
Transient outward current (Ito) gain-of-function mutations ...
9
Transient outward current (I to ) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome John R. Giudicessi, BA,* † Dan Ye, MD,* David J. Tester, BS,* Lia Crotti, MD, PhD, ‡§¶ Alessandra Mugione, PhD, ¶ Vladislav V. Nesterenko, PhD,** Richard M. Albertson, BS,* Charles Antzelevitch, PhD,** Peter J. Schwartz, MD, ‡§¶# Michael J. Ackerman MD, PhD* From the *Department of Medicine (Division of Cardiovascular Diseases), Department of Pediatrics (Division of Pediatric Cardiology), and Department of Molecular Pharmacology and Experimental Therapeutics, Windland Smith Rice Sudden Death Genomics Laboratory, and † Mayo Medical School, Mayo Clinic, Rochester, Minnesota, ‡ Department of Lung, Blood, and Heart, University of Pavia, and § Department of Cardiology, Fondazione IRCCS Policlinico San Matteo, Pavia, and ¶ Laboratory of Cardiovascular Genetics, IRCCS Istituto Auxlogico, Milan, Italy, Cardiovascular Genetics Laboratory, Hatter Institute for Cardiovascular Research, Department of Medicine, University of Cape Town, South Africa, # Chair of Sudden Death, Department of Family and Community Medicine, College of Medicine, King Saud University, Riyadh, Saudi Arabia, and **Masonic Medical Research Laboratory, Utica, New York. BACKGROUND Brugada syndrome (BrS) is a sudden death–pre- disposing genetic condition characterized electrocardiographi- cally by ST segment elevation in the leads V 1 –V 3 . Given the prominent role of the transient outward current (I to ) in BrS patho- genesis, we hypothesized that rare gain-of-function mutations in KCND3 may serve as a pathogenic substrate for BrS. METHODS Comprehensive mutational analysis of KCND3-encoded Kv4.3 (I to ) was conducted using polymerase chain reaction, de- naturing high performance liquid chromatography, and direct se- quencing of DNA derived from 86 unrelated BrS1-8 genotype- negative BrS patients. DNA from 780 healthy individuals was examined to assess allelic frequency for nonsynonymous variants. Putative BrS-associated Kv4.3 mutations were engineered and coexpressed with wild-type KChIP2 in HEK293 cells. Wild-type and mutant I to ion currents were recorded using whole-cell patch clamp. RESULTS: Two BrS1-8 genotype-negative cases possessed novel Kv4.3 missense mutations. Both Kv4.3-L450F and Kv4.3-G600R were absent in 1,560 reference alleles and involved residues highly conserved across species. Both Kv4.3-L450F and Kv4.3-G600R demonstrated a gain-of-function phenotype, increasing peak I to current density by 146.2% (n 15, P .05) and 50.4% (n 15, P .05), respectively. Simulations using a Luo-Rudy II action potential (AP) model demonstrated the stable loss of the AP dome as a result of the increased I to maximal conductance associated with the heterozygous expression of either L450F or G600R. CONCLUSIONS These findings provide the first molecular and functional evidence implicating novel KCND3 gain-of-function mu- tations in the pathogenesis and phenotypic expression of BrS, with the potential for a lethal arrhythmia being precipitated by a genetically enhanced I to current gradient within the right ventri- cle where KCND3 expression is the highest. KEYWORDS Brugada syndrome; Genetic diseases; Ion channels; I to current; J-wave syndromes; Kv4.3 channels; Sudden cardiac death ABBREVIATIONS AP action potential; APD action potential duration; BrS Brugada Syndrome; I to transient outward cur- rent; DHPLC denaturing high performance liquid chromatogra- phy; ECG electrocardiogram; G Kv4.3 I to /Kv4.3 maximal con- ductance; Kv4.3 voltage-gated potassium channel; LQTS long QT syndrome; LV left ventricle; PCR polymerase chain reac- tion; RV right ventricle; SCD sudden cardiac death; SEM standard error of the mean (Heart Rhythm 2011;8:1024 –1032) © 2011 Published by Elsevier Inc. on behalf of Heart Rhythm Society. Introduction Sudden cardiac death (SCD) accounts for approximately 300,000 deaths in the United States annually. 1 While struc- tural heart disease represents the primary etiology of SCD in the general population, it is estimated that as many as one-third of young cases and 5%–10% of all SCDs occur The first two authors contributed equally to this study. M.J.A. is a consultant for Transgenomic/FAMILION. Intellectual property derived from M.J.A.’s research program resulted in license agreements in 2004 between Mayo Clinic Health Solutions (formerly Mayo Medical Ventures) and PGxHealth (recently acquired by Transgenomic). Address reprint requests and correspondence: Michael J. Ackerman, M.D., Ph.D., Mayo Clinic Windland Smith Rice Sudden Death Genomics Laboratory, Gug- genheim 501, Mayo Clinic, Rochester, Minnesota 55905. Email address: [email protected]. (Received August 20, 2010; accepted Feb- ruary 15, 2011.) 1547-5271/$ -see front matter © 2011 Published by Elsevier Inc. on behalf of Heart Rhythm Society. doi:10.1016/j.hrthm.2011.02.021
Transcript of Transient outward current (Ito) gain-of-function mutations ...

ts
C
L
LA
p
KnqnePcm
P
Transient outward current (Ito) gain-of-function mutations inhe KCND3-encoded Kv4.3 potassium channel and Brugadayndrome
John R. Giudicessi, BA,*† Dan Ye, MD,* David J. Tester, BS,* Lia Crotti, MD, PhD,‡§¶
Alessandra Mugione, PhD,¶ Vladislav V. Nesterenko, PhD,** Richard M. Albertson, BS,*harles Antzelevitch, PhD,** Peter J. Schwartz, MD,‡§¶�# Michael J. Ackerman MD, PhD*
From the *Department of Medicine (Division of Cardiovascular Diseases), Department of Pediatrics (Division ofPediatric Cardiology), and Department of Molecular Pharmacology and Experimental Therapeutics, Windland Smith RiceSudden Death Genomics Laboratory, and †Mayo Medical School, Mayo Clinic, Rochester, Minnesota,‡Department of
ung, Blood, and Heart, University of Pavia, and §Department of Cardiology, Fondazione IRCCS Policlinico San Matteo,Pavia, and ¶Laboratory of Cardiovascular Genetics, IRCCS Istituto Auxlogico, Milan, Italy, �Cardiovascular Genetics
aboratory, Hatter Institute for Cardiovascular Research, Department of Medicine, University of Cape Town, Southfrica, #Chair of Sudden Death, Department of Family and Community Medicine, College of Medicine, King Saud
University, Riyadh, Saudi Arabia, and **Masonic Medical Research Laboratory, Utica, New York.
pa
c
K
d
BACKGROUND Brugada syndrome (BrS) is a sudden death–pre-disposing genetic condition characterized electrocardiographi-cally by ST segment elevation in the leads V1–V3. Given therominent role of the transient outward current (Ito) in BrS patho-
genesis, we hypothesized that rare gain-of-function mutations inKCND3 may serve as a pathogenic substrate for BrS.
METHODS Comprehensive mutational analysis of KCND3-encodedv4.3 (Ito) was conducted using polymerase chain reaction, de-aturing high performance liquid chromatography, and direct se-uencing of DNA derived from 86 unrelated BrS1-8 genotype-egative BrS patients. DNA from 780 healthy individuals wasxamined to assess allelic frequency for nonsynonymous variants.utative BrS-associated Kv4.3 mutations were engineered andoexpressed with wild-type KChIP2 in HEK293 cells. Wild-type andutant Ito ion currents were recorded using whole-cell patch
clamp.
RESULTS: Two BrS1-8 genotype-negative cases possessed novelKv4.3 missense mutations. Both Kv4.3-L450F and Kv4.3-G600Rwere absent in 1,560 reference alleles and involved residues highlyconserved across species. Both Kv4.3-L450F and Kv4.3-G600Rdemonstrated a gain-of-function phenotype, increasing peak Ito
current density by 146.2% (n � 15, P �.05) and 50.4% (n � 15,
and PGxHealth (recently acquired by Transgenomic). Address reprint
1547-5271/$ -see front matter © 2011 Published by Elsevier Inc. on behalf of H
otential (AP) model demonstrated the stable loss of the AP domes a result of the increased Ito maximal conductance associated
with the heterozygous expression of either L450F or G600R.
CONCLUSIONS These findings provide the first molecular andfunctional evidence implicating novel KCND3 gain-of-function mu-tations in the pathogenesis and phenotypic expression of BrS,with the potential for a lethal arrhythmia being precipitated by agenetically enhanced Ito current gradient within the right ventri-le where KCND3 expression is the highest.
EYWORDS Brugada syndrome; Genetic diseases; Ion channels; Itocurrent; J-wave syndromes; Kv4.3 channels; Sudden cardiac death
ABBREVIATIONS AP � action potential; APD � action potentialduration; BrS � Brugada Syndrome; Ito � transient outward cur-rent; DHPLC � denaturing high performance liquid chromatogra-phy; ECG � electrocardiogram; GKv4.3 � Ito/Kv4.3 maximal con-uctance; Kv4.3 � voltage-gated potassium channel; LQTS � long
QT syndrome; LV � left ventricle; PCR � polymerase chain reac-tion; RV � right ventricle; SCD � sudden cardiac death;SEM � standard error of the mean
(Heart Rhythm 2011;8:1024–1032) © 2011 Published by Elsevier
�.05), respectively. Simulations using a Luo-Rudy II action Inc. on behalf of Heart Rhythm Society.IntroductionSudden cardiac death (SCD) accounts for approximately300,000 deaths in the United States annually.1 While struc-
The first two authors contributed equally to this study. M.J.A. is aconsultant for Transgenomic/FAMILION. Intellectual property derivedfrom M.J.A.’s research program resulted in license agreements in 2004between Mayo Clinic Health Solutions (formerly Mayo Medical Ventures)
tural heart disease represents the primary etiology of SCD inthe general population, it is estimated that as many asone-third of young cases and 5%–10% of all SCDs occur
requests and correspondence: Michael J. Ackerman, M.D., Ph.D., MayoClinic Windland Smith Rice Sudden Death Genomics Laboratory, Gug-genheim 501, Mayo Clinic, Rochester, Minnesota 55905. Email address:[email protected]. (Received August 20, 2010; accepted Feb-
ruary 15, 2011.)eart Rhythm Society. doi:10.1016/j.hrthm.2011.02.021

oale
msmaediw
I
igs
ialpt
cait
fpBppwtsc
cq
1025Giudicessi et al KCND3 Mutations in Brugada Syndrome
within individuals with no detectable structural abnormali-ties upon postmortem investigation.2,3 Tragically, thousandsf individuals under the age of 40 with otherwise structur-lly normal hearts die suddenly each year, representing aoss of life-years that rivals ischemic-related heart dis-ase.2,4 Cardiac channelopathies such as catecholaminergic
polymorphic ventricular tachycardia, long QT syndrome(LQTS), and Brugada syndrome (BrS), which arise fromheritable defects in cardiac ion channel function, representthe most common identifiable causes underlying autopsy-negative sudden unexplained death.5
BrS affects as many as 1 in 2,500 individuals and ischaracterized electrocardiographically by dynamic coved-type ST segment elevation in the right precordial leads inthe absence of ischemia, structural heart disease, and phar-macologic agents known to induce a BrS-like electrocardio-gram (ECG) pattern.6,7 Affected individuals are typically
ales in the fourth decade of life with a high incidence ofyncope and sudden death secondary to ventricular arrhyth-ias that often develop during times of increased vagal
ctivity such as sleep or rest. Owing to the variable pen-trance and expressivity of this autosomal-dominant disor-er, the clinical course of BrS assumes a spectrum thatncludes lifelong asymptomatic individuals as well as thoseho die suddenly in early life, including infancy.8 It is
estimated that BrS accounts for nearly 5% of all suddendeaths and up to 20% of sudden deaths in individuals withotherwise structurally normal hearts.7
Since its seminal description as a clinical entity in 1992,BrS has been linked to mutations in genes that perturbcardiac sodium (INa), calcium (ICa.L), or potassium (Ito and
K-ATP) channel function. At the cellular and ionic level,insufficient sodium (INa) or calcium (ICa) inward depolariz-ng current coupled with a right ventricular (RV) transmuralradient (epicardium � endocardium) involving the tran-ient outward repolarizing current (Ito) conducted by the
KCND3-encoded Kv4.3 �-subunit is hypothesized to resultn an outward shift in current, heterogenous loss of thection potential (AP) dome, ST segment elevation on ECG,ocal reexcitation via phase II reentry, and the initiation ofolymorphic ventricular tachycardia or ventricular fibrilla-ion.9 The recent identification of (1) a BrS-associated loss-
of-function missense mutation in the negative modulatingKCNE3-encoded MiRP2 Kv4.3 �-subunit10 that signifi-antly increases Ito current and (2) a J-wave syndrome-ssociated pleiotropic gain-of-function missense mutationn the KCNJ8-encoded Kir6.1 �-subunit of the ATP-sensi-ive potassium (IK-ATP) channel11 have strengthened the
hypothesis that a gain-of-function in outward potassiumcurrents (Ito and IK-ATP) active during early repolarizationcan contribute to the pathogenesis of BrS and other disor-ders of early repolarization. Despite major advances in theunderstanding of the cellular, ionic, and genetic basis ofBrS, an estimated 60%–70% of BrS cases remain geneti-
cally elusive. fEven though no mutations in the KCND3-encoded Kv4.3channel have been identified to date, we hypothesized thatrare gain-of-function mutations in KCND3 may confer riskor lethal ventricular arrhythmias and thus may serve as theathogenic substrate for some genetically elusive cases ofrS. In this study, we sought to determine the spectrum andrevalence of KCND3 mutations in a cohort of 86 BrSatients previously screened for mutations in BrS1-BrS8ith spontaneous or flecainide-induced ST segment eleva-
ion in the right precordial leads in the absence of anytructural or ischemic heart disease by echocardiogram andoronary angiogram.
MethodsStudy populationThe study population consisted of 86 unrelated patientsdiagnosed with BrS on established clinical criteria whowere referred to the Windland Smith Rice Sudden DeathGenomics Laboratory at Mayo Clinic, Rochester, Minne-sota, or the Molecular Cardiology Laboratory, FondazioneIRCCS Policlinico San Matteo, Pavia, Italy, for laboratory-based genetic testing. All BrS cases included in this studywere screened previously for mutations in the eight knownBrS-susceptibility genes (SCN5A, GPD1L, CACNA1C,CACNB2, SCN1B, KCNE3, SCN3B, and KCNJ8).
This study was approved by both the Mayo FoundationInstitutional Review Board and the Medical Ethical Com-mittee of Fondazione IRCCS Policlinico San Matteo. In-formed consent was obtained for all patients.
Control populationDNA from a cohort of 780 (680 Caucasian and 100 AfricanAmerican) ostensibly healthy individuals was obtained fromthe Coriel Cell Repository and the European Collection ofCell Cultures. These samples were used to assess allelicfrequency for all identified nonsynonymous variants and toindependently determine the rate of background geneticvariation within KCND3 among healthy blood donors.
Mutational analysisGenomic DNA was extracted from peripheral blood lym-phocytes using Purgene DNA extraction kits (Gentra Sys-tems Inc., Minneapolis, MN). Comprehensive mutationalanalysis of KCND3 was accomplished in cases and controlsusing polymerase chain reaction (PCR), denaturing high-performance liquid chromatography (DHPLC), and directDNA sequencing as described elsewhere.12 The flankingprimers used to amplify KCND3 were designed using thePrimer3 Web-based interface.13 Primer sequences, PCRonditions, and DHPLC conditions are available upon re-uest.
KCND3 and KCNIP2 mammalian expression vectorsand mutagenesisWild-type human KCND3 cDNA was provided by CharlesAntzelevitch. The wild-type human KCNIP2 cardiac iso-
orm cDNA was synthesized as a custom minigene con-
Tp
K
((c(tm
cm2CvepFIfiM
a
ssmau
ers5
s
1026 Heart Rhythm, Vol 8, No 7, July 2011
tained in the pIDTSMART vector backbone (IntegratedDNA Technologies, Coralville, IA). Wild-type KCND3cDNA was subcloned into pIRES2-EGFP (Clontech, Moun-tain View, CA) to produce pIRES2-KCND3WT-EGFP, andwild-type KCNIP2 cDNA was subcloned into pIRES2-dsRed2 (Clontech) to produce pIRES2-KCNIP2WT-dsRed2.
he L450F and G600R mutations were engineered intoIRES2-KCND3WT-EGFP using the Quikchange XL Site-
Directed Mutagenesis Kit (Stratagene, La Jolla, CA). DNAsequencing was used to confirm the integrity of all vectors.
HEK293 cell culture and transfectionHEK293 cells were cultured in minimum essential mediumsupplemented with 1% nonessential amino acid solution,10% horse serum, 1% sodium pyruvate solution, and 1.4%penicillin/streptomycin solution. All cells were plated inT25 flasks and stored in a 5% CO2 incubator at 37oC for 24hours. Heterologous expression of Kv4.3 and KChIP2was accomplished by cotransfecting 0.5 �g of pIRES2-
CND3WT-EGFP, pIRES2-KCND3L450F-EGFP, or pIRES2-KCND3G600R-EGFP with 1.5 �g pIRES2-KCNIP2WT-dsRed2 using 5 �L of lipofectamine transfection reagentInvitrogen, Carlsbad, CA) in Gibco OPTI-MEM mediaInvitrogen). Cells exhibiting both green fluorescence (ex-itation 488 nm, emission 507 nm) and red fluorescenceexcitation 558 nm, emission 583 nm) at 24 hours post-ransfection were selected for electrophysiological experi-ents.
Electrophysiological measurements and dataanalysisThe standard whole-cell patch clamp technique was used tomeasure Kv4.3-WT, Kv4.3-L450F, or Kv4.3-G600R plusKChIP2-WT currents at room temperature (22–24°C) withthe use of an Axopatch 200B amplifier, Digidata 1440A,and pClamp version 10.2 software (Axon Instruments, Fos-ter City, CA). The extracellular (bath) solution contained(mmol/L) 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, and 10HEPES, pH adjusted to 7.4 with NaOH. The pipette solutioncontained (mmol/L) 110 KCl, 31 KOH, 10 EDTA, 5.17CaCl2, 1.42 MgCl2, 4 MgATP, and 10 HEPES, pH adjustedto 7.2 with KOH following established protocols.10,14 Mi-roelectrodes were pulled on a P-97 puller (Sutter Instru-ents, Novato, CA) and fire polished to a final resistance of
–3 M�. Series resistance was compensated by 80%–85%.urrents were filtered at 5 kHz and digitized at 10 kHz. Theoltage-dependence of activation, inactivation, and recov-ry from inactivation were determined using voltage-clamprotocols described in the figure legends and insets forigures 3 and 4. Data were analyzed using Clampfit (Axonnstruments) and Excel (Microsoft, Redmond, WA) andtted with Origin 8 (OriginLab Corporation, Northampton,A).The voltage-dependent inactivation curve was fitted with
Boltzmann function: I/I, max� {1�exp [(V � V1/2)/k]}�1,where V1/2 and k are the half-maximal voltage of inactiva-
tion and the slope factor, respectively. Recovery from inac-tivation was fitted with a one-exponential function: y �y0�[A1 exp(�x/�1)], where A1 indicates the fractions ofrecovery from inactivation and �1 indicates the time con-tant for recovery from inactivation. Inactivation time con-tants for each voltage step were determined by fitting aonoexponential function to current decay. Total charge asfunction of voltage was obtained by measuring the area
nder curve during the first 50 ms of each voltage step.
Simulated ventricular epicardial APsRV and left ventricular (LV) epicardial APs were simulatedusing a Luo-Rudy II multicellular one-dimensional fiber APmodel, modified to include the Ito current, as describedelsewhere.15,16 The incorporation of Ito in the Luo-Rudy IIAP model requires decreasing maximal conductance of ICaL
by 20% to compensate for the significant increase of ICaL
during the notch of the AP. The maximal conductance ofRV Ito was set to 1.3 mS/�F for wild type, 2.32 mS/�F forL450F, and 1.56 mS/�F for G600R in accordance withxperimentally derived values across the 0 to �40 mVange that reflect the assumption that heterozygous expres-ion of L450F and G600R in the heart results in at least a0% reduction in the observed Ito gain of function. The time
constant of inactivation of the G600R mutant was increasedby 19% in agreement with experimental data.
Statistical analysisAll data points represent the mean value, and bars representthe standard error of the mean (SEM). Determination ofstatistical significance between two groups was accom-plished using Student’s t-test. One-way analysis of variancewas used for multiple-group comparison. P �.05 was con-idered statistically significant.
Statement of responsibilityThe authors had full access to and take full responsibilityfor the integrity of the data. All authors have read and agreeto the manuscript as written.
ResultsMutation analysis and clinical dataWe molecularly interrogated KCND3 in 86 unrelated geno-type-negative/phenotype-positive patients with a referral di-agnosis of BrS who had been previously analyzed for mu-tations in the eight known BrS-susceptibility genes. Overall,96% of patients were of Caucasian descent, 84% were male,the average age at diagnosis was 45 � 14 years, and 34%exhibited a spontaneous type I Brugada ECG pattern. Com-prehensive mutational analysis of KCND3 yielded two pu-tative pathogenic mutations in two genetically elusive BrScases.
Abnormal DHPLC elution profiles (Figure 1A) and sub-sequent direct DNA sequencing (Figure 1B) led to theidentification of a KCND3 c.348 C¡T nucleotide substitu-tion and a KCND3 c.1798 G¡C nucleotide substitution intwo distinct genetically elusive BrS cases, which resulted in
a L450F missense mutation (lysine [L] for phenylalanine
wotd
owWe
6
c
1027Giudicessi et al KCND3 Mutations in Brugada Syndrome
[F] at position 450) and G600R missense mutation (glycine[G] for arginine [R] at position 600), respectively. Both theL450F and G600R missense mutations were absent from1,560 reference alleles (P �.001) and involve highly con-served residues located in the Kv4.3 carboxyl (C)-terminus(Figure 1C, 1D). The L450F- and G600R-positive BrS in-dex cases were genotype-negative for mutations in the eightknown BrS-susceptibility genes.
In addition, the analysis of KCND3’s seven translatedexons in 780 ostensibly healthy controls revealed only twoamino acid–altering genetic variants (K214R and T486A),suggesting that KCND3 harbors minimal background aminoacid–altering genetic variation in comparison with otherchannelopathic genes.
The first BrS index case (Kv4.3-L450F) was a 45-year-old male with a history of heart palpitations at rest. Noarrhythmias have been documented, but ST segment eleva-tion in leads V1 and V2 of the patient’s resting Holter ECG
ere suspicious for BrS. There was no documented historyf syncope or cardiac events, and the patient’s family his-ory was negative. He underwent a flecainide test that in-uced a type 1 ECG pattern (Figure 2A).
The second BrS index case (Kv4.3-G600R), a 22-year-ld male with a history of heart palpitations and presyncope,as discovered unconscious and unresponsive in bed.hile hospitalized, a 12-lead ECG revealed ST segment
levation in V1, V2, and V3 and a QTc of 523 ms (data notshown). Cardiac enzymes were borderline normal, with a
Figure 1 Mutations in KCND3 are associated with BrS. Depicted are (Asequencing chromatograms. C: Predicted protein topology schematic of Konservation across species for the L450F and G600R mutations.
creatine kinase of 370, a troponin I of 0.32, and a relative c
index of 1.3. A complete cardiac workup including echo-cardiogram and coronary angiogram was performed andfound to be normal. All drug/toxicology screens were neg-ative.
A follow-up ECG several days later revealed down-sloping ST segment elevation in the right precordial leads(V1–V3) with a normal PR interval of 150 ms (Figure 2B).The patient’s QT interval remained slightly prolonged.Given the patient’s significant paternal family history ofsudden cardiac death (paternal grandfather died suddenly atage 39 and a paternal uncle died suddenly at age 35) and theabnormal ECG findings suggestive of BrS, an internal car-diac defibrillator was implanted as a secondary prevention.
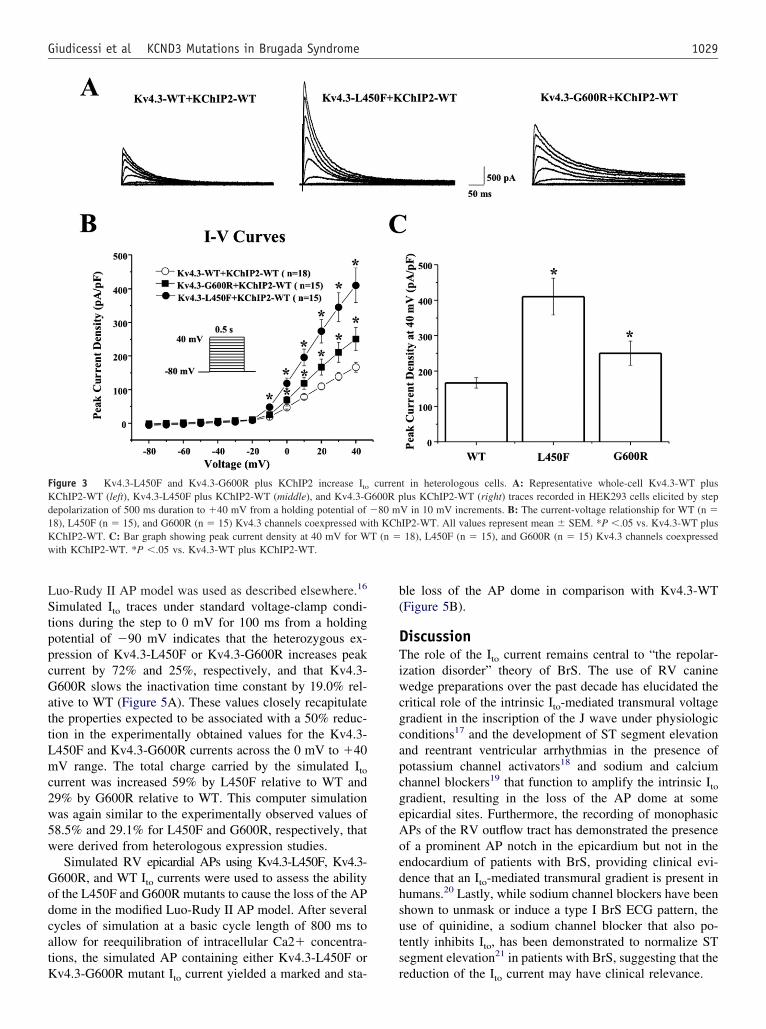
Heterologous expression and functionalcharacterization of Kv4.3-L450F and Kv4.3-G600RTo recapitulate the Ito current in vitro, Kv4.3-WT, Kv4.3-L450F, or Kv4.3-G600R was heterologously coexpressedwith KChIP2-WT in HEK293 cells (Figure 3A). Analysis ofthe current-voltage relationship indicated that Kv4.3-L450For Kv4.3-G600R plus KChIP2-WT significantly increa-sed Ito current density from 0 mV to �40 mV (n � 15 forboth mutations, P �.05) compared with Kv4.3-WT plusKChIP2-WT (n � 18, Figure 3B). Kv4.3-L450F and Kv4.3-G600R significantly increased current density at 0 mV by154.3% and 48.1% percent, respectively, from 46.6 � 4.6(WT, n � 18) to 118.5 � 16.0 (L450F, n � 15, P �.05) and9.0 � 10.6 (G600R, n � 15, P �.05) and similarly in-
LC profiles (normal, red trace, and abnormal, green trace) and (B) DNAndicating the location of the L450F and G600R mutations. D: Sequence
) DHPv4.3 i
reased the peak current density at 40 mV by 146.2% and

t4t
cu
�0
af
index
1028 Heart Rhythm, Vol 8, No 7, July 2011
50.4%, respectively, from 166.6 � 14.8 (WT, n � 18) to410.2 � 51.4 (L450F, n � 15, P �.05) and 250.5 � 34.2(G600R, n � 15, P �.05, Figure 3C). Kv4.3-G600R plusKChIP2-WT exhibited significantly slower inactivation �across the 0–40 mV range compared with Kv4.3-WT plusKChIP2-WT from 71.9 � 4.5 ms (WT at 40 mV, n � 18)o 89.3 � 3.0 ms (G600R at 40 mV, n � 15, P �.05, FigureA). Kv4.3-L450F plus KChIP2-WT significantly increasedhe Ito total charge by 117.0% at 40 mV (n � 15, P �.05),
and Kv4.3-G600R plus KChIP2-WT significantly increasedthe Ito total charge by 58.3% at 40 mV (n � 15, P �.05)ompared with Kv4.3-WT plus KChIP2-WT (n � 18, Fig-re 4B).
Kv4.3-L450F significantly shifted V1/2 of inactivation5.2 mV from �21 � 0.7 mV (WT, n � 12) to �26.2 �
Figure 2 Representative ECGs for the L450F- and G600R-positive BrSsegment elevation in leads V2 and V3 and flecainide challenge 12-lead ECGcase. B: Representative baseline 12-lead ECG of the G600R-positive BrS
.7 mV (L450F, n � 7, P �.05, Figure 4C), while the
Kv4.3-G600R mutation did not significantly alter the inac-tivation kinetics of Ito compared with Kv4.3-WT. The re-spective k (slope factor) remains unchanged for both muta-tions. The time constants for the recovery from inactivation,assessed using a two-pulse protocol (see inset Figure 4Dnd figure legend) were 57.4 � 1.7 ms for cells cotrans-ected with Kv4.3-WT plus KChIP2 (n � 7), 55.2 � 1.6 ms
for cells cotransfected with Kv4.3-L450F plus KChIP2 (n �5), and 61.6 � 1.8 ms for cells cotransfected with Kv4.3-G600R plus KChIP2 (n � 6). There was no significantdifference among the three groups.
Simulated effect of Kv4.3-L450F andKv4.3-G600R on the RV epicardial APTo simulate the experimentally observed effects of the
ases. A: Representative basal Holter ECG demonstrating spontaneous STunmasked a coved-type BrS ECG pattern in the L450F-positive BrS index
case with V1–V3 leads in the normal position.
index c, which
L450F and G600R mutations on the RV AP, a modified
b(
s
w
1029Giudicessi et al KCND3 Mutations in Brugada Syndrome
Luo-Rudy II AP model was used as described elsewhere.16
Simulated Ito traces under standard voltage-clamp condi-tions during the step to 0 mV for 100 ms from a holdingpotential of �90 mV indicates that the heterozygous ex-pression of Kv4.3-L450F or Kv4.3-G600R increases peakcurrent by 72% and 25%, respectively, and that Kv4.3-G600R slows the inactivation time constant by 19.0% rel-ative to WT (Figure 5A). These values closely recapitulatethe properties expected to be associated with a 50% reduc-tion in the experimentally obtained values for the Kv4.3-L450F and Kv4.3-G600R currents across the 0 mV to �40mV range. The total charge carried by the simulated Ito
current was increased 59% by L450F relative to WT and29% by G600R relative to WT. This computer simulationwas again similar to the experimentally observed values of58.5% and 29.1% for L450F and G600R, respectively, thatwere derived from heterologous expression studies.
Simulated RV epicardial APs using Kv4.3-L450F, Kv4.3-G600R, and WT Ito currents were used to assess the abilityof the L450F and G600R mutants to cause the loss of the APdome in the modified Luo-Rudy II AP model. After severalcycles of simulation at a basic cycle length of 800 ms toallow for reequilibration of intracellular Ca2� concentra-tions, the simulated AP containing either Kv4.3-L450F or
Figure 3 Kv4.3-L450F and Kv4.3-G600R plus KChIP2 increase Ito
KChIP2-WT (left), Kv4.3-L450F plus KChIP2-WT (middle), and Kv4.3-Gdepolarization of 500 ms duration to �40 mV from a holding potential of18), L450F (n � 15), and G600R (n � 15) Kv4.3 channels coexpressed wKChIP2-WT. C: Bar graph showing peak current density at 40 mV for W
ith KChIP2-WT. *P �.05 vs. Kv4.3-WT plus KChIP2-WT.
Kv4.3-G600R mutant Ito current yielded a marked and sta- r
le loss of the AP dome in comparison with Kv4.3-WTFigure 5B).
DiscussionThe role of the Ito current remains central to “the repolar-ization disorder” theory of BrS. The use of RV caninewedge preparations over the past decade has elucidated thecritical role of the intrinsic Ito-mediated transmural voltagegradient in the inscription of the J wave under physiologicconditions17 and the development of ST segment elevationand reentrant ventricular arrhythmias in the presence ofpotassium channel activators18 and sodium and calciumchannel blockers19 that function to amplify the intrinsic Ito
gradient, resulting in the loss of the AP dome at someepicardial sites. Furthermore, the recording of monophasicAPs of the RV outflow tract has demonstrated the presenceof a prominent AP notch in the epicardium but not in theendocardium of patients with BrS, providing clinical evi-dence that an Ito-mediated transmural gradient is present inhumans.20 Lastly, while sodium channel blockers have beenshown to unmask or induce a type I BrS ECG pattern, theuse of quinidine, a sodium channel blocker that also po-tently inhibits Ito, has been demonstrated to normalize STegment elevation21 in patients with BrS, suggesting that the
t in heterologous cells. A: Representative whole-cell Kv4.3-WT pluslus KChIP2-WT (right) traces recorded in HEK293 cells elicited by step
V in 10 mV increments. B: The current-voltage relationship for WT (n �IP2-WT. All values represent mean � SEM. *P �.05 vs. Kv4.3-WT plus18), L450F (n � 15), and G600R (n � 15) Kv4.3 channels coexpressed
curren600R p�80 mith KChT (n �
eduction of the Ito current may have clinical relevance.

c
eati
goK
omK*hda
own rep
1030 Heart Rhythm, Vol 8, No 7, July 2011
Despite the growing body of evidence linking the Ito
current to the pathogenesis of BrS, until recently there wasminimal experimental data to directly implicate perturba-tions in Ito to disease. The identification of a BrS-associatedloss-of-function missense mutation in KCNE3 provided thefirst evidence that mutations resulting in an increase in Ito
could produce a type I BrS ECG pattern.10 Furthermore,anine wedge preparations treated with the Ito-specific ac-
tivator NS5806 increased the Ito-mediated AP notch in thepicardium but not in the endocardium, leading to J-waveccentuation, development of phase 2 reentry, and the ini-iation of ventricular tachycardia in both ventricles provid-ng further evidence that Ito gain of function can recapitulate
Figure 4 Kinetic alteration for Kv4.3-WT, Kv4.3-L450F, or Kv4.3-G60r Kv4.3-G600R plus KChIP2 currents plotted as a function of voltage.onoexponential function to current decay. All values represent mean �v4.3-L450F, or Kv4.3-G600R with KChIP2 as a function of voltage obtaiP �.05 vs. Kv4.3-WT plus KChIP2. C: Steady-state inactivation curvesolding potential of �100 mV to prepulse of �5 mV in 5-mV incrementuration and fitted with a Boltzmann function. D: Recovery from inactivatholding potential of �80 mV to prepulse of �20 mV with 0.5-second du
500-ms duration and fitted with a one-exponential function. All values sh
the hallmarks of BrS.14
Consistent with the established role of Ito in BrS patho-enesis, our present study demonstrates the first associationf gain-of-function mutations in the KCND3-encodedv4.3 �-subunit that conducts Ito in humans with BrS. Here
we present two unrelated cases, one an asymptomatic 45-year-old male with documented ST segment elevation in theright precordial leads on Holter ECG and a positive flecain-ide test, and the second, a 22-year-old male with a docu-mented type I BrS ECG pattern and a family history ofsudden death who was discovered unresponsive in bed.Both were found to harbor novel missense mutations inKCND3. In addition, these mutations involved highly con-served residues in the Kv4.3 C-terminus and were both
KChIP2. A: Inactivation time constants (�) for Kv4.3-WT, Kv4.3-L450F,ation time constants for each voltage step were determined by fitting a. *P �.05 vs. Kv4.3-WT plus KChIP2. B: Total charge of Kv4.3-WT,measuring the area under curve during the first 50 ms of each voltage step..3-WT, Kv4.3-L450F, or Kv4.3-G600R with KChIP2 determined from a.5-second duration followed by a test pulse of �20 mV with 0.5-secondv4.3-WT, Kv4.3-L450F, or Kv4.3-G600R with KChIP2 determined fromith increased recovery interval, followed by a test pulse of �20 mV withresent mean � SEM.
0R plusInactiv
SEMned byof Kv4
s with 0ion of Kration, w
absent in 1,560 reference alleles.

G7a1GA
R
tw
hce
aAtultswhstisAedt
bGits
haLo
msepm
1031Giudicessi et al KCND3 Mutations in Brugada Syndrome
The heterologous coexpression of both Kv4.3-L450F andKv4.3-G600R with KChIP2 resulted in a gain-of-functionin the Ito current. Further, simulations using a modifiedLuo-Rudy II AP model that incorporates an increase in Ito
maximal conductance (GKv4.3) consistent with experimen-tally derived data (1.30, 1.56, and 2.32 mS/�F for WT,
600R, and L450F, respectively) demonstrated that the3.1% increase in Ito current associated with Kv4.3-L450Fnd the modest 25.2% increase in Ito current together with a9% slowing of Ito inactivation associated with Kv4.3-600R are both capable of producing the stable loss of theP dome in RV epicardial tissue.Interestingly, simulations using the same modified Luo-
udy II model that incorporates a smaller GKv4.3 intended tomodel the LV epicardial AP (0.5 and 0.7 mS/�F for WT andG600R, respectively) did not demonstrate loss of the APdome. The use of a smaller wild-type GKv4.3 (0.25 mS/�F)hat fails to fully reproduce the deep RV AP likely explainshy a 3.5-fold acceleration of INa inactivation and a seven-
fold increase in GKv4.3 (to 1.75 mS/�F) were required tosimulate the ionic basis of BrS in a previous study that used
Figure 5 Effects of Kv4.3-L450F and Kv4.3-G600R on the RV epicar-dial AP. A: Simulated Ito current traces during step depolarization for 100
s to 0 mV from a holding potential of �90 mV. Only the first 60 ms arehown. Solid line, WT; dotted line, L450F; dashed line, G600R. B: RVpicardial APs simulated using WT, L450F, and G600R mutant Ito incor-orated into a modified Luo-Rudy II AP model. Basic cycle length � 800s (75 bpm). Solid line, WT; dotted line, L450F; dashed line, G600R.
Only the fifth AP in the equilibration chain is displayed here; the AP shapedoes not change in subsequent cycles.
a Luo-Rudy II AP model.16 By setting wild-type Ito to a
more physiologically relevant GKv4.3, the in silico modelingincluded in this study demonstrates that even the modestincrease in Ito that is expected to be associated with theeterozygous expression of Kv4.3-G600R in the heart isapable of causing the loss of the AP dome in the RVpicardium.
While the role of the Ito current in producing the char-cteristic “spike and dome” morphology of the epicardialP is well established, the precise role of Ito in regulating
he AP duration (APD) in large mammals remains largelyndefined. In both the Winslow-Rice-Jafri canine ventricu-ar cell and the Luo-Rudy II multicellular fiber AP models,he effect of increased Kv4.3 current density on canine APhape and duration demonstrates a bimodal phenomenon,hereby increasing Ito current density in the presence ofigh baseline densities is predicted to shift the AP from apike-and-dome morphology to a shortened AP that lackshe plateau phase.22 Interestingly, in both models, increas-ng Ito current density in the presence of low baseline den-ities is predicted to result in a modest prolongation of thePD. Based on both single-cell and multicellular AP mod-
ling, the effect of Ito perturbations appears to be largelyependent on the strength of the underlying Ito current thathe Ito alterations are superimposed on.
Thus, one could envision a scenario wherein the roughly73.1% and 25.2% increase in Ito current density conferredy heterozygous expression of Kv4.3-L450F and Kv4.3-600R, respectively, in the heart might be expected to
ncrease APD in the LV where Ito current density is rela-ively low, as was observed for Kv4.3-G600R (data nothown) and the Ito activator NS5806,14 resulting in a pro-
longed QT interval and the heterogeneous loss of the APdome in RV epicardium where Ito current densities are theighest. While this scenario certainly fits with the clinicalnd electrophysiological phenotype associated with Kv4.3-450F and Kv4.3-G600R, drawing a definitive conclusionn the impact of Ito gain-of-function mutations on the APD
and thus QT interval based on existing modeling is compli-cated by the existing disagreement between various strandand single myocyte computational models in regards to thetrue role the Ito current plays in determining cardiac APD.23
Even though the exact effect of loss- and gain-of-func-tion mutations in KCND3 on the APD and QT interval inhumans is not thoroughly understood, prolongation of theQT interval in the presence of ST segment elevation appearsto be a clinical hallmark in some BrS cases.24 Thus, thepresence of QT prolongation in the G600R-positive BrSindex case is consistent with the clinical scenario of BrS andnot necessarily an indication of an additional underlyingchannelopathy such as LQTS. The functional investigationof rare mutations in the KCND3 may provide an importantopportunity to deepen our understanding of the physiologicand pathophysiologic roles of the Ito current in shaping thehuman cardiac AP.
Given the modest increase in Ito current associated with
Kv4.3-L450F and Kv4.3-G600R, we speculate that the
ow
miiGpciigl
BsrKw
1032 Heart Rhythm, Vol 8, No 7, July 2011
gain-of-function mutations in KCND3 may leave affectedindividuals susceptible to modest physiologic changes in theinward (INa and IL,Ca) or outward (Ito and IK-ATP) currentsactive during early repolarization. An increase in vagal toneduring sleep or the activation of epicardial predominantIK-ATP currents as a result of metabolic stress could furtherexacerbate the genetically enhanced Ito-mediated transmuralvoltage gradient associated with Kv4.3-L450F or Kv4.3-G600R in the RV epicardium, pushing the AP to morenegative potentials and resulting in the failure of the L-typecalcium channel to activate and the heterogenous loss of APdome at some epicardial sites.9 Due to higher baseline levelsf Ito expression in the RV epicardium of males comparedith females,25 males harboring Ito gain-of-function muta-
tions are likely more sensitive to physiologic changes thatcan precipitate the arrhythmic manifestations of BrS thanfemales, thus the identification of Kv4.3-L450F and Kv4.3-G600R in two male BrS index cases is not surprising.
The discovery of rare BrS-associated Ito gain-of-functionutations in KCND3 supports the hypothesis that a genet-
cally enhanced transmural dispersion of repolarizationn the RV may directly contribute to BrS pathogenesis.iven that loss-of-function mutations in Nav1.5-interactingroteins as well as in the Cav1.2-interacting proteins mayontribute to electrocardiographic and arrhythmogenic man-festations of BrS by exposing a large unopposed Ito currentn the RV epicardium, it stands to reason that mutations inenes encoding Kv4.3-interacting proteins or genetic regu-ators of KCND3 expression that enhance the Ito-mediated
transmural voltage gradient could function as novel patho-genic substrates for BrS. Further interrogation of the Kv4.3macromolecular complex and regulators of KCND3 expres-sion is needed to elucidate the role of the Ito current in thepathogenesis of BrS and other J-wave syndromes.
ConclusionIn conclusion, this study provides the molecular and func-tional evidence necessary to implicate KCND3 as a novel
rS-susceptibility gene (KCND3-BrS or BrS9). Furthertudies are warranted to fully elucidate the mechanismsesponsible for the increase in Ito current observed withv4.3-L450F and Kv4.3-G600R and the full extent tohich rare Ito gain-of-function mutations contribute to BrS
pathogenesis.
AcknowledgmentWe acknowledge support for this work from the MayoClinic Windland Smith Rice Comprehensive Sudden Car-diac Death Program (to M.J.A.), the Dr. Scholl Foundation(to R.M.A., M.J.A.), the Hannah Wernke Memorial Foun-
dation (to M.J.A.), and the National Institutes of Health(R01-HD42569 to M.J.A., R01-HL47678 to C.A., and F30-HL106993 to J.R.G).
References1. Zheng ZJ, Croft JB, Giles WH, Mensah GA. Sudden cardiac death in the United
States, 1989 to 1998. Circulation 2001;104:2158–2163.2. Ellsworth EG, Ackerman MJ. The changing face of sudden cardiac death in the
young. Heart Rhythm 2005;2:1283–1285.3. Wren C. Sudden death in children and adolescents. Heart 2002;88:426–431.4. Liberthson RR. Sudden death from cardiac causes in children and young adults.
N Engl J Med 1996;334:1039–1044.5. Tester DJ, Ackerman MJ. Postmortem long QT syndrome genetic testing for
sudden unexplained death in the young. J Am Coll Cardiol 2007;49:240–246.6. Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the
second consensus conference: endorsed by the Heart Rhythm Society and theEuropean Heart Rhythm Association. Circulation 2005;111:659–670.
7. Chen PS, Priori SG. The Brugada syndrome. J Am Coll Cardiol 2008;51:1176–1180.
8. Kapplinger JD, Tester DJ, Alders M, et al. An international compendium ofmutations in the SCN5A-encoded cardiac sodium channel in patients referred forBrugada syndrome genetic testing. Heart Rhythm 2010;7:33–46.
9. Antzelevitch C, Yan GX. J wave syndromes. Heart Rhythm 2010;7:549–558.10. Delpon E, Cordeiro JM, Nunez L, et al. Functional effects of KCNE3 mutation
and its role in the development of Brugada syndrome. Circ Arrhythm Electro-physiol 2008;1:209–218.
11. Medeiros-Domingo A, Tan BH, Crotti L, et al. Gain-of-function mutation S422Lin the KCNJ8-encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substratefor J-wave syndromes. Heart Rhythm 2010;7:1466–1471.
12. Ackerman MJ, Tester DJ, Jones GS, Will ML, Burrow CR, Curran ME. Ethnicdifferences in cardiac potassium channel variants: implications for genetic sus-ceptibility to sudden cardiac death and genetic testing for congenital long QTsyndrome. Mayo Clin Proc 2003;78:1479–1487.
13. Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologistprogrammers. Methods Mol Biol 2000;132:365–386.
14. Calloe K, Cordeiro JM, Di Diego JM, et al. A transient outward potassiumcurrent activator recapitulates the electrocardiographic manifestations of Bru-gada syndrome. Cardiovasc Res 2009;81:686–694.
15. Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for theelectrocardiographic phenotype of the Brugada syndrome are temperature de-pendent. Circ Res 1999;85:803–809.
16. Gima K, Rudy Y. Ionic current basis of electrocardiographic waveforms: amodel study. Circ Res 2002;90:889–896.
17. Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave.Circulation 1996;93:372–379.
18. Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and othermechanisms of arrhythmogenesis associated with ST-segment elevation. Circu-lation 1999;100:1660–1666.
19. Fish JM, Antzelevitch C. Role of sodium and calcium channel block in unmask-ing the Brugada syndrome. Heart Rhythm 2004;1:210–217.
20. Kurita T, Shimizu W, Inagaki M, et al. The electrophysiologic mechanism ofST-segment elevation in Brugada syndrome. J Am Coll Cardiol 2002;40:330–334.
21. Hermida JS, Denjoy I, Clerc J, et al. Hydroquinidine therapy in Brugadasyndrome. J Am Coll Cardiol 2004;43:1853–1860.
22. Greenstein JL, Wu R, Po S, Tomaselli GF, Winslow RL. Role of the calcium-independent transient outward current I(to1) in shaping action potential mor-phology and duration. Circ Res 2000;87:1026–1033.
23. Decker KF, Heijman J, Silva JR, Hund TJ, Rudy Y. Properties and ionicmechanisms of action potential adaptation, restitution, and accommodation incanine epicardium. Am J Physiol Heart Circ Physiol 2009;296:H1017–1026.
24. Pitzalis MV, Anaclerio M, Iacoviello M, et al. QT-interval prolongation in rightprecordial leads: an additional electrocardiographic hallmark of Brugada syn-drome. J Am Coll Cardiol 2003;42:1632–1637.
25. Di Diego JM, Cordeiro JM, Goodrow RJ, et al. Ionic and cellular basis for the
predominance of the Brugada syndrome phenotype in males. Circulation 2002;106:2004–2011.

















