévérité de la
résistance à l’insuline, de la même manière qu’elles en ont une sur toutes les complications
liées à l’obésité. Ainsi, chez l’humain, on a pu prouver que c’est l’obésité centrale qui est
davantage impliquée dans les cas les plus graves de résistance à l’insuline et de diabète de
type 2 par rapport à l’obésité périphérique (KISSEBAH, 1996).
Cela est expliqué par le fait que la graisse viscérale est plus sensible que la graisse sous-
cutanée à la lipolyse induite par les catécholamines, et moins sensible aux effets anti-
lipolytiques de l’insuline. Cela augmente alors le taux d’acides gras libres dans la circulation
portale et systémique. S’en suivent des désordres métaboliques, dont la diminution de la
tolérance au glucose et de la sensibilité à l’insuline, d’où un risque élevé de développer un
diabète de type 2 (HUXLEY, et al., 2010).
On peut noter que la distribution périphérique des graisses est pour sa part associée à une
amélioration de la sensibilité à l’insuline (GEER, et al., 2009).
Ce type d’obésité étant plus fréquent chez la femme, cette donnée corrobore le fait que le
diabète de type 2 présente une incidence supérieure dans la population de sexe masculin.
Une telle observation quant au type d’obésité n’a pu être publiée dans l’espèce féline,
cependant, on remarque que les chats Birmans en surpoids ont tendance à stocker de la graisse
abdominale, alors que chez les chats européens en surpoids, il s’agira plutôt de graisse
inguinale sous-cutanée (RAND, et al., 2004).
Le lien entre obésité et diabète est si fort et indiscutable que l’on a vu apparaître dans la
littérature médicale humaine le terme de « diabésité » (« diabesity »).
Enfin, l’absence d’activité physique est très importante à prendre en compte
puisqu’elle augmente de manière significative le risque de développer un diabète de type 2
chez l’homme. Deux mécanismes sont distincts, tout d’abord une action directe en diminuant
la sensibilité à l’insuline, puis de manière indirecte en agissant sur le poids corporel (HU, et
al., 2001).
En ce qui concerne les autres espèces, on sait que les chiens sédentaires sont aussi
insulinorésistants.
De plus, on a constaté que chez les chats Birmans, le mode de vie à l’intérieur avec peu
d’activité physique est un facteur de risque significatif pour le diabète de type 2 (LEDERER,
et al., 2003). De manière générale, le style de vie moderne des félins de toutes races est peu
propice à l’activité physique dans la mesure où ils n’ont plus besoin de chasser et de se battre

37
pour s’alimenter et vivent enfermés, contrairement aux chats sauvages. Ils sont de ce fait
prédisposés à développer une résistance à l’insuline.
d- Maladies et facteurs iatrogéniques
Dans l’espèce humaine, on sait que l’inflammation joue un rôle dans la mise en place du
diabète de type 2 (cf. I-2-c). Les maladies parodontales favorisent le diabète et de la même
manière le diabète aggrave ces affections dentaires (MEALEY, et al., 2003).
On pense que le fait d’être malade diminue la sensibilité à l’insuline, et il a été prouvé que la
thérapie à l’insuline dans le cadre d’une maladie aigue (infarctus du myocarde, accident
vasculaire cérébral…) permet de maintenir une glycémie dans les normes, même chez les
patients non diabétiques (GROENEVELD, et al., 2002).
Chez les chats Birmans, les maladies récurrentes ou chroniques, et plus particulièrement
les affections dentaires, sont décrites comme des facteurs de risque significatifs pour le
diabète de type 2 (LEDERER, et al., 2003).
Dans l’espèce féline en général, le diabète peut être également secondaire à diverses
affections faisant diminuer la sécrétion d’insuline par les cellules ß pancréatiques, ou
réduisant la sensibilité à l’insuline. On peut citer d’une part les pancréatites et les tumeurs
pancréatiques. Certaines dysendocrinies peuvent aussi être à l’origine du diabète sucré, bien
qu’assez rarement chez le chat. On retrouve le syndrome de Cushing et l’acromégalie suite à
hypersomatotropisme d’origine hypophysaire, ou excès d’hormone de croissance GH
(COURTIN-DONAS, 2009 ; FELDMAN, et al., 2004, a).
La perte de la sensibilité à l’insuline peut parfois être consécutive à l’administration de
certaines molécules à l’origine de résistance à l’insuline, surtout lors de traitement au long
cours ou lors de l’utilisation de formes à longue action.
Chez l’homme, on connaît de nombreuses molécules qui induisent le diabète, notamment
les corticostéroïdes.
Dans l’espèce féline, les médicaments les plus utilisés entraînant une résistance à
l’insuline sont également les corticostéroïdes et les progestatifs de type acétate de mégestrol
(FELDMAN, et al., 2000). Ils diminuent la sensibilité à l’insuline, d’autant plus lors de
traitements au long cours ou d’administration des formes à longue action, chez le chat aussi.
Ils entrainent une augmentation de l’appétit, d’où une prise de poids, réduisant encore
davantage la sensibilité à l’insuline (LEDERER, et al. 2003).

38
Une étude a montré que deux traitements ou plus à base de corticostéroïdes dans les 2 ans
précédant le diagnostic de diabète de type 2, étaient un facteur de risque significatif pour cette
maladie chez les chats Birmans (LEDERER, et al., 2003). Ce fait a également été rapporté
pour les chats européens (HOENIG, et al., 2000).
La question se pose encore sur le rôle significatif ou non de l’administration d’acétate de
mégestrol dans le développement du diabète chez les chats Birmans (RAND, et al., 2004).
A l’inverse, on remarque que le traitement pour les maladies parodontales s’accompagne
souvent d’un meilleur contrôle de la glycémie chez les chats diabétiques, et conduit parfois à
une réduction de la dose d’insuline nécessaire.
3) Pathogénie du diabète sucré
De manière synthétique, on considère que le diabète de type 2 est la conséquence chez les
deux espèces étudiées d’une résistance à l’insuline et d’un état d’hyper insulinémie. Ceux-ci
augmentent la production d’insuline et conduisent à une situation irréversible dans laquelle les
cellules ß pancréatiques sont détruites, cessant de produire de l’insuline (DIXON, 2009).
En effet, le diabète non insulinodépendant est la résultante d’une sécrétion d’insuline altérée,
corrélée à une sensibilité à l’insuline diminuée, aussi appelée résistance à l’insuline. On
définit la sensibilité à l’insuline comme la diminution du taux de glucose pour une quantité
donnée d’insuline. Dès lors, la résistance à l’insuline correspond à une diminution importante
de la sensibilité à l’insuline (PORTE, 1991).
a- La résistance à l’insuline
On peut imputer certains cas de résistance à l’insuline et donc de diabète à un état
inflammatoire chez le chat, mais les mécanismes exacts sont encore inconnus. Les chats de
race birmane présentant des maladies parodontales récurrentes ont un contrôle glycémique
altéré et sont plus prompts à développer du diabète (RAND, et al., 2004).
D’autre part, les chats obèses ont une sensibilité à l’insuline de moitié diminuée par
rapport aux chats avec une masse corporelle normale (HOENIG, et al., 2006). De plus, avec
une prise de 44% de poids corporel en moyenne, presque 50% de chats sains développent une
intolérance au glucose. Les chats minces dont la tolérance au glucose s’amenuise avec la prise
de poids ont présenté une sensibilité à l’insuline diminuée de 35% en moyenne par rapport

39
aux chats ne subissant pas de modification de leur tolérance au glucose après la prise de poids
(APPLETON, et al., 2001).
Lorsque l’index de sensibilité à l’insuline est inférieur à la médiane de référence chez le chat,
le risque de développer une intolérance au glucose avec la prise de poids est triplé. Donc les
chats minces avec une faible sensibilité à l’insuline sous-jacente ont un risque plus important
de présenter une tolérance au glucose altérée s’ils deviennent en surpoids ou obèses. Cela
conduit par la suite à un risque très élevé de développer un diabète de type 2 avéré chez ces
chats obèses (RAND, et al., 2004).
De plus, on a constaté que les chats diabétiques ont une sensibilité à l’insuline six fois
moindre par rapport aux chats sains. Chaque kilogramme de poids corporel supplémentaire
réduit la sensibilité à l’insuline et l’efficacité du glucose de 30% (HOENIG, et al., 2007).
Chez le patient humain présentant une tolérance au glucose diminuée, l’évolution vers
le diabète se fait à un taux de 6% par an (HARRIS, 1995).
Environ 25% des personnes non obèses avec une tolérance au glucose normale présentent une
résistance à l’insuline (HOLLENBECK, et al., 1987). En cas de prise de poids chez ces
individus, certains ne pourront plus sécréter suffisamment d’insuline pour compenser la
péjoration de la résistance à l’insuline due à cette prise de masse corporelle. Alors, une
décompensation du métabolisme du glucose survient, menant à l’intolérance au glucose.
Cependant, chez le chat, la résistance à l’insuline est réversible lorsque survient une
perte de poids. Ces observations ont été réalisées sur des chats non diabétiques et obèses
(BIOURGE, et al., 1997).
De plus, les chats mâles obèses ont de manière innée une sensibilité moindre à l’insuline
et des concentrations d’insuline basales supérieures, ce qui pourrait expliquer la tendance des
mâles obèses à développer un diabète par rapport aux femelles obèses (APPLETON, et al.,
2001).
Chez l’homme comme chez le chat, la capacité d’un individu à compenser la diminution
de la sensibilité à l’insuline en augmentant sa sécrétion d’insuline détermine de manière
décisive de quelle manière sa tolérance au glucose sera détériorée ou non (REAVEN, 1988).
Ainsi, chez les deux espèces étudiées, le diabète est la conséquence de la détérioration des
cellules ß pancréatiques, et la cause principale mise en évidence est la demande trop
importante en sécrétion d’insuline par ces cellules ß secondairement à la résistance à
l’insuline. Les cellules ß sont endommagées suite à cette surproduction, en partie par

40
oxydation, ce qui conduit à l’apoptose ou mort cellulaire programmée (PORTE, 1991 ;
RAND, et al., 2004).
Si ce mécanisme semble véritablement être au cœur de la pathogénie du diabète de
type 2, puisque la résistance à l’insuline implique l’augmentation de la sécrétion d’insuline,
donc la destruction des cellules ß pour maintenir l’euglycémie ; de nombreux facteurs
supplémentaires sont à considérer dans la mise en place et le développement de cette maladie.
b- Les dépôts amyloïdes
Chez l’homme, en conséquence de l’hyperglycémie et de l’hyper insulinémie, on retrouve
une sécrétion d’amyline modifiée. Cette modification altère plus particulièrement le
cheminement normal de l’amyline dans l’organisme et son dépôt localement dans les îlots de
Langerhans sous forme de substance amyloïde. Ce fait pourrait engendrer la destruction
progressive des cellules ß dans le diabète de type 2 (OSTO, et al., 2013).
Les dépôts amyloïdes sont retrouvés chez près de 90% des patients atteints de diabète de type
2 (LUTZ, et al., 1993).
Chez l’homme, le singe et le chat, contrairement à la souris et au rat, l’amyline possède
une région promoteur amyloïdogénique, constituée des acides aminés 20 à 29 du peptide de
l’hormone, et qui prédispose l’amyline à s’agréger et à former des îlots pancréatiques
amyloïdes dans ces espèces-là. L’amyloïdogénèse des îlots implique l’agrégation petit à petit
de monomères d’amyline en oligomères, puis fibrilles, pour finir en dépôts amyloïdes matures
observables au microscope.
Cependant, il semble que ce soit davantage les petits oligomères d’amyline que les gros,
formés au début de l’agrégation des fibrilles, qui sont responsables principalement de la
cytotoxicité pour les cellules ß et de leur mort. La toxicité de ces oligomères semble provenir
de la perturbation et de la destruction des membranes des cellules ß, par la formation de pores
ioniques dans celles-ci (KHEMTEMOURIAN, et al., 2008 ; MIRZABEKOV, et al., 1996).
Par conséquent, l’expansion des fibrilles d’amyline dans les espaces extracellulaires réduit
l’absorption de nutriments et d’oxygène par les cellules ß. Cela entraîne à son tour un stress
du réticulum endoplasmique et ainsi l’apoptose cellulaire (LORENZO, et al., 1994).

41
Cette cascade pathogénique est également présente chez le chat puisque l’on retrouve des
dépôts pancréatiques amyloïdes chez plus de 80% des chats. Selon le degré d’extension de
cette amyloïdose, elle est associée à environ 50% de perte des cellules ß (MA, et al., 1998).
Ainsi, chez l’homme comme chez le chat, la quantité de dépôts amyloïdes est directement
corrélée à la sévérité clinique du diabète mellitus (LUTZ, et al., 1993).
On retrouve également fréquemment à l’histologie chez les chats diabétiques une
dégénérescence vacuolaire des cellules ß, une pancréatite chronique et un nombre réduit
d’îlots ou de cellules ß (NELSON, et al., 1999 ; RAND, 1999).
Cependant, tous les chats présentant des dépôts amyloïdes ne développent pas de diabète
(LUTZ, et al, 1997) et à l’inverse on retrouve des dépôts amyloïdes chez des chats non
diabétiques (LUTZ, et al, 1994 ; YANO, et al, 1981).
De plus, on a observé récemment que la principale zone de section des échantillons
pancréatiques de 37 chats diabétiques positive à l’amyloïde, ne variait pas en fonction du
sexe, de la race, et de l’état corporel de ces chats (ZINI, et al., 2011 ; OSTO, et al, 2013).
c- L’inflammation et la résistance à l’insuline
On sait aujourd’hui que le tissu adipeux produit et sécrète de nombreuses protéines qui
modulent un grand nombre de processi métaboliques. Ce sont les adipokines ou
adipocytokines, elles contrôlent directement ou indirectement l’accumulation des
triglycérides, la différenciation des pré-adipocytes, et initient la réponse inflammatoire dans le
tissu adipeux.
Cette sécrétion d’adipokines, notamment de leptine et d’adiponectine, a également été
démontrée chez le chat (RADIN, et al., 2009).
i. Les adipokines
Les adipokines primaires, chez le chat comme chez l’homme, telles que la leptine et
l’adiponectine, sont produites primairement voire exclusivement par le tissu adipeux.
Chez l’homme, la concentration plasmatique en leptine est corrélée à la masse
graisseuse de l’individu et diminue ou augmente selon qu’il perd ou prend du poids

42
respectivement. Il a été également montré que l’augmentation de leptine est d’autant plus
marquée en réponse à la prise alimentaire (HARRIS, 2000).
La leptine stimule la ß-oxydation des acides gras et inhibe la lipogenèse dans les tissus
périphériques. Dès lors, la leptine permet de diminuer le contenu en triglycérides dans ces
tissus et de faire baisser le niveau d’acides gras circulant.
Dans un cas d’obésité, les réponses physiologiques à la leptine sont diminuées au
moins dans l’hypothalamus, et les cellules-cibles de la leptine deviennent résistantes à ses
effets. La baisse du métabolisme énergétique qui s’en suit dans le corps entraîne une prise de
poids supplémentaire chez les sujets obèses. La résistance aux actions de la leptine est souvent
associée à une sévère résistance à l’insuline, suite à une accumulation majorée de gras
ectopique, et à la lipotoxicité sur les tissus sensibles à l’insuline.
De plus, lorsque le corps est exposé de manière chronique à des concentrations élevées en
leptine, s’en suivent des réactions inflammatoires et l’apoptose des cellules ß. Cette apoptose,
qu’elle soit induite par le glucose ou la leptine, est liée à l’activation de la voie de la c-jun-
NH2-terminal kinase, dans les îlots humains et les cellules à insuline (MAEDLER, et al.,
2008).
L’adiponectine, quant à elle, n’est produite que par les adipocytes matures, et circule dans
le plasma sous forme multimérique, la forme de haut poids moléculaire étant la plus active
biologiquement. Sa sécrétion est stimulée par l’insuline et des éléments du régime alimentaire
comme certains acides aminés (CHANDRAN, et al., 2003).
Les taux d’adiponectine sont inversement corrélés à la masse graisseuse, au contenu
lipidique hépatique, à la dyslipidémie et à la résistance à l’insuline. Cette protéine augmente
la sensibilité à l’insuline et son rôle dans le métabolisme du glucose est d’augmenter la
glycolyse et l’oxydation des acides gras.
Ainsi, le taux d’adiponectine dans le plasma est plus bas chez les sujets obèses, ou présentant
un diabète de type 2, une maladie cardiovasculaire, de l’hypertension, ou un syndrome
métabolique, que chez des individus sains (WHITEHEAD, et al., 2006).
ii. Les adipocytokines
Parmi les nombreuses cytokines sécrétées par les adipocytes hypertrophiés dans le
cadre d’obésité, la protéine-1 chimiotactique pour les monocytes (MCP-1) a pour rôle d’attirer
les monocytes circulant afin qu’ils migrent dans le tissu adipeux. Ces monocytes recrutés

43
mûrissent jusqu’à devenir des macrophages activés classiquement. En sécrétant des TNF-α et
d’autres cytokines telles que IL-1ß, ils continuent à attirer encore plus de macrophages.
Les taux de TNF-alpha et d’IL-6 dans le plasma des hommes et des animaux obèses
sont élevés, et leur production dans la graisse viscérale semble supérieure à celle dans le tissu
adipeux sous-cutané. Cela est cohérent avec le fait que l’accumulation de graisse viscérale a
des effets bien plus délétères que celle de graisse sous-cutanée. Ces deux types de cytokines
inhibent l’action de l’insuline dans les adipocytes, notamment via la phosphorylation des
protéines substrats des récepteurs à l’insuline (IRS). Celle-ci diminue leur activité grâce à
l’inhibition du transport de glucose par GLUT4 stimulé par l’insuline.
De plus, l’activation des voies de l’inflammation contribue à provoquer et propager le
stress du réticulum endoplasmique, avec des conséquences délétères sur la sensibilité à
l’insuline et le métabolisme systémique (HOTAMISLIGIL, 2010).
iii. Les médiateurs lipidiques de l’inflammation
Lors d’une surcharge calorique prolongée, l’accumulation anormale de lipides dans
des tissus autres qu’adipeux, comme le foie, les muscles striés squelettiques, le cœur et les
cellules ß pancréatiques, survenant rarement dans des conditions physiologiques, dépasse les
capacités des cellules à oxyder la graisse, et détériore la réponse métabolique normale et
l’intégrité fonctionnelle du réticulum endoplasmique. En se liant au récepteur 4 toll-like
(TLR4) sur les cellules adipeuses et les macrophages, les acides gras libres peuvent
directement induire l’activation de la cascade de l’inflammation via les JNK et NF-kß-Ikß
kinase (IKKß). Celle-ci est directement liée à la phosphorylation d’IRS-1 et IRS-2, et à la
détérioration de la sensibilité à l’insuline et du transport du glucose (SHI, et al., 2006).
Par conséquent, la sensibilisation des acides gras saturés par TLR4 entraîne une
activation classique des voies de l’inflammation et contribue au développement de la
résistance à l’insuline, impliquant un couplage entre l’inflammation et la résistance à
l’insuline, dans le cadre de l’obésité.
Une étude a été réalisée chez une population féline (OSTO, et al., 2011), dans laquelle
des chats non obèses recevaient une perfusion de lipopolysaccharide (LPS) pendant 10 jours
afin de provoquer expérimentalement une inflammation subaigüe. Cela a suffi à entrainer un
état transitoire de résistance à l’insuline à l’échelle du corps entier, ainsi qu’une résistance à
l’insuline périphérique et spécifique des tissus durable. Ces deux effets ont pu être observés
en l’absence d’effets significatifs sur les cellules ß du pancréas. Cela concorde avec les

44
données obtenues chez l’homme, chez qui l’administration massive de LPS détériore la
sensibilité à l’insuline mais pas la fonction des cellules ß pancréatiques (CANI, et al, 2007).
La perfusion de LPS entraîne également chez les chats une augmentation des
marqueurs de l’inflammation, circulants et dans les tissus (OSTO, et al., 2011).
De plus, l’analyse d’ARNm et de protéines a montré que l’expression de gènes clés du
métabolisme du glucose, des lipides et de l’insuline était altérée considérablement avec une
réduction de la sensibilité à l’insuline spécifique aux tissus. Les changements les plus
importants ont été observés dans le tissu adipeux, plus particulièrement dans le tissu gras
sous-cutané. A cela est liée une taille réduite des adipocytes due à une augmentation de
l’activité lipasique, une diminution des taux de cholestérol de haute densité, ainsi qu’une
augmentation des niveaux de triglycérides dans le plasma et le foie, ces différents acteurs
conduisant à une dyslipidémie sévère.
Il est ainsi remarquable que la taille des adipocytes dans les dépôts de gras des chats perfusés
au LPS est diminuée de façon significative et que la graisse sous-cutanée exprime, par rapport
à la graisse viscérale, beaucoup plus de facteurs pro-inflammatoires.
Des découvertes récentes chez l’homme (MCLAUGHLIN, et al., 2007) montrent
qu’en plus de la masse et de la distribution adipeuse, la taille moyenne de la cellule adipeuse
est associée à des complications métaboliques, comme la résistance à l’insuline et
l’inflammation du tissu adipeux.
De plus, on a démontré qu’une proportion accrue de petites cellules adipeuses dans le tissu
adipeux sous-cutané chez l’homme est associée à une inflammation, indépendamment de
l’indice de masse corporelle et de la résistance à l’insuline (MCLAUGHLIN, et al., 2010). On
ne sait pas actuellement quel rôle jouent ces différents facteurs chez le chat.
d- L’inflammation et la fonction des cellules bêta
Chez l’humain, au cours d’infections et de réponses immunitaires comme dans le cas du
syndrome métabolique, le stress métabolique chronique peut induire un processus d’auto-
inflammation délétère dans les îlots pancréatiques, pouvant aller jusqu’à une panne de la
sécrétion d’insuline et au diabète de type 2. Des découvertes récentes (DONATH, et al., 2010)
orientent vers un rôle causal de l’auto-inflammation induite par des niveaux élevés de
glucose, acides gras libres et IL-1ß, dans la pathogénie du diabète de type 2.

45
Lors d’états inflammatoires ou d’obésité, la synthèse d’IL-1 par les macrophages résidents
est diminuée. Les IL-1ß, la leptine et les acides gras agissent directement sur les îlots
pancréatiques de cellules ß, induisant par la suite la transcription, la traduction et la libération
locale d’IL-1ß (DONATH, et al., 2005). Un des principaux effets des IL-1ß est d’entraîner la
production de cytokines et des chemokines depuis les cellules épithéliales, endothéliales et
immunocompétentes, et ainsi contribuer à l’augmentation du recrutement des macrophages
dans les îlots.
Par conséquent, les macrophages infiltrants renforcent la production locale d’IL-1ß, qui, via la
voie paracrine, contribue à l’induction du circuit de l’auto-inflammation dans l’inflammation
de cellules ß. L’augmentation des réponses inflammatoires locales entraîne la déchéance des
cellules ß et la destruction morphologique du pancréas endocrine (DINARELLO, et al.,
2010).
Une autre des cytokines impliquées est l’interleukine IL-6. Cependant, contrairement à
ses effets systémiques délétères, l’IL-6 locale favorise la production et la sécrétion du peptide
1 glucagon-like (GLP-1) par les cellules intestinales L ainsi que la prolifération des cellules
pancréatiques alpha en réponse à un régime riche en graisses (ELLINGSGAARD, et al.,
2008). En stimulant la sécrétion de GPL-1, l’IL-6 produite par les tissus insulinosensibles peut
mener jusqu’à l’amélioration de la sécrétion d’insuline et de la tolérance au glucose dans les
cellules ß (ELLINGSGAARD, et al., 2011).
On a observé chez le chat (ZINI, et al., 2009) que l’hyperglycémie et l’hyperlipidémie
induites expérimentalement entraînent une inflammation systémique similaire à celle
rencontrée chez l’homme. Cependant, la différence réside dans le fait que l’on n’a pu mettre
en évidence de réaction inflammatoire locale dans les îlots après l’hyperglycémie ou
l’hyperlipidémie. De ce fait, même si l’hyperglycémie (mais pas l’hypertriglycéridémie) a des
effets délétères sur le pancréas endocrine apparentés à ceux observés lors de diabète, ces
effets ne semblent pas découler de l’activation de réponses inflammatoires locales, du moins
les perfusions de 10 jours de glucose ou de lipides respectivement. Il a aussi été montré que la
fonction des cellules ß était altérée durant les premiers jours de la perfusion d’endotoxine
mais l’on ne pouvait plus constater cet effet à partir du 10ème jour (OSTO, et al., 2011).
Une étude (ZINI, et al., 2012) a récemment cherché à estimer si les chats diabétiques
présentaient des preuves anatomo-pathologiques d’inflammation des îlots ou de pancréatite.
Celle-ci a montré le nombre moyen de neutrophiles, lymphocytes T et B dans les îlots était

46
semblable entre les chats sains et les chats diabétiques bien que la présence des lymphocytes
en général semblât plus fréquente chez les chats diabétiques que ceux contrôlés. De plus, un
sous-ensemble de chats diabétiques a présenté une infiltration lymphocytaire des îlots ayant
pu contribuer à la perte des cellules ß. Ces résultats sont en accord avec les observations
précédentes, soit que les chats diabétiques perdent des cellules ß, et suggèrent que
l’augmentation de la nécrose et de la fibrose du tissu exocrine peut être associée à une
pancréatite chez quelques chats diabétiques au moins (MA, et al. 1998).
e- Cas particulier de la glucotoxicité réversible chez le chat
On sait que l’exposition chronique à une hyperglycémie entraîne l’hypertrophie, jusqu’à
l’apoptose, des cellules ß. L’exposition prolongée des cellules ß d’humain à des hauts niveaux
de glucose entraîne l’augmentation de la production et du relargage de l’IL-1ß, suivie par une
fine régulation du taux d’apoptose des cellules ß (MAEDLER, et al., 2001, 2002).
Lors de l’augmentation du taux de glucose, la sécrétion d’insuline est supprimée, c’est ce
que l’on appelle la glucotoxicité.
Chez le chat, cet arrêt de la sécrétion d’insuline est initialement réversible, mais elle finit par
conduire à la perte des cellules ß si le taux de glucose n’est pas régulé (UNGER, et al., 1995 ;
RAND, et al., 2005). Lorsqu’un traitement adapté est mis en place précocement, de nombreux
chats retrouvent la fonction de leurs cellules ß et ne nécessitent plus d’injections d’insuline,
c’est ce que l’on appelle la rémission diabétique (RAND, et al., 2005).
15% des chats diabétiques présentent ce caractère transitoire de la maladie, et
retournent effectivement spontanément à un état euglycémique après un traitement
correctement ajusté de plusieurs semaines à plusieurs mois (NELSON, et al., 1992).
Ce phénomène n’est pas décrit chez l’homme.

47
On constate ainsi que les premiers aspects du diabète de type 2, particulièrement son
épidémiologie et ses facteurs de risque, même si les critères de définition de ces derniers ne
sont pas tous identiques, sont très similaires chez le chat et l’homme.
De plus, si quelques mécanismes connus en médecine humaine ne sont pas encore
élucidés chez les félins, nous nous apercevons que la pathophysiologie du diabète sucré est
très similaire dans les deux espèces.
Malgré quelques différences, notamment pour ce qui est de la réversibilité de la
glucotoxicité, cette première étude nous permet réellement d’envisager la maladie chez le chat
comme approche chez l’homme, afin d’élucider les éléments encore obscurs concernant
l’étiologie et la pathogénie du diabète de type 2 chez ce dernier.

48
II) Le diabète sucré : démarche diagnostique et tableau clinique
1) Une évolution à bas bruit
Dans de nombreux cas, la découverte du diabète de type 2 est fortuite, au détour
d’analyses de routine, ou bien on le recherche lors de décompensation, c’est-à-dire lorsque
des signes cliniques s’expriment.
a. Le syndrome métabolique
C’est un syndrome qui est décrit en médecine humaine pour la première fois en 1988
sous le nom de « Syndrome X » par le professeur Gerald Reaven (REAVEN, 1988). Il fait
l’objet de consensus réguliers dans le monde médical quant à sa définition et sa contenance.
Nous nous baserons sur le plus récent (ALBERTI, et al., 2009), établi conjointement par
différentes institutions suivantes : International Diabetes Federation Task Force on
Epidemiology and Prevention ; National Heart, Lung, and Blood Institute ; American Heart
Association ; World Heart Federation ; International Atherosclerosis Society ; et
International Association for the Study of Obesity ; et publié par l’American Heart
Association en 2009.
Il s’agit en effet d’un panel de facteurs de risques pour les maladies cardiovasculaires
et le diabète de type 2, souvent étroitement liés. Ces facteurs de risques comprennent
l’hyperglycémie, l’hypertension artérielle, la dyslipidémie (un taux trop important de
triglycérides par rapport à un taux trop faible de cholestérol à lipoprotéine de haute densité),
et l’obésité (en particulier centrale) (tab III).
Si l’on décrit une telle association de facteurs depuis des décennies, la question se pose encore
quant au rôle éventuel de la résistance à l’insuline comme facteur liant, même si la pathogénie
exacte reste encore très mal connue.
La première définition officielle du syndrome métabolique a été établie en 1998 par un
groupe de consultation sur la définition du diabète pour la World Health Organization
(WHO). Ce groupe a alors pointé du doigt la résistance à l’insuline comme étant un des
facteurs de risque sous-jacents les plus importants (ALBERTI, et al., 1998).
Les autres critères majeurs ont été émis par le National Cholesterol Education Program Adult
Treatment Panel III (ATPIII) en 2001 (NATIONAL CHOLESTEROL EDUCATION

49
PROGRAM EXPERT PANEL ON DETECTION, EVALUATION, AND TREATMENT OF
HIGH BLOOD CHOLESTEROL IN ADULTS, 2002). Ces critères n’incluaient pas la
présence de résistance à l’insuline elle-même. Le diagnostic devait s’établir non plus grâce à
la présence d’un seul facteur, mais de trois des cinq facteurs suivants : l’obésité abdominale,
l’élévation de la triglycéridémie, la diminution du cholestérol à lipoprotéine de haute densité,
l’hyperglycémie, et l’hypertension artérielle. En l’absence de maladie cardiovasculaire ou de
diabète, le syndrome métabolique est prédicteur de ces affections. On remarque que
lorsqu’elles sont déclarées, le syndrome métabolique est souvent présent, et le nombre des
facteurs que l’on retrouve contribue à la progression de la maladie.
Tableau III : Critères pour le diagnostic clinique du syndrome métabolique, d'après ALBERTI, et al., 2009

50
Ce syndrome est courant et il est de plus en plus décrit dans le monde entier. Cette
expansion est imputée à l’augmentation de l’obésité ainsi qu’au mode de vie sédentaire. Le
syndrome métabolique est ainsi devenu un problème de santé publique autant qu’un problème
clinique.
Cependant, le critère faisant le plus débat au sein de la communauté scientifique à
propos du syndrome métabolique est justement l’obésité. En effet, s’il est reconnu que
l’obésité et ses complications médicales, dont le syndrome métabolique, méritent une
attention très particulière, sa caractérisation et sa place parmi les critères de ce syndrome sont
discutées.
En 2005, deux associations médicales tentent de recouper les différentes définitions
cliniques, l’International Diabetes Federation (IDF) et l’American Heart
Association/National Heart, Lung and Blood Institute (AHA/NHLBI) (ALBERTI, et al.,
2005 ; GRUNDY, et al., 2005).
Cependant, ils ne parviennent pas à s’accorder sur tout, notamment la présence ou non
de l’obésité parmi les 5 facteurs de risque (indispensable pour l’IDF, non requise pour
l’AHA/NHBLI), alors que les 4 autres font consensus. D’autre part, les seuils de tour de taille
définissant l’obésité ne sont pas les mêmes : > 94 cm pour les hommes et > 80 cm pour les
femmes pour l’IDF, contre > 102 cm pour les hommes et > 88 cm pour les femmes pour
l’AHA/NHBLI. Cela correspond à un indice de masse corporelle d’environ 30 kg/m² d’après
les définitions apportées par le National Institutes of Health concernant l’obésité
(NATIONAL INSTITUTES OF HEALTH, 1998). Ces données sont valables pour une
population européenne et il est important de préciser qu’elles doivent être adaptées selon les
différents groupes ethniques (tab IV).
Finalement, l’obésité n’a pas été considérée comme un prérequis pour le diagnostic
mais comme un des 5 critères, de telle façon que la présence de n’importe lequel de 3 sur les 5
facteurs de risque soit diagnostique pour le syndrome métabolique.

51
Tableau IV : Recommandations actuelles, par organisme, des seuils de tour de taille pour l'obésité abdominale, d'après ALBERTI, et al., 2009
D’autre part, le syndrome métabolique est-il à proprement parler un syndrome ? Ou un
mélange de phénotypes sans lien ? Si l’on considère la définition d’un syndrome, c’est-à-dire
le rassemblement de facteurs qui surviennent ensemble plus souvent qu’isolément au hasard
et dont la cause est souvent inconnue ou incertaine, le syndrome métabolique remplit ces
critères. Pourtant, il n’est pas un indicateur de risque absolu.
Les études montrent que les patients présentant le syndrome métabolique ont deux fois plus
de chances de développer une maladie cardiovasculaire dans les 5 à 10 ans que des individus
sans ce syndrome. Ce risque est sans aucun doute encore plus élevé à l’échelle d’une vie. De
plus, il confère un risque 5 fois supérieur pour le diabète mellitus de type 2.
Les paramètres les plus reconnus à travers le monde comme facteurs de risque pour le
syndrome métabolique sont la dyslipidémie athérogénique, l’hypertension artérielle, et
l’hyperglycémie plasmatique. Les individus présentant ces caractéristiques présentent un état
pro-thrombotique et pro-inflammatoire.
La dyslipidémie athérogénique consiste en l’association d’anomalies des lipoprotéines.
On retrouve une hypertriglycéridémie et une élévation de l’apolipoprotéine B, une
augmentation des petites lipoprotéines de faible densité, et une diminution du taux de
cholestérol à lipoprotéine de haute densité. La plupart des personnes avec un syndrome
métabolique présentent une obésité abdominale et une résistance à l’insuline. A nouveau, ces

52
paramètres sont indéniablement liés au développement de risques métaboliques, mais les
mécanismes sous-jacents de leur implication sont méconnus.
L’hyperglycémie est un facteur de risque qui n’a jamais été remis en cause parmi les 5
critères du syndrome métabolique. Dès lors, la plupart des patients souffrant de diabète
mellitus de type 2 ont un syndrome métabolique. Ces derniers présentent un risque plus élevé
à long terme de développer une maladie cardio-vasculaire. Tout patient ayant un diabète
mellitus ajouté à d’autres facteurs de risque du syndrome métabolique devra être suivi de près,
traité par des mesures hygiéniques voire médicales pour modifier ces autres composantes.
On ne retrouve pas de telle entité chez le chat, aucun syndrome métabolique n’a jamais
été défini ou identifié. Cependant, est-il possible d’extrapoler ?
La question a été étudiée chez le chien (VERKEST, 2014). Chez le chien obèse comme chez
l’homme obèse, on remarque l’apparition d’une résistance à l’insuline, ainsi qu’une
augmentation de la concentration des triglycérides plasmatiques, du cholestérol et de la
pression artérielle. Cependant, aucune hyperglycémie ou hyperlipidémie athérogénique n’ont
été mises en évidence dans cette étude. Les chiens ne semblent pas développer de diabète de
type 2. Quant aux maladies cardio-vasculaires, elles sont rares chez le chien et ne sont pas
associées à l’obésité. Ainsi, la notion de syndrome métabolique paraît peu appropriée chez
cette espèce.
Contrairement aux chiens, cochons, moutons et primates non humains, le chat n’est
pour l’instant pas utilisé comme modèle pour l’étude des différents volets du syndrome
métabolique (notamment dans l’identification des gènes candidats à sa mise en place)
(LAWSON, 2013). Si aucune étude n’est à ce jour réalisée dans l’espèce féline, on sait que les
maladies cardio-vasculaires restent rares et que, comme décrit précédemment, l’obésité
prédispose très fortement au diabète de type 2. Comme chez le chien, ce terme est
probablement mal approprié à cet égard, mais il faut considérer un type d’individu à risque,
avant d’établir une maladie au vu des signes cliniques, et mettre en place une surveillance
rapprochée entraînant un diagnostic précoce.
b. Les signes cliniques
Chez l’homme, le diabète de type 2 s’avère très souvent asymptomatique et l’on ne
retrouve que peu fréquemment les signes cliniques décrits. C’est une maladie insidieuse qui
progresse lentement et sur de nombreuses années (tab V).

53
Lors de l’apparition de signes cliniques, on peut observer classiquement de la soif, de
la polyurie, de la fatigue et des malaises, des infections (particulièrement des candidoses
génitales), et une vision floue. On les retrouve en général chez des individus âgés et ils ne sont
en général ni aussi sévères ni aussi soudains que chez les jeunes souffrant de diabète de type 1
(ou insulinodépendant), chez qui c’est une destruction des cellules ß à médiation immunitaire
qui est en cause (CAMPBELL, 2000).
Tableau V : Caractéristiques du diabète de type 2 chez l’homme, d'après CAMPBELL, 2000
Chez le chat, il n’existe aucun consensus international sur les critères diagnostiques du
diabète mellitus (LEDERER, et al., 2003).
Un des symptômes classiques est, comme chez l’homme, la polyurie-polydipsie, dans
70% des cas. En effet, lorsque la glycémie dépasse 1,8 à 2 g/L, on constate une glycosurie
(présence de glucose dans les urines). Ce glucose va être à l’origine d’un appel osmotique et
va entraîner une sortie d’eau en parallèle. Le volume des urines augmente alors, c’est la
polyurie. La polydipsie quant à elle intervient comme mécanisme compensateur des pertes
engendrées, afin d’éviter la déshydratation.
Le stéréotype du chat diabétique correspond à un animal obèse et dysorexique à
anorexique. On peut ainsi constater un amaigrissement suite à l’établissement du diabète dans
68% des cas. Cette caractéristique le rend comparable à l’humain, toutes les espèces ne
présentant pas une telle stéréotypie. Chez le chien par exemple, un sujet diabétique sera au
contraire maigre et boulimique dans la majorité des cas (COURTIN-DONAS, 2009). Le chat
pourra être polyphage, mais seulement dans 20% des cas.
On constate ainsi que la clinique est également très limitée et non pathognomonique
chez cette espèce, et ce sont davantage les complications de cette maladie qui vont s’exprimer
(cf. II-2).

54
c. Méthodes diagnostiques
i. Démarche diagnostique
Comme cela a été décrit précédemment, le diagnostic est souvent tardif du fait de
l’évolution silencieuse de la maladie, et dans de nombreux cas, le diabète est révélé via les
complications cliniques qu’il entraîne.
Fréquemment, la découverte du diabète de type 2 est fortuite. En effet, chez les personnes
âgées, le dosage du glucose dans le sang et les urines est souvent réalisé dans le cadre d’une
maladie intercurrente, au premier ou au second niveau de soins, ou à travers un bilan annuel
de santé. Les études décrivent que 50 % des patients atteints de diabète de type 2 sont
asymptomatiques au moment du diagnostic (CAMPBELL, 2000).
Les recommandations de l’OMS (1999) et de l’ADA (2003) pour les valeurs usuelles
de la glycémie sanguine sont les suivantes :
A jeun, la concentration en glucose plasmatique doit être inférieure à 6,1 mmol/L pour l’OMS
et inférieure à 5,6 mmol/L pour l’ADA. Pour les deux organismes, une valeur supérieure à 6,9
mmol/L est diagnostique d’un diabète.
Deux heures après l’ingestion de 75 g de glucose, la glycémie ne doit pas dépasser 7,8
mmol/L pour l’OMS et l’ADA et l’on parle de diabète au-delà de 11 mmol/L.
Chez le chat, le diagnostic répond aux mêmes contraintes de découverte tardive et
souvent fortuite mais est encore compliqué par une particularité de l’espèce féline. En effet,
ces animaux présentent ce que l’on appelle une hyperglycémie de stress, soit une
augmentation de la concentration plasmatique en glucose artéfactuelle puisqu’elle est liée au
stress engendré par la manipulation. Le chat étant rapidement stressé en consultation, l’acte
de la prise de sang se rajoutant, engendre systématiquement cette élévation immédiate du taux
de glucose. De plus, nous sommes confrontés à une quantité non négligeable de chats peu
coopératifs dès leur contention, ce qui complique encore cette mesure.
La glycémie normale d’un chat non diabétique et non stressé est inférieure à 9,5
mmol/L (LINK, et al., 1998).
Le vétérinaire devra bien veiller à être le moins agressif possible dans la manipulation de
l’animal avant et au moment du prélèvement. Ainsi, l’hyperglycémie de stress transitoire peut
atteindre une valeur de 16 mmol/L chez un chat normal (RAND, et al., 2002).

55
Pour cette raison, il faudra mesurer à nouveau le glucose sanguin quelques heures après le
premier prélèvement pour confirmer l’hyperglycémie, surtout lorsque la glycémie était
inférieure à 20 mmol/L (RAND, et al., 2005).
Il est possible de mesurer un autre paramètre sanguin, la fructosamine, qui représente
l’ensemble des protéines glyquées présentes dans le sérum, en particulier l’albumine glyquée
(80%).
Le dosage de cette dernière est également intéressant chez l’homme, notamment en l’absence
de signes cliniques, puisqu’il s’agit du reflet de la glycémie durant les 10-15 jours précédant
la prise de sang (COURTIN-DONAS, 2009). Donc lorsque cette mesure est majorée, cela
signifie que la glycémie est supérieure aux valeurs usuelles depuis plus de 10 jours.
Chez le chat, la fructosamine doit normalement être inférieure à 370-400 µmol/L. Pour
l’homme, la norme supérieure est de 290 µmol/L, d’après les recommandations de l’OMS.
Une élévation de ce paramètre est très en faveur du diagnostic du diabète (LUTZ, et al.,
1995).
On peut également procéder à des contrôles de la concentration en glucose plasmatique
sur 12 à 24 heures. Cela nécessite une hospitalisation de l’animal et permet de réaliser des
courbes de glycémie, beaucoup plus représentatives, et corrélables à l’administration des
repas ou du traitement.
Ces courbes seront surtout importantes pour l’évaluation de l’efficacité du traitement
antidiabétique et son ajustement (cf. III). On s’intéressera à la valeur initiale de la glycémie et
l’on utilisera en particulier le nadir. Il correspond à la valeur la plus base de la glycémie, le
but étant de calculer le temps au bout duquel il apparaît suite à l’administration de la molécule
hypoglycémiante, ainsi que le temps pour revenir à l’état glycémique initial (RAND, et al.,
2005).
Il est également important dans les deux espèces étudiées de constater l’apparition d’une
glucosurie, à l’aide d’une simple bandelette urinaire, et de l’évaluer par la suite. L’apparition
d’un tel signe met en évidence le fait que la concentration plasmatique en glucose dépasse le
seuil rénal, qui laisse alors passer du glucose dans les urines.
Cependant, il faut à nouveau se méfier de l’hyperglycémie de stress chez les félins, qui est à
l’origine d’artefacts également pour cette mesure (RAND, et al. 2005).

56
Enfin, on peut doser l’hémoglobine glyquée A1c (HbA1c), reflétant les valeurs de la
glycémie sur trois mois. Il s’agit du gold standard pour la détection et la surveillance du
diabète chez l’homme.
L’OMS et l’ADA ont établi un seuil diagnostic pour le diabète : il faut que la concentration
d’HbAc1 soit supérieure ou égale à 6,5% (WHO, 2011 ; AMERICAN DIABETES
ASSOCIATION, 2010).
Ce dosage est encore peu réalisé en médecine vétérinaire courante. De plus, c’est un
paramètre qui ne semble pas être plus sensible ou spécifique que la fructosamine pour le
diagnostic du diabète sucré. Les valeurs de référence qui ont pu être établies dans cette espèce
sont très variables, et dépendantes du laboratoire qui effectue l’analyse (ELIOTT, et al.,
1997 ; AKOL, et al., 1992).
ii. Méthodes complémentaires
Les méthodes évoquées précédemment pour évaluer la glycémie consistent malgré
tout en des prélèvements sanguins très régulièrement, que ce soit à intervalles de quelques
semaines ou au cours de la journée pour la réalisation de courbes de glycémie. Elles s’avèrent
donc invasives et stressantes pour l’animal.
On soulignera alors l’intérêt du holter glycémique (fig. 5). Déjà utilisé en médecine
humaine depuis le début du 20ème siècle, il offre un suivi glycémique très complet. Il s’agit
d’un capteur qui relève toutes les 5 à 10 minutes les valeurs de la glycémie. Les résultats sont
donc nombreux et surtout, n’ayant pas engendré de stress chez l’animal, ils sont plus fiables.
Cela constitue aussi une technique particulièrement intéressante pour la prise en charge des
chats agressifs, le but étant toujours de ne pas passer à côté d’épisodes d’hypoglycémie, voire
d’hyperglycémie.
Chez l’homme, cette technique présente également des avantages particulièrement
pertinents chez certains types d’individus, notamment les jeunes à très jeunes patients, qui
tolèrent difficilement les prélèvements répétés (TRIBOULIN, 2010).

57
Figure 5 : Holter glycémique chez un chat, photo de TRIBOULIN, 2010
Il faut noter que la mesure de la prise de boisson est un moyen gratuit et utile pour
confirmer la polydipsie lorsque la concentration en glucose sanguin est plus élevée que le
seuil rénal.
Chez un chat normal, la consommation totale d’eau par 24 heures, incluant celle présente dans
l’alimentation, va de 60 à 100 ml/kg. La prise d’eau par la boisson exclusivement est estimée
à 20 ml/kg toutes les 24 heures. Cette mesure présente une utilité quasiment uniquement pour
des sujets ne présentant aucun symptôme (DONAGHUE, et al., 1993 ; RAND, et al., 2005).
Chez l’homme, il est plus facile de mettre une évidence une polydipsie, le patient pouvant
évaluer seul sa consommation d’eau, ou du moins l’augmentation de celle-ci.
iii. Diagnostic étiologique
Enfin, en ce qui concerne le diagnostic de l’étiologie du diabète sucré, on peut rechercher
les dysendocrinies parfois responsables, en médecine féline, du diabète de type 2.
Le syndrome de Cushing se traduira surtout dans cette espèce par une peau fragile et
élastique. Il faut rappeler que cette maladie reste race chez le chat.
Quant à l’acromégalie, dont la prévalence réelle est certainement supérieure à celle décrite
dans la littérature du fait de la difficulté diagnostique, on notera majoritairement de

58
l’hyperostose. Cette hypertrophie osseuse touchera les os plats de la boîte crânienne
notamment.
2) Les complications du diabète sucré
Tous les patients diabétiques de type 2 présentent un risque de développer des
complications et ce sont souvent elles qui sont les plus graves et s’avèrent fatales dans cette
affection.
En 2000, la mortalité mondiale imputable au diabète en général, et dans la plupart des cas
au diabète de type 2 s’exprime en millions d’individus. 2,9 millions de morts, soit 5,2%, sont
recensées (ROGLIC, et al., 2005).
Les maladies cardiovasculaires sont ainsi la cause principale du décès chez 70 % des
patients (CAMPBELL, 2000). Le développement d’athérosclérose (perte d’élasticité des
artères due à la sclérose, elle-même provoquée par l’accumulation de corps gras au niveau de
l’intima des artères ; définition de la Société de Chirurgie Vasculaire de Langue Française)
peut s’accélérer plusieurs années avant la mise en place des signes cliniques.
Les effets délétères du diabète sur les autres organes dépendent de la durée de la maladie ainsi
que de sa prise en charge.
En médecine humaine, les complications du diabète de type 2 sont séparées en deux
grands groupes (tab VI).
D’une part, les complications microvasculaires, spécifiques au diabète mellitus et que
l’on ne rencontre pas chez des individus non diabétiques. Les organes principalement touchés
sont les yeux (rétinopathie), les reins (néphropathie), et le système nerveux (neuropathie).
Cliniquement, on observera une cécité, une insuffisance rénale ou des affections podales avec
un risque d’amputation.
D’autre part, les complications macrovasculaires, non spécifiques au diabète mais plus
fréquentes chez des sujets diabétiques. Elles concernent les gros vaisseaux desservant le cœur,
le cerveau et les jambes. Les conséquences cliniques sont de ce fait la crise cardiaque,
l’attaque et la gangrène.

59
Tableau VI : Complications chroniques du diabète de type 2 chez l'homme, d'après CAMPBELL, 2000
Toujours chez l’homme, on a aussi pu associer le diabète à des hépatopathies par
surcharge lipidique d’origine non alcoolique, dont la stéatose hépatique non alcoolique ; des
syndromes ovariens polykystiques ; ainsi qu’un certain nombre de cancers. Ces derniers n’ont
pu être reliés avec certitude à cette endocrinopathie (MORAN, et al., 2010. ; LARTER, et al.,
2010).
En médecine féline, le panel des complications est beaucoup plus fruste.
Comme chez l’homme, le diabète de type 2 est à l’origine d’une baisse d’immunité et donc
entraîne l’apparition de diverses infections. On retrouve en priorité les infections urinaires et
les infections cutanées. Les premières sont à corréler à la dilution des urines associée à la
baisse d’immunité locale. Quant aux secondes, elles conduisent à des retards de cicatrisation.
L’autre type de complication que l’on retrouve chez les félins est, comme chez
l’humain la neuropathie périphérique. Cette dernière va affecter plus particulièrement les
zones déclives du corps et s’exprimera par une plantigradie (COURTIN-DONAS, 2009).
Chez le chat, on retrouve donc principalement des complications que l’on pourrait
classer dans la catégorie des complications microvasculaires. En effet, ces animaux ne sont
que très peu concernés par les conséquences cardiovasculaires et cérébrovasculaires du
diabète de type 2.
Si certaines maladies prédisposent au diabète, le diabète prédispose à l’apparition de
certaines maladies dans l’espèce féline aussi, et l’on retrouve parfois les pathologies suivantes
au moment du diagnostic : hyperthyroïdie, maladie inflammatoire intestinale, complexe
granulome éosinophilique félin, anémie, néoplasie et insuffisance rénale (CRENSHAW, et al.,
1996).

60
a. Les complications microvasculaires
i. Les rétinopathies diabétiques
Dans les pays occidentaux, le diabète mellitus est la cause la plus importante de cécité
chez les humains de 20 à 60 ans (CAMPBELL, 2000).
5 à 10% des patients diabétiques de type 2 auraient une rétinopathie au moment du diagnostic,
de par l’installation insidieuse de la maladie. Ce chiffre s’élève à 30% dix ans après le
diagnostic.
Le risque que présentent ces patients est de perdre la vue par maculopathie, affectant la vision
centrale.
Le traitement de choix est la photocoagulation par laser, il prévient la cécité dans 60 à
70% des cas, si la rétinopathie est détectée précocement. Un dépistage annuel des
rétinopathies est indispensable dans le cadre d’un individu diabétique.
ii. Les néphropathies diabétiques
La maladie rénale concerne 15 à 20% des sujets humains atteints de diabète de type 2,
et chez près de la moitié d’entre eux (44%), elle ira jusqu’à l’insuffisance rénale
(CAMPBELL, 2000 ; NOLAN, et al., 2011).
La néphropathie diabétique est ainsi en train de devenir la cause la plus importante
d’insuffisante rénale au stade terminal.
En Europe, plus de 10% des patients nécessitant des dialyses ou des transplantations
présentent un diabète mellitus. Ce chiffre est doublé aux Etats-Unis d’Amérique.
On retrouve une insuffisance rénale chez certains chats diabétiques, mais on se sait s’il
s’agit véritablement d’une complication, ou si la maladie rénale constitue une prédisposition
de cette endocrinopathie, ou encore si les deux phénomènes coexistent (CRENSHAW, et al.,
1996).
iii. Les neuropathies diabétiques
La forme la plus courante des atteintes nerveuses chez l’homme dans le cadre du
diabète de type 2 est la neuropathie périphérique chronique. L’appréciation du toucher et de la
douleur disparaissent tout d’abord, puis il y a perte de la proprioception des membres

61
inférieurs. Les pieds sont engourdis, blessés beaucoup plus facilement par la chaleur ou les
traumatismes mineurs, jusqu’à l’ulcération.
C’est une affection difficile à traiter et la prise en charge repose essentiellement sur
des mesures hygiéniques et l’apprentissage du patient pour des soins de pieds corrects.
Chez le chat, on retrouve donc également des neuropathies périphériques, s’exprimant
cliniquement par des plantigradies (pose de toute l’extrémité digitée sur le chat). Ce
symptôme est particulièrement évocateur du diabète sucré dans cette espèce.
Ce symptôme est réversible si le traitement hypoglycémiant mis en place est correctement
ajusté (COURTIN-DONAS, 2009).
iv. Les affections des pieds liés au diabète
Les affections podales sont une cause majeure de morbidité chez les patients
diabétiques de type 2 et l’un des principaux motifs d’hospitalisation dans ce cadre
(CAMPBELL, 2000).
On rencontre une variété de désordres au niveau du pied, depuis l’insuffisance vasculaire, la
neuropathie et l’infection, jusqu’à la gangrène.
La neuropathie sensorielle chronique est un des déclencheurs majeurs dans 70 à 80 %
des cas d’ulcères podaux, et elle est en général toujours présente. Le diabète de type 2 est
responsable de 60% des amputations d’origine atraumatiques des membres inférieurs aux
Etats-Unis (NOLAN, et al., 2011).
Par conséquent, la prévention de ce type de complications par des soins de pieds
appropriés est essentielle. Il faut éduquer le patient et ce dernier doit effectuer des contrôles
réguliers chez le podologue.
b. Les complications macrovasculaires
i. Les maladies cardiovasculaires
Ce sont les principales causes de décès et d’hospitalisation chez les patients humains
atteints de diabète de type 2 (CAMPBELL, 2000). L’infarctus aigu du myocarde, ou crise
cardiaque, est deux à quatre fois plus fréquent chez les patients atteints de diabète de type 2
que dans la population globale.

62
De plus, la mortalité suite aux défaillances ou crises cardiaques est également plus élevée. En
2004, les maladies cardiaques et attaques ont constitué respectivement 68% et 16% des décès
reliés au diabète aux Etats-Unis (NOLAN, et al., 2011).
Une étude finlandaise (HAFFNER, et al., 1998), a montré que le risque était identique
entre un individu atteint de diabète de type 2 sans antécédent de crise cardiaque et un sujet
non diabétique ayant déjà présenté un infarctus du myocarde (tab VII, VIII ; fig. 6). On en
conclut ainsi que le diabète de type 2 est un important facteur de risque cardiovasculaire.
Tableau VII : Facteurs de risque pour les maladies cardiovasculaires relativement aux antécédents d'infarctus du myocarde, chez des sujets avec diabète de type 2 et non diabétiques, d'après HAFFNER, et al., 1998

63
Tableau VIII : Taux d’affections cardiovasculaires lors d'un suivi sur 7 ans, relativement aux antécédents d'infarctus du myocarde, chez des sujets avec diabète de type 2 et non diabétiques, d'après HAFFNER, et al., 1998
Figure 6 : Estimations de Kaplan-Meier de la probabilité de décès suite à une maladie cardiovasculaire, chez 1059 sujets ayant un diabète de type 2 et 1378 sujets non diabétiques, avec et sans antécédent d'infarctus du myocarde, d'après HAFFNER, et al., 1998

64
La United Kingdom Prospective Diabetes Study (UKPDS) a défini plusieurs facteurs
de risques significatifs pour les maladies cardiovasculaires chez les patient diabétiques de
type 2 (TURNER, et al., 1998) : le vieillissement de l’individu, le contrôle de la glycémie,
l’élévation de la pression artérielle, un cholestérol LDL élevé avec un cholestérol HDL
diminué, et enfin le tabac.
ii. Les maladies cérébrovasculaires
On retrouve 3 à 4 fois plus d’attaques ischémiques aigües et cérébrales transitoires
chez des patients diabétiques de type 2 que dans le reste de la population (CAMPBELL,
2000).
iii. Les maladies vasculaires périphériques
L’incidence des maladies vasculaires périphériques est jusqu’à 6 fois plus importante
chez les humains atteints de diabète non insulinodépendant (CAMPBELL, 2000).
Les signes cliniques peuvent être les suivants : boiterie intermittente, ulcération ou gangrène.
L’amputation d’un membre inférieur est alors 20 fois plus courante chez les patients
diabétiques de type 2.
En pratique, les techniques d’intervention pour les maladies macrovasculaires
diabétiques sont souvent décevantes, de par la sévérité et l’extension de ces affections
vasculaires athérosclérotiques.
La plupart des patients humains atteints de diabète de type 2 meurent prématurément de
maladies macrovasculaires. Cette mortalité est évaluée à 50% 10 ans après la mise en
évidence du diabète.
La prévention semble la meilleure des solutions face à ces complications. Cependant,
se pose à nouveau le problème de la détection tardive du diabète chez les individus
d’âge moyen. Le diagnostic est en effet souvent posé plusieurs années après l’apparition
d’une intolérance au glucose, et alors qu’une importante maladie macrovasculaire est déjà
présente.
De ce fait, de récentes publications (AMERICAN DIABETIC ASSOCIATION, 2000)
recommandent une surveillance accrue de la glycémie, de la pression artérielle et de la
lipidémie chez les sujet diabétiques de type 2. Un bon contrôle de la glycémie et de la

65
pression artérielle pourrait ainsi prévenir ces complications ou au moins limiter leur
progression (UK PROSPECTIVE DIABETES STUDY GROUP, 1998).
La détection précoce des rétinopathies, néphropathies et neuropathies pourrait réduire
l’incidence de la cécité, l’insuffisance rénale et l’amputation à cause du diabète.
3) La crise acidocétosique
a. Pathogénie
Chez l’homme, on décrit cette forme de coma chez les patients diabétiques de type 2
les plus âgés.
Chez le chat, c’est l’acidose cétosique qui survient dans 12 à 37% des cas au moment
du diagnostic, un faible pourcentage présentant uniquement de la cétose (sans acidose)
(GOOSSENS, et al., 1998 ; CRENSHAW, et al., 1996). Cette crise peut être favorisée par des
maladies concomitantes, notamment des infections (FELDMAN, et al., 2004, b).
Chez les chats qui deviennent fortement insulinopéniques, même s’ils étaient sains
auparavant, l’évolution vers la cétose a lieu en 10 à 30 jours, même en l’absence de maladie
favorisant ce phénomène. Si cet état n’est pas traité, la progression vers l’acidose est
inéluctable (LINK, 2001).
Dans les deux espèces, le mécanisme de la crise acidocétosique est comparable. Ainsi,
lors de diabète de type 2, il y a un stockage important d’acides gras dans les hépatocytes, dû à
l’hyperlipémie, elle-même liée à l’absence de stockage dans les tissus périphériques ainsi qu’à
la lipolyse importante. Les cellules hépatiques synthétisent de ce fait des corps cétoniques à
partir de ces acides gras, utilisés par les tissus périphériques carencés en glucose comme
source d’énergie. Si le diabète n’est pas correctement pris en charge, ou s’il est associé à une
affection intercurrente, notamment une de ses complications ; alors les taux plasmatiques de
catécholamines, glucagon et cortisol vont fortement augmenter. L’élévation des
concentrations de ces hormones de stress va induire une forte lipolyse, et donc une cétogenèse
encore plus importante. C’est lorsque les corps cétoniques sont trop nombreux pour être pris
en charge par l’organisme que survient la crise acidocétosique (COURTIN-DONAS, 2009).

66
Chez l’homme, la crise acidocétosique survient souvent à la suite de l’initiation d’un
traitement à base de thiazide ou de stéroïdes, ou après une affection vasculaire majeure tels un
infarctus du myocarde ou une attaque (CAMPBELL, 2000).
b. Présentation clinique
Le patient présentant une crise acidocétosique est en général somnolent voire
inconscient et fortement déshydraté (CAMPBELL, 2000).
Les chats subissant cette crise sont également en état de choc, déshydratés, très abattus
et souvent en tachypnée, l’acétone augmentant la fréquence respiratoire. On peut également
constater la présence de troubles digestifs tels que de l’anorexie, des vomissements et de la
diarrhée, ainsi qu’une odeur buccale caractéristique de pomme verte (COURTIN-DONAS,
2009 ; FELDMAN, et al., 2004, b).
c. Prise en charge
C’est une prise en charge d’urgence qui doit être effectuée, dans les deux espèces
étudiées ; avec une hospitalisation indispensable.
L’administration d’une fluidothérapie par voie intraveineuse sera réalisée pour corriger
les désordres électrolytiques et la déshydratation. Le type de fluide n’est pas très important en
soi, on choisira une solution équilibrée en électrolytes, de préférence avec des lactates.
La vitesse de perfusion peut aller de 100 à 150 ml/kg/24h.
Dans le cas d’hyperglycémies sévères (au-delà de 32 mmol/L pour les chats), on préfèrera
utiliser du chlorure de sodium NaCl à 0,45% pour les premières heures, pour son pouvoir
hyper osmotique.
Il est très important de monitorer, tout au cours de la prise en charge d’urgence, la
glycémie et les électrolytes, notamment le potassium. En effet, les patients passent souvent
d’une hyperkaliémie initiale du fait de l’acidose, à une hypokaliémie dans les 12 à 24 heures
suivant la prise en charge pour les félins. De la même manière, une hypophosphatémie
survient souvent avec la résolution de la crise.
Une complémentation en bicarbonates est peu recommandée, l’acidose se résout
rapidement avec la mise en place de la fluidothérapie et de l’insulinothérapie (MARTIN, et
al., 2000).

67
L’autre élément essentiel dans la gestion de la crise acidocétosique est donc la mise en
place d’une insulinothérapie afin de faire baisser la glycémie. Il faudra alors utiliser une
insuline à action rapide, Actrapid étant une formulation de choix chez l’homme et chez le
chat, même si le but est de faire diminuer assez lentement le niveau de glucose, en résolvant
progressivement l’acidocétose. Si la concentration en glucose chute trop rapidement, cela peut
être délétère puisque les individus concernés se sont adaptés à un état d’hyper osmolarité et
d’hyperglycémie prolongé.
Chez le chat, le but est de faire diminuer le glucose de 3 à 4 mmol/L/h et de ne pas
dépasser 6 mmol/L/h.
Pour les animaux déshydratés et comateux en acidocétose, des injections intramusculaires
régulières d’insuline sont recommandées avec une dose de charge de 0.2 UI/kg, puis toutes les
heures à 0.1 UI/kg. Afin d’obtenir le rythme de diminution du glucose correct, la dose
d’insuline peut être ajustée jusqu’à 0.2 UI/kg/h ou diminuée jusqu’à 0.05 UI/kg/h
(FELDMAN, et al., 2004, b).
Les injections aux heures doivent être continuées jusqu’à ce que la concentration
sanguine de glucose atteigne 16 mmol/L, puis toutes les 4 à 6 heures à une dose de 0.1 à 0.4
UI/kg jusqu’à obtenir une glycémie entre 11 et 14 mmol/L (MARTIN, et al., 2000).
La voie d’administration est préférentiellement intraveineuse ou intramusculaire.
A nouveau, il conviendra de monitorer rigoureusement la concentration sanguine en glucose.
Après rétablissement, on considère que 50% des patients humains présenteront une
glycémie normale sans traitement à l’insuline, mais simplement grâce à un régime alimentaire
adapté et des hypoglycémiants oraux (cf. III) (CAMPBELL, 2000).
Enfin, même si les chats présentant une cétose ont en général de faibles concentrations
en insuline, une thérapie adaptée pour lutter contre la glucotoxicité et la lipotoxicité permet de
retrouver la fonction d’un certain nombre de cellules ß pancréatiques (RAND, et al., 2005).

68
Si la notion de syndrome métabolique stricto sensu -ou pré-diabète- décrite chez l’homme n’existe pas chez le chat, les mécanismes d’apparition du diabète de type 2 sont, comme nous l’avons vu précédemment, très similaires dans les deux espèces.
D’autre part, les méthodes diagnostiques, orientées par la clinique fruste chez l’un comme chez l’autre, sont tout à fait comparables, et l’on tend à développer en médecine féline les techniques qui s’avèrent les plus discriminantes chez l’homme pour la mise en évidence du diabète de type 2. Cependant, le diagnostic est compliqué chez les félins par une de leurs particularités, soit l’hyperglycémie transitoire de stress, non retrouvée parmi les patients humains. Ces derniers présentent de nombreuses complications liées à cette affection, et ce sont principalement celles dénommées microvasculaires, qui font du chat un modèle en pathologie comparée pour ce qui est de la clinique du diabète sucré. En effet, les complications cardiovasculaires et cérébrovasculaires, mais aussi les rétinopathies notamment, constituent une spécificité de l’espèce humaine.
Enfin, nous constatons que la crise acidocétosique, seule véritable urgence dans le cadre du diabète de type 2, est en tout point identique dans les deux espèces étudiées : la présentation clinique, la physiopathologie, les facteurs déclenchants ainsi que la prise en charge.
Malgré quelques discordances entre le diabète sucré de l’homme et celui du chat dans la suite de notre étude, la médecine comparative semble s’avérer véritablement pertinente dans le cadre de cette maladie.
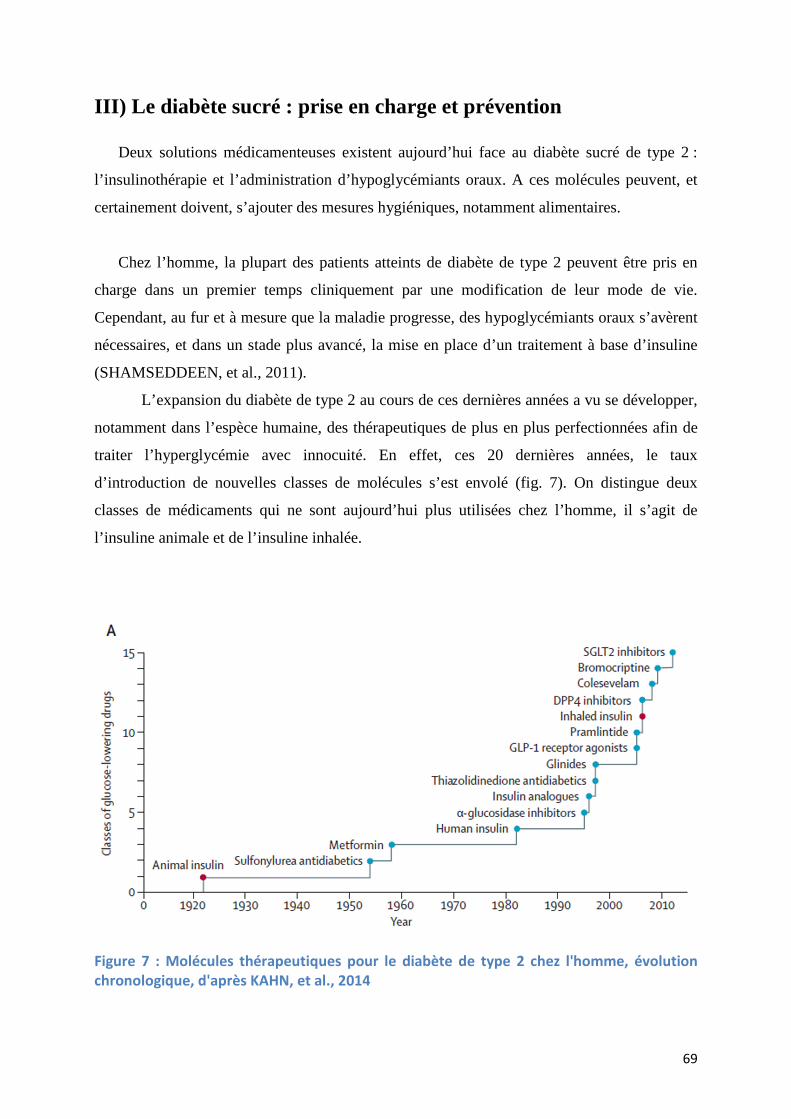
69
III) Le diabète sucré : prise en charge et prévention
Deux solutions médicamenteuses existent aujourd’hui face au diabète sucré de type 2 :
l’insulinothérapie et l’administration d’hypoglycémiants oraux. A ces molécules peuvent, et
certainement doivent, s’ajouter des mesures hygiéniques, notamment alimentaires.
Chez l’homme, la plupart des patients atteints de diabète de type 2 peuvent être pris en
charge dans un premier temps cliniquement par une modification de leur mode de vie.
Cependant, au fur et à mesure que la maladie progresse, des hypoglycémiants oraux s’avèrent
nécessaires, et dans un stade plus avancé, la mise en place d’un traitement à base d’insuline
(SHAMSEDDEEN, et al., 2011).
L’expansion du diabète de type 2 au cours de ces dernières années a vu se développer,
notamment dans l’espèce humaine, des thérapeutiques de plus en plus perfectionnées afin de
traiter l’hyperglycémie avec innocuité. En effet, ces 20 dernières années, le taux
d’introduction de nouvelles classes de molécules s’est envolé (fig. 7). On distingue deux
classes de médicaments qui ne sont aujourd’hui plus utilisées chez l’homme, il s’agit de
l’insuline animale et de l’insuline inhalée.
Figure 7 : Molécules thérapeutiques pour le diabète de type 2 chez l'homme, évolution chronologique, d'après KAHN, et al., 2014

70
Nous choisirons dans cet exposé un classement de ces molécules hypoglycémiantes
par organes cibles. Chaque traitement possède une action bien précise et concerne en général
un organe en particulier.
Classiquement, les cibles sont les îlots pancréatiques, le foie, les muscles et le tissu adipeux.
Plus récemment, ce sont des molécules à visée digestive (intestins), rénale et cérébrale qui ont
été développées. Chaque médication présente un mode d’action différent, même si plusieurs
peuvent cibler le même organe (fig. 8).
L’insuline quant à elle, a pour but de remplacer le produit naturel des cellules ß des
îlots pancréatiques (KAHN, et al., 2014).
Figure 8 : Molécules thérapeutiques pour le diabète de type 2 chez l'homme, classement par organe-cible, d'après KAHN, et al., 2014 Chez l’homme, le but de ces différentes thérapies est de réduire la glycémie et de
maintenir cette concentration en glucose au plus proche de la normale pendant aussi
longtemps que possible après le diagnostic. De ce fait, elles ont également pour visée de
prévenir l’apparition des complications (cf. II-2).
Certains des traitements qui ont été développés ne se sont pas avérés concluants car
l’efficacité thérapeutique était mineure et de nombreux effets secondaires étaient observables.
Cependant, beaucoup d’entre eux sont très efficaces et bien tolérés, ainsi utilisés dans le

71
monde entier, malgré la diversité individuelle des réponses au traitement qui peut survenir.
Cela est imputable à la pathophysiologie très hétérogène du diabète de type 2 (KAHN, et al.
2014).
Les molécules sont administrables autant par voie orale que par voie injectable, même
si les comprimés seront préférés pour la gestion quotidienne d’un diabète peu évolué (tab IX,
X).
Tableau IX : Molécules injectables pour le traitement de l'hyperglycémie dans le diabète de type 2 chez l'homme, d'après KAHN, et al., 2014

72
Tableau X : Molécules per os pour le traitement de l'hyperglycémie dans le diabète de type 2 chez l'homme, d'après KAHN, et al., 2014
Chez le chat, on constate que le but du traitement du diabète de type 2 n’est pas le
même que dans l’espèce humaine. En effet, on ne cherche pas à atteindre l’euglycémie à tout
prix, mais bien à stabiliser cliniquement le sujet diabétique. Cet objectif est en général atteint

73
pour des valeurs de glucose plasmatique comprises entre 0.8 et 2.5 g/L (COURTIN-DONAS,
2009).
Il faut noter que la mise en place du traitement chez le chat doit être très précoce, notamment
chez ceux qui sont atteints d’hyperadrénocorticisme -ou acromégalie-. Dès la détection d’une
glycémie supérieure ou égale à 15 mmol/L, même si le doute persiste quant à son origine,
c’est-à-dire si elle est transitoire et liée au stress, ou alors diagnostique d’un diabète, il faut
initier la thérapie à l’insuline. En effet, la glucotoxicité peut réduire la sécrétion d’insuline
chez des chats normaux, jusqu’à atteindre le même stade que les chats diabétiques
insulinodépendants, en seulement 3 à 7 jours.
Si un doute était présent en ce qui concerne la cause de l’hyperglycémie constatée, il faudra
bien sûr monitorer attentivement les concentrations plasmatiques en glucose les jours suivants
(RAND, et al., 2005)
1) L’insuline
Ces dernières années, on a pu assister à une nette évolution au sein des thérapies pour le
diabète de type 2 à base d’insuline, que ce soit dans l’espèce humaine ou l’espèce féline. On
voit ainsi de plus en plus disparaître les insulines d’origine animale, notamment en
thérapeutique humaine, pour laisser place à des formes d’insuline humaines.
Administrer cette hormone, qu’elle soit naturelle ou synthétique a pour but de suppléer la
fonction des cellules ß pancréatiques qui ne peuvent plus assumer sa sécrétion. C’est
pourquoi, chez l’homme, on s’oriente vers un tel traitement dans des stades assez avancés du
diabète de type 2, lorsque la destruction fonctionnelle de ces cellules est importante.
Les études visant à modifier les formes d’insuline administrables s’appuient sur les
changements de pharmacocinétique, le but étant d’obtenir une action plus rapide ou au
contraire plus prolongée. Une insuline à action rapide est en effet très intéressante pour
stimuler au mieux la diminution de la glycémie en période postprandiale, alors qu’une
insuline à action longue permettra de diminuer le nombre d’administrations quotidiennes et
d’adapter plus facilement les doses (DEWITT, et al., 2003).
La difficulté reste toujours de trouver le meilleur compromis entre ces différentes
formes d’insuline, chacune ayant bien sûr ses avantages mais aussi ses effets péjoratifs.

74
Chez le chat, à l’apparition d’un diabète de type 2, c’est dans la plupart des cas une
insulinothérapie qui sera proposée d’office. De nombreuses formulations de l’insuline sont
aussi disponibles dans cette espèce, toujours sous forme injectable, et particulièrement sous-
cutanée pour l’usage quotidien (tab XI).
L’un des enjeux, dans le cadre de cette maladie chez les félins, est l’observance du
traitement. En effet, le propriétaire du chat joue un rôle crucial dans la réussite de la thérapie.
C’est ainsi lui qui compose et administre les repas, qui réalise les injections d’insuline, et qui
conserve la molécule dans de bonnes conditions. Enfin et surtout, il doit surveiller l’apparition
de signes cliniques chez son animal. Ce dernier point est très important dans la mesure où il
représente, en dehors des contrôles, le seul véritable signe d’alerte que le traitement n’est plus
adapté.
En l’absence de symptômes inquiétants, révélateurs d’hyperglycémie ou surtout
d’hypoglycémie, tels que des syncopes ou simplement de la fatigue, un contrôle de la
glycémie très régulier est tout de même recommandé, soit toutes les 4 à 6 semaines.
En effet, on peut assister dans l’espèce féline à une levée soudaine de la glucotoxicité (cf. I-3),
donc des besoins en insuline nettement diminués, associés à une importante et dangereuse
hypoglycémie.
C’est donc un traitement qui devra, pour la plupart des individus, être ajusté à plusieurs
reprises dans l’espèce féline, de par cette réversibilité de la glucotoxicité, caractéristique par
rapport à l’humain.

75
Tableau XI : Exemples d'insulines pour les chats diabétiques, d'après MARTIN, et al., 2000

76
a. Les analogues de l’insuline à action rapide
En médecine humaine, on a commencé par utiliser un mélange d’insuline standard et
d’insuline à action intermédiaire pour remplacer l’insuline postprandiale lors de diabète.
Cependant, l’absorption de l’insuline standard est très lente après injection sous-cutanée, ce
qui la rend peu adéquate au traitement du diabète de type 2. De plus, sa durée d’action est trop
longue. Elle agit 5 à 8 heures chez les humains et environ 5 heures chez les chats (RAVE, et
al., 2009 ; GILOR, et al., 2008).
Le premier analogue de l’insuline à action rapide à avoir été mis en place chez
l’homme est l’insuline lispro. Son action est rapide à se mettre en place, de 30 minutes à 1
heure ; et le pic d’activité est assez élevé. De plus, la durée de son action est brève, de 2 à 3
heures. Des injections sous-cutanées de ce type d’insuline semblent ainsi imiter plus
correctement la sécrétion postprandiale d’insuline que les injections sous-cutanées d’insuline
standard.
On retrouve également l’insuline aspart et l’insuline glulisine, qui présentent des
profils pharmacocinétiques et pharmacodynamiques comparables à ceux de l’insuline lispro
(SHELDON, et al., 2009).
Ces trois médicaments sont utilisés chez l’homme pour le traitement du diabète de
type 1 et de type 2.
Dans le cadre du diabète de type 2, ces analogues à l’insuline, combinés à l’insuline basale,
confèrent un meilleur contrôle glycémique que l’insuline standard, en limitant les épisodes
d’hypoglycémie (MANNUCCI, et al., 2009).
L’utilisation de ces analogues de l’insuline à action rapide n’a pas encore été étudiée
dans le traitement chronique du diabète chez le chat.
Ces molécules ont été créées pour remplacer le bolus normal correspondant à une
phase de sécrétion d’insuline, qui a une durée d’action de 3 heures ou moins chez l’homme.
Deux études montrent que chez le chat non diabétique, cette action est plus longue que
chez l’humain. Selon les auteurs, la durée d’action varie entre 6 et 12 heures (APPLETON, et
al., 2001 ; MORI, et al., 2009). Le pic de concentration d’insuline intervient plus tard, soit au
bout de 6 heures ; et il est environ 2 fois moins important.

77
Dès lors, en se basant sur ces résultats concernant le profil insulinique postprandial
dans l’espèce féline, les analogues de l’insuline à action rapide ne semblent pas présenter un
intérêt dans le traitement du diabète de type 2 félin (GILOR, et al., 2010)
b. Les analogues de l’insuline à action intermédiaire à lente
i. L’insuline glargine
La glargine est un analogue synthétique récent de l’insuline, d’action longue, qui a
montré des résultats très probants chez les chats sains comme les chats diabétiques.
Elle est utilisée en médecine humaine, et permet un abaissement de la glycémie
régulier, elle est décrite comme une insuline « sans pic ».
Paradoxalement, des études effectuées sur des chats sains ont montré la présence d’un pic
d’action de cette insuline, au bout d’environ 16 heures. La diminution de la concentration
sanguine de glucose était particulièrement marquée après 24 heures chez la plupart des chats
(MARSHALL, et al., 2002).
Cependant, il a été prouvé que la glargine est plus intéressante que l’insuline lente chez les
chats diabétiques, de par un meilleur contrôle glycémique, et des taux de rémission
diabétiques supérieurs (MARSHALL, et al., 2004).
Avec une telle molécule, une seule injection par jour est normalement nécessaire.
Cependant, des études récentes ont prouvé que l’insuline glargine, ainsi qu’une autre
formulation à action lente, l’insuline zinc protamine (PZI), apportent un meilleur contrôle de
la glycémie et un risque moins important d’hypoglycémie clinique lorsqu’elles sont
administrées deux fois par jour, en association avec un régime pauvre en hydrates de carbone
(MARSHALL, RAND, données non publiées, 2004 ; RAND, et al., 2005 ; MARSHALL et
al., 2002).
Donc, même si de nombreux chats pourraient certainement être stabilisés ou contrôlés
avec une injection d’insuline glargine une seule fois par jour, les données recueillies, bien que
peu nombreuses, nous orientent vers un contrôle de la glycémie optimal avec peu de risques
d’hypoglycémie avec une administration biquotidienne.
Le dosage de l’insuline glargine doit être adapté à la glycémie initiale et calculé pour
le poids idéal du patient.

78
Chez le chat, on commencera avec 0.5 UI/kg deux fois par jour lorsque la concentration en
glucose est supérieure à 20 mmol/L. Si elle est en-deçà de 20 mmol/L, la dose initiale sera de
0.25 UI/kg deux fois par jour.
La dose ne doit pas être augmentée au cours de la première semaine de traitement, en raison
de la longue action de cette insuline, et du fait que les effets à chaque injection peuvent se
chevaucher. Il faut par contre baisser la dose si la glycémie descend en-dessous des valeurs
normales.
Dans l’espèce féline, on recommande vivement un suivi rapproché de la concentration
plasmatique en glucose pendant les trois jours suivant la mise en place du traitement, par la
réalisation de courbes de glycémie (via une hospitalisation, ou chez le propriétaire s’il s’en
sent capable).
En général, une légère hypoglycémie est notable pendant ces trois premiers jours, de
manière physiologique. Puis, les contrôles doivent être effectués à 1, 2 et 4 semaines de
l’initiation de la thérapie (RAND, et al., 2005). La dose doit ainsi être surveillée et adaptée
très régulièrement chez le chat (tab XII), alors que cela est moins contraignant chez l’homme.

79
Tableau XII: Paramètres de modifications du dosage d'insuline pour la glargine ou la zinc protamine chez les chats diabétiques, d'après RAND, et al., 2005
L’insuline glargine ne peut être mélangée ou diluée car sa lente absorption repose sur
son pH bas et son interaction avec la graisse sous-cutanée.
Elle doit être maintenue au réfrigérateur, car sa durée de vie maximale est de 4 semaines
lorsqu’elle est conservée à température ambiante (RAND, et al., 2005).
Ce type d’insuline n’est a priori pas intéressant dans le cadre de la prise en charge du
diabète acidocétosique, car elle abaisse peu la glycémie dans un premier temps (RAND, et al.,
2005).

80
ii. L’insuline detemir
Contrairement à l’insuline glargine, l’insuline detemir présente une pharmacocinétique
très prévisible, avec des variations inter- et intra-spécifiques quasi nulles.
Grâce à cette prédictibilité, cette molécule semble être la clé pour réduire les épisodes
d’hypoglycémie au maximum, comme l’ont montré des études réalisées chez l’homme
(HEISE, et al., 2007 ; MONAMI, et al., 2008).
De plus, l’interaction de l’insuline detemir avec l’albumine augmente sa disponibilité pour les
organes possédant des capillaires fenêtrés comme le foie. C’est dans cet organe que l’on
retrouvera les concentrations les plus importantes d’insuline detemir par rapport aux autres
tissus.
Cette insuline inhibe donc la néoglucogenèse hépatique avec encore plus d’efficacité que la
glargine, elle diminue la lipolyse dans les tissus adipeux, et réduit la prise de poids (GILOR,
et al., 2010).
A partir d’études sur des hommes sains, on a pu constater que l’insuline detemir avait
une durée d’action d’environ 20 heures, et elle est administrée normalement une fois par jour
chez les individus diabétiques (HEISE, et al., 2007).
Les résultats cliniques pour l’espèce humaine semblent toutefois identiques en tous
points entre l’insuline detemir et l’insuline glargine. On observe la même diminution du taux
d’hémoglobine glyquée, avec un nombre réduit d’épisodes d’hypoglycémie. Un doute persiste
sur le fait que la glargine soit légèrement plus efficace dans la réduction du glucose et la
detemir moins favorable aux hypoglycémies.
Il est cependant certain que l’insuline detemir est systématiquement associée à une prise de
poids moindre chez les patients (DANNE, et al., 2008 ; MONAMI, et al., 2008).
Une étude a comparé chez le chat la pharmacodynamique d’injections uniques de 0.5
UI/kg d’insuline glargine et d’insuline detemir. La durée d’action de la glargine était de 11,3
plus ou moins 4,5 heures, alors que celle de la detemir était de 13,5 plus ou moins 3,5 heures.
Une variation significative des profils « temps-action » entre les différents individus était
remarquable pour les deux types d’insuline (GILOR, et al., 2010).

81
Une autre étude a comparé ces deux insulines au niveau de la régulation du glucose sur
des chats nourris avec un régime alimentaire optimal pour le diabète sucré, monitorés pour la
glycémie, et traités chez leurs propriétaires.
Le taux de rémission a atteint 67% pour l’insuline detemir et 64% pour l’insuline glargine. On
a noté des épisodes d’hypoglycémie ; cependant très peu de signes cliniques ont pu être mis
en évidence.
La médiane de la dose maximale d’insuline glargine était de 2,5 UI/kg, alors qu’elle était de
1,75 UI pour la forme detemir, avec une administration biquotidienne pour ces deux
molécules (ROOMP, et al., 2009).
iii. Autres formulations
On retrouve chez l’homme l’insuline degludec, qui forme des multi hexamères solubles
par administration par voie sous-cutanée. Sa durée d’action est plus longue que l’insuline
glargine, elle autorise le même contrôle du glucose en réduisant les hypoglycémies nocturnes,
et a été approuvée en Europe ainsi que dans plusieurs autres pays (GARBER, et al., 2012).
Cependant, l’US Food and Drug Administration a émis des doutes sur les potentiels effets
néfastes de cette molécule sur le plan cardiovasculaire, et requiert une étude sur la sécurité
d’un point de vue cardiaque de l’insuline degludec avant d’approuver son utilisation (KAHN,
et al., 2014).
Une autre formulation récente encore en cours de développement pour l’espèce
humaine est l’insuline couplée au polyéthylène glycol. Cela permettrait de retarder
l’absorption et la clairance de cette molécule et ainsi d’obtenir une thérapie longue action
(ROSENSTOCK, et al., 2013).
Des formulations concentrées d’insuline (500 unités par millilitre), sont efficaces chez
quelques patients fortement résistants à l’insuline. Des études complémentaires sur son
efficacité lors d’administrations bi- à tri- quotidiennes sont encore en cours, avec un nombre
élevés d’individus surveillés (ZIESMER, et al., 2012 ; KAHN, et al., 2014).
La voie d’administration de l’insuline est également un des domaines d’étude actifs, afin
d’empêcher au maximum l’apparition d’épisodes d’hypoglycémie. On a envisagé il y a
quelques années que l’insuline inhalée pouvait être une solution très intéressante. Cependant,
sa mise en place sur le plan pratique s’est avérée très compliquée, et son développement a été

82
arrêté lorsqu’un risque de carcinome pulmonaire, suite à son administration, a été soulevé
(SIEKMEIER, et al., 2008).
En ce qui concerne les galéniques par voie orale, la difficulté se situe au niveau du tractus
digestif. En effet, il faut éviter la destruction de l’insuline par les sécrétions intestinales tout
en délivrant une quantité fixe d’insuline depuis le tractus intestinal jusqu’au plasma (CARD,
et al., 2011).
On voit apparaître de plus en plus de formes d’insuline qui réduisent de manière très
agressive la glycémie, ces dernières sont appelées « insulines intelligentes », et sont
dépendantes de la concentration en glucose de l’organisme. Ainsi, ces formulations
deviennent actives lors de l’élévation de la glycémie. Le glucose en trop forte quantité rentre
en compétition avec l’insuline glycosylée pour se lier à la lectine, libérant ainsi l’insuline.
Nous comprenons bien que l’insuline n’est pas libérée, donc est inactive, si la concentration
en glucose est en-dessous ou égale à la normale (BROWNLEE, et al., 1979).
Cependant, cette technique est très peu développée alors qu’elle pourrait représenter une
alternative très intéressante aux traitements actuels, que ce soit pour l’homme ou pour le chat.
Enfin, sont à l’étude, pour l’espèce humaine, des molécules d’insuline modifiées pour
être spécifiquement ciblées vers le foie. Le contrôle de la glycémie semble réellement bon
avec un tel traitement, et il présenterait surtout peu d’effets secondaires, notamment peu
d’hypoglycémies (BERGENSTAL, et al., 2012).
2) Les hypoglycémiants oraux
Ce sont les molécules qui sont administrées dans la grande majorité des cas de diabète de
type 2 chez l’homme, puisqu’il reste en général une sécrétion d’insuline résiduelle par les
cellules ß pancréatiques. On considère que 75% des patients humains souffrant de cette
maladie peuvent être traités avec succès grâce à des hypoglycémiants oraux et une perte de
poids (cf. III-3) (LUTZ, et al., 1993).
Pourtant, on constate que les félins, bien qu’atteints de diabète dit non insulinodépendant,
ne sont que très rarement traités avec cette catégorie de molécules, le traitement de choix pour
cette espèce étant jusqu’alors l’insuline. En effet, 50 à 75% des chats sont en réalité
insulinodépendants. On ne peut encore évaluer avec certitude si ces individus présentent un
diabète de type 2 très avancé avec un remplacement massif des cellules ß pancréatiques par

83
des îlots amyloïdes ; ou si cette déficience en insuline correspond à une destruction à
médiation immune des cellules ß (comme dans le cas du diabète de type 1) ; ou encore un
autre processus pathologique indéterminé (LUTZ, et al., 1993). Ainsi, les médicaments
stimulant la sécrétion d’insuline nécessitent une quantité suffisante de cellules ß
fonctionnelles à cet effet. Si l’abaissement du taux de glucose est insuffisant, l’hyperglycémie
résiduelle continuera à détruire les cellules ß à travers la toxicité du glucose et des lipides.
On pense aussi que ces hypoglycémiants pourraient accélérer les dépôts de substance
amyloïde dans les îlots de Langerhans, aggravant encore la détérioration des cellules ß.
D’autre part, de nombreux propriétaires de chats ont plus de difficultés à administrer des
comprimés à leur animal, que de lui injecter de l’insuline (RAND, et al., 2005).
a. Hypoglycémiants à action dépendante du tractus gastro-intestinal
On retrouve 3 médicaments chez l’homme dans la catégorie des molécules agissant sur
le tractus digestif.
Tout d’abord les inhibiteurs de l’α-glucosidase, soit l’acarbose, qui ralentissent
l’absorption du glucose en retardant la dégradation du complexe glucidique dans le tractus
gastro-intestinal (VAN DE LAAR, et al., 2005).
Ces molécules ne sont en général pas efficaces pour le traitement du diabète félin si elles sont
administrées seules, mais en association avec de l’insuline ou un autre agent hypoglycémiant
oral on peut obtenir un contrôle de la glycémie satisfaisant. Ainsi, les chats à qui l’on a
administré de l’acarbose et fait suivre un régime pauvre en hydrates de carbone ont présenté
des besoins en insuline diminués et une glycémie mieux contrôlée.
Cependant, on s’est aperçu que les résultats étaient enfait identiques avec ce régime
alimentaire seul (cf. III-3) (RAND, et al., 2005).
On retrouve ensuite la pramlintide, qui ralentit la vidange gastrique, et par conséquent
diffère l’absorption du glucose (YOUNK, et al., 2011).
Enfin, le colevesam, résine se liant aux acides biliaires, qui diminue le cholestérol et
modifie le relargage d’autres peptides gastro-intestinaux, permettant de réduire le taux de
glucose plasmatique (MARINA, et al., 2012).

84
b. Les inhibiteurs du co-transporteur 2 sodium-glucose
Il faut savoir que les reins sont des organes centraux dans le métabolisme du glucose,
dans la mesure où ils en absorbent et en excrètent, mais ils en produisent également via la
néoglucogenèse. En général, la quantité de glucose filtrée ne dépasse pas le seuil rénal de
réabsorption. De ce fait, on en retrouve peu dans les urines (GERICH, 2010).
On a découvert que le co-transporteur sodium-glucose 2 (SGLT2) réabsorbait du
glucose à partir de l’urine. Cela a motivé le développement d’inhibiteurs de ce transporteur
afin d’augmenter l’excrétion urinaire du glucose (WRIGHT, et al., 2011 ; LIU, et al., 2012).
Deux de ces molécules ont été récemment introduites sur le marché, la dapaglifozine
et la canaglifozine, certaines sont encore à l’essai clinique. Ces molécules réduisent la
glycémie de manière efficace, mais aussi le poids corporel et la pression artérielle.
Cependant, l’augmentation de la glycosurie est associée à taux de mycoses génitales
cinq fois supérieur, et 40% d’infections du bas tractus urinaire en plus qu’en l’absence de ces
molécules (VASILAKOU, et al., 2013).
On a également constaté, mais pas encore expliqué, une augmentation des taux de cholestérol
LDL et HDL (LAMOS, et al., 2013).
L’augmentation des infections et les potentielles prédispositions aux maladies
cardiovasculaires, si le cholestérol LDL augmente et le HDL diminue, peuvent freiner
l’acceptation de ces médicaments par les patients et les professionnels de santé. Des études
sont actuellement en cours afin de déterminer précisément quels sont les risques encourus,
notamment sur le plan cardiovasculaire, avec l’utilisation de cette classe d’hypoglycémiants
(KAHN, et al., 2014).
c. Hypoglycémiants à action sur le système nerveux central
C’est certainement chez l’homme l’approche thérapeutique la plus complexe et la
moins aboutie, alors que le cerveau joue un rôle central dans la régulation du métabolisme du
glucose.
Le seul médicament mis sur le marché et reconnu comme ayant une action régulatrice
centrale de la glycémie est la bromocriptine, un agoniste des récepteurs à la dopamine.
Le mécanisme de cette molécule s’appuie sur le rythme circadien, qu’elle a pour but de
restaurer (HOLT, et al., 2010). En effet, le rythme circadien est en partie établi par des gènes

85
« horloge » exprimés centralement et dans les tissus périphériques. À partir de là, il affecte
plusieurs des organes impliqués dans le métabolisme (BASS, et al., 2010).
On connaît d’autres molécules qui réduisent la glycémie grâce à des effets centraux, et
la plupart du temps le mode d’action passe par la réduction de la prise alimentaire et du poids
corporel. On compte parmi celles-ci les agonistes des récepteurs GLP-1, qui induisent une
perte de poids s’ils traversent la barrière hémato-méningée (DRUCKER ; et al., 2006).
d. Nouvelles options thérapeutiques
i. Chez l’homme
Les molécules corrélées aux incrétines ont pour but d’imiter ou d’accroître l’action des
GLP-1 et GIP, libérés par les intestins. Les agonistes aux récepteurs GLP-1 sont des peptides
avec une demi-vie plus longue que GLP-1, tandis que les inhibiteurs des dipeptidyl peptidase
4 (DPP4) bloquent l’action des DPP4. Cela conduit à la dégradation rapide de GLP-1 et GIP
(DRUCKER, et al., 2006).
Actuellement, ces médicaments à base d’incrétines sont encore à l’étude sur les plans
pharmacocinétique et pharmacodynamique afin de réduire les doses nécessaires et d’améliorer
le contrôle de la glycémie (DRUCKER, et al., 2008).
Les mécanismes sont encore mal connus mais l’on a constaté qu’une perfusion intraveineuse
de fortes doses de GPL-1 permet de normaliser le taux de glucose en entraînant moins de
nausées ou de vomissements que par voie sous-cutanée. Ce fait est intéressant dans la mesure
où ces effets secondaires, lorsqu’ils se déclarent, empêchent l’administration de la totalité de
la dose, donc une normalisation de la glycémie (RACHMAN, et al., 1997 ; ZANDER, et al,
2002).
Les dernières études tendent également à prouver que cette thérapie à base d’incrétines
pourrait présenter des effets bénéfiques sur le système cardio-vasculaire, fait intéressant
lorsque l’on sait que ce sont des organes particulièrement touchés par les complications du
diabète de type 2 (SCHEEN, 2013).
D’autre part, les agonistes aux récepteurs GLP-1 et inhibiteurs des DPP4 présenteraient un
risque de pancréatite aigüe, ou encore de tumorisation du pancréas. Cependant, les études
ayant abouti à de telles observations ont pu être réfutées pour cause d’un trop faible nombre
d’individus inclus dans l’expérimentation, ou d’un biais dans le recrutement des échantillons

86
(KAHN, et al., 2014). Le risque de développer un cancer du pancréas corrélé à
l’administration de ces médicaments a été officiellement réfuté par l’Agence Européenne du
Médicament en juillet 2013.
Si de nombreuses molécules semblent prometteuses et sont encore à l’étude, le contrôle de
la glycémie reste encore incertain lorsque la fonction des cellules ß pancréatiques est de plus
en plus détériorée. Cela est surtout valable chez l’homme, pour qui, nous le rappelons, on
cherche véritablement à atteindre l’euglycémie, alors qu’on s’attachera plutôt à la stabilisation
clinique chez le chat.
Ainsi, on constate que de nombreux mécanismes du diabète de type 2 n’ont pas encore été
exploités, mais représentent des approches potentiellement très intéressantes dans le
développement de nouvelles molécules, dans le cadre du traitement de cette maladie (tab
XIII) (KAHN, et al., 2014).
Forts du fait que la pathogénie du diabète de type 2 est très semblable dans les espèces
féline et humaine, on peut, malgré des objectifs thérapeutiques différents, se demander si
l’extrapolation de ces traitements ne serait pas intéressante chez le chat.

87
Tableau XIII : Cibles thérapeutiques et mécanismes non encore testés pour le diabète de type 2, d'après KAHN, et al., 2014
ii. Chez le chat Des études rétrospectives se sont avérées concluantes quant à l’utilisation d’un
sulfamide hypoglycémiant dans le cadre du traitement du diabète de type 2 chez les félins.
Il s’agit du glipizide, molécule qui ne présente pas d’AMM chez le chat, mais qui peut être
utilisée d’après le principe de la cascade de la pharmacopée européenne sous deux formes
commerciales : Glibénèse® et Minidiab®.

88
25 à 50% des chats diabétiques ont ainsi été traités avec succès grâce à l’utilisation de cette
molécule (MILLER, et al., 1991 ; NELSON, et al., 1991).
La posologie est alors de 2,5 à 5 mg/kg.
Les effets secondaires rapportés sont des vomissements, de l’ictère et de l’hypoglycémie.
Les molécules sensibilisantes à l’insuline, telles que les thiazolidinediones et les
biguanides, intensifient la réponse des muscles, du foie et du tissu adipeux à l’insuline.
Elles diminuent en priorité la résistance des tissus périphériques à l’insuline, et augmentent la
consommation de glucose stimulée par l’insuline par les muscles. Elles réduisent également la
néoglucogenèse hépatique.
Toutefois, malgré ces mécanismes d’action aptes à contrecarrer la résistance à l’insuline, les
études n’ont montré aucune efficacité de ce traitement chez les chats diabétiques,
contrairement à ce que l’on constate chez l’homme (SALTIEL, et al., 1996 ; KAHN, et al.,
1990).
Cependant, la sensibilité à l’insuline ainsi que le métabolisme lipidique peuvent être
améliorés chez les chats obèses présentant un diabète de type 2 par la darglitazone, molécule
sensibilisante à l’insuline (HOENIG, et al., 2003).
Enfin, on a récemment établi que les thérapies hormonales à base d’incrétines peuvent
s’avérer pertinentes chez le chat, au même titre que ce qui a déjà été constaté en médecine
humaine.
Pour l’instant, les agonistes de GLP-1 et les inhibiteurs de DPP-4 n’ont été étudiés que chez
des chats sains (GILOR, et al., 2011 ; PADRUTT, et al., 2012 ; HUGUES T.E., 1999 ;
FURRER, et al., 2010).
Les différents auteurs ont pu constater une élévation conséquente de la sécrétion d’insuline
dans tous les cas, et cela suite à l’administration de ces deux types d’incrétines.
.
Les études cliniques sur le possible traitement du diabète félin par ces molécules sont
encore en cours, mais leur coût particulièrement élevé rend leur achèvement complexe.
Pourtant, il paraît clair que la thérapie hormonale par les incrétines est une voie d’avenir dans
le traitement du diabète de type 2 chez le chat (REUSCH, et al., 2013).

89
3) Intérêt des mesures diététiques a. Perte de poids et activité physique
Chez l’homme, lors de pré-diabète -syndrome métabolique-, ou de diabète débutant, il est
vivement recommandé dans un premier temps de pratiquer une activité physique, et de
procéder à un changement de régime alimentaire, afin de perdre du poids.
En effet, de simples changements dans le mode de vie peuvent retarder de façon significative
la progression de la maladie, ainsi que de ses complications. Une perte de poids modeste peut
déjà réduire de 20% tous les effets délétères de l’obésité pour la santé de l’obésité ainsi que la
mortalité (SHAMSEDDEEN, et al., 2011).
Une prise de poids de 5 à 7 kg peut augmenter le risque de diabète de 50%, alors qu’une perte
de 5 kg le diminue de moitié (WILLIAMSON, et al., 1995).
A cet égard, et en considérant toutes les données exposées au préalable liant diabète de
type 2 et obésité, on note une rémission clinique de cette maladie chez 64% à 83% des
patients après une chirurgie bariatrique (fig. 9) (DIXON, 2009).
L’effet bénéfique sur le fonctionnement des cellules ß est positivement corrélé à la précocité
de l’intervention. Ainsi, le bypass gastrique entraîne une rémission du diabète chez 95% des
patients qui présentent la maladie depuis moins de 5 ans, alors que seuls 54% des patients
bénéficient de cette rémission parmi les diabétiques depuis plus de 10 ans (SCHAUER, et al.,
2003).

90
Figure 9 : Bypass gastrique Roux en-Y par laparoscopie : technique de l'université de Pittsburgh, d’après SCHAUER, et al., 2003
Un patient obèse doit être pris en charge par le biais d’une gestion du poids, associée à
un contrôle sérieux de la glycémie. Dès que le diabète est diagnostiqué, la perte de poids est
indispensable. De la même façon, chez les patients diabétiques ou souffrant d’intolérance au
glucose, la prévention de la prise de poids doit constituer le pilier de la thérapie mise en place
(COLAGIURI, 2010).
La perte de poids est recommandée pour des patients présentant un IMC supérieur ou égal à
30 kg/m² ; ou pour des patients avec un IMC situé entre 25 kg/m² et 29 kg/m² ou un tour de
taille à risque, et 2 facteurs de risque ou plus.
Le régime consiste en une alimentation à base de nutriments pauvres en calories et
pauvres en graisses, le but étant d’atteindre 1000 à 1200 kilocalories par jour pour les femmes
et de 1200 à 1600 kilocalories par jour pour les hommes.
Une ingestion chronique d’aliments riches en hydrates de carbone concourt au
développement de l’obésité et augmente la demande d’insuline à sécréter par les cellules ß, ce

91
qui entraîne une forte prédisposition à l’hyper insulinémie, l’apoptose, la détérioration des
cellules ß, et enfin la mise en place du diabète de type 2 (PORTE, 1991).
Chez le chat également, l’ingestion d’hydrates de carbone via différents aliments,
comme le riz par exemple, augmente la demande de l’organisme en insuline. Ainsi, elle
contribue à la destruction des cellules ß chez les chats prédisposés, lorsque ce régime
alimentaire se prolonge (RAND, et al., 2004).
De plus, les chats sauvages, en tant que carnivores, consomment des protéines alimentaires en
grande quantité, et ne sont pas adaptés à l’ingestion de nourriture à haute teneur en glucides
(DROCHNER, et al., 1980).
Une étude a cherché à évaluer les effets de différents régimes chez les félins, en
comparant l’effet de trois macronutriments sur des chats sains.
Un régime était riche en protéines, un autre riche en gras, et le dernier riche en hydrates de
carbone. Les chats nourris avec le régime riche en hydrates de carbone présentaient des
moyennes et des pics de glycémie significativement plus élevés (23 à 32%), ainsi que des taux
d’insuline plus importants que ceux nourris avec les régimes à haute teneur en lipides ou en
protéines (FARROW, et al., 2002).
Dès lors, le régime alimentaire qui semble le plus intéressant dans l’espèce féline pour
prévenir l’apparition du diabète, notamment chez les chats à risque, serait très pauvre en
hydrates de carbone, riche en protéines et modérément riche en lipides. Une telle alimentation
permettrait d’améliorer l’état d’hyperglycémie, de réduire la dose d’insuline nécessaire, et
d’augmenter le taux de rémission diabétique (FRANCK, et al., 2001).
Sur un échantillon de 66 chats Birmans, on a montré qu’un régime supplémenté en
aliments riches en protéines et pauvres en hydrates de carbone, comme la viande ou le
poisson, offrait une protection significative contre le diabète de type 2 (LEDERER, et al.,
2003).
On peut rapporter de manière anecdotique que chez cette race féline, de nombreux cas de
diabète ont été associés au traitement de maladies rénales, qui comprend notamment un
régime pauvre en protéines et riche en hydrates de carbone.
Différents types de glucides sont discernables et les conséquences de leur ingestion ont
également été étudiées dans les espèces qui nous intéressent.
On a nourri des chats en surpoids, présentant une diminution de la sensibilité à l’insuline
avérée, avec deux types d’aliments. Chacun d’entre eux contenait le même pourcentage

92
d’amidon (33%), mais provenant de céréales différents. Un régime était à base de maïs et de
sorgho, tandis que l’autre était constitué de riz. On a alors constaté que les chats nourris à
volonté avec du riz consommaient davantage de calories et prenaient plus de poids que ceux
qui l’étaient, dans les mêmes conditions de libre accès, avec le maïs et le sorgho.
S’ajoute à cela une tendance des animaux qui subissent le régime à base de riz à présenter des
taux de glycémie et une sécrétion d’insuline plus importants, en réponse à une charge de
glucose ou un repas test (APPLETON, et al., 2004).
Chez certains chats, en particulier ceux qui présentent une faible sensibilité à l’insuline
sous-jacente, la consommation tout au cours de la vie d’un aliment à haut index glycémique,
comme le riz, peut contribuer au « burnout » prématuré des cellules ß pancréatiques. Par la
suite, on assiste alors à la mise en place d’un diabète de type 2, à cause de la demande trop
importante en insuline par l’organisme (RAND, et al., 2004).
Chez l’homme, on a prouvé que les aliments induisant des réponses glycémiques et
insuliniques importantes et rapides en période postprandiale, soient les aliments à haute teneur
glycémique, entraînent une satiété moindre et par conséquent une prise alimentaire plus
conséquente. Dès lors, le fait de consommer des aliments à haut index glycémique entraîne
l’augmentation de la faim, donc une prise de nourriture excessive et finalement une prise de
poids (HOLT, et al., 1992 ; BRAND, et al., 1985).
Un autre aspect des mesures hygiéniques est à considérer attentivement.
En effet, l’activité physique augmente la dépense d’énergie, permet de maintenir son poids, et
réduit les risques de maladies cardiovasculaires (NIH-NHLBI, 2000).
Dans l’espèce féline, il a été prouvé que seules 10 minutes de jeu quotidien afin
d’augmenter l’activité physique permettent de perdre du poids de manière aussi efficace
qu’avec un régime alimentaire restrictif en calories (GILES, et al., 2003).
Le seul médicament approuvé pour le traitement à long terme de l’obésité chez
l’homme est orlistat, inhibant la lipase pancréatique et, de ce fait, diminuant l’absorption de
graisses par le tractus gastro-intestinal (IOANNIDES-DEMOS, et al., 2011).
Ce traitement propose une perte de poids insuffisante pour faire une différence clinique ;
soient 2,9 kg par an perdus en moyenne (PADWAL, et al., 2007) ; il est alors recommandé en
adjonction à un traitement chirurgical (MONKHOUSE, et al., 2009).

93
Dans la mesure où ces mesures diététiques et hygiéniques suffisent rarement à acquérir
une perte de poids suffisante et durable, la chirurgie bariatrique semble être une bonne
solution pour obtenir une perte de poids réduisant voire résolvant la morbidité relative à
l’obésité.
Une étude randomisée (BUCHWALD, et al., 2004) a montré une perte moyenne de 61% du
poids excessif avec cette chirurgie, et une nette amélioration voire une résolution du diabète
de type 2 entre autres maladies consécutives à l’obésité.
De plus, il a été montré que la chirurgie bariatrique diminue surtout la mortalité sur les
individus obèses à long terme, et cela à hauteur de 40%. Cet effet est particulièrement vérifié
dans le cadre du diabète mellitus (ADAMS, et al., 2007).
Ces solutions chirurgicales ne sont actuellement pas encore envisagées dans l’espèce féline.
b. Intérêt de certaines complémentations alimentaires
Enfin, on peut se pencher sur l’intérêt du chrome dans la prévention voire le traitement
du diabète de type 2.
Cet élément est physiologiquement présent chez l’homme et l’animal et contribue au
métabolisme normal des glucides et des lipides (ANDERSON, 1986).
Chez les sujets humains, le chrome améliorerait la tolérance au glucose en augmentant
la sensibilité à l’insuline. En effet, si l’efficacité de l’insuline est meilleure, la glycémie
diminue (ANDERSON, et al., 1983).
Une étude de 2002 a cherché à investiguer l’intérêt d’une complémentation alimentaire
en chrome chez des chats sains et non obèses, en regard du métabolisme du glucose et de
l’insuline. L’incorporation de chrome tripicolinate au 300 à 600 milliardième dans la ration de
ces chats induisit de légères mais significatives améliorations de la tolérance au glucose. Ces
résultats ont été estimés via des mesures de demi-vie du glucose, d’aires sous la courbe
glycémique et de concentrations absolues en glucose (APPLETON, et al., 2002).
La supplémentation en chrome présente-elle alors un intérêt dans les deux espèces ?
Cet élément n’est pas un médicament, mais bien un nutriment essentiel normalement présent
dans l’organisme. Il semblerait donc intéressant de ne supplémenter que les individus
présentant une carence en chrome, notamment d’origine alimentaire (ANDERSON, 1987).

94
Or aux Etats-Unis, on considère que plus de 90% des repas classiques des habitants
contiennent un taux de chrome inférieur aux recommandations journalières pour cet élément
(ANDERSON, et al., 1985).
De telles données concernant la quantité de chrome nécessaire par jour ne sont pas
encore établies dans l’espèce féline. Cependant, une étude réalisée en 2004 a montré qu’en
nourrissant des chats avec une alimentation complète et équilibrée, constituée d’après les
standards de l’Association of American Feed Control Officials, ces derniers n’avaient qu’un
apport très modéré de chrome. Ainsi, il pourrait être intéressant de mettre en place une
supplémentation chez tous les chats adultes.
Pourtant, ce sont bien les sujets présentant une intolérance au glucose et une résistance à
l’insuline liées au manque d’exercice, à l’obésité et au vieillissement pour qui cet apport en
chrome serait le plus bénéfique. De même pour ceux qui ont une moindre sensibilité à
l’insuline ; ou encore génétiquement prédisposés, c’est-à-dire les chats Birmans (RAND, et
al., 2004).
Le vanadium est également un métal de transition qui potentialise l’action de
l’insuline. Tout comme le chrome, si l’on connaît bien son intérêt chez les patients
diabétiques humains, des investigations supplémentaires s’avèrent nécessaires chez le chat
pour conclure sur ses effets et mettre en place des recommandations (RAND, et al., 2005).

95
Les prises en charge du diabète de type 2 chez l’homme et chez le chat sont
aujourd’hui relativement différentes, de par l’objectif thérapeutique qui n’est pas le même, et
du fait de quelques spécificités des félins, qui semblent finalement insulinodépendants pour
un certain nombre.
Cependant, de nouvelles thérapies à l’étude montrent que certains hypoglycémiants
oraux notamment peuvent trouver leur intérêt dans le traitement de cette endocrinopathie chez
le chat, en particulier les analogues des incrétines, la pathogénie de la maladie étant
finalement quasiment identique chez l’homme.
Enfin, chez l’homme comme chez le chat, il est indéniable que le traitement
médicamenteux ne peut être dissocié de certaines mesures hygiéniques, à savoir un régime
alimentaire adapté, voire une chirurgie bariatrique chez les patients humains, associés à une
l’activité physique.
On constate une fois de plus la réciprocité du bénéfice que peut engendrer la médecine
comparative. En effet, chaque espèce peut profiter des découvertes effectuées chez l’espèce
qui lui est assimilée pour une pathologie donnée, à savoir dans notre étude le diabète sucré de
l’homme et du chat.

96
CONCLUSION Le diabète sucré, encore appelé de type 2, non insulinodépendant, connaît ces
dernières années une importante prévalence dans les populations humaine et féline. La comparaison des différents aspects de la maladie au sein des deux espèces permet de mettre en évidence de nombreux points communs.
Les patients et chats diabétiques sont obèses pour la majorité. Cette obésité semble largement contribuer à la résistance à l’insuline, pilier de la pathogénie de cette maladie. Une origine génétique a également été démontrée chez ces deux espèces. De plus, contrairement aux autres modèles animaux utilisés, le chat et l’homme présentent une altération fonctionnelle et une diminution du nombre de cellules ß, ainsi que des dépôts amyloïdes pancréatiques, conduisant conjointement à l’intolérance au glucose. Chez les deux espèces, le diagnostic est dans la plupart des cas une découverte fortuite, dans la mesure où les symptômes sont soit très frustes soit absents, et toujours peu spécifiques pendant plusieurs années. Cependant, les conséquences peuvent être graves à mortelles. Chez le chat, on retrouvera principalement l’hypertension, les neuropathies, les endocrinopathies et la crise acidocétosique alors que chez l’homme on rapporte en plus des rétinopathies, néphropathies et de nombreuses complications vasculaires supplémentaires (cardiaques, cérébrales ou périphériques).
Si l’insuline représente le traitement largement prédominant du diabète sucré chez le chat, trois-quarts des patients humains stabilisent leur diabète à l’aide d’hypoglycémiants oraux et d’un régime alimentaire adapté (pauvre en sucres et en graisses). Dans l’espèce féline ou humaine, l’alimentation semble être l’une des pierres angulaires de la thérapeutique, depuis la prévention jusqu’à la prise en charge du diabète mellitus. Une rémission de la maladie et une réversibilité de la glucotoxicité sur les cellules ß, après quelques semaines à quelques mois d’une thérapeutique mise en place précocement, représente une particularité féline.
Ainsi, le chat peut probablement être considéré comme le meilleur modèle animal non primate pour le diabète sucré chez l’homme, par les nombreuses similitudes mises en évidence. Certaines questions restent encore à élucider, tels que les fondements génétiques complets du diabète de type 2, ou encore les mécanismes précis aboutissant au dysfonctionnement et à la disparition des cellules ß. On pourrait ainsi envisager de nouveaux traitements plus ciblés pour l’homme, et à l’inverse, s’appuyer sur les connaissances thérapeutiques chez l’humain pour envisager des traitements alternatifs à l’insuline chez le chat.

97
BIBLIOGRAPHIE ADAMS T.D., GRESS R.E., SMITH S.C., HALVERSON R.C., SIMPER S.C.,
ROSAMOND W.D. et al. (2007)
Long-term mortality after gastric bypass surgery.
The New England Journal of Medicine, vol 357, p. 753-761.
AKOL K.G., WADDLE J.R., WILDING P. (1992)
Glycated hemoglobin and fructosamine in diabetic and non-diabetic cats.
Journal of the American Animal Hospital Association, vol 28, p. 227-231.
ALBERTI K.G., ZIMMET P.Z. (1998)
Definition, diagnosis and classification of diabetes mellitus and its complications, part 1:
diagnosis and classification of diabetes mellitus provisional report of a WHO
consultation.
Diabetic Medicine, vol 15, p. 539-553.
ALBERTI K.G., ZIMMET P., SHAW J.; IDF Epidemiology Task Force Consensus Group
(2005)
The metabolic syndrome: a new worldwide definition.
Lancet, vol 366, p. 1059-1062.
ALBERTI K.G., ECKEL R.H., GRUNDY S.M., et al. (2009)
Harmonizing the metabolic syndrome: a joint interim statement of the International
Diabetes Federation Task Force on Epidemiology and Prevention; National Heart,
Lung, and Blood Institute; American Heart Association; World Heart Federation;
International Atherosclerosis Society; and International Association for the Study of
Obesity.
Circulation, vol 120, p. 1640-1645.
AMERICAN DIABETES ASSOCIATION (2000)
Clinical practice recommendations.
Diabetes Care, S16-S116.

98
AMERICAN DIABETES ASSOCIATION (2010).
Diagnosis and classification of diabetes mellitus.
Diabetes Care, vol 33 (supplement 1), p. S62–S69.
AMOS A., MCCARTY D., ZIMMET P. (1997)
The rising global burden of diabetes and its complications: Estimates and projections to
the year 2010.
Diabetic Medicine, vol 14, p. S1-S85.
ANDERSON R.A. (1986)
Chromium metabolism and its role in disease processes in man.
Clinical Physiology and Biochemistry, vol 4, p. 31-41.
ANDERSON R.A. (1987)
Chromium.
In: Trace Elements in Human and Animal Nutrition, 5th ed. (Mertz W.).
Academic Press, San Diego, CA, p.225-244.
ANDERSON R.A., POLANSKY M., BRYDEN N.A., ROGINSKI E.E., MERTZ W.,
GLINSMAN W. (1983)
Chromium supplementation of human subjects: effects on glucose insulin and lipid
variables.
Metabolism, vol 32, p. 894-899.
ANDERSON R.A., KOZLOVSKY A.S. (1985)
Chromium intake, absorption and excretion of subjects consuming self-selected diets.
American Journal of Clinical Nutrition, vol 41, p. 1177-1183.
ANONYMOUS (2004)
Appropriate body-mass index for Asian populations and its implications for policy and
intervention strategies.
Lancet, vol 363, p.157-163.

99
APPLETON D.J., RAND J.S., SUNVOLD G.D. (2001)
Insulin sensitivity decreases with obesity, and lean cats with low insulin sensitivity are at
greatest risk of glucose intolerance with weight gain.
Journal of Feline Medicine and Surgery, vol 3, p. 211-228.
APPLETON D.J., RAND J.S., SUNVOLD G.D.., PRIEST J. (2002)
Dietary chromium tripicolinate supplementation reduces glucose concentrations and
improves glucose tolerance in normal-weight cats.
Journal of Feline Medicine and Surgery, vol 4, p. 13-25.
APPLETON D.J., RAND J.S., PRIEST J., SUNVOLD G.D., VICKERS J.R. (2004)
Dietary carbohydrate source affects glucose concentrations, insulin secretion and food
intake in overweight cats.
Nutrition Research, vol 24 (issue 6), p. 447-467.
BARAL R., RAND J., CATT M., FARROW H. (2003)
Prevalence of feline diabetes mellitus in a feline private practice (abstract).
Journal of Veterinary Internal Medicine, vol 17 (issue 3), p. 433-434.
BASS J., TAKAHASHI J.S. (2010)
Circadian integration of metabolism and energetics.
Science, vol 330, p. 1349-1354.
BERGMAN R.N., PRAGER R., VOLUND A., OLEFSKY J.M. (1987)
Equivalence of the insulin sensitivity index in man derived by the minimal model
method and the euglycaemic glucose clamp.
Journal of Clinical Investigation, vol 79, p. 790-800.
BIOURGE V., NELSON R.W., FELDMAN E.C., WILLITS N.H., MORRIS J.G., ROGERS
Q.R. (1997)
Effect of weight gain and subsequent weight loss on glucose tolerance and
insulin response in healthy cats.
Journal of Veterinary Internal Medicine/ American College of Veterinary Internal Medicine,
vol 11, p. 86-91.

100
BRAND J.C., NICHOLSON P.L., THORBURN P.W., TRUSWELL A.S. (1985)
Food processing and the glycaemic index.
American Journal of Clinical Nutrition, vol 42, p. 1192-1196.
BRAND MILLER J.C, COLAGIURI S. (1994)
The carnivore connection: dietary carbohydrate in the evolution of NIDDM.
Diabetologia, vol 37, p. 1280-1286.
BUCHWALD H., AVIDOR Y., BRAUNWALD E., JENSEN M.D., PORIES W.,
FAHRBACH K. et al. (2004)
Bariatric surgery: a systematic review and meta-analysis.
The Journal of the American Medical Association, vol 292, p. 1724-1737.
CAMPBELL I.W. (2000)
Epidemiology and Clinical Presentation of Type 2 Diabetes.
Value in Health, vol 3 (supplement 1), p. S3-S6.
CANI P.D., AMAR J., IGLESIAS M.A., POGGI M., KNAUF C., BASTELICA D., et al.
(2007)
Metabolic endotoxemia initiates obesity and insulin resistance.
Diabetes, vol 56, p. 1761-1772.
CARD J.W., MAGNUSON B.A. (2011)
A review of the efficacy and safety of nanoparticle-based oral insulin delivery systems.
American Journal of Physiology Gastrointestinal and Liver Physiology, vol 301, p. 956-967.
CAVE N.J., BACKUS R.C., MARKS S.L., KLASING K.C. (2007)
Oestradiol, but not genistein, inhibits the rise in food intake following gonadectomy in
cats, but genistein is associated with an increase in lean body mass.
Journal of Animal Physiology and Animal Nutrition, vol 91, p. 400-410.

101
CHANDRAN M., PHILLIPS S.A., CIARALDI T., HENRY R.R. (2003)
Adiponectin: more than just another fat cell hormone?
Diabetes Care, vol 26, p. 2442-2450.
CHIU K., LEE N., COHAN P., CHUANG L. (2000)
Beta cell function declines with age in glucose tolerant Caucasians.
Clinical Endocrinology, vol 53, p. 569-575.
COLAGIURI S. (2010)
Diabesity: therapeutic options.
Diabetes, Obesity and Metabolism, vol 12, p. 463-473.
COLDITZ G.A., WILLETT W.C., ROTNITZKY A., MANSON J.E. (1995)
Weight gain as a risk factor for clinical diabetes in women.
Annals of Internal Medicine, vol 122, p. 481-486.
COLLIARD L., PARAGON B.M., LEMUET, BENET J.J., BLANCHARD G. (2009)
Prevalence and risk factors of obesity in an urban population of healthy cats.
Journal of Feline Medicine and Surgery, vol 11, p. 135–140.
COURTIN-DONAS S. (2011)
Le diabète chez le chien et le chat.
Actualités pharmaceutiques, vol 509, p. 45-47.
CRENSHAW K.L., PETERSON M.E. (1996)
Pretreatment clinical and laboratory evaluation of cats with diabetes mellitus: 104 cases
(1992–1994).
Journal of American Veterinary Medical Association, vol 209, p. 943-949.
DANIELS J. (2006)
Obesity: America’s epidemic: What goes up does not always come down. Is there a
solution?
American Journal of Nursing, vol 106, p. 40-49 [quiz: 9–50].

102
DANNE T., DATZ N., ENDAHL L., HAAHR H., NESTORIS C., WESTERGAARD L. et
al. (2008)
Insulin detemir is characterized by a more reproducible pharmacokinetic profile than
insulin glargine in children and adolescents with type 1 diabetes: results from a
randomized, double-blind, controlled trial.
Pediatric Diabetes, vol 9, p. 554-560.
DEWITT D.E., HIRSCH I.B. (2003)
Outpatient insulin therapy in type 1 and type 2 diabetes mellitus: scientific review.
The Journal of the American Medical Association, vol 289, p. 2254-2264.
DINARELLO C.A., DONATH M.Y., MANDRUP-PULSEN T. (2010)
Role of IL-1beta in type 2 diabetes.
Current Opinion in Endocrinology, Diabetes, and Obesity, vol 17, p. 314-321.
DIXON J.B. (2009)
Obesity and diabetes: the impact of bariatric surgery on type-2 diabetes.
World Journal of Surgery, vol 33 (10), p. 2014-2021.
DONAGHUE S., KRONFELD D.S. (1993)
Feeding the hospitalised cat.
In: Wills J, Wolf A (ed.), Handbook of feline medicine.
Oxford: Pergamon Press, p. 42-44.
DONATH M.Y., EHSES J.A., MAEDLER K., SCHUMANN D.M., ELLINGSGAARD H.,
EPPLER E., et al. (2005)
Mechanisms of beta-cell death in type 2 diabetes.
Diabetes, vol 54 (supplement 2), p. S108-S113.
DONATH M.Y., BONI-SCHNETZLER M., ELLINGSGAARD H., HALBAN P.A., EHSES
J.A. (2010)
Cytokine production by islets in health and diabetes: cellular origin, regulation and
function.
Trends in Endocrinology and Metabolism, vol 21, p. 261-267.

103
DROCHNER W., MULLER-SCHLOSSER S. (1980)
Digestibility and tolerance of various sugars in cats.
Proceedings of the International Symposium on the Nutrition of the Dog and Cat, 1978,
Hannover, Germany.
DRUCKER D.J., NAUCK M.A. (2006)
The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-
4 inhibitors in type 2 diabetes.
Lancet, vol 368, p. 1696-1705.
DRUCKER D.J., BUSE J.B., TAYLOR K., KENDALL D.M., TRAUTMANN M.,
ZHUANG D. et al. (2008)
Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: a
randomised, open-label, non-inferiority study.
Lancet, vol 372, p. 1240-1250.
DUNCAN C., CHALMERS J., CAMPBELL I.W., JONES I.G. (1992)
An audit of non-insulin-dependent diabetes attending a district general hospital diabetic
clinic: implications for shared care between hospital and general practice.
Health Bulletin, vol 50 (issue 4), p. 302-308.
ELLINGSGAARD H., EHSES J.A., HAMMAR E.B., VAN LOMMEL L., QUINTENS R.,
MARTENS G., et al. (2008)
Interleukin-6 regulates pancreatic alpha-cell mass expansion.
Proceedings of the National Academy of Sciences of the United States of America, vol 105, p.
13163-13168.
ELLINGSGAARD H., HAUSELMANN I., SCHULER B., HABIB A.M., BAGGIO L.L.,
MEIER D.T., et al. (2011)
Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion
from L cells and alpha cells.
Nature Medicine, vol 17, p. 1481-1489.

104
ELLIOTT D.A., NELSON R.W., FELDMAN E.C., NEA L.A. (1997)
Glycosylated hemoglobin concentration for assessment of glycemic control in diabetic
cats.
Journal of Veterinary Internal Medicine, vol 11, p. 161-165.
FARROW H. A., RAND J.S., SUNVOLD G.D. (2002)
Diets high in protein are associated with lower postprandial glucose and insulin
concentrations than diets high in either fat or carbohydrate in normal cats.
Journal of Veterinary Internal Medicine, vol 16, p. 360.
FELDHAHN J., RAND J.S., MARTIN G.M. (1999)
Insulin sensitivity in normal and diabetic cats.
Journal of Feline Medicine and Surgery, vol 1, p. 107-115.
FELDMAN E.C., NELSON R.W. (2004) (a)
Feline diabetes mellitus.
In: Feldman E.C., Nelson R.W., Canine and feline endocrinology and reproduction.
St. Louis (MO): WB Saunders; p. 539–579.
FELDMAN E.C., NELSON R.W. (2004) (b)
Diabetic ketoacidosis.
In: Feldman E.C., Nelson R.W., Canine and feline endocrinology and reproduction.
St. Louis (MO): WB Saunders; p. 580–615.
FIELD A.E., COAKLEY E.H., MUST A., SPADANO J.L., LAIRD N., DIETZ W.H., et al.
(2001)
Impact of overweight on the risk of developing common chronic diseases during a 10-
year period.
Archives of Internal Medicine, vol 161, p. 1581-1586.

105
FORCADA Y., HOLDER A., JEPSON R., CHURCH D., CATCHPOLE B. (2010)
A Missense Mutation in the Coding Sequence of MC4R (MC4R:c.92 C>T) is Associated
with Diabetes Mellitus in DSH Cats.
Proceedings, European College of Veterinary Internal Medicine – Companion Animals,
Toulouse, France, 2010.
FRANK G., ANDERSON W., PAZAK H., HODGKINS E., BALLAM J., LAFLAMME D.
P. (2001)
Use of a high-protein diet in the management of feline diabetes mellitus.
Veterinary Therapeutics, vol 2, p. 238-246.
FURRER D., KAUFMANN K., TSCCHUOR F., REUSCH C., LUTZ T.A. (2010)
The dipeptidyl peptidase IV inhibitor NVPDPP728 reduces plasma glucagon
concentration in cats.
Veterinary Journal, vol 183, p. 355-357.
GARBER A.J., KING A.B., DEL PRATO S., SREENAN S., BALCI M.K., MUNOZ-
TORRES M. et al. (2012)
Insulin degludec, an ultra-long acting basal insulin, versus insulin glargine in basal-
bolus treatment with mealtime insulin aspart in type 2 diabetes (BEGIN Basal-Bolus
Type 2): a phase 3 randomized, open-label, treat-to-target non-inferiority trial.
Lancet, vol 379, p. 1498-1507.
GEARY N., TRACE D., MCEWEN B., SMITH G.P. (1994)
Cyclic estradiol replacement increases the satiety effect of CCK-8 in ovariectomized
rats.
Physiology & Behavior, vol 56, p. 281–289.
GEER E.B., SHEN W. (2009)
Gender differences in insulin resistance, body composition, and energy balance.
Gender Medicine, vol 6 (supplement 1), p. 60-75.

106
GERICH J.E. (2010)
Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes
mellitus: therapeutic implications.
Diabetic Medicine, vol 27, p. 136-142.
GILES R., GRUFFYDD-JONES T, STURGESS C. (2003)
A preliminary investigation into the effect of different strategies for achieving weight
loss in cats.
Proceedings of the 46th Annual Congress of the British Small Animal Veterinary Association.
Birmingham, UK, 2003, p. 546.
GILOR C., KEEL T., ATTERMEIER K.J., et al. (2008)
Hyperinsulinemic-euglycemic clamps using insulin detemir and insulin glargine in
healthy cats (abstract).
Journal of Veterinary Internal Medicine, vol 22 (issue 3), p. 729.
GILOR C., GRAVES T.K. (2010)
Synthetic insulin analogs and their use in dogs and cats.
The Veterinary clinics of North America. Small animal practice, vol 40 (issue 2), p. 297-307.
GILOR C., RIDGE T.K., ATTEMEIER K.J., GRAVES T.K. (2010)
Pharmacodynamics of insulin detemir and insulin glargine assessed using an isoglycemic
clamp method in healthy cats.
Journal of Veterinary Internal Medicine, vol 24 (issue 4), p. 870-874.
GILOR C., GRAVES T.K., GILOR S., R IDGE T.K., RICK M. (2011)
The GLP-1 mimetic exenatide potentiates insulin secretion in healthy cats.
Domestic Animal Endocrinology, vol 41, p. 42-49.
GOOSSENS M.M.C., NELSON R.W., FELDMAN E.C., GRIFFEY S.M. (1998)
Response to treatment and survival in 104 cats with diabetes mellitus (1985–1995).
Journal of Veterinary Internal Medicine, vol 12, p. 1-6.

107
GROENEVELD J., BEISHUIZEN A., VISSER F. (2002)
Insulin: a wonder drug in the critically ill?
Critical Care, vol 6, p. 102-105.
GRUNDY S.M., CLEEMAN J.I., DANIELS S.R., et al. (2005)
Diagnosis and management of the metabolic syndrome: an American Heart
Association/National Heart, Lung, and Blood Institute Scientific Statement.
Circulation, vol 112, p. 2735-2752.
HAFFNER S.M., LEHTO S., RONNEMAA T., PYORALA K., LAAKSO M. (1998)
Mortality from coronary heart disease in subjects with type 2 diabetes and in non-
diabetic subjects with and without prior myocardial infarction.
The New England Journal of Medicine, vol 339, p. 229-234.
HARRIS M.I. (1995)
Epidemiological studies on the pathogenesis of non-insulin dependent diabetes mellitus
(NIDDM).
Clinical and Investigative Medicine, vol 18, p. 231-239.
HARRIS R.B. (2000)
Leptin, much more than a satiety signal.
Annual Review of Nutrition, vol 20, p. 45-75.
HEISE T., PIEBER T.R. (2007)
Towards peakless, reproducible and long-acting insulins. An assessment of the basal
analogues based on isoglycaemic clamp studies.
Diabetes, Obesity and Metabolism, vol 9, p. 648–659.
HENSON M.S., O’BRIEN T.D. (2006)
Feline models of type 2 diabetes mellitus.
Institute for Laboratory Animal Research Journal/National Research Council, Institute of
Laboratory Animal Resources, vol 47, p. 234-242.

108
HOENIG M., FERGUSON D.C. (2003)
Effects of darglitazone on glucose clearance and lipid metabolism in obese cats.
American Journal of Veterinary Research, vol 64 (issue 11), p.1409-1413.
HOENIG M., THOMASETH K., BRANDAO J., WALDRON M., FERGUSON D.C. (2006)
Assessment and mathematical modeling of glucose turnover and insulin sensitivity in
lean and obese cats.
Domestic Animal Endocrinology, vol 31, p. 373-389.
HOENIG M., THOMASETH K., WALDRON M., FERGUSON D.C. (2007)
Insulin sensitivity, fat distribution, and adipocytokine response to different diets in lean
and obese cats before and after weight loss.
American Journal of Physiology, vol 292, p. R227-R234.
HOLLENBECK C., REAVEN G.M. (1987)
Variations in insulin-stimulated glucose uptake in healthy individuals with normal
glucose tolerance.
The Journal of Clinical Endocrinology and Metabolism, vol 64, p. 1169-1173.
HOLT S., BRAND J., SOVENY C., HANSKY J. (1992)
Relationship of satiety to postprandial glycaemic, insulin and cholecystokinin responses.
Appetite, vol 18, p. 129-141.
HOLT R.I., BARNETT A.H., BAILEY C.J. (2010)
Bromocriptine: old drug, new formulation and new indication.
Diabetes, Obesity and Metabolism, vol 12, p. 1048-1057.
HOTAMISLIGIL G.S. (2010)
Endoplasmic reticulum stress and the inflammatory basis of metabolic disease.
Cell, vol 140, p. 900-917.
HU F., MANSON J., STAMPFER M., COLDITZ G., LIU S., SOLOMON C. et al. (2001)
Diet, lifestyle and the risk of type 2 diabetes mellitus in women.
The New England Journal of Medicine, vol 13, p. 790-797.

109
HUDSON L.C., HAMILTON W.P. (1993)
Pancreas.
In: Hudson L.C., Hamilton W.P., Atlas of feline anatomy for veterinarians.
W.B. Saunders Company, Philadelphia, p. 165-166.
HUGUES T.E., MONE M.D., RUSSELL M.E., WELDON S.C., VILHAUER E.B. (1999)
NVP-DPP728 (1-[[[2-[(5-cyanopyridin-2-yl) amino] ethyl] amino] acetyl]-2cyano-(S)-
pyrrolidine), a slow-binding inhibitor of dipeptidy l peptidase IV.
Biochemistry, vol 38 (issue 36), p. 11597-11603.
HUXLEY R., MENDIS S., ZHELEZNYAKOV E, REDDY S., CHAN J. (2010)
Body mass index, waist circumference and waist: hip ratio as predictors of
cardiovascular risk–a review of the literature.
European Journal of Clinical Nutrition, vol 64, p. 16-22.
IOANNIDES-DEMOS L.L., PICCENNA L., MCNEIL J.J. (2011)
Pharmacotherapies for obesity: past, current, and future therapies.
Journal of Obesity, vol 2011: 179674.
INTERNATIONAL DIABETES FEDERATION (2009)
International Diabetes Federation diabetes atlas 4th edition.
Brussels (Belgium): International Diabetes Federation; 2009.
KAHN C., MEYEROVITCH J., SHETCHTER Y. (1990)
A family of polypeptide substrates and inhibitors of insulin receptor kinase.
Biochemistry, vol 29 (issue 15), p. 3654-3660.
KAHN S.E., COOPER M.E., DEL PRATO S. (2014)
Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and
future.
The Lancet, vol 383, p. 1068-1083.

110
KANCHUK M.L., BACKUS R.C., CALVERT C.C., MORRIS J.G., ROGERS Q.R. (2003)
Weight gain in gonadectomized normal and lipoprotein lipase-deficient male domestic
cats results from increase food intake and not decreased energy expenditure.
The Journal of Nutrition, vol 133, p. 1866-1874.
KELLY T., YANG W., CHEN C.S., REYNOLDS K., HE J. (2008)
Global burden of obesity in 2005 and projections to 2030.
Internal Journal of Obesity (London), vol 32, p. 1431-1437.
KHEMTEMOURIAN L., KILLIAN J.A., HOPPENER J.W., ENGEL M.F. (2008)
Recent insights in islet amyloid polypeptide-induced membrane disruption and its role
in beta cell death in type 2 diabetes mellitus.
Experimental Diabetes Research, vol 2008: 421287.
KISSEBAH A.H. (1996)
Intra-abdominal fat: is it a major factor in developing diabetes and coronary artery
disease?
Diabetes Research and Clinical Practice, vol 30, p. 25-30.
KRUTH S.A., COWGILL L.D. (1982)
Renal glucose transport in the cat (abstract).
Proceedings of the American College of Veterinary Internal Medicine Forum, p. 78.
LAMOS E.M., YOUNK L.M., DAVIS S.N. (2013)
Canagliflozin, an inhibitor of sodium-glucose cotransporter 2, for the treatment of type
2 diabetes mellitus.
Expert Opinion on Drug Metabolism and Toxicology, vol 9, p. 763-775.
LARTER C.Z., CHITTURI S., HEYDET D., FARRELL G.C. (2010)
A fresh look at NASH pathogenesis. Part 1: the metabolic movers.
Journal of Gastroenterology and Hepatology, vol 25, p. 672-690.

111
LAWSON H.A. (2013)
Chapter 11 – Animal Models of Metabolic Syndrome.
In: Conn P.M. (ed.), Animal Models for the Study of Human Disease.
Academic Press, p. 243-264.
LEDERER R., RAND J.S., HUGHES I., FLEEMAN L.M. (2003)
Chronic or recurring medical problems, dental disease, repeated corticosteroid
treatment, and lower physical activity are associated with diabetes in Burmese cats.
Journal of Veterinary Internal Medicine, vol 17, p. 433.
LI S., ZHAO J.H., LUAN J., LANGENBERG C., LUBEN R.N., KHAW K.T. et al. (2011)
Genetic predisposition to obesity leads to increased risk of type 2 diabetes.
Diabetologia, vol 54, p. 776-782.
LINK K.R.J., RAND J.S. (1998)
Reference values for glucose tolerance and glucose tolerance status in cats.
Journal of the American Veterinary Medical Association, vol 213, p. 492-496.
LINK K.R.J. (2001)
Feline diabetes: diagnostics and experimental modelling.
Doctoral thesis, Queensland: The University of Queensland, 2001. p. 58-159.
LIU J.J., LEE T., DEFRONZO R.A. (2012)
Why Do SGLT2 inhibitors inhibit only 30–50% of renal glucose reabsorption in
humans?
Diabetes, vol 61, p. 2199-2204.
LORENZO A., RAZZABONI B., WEIR G.C., YANKNER B.A. (1994)
Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus.
Nature, vol 368, p. 756-760.
LUTZ T.A., RAND J.S. (1993)
A review of new developments in type 2 diabetes in human beings and cats.
British Veterinary Journal, vol 149 (issue 6), p. 527-536.

112
LUTZ T.A., RAND J.S, WATT P., GALLOWAY A. (1994)
Pancreatic biopsy in normal cats.
Australian Veterinary Journal, vol 71, p. 223-225.
LUTZ T.A., RAND J.S. (1995)
Pathogenesis of feline diabetes mellitus.
The Veterinary Clinics of North America, vol 25, p. 527-552.
LUTZ T.A., RAND J.S., RYAN E. (1995)
Fructosamine concentrations in hyperglycemic cats.
The Canadian Veterinary Journal, vol 36, p. 155-159.
LUTZ T.A., RAND J.S. (1997)
Detection of amyloid deposition in various regions of the feline pancreas by different
staining techniques.
Journal of Comparative Pathology, vol 116, p. 157-170.
MA Z., WESTERMARK G.T., JOHNSON K.H., O’BRIEN T.D., WESTERMARK P. (1998)
Quantitative immunohistochemical analysis of islet amyloid polypeptide (IAPP) in
normal, impaired glucose, tolerant and diabetic cats.
Amyloid: The International Journal of Experimental and Clinical Investigation, vol 5, p. 255-
261.
MAEDLER K., SPINAS G.A., LEHMANN R., SERGEEV P., WEBER M., FONTANA A.,
et al. (2001)
Glucose induces beta-cell apoptosis via upregulation of the Fas receptor in human islets.
Diabetes, vol 50, p. 1683-1690.
MAEDLER K., SERGEEV P., RIS F., OBERHOLZER J., JOLLER-JEMELKA H.I.,
SPINAS G.A., et al. (2002)
Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human
pancreatic islets.
The Journal of Clinical Investigation, vol 110, p. 851-860.

113
MAEDLER K., SCHULTHESS F.T., BIELMAN C., BERNEY T., BONNY C., PRENTKI
M., et al. (2008)
Glucose and leptin induce apoptosis in human beta-cells and impair glucose stimulated
insulin secretion through activation of c-Jun N-terminal kinases.
The Federation of American Societies for Experimental Biology Journal, vol 22, p. 1905-
1913.
MANNUCCI E., MONAMI M., MARCHIONNI N. (2009)
Short-acting insulin analogues vs. regular human insulin in type 2 diabetes: a meta-
analysis.
Diabetes, Obesity and Metabolism, vol 11, p. 53-59.
MARINA A.L., UTZSCHNEIDER K.M., WRIGHT L.A., MONTGOMERY B.K.,
MARCOVINA S.M., KAHN S.E. 2012)
Colesevelam improves oral but not intravenous glucose tolerance by a mechanism
independent of insulin sensitivity and β-cell function.
Diabetes Care, vol 35, p. 1119-1125.
MARSHALL R., RAND J. (2002)
Comparison of the pharmacokinetics and pharmacodynamics of glargine, protamine
zinc and porcine lente insulins in normal cats (abstract).
Journal of Veterinary Internal Medicine, vol 16 (issue 3), p. 358.
MARSHALL R., RAND J. (2004)
Insulin glargine and a high protein–low carbohydrate diet are associated with high
remission rates in newly diagnosed diabetic cats (abstract).
Journal of Veterinary Internal Medicine, vol 18, p. 401.
MARTIN L.J., SILIART B., DUMON H.J., NGUYEN P. (2006)
Spontaneous hormonal variations in male cats following gonadectomy.
Journal of Feline Medicine and Surgery, vol 8, p. 309-314.

114
MARTIN G., RAND J. (2000)
Current understanding of feline diabetes: Part 2, treatment.
Journal of Feline Medicine & Surgery, vol 2 (issue 1), p. 3-17.
MCLAUGHLIN T., SHERMAN A., TSAO P., GONZALEZ O., YEE G., LAMENDOLA C.,
et al. (2007)
Enhanced proportion of small adipose cells in insulin-resistant vs insulin sensitive obese
individuals implicates impaired adipogenesis.
Diabetologia, vol 50, p. 1707-1715.
MCLAUGHLIN T., DENG A., YEE G., LAMENDOLA C., REAVEN G., TSAO P.S., et al.
(2010)
Inflammation in subcutaneous adipose tissue: relationship to adipose cell size.
Diabetologia, vol 53, p. 369-377.
MEALEY B., RETHMAN M. (2003)
Periodontal disease and diabetes mellitus. Bidirectional relationship.
Dentistry Today, vol 22, p. 107-113.
MILLER A.B., NELSON R.W., KIRK C.A., FELDMAN E.C. (1991)
Effect of glipizide on serum insulin and glucose concentrations in healthy cats.
Proceedings of the 9th Forum of the American College of Veterinary Internal Medicine, New
Orleans, May 1991, p. 880.
MICHELL A.R. (2005)
Comparative clinical science: The medicine of the future.
The Veterinary Journal, vol 170, p. 153-162.
MIRZABEKOV T.A., LIN M.C., KAGAN B.L. (1996)
Pore formation by the cytotoxic islet amyloid peptide amylin.
The Journal of Biological Chemistry, vol 271, p. 1988-1992.

115
MOKDAD A.H., FORD E.S., BOWMAN B.A., DIETZ W.H., VINICOR F., BALES V.F. et
al. (2003)
Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001.
The Journal of the American Medical Association, vol 289, p. 76-79.
MONAMI M., MARCHIONNI N., MANNUCCI E. (2008)
Long-acting insulin analogues versus NPH human insulin in type 2 diabetes: a meta-
analysis.
Diabetes Research and Clinical Practice, vol 81, p. 184-189.
MONKHOUSE S.J., MORGAN J.D., BATES S.E., NORTON S.A. (2009)
An overview of the management of morbid obesity.
Postgraduate Medical Journal, vol 85, p. 678-681.
MORAN L.J., MISSO M.L., WILD R.A., NORMAN R.J. (2010)
Impaired glucose tolerance, type 2 diabetes and metabolic syndrome in polycystic ovary
syndrome: a systematic review and meta-analysis.
Hum Reproduction Update, vol 16, p. 347-363.
MORI A., SAKO T., LEE P., NISHIMAKI Y., FUKUTA H., MIZUTANI H., et al. (2009)
Comparison of three commercially available prescription diet regimens on short-term
post-prandial serum glucose and insulin concentrations.
Veterinary Research Communications, vol 33, p. 669-680.
NATIONAL CHOLESTEROL EDUCATION PROGRAM (NCEP) EXPERT PANEL ON
DETECTION, EVALUATION, AND TREATMENT OF HIGH BLOOD CHOLESTEROL
IN ADULTS (ADULT TREATMENT PANEL III) (2002)
Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on
Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult
Treatment Panel III) final report.
Circulation, vol 106, p. 3143-3421.

116
NATIONAL INSTITUTES OF HEALTH (1998)
Clinical guidelines on the identification, evaluation, and treatment of overweight and
obesity in adults: the evidence report.
Obesity Research, vol 6 (supplement 2), p. S51-S209.
NEEL I.V. (1962)
Diabetes mellitus: a ‘‘thrifty’’ genotype rendered detrimental by ‘‘progress”.
American Journal of Human Genetics, vol 14, p. 353-362.
NELSON R.W., FELDMAN E.C., FORD S. (1991)
Glipizide therapy for feline diabetes mellitus.
Proceedings of the 10th Forum of the American College of Veterinary Internal Medicine, New
Orleans, May 1991, p. 880.
NELSON R.W., FELDMAN E.C. (1992)
Non-insulin-dependent diabetes mellitus in the cat.
Proceedings of the 10th Forum of the American College of Veterinary Internal Medicine, San
Diego, May 1992, p. 351-353.
NELSON R.W., GRIFFEY S.M., FELDMAN E.C., FORD S.L. (1999)
Transient clinical diabetes mellitus in cats: 10 cases (1989–1991).
Journal of Veterinary Internal Medicine/American College of Veterinary Internal Medicine,
vol 13, p. 28-35.
NOLAN C.J., DAMM P., PRENTKI M. (2011)
Type 2 diabetes across generations: from pathophysiology to prevention and
management.
The Lancet, vol 378, p. 169-181.
NIH-NHLBI (2000)
North American Association for the Study of Obesity. The practical guide:
identification, evaluation, and treatment of overweight and obesity in adults.
Bethesda (MD): National Institutes of Health; 2000.

117
OSTO M., ZINI E., FRANCHINI M., WOLFRUM C., GUSCETTI F., HAFNER M., et al.
(2011)
Subacute endotoxemia induces adipose inflammation and changes in lipid and
lipoprotein metabolism in cats.
Endocrinology, vol 152, p. 804-815.
OSTO M., ZINI E., REUSH C.E., LUTZ T.A. (2013)
Diabetes from humans to cats.
General and comparative endocrinology, vol 182, p. 48-53.
PADRUTT I., ZINI E., KAUFMANN K., et al. (2012)
Comparison of the GLP-1 analogues exenatide short-acting, exenatide long-acting and
the DPP-4 inhibitor sitagliptin to increase insulin secretion in healthy cats.
Journal of Veterinary Internal Medicine, vol 26, p. 1505-1538.
PADWAL R.S., MAJUMDAR S.R. (2007)
Drug treatments for obesity: orlistat, sibutramine, and rimonabant.
Lancet, vol 369, p. 71-77.
PANCIERA D.L., THOMAS C.B., EICKER S.W., ATKINS C.E. (1990)
Epizootiological patterns of diabetes mellitus in cats: 333 cases (1980–1986).
Journal of the American Veterinary Medical Association, vol 197, p. 1504-1508.
PORTE D. (1991)
ß-cells in type II diabetes mellitus.
Diabetes, vol 40, p. 166-180.
PRAHL A., GLICKMAN N.W., GUPTILL L., TETRICK M., GLICKMAN L.T. (2007)
Time trends and risk factors for diabetes mellitus in cats.
Journal of Feline Medicine and Surgery, vol 9, p. 351-358.

118
RACHMAN J., BARROW B.A., LEVY J.C., TURNER R.C. (1997)
Near-normalization of diurnal glucose concentrations by continuous administration of
glucagon-like peptide-1 (GLP-1) in subjects with NIDDM.
Diabetologia, vol 40, p. 205-211.
RADIN M.J, SHARKEY L.C., HOLYCROSS B.J. (2009)
Adipokines: a review of biological and analytical principles and an update in dogs, cats,
and horses.
Veterinary Clinical Pathology/American Society for Veterinary Clinical Pathology, vol 38, p.
136-156.
RAND J. (1999)
Current understanding of feline diabetes: part 1, pathogenesis.
Journal of Feline Medicine and Surgery, vol 1, p. 143-153.
RAND J.S., MARTIN G.J.W. (2001)
Management of feline diabetes.
The Veterinary Clinics of North America, Small Animal Practice, vol 31, p. 881-913.
RAND J.S., KINNAIRD E.R., BAGLIONI A., BLACKSHAW J., PRIEST J. (2002)
Acute stress hyperglycaemia in cats is associated with struggling and increased
concentrations of lactate and norepinephrine.
Journal of Veterinary Internal Medicine, vol 16, p. 123-132.
RAND J.S., FLEEMAN L.M., FARROW H.A., APPLETON D.J., LEDERER R. (2004)
Canine and feline diabetes mellitus: nature or nurture?
The Journal of Nutrition, vol 134, p. S2072–S2080.
RAND J.S., MARSHALL R.D. (2005)
Diabetes Mellitus in Cats.
The Veterinary Clinics of North America, Small Animal Practice, vol 35, p. 211-224.

119
RAVE K., POTOCKA E., BOSS A.H., MARINO M., COSTELLO D., CHEN R. (2009)
Pharmacokinetics and linear exposure of AFRESA compared with the subcutaneous
injection of regular human insulin.
Diabetes, Obesity and Metabolism, vol 11, p. 715-720.
RAVUSSIN E., VALENCIA M., ESPARZA J., BENNETT P., SCHULZ L. (1994)
Effects of a traditional lifestyle on obesity in Pima Indians.
Diabetes Care, vol 17, p. 1067-1074.
REAVEN G.M. (1988)
Role of insulin resistance in human disease.
Diabetes, vol 37, p. 1595-1607.
REUSCH C.E., PADRUTT I. (2013)
New incretin hormonal therapies in humans relevant to diabetic cats.
The Veterinary clinics of North America. Small animal practice, vol 43 (issue 2), p. 417-433.
ROBERTSON I.D. (1999)
The influence of diet and other factors on owner-perceived obesity in privately owned
cats from metropolitan Perth, Western Australia.
Preventive Veterinary Medicine, vol 40, p. 75-85.
ROGLIC G., UNWIN N., BENNETT P.H., MATHERS C., TUOMILEHTO J., NAG S. et al.
(2005)
The burden of mortality attributable to diabetes: realistic estimates for the year 2000.
Diabetes Care, vol 28, p. 2130-2135.
ROOMP K., RAND J.S. (2009)
Evaluation of detemir in diabetic cats managed with a protocol for intensive blood
glucose control (abstract).
Journal of Veterinary Internal Medicine, vol 23 (issue 3), p. 697.

120
ROSENSTOCK J., BERGENSTAL R.M., BLEVINS T.C., MORROW L.M., PRINCE M.J.,
QU Y. et al. (2013)
Better glycemic control and weight loss with the novel long-acting basal insulin
LY2605541 compared with insulin glargine in type 1 diabetes: a randomized, crossover
study.
Diabetes Care, vol 36, p. 522-528.
SALGADO D., REUSCH C., SPIESS B. (2000)
Diabetic cataracts: different incidence between dogs and cats.
Schweiz Arch Tierheilkd, vol 142, p. 349-353.
SAXENA R., VOIGHT B.F., LYSSENKO V., et al. (2007)
Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride
levels.
Science (New York, NY), vol 316, p. 1331-1336.
SALTIEL A., OLEFSKYl J. (1996)
Thiazolidinediones in the treatment of insulin resistance and type II diabetes.
Diabetes, vol 45 (issue 12), p. 1661-1669.
SCARLETT J.M., DONOGHUE S., SAIDLA J., WILLS J. (1994)
Overweight cats: prevalence and risk factors.
International Journal of Obesity and Related Metabolic Disorders, vol 18 (supplement 1), p.
S22–S28.
SCHEEN A.J. (2013)
Cardiovascular effects of gliptins.
Nature Reviews. Cardiology, vol 10, p. 73-84.
SHAMSEDDEEN H., GETTY J.Z., HAMDALLAH I.N., ALI M.R. (2011)
Epidemiology and economic impact of obesity and type 2 diabetes
Surgical Clinics of North America, vol 91 (issue 6), p. 1163-1172.

121
SCHAUER P.R., BURGUERA B., IKRAMUDDIN S., COTTAM D., GOURASH W.,
HAMAD G. et al. (2003)
Effect of laparoscopic Roux-en Y gastric bypass on type 2 diabetes mellitus.
Annals of Surgery, vol 238, p. 467-484.
SHAW J.E., SICREE R.A., ZIMMET P.Z. (2010)
Global estimates of the prevalence of diabetes for 2010 and 2030.
Diabetes Research and Clinical Practice, vol 87, p. 4-14.
SHELDON B., RUSSELL-JONES D., WRIGHT J. (2009)
Insulin analogues: an example of applied medical science.
Diabetes, Obesity and Metabolism, vol 11, p. 5-19.
SHI H., KOKOEVA M.V., INOUYE K., TZAMELI I., YIN H., FLIER J.S. (2006)
TLR4 links innate immunity and fatty acid-induced insulin resistance.
The Journal of Clinical Investigation, vol 116, p. 3015-3025.
SIEKMEIER R., SCHEUCH G. (2008)
Inhaled insulin - does it become reality?
Journal of Physiology and Pharmacology, vol 59 (supplement 6), p. 81-113.
SPELLMAN C.W. (2010)
Pathophysiology of type 2 diabetes: targeting islet cell dysfunction.
The journal of the American Osteopathic Association, vol 110, p. S2-S7.
TANIGUCHI A., NAKAI Y., DOI K., FUKUZAWA H., FUKUSHIMA M., KAWAMURA
H., et al. (1995)
Insulin sensitivity, insulin secretion, and glucose effectiveness in obese subjects: a
minimal model analysis.
Metabolism, vol 44, p. 1397-1400.
TRIBOULIN, C. (2010)
Apport du holter glycémique dans la prise en charge des chiens et des chats diabétiques.
Thèse de Doctorat Vétérinaire, Faculté de Médecine, Créteil, 102 p.

122
TURNER R.C., MILLNS H., NEIL H.A., STRATTON I.M., MANLEY S.E., MATTHEWS
D.R., et al. (1998)
Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus:
United Kingdom Prospective Diabetes Study (UKPDS 23).
British Medical Journal, vol 316, p. 823-828.
UNGER R.H., GRUNDY S. (1995)
Hyperglycaemia as an inducer as well as a consequence of impaired islet cell function
and insulin resistance: implications for the management of diabetes.
Diabetologia, vol 28, p.119-121.
UNITED KINGDOM PROSPECTIVE DIABETES STUDY GROUP (1998)
Intensive blood glucose control with sulphonylureas or insulin compared with
conventional treatment and risk of complications in patients with type 2 diabetes
(UKPDS 33).
Lancet, vol 352, p. 837-853.
UNITED KINGDOM PROSPECTIVE DIABETES STUDY GROUP (1998)
Tight blood pressure control and risk of macrovascular disease and microvascular
complications in type 2 diabetes (UKPDS 38).
British Medical Journal, vol 317, p. 703-713.
VAN DE LAAR F.A., LUCASSEN P.L., AKKERMANS R.P., VAN DE LISDONK E.H.,
RUTTEN G.E., VAN WEEL C. (2005)
α-glucosidase inhibitors for patients with type 2 diabetes: results from a Cochrane
systematic review and meta-analysis.
Diabetes Care, vol 28, p. 154-163.
VASILAKOU D., KARAGIANNIS T., ATHANASIADOU E., MAIN OU M., LIAKOS A.,
BEKIARI E. et al. (2013)
Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: a systematic review and
meta-analysis.
Annals of Internal Medicine, vol 159, p. 262-274.

123
VENEL L. (2009)
Les affections pancréatiques du chat.
Thèse de Doctorat Vétérinaire, Faculté de Médecine, Lyon, 145 p.
VERKEST K.R. (2014)
Is the metabolic syndrome a useful clinical concept in dogs? A review of the evidence.
The Veterinary Journal, vol 199 (issue 1), p. 24-30.
WADE C., GETHING M., RAND J.S. (1999)
Evidence of a genetic basis for diabetes mellitus in Burmese cats.
Journal of Veterinary Internal Medicine, vol 13, p. 269.
WALKER A.D., WEAVER A.D., ANDERSON R.S., CRIGHTON G.W., FENNELL C.,
GASKELL C.J. (1977)
An epidemiological survey of the feline urological syndrome.
Journal of Small Animal Practice, vol 18, p. 283.
WANG Y., BEYDOUN M.A., LIANG L., CABALLERO B., KUMANYIKA S.K. (2008)
Will all Americans become overweight or obese? Estimating the progression and cost of
the US obesity epidemic.
Obesity (Silver Spring), vol 16, p. 2323-2330.
WHITEHEAD J.P., RICHARDS A.A., HICKMAN I.J., MACDONALD G.A., PRINS J.B.
(2006)
Adiponectin, a key adipokine in the metabolic syndrome.
Diabetes, Obesity & Metabolism, vol 8, p. 264-280.
WHO (2006)
Definition and diagnosis of diabetes mellitus and intermediate hyperglycemia: Report of
World Health Organization / International Diabetes Federation.
WK 810.

124
WHO (2011)
Use of glycated hemoglobin (HbA1c) in the diagnosis of diabetes mellitus: abbreviated
report of a WHO consultation.
Geneva: World Health Organization, 2011.
WHO/IASO/IOTF (2000)
The Asia-Pacific perspective: redefining obesity and its treatment.
Melbourne (Australia): Health Communications Australia; 2000.
WILLIAMSON D.F., PAMUK E., THUN M., FLANDERS D., BYERS T., HEATH C.
(1995)
Prospective study of intentional weight loss and mortality in never-smoking overweight
US white women aged 40–64 years.
American Journal of Epidemiology, vol 141, p. 1128-1141.
WRIGHT E.M., LOO D.D., HIRAYAMA B.A. (2011)
Biology of human sodium glucose transporters.
Physiological Reviews, vol 91, p. 733-794.
YANO B.L., HAYDEN D.W., JOHNSON K.H. (1981)
Feline insular amyloid: incidence in adult cats with no clinicopathologic evidence of
overt diabetes mellitus.
Veterinary Pathology, vol 18, p. 310-315.
YOUNK L.M., MIKELADZE M., DAVIS S.N. (2011)
Pramlintide and the treatment of diabetes: a review of the data since its introduction.
Expert Opinion on Pharmacotherapy, vol 12, p. 1439-1451.
ZANDER M., MADSBAD S., MADSEN J.L., HOLST J.J. (2002)
Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin
sensitivity, and β-cell function in type 2 diabetes: a parallel-group study.
Lancet, vol 359, p. 824-830.

125
ZIESMER A.E., KELLY K.C., GUERRA P.A., GEORGE K.G., DUNN F.L. (2012)
U500 regular insulin use in insulin-resistant type 2 diabetic veteran patients.
Endocrine Practice, vol 18, p. 34-38.
ZINI E., OSTO M., FRANCHINI M., GUSCETTI F., DONATH M.Y., PERREN A., et al.
(2009)
Hyperglycaemia but not hyperlipidaemia causes beta cell dysfunction and beta cell loss
in the domestic cat.
Diabetologia, vol 52, p. 336-346.
ZINI E., ZANETTI R., COPPOLA L., ACKERMANN M., LUTZ T.A., REUSCH C.E.,
CAVICCHIOLI L. (2011)
Histological investigation of endocrine and exocrine pancreas in cats with diabetes
mellitus.
Proceedings, European College of Veterinary Internal Medicine – Companion Animals.
Maastricht, 6-8 September 2011
Maastricht Exhibition and Congress Centre (MECC) in Maastricht, 2012.
ZORAN D.L. (2010)
Obesity in dogs and cats: a metabolic and endocrine disorder.
The Veterinary Clinics of North America, vol 40, p. 221–239.

CHENIVESSE CELIA
TITRE : LE DIABÈTE SUCRÉ DU CHAT : UN MODÈLE EN PATHOLOGIE COMPARÉE ?
Thèse d’État de Doctorat Vétérinaire : Lyon, 26 septembre 2014
RÉSUMÉ : Le diabète sucré, encore appelé de type 2, non insulinodépendant, connaît ces dernières années une importante prévalence dans les populations humaine et féline. La comparaison des différents aspects de la maladie au sein des deux espèces permet de mettre en évidence de nombreux points communs.
Les patients et chats diabétiques sont obèses pour la majorité. Cette obésité semble largement contribuer à la résistance à l’insuline, pilier de la pathogénie de cette maladie. Une origine génétique a également été démontrée chez ces deux espèces. De plus, contrairement aux autres modèles animaux utilisés, le chat et l’homme présentent une altération fonctionnelle et une diminution du nombre de cellules ß, ainsi que des dépôts amyloïdes pancréatiques, conduisant conjointement à l’intolérance au glucose. Chez les deux espèces, le diagnostic est dans la plupart des cas une découverte fortuite, dans la mesure où les symptômes sont soit très frustes soit absents, et toujours peu spécifiques pendant plusieurs années. Cependant, les conséquences peuvent être graves à mortelles.
Si l’insuline représente le traitement largement prédominant du diabète sucré chez le chat, trois-quarts des patients humains stabilisent leur diabète à l’aide d’hypoglycémiants oraux et d’un régime alimentaire adapté (pauvre en sucres et en graisses). Dans l’espèce féline ou humaine, l’alimentation semble être l’une des pierres angulaires de la thérapeutique, depuis la prévention jusqu’à la prise en charge du diabète mellitus.
Ainsi, le chat peut probablement être considéré comme le meilleur modèle animal non primate pour le diabète sucré chez l’homme, par les nombreuses similitudes mises en évidence.
MOTS CLÉS : • diabète non-insulinodépendant • chats
• homme • modèle
JURY : Président : Monsieur le Professeur Charles THIVOLET 1er Assesseur : Monsieur le Professeur Jean-Luc CADORE 2ème Assesseur : Madame le Docteur Marine HUGONNARD
DATE DE SOUTENANCE : 26 septembre 2014
ADRESSE DE L’AUTEUR : 413, avenue Bourgelat 69280 MARCY L’ETOILE


















