
Rapport - Béatrice Nollet et Lucas Gaudissart - Mexico - Michel Calvino
Upload
vuongkhanhCategory
view
217download
0
1
Faculteit Bio-ingenieurswetenschappen
Academiejaar 2013 – 2014
Opsporen van genmutaties door middel van Sanger sequencing bij leukemieën en lymfomen
Tessa Dickele Promotor: Prof. dr. ir. Van Landschoot Anita Tutor: Dr. Sc. Nollet Friedel
Masterproef voorgedragen tot het behalen van de graad van Master of Science in de industriële wetenschappen: biochemie

2

3
Faculteit Bio-ingenieurswetenschappen
Academiejaar 2013 – 2014
Opsporen van genmutaties door middel van Sanger sequencing bij leukemieën en lymfomen
Tessa Dickele Promotor: Prof. dr. ir. Van Landschoot Anita Tutor: Dr. Sc. Nollet Friedel
Masterproef voorgedragen tot het behalen van de graad van Master of Science in de industriële wetenschappen: biochemie

iv
The author and supervisor give the permission to use this thesis for consultation and to copy parts of
it for personal use. Every other use is subject to the copyright laws, more specifically the source must
be extensively specified when using results from this thesis.
De auteur en de promotor geven de toelating deze scriptie voor consultatie beschikbaar te stellen en delen ervan te kopiëren voor persoonlijk gebruik. Elk ander gebruik valt onder de beperkingen van het auteursrecht, in het bijzonder met betrekking tot de verplichting uitdrukkelijk de bron te vermelden bij het aanhalen van resultaten uit deze scriptie.
Gent, juni 2014

v
Woord vooraf
Deze masterproef is het sluitstuk van een vierjarige opleiding in de industriële wetenschappen,
afstudeerrichting biochemie. Dit eindresultaat zou echter niet mogelijk zijn geweest zonder de hulp
van een aantal mensen. Ik wil dan ook van deze gelegenheid gebruik maken om hen hiervoor te
bedanken.
Mijn dank gaat in de eerste plaats uit naar het ziekenhuis A.Z. Sint-Jan Brugge-Oostende AV waar ik
de kans kreeg om dit onderzoek uit te voeren. Dit maakte het voor mij mogelijk om heel wat
technieken en theoretische inzichten die mij tijdens de opleiding zijn meegegeven nu in de praktijk
aan te wenden voor het uitvoeren van een wetenschappelijke studie. In het bijzonder wens ik
Dr. Sc. Friedel Nollet te bedanken voor zijn tijd, zijn nuttige inzichten, zijn kritische bemerkingen en
zijn uitstekende begeleiding. Zijn deskundige kennis was zonder twijfel een meerwaarde voor deze
paper. Daarnaast ook een woordje van dank voor ing Stefanie Vermeire. Haar additionele inzichten
zijn zeer hulpvol geweest voor de totstandkoming van deze masterproef.
Mijn onderzoek was voornamelijk verbonden aan de afdeling moleculaire biologie van het A.Z.
Sint-Jan Brugge-Oostende AV. Daar wens ik in het bijzonder de laboranten Bernadette, Astrid,
Nadine, Ellen en Rik te bedanken voor hun begeleiding. Ook de laboranten van de afdelingen HLA en
microbiologie waren steeds bereid om bij te springen waar nodig.
Ik wens ook van deze gelegenheid gebruik te maken om mijn ouders te bedanken van wie ik de kans
kreeg om deze vierjarige studie te vervolmaken. Een succesvolle beëindiging zou niet mogelijk zijn
geweest zonder hun onvoorwaardelijke steun.
Ten slotte wens ik nog mijn zus en vrienden te bedanken voor hun steun en aanmoedigingen.

vi
Abstract - English
In recent literature a lot of recurrent mutations which appear with leukemias and lymphomas are
described. The purpose of this thesis consists of developping a method to search for pointmutations
in a more efficient way.
Mutationhotspot regions have been amplified by means of PCR and a DNA sequence was achieved
via direct sanger sequencing. The gene-specific primers were prolonged with a universal
M13-sequence to facilitate the execution of the sequence analysis.
The method was validated by means of reanalyzing MPL, IDH1 and IDH2 genemutations. This
confirmed the analyses executed in the AZ Sint-Jan. Bonemarrow and bloodsamples of patient
diagnosed with MDS, MPN and MDS/MPN were examined for SETBP1-, CSF3R- en ASXL1- mutations.
A mutationfrequence of respectively 28,6% (6/21), 0% (0/38) and 19,1% (9/47) was achieved. In case
of CLL-patients a respective mutationfrequence of 12,5% (4/32) and 2,9% (1/34) was achieved for the
TP53- en NOTCH1-gene. NOTCH1-mutations were also tested with T-ALL (40%, 4/10) and MCL (0%,
0/13). In case of AML-patients c-KIT (5%, 2/40), ASXL1 (14,3%, 3/21) en DNMT3A (13,2%, 7/53)
mutations were being looked for. Patients diagnosed with LGL were examined for the STAT3-gene, in
which case a mutationfrequence of 57,1% (4/7) was achieved.
An efficient method was developed to detect pointmutations in mutationhotspots. However, the
method has a limited sensitivity so that approximately 20 % of mutant DNA has to be present before
it can be detected. Furthermore, the method does not allow one to look for genemutations outside
mutationhotspots. This optimalized and validated mutationanalyses are from now onwards routinely
performed in the lab, pending the introduction of a new method for extended mutationanalysis by
means of Next Generation Sequencing.
KEY WORDS: Mutation analysis, Sanger sequencing, leukemias and lymphomas, gene mutations

vii
Abstract
In de recente literatuur werden heel wat recurrente mutaties beschreven die voorkomen bij
leukemieën en lymfomen. Het doel van deze thesis is om een methode te ontwikkelen om
puntmutaties op een efficiënte manier op te sporen.
Mutatiehotspot regio’s worden met PCR geamplificeerd, en een DNA-sequentie wordt bekomen door
directe Sanger sequencing. De gen-specifieke primers werden met een universele 5’ M13-sequentie
verlengd om de uitvoering van de sequentie analyse te vereenvoudigen.
De methode werd gevalideerd door heranalyse van MPL, IDH1 en IDH2 genmutaties, als bevestiging
van de analyses uitgevoerd in het AZ Sint-Jan. Beenmerg of bloedstalen van patiënten met een
diagnose van MDS, MPN en MDS/MPN werden onderzocht naar SETBP1-, CSF3R- en ASXL1-mutaties.
Een respectievelijke mutatiefrequentie van 28,6% (6/21), 0% (0/38) en 19,1% (9/47) werd bekomen.
Bij CLL-patiënten werd voor het TP53- en NOTCH1-gen een mutatiefrequentie van respectievelijk
12,5% (4/32) en 2,9% (1/34) bekomen. NOTCH1-mutaties werden eveneens getest bij T-ALL (40%,
4/10) en MCL (0%, 0/13). Bij AML-patiënten werden c-KIT (5%, 2/40), ASXL1 (14,3%, 3/21) en
DNMT3A (13,2%, 7/53) mutaties opgespoord. Het STAT3-gen werd onderzocht bij de diagnose van
LGL, waarbij een mutatiefrequentie bekomen werd van 57,1% (4/7).
Een efficiënte methode werd ontwikkeld voor het opsporen van puntmutaties in mutatiehotspots.
De methode heeft echter een beperkte gevoeligheid, ongeveer 20% mutant DNA dient aanwezig te
zijn. Bovendien laat de methode niet toe genmutaties op te sporen buiten mutatiehotspots. Deze
geoptimaliseerde en gevalideerde mutatieanalyses worden vanaf heden routinematig uitgevoerd in
het laboratorium, dit in afwachting van de introductie van een nieuwe methode voor uitgebreide
mutatieanalyse door middel van Next Generation Sequencing.
SLEUTELWOORDEN: Mutatie-analyse, Sanger sequencing, leukemieën en lymfomen, genmutaties

1
Inhoudsopgave
Woord vooraf ........................................................................................................................................................... v
Abstract – Engels ..................................................................................................................................................... vi
Abstract ................................................................................................................................................................. vii
Lijst met gebruikte afkortingen ............................................................................................................................... 3
Lijst met figuren ...................................................................................................................................................... 6
Lijst met tabellen ..................................................................................................................................................... 8
Inleiding ................................................................................................................................................................. 10
Hoofdstuk I: Literatuurstudie
1 Mutaties ....................................................................................................................................................... 12
1.1 Soorten mutaties ................................................................................................................................. 12
1.2 Gevolgen van mutaties ........................................................................................................................ 12
1.3 Opsporen van mutaties ....................................................................................................................... 13
1.4 Nomenclatuur ...................................................................................................................................... 13
2 Mutaties en kanker ...................................................................................................................................... 14
3 World Health Organization (WHO) classificatie van hematologische neoplasma’s ..................................... 16
4 Genen die in verband kunnen gebracht worden met leukemieën en lymfomen ........................................ 17
4.1 Genmutaties bij MPN, MDS en MDS/MPN .......................................................................................... 17
4.2 Genmutaties bij chronische lymfatische leukemie .............................................................................. 33
4.3 Genmutaties bij T-cel acute lymfoblastaire leukemie ......................................................................... 39
4.4 Genmutaties bij mantelcellymfoom .................................................................................................... 40
4.5 Genmutaties bij acute myeloïde leukemie .......................................................................................... 42
4.6 Genmutaties bij large granulaire lymfocytaire leukemie .................................................................... 49
4.7 Genmutaties bij lymfoplasmocytair lymfoom ..................................................................................... 51
5 Implementatie van nieuwe mutaties als merkers ........................................................................................ 55

2
Hoofdstuk II: Materialen en methoden
1 Algemeen ..................................................................................................................................................... 57
2 PCR-reactie ................................................................................................................................................... 58
3 Controle PCR-amplificatie – agarosegelelektroforese.................................................................................. 61
4 Zuivering van het PCR-product - ExoSAP-it® ................................................................................................. 63
5 Sequentiereactie .......................................................................................................................................... 64
6 Zuivering van de sequentieproducten - Sephadex kolomzuivering ............................................................. 66
7 Capillaire Elektroforese ................................................................................................................................ 67
8 Interpretatie van de sequenties ................................................................................................................... 68
Hoofdstuk III: Resultaten en bespreking
1 Screening van het MPL-gen .......................................................................................................................... 70
2 Screening van het IDH1- en IDH2-gen .......................................................................................................... 71
3 Screening van het TP53-gen ......................................................................................................................... 72
4 Screening van het SETBP1-gen ..................................................................................................................... 74
5 Screening van het STAT3-gen ....................................................................................................................... 75
6 Screening van het DNMT3A-gen .................................................................................................................. 76
7 Screening van het CSF3R–gen ...................................................................................................................... 77
8 Screening van het NOTCH1-gen ................................................................................................................... 78
9 Screening van het ASXL1–gen ...................................................................................................................... 79
10 Screening van het c-KIT–gen .................................................................................................................... 83
11 Screening van het MYD88–gen ................................................................................................................ 84
12 Vergelijkende studie met de literatuur .................................................................................................... 85
Algemeen besluit .................................................................................................................................................. 89
Referentielijst ........................................................................................................................................................ 93
Bijlage I: Overzicht positieve stalen .................................................................................................................... 103
Bijlage II: Overzicht vergelijkende studie MPL .................................................................................................... 117
Bijlage III: Overzicht alle stalen ........................................................................................................................... 123

3
Lijst met gebruikte afkortingen
A
α-KG α-ketoglutaraat
ABL Abelson-gen
aCML Atypische chronische myeloïde leukemie
ALL Acute lymfatische leukemie
AML Acute myeloïde leukemie
ANK-domein Ankyrin repeat domein
ASX Additional sex combs
ASXL1 Additional seks comb-like 1
B
BCR Breekpunt cluster regio
Bp Basenparen
C
CBF Core binding factor
CD Cluster of differentiation
CLL Chronische lymfatische leukemie
CML Chronische myeloïde leukemie
CMML Chronische myelomonocytaire leukemie
CNL Chronische neutrofiele leukemie
CSF3R Colony Stimulerende Factor 3-Receptor
D
DNMT3A DNA (cytosine-5)-methyltransferase 3A
E
EGF Epidermale groeifactor
ET Essentiële thrombocytemie
F
FN Fibronectine
G
GDH Glutamaat dehydrogenase
H
HG Hydroxyglutaraat
HD-domein Heterodimerisatie domein
I
IDH1 Isocitraat dehydrogenase 1
IDH2 Isocitraat dehydrogenase 2
Ig Immunoglobuline
IL Interleukinen

4
IRAK Interleukine receptor geassocieerd kinase
ISM Indolente systemische mastocytose
J
JAK Janus kinase
K
KDM Histon lysine demethylase
L
LGL Large granulaire lymfocytaire leukemie
LNR Lin-12-Notch repeats
LPL Lymfoplasmocytair lymfoom
M
MCL Mantelcellymfoom
MDS Myelodysplastisch syndroom
MDS/MPN Myelodysplastische/myeoloproliferatieve neoplasma’s
MPL Myeloproliferatief leukemie virus oncogene
MPN Myeloproliferatieve neoplasma
MYD88 Myeloid differentiation primary response gene 88
MYH Myosine heavy chain
N
NHL Non-Hodgkin-lymfoom
NK-cel Natural killer cel
NLS-domein Nuclear localization sequence domein
O
OPA-domein opposite paired domein
P
PCR Polymerase kettingreactie
PHD Plant homeodomein
PMF Primaire myelofibrose
PP2A Proteïne fosfatase 2
PV Polycythemia vera
R
RAM-domein RBP-Jkappa-associated module domein
S
SCF Stamcel groeifactor
SETBP1 SET binding proteïne 1
SH2-domein Scr Homology 2-domein
STAT3 Signal Transducer and Activator of Transcription
SM Systemische mastocytose
SNP Single nucleotide polymorphism

5
T
TET Ten-eleven-translocatie
T-ALL T-cel acute lymfoblastaire leukemie
TIR Toll-interleukine-1-receptor
TLR Toll-like receptoren
W
WHO World Health Organisation
WM Waldenström macroglobulinemie

6
Lijst met figuren
Hoofdstuk I: Literatuurstudie
Figuur 1: Voorstelling van de hematopoëse (All Things Stem Cell 2013) 16
Figuur 2: Chronische Neutrofiele Leukemie – Perifeer bloed (Scholten 2013) 18
Figuur 3: Chronische Neutrofiele Leukemie – Beenmerg (Scholten 2013) 18
Figuur 4: Dysplasie perifeer bloed (Scholten 2013) 19
Figuur 5: Dysplasie perifeer bloed: reuzentrombocyten (Scholten 2013) 19
Figuur 6: Atypische Chronische Myeloïde Leukemie- Perifeer bloed (Scholten 2013) 20
Figuur 7: Voorstelling van het ASXL1-eiwit en het mutatiespectrum (Schnittger et al. 2013) 22
Figuur 8: Vergelijking tussen de prognose van MDS-patiënten met wild type ASXL1 en
mutant ASXL1 (Chen et al. 2014) 23
Figuur 9: c-KIT receptor dimeriseert in de aanwezigheid van het ligand SCF en initieert op
deze manier de signaalcascade (Royster 2010) 24
Figuur 10: Voorstelling van de ligand-afhankelijke en ligand-onafhankelijke activatie van
c-KIT (Verstovsek 2013) 25
Figuur 11: Hematopoietische groeifactor signaaltransductie (Kaushansky 2006; Vainchenker and
Constantinescu 2013) 26
Figuur 12: Opbouw van het CSF3R-gen met het mutatiespectrum (Maxson et al. 2013) 27
Figuur 13: Voorstelling twee grote groepen mutaties bij CSF3R (Maxson et al. 2013) 27
Figuur 14: MPL-signaalpathway en negatieve regulatoren (Modlich et al. 2013) 29
Figuur 15: Mutatiespectrum van het MPL-gen (gebaseerd Chaligne et al. 2008) 30
Figuur 16: Vergelijking van de overall survival van MPN-patiënten met en zonder MPL- en
JAK2-mutaties (Paradanani et al. 2011) 30
Figuur 17: Voorstelling mutaties bij het SETBP1-eiwit (Cristobal et al. 2010) 31
Figuur 18: Opbouw van het SETBP1-eiwit met het mutatiespectrum (Makishima et al. 2013) 32
Figuur 19: Vergelijking overlevingskansen tussen aCML-patiënten SETBP1 wild type en
gemuteerd en gemuteerd (Piazza et al. 2013) 33
Figuur 20: Vergelijking overlevingskansen tussen CMML-patiënten met wild type en mutant
SETBP1 (Damm et al. 2013) 33
Figuur 21: Chronische lymfatische leukemie (Scholten 2013) 34
Figuur 22: Chronische lymfatische leukemie (Scholten 2013) 34
Figuur 23: Voorstelling van het TP53-gen met mutatiespectrum (Soussi 2012a) 35
Figuur 24: Vergelijking van progressie vrije fractie tussen CLL-patiënten met mutant en
wild type TP53 (Zenz et al. 2010) 36
Figuur 25: Vergelijking tussen de overlevingskansen van CLL-patiënten met en zonder
mutaties in het P53-gen (Zenz et al. 2010) 36
Figuur 26: Schematische voorstelling van de NOTCH1-signalering in T-cel voorlopers
(Van Vlierberghe en Ferrando 2012) 37
Figuur 27: Schematische voorstelling van de domein architectuur van een humane
NOTCH-receptor (Sala et al. 2012) 38
Figuur 28: Voorstelling NOTCH1-gen en -eiwit met mutatiespectrum bij CLL (Fabbri et al. 2011) 39

7
Figuur 29: Voorstelling NOTCH1-eiwit met mutatiespectrum bij T-ALL (Zhu et al. 2006) 40
Figuur 30: Voorstelling NOTCH1-eiwit met mutatiespectrum bij MCL (Parekh 2012) 41
Figuur 31: Prognostisch effect van NOTCH1-mutaties bij MCL (Parekh 2012) 41
Figuur 32: Mutatiespectrum van het DNMT3A-gen (Roller et al. 2013) 45
Figuur 33: Schematische voorstelling van de productie en het gebruik van α-ketoglutaraat
in humane cellen (Yang et al. 2012) 46
Figuur 34: IDH1/2 mutaties inhiberen zowel histon- als DNA-demethylaties (Yang et al. 2012) 47
Figuur 35: Vergelijking overlevingskansen tussen AML-patiënten WT en gemuteerd ASXL1-gen
(Schnittger et al. 2013) 48
Figuur 36: STAT3-signaalpathway (Yu et al. 2007) 50
Figuur 37: Opbouw van het STAT3-eiwit met het mutatiespectrum (Makishima et al. 2013) 51
Figuur 38: MYD88-signaalpathway (O'Neill en Bowie 2007) 53
Hoofdstuk II: Materialen en methoden
Figuur 39: Opeenvolging van de verschillende stappen voor de mutatieanalyse 58
Figuur 40: Principe ExoSAP-IT® (Affymetrix 2013) 63
Figuur 41: Voorstelling voor het aanmaken van Sephadex-kolommen (Sheer et al. 1997) 67
Figuur 42: Multiscreen HV plaat (Millipore 2014) 67
Figuur 43: Voorstelling Multiscreen Align Frame Blue (Sheer et al. 1997) 67
Figuur 44: 3500xL Genetic Analyzer (Life Technologies 2014) 67
Figuur 45: ABI 3130 Genetic Analyzer (Life Technologies 2014) 67
Hoofdstuk III: Resultaten en bespreking
Figuur 46: Overzicht controle van de PCR-amplificatie d.m.v. agarosegelektroforese 69
Figuur 47: Voorstelling c.1934dupG (p.Gly646TrpfsX12) - patiënt 9L17 0765 80
Figuur 48: Voorstelling van het verdwijnen van de p.Gly646TrpfsX12-mutatie bij remissie
(gebaseerd op Schnittger et al. 2013) 81
Figuur 49: Voorstelling p.Gly646TrpfsX12-mutatie bij diagnose (2009) –
Patiënt VM - Staal 9C23 1163 81
Figuur 50: Voorstelling p.Gly646TrpfsX12-mutatie bij follow up (2014) –
Patiënt VM - Staal DD30 0723 81
Figuur 51: Vergelijking van de gevonden mutatiefrequentie en de mutatiefrequenties
beschreven in de literatuur per gen en per diagnose 85

8
Lijst met tabellen
Hoofdstuk I: Literatuurstudie
Tabel 1: Nomenclatuur sequentievariaties op DNA-niveau (op basis van den Dunnen
en Antonarakis 2000; Ogino et al. 2007) 14
Tabel 2: Vergelijking voorkomen van aanwezigheid van een ASXL1-mutatie voornamelijk
bij MPN, AML, MDS en MDS/MPN in verschillende publicaties 23
Tabel 3: Vergelijking voorkomen van aanwezigheid van een c-KIT-mutatie bij mastocytose
in verschillende publicaties 25
Tabel 4: Vergelijking voorkomen van aanwezigheid van een CSF3R-mutatie voornamelijk
bij CNL, aCML en CMML in verschillende publicaties 28
Tabel 5: Vergelijking voorkomen van aanwezigheid van een MPL-mutatie voornamelijk
bij myeloproliferatieve aandoeningen in verschillende publicaties 30
Tabel 6: Vergelijking voorkomen van aanwezigheid van een SETBP1-mutatie bij specifieke
ziektebeelden in verschillende publicaties 32
Tabel 7: Vergelijking voorkomen van aanwezigheid van een TP53-mutatie bij Chronische
Lymfatische Leukemie in verschillende publicaties 36
Tabel 8: Vergelijking voorkomen van aanwezigheid van een NOTCH1-mutatie bij CLL in
verschillende publicaties 39
Tabel 9: Vergelijking voorkomen van aanwezigheid van een NOTCH1-mutatie bij T-ALL in
verschillende publicaties 40
Tabel 10: Vergelijking voorkomen van aanwezigheid van een NOTCH1-mutatie bij MCL in
verschillende publicaties 41
Tabel 11: Vergelijking voorkomen van aanwezigheid van een c-KIT-mutatie voornamelijk
bij AML in verschillende publicaties 42
Tabel 12: Vergelijking voorkomen van aanwezigheid van een DNMT3A-mutatie bij MPN en
AML in verschillende publicaties 44
Tabel 13: Vergelijking voorkomen van aanwezigheid van een IDH1/IDH2-mutatie
voornamelijk bij AML in verschillende publicaties 47
Tabel 14: De meest voorkomende mutaties in het IDH1- en IDH2-gen met betrekking tot
AML, met in het vet aangeduid de meest voorkomende mutaties
(Paschka et al. 2010; Patel et al. 2011a; Patel et al. 2011b; Ashraf et al. 2013) 47
Tabel 15: Vergelijking voorkomen van aanwezigheid van een ASXL1-mutatie bij AML in
verschillende publicaties 48
Tabel 16: Vergelijking voorkomen van aanwezigheid van een CSF3R-mutatie bij AML
in verschillende publicaties 49
Tabel 17: Vergelijking voorkomen van aanwezigheid van een STAT3- mutatie bij LGL en
NK-LGL in verschillende publicaties 51
Tabel 18: Vergelijking voorkomen van een MYD88-mutatie bij Lymfoplasmocytair Lymfoom
in verschillende publicaties 54
Tabel 19: Overzichtstabel van de reeds uitgevoerde testen en met implementatie van de
voorgestelde nieuwe mutatieanalyses 55
Tabel 20: Overzichtstabel van de reeds uitgevoerde testen en met implementatie van de
voorgestelde nieuwe mutatieanalyses - vervolg 56

9
Hoofdstuk II: Materialen en methoden
Tabel 21: Overzicht gebruikte primers 60
Tabel 22: Gebruikte primers tijdens de sequentiereacties 65
Hoofdstuk III: Resultaten en bespreking
Tabel 23: Overzicht gevonden mutaties in het MPL-gen 70
Tabel 24: Overzicht gevonden mutaties in het IDH1- en IDH2-gen 72
Tabel 25: Overzicht gevonden mutaties in het TP53-gen 73
Tabel 26: Overzicht gevonden mutaties in het SETBP1-gen 74
Tabel 27: Overzicht gevonden mutaties in het STAT3-gen 75
Tabel 28: Overzicht gevonden mutaties in het DNMT3A-gen 76
Tabel 29: Overzicht gevonden mutaties in het CSF3R-gen 78
Tabel 30: Overzicht gevonden mutaties in het NOTCH1-gen 78
Tabel 31: Overzicht gevonden mutaties in het ASXL2-gen 80
Tabel 32: Overzicht gevonden mutaties in het c-KIT-gen 83
Tabel 33: Overzicht gevonden mutaties in het MYD88-gen 84
Tabel 34: Overzicht van de gevonden mutatiefrequentie en de in de literatuur beschreven
mutatiefrequenties met eventueel een verklaring voor het verschil tussen beide
mutatiefrequenties 86
Tabel 35: Overzicht van de gevonden mutatiefrequentie en de in de literatuur beschreven
mutatiefrequenties met eventueel een verklaring voor het verschil tussen beide
mutatiefrequenties - vervolg 87
Tabel 36: Overzicht van de gevonden co-mutaties 88

10
Inleiding
Kanker heeft een enorme impact op het leven van patiënten en hun omgeving. Het is na hart- en
vaatziekten de belangrijkste natuurlijke doodsoorzaak in België (Nationaal Instituut voor de
Statistiek). Jaarlijks overlijden zo’n acht miljoen personen aan de verscheidene vormen van kanker
(WHO 2012). Daarnaast vergen de courante behandelingsmethoden van kanker (chemotherapie,
bestraling en chirurgie) het uiterste van de patiënt op zowel fysiek als mentaal vlak. Desalniettemin is
de overlevingskans bij vele kankers nog steeds gering. Een cruciale factor hierbij is het tijdstip waarop
de ziekte wordt vastgesteld. Hoe sneller de correcte diagnose gesteld wordt, des te sneller kan de
gepaste behandeling gestart worden. Dit resulteert in verhoogde overlevingskansen voor de patiënt
(WHO 2012).
Kanker valt niet onder één noemer te brengen, het is een verzamelnaam voor meer dan honderd
verschillende ziekten, die één gemeenschappelijk kenmerk delen: een ongecontroleerde en
ongeremde deling van lichaamscellen. Momenteel zijn de belangrijkste types van kwaadaardige
kankers carcinomen, sarcomen, melanomen, lymfomen en leukemieën, waarbij het onderscheid
wordt gemaakt op basis van welke cel zich differentieert tot een kankercel. Leukemieën en lymfomen
vormen de focus van deze masterproef. Het zijn beiden bloedkankers waarbij geen echte
tumorvorming plaatsvindt, maar een overdreven deling van de witte bloedcellen, die zich
onmiddellijk in het hele lichaam kunnen verspreiden (Movva 2013).
Centraal in het ontstaan van kanker staan mutaties in het DNA, meer bepaald in genen die betrokken
zijn bij het reguleren en controleren van de celdeling. Het zijn combinaties van dit soort mutaties die
uiteindelijk leiden tot de ontwikkeling van kanker. Verschillen in gemuteerde gencombinaties leiden
tot verschillende ziektebeelden.
Er is bovendien een duidelijk verband tussen deze mutaties en het voorkomen van een welbepaalde
vorm van kanker, wat perspectieven biedt voor het (voortijdig) opsporen van de ziekte. Tot op
vandaag worden de meeste kankers immers gediagnosticeerd op basis van morfologische,
immunofenotypische of cytogenische principes. Dit maakt het vaak moeilijk om onderscheid te
maken tussen de verschillende ziektebeelden. Bovendien is het stellen van dergelijke diagnoses vaak
gebaseerd op exclusie, wat soms kostbare tijd in beslag neemt. Wanneer de ziekte echter gelinkt kan
worden aan het voorkomen van een bepaalde mutatie, een moleculaire merker, kan het stellen van
de diagnose mogelijk versneld worden. Dit leidt vervolgens tot een snellere start van een effectieve
behandeling, wat de overlevingskans van de patiënt drastisch vergroot. Anderzijds kan de
aanwezigheid van een specifieke mutatie ook als extra controle dienen.
Het AZ Sint-Jan Brugge maakt voor een aantal gevallen reeds gebruik van moleculaire merkers. Toch
worden een aantal diagnosen nog steeds gesteld op basis van exclusie. Om deze reden is het
interessant voor het ziekenhuis om op zoek te gaan naar nieuwe moleculaire merkers. Het doel van
deze masterproef is dan ook om een methode te ontwikkelen die in staat is om de in de literatuur
beschreven puntmutaties op een efficiënte manier te kunnen opsporen. Voor deze mutatieanalyse
wordt gebruik gemaakt van Sanger sequencing.
Deze thesis is opgebouwd uit drie hoofdstukken. Het eerste hoofdstuk handelt over de theoretische
achtergrond. Hierin wordt dieper ingegaan op mutaties in het algemeen en het verband tussen het

11
voorkomen van mutaties en kanker. Er wordt beschreven welke genen in verband gebracht worden
met een specifieke vorm van bloedkanker. Er wordt telkens wat dieper ingegaan op de ziekte en op
de genen. De nadruk hierbij ligt vooral op het belang van het opsporen van de mutatie. In een
tweede hoofdstuk worden de materialen en methoden beschreven die tijdens het uitvoeren van dit
onderzoek gebruikt werden. In een laatste hoofdstuk worden de bekomen resultaten van de
mutatieanalyses besproken.

12
Hoofdstuk I: Literatuurstudie
1 Mutaties
Een mutatie is een verandering in het erfelijk materiaal van een organisme, dit kan zowel in het DNA
als in het RNA. De nucleotidevolgorde wordt een sequentie genoemd. Met een mutatie wordt een
verandering in deze nucleotidevolgorde of sequentie bedoeld. Er bestaan verschillende soorten
mutaties, deze worden hieronder verduidelijkt.
1.1 Soorten mutaties
Een eerste soort zijn de genmutaties. Er wordt gesproken over genmutaties als de mutatie slechts
één of slechts enkele nucleotiden betreft. Tot deze groep behoren als eerste de puntmutaties. Hierbij
wordt het ene nucleotide uitgewisseld voor een ander nucleotide. Hiertoe behoren eveneens kleine
deleties en inserties. Bij een deletie worden één of enkele nucleotiden uit de sequentie verwijderd,
terwijl bij een insertie net het omgekeerde gebeurt, één of enkele nucleotiden worden aan de
sequentie toegevoegd. Als laatste behoren tot de genmutaties eveneens de inversie mutaties.
Inversie houdt in dat een stukje van de DNA-streng is losgeraakt en achterstevoren terug is
geïntegreerd (Loewe 2008).
Als tweede soort zijn er de chromosoommutaties. Een chromosoommutatie treedt op als een
chromosoom van structuur verandert. Dit kan onder andere door een verkeerde deling van het
chromosoom, door een deletie waarbij een deel van het chromosoom verloren gaat, door een
translocatie of door een duplicatie (Loewe 2008).
Als laatste zijn er de genoommutaties. Bij een genoommutatie zijn er meer of minder chromosomen
dan het normale aantal van 46 aanwezig (Loewe 2008).
1.2 Gevolgen van mutaties
Grote delen van het DNA, genaamd junk DNA, hebben (nog) geen duidelijke functie. Mutaties in deze
delen hebben meestal weinig gevolgen: ze veranderen het genotype, maar niet het fenotype. Een
dergelijke onschuldige mutatie wordt ook wel een polymorfisme genoemd. Wanneer een mutatie
echter optreedt in een coderend gedeelte van een gen, kunnen er wel duidelijke gevolgen zijn.
Mutaties kunnen een gevolg hebben voor de structuur van eiwitten. Puntmutaties kunnen een codon
veranderen, met als mogelijk gevolg een silent mutatie, nonsense mutatie of missense mutatie. Bij
een silent mutatie codeert het nieuwe codon voor hetzelfde aminozuur, zodat deze mutatie geen
gevolgen heeft voor de structuur en functie van het eiwit. Dit soort puntmutatie wordt ook wel een
single nucleotide polymorfisme (SNP) genoemd. Bij een nonsense mutatie codeert het nieuwe codon
voor een stopcodon, waardoor het eiwit ingekort wordt en al dan niet zijn functie verliest. Als laatste
zijn er de missense mutaties. Hierbij codeert het nieuwe codon voor een ander aminozuur. De
gevolgen zijn niet altijd even duidelijk, afhankelijk van het soort aminozuur dat gemuteerd wordt,
kan de functie van het eiwit al dan niet veranderen. Wanneer deze functie niet verandert, wordt
gesproken van een SNP, terwijl een mutatie duidt op een functieverandering ten gevolge van een

13
mutatie. Bij een deletie of een insertie kan het gaan over een verwijdering of toevoeging van één of
meerdere nucleotiden of aminozuren. Een insertie of deletie kan eveneens een verschuiving van het
reading frame tot gevolg hebben, waardoor de eigenschappen van het al dan niet ontstane eiwit
volledig veranderen (Loewe 2008).
1.3 Opsporen van mutaties
Vaak wordt de aanwezigheid van een specifieke mutatie in verband gebracht met een welbepaald
ziektebeeld. Het opsporen van deze mutaties kan dus helpen bij het stellen van een diagnose.
Momenteel worden in het AZ Sint-Jan Brugge reeds verschillende mutaties opgespoord die in
verband kunnen gebracht worden met een specifiek ziektebeeld. De gebruikte techniek is afhankelijk
van het soort mutatie dat dient gelokaliseerd te worden. Hierbij wordt een onderscheid gemaakt
tussen drie verschillende types. Ten eerste zijn er de puntmutaties. Deze worden meestal
opgespoord met behulp van Sanger sequencing gecombineerd met capillaire elektroforese. Als
tweede type mutaties zijn er de deleties en inserties. Deze kunnen opgespoord worden door middel
van fragmentanalyse m.b.v. capillaire elektroforese. Fragmentanalyse is de overkoepelende naam
voor de technologieën waarbij DNA-fragmenten volgens lengte gescheiden worden en waarbij hun
grootte en hoeveelheid bepaald wordt. Als laatste type mutaties die reeds onderzocht worden zijn er
de chromosoommutaties. Deze mutaties kunnen opgespoord worden via karyotypering en FISH
(Fluorescent In Situ Hybridization). Deze laatste is een techniek waarbij chromosoomdelen of
chromosomen fluorescent gekleurd worden en vervolgens onder een fluorescentie microscoop
bestudeerd worden.
In tabel 19 en 20 op het einde van de literatuurstudie wordt een overzicht weergegeven van de
testen die uitgevoerd worden in het AZ Sint-Jan Brugge bij het vermoeden van een specifieke
diagnose. Uit de tabel kan besloten worden dat voor een aantal diagnosen nog geen moleculaire
merkers gevonden zijn, bijvoorbeeld voor atypische chronische myeloïde leukemie, chronische
myelomonocytaire leukemie en large granulaire lymfocytaire leukemie. Het valt tevens op dat de
meeste merkers die onderzocht worden niet specifiek zijn voor een bepaalde diagnose. Het is dus
belangrijk om op zoek te gaan naar nieuwe moleculaire merkers die in verband kunnen gebracht
worden met een specifieke diagnose, zodat op een snellere en efficiënte manier de correcte
diagnose kan gesteld worden.
1.4 Nomenclatuur
Een uniforme beschrijving van mutaties is ten eerste nodig om een efficiënte en nauwkeurige
rapportering te verzekeren en ten tweede om de vastgestelde stijging in sequentievariatie correct te
controleren, te documenteren en op te slaan om toekomstige raadpleging te vereenvoudigen
(Whitfield 2013).
Sequentievariaties worden het best beschreven op DNA-niveau. Om verwarring te vermijden wordt
het nucleotidenummer voorafgegaan door ‘g.’ wanneer een genomische referentiesequentie
gebruikt is, door een ’c.’ wanneer een coderende DNA-sequentie gebruikt is of door een ‘m.’
wanneer een mitochondriale sequentie gebruikt is als referentie. De A van de ATG-initiator
methionine wordt nucleotide +1 genoemd. De nucleotide in de 5’-richting ten opzichte van dit
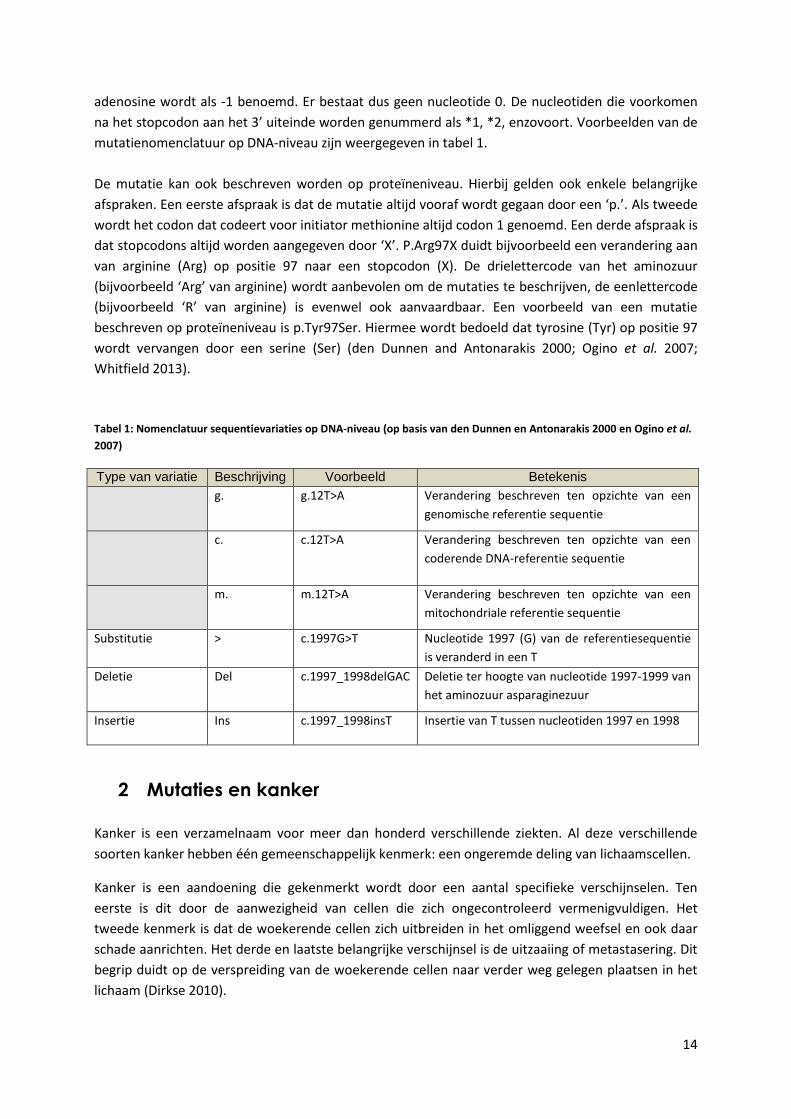
14
adenosine wordt als -1 benoemd. Er bestaat dus geen nucleotide 0. De nucleotiden die voorkomen
na het stopcodon aan het 3’ uiteinde worden genummerd als *1, *2, enzovoort. Voorbeelden van de
mutatienomenclatuur op DNA-niveau zijn weergegeven in tabel 1.
De mutatie kan ook beschreven worden op proteïneniveau. Hierbij gelden ook enkele belangrijke
afspraken. Een eerste afspraak is dat de mutatie altijd vooraf wordt gegaan door een ‘p.’. Als tweede
wordt het codon dat codeert voor initiator methionine altijd codon 1 genoemd. Een derde afspraak is
dat stopcodons altijd worden aangegeven door ‘X’. P.Arg97X duidt bijvoorbeeld een verandering aan
van arginine (Arg) op positie 97 naar een stopcodon (X). De drielettercode van het aminozuur
(bijvoorbeeld ‘Arg’ van arginine) wordt aanbevolen om de mutaties te beschrijven, de eenlettercode
(bijvoorbeeld ‘R’ van arginine) is evenwel ook aanvaardbaar. Een voorbeeld van een mutatie
beschreven op proteïneniveau is p.Tyr97Ser. Hiermee wordt bedoeld dat tyrosine (Tyr) op positie 97
wordt vervangen door een serine (Ser) (den Dunnen and Antonarakis 2000; Ogino et al. 2007;
Whitfield 2013).
Tabel 1: Nomenclatuur sequentievariaties op DNA-niveau (op basis van den Dunnen en Antonarakis 2000 en Ogino et al.
2007)
Type van variatie Beschrijving Voorbeeld Betekenis
g. g.12T>A Verandering beschreven ten opzichte van een
genomische referentie sequentie
c. c.12T>A Verandering beschreven ten opzichte van een
coderende DNA-referentie sequentie
m. m.12T>A Verandering beschreven ten opzichte van een
mitochondriale referentie sequentie
Substitutie > c.1997G>T Nucleotide 1997 (G) van de referentiesequentie
is veranderd in een T
Deletie Del c.1997_1998delGAC Deletie ter hoogte van nucleotide 1997-1999 van
het aminozuur asparaginezuur
Insertie
Ins c.1997_1998insT Insertie van T tussen nucleotiden 1997 en 1998
2 Mutaties en kanker
Kanker is een verzamelnaam voor meer dan honderd verschillende ziekten. Al deze verschillende
soorten kanker hebben één gemeenschappelijk kenmerk: een ongeremde deling van lichaamscellen.
Kanker is een aandoening die gekenmerkt wordt door een aantal specifieke verschijnselen. Ten
eerste is dit door de aanwezigheid van cellen die zich ongecontroleerd vermenigvuldigen. Het
tweede kenmerk is dat de woekerende cellen zich uitbreiden in het omliggend weefsel en ook daar
schade aanrichten. Het derde en laatste belangrijke verschijnsel is de uitzaaiing of metastasering. Dit
begrip duidt op de verspreiding van de woekerende cellen naar verder weg gelegen plaatsen in het
lichaam (Dirkse 2010).

15
Momenteel zijn de belangrijkste types van kwaadaardige kankers carcinomen, sarcomen,
melanomen, lymfomen en leukemieën. Het onderscheid wordt gemaakt op basis van welke cel zich
differentieert tot een kankercel. Sommige kankers vormen niet echt tumoren, maar er treedt wel
overdreven veel deling van sommige cellen op, zoals bij de leukemieën en lymfomen. Dit zijn
bloedkankers waarbij een overmatige deling van de witte bloedcellen voorkomt (Movva 2013). In dit
werk wordt enkel dieper ingegaan op de leukemieën en lymfomen, dewelke kwaadaardige
hematologische kankers zijn.
Centraal in het ontstaan van kanker staan defecten, zijnde mutaties, in het DNA. Deze mutaties
kunnen op twee verschillende manieren verkregen worden. Als eerste zijn er de erfelijke mutaties en
als tweede zijn er de verworven mutaties door bijvoorbeeld infecties (bv. humaan popillomavirus),
fysische factoren (bv. UV- en ioniserende straling) en/of chemische stoffen (bv. asbest,
benzopyreen).
Om daadwerkelijk kanker te krijgen moeten de mutaties optreden in genen die betrokken zijn bij het
reguleren en controleren van de celdeling. Enkele belangrijke genen zijn proto-oncogenen,
tumorsuppressorgenen, genen die apoptose reguleren en genen die DNA-herstel reguleren.
Proto-oncogenen komen steeds normaal voor in het lichaam. Het zijn genen die coderen voor
eiwitten die een actieve rol spelen in de celregulatie. Ze zijn gewoonlijk betrokken bij het stimuleren
van normale celdelingen. Indien een mutatie optreedt in een proto-oncogen kan een oncogen
gevormd worden die de cel aanzet tot overmatige deling of zelfs onbeperkte groei.
Tumorsuppressorgenen bezitten als het ware de omgekeerde functie als proto-oncogenen en zorgen
er bijgevolg voor dat de groei van de cel onder controle wordt gehouden. Ze voorkomen de
onbeperkte deling van de cel en verhinderen zo het ontstaan van een tumor. Wanneer ter hoogte
van dit type gen een mutatie optreedt kan de controle op de deling van de cel verdwijnen, waardoor
het mogelijk wordt dat de cel ongecontroleerd kan verder gaan met delen.
Wanneer een cel niet meer op de normale wijze functioneert, treedt er apoptose op, waardoor de
cel vernietigd wordt. Dit verschijnsel wordt gereguleerd door apoptose regulerende genen. Bij kanker
zijn deze genen vaak uitgeschakeld.
Als laatste zijn er de genen die het DNA-herstel reguleren. Aan de hand van dit systeem kunnen
afwijkingen in het DNA hersteld worden. Wanneer ter hoogte van deze genen een mutatie optreedt,
worden fouten in het DNA niet meer voldoende hersteld waardoor er steeds meer defecten ontstaan
in het DNA.
Het is belangrijk te vermelden dat kanker pas optreedt wanneer in meerdere van bovengenoemde
genen mutaties zijn opgetreden (multi-step proces). Verder is het zo dat bij iedere mutatie de kans
op nieuwe mutaties steeds verder toeneemt. Mutaties in bijvoorbeeld proto-oncogenen en
tumorsuppressorgenen maken het mogelijk dat cellen ongeremd kunnen delen. Bij iedere deling is er
altijd een kans op nieuwe mutaties. Mutaties in DNA-herstel genen verhogen eveneens de kans op
mutaties. Dankzij de onderdrukking van de apoptose wordt de cel niet vernietigd (Dirkse 2010).

16
3 World Health Organization (WHO) classificatie van
hematologische neoplasma’s
Leukemieën en lymfomen zijn, zoals eerder vermeld, bloedkankers waarbij een overmatige deling
van de witte bloedcellen voorkomt. Bij deze ziektes is de hematopoëse ernstig verstoord.
Tijdens de hematopoëse differentieert de pluripotente hematopoietische stamcel naar de myeloïde
of lymfoïde cellijn (zie figuur 1). Uit de myeloïde lijn (links) ontstaan de granulocyten, de monocyten,
de erytrocyten en de bloedplaatjes, terwijl uit de lymfoïde lijn (rechts) de lymfocyten ontstaan. Deze
groep bestaat uit B-lymfocyten (B-cellen), T-lymfocyten (T-cellen) en Natural Killer cellen (NK-cellen).
Vanuit de pluripotente stamcel ontstaat dus een myeloïde of lymfoïde voorloper- of precursorcel.
Telkens wordt van één van deze twee voorlopercellen vertrokken om een bepaald type bloedcel aan
te maken.
Op elk niveau in de hematopoëse kan er iets misgaan waardoor er een kanker of neoplasma ontstaat.
Elk differentiatiestadium kan aanleiding geven tot een specifiek hematologisch neoplasma. Het
aantal mogelijke types van hematologische neoplasma’s is zeer groot en ze hebben vaak
overlappende kenmerken. Niettemin is het van belang om alle subtypes correct te kunnen
identificeren om een correcte behandeling te kunnen starten. Daarom heeft de WHO deze
verschillende soorten neoplasma’s geordend in de WHO-classificatie. Er kan binnen de
hematologische maligniteiten een onderscheid gemaakt worden tussen myeloïde neoplasma’s en
lymfoïde neoplasma’s. Dit onderscheid wordt gemaakt op basis van de origine van de maligne cellen
(Swerdlow et al. 2008). Myeloïde neoplasma’s zijn tumoren van de myeloïde cellijn. Terwijl de
lymfoïde neoplasma’s (meestal lymfomen) tumoren zijn van mature of immature B-, T- of
NK-lymfocyten. Ze komen voort uit verschillende differentiatiestadia van deze lymfocyten. De T-cel
neoplasma’s en NK-cel neoplasma’s worden gezien als één groep omdat de T- en NK-cellen veel
gelijkenissen vertonen met elkaar wat betreft immunofenotype en functie. De lymfoïde neoplasma’s
kunnen ruim gezien in twee subgroepen worden onderverdeeld: de T-cel lymfomen en de B-cel
lymfomen (Swerdlow et al. 2008).
Figuur 1: Voorstelling van de hematopoëse. Een pluripotente hematopoëtische stamcel wordt gedifferentieerd naar de myeloïde cellijn (links) of de lymfoïde cellijn (rechts) (All Things Stem Cell 2013).

17
4 Genen die in verband kunnen gebracht worden met
leukemieën en lymfomen
Er is een duidelijk verband tussen bepaalde puntmutaties en het voorkomen van een welbepaalde
vorm van bloedkanker. Vaak zijn de diagnoses van bepaalde ziekten op zowel morfologisch,
immunofenotypisch als cytogenisch vlak moeilijk te onderscheiden van andere ziektebeelden. Het
stellen van dergelijke diagnoses is dan ook vaak gebaseerd op exclusie. Wanneer de ziekte echter in
verband kan gebracht worden met het voorkomen van een specifieke mutatie, kan het stellen van de
diagnose versneld worden, wat leidt tot een snellere start van een effectieve behandeling. In andere
gevallen kan de aanwezigheid van een specifieke mutatie als extra controle dienen voor de diagnose.
Zoals eerder vermeld worden voor een aantal diagnosen reeds specifieke testen uitgevoerd in het AZ
Sint-Jan Brugge (zie tabel 19 en 20). Aangezien niet voor alle diagnosen dergelijke moleculaire
merkers getest worden en bepaalde diagnosen nog steeds gesteld worden op basis van exclusie, is
het nuttig om op zoek te gaan naar nieuwe merkers. Hieronder wordt een overzicht gegeven van de
mutaties die in verband kunnen gebracht worden met een specifieke vorm van bloedkanker. Hierbij
wordt telkens wat dieper ingegaan op het gen zelf, de functie en het eiwit, maar de nadruk ligt vooral
op het belang van het opsporen van de mutaties.
De genen worden hier uit praktische overweging ingedeeld in verschillende groepen. Deze indeling
gebeurt op basis van de soort leukemie waarmee de mutatie van het gen in verband kan gebracht
worden.
4.1 Genmutaties bij myeloproliferatieve neoplasma’s (MPN),
myelodysplastisch syndroom (MDS) en
myelodyplastische/myeloproliferatieve neoplasma’s (MDS/MPN)
4.1.1 Indeling neoplasma’s en hun kenmerken Volgens de WHO-classificatie kunnen neoplasma’s ingedeeld worden in verschillende grote groepen.
Drie van deze groepen zijn MPN, MDS en MDS/MPN.
4.1.1.1 Myeloproliferatieve neoplasma’s
Myeloproliferatieve aandoeningen zijn hematopoiëtische stamcelaandoeningen die gekenmerkt
worden door proliferatie van een of meer myeloïde lijnen (zie figuur 1), bijvoorbeeld proliferatie van
granulocyten, mestcellen of megakaryocyten. Tot deze groep van aandoeningen behoren onder
andere chronische myeloïde leukemie (CML), chronische neutrofiele leukemie (CNL), polycythemia
vera (PV), primaire myelofibrose (PMF), essentiële thrombocytemie (ET), chronische eosinofiele
leukemie (CEL) en mastocytose. Het zijn chronische aandoeningen waarbij het beenmerg te veel rode
bloedcellen, witte bloedcellen en/of bloedplaatjes aanmaakt. MPN komt het meest voor bij
volwassenen tussen de 50 en 70 jaar. Enkele subtypes, voornamelijk CML en ET worden eveneens
teruggevonden bij kinderen. Elk jaar wordt bij 6 tot 10 personen op 100 000 de diagnose van MPN
vastgesteld (Swerdlow et al. 2008). De belangrijke diagnoses in dit werk worden hieronder uitvoerig
besproken.

18
4.1.1.1.1 Chronische neutrofiele leukemie
CNL is een uiterst zeldzame MPN waarbij een blijvend hoog aantal neutrofiele granulocyten in de
bloedsomloop bestaat (Swerdlow et al. 2008).
a) Epidemiologie
Het voorkomen van CNL is onbekend, maar er zijn slechts ongeveer 150 gevallen gemeld volgens de
WHO-classificatie. In een onderzoek van 660 gevallen van chronische leukemie van myeloïde
oorsprong, werd geen enkel geval van CNL waargenomen. CNL treft in het algemeen meestal oudere
volwassenen, maar werd ook reeds gemeld bij adolescenten. De ratio man:vrouw is ongeveer gelijk
aan 1:1 (Swerdlow et al. 2008).
b) Klinische kenmerken
De meest voorkomende klinische kenmerken bij CNL-patiënten zijn splenomegalie (vergroting van de
milt) en hepatomegalie (vergroting van de lever) (Swerdlow et al. 2008).
In het perifere bloed treedt leukocytose op (verhoging van het aantal witte bloedcellen in het bloed,
meestal >25x109 leukocyten/l), bestaande uit overwegend neutrofiele granulocyten. Deze
granulocyten kunnen toxische en grove korreling bevatten (zie figuren 2 en 3) (Swerdlow et al. 2008).
Het beenmerg toont een hyperplastisch wit systeem zonder morfologische afwijkingen. Hyperplasie
duidt op bovenmatige vermeerdering van weefsel (Swerdlow et al. 2008).
c) Diagnose
CNL is een zeer zeldzaam MPN dat vooral gekenmerkt wordt door leukocytose. Aangezien specifieke
klinische en moleculaire merkers ontbreken, is de diagnose vaak gebaseerd op exclusie. Het is
bijgevolg belangrijk dat er gezocht wordt naar nieuwe moleculaire merkers die in tegenstelling tot de
reeds bestaande merkers niet overlappen met andere neoplasma’s, maar specifiek zijn voor CNL. Op
deze manier zullen de moleculaire merkers niet alleen een belangrijke rol spelen in het stellen van de
juiste diagnose, maar zullen ze eveneens aanleiding geven tot een snellere start van een effectieve
behandeling (Ziai et al. 2010).
Figuur 2: Chronische Neutrofiele Leukemie -
Perifeer bloed: zeer veel neutrofiele
granulocyten met grove korreling (Scholten
2013)
Figuur 3: Chronische Neutrofiele Leukemie -
Beenmerg: hyperplastisch beenmerg met
hyperplasie van de myelopoëse (Scholten
2013)

19
4.1.1.2 Myelodysplastisch syndroom
MDS staat voor een aantal stoornissen van het beenmerg waardoor de productie van bloedcellen is
verstoord. Het ontstaat door een beschadiging in het erfelijk materiaal van een myeloïde
voorlopercel van één of meer verschillende types bloedcellen (rode, witte en bloedplaatjes). Het
resultaat van deze gestoorde aanmaak is dat er misvormde en niet goed uitgegroeide bloedcellen
ontstaan (zie figuren 4 en 5). Deze misvormingen worden dysplasie genoemd (vandaar de naam
myelodysplasie). Door de slechte kwaliteit van de bij MDS geproduceerde bloedcellen, wordt een
belangrijk deel van de bloedcellen vernietigd voor het verlaten van het beenmerg, waardoor een
tekort aan bloedcellen kan ontstaan. Deze aandoeningen komen frequenter voor bij mannen en
komt vooral voor bij ouderen tussen 60 en 80 jaar (Swerdlow et al. 2008).
4.1.1.3 Myelodysplastische/myeoloproliferatieve neoplasma’s
De MDS/MPN groep bestaat uit myeloïde neoplasma’s die klinische en morfologische kenmerken
bevatten die zowel de diagnose van MDS als MPN ondersteunen. Tot deze groep behoren onder
andere atypische chronische myeloïde leukemie (aCML) en chronische myelomonocytaire leukemie
(CMML).
4.1.1.3.1 Atypische chronische myeloïde leukemie
ACML heeft zowel dysplastische als proliferatieve kenmerken. Het lijkt op CML, maar mist het
Philadelphia-chromosoom en de voor CML typische toename van basofielen. Dysplasie staat meer op
de voorgrond dan bij CML.
a) Epidemiologie
Het exacte voorkomen van aCML is niet bekend, maar is naar verluidt slecht 1 à 2 gevallen per 100
gevallen van BCR-ABL positieve CML. Met BCR-ABL positieve CML wordt bedoeld dat er een
translocatie heeft plaatsgevonden. Deze translocatie leidt tot de overdracht van het
Abelson(ABL)-gen op chromosoom 9 naar een gebied van chromosoom 22 dat de breekpunt cluster
Figuur 4: Dysplasie perifeer bloed: granulocyt met slechte segmentatie, hypogranulatie en afwijkende chromatinecondensatie (Scholten 2013)
Figuur 5: Dysplasie perifeer bloed: reuzen-
trombocyten (Scholten 2013)

20
regio (BCR) wordt genoemd. Dit resulteert op zijn beurt in een gefuseerd BCR-ABL gen en in de
productie van een abnormaal ABL-tyrosine kinase eiwit. De gemiddelde leeftijd bij diagnose is 70 à 80
jaar, maar de ziekte werd ook gemeld bij tieners. De man:vrouw ratio varieert, maar is in de meeste
gevallen ongeveer 1:1 (Swerdlow et al. 2008).
b) Klinische kenmerken
Er zijn slechts een paar verslagen van de klinische kenmerken van patiënten met aCML. De meeste
patiënten vertonen symptomen die verband houden met bloedarmoede of soms trombocytopenie,
terwijl in andere landen de belangrijkste klacht is gerelateerd aan splenomegalie (vergroting van de
milt). Met trombocytopenie bedoelt men het symptoom waarbij er te weinig bloedplaatjes
(trombocyten) in het bloed aanwezig zijn (Swerdlow et al. 2008).
c) Morfologie
In het perifere bloedbeeld is een leukocytose altijd > 13 x 109 leukocyten/l, variërend van 30x109 tot
100x109 leukocyten/l, met hierin vele onrijpe vormen. Het aantal blasten is meestal kleiner dan 5%,
maar altijd kleiner dan 20%. De cellen van de myelopoëse vertonen een bizarre segmentatie en
afwijkende granulatie (zie figuur 6) (Swerdlow et al. 2008).
Het beenmerg is hyperplastisch waarbij er een proliferatie van de myelopoëse zichtbaar is. Het aantal
myeloblasten varieert van 3 tot 10%. Ook in het beenmerg is dysplasie in de myelopoëse aanwezig
(Swerdlow et al. 2008).
d) Diagnose
ACML vertoont heel wat gemeenschappelijke kenmerken met andere MDS/MPN. Zowel CMML en
aCML worden bijvoorbeeld beide gekenmerkt door een verhoogd leukocytgehalte, trombocytopenie
en splenomegalie. Er kunnen heel wat abnormaliteiten gevonden worden bij patiënten met aCML,
maar geen enkel is specifiek. Er dient dus gezocht te worden naar een nieuwe merker die specifiek is
voor aCML, zodat een snelle diagnose kan gesteld worden.
Figuur 6: atypische Chronische Myeloïde Leukemie – Perifeer bloed: myeloïde voorlopers en twee segmenten met dysplasie: slechte segmentatie en afwijkende chromatinestructuur (pijlen) (Scholten 2013).

21
4.1.1.3.2 Chronische myelomonocytaire leukemie
CMML wordt eveneens zowel gekenmerkt door dysplastische als proliferatieve kenmerken. Patiënten
met CMML hebben een te veel aan monocyten in hun bloed (>1x109/l). De meest voorkomende
symptomen zijn vermoeidheid, gewichtsverlies en koorts. De gemiddelde leeftijd van diagnose ligt
tussen de 65 en 75 jaar. Het precieze voorkomen is nog niet gekend. Dit blijkt eveneens duidelijk uit
het feit dat verschillende studies CMML classificeren onder verschillende groepen, bijvoorbeeld bij
CML en een andere bij MDS. Er wordt geschat dat ongeveer bij dertien patiënten per 100 000 de
diagnose CMML wordt gesteld per jaar. Voor het stellen van de diagnose CMML gebeuren
momenteel nog geen specifieke testen in het AZ Sint-Jan Brugge. De diagnose is dus gebaseerd op
exclusie, waardoor het belangrijk is om te zoeken naar moleculaire merkers die het stellen van de
diagnose CMML kunnen versnellen.
4.1.1.3.3 Mastocytose
Mastocytose wordt volgens het WHO-criterium beschouwd als een subcategorie van MPN. De
aandoening wordt gekenmerkt door een abnormale groei van de mestcellen. Mestcellen zijn witte
bloedcellen die zich niet vrij doorheen de bloedbaan bewegen, maar aanwezig zijn in de meeste
organen. Ze spelen een belangrijke rol in het immuunsysteem. Mestcellen zijn betrokken bij
allergische reacties en het bestrijden van bepaalde infecties.
Mastocytose wordt ingedeeld in twee grote groepen, namelijk cutane mastocytose en systemische
mastocytose. Cutane mastocytose is de meest voorkomende mestcel ziekte. Hierbij zijn geen andere
organen betrokken dan de huid. In deze studie wordt enkel dieper ingegaan op de tweede groep van
mastocytose, namelijk systemische mastocytose (SM). Bij SM infiltreren de mestcellen eveneens in
extracutane organen zoals het beenmerg, de milt en de lever. Het klinisch verloop van SM kan
variëren van indolente SM (ISM) tot een meer agressieve, levensbedreigende SM. SM is een
zeldzame aandoening van de mestcel, gekenmerkt door abnormale proliferatie en accumulatie van
mestcellen. De meest voorkomende symptomen zijn gewichtsverlies, pijn, misselijkheid, hoofdpijn en
vermoeidheid. De meeste patiënten met SM hebben ISM. Hun levensverwachting loopt gelijkaardig
aan deze van de algemene bevolking. Het bestaat evenwel dat de indolente vorm van de ziekte zich
ontwikkeld tot de agressieve vorm. In tegenstelling tot de goedaardige vorm van SM, is de
levensverwachting bij agressieve SM aanzienlijk korter dan deze van de algemene bevolking,
variërend van 2 tot 41 maanden afhankelijk van het subtype. De diagnose van mastocytose wordt
gemakkelijk gemist en de ziekte wordt vaak slechts ontdekt jaren na de beginnende klachten wat
soms leidt tot een rampzalige uitkomst. Mastocytose kan voorkomen op iedere leeftijd. De kliniek
van een patiënt met SM wordt sterk bepaald door enerzijds de uitscheiding van de vele mediatoren,
waaronder onder andere histamine en prostaglandines na mestceldegranulatie, en anderzijds door
het soms tumorvormende gedrag van de mestcellen (Alto and Clarcq 1999; Verstovsek 2013).
Voor het stellen van de diagnose SM worden geavanceerde technieken ingezet en is specifiek
expertise nodig van de afdelingen hematologie, allergologie, dermatologie, pathologie, klinische
chemie (flowcytometrie en moleculaire biologische technieken) en radiodiagnostiek. Bij een
laboratoriumonderzoek wordt het tryptasegehalte in het bloed evenals afbraakproducten van
histamine in de urine bepaald. Tryptase is een stof die uitgescheiden wordt door de mestcellen en
die bij SM verhoogd is. Bij een lichamelijk onderzoek wordt gelet op huidafwijkingen, een vergrote

22
lever, milt of lymfeklieren. Bij flowcytometrie worden de mestcellen specifiek gekleurd en verder
onderzocht. Bij de moleculair biologische analyse wordt het beenmergmonster onderzocht op
mutaties die specifiek zijn voor de verschillende types mastocytose. In het AZ Sint-Jan Brugge worden
mastocytose-patiënten reeds getest op de aanwezigheid van c-KIT-mutaties, waardoor het hier dus
niet gaat om een nieuwe merker. Deze testen worden momenteel nog buitenshuis uitgevoerd
(kwantitatief en gevoeliger), maar het is de bedoeling om deze test ook binnenhuis te kunnen
uitvoeren.
4.1.2 Belangrijke mutaties aanwezig in MPN, MDS en MDS/MPN
4.1.2.1 Additional Sex Combs Like (ASXL1)-mutatie
Het ASXL1-gen codeert voor een proteïne die deel uitmaakt van de polycomb-groep eiwitten. Dit is
een groep van eiwitten die in staat is chromatine te remodelleren zodanig dat epigenetische silencing
van genen kan plaatsvinden. Het is de menselijke homoloog van het additional sex combs (ASX)-gen
van Drosophila. Het gen is bij verschillende soorten zeer goed geconserveerd. Bij de mens is de
exacte functie nog niet achterhaald, maar het kan wel functioneren als een ligandafhankelijke
co-activator voor een retinoïnezuurreceptor door binding met een steroïde receptor coactivator-1.
Bovendien is ASXL1 ook betrokken bij de regulatie van de histonen-methylatie. Dit gebeurt door de
coöperatie met het heterochromatine proteïne-1 (Weizmann Institute of Science 2013).
4.1.2.1.1 ASXL1-eiwit en genstructuur
Het expressieproduct van het ASXL1-gen is een eiwit dat bestaat uit 1541 aminozuren dat
voornamelijk voorkomt in de kern en heeft een moleculaire massa van ongeveer 165 kDa (Weizmann
Institute of Science 2013). Het humane eiwit bestaat uit een sterk geconserveerd N-terminaal
ASX-homoloog domein (ASXN) en een C-terminaal plant homeodomein (PHD) zinc finger domein, een
nucleair proteïne interactie domein. Het ASXL1-eiwit bevat eveneens een geconserveerd domein in
het midden van het eiwit, namelijk het ASXM-domein en een nucleair receptor domein (NR box) (zie
figuur 7) (Mozziconacci and Birnbaum 2010). Het humane ASXL1-gen is gelokaliseerd op
chromosoom 20 (20q11.21). Het ASXL1-gen bestaat uit 80 976 bp en 13 exonen (Weizmann Institute
of Science 2013).
Figuur 7: Voorstelling van het ASXL1-eiwit en het mutatiespectrum van het ASXL1-eiwit (Schnittger et al. 2013)

23
4.1.2.1.2 Belang van de ASXL1-mutatie
In heel wat studies worden mutaties in het ASXL1-gen in verband gebracht met MPN, MDS/MPN,
MDS en eveneens met acute myeloïde leukemie (AML) dat verder aan bod komt. In tabel 2 wordt
een overzicht gegeven van verschillende publicaties waarin dit verband wordt bestudeerd. De
verschillende studies zijn het erover eens dat de mutatiehotspot zich bevindt ter hoogte van exon 12
(zie figuur 7) net voor het PHD-domein. Alle mutaties die optreden in deze mutatiehotspot resulteren
in een verstoring van dit domein. Het PHD-domein is een structureel motief dat aanwezig is in
nucleaire eiwitten die betrokken zijn bij modificaties van het chromatine, zoals histondemethylasen.
Mutaties in het ASXL1-gen leiden bijgevolg tot afwijkende DNA-methylaties en histon wijzigingen.
Mutaties in het ASXL1-gen worden geassocieerd met een slechte prognose voor de patiënt. Dit wordt
geïllustreerd voor de diagnose MDS in figuur 8, maar dit geldt eveneens voor de andere diagnosen
waarmee ASXL1-mutaties in verband gebracht worden (Gelsi-Boyer et al. 2009).
Tabel 2: Vergelijking voorkomen van aanwezigheid van een ASXL1-mutatie voornamelijk bij MPN, AML, MDS en MDS/MPN in verschillende publicaties
Publicatie Gebruikte techniek voor het
opsporen van mutaties
Ziekte Voorkomen
Tefferi et al. (2011) Niet vermeld (Review) PMF
MPN
13%
18%
Schnittger et al. (2013) Sanger sequencing AML 127/740 (17,2%)
Gelsi-Boyer et al. (2012)
(vergelijking van 19 studies)
MDS
CMML
PMF
AML (secundair)
AML (de novo)
148/914 (16,2%)
124/274 (45%)
41/119 (34,5%)
30/99 (30,3%)
130/2000 (6,5%)
Abdel-Wahab et al. (2013) Niet vermeld (Review) MDS 10-30%
Meggendorfer et al. (2013) Sanger sequencing CNL aCML CMML
8/11 (73%) 38/59 (64%) 110/251(44%)
Figuur 8: Vergelijking tussen de prognose van MDS-patiënten
met wild type ASXL1 en mutant ASXL1 (Chen et al. 2014)

24
4.1.2.2 Mest-/stamcel groeifactor receptor KIT-mutatie
C-KIT is een proto-oncogen dat codeert voor een type 3 transmembraan receptor voor mestcel
groeifactoren. Synoniemen voor het proto-oncogen c-KIT zijn CD117, mest- of stamcel groeifactor
(SCF) en tyrosine-proteïne kinase Kit. Cluster of differentation (CD)-molecules zijn merkers die
aanwezig zijn op het celoppervlak. Deze merkers kunnen herkend worden door specifieke
antilichamen. CD117 is een cytokine receptor die zowel voorkomt op het oppervlak van
hematopoietische stamcellen als op het oppervlak van andere celtypes. Wijzigende vormen van deze
receptor kunnen geassocieerd worden met sommige vormen van kanker. CD117 is een tyrosine
kinase type III-receptor die bindt met een stamcel groeifactor. Wanneer deze receptor bindt met een
SCF, wordt een dimeer gevormd dat de intrinsieke tyrosine kinase activiteit activeert. Hierdoor
ontstaat een signaalcascade waardoor het signaal zich kan verderzetten in de cel (zie figuur 9). CD117
speelt een rol in proliferatie, differentiatie en overleving van de cel (Royster 2010).
4.1.2.2.1 C-KIT-eiwit en genstructuur
Het c-KIT eiwit is opgebouwd uit 976 aminozuren en heeft een moleculair gewicht van ongeveer
110 kDa (Weizmann Institute of Science 2013). Het eiwit bestaat uit zes verschillende domeinen, vijf
Ig-like domeinen en één proteïne kinase domein (aminozuren 589-937) (UniProt 2014). Het humane
c-KIT-gen is gelokaliseerd op chromosoom 4 (4q12) en bestaat uit 82 797 bp en 21 exonen
(Weizmann Institute of Science 2013).
4.1.2.2.2 Belang van de mutatie
Mutaties in de c-KIT receptor zijn geassocieerd met SM. Vooral de puntmutatie p.Asp816Val
(c.2447A>T) blijkt sterk in verband te kunnen worden gebracht met SM. Door deze mutatie wordt de
mestcel voortdurend gestimuleerd, ook zonder dat binding met de mestcelgroeifactor heeft plaats
gevonden (zie figuur 10). Het gaat hier om een activerende puntmutatie die wordt gevonden in het
tyrosine kinase domein, wat leidt tot een conformationele verandering die resulteert in een
ligand-onafhankelijke constitutieve activatie van c-KIT. Dit leidt op haar beurt tot een verhoogde
proliferatie en een vermindering van apoptose (Verstovsek 2013).
Figuur 9: c-KIT receptor dimeriseert in de aanwezigheid van het
ligand SCF en initieert op deze manier de signaalcascade (Royster
2010)

25
Volgens de studie van Bunimovich et al. (2009) wordt de mutatie p.Asp816Val bij meer dan 80% van
de patiënten met ISM gevonden terwijl andere mutaties, zoals p.Asp816Gly, p.Lys509Ile, p.Asp816His
en p.Val530Ile, worden gevonden bij de meer agressieve varianten van mastocytose. In tabel 3 wordt
een overzicht gegeven van de verschillende studies die het verband tussen specifieke mutaties en SM
onderzocht hebben. Over de prognostische invloed van deze mutaties is nog weinig gekend.
Tabel 3: Vergelijking voorkomen van aanwezigheid van een c-KIT-mutatie bij mastocytose in verschillende publicaties
Publicatie Gebruikte techniek voor het opsporen van mutaties
Voorkomen
Kristensen et al. (2011) Sanger sequencing 19/20 (95%)
Bunimovich et al. (2009) Niet vermeld (Review) > 80 %
Lim et al. (2009)
Allel-specifieke PCR
112/165 (68%)
Figuur 10: Voorstelling van de ligand-afhankelijke en
ligand-onafhankelijke activatie van c-KIT (Verstovsek 2013).

26
4.1.2.3 Colony Stimulerende Factor 3-Receptor (CSF3R)-mutatie
Het eiwit dat gecodeerd wordt door het CSF3R-gen is de transmembraanreceptor voor de colony
stimulerende factor 3. Dit is een cytokine dat de productie, differentiatie en functie van granulocyten
controleert. Het gecodeerde proteïne is lid van de familie van de cytokinereceptors en heeft ook
functies in bijvoorbeeld herkenningsprocessen. Cytokinereceptoren zijn gekoppeld aan de STAT,
Ras-MAPK en fosfatidylinositol-3’-kinase(PI3K)-AKT routes die convergeren in de kern en daar de
genenexpressie reguleren (zie figuur 11) (Kaushansky 2006; Vainchenker and Constantinescu 2013).
Het CSF3R-gen is een proto-oncogen, dat kan omgezet worden naar een oncogen dat op zijn beurt de
vorming van kanker bevordert.
4.1.2.3.1 CSF3R-eiwit en genstructuur
Het humaan CSF3R-eiwit is opgebouwd uit 836 aminozuren en heeft een moleculair gewicht van
ongeveer 92 kDa. Het eiwit bevindt zich ter hoogte van het celmembraan en bestaat uit twee
belangrijke domeinen: het immunoglobuline(Ig)-like domein en het fibronectine type-III domein
(UniProt 2014) (figuur 12). Er zijn twee belangrijke motieven in het CSF3R-eiwit aanwezig. Een eerste
is het WSXWS motief, dat noodzakelijk lijkt voor een goede eiwitvorming, wat op zijn beurt leidt tot
een efficiënt intracellulair transport en een efficiënte celoppervlak receptorbinding. Een tweede
motief is het box-1 motief, dat nodig is voor de JAK-interactie en/of -activatie (UniProt 2014). Het
humane CSF3R-gen is gelokaliseerd op chromosoom 1 (1p34.3) en bestaat uit 17 272 bp en 14
exonen (Weizmann Institute of Science 2013).
Figuur 11: Hematopoietische groeifactor signaaltransductie. Iedere hematopoietische groeifactor-receptor is
samengesteld uit twee subunits die twee moleculen Janus kinase 2 (JAK2) kunnen binden. De binding van een ligand
induceert een conformationele verandering in het dimeer, wat leidt tot tyrosinefosforylering en een cross-activatie van
JAK2, die de intracellulaire receptor tyrosine residuen fosforyleren. Deze trekken op hun beurt adaptereiwitten aan die de
specifieke tyrosine gefosforyleerde sequenties herkennen. Verschillende adaptereiwitten worden substraat van JAK’s
triggering signaleringscascade. Cytokinereceptoren zijn gekoppeld aan de STAT, Ras-MAPK en fosfatidylinositol-3’-
kinase(PI3K)-AKT routes die convergeren in de kern en daar de genenexpressie reguleren (Kaushansky 2006; Vainchenker
and Constantinescu 2013)

27
4.1.2.3.2 Belang van de mutatie
De CSF3R-mutaties kunnen ingedeeld worden in twee grote groepen (zie figuren 12 en 13). Als eerste
zijn er de CSF3R-mutaties die een ingekort eiwit leveren. Dit belemmert het vermogen om signalen
door te geven die nodig zijn voor de neutrofiele differentiatie (Kosmider et al. 2013). Als tweede zijn
er de CSF3R membraan proximale mutaties. Deze eerste soort mutaties resulteren in verhoogde
niveaus van CSF3R. De tweede soort daarentegen activeren sterk de JAK/STAT-route. Beide soorten
mutaties zetten het oorspronkelijke proto-oncogen om in een oncogen die de vorming van kanker
stimuleert door overactivatie (Gotlib 2013).
Verschillende studies hebben het verband aangetoond tussen het voorkomen van CSF3R-mutaties en
bepaalde specifieke vormen van bloedkanker, voornamelijk MDS/MPN, CNL en AML (zie later)
(zie tabel 4).
Figuur 12: Opbouw van het CSF3R-gen met het mutatiespectrum (Maxson et al. 2013)
Figuur 13: Voorstelling twee grote groepen mutaties bij CSF3R (Maxson et al. 2013)
28
Tabel 4: Vergelijking voorkomen van aanwezigheid van een CSF3R-mutatie voornamelijk bij CNL, aCML en CMML in
verschillende publicaties
Publicatie Gebruikte techniek voor het opsporen van mutaties
Ziekte Voorkomen
Maxson et al. (2013)
Next generation sequencing CNL/aCML AML T-ALL
16/27 (59%) 3/292 (1%) 1/3 (33%)
Pardanani et al. (2013) Sanger sequencing CNL aCML
12/12 (100%) 0/9 (0%)
Meggendorfer et al. (2013) Sanger sequencing CMML aCML CNL
2/252 (0,8%) 2/60 (3,3%) 7/20 (35%)
Kosmider et al. (2013) Sanger sequencing CMML 6/196 (3%)
Figuur 12 geeft het mutatiespectrum weer van het CSF3R-gen. Uit deze figuur kan besloten worden
dat er twee mutatiehotspots aanwezig zijn, namelijk ter hoogte van exon 14 (membraan proximale
mutaties) en ter hoogte van exon 17 (truncation mutations). Volgens Maxson et al. (2013) bevinden
60 tot 80% van de mutaties zich ter hoogte van aminozuur 618, waarbij het oorspronkelijke
threonine verandert in isoleucine. Volgens Pardanani et al. (2013) is er geen verband tussen de
aanwezigheid van CSF3R-mutaties en de prognose voor de patiënt.
4.1.2.4 Myeloproliferatief leukemie virus oncogene (MPL)-mutatie
De functies van hematopoiëtische stamcellen worden gecontroleerd door veel verschillende
factoren, onder andere door cytokines en hun receptoren, zoals de thrombopoëtine receptor MPL
(zie figuur 14). MPL werd geïdentificeerd als de cellulaire homoloog van het myeloproliferatieve
leukemie virus oncogen (v-MPL). De MPL-signaalpathway is betrokken bij zowel de ontwikkeling als
de instandhouding van de hematopoiëtische stamcellen. Bovendien is de signaalpathway via MPL
cruciaal voor de megakaryocyt differentiatie en de vorming van de bloedplaatjes. Ontregeling van
deze pathway kan leiden tot hematologische aandoeningen (Modlich et al. 2013).

29
4.1.2.4.1 MPL-eiwit en genstructuur
Het proteïne dat gecodeerd wordt door het MPL-gen is opgebouwd uit 635 aminozuren en heeft een
moleculair gewicht van ongeveer 71 kDa. Het bestaat uit twee extracellulaire cytokinereceptor
domeinen en twee intracellulaire cytokine receptor box motieven (Weizmann Institute of Science
2013). Het humane MPL-gen is gelokaliseerd op chromosoom 1 (1p34.2) en bestaat uit 16 661 bp en
12 exonen (Weizmann Institute of Science 2013).
4.1.2.4.2 Belang van de mutatie
In heel wat studies worden mutaties in het MPL-gen in verband gebracht met MPN, voornamelijk
met PMF en ET en in mindere mate met MDS en MDS/MPN (zie tabel 5).
Figuur 14: MPL-signaalpathway en negatieve regulatoren. Na het binden van het trombopoëtine aan de transmembraan receptor MPL, wordt JAK gerekruteerd en geactiveerd, wat leidt tot de inductie van drie belangrijke downstream signaalwegen (Modlich et al. 2013).

30
Tabel 5: Vergelijking voorkomen van aanwezigheid van een MPL-mutatie voornamelijk bij myeloproliferatieve aandoeningen in verschillende publicaties
Publicatie Gebruikte techniek voor het opsporen van mutaties
Ziekte Voorkomen
Tefferi et al. (2011) Niet vermeld (Review)
ET PMF MPN
3% 10% 5%
Integrated Oncology (2013) Niet vermeld (Review)
PMF ET
5% 1%
Chaligne et al. (2008) Niet vermeld (Review)
PMF 5-10 %
Vannucchi and Guglielmelli (2008) Niet vermeld (Review)
ET 3-7 %
Schmitt-Graeff et al. (2008) Allel-specifieke PCR MDS 1/23 (4,35%)
Schnittger et al. (2008) Sequencing MDS 1/1 (100%)
Opmerking: Het onderscheid tussen MPN, MDS en MDS/MPN is vaak moeilijk (Olsen et al. 2008)
Tot de mutatiehotspot van het MPL-gen behoren twee aminozuren in exon 10. Een eerste
aminozuur, tryptofaan 515, wordt vaak gemuteerd met de vorming van leucine, lysine of alanine. Een
tweede aminozuur dat behoort tot de mutatiehotspot is serine 505, dat meestal omgevormd wordt
tot asparagine (figuur 15) (Chaligne et al. 2008). Beide aminozuren behoren tot het transmembranair
domein. De mechanismen van de MPL-activering blijven onduidelijk. Aan de hand van specifieke
software werden de mechanismen van de MPL-activatie bestudeerd voor verschillende mutaties. De
simulatieresultaten suggereren dat serine 505 en tryptofaan 515 belangrijk zijn in het behouden van
de correcte positie van het transmembranair domein van MPL. Mutaties in een van deze twee
posities leidt tot een verandering van dit domein, waardoor de conformatie van nabijgelegen
intracellulaire domeinen op een onverwachte manier wijzigt. Deze structuurveranderingen kunnen
op hun beurt leiden tot een constitutieve activatie van JAK2, de MPL kinase partner, wat leidt tot
overexpressie (Lee et al. 2011). Volgens Pardanani et al. (2011) heeft een MPL-mutatie slechts een
geringe prognostische waarde (zie figuur 16).
Figuur 15: Mutatiespectrum van het MPL-gen
(gebaseerd Chaligne et al. 2008).
Figuur 16: Vergelijking van de overall survival van
MPN-patiënten met en zonder MPL- en
JAK2-mutaties (Pardanani et al. 2011).

31
4.1.2.5 SET binding proteïne 1-mutatie
Het SET binding proteïne 1 (SETBP1)-gen codeert voor het eiwit genaamd SET bindend eiwit 1. Dit is
een slecht gekarakteriseerd eiwit dat wordt verondersteld om PP2A fosfatase-activiteit te remmen
door zich aan een ander eiwit (SET) te binden. SET is een multitasking proteïne dat betrokken is in
verschillende cellulaire processen. SET speelt onder andere een rol in apoptose, in de regulering van
de celcyclus, in de samenstelling van het nucleosoom en in de migratie. Het SET-eiwit is een nucleair
oncogen in tegenstelling tot het PP2A eiwit, dat een tumorsupressorgen is (Perrotti and Neviani
2008). PP2A staat voor proteïne fosfatase 2 en is een sterk geconserveerd en alomtegenwoordig
serine-threonine fosfatase met een brede substraatspecificiteit en diverse cellulaire functies. Over de
functie van het SETBP1-eiwit en het effect van de binding ervan aan het SET-eiwit is nog weinig
bekend. Mutaties in het SETBP1-gen zorgen voor een overexpressie van het SETBP1-eiwit, waarbij
het tumorsuppressor eiwit fosfatase 2A wordt onderdrukt (zie figuur 17) (Piazza et al. 2013).
4.1.2.5.1 SETBP1-eiwit en genstructuur
Het SETBP1-eiwit is opgebouwd uit 1596 aminozuren en heeft een moleculair gewicht van 175 kDa.
De SETBP1 sequentie bevat drie AT-hook domeinen (zie figuur 18). Deze AT-hook domeinen zijn
kleine DNA-bindende motieven met een voorkeur voor AT-rijke gebieden. De sequentie bevat
eveneens een SKI homologe regio (aminozuren 706-917). Het SKI-eiwit is een nucleaire
proto-oncoproteïne die wordt geassocieerd met tumoren bij hoge cellulaire concentraties. SKI
interfereert met de normale cellulaire werking door zowel de directe belemmering van de expressie
van bepaalde genen binnen de kern van de cel, als door de verstoring van de signalerende eiwitten
die genen activeren. Als laatste twee domeinen van het SETBP1-eiwit zijn er een SET-binding domein
(aminozuren 1292-1488) en een repeat domein (aminozuren 1520-1543) (Makishima et al. 2013).
Het humane SETBP1-gen is gelokaliseerd op de lange arm van chromosoom 18 (18q12.3) en bestaat
uit 388 338 bp en vijf exonen (Weizmann Institute of Science 2013).
Figuur 17: SETBP1-eiwit bindt met het SET-eiwit, wat leidt tot de vorming van een
SETBP1-SET-PP2A complex dat resulteert in de inhibitie van PP2A, wat de proliferatie van
leukemiecellen bevordert (Cristobal et al. 2010)

32
4.1.2.5.2 Belang van de mutatie
In 2012 werd SETBP1 geïdentificeerd als een nieuw oncogen. Specifieke somatische mutaties van dit
gen werden voornamelijk ontdekt bij patiënten die lijden aan aCML en CNL en in mindere mate bij
CMML, MDS en MPN. De mutatiehotspot bevindt zich ter hoogte van codon 858 tot codon 871
(zie figuur 18). Volgens Piazza et al. (2013) komen hier 92% van de mutaties voor. Volgens Makishima
et al. (2013) komen deze somatische SETBP1-mutaties voornamelijk voor ter hoogte van p.Asp868,
p.Ser869, p.Gly870, p.Ile871 en p.Asp880. Zoals eerder vermeld leiden mutaties in het SETBP1-gen
tot overexpressie van het SETBP1-eiwit, wat op zijn beurt leidt tot onderdrukking van het
tumorsuppressor eiwit fosfatase 2A. In tabel 6 is een overzicht te zien van de reeds uitgevoerde
studies over dit onderwerp. In figuren 19 en 20 wordt het prognostisch belang weergegeven voor
aCML- en CMML-patiënten van de SETBP1-mutatie, maar dit geldt eveneens voor de andere
ziektebeelden waarmee SETBP1-mutaties gelinkt zijn. Er kan uit deze figuren besloten worden dat
patiënten met het mutante gen een duidelijk slechtere prognose hebben (Piazza et al. 2013).
Tabel 6: Vergelijking voorkomen van aanwezigheid van een SETBP1-mutatie bij specifieke ziektebeelden in verschillende publicaties
Publicatie Gebruikte techniek voor het opsporen van mutaties
Ziekte Voorkomen
Piazza et al. (2013) Sanger sequencing aCML CNL MDS/MPN CMML
17/70 (24,3%) 1/4 (25%) 3/30 (10%) 3/82 (3,7%)
Meggendorfer et al. (2013) Sanger sequencing aCML MDS/MPN
19/60 (31,7%) 20/240 (9,3%)
Laborde et al. (2013) Sanger sequencing CMML PMF
8/179 (4,5%) 6/236 (2,5%)
Damm et al. (2013) Sanger sequencing CMML MDS AML
12/195 (6,2%) 5/222 (2,2%) 4/241 (1,7%)
Figuur 18: Opbouw van het SETBP1-eiwit met het mutatiespectrum (Makishima et al. 2013)

33
4.2 Genmutaties bij chronische lymfatische leukemie
4.2.1 Chronische lymfatische leukemie
Chronische lymfatische leukemie (CLL) wordt gekenmerkt door de accumulatie van min of meer rijpe
B-lymfocyten. Het is een neoplasma bestaande uit monomorfe kleine, ronde tot licht onregelmatige
B-lymfocyten in het perifere bloed, het beenmerg, de milt en de lymfeklieren. Hoewel de maligne
lymfocyten bij CLL er normaal uitzien, zijn ze dat niet waardoor ze infecties niet effectief kunnen
bestrijden (Swerdlow et al. 2008).
4.2.1.1 Epidemiologie
CLL is de meest voorkomende vorm van leukemie bij volwassenen in de Westerse landen en komt
zelden voor in de Oosterse landen. Elk jaar wordt bij 2 tot 6 personen op 100 000 de diagnose van
CLL vastgesteld. De gemiddelde leeftijd bij diagnose schommelt tussen de 65 en 75 jaar. CLL wordt
zelden gezien bij patiënten jonger dan 30 jaar. Slechts 20 à 30% van de patiënten zijn jonger dan 55
jaar. CLL heeft een man:vrouw ratio van 1,5-2:1. Er kan bijgevolg gesteld worden dat de ziekte
ongeveer twee keer zoveel voorkomt bij mannen als bij vrouwen (Swerdlow et al. 2008).
4.2.1.2 Klinische kenmerken
In het begin van de ziekte zijn er meestal geen klachten. Bij 70-80% van de patiënten wordt CLL pas
vastgesteld naar aanleiding van een toevallig bloedonderzoek. De klachten ontstaan pas na verloop
van tijd en kunnen onder andere bleekheid, onverklaarbare vermoeidheid, herhaalde infecties,
nachtelijke koorts en zweten, gewichtsverlies, botpijnen, spontane blauwe plekken en bloedingen
omvatten (Swerdlow et al. 2008).
Figuur 20: Vergelijking overlevingskansen tussen CMML-patiënten wild type en mutant SETBP1 (Damm et al. 2013)
Figuur 19: Vergelijking overlevingskansen tussen aCML-patiënten SETBP1 wild type en gemuteerd (Piazza et al. 2013)

34
4.2.1.3 Morfologie
Het perifere bloed vertoont een karakteristieke morfologie. Enkele typische kenmerken zijn kleine
rijpe lymfocyten, geblokt kernchromatine, gumprechtse schaduwen en minder dan 10%
prolymfocyten (figuur 21 en 22) (Swerdlow et al. 2008). Het beenmerg wordt gekenmerkt door het
voorkomen van meer dan 30% lymfocyten (Swerdlow et al. 2008).
4.2.1.4 Diagnose
De diagnose van CLL wordt gebaseerd op de combinatie van het aantal lymfocyten (lymfocytose),
cytomorfologisch onderzoek van het bloeduitstrijkje en immunofenotypering van het perifere bloed
(Swerdlow et al. 2008). Er zijn echter ziektebeelden die heel erg op CLL kunnen lijken, bijvoorbeeld
small lymfatisch lymfoom (Hallek et al. 2008). Het is dus belangrijk om te zoeken naar een nieuwe
manier om een vlugge diagnose te stellen zodat een effectieve behandeling gestart kan worden.
4.2.2 Mutaties die kunnen voorkomen bij CLL
4.2.2.1 TP53-mutatie
Bij de regulering van de celgroei en –differentiatie zijn zoals eerder vermeld meestal twee groepen
genen betrokken, namelijk de proto-oncogenen en de tumorsuppressorgenen. Deze eerste hebben
een stimulerende werking in tegenstelling tot de tumorsuppressorgenen, die een remmende werking
hebben. Kanker wordt meestal veroorzaakt door mutaties in deze genen. P53, ook gekend als TP53,
heeft de functie van een tumorsuppressor. Naast het repareren van fouten in het DNA bij de
celdeling, brengt het TP53-gen ook de apoptose op gang als de schade niet meer hersteld kan
worden. TP53 stimuleert P21, het BAX-gen en MDM2. Het gen P21 stimuleert de remming van de
celcyclus. Wanneer TP53 gemuteerd is en daardoor niet meer aan P21 kan binden, wordt de
celcyclus niet meer geremd waardoor er een ongeremde celdeling gaat plaatsvinden. BAX zorgt voor
apoptose van cellen met mutaties. Zonder een ongemuteerd TP53-eiwit komt er dus een overvloed
aan gemuteerde cellen die niet opgeruimd worden. MDM2 zorgt voor negatieve terugkoppeling. Het
breekt TP53-eiwitten af. Wanneer MDM2 een mutatie heeft waardoor het beter of sneller werkt,
Figuur 22: Chronische lymfatische leukemie - Woekering bij rijpcellige lymfocyten bij CLL (Scholten 2013)
Figuur 21: Chronische lymfatische leukemie - Toename
van rijpcellige lymfocyten met kenmerkende
marmerstructuur in de kern en verspreid meerdere kapot
gestreken cellen: gumprechtse schollen (Scholten 2013)

35
blijven er nauwelijks TP53-eiwitten over. Dit doet de kans op kankerontwikkeling stijgen (Lodish et al.
2013, Ryan et al. 2001, Soussi 2012b, Vogelstein et al. 2000).
4.2.2.1.1 TP53-eiwit en genstructuur
Het TP53-proteïne is een relatief klein fosfoproteïne dat opgebouwd is uit 393 aminozuren en kan in
ingedeeld worden in vijf domeinen (zie figuur 23). Het N-terminale einde (aminozuren 1-42) bevat
het domein dat het TP53-eiwit als transcriptiefactor activeert en bevat een bindingsplaats voor het
MDM2-eiwit. Daarna bevindt zich een proline-rijke regio die een belangrijke functie heeft in de
apoptose. De centrale regio (aminozuren 102-292) bevat het domein dat een specifieke
DNA-sequentie herkent en eraan bindt. De volgende regio (aminozuren 323-356) bevat het domein
dat verantwoordelijk is voor de tetramerisatie van het TP53-eiwit. Tenslotte is er het C-terminale
einde van TP53 (aminozuren 363-393) dat een niet-specifiek DNA-domein bevat dat kan binden aan
beschadigd DNA.
Het humane TP53-gen is gelokaliseerd op de korte arm van chromosoom 17 (17p13) en bestaat uit
22 000 bp en 11 exonen die coderen voor een eiwit met een moleculair gewicht van 53 kDa, die
betrokken is bij de regulering van de celcyclus.
4.2.2.1.2 Belang van de TP53-mutatie
Het TP53-eiwit bevat vijf aminozuren die een hoge mutatiefrequentie (28%) vertonen (zie figuur 23).
Deze hotspot regio’s situeren zich van exon 5 tot exon 8 (van aminozuur 126 tot 306). De meest
frequente mutaties komen voor op de plaatsen waar de lussen van het TP53-eiwit contact maken
met het DNA (Lodish et al. 2013). TP53-mutaties worden teruggevonden in ongeveer 50% van de
humane kankers (Soussi 2012a). Deze hoge frequentie is logisch aangezien TP53 een erg belangrijk
tumorsuppressorgen is dat het herstel van fouten in het DNA reguleert en de apoptose op gang
brengt als de schade niet hersteld kan worden. Als het TP53-gen gemuteerd wordt, kan deze functie
wegvallen waardoor een overvloed aan beschadigde cellen niet opgeruimd wordt, wat leidt tot
kanker. Tabel 7 geeft een overzicht van verschillende studies die het verband onderzocht hebben
tussen CLL en TP53-mutaties.
Figuur 23: Voorstelling van het TP53-gen met aanduiding van de hotspotregio's (Soussi 2012a).
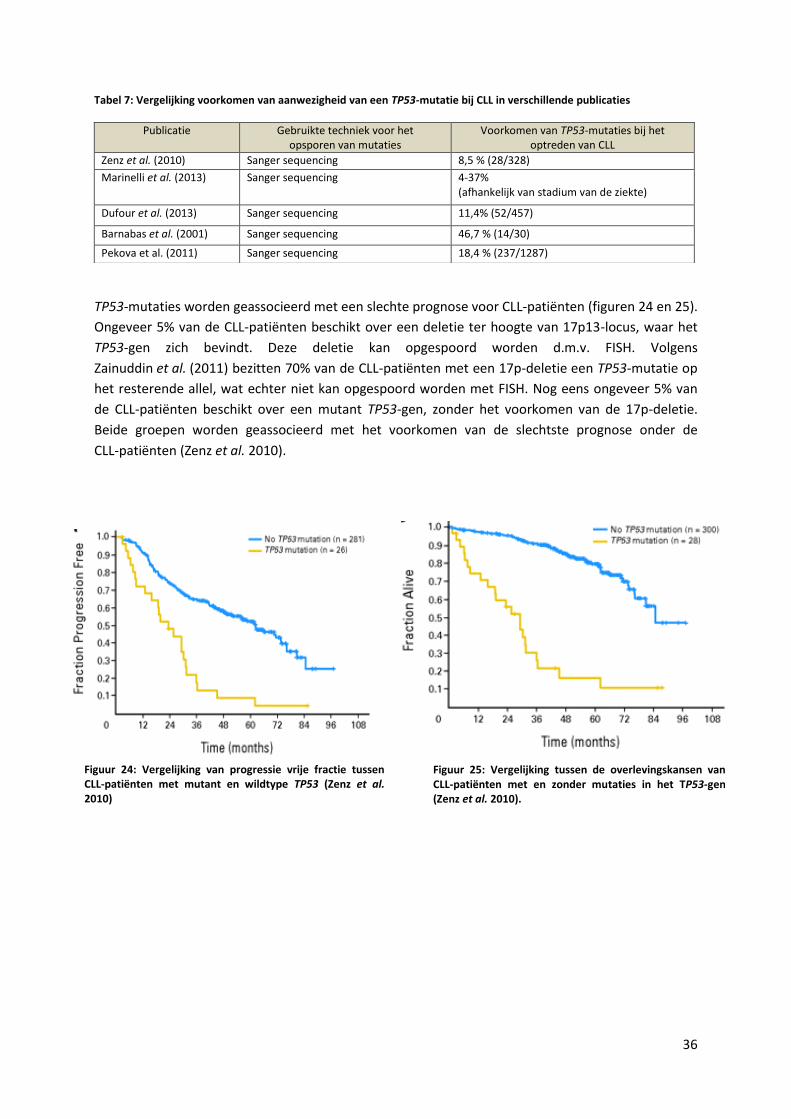
36
Tabel 7: Vergelijking voorkomen van aanwezigheid van een TP53-mutatie bij CLL in verschillende publicaties
TP53-mutaties worden geassocieerd met een slechte prognose voor CLL-patiënten (figuren 24 en 25).
Ongeveer 5% van de CLL-patiënten beschikt over een deletie ter hoogte van 17p13-locus, waar het
TP53-gen zich bevindt. Deze deletie kan opgespoord worden d.m.v. FISH. Volgens
Zainuddin et al. (2011) bezitten 70% van de CLL-patiënten met een 17p-deletie een TP53-mutatie op
het resterende allel, wat echter niet kan opgespoord worden met FISH. Nog eens ongeveer 5% van
de CLL-patiënten beschikt over een mutant TP53-gen, zonder het voorkomen van de 17p-deletie.
Beide groepen worden geassocieerd met het voorkomen van de slechtste prognose onder de
CLL-patiënten (Zenz et al. 2010).
Publicatie Gebruikte techniek voor het opsporen van mutaties
Voorkomen van TP53-mutaties bij het optreden van CLL
Zenz et al. (2010) Sanger sequencing 8,5 % (28/328)
Marinelli et al. (2013) Sanger sequencing 4-37% (afhankelijk van stadium van de ziekte)
Dufour et al. (2013) Sanger sequencing 11,4% (52/457)
Barnabas et al. (2001) Sanger sequencing 46,7 % (14/30)
Pekova et al. (2011) Sanger sequencing 18,4 % (237/1287)
Figuur 24: Vergelijking van progressie vrije fractie tussen CLL-patiënten met mutant en wildtype TP53 (Zenz et al. 2010)
Figuur 25: Vergelijking tussen de overlevingskansen van CLL-patiënten met en zonder mutaties in het TP53-gen (Zenz et al. 2010).

37
4.2.2.2 NOTCH1-mutatie
NOTCH1 is een humaan gen dat codeert voor een transmembraan receptor. Deze NOTCH1-receptor
functioneert als een ligand geactiveerde transcriptiefactor die extracellulaire signalen overbrengt van
het celoppervlak, wat leidt tot verandering van genenexpressie in de kern (figuur 26) (Ferrando
2009). De NOTCH-signaleringspathway wordt geactiveerd door een ligand op een naburige cel en
speelt een essentiële rol in het controleren van de proliferatie, differentiatie en overleving van cellen
(Willander et al. 2013). De NOTCH1-receptor is een membraangebonden proteïne die bestaat uit een
extracellulair, transmembraan en intracellulair domein. NOTCH1-eiwitten spelen een rol bij een
waaier aan ontwikkelingsprocessen, bijvoorbeeld bij de regulering van de differentiatie van T-helper
cellen. NOTCH1 is een oncogen waarvan constitutieve activering van NOTCH1-signalering de meest
prominente oncogene pathway is in T-cel transformatie (Ferrando 2009; Van Vlierberghe en
Ferrando 2012).
Figuur 26: Schematische voorstelling van de NOTCH1-signalering in T-cel voorlopers. Interactie van de NOTCH1-receptor met het NOTCH ligand delta-like-4. De delta gen familie codeert voor NOTCH liganden die worden gekarakteriseerd door een DSL domein, EGF repeats en een transmembraan domein. Deze NOTCH ligand delta-like 4 zijn aanwezig op het oppervlak van stroma cellen van de thymus. Interactie tussen ligand en receptor leidt tot een dubbele proteolytische splitsing van de receptor in T-cel voorlopers. De eerste splitsing gebeurt door ADAM 10, een metalloprotease en een tweede splitsing gebeurt door de aanwezigheid van een gamma-secretase complex. Vrijstelling van de intracellulaire domeinen van NOTCH1 van het membraan, leidt tot de activatie van de expressie van NOTCH doelgenen in de kern (Van Vlierberghe en Ferrando 2012).

38
4.2.2.2.1 NOTCH1-eiwit en genstructuur
Het NOTCH1-eiwit is opgebouwd uit 2555 aminozuren en heeft een moleculair gewicht van
272,5 kDa (Weizmann institute of Science 2013). Gaande van de N- naar de C-terminus bevat de
humane NOTCH-receptor vijf regio’s (zie figuur 27). Een eerste regio is een variabel aantal (25 tot 36)
van epidermale groeifactor(EGF)-achtige domeinen. Vele van deze domeinen bevatten calcium
bindingsplaatsen. Een tweede regio is de LNR-regio (Lin-12-Notch repeats). De volgende regio is het
HD-domein (niet aangegeven op figuur 27), het receptor heterodimerisatie domein. Na dit domein
aan het C-terminale uiteinde bevindt zich een specifieke knipplaats. Deze knipplaats wordt zoals
eerder vermeld herkend door ADAM 10, een metallo-protease. Daarna volgt het transmembraan
(TM) domein. Dit domein bevat eveneens een knipplaats. Deze plaats wordt specifiek herkend door
het secretasecomplex, waardoor het intracellulaire domein van de receptor vrijgesteld kan worden.
De laatste regio bevat een RAM-, ANK-, NLS-, OPA- en PEST-domein. Het PEST-domein (rijk aan
proline (P), glutaminezuur (E), serine (S) en threonine (T)) speelt een belangrijke rol in de
eiwitafbraak. Mutaties van dit domein leiden tot een grotere stabiliteit van de receptor, wat
samenhangt met kanker (Ranganathan et al. 2011; Sala et al. 2012). Het humane NOTCH1-gen is
gelokaliseerd op de lange arm van chromosoom 9 (9q34.3) en bestaat uit 51 419 bp en 34 exonen
(Weizmann institute of Science 2013).
4.2.2.2.2 Belang van de mutatie
In heel wat studies worden mutaties in het NOTCH1-gen in verband gebracht met CLL (zie tabel 8).
Volgens de studies van Willander et al. (2013) en Fabbri et al. (2011) komen de mutaties die gelinkt
worden met CLL enkel voor in het PEST-domein (exon 34) (zie figuur 28). Deze heeft een belangrijke
rol in de eiwitafbraak. Mutaties in dit domein leiden tot een grotere stabiliteit van de receptor, wat in
verband kan gebracht worden met kanker, in dit geval bloedkanker. NOTCH1-mutaties kunnen een
belangrijke merker zijn van een ongunstige prognose bij CLL-patiënten (Fabbri et al. 2011).
Figuur 27: Schematische voorstelling van de domein architectuur van een
humane NOTCH1-receptor (Sala et al. 2012).
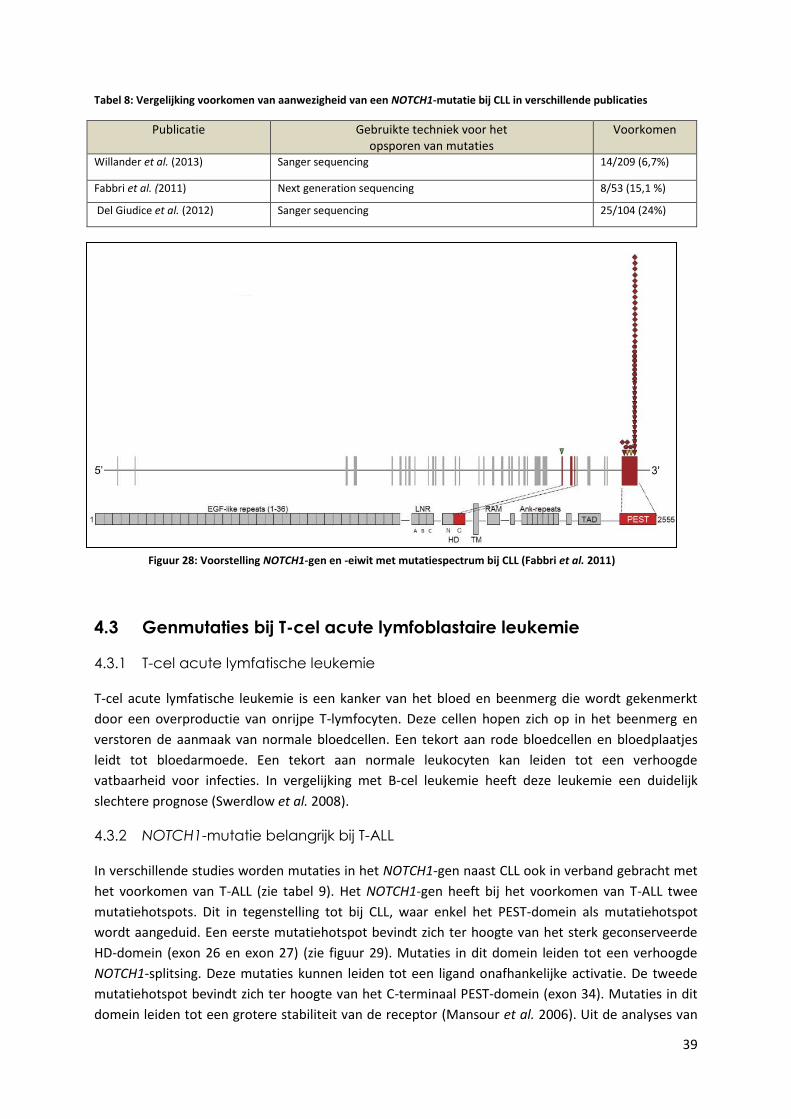
39
Tabel 8: Vergelijking voorkomen van aanwezigheid van een NOTCH1-mutatie bij CLL in verschillende publicaties
Publicatie Gebruikte techniek voor het opsporen van mutaties
Voorkomen
Willander et al. (2013) Sanger sequencing 14/209 (6,7%)
Fabbri et al. (2011) Next generation sequencing 8/53 (15,1 %)
Del Giudice et al. (2012) Sanger sequencing 25/104 (24%)
4.3 Genmutaties bij T-cel acute lymfoblastaire leukemie
4.3.1 T-cel acute lymfatische leukemie
T-cel acute lymfatische leukemie is een kanker van het bloed en beenmerg die wordt gekenmerkt
door een overproductie van onrijpe T-lymfocyten. Deze cellen hopen zich op in het beenmerg en
verstoren de aanmaak van normale bloedcellen. Een tekort aan rode bloedcellen en bloedplaatjes
leidt tot bloedarmoede. Een tekort aan normale leukocyten kan leiden tot een verhoogde
vatbaarheid voor infecties. In vergelijking met B-cel leukemie heeft deze leukemie een duidelijk
slechtere prognose (Swerdlow et al. 2008).
4.3.2 NOTCH1-mutatie belangrijk bij T-ALL In verschillende studies worden mutaties in het NOTCH1-gen naast CLL ook in verband gebracht met
het voorkomen van T-ALL (zie tabel 9). Het NOTCH1-gen heeft bij het voorkomen van T-ALL twee
mutatiehotspots. Dit in tegenstelling tot bij CLL, waar enkel het PEST-domein als mutatiehotspot
wordt aangeduid. Een eerste mutatiehotspot bevindt zich ter hoogte van het sterk geconserveerde
HD-domein (exon 26 en exon 27) (zie figuur 29). Mutaties in dit domein leiden tot een verhoogde
NOTCH1-splitsing. Deze mutaties kunnen leiden tot een ligand onafhankelijke activatie. De tweede
mutatiehotspot bevindt zich ter hoogte van het C-terminaal PEST-domein (exon 34). Mutaties in dit
domein leiden tot een grotere stabiliteit van de receptor (Mansour et al. 2006). Uit de analyses van
Figuur 28: Voorstelling NOTCH1-gen en -eiwit met mutatiespectrum bij CLL (Fabbri et al. 2011)

40
de prognostische betekenis van NOTCH1-mutaties in T-ALL is gebleken dat deze mutaties niet
geassocieerd worden met een ongunstige prognose. In bepaalde gevallen leiden NOTCH1-mutaties
zelfs tot betere prognoses voor de patiënt (Ferrando 2009).
Tabel 9: Vergelijking voorkomen van aanwezigheid van een NOTCH1-mutatie bij T-ALL in verschillende publicaties
4.4 Genmutaties bij mantelcellymfoom
4.4.1 Mantelcellymfoom Een non-Hodgkin-lymfoom (NHL) is een vorm van lymfeklierkanker. Het is een kanker die begint in
het lymfestelsel. Het lymfestelsel speelt een belangrijke rol in de afweer tegen ziekteverwekkers. Bij
NHL zijn de lymfeklieren groter geworden door een overdreven celwoekering. De toename van het
aantal cellen zorgt er voor dat de lymfocyten niet meer optimaal werken, waardoor het lichaam dus
een deel van zijn afweersysteem verliest. Er bestaan meer dan 30 verschillende NHL’s die zeer
verschillend zijn qua gedrag, presentatie in het lichaam en gevoeligheid voor therapie.
Een van de vele soorten NHL’s is het mantelcellymfoom (MCL). Dit is een zeldzaam voorkomende
groep van NHL’s (ongeveer 5%). MCL is een B-cel NHL, wat duidt op het feit dat het de B-lymfocyten
zijn die ongecontroleerd gaan vermenigvuldigen met de vorming van een lymfoom als gevolg. Het
komt meest voor bij mannen ouder dan 60 jaar. De ziekte heet mantelcellymfoom omdat de
tumorcellen afkomstig zijn uit de mantelzone van de lymfeklier (Swerdlow et al. 2008).
Publicatie Gebruikte techniek voor het opsporen van mutaties
Voorkomen
Van Vlierberghe and Ferrando (2012) Niet vermeld (Review) 60 %
Ferrando (2009)
Niet vermeld (Review) 50%
Figuur 29: Voorstelling NOTCH1-eiwit met mutatiespectrum bij T-ALL (Zhu et al. 2006)

41
4.4.2 NOTCH1-mutatie belangrijk bij MCL Verschillende studies hebben het verband bestudeert tussen het voorkomen van NOTCH1-mutaties
en MCL (zie tabel 10). De studies zijn het er allemaal over eens dat de mutatiehotspot zich net zoals
bij CLL bevindt ter hoogte van het PEST-domein (exon 34) (zie figuur 30). De meest voorkomende
mutaties zijn nonsense mutaties. Hierbij verandert het oorspronkelijke aminozuur in een stopcodon,
wat resulteert in een ingekort eiwit met een verhoogde oncogene activiteit. Het PEST-domein speelt
een belangrijke rol in de eiwitafbraak. Mutaties in dit domein leiden tot een grotere stabiliteit van de
receptor, wat in verband kan gebracht worden met kanker. De aanwezigheid van een
NOTCH1-mutatie bij patiënten met MCL wordt geassocieerd met een slechte prognose (zie figuur 31)
(Parekh 2012).
Tabel 10: Vergelijking voorkomen van aanwezigheid van een NOTCH1-mutatie bij MCL in verschillende publicaties
Publicatie Gebruikte techniek voor het opsporen van mutaties
Voorkomen
Parekh (2012)
Sanger sequencing 14/121 (12%)
Bea et al. (2013) Sanger sequencing 8/172 (5%)
Figuur 30: Voorstelling NOTCH1-eiwit met mutatiespectrum bij MCL (Parekh
2012)
Figuur 31: Prognostisch effect van
NOTCH1-mutaties bij MCL (Parekh 2012)
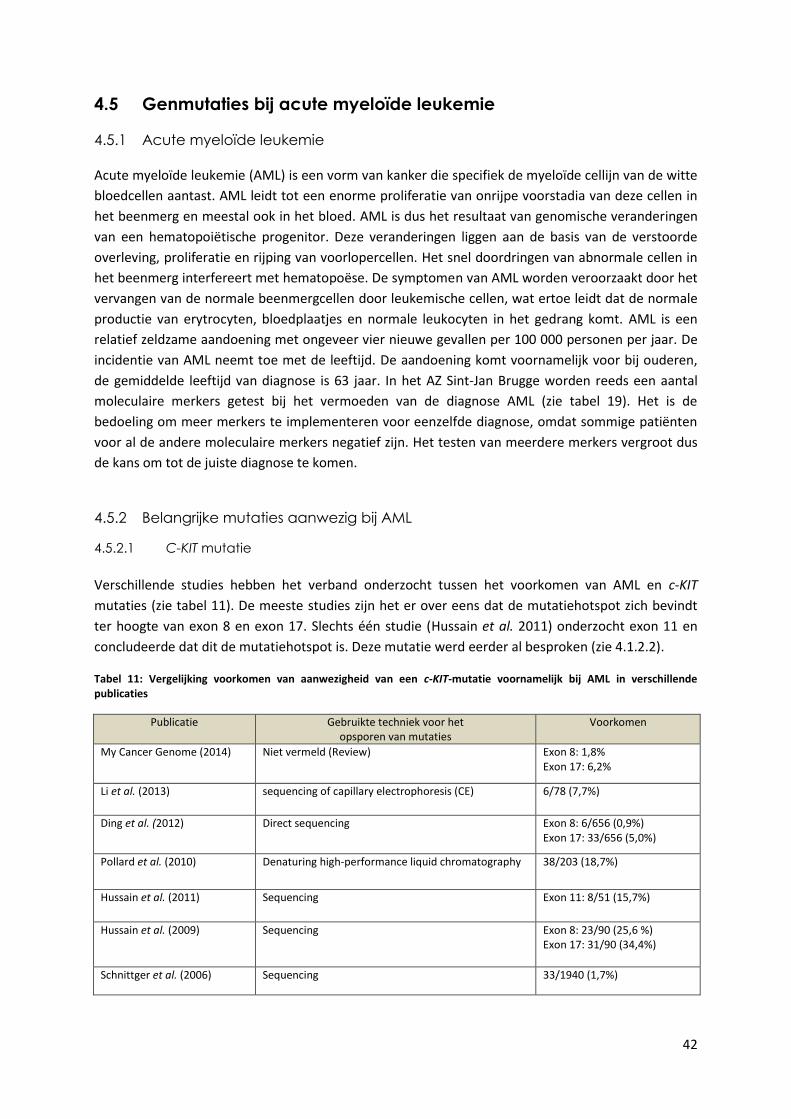
42
4.5 Genmutaties bij acute myeloïde leukemie
4.5.1 Acute myeloïde leukemie Acute myeloïde leukemie (AML) is een vorm van kanker die specifiek de myeloïde cellijn van de witte
bloedcellen aantast. AML leidt tot een enorme proliferatie van onrijpe voorstadia van deze cellen in
het beenmerg en meestal ook in het bloed. AML is dus het resultaat van genomische veranderingen
van een hematopoiëtische progenitor. Deze veranderingen liggen aan de basis van de verstoorde
overleving, proliferatie en rijping van voorlopercellen. Het snel doordringen van abnormale cellen in
het beenmerg interfereert met hematopoëse. De symptomen van AML worden veroorzaakt door het
vervangen van de normale beenmergcellen door leukemische cellen, wat ertoe leidt dat de normale
productie van erytrocyten, bloedplaatjes en normale leukocyten in het gedrang komt. AML is een
relatief zeldzame aandoening met ongeveer vier nieuwe gevallen per 100 000 personen per jaar. De
incidentie van AML neemt toe met de leeftijd. De aandoening komt voornamelijk voor bij ouderen,
de gemiddelde leeftijd van diagnose is 63 jaar. In het AZ Sint-Jan Brugge worden reeds een aantal
moleculaire merkers getest bij het vermoeden van de diagnose AML (zie tabel 19). Het is de
bedoeling om meer merkers te implementeren voor eenzelfde diagnose, omdat sommige patiënten
voor al de andere moleculaire merkers negatief zijn. Het testen van meerdere merkers vergroot dus
de kans om tot de juiste diagnose te komen.
4.5.2 Belangrijke mutaties aanwezig bij AML
4.5.2.1 C-KIT mutatie
Verschillende studies hebben het verband onderzocht tussen het voorkomen van AML en c-KIT
mutaties (zie tabel 11). De meeste studies zijn het er over eens dat de mutatiehotspot zich bevindt
ter hoogte van exon 8 en exon 17. Slechts één studie (Hussain et al. 2011) onderzocht exon 11 en
concludeerde dat dit de mutatiehotspot is. Deze mutatie werd eerder al besproken (zie 4.1.2.2).
Tabel 11: Vergelijking voorkomen van aanwezigheid van een c-KIT-mutatie voornamelijk bij AML in verschillende publicaties
Publicatie Gebruikte techniek voor het opsporen van mutaties
Voorkomen
My Cancer Genome (2014) Niet vermeld (Review) Exon 8: 1,8% Exon 17: 6,2%
Li et al. (2013) sequencing of capillary electrophoresis (CE) 6/78 (7,7%)
Ding et al. (2012) Direct sequencing Exon 8: 6/656 (0,9%) Exon 17: 33/656 (5,0%)
Pollard et al. (2010) Denaturing high-performance liquid chromatography 38/203 (18,7%)
Hussain et al. (2011) Sequencing Exon 11: 8/51 (15,7%)
Hussain et al. (2009) Sequencing Exon 8: 23/90 (25,6 %) Exon 17: 31/90 (34,4%)
Schnittger et al. (2006) Sequencing 33/1940 (1,7%)

43
Er zijn twee chromosomale afwijkingen die voorkomen bij AML die een aparte groep AML definiëren.
Een eerste afwijking is de translocatie t(8;21)(q22;q22). Het resultaat hiervan is het fusiegen
AML1-ETO. Er werd aangetoond dat dit gen de differentiatie, proliferatie en apoptose beïnvloedt
(Schnittger et al. 2006).
Een tweede belangrijke chromosomale afwijkingen is een inversie, namelijk inv(16)(p13q22). De
inversie bevindt zich ter hoogte van chromosoom 16, waar het DNA-segment vanaf p13 tot q22
volledig omgekeerd ingebouwd is. Deze herschikking leidt tot de verstoring van het MYH11-gen op
16p13 en het CBFβ-gen op 16q22.
In beide subgroepen van AML treden veranderingen op ter hoogte van de CBF-transcriptiefactor:
AML1 (CBFα2) in t(8;21) en CBFβ in inv(16). Dit leidt tot de gemeenschappelijke nomenclatuur van
‘Core-binding-factor’ (CBF)-leukemieën voor inv(16)- en t(8;21)-positieve AML (Schnittger et al.
2006). Deze twee subgroepen worden beiden in verband gebracht met een relatief goede prognose
voor de patiënt (Cho et al. 2003; Van der Reijden et al. 2010).
Volgens de studie van Schnittger et al. (2006) worden c-KIT-mutaties in exon 8 vooral teruggevonden
bij CBFβ-MYH11 positieve AML-patiënten. Terwijl c-KIT-mutaties in exon 17, voornamelijk ter hoogte
van asparaginezuur 816, hoofdzakelijk worden teruggevonden bij AML1-ETO positieve
AML-patiënten. In deze studie werd eveneens aangetoond dat AML1-ETO-positieve AML-patiënten
met een c-KIT-Asp816 mutatie behoren tot een subgroep met een ongunstige prognose. Er kan dus
gesteld worden dat een c-KIT-Asp816 mutatie een betrouwbare moleculaire merker is voor het
identificeren van patiënten met een slechte prognose binnen een prognostisch gunstige AML-groep.
Dit wordt eveneens aangetoond in de studie van Boissel et al. (2006).
In de studie van Schwind et al.(2013) wordt de prognostische waarde onderzocht van c-KIT-mutaties
bij CBFβ-MYH11 positieve AML-patiënten. Hier wordt net zoals bij AML1-ETO-positieve
AML-patiënten, besloten dat deze subgroep van AML geassocieerd wordt met een slechte prognose.
Terwijl in de studie van Boissel et al. (2006) wordt aangetoond dat er geen prognostisch verschil
bestaat tussen CBFβ-MYH11 positieve AML-patiënten met en zonder c-KIT-mutatie.
4.5.2.2 DNA (cytosine-5)-methyltransferase 3A (DNMT3A)-mutatie
DNA-methylatie is een epigenetische modificatie die een cruciale rol speelt in diverse biologische
processen, zoals transcriptionele repressie van genen en transposon silencing. Bovendien zijn
afwijkende DNA-methylatie patronen gerapporteerd in vele tumoren en ziekten bij mensen. In het
humaan genoom worden cytosine residuen van CpG dinucleotide sequenties gemodificeerd tijdens
de post-replicatie door DNA-methylatie. Ongeveer 70%-80% van de CpG dinucleotiden zijn
gemethyleerd. CpG dinucleotide sequenties zijn regio’s van het DNA waar een cytosine nucleotide
voorkomt naast een guanine nucleotide. Het DNMT3A-gen codeert voor een DNA-methyltransferase
waarvan gedacht wordt vooral te functioneren in de novo methylatie (Umehara et al. 2013).

44
4.5.2.2.1 DNMT3A-eiwit en genstructuur
Het DNMT3A-eiwit is opgebouwd uit 912 aminozuren en heeft een moleculair gewicht van ongeveer
102 kDa. Het eiwit is een heterotetrameer dat samengesteld is uit één DNMT3A-homodimeer en
twee DNMT3L-subunits. Het eiwit is terug te vinden in de nucleus en in het cytoplasma (Weizmann
Institute of Science 2013). Het humane DNMT3A-gen is gelokaliseerd op de korte arm van
chromosoom 2 (2p23.3) en bestaat uit 109 615 bp en 23 exonen (Weizmann Institute of Science
2013).
4.5.2.2.2 Belang van de mutatie
DNMT3A-mutaties worden vooral in verband gebracht met AML en in mindere mate met MPN. In
tabel 12 wordt een overzicht gegeven van verschillende publicaties waarin dit verband wordt
bestudeerd.
De verschillende studies zijn het erover eens dat de mutatiehotspot zich bevindt ter hoogte van
aminozuur 882 (zie figuur 32). Hier bevinden zich 50 tot 60% van de mutaties. De meest
voorkomende mutatie is p.Arg882His. Dit aminozuur maakt deel uit van het katalytisch centrum. Er
kan dus gesteld worden dat DNMT3A-mutaties de katalytische activiteit aantasten, maar wat hun
effect juist is op de DNA-methylatie in de cel is nog niet duidelijk (Kim et al. 2013). Patiënten met een
DNMT3A-mutatie hebben een duidelijk slechtere prognose dan patiënten zonder mutatie (Ley et al.
2010).
Tabel 12: Vergelijking voorkomen van aanwezigheid van een DNMT3A-mutatie bij MPN en AML in verschillende publicaties
Publicatie Gebruikte techniek voor het opsporen van mutaties
Ziekte Voorkomen
Tefferi et al. (2011) Niet vermeld (Review)
PV PMF MPN
7% 6% 14%
Hou et al. (2012) Direct sequencing AML 70/500 (14%)
Shivarov et al. (2013) Niet vermeld (vergelijking van negen studies)
AML 22,5 % (variërend van 14% tot 34,2%)

45
4.5.2.3 Isocitraat dehydrogenase – mutatie
Er bestaan meerdere isovormen van het enzym isocitraat dehydrogenase (IDH). Isocitraat
dehydrogenasen katalyseren de oxidatieve decarboxylering van isocitraat naar 2-oxoglutaraat, wat
op zijn beurt verder omgezet wordt tot α-ketoglutaraat (α-KG) en CO2 (zie figuur 33). Deze enzymen
kunnen ingedeeld worden in twee verschillende subklassen. De eerste subklasse maakt gebruik van
NAD+ als elektronacceptor, terwijl de tweede subklasse gebruik maakt van NADP+ als
elektronacceptor. Het humane isocitraat dehydrogenase bestaat in drie isovormen namelijk IDH1,
IDH2 en IDH3. IDH1 en IDH2 zijn twee NADP+-afhankelijke isocitraat dehydrogenasen, waarvan één
terug te vinden is in de mitochondriën (IDH2) en de andere hoofdzakelijk terug te vinden is in het
cytoplasma (IDH1). De isovorm IDH3 is een NAD+-afhankelijke isocitraat dehydrogenasen dat
voorkomt in de mitochondriën (Yang et al. 2012).
Figuur 32: Mutatiespectrum van het DNMT3A-gen (Roller et al. 2013)

46
4.5.2.3.1 Isocitraat dehydrogenase-eiwit en genstructuur
Het IDH1-eiwit is opgebouwd uit 414 aminozuren en heeft een moleculair gewicht van ongeveer
46,6 kDa. Het is een homodimeer waarvan elke subunit bindt met één magnesium- of mangaan-ion.
Het eiwit komt voor in het cytoplasma en in de peroxisomen (Weizmann Institute of Science 2013).
Het humane IDH1-gen is gelokaliseerd op de lange arm van chromosoom 2 (2q34) en bestaat uit
29 848 bp en 10 exonen (Weizmann Institute of Science 2013).
Het IDH2-eiwit is opgebouwd uit 452 aminozuren en heeft een moleculair gewicht van ongeveer
51 kDa. Het is een homodimeer waarvan elke subunit eveneens bindt met één magnesium- of
mangaan-ion. Het eiwit komt in tegenstelling tot het IDH1-eiwit voor in de mitochondriën
(Weizmann Institute of Science 2013). Het humane IDH2-gen is gelokaliseerd op de lange arm van
chromosoom 15 (15q26.1) en bestaat uit 19 460 bp en 11 exonen (Weizmann Institute of Science
2013).
4.5.2.3.2 Belang van de mutatie
Mutante IDH1- en IDH2-enzymen beschikken over de capaciteit om α-KG om te zetten in
2-hydroxyglutaraat (2-HG), een klein onco-metaboliet. De aanwezigheid van mutante IDH1- of
IDH2-eiwitten resulteert in verhoogde hoeveelheden 2-HG. Deze verhoging leidt op zijn beurt tot een
verandering van een aantal downstream cellulaire activiteiten. 2-HG is bijvoorbeeld een competitieve
inhibitor voor α-KG voor de binding aan verschillende histon demethylasen, bijvoorbeeld KDM
(histon lysine demethylase). Door deze inhibitie ontstaat een sterk afwijkend histon methylatie
profiel. Het metaboliet 2-HG remt eveneens de activiteit van de TET1 en TET2 hydroxymethylasen,
wat opnieuw resulteert in een verandering van de epigenetische regulatie. Deze epigenetische
veranderingen kunnen leiden tot een verandering in differentiatie en bijgevolg tot tumorvorming
(zie figuur 34) (Prensner and Chinnaiyan 2011; Yang et al. 2012).
Figuur 33: Schematische voorstelling van de productie en het gebruik van α-ketoglutaraat
(α-KG) in humane cellen. Vier enzymen (IDH1, IDH2, IDH3 en glutamaat dehydrogenase (GDH))
kunnen α-KG produceren. Dit α-KG wordt gebruikt in vier afzonderlijke pathways (Yang et al.
2012).

47
Verschillende studies hebben het verband onderzocht tussen het voorkomen van deze mutaties en
het voorkomen van bepaalde specifieke bloedkankers (zie tabel 13).
Tabel 13: Vergelijking voorkomen van aanwezigheid van een IDH1/IDH2-mutatie voornamelijk bij AML in verschillende publicaties
Publicatie Gebruikte techniek voor het opsporen van mutaties
Mutatie Voorkomen
Ashraf et al. (2013) allele-specifieke oligonucleotide-PCR IDH2 22/120 (18%)
Shih et al. (2012) Niet vermeld (Review) IDH1/IDH2 15-33 %
Patel et al. (2011) Sanger sequencing IDH1 IDH2
12/199 (6%) 4/199 (4%)
Paschka et al. (2010) Denaturerende HPLC IDH1 IDH2
61/806 (7,6%) 70/806 (8,7%)
De meest voorkomende mutaties in het IDH1-gen bevinden zich ter hoogte van het arginineresidu
132. Dit oorspronkelijke arginine wordt vaak omgezet in histidine, cytosine, serine, glycine of leucine.
De meest voorkomende IDH2-mutatie is het arginineresidu 172 dat verandert in een lysine. Andere
vaak voorkomende mutaties worden weergegeven in tabel 14. Het voorkomen van een IDH1- en/of
IDH2-mutatie wordt geassocieerd met een slechte prognose (Paschka et al. 2010).
Tabel 14: De meest voorkomende mutaties in het IDH1- en IDH2-gen met betrekking tot AML, met in het vet aangeduid
de meest voorkomende mutaties (Paschka et al. 2010; Patel et al. 2011a; Patel et al. 2011b; Ashraf et al. 2013)
IDH1 IDH2
c.368G>A (p.Gly123Glu) c.392_296delinsTCCTG (p.Gly131_Arg132delinsValLeu) c.395G>A (p.Arg132His) c.394C>T (p.Arg132Cys) c.394C>A (p.Arg132Ser) c.394C>G (p.Arg132gly) c.395G>T (p.Arg132Leu) c.394_395delinsGA (p.Arg132Glu)
c.419G>A (p.Arg140Gln) c.515G>A (p.Arg172Lys)
Figuur 34: IDH1/2-mutaties inhiberen zowel histon- als DNA-demethylaties en leiden tot epigenetische veranderingen, wat op hun beurt kan leiden tot verandering in differentiatie en tumorvorming (Yang et al. 2012).
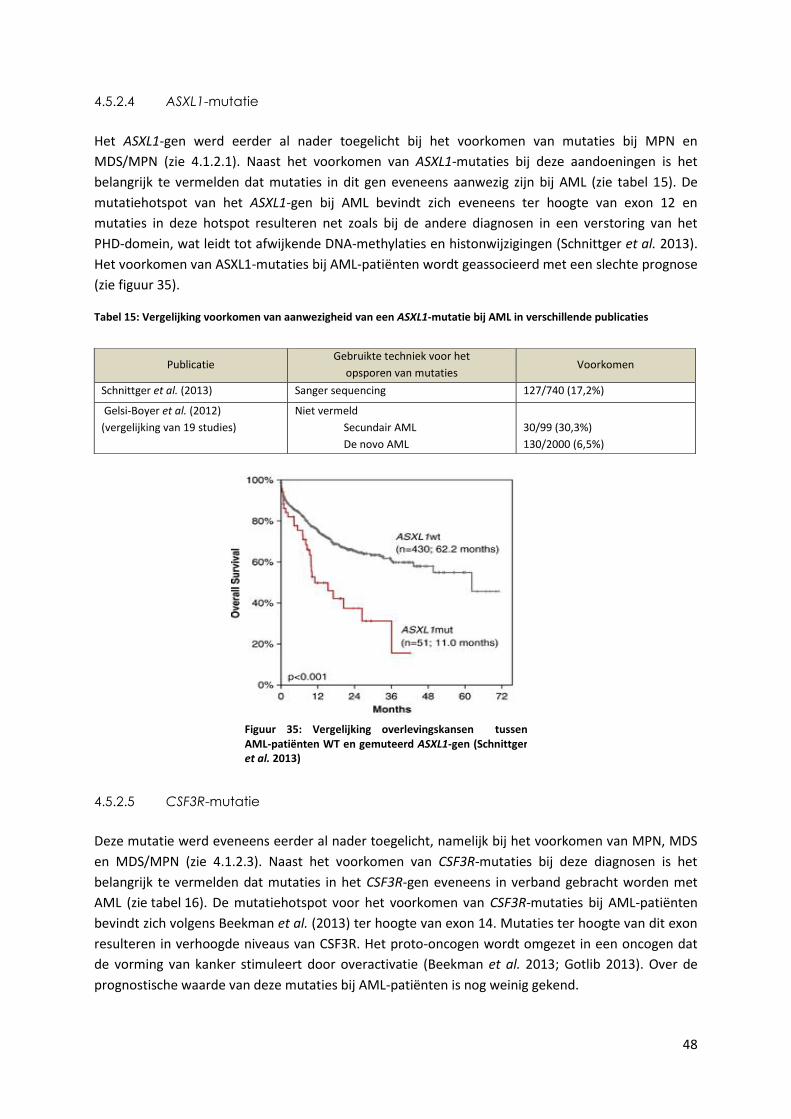
48
4.5.2.4 ASXL1-mutatie
Het ASXL1-gen werd eerder al nader toegelicht bij het voorkomen van mutaties bij MPN en
MDS/MPN (zie 4.1.2.1). Naast het voorkomen van ASXL1-mutaties bij deze aandoeningen is het
belangrijk te vermelden dat mutaties in dit gen eveneens aanwezig zijn bij AML (zie tabel 15). De
mutatiehotspot van het ASXL1-gen bij AML bevindt zich eveneens ter hoogte van exon 12 en
mutaties in deze hotspot resulteren net zoals bij de andere diagnosen in een verstoring van het
PHD-domein, wat leidt tot afwijkende DNA-methylaties en histonwijzigingen (Schnittger et al. 2013).
Het voorkomen van ASXL1-mutaties bij AML-patiënten wordt geassocieerd met een slechte prognose
(zie figuur 35).
Tabel 15: Vergelijking voorkomen van aanwezigheid van een ASXL1-mutatie bij AML in verschillende publicaties
4.5.2.5 CSF3R-mutatie
Deze mutatie werd eveneens eerder al nader toegelicht, namelijk bij het voorkomen van MPN, MDS
en MDS/MPN (zie 4.1.2.3). Naast het voorkomen van CSF3R-mutaties bij deze diagnosen is het
belangrijk te vermelden dat mutaties in het CSF3R-gen eveneens in verband gebracht worden met
AML (zie tabel 16). De mutatiehotspot voor het voorkomen van CSF3R-mutaties bij AML-patiënten
bevindt zich volgens Beekman et al. (2013) ter hoogte van exon 14. Mutaties ter hoogte van dit exon
resulteren in verhoogde niveaus van CSF3R. Het proto-oncogen wordt omgezet in een oncogen dat
de vorming van kanker stimuleert door overactivatie (Beekman et al. 2013; Gotlib 2013). Over de
prognostische waarde van deze mutaties bij AML-patiënten is nog weinig gekend.
Publicatie Gebruikte techniek voor het
opsporen van mutaties Voorkomen
Schnittger et al. (2013) Sanger sequencing 127/740 (17,2%)
Gelsi-Boyer et al. (2012)
(vergelijking van 19 studies)
Niet vermeld
Secundair AML
De novo AML
30/99 (30,3%)
130/2000 (6,5%)
Figuur 35: Vergelijking overlevingskansen tussen AML-patiënten WT en gemuteerd ASXL1-gen (Schnittger et al. 2013)

49
Tabel 16: Vergelijking voorkomen van aanwezigheid van een CSF3R-mutatie bij AML in verschillende publicaties
4.6 Genmutaties bij large granulaire lymfocytaire leukemie
4.6.1 Large granulaire lymfocytaire leukemie Large granulaire lymfocytaire (LGL) leukemie is een type chronische leukemie van de lymfocyten. LGL
wordt gekarakteriseerd door grotere lymfocyten, die duidelijke korrels bevatten. Er zijn twee types
van LGL-leukemie: T-LGL waarbij T-lymfocyten uitgroeien tot kankercellen en NK-LGL waarbij
NK-lymfocyten uitgroeien tot kankercellen. Ongeveer 2 tot 5 procent van de chronische
lymfoproliferatieve aandoeningen betreft LGL. De ziekte treft zowel mannen als vrouwen met een
gemiddelde leeftijd van 60 jaar. Bij het optreden van LGL neemt het aantal abnormale cellen in het
bloed toe, terwijl het aantal normale witte bloedcellen daalt. Hierdoor kan het lichaam minder goed
infecties bestrijden (Loughran 2012). Voor het stellen van de diagnose LGL worden momenteel nog
geen testen uitgevoerd in het AZ Sint-Jan Brugge. Er dient dus gezocht te worden naar moleculaire
merkers die in aanmerking komen om op een snelle en efficiënte manier de diagnose van LGL vast te
stellen.
4.6.2 Signaaltransducer en activator van transcriptie 3 gen (STAT3)-mutatie
belangrijk bij LGL
Het proteïne gecodeerd door het STAT3-gen behoort tot de STAT-eiwitfamilie. STAT staat voor ‘Signal
Transducer and Activator of Transcription’. Deze naam geeft de dubbele functie van deze moleculen
weer, namelijk het doorgeven van signalen in een cel en het initiëren van transcriptie van genen. Als
reactie op de werking van cytokinen en groeifactoren, worden de STAT-moleculen gefosforyleerd
door de receptor geassocieerde kinasen. Het STAT3-eiwit wordt geactiveerd door fosforylatie van
tyrosine 705 als reactie op verschillende groei- en transcriptiefactoren. Het reguleert de expressie
van diverse genen en speelt dus een belangrijke rol in vele cellulaire processen zoals celgroei en
apoptose. STAT3 is een oncogen en kan oncogenesis bevorderen wanneer het constitutief actief is
(zie figuur 36) (Weizmann Institute of Science 2013).
Publicatie Gebruikte techniek voor het
opsporen van mutaties Voorkomen
Sano et al. (2013)
Next generation sequencing
De novo AML
5/376 (1,3%)
Aref et al. (2014) Sequencing
De novo AML
Secundair AML
2/159 (1,2%)
19/23 (78,2%)
Beekman et al. (2013)
Niet vermeld (Review)
De novo AML
Secundair AML
0%
80%

50
4.6.2.1 STAT3-eiwit en genstructuur
Het STAT3-eiwit bestaat uit 770 aminozuren en heeft een moleculair gewicht van 88 kDa. Het gen is
opgebouwd uit vijf verschillende domeinen (zie figuur 37). De twee belangrijkste domeinen zijn het
DNA-bindingsdomein en het Scr Homology 2 (SH2)-domein. Dit eerste is een onafhankelijk gevouwen
eiwitdomein met ten minste één motief dat dubbel- of enkelstrengig-DNA herkent. Het
DNA-bindingsdomein herkent een specifieke DNA-sequentie of heeft een algemene
aantrekkingskracht tot DNA. Het tweede belangrijke domein (aminozuren 584-674) is een
fosfotyrosine bindende module die in veel signaalmoleculen aanwezig is. Het behoort tot de groep
eiwitten die een rol spelen in de signaalcascade. Het SH2-domein maakt het mogelijk om de
proteïnen die deze domeinen bevatten te laten migreren naar een gefosforyleerd tyrosineresidu van
andere proteïnen. Het domein interageert met eiwitten via de gefosforyleerde tyrosine van het eiwit.
Er is bijgevolg een associatie tussen die eiwitten van het SH2-domein wat resulteert in een verdere
signaalcascade (Makishima et al. 2013). Het humane STAT3-gen is gelokaliseerd op de lange arm van
chromosoom 17 (17q21.2) en bestaat uit 5 047 bp en 24 exonen (Weizmann Institute of Science
2013).
Figuur 36: Autocriene of paracriene signalen activeren tyrosine kinase groeifactorreceptoren, cytokinereceptor
geassocieerde Janus-familie kinasen (JAKs) en SRC tyrosine kinasen. Deze activeren op hun beurt STAT3 door fosforylatie
van tyrosine 705. In getransformeerde cellen kan STAT3 ook geactiveerd worden door constitutief actieve niet-
receptortyrosinekinasen zoals SRC en ABL. Na de tyrosinefosforylering gaan de STAT3-moleculen dimeriseren, waarna ze
zich verplaatsen naar de kern, waar ze de transcriptie kunnen activeren. De STAT3-signalering wordt normaal sterk
gereguleerd door een aantal remmende moleculen (vb. SOCS-eiwitten). In kankercellen gaat deze sterke controle echter
verloren, waardoor een overactieve receptor of niet-receptor tyrosinekinasen continu STAT3 fosforyleren en activeren.
Wat resulteert in continue stimulatie van de genenexpressie (Yu et al. 2007).

51
4.6.2.2 Belang van de mutatie
Recent werd aangetoond dat mutaties in het STAT3-gen in verband kunnen gebracht worden met het
voorkomen van T-LGL en NK-LGL (zie tabel 17). STAT3 is een oncogen waarvan de activatie een
centrale rol speelt in de celsignalering van verschillende kankers. Bij LGL komen mutaties in het
STAT3-gen voor in een zeer nauwe regio, namelijk het SH2-domein (aminozuur 584-674)
(zie figuur 37). Dit domein speelt een sleutelrol in de functie van het gen. Mutaties in deze regio
kunnen leiden naar constitutieve activiteit van STAT3 en wat leidt tot overactivatie, wat op zijn beurt
oncogenese bevordert. Uit de studie van Jerez et al. (2012) kan besloten worden dan een
STAT3-mutatie bij LGL-patiënten niet als een belangrijke prognostische merker kan beschouwd
worden.
Tabel 17: Vergelijking voorkomen van aanwezigheid van een STAT3- mutatie bij LGL en NK-LGL in verschillende publicaties
Publicatie Gebruikte techniek voor het opsporen van mutaties
Ziekte Voorkomen
Koskela et al. (2012) Next generation sequencing LGL 31/77 (40%)
Jerez et al. (2012)
Sanger sequencing T-LGL NK-LGL
32/120 (27%) 15/50 (30%)
Fasan et al. (2013) Niet vermeld (Review)
T-LGL NK-LGL
40% 30%
Qiu et al. (2013) Sequencing T-LGL 6/28 (21,4 %)
4.7 Genmutaties bij lymfoplasmocytair lymfoom
4.7.1 Lymfoplasmocytair lymfoom
Het lymfoplasmocytair lymfoom (LPL) is een subtype van de mature B-cel neoplasieën. Mature B-cel
neoplasieën zijn tumoren van de rijpe of mature B-cellen. Ze ontstaan doordat er een fout optreedt
in de celdeling van de B-lymfocyten. De cellen zullen niet meer uitrijpen maar blijven
ongecontroleerd delen of sterven niet meer af. Er is een ongecontroleerde proliferatie van
lymfocyten in het beenmerg en ook in de lymfeklieren. De maligne cellen verplaatsen zich soms via
lymfe naar andere plaatsen in het lichaam. Vaak gaat de ziekte gepaard met een paraproteïnemie
van het IgM-type, waarbij er een zeer grote hoeveelheid antistoffen van het type IgM in het bloed
Figuur 37: Opbouw van het STAT3-eiwit met het mutatiespectrum (Makishima et al. 2013)

52
circuleren. Deze antistoffen, ook wel M-proteïnen genoemd, worden geproduceerd door de
tumorcellen. Wanneer er dergelijke monoklonale IgM gammopathie aanwezig is, spreekt men van
Waldenström macroglobulinemie (WM), ook wel de ziekte van Waldenström genoemd (Swerdlow et
al. 2008).
4.7.1.1 Epidemiologie
LPL komt voornamelijk voor bij volwassenen met een gemiddelde leeftijd van 60 jaar. De ziekte komt
vaker voor bij mannen dan bij vrouwen (Swerdlow et al. 2008).
4.7.1.2 Klinische kenmerken
Door de ontwikkeling van maligne cellen en door de productie van het IgM ontwikkelen veel
patiënten allerhande symptomen. De meest voorkomende symptomen zijn algemene zwakte en
vermoeidheid, die beide het gevolg zijn van een onderliggende anemie. Hyperviscositeit van het
bloed komt voor in 30% van de gevallen. Door de grote hoeveelheden paraproteïnen kan het bloed
stroperig worden waardoor het minder goed circuleren in de kleine bloedvaatjes. Dit kan leiden tot
onder andere hartfalen, moeheid, daling van de eetlust en kortademigheid (Swerdlow et al. 2008).
4.7.1.3 Morfologie
Het bloedbeeld wordt meestal gekenmerkt door agglutinatie van de erytrocyten in samenhang met
een M-proteïne, terwijl ook afwijkende circulerende lymfatische cellen aangetroffen kunnen worden.
Vaak is er dan sprake van een mengbeeld van kleine lymfocyten met een grof chromatine en
middelgrote lymfocyten met veel meer cytoplasmaontwikkeling.
4.7.1.4 Diagnose
De diagnose van LPL/WM is zowel op morfologisch, immunofenotypisch en cytogenische
eigenschappen moeilijk te onderscheiden van andere B-cel lymfomen en andere celplasma
myelomen zoals milt marginale zone lymfomen. Een specifieke merker om LPL immunofenotypisch te
definiëren ontbreekt dus. Een andere manier om de diagnose van LPL te stellen zou de aanwezigheid
van het IgM-paraproteïne kunnen zijn, maar ook dit kenmerk is niet specifiek voor LPL, aangezien het
in veel lymfoïde neoplasma’s voorkomt. Hieruit kan besloten worden dat het stellen van diagnose
LPL vaak gebaseerd is op exclusie (Ondrejka et al. 2013 , Xu et al. 2011). Daardoor is het belangrijk
om te zoeken naar een nieuwe manier om een vlugge diagnose te stellen zodat een effectieve
behandeling gestart kan worden.

53
4.7.2 Myeloid differentiation primary response gene 88 (MYD88) –mutatie
Het MYD88-gen codeert voor een cytolisch adaptereiwit dat een centrale rol speelt bij het
aangeboren en adaptief immuunsysteem. Dit eiwit functioneert als een essentiële signaalomvormer
in de interleukine-1 en Toll-like receptor signaleringstrajecten (zie figuur 38). Deze trajecten regelen
de activatie van de talrijke pro-inflammatoire genen (Medicine 2013).
Interleukinen (IL) zijn een groep cytokinen die geproduceerd worden door geactiveerde macrofagen
en lymfocyten gedurende een immuunrespons. Cytokinen zijn een groep van signaalmoleculen die
op grote schaal worden gebruikt voor de mobiele communicatie doorheen het lichaam. Het doel van
de interleukineproductie is dan ook om met andere leukocyten te communiceren. Interleukinen
stimuleren leukocyten om tot proliferatie en differentiatie over te gaan.
Toll-like receptoren (TLRs) zijn een klasse van eiwitten die een rol spelen in het aangeboren
afweersysteem. De TLRs bevinden zich op het celoppervlak en functioneren als
patroonherkenningsmoleculen voor micro-organismen. TLRs die een ziekteverwekker tegenkomen,
kunnen na herkenning een reactie in de cel opwekken wat leidt tot de productie van een heel scala
aan eiwitten die een functie hebben in de afweer.
Figuur 38: MYD88 is het belangrijkste signalerende adaptoreiwit voor Toll-like receptoren (TLRs)
(met uitzondering van TLR3 en bepaalde TLR4 signalen), IL-1R en IL-18R. De belangrijkste taak van
MYD88 is de activatie van NFκB (nuclear factor-kappaB). NFκB is een complex eiwit dat de
transcriptie van DNA regelt en speelt eveneens een belangrijke rol in het reguleren van de
immuunrespons op infecties (O'Neill and Bowie 2007)
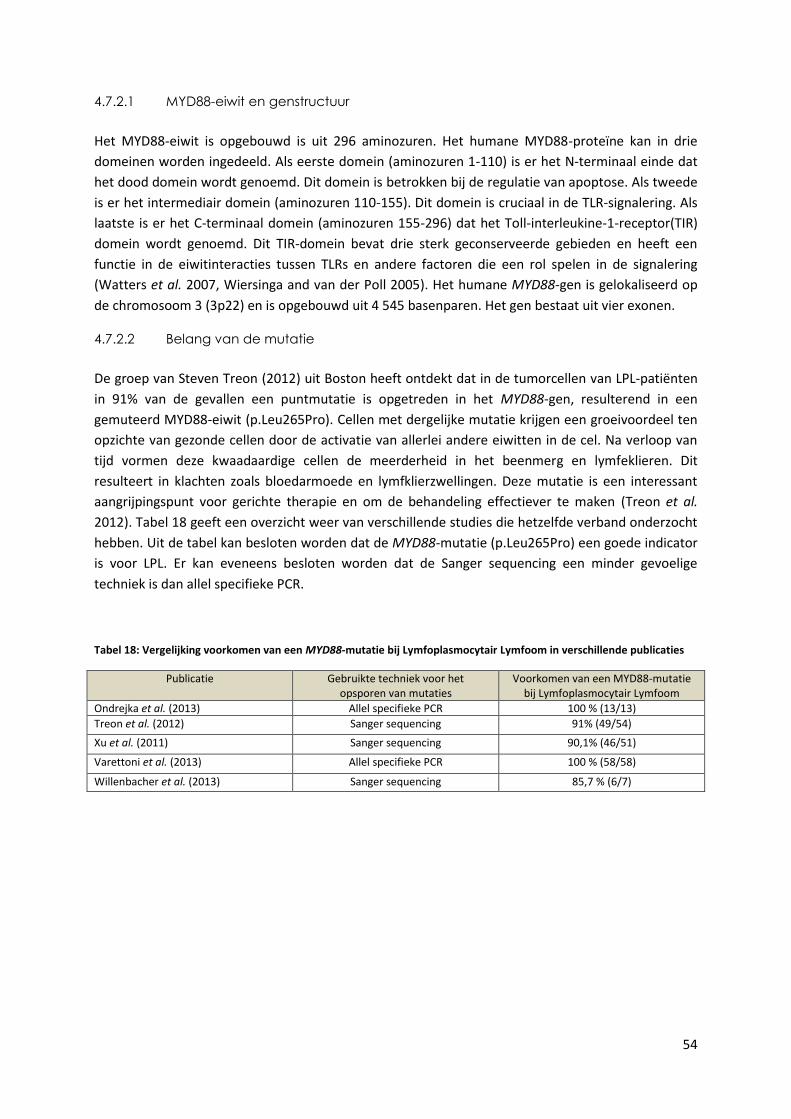
54
4.7.2.1 MYD88-eiwit en genstructuur
Het MYD88-eiwit is opgebouwd is uit 296 aminozuren. Het humane MYD88-proteïne kan in drie
domeinen worden ingedeeld. Als eerste domein (aminozuren 1-110) is er het N-terminaal einde dat
het dood domein wordt genoemd. Dit domein is betrokken bij de regulatie van apoptose. Als tweede
is er het intermediair domein (aminozuren 110-155). Dit domein is cruciaal in de TLR-signalering. Als
laatste is er het C-terminaal domein (aminozuren 155-296) dat het Toll-interleukine-1-receptor(TIR)
domein wordt genoemd. Dit TIR-domein bevat drie sterk geconserveerde gebieden en heeft een
functie in de eiwitinteracties tussen TLRs en andere factoren die een rol spelen in de signalering
(Watters et al. 2007, Wiersinga and van der Poll 2005). Het humane MYD88-gen is gelokaliseerd op
de chromosoom 3 (3p22) en is opgebouwd uit 4 545 basenparen. Het gen bestaat uit vier exonen.
4.7.2.2 Belang van de mutatie
De groep van Steven Treon (2012) uit Boston heeft ontdekt dat in de tumorcellen van LPL-patiënten
in 91% van de gevallen een puntmutatie is opgetreden in het MYD88-gen, resulterend in een
gemuteerd MYD88-eiwit (p.Leu265Pro). Cellen met dergelijke mutatie krijgen een groeivoordeel ten
opzichte van gezonde cellen door de activatie van allerlei andere eiwitten in de cel. Na verloop van
tijd vormen deze kwaadaardige cellen de meerderheid in het beenmerg en lymfeklieren. Dit
resulteert in klachten zoals bloedarmoede en lymfklierzwellingen. Deze mutatie is een interessant
aangrijpingspunt voor gerichte therapie en om de behandeling effectiever te maken (Treon et al.
2012). Tabel 18 geeft een overzicht weer van verschillende studies die hetzelfde verband onderzocht
hebben. Uit de tabel kan besloten worden dat de MYD88-mutatie (p.Leu265Pro) een goede indicator
is voor LPL. Er kan eveneens besloten worden dat de Sanger sequencing een minder gevoelige
techniek is dan allel specifieke PCR.
Tabel 18: Vergelijking voorkomen van een MYD88-mutatie bij Lymfoplasmocytair Lymfoom in verschillende publicaties
Publicatie Gebruikte techniek voor het opsporen van mutaties
Voorkomen van een MYD88-mutatie bij Lymfoplasmocytair Lymfoom
Ondrejka et al. (2013) Allel specifieke PCR 100 % (13/13)
Treon et al. (2012) Sanger sequencing 91% (49/54)
Xu et al. (2011) Sanger sequencing 90,1% (46/51)
Varettoni et al. (2013) Allel specifieke PCR 100 % (58/58)
Willenbacher et al. (2013) Sanger sequencing 85,7 % (6/7)

55
5 Implementatie van nieuwe mutaties als merkers
Uit bovenstaande literatuurstudie kan besloten worden dat een aantal nieuwe moleculaire merkers
gevonden zijn die specifiek in verband kunnen gebracht worden met een bepaalde diagnose. Het is
belangrijk dat deze merkers opgenomen worden in het routine onderzoek aangezien deze aanleiding
kunnen geven tot een versnelde diagnose. Tabellen 19 en 20 geven een finaal overzicht, dus met
implementatie van de voorgestelde nieuwe mutaties, van de verschillende testen die bij een
specifieke diagnose dienen uitgevoerd te worden.
Tabel 19: Overzichtstabel van de reeds uitgevoerde testen en met implementatie van de voorgestelde nieuwe mutatieanalyses die in het AZ Sint-Jan Brugge opgespoord worden bij het vermoeden van specifieke diagnose. De leukemieën en overeenkomstige diagnoses besproken in de tekst zijn in vet aangeduid.
Diagnose Reeds uitgevoerde testen Voorstel nieuwe testen
ALL Acute lymfatische leukemie
B-ALL B-cel acute lymfatische leukemie
IGH/IGK klonaliteitsanalyse TCR klonaliteitsanalyse Hemavision kit
T-ALL T-cel acute lymfatische leukemie
IGH/IGK klonaliteitsanalyse TCR Hemavision kit HOX11 overexpressieanalyse BCR-ABL translocatie
NOTCH1-mutatieanalyse
AML Acute myeloïde leukemie
Alle AML Hemavision kit EVI 1 overexpressieanalyse
IDH1- en IDH2-mutatieanalyse
M3 HEVI/EVI1 BCR (PMC-RARa)
CLL Chronische lymfatische leukemie
Alle CLL IGH/IGK klonaliteitsanalyse IgHV hupermutatieanalys
TP53-mutatieanalyse
MDS Myelodysplastisch syndroom
RA, RARS, RAEBI, SQ- JAK2 V617F mutatieanalyse MPL mutatieanalyse
RAEB II AML EVI 1 overexpressieanalyse JAK2 V617F mutatieanalyse
MDS/MPN Myelodysplastische/myeoloproliferatieve neoplasma’s
Alle MDS/MPN BCR ABL translocatie JAK2 V617F mutatieanalyse Calreticuline mutatieanalyse
aCML Atypische chronische myeloïde leukemie
SETBP1-mutatieanalyse ASXL1-mutatieanalyse
CMML Chronische myelomonocytaire leukemie
ASXL1-mutatieanalyse
JMML Juveniele myelomonocytaire leukemie
MPN Myeloproliferatieve neoplasma
CML Chronische myeloïde leukemie
BCR ABL translocatie
CNL Chronische neutrofiele leukemie
BCR ABL translocatie JAK2 V617F mutatieanalyse
SETBP1-mutatieanalyse CSF3R-mutatieanalyse ASXL1-mutatieanalyse
CEL/HES Chronische eosinofiele leukemie /hypereosinofiel syndroom
BCR ABL translocatie TCR FIP1L1/PDGFRα translocatie
ET Essentiële trombocytemie
BCR ABL translocatie JAK2 V617F mutatieanalyse Calreticuline mutatieanalyse
MPL-mutatieanalyse

56
Tabel 20: Overzichtstabel van de reeds uitgevoerde testen en met implementatie van de voorgestelde nieuwe mutatieanalyses die in het AZ Sint-Jan Brugge opgespoord worden bij het vermoeden van specifieke diagnose - vervolg
Diagnose Reeds uitgevoerde testen Voorstel nieuwe testen
MPN Myeloproliferatieve neoplasma
PMF Primaire myelofibrose
BCR ABL translocatie JAK2 V617F mutatieanalyse Calreticuline mutatieanalyse
MPL-mutatieanalyse
PV Polytemia vera
JAK2 V617F mutatieanalyse JAK2 exon 12 mutatieanalyse
NHL (B) B-cel Non-Hodgkin-lymfoom
Alle B-NHL IGH/IGK klonaliteitsanalyse
ALCL Anaplastic Large Cell Lymphoma
TCR NPM-ALK translocatie
Burkitt
DLBCL Diffuse large B-cell lymphoom
IgH-BCL2 translocatie
FL Folliculair lymfoom
IgH-BCL2 translocatie
HCL Haarcelleukemie
BRAF V600E mutatieanalyse
LPL Lymfoplasmocytair lymfoom
MALT Mucosa associated lymphoid tissue lymfoom
MCL Mantelcellymfoom
SOX11 overexpressieanalyse
SLVL Splenic lymphoma with villous lymphocytes
NHL (T) T-cel Non-Hodgkin-lymfoom
Alle TCR
T-LGL T-cel large granulaire lymfocytaire leukemie
STAT3-mutatieanalyse
Opmerking: niet alle besproken moleculaire merkers zijn opgenomen in de tabel, de verklaringen
hiervoor zijn te vinden in hoofdstuk III.

57
Hoofdstuk II: Materialen en methoden
1 Algemeen
DNA-sequencing is het bepalen van de volgorde aan nucleotiden, aangeduid als de basen (adenine,
guanine, cytosine en thymine) in een DNA-molecule (zie figuur 39). Hiertoe vertrekt men van het te
analyseren DNA-staal waarvan, met behulp van een polymerase ketting reactie (PCR), miljoenen
kopieën worden gemaakt van de regio waarin men geïnteresseerd is. Tijdens de voorbereidende PCR
wordt een watercontrole meegenomen om contaminatie uit te sluiten. Daarna wordt de amplificatie
gecontroleerd met behulp van agarosegelelektroforese, waarna de PCR-producten opgezuiverd
worden met ExoSAP-IT®.
Na het opzuiveren van het bekomen reactieproduct voegt men een mengsel toe van
sequentieprimers, een polymerase en zowel fluorescerende dideoxynucleotiden (ddNTP) als niet
fluorescerende deoxynucleotiden (dNTP). Deze ddNTP’s zijn nucleotiden zonder –OH groep op het
derde koolstofatoom van deoxyribose. Hierdoor kan het DNA polymerase geen volgende
fosfodiësterbinding vormen en kan er dus ook geen nieuw nucleotide worden ingebouwd, waardoor
verdere DNA synthese onmogelijk is. Bij het principe van cycle sequencing vindt de sequentiereactie
verschillende keren plaats door een opeenvolging van denaturatie, annealing en verlenging in een
PCR-toestel. Na een aantal cycli ontstaan er DNA-strengen die telkens 1 bp van elkaar verschillen in
lengte. Om vlotte detectie mogelijk te maken worden de dideoxynucleotiden fluorescent gemerkt.
In een volgende stap worden de sequentieproducten gezuiverd met behulp van Sephadex
kolomzuivering. Hierbij wordt gebruik gemaakt van 96-well gelfiltratie chromatografie met een
Sephadex gel. Sephadex wordt gevormd door het crosslinken van dextrine en epichlorohydrine en
wordt geleverd als een fijn poeder dat moet worden opgelost in water om een gel te vormen. Deze
chromatografie wordt ook wel size exclusion chromatografie genoemd. Tussen de DNA-moleculen en
de primers, dNTP’s of ddNTP’s is er een sterk verschil in grootte. Hierdoor kunnen de DNA moleculen
niet migreren in de poriën van de Sephadexkorrels en migreren ze snel doorheen de gevormde
kolom. De kleinere moleculen, zoals primers, dNTP’s en ddNTP’s, blijven achter in de poriën.
In de laatste stap worden de sequentieproducten via capillaire elektroforese van elkaar gescheiden,
waarna de detector telkens het fluorochroom detecteert. Op die manier wordt het bijhorende
nucleotide herkend en kan de volledige sequentie van de DNA-streng achterhaald worden. De
bekomen resultaten worden geanalyseerd aan de hand van software van DNA-star.

58
2 PCR-reactie
PCR is een snelle amplificatietechniek die door nabootsing van een natuurlijk proces, namelijk de
DNA-replicatie, enorme hoeveelheden DNA kan aanmaken. Met behulp van twee geselecteerde
primers, voldoende dNTP’s en een thermostabiele DNA-polymerase kan een cyclische synthese van
DNA worden opgezet, uitgaande van in theorie slechts één enkelstreng. Opeenvolgend wordt de
temperatuur ingesteld voor de denaturatie van de DNA-modelsequentie, het hybridiseren van de
primers op de enkelstrengen en de werking van de DNA-polymerase. Het gebruik van een
thermostabiel polymerase laat toe het proces in een dertigtal cycli te automatiseren.
2.1 Principe
De PCR-reactie bestaat uit drie fases. In een eerste fase wordt het dubbelstrengig DNA
gedenatureerd, met twee enkelstrengen als resultaat. Dit gebeurt door een temperatuursverhoging
tot 95°C. Bij deze temperatuur worden de waterstofbruggen tussen beide ketens verbroken, terwijl
de covalente bindingen onaangetast blijven. Deze twee enkelstrengige DNA-ketens dienen als matrijs
voor de aanmaak van twee complementaire DNA-ketens. Het enzym dat verantwoordelijk is voor
deze aanmaak is het DNA-polymerase. Als polymerase wordt hier gebruik gemaakt van
AmpliTaq Gold®. Dit is een recombinant, thermostabiel 94 kDa DNA-polymerase dat gecodeerd
wordt door een gemodificeerde vorm van het Thermus aquaticus DNA polymerase gen dat in een
Figuur 39: Opeenvolging van de verschillende stappen voor het opsporen van mutaties door DNA sequencing.

59
E.coli gastheer werd ingebracht (Lawyer et al. 1989). AmpliTaq Gold is een chemisch gemodificeerd
AmpliTaq DNA polymerase. De reversibele modificaties houden het enzym inactief op
kamertemperatuur. Hoge temperaturen en een lage pH bevorderen de activatie van de
enzymactiviteit. De activatie van AmpliTaq Gold DNA Polymerase is afhankelijk van drie parameters,
namelijk incubatietemperatuur, incubatietijd en pH. De activatie van het enzym gebeurt door het
opwarmen van de enzymreactiemix voor tien minuten bij 95°C. Om het enzym te activeren wordt
dus een hotstart toegevoegd aan de PCR-reactie, 95°C gedurende tien minuten (Chen and Janes
2002). Het DNA-polymerase is slechts werkzaam indien het een startpunt vindt in de vorm van een
stukje dubbelstrengs DNA. In de PCR wordt hiervoor gebruik gemaakt van primers. Dit zijn kleine
stukjes enkelstrengig DNA, meestal ongeveer 20 tot 40 basen lang, die precies complementair zijn
aan de uiteinden van het stuk DNA waarin we geïnteresseerd zijn. Deze primers binden tijdens de
annealing stap aan het gedenatureerde DNA. Na deze annealing kan het DNA-polymerase beginnen
met de aanmaak van een complementaire DNA-streng, met als startpunt het kleine stukje
dubbelstrengig DNA ter hoogte van de primer. Zo ontstaan, vanaf de primer, twee nieuwe
dubbelstrengige DNA-ketens, volledig identiek aan het oorspronkelijke moedermolecule.
Dit hele proces van denaturatie, annealing en verlenging wordt een aantal keren herhaald. De nieuwgevormde DNA-strengen fungeren hierbij in de volgende cyclus als matrijs voor de productie van nieuwe strengen. Bij elke cyclus verdubbelt de hoeveelheid DNA. Er is dus een exponentiële vermenigvuldiging van het op te sporen stukje DNA. Na de laatste PCR-cyclus wordt een extra lange elongatiestap uitgevoerd, die er voor zorgt dat alle geamplificeerde PCR-producten volledig geëlongeerd worden.
2.2 Benodigdheden
- Geëxtraheerd template DNA; - Forward en reverse primers (IDT); - Deoxynucleotiden trifosfaten (5 mM per
dNTP) ; - 10 x PCR-buffer II (zonder MgCl2)
(Applied Biosystems); - MgCl2 (Applied Biosystems, 25 mM);
- AmpliTaq Gold® DNA Polymerase (Applied Biosystems, 5U/µl);
- RNase-vrij water; - Vortex; - Centrifuge (Biofuge); - 96-well plaat; - Thermocycler (GeneAmp PCR system
9700).
2.2.1 Gebruikte primers
Opmerking: Aan elke specifieke primer van iedere mutatie wordt een universele M13-adapter
gekoppeld (tgtaaaacgacggccagt (forward) en caggaaacagctatgac (reverse)). Deze M13-staart is zo
ontwikkeld dat hij niet kan binden aan humaan DNA.
Hierdoor wordt het mogelijk om bij de sequentiereactie gebruik te maken van een primer homoloog
aan de adapteruiteinden, waardoor voor iedere mutatie gebruik gemaakt kan worden van eenzelfde
primer. De primers voor het uitvoeren van de mutatieanalyses werden gehaald uit de literatuur met
daaraan de adaptersequentie M13 gekoppeld. De primers zoals aangegeven in de literatuur worden
weergegeven in tabel 21.

60
Tabel 21: Overzicht gebruikte primers
Gen Primers Sequentie (5’ -> 3’)
ASXL1-gen (Gelsi-Boyer et al. 2012)
Forward primer aggtcagatcacccagtcagtt
Reverse primer tagcccatctgtgagtccaactgt
CSF3R-gen (Maxson et al. 2013)
Forward primer (exon 14) ccacggaggcagctttac
Reverse primer (exon 14) aaatcagcatcctttgggtg
Forward primer (exon 17) ctgtcacttccggcaacat
Reverse primer (exon 17) tggcccaaagacacagtcgt
MPL-gen Forward primer agtctgaccctttttgtctcctag
Reverse primer cacagagcgaaccaagaatg
SETBP1-gen (Piazza et al. 2013)
Forward primer ccactttcaacacagttaggtg
Reverse primer tctcgtggtagaaggtgtaactc
TP53-gen (Soussi 2012)
Forward primer exon 5 cacttgtgccctgactttcaac
Reverse primer exon 5 cgcaaccagccctgtcgtctc
Forward primer exon 6 ggttgcccagggtccccag
Reverse primer exon 6 cggccactgacaaccacccttaacc
Forward primer exon 7 gccacaggtctccccaaggc
Reverse primer exon 7 ctggggcacagcaggccagtg
Forward primer exon 8 ggaacctgatttccttactgcc
Reverse primer exon 8 cgaatctgaggcataactgcacc
NOTCH1-gen (Fabbri et al. 2011; Willander et al. 2013)
Forward primer exon 26a agccccctgtacgaccagta
Reverse primer exon 26a cttgcgcagctcctcctc
Forward primer exon 26b acacggccagcagatgat
Reverse primer exon 26b gagagttgcggggattgac
Forward primer exon 27 gtggcgtcatgggcctca
Reverse primer exon 27 tagcaactggcacaaacagc
Forward primer exon 34 acagctactcctcgcctgtg
Reverse primer exon 34 aaggcttgggaaaggaagc
c-KIT-gen (Pollard et al. 2010; Kong et al. 2011)
Forward primer (exon 8) ttcagattctgccctttgaacttg
Reverse primer (exon 8) tgaaattcaagtgaattgcagtcc
Forward primer (exon 17) agttagttttcactctttacaa
Reverse primer (exon 17) taaaatgtgtgatatccctaga
DNMT3A-gen (Neumann et al. 2012)
Forward primer gtgtggttagacggcttccg
Reverse primer cccatgtcccttacacacacg
IDH1- en IDH2-gen (Paschka et al. 2010)
Forward primer - IDH1 ccatttgtctgaaaaactttgct
Reverse primer - IDH1 tcacattattgccaacatgactt
Forward primer – IDH2 gctgcagtgggaccactatt
Reverse primer - IDH2 tgtggccttgtactgcagag
STAT3-gen (Koskela et al. 2012)
Forward primer tcccatcggtcaccccaaca
Reverse primer gccaggccactgaacagggtg
MYD88-gen (Treon et al. 2012)
Forward primer gggatatgctgaactaagttgccac
Reverse primer gacgtgtctgtgaagttggcatctc

61
2.3 Methode
Als eerste stap wordt een mastermix gemaakt. Het volume van de mastermix is afhankelijk van het
aantal reacties dat dient uitgevoerd te worden. Er wordt ook altijd rekening gehouden met eventuele
pipetteerfouten door ongeveer 10% meer mastermix te maken. De PCR-reactie wordt uitgevoerd in
een reactievolume van 25 µl.
De mastermix bestaat uit:
16,9 µl RNase-vrij water (of 17.4µl);
2,5 µl 10x PCR-buffer II (zonder MgCl2) ;
2,5 µl MgCl2 (of 2 µl) ;
1 µl dXTP’s;
0,1 µl AmpliTaq Gold.
(hoeveelheden vermenigvuldigen met aantal reacties x 1.1)
Alle stockoplossingen worden vooraleer ze toegevoegd worden aan de mastermix gevortext en
afgecentrifugeerd, uitgezonderd de enzymmix, dewelke enkel wordt afgedraaid. De mastermix wordt
bereid in een eppendorf van 1,5 ml. Wanneer de mastermix bereid is, wordt deze opnieuw kort
gevortext en afgecentrifugeerd. Vervolgens wordt telkens 23 µl van de mastermix overgebracht in
een 96-well plaat waarbij telkens nog 1 µl primer en 1 µl template DNA wordt gevoegd. Tenslotte
wordt de plaat met de stalen in het PCR-toestel geplaatst en wordt het gewenste programma
gestart.
PCR-programma:
Afhankelijk van de mutatie wordt gebruik gemaakt tijdens de annealing van 55°C of 60°C.
95°C gedurende 10 minuten
95°C gedurende 30 seconden
60°C gedurende 30 seconden 35 cycli Of 55°C gedurende 30 seconden
72°C gedurende 60 seconden
72°C gedurende 7 minuten
4°C tot gebruik
3 Controle PCR-amplificatie – agarosegelelektroforese
3.1 Principe
Agarosegelelektroforese wordt in dit werk gebruikt als controle van de PCR-amplificatie.
Gelelektroforese is een techniek die macromoleculen van elkaar scheidt op basis van grootte en
elektrische lading. Aangezien alle DNA-moleculen eenzelfde massa/lading verhouding hebben
gebeurt de scheiding hier enkel op basis van de grootte. Onder invloed van een elektrisch veld gaan
de moleculen bewegen in een gel. Negatief geladen moleculen, zoals DNA, bewegen van de

62
negatieve pool naar de positieve pool. Grote fragmenten migreren trager door de gel dan kleine
fragmenten. Het DNA op gel wordt finaal gevisualiseerd met SybrSafe. Wanneer een 100 bp ladder
eveneens aan de elektroforese wordt onderworpen, kan de lengte van de nucleïnezuurfragmenten
worden afgeleid.
3.2 Benodigdheden
3.2.1 Hulpmiddelen en toestellen
- Balans; - Microgolfoven; - Warmwaterbad; - Pipetten en filtertips (Post-PCR); - Gelhouder; - Kam; - Erlenmeyer (250ml);
- Maatcilinder (100ml); - Kleefband ; - Stroombron; - Elektroforesekamer; - UV illuminator (Genosmart 2); - Microtiterplaat; - Schaar.
3.2.2 Reagentia en producten
- Agarose (Sigma); - Sybr Safe (Invitrogen); - TBE-buffer 0.5x (Sigma); - PCR-product;
- 100 bp ladder (Bio-Rad); - Molecular grade water (Sigma); - 5x ladings buffer (Sigma).
3.3 Methode
Als eerste stap wordt een agarosegel van 1% gemaakt. Hiervoor wordt 0,8 gram agarose afgewogen
in een erlenmeyer, waarna 80 ml TBE 0,5 x buffer wordt toegevoegd. Als laatste wordt 8 µl Sybr Safe
toegevoegd. Daarna wordt dit mengsel verwarmd in de microgolfoven tot het kookt en een heldere,
homogene oplossing wordt bekomen. Deze oplossing wordt enkele minuten afgekoeld in een
warmwaterbad. De erlenmeyer wordt voorzichtig opgemengd, waarna een 3-4 mm dikke gel wordt
gegoten in de gelhouder. Als laatste stap worden 1 of 2 brede kammen geplaatst in de gegoten gel.
Tenslotte wordt de gel gedurende minimum 20 minuten gestold. Wanneer de gel volledig gestold is,
kunnen de kammen en de tape verwijderd worden.
Om de elektroforese te starten wordt de gelhouder met gel in de elektroforesekamer, die gevuld is
met 0,5x TBE-buffer, gebracht. De te laden reactieproducten worden verdund (1 µl loading buffer +
5 µl reactieproduct) in een microtiterplaat.
Na deze verdunning wordt de gel geladen. Dit gebeurt door 6 µl van de verdunde reactieproducten
en 5 µl van de 100 bp ladder in de slotjes van de gel te pipetteren. Tenslotte wordt het deksel op de
elektroforesekamer geplaatst en de tijdsduur (20 minuten) en het voltage (150 V) ingesteld. Na de
elektroforese wordt een foto genomen van het gel met behulp van UV-licht en een digitale camera.

63
4 Zuivering van het PCR-product - ExoSAP-it®
4.1 Principe
Na amplificatie van het DNA is er in het PCR product naast geamplificeerd DNA ook een overschot
aan primers en dNTP’s aanwezig. Deze overschotten hebben een nadelig effect op de
sequentiereactie en moeten verwijderd worden. Dit kan met behulp van ExoSAP-IT®. Bij deze
opzuivering wordt gebruik gemaakt van twee enzymes: Exonuclease I en Shrimp Alkalisch Fosfatase.
Het exonuclease I hydrolyseert enkelstrengig DNA, zoals primers, in een 3’-5’ richting. Hierdoor
worden de primers afgebroken tot dNTP’s. Het Shrimp Alkalisch Fosfatase inactiveert de dNTP’s door
de 5’ uiteinden te defosforyleren (zie figuur 40).
4.2 Benodigdheden
- PCR-product; - PCR cooler (96 x 0.2 ml); - ExoSAP-IT® (100 reactions)(glycerol
solutions) (Affymetrix); - centrifuge;
- 96-well plaat; - pipetten en filtertips; - Thermocycler (GeneAmp PCR system
9700).
Figuur 40: Principe ExoSAP-IT® (Affymetrix 2013)

64
4.3 Methode
Voor deze procedure wordt een 96-well plaat gebruikt die gedurende de ganse procedure in een
koelblok wordt geplaatst. In de 96-well plaat wordt telkens 7 µl PCR-product gevoegd. Daarna wordt
3,5 µl ExoSAP-IT toegevoegd. Tijdens de ganse procedure dient ExoSAP-it® in een koelblok gebruikt
te worden, aangezien dit product zeer thermogevoelig is. Na het toevoegen van het ExoSAP-IT®,
wordt de plaat meteen in de thermocycler geplaatst, waar het programma ‘EXOSAP’ wordt gestart.
Programma EXOSAP:
37°C gedurende 30 minuten
80°C gedurende 20 minuten
4°C tot gebruik
5 Sequentiereactie
5.1 Principe
Het bepalen van een DNA-sequentie is in dit werk gebaseerd op de enzymatische sequenering
volgens Sanger (Sanger en Coulson 1975). De methode is ontwikkeld op de werking van
DNA-polymerasen en maakt gebruik van het feit dat het reactiemechanisme van DNA-replicatie de
vrije 3’-OH-groep van de DNA-keten nodig heeft voor het inbouwen van een nieuw nucleotide. In de
reactiemix zijn zowel niet fluorescerende dNTP’s als fluorescerende ddNTP’s aanwezig en dit in een
concentratie ten opzichte van elkaar die toelaat dat een keuze tussen de twee types van nucleotides
mogelijk is. De sequentiereactie is een PCR-reactie die uitgevoerd wordt met slechts één primer. Er
ontstaan PCR-producten van verschillende lengtes, doordat er competitie is tussen deoxynucleotiden
en dideoxynucleotiden. Inbouw van een dideoxynucleotide laat geen verdere polymerisatie toe. Bij
de annealingtemperatuur van 50°C gaat de primer zich vasthechten aan de template-DNA streng. Het
AmpliTaq DNA-polymerase zorgt voor de verlenging van de primer door het inbouwen van dNTP’s of
ddNTP’s. Wanneer een ddNTP wordt ingebouwd valt de ketenverlenging stil omdat er geen
fosfaatbrug meer kan gevormd worden met het volgende (di)deoxynulcleotide.
5.2 Benodigdheden
- Big Dye® terminator v1.1 Ready Reaction
premix (2.5 x) (Applied Biosystems); - Big Dye® terminator v1.1 5x sequencing
buffer (Applied Biosystems); - RNase-vrij water; - forward en reverse primers;
- 96-well plaat; - opgezuiverd PCR-product; - thermocycler (GeneAmp PCR system
9700); - pipetten en filtertips.

65
Tabel 22: Gebruikte primers tijdens de sequentiereactie
Door de keuze om gebruik te maken van universele M13 primeruiteinden, kunnen voor iedere
mutatie-analyse dezelfde primers gebruikt worden tijdens de sequentiereactie. Alle amplicons
worden zowel forward als reverse gesequeneerd.
5.3 Methode
Als eerste stap in de sequentiereactie wordt een mastermix gemaakt. Het volume van de mastermix
is afhankelijk van het aantal reacties die dienen uitgevoerd te worden. Er wordt ook altijd rekening
gehouden met eventuele pipetteerfouten door ongeveer 10% meer mastermix te maken. De
sequentiereactie wordt uitgevoerd in een reactievolume van 10 µl.
De mastermix bestaat uit :
1 µl BDT Ready Reaction premix (2,5x);
1,5 µl BDT buffer (5x);
1 µl primer (forward of reverse);
5,5 µl RNase vrij water.
(hoeveelheden vermenigvuldigen met aantal reacties x 1.1 )
Vervolgens wordt telkens 9 µl van deze mastermix overgebracht in een 96-well plaat. Bij deze
mastermix wordt telkens nog 1 µl opgezuiverd PCR-product gevoegd. Waarna de 96-well plaat in de
thermocycler wordt geplaatst en het programma wordt gestart.
Programma :
96°C gedurende 10 seconden
96°C gedurende 10 seconden
50°C gedurende 10 seconden
60°C gedurende 2 minuten
4°C tot gebruik
Primers Sequentie ( 5’ -> 3’ )
Forward primer Tgtaaaacgacggccagt
Reverse primer Caggaaacagctatgacc
25 cycli

66
6 Zuivering van de sequentieproducten - Sephadex
kolomzuivering
6.1 Principe
Na de sequentiereactie moeten resterende dNTP’s, ddNTP’s, zouten en andere overschotten van het
sequentiereagens verwijderd worden uit het DNA-sequentiereagens. Deze zuivering wordt
uitgevoerd door middel van gelfiltratie met Sephadex 50 (Separation Pharmacia Dextran) kolommen.
Bij deze vorm van gelfiltratie - ook wel size-exclusion chromatography genoemd- bestaat de kolom
uit gecrosslinkte dextraandeeltjes of met andere woorden een gel bestaande uit met poriën bedekte
korreltjes. De poriën van dergelijke gelkolommen hebben een zekere diameter. Kleinere moleculen
zullen in de poriën vastgehouden worden, terwijl grotere moleculen zoals DNA niet in de poriën
passen en de kortste weg langs de met poriën bedekte korreltjes nemen om de kolom te verlaten
waarna ze opgevangen worden. Sephadex G-50 Superfine is een gelfiltratiemedium voor onder
andere het ontzouten en het verwijderen van eiwitten. Het heeft een cutoff-waarde van 30 000
molecuulgewicht (Healthcare 2014). Dus moleculen met een moleculair gewicht groter dan 30 000
Da zullen niet in de poriën passen en zullen dus het snelst elueren.
6.2 Benodigdheden
- multiscreen 45µl Column Loader
(Millipore); - Sephadex G-50 Superfine; - multiscreen Column Loader Scraper
Centrifuge Clear (Millipore); - sequencingproducten; - pipetten en filtertips;
- multiscreen HV plaat (Millipore); - lichrosolv water voor chromatografie
(Merck); - 96-well microtiterplaat; - 96-well PCR-plaat; - Multiscreen centrifuge Align Frame Blue
(Millipore).
6.3 Methode
Als eerste stap worden de Sephadex-kolommen aangemaakt. Dit gebeurt door de wellen van de
Multiscreen 45 µl Column Loader te vullen met Sephadex G-50. Het teveel aan Sephadex wordt met
behulp van de Multiscreen Column Loader Scraper Clear verwijderd (zie figuur 41). Daarna wordt een
Multiscreen HV plaat (zie figuur 42) ondersteboven op de gevulde Column Loader geplaatst, waarna
het geheel wordt omgedraaid zodat alle Sephadex in de Multiscreen HV plaat is gevallen. Als laatste
stap in de vorming van de Sephadex kolommen wordt 300 µl lichrosolv water toegevoegd aan de
verschillende wellen. Na 3 uur wellen zijn de kolommen klaar voor gebruik.
Om de zuivering te starten wordt de Multiscreen HV plaat op een 96-well microtiterplaat geplaatst
met een Multiscreen Align Frame Blue er tussenin (zie figuur 43). Dit geheel wordt gecentrifugeerd
bij 2500 rpm gedurende 5 minuten. Daarna wordt de Multiscreen HV plaat op een gelabelde 96-well
PCR-plaat geplaatst. Vooraleer de sequencing-producten op de kolommen worden gebracht, wordt
aan elk van deze producten 10 µl AD water toegevoegd. Het volledige staal (20 µl) wordt daarna op
de overeenkomstige Sephadex kolom gebracht, waarna alles nog eens gecentrifugeerd wordt bij
2500 rpm gedurende 5 minuten.

67
7 Capillaire Elektroforese
7.1 Principe
Het gezuiverd product van de sequentiereactie wordt geanalyseerd met capillaire elektroforese. De
verschillende basen zijn gemerkt met een verschillende dideoxyterminator en zullen bij
sequentiebepaling met de ABI 3500xL Genetic Analyzer of ABI3130 Genetic Analyzer licht van een
verschillende golflengte fluoresceren (zie figuren 44 en 45).
Figuur 44: 3500xL Genetic Analyzer (Life Technologies 2014)
Figuur 45: ABI 3130 Genetic Analyzer (Life Technologies 2014)
Figuur 41: Voorstelling voor het aanmaken
van de Sephadex-kolommen (Sheer et al.
1997)
Figuur 43: Om de zuivering te starten
wordt de Multiscreen HV plaat op een 96-
well microtiterplaat geplaatst met een
Multiscreen Align Frame Blue er tussenin
(Sheer et al. 1997)
Figuur 42: Multiscreen HV plaat (Millipore
2014)

68
7.2 Benodigdheden
- gezuiverd sequentieproduct; - plate septa 96-well; - plaathouder; - 3500xL Genetic Analyzer of ABI 3130 Genetic Analyzer.
7.3 Methode
Op de 96-well plaat met het gezuiverde sequentieproduct wordt een septa geplaatst. De plaat met
septa wordt vervolgens in een plaathouder gebracht, waarna de volledige plaat op het toestel wordt
geladen.
8 Interpretatie van de sequenties
8.1 Aligneren forward en reverse sequentie
Het aligneren van de sequenties gebeurt met het programma SeqMan van DNA-star. De software
vergelijkt de bekomen sequenties van de forward en reverse primer met elkaar, zodat één consensus
sequentie bekomen wordt. Het programma neemt van de reverse sequentie het complement zodat
een sequentie bekomen wordt die identiek zou moeten zijn aan de forward sequentie. Het
sequeneren met zowel de forward als de reverse primer leidt meteen tot een extra controle van de
reactie. De forward en reverse streng dienen identiek te zijn. Wanneer dit niet het geval is, kan
besloten worden dat de reactie niet correct is verlopen.
8.2 Opsporen van mutaties
Het opsporen van mutaties gebeurt met dezelfde software als het aligneren van de forward en
reverse sequentie, namelijk met SeqMan van DNA-star. Deze software is in staat om de mutaties te
detecteren vanaf het moment dat 20% van het gesequeneerde materiaal gemuteerd is. Wanneer dit
niet het geval is, wordt de mutatie niet weergegeven. Er kan dus besloten worden dat de gebruikte
Sanger sequencing-methode voor de mutatie-analyse een gevoeligheid heeft van 20%.

69
Hoofdstuk III: Resultaten en bespreking
De resultaten worden besproken per gen waarin mutaties optreden die gelinkt worden met een
specifieke vorm van leukemie. De patiëntenstalen werden telkens geselecteerd op basis van hun
diagnose zodat de gestelde diagnose overeenstemt met de diagnose waarmee het gen in verband
wordt gebracht in de literatuur. Als template DNA werd steeds geïsoleerd DNA uit witte bloedcellen
of beenmergcellen gebruikt. De PCR-condities zijn degene zoals beschreven bij ‘Materialen en
methoden’ met een annealingtemperatuur van 60°C, tenzij anders vermeld. In figuur 46 wordt het
resultaat gegeven van de agarosegelelektroforese, waarop alle primermixen getest worden op een
normaal staal (CG22 1103) en waarbij voor iedere mix de geoptimaliseerde PCR-condities gebruikt
werden. Bij ieder gen worden de gevonden mutaties besproken. Een overzicht van alle onderzochte
stalen en alle figuren van de gevonden mutaties zijn terug te vinden in de bijlages I en III.
55°C 2µl MgCl2 2µl MgCl2
Figuur 46: Overzicht controle van de PCR-amplificatie d.m.v. agarosegelektroforese met optimale PCR-condities voor alle genen. Voor alle genen werd gebruik gemaakt van een annealingtemperatuur van 60°C en een 2,5µl MgCl2 per reactie, tenzij anders vermeld.

70
1 Screening van het MPL-gen 1.1 Methode
Het opsporen van MPL-mutaties wordt al uitgevoerd in het labo van AZ Sint-Jan Brugge. Er is dus
reeds een methode beschikbaar. Deze methode is volledig gebaseerd op hetzelfde principe, maar
aan de gebruikte primers zijn geen M13-sequenties gekoppeld. Dit heeft als nadeel dat bij de
sequentiereactie voor ieder gen verschillende forward en reverse primers dienen gebruikt te worden.
Aangezien het de bedoeling is om een zo uniform mogelijke methode te ontwikkelen voor het
onderzoeken van meerdere mutaties, is het de bedoeling om de methode voor het opsporen van
MPL-mutaties ook aan te passen zodat gebruik kan gemaakt worden van de universele forward en
reverse primer. Dit wordt mogelijk door aan de reeds gebruikte primers de M13-sequentie te
koppelen. Aangezien hier dus met nieuwe primers wordt gewerkt, is het nodig dat deze getest
worden en dat de bekomen resultaten vergeleken worden met de resultaten verkregen met de oude
primers.
De stalen werden uitgezocht op basis van de diagnosen MPN, MDS en AML. Aangezien het hier gaat
om een reeds bestaande test, werden voornamelijk de reeds geteste positieve stalen getest met de
nieuwe primers. Exon 10 wordt gesequeneerd vanaf p.Trp491 tot p.Tyr521.
1.2 Resultaten
Er werden achttien stalen onderzocht op de aanwezigheid van mutaties in het MPL-gen. Hiervan zijn
15 stalen afkomstig van MPN-patiënten, 2 van MDS-patiënten en 1 staal afkomstig van een
MDS/MPN-patiënt. In deze stalen werden dertien mutaties gevonden (zie tabel 23). Aangezien hier
bewust getest werd op positieve stalen, heeft de mutatiefrequentie weinig betekenis.
Tabel 23: Overzicht gevonden mutaties bij het MPL-gen
Mutatie Type mutatie Aantal Effect op eiwit Beschreven in literatuur
c.1544G>T (p.Trp515Leu)
Missense mutatie
8 MPN 1 MDS/MPN
Tryptofaan 515 is belangrijk in het behouden van de correcte positie van het transmembranaire domein van MPL (Lee et al. 2011). Vooral in verband gebracht met ET en PMF: Resulteert in een cytokine onafhankelijke groei, wat resulteert in een constitutieve activatie van de JAK-STAT signaleringspathway (ExPASy 2014).
Ja, zowel bij MPN-, MDS- als AML-patiënten (He et al. 2013)
c.1514G>A (p.Ser505Asn)
Missense mutatie
3 MPN 1 MDS
Serine 505 heeft eveneens een belangrijke functie in het behouden van de correcte positie van het transmembranaire domein van MPL (Lee et al. 2011). Voornamelijk in verband gebracht met ET: activerende mutatie die MPL-autonome dimerisatie induceert en signaalactivering in afwezigheid van een ligand (ExPASy 2014).
Ja, zowel bij MPN, MDS- als AML-patiënten (He et al. 2013)
c.1502T>C (p.Val501Ala)
Missense mutatie
1 MPN Niet gekend Slechts in 1 studie, nl. Pietra et al. (2011)

71
1.3 Besluit Alle studies, onder andere Lee et al. (2011), zijn het er over eens dat de twee meest voorkomende
mutaties zich bevinden ter hoogte van tryptofaan 515 en serine 505. Deze twee mutaties werden in
deze studie eveneens gevonden. De derde mutatie, namelijk p.Val501Ala werd slechts één keer
gevonden bij een MPN-patiënt, en dit samen met de p.Ser505Asn-mutatie (patiënt CI30 1693). De
mutatie wordt beschreven in de studie van Pietra et al. (2011). Hier werd deze tweemaal gevonden
en dit twee keer in combinatie met een andere mutatie, namelijk samen met p.Tyr515Leu en
p.Tyr515Arg, maar niet in combinatie met de hier gevonden mutatie. Het verband tussen het samen
voorkomen van zowel p.Ser505Asn en p.Val501Ala wordt niet beschreven in de literatuur.
De bedoeling van deze proefopzet was het nagaan of de nieuwe primers tot dezelfde resultaten
leidden als de oude, reeds gebruikte primers. Het resultaat van deze vergelijking is te zien in bijlage II.
Er kan besloten worden dat de nieuwe primers dezelfde resultaten opleveren als de oude primers en
dat de oude primers, zonder universele M13-sequentie, dus kunnen vervangen worden door de
nieuwe primers waaraan wel een universele M13-sequentie gekoppeld is. Er dient wel opgemerkt te
worden dat twee van de achttien geteste stalen niet konden vergeleken worden met de oude
methode aangezien door een computercrash de oude resultaten niet meer beschikbaar waren.
2 Screening van het IDH1- en IDH2–gen 2.1 Methode
Mutaties in het IDH1- en IDH2-gen worden voornamelijk gelinkt met AML. De stalen werden dan ook
uitgezocht op deze diagnose. Het IDH1-gen werd gesequeneerd vanaf p.Tyr42 tot p.Gln138 en het
IDH2-gen vanaf p.Phe126 tot p.Gln178. Het gaat hier net zoals bij het MPL-gen om een vergelijkende
studie, namelijk een vergelijking met een eindwerk die eerder al uitgevoerd werd op IDH1 en IDH2
(Genête 2011).
2.2 Resultaten
In totaal werden zes stalen afkomstig van AML-patiënten getest op de aanwezigheid van IDH1- en
IDH2-mutaties. Twee van deze zes stalen vertonen een mutatie, waarvan één staal eveneens
beschikt over een SNP (zie tabel 24). De mutatiefrequentie heeft hier opnieuw weinig betekenis,
aangezien het hier opnieuw gaat om een vergelijkende studie.

72
Tabel 24: Overzicht gevonden mutaties en SNP’s in het IDH1- en IDH2-gen
Opmerking:
- Voor dit gen werden slechts zes stalen onderzocht aangezien vroeger al een eindwerk
hierover gemaakt werd in het AZ Sint-Jan Brugge (Genête 2011). Hier wordt enkel gebruik
gemaakt van andere primers, namelijk de genspecifieke primers met daaraan M13
gekoppeld.
- Er werd bij één patiënt zowel de mutatie c.394C>T(p.Arg132Cys) gevonden als de SNP
c.315C>T(p.Gly106Gly). Deze combinatie werd in de literatuur eveneens beschreven bij een
AML-patiënt (Wagner et al. 2010). Volgens deze studie heeft een Arg132-mutatie geen
invloed op de prognose. Dit in tegenstelling tot de IDH1-SNP c.315C>T(p.Gly106Gly). Het
voorkomen van deze SNP duidt volgens deze studie op een ongunstige prognose. Dit is een
merkwaardige vaststelling aangezien de eiwitsequentie ongewijzigd blijft.
3 Screening van het TP53-gen 3.1 Methode
De patiëntenstalen werden gescreend naar mutaties in exon 5 tot exon 8. Hiervoor werden vier
reacties per staal opgezet. Exon 5 wordt gesequeneerd van aminozuur p.Tyr126 tot p.Asp186, exon 6
vanaf p.Leu188 tot p.Glu224, exon 7 vanaf p.Val225 tot p.Ser260 en exon 8 vanaf p.Gly262 tot
p.Arg306. De stalen werden uitgezocht op basis van de diagnose CLL.
3.2 PCR-optimalisatie
Initieel werd de PCR-reactie uitgevoerd bij een hybridisatietemperatuur van 60°C. Bij de controle van
de amplificatiereactie werd besloten dat de amplificatie van exon 7 niet optimaal was, aangezien bij
de meeste reacties geen bandjes te zien waren. Om dit probleem op te lossen werd de PCR-reactie
geoptimaliseerd door de geschikte annealingtemperatuur te bepalen. Het PCR-programma werd
Mutatie Type mutatie Aantal Effect op eiwit Beschreven in literatuur
IDH1: c.394C>T (p.Arg132Cys)
Missense mutatie
1 AML De aanwezigheid van dergelijke mutatie
resulteert in verhoogde hoeveelheden
2-hydroxyglutaraat, wat leidt tot een
verandering van een aantal downstream
cellulaire activiteiten (Prensner en Chinnaiyan
2011; Yang et al. 2012).
Ja, 34,5% van de mutaties in het IDH1-gen bij AML-patiënten (Trust Sanger Institute 2014)
IDH1 : c.315C>T (p.Gly106Gly)
SNP (Patel et al. 2011)
1 AML Opmerking: volledig homozygoot aanwezig
Geen effect Ja (Patel et al. 2011)
IDH2: c.419G>A (p.Arg140Gln)
Missense mutatie
1 AML De aanwezigheid van dergelijke mutatie
resulteert in verhoogde hoeveelheden
2-hydroxyglutaraat, wat leidt tot een
verandering van een aantal downstream
cellulaire activiteiten (Prensner en Chinnaiyan
2011; Yang et al. 2012).
Ja, 74,8% van de IDH2-gemuteerde AML-patiënten (Trust Sanger Institute 2014)

73
zowel uitgevoerd met 55°C als 60°C als annealingtemperatuur. Aangezien dit niet leidde tot betere
resultaten, werd de reactie geoptimaliseerd door het aanpassen van de concentratie MgCl2. De
optimale reactiecondities bleken degene te zijn met een annealingstemperatuur van 60°C en 2 µl
MgCl2, in plaats van 2,5µl MgCl2.
3.3 Resultaten
Van de 32 geteste stalen werd in vier stalen (12,5%) een mutatie gevonden. Het gaat hier om vier
verschillende mutaties (zie tabel 25).
Tabel 25: Overzicht gevonden mutaties in het TP53-gen
3.4 Besluit
Zoals in de literatuurstudie reeds besproken werd, hebben patiënten met een deletie ter hoogte van
de 17p13-locus, waar het TP53-gen zich bevindt, de slechtste prognose onder de CLL-patiënten.
Volgens Zainuddin et al. (2011) bezitten 70% van de CLL-patiënten met een 17p-deletie een
TP53-mutatie op het resterende allel. In de hier uitgevoerde studie bedraagt dit 40% (2/5). Dit
percentage is niet volledig representatief aangezien er te weinig stalen met een 17p-deletie
onderzocht werden.
Zainuddin et al. vermelden eveneens in hun studie dat recente studies aantonen dat 3-5% van de
CLL-patiënten beschikken over een mutant TP53-gen zonder een 17p-deletie en dat deze
TP53-mutaties eveneens leiden tot eenzelfde slechte prognose. In de hier uitgevoerde studie
behoren 7,6% (2/26) van de CLL-patiënten tot deze groep, wat hoger is dan de beschreven 3-5%.
Mutatie Type mutatie Aantal Effect op eiwit Beschreven in literatuur
c.733G>A (p.Gly245Ser)
Missense mutatie 1 CLL Leidt tot een structurele verandering waardoor de TP53-bindingsactiviteit veranderd (Pospisilova et al. 2006).
Nog maar 1 keer beschreven bij CLL (Pospisilova et al. 2006)
c.517G>C (p.Val173Leu)
Missense mutatie 1 CLL Onduidelijk (ExPASy 2014) Ja (ExPASy 2014)
c.743G>A (p.Arg248Gln)
Missense mutatie 1 CLL Leidt tot een structurele verandering waardoor de TP53-bindingsactiviteit veranderd (Pospisilova et al. 2006).
Ja (Pospisilova et al. 2006) Eén van de meest voorkomende TP53-mutaties bij CLL
(Hagenkord et al. 2010)
c.734G>T (p.Gly245Val)
Missense mutatie 1 CLL Ongekend Ja maar niet specifiek bij CLL (ExPASy 2014)

74
4 Screening van het SETBP1-gen 4.1 Methode
De mutatiehotspot van het SETBP1-gen bevindt zich ter hoogte van exon 4. Er wordt gesequeneerd
van p.Leu790 tot p.Gln965. De stalen werden uitgezocht op basis van de diagnosen MPN, MDS en
MDS/MPN.
4.2 Resultaten
Er werden 21 stalen onderzocht op aanwezigheid van mutaties in het SETBP1-gen. Waarvan 16 stalen
afkomstig zijn van MDS/MPN-patiënten, 1 staal van een MDS-patiënt en 4 stalen van een
MPN-patiënt. Van deze stalen bezitten zes stalen een mutatie in het SETBP1-gen (28,6%). Er werden
twee verschillende mutaties gevonden, namelijk p.Gly870Ser en p.Asp868Asn (zie tabel 26).
Tabel 26: Overzicht gevonden mutaties bij het SETBP1-gen
4.3 Besluit
Volgens Piazza et al. (2013) is één derde van de gevonden mutaties in het SETBP1-gen p.Gly870Ser
en één derde p.Asp868Asn. De p.Gly870Ser- en p.Asp868Asn-mutaties werden hier in de verhouding
4/6 (66,6%) en 2/6 (33,3%) gevonden. Het hier gevonden percentage p.Gly870Ser is bijgevolg hoger
dan in de literatuur beschreven wordt. Er dient opgemerkt te worden dat er slechts een beperkt
aantal stalen getest werden, waardoor deze mutatiefrequenties niet volledig representatief zijn.
Mutatie Type mutatie Aantal Effect op eiwit Beschreven in literatuur
c.2608G>A (p.Gly870Ser)
Missense mutatie 3 aCML 1 CMML
Leidt tot overexpressie van het SETBP1-eiwit, wat leidt tot onderdrukking van het tumorsuppressoreiwit fosfatase 2A (Piazza et al. 2013).
Ja, zowel bij aCML als bij CMML (Makishima et al. 2013)
c.2602G>A (p.Asp868Asn)
Missense mutatie 1 MPN 1 MDS
Idem Ja, zowel bij MPN als bij MDS (Piazza et al. 2013)

75
5 Screening van het STAT3–gen 5.1 Methode
Mutaties in het STAT3-gen worden gelinkt aan LGL. De te onderzoeken stalen werden dan ook
gezocht op deze diagnose. Van deze stalen werden mutaties gezocht door het sequeneren van
exon 21, vanaf p.Gly630 tot p.Gly700.
5.2 PCR-optimalisatie
Initieel werd de PCR-reactie uitgevoerd bij een annealingtemperatuur van 60°C. Bij het uitvoeren van
gelelektroforese werden op het gel per laan twee bandjes waargenomen. Dit duidt op het optreden
van niet-specifieke bindingen van de primers aan de doelsequentie. Er kan dus besloten worden dat
de PCR-reactie dient geoptimaliseerd te worden. Voor het optimaliseren komen een aantal
parameters in aanmerking onder andere de magnesiumconcentratie en de cyclustemperaturen.
Aangezien volgens de enzymfiche een annealingtemperatuur van 60°C maximaal is en aangezien
deze temperatuur hier reeds gehanteerd werd, kan de temperatuur niet als aanpasbare parameter
beschouwd worden (Applied Biosystems 2010).
Een parameter die wel kan aangepast worden is de magnesiumconcentratie. De initiële reactie werd
uitgevoerd met een magnesiumconcentratie van 2,5µl per reactie. Deze reacties werden opnieuw
uitgevoerd, maar nu met 1 µl, 1,5 µl en 2 µl magnesiumchloride per reactie. Uit het resultaat van de
agarosegelelektroforese kon besloten worden dat bij alle concentraties het niet-specifieke bandje
verdwenen was en dat enkel bij de reacties met 2 µl magnesiumchloride alle reacties zijn opgegaan.
Voor het uitvoeren van de amplificatiereactie wordt dus gebruik gemaakt van een
annealingtemperatuur van 60°C en 2 µl magnesiumchloride per reactie.
5.3 Resultaten
In totaal werden zeven stalen onderzocht, allemaal met diagnose LGL. Van deze stalen bleken vier
stalen positief te zijn voor mutaties in het STAT3-gen (57,1%) (zie tabel 27).
Tabel 27: Overzicht gevonden mutaties in het STAT3-gen
Mutatie Type mutatie Aantal Effect op eiwit Beschreven in literatuur
c.1919A>T (p.Tyr640Phe)
Missense mutatie
3 LGL De mutatie bevindt zich in het SH2-domein. Dit domein speelt een sleutelrol in de functie van het gen. Mutaties in deze regio kunnen leiden naar constitutieve activiteit van STAT3, wat leidt tot overactivatie, wat op zijn beurt oncogenese bevordert (Jerez et al. 2012).
Ja, een van de meest frequent voorkomende STAT3-mutatie bij LGL (samen met p.Asp661Tyr: 80% van alle gevonden mutaties) (Jerez et al. 2012)
c.1981G>T (p.Asp661Tyr)
Missense mutatie
1 LGL Idem Ja (Jerez et al. 2012)

76
6 Screening van het DNMT3A–gen 6.1 Methode
Aangezien mutaties in het DNMT3A-gen voornamelijk in verband worden gebracht met AML en in
mindere mate met MPN, werden de stalen op basis van deze diagnosen uitgezocht. De verschillende
studies zijn het erover eens dat de mutatiehotspot van het DNMT3A-gen zich bevindt ter hoogte van
aminozuur 882. Er werd dan ook gesequeneerd vanaf p.Val867 tot p.Val912, deze sequentie maakt
deel uit van exon 23 van het DNMT3A-gen.
6.2 Resultaten
In totaal werden 83 stalen getest op de aanwezigheid van mutaties in het DNMT3A-gen. Drie stalen
afkomstig van MDS-patiënten, 2 van MDS/MPN-patiënten, 22 van MPN-patiënten, 1 van een
CLL-patiënt, 53 van AML-patiënten en 2 afkomstig van andere diagnosen. Zeven van deze 83 stalen
testten positief op een mutatie in het DNMT3A-gen (8,4%) (zie tabel 28).
Tabel 28: Overzicht gevonden mutaties in het DNMT3A-gen
Mutatie Type mutatie Aantal Effect op eiwit Beschreven in literatuur
c.2645G>A (p.Arg882His)
Missense mutatie 6 AML
Aminozuur 882 is de mutatiehotspot van DNMT3A. Dit aminozuur maakt deel uit van het katalytisch centrum. Er kan dus gesteld worden dan DNMT3A-mutaties de katalytische activiteit aantasten, maar wat hun effect juist is op de DNA-methylatie in de cel is nog niet duidelijk (Kim et al. 2013).
Ja, 39,6% van de mutaties in het DNMT3A-gen bij AML-patiënten (Trust Sanger Institute 2014)
c.2644C>T (p.Arg882Cys)
Missense mutatie 1 AML Mutaties ter hoogte van dit aminozuur tasten de katalytische activiteit aan, maar wat hun effect juist is op de DNA-methylatie in de cel is nog niet duidelijk (Kim et al. 2013).
Ja, 18,5% van de mutaties in het DNMT3A-gen bij AML-patiënten (Trust Sanger Institute 2014)

77
7 Screening van het CSF3R-gen 7.1 Methode
Aangezien het CSF3R-gen voornamelijk gelinkt wordt aan MPN, MDS, MPN/MDS en AML, worden
stalen uitgezocht van patiënten met een van deze diagnosen. Van de verschillende stalen werd zowel
exon 14 (van p.Ala576 tot p.Pro621) en exon 17 (van p.Asp681 tot p.Glu808) onderzocht, aangezien
deze twee exonen beschreven worden als mutatiehotspots.
7.2 PCR-optimalisatie
Bij het testen van de stalen met de standaardinstellingen van het PCR-programma, namelijk met een
annealingtemperatuur van 60°C, werd besloten dat met beide primermixen, zowel met die voor het
amplificeren van exon 14 als voor het amplificeren van exon 17, geen goede amplificatie plaatsvond.
Dit kon besloten worden na controle van de PCR-reactie met agarose gelelektroforese. Een belangrijk
onderdeel van de optimalisatie van de PCR-reactie is het analyseren bij welke annealingtemperatuur
de DNA-amplificatie optimaal is. Bij een hoge annealingtemperatuur zal de selectiviteit van de
primers toenemen, maar bij een te hoge annealingtemperatuur zullen de primers uiteindelijk niet
meer optimaal binden aan het DNA. Een te hoge temperatuur kan eveneens leiden tot inactivatie van
het gebruikte polymerase. In de ideale situatie is de annealingtemperatuur laag genoeg om efficiënte
primerhybridisatie met de doelsequentie te garanderen, maar ook hoog genoeg om niet-specifieke
bindingen te reduceren, zodat de kans op onspecifieke producten zo klein mogelijk is.
Aangezien bij de controle door middel van agarosegelelektroforese geen bandjes werden
gedetecteerd bij een annealingtemperatuur van 60°C, kan besloten worden dat de primers niet
optimaal binden aan het DNA, zodat geen verlenging kan optreden. De reactie werd opnieuw
uitgevoerd maar nu bij een lagere annealingtemperatuur van 55°C. Na het amplificeren werd
opnieuw een controle uitgevoerd waaruit kon besloten worden dat bij deze temperatuur wel een
efficiënte primerhybridisatie met de doelsequentie is opgetreden. Deze annealingtemperatuur is
onlogisch aangezien volgens PCR3 de ideale annealingtemperatuur 60°C is.
7.3 Resultaten
Er werden 76 stalen getest op de aanwezigheid van mutaties in het CSF3R-gen. Hiervan zijn 2 stalen
afkomstig van MDS-patiënten, 31 van MDS/MPN-patiënten, 5 van MPN-patiënten, 1 van een
CLL-patiënt, 35 AML en nog twee afkomstig van andere diagnose. In één van deze stalen werd een
mutatie gevonden (1,3%) en in twee stalen werd eenzelfde mogelijks eiwit-polymorfisme (2,6%)
gevonden (zie tabel 29).
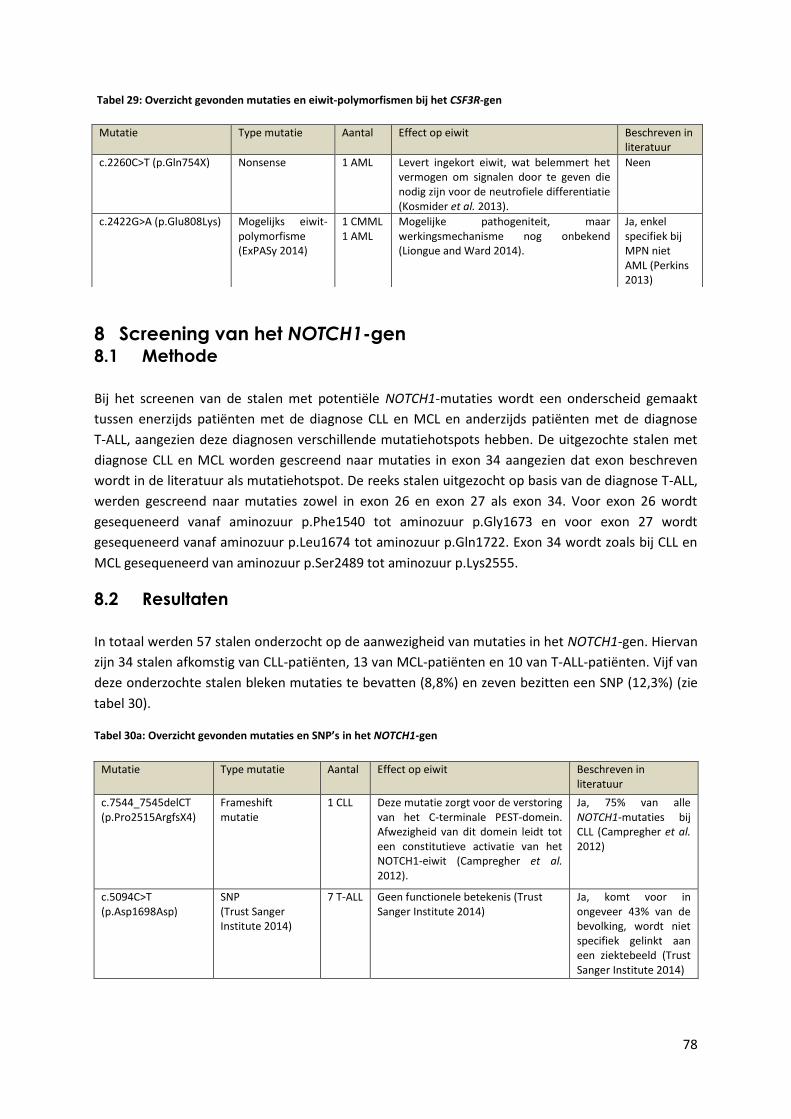
78
Tabel 29: Overzicht gevonden mutaties en eiwit-polymorfismen bij het CSF3R-gen
8 Screening van het NOTCH1-gen 8.1 Methode
Bij het screenen van de stalen met potentiële NOTCH1-mutaties wordt een onderscheid gemaakt
tussen enerzijds patiënten met de diagnose CLL en MCL en anderzijds patiënten met de diagnose
T-ALL, aangezien deze diagnosen verschillende mutatiehotspots hebben. De uitgezochte stalen met
diagnose CLL en MCL worden gescreend naar mutaties in exon 34 aangezien dat exon beschreven
wordt in de literatuur als mutatiehotspot. De reeks stalen uitgezocht op basis van de diagnose T-ALL,
werden gescreend naar mutaties zowel in exon 26 en exon 27 als exon 34. Voor exon 26 wordt
gesequeneerd vanaf aminozuur p.Phe1540 tot aminozuur p.Gly1673 en voor exon 27 wordt
gesequeneerd vanaf aminozuur p.Leu1674 tot aminozuur p.Gln1722. Exon 34 wordt zoals bij CLL en
MCL gesequeneerd van aminozuur p.Ser2489 tot aminozuur p.Lys2555.
8.2 Resultaten
In totaal werden 57 stalen onderzocht op de aanwezigheid van mutaties in het NOTCH1-gen. Hiervan
zijn 34 stalen afkomstig van CLL-patiënten, 13 van MCL-patiënten en 10 van T-ALL-patiënten. Vijf van
deze onderzochte stalen bleken mutaties te bevatten (8,8%) en zeven bezitten een SNP (12,3%) (zie
tabel 30).
Tabel 30a: Overzicht gevonden mutaties en SNP’s in het NOTCH1-gen
Mutatie Type mutatie Aantal Effect op eiwit Beschreven in literatuur
c.2260C>T (p.Gln754X)
Nonsense 1 AML Levert ingekort eiwit, wat belemmert het vermogen om signalen door te geven die nodig zijn voor de neutrofiele differentiatie (Kosmider et al. 2013).
Neen
c.2422G>A (p.Glu808Lys)
Mogelijks eiwit-polymorfisme (ExPASy 2014)
1 CMML 1 AML
Mogelijke pathogeniteit, maar werkingsmechanisme nog onbekend (Liongue and Ward 2014).
Ja, enkel specifiek bij MPN niet AML (Perkins 2013)
Mutatie Type mutatie Aantal Effect op eiwit Beschreven in literatuur
c.7544_7545delCT (p.Pro2515ArgfsX4)
Frameshift mutatie
1 CLL Deze mutatie zorgt voor de verstoring van het C-terminale PEST-domein. Afwezigheid van dit domein leidt tot een constitutieve activatie van het NOTCH1-eiwit (Campregher et al. 2012).
Ja, 75% van alle NOTCH1-mutaties bij CLL (Campregher et al. 2012)
c.5094C>T (p.Asp1698Asp)
SNP (Trust Sanger Institute 2014)
7 T-ALL Geen functionele betekenis (Trust Sanger Institute 2014)
Ja, komt voor in ongeveer 43% van de bevolking, wordt niet specifiek gelinkt aan een ziektebeeld (Trust Sanger Institute 2014)

79
Tabel 30b: Overzicht gevonden mutaties en SNP’s in het NOTCH1-gen
8.3 Besluit In het NOTCH1-gen werd een zeer frequente SNP gevonden bij T-ALL-patiënten, namelijk
c.5094C>T(p.Asp1698Asp). Er kan opgemerkt worden dat T-ALL-patiënten frequenter beschikken
over een gemuteerd NOTCH1-gen dan CLL- en MCL-patiënten, respectievelijk 40% (4/10), 2,9% (1/34)
en 0% (0/13). Deze dalende mutatiefrequenties worden eveneens zo beschreven in de literatuur,
respectievelijk 55%, 15,3% en 8,5% (zie figuur 51).
9 Screening van het ASXL1-gen 9.1 Methode
De verschillende stalen werden gescreend naar mutaties in exon 12 aangezien dit exon beschreven
wordt in de literatuur als mutatiehotspot. De ASXL1-stalen werden uitgezocht op basis van de
diagnosen MPN, MDS, MPN/MDS en AML. De stalen worden gesequeneerd van p.Ile574 tot
p.Leu743.
9.2 Resultaten
Er werden 79 stalen getest op de aanwezigheid van mutaties in het ASXL1-gen. Hiervan zijn 7 stalen
afkomstig van MDS-patiënten, 17 van MDS/MPN-patiënten, 23 van MPN-patiënten, 9 van
CLL-patiënten, 21 van AML-patiënten, 1 van een B-NHL-patiënt en 1 van een ALL patiënt. In twaalf
van deze stalen werd een mutatie gevonden (15,2%) en in één staal (1,3%) werd een
eiwit-polymorfisme gevonden (zie tabel 31).
Mutatie Type mutatie Aantal Effect op eiwit Beschreven in literatuur
c.4754T>C (p.Leu1585Pro)
Missense mutatie 1 T-ALL Stimuleert de NOTCH1-signalering door de stimulatie van het gamma-secretase complex, wat leidt tot een verhoogde NOTCH1-splitsing (Malecki et al. 2006).
Ja (Park et al. 2009)
c.4821G>C (p.Lys1607Asn)
Missense mutatie 1 T-ALL Niet gekend Nee
c.4735_4737delGTG (p.Val1578del )
Deletie 1 T-ALL Deze mutatie leidt tot een verandering in de conformatie van het HD-domein, welke de NOTCH1-splitsing bevordert, resulterend in verhoogde intracellulaire NOTCH1-concentraties (Mansur et al. 2010).
Ja (Mansour et al. 2006)
c.4778T>A (p.Leu1593Pro)
Missense mutatie 1 T-ALL Stimuleert de NOTCH1-signalering door de stimulatie van het gamma-secretase complex, wat leidt tot een verhoogde NOTCH1-splitsing (Malecki et al. 2006).
Ja (Mansour et al. 2006)
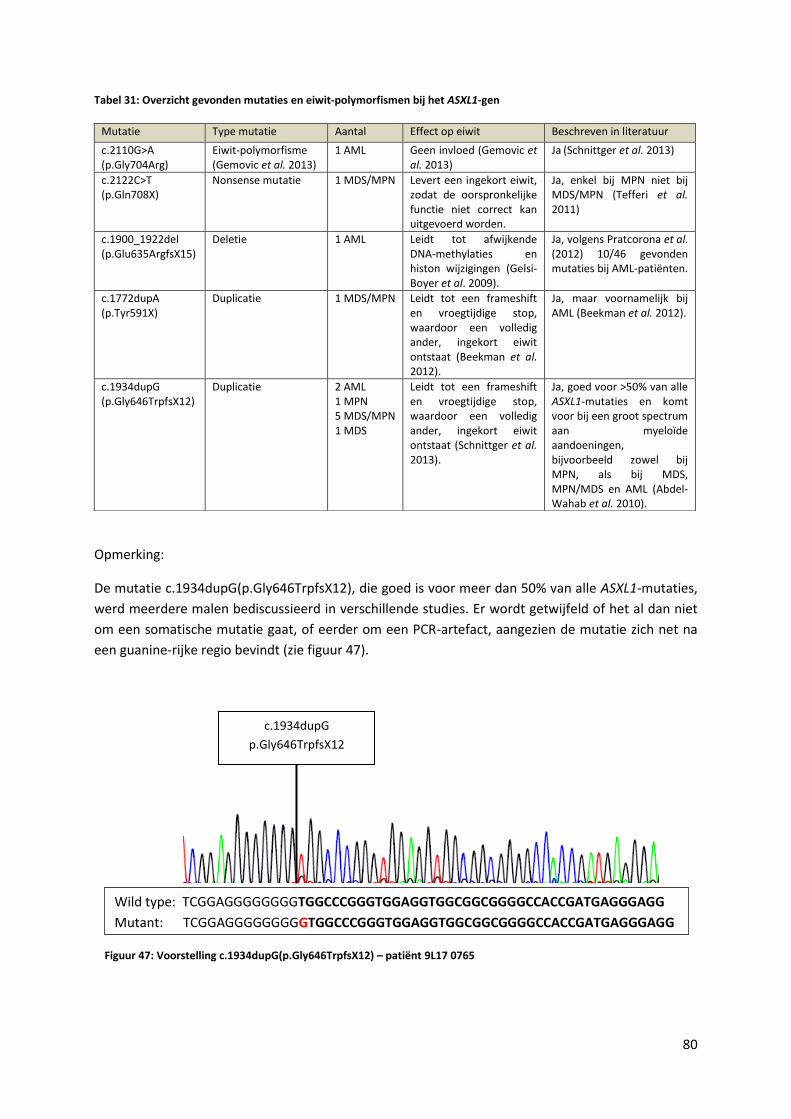
80
Tabel 31: Overzicht gevonden mutaties en eiwit-polymorfismen bij het ASXL1-gen
Opmerking:
De mutatie c.1934dupG(p.Gly646TrpfsX12), die goed is voor meer dan 50% van alle ASXL1-mutaties,
werd meerdere malen bediscussieerd in verschillende studies. Er wordt getwijfeld of het al dan niet
om een somatische mutatie gaat, of eerder om een PCR-artefact, aangezien de mutatie zich net na
een guanine-rijke regio bevindt (zie figuur 47).
Mutatie Type mutatie Aantal Effect op eiwit Beschreven in literatuur
c.2110G>A (p.Gly704Arg)
Eiwit-polymorfisme (Gemovic et al. 2013)
1 AML Geen invloed (Gemovic et al. 2013)
Ja (Schnittger et al. 2013)
c.2122C>T (p.Gln708X)
Nonsense mutatie 1 MDS/MPN Levert een ingekort eiwit, zodat de oorspronkelijke functie niet correct kan uitgevoerd worden.
Ja, enkel bij MPN niet bij MDS/MPN (Tefferi et al. 2011)
c.1900_1922del (p.Glu635ArgfsX15)
Deletie 1 AML Leidt tot afwijkende DNA-methylaties en histon wijzigingen (Gelsi-Boyer et al. 2009).
Ja, volgens Pratcorona et al. (2012) 10/46 gevonden mutaties bij AML-patiënten.
c.1772dupA (p.Tyr591X)
Duplicatie 1 MDS/MPN Leidt tot een frameshift en vroegtijdige stop, waardoor een volledig ander, ingekort eiwit ontstaat (Beekman et al. 2012).
Ja, maar voornamelijk bij AML (Beekman et al. 2012).
c.1934dupG (p.Gly646TrpfsX12)
Duplicatie 2 AML 1 MPN 5 MDS/MPN 1 MDS
Leidt tot een frameshift en vroegtijdige stop, waardoor een volledig ander, ingekort eiwit ontstaat (Schnittger et al. 2013).
Ja, goed voor >50% van alle ASXL1-mutaties en komt voor bij een groot spectrum aan myeloïde aandoeningen, bijvoorbeeld zowel bij MPN, als bij MDS, MPN/MDS en AML (Abdel-Wahab et al. 2010).
Wild type: TCGGAGGGGGGGGTGGCCCGGGTGGAGGTGGCGGCGGGGCCACCGATGAGGGAGG
Mutant: TCGGAGGGGGGGGGTGGCCCGGGTGGAGGTGGCGGCGGGGCCACCGATGAGGGAGG
c.1934dupG
p.Gly646TrpfsX12
Figuur 47: Voorstelling c.1934dupG(p.Gly646TrpfsX12) – patiënt 9L17 0765

81
In de studie van Schnittger et al. (2013) werd op basis van een aantal bevindingen besloten dat het
niet gaat om een artefact. Er werden twintig monsters elk tien keer onderzocht, alle positieve
monsters bleven positief terwijl alle negatieve monsters negatief bleven. Bovendien verdwijnt deze
afwijking na remissie (zie figuur 48).
Bij de studie van Pratcorona et al. (2012) werd een tweede onafhankelijk staal onderzocht van iedere
patiënt, waarbij identieke resultaten bekomen werden. Er werd eveneens geconstateerd dat bij
remissie de mutatie volledig verdwenen was. Bovendien werden er 91 stalen onderzocht van
gezonde personen, waarbij geen enkele mutatie in exon 12 van het ASXL1-gen gevonden werd. Hier
werd dus eveneens besloten dat het om een somatische mutatie gaat en niet om een artefact.
Thol et al. (2011) sequeneerden eveneens 65 gezonde stalen, waarbij geen enkel een mutatie
vertoonde. De groep van West et al. (2014) herhaalde de PCR-reacties minstens drie keer voor ieder
positief staal, waarbij telkens hetzelfde resultaat bekomen werd. Deze PCR-reacties werden
uitgevoerd door gebruik te maken van verschillende enzymen en verschillende primers. Ook in de
studie van Gelsi-Boyer et al. (2012) werd besloten dat het om een somatische mutatie gaat door
onder andere het onderzoeken van stalen van patiënten met andere vormen van kanker, waar de
mutatie telkens afwezig was.
Figuur 48: Voorstelling van het verdwijnen van de
p.Gly646TrpfsX12-mutatie bij remissie (gebaseerd op Schnittger
et al. 2013)
Figuur 50: Voorstelling p.Gly646TrpfsX12-mutatie bij follow up (2014) – Patiënt VM - Staal DD30 0723
Figuur 49: Voorstelling p.Gly646TrpfsX12-mutatie bij diagnose (2009) – Patiënt VM - Staal 9C23 1163

82
In de studie van Abdel-Wahab et al. (2010) wordt het tegenovergestelde aangetoond. Een eerste
punt dat hier aangehaald wordt is het feit dat de mutatie net voorafgegaan wordt door een
guanine-rijke regio, wat kan leiden tot problemen tijdens de amplificatie. De verklaring voor het feit
dat er eveneens stalen zijn waarbij deze mutatie afwezig is, is volgens hen dat niet bij iedere
amplificatie deze ‘slipped-strand mispairing’ optreedt. Aangezien Sanger sequencing slechts een
beperkte nauwkeurigheid heeft, werden de stalen eveneens geanalyseerd m.b.v.
massaspectroscopie, om zo een onderscheid te kunnen maken tussen de aanwezigheid van acht of
negen guanine-nucleotiden. Bij het uitvoeren van de PCR-reactie gevolgd door massaspectroscopie,
werd in ieder staal de mutatie teruggevonden. Finaal werden 96 gezonde stalen onderzocht d.m.v.
Sanger sequencing, waarvan 25% positief bleek te zijn voor p.Gly646TrpfsX12. In deze studie werd
dus besloten dat het niet om een somatische mutatie gaat, maar wel om een PCR-artefact.
In de hier uitgevoerde studie werd deze mutatie, net zoals in de literatuur, ook het meest gevonden.
Om zeker te zijn dat het effectief om een mutatie gaat en niet om een PCR-artefact werden alle
positieve stalen een tweede keer gescreend op mutaties. Telkens werd exact hetzelfde resultaat
bekomen als bij de eerste analyse. Er werd hier net zoals in de studie van Schnittger et al. (2013)
gevonden dat de mutatie verminderd naarmate de ziekte gunstig evolueert (zie figuren 49 en 50).
Een derde argument is het feit dat hier ook stalen onderzocht werden waar deze mutatie duidelijk
afwezig bleek te zijn. Op basis van deze bevindingen werd hier besloten dat het weldegelijk om een
mutatie gaat en niet om een PCR-artefact zoals in de studie van Abdel-Wahab et al. (2010)
beschreven wordt.
9.3 Besluit
De meest voorkomende mutaties bij het ASXL1-gen zijn volgens de literatuur frameshift mutaties
(Trust Sanger Institute 2014), wat hier eveneens het geval is (11/12). Volgens de meeste studies,
onder andere volgens Schnittger et al. (2013) is de meest voorkomende mutatie p.Gly646TrpfsX12
(53,9%, 69/128). In de hier uitgevoerde studie werd dit ook gevonden, namelijk negen van de twaalf
(75%) gevonden mutaties betreffen deze mutatie. De tweede meest frequente mutatie is
p.Gln635ArgfsX15, gevolgd door p.Tyr591X, p.Arg693X en p.Gln733X. Er kan dus besloten worden dat
hier drie van de meest voorkomende mutaties gevonden werden en één minder voorkomende,
namelijk p.Gln708X. Alle gevonden mutaties worden in de literatuur eveneens beschreven bij de
specifieke diagnosen van de stalen, uitgezonderd p.Tyr591X. Deze mutatie wordt in de literatuur
enkel in verband gebracht met AML en niet met MDS/MPN, zoals hier gevonden werd. In de studie
van Gelsi-Boyer et al. (2012) wordt deze mutatie zeven keer gevonden, maar hier wordt geen
onderscheid gemaakt tussen de verschillende diagnosen MPN, MDS, MDS/MPN en AML. Het is dus
onduidelijk of deze mutatie al dan niet in verband wordt gebracht met de specifieke diagnosen van
de patiënt, namelijk MDS/MPN.

83
10 Screening van het c-KIT–gen 10.1 Methode
C-KIT-mutaties worden voornamelijk gelinkt aan AML en SM. De stalen werden uitgezocht op basis
van deze diagnosen en werden gescreend naar zowel exon 8 (van p.Lys412 tot p.Gln448) als exon 17
(van p.Cys788 tot p.Asn828). Voor SM worden de stalen reeds opgestuurd naar CME Leuven voor een
c-KIT-screening. De daar uitgevoerde screening is een zeer gevoelige, kwantitatieve test. Er kan dus
gesteld worden dat de screening van het c-KIT-gen geen nieuwe moleculaire merker is bij SM, maar
dat het hier eerder gaat om een vergelijkende methode, dit in tegenstelling tot de screening bij de
diagnose AML, wat wel een nieuwe moleculaire merker is.
10.2 Resultaten
Er werden 49 stalen onderzocht, waarvan 40 afkomstig van AML-patiënten, 6 van SM-patiënten, 1
MDS-patiënt, 1 MDS/MPN-patiënt en 1 afkomstig van een patiënt met een andere diagnose.
Aangezien het bij de diagnose SM gaat om een vergelijkende studie wordt hier geen
mutatiefrequentie weergegeven aangezien bewust getest werd op positieve stalen. Van de
onderzochte AML-stalen vertonen twee stalen een mutatie (5%) en twee stalen beschikken over een
SNP (5%) (zie tabel 32).
Tabel 32: Overzicht gevonden mutaties en SNP’s in het c-KIT-gen
Mutatie Type mutatie Aantal Effect op eiwit Beschreven in literatuur
c.2394C>T(p.Ile798Ile)
SNP (Colarossi et al. 2007)
2 SM 2 AML
Geen effect Ja, zowel bij SM als AML (Colarossi et al. 2007; Riera et al. 2013)
c.1255_1257delGAC (p.Asp419del)
Deletie 1 AML Exon 8 codeert voor het extracellulaire juxtamembraan domein, dit is een functioneel gebied dat instaat voor de c-KIT-activiteit. Mutaties in dit gebied leiden tot een ligand-onafhankelijke constitutieve activatie (Casteran et al. 2003).
Ja (Matthews and Gerritsen 2010)
c.2466T>G (p.Asn822Lys)
Missense mutatie 1 AML Onduidelijk (ExPASy 2014) Ja (Hussain et al. 2009)
c.2447A>T (p.Asp816Val)
Missense mutatie 1 SM Het gaat hier om een activerende puntmutatie die wordt gevonden in het tyrosine kinase domein, wat leidt tot een conformationele verandering die resulteert in een ligand-onafhankelijke constitutieve activatie van c-KIT, wat leidt tot een verhoogde proliferatie en een vermindering van apoptose (Verstovsek 2013).
Ja, bij >80% van de ISM-patiënten (Verstovsek 2013)

84
10.3 Besluit
De screening van het c-KIT-gen bij SM gebeurde als vergelijkende studie. Het gaat hier in dit geval dus
niet om een nieuwe merker. Deze test wordt reeds buitenhuis (CME Leuven) uitgevoerd door middel
van een kwantitatieve, zeer gevoelige test. Het is de bedoeling dat deze test kan uitgevoerd worden
in het labo van het AZ Sint-Jan Brugge. Dit is momenteel nog niet mogelijk aangezien de gewenste
gevoeligheid niet behaald kan worden met Sanger sequencing. Er zijn namelijk drie patiënten
CL05 0980, CJ31 0746 en CI17 0517 (zie bijlage 3) waar een c-KIT-mutatie aanwezig is en die d.m.v.
Sanger sequencing niet kan gedetecteerd worden aangezien respectievelijk slechts 15,59%, 0,06% en
0,09% gemuteerde cellen aanwezig zijn, terwijl de gevoeligheid van Sanger sequencing hier 20%
bedraagt. De screening van het c-KIT-gen bij AML is echter wel een nieuwe merker. Hierbij wordt een
mutatiefrequentie van 5% (2/40) bekomen. Beide mutaties werden gevonden bij
AML1-ETO-positieve AML-patiënten.
11 Screening van het MYD88–gen 11.1 Methode
Mutaties in het MYD88-gen worden in verband gebracht met LPL of ook wel de ziekte van
Waldenström genoemd. De stalen worden dan ook uitgezocht op basis van deze diagnose. De
mutatiehotspot van het MYD88-gen bevindt zich ter hoogte van aminozuur 265 (leucine).
11.2 Resultaten
Er werden 31 LPL-stalen gescreend naar mutaties in het MYD88-gen. Van deze stalen vertoonden
slechts twee stalen een mutatie (6,5%) (zie tabel 33).
Tabel 33: Overzicht gevonden mutaties in het MYD88-gen
11.3 Besluit In de literatuur wordt beschreven dat ongeveer 95% van de LPL-patiënten beschikken over een
mutatie ter hoogte van aminozuur 265 in het MYD88-gen (Treon et al. 2012). Er kan dus besloten
worden dat de gevonden mutatiefrequentie veel lager is, namelijk 6,5% (2/31). Dit kan verklaard
worden omdat bij de diagnose LPL, maximaal 30% van de cellen maligne cellen zijn en aangezien hier
slechts een gevoeligheid is van 20%, worden heel wat mutaties niet gedetecteerd. Dit probleem kan
opgelost worden bijvoorbeeld door het ontwerpen van een specifiekere PCR-reactie waardoor de
gevoeligheid aanzienlijk zal stijgen. Dit is mogelijk aangezien de mutatiehotspot slechts één
aminozuur betreft. Een andere mogelijkheid om dit probleem op te lossen is om voorafgaandelijk
Mutatie Type mutatie Aantal Effect op eiwit Beschreven in literatuur
c.794T>C (p.Leu265Pro)
Missense mutatie
2 LPL Cellen met dergelijke mutatie krijgen een groeivoordeel ten opzichte van gezonde cellen door de activatie van allerlei andere eiwitten in de cel (Treon et al. 2012)
Meest frequent voorkomende mutatie in het MYD88-gen die in verband kan gebracht worden met LPL (Treon et al. 2012)

85
aan de amplificatie het staal te onderwerpen aan een celscheiding. Waldenström B-cellen vertonen
in ongeveer 90% van de B-cellen expressie van CD19 (Owen et al. 2001). Wanneer deze cellen
voorafgaandelijk geïsoleerd worden, is er een veel grotere kans om mutaties te vinden.
12 Vergelijkende studie met de literatuur
In onderstaande figuur wordt een vergelijking gemaakt tussen het aantal mutaties per gen en per
diagnose gevonden in deze studie en beschreven in de literatuur (zie literatuurstudie). Een
belangrijke opmerking die dient gemaakt te worden bij deze vergelijking is het feit dat in de literatuur
geen onderscheid gemaakt wordt tussen mutaties en eiwit-polymorfismen, waar in tegenstelling tot
een SNP het aminozuur wel verandert. Zowel mutaties als eiwit-polymorfismen worden gerekend
onder de noemer mutaties, terwijl in deze studie wel degelijk een verschil gemaakt wordt tussen
dergelijke polymorfismen en mutaties. In tabellen 34 en 35 wordt een overzicht gegeven van de
gevonden mutatiefrequenties en de in de literatuur beschreven mutatiefrequenties met eventueel
een verklaring voor het verschil tussen beide mutatiefrequenties.
Er kan opgemerkt worden dat de vergelijkende studies, namelijk IDH1- en IDH2-, MPL en c-KIT bij SM,
evenals de totaal niet representatieve waarden in deze figuur niet weergegeven worden, aangezien
hierbij geen representatieve mutatiefrequenties bekomen worden.
Figuur 51: Vergelijking van de gevonden mutatiefrequenties en de mutatiefrequenties beschreven in de literatuur (zie literatuurstudie) per gen en per diagnose

86
Tabel 34: Overzicht van de gevonden mutatiefrequentie en de in de literatuur beschreven mutatiefrequenties met eventueel een verklaring voor het verschil tussen beide mutatiefrequenties (voor de in de literatuur beschreven mutatiefrequenties wordt verwezen naar de voorafgaande literatuurstudie).
Diagnose Genen Gevonden Literatuur (gemiddelde)
Besluit
MDS SETBP1 100% (1/1) 2,2% De mutatiefrequentie is niet representatief aangezien slechts 1 staal getest werd.
ASXL1 14,3% (1/7) 16% Beide mutatiefrequenties komen ongeveer overeen. Er dient wel opgemerkt te worden dat slechts 7 stalen getest werden.
MPL 50% (1/2) 4,4% Gevonden mutatiefrequentie heeft weinig betekenis aangezien hier bewust getest werd op positieve stalen.
MDS/MPN SETBP1 25% (4/16) 7,5% Voor SETBP1 wordt een hogere mutatiefrequentie bekomen dan in de literatuur beschreven wordt. Dit kan verklaard worden door het feit dat bij aCML een hogere mutatiefrequentie bekomen wordt in de literatuur dan CMML, respectievelijk 28% en 4,8%. Hier werden voornamelijk aCML-patiënten onderzocht (14 stalen) en slechts 2 CMML-stalen. Voor aCML wordt dus een mutatiefrequentie bekomen van 21,4%, wat de beschreven mutatiefrequentie benadert, namelijk 28%. Voor CMML werden slechts twee stalen getest waardoor de verkregen mutatiefrequentie weinig betekenis heeft.
CSF3R 0% (0/31) 3,6% Er werd bij 1 van de 31 (3,23%) onderzochte stalen een eiwit-polymorfisme gevonden. Aangezien in de literatuur vaak geen onderscheid gemaakt wordt tussen deze eiwit-polymorfismen en mutaties kan het verschil in beide mutatiefrequenties hierdoor verklaard worden.
ASXL1 41,2% (7/17) 51% Alle mutaties werden gevonden bij aCML-patiënten. In de literatuur hebben deze een hogere mutatiefrequentie dan CMML-patiënten (respectievelijk 64% vs. 44%). Er werden slechts 5 CMML-patiëntenstalen onderzocht. Door het beperkt aantal CMML-stalen ter beschikking worden geen genmutaties gevonden. Er kan dus geen uitgebreid onderzoek gebeuren. Voor aCML wordt hier een vergelijkbare mutatiefrequenties bekomen t.o.v. de literatuur, respectievelijk 58,3% en 64%.
MPL 100% (1/1) Wordt niet specifiek beschreven in de literatuur
Gevonden mutatiefrequentie heeft weinig betekenis aangezien hier bewust getest werd op positieve stalen. Het onderscheid tussen MPN, MDS en MDS/MPN is vaak moeilijk te onderscheiden.
MPN SETBP1 25% (1/4) 13,8% Te weinig stalen werden getest om een representatieve mutatiefrequentie te bekomen.
CSF3R 0% (0/5) 64,7% Mutaties in het CSF3R-gen bij MPN worden voornamelijk beschreven bij CNL. Aangezien dit een uiterst zeldzame aandoening is, werd deze specifieke diagnose hier niet getest, maar werden andere diagnosen van de groep MPN getest. Er werden eveneens slechts vijf stalen getest, wat geen representatieve mutatiefrequentie oplevert.
DNMT3A 0% (0/22) 14% Geen verklaring
ASXL1 4,3% (1/23) 41,8% Mutaties in het ASXL1-gen bij MPN worden voornamelijk beschreven bij CNL. Aangezien dit een uiterst zeldzame aandoening is, werd deze specifieke diagnose hier niet getest, maar werden andere diagnosen van de groep MPN getest.
MPL 73,3% (11/15) 5% Gevonden mutatiefrequentie heeft weinig betekenis aangezien hier bewust getest werd op positieve stalen.
87
Tabel 35: Overzicht van de gevonden mutatiefrequentie en de in de literatuur beschreven mutatiefrequenties met
eventueel een verklaring voor het verschil tussen beide mutatiefrequenties - vervolg
c-KIT 5% (2/40) 11,8% Geen verklaring
T-ALL NOTCH1 40% (4/10) 55% Geen verklaring
SM c-KIT 16,7% (1/6) 80% Gevonden mutatiefrequentie heeft weinig betekenis aangezien hier bewust getest werd op positieve stalen.
LPL MYD88 6,5% (2/31) 93,4% Maximaal 30% van de cellen bij LPL zijn maligne cellen, aangezien de hier gebruikte methode slechts een gevoeligheid heeft van 20%, worden veel mutaties niet gedetecteerd. Oplossing 1: ontwerpen van een specifieke PCR-reactie, aangezien de mutatiehotspot slechts één aminozuur betreft. Oplossing 2: CD19-cellen die in een laag percentage aanwezig zijn voorafgaandelijke isoleren. Deze tweede oplossing wordt ook uitgevoerd in de literatuur, vandaar de hogere mutatiefrequentie.
LGL STAT3 57,1% (4/7) 33,4% Er werden te weinig stalen getest om een representatieve mutatiefrequentie te bekomen.
MCL NOTCH1 0% (0/13) 8,5% Geen verklaring
Er werden eveneens patiënten gevonden die meerdere malen gemuteerd zijn. Er werd in de
literatuur op zoek gegaan of deze co-mutaties reeds beschreven werden (zie tabel 36).
Diagnose Genen Gevonden Literatuur (gemiddelde)
Besluit
CLL TP53 12,5% (4/32) 21,3% Geen verklaring
NOTCH1 2,9% (1/34) 15,3% Geen verklaring
AML CSF3R 2,9% (1/35) De novo AML 0,8% Secundaire AML 79%
In de literatuur wordt meestal onderscheid gemaakt tussen secundaire AML en de novo AML, wat hier niet gedaan werd. Uit de literatuur blijkt duidelijk dat secundaire AML-patiënten een veel hogere mutatiefrequentie vertonen, namelijk (respectievelijk 79% vs. 0,83%).
IDH1 en IDH2
33,3% (2/6) 11,4% Gevonden mutatiefrequentie heeft weinig betekenis aangezien hier bewust getest werd op positieve stalen.
DNMT3A 13,2% (7/53) 18,3% Ongeveer gelijke mutatiefrequenties werden bekomen.
ASXL1 14,3% (3/21) De novo AML 6,5% Secundaire AML 30,3%
In de literatuur wordt meestal onderscheid gemaakt tussen secundaire AML en de novo AML, wat hier niet gedaan werd. Uit de literatuur blijkt duidelijk dat secundaire AML-patiënten een veel hogere mutatiefrequentie vertonen, namelijk (30,3% vs. 6,5%). Er werd eveneens bij 1 van de 21 stalen een eiwit-polymorfisme gevonden.
88
Tabel 36: Overzicht van de gevonden co-mutaties
Patiënt Diagnose Mutaties Literatuur
BD12 0806 MDS/MPN aCML
ASXL1: c.1934dupG(p.Gly646TrpfsX12) CEBPA
26% van de mutante ASXL1-patiënten bezitten eveneens een CEBPA-mutatie, terwijl slechts 9% van de WT-ASXL1-patiënten over een CEBPA-mutatie beschikt. Patiënten die zowel beschikken over een ASXL1-mutatie en een CEBPA-mutatie behoren tot de groep met een gunstige prognose (Metzeler et al. 2011).
9L17 0765 MDS/MPN aCML
ASXL1: c.1934dupG(p.Gly646TrpfsX12) SETBP1: c.2608G>A(p.Gly870Ser)
SETBP1-mutaties worden sterk geassocieerd met ASXL1-mutaties. SETBP1-mutaties komen frequenter voor bij mutante ASXL1-patiënten dan bij wild type ASXL1-patiënten. Bijna één derde van de ASXL1-gemuteerde gevallen bezit eveneens een mutatie in SETBP1 (63/208, 30,3%). In deze studie van Meggendorfer et al. (2013) wordt hieruit besloten dat een SETBP1-mutatie eerder een mutatie is die later optreedt tijdens de ziekte i.p.v. een initiële mutatie. In de studie van Hou et al. (2014) wordt eveneens besloten dat de SETBP1-muatie geen initiële mutatie is, maar dat de mutatie verkregen wordt gedurende het klinisch verloop en dat deze dus een rol speelt bij de progressie van de ziekte. Hier werd ook besloten dat het voorkomen van een SETBP1-mutatie en een ASXL1-mutatie zowel bij MDS-patiënten als MDS/MPN-patiënten geassocieerd wordt met een slechte prognose.
8J13 05990 MDS/MPN aCML
ASXL1: c.1934dupG(p.Gly646TrpfsX12) SETBP1: c.2608G>A(p.Gly870Ser)
BI13 0650 MDS ASXL1: c.1934dupG(p.Gly646TrpfsX12) SETBP1: c.2602G>A(p.Asp868Asn)
9C23 1163 MPN PMF
ASXL1: c.1934dupG(p.Gly646TrpfsX12) JAK2: c.1849G>T (p.Val617Phe)
In de literatuur wordt het gelijktijdig voorkomen van deze twee mutaties niet beschreven.
9E08 0454 AML CSF3R: c.2422G>A(p.Glu808Lys) FLT3-ITD-mutatie DNMT3A: c.2645G>A(p.Arg882His)
FLT3-ITD-mutaties komen vaak (40%) voor bij DNMT3A-gemuteerde AML-patiënten. Uit de studie van Shlush et al. (2014) wordt besloten dat de mutatie in DNMT3A eerder aanwezig is dan de FLT3-ITD-mutaties. FLT3-mutante AML-patiënten met een DNMT3A-mutatie worden geassocieerd met een zeer slechte prognose (Testa and Pelosi 2013). Er wordt geen verband beschreven tussen deze mutaties en het voorkomen van het CSF3R-polymorfisme.
CI30 1693 MPN MPL: c.1514G>A(p.Ser505Asn) en c.1502T>C(p.Val501Ala) JAK2: V617F
Mutaties in het MPL-gen leiden tot constitutieve activatie van JAK2. Beide mutaties leiden dus tot een constitutieve activering van de JAK2-signaalpathway. Er worden geen specifieke verbanden beschreven tussen de hier voorkomende co-mutaties.
CL02 0676 AML DNMT3A: c.2645G>A(p.Arg882His) FLT3-ITD mutatie NPM1-mutatie
DNMT3A-mutaties worden significant geassocieerd met NPM1-mutaties en FLT3-ITD-mutaties (Ribeiro et al. 2012). In de studie van Hou et al. (2012) werd gevonden dat van de 70 patiënten met mutant DNMT3A-gen er 68 (97,1%) beschikken over additionele moleculaire abnormaliteiten, waaronder voornamelijk NMP1-mutaties (55,9% (38/68)) en FLT3-ITD-mutaties (44,1% (30/68)). Mutante DNMT3A-AML-patiënten met een NPM1-mutatie en wild type FLT3-ITD beschikken over een gunstige prognose binnen de groep van mutante DNMT3A-AML-patiënten die geassocieerd worden met een slechte prognose. Aanwezigheid van zowel een NPM1-mutatie en een FLT3-ITD-mutatie bij mutante DNMT3A-AML-patiënten leiden tot een slechte prognose.
CF18 1132 AML DNMT3A: c.2645G>A(p.Arg882His) NPM1-mutatie
CG23 0588 AML DNMT3A: c.2645G>A(p.Arg882His) NPM1-mutatie FLT3-ITD-mutatie

89
Algemeen besluit
Er is een duidelijk verband tussen mutaties en het voorkomen van een welbepaalde vorm van
kanker, wat perspectieven biedt voor het (voortijdig) opsporen van de ziekte en het stellen van de
juiste diagnose. Tot op vandaag worden de meeste kankers gediagnosticeerd op basis van
morfologische, immunofenotypische of cytogenische kenmerken. In bepaalde gevallen is het stellen
van de juiste diagnose echter moeilijk en is het stellen ervan vaak gebaseerd op exclusie, wat
kostbare tijd in beslag neemt. Wanneer de ziekte echter gelinkt kan worden aan het voorkomen van
een bepaalde mutatie, een moleculaire merker, kan het stellen van de diagnose mogelijk versneld
worden. Dit leidt vervolgens tot een snellere start van een effectieve behandeling, wat de
overlevingskans van de patiënt vergroot. In de literatuur werden recent heel wat recurrente mutaties
beschreven dankzij het fundamenteel kankeronderzoek m.b.v. Next Generation Sequencing. Het doel
van deze thesis was om een methode te ontwikkelen die in staat is om de in de literatuur beschreven
puntmutaties op een efficiënte manier te kunnen opsporen.
De gebruikte methode is Sanger sequencing, dit omdat deze techniek in het AZ Sint-Jan Brugge reeds
wordt toegepast o.a. voor HLA-typering. In deze studie wordt gebruik gemaakt van de in de literatuur
beschreven gen-specifieke primers die tijdens de synthese verlengd werden met de universele
M13-sequentie, zodat het mogelijk wordt om voor iedere analyse gebruik te maken van de
universele M13-primers als sequentieprimer. Dit gebeurde om een efficiënte screening mogelijk te
maken, wat het doel was van deze thesis. De gevoeligheid van Sanger sequencing is echter ongeveer
20%, waarmee dient rekening gehouden te worden tijdens de interpretatie.
Het MPL-, IDH1- en IDH2-gen worden momenteel in het laboratorium van het AZ Sint-Jan Brugge
gescreend naar mutaties, maar om deze analyses te integreren in de efficiëntere werkwijze, werd
een vergelijkende studie uitgevoerd met de gen-specifieke primers gekoppeld aan de
M13-sequentie. In totaal werden zestien stalen met gekende MPL-mutatiestatus onderzocht naar
mutaties in het MPL-gen. Het IDH1- en IDH2-gen werden gescreend bij zes verschillende stalen. Bij de
drie onderzochte genen werden telkens dezelfde resultaten bekomen als bij de huidige methode. Er
kan dus besloten worden dat deze huidige methode in de routineanalyses kan vervangen worden
door de efficiëntere nieuwe methode.
Aangezien er een grote noodzaak is om nieuwe diagnostische merkers te vinden bij diagnose van
MDS, MPN en MDS/MPN, werd een set van drie genen geselecteerd, namelijk SETBP1, CSF3R en
ASXL1. Er werden in totaal 21 stalen gescreend voor het SETBP1-gen, waarbij bij zes stalen een
mutatie werd gevonden (28,6%). Bij het CSF3R-gen werd geen enkele mutatie gevonden bij het
screenen van 38 stalen. Voor het ASXL1-gen werden 47 stalen gescreend waarbij bij negen stalen een
mutatie werd gevonden (19,1%). Het CSF3R- en ASXL1-gen werden naast het screenen bij
voorgaande diagnosen ook nog onderzocht bij AML. Hierbij werden mutatiefrequenties bekomen van
respectievelijk 2,9% (1/35) en 14,3% (3/21). Bij het CSF3R-gen werd slechts één mutatie gevonden bij
de 35 onderzochte AML-stalen. Het gaat hier om een niet eerder beschreven nonsense mutatie,
namelijk c.2260C>T(p.Gln754X).

90
Volgende vijf genen met gekende mutatiehotspots werden eveneens onderzocht:
Bij het TP53-gen werden 32 stalen afkomstig van CLL-patiënten getest waarbij bij vier
patiënten (12,5%) een mutatie werd gevonden.
NOTCH1-mutaties werden opgespoord bij CLL, MCL en T-ALL. Belangrijk op te merken bij het
NOTCH1-gen is dat T-ALL-patiënten frequenter beschikken over een gemuteerd NOTCH1-gen
dan CLL- en MCL-patiënten, respectievelijk 40% (4/10), 2,9% (1/34) en 0% (0/13), wat
overeenstemt met wat in de literatuur beschreven wordt, namelijk 55%, 15,3% en 8,5%.
Bij het STAT3-gen werden zeven stalen onderzocht, allemaal met de diagnose LGL. Vier van
deze stalen bleken een mutatie te bevatten (57,1%), dit in vergelijking met 33,4% beschreven
in de literatuur.
Het DNMT3A-gen werd voornamelijk getest bij MPN- en AML-patiënten, waarbij
mutatiefrequenties bekomen werden van respectievelijk 0% (0/22) en 13,2% (7/53). Deze
gevonden waarden zijn lager dan de in de literatuur beschreven frequenties, namelijk
respectievelijk 14% en 18,3%.
Het c-KIT-gen werd onderzocht bij AML-patiënten. Veertig stalen werden onderzocht,
waarbij bij twee stalen (5%) een mutatie werd gevonden. In de literatuur wordt beschreven
dat bij deze diagnose 11,8% van de patiënten beschikken over een mutant c-KIT-gen.
Tijdens het uitvoeren van de vele mutatieanalyses werden een aantal technische beperkingen en
moeilijkheden bij de interpretatie ondervonden.
Een eerste technische beperking situeert zich bij de gevoeligheid van de Sanger sequencing methode
die slechts ongeveer 20% bedraagt. Deze gevoeligheid veroorzaakt problemen bij het screenen van
onder andere het MYD88-gen bij LPL. In de literatuur wordt beschreven dat ongeveer 95% van de
LPL-patiënten beschikken over de mutatie p.Leu265Pro in het MYD88-gen, terwijl hier slechts een
mutatiefrequentie bekomen wordt van 6,5% (2/32), wat verklaard kan worden door de beperkte
gevoeligheid. Dit probleem kan opgelost worden door bijvoorbeeld een specifiekere PCR-reactie te
ontwerpen zodat de gevoeligheid stijgt of door de stalen te onderwerpen aan een voorafgaandelijke
celscheiding, namelijk een CD19-celscheiding. Bij de analyse van het c-KIT-gen bij de diagnose SM
werden eveneens problemen ondervonden door de beperkte gevoeligheid. Dit gen wordt reeds
buitenhuis getest bij de diagnose SM d.m.v. een kwantitatieve, gevoelige test. Het is de bedoeling dat
deze test kan uitgevoerd worden in het labo van het AZ Sint-Jan Brugge, maar na onderzoek blijkt dat
dit momenteel nog niet mogelijk is aangezien de nodige gevoeligheid niet behaald kan worden met
Sanger sequencing.
Een tweede technisch aandachtspunt is het frequent voorkomen van een mutatie in het ASXL1-gen,
namelijk c.1934dupG(p.Gly646TrpfsX12). De literatuur is het er niet over eens of het om een
somatische mutatie gaat dan wel om een PCR-artefact. De reden ligt in het feit dat de mutatie net
vooraf gegaan wordt door acht guanine-residuen. In één studie wordt beweerd dat het hier gaat om
‘slipped strand mispairing’, terwijl men er in de meeste studies van overtuigd is dat dit weldegelijk
een mutatie is. Ook in deze studie wordt dit laatste besloten.

91
Als laatste werd een moeilijkheid ondervonden gedurende de interpretatie van de resultaten,
namelijk bij het onderscheid tussen eiwit-polymorfismen en mutaties. Bij het uitvoeren van de
verschillende mutatieanalyses werd een eiwit-polymorfisme gevonden bij het ASXL1-gen
(c.2210G>A(p.Gly704Arg)) en een mogelijks eiwit-polymorfisme bij het CSF3R-gen, namelijk
c.2422G>A(p.Glu808Lys). Deze mutaties hebben dus geen invloed op het eiwit, waardoor ze niet in
verband kunnen gebracht worden met een specifieke diagnose met als gevolg dat ze niet bruikbaar
zijn als moleculaire merker.
Het doel van deze thesis was om een methode te ontwikkelen die in staat is om de in de literatuur
beschreven puntmutaties op een efficiënte manier te kunnen opsporen. Er kan besloten worden dat
de methode op punt gesteld werd en dat het mogelijk is om op een efficiënte manier verschillende
mutaties te onderzoeken. Deze methode heeft echter een beperkte gevoeligheid, ongeveer 20%
mutant DNA dient aanwezig te zijn. Bovendien laat de methode niet toe genmutaties op te sporen
buiten mutatiehotspots. Het is bijna onmogelijk om bijvoorbeeld alle exonen van een gen te testen,
wat bijvoorbeeld belangrijk kan zijn bij het DNMT3A-gen, waarbij nu enkel exon 23 gescreend werd,
waar zich 50% van de mutaties bevinden, terwijl ook nog mutatiehotspots aanwezig zijn van exon 11
tot exon 24 (Roller et al. 2013). Ook het onderzoeken van zeer grote genen, zoals het TET2-gen,
vormen een probleem om m.b.v. Sanger sequencing op een efficiënte manier te onderzoeken.
Deze geoptimaliseerde en gevalideerde mutatieanalyses worden vanaf heden routinematig
uitgevoerd in het laboratorium, dit in afwachting van de introductie van een nieuwe methode voor
uitgebreide mutatieanalyse door middel van Next Generation Sequencing (NGS). Met NGS wordt het
mogelijk om de gevoeligheid sterk te verhogen en om op een efficiënte manier meer exonen en
grote genen zonder duidelijke mutatiehotspot te screenen.

92
Referentielijst
Abdel-Wahab, O., J. Gao, M. Adli, A. Dey, T. Trimarchi, Y. R. Chung, C. Kuscu, T. Hricik, D. Ndiaye-Lobry, L. M. Lafave, R. Koche, A. H. Shih, O. A. Guryanova, E. Kim, S. Li, S. Pandey, J. Y. Shin, L. Telis, J. Liu, P. K. Bhatt, S. Monette, X. Zhao, C. E. Mason, C. Y. Park, B. E. Bernstein, I. Aifantis and R. L. Levine (2013) Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. The Journal of experimental medicine. 210, 2641-59.
Abdel-Wahab, O., O. Kilpivaara, J. Patel, L. Busque and R. L. Levine (2010) The most commonly reported variant in ASXL1 (c.1934dupG;p.Gly646TrpfsX12) is not a somatic alteration. Leukemia. 24, 1656-1657.
Affymetrix (2013) ExoSAP-IT® For PCR Product Cleanup Alto, W. A. and L. Clarcq (1999) Cutaneous and systemic manifestations of mastocytosis. American family
physician. 59, 3047-54, 3059-60. Alves (2013) The genetic code Arcaini, L., S. Zibellini, E. Boveri, R. Riboni, S. Rattotti, M. Varettoni, M. L. Guerrera, M. Lucioni, A. Tenore,
M. Merli, S. Rizzi, L. Morello, C. Cavalloni, M. C. Da Via, M. Paulli and M. Cazzola (2012) The BRAF V600E mutation in hairy cell leukemia and other mature B-cell neoplasms. Blood. 119, 188-191.
Aref, S., M. El-Ghonemy, T. Abouzeid, A. El-Sabbagh and M. El-Baiomy (2014) Prevalence and impact of colony stimulating factor 3 receptor (CSF3R) mutations among Egyptian acute myeloid leukemia patients. Leukemia research.
Ashraf, S., N. I. Noguera, J. Di Giandomenico, S. Zaza, S. K. Hasan and F. Lo-Coco (2013) Rapid detection of IDH2 (R140Q and R172K) mutations in acute myeloid leukemia. Annals of hematology. 92, 1319-23.
Bai, L. and W.-G. Zhu (2006) p53: Structure, Function and Therapeutic Applications. Journal of Cancer Molecules.
Balsat, M. and J. Cornillon (2013) Molecular and therapeutic advances in Hairy cell leukemia. Bulletin du cancer. 100, 1043-1047.
Barnabas, N., M. Shurafa, D. L. Van Dyke, S. R. Wolman, D. Clark and M. J. Worsham (2001) Significance of p53 mutations in patients with chronic lymphocytic leukemia: a sequential study of 30 patients. Cancer. 91, 285-93.
Bea, S., R. Valdes-Mas, A. Navarro, I. Salaverria, D. Martin-Garcia, P. Jares, E. Gine, M. Pinyol, C. Royo, F. Nadeu, L. Conde, M. Juan, G. Clot, P. Vizan, L. Di Croce, D. A. Puente, M. Lopez-Guerra, A. Moros, G. Roue, M. Aymerich, N. Villamor, L. Colomo, A. Martinez, A. Valera, J. I. Martin-Subero, V. Amador, L. Hernandez, M. Rozman, A. Enjuanes, P. Forcada, A. Muntanola, E. M. Hartmann, M. J. Calasanz, A. Rosenwald, G. Ott, J. M. Hernandez-Rivas, W. Klapper, R. Siebert, A. Wiestner, W. H. Wilson, D. Colomer, A. Lopez-Guillermo, C. Lopez-Otin, X. S. Puente and E. Campo (2013) Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 110, 18250-5.
Beekman, R., M. Valkhof, P. van Strien, P. J. Valk and I. P. Touw (2013) Prevalence of a new auto-activating colony stimulating factor 3 receptor mutation (CSF3R-T595I) in acute myeloid leukemia and severe congenital neutropenia. Haematologica. 98, e62-3.
Beekman, R., M. G. Valkhof, M. A. Sanders, P. M. van Strien, J. R. Haanstra, L. Broeders, W. M. Geertsma-Kleinekoort, A. J. Veerman, P. J. Valk, R. G. Verhaak, B. Lowenberg and I. P. Touw (2012) Sequential gain of mutations in severe congenital neutropenia progressing to acute myeloid leukemia. Blood. 119, 5071-7.
Biosystems, A. (2010) AmpliTaq Gold® DNA Polymerase - Protocol. Blombery, P. A., S. Q. Wong, C. A. Hewitt, A. Dobrovic, E. L. Maxwell, S. Juneja, G. Grigoriadis and D. A.
Westerman (2012) Detection of BRAF mutations in patients with hairy cell leukemia and related lymphoproliferative disorders. Haematologica. 97, 780-3.
Boissel, N., H. Leroy, B. Brethon, N. Philippe, S. de Botton, A. Auvrignon, E. Raffoux, T. Leblanc, X. Thomas, O. Hermine, B. Quesnel, A. Baruchel, G. Leverger, H. Dombret, C. Preudhomme, A. Acute Leukemia French and G. Leucemies Aigues Myeloblastiques de l'Enfant Cooperative (2006)

93
Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia. 20, 965-70.
Boyd, E. M., A. J. Bench, M. B. van 't Veer, P. Wright, D. M. Bloxham, G. A. Follows and M. A. Scott (2011) High resolution melting analysis for detection of BRAF exon 15 mutations in hairy cell leukaemia and other lymphoid malignancies. Br J Haematol. 155, 609-12.
Bunimovich, O., M. Grassi and M. R. Baer (2009) Systemic Mastocytosis: Classification, Pathogenesis, Diagnosis, and Treatment. Cutis. 83, 29-36.
Campregher, P. V., R. C. Petroni, N. Muto, R. Santana, R. Sitnik, O. P. S. Ramos, N. Bacal, C. B. Bub, F. P. S. Santos, P. Guilherme, H. Ricardo, C. Souza, R. A. de Assis, G. B. Oliveira, I. L. Metze and N. Hamerschlak (2012) Novel Assay for the Identification of NOTCH1 PEST Domain Mutations Using Fragment Analysis and Allele Specific PCR. Blood. 120.
Casteran, N., P. De Sepulveda, N. Beslu, M. Aoubala, S. Letard, E. Lecocq, R. Rottapel and P. Dubreuil (2003) Signal transduction by several KIT juxtamembrane domain mutations. Oncogene. 22, 4710-4722.
Cell, A. T. S. (2013) Hematopoiesis Chaligne, R., C. Tonetti, R. Besancenot, L. Roy, C. Marty, P. Mossuz, J. J. Kiladjian, G. Socie, D. Bordessoule,
M. C. Le Bousse-Kerdiles, W. Vainchenker and S. Giraudier (2008) New mutations of MPL in primitive myelofibrosis: only the MPL W515 mutations promote a G1/S-phase transition. Leukemia. 22, 1557-66.
Chen, B. and W. Janes (2002) PCR Cloning Protocols, pp 439 Chen, T. C., H. A. Hou, W. C. Chou, J. L. Tang, Y. Y. Kuo, C. Y. Chen, M. H. Tseng, C. F. Huang, Y. J. Lai, Y. C.
Chiang, F. Y. Lee, M. C. Liu, C. W. Liu, C. Y. Liu, M. Yao, S. Y. Huang, B. S. Ko, S. C. Hsu, S. J. Wu, W. Tsay, Y. C. Chen and H. F. Tien (2014) Dynamics of ASXL1 mutation and other associated genetic alterations during disease progression in patients with primary myelodysplastic syndrome. Blood cancer journal. 4, e177.
Chin, E. L., C. da Silva and M. Hegde (2013) Assessment of clinical analytical sensitivity and specificity of next-generation sequencing for detection of simple and complex mutations. BMC genetics. 14, 6.
Cho, E. K., S. M. Bang, J. Y. Ahn, S. M. Yoo, P. W. Park, Y. H. Seo, D. B. Shin and J. H. Lee (2003) Prognostic value of AML 1/ETO fusion transcripts in patients with acute myelogenous leukemia. The Korean journal of internal medicine. 18, 13-20.
Colarossi, S., S. Soverini, A. Gnani, M. Rondoni, R. Zanotti, S. Merante, S. Gatto, M. Tiribelli, F. Musto, E. Gottardi, D. Cilloni, G. Martinelli and M. Baccarani (2007) A novel denaturing-high performance liquid chromatography (D-HPLC)based method for kit mutation screening of patients (PTS) with systemic mastocytosis (SM). Haematol-Hematol J. 92, 41-41.
Cristobal, I., F. J. Blanco, L. Garcia-Orti, N. Marcotegui, C. Vicente, J. Rifon, F. J. Novo, E. Bandres, M. J. Calasanz, C. Bernabeu and M. D. Odero (2010) SETBP1 overexpression is a novel leukemogenic mechanism that predicts adverse outcome in elderly patients with acute myeloid leukemia. Blood. 115, 615-625.
Damm, F., R. Itzykson, O. Kosmider, N. Droin, A. Renneville, V. Chesnais, V. Gelsi-Boyer, S. de Botton, N. Vey, C. Preudhomme, A. Clavert, E. Delabesse, S. Park, D. Birnbaum, M. Fontenay, O. A. Bernard and E. Solary (2013) SETBP1 mutations in 658 patients with myelodysplastic syndromes, chronic myelomonocytic leukemia and secondary acute myeloid leukemias. Leukemia. 27, 1401-3.
Del Giudice, I., D. Rossi, S. Chiaretti, M. Marinelli, S. Tavolaro, S. Gabrielli, L. Laurenti, R. Marasca, S. Rasi, M. Fangazio, A. Guarini, G. Gaidano and R. Foa (2012) NOTCH1 mutations in +12 chronic lymphocytic leukemia (CLL) confer an unfavorable prognosis, induce a distinctive transcriptional profiling and refine the intermediate prognosis of +12 CLL. Haematologica. 97, 437-41.
den Dunnen, J. T. and S. E. Antonarakis (2000) Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Human mutation. 15, 7-12.
Ding, Z. X., H. J. Shen, J. C. Miao, S. N. Chen, Q. C. Qiu, X. F. Qi, Z. M. Jin, D. P. Wu and J. He (2012) [C-kit, NPM1 and FLT3 gene mutation patterns and their prognostic significance in 656 Chinese patients with acute myeloid leukemia]. Zhonghua xue ye xue za zhi = Zhonghua xueyexue zazhi. 33, 829-34.
Dirkse, K. (2010) neoplasma malignum, kanker, carcinoom

94
Dufour, A., G. Palermo, E. Zellmeier, G. Mellert, G. Duchateau-Nguyen, S. Schneider, T. Benthaus, P. M. Kakadia, K. Spiekermann, W. Hiddemann, J. Braess, S. Truong, N. Patten, L. Wu, S. Lohmann, D. Dornan, D. GuhaThakurta, R. F. Yeh, G. Salogub, P. Solal-Celigny, A. Dmoszynska, T. Robak, M. Montillo, J. Catalano, C. H. Geisler, M. Weisser and S. K. Bohlander (2013) Inactivation of TP53 correlates with disease progression and low miR-34a expression in previously treated chronic lymphocytic leukemia patients. Blood. 121, 3650-7.
ExPASy (2014) UniProtKB/Swiss-Prot variant pages Fabbri, G., S. Rasi, D. Rossi, V. Trifonov, H. Khiabanian, J. Ma, A. Grunn, M. Fangazio, D. Capello, S. Monti,
S. Cresta, E. Gargiulo, F. Forconi, A. Guarini, L. Arcaini, M. Paulli, L. Laurenti, L. M. Larocca, R. Marasca, V. Gattei, D. Oscier, F. Bertoni, C. G. Mullighan, R. Foa, L. Pasqualucci, R. Rabadan, R. Dalla-Favera and G. Gaidano (2011) Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. The Journal of experimental medicine. 208, 1389-401.
Fasan, A., W. Kern, V. Grossmann, C. Haferlach, T. Haferlach and S. Schnittger (2013) STAT3 mutations are highly specific for large granular lymphocytic leukemia. Leukemia. 27, 1598-600.
Ferrando, A. A. (2009) The role of NOTCH1 signaling in T-ALL. Hematology / the Education Program of the American Society of Hematology. American Society of Hematology. Education Program, 353-61.
Gambacorti-Passerini, C., S. Valletta, N. Winkelmann, S. Redaelli, R. Spinelli, A. Pirola, L. Antolini, L. Mologni, C. Donadoni, E. Papaemmanuil, S. Schnittger, K. Dong-Wook, J. Boultwood, F. Rossi, G. Gaipa, G. De Martini, P. F. di Celle, H. G. Jang, V. Fantin, G. R. Bignell, V. Magistroni, T. Haferlach, E. M. Pogliani, P. Campbell, A. J. Chase, W. J. Tapper, N. C. P. Cross and R. Piazza (2012) Recurrent SETBP1 Mutations in Atypical Chronic Myeloid Leukemia Abrogate an Ubiquitination Site and Dysregulate SETBP1 Protein Levels. Blood. 120.
Gelsi-Boyer, V., M. Brecqueville, R. Devillier, A. Murati, M. J. Mozziconacci and D. Birnbaum (2012) Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. Journal of hematology & oncology. 5, 12.
Gelsi-Boyer, V., V. Trouplin, J. Adelaide, J. Bonansea, N. Cervera, N. Carbuccia, A. Lagarde, T. Prebet, M. Nezri, D. Sainty, S. Olschwang, L. Xerri, M. Chaffanet, M. J. Mozziconacci, N. Vey and D. Birnbaum (2009) Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 145, 788-800.
Gemovic, B., V. Perovic, S. Glisic and N. Veljkovic (2013) Feature-based classification of amino acid substitutions outside conserved functional protein domains. TheScientificWorldJournal. 2013, 948617.
Genête, T. (2011) Detection of IDH1 and IDH2 mutations in acute lmyeloid leukemia Genetica, C. v. M. (2013) Fragment analyse Genome, M. C. (2010-2014) KIT in Acute Myeloid Leukemia Gotlib, J. (2013) Activating G-CSF Receptor Mutations in Neutrophilic Leukemias. American Society of
Hematology. Gotlib, J., J. E. Maxson, T. I. George and J. W. Tyner (2013) The new genetics of chronic neutrophilic
leukemia and atypical CML: implications for diagnosis and treatment. Blood. 122, 1707-11. Hagenkord, J. M., F. A. Monzon, S. F. Kash, S. Lilleberg, Q. M. Xie and J. A. Kant (2010) Array-Based
Karyotyping for Prognostic Assessment in Chronic Lymphocytic Leukemia Performance Comparison of Affymetrix 10K2.0, 250K Nsp, and SNP6.0 Arrays. Journal of Molecular Diagnostics. 12, 184-196.
Hallek, M., B. D. Cheson, D. Catovsky, F. Caligaris-Cappio, G. Dighiero, H. Dohner, P. Hillmen, M. J. Keating, E. Montserrat, K. R. Rai, T. J. Kipps and L. International Workshop on Chronic Lymphocytic (2008) Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 111, 5446-56.
He, X., Z. G. Chen, Y. Y. Jiang, X. Qiu and X. Y. Zhao (2013) Different mutations of the human c-mpl gene indicate distinct haematopoietic diseases. Journal of hematology & oncology. 6.
Healthcare (2014) Sephadex G-50 Superfine Hou, H. A., Y. Y. Kuo, C. Y. Liu, W. C. Chou, M. C. Lee, C. Y. Chen, L. I. Lin, M. H. Tseng, C. F. Huang, Y. C.
Chiang, F. Y. Lee, M. C. Liu, C. W. Liu, J. L. Tang, M. Yao, S. Y. Huang, B. S. Ko, S. C. Hsu, S. J. Wu, W.

95
Tsay, Y. C. Chen and H. F. Tien (2012) DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood. 119, 559-68.
Hou, H. A., Y. Y. Kuo, J. L. Tang, W. C. Chou, M. Yao, Y. J. Lai, C. C. Lin, C. Y. Chen, C. Y. Liu, M. H. Tseng, C. F. Huang, Y. C. Chiang, F. Y. Lee, M. C. Liu, C. W. Liu, S. Y. Huang, B. S. Ko, S. J. Wu, W. Tsay, Y. C. Chen and H. F. Tien (2014) Clinical implications of the SETBP1 mutation in patients with primary myelodysplastic syndrome and its stability during disease progression. American journal of hematology. 89, 181-6.
Hussain, S., F. Ahmed, H. Naqvi, P. Singh, F. Mahdi and S. Babu (2009) C-kit proto-oncogene deletion and pointmutation at exon 8, 17 in human acute myeloid leukemia Turkish Journal of Cancer. 39.
Hussain, S., S. Raza, S. Babu, P. Singh, H. Naqvi and F. Mahdi (2011) Screening of C-kit gene Mutation in Acute Myeloid Leukaemia in Northern India. Iran J Cancer Prev.
Institute, T. S. (2014) Catalogue Of Somatic Mutations In Cancer (COSMIC) Jenkinson, S., K. Koo, M. R. Mansour, N. Goulden, A. Vora, C. Mitchell, R. Wade, S. Richards, J. Hancock, A.
V. Moorman, D. C. Linch and R. E. Gale (2013) Impact of NOTCH1/FBXW7 mutations on outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on the MRC UKALL 2003 trial. Leukemia. 27, 41-7.
Jerez, A., M. J. Clemente, H. Makishima, H. Koskela, F. Leblanc, K. Peng Ng, T. Olson, B. Przychodzen, M. Afable, I. Gomez-Segui, K. Guinta, L. Durkin, E. D. Hsi, K. McGraw, D. Zhang, M. W. Wlodarski, K. Porkka, M. A. Sekeres, A. List, S. Mustjoki, T. P. Loughran and J. P. Maciejewski (2012) STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood. 120, 3048-57.
Kaushansky, K. (2006) Lineage-specific hematopoietic growth factors. The New England journal of medicine. 354, 2034-45.
Kim, S. J., H. Zhao, S. Hardikar, A. K. Singh, M. A. Goodell and T. Chen (2013) A DNMT3A mutation common in AML exhibits dominant-negative effects in murine ES cells. Blood. 122, 4086-9.
Kong, Y., L. Si, Y. Zhu, X. Xu, C. L. Corless, K. T. Flaherty, L. Li, H. Li, X. Sheng, C. Cui, Z. Chi, S. Li, M. Han, L. Mao, A. Lu and J. Guo (2011) Large-scale analysis of KIT aberrations in Chinese patients with melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 17, 1684-91.
Koskela, H. L., S. Eldfors, P. Ellonen, A. J. van Adrichem, H. Kuusanmaki, E. I. Andersson, S. Lagstrom, M. J. Clemente, T. Olson, S. E. Jalkanen, M. M. Majumder, H. Almusa, H. Edgren, M. Lepisto, P. Mattila, K. Guinta, P. Koistinen, T. Kuittinen, K. Penttinen, A. Parsons, J. Knowles, J. Saarela, K. Wennerberg, O. Kallioniemi, K. Porkka, T. P. Loughran, Jr., C. A. Heckman, J. P. Maciejewski and S. Mustjoki (2012) Somatic STAT3 mutations in large granular lymphocytic leukemia. The New England journal of medicine. 366, 1905-13.
Kosmider, O., R. Itzykson, V. Chesnais, T. Lasho, R. Laborde, R. Knudson, A. Gauthier, J. Merlevede, L. Ades, M. Morabito, M. Fontenay, A. Tefferi, N. Droin and E. Solary (2013) Mutation of the colony-stimulating factor-3 receptor gene is a rare event with poor prognosis in chronic myelomonocytic leukemia. Leukemia. 27, 1946-1949.
Kristensen, T., H. Vestergaard and M. B. Moller (2011) Improved Detection of the KIT D816V Mutation in Patients with Systemic Mastocytosis Using a Quantitative and Highly Sensitive Real-Time qPCR Assay. Journal of Molecular Diagnostics. 13, 180-188.
Laborde, R. R., M. M. Patnaik, T. L. Lasho, C. M. Finke, C. A. Hanson, R. A. Knudson, R. P. Ketterling, A. Pardanani and A. Tefferi (2013) SETBP1 mutations in 415 patients with primary myelofibrosis or chronic myelomonocytic leukemia: independent prognostic impact in CMML. Leukemia. 27, 2100-2.
Laurini, J. A., P. Aoun, J. Iqbal, W. Chan and T. C. Greiner (2012) Investigation of the BRAF V600E mutation by pyrosequencing in lymphoproliferative disorders. Am J Clin Pathol. 138, 877-83.
Lawyer, F. C., S. Stoffel, R. K. Saiki, K. Myambo, R. Drummond and D. H. Gelfand (1989) Isolation, characterization, and expression in Escherichia coli of the DNA polymerase gene from Thermus aquaticus. The Journal of biological chemistry. 264, 6427-37.
Lee, T. S., H. Kantarjian, W. L. Ma, C. H. Yeh, F. Giles and M. Albitar (2011) Effects of Clinically Relevant MPL Mutations in the Transmembrane Domain Revealed at the Atomic Level through Computational Modeling. Plos One. 6.

96
Ley, T. J., L. Ding, M. J. Walter, M. D. McLellan, T. Lamprecht, D. E. Larson, C. Kandoth, J. E. Payton, J. Baty, J. Welch, C. C. Harris, C. F. Lichti, R. R. Townsend, R. S. Fulton, D. J. Dooling, D. C. Koboldt, H. Schmidt, Q. Zhang, J. R. Osborne, L. Lin, M. O'Laughlin, J. F. McMichael, K. D. Delehaunty, S. D. McGrath, L. A. Fulton, V. J. Magrini, T. L. Vickery, J. Hundal, L. L. Cook, J. J. Conyers, G. W. Swift, J. P. Reed, P. A. Alldredge, T. Wylie, J. Walker, J. Kalicki, M. A. Watson, S. Heath, W. D. Shannon, N. Varghese, R. Nagarajan, P. Westervelt, M. H. Tomasson, D. C. Link, T. A. Graubert, J. F. DiPersio, E. R. Mardis and R. K. Wilson (2010) DNMT3A mutations in acute myeloid leukemia. The New England journal of medicine. 363, 2424-33.
Li, L., X. D. Lyu, R. H. Mi, J. Ding, L. Chen, Q. Wang, Q. S. Yin, J. Y. Hu, R. H. Fan and X. D. Wei (2013) [Detection of NPM1, FLT3 and C-KIT mutations in acute myeloid leukemia and their prognostic analysis]. Zhongguo shi yan xue ye xue za zhi / Zhongguo bing li sheng li xue hui = Journal of experimental hematology / Chinese Association of Pathophysiology. 21, 601-6.
Lim, K. H., A. Tefferi, T. L. Lasho, C. Finke, M. Patnaik, J. H. Butterfield, R. F. McClure, C. Y. Li and A. Pardanani (2009) Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood. 113, 5727-36.
Liongue, C. and A. Ward (2014) Granulocyte Colony-Stimulating Factor Receptor mutations in myeloid malignancy. Frontiers in Oncology.
Lodish, H. F. (2013) Molecular cell biology, 7th ed., W.H. Freeman and Co., New York Loewe, L. (2008) Genetic Mutation. Nature Education. Loughran, T. (2012) Large Granular Lymphocytic Leukemia Makishima, H., K. Yoshida, N. Nguyen, B. Przychodzen, M. Sanada, Y. Okuno, K. P. Ng, K. O. Gudmundsson,
B. A. Vishwakarma, A. Jerez, I. Gomez-Segui, M. Takahashi, Y. Shiraishi, Y. Nagata, K. Guinta, H. Mori, M. A. Sekeres, K. Chiba, H. Tanaka, H. Muramatsu, H. Sakaguchi, R. L. Paquette, M. A. McDevitt, S. Kojima, Y. Saunthararajah, S. Miyano, L. Y. Shih, Y. Du, S. Ogawa and J. P. Maciejewski (2013) Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 45, 942-U298.
Malecki, M. J., C. Sanchez-Irizarry, J. L. Mitchell, G. Histen, M. L. Xu, J. C. Aster and S. C. Blacklow (2006) Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Mol Cell Biol. 26, 4642-4651.
Mansour, M. R., D. C. Linch, L. Foroni, A. H. Goldstone and R. E. Gale (2006) High incidence of Notch-1 mutations in adult patients with T-cell acute lymphoblastic leukemia. Leukemia. 20, 537-9.
Mansur, M. B., M. Emerenciano, A. Splendore, L. Brewer, R. Hassan, M. S. Pombo-De-Oliveira and B. C. S. Grp (2010) T-cell lymphoblastic leukemia in early childhood presents NOTCH1 mutations and MLL rearrangements. Leukemia research. 34, 483-486.
Marinelli, M., N. Peragine, V. Di Maio, S. Chiaretti, M. S. De Propris, S. Raponi, S. Tavolaro, F. R. Mauro, I. Del Giudice, A. Guarini and R. Foa (2013) Identification of molecular and functional patterns of p53 alterations in chronic lymphocytic leukemia patients in different phases of the disease. Haematologica. 98, 371-5.
Matthews, D. J. and M. E. Gerritsen (2010) Targeting protein kinases for cancer therapy, John Wiley & Sons, Hoboken, N.J.
Maxson, J. E., J. Gotlib, D. A. Pollyea, A. G. Fleischman, A. Agarwal, C. A. Eide, D. Bottomly, B. Wilmot, S. K. McWeeney, C. E. Tognon, J. B. Pond, R. H. Collins, B. Goueli, S. T. Oh, M. W. Deininger, B. H. Chang, M. M. Loriaux, B. J. Druker and J. W. Tyner (2013) Oncogenic CSF3R Mutations in Chronic Neutrophilic Leukemia and Atypical CML. New Engl J Med. 368, 1781-1790.
Medicine, U. S. N. L. o. (2013) MYD88 myeloid differentiation primary response 88 [Homo sapiens (human)]
Meggendorfer, M., T. Alpermann, T. Haferlach, C. Schrauder, R. Konietschke, C. Haferlach, W. Kern and S. Schnittger (2013a) Mutational Screening Of CSF3R, ASXL1, SETBP1, and SRSF2 In Chronic Neutrophilic Leukemia (CNL), Atypical CML and CMML Cases. American Society of Hematology.
Meggendorfer, M., U. Bacher, T. Alpermann, C. Haferlach, W. Kern, C. Gambacorti-Passerini, T. Haferlach and S. Schnittger (2013b) SETBP1 mutations occur in 9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i(17)(q10), ASXL1 and CBL mutations. Leukemia. 27, 1852-60.
Metzeler, K. H., H. Becker, K. Maharry, M. D. Radmacher, J. Kohlschmidt, K. Mrozek, D. Nicolet, S. P. Whitman, Y. Z. Wu, S. Schwind, B. L. Powell, T. H. Carter, M. Wetzler, J. O. Moore, J. E. Kolitz, M.

97
R. Baer, A. J. Carroll, R. A. Larson, M. A. Caligiuri, G. Marcucci and C. D. Bloomfield (2011) ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood. 118, 6920-6929.
Millipore (2014) MultiScreen-HV Modlich, U., N. Heinz, S. Kohlscheen, L. Latorre and F. Schenk (2013) Thrombopoietin-induced genes and
pathways for the regeneration of hematopoietic stem cells. Movva, S. (2013) Understanding Cancer -- the Basics Mozziconacci, M. and D. Birnbaum (2010) ASXL1 (additional sex combs like 1 (Drosophila)). Atlas Genet
Cytogenet Oncol Haematol. Neumann, M., S. Heesch, C. Schlee, S. Schwartz, N. Goekbuget, D. Hoelzer, N. P. Konstandin, B. Ksienzyk,
A. Graf, S. Krebs, H. Blum, T. Raff, M. Brueggemann, W. K. Hofmann, S. K. Bohlander, P. A. Greif and C. D. Baldus (2012) Exome Sequencing Identifies Recurring DNMT3A Mutations in Adult ETP-ALL. Blood. 120.
O'Neill, L. A. and A. G. Bowie (2007) The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nature reviews. Immunology. 7, 353-64.
Ogino, S., M. L. Gulley, J. T. den Dunnen, R. B. Wilson, T. Association for Molecular Patholpogy and C. Education (2007) Standard mutation nomenclature in molecular diagnostics: practical and educational challenges. The Journal of molecular diagnostics : JMD. 9, 1-6.
Olivier, M., Eeles, R., Hollstein, M., Khan, M.A., Harris, C.C. & Hainaut, P. (2005) The IARC TP53 Database: new online mutation analysis and recommendations to users.
Olsen, R. J., C. H. Dunphy, D. P. O'Malley, L. Rice, A. A. Ewton and C. C. Chang (2008) The implication of
identifying JAK2 ( V617F ) in myeloproliferative neoplasms and myelodysplastic syndromes with bone marrow fibrosis. Journal of hematopathology. 1, 111-7.
Oncology, I. (2013) MPL Mutation Analysis Ondrejka, S. L., J. J. Lin, D. W. Warden, L. Durkin, J. R. Cook and E. D. Hsi (2013) MYD88 L265P Somatic
Mutation Its Usefulness in the Differential Diagnosis of Bone Marrow Involvement by B-Cell Lymphoproliferative Disorders. Am J Clin Pathol. 140, 387-394.
Owen, R. G., S. L. Barrans, S. J. Richards, S. J. M. O'Connor, J. A. Child, L. A. Parapia, G. J. Morgan and A. S. Jack (2001) Waldenstrom macroglobulinemia - Development of diagnostic criteria and identification of prognostic factors. Am J Clin Pathol. 116, 420-428.
Pardanani, A., P. Guglielmelli, T. L. Lasho, A. Pancrazzi, C. M. Finke, A. M. Vannucchi and A. Tefferi (2011) Primary myelofibrosis with or without mutant MPL: comparison of survival and clinical features involving 603 patients. Leukemia. 25, 1834-1839.
Pardanani, A., T. L. Lasho, R. R. Laborde, M. Elliott, C. A. Hanson, R. A. Knudson, R. P. Ketterling, J. E. Maxson, J. W. Tyner and A. Tefferi (2013) CSF3R T618I is a highly prevalent and specific mutation in chronic neutrophilic leukemia. Leukemia. 27, 1870-1873.
Parekh, S. (2012) Taking it up a notch in MCL. Blood. 119, 1957-8. Park, M. J., T. Taki, M. Oda, T. Watanabe, K. Yumura-Yagi, R. Kobayashi, N. Suzuki, J. Hara, K. Horibe and Y.
Hayashi (2009) FBXW7 and NOTCH1 mutations in childhood T cell acute lymphoblastic leukaemia and T cell non-Hodgkin lymphoma. Brit J Haematol. 145, 198-206.
Park, S. H., H. S. Chi, S. K. Min, B. G. Park, S. Jang and C. J. Park (2011) Prognostic impact of c-KIT mutations in core binding factor acute myeloid leukemia. Leukemia research. 35, 1376-1383.
Paschka, P., R. F. Schlenk, V. I. Gaidzik, M. Habdank, J. Kronke, L. Bullinger, D. Spath, S. Kayser, M. Zucknick, K. Gotze, H. A. Horst, U. Germing, H. Dohner and K. Dohner (2010) IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 28, 3636-43.
Patel, K. P., B. A. Barkoh, Z. Chen, D. Q. Ma, N. Reddy, L. J. Medeiros and R. Luthra (2011a) Diagnostic Testing for IDH1 and IDH2 Variants in Acute Myeloid Leukemia An Algorithmic Approach Using High-Resolution Melting Curve Analysis. Journal of Molecular Diagnostics. 13, 678-686.

98
Patel, K. P., F. Ravandi, D. Ma, A. Paladugu, B. A. Barkoh, L. J. Medeiros and R. Luthra (2011b) Acute myeloid leukemia with IDH1 or IDH2 mutation: frequency and clinicopathologic features. Am J Clin Pathol. 135, 35-45.
Pavletich, N. P., K. A. Chambers and C. O. Pabo (1993) The DNA-Binding Domain of P53 Contains the 4 Conserved Regions and the Major Mutation Hot-Spots. Gene Dev. 7, 2556-2564.
Pekova, S., O. Mazal, R. Cmejla, D. W. Hardekopf, R. Plachy, L. Zejskova, R. Haugvicova, T. Jancuskova, M. Karas, V. Koza, L. Smolej, L. Bezdickova and T. Kozak (2011) A comprehensive study of TP53 mutations in chronic lymphocytic leukemia: Analysis of 1287 diagnostic and 1148 follow-up CLL samples. Leukemia research. 35, 889-98.
Perkins, A. (2013) Harnessing tumour banks to improve patient outcomes. Perrotti, D. and P. Neviani (2008) Protein phosphatase 2A (PP2A), a drugable tumor suppressor in Ph1(+)
leukemias. Cancer metastasis reviews. 27, 159-68. Piazza, R., S. Valletta, N. Winkelmann, S. Redaelli, R. Spinelli, A. Pirola, L. Antolini, L. Mologni, C. Donadoni,
E. Papaemmanuil, S. Schnittger, D. W. Kim, J. Boultwood, F. Rossi, G. Gaipa, G. P. De Martini, P. F. di Celle, H. G. Jang, V. Fantin, G. R. Bignell, V. Magistroni, T. Haferlach, E. M. Pogliani, P. J. Campbell, A. J. Chase, W. J. Tapper, N. C. Cross and C. Gambacorti-Passerini (2013) Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet. 45, 18-24.
Pietra, D., A. Brisci, E. Rumi, S. Boggi, C. Elena, A. Pietrelli, R. Bordoni, M. Ferrari, F. Passamonti, G. De Bellis, L. Cremonesi and M. Cazzola (2011) Deep sequencing reveals double mutations in cis of MPL exon 10 in myeloproliferative neoplasms. Haematologica. 96, 607-11.
Pollard, J. A., T. A. Alonzo, R. B. Gerbing, P. A. Ho, R. Zeng, Y. Ravindranath, G. Dahl, N. J. Lacayo, D. Becton, M. Chang, H. J. Weinstein, B. Hirsch, S. C. Raimondi, N. A. Heerema, W. G. Woods, B. J. Lange, C. Hurwitz, R. J. Arceci, J. P. Radich, I. D. Bernstein, M. C. Heinrich and S. Meshinchi (2010) Prevalence and prognostic significance of KIT mutations in pediatric patients with core binding factor AML enrolled on serial pediatric cooperative trials for de novo AML. Blood. 115, 2372-9.
Pospisilova, S., D. Gonzalez, J. Malcikova, M. Trbusek, D. Rossi, A. P. Kater, F. Cymbalista, B. Eichhorst, M. Hallek, H. Dohner, P. Hillmen, M. van Oers, J. Gribben, P. Ghia, E. Montserrat, S. Stilgenbauer, T. Zenz and C. L. L. European Research Initiative on (2012) ERIC recommendations on TP53 mutation analysis in chronic lymphocytic leukemia. Leukemia. 26, 1458-61.
Pospisilova, S., J. Malcikova, J. Damborsky, J. Smardova, M. Trbusek, B. Tichy, Y. Brychtova, M. Doubek, J. Vorlicek and J. Mayer (2006) Role of p53 gene mutation type in B-CLL prognosis. Blood. 108, 159b-160b.
Pratcorona, M., S. Abbas, M. A. Sanders, J. E. Koenders, F. G. Kavelaars, C. A. Erpelinck-Verschueren, A. Zeilemakers, B. Lowenberg and P. J. Valk (2012) Acquired mutations in ASXL1 in acute myeloid leukemia: prevalence and prognostic value. Haematologica. 97, 388-92.
Prensner, J. R. and A. M. Chinnaiyan (2011) Metabolism unhinged: IDH mutations in cancer. Nature medicine. 17, 291-3.
Qiu, Z. Y., L. Fan, L. Wang, C. Qiao, Y. J. Wu, J. F. Zhou, W. Xu and J. Y. Li (2013) STAT3 mutations are frequent in T-cell large granular lymphocytic leukemia with pure red cell aplasia. Journal of hematology & oncology. 6, 82.
Ranganathan, P., K. L. Weaver and A. J. Capobianco (2011) Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 11, 338-351.
Ribeiro, A. F. T., M. Pratcorona, C. Erpelinck-Verschueren, V. Rockova, M. Sanders, S. Abbas, M. E. Figueroa, A. Zeilemaker, A. Melnick, B. Lowenberg, P. J. M. Valk and R. Delwel (2012) Mutant DNMT3A: a marker of poor prognosis in acute myeloid leukemia. Blood. 119, 5824-5831.
Riera, L., F. Marmont, D. Toppino, C. Frairia, F. Sismondi, E. Audisio, C. Di Bello, S. D'Ardia, P. F. Di Celle, E. Messa, G. Inghirami, U. Vitolo and A. Pich (2013) Core binding factor acute myeloid leukaemia and c-KIT mutations. Oncology reports. 29, 1867-72.
Roller, A., V. Grossmann, U. Bacher, F. Poetzinger, S. Weissmann, N. Nadarajah, L. Boeck, W. Kern, C. Haferlach, S. Schnittger, T. Haferlach and A. Kohlmann (2013) Landmark analysis of DNMT3A mutations in hematological malignancies. Leukemia. 27, 1573-8.
Royster, J. (2010) Diagnosing GIST Ryan, K. M., A. C. Phillips and K. H. Vousden (2001) Regulation and function of the p53 tumor suppressor
protein. Current opinion in cell biology. 13, 332-7.

99
Sala, E., L. Ruggiero, G. Di Giacomo and O. Cremona (2012) Endocytosis in Notch Signaling Activation Sanger, F. and A. R. Coulson (1975) A rapid method for determining sequences in DNA by primed
synthesis with DNA polymerase. Journal of molecular biology. 94, 441-8. Sano, H., K. Ohki, M. Park, Y. Hara, N. Shiba, D. Tomizawa, T. Taga, A. Saito, J. Fujimoto, A. Tawa, K. Horibe,
S. Adachi and Y. Hayashi (2013) CSF3R Gene Mutations In Myeloid Malignancy Of Childhood. American Society of Hematology.
Schmitt-Graeff, A. H., S. S. Teo, M. Olschewski, F. Schaub, S. Haxelmans, A. Kim, P. Reinecke, U. Germing and R. C. Skoda (2008) JAK2(V617F) mutation status identifies subtypes of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Haematol-Hematol J. 93, 34-40.
Schnittger, S., U. Bacher, C. Haferlach, R. Dengler, A. Krober, W. Kern and T. Haferlach (2008) Detection of an MPLW515 mutation in a case with features of both essential thrombocythemia and refractory anemia with ringed sideroblasts and thrombocytosis. Leukemia. 22, 453-455.
Schnittger, S., C. Eder, S. Jeromin, T. Alpermann, A. Fasan, V. Grossmann, A. Kohlmann, T. Illig, N. Klopp, H. E. Wichmann, K. A. Kreuzer, C. Schmid, P. Staib, R. Peceny, N. Schmitz, W. Kern, C. Haferlach and T. Haferlach (2013) ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia. 27, 82-91.
Schnittger, S., T. M. Kohl, T. Haferlach, W. Kern, W. Hiddemann, K. Spiekermann and C. Schoch (2006) KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired event-free and overall survival. Blood. 107, 1791-1799.
Scholten, L. (2013) Morfologie van bloed en beenmerg Deventer Schuit, F. C. (2000) Medische biochemie, Bohn Stafleu Van Loghum Schwind, S., C. G. Edwards, D. Nicolet, K. Mrozek, K. Maharry, Y. Z. Wu, P. Paschka, A. K. Eisfeld, P.
Hoellerbauer, H. Becker, K. H. Metzeler, J. Curfman, J. Kohlschmidt, T. W. Prior, J. E. Kolitz, W. Blum, M. J. Pettenati, P. Dal Cin, A. J. Carroll, M. A. Caligiuri, R. A. Larson, S. Volinia, G. Marcucci, C. D. Bloomfield and O. Alliance for Clinical Trials in (2013) inv(16)/t(16;16) acute myeloid leukemia with non-type A CBFB-MYH11 fusions associate with distinct clinical and genetic features and lack KIT mutations. Blood. 121, 385-91.
Science, W. I. o. (2013) The GeneCards Human Gene Database Sheer, D., M. Lurantos and A. Pitt (1997) High Throughput Sample Preparation for Protein/Peptide
Structural Characterization. Shih, A. H., O. Abdel-Wahab, J. P. Patel and R. L. Levine (2012) The role of mutations in epigenetic
regulators in myeloid malignancies. Nat Rev Cancer. 12, 599-612. Shivarov, V., R. Gueorguieva, A. Stoimenov and R. Tiu (2013) DNMT3A mutation is a poor prognosis
biomarker in AML: Results of a meta-analysis of 4500 AML patients. Leukemia research. 37, 1445-1450.
Shlush, L. I., S. Zandi, A. Mitchell, W. C. Chen, J. M. Brandwein, V. Gupta, J. A. Kennedy, A. D. Schimmer, A. C. Schuh, K. W. Yee, J. L. McLeod, M. Doedens, J. J. Medeiros, R. Marke, H. J. Kim, K. Lee, J. D. McPherson, T. J. Hudson, H. P.-L. G. P. Consortium, A. M. Brown, F. Yousif, Q. M. Trinh, L. D. Stein, M. D. Minden, J. C. Wang and J. E. Dick (2014) Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 506, 328-33.
Sigma-Aldrich (2014) Gel Filtration Chromatography Silva, A., P. Y. Jotta, A. B. Silveira, D. Ribeiro, S. R. Brandalise, J. A. Yunes and J. T. Barata (2010) Regulation
of PTEN by CK2 and Notch1 in primary T-cell acute lymphoblastic leukemia: rationale for combined use of CK2-and gamma-secretase inhibitors. Haematol-Hematol J. 95, 674-678.
Soussi, T. (2012a) TP53 Mutations In Human Cancer Soussi, T. (2012b) The TP53 Web Site Swerdlow, S. H., E. Campo, N. L. Harris, E. S. Jaffe, S. A. Pileri, H. Stein, J. Thiele and J. W. Vardiman (2008)
WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed., International Agency for Research on Cancer (IARC), Lyon
Technologies, L. (2014) 3500xL Genetic Analyzer for Human Identification Tefferi, A., P. Noel and C. A. Hanson (2011) Uses and abuses of JAK2 and MPL mutation tests in
myeloproliferative neoplasms a paper from the 2010 William Beaumont hospital symposium on molecular pathology. The Journal of molecular diagnostics : JMD. 13, 461-6.

100
Testa, U. and E. Pelosi (2013) The Impact of FLT3 Mutations on the Development of Acute Myeloid Leukemias. Leukemia research and treatment. 2013, 275760.
Thol, F., F. Damm, A. Ludeking, C. Winschel, K. Wagner, M. Morgan, H. Y. Yun, G. Gohring, B. Schlegelberger, D. Hoelzer, M. Lubbert, L. Kanz, W. Fiedler, H. Kirchner, G. Heil, J. Krauter, A. Ganser and M. Heuser (2011a) Incidence and Prognostic Influence of DNMT3A Mutations in Acute Myeloid Leukemia. Journal of Clinical Oncology. 29, 2889-2896.
Thol, F., I. Friesen, F. Damm, H. Yun, E. M. Weissinger, J. Krauter, K. Wagner, A. Chaturvedi, A. Sharma, M. Wichmann, G. Gohring, C. Schumann, G. Bug, O. Ottmann, W. K. Hofmann, B. Schlegelberger, M. Heuser and A. Ganser (2011b) Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 29, 2499-506.
Thol, F., K. J. Suchanek, C. Koenecke, M. Stadler, U. Platzbecker, C. Thiede, T. Schroeder, G. Kobbe, S. Kade, P. Loffeld, S. Banihosseini, G. Bug, O. Ottmann, W. K. Hofmann, J. Krauter, N. Kroger, A. Ganser and M. Heuser (2013) SETBP1 mutation analysis in 944 patients with MDS and AML. Leukemia. 27, 2072-2075.
Tiacci, E., V. Trifonov, G. Schiavoni, A. Holmes, W. Kern, M. P. Martelli, A. Pucciarini, B. Bigerna, R. Pacini, V. A. Wells, P. Sportoletti, V. Pettirossi, R. Mannucci, O. Elliott, A. Liso, A. Ambrosetti, A. Pulsoni, F. Forconi, L. Trentin, G. Semenzato, G. Inghirami, M. Capponi, F. Di Raimondo, C. Patti, L. Arcaini, P. Musto, S. Pileri, C. Haferlach, S. Schnittger, G. Pizzolo, R. Foa, L. Farinelli, T. Haferlach, L. Pasqualucci, R. Rabadan and B. Falini (2011) BRAF mutations in hairy-cell leukemia. The New England journal of medicine. 364, 2305-15.
Treon, S. P., L. Xu, G. Yang, Y. Zhou, X. Liu, Y. Cao, P. Sheehy, R. J. Manning, C. J. Patterson, C. Tripsas, L. Arcaini, G. S. Pinkus, S. J. Rodig, A. R. Sohani, N. L. Harris, J. M. Laramie, D. A. Skifter, S. E. Lincoln and Z. R. Hunter (2012) MYD88 L265P somatic mutation in Waldenstrom's macroglobulinemia. The New England journal of medicine. 367, 826-33.
Umehara, Y., K. Hanaoka and D. Watanabe (2013) Distinct functions of Dnmt3a and Dnmt3b de novo DNA methyltransferases in ES cell proliferation and differentiation. Stem Cell Discovery. 3, 127-132.
UniProt (2014) Protein knowledgebase Vainchenker, W. and S. N. Constantinescu (2013) JAK/STAT signaling in hematological malignancies.
Oncogene. 32, 2601-13. Van der Reijden, B. A., M. Massop, A. Simons, T. de Witte, M. Breuning and J. H. Jansen (2010) The NDE1
gene is disrupted by the inv(16) in 90% of cases with CBFB-MYH11-positive acute myeloid leukemia. Leukemia. 24, 857-859.
Van Vlierberghe, P. and A. Ferrando (2012) The molecular basis of T cell acute lymphoblastic leukemia. The Journal of clinical investigation. 122, 3398-406.
Vanderbilt (2013) My Cancer Genome Vannucchi, A. M. and P. Guglielmelli (2008) Molecular pathophysiology of Philadelphia-negative
myeloproliferative disorders: beyond JAK2 and MPL mutations. Haematologica. 93, 972-6. Varettoni, M., L. Arcaini, S. Zibellini, E. Boveri, S. Rattotti, R. Riboni, A. Corso, E. Orlandi, M. Bonfichi, M.
Gotti, C. Pascutto, S. Mangiacavalli, G. Croci, V. Fiaccadori, L. Morello, M. L. Guerrera, M. Paulli and M. Cazzola (2013) Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenstrom's macroglobulinemia and related lymphoid neoplasms. Blood. 121, 2522-8.
Verma, S., W. O. Greaves, F. Ravandi, N. Reddy, C. E. Bueso-Ramos, S. O'Brien, D. A. Thomas, H. Kantarjian, L. J. Medeiros, R. Luthra and K. P. Patel (2012) Rapid Detection and Quantitation of BRAF Mutations in Hairy Cell Leukemia Using a Sensitive Pyrosequencing Assay. Am J Clin Pathol. 138, 153-156.
Verstovsek, S. (2013) Advanced systemic mastocytosis: the impact of KIT mutations in diagnosis, treatment, and progression. European journal of haematology. 90, 89-98.
Vogelstein, B., D. Lane and A. J. Levine (2000) Surfing the p53 network. Nature. 408, 307-310. W.J.Wiersinga and T. v. d. Poll (2005) ‘Toll-like’ receptoren en de betekenis voor de kliniek. Ned Tijdschr
Geneeskd. 149, 1150-1555. Wagner, K., F. Damm, G. Gohring, K. Gorlich, M. Heuser, I. Schafer, O. Ottmann, M. Lubbert, W. Heit, L.
Kanz, G. Schlimok, A. A. Raghavachar, W. Fiedler, H. H. Kirchner, W. Brugger, M. Zucknick, B.

101
Schlegelberger, G. Heil, A. Ganser and J. Krauter (2010) Impact of IDH1 R132 Mutations and an IDH1 Single Nucleotide Polymorphism in Cytogenetically Normal Acute Myeloid Leukemia: SNP rs11554137 Is an Adverse Prognostic Factor. Journal of Clinical Oncology. 28, 2356-2364.
Watters, T. M., E. F. Kenny and L. A. O'Neill (2007) Structure, function and regulation of the Toll/IL-1 receptor adaptor proteins. Immunol Cell Biol. 85, 411-419.
West, R. R., A. P. Hsu, S. M. Holland, J. Cuellar-Rodriguez and D. D. Hickstein (2014) Acquired ASXL1 mutations are common in patients with inherited GATA2 mutations and correlate with myeloid transformation. Haematologica. 99, 276-81.
Whitfield, E. (2013) Mutation nomenclature WHO (2012) Cancer Prevention and Control Willander, K., R. K. Dutta, J. Ungerback, R. Gunnarsson, G. Juliusson, M. Fredrikson, M. Linderholm and P.
Soderkvist (2013) NOTCH1 mutations influence survival in chronic lymphocytic leukemia patients. BMC cancer. 13, 274.
Willenbacher, W., E. Willenbacher, A. Brunner and C. Manzl (2013) Improved accuracy of discrimination between IgM Multiple Myeloma and Waldenstrom Macroglobulinaemia by testing for MYD88 L265P mutations. Brit J Haematol. 161, 902-904.
Wong, K. B., B. S. DeDecker, S. M. V. Freund, M. R. Proctor, M. Bycroft and A. R. Fersht (1999) Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proceedings of the National Academy of Sciences of the United States of America. 96, 8438-8442.
Xu, L., A. R. Sohani, L. Arcaini, Z. Hunter, G. Yang, Y. S. Zhou, X. Liu, Y. Cao, R. Manning, C. J. Patterson, L. Ioakimidis, C. Tripsas, G. S. Pinkus, N. L. Harris, S. J. Rodig and S. Treon (2011) A Somatic Variant in MYD88 (L265P) Revealed by Whole Genome Sequencing Differentiates Lymphoplasmacytic Lymphoma From Marginal Zone Lymphomas. Blood. 118, 120-120.
Yang, H., D. Ye, K. L. Guan and Y. Xiong (2012) IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clinical cancer research : an official journal of the American Association for Cancer Research. 18, 5562-71.
Yu, H., M. Kortylewski and D. Pardoll (2007) Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nature Reviews Immunology. 7, 41-51.
Zainuddin, N., F. Murray, M. Kanduri, R. Gunnarsson, K. E. Smedby, G. Enblad, J. Jurlander, G. Juliusson and R. Rosenquist (2011) TP53 Mutations are infrequent in newly diagnosed chronic lymphocytic leukemia. Leukemia research. 35, 272-4.
Zenz, T., B. Eichhorst, R. Busch, T. Denzel, S. Habe, D. Winkler, A. Buhler, J. Edelmann, M. Bergmann, G. Hopfinger, M. Hensel, M. Hallek, H. Dohner and S. Stilgenbauer (2010) TP53 mutation and survival in chronic lymphocytic leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 28, 4473-9.
Zhu, Y. M., W. L. Zhao, J. F. Fu, J. Y. Shi, Q. Pan, J. Hu, X. D. Gao, B. Chen, J. M. Li, S. M. Xiong, L. J. Gu, J. Y. Tang, H. Liang, H. Jiang, Y. Q. Xue, Z. X. Shen, Z. Chen and S. J. Chen (2006) NOTCH1 mutations in T-cell acute lymphoblastic leukemia: prognostic significance and implication in multifactorial leukemogenesis. Clinical cancer research : an official journal of the American Association for Cancer Research. 12, 3043-9.
Ziai, J., R. Torres and C. Tormey (2010) Chronic Neutrophilic Leukemia: A Rare and Difficult Diagnosis of Exclusion. Journal of Blood Disorders & Transfusion.

102
Bijlage I: Overzicht positieve stalen
Tabel 37: Overzicht positieve stalen ASXL1-gen
Patiënt Mutatie Staaltype Diagnose Moleculaire testen (Mutaties)
FISH Verwijzing
CI26 0602 c.2110G>A (p.Gly704Arg)
Beenmerg AML EVI1 overexpressie HEVI afwezig FLT3-ITD DNA afwezig NPM1 afwezig CEBPA afwezig
FICSF1R afwezig
Figuur 52
DA13 0417 c.1934dupG (p.Gly646TrpfsX12)
Beenmerg
AML EVI1 geen overexpressie HEVI afwezig FLT3-ITD DNA afwezig NPM1 type A aanwezig CEBPA afwezig
Leuven: geen breukpunten vastgesteld, kleine celfractie met trisomie 3
Figuur 58
CI30 0872 c.1900_1922del (p.Glu635ArgfsX15)
Beenmerg AML EVI1 geen overexpressie HEVI afwezig FLT3-ITD DNA afwezig NPM1 afwezig CEBPA afwezig
/ Figuren 54 en 55
DB03 0771 c.2122C>T (p.Gln708X)
Beenmerg MDS/MPN BCRABL afwezig JAK2 V617F afwezig CALR afwezig MPL afwezig SETBP1 afwezig
/ Figuur 53
CG26 0868 c.1934dupG (p.Gly646TrpfsX12)
Beenmerg MDS/MPN BCRABL afwezig EVI1 geen overexpressie HEVI afwezig FLT3-ITD DNA afwezig NPM1 mutatie afwezig CEBPA mutatie afwezig
/ Figuur 58
CF12 0791 c.1934dupG (p.Gly646TrpfsX12)
Perifeer bloed
AML BCRABL afwezig JAK2 V614F afwezig CALR afwezig HEVI afwezig
/ Figuur 58
BD12 0806 c.1934dupG (p.Gly646TrpfsX12)
Beenmerg MDS/MPN BCRABL afwezig JAK2 V617F afwezig FLT3-ITD DNA afwezig NPM1 afwezig CEBPA aanwezig 40,9% mutant TAD1 & BZIP
/ Figuur 58
BB06 0774 c.1772dupA (p.Tyr591X)
Beenmerg MDS/MPN BCRABL afwezig JAK2 V617F afwezig
/ Figuren 56 en 57
OF04 0388 c.1934dupG (p.Gly646TrpfsX12)
Beenmerg MDS/MPN BCRABL afwezig JAK2 V617F afwezig
/ Figuur 58
9L17 0765 c.1934dupG (p.Gly646TrpfsX12)
Beenmerg MDS/MPN BCRABL afwezig JAK2 V617F afwezig SETBP1 aanwezig
/ Figuur 58
8J13 0599 c.1934dupG (p.Gly646TrpfsX12)
Beenmerg MDS/MPN JAK2V617F afwezig SETBP1 aanwezig
/ Figuur 58
BI13 0650 c.1934dupG (p.Gly646TrpfsX12)
Beenmerg MDS SETBP1 aanwezig FICSF1R afwezig
Figuur 58
9C23 1163 c.1934dupG (p.Gly646TrpfsX12)
Beenmerg Fibrose JAK2 V614F aanwezig / Figuur 58

103
Figuur 52: Patiënt CI26 0602 - c.2110G>A (p.Gly704Arg)
Figuur 53: Patiënt DB03 0771 - c.2122C>T (p.Gln708X)
Figuur 54: Patiënt CI30 0872 - c.1900_1922del (p.Glu635ArgfsX15)
c.1900_1922del
p.Glu635ArgfsX15
Wild type: GAGAGGTCACCACTGCCATAGAGAGGCGGCCACCACTGCCATCGGAGGGGGGGGTGGCCCGGG
Mutant: GAGAGGTCACCACTGCCA TCGGAGGGGGGGGTGGCCCGGG
Figuur 55: Voorstelling c.1900_1922del (p.Glu635ArgfsX15) – patiënt CI30 0872

104
Figuur 56: patient BB06 0774 - c.1772dupA (p.Tyr591X)
Wild type:CCCACTTACCAGATATGCCCCCGGATCATCCCCACCACGGAGTCCTCCTGCCGGGGTTGGACTGG Mutant: CCCACTTAACCAGATATGCCCCCGGATCATCCCCACCACGGAGTCCTCCTGCCGGGGTTGGACTGG
c.1772dupA
p.Tyr591X
Figuur 57: Voorstelling van c.1772dupA (p.Tyr591X) – patiënt BB06 0774
Figuur 58: patiënt 9L17 0765 – c.1934dupG (p.Gly646TrpfsX12)

105
Tabel 38: Overzicht positieve stalen CSF3R-gen
Patiënt Mutatie Staaltype Diagnose Moleculaire testen (Mutaties)
FISH Verwijzing
CC06 0611 c.2260C>T (p.Gln754X)
Beenmerg AML EVI geen overexpressie HEVI afwezig FLT3-ITD DNA afwezig NPM1 mutatie afwezig CEBPA afwezig
/ Figuur 59
AE06 0453 c.2422G>A (p.Glu808Lys) Eiwit-olymorfisme
Beenmerg MDS/MPN BCRABL niet gedetecteerd JAK2 V614F niet gedetecteerd
/ Figuur 60
9E08 0454 c.2422G>A (p.Glu808Lys) Eiwit-polymorfisme
Beenmerg AML FLT3-ITD RNA mutatie aanwezig FIEVI1 aanwezig in 15% van de cellen DNMT3A mutatie aanwezig HEVI niet gedetecteerd NPM niet gedetecteerd
FIEVI 15% van de cellen vertoont een deletie van een fusie-signaal, suggestief voor verlies van 3q materiaal
Figuur 60
Figuur 59: Patiënt CC06 0611 - c.2260C>T (p.Gln754X)
Figuur 60: Patiënt AE06 0453 - c.2422G>A (p.Glu808Lys)
Figuur 61: Patiënt DA14 1034 - c.1544G>T (p.Trp515Leu)
c.1514G>A
(p.Ser505Asn)
c.1502T>C
(p.Val501Ala)
Figuur 62: Patiënt CI30 1693 - c.1514G>A (p.Ser505Asn) en c.1502T>C (p.Val501Ala)

106
Tabel 39: Overzicht positieve stalen MPL-gen
Patiënt Mutatie Staaltype Diagnose Moleculaire testen (Mutaties)
FISH Verwijzing
DA14 1034 c.1544G>T (p.Trp515Leu)
Perifeer bloed
MPN BCRABL afwezig CALR afwezig MPL aanwezig
/ Figuur 61
CI30 1693 c.1514G>A (p.Ser505Asn) en c.1502T>C (p.Val501Ala)
Perifeer bloed
MPN BCRABL afwezig JAK2 V617F zwak aanwezig, niet kwantificeerbaar CALR afwezig MPL aanwezig
/ Figuur 62
CJ22 0569 c.1544G>T (p.Trp515Leu)
Beenmerg MPN BCRABL afwezig JAK2 V617F afwezig CALR afwezig MPL aanwezig
/ Figuur 61
CG29 0705 c.1544G> (p.Trp515Leu)
Perifeer bloed
MPN BCRABL afwezig JAK2 V617F afwezig CALR afwezig MPL aanwezig
/ Figuur 61
CA18 0555 c.1544G>T (p.Trp515Leu)
Beenmerg MDS/MPN EVI1 geen overexpressie MPL aanwezig HEVI afwezig FLT3-ITD DNA afwezig NPM1 afwezig CEBPA afwezig
/ Figuur 61
DA30 0711 c.1544G>T (p.Trp515Leu)
Perifeer bloed
MPN JAK2 V617F afwezig CALR afwezig MPL W515L zwak aanwezig
/ Figuur 61
BA26 0615 c.1514G>A (p.Ser505Asn)
Beenmerg MDS JAK2 V617F zeer zwak aanwezig MPL mutatie s505N aanwezig
Karyotypering Kloon 1: 46,XY,DEL(20)(Q11Q12) (10), niet kwantificeerbaar
Figuur 62
BB03 0653 c.1544G>T (p.Trp515Leu)
Beenmerg MPN JAK2 V617F afwezig MPL W515L aanwezig
/ Figuur 61
AC17 0722 c.1514G>A (p.Ser505Asn)
Beenmerg MPN BCRABL afwezig JAK2 V617F afwezig MPl s505N aanwezig
/ Figuur 62
AC29 0788 c.1514G>A (p.Ser505Asn)
Perifeer bloed
MPN BCRABL afwezig JAK2 V617F afwezig MPL S505N aanwezig
/ Figuur 62
AH24 0586 c.1544G>T (p.Trp515Leu)
Beenmerg MPN JAK2 V617F afwezig MPL W515L mutatie aanwezig
/ Figuur 61
0F21 0450 c.1544G>T (p.Trp515Leu)
Perifeer bloed
MPN BCRABL afwezig JAK2 V617F afwezig MPL mutatie W515L aanwezig
/ Figuur 61
0K25 0255 c.1544G>T (p.Trp515Leu)
Beenmerg MPN BCRABL afwezig JAK2 V617F afwezig MPL mutatie W515L aanwezig
/ Figuur 61
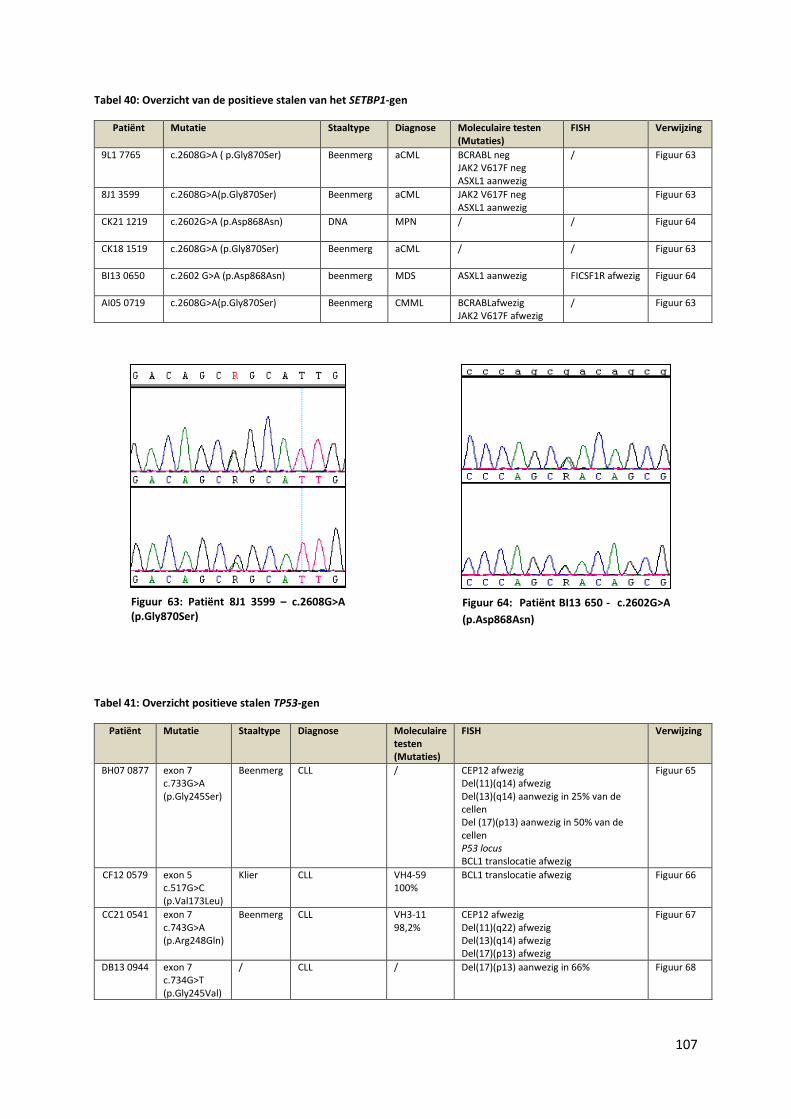
107
Tabel 40: Overzicht van de positieve stalen van het SETBP1-gen
Patiënt Mutatie Staaltype Diagnose Moleculaire testen (Mutaties)
FISH Verwijzing
9L1 7765 c.2608G>A ( p.Gly870Ser) Beenmerg aCML BCRABL neg JAK2 V617F neg ASXL1 aanwezig
/ Figuur 63
8J1 3599 c.2608G>A(p.Gly870Ser) Beenmerg aCML JAK2 V617F neg ASXL1 aanwezig
Figuur 63
CK21 1219 c.2602G>A (p.Asp868Asn) DNA MPN / / Figuur 64
CK18 1519 c.2608G>A (p.Gly870Ser) Beenmerg aCML / / Figuur 63
BI13 0650
c.2602 G>A (p.Asp868Asn) beenmerg MDS ASXL1 aanwezig FICSF1R afwezig Figuur 64
AI05 0719 c.2608G>A(p.Gly870Ser) Beenmerg CMML BCRABLafwezig JAK2 V617F afwezig
/ Figuur 63
Tabel 41: Overzicht positieve stalen TP53-gen
Patiënt Mutatie Staaltype Diagnose Moleculaire testen (Mutaties)
FISH Verwijzing
BH07 0877 exon 7 c.733G>A (p.Gly245Ser)
Beenmerg CLL / CEP12 afwezig Del(11)(q14) afwezig Del(13)(q14) aanwezig in 25% van de cellen Del (17)(p13) aanwezig in 50% van de cellen P53 locus BCL1 translocatie afwezig
Figuur 65
CF12 0579 exon 5 c.517G>C (p.Val173Leu)
Klier CLL VH4-59 100%
BCL1 translocatie afwezig Figuur 66
CC21 0541 exon 7 c.743G>A (p.Arg248Gln)
Beenmerg CLL VH3-11 98,2%
CEP12 afwezig Del(11)(q22) afwezig Del(13)(q14) afwezig Del(17)(p13) afwezig
Figuur 67
DB13 0944 exon 7 c.734G>T (p.Gly245Val)
/ CLL / Del(17)(p13) aanwezig in 66% Figuur 68
Figuur 63: Patiënt 8J1 3599 – c.2608G>A (p.Gly870Ser)
Figuur 64: Patiënt BI13 650 - c.2602G>A
(p.Asp868Asn)

108
Figuur 65: Patiënt BH07 0877 - c.733G>A (p.Gly245Ser)
Figuur 66: Patiënt CF12 0579 – c.517G>C
(p.Val173Leu)
Figuur 67: Patiënt CC21 0541 – c.743G>A
(p.Arg248Gln) Figuur 68: Patiënt DB13 0944 - c.734G>T (p.Gly245Val)

109
Tabel 42: Overzicht positieve stalen NOTCH1-gen
Patiënt Mutatie Staaltype Diagnose Moleculaire testen (Mutaties)
FISH Verwijzing
CA03 0949 c.7544_7545delCT (p.Pro2515ArgfsX4)
Beenmerg CLL VHSEQ VH3-23 100% CEP12 afwezig Del(11)(q22) afwezig Del(13)(q14) aanwezig in 40% Del(17)(p13) afwezig
Figuren 69 en 70
CJ29 0729 c.5094C>T (p.Asp1698Asp) SNP
EN
c.4754T>C (p.Leu1585Pro)
Beenmerg T-ALL HOX11L2 en HOX11 zwak aanwezig, niet kwantificeerbaar HEVI niet gedetecteerd FLT3-ITD DNA niet gedetecteerd NPM1 niet gedetecteerd CEBPA mutatie niet gedetecteerd EVI1 geen overexpressie
/ Figuur 74 Figuur 71
CK07 0664 c.5094C>T (p.Asp1698Asp) SNP
Beenmerg T-ALL
HOX11L2, HOX11 zwak aanwezig, niet kwantificeerbaar HEVI niet gedetecteerd FLT3-ITD RNA niet gedetecteerd
Figuur 74
CK29 0682 c.5094C>T (p.Asp1698Asp) SNP
Beenmerg T-ALL of B-ALL?
HEVI aanwezig BCR/ABL-t(9;22) p190 aanwezig HOX11L2 overexpressie niet gedetecteerd
Kloon 1 : 46,XY,T(9;22)(Q34;Q11) (7) FISH onderzoek bevestigt uw diagnose van B-ALL met t(9;22)BCR-ABL1.
Figuur 74
BF28 0490 c.5094C>T (p.Asp1698Asp) SNP
Beenmerg T-ALL HOX11L2 en HOX11 zijn zwak aanwezig HEVI niet gedetecteerd
/ Figuur 74
OG27 0786 c.5094C>T (p.Asp1698Asp) SNP c.4821G>C (p.Lys1607Asn)
Beenmerg T-ALL HOX 11L2 zwak aanwezig HOX11 aanwezig HEVI niet gedetecteerd
/ Figuur 74 Figuur 72
CE03 0231 c.5094C>T (p.Asp1698Asp) SNP
Beenmerg T-ALL
HOX11L2, HOX11 zwak aanwezig, niet kwantificeerbaar HEVI niet gedetecteerd FLT3-ITD DNA niet gedetecteerd
/ Figuur 74
AA21 0340 c.5094C>T (p.Asp1698Asp)
Beenmerg T-ALL HOX11 zwak aanwezig, niet kwantificeerbaar HOX 11L2 geen overexpressie aanwezig HEVI niet gedetecteerd
/ Figuur 74
CH20 0654 c.4732_4734delGTG (p.Val1578del)
Beenmerg T-ALL HEVI niet gedetecteerd HOX 11L2 geen overexpressie gedetecteerd HOX 11 overexpressie aanwezig
Karyotypering Kloon 1: 47,XY,+MAR (1) FISH:5q35(HOX11L2),7p14(TCRG),7q34(TCRB),9p21(p16,CDKN2A),CEP9,9q34(ABL1),11q23(MLL),14q11(TCRAD), 22q11(BCR). Besluit: T-ALL met een breukpunt in de regio 14q11(TCRAD) en verlies van beide regio's 9p21:recurrente afwijkingen in T-ALL.
Figuur 75
CE07 0710 c.4733T>C (p.Leu1593Pro)
Beenmerg T-ALL HOX11L2 zwak aanwezig, niet kwantificeerbaar HOX11 aanwezig HEVI niet gedetecteerd
/ Figuur 73

110
c.7544_7545delCT
p.Pro2515ArgfsX4
Wild type: AGCACCCCTTCCTCACCCCGTCCCCTGAGTCCCCTGACCAGTGGTCCAGCTCGTCCCC
Mutant: AGCACCCCTTCCTCACCCCGTCCCGAGTCCCCTGACCAGTGGTCCAGCTCGTCCCC
Figuur 70: Voorstelling c.7544_7545delCT (p.Pro2515ArgfsX4) –patiënt CA03 0949
Figuur 69: Patiënt CA03 0949 - c.7544_7545delCT (p.Pro2515ArgfsX4)

111
Figuur 72: Patiënt 0G27 0786 - c.4821G>C (p.Lys1607Asn)
Figuur 71: Patiënt CJ29 0729 - c.4754T>C (p.Leu1585Pro)
Figuur 75: Patiënt CH20 0654- c.4735_4737delGTG (p.Val1579del)
Figuur 73: Patiënt CE07 0710 - c.4733T>C (p.Leu1593Pro)
Figuur 74: Patiënt CE03 0231 - c.5094C>T (p.Asp1698Asp)

112
Tabel 43: Overzicht positieve stalen c-KIT-gen
Opmerking: Bij patiënt CI17 0517 werd naast het gevonden polymorfisme in het AZ Sint-Jan Brugge,
eveneens nog een tweede mutatie gevonden bij analyse door CME Leuven. De mutatie werd slechts
vastgesteld in 0.09% van de cellen, wat meteen ook verklaart waarom dit niet waargenomen werd
tijdens de analyse d.m.v. Sanger sequencing, aangezien de gevoeligheid hier 20% bedraagt.
Patiënt Mutatie Staaltype Diagnose Moleculaire testen (Mutaties)
FISH Verwijzing
CG31 0134 c.2394C>T(p.Ile798Ile) SNP
Perifeer bloed
AML EVI1 overexpressie afwezig HEVI afwezig FLT3-ITD DNA aanwezig NPM1 aanwezig type A CEBPA afwezig
/ Figuur 79
CG23 1140 c.2394C>T(p.Ile798Ile) SNP
Beenmerg AML EVI1 overexpressie afwezig HEVI afwezig FLT3-ITD DNA aanwezig NPM1 afwezig CEBPA afwezig
/ Figuur 79
8L15 0440 c.1255_1257delGAC (p.Asp419del)
Beenmerg AML HEVI type AML1-ETO aanwezig FLT3-ITD RNA afwezig NPM mutatie afwezig QAML1ETO aanwezig QINV16A afwezig QINV16E afwezig
kloon 1: 45,X,-Y,del(7)(p12p13 of p14p15), t(8;21)(q22;q22) (11)
Figuren 76 en 78
BA25 1050 c.2466T>G (p.Asn822Lys)
Beenmerg AML AML1/ETO-t(8;21) aanwezig EVI1 geen overexpressie FLT3-ITD DNA afwezig NPM1 mutatie afwezig CEBPA mutatie afwezig
Karyotypering 45,X,-Y,t(8;21)(q22;q22) (7)
Figuur 77
AB22 0391 c.2394C>T (p.Ile798Ile) SNP
Beenmerg Mastocytose CKIT afwijkende sequentie in exon 10 (codon 541) en exon 17 (codon 798); het betreft polymorfismen. uitgevoerd in CME Leuven BCRABL afwezig
/ Figuur 79
0L30 0418 c.2447A>T (p.Asp816Val)
Beenmerg Mastocytose Mutatie aanwezig in codon 816 van exon 17 van cKIT uitgevoerd in CME BCRABL afwezig FIP1L1 afwezig JAK2 V617F afwezig
/ Figuur 78
CI17 0517 c.2394C>T (p.Ile798Ile) SNP
Beenmerg Mastocytose Er werd een KIT p.D816V mutatie vastgesteld in 0.09 % van de cellen in dit staal. CME Leuven BCRABL afwezig
/ Figuur 79

113
Figuur 76: Patiënt 8L15 0440 - c.1255_1257delGAC (p.Asp419del)
Figuur 77: Patiënt BA25 1050 - p.Asn822Lys of c.2466T>G
Figuur 79: Patiënt 0L30 0418 - p.Asp816Val of c.2447A>T
Figuur 80: Patiënt CI17 0517 - c.2394 C>T (p.Ile798Ile)
c.1255_1257delGAC
p.Asp419del
Wild type: GAAATCCTGACTTACGACAGGCTCGTGAATGGCATGCTCCAATGTGTGGCA
Mutant: GAAATCCTGACTTACAGGCTCGTGAATGGCATGCTCCAATGTGTGGCA
Figuur 78: Voorstelling c.1255_1257delGAC (p.Asp419del) – patiënt 8L15 0440

114
Tabel 44: Overzicht positieve stalen DNMT3A-gen
Patiënt Mutatie Staaltype Diagnose Moleculaire testen (Mutaties) FISH Verwijzing
CL02 0676 c.2645G>A (p.Arg882His)
Beenmerg AML EVI1 overexpressie afwezig HEVI afwezig FLT3-ITD DNA aanwezig NPM1 mutatie aanwezig NPM1 mutatie type A aanwezig CEBPA afwezig
/ Figuur 81
CC27 1152 c.2644C>T (p.Arg882Cys)
Beenmerg AML EVI1 overexpressie afwezig HEVI afwezig FLT3-ITD DNA afwezig NPM1 mutatie afwezig CEBPA afwezig Chimerisme 26,8% donor
/ Figuur 82
CD17 1140 c.2645G>A (p.Arg882His)
Beenmerg AML EVI1 overexpressie afwezig HEVI afwezig FLT3-ITD DNA afwezig NPM1 afwezig CEBPA afwezig
/ Figuur 81
9D27 0574 c.2645G>A (p.Arg882His)
Beenmerg AML HEVI afwezig FLT3-ITD DNA afwezig NPM1 afwezig
/ Figuur 81
CF18 1132 c.2645G>A (p.Arg882His)
Beenmerg AML EVI1 overexpressie afwezig HEVI afwezig FLT3-ITD afwezig NPM1 aanwezig nieuw type CEBPA afwezig QNPMA afwezig
/ Figuur 81
CG23 0588 c.2645G>A (p.Arg882His)
Beenmerg AML EVI1 overexpressie afwezig HEVI afwezig FLT3-ITD DNA aanwezig NPM1 aanwezig CEBPA afwezig
/ geen structurele noch numerische afwijkingen aan van de regio 11q23, noch van de regio's 3q21 en 3q26.
Figuur 81
9E08 0454 c.2645G>A (p.Arg882His)
Beenmerg AML FLT3-ITD RNA mutatie aanwezig HEVI niet gedetecteerd NPM mutatie niet gedetecteerd
FIEVI1 aanwezig in 15% van de cellen
Figuur 81
Figuur 81: Patiënt CL02 0676 - c.2645G>A (p.Arg882His)
Figuur 82: Patiënt CC27 1152 - c.2644C>T (p.Arg882Cys)

115
Tabel 45: Overzicht positieve stalen IDH1- en IDH2-gen
Patiënt Mutatie Staaltype Diagnose Moleculaire testen (Mutaties)
FISH Verwijzing
0I30 447 IDH1: c.394C>T (p.Arg132Cys) c.315C>T (p.Gly106Gly)
Beenmerg AML HEVI afwezig FLT3-ITD RNA afwezig NPM1 mutatie afwezig CEBPA afwezig QNMPA afwezig
/ Figuur 83
0G29 429 IDH2: c.419G>A (p.Arg140Gln)
Beenmerg AML/RAEB(t) HEVI afwezig FLT3-ITD RNA afwezig NPM1 mutatie afwezig QNPMa afwezig
/ Figuur 84
Tabel 46: Overzicht positieve stalen STAT3-gen
Patiënt Mutatie Staaltype Diagnose Moleculaire testen (Mutaties)
FISH Verwijzing
CB11 0545 c.1919A>T (p.Tyr640Phe) Beenmerg T-LGL TCR monoclonaal / Figuur 85
BD20 0766 c.1919A>T (p.Tyr640Phe) Beenmerg T-LGL TCR monoclonaal / Figuur 85
AJ24 0758 c.1919A>T (p.Tyr640Phe) Beenmerg T-LGL TCR monoclonaal / Figuur 85
BE03 0656 c.1981G>T (p.Asp661Tyr) Beenmerg T-LGL TCR monoclonaal / Figuur 86
Figuur 83: Patiënt OI30 0447 - c.394C>T (p.Arg132Cys)
Figuur 84: Patiënt 0G29 0429 - c.419G>A (p.Arg140Gln)
Figuur 85 : Patiënt AJ24 0758 - c.1919A>T (p.Tyr640Phe)
Figuur 86: Patiënt BE03 0656 - c.1981G>T (p.Asp661Tyr)

116
Tabel 47: Overzicht positieve stalen MYD88-gen
Patiënt Mutatie Staaltype Diagnose Moleculaire testen (Mutaties)
FISH Verwijzing
BF21 0608 c.794T>C (p.Leu265Pro) Beenmerg Waldenström IGHFR3, IGHKA, IGKB, IGK, IGH monoclonaal
/ Figuur 87
BD25 1062 c.794T>C (p.Leu265Pro) Beenmerg Waldenström IGHFR1 polyclonaal / Figuur 87
Figuur 87: Patiënt BD25 1062 - c.794T>C (p.Leu265Pro)
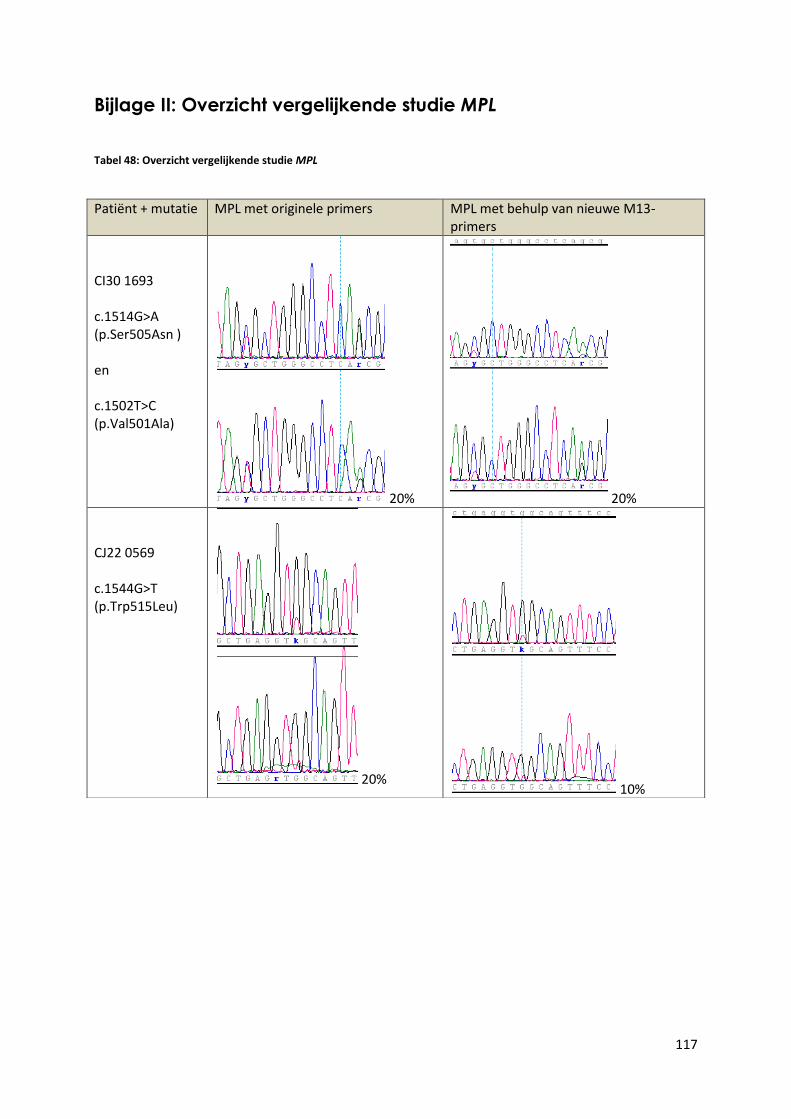
117
Bijlage II: Overzicht vergelijkende studie MPL
Tabel 48: Overzicht vergelijkende studie MPL
Patiënt + mutatie MPL met originele primers MPL met behulp van nieuwe M13-primers
CI30 1693 c.1514G>A (p.Ser505Asn ) en c.1502T>C (p.Val501Ala)
20% 20%
CJ22 0569 c.1544G>T (p.Trp515Leu)
20% 10%

118
CG29 0705 c.1544G>T (p.Trp515Leu)
20% 10%
CA18 0555 c.1544G>T (p.Trp515Leu)
100% 100%
DA14 1034 c.1544G>T (p.Trp515Leu)
20% 20%

119
DA30 0711 c.1544G>T (p.Trp515Leu)
10% <1%
BA26 0615 c.1514G>A (p.Ser505Asn)
20% 20%
BB03 0653 c.1544G>T (p.Trp515Leu)
20% 10%

120
AC17 0722 c.1514G>A (p.Ser505Asn)
20% 20%
AC29 0788 c.1514G>A (p.Ser505Asn)
20% 15%
AH24 0586 c.1544G>T (p.Trp515Leu)
100% 100%

121
0F21 0450 c.1544G>T (p.Trp515Leu)
Resultaten verloren door computercrash
20%
0K25 0255 c.1544G>T (p.Trp515Leu)
Resultaten verloren door computercrash
100%

122
Bijlage III: Overzicht alle stalen Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
BD20 919 MDS/ MPN
aCML SETBP1 WT
BD12 806 MDS/ MPN
aCML aCML/CMML-2
SETBP1 WT
BB06 774 MDS/ MPN
aCML SETBP1 WT
0F04 388 MDS/ MPN
aCML SETBP1 WT
9L17 0765 MDS/ MPN
aCML SETBP1 MUT
8J13 0599 MDS/ MPN
aCML SETBP1 MUT
CK06 0551 MDS/ MPN
aCML SETBP1 WT
CK21 1219 MPN MPN SETBP1 MUT
CK18 1519 MDS/ MPN
aCML SETBP1 MUT
DA21 1065 MDS/ MPN
aCML SETBP1 WT
DA02 0599 MPN CML SETBP1 WT
CC25 0713 MDS/ MPN
CMML CMML-1
SETBP1 WT
BI13 0650 MDS RCMD SETBP1 MUT
CB20 1255 MPN MPN SETBP1 WT
AI05 0719 MDS/ MPN
CMML CMML-1
SETBP1 MUT
DB19 592 MDS/ MPN
aCML SETBP1 WT
DB11 703 MDS/ MPN
aCML SETBP1 WT
DB14 584 MPN MPN SETBP1 WT
BK22 914 MDS/ MPN
aCML CSF3R WT
CG26 868 MDS/ MPN
aCML aCML/RAEBII
CSF3R WT
CF12 791 AML AML-? CSF3R WT
0L22 1010 MDS/ MPN
aCML aCML/PMF
CSF3R WT
BD20 919 MDS/ MPN
aCML CSF3R WT
BD12 806 MDS/ MPN
aCML aCML/CMML-2
CSF3R WT
BB06 774 MDS/ MPN
aCML CSF3R WT
OF 04388 MDS/ MPN
aCML CSF3R WT
9L1 7765 MDS/ MPN
aCML CSF3R WT
8J1 3599 MDS/ MPN
aCML CSF3R WT
CK06551 MDS/ MPN
aCML CSF3R WT
CK21 1219 MPN MPN CSF3R WT
CK18 1519 MDS/ MPN
aCML CSF3R WT
DA21 1065 MDS/ MPN
aCML CSF3R WT
DA02 599 MPN CML CSF3R WT
Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
CF241160 CLL Typisch TP53 WT
CB22676 CLL Typisch TP53 WT
BH07877 CLL Typisch TP53 MUT
CJ071113 CLL Typisch TP53 WT
CJ07792 CLL Typisch cat 3/5
TP53 WT
CJ07774 CLL Typisch TP53 WT
CJ03675 CLL Typisch cat 5/5
TP53 WT
CI24578 CLL Typisch cat 5/5
TP53 WT
CI161108 CLL Typisch cat 4,5/5
TP53 WT
CI111117 CLL Typisch cat 4/5
TP53 WT
CI10995 CLL Typisch cat 4/5
TP53 WT
CI10388 CLL Typisch cat 4,5/5
TP53 WT
CI03666 CLL Typisch TP53 WT
CH261179 CLL Typisch cat 2/5
TP53 WT
CH191020 CLL Typisch cat 5/5
TP53 WT
CH08394 CLL Typisch TP53 WT
CH01579 CLL Typisch TP53 WT
CG151005 CLL Typisch TP53 WT
CF181031 CLL Typisch cat 5/5
TP53 WT
CF12579 CLL Typisch TP53 MUT
CF101104 CLL Typisch TP53 WT
CE281263 CLL Typisch TP53 WT
CE15585 CLL Atypisch cat 3,5/5
TP53 WT
CE07658 CLL Typisch cat 3/5
TP53 WT
CC18669 CLL Typisch TP53 WT
CD22878 CLL Typisch cat 5/5
TP53 WT
CC21541 CLL Typisch TP53 MUT
DB13944 CLL Typisch TP53 MUT
DB251304 CLL Typisch TP53 WT
CL03699 CLL Typisch TP53 WT
CI10388 CLL Typisch cat 4,5/5
TP53 WT
CK19822 CLL Typisch cat 5/5
TP53 WT
BK22 914 MDS/ MPN
aCML SETBP1 WT
CG26 868 MDS/ MPN
aCML aCML/RAEBII
SETBP1 WT
0L22 1010 MDS/ MPN
aCML aCML/PMF
SETBP1 WT
Tabel 49: Overzicht alle geteste stalen

123
Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
DA20 743 MDS/ MPN
aCML na alloSCT
CSF3R WT
CC25 713 MDS/ MPN
CMML CMML-1
CSF3R WT
BI13 650 MDS RCMD CSF3R WT
CB20 1255 MPN MPN CSF3R WT
DB19 592 MDS/ MPN
aCML CSF3R WT
DB03 771 CLL Typisch cat 5/5
CSF3R WT
DB04 856 Andere LPL evolutie naar MDS
CSF3R WT
DB11 703 MDS/ MPN
aCML CSF3R WT
DB14 584 MPN MPN CSF3R WT
CH010838 AML AML-? CSF3R WT
CB111115 AML AML-? CSF3R WT
CB271191 AML AML-? CSF3R WT
CC060611 AML AML-? CSF3R MUT
CC07 0681 AML AML-? CSF3R WT
CC27 0693 AML AML-? CSF3R WT
CA17 0770 MDS/ MPN
CMML CMML-1
CSF3R WT
BB14 0933 Andere normaal CSF3R WT
BI11 0914 MDS/ MPN
CMML CMML-1
CSF3R WT
BL04 1183 MDS/ MPN
CMML CMML-2
CSF3R WT
AA24 0888 MDS/ MPN
CMML CSF3R WT
AB01 0400 MDS/ MPN
aCML CSF3R WT
AE06 0453 MDS/ MPN
CMML CSF3R SNP
CB18 0775 MDS/ MPN
CMML CMML-1
CSF3R WT
CI30 0872 AML AML-? met MDS
CSF3R WT
OK22 1128 AML AML-? AML M5 of CMML
CSF3R WT
CK14 0552 MDS/ MPN
CMML CMML-1
CSF3R WT
OD07 0693 MDS/ MPN
CMML CSF3R WT
Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
OJ18 0518 MDS/ MPN
MDS/ MPN
CSF3R WT
9G16 0412 MDS/ MPN
CMML CSF3R WT
BB06 0774 MDS/ MPN
aCML CSF3R WT
AH09 0799 MDS/ MPN
aCML aCML of CMML
CSF3R WT
9L17 0765 MDS/ MPN
aCML CSF3R WT
CE02 0302 AML AML-M5 CSF3R WT
CA16 0697 AML AML-? CSF3R WT
CG31 0134 AML AML-? CSF3R WT
CA14 1253 AML AML-M2 CSF3R WT
CA22 1193 AML AML-? sec AML uit MDS
CSF3R WT
CA18 0555 AML Andere uit MDS
CSF3R WT
CA15 1035 AML Andere met MDS; M2
CSF3R WT
BD16 0598 AML Andere AML/AEL
CSF3R WT
9E08 0454 AML AML-M0 CSF3R SNP
BH13 1100 AML AML-? CSF3R WT
CC27 1152 AML Recidief na alloSCT
CSF3R WT
BL03 0654 AML AML-M1 CSF3R WT
8J29 0696 AML AML-M1 /MDS CSF3R WT
CD26 0417 AML MDS met MDS related changes
CSF3R WT
AG22 0714 AML AML-M5 CSF3R WT
CD17 0622 AML AML-? CSF3R WT
CD17 1140 AML MDS met MDS related changes
CSF3R WT
CE07 0655 MDS RAEBII CSF3R WT
CE10 0643 AML AML-? CSF3R WT
9D27 0574 AML AML-M2 CSF3R WT
BJ04 868 AML AML-NOS CSF3R WT
CF13 0691 AML Andere MDS/AML overgang naar RAEBII
CSF3R WT

124
Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
CF18 1132 AML MDS met MDS related changes
CSF3R WT
CG15 0603 AML MDS met MDS related changes
CSF3R WT
AI05 719 MDS/ MPN
CMML CMML-1
CSF3R WT
CG16 1112 AML AML-M2 CSF3R WT
CG23 0588 AML AML-? CSF3R WT
CD11 0527 MPN Andere MPN CML/CMML?
CSF3R WT
CG23 1140 AML MDS met MDS related changes (CMML)
CSF3R WT
0I02 549 NHL(T) T-LGL STAT3 WT
CB11 545 NHL(T) T-LGL STAT3 MUT
CG26 603 NHL(T) T-LGL STAT3 WT
BD20 766 NHL(T) T-LGL STAT3 MUT
AC14 747 NHL(T) T-LGL STAT3 WT
AJ24 758 NHL(T) T-LGL STAT3 MUT
BE03 656 NHL(T) T-LGL STAT3 MUT
CL30 789 Andere NK-cel leukemie?
STAT3 WT
OK05 569 AML AML-? IDH1/2 WT
0I30 447 AML AML-? IDH1/2 MUT
0G29 429 AML AML-? IDH1/2 MUT
J26 125 AML Andere MDS of AML
IDH1/2 WT
F23 480 AML AML-? IDH1/2 WT
DB26 0633 AML AML-? IDH1/2 WT
DA13 417 AML MDS met MDS related changes (CMML)
DNMT3A
WT
DA14 597 AML MDS met MDS related changes
DNMT3A
WT
DA07 817 MDS RAEBII DNMT3A
WT
Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
DA13 790 AML MDS met MDS related changes
DNMT3A
WT
DA15 1102 MDS RAEBII DNMT3A
WT
DA20 1338 AML AML-? DNMT3A
WT
DA21 798 AML MDS met MDS related changes
DNMT3A
WT
CL02 979 MPN MPN DNMT3A
WT
CL04 941 MPN RAEBII DNMT3A
WT
CL13 727 MPN MPN DNMT3A
WT
DA02 522 MPN MPN DNMT3A
WT
DA09 616 MPN MPN DNMT3A
WT
DA09 1051 MPN MPN DNMT3A
WT
DA14 1032 MPN MPN DNMT3A
WT
DA31 666 MPN MPN DNMT3A
WT
DB03 771 CLL Typisch cat 5/5, MBL
DNMT3A
WT
DB06 566 MPN MPN DNMT3A
WT
CL03 469 MPN MPN DNMT3A
WT
CL05 788 MPN MPN DNMT3A
WT
CL11 524 MPN MPN DNMT3A
WT
CL11 801 MDS/ MPN
CMML DNMT3A
WT
8K26 0904 Fibrose Fibrose DNMT3A
WT
CL17 752 MPN MPN DNMT3A
WT
9C23 1163 Fibrose Fibrose DNMT3A
WT
CL19 598 MPN MPN DNMT3A
WT
CL19 799 MPN MPN DNMT3A
WT
CL20 664 MPN MPN DNMT3A
WT
CL23 608 MPN MPN DNMT3A
WT
CL24 234 MPN MPN DNMT3A
WT
CL27 678 MPN MPN DNMT3A
WT
CL27 708 MPN MPN DNMT3A
WT
CH16 0692 AML MDS MDS related changes
DNMT3A
WT

125
Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
CH21 0500 AML andere hypogranulaire variant
DNMT3A
WT
CH22 0578 AML MDS MDS related changes
DNMT3A
WT
CI25 0770 AML MDS MDS related changes
DNMT3A
WT
CI26 0602 AML MDS MDS related changes
DNMT3A
WT
CI30 0872 AML AML-M5a CMML met evolutie naar AML met MDS gerelateerde veranderingen
DNMT3A
WT
CI30 0199 AML AML-M3 DNMT3A
WT
AG28 0472 andere B-NHL cat 2,5/5
DNMT3A
WT
CJ11 1004 AML Andere AML/APL
DNMT3A
WT
CK06 1099 AML MDS MDS-related changes
DNMT3A
WT
CK14 0614 cALL recidief evolutie naar MDS
DNMT3A
WT
CK22 0377 AML AML-? DNMT3A
WT
CL02 0676 AML AML-? DNMT3A
MUT
DA13 0417 AML MDS MDS related changes (CMML)
DNMT3A
WT
DA13 0790 AML MDS MDS-related changes
DNMT3A
WT
DA20 1338 AML AML-? DNMT3A
WT
DA30 0857 AML AML-? DNMT3A
WT
DB26 0633 AML AML-? DNMT3A
WT
Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
CH01 0838 AML AML-? DNMT3A
WT
CB11 1115 AML AML-? DNMT3A
WT
CB27 1191 AML AML-? DNMT3A
WT
CC06 0611 AML AML-? DNMT3A
WT
CC07 0681 AML AML-? DNMT3A
WT
CE02 0302 AML AML-M5 DNMT3A
WT
CA16 0697 AML AML-? DNMT3A
WT
CA14 1253 AML AML-M2 DNMT3A
WT
CA22 1193 AML AML-? of sec AML uit MDS
DNMT3A
WT
CA18 0555 AML AML-? MDS DNMT3A
WT
CA15 1035 AML AML-M2 met MDS
DNMT3A
WT
BD16 0598 AML Andere AML/AEL
DNMT3A
WT
9E08 0454 AML AML-M0 DNMT3A
MUT
CC27 1152 AML recidief na alloSCT
DNMT3A
MUT
BL03 0654 AML AML-M1 DNMT3A
WT
8J29 0696 AML Andere AML M1/MDS
DNMT3A
WT
CD26 0417 AML MDS MDS related changes
DNMT3A
WT
AG22 0714 AML AML-M5 DNMT3A
WT
CD170622 AML AML-? DNMT3A
WT
CD17 1140 AML MDS met MDS related changes
DNMT3A
MUT
CE07 0655 MDS RAEBII DNMT3A
WT
AI05 0719 MDS/MPN
CMML CMML-1
DNMT3A
WT
CG31 0134 AML AML-? DNMT3A
WT
CC27 0693 AML AML-? DNMT3A
WT
CE10 0643 AML AML-? DNMT3A
WT
9D27 0574 AML AML-M2 DNMT3A
MUT
BJ04 868 AML AML-NOS DNMT3A
WT

126
Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
CF13 0691 AML Andere MDS/AML overgang naar RAEBII
DNMT3A
WT
CF18 1132 AML MDS MDS related changes
DNMT3A
MUT
BH13 1100 AML AML-? DNMT3A
WT
CG15 0603 AML MDS MDS related changes
DNMT3A
WT
CG16 1112 AML MDS MDS related changes
DNMT3A
WT
CG23 0588 AML AML-? DNMT3A
MUT
CG23 1140 AML MDS MDS related changes (CMML)
DNMT3A
WT
DA02 0443 CLL Atypisch cat 2,5/5
NOTCH1
WT
CI02 1264 CLL Typisch cat 5/5
NOTCH1
WT
CI03 0666 CLL Typisch cat 5/5
NOTCH1
WT
CI10 0995 CLL Typisch cat 4/5
NOTCH1
WT
CI11 1117 CLL Typisch cat 4/5
NOTCH1
WT
CI16 1108 CLL Typisch cat 4,5/5
NOTCH1
WT
CH20 0654 ALL T-ALL NOTCH1
MUT
CI17 0760 ALL T-ALL NOTCH1
WT
CL19 0620 CLL Typisch cat 5/5
NOTCH1
WT
DB04 0858 CLL Typisch cat 4/5
NOTCH1
WT
DA27 0708 CLL Typisch cat 5/5
NOTCH1
WT
CJ29 0729 ALL T-ALL NOTCH1
MUT
CJ22 1279 CLL Typisch cat 4/5
NOTCH1
WT
CL03 699 CLL Typisch NOTCH1
WT
CG15 1005 CLL Typisch NOTCH1
WT
CI10 388 CLL Typisch cat 4,5/5
NOTCH1
WT
CI24 578 CLL Typisch cat 5/5
NOTCH1
WT
CJ03 675 CLL Typisch cat 5/5
NOTCH1
WT
CJ07 774 CLL Typisch NOTCH1
WT
Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
CJ07 1113 CLL Typisch NOTCH1
WT
CK19 822 CLL Typisch cat 5/5
NOTCH1
WT
CF10 1104 CLL Typisch NOTCH1
WT
CK07 0664 ALL T-ALL NOTCH1
SNP
CK29 0682 ALL T-ALL of B-ALL?
NOTCH1
SNP
CK25 0884 CLL Typisch cat 5/5
NOTCH1
WT
CE07 0710 ALL T-ALL NOTCH1
MUT
CE03 0231 ALL T-ALL NOTCH1
SNP
CH22 0489 CLL Typisch NOTCH1
WT
DB12 0670 CLL Typisch NOTCH1
WT
BF28 0490 ALL T-ALL NOTCH1
SNP
OG27 0786 ALL T-ALL NOTCH1
MUT
AA21 0340 ALL T-ALL NOTCH1
SNP
CA03 0949 CLL Typisch cat 5/5
NOTCH1
MUT
CA07 1413 CLL Typisch NOTCH1
WT
CA17 1011 CLL Typisch NOTCH1
WT
CA31 0756 CLL Typisch cat 5/5
NOTCH1
WT
CB05 1127 CLL Typisch NOTCH1
WT
CC04 0663 CLL Typisch NOTCH1
WT
CC07 0993 CLL Typisch cat 4/5
NOTCH1
WT
CC14 0776 CLL Typisch cat 3/5
NOTCH1
WT
CC21 0541 CLL Typisch NOTCH1
WT
CC21 1079 CLL Typisch NOTCH1
WT
CH01 0579 CLL Typisch cat 4,5/5
NOTCH1
WT
DA02 0747 CLL Typisch NOTCH1
WT
8K20 0370 MCL MCL MCL NOTCH1
WT
9C06 0846 MCL MCL MCL NOTCH1
WT
0A22 0634 MCL MCL MCL NOTCH1
WT
CG25 0979 MCL MCL MCL NOTCH1
WT
9L21 0680 MCL MCL MCL NOTCH1
WT
0G05 0609 MCL MCL MCL NOTCH1
WT
0J25 0842 MCL MCL MCL NOTCH1
WT
0L02 0464 MCL MCL MCL NOTCH1
WT
0F24 0745 MCL MCL MCL NOTCH1 WT

127
Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
CJ08 0746 MCL MCL MCL NOTCH1
WT
9A15 0491 MCL MCL MCL NOTCH1
WT
CJ29 0645 MCL MCL MCL NOTCH1
WT
0L22 0644 MCL MCL MCL NOTCH1
WT
CL03 699 CLL Typisch ASXL1 WT
CG15 1005 CLL Typisch ASXL1 WT
CI10 388 CLL Typisch cat 4,5/5
ASXL1 WT
CI24 578 CLL Typisch cat 5/5
ASXL1 WT
CI03 675 CLL Typisch cat 5/5
ASXL1 WT
CJ07 774 CLL Typisch ASXL1 WT
CJ07 1113 CLL Typisch ASXL1 WT
CK19 822 CLL Typisch cat 5/5
ASXL1 WT
CF10 1104 CLL Typisch ASXL1 WT
AG28 0472 B-NHL cat 2,5/5 ASXL1 WT
BE23 0979 MDS MDS ASXL1 WT
BD19 0550 MDS RAEBI ASXL1 WT
BJ31 0997 AML Andere AML/ALL/MDS
ASXL1 WT
CD11 0714 AML Andere AML/RAEB
ASXL1 WT
CF12 0663 MDS MDS ASXL1 WT
CJ11 1004 AML AML-M5 PML-RARA
ASXL1 WT
CI26 0602 AML MDS MDS related changes
ASXL1 SNP
CK22 0377 AML AML-? ASXL1 WT
CK14 0614 cALL recidief evolutie naar MDS
ASXL1 WT
CK06 1099 AML MDS MDS related changes
ASXL1 WT
CL02 0676 AML AML-? ASXL1 WT
DA13 0790 AML MDS MDS related changes
ASXL1 WT
DA13 0417 AML MDS MDS related changes (CMML)
ASXL1 MUT
DA20 1338 AML AML-? ASXL1 WT
DA30 0857 AML AML-? ASXL1 WT
Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
DA30 1270 AML Andere MDS of AML
ASXL1 WT
DA30 0739 MDS/MPN
CMML CMML-1
ASXL1 WT
DB03 0671 MDS/MPN
CMML CMML-1
ASXL1 WT
DA07 0817 MDS RAEBII ASXL1 WT
CC07 0681 MDS RAEBI /RAEBII
ASXL1 WT
CC21 0966 AML MDS MDS related changes
ASXL1 WT
CH16 0692 AML MDS MDS related changes
ASXL1 WT
CH21 0500 AML andere hypogranulaire variant
ASXL1 WT
CH22 0578 AML MDS MDS related changes
ASXL1 WT
CI25 0770 AML MDS MDS related changes
ASXL1 WT
CI30 0872 AML Andere AML M5a, CMML met evolutie naar AML met MDS gerelateerde veranderingen
ASXL1 MUTANT
CI30 0199 AML AML-M3 ASXL1 WT
CL02 0579 MPN MPN ASXL1 WT
CL04 0941 MDS RAEBII ASXL1 WT
CL03 0469 MPN MPN ASXL1 WT
DA02 0522 MPN MPN ASXL1 WT
DA09 0616 MPN MPN ASXL1 WT
DA09 1051 MPN MPN ASXL1 WT
8K26 0904 Fibrose Fibrose ASXL1 WT
CL17 0752 MPN MPN ASXL1 WT
9C23 1163 Fibrose Fibrose ASXL1 MUT
128
Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
CL19 0598 MPN MPN ASXL1 WT
CL19 0799 MPN MPN ASXL1 WT
CL20 0664 MPN MPN ASXL1 WT
CL23 0608 MPN MPN ASXL1 WT
DA14 1032 MPN MPN ASXL1 WT
CL11 0524 MPN MPN ASXL1 WT
CL11 0801 MDS/ MPN
CMML ASXL1 WT
CL05 0788 MPN MPN ASXL1 WT
CL13 0727 MPN MPN ASXL1 WT
DA31 0666 MPN MPN ASXL1 WT
DB03 0771 MDS/ MPN
ASXL1 MUT
DB06 0566 MPN MPN ASXL1 WT
CL24 0234 MPN MPN ASXL1 WT
CL27 0678 MPN MPN ASXL1 WT
CL27 0708 MPN MPN ASXL1 WT
BK22 0914 MDS/ MPN
aCML ASXL1 WT
OL22 1010 MDS/ MPN
aCML aCML/PMF
ASXL1 WT
BD20 0919 MDS/ MPN
aCML ASXL1 WT
CK06 0551 MDS/ MPN
aCML ASXL1 WT
CK21 1219 MPN MPN ASXL1 WT
CK18 1519 MDS/ MPN
aCML ASXL1 WT
CG26 0868 MDS/ MPN
aCML aCML, RAEBII
ASXL1 MUT
CF12 0791 AML AML-? ASXL1 MUT
BD12 0806 MDS/ MPN
aCML aCML/ CMML-2
ASXL1 MUT
BB06 0774 MDS/ MPN
aCML ASXL1 MUT
OF04 0388 MDS/ MPN
aCML ASXL1 MUT
9L17 0765 MDS/ MPN
aCML ASXL1 MUT
8J13 0599 MDS/ MPN
aCML ASXL1 MUT
CC25 0713 MDS/ MPN
CMML CMML-1
ASXL1 WT
BI13 0650 MDS RCMD ASXL1 MUT
CB20 1255 MPN MPN ASXL1 WT
AI05 0719 MDS/ MPN
CMML CMML-1
ASXL1 WT
9E08 0454 AML AML-M0 ASXL1 WT
CK05 1152 AML AML-M2 C-KIT WT
CH01 0838 AML AML-? C-KIT WT
CB11 1115 AML AML-? C-KIT WT
Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
CB27 1191 AML AML-? C-KIT WT
CC06 0611 AML AML-? C-KIT WT
CC07 0681 AML AML-? C-KIT WT
CE02 0302 AML AML-M5 C-KIT WT
CA16 0697 AML AML-? C-KIT WT
CA14 1253 AML AML-M2 C-KIT WT
CA22 1193 AML AML-? of sec AML uit MDS
C-KIT WT
CA18 0555 AML MDS uit MDS
C-KIT WT
CA15 1035 AML AML-M2 met MDS
C-KIT WT
BD16 0598 AML Andere AML/AEL
C-KIT WT
9E08 0454 AML AML-M0 C-KIT WT
CC27 1152 AML recidief na alloSCT
C-KIT WT
BL03 0654 AML AML-M1 C-KIT WT
8J29 0696 AML AML-M1 AML M1/MDS
C-KIT WT
CD26 0417 AML MDS MDS related changes
C-KIT WT
AG22 0714 AML AML-M5 C-KIT WT
CD170622 AML AML-? C-KIT WT
CD17 1140 AML MDS MDS related changes
C-KIT WT
CE07 0655 MDS RAEBII C-KIT WT
AI05 0719 MDS/MPN
CMML CMML-1
C-KIT WT
CG31 0134 AML AML-? C-KIT SNP
CC27 0693 AML AML-? C-KIT WT
CE10 0643 AML AML-? C-KIT WT
9D27 0574 AML AML-M2 C-KIT WT
BJ04 868 AML AML-NOS C-KIT WT
CF13 0691 AML Andere MDS/AML overgang naar RAEBII
C-KIT WT
CF18 1132 AML MDS MDS related changes
C-KIT WT
BH13 1100 AML AML-? C-KIT WT

129
Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
CG15 0603 AML MDS MDS related changes
C-KIT WT
CG16 1112 AML MDS MDS related changes
C-KIT WT
CG23 0588 AML AML-? C-KIT WT
CG23 1140 AML MDS MDS related changes (CMML)
C-KIT SNP
DA20 1338 AML AML-? C-KIT WT
AE12 0388 AML AML-? in remissie
C-KIT WT
BE18 0676 AML MDS MDS related changes
C-KIT WT
BA25 1050 AML AML-? C-KIT MUT
8L15 0440 AML AML-M2 C-KIT MUT
AG22 0714 AML AML-M5 C-KIT WT
CD17 1140 AML MDS MDS related changes
C-KIT WT
DB27 0904 Mastocytose
mastocytose C-KIT WT
AB22 0391 Mastocytose
mastocytose C-KIT SNP
0L30 0418 Mastocytose
mastocytose C-KIT MUT
9G16 0610 Andere Normaal, mastocytose?
C-KIT WT
CI17 0517 Mastocytose
mastocytose C-KIT SNP
CJ31 0746 Mastocytose
mastocytose C-KIT WT
CL05 0980 Mastocytose
mastocytose C-KIT WT
DA14 1034 MPN MPN MPL MUT
DA14 0238 MPN MPN MPL WT
DA09 1053 MPN MPN MPL WT
CI30 1693 MPN MPN MPL MUT
CJ14 0491 MDS LPL MPL WT
CJ22 0569 MPN MPN MPL MUT
CH01 0838 MPN PMF AML MPL WT
CG29 0705 MPN MPN MPL MUT
CA18 0555 MDS/MPN
AML uit MDS MPL MUT
CA24 0825 MPN MPN MPL WT
Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
DA30 0711 MPN MPN MPL MUT
BA26 0615 MDS RAEBII MPL MUT
BB03 0653 MPN MPN MPL MUT
AC17 0722 MPN MPN MPL MUT
AC29 0788 MPN MPN MPL MUT
AH24 0586 MPN MPN MPL MUT
0F21 0450 MPN MPN MPL MUT
0K25 0255 MPN MPN MPL MUT
CH26 1215 Normaal beenmerg
MYD88 WT
CF13 0608 LPL LPL 8% van de cellen
MYD88 WT
BL13 0615 LPL LPL MYD88 WT
BL04 1296 LPL LPL MYD88 WT
BI19 0930 LPL LPL lymfoominfiltratie
MYD88 WT
BH16 0861 LPL LPL MYD88 WT
BF28 0858 LPL MYD88 WT
BF21 0608 LPL LPL MYD88 MUT
BD25 1062 LPL LPL MYD88 MUT
BD24 0416 LPL LPL MYD88 WT
BD19 0704 LPL LPL MYD88 WT
BC14 1091 LPL LPL MYD88 WT
BA19 0549 LPL LPL MYD88 WT
AF17 0574 LPL Onvoldoende argumenten voor infiltratie door NHL
MYD88 WT
AE23 0324 LPL LPL MYD88 WT
AD21 0554 LPL LPL Beginnende ziekte van LPL
MYD88 WT
AD05 1020 LPL LPL MYD88 WT

130
Patiënt Diagnose-1
Diagnose-2
overige
Gen Resul-taat
AB24 0552 LPL LPL MYD88 WT
0L16 0554 LPL LPL MYD88 WT
0L08 0737 LPL LPL Lymfoplasmocytair lymfoom
MYD88 WT
0L06 0530 LPL LPL MYD88 WT
0I08 0632 LPL LPL MYD88 WT
0G29 0468 LPL LPL MYD88 WT
0F24 0567 LPL LPL MYD88 WT
0C30 0593 LPL LPL MYD88 WT
0C05 0631 Normaal LPL? MYD88 WT
0B17 0965 LPL LPL MYD88 WT
0A26 0681 LPL LPL MYD88 WT
0A22 0741 LPL LPL MYD88 WT
0A15 0453 Normaal MYD88 WT
0A14 0921 Normaal MYD88 WT
9K09 0958 LPL LPL Lymfoplasmocytair lymfoom
MYD88 WT
9K04 0348 LPL LPL MYD88 WT
9J29 0679 LPL LPL MYD88 WT
9D30 0490 Normaal MYD88 WT
9C12 0628 Normaal MYD88 WT
9B17 0461 LPL LPL MYD88 WT
9B13 0537 LPL LPL MYD88 WT
9B05 0709 Normaal MYD88 WT
9B04 0635 LPL LPL MYD88 WT
9A21 0491 Normaal Geen argumenten voor lymfoom LPL?
MYD88 WT


















