Tree Confidence Have we got the true tree? Use known phylogenies Unfortunately, very rare Hillis et...
32
Tree Confidence • Have we got the true tree? • Use known phylogenies • Unfortunately, very rare • Hillis et al. (1992) created experimental phylogenies using phage cultures and mutagens
-
Upload
bertram-harris -
Category
Documents
-
view
230 -
download
1
Transcript of Tree Confidence Have we got the true tree? Use known phylogenies Unfortunately, very rare Hillis et...

Tree Confidence• Have we got the true tree?• Use known phylogenies• Unfortunately, very rare• Hillis et al. (1992) created experimental
phylogenies using phage cultures and mutagens

Tree Confidence• Created a phylogeny of nine taxa from
T7 phage cultures – ~135,000 possible topologies
• Divided cultures in the presence of a mutagen at predetermined intervals
• True phylogeny is symmetric and has equal branch lengths
• Obtained restriction maps and sequences for all cultures (and the internodes) and inferred phylogenies using UPGMA, NJ, parsimony

Tree Confidence• Correct phylogeny inferred 100%• Ancestral states (via parsimony)
correctly inferred 97.3%• Correlations between predicted and
actual branch lengths used to compare the methods
• Parsimony > NJ > UPGMA• “The results… directly support the
legitimacy of methods for phylogenetic estimation… with regard to branching relationships…, branch lengths, and ancestral genotypes.”
• Unfortunately, the tree was relatively simple with plenty of informative changes along each branch

Tree Confidence• Use simulated data sets• Supply a computer with sequences and ‘evolve’ the sequences
according to some model• Analyze the resulting sequences using various phylogenetic methods• Advantage – we can test a wide variety of parameters• Disadvantage – are the models biologically accurate?• Hillis et al. (1993) did this also

Tree Confidence• Attempted to find the correct
unrooted tree for four sequences, a relatively simple problem
• Varied rates of nucleotide substitution at sites and along branches
• Tested performance using UPGMA and parsimony

Tree Confidence• Note the relative performances• Why does UPGMA do so well along
the diagonal?• Note the failure of both methods in
the ‘Felsenstein zone’ where long branch attraction tends to occur
UPGMA MP

Tree Confidence

Tree Confidence• What about the amount of data
available? (Huelsenbeck et al., 1996)• Note that some methods converge
on the correct tree with less data necessary than others… if all branches evolve equally
• What if some branches evolve more quickly?

Tree Confidence• Congruence among
alternative/independent data sets

Tree Confidence• Putting confidence estimates on nodes• We usually have only one data set. • How do we obtain information on the statistical support of the nodes
in our tree if we don’t have replicate data?• Two most common techniques are resampling methods,
bootstrapping and jackknifing

Tree Confidence• Bootstrap analysis (non-
parametric)• First used for phylogenetics
by Felsenstein in 1985• “Resampling with
replacement” to generate pseudo replicates
• Typically repeated 100 – 5000 times
• Useful and widely implemented for phenetic and likelihood methods

Tree Confidence• Bootstrap analysis
1 2 3 4 5 6 7 8 9 10 A – A T G G A T T T C GB – A T G G C G T T C GC – G C G G A G T T C GD – G C G G C G T T T G
4GGGG
2 TTCC
9 CCCT
4GGGG
4 2 9 4 8 7 5 1 3 1 A – G T C G T T A A G AB – G T C G T T C A G AC – G C C G T T A G G GD – G C T G T T C G G G
1
1 3 109 2 7 5 7 3 4 A – A G G C T T A T G GB – A G G C T T C T G GC – G G G C C T A T G GD – G G G T C T C T G G
2
10104 5 2 7 9 2 3 9 A – G G G A T T C T G CB – G G G C T T C T G CC – G G G A C T C C G CD – G G G C C T T C G T
3

Tree Confidence
4 2 9 4 8 7 5 1 3 1 A – G T C G T T A A G AB – G T C G T T C A G AC – G C C G T T A G G GD – G C T G T T C G G G
1
1 3 109 2 7 5 7 3 4 A – A G G C T T A T G GB – A G G C T T C T G GC – G G G C C T A T G GD – G G G T C T C T G G
2
10104 5 2 7 9 5 3 9 A – G G G A T T C A G CB – G G G C T T C C G CC – G G G A C T C A G CD – G G G C C T T C G T
3
1 2 3 4 5 6 7 8 9 10 A – A T G G A T T T C GB – A T G G C G T T C GC – G C G G A G T T C GD – G C G G C G T T T G
OA
D
BC
((A,B),(C,D)) A
D
BC
((A,B),(C,D))
A
D
BC
((A,B),(C,D))
A
D
CB
((A,C),(B,D))

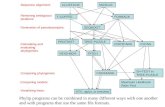
Tree Confidence• Consensus trees• Majority-rule consensus trees
only display branches with 50% support or more
• A majority-rule consensus tree may or may not be congruent with any of the pseudoreplicate topologies
• Other people and software will superimpose the branch support on the tree obtained from the original data set

Tree Confidence• Consensus trees• In this example, ¾ trees contain the branch
linking AB and CD• They get 75% bootstrap support
A
D
BC
((A,B),(C,D)) A
D
BC
((A,B),(C,D))
A
D
BC
((A,B),(C,D))
A
D
CB
((A,C),(B,D))
A
D
BC
((A,B),(C,D))
75

Tree Confidence• What does the bootstrap really tell us?• It only reflects the strength of the phylogenetic signal in the data. • Tells us nothing about the accuracy of the method we chose• If the data set is biased, the bootstrap tree will be also• If evolutionary rates are unequal, long branch attraction will likely
influence the consensus tree as much as the original tree• Sites may not evolve independently – if that is true, randomly
sampling them is invalid– Block-bootstrapping – sample n/b blocks of b adjacent sites (to correct for
correlation among adjacent sites) (Künsch, 1989)

Tree Confidence• Parametric bootstrap• Here, we are trying to determine if the data set is typical of the
parameters we have estimated for it.• For example, we may find the ML tree, with estimates of branch
lengths and substitution rates.• We can now construct alignments by simulating sequences following
the parameters: topology, branch lengths, substitution rates.• Does the original alignment resemble the bootstraps?

Tree Confidence• Parametric bootstrap - application
• Say that we obtain tree T1 in a phylogenetic analysis• We were expecting T2. We can test the null hypothesis that the data
were actually generated on treeT2 but that stochasticity (or some other process) resulted in T1 being preferred.
• Estimate all model parameters on T2 and generate a set of reference data sets using those values and parametric bootstrapping.
• On each of the generated data sets, measure the difference in likelihood score of the two trees.
• Use this reference distribution in evaluating whether the preference for T1 in the original data set could be due to chance alone causing a deviation in data actually generated on tree T2.
• Extremely computationally intensive.

Tree Confidence• Parametric bootstrap – the
placement of Strepsiptera• Two topologies proposed
– Classical (bottom) Strepsiptera is sister to the beetles
– MP based analysis of rRNA suggested Strepsiptera is sister to the flies
• Huelsenbeck (1997) performed a parametric bootstrap test
• 1. created a “constrained” tree, forcing the relationship with

Tree Confidence• Huelsenbeck (1997) performed a
parametric bootstrap test– created a “constrained” tree, forcing the
relationship with beetles– Identified the best tree under this
constraint– created many simulated data sets using
a parametric bootstrap and allowed them to evolve under the constraints
– analyzed the resulting data sets under parsimony criterion
– 92% of the trees had topologies that included a sister relationship between flies and Strepsiptera, despite the fact that we had stacked the deck in favor of the classical topology

Tree Confidence• Jackknifing – the most common variation is the delete-half jackknife• Randomly purge half of the sites from the original data set• Not commonly used anymore

Tree Confidence• Decay indices – aka Bremer support• The decay index is the length difference between the shortest trees
including a group and the shortest trees that exclude the group (the extra steps required to overturn a group)
• Generally, the higher the decay index the better the relative support for a group
• Like bootstrap proportions (BP), decay indices may be misleading if the data is misleading
• Unlike BPs decay indices are not scaled (0-100) and it is less clear what is an acceptable decay index
• Magnitude of decay indices and BPs generally correlated• The higher the number of terminal taxa, the higher the index

Tree Confidence• The approximate Likelihood Ratio Test
(aLRT)• For any strictly bifurcating tree, any
branch connects to four other branches• The tree can be simplified• We can also hypothesize that the
internal branch does not exist• The likelihoods of all three possible
internal nodes can be calculated (and are as a part of standard ML inference)
• These likelihoods can be compared via a modified LRT to determine whether any one alternative is significantly better than the original
A
D
B
C
A
D
BC
A
D
B
C

Tree Confidence• Permutation testing• Sometimes it is desirable to test if various rearrangements of a
phylogeny are significantly different from others
• Several such tests allow us to determine if one tree is statistically significantly worse than another:
– Kishino-Hasegawa (KH) test, Shimodaira-Hasegawa (SH) test
• Null hypothesis for all tests is that the trees are no different than would be expected from random sampling error

Tree Confidence• Distributions and hypothesis testing• Typical procedure
– Generate a sampling distribution which consists of many values of a test statistic generated from the data or from some other distribution of values.
– Generate a test statistic for your particular situation– Find out where your test statistic falls in the overall distribution– Is it in the acceptance region or the rejection region, p-value set a priori– One-sided test – we know directionality of the effect we are expecting– Two-sided test – we don’t know directionality of the effect

Tree Confidence• Distributions and hypothesis testing• Typical procedure
– Generate a sampling distribution which consists of many values of a test statistic generated from the data or from some other distribution of values.
– Generate a test statistic for your particular situation– Find out where your test statistic falls in the overall distribution– Is it in the acceptance region or the rejection region, p-value set a priori– One-sided test – we know directionality of the effect we are expecting– Two-sided test – we don’t know directionality of the effect
– Ideally, we would be able to generate distributions by sampling from the real world. We can’t re-run evolution, so this isn’t possible – generate distributions by resampling from the data

Tree Confidence• The Kishino-Hasegawa test uses differences in the support provided
by individual sites for two trees (T0, T1) to determine if the overall differences between the trees are significantly greater than expected from random sampling error
• Valid only if the trees to be tested are identified a priori– Infer likelihoods of each tree l0, l1– Generate many bootstrap replicates (i) for each tree and calculate l for all of them– Generate a distribution of differences between all of the trees – this is the null
distribution– Use the distribution to test your hypothesis – does the difference you originally
calculated fall into one of the tails?

Tree Confidence• The Kishino-Hasegawa test• H0 – the trees are not different• H1 – the trees are different

Tree Confidence• The Kishino-Hasegawa test• The test statistic is the score (likelihood) difference between the trees• The null distribution is all of the possible differences if the trees are
not different (generated by bootstrapping the data)• Assumptions:
– Trees selected a priori– Sites are independent– Sites are identically distributed– Large numbers of sites are sampled

Tree Confidence• The Shimodaira-Hasegawa (SH) test is an alternative test involving
bootstrapping to test whether the best tree is better than other trees identified a posteriori
– The test statistic in this case is the score difference between the best tree and all other trees to be compared
– H0 – all trees to be compared are equally good– H1 – some or all threes are not equally good explanations of the data

Tree Confidence• If there is a lot of homoplasy, trees derived using parsimony could be
unreliable. Values are available to evaluate the reliability of parsimony-based trees.
• Consistency Index (CI) = the minimum number of changes/tree length
• ci = mi/si (for any i-th site in the alignment)
– mi = the minimum possible number of substitutions for any conceivable topology
– si = the minimum number of substitutions required for the topology being considered
– The overall CI for the entire tree is Σmi/ Σsi
• Homoplasy Index (HI) = 1 – CI• Both will provide an idea of the relative value of the data with regard
to the given tree. • But random data can give CI’s with values between 0.4 and 0.6

Tree Confidence• What can go wrong in phylogenetic analysis?• Sampling error
– All inferred trees can only represent the sequences used– Improper sampling of the taxa will yield incorrect trees– Improperly selected sequences will yield incorrect trees
• Incorrect evolutionary model– Choosing the wrong model of sequence evolution will likely lead to the wrong tree
• Evolutionary history– Sometimes, despite our best efforts, finding the best tree may just be impossible– Rapid radiations, widely differing rates of evolution, extinction