
Think - c4oh.org acidosis cardiac disease liver disease seizures susceptibility to infections muscle...
70
UNITED MITOCHONDRIAL DISEASE FOUNDATION Think Mitochondria
Transcript of Think - c4oh.org acidosis cardiac disease liver disease seizures susceptibility to infections muscle...

Could it be Mitochondrial Disease?
developmental delays
visual and hearing problems
lactic acidosis
cardiac disease
liver disease
seizures
susceptibility to infections
muscle weakness
diabetes
respiratory complications
loss of motor control
gastro-intestinal disorders and swallowing difficulties
poor growth
Think MitochondriaVolume 1, June 2002 This publication serves to
increase awareness and informmedical professionals, families
and individuals aboutMitochondrial Disease.
UMDF MissionTo promote research for curesand treatments of MitochondrialDisorders and to provide support
to affected families.
UNITED MITOCHONDRIAL DISEASE FOUNDATION
8085 Saltsburg Road, Suite 201Pittsburgh, PA 15239
Voice: 412-793-8077Fax: 412-793-6477
Email: [email protected] Site: www.umdf.org
UNITED MITOCHONDRIAL DISEASE FOUNDATION
ThinkMitochondria

Board of TrusteesCharles A. Mohan, Jr. - ChairmanMark Fleming - Vice Chairman
Stan Davis - SecretaryJohn DiCecco - Treasurer
Bruce H. Cohen, M.D.Charles L. Hoppel, M.D.
Jennifer LymanJane Clarke McManus
Nick RilloRand Wortman
Scientific Advisory BoardMichael J. Bennett, Ph.D., FRCPath, DABCC
Gerard T. Berry, M.D.Richard G. Boles, M.D.
Salvatore DiMauro, M.D.Carol Greene, M.D.
Richard H. Haas, M.B., B.Chir.Richard Kelly, M.D., Ph.D.
Douglas S. Kerr, M.D., Ph.D.Nicolas Krawiecki, M.D.
Arnold Munnich, M.D., Ph.D.Robert K. Naviaux, Ph.D., M.D.
William Nyhan, M.D., Ph.D.Brian Robinson, Ph.D.
Eric Schon, Ph.D.John Shoffner, M.D.
Eric A. Shoubridge, Ph.D.Keshav Singh, Ph.D.
David Thorburn, Ph.D.D.M. Turnbull, M.D., Ph.D.
Rajiv R. Varma, M.D.Georgirene Vladutiu, Ph.D.Douglas C. Wallace, Ph.D.
National OfficeGeorgette Demes, Ph.D.
Director of Development and Programs
Support StaffBetsy AhearnJean Bassett
Antoinette R. BeasleyRobert BolewitzDoug BeckettJulie Hughes
Melinda O’TooleKara Strittmatter
Sandy Turi
© The United Mitochondrial Disease Foundation.All rights reserved.
UMDF’s intent is to keep you informed - we ask thatyou always discuss any diagnoses, treatments, or
medications with your personal physician.UMDF assumes no liability for any information in
this publication.
Afew years ago, I had the opportunity to speak to agroup of parents and doctors about the goals ofthe UMDF. Afterwards, one of the doctors
approached me and wanted to know what medical back-ground I had qualifying me to discuss the approaches nec-essary to reach a cure.
He really got my attention when he said, “You volun-teers are all alike.” I told him I consider myself a donkeynot a volunteer. A donkey harnessedto a large wagon. I told him not tospend any time thinking about thedonkey, but tell the donkey what isneeded to find a cure for mitochondr-ial diseases. I told him I couldn’tdevelop the complex formulas fortreatments and cures and I certainlycouldn’t design the equipment thatwould test for the diagnosis but I could get the test tubes,the computers, and the nuts and bolts that would be essen-tial for research. I told him to load the wagon and don’tworry about the donkey. When the wagon gets heavy wewill find others that will help push and together we couldbe part of the “quest” toward the cure.
UMDF has been pulling that wagon for the past 7 yearsand it sure has gotten bigger and loaded with morerequests than we ever expected, but it hasn’t gotten anyheavier. Every time we look around, we see more andmore people pushing. That’s volunteerism!
This first edition compendium of articles on mitochon-drial disease has been created as a result of many requestsfrom physicians, and families. We hope this compendiumof information will be an aid in increasing awareness ofmitochondrial disease.
UMDF invites you to join us in expanding the field ofmitochondrial medicine. After all, there is plenty of roombehind, and in, our wagon.
Yours toward a cure!
Charles A. Mohan, Jr.Chairman, UMDF
UMDF MissionTo promote research for cures and treatments of
Mitochondrial Disorders and to provide support toaffected individuals and families.

Presentation and Management ofMitochondrial DiseaseMitochondrial Cytopathies: A Primer 2000,Bruce Cohen, M.D., Cleveland Clinic Foundation,Department of Neurology. Provides a review ofbasic biochemistry of oxidative Metabolism,describes mitochondrial molecular genetics andBiochemistry of mito disorders, info on recognizingpatients at risk, assessing patients for furtherevaluation, and organizing a care plan. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
Treatment of Mitochondrial Cytopathies,Deborah Gold M.D., and Bruce H. Cohen, M.D.,Seminars in Neurology, Volume 21, Number 3, 2001Summarizes current treatment options for patientswith mitochondrial disorders. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
Adult Presentations of Mitochondrial Diseases,Robert K. Naviaux, M.D., Spring 2000, UMDFNewsletter. Reviews some of the hallmarks ofmitochondrial disorders in adults, and outlines someof the tests that are required for diagnosis. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
Management Strategy for Acute Illness in Patientswith Mitochondrial Cytopathy,Russell Saneto, D.O., and Bruce Cohen, M.D.,Winter 2000, UMDF Newsletter. Based on experienceand understanding of some of the practical and theoreticalimplications of how the body’s biochemistry affects thebioenergetic health of a mitochondrial patient. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
Anesthesia and Mitochondrial Cytopathies,Bruce Cohen, M.D., John Shoffner, M.D., and GlennDeBoer, M.D., Spring 1998, UMDF Newsletter .Outlines some basic aspects of anesthesia and addressesthe issue of the special risks of anesthesia in patientswith mitochondrial cytopathies. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
UNITED MITOCHONDRIAL DISEASE FOUNDATION
Table of ContentsThink Mitochondria

Selected Topics in Mitochondrial MedicineLeigh Syndrome: Clinical Features andBiochemical and DNA Abnormalities,David R. Thorburn, Ph.D., Summer 1998, UMDFNewsletter. A layman version of Dr. Thorburn’s 1996article from the Annals of Neurology, volume 39,pages 343-351. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
Strokes and Transient Events in Mitochondrial Cytopathies,Bruce Cohen, M.D., Fall 1998, UMDF Newsletter. Providesa brief historical review and basics of mitochondrial genetics.It also addresses MELAS, strokes outside setting of MELASand prevention, diagnosis and treatment. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
Prenatal Diagnosis of Mitochondrial Disease,Brian Robinson, Ph.D., Spring 1999, UMDFNewsletter. Provides insight on prenatal diagnoseswith detailed information divided into discussion ofnuclear DNA defects and mtDNA encoded defects. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
Cyclic Vomiting,Richard Boles, Spring 2001, UMDF Newsletter.Cyclic Vomiting is discussed in this article as onetype of gastrointestinal symptom encountered in patientswith mitochondrial disease. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61
Lab Site Resource (www.geneclinics.org)
UMDF Membership Form
Table of ContentsThink Mitochondria
UNITED MITOCHONDRIAL DISEASE FOUNDATION

Think Mitochondria UMDF1
Objectives1. Review of the basic biochemistry of oxidativemetabolism
2. Describe mitochondrial molecular genetics andbiochemistry of mitochondrial disorders
3. Acquire information needed to recognize thepatient at risk for a mitochondrial cytopathy
4. Assess which patient needs referral for furtherevaluation
5. Organize a care plan6. Package includes: Primer (Biology, Geneticsand Overview of Evaluation, Details ofEvaluation Guidelines, Primer for Management)
Introduction:• Heterogeneous group of disorders• Primary defect is a deficit of energy output• Genetic defects are due to alteration ofmitochondrial enzyme function, due to eithernuclear DNA (nDNA) or mitochondrial DNA(mtDNA) mutations• diagnosis is clinical and usually should be con-firmed with laboratory testing
Pediatric Case Reports:1. 1982 DF was born at term and had no prob-
lems until she was 4 months old when it was foundshe was anemic during an evaluation for fever. Shehad multiple courses of antibiotics for UTIs andotitis media. She developed a picture of over-whelming sepsis and her lactic acid was elevated at12 mM. She subsequently went on to have achronic sideroblastic anemia, pancreatic failure,cardiac dysfunction, hearing loss, ptosis, myopathyand retinal degeneration. She died at the age of 15from cardiac failure. Her mtDNA showed a typicaldeletion seen in Kearn Sayre syndrome.2. 1984 CP was born at term weighing 1900 gm
after an uncomplicated pregnancy, labor and deliv-ery. He had mild dysmorphic features, along withthe fact he was SGA, with rocker bottom feet,
small upturned nose and epicanthal folds. He wasnoted to be hypotonic at birth and at 19 hours oflife had his first seizure. His initial evaluationshowed a metabolic acidosis with a lactic acid of 6mM, with a lactate to pyruvate ratio of 10:1.Seizures continued and he was transferred to ourhospital. Over the next several days his acidosisworsened, and fluids of D10+ 1⁄2 Normal bicarbon-ate were used to attempt to correct his acidosis. Aurine organic acid showed enormous lactateexcretion. A tentative diagnosis of pyruvatedehydrogenase deficiency was made, and the fluidswere changed to a D2.5 solution with amino acidsand intralipids. Within hours, the acidosis reversedand the child was extubated 2 days later. He wastreated with high dose B1 and lipoic acid (stimula-tors of PDH), as well as with polycitra. He wasgiven a regular infant formula with MCT oil andthe lactic acid remained in the 4-6 mM range. Hedied in his sleep at 8 months of life. The E1-αsubunit of PDH was absent. This is an X-linkeddisease.3. 1984 A 7 day old boy was transferred to our
hospital for jaundice. He was born at term withoutproblems. He began regular infant feeds and atewell initially. He became jaundiced at 3 days oflife and was placed under bililights. He becameincreasingly lethargic and developed diarrhea. Hewas placed in an incubator for hypothermia, despitebundling him in blankets. He continued to bottlefeed, despite his increasing lethargy. His bilirubinwas as high as 15 with about 50% conjugated. Onarrival the transport team checked his urine forreducing substances (positive) and glucose (nega-tive). A tentative diagnosis of galactosemia wasmade and subsequently confirmed. He was treatedwith IV glucose and prophylactic antibiotics for afew days and recovered. He was started on asoy-based formula and did well until 3 weeks oflife when he developed e. coli. sepsis. At this time(1999) he is alive and aside from learning
Mitochondrial Cytopathies: A PrimerMitochondrial Cytopathies 2000
Reprinted with permission ofBruce H. Cohen, MD
Cleveland Clinic Foundation, Staff, Department of NeurologyUMDF Conference, Cleveland, OH, 2000

UMDF Think Mitochondria2
problems, doing well on a lactose free diet.4. 1984 A 5 week old girl was seen in the emer-
gency room for dehydration. She was born at termweighing 3100 grams and had been a poor feedersince discharge. She had not gained any weight byher two-week pediatric visit and at the time of theER visit had weighed 2800 grams. She was thoughtto be 5-10% dehydrated. A sepsis work up wasperformed in the ER and antibiotics started. Herinitial bloods tests showed glucose of 46, Na 148,CO2 15, Cl 113, K 4, BUN 1, WBC 2.0, Hb 8.8,Plts 49K. Before a bed was available a ammoniawas drawn (2200 mg/dl) and organic acids (hugeproprionic acid peak).5. 1987 SC was born weighing 3250 gm at term
following a normal pregnancy, labor and delivery.A rapid respiratory rate was noted on the first dayof life and initial labs showed an anion gap of 22.A sepsis work-up was initiated and antibioticsbegun. The initial lactic acid was 20 mM (normal< 2.0 mM). The child was ventilated and bicarbon-ate was begun. The child was transferred to ourhospital where the lactate increased to 60 mM overthe next few days, with a lactate to pyruvate ratioof 40:1, and the child died on day 3 of life. Livertissue was analyzed and there was no COX (com-plex IV) activity present.6. Circa 1990 A 3 week-old infant was admitted
to a Cleveland hospital with e. coli. sepsis andmeningitis. He responded to antibiotics and wasdischarged. Over the next several years he wasadmitted repeatedly for vomiting, dehydration andpresumed sepsis. During a legal review, a positiveState infant screening test was found in the doctor’sfiles, positive for galactosemia.7. 1998 An infant girl was born at CCF follow-
ing a normal pregnancy, labor and delivery. Shewas very hypotonic at birth and was transferred toMetro for respiratory support. She underwent amuscle biopsy procedure, which showed giantabnormal mitochondria. After discharge, shereturned to CCF for ongoing neurologic care. Thesubsequent muscle biopsy determined the etiologyto be a complex III ETC deficiency.8. 1999 A 12 week old was admitted to CCF
with intractable seizures. She had been healthy atbirth but developed seizures in the first month oflife. Her initial laboratory work-up showed a peakof 3-OH isobutyric acid.
Adult Case Reports:1. R.L. 71 year old man with repeated ER visits
and hospitalizations due to altered mental status.The evaluation determined a cirrhotic liver and itwas assumed his alteration in mental status was dueto alcohol ingestion, despite ethanol levels of zero.He was treated with IV fluids and seemed to be inhis normal state within hours. During one ER visit,the neurology consult was a resident that just rotat-ed off pediatric neurology and performed a mini-metabolic evaluation in the ER (elevated NH3,muscle CK and lactic acid and no ketones). Dx:Long-chain acylCoA dehydrogenase deficiency.Comments: LCAD is the first step in the sequentialbeta-oxidation of long chain fats (dietary or stored).With LCAD deficiency, one cannot rely on fats forenergy. R.L. relied on his wife to cook his meals,and when she died, R.L was on his own for the firsttime in his life. He began skipping meals, and ifthe fasts were long enough, glycogen stores weredepleted and he was not able to generate energyfrom fat stores. Hypoglycemic hypoketonuria is animportant clue to detecting disorders of fat metabo-lism. Interviews with the family indicated that R.L.never consumed more than a glass of red wine aday. He has responded to a diet very low in fats,with frequent meals (including bedtime snacks)high in complex carbohydrates. Supplementsinclude levo-carnitine & multivitamins.2. S.K. 57 year old man came from India for
evaluation of his cardiac conduction defect. Hisfamily history is significant for his mother dying ata young age of a cardiomyopathy, and that hisbrothers have similar problems to his. He was thepresident of a successful business in India. As ayoung man he was diagnosed with calcifyingpancreatitis, and had been on replacement digestiveenzymes for years. He has had numerousadmissions for non-surgical bowel obstruction andhas had several exploratory laporatomies, the etiol-ogy was never determined. Over the last 15 yearshe lost 75 pounds, and developed aching in hislimbs. In January 1996 he was admitted for astroke, and a cardiac evaluation determined he wasin A-fib. He was placed on Coumadin. He wasreadmitted for a stroke several months later, andwas again found to be in A-fib. His neurologist andcardiologist referred him to CCF Cardiology forplacement of a pacemaker. In October 1996, after a

Think Mitochondria UMDF3
24 hour journey, he visited our cardiology depart-ment, and was told that a pacemaker was notindicated. While resting in the waiting room, try-ing to gather the energy to walk back to the hotel,he collapsed. He spent the next two weeks in astupor in the NICU, and again, another stroke wasdetected, along with his A-fib. The patient wasfound to have an elevated lactate, ammonia and CK(MM). Polarography revealed decreased oxidationof substrates that donate reducing equivalants tocomplex I and beta oxidation. An evaluationdetermined that he had a deficiency in carnitinepalmitoyl transferase II activity. Treatment wasstarted and included a low fat diet with frequentmeals rich in complex carbohydrates, along withlevo-carnitine, CoQ10 and other vitamins. He hashad no further events since his hospitalization inOctober 1996. Comments: In order for the mito-chondria to burn fats, the free fatty acid must firstenter the mitochondrial inner membrane. CPT Icatalyzes the conversion of the activated free FA(acyl CoA) + carnitine to the acyl-carnitine. Acarnitine translocase exchanges the acyl-carnitineacross the inner membrane for a free carnitinemolecule. CPT II catalyzes the conversion of theacyl-carnitine to the acyl-CoA and free carnitine.The acyl-CoA can then enter the beta-oxidationspiral. CPT II deficiency usually results in exerciseintolerance, muscle cramping and fatigue inyoung adults, but is also known to cause an earlycardiomyopathy.3. P.L. is a 40 year old woman with “CFS”. She
has been evaluated by dozens of CCF doctors andseems to have secondary gain issues. After asurgical procedure she did not recover normallyfrom anesthesia, and remained apneic for 30minutes, and was referred for evaluation. Anextensive laboratory evaluation was not helpful. Amuscle biopsy was performed and demonstratedmild mitochondrial proliferation and mild abnor-malities in electron transport chain activity. It isnot clear if she does or does not have a genuinedisorder.
Catastrophic Presentations of Metabolic Diseasein the Newborn• nonspecific finding• lethargy, irritability, hyperactivity• failure to feed well
• hypothermia or fever• cyanosis• seizures• vomiting• RTA• jaundice (early and/or prolonged)• diarrhea or abdominal distension
Brief Differential Diagnosis• organic acidemias: MSUD, propionic, isovaleric,methylmalonic, others• urea acid cycle defects: carbamyl phosphate syn-thetase deficiency, OTC, citrullinemia, argini-nosuccinic aciduria• carbohydrate disorders: galactosemia, hereditaryfructose intolerance• aminoacidopathies: homocystinuria, tyrosinemia,nonketotic hyperglycinemia• endocrinopathies: “CAH, congenital diabetes”
Exam• odor• neurologic: tone, level of alertness, DTRs• general: dysmorphic features
Lab Evaluation• glucose, glucose, glucose• electrolytes, calculate anion gap• CBC (look for low counts)• BUN (low BUN indicates failure of urea acidcycle, either primary or secondary)• Lactate, pyruvate, and L/P ratio• ⇑ lactate with L/P 10-20 indicates a disorder ofpyruvate metabolism such as PDH deficiency• ⇑ lactate with L/P of > 20 indicates a disorder ofoxidative phosphorylation• Ammonia• CK• Biotinidase level (usually causes problems after 6months)• VLCFA (neonatal paroxysmal disorders)• Amino Acids (blood and urine)• Organic Acids (quantitative)• Acyl carnitines (blood and urine)• Skin biopsy for EM and fibroblast culture• Muscle biopsy

UMDF Think Mitochondria4
Treatment(Supportive and varies according to diagnosis)
Presentation of Mitochondrial Disease in AdultsAs varied as in children, more complicated to diag-nosis because adults have acquired other diseases• Childhood onset mitochondrial diseases• Muscle: new muscle weakness, cramping• Brain: migraine, stroke or stroke-like events,dementia, MS-like presentation• Endocrine: diabetes (~5% of DM in Great Britainmay be due to the mtDNA 3243 mutation)• Cardiac: early cardiomyopathy, cardiacconduction defects (association of LHON withWPW, etc).• Systemic: CFS-like illness
Brief Differential Diagnosis:primary endocrine diseasevitamin deficiency: B12homocystinuria and associated disordersprimary muscle disease: polymyositis,dystrophin associated glycoprotein musculardystrophies“chronic fatigue syndrome”autoimmune disordersglycogen storage disordersdepression and related psychosomatic disordersother neurodegenerative disorders (MS, ALS, HD,combined systems degeneration)
History of Mitochondrial Diseases:• 1962 Luft describes first case of a euthyroidwoman with extreme hypermetabolism andgigantic mitochondria in muscle• 1962 Milton Shy describes mitochondrialproliferation in myopathic patients• 1962 W. King Engel applies histochemical tech-niques to muscle, uses modified Gomoritrichrome stain• 1975 L.P. Rowland lumper/splitter debate regard-ing KSS/progressive ophthalmoplegia• 1975 Koenigsberger describe a case of MELAS• 1981 MtDNA genome mapped• 1982 Rowland and Fukuhara presentindependent papers regarding KSS and MERRF• 1984 MERRF, MELAS, KSS paper in AnnNeurol by Pavlakas, Phillips, DiMauro, DeVivoand Rowland
• 1985 Carnitine palmitoyltransferase (CPT) defi-ciency described by DiMauro• 1991 Biochemical and molecular analysisbecomes commercially available• 1995 Review articles appear in major medicaljournals
When To Suspect Mitochondrial Dysfunction:There is no one identifying feature of mitochon-
drial disease. Patients can have combinations ofproblems whose onset may occur from before birthto late adult life. Mitochondrial diseases should beconsidered in the differential diagnosis when thereare these unexplained features, especially whenthese occur in combination.• Encephalopathy
SeizuresDevelopmental Delay or Regression(including early and late-onset dementia)MyoclonusMovement Disorders (dystonia, dyskinesias,chorea, etc.)Complicated MigraineStroke
• Neuropathy• Cardiac Conduction Defects or Cardiomyopathy• Hearing Deficits• Short Stature• Disorders of Extraoccular muscles includingptosis, acquired strabismus and ophthalmoplegia• Diabetes• Renal tubular disease• Visual Loss (retinitis)• Lactic acidosis, which may be mild
Ultrastructure & Function• mitochondria are intracellular double-membraneorganelles• role is to generate ATP (the universal currency orfuel)• Defects include:
1. mitochondrial transport2. substrate utilization3. citric acid cycle4. oxidative-phosphorylation coupling5. respiratory chain defects
• From a molecular “point of view” the defects ofthese genes include:
1. defects in transcription, translation or

Think Mitochondria UMDF5
post-transitional processing of mitochondrialproteins coded for by nuclear genes(Complex II , PDH, or CPT-II deficiency)2. defects in mtDNA genes (includingprotein, rRNA or tRNA)3. defects of nuclear-encoded factors thatmodulate mtDNA genes4. Defects in non-protein parts of themitochondria (CoQ10 deficiency, prostheticgroups, Menkes)
Mitochondrial DNA• Circular gene• 16,569 base pairs (exactly)....these arenumbered 1 to 16,569• Heavy and Light strand, each with its ownorigin of replication• All coding sequences are contiguous (no introns)• Each mitochondria contains 2 to 10 copies of themtDNA• Each cell can have hundreds of mitochondria• mtDNA mutates 6 - 17 times faster than nuclearDNA• The only non-coding region is a 1 kB regionwhich contains the origin of replication of the Hstrand and the promoters for the L and H strandtranscription• The mitochondrial genetic code differs from theuniversal code....therefore the mitochondrial pro-tein synthesis relies on nuclear encoded transcrip-tional and translations factors with tRNA andrRNA derived from mitochondrial genes• mit genes: 13 protein-encoding genes7 subunits for NADH dehydrogenase (complex I)[25 total subunits]3 subunits of cytochrome oxidase (COX)(complex IV) [13 subunits]2 subunits of ATP synthetase (complex V) [12subunits]apocytochrome b (complex III) [9-10 subunits]• syn genes: protein-synthesis genes2 rRNA (12 and 16s)22 tRNAs
Genetics of mtDNADuring fertilization the sperm “donates” its
nuclear DNA. The sperm contributes little to nomitochondria and therefore no mtDNA. Therefore,our mitochondria are our mothers.
• 100s of mitochondria per cell• 2 - 10 copies of mtDNA per mitochondria• 1000s of mtDNAs contribute to the mitochondrialgenotype of each cell. Remember that one (ortwo) copies of nDNA contribute to the genotypeof a cell, and except for those diseases withmosaicism, all tissues in nDNA disorders aregenotypically identical.• Heteroplasmy: During mitosis the mitochondriaare randomly distributed to daughter cells.Therefore if the original cell has a mixture of dif-ferent mtDNAs (an example is the situation wherea single mitochondria has one mutant mtDNA and9 wild mtDNAs in the fertilized ovum), the dis-tribution of the mutant mtDNA will vary widelyfrom cell to cell and organ to organ. i.e.: Thenewly formed mtDNA will be distributed random-ly into the newly formed mitochondria of thesedaughter cells. What ends up happening is thatvariable ratios of normal to mutant mtDNA arefound in each cell. Because of heteroplasmy, thenumber of mitochondrial genotypes is enormous.The degree of heteroplasmy can be quantitated ifa mtDNA mutation can be detected.• Homoplasmy: The mtDNA is all one type withina tissue....this is the normal state.• Threshold expression: All tissues require ATP tosurvive. Some tissues require a greater flux ofATP production and utilizatioin, and thereforerequire the integrity of the ox-phos enzyme sys-tem. Cellular dysfunction will occur if notenough ATP can be generated. The tissues mostaffected are those where there is little post-birthmitotic activity (which would cause a selectionbias towards cells with healthy mitochondria), i.e.:brain, type I skeletal muscle, cardiac muscle,nerve, liver, proximal tubule of the kidney. Mosttissues do not require the ox-phos engine toalways be functioning at 100%. In fact, most tis-sues probably can get by on much less than 100%activity. Therefore, whether or not a tissue isaffected depends on the metabolic needs of thetissue and the ability of the mitochondria to meetthat need. The phenotypic expression onlybecomes evident when a proportion of the corre-sponding mtDNA reaches a critical level.• Phenotype Variability: The segregated mtDNAleads to graded biochemical defects. Not everyaffected family member has the same exact phe-

UMDF Think Mitochondria6
notype. The genetic defect may not have reacheda threshold in the mother, for example.• mtDNA and Aging: OXPHOX activity declineswith age in muscle, liver and brain. In heart mus-cle, the cytochrome c oxidase activity decreaseswith age, seemingly due to an accumulation ofmtDNA mutations and base substitutions. Theextent of damage is tissue specific; for example inthe brain, the damage seems most severe in thebasal ganglia, followed by the cortex. The cere-bellum does not accumulate mtDNA mutations asa function of age. OXPHOS defects are reportedin PD, Huntingtons, AD, dystonia. It is unknownif OXPHOS activity diminution is the cause of“aging” (the makers of vitamins want us to thinkso anyway.)
To complicate matters:• -environmental factors play a role (EtOH andtobacco accelerate the optic nerve damage inLebers Hereditary Optic Neuropathy)• -point mutations may be pathologic or non-patho-logic• -immunologic factors may play a role...for exam-ple there is an association between MS andLHON in A11778 - positive females....optic nervedamage in LHON may be immunologicallymediated and mtDNA may play a role in MS• -a specific phenotype may have many differentgenotypes associated with it (LHON has 17different known mutations, some forms of LHONare more severe than others and in one form, 28%recover vision
Biochemistry of the Respiratory Chain andOxidative Phosphorylation (OXPHOX)• Complex I: Transfers e- from NADH tocoenzyme Q.• Complex II: Transfers e- from FADH or FMNHto coenzyme Q. Succinate dehydrogenase (Krebscycle) is part of the complex. This is the only partof the chain that is not coded in part by mtDNA,and succinate dehydrogenase deficiency is theonly identified nDNA disorder causing anOXPHOS disorder.• Complex III (coenzyme Q-cytochrome c reduc-tase): Transfers e- from reduced CoQ tocytochrome c. The apoprotein of cytochrome b isa mtDNA coded polypeptide.
• Complex IV (cytochrome c oxidase or COX):reduces molecular oxygen to water, using the e-
donated from cytochrome c.• Complex V (ATPase): Converts ADP and Pi toATP.• Coenzyme Q10 and cytochrome c act as shuttlesbetween complex I and III and II and III.Coenzyme Q10 also is a potent antioxidant.
Evaluation of a patient with suspectedmitochondrial disease:• History• Physical Exam• Lactate, Pyruvate (blood and CSF)• Amino Acids (serum, urine and CSF)• Organic Acids (urine, CSF)• Carnitine & Acyl Carnitine• Audiogram• ECG• Eye exam• Blood for mtDNA (if you know what you arelooking for....search and detect missions blindlyhave less of a chance in finding the mutation)• Blood for nuclear DNA (limited availability, onlya few defects have been identified)• Muscle for mtDNA (same as above)• Muscle of OXPHOX analysis (spectrophotometryor polarography)• Muscle of immunologic staining of mtCOX sub-units and nCOX subunits• Fibroblast Culture for OXPHOS analysis
Pearls1. Mitochondrial cytopathies are not one disease.2. Keep in mind that there are mitochondrial dis-eases that are due to inherited mutations(germline mutations) and those due to acquiredmutations (somatic mutations). Furthermore it isreasonable to think that there are those that areprimary (something inherently wrong with mito-chondrial function) and those that are secondary(the mitochondria is injured as a bystander toanother process).
3. Not all patients with mitochondrial cytopathieshave systemic lactic acidosis. As a general rule,aside from certain mtDNA defects such asMELAS, MERRF, and KSS, lactic acid levelsoften decrease to normal as the child gets older.
4. A single normal blood or urine lab test does not

Think Mitochondria UMDF7
rule out mitochondrial disease. This is true fororganic acids, lactic acid, carnitine analysis andamino acid analysis.
5. Brain dysmorphology (agenesis of the corpuscallosum, migrational defects) does not rule outmitochondrial disease.
6. Think of mitochondrial cytopathy or other meta-bolic disease in the setting of atypical white mat-ter disease (atypical multiple sclerosis, work-upnegative leukodystrophy)
7. Think of mitochondrial cytopathy when there area number of organ systems involved.
8. The majority of mitochondrial diseases are prob-ably not due to mutations in the mitochondrialDNA genome.
9. It is not possible to chart the future of a personwith a mitochondrial cytopathy. Those with ahigh degree of pathologic mtDNA heteroplasmydo worse, on average, than those with a lessordegree, but this is only valid for populations ofpatients and cannot be used to predict what willoccur in any one patient. It is not possible topredict the response to vitamins, supplements ordiet changes before they are tried. It is not pos-sible predict the course of siblings or other firstdegree family members based on what happenedwith the first family member identified.Remember the literature that is available early inthe description of a particular disease (such asexists today with mitochondrial cytopathies)reflects what happens with the sickest patients.Many of those that are not critically ill haveescaped detection by doctors, and thereforemany of those people that have been diagnosedwith a mitochondrial disease in the last 5 years,free of identifiable mtDNA mutations, may havea better overall prognosis than what the literaturesuggests.
10. Mitochondrial diseases that impact on thedeveloping fetus may cause permanent problemswith brain development. During the process ofembryogenesis the brain cells 1) undergo rapidcell division, 2) begin migrating to their finaldestination in the brain, 3) begin connecting toeach other and 4) myelinate (the white mattersurrounds parts of the nerve cells). The firstthree processes are finished before a baby isborn. The fourth process begins before birth andcontinues until the 40s. Although the brain con-
tinues to develop in some respects after birth,some injuries that occur before birth are notrepairable by normal development or by medica-tion or treatments. For example, any metabolicdisease that interferes with processes 1,2 or 3may result in inevitable mental retardation.These injuries have been labeled mitochondrialembryopathies. Although treatment may helpother aspects of mitochondrial dysfunction, thepart of the illness that results in damage to thenon-plastic brain functions will not improve.Predicting potential improvement in childrenunder five years of age can be difficult in somecircumstances.
11. This is an evolving field. 20 years ago therewere fewer identified patients than there are peo-ple at this conference. Expect to relearn thisyear’s truth next year.
Laboratory Evaluation for Disorders of EnergyMetabolismLaboratory testing is the usual method physi-
cians go about evaluating patients for disorders ofenergy metabolism (which include mitochondrialdisorders, disorders of oxidative phosphorylationand β-oxidation). Most hospitals do not have ametabolic laboratory and therefore can run only themost basic tests. However, most hospitals will sendspecimens to any laboratory in the country. Not alllaboratory tests are required for all patients, andyour physician may decide that some of these testsare not necessary. The lists are authoritative, butare meant to serve as a general guide for evalua-tion. Not all metabolic disorders primarily affectenergy metabolism, but the clinical features mayoverlap. Testing for these metabolic disorders arelisted in a separate table. There is no substitute forgood clinical judgement.The initial laboratory evaluation is generally
used as a non-invasive screening for inborn errorsof energy metabolism. If the results of this evalua-tion are suggestive of a specific disorder, a directtest for the disease in question may be able to beperformed. If the results of the initial evaluationare normal and there is a strong suspicion of a dis-order of a mitochondrial disease, a more intensiveevaluation is performed.The secondary tests are more invasive (and may
include a spinal tap) and because some of the tests

UMDF Think Mitochondria8
may require urine specimens collected over time, abladder catheter may be required in young children.Many of these tests require the specimens to besent to a special laboratory. Abnormalities foundon the secondary tests will guide the physician as tothe direction of further testing. However, as withthe initial testing, normal results do not eliminatethe possibility of a mitochondrial disease, but makeit less likely.The tertiary tests are invasive and/or expensive,
and may carry with them some risks, such as meta-bolic decompensation during a fast. However, ifthe physician strongly suspects a metabolic illness,these tests may be diagnostic. The muscle biopsyis a tertiary test, but is listed separately because it isthe most complicated and invasive of all tests, andin children requires a general anesthesia. Althougha muscle biopsy can be performed at any medicalcenter, very few centers have the ability to do allthe testing necessary to make a diagnosis.Therefore, the physician must be very conscien-tious in planning before the biopsy is done.
A lists of tests and centers performing thesetests can be found at the following web address:http://biochemgen.ucsd.edu/wbgtests/wbgtests.htm
Muscle BiopsyMuscle tissue can be used for tests that can be
diagnostic, even when the above tests are normal.Because this is the most invasive test, the risks andcosts of the procedure must be weighed against thechance the biopsy will yield positive results and thebenefits gained by a diagnosis (treatment decisions,family planning). Before a muscle biopsy is done aplan needs to be arranged for how the muscle isdistributed. References labs should be contactedbefore the biopsy is done so that preparation of the
muscle is done correctly. Muscle can be sent for:• Routine light microscopy including modifiedGomori Trichrome stain (checking for ragged redfibers)• Specific immunohistochemistry (cytochromeoxidase and COX subunits), succinatedehydrogenase, etc.• Electron microscopy (useful to view the structureof the mitochondria, evaluate for accumulation ofexcessive mitochondria in the subsarcolemmaregion and evaluate for mitochondrialproliferation.• Electric Transport Chain Activity (photometricanalysis), preferable performed on fresh musclebut can be done on frozen muscle.• Oxidative Phosphorylation Activity (oxygenuptake), which can determine the activity of allfive complexes, state iii and state iv respiration,respiratory control ratio and estimate efficiency ofcoupling of electron transport and oxidativephosphorylation. This can be run on fresh muscleonly.• Enzyme activity for β-oxidation disordersincluding the enzymes of the β-oxidation spiraland carnitine transport.• Determination of carnitine and acyl-carnitinelevels, Co-Enzyme Q10 levels.
Testing That May Be Necessary in Patients withMitochondrial CytopathiesBrain MRI, MRSEye: Retinal exam, electroretinogramHeart: EKG and echocardiogramThyroid Function Tests (blood)Ears: Audiogram or BAEP
Initial Laboratory EvaluationTest Tissue
* Comment
Glucose BElectrolytes BBlood Counts B
Lactate B Proper technique must be used, tourniquet must be released before blood is sampledAmmonia B
Metabolic Screen B,U The metabolic screen varies between hospitals, but may include screening testing for avariety of disorders as well as urine and blood amino acid profile, and screening organicacid testing
Ketones B,U Most valuable if collected at the time of an illness*B = blood, U = Urine
S

Think Mitochondria UMDF9
Treatment:• At this time, there is no cure for these disorders.• Purposes for treatment• alleviate symptoms• slow down the progression of the disease
• Effectiveness of treatment• varies from patient to patient, depending onthe exact disorder and the severity of thedisorder• as a general rule patients with mild disorderstend to respond to treatment better than thosewith severe disorders.• in some circumstances, the treatment can betailored specifically to the patient, and thattreatment is effective, whereas in other circum-stance, the treatment is “emperic”, meaning thatthe treatment makes sense, but that the benefitof treatment is not obvious or proven to beeffective
• Benefits of Treatment/Effectiveness of Therapies• Vary• treatment may be beneficial and notedimmediately in some disorders• benefit of treatment may take a few months tonotice• benefit of treatment may never be noticed, butthe treatment may be effective in delaying orstopping the progression of the disease• some patients may not benefit from therapy
• Key Points to Treatment• dietary• vitamins and supplements• avoidance of stressful factors• These recommendations must be tailored bythe patient’s physician to meet that patient’sneed. Many of these therapies are totallyineffective in some mitochondrial disorders andwould be a waste of time, money and effort. Insome cases, the treatment could be dangerous.
Dietary TherapyMany patients, including young children or men-
tally impaired persons have already “self-adjusted”their diet because they know what foods their bodyseem to tolerate. The points below are not meant tobe suggested therapies for all patients withOXPHOS disorders, and some of the points aredangerous for patients with other disorders(4b could be lethal in pyruvate dehydrogenase defi-ciency for example). Do not make any of thesedietary changes without consulting a physician. Adietitian experienced in metabolic disorders may behelpful.1. Avoiding fasting is perhaps the most importantpart of treatment. This means avoid prolongedperiods without a meal (even an overnight “fast”from 8 pm to 8 am may be dangerous in somepatients). This also means that some patientsshould not intentionally try to lose weight. In
Secondary Laboratory EvaluationTest Tissue
* Comment
Lactate B,CSF see abovePyruvate B Proper determination of pyruvate requires the specimen be instantly deprotinized.L/P Ratio B The ratio of lactate to pyruvate can be very helpful in determing which type of disorder
may be presentAmino Acids B,U, CSF Urine collections may be random or timed; and may be collected after a meal or after a
fasting period, depending on the clinical situation. “Generalized aminoaciduria” mayindicate the presence of a mitochondrial cytopathy, as well as other medical conditions.
Organic Acids U, CSF Samples must be kept refrigerated or frozen. Different techniques, some more sensitiveare used by certain laboratories. Urine collections may be random or timed, and may becollected after a fasting period, depending on the clinical situation.
Carnitine Analysis B,U Most laboratories determine the free carnitine and total carnitine. Fractionation intospecific acyl carnitines may be helpful in some situations. Urine collections may berandom or timed, and may be collected after a fasting period, depending on the clinicalsituation.
Ketones B,U Fractionation of ketones into β-hydroxybuterate and acetoacetate may be helpful. Thistest is most valuable if collected during an acute illness or after a fast.
Free Fatty Acids BMitochondrial DNAPoint Mutations
B If a patient fits into a specific, well-described mitochondrial phenotype, testing forspecific, known point mutations may be helpful at this stage.
Mitochondrial DNASouthern Blot
B If a patient fits into a specific, well-described mitochondrial phenotype, Southern blottesting may be helpful at this stage.
*B = blood, U = Urine, CSF = Cerebral Spinal Fluid
T

UMDF Think Mitochondria10
Other Metabolic Tests That May Be Indicated in Certain SituationsTest Tissue Disease(s) Comment
Uric Acid, Creatinine B,U Lesch-Nyhan These patients often have lactic acidosisCopper, Ceruloplasm B,U Menkes Kinky Hair Disease,
Wilsons Disease, othermovement disorders anddementias
Very Long ChainFatty Acids
B Adrenoleukodystrophy and otherdisorders of peroxisomalmetabolism
Lysozomal Enzymes B.U variety of storage diseases andleukodystrophies
Table 1: Problems Associated with Mitochondrial CytopathiesOrgan System Possible ProblemsBrain developmental delays, mental retardation, dementia, seizures, neuro-
psychiatric disturbances, atypical cerebral palsy, migraines, strokesNerves weakness (which may be intermittent), neuropathic pain, absent reflexes,
gastrointestinal problem (ge reflux, constipation, pseudo-obstruction),fainting, absent or excessive sweating resulting in temperature regulationproblems
Muscles weakness, hypotonia, cramping, muscle painKidneys proximal renal tubular wasting resulting in loss of protein, magnesium,
phosphorous, calcium and other electrolytesHeart cardiac conduction defects (heart blocks), cardiomyopathyLiver hypoglycemia (low blood sugar), liver failureEyes visual loss and blindnessEars hearing loss and deafnessPancreas diabetes and exocrine pancreatic failure (inability to make digestive
enzymes)Systemic failure to gain weight, short statue, fatigue, respiratory problems
including intermittent air hunger
Tertiary Laboratory Testing
Test CommentRepeat Testing Repeating some of the above listed tests, sometimes under different conditions (such as during an
illness), may be helpful.Provocative Testing Under monitored conditions, usually in the hospital, repeating some of the above tests after a fast or
after a specific meal or intravenous infusion, may be helpful.Skin Biopsy A skin (also known as a fibroblast) culture can be established with the skin obtained from a biopsy.
This can be sent for testing electron transport chain activity, β-oxidation disorders, as well as for avariety of other specific diseases.
Mitochondrial DNA PointMutations
If a patient fits into a specific, well-described mitochondrial phenotype, testing for specific, knownpoint mutations may be helpful at this stage.
Mitochondrial DNASouthern Blot
If a patient fits into a specific, well-described mitochondrial phenotype, Southern blot testing may behelpful at this stage.
Coenzyme Q10 Blood Test

Think Mitochondria UMDF11
some patients an unintended fast resulting froman illness that causes vomiting or loss ofappetite (like the flu) should be hospitalized toensure continuous nutrition (intraveneous glu-cose for example).
2. Small frequent meals may be better than a typi-cal 3-meal-a-day routine for some patients.
3. A snack before bedtime may be helpful in somepatients. This snack should not be mainly“sugar”, like a candy bar, jello or sweetenedcereal. It is usually best if the snack consists of acomplex carbohydrate. Cornstarch is the bestcomplex carbohydrate, but this is not very tasty.There is a cornstarch bar called ZBar which isnot bad. Theoretically, the best snack would bea homemade low-sugar rice pudding thickenedwith a lot of cornstarch. If you come up with atasty recipe, let the UMDF know. Pasta, breadand butter, unsweetened cereal (oatmeal) or asandwich are acceptable.
4a. In patients with complex I deficiency, the addi-tion of extra fat (fats include added oil, butter, &margarine, as well as other “fatty foods”) to thediet should theoretically result in more energyproduction. This is because the metabolism ofprotein and carbohydrate produces electrons thatmust flow through complex I, which is obvious-ly not working properly in complex I deficiency,but fats produce electrons that in addition toflowing through complex I, also produces elec-trons that can flow through complex II (bypass-ing complex I). Therefore, if complexes II, III,IV and V are working properly, fats should beslightly more effective in producing energy. Asmall clinical study yielded mixed results, withsome patients improving and others not.
4b. In some patients with OXPHOS disorders,reducing fat may be helpful. This includesreducing added oil, butter, & margarine, and cut-ting down on cheese and fatty meats. This rec-ommendation is not meant to avoid fats alto-gether. A defect in the OXPHOS can create an“energy backup”, as the respiratory chain cannothandle the flow of electrons coming into it. Thisbackup may result in the formation of excessfree fatty acids (fats waiting to be burned),which can poison the enzyme (adenosinenucleotide translocase) that exchanges the low-energy ADP located outside the mitochondria
for the high-energy ATP formed at complex V.If you take the approach of limiting fats, extraeffort needs to be made to increase the total car-bohydrate (in the form of complex carbohy-drates) in the diet.
4c.In some patients (see #4a and #4b above),adding fat in the form of medium chain triglyc-erides (MCT), may be helpful. Medium chaintriglycerides of 8 to 10 carbons long are easierto metabolize (turn into energy) than the longerchain triglycerides (those with 12-18 carbons)because they do not require carnitine to be trans-ported into the mitochondria. MCT Oil ismainly made of 8 and 10 carbon triglyceridesand this type of oil does not occur in nature, butis made from coconut oil. MCT Oil is madeby the baby formula company Mead-Johnson. Itcomes in quart bottles, available by prescriptionand runs about $70 a quart. It can be added likeoil over pasta and rice. You can cook with it,but this is a light oil and burns easily. The spe-cial rules are explained in a recipe book that youcan request from the pharmacist. Depending onthe situation, a patient may benefit from a fewteaspoons to a few tablespoons a day. There areoils sold in health food stores called “MCT Oil”or “medium chain triglyceride oil”. Many ofthese contain unprocessed coconut oil, which isa 12 carbon triglyceride that requires carnitinefor entry into the mitochondria. This would be awaste of money. Unless there is a certifiedanalysis on the label, stay away from these prod-ucts and stick with the Mead-Johnson brand.
5. Iron generates free radicals under certain condi-tions, which is especially bad in mitochondrialdiseases because the free radicals injuremitochondrial DNA and “poke holes” in themitochondria, making a bad problem worse.Therefore, iron is theoretically harmful inexcess. There is no need to give supplementaliron in vitamins, nor is there a reason to eatfoods rich in iron, such as extra red meat, for thepurpose of eating foods rich in iron. This doesnot mean that the person should not eat redmeat, especially if they enjoy it. There is no rea-son to take vitamins with added iron. In addi-tion, vitamin C enhances the absorption of ironfrom the intestines, and vitamin C should not begiven around a meal rich in iron. This is impor-

UMDF Think Mitochondria12
tant to remember because some experts feel thatvitamin C is a good antioxidant, and also may behelpful in some disorders of OXPHOS.
Vitamins and CofactorsVitamins and cofactors are compounds that are
required in order for the chemical reactions, whichmake energy, to run efficiently. By definition, acofactor can be made by the body, whereas a vitamincannot, and therefore must be eaten. For most peo-ple, a regular diet contains all the vitamins one couldpossibly need and their bodies can make as much ofany specific cofactor that it needs. For those withmitochondrial disorders, added vitamins and cofac-tors can be useful. The use of supplemental vitaminsand cofactors is controversial in that there are noproven benefits to some of these therapies. For dis-orders of OXPHOS, coenzyme Q10 is considered asa generally accepted effective therapy, although it
may not ultimately be effective for an individualpatient. Other treatments are proven therapy inspecific disorders, but in other disorders cannot beconsidered as “proven and effective” but still maybe helpful. Some treatments should only be under-taken under the specific guidance of your physi-cian. For specific information about the controver-sy, as it relates to you or your child's situation, askyour physician. Most of these vitamins can be pur-chased from many sources, including the drugstore.The sources listed above have been found to be
fairly priced (often significantly less than the drug-store) and sell very high quality products. Thesesupplemental compounds can serve two functions:-POSSIBLY ENHANCE ENZYME FUNCTION
AND RESULT IN IMPROVED EFFICIENCY OFENERGY GENERATION-SERVE AS ANTIOXIDANTS, WHICH MAY
SLOW THE PROGRESSION OF THE DISEASE
Table 2: Vitamins and Supplements That May be Helpful
Table 2a: Suggested to most of my patientsSupplement Dose Range Patient Dose
CoQ10 5 – 15 mg/kg/day
levo-carnitine (Carnitor
)Variable, starting dose of 30mg/kg/day, typical max 100 mg/kg/day
Riboflavin (B2) 50-100+ mg a day
Table 2b: Second Tier SupplementsSupplement Dose Range Patient Dose
Acetyl-L-Carnitine 250 – 1000 mg per dayThiamine (B1) 50-100 mg a dayRiboflavin (B2) 50-100+ mg a dayNicotinamide (B3) 50-100 mg a dayVitamin E 200-400 IU; 1 - 3 times a dayVitamin C 100-500 mg; 1 - 3 times a day
Lipoic Acid (α-lipoate) 60-200 mg; 3 times a day
Selinium 25-50 micrograms a day
β-carotene 10,000 IU; every other day to daily
Biotin 2.5 – 10 mg a dayFolic Acid 1 – 10 mg a day
Table 3: Medication, Minerals, Vitamins, Substrates that May be Helpful (only to be usedunder a physicians direction)Supplement Dose Range Your Dose
Calcium VariableMagnesium VariablePhosphorous VariableVitamin K3 5 - 30 mg per day (1-800-266-9583)
Succinate 6 gm per dayCreatine 5 gm bid after initial load (adults)Uridine To be determinedCitrates variablePrednisone variable

Think Mitochondria UMDF13
Avoidance of Physiologic “Stress”Physiologic stress is an external factor that may
result in worsening the metabolic situation, whichmay result in temporary, or in sometimes, perma-nent worsening of the condition. It is impossible toavoid all physiologic stressful conditions, so oneshould not attempt to do so. However, recognizingwhat may be stressful for a patient allows one toadjust the lifestyle. Many patients and their parentshave already identified these stresses, despite not
knowing why the stresses were important, andavoid them.• Cold Stress is extremely important. Thermal reg-ulation (temperature control) is not always normalin people with mitochondrial diseases and expo-sure to cold can result in severe heat loss and trig-ger an energy crisis. When going out into thecold, all exposed body parts should becovered, and exposure to extreme cold be avoidedfor anything more than a short period. Over
Mitochondrial Evaluation WorksheetName:
# DOBTest Laboratory Date ResultsCKLactatePyruvateL/P RatioAmmoniaFree T4, TSHElectrolytesGlucoseKetones; urineKetones; blood
Amino Acids; bloodstate:Amino Acids; urinestate:Organic Acidsstate:Carnitine; urinestate:Carnitine; bloodstate:Acylglycines
EKGCardiac Echo
Eye ExamERG
MRI
AudiogramBAEP
Molecular GeneticsBlood
Southern Blot
Point Mutations
Skin EM
Fibroblast CultureFibroblast Studies
Muscle HistologyMuscle EMMito YieldMuscle OXPHOS

UMDF Think Mitochondria14
bundling can be a problem too (see below).• Heat Stress can be a problem in some people.This is especially true of those with an inability tosweat normally. Heat exhaustion and heat strokemay occur on hot days. An example of a typicalscenario for this situation would be a child thatseems to “wilt” in situations like hot classrooms,whereas the other students function normally.Light clothing is important. Patients should avoiddirect sunlight on hot days and stay indoors if it istoo warm outside. An air conditioned environ-ment may be needed.• Starvation....see previous sections about fasting• Lack of sleep may be harmful.• Individual distinctive stresses
Avoidance of Toxins• Alcohol has been known to hasten the progressionof some conditions.• Cigarette smoke, probably due to the carbonmonoxide, is known to hasten the progression ofsome conditions. Remember that carbon monox-ide kills by inhibiting complex IV of theOXPHOS chain. If there is already a dysfunctionof OXPHOS, why make it worse. Cigarettesmoke will make it worse.• MSG (monosodium glutamate) has for years beenknown to cause migraine headaches in otherwisehealthy individuals, and may trigger these eventsin susceptible people with mitochondrial diseases.MSG is frequently added to Chinese (and otherAsian) foods, and is also found in high levels indried and canned soups. Read the label and avoidMSG.
BibliographyMainstream Journals:1. Sokol RJ. Expanding spectrum of mitochondri-al disorders. J Peds 1996;128:597-9.
2. Johns DR. Mitochondrial DNA and disease.NEJM 1995;333:638-44.
3. Munnich A et al. Clinical presentation of mito-chondrial disorders in childhood. J Inher MetabDis 1996;19:521-527.
Lay Press:4. Wallace DC. Scientific American. August1997. Expanding Spectrum into Aging andCommon Degenerative Diseases (Alzheimers,Parkinsons, etc.)
5. Beal MF, et al. Do defects in mitochondrialenergy metabolism underlie the pathology ofneurodegenerative diseases? Treds Neurosci1993;16:125-131.
6. Wallace DC. Mitochondrial genetics: a para-digm for aging and degenerative diseases?Science 1992;256:628-632.
What is next?!7. Priller J et al. Frataxin gene of Friedreich’sAtaxia is targeted to mitochondria. Ann Neurol1997;42:265-269.
Best Overview of Subject:8. Shoffner JM, Wallace DC. Oxidative phospho-rylation diseases and mitochondrial DNA muta-tions: diagnosis and treatment. Annu Rev Nutr1994;14:535-568.

Think Mitochondria UMDF15
AbstractMitochondrial cytopathies are clinically andbiochemically heterogeneous disorders affectingenergy production. Because of the diversesymptoms spanning organ systems, the largenumber of biochemical and genetic defects, and anunpredictable clinical course, there are limited dataregarding proven effective therapies. In general,treatments for mitochondrial cytopathies are intend-ed to augment energy production as well as reducethe production of free radicals and other toxicmetabolites that further limit the generation ofcellular energy. Theoretically, treatment can beaimed at increasing respiratory chain activity bysupplementing relative deficiencies of cofactorsrequired for proper functioning. Possible strategiesto consider may include dietary management, sup-plemental vitamins and cofactors, and/or specificmedications aimed at a particular symptom.
Keywords: Mitochondrial encephalomyopathy,coenzyme Q10, carnitine, experimental treatment.
Objectives: On completion of this article the readerwill be able to summarize the current treatmentoptions for patients with mitochondrial disorders.Accreditation: The Indiana University School ofMedicine is accredited by the AccreditationCouncil for Continuing Medical Education to pro-vide continuing medical education for physicians.Credit: The Indiana University School of Medicinedesignates this educational activity for a maximumof 1.0 hours in category one credit toward the AMAPhysicians Recognition Award. Each physicianshould claim only those hours of credit that he/sheactually spent in the educational activity.
Disclosure: Statements have been obtained regard-ing the authors’ relationships with financial sup-
porters of this activity. There is no apparent conflictof interest related to the context of participation ofthe authors of this article.Understanding therapy for those with mitochon-
drial disease requires knowledge of the underlyingpathogenesis. The term mitochondrial cytopathiesrefers to the human illnesses resulting from primaryand secondary mitochondrial dysfunction. Themitochondria are responsible for energy production,which is generated in the form of adenosinetriphosphate (ATP). A series of well-orchestratedchemical reactions culminate in the phosphoryla-tion of adenosine diphosphate (ADP) by theprocess of oxidative phosphorylation (OXPHOS),which occurs in the five enzyme complexesimbedded in the inner mitochondrial membranethat comprise the electron transport chain (ETC). Inaddition to energy generation, the mitochondriaalso play pivotal roles in both the generation of freeradicals and the process of apoptosis, or “pro-grammed” cell death. Although therapy primarilyfocuses on improving energy production, the otherfunctions of the mitochondria may be important infuture consideration of treatment options.Physicians caring for those with mitochondrialcytopathies are faced with a new challenge. Thecurrent practice of specialized medical carestratifies physicians and their patients by diseasesof organs and organ systems. Although dysfunctionof one organ can affect another adjacent organ,such as congestive heart failure causing pulmonaryedema, it is usually observed that successfultreatment of the primary disease will result inimprovement of other organ dysfunction.Mitochondrial cytopathies are not diseases ofparticular organs, but a disease or disease state ofan organelle. The consequences of faulty ATPproduction are more severe in those tissues with ahigh-energy requirement, which may impact on the
Treatment of Mitochondrial CytopathiesDeborah R. Gold, M.D.1 and Bruce H. Cohen, M.D.1
Reprinted with permission of Thieme Medical Publishers, Inc., 333 Seventh Avenue, New York, NY 10001
Seminars in Neurology, Volume 21, Number 3, 2001. Address for correspondence and reprint requests: Dr. Bruce H. Cohen, Chief,1Section of Pediatric Neurology, Cleveland Clinic Foundation, Desk S-80, 9500 Euclid Avenue, Cleveland, OH 44195. 1Section ofPediatric Neurology, Cleveland Clinic Foundation, Cleveland, Ohio. Copyright © 2001 by Thieme Medical Publishers, Inc.,333 Seventh Avenue, NewYork, NY 10001, USA.Tel: +1(212) 584-4662. 02718235,p;2001,21,03,309,326,ftx,en;sin00149x.

UMDF Think Mitochondria16
function of only a few selected organs or causewidespread damage affecting most organ systems.Successful management of an ill person with amitochondrial cytopathy requires the orchestratedefforts of a primary care physician, medical spe-cialists, and a physician comfortable with theintricacies of mitochondrial disorders. Because ofthe diverse nature of affected organ systems,evaluation of any given therapy can be quite achallenge.In spite of the multiplicity of clinical presenta-
tions and underlying pathophysiology, there areseveral well-described phenotypes that have beeninstrumental in the evolution of our knowledge ofmitochondrial diseases. Kearns-Sayre syndrome(KSS), typically seen in conjunction with a defectin metabolism of coenzyme Q10, usually presentswith ophthalmoplegia, retinopathy, cardiacconduction defects, ataxia, and short stature.Episodic vomiting, lactic acidosis, myopathy,seizures, strokelike events, and short stature tend tocharacterize mitochondrial encephalomyopathy,lactic acidosis, and strokelike episodes (MELAS).Myoclonic epilepsy with ragged-red fibers(MERRF) is distinguished by the presence ofsevere myoclonus, epilepsy, ataxia, and myopathywith ragged-red fibers. Leber hereditary opticneuropathy (LHON) is characterized primarily byblindness in men. Respiratory irregularities,myopathy/weakness, and visual and auditoryimpairments comprise Leigh’s syndrome. Despitethese well-defined syndromes, their clinical expres-sion often overlaps.A number of factors make it difficult to assess
whether a given treatment may be effective. Theseinclude:1. Mitochondrial cytopathies represent literallyhundreds of different disease states. They may becaused by genetic mutations that result in deficientquantity or function of an enzyme, assembly ofmultisubunit enzymes, disorders of mitochondrialmembrane structure, defects in substrate transport,or vitamin and cofactor deficiencies. The mutationsthemselves may involve nuclear DNA (nDNA) ormitochondrial DNA (mtDNA); point mutations,deletions, or rearrangements. It is not reasonable tobelieve that any one treatment would have a similareffect on all mitochondrial diseases.2. Mitochondrial diseases affect an unpredictable
combination of a number of organs or organsystems. This is a result of the process known assegregative replication, in which the abnormal mito-chondria may be “compartmentalized” within agiven organ (i.e., muscle, brain) and not others.There may be a “threshold” effect in which acertain level of mutant mitochondrial genomes isrequired for disease to be evident clinically and/orbiochemically.1 Despite the existence of this criticalthreshold, the genetic burden or measured biochemi-cal deficiency does not necessarily correlate with theseverity or rapidity of progression of thedisease. The variability of clinical features amongaffected family members is enormous,even if theunderlying genetic or biochemical defect is thesame. In addition, exacerbations and remissions arecharacteristic of these disorders, potentially cloudingevaluation of the efficacy of a particular intervention.3. Mitochondrial diseases can be classified on thebasis of a genetic defect, biochemical defect, orpathologic finding. Based on this classification, thereare no defined methods of defining severity of ill-ness, nor is there any understanding or consistentability to predict the natural history of any onepatient’s illness. Therefore, treatment trials that arenot conducted over a sufficient time period couldreject a potentially adequate treatment.4. Given the potentially systemic nature of the mito-chondrial cytopathies, developing a treatment triallooking at efficacy of a particular medication or sup-plement by evaluating the response of all possibleaffected organ systems would be quite cumbersomeand expensive and would require an unacceptablenumber of patients. On the other hand, trials thatlook at the response of only one organ system totherapy may miss an existent benefit to other organsystems.5. The commonly investigated biochemicalparameters (i.e., serum or cerebrospinal fluidlactate, pyruvate, enzyme assays) in isolation maynot be a full indicator of therapeutic efficacy for anygiven supplement or medication. Monitoringprogress via neurophysiologic studies, magnetic res-onance spectroscopy (MRS), and/or objective mus-cle strength testing will likely add to the overallassessment of patients maintained on specific treat-ment regimens.For these reasons, it is very unlikely that there
will be class 1 proof that any specific medication orsupplement will be effective in the treatment of

Think Mitochondria UMDF17
mitochondrial cytopathies. There is good reason forthis skepticism. At this time mitochondrialcytopathies are still considered by most to berelatively rare disorders. There are limitedpatients with any one specific mutation, and theclinical variability of those with a specific mutationis tremendous. Even if mitochondrial disorders areultimately shown to be common, the vastphenotypic variability in terms of distribution oforgan dysfunction and severity even among familymembers with identical genotypic disorders makesit impossible to know the natural history of diseaseprogression (and unexplained occasional temporaryremissions). Trying to collect class 1 data in agroup of diseases with varied molecular geneticsand biochemical defects is not likely to be possible.Although there may be one best treatment
approach for one individual with mitochondrialdisease, it is naïve to think that there can be aunified treatment strategy for groups of patientsidentified as having a mitochondrial cytopathy. Asmitochondrial diseases are often considered to bedegenerative in nature, familiarity with the underly-ing pathophysiology of these disease processes canaid the clinician in developing potentially effectivetreatment regimens that can result in an improvedquality of life. Despite this knowledge, therapy/amelioration of these disorders continues to posequite a challenge. In general, therapeutic approach-es are principally based on the use of antioxidants,vitamins and supplements (Table 1), replacement ofrespiratory chain cofactors, dietary management,and medications aimed at reduction of a particularsymptom (i.e., seizures, neuropathic pain, cardiacdysfunction).Consulting Management
Given the multisystem involvement commonlyobserved in patients with mitochondrialcytopathies, the coordinating physician (typically aneurologist) often needs to work in conjunctionwith additional subspecialists. Members of thisintegrated team are determined by the systemicmanifestations of a given patient. Because cardiacinvolvement is fairly common, it is reasonablefor a cardiologist to evaluate patients with a docu-mented or suspected disorder of mitochondrialfunction. At minimum a 12-lead electrocardiogram(EKG) should be performed on an annual basiswith echocardiography reserved for those patientswho demonstrate cardiorespiratory symptoms or anabnormal EKG, or in the setting of Kearns-Sayresyndrome. The cardiologist will provide treatmentfor conduction defects or cardiac failure. Inaddition, patients should be evaluated by a neuro-ophthalmologist to document the presence ofpigmentary retinopathy, optic atrophy, and/or ptosis(which may be amenable to surgical intervention).Surveillance for commonly associated eye findingsshould be done at least every few years. Theinvolvement of additional specialists will be guidedby patient symptoms.In the presence of motor dysfunction from cen-
tral and/or peripheral nervous system disease, anorthopedic evaluation and a combination of physi-cal and occupational therapy may be crucial interms of improving a patient’s level of functioning.The role of the physical therapist (PT) and occupa-tional therapist (OT) encompasses a vast range ofpotential areas of dysfunction but, in general, thegoal should be to preserve or restore mobility andmuscle strength. For a patient who has recentlybeen started on supplemental vitamins and/orcofactors, the PT or OT may assist in a supervisedexercise regimen and monitor strength changes.The PT and OT play instrumental roles in assistingwith wheelchair fitting or assistive walking devices.Speech therapy would be beneficial for a hearingimpaired child by teaching alternative means ofcommunication. For the patient whose primarymanifestation of mitochondrial dysfunction ispervasive developmental delay, the languagetherapist could focus on improvement in thepragmatic use of language.Once the diagnosis of mitochondrial disease is
established, genetic counseling should be made
Table 1 Commonly Used Supplements forMitochondrial Cytopathies
Supplement Dose rangeCoenzyme Q10 4.3–15 mg/kg/d, 200 mg tid maximumlevo-carnitine 100 mg/kg/d, 1000 mg tid maximumThiamine (B1) 50–200 mg/dRiboflavin (B2) 50–600 mg/dVitamin K3 5–80 mg/dFolate 1–10 mg/dLipoic acid 12.5mg/kg/d, 400 mg tid maximumVitamin E 200–1200 IU/d in divided dosesVitamin C 100–2000 mg in divided dosesSelenium 25–50 mcg/d

UMDF Think Mitochondria18
available to patients and their families. Providingpatients with an accurate prognosis is difficultbecause of the phenotypic variability and theunpredictable nature of the underlying diseaseprocess. For those patients with the more commonpoint mutations (A3243G, A8344G), it may be pos-sible to predict potential associated complications.In one study of 245 patients with either of thesemutations the frequency of clinical findings wasestablished. There was a statistically significantdifference in that the following were morecommonly seen in the patients with a 3243mutation: recurrent strokes, chronic progressiveexternal ophthalmoplegia (CPEO), diabetesmellitus, pigmentary retinopathy. In contrast, opticatrophy, neuropathy, ataxia, and myoclonus weremore frequently observed in those patients with the8344 mutation; this also reached statisticalsignificance. A clear relationship existed betweenthe degree of heteroplasmy of mutant mtDNA inmuscle and the occurrence of the more commonsymptoms for both mutation types. However, therewas no relationship between the absolute level ofA3243G or A8433G in blood and the frequency ofany of these clinical features.1
Global precautions and recommendations shouldbe considered and relayed to caregivers. Some ofthese recommendations are not relevant to manypatients, and therefore these should be individual-ized to the particular needs of the affected person.Patients should be instructed to avoid temperatureextremes, as exposure to extremes of cold or heatthat most people can tolerate can exacerbate symp-toms. Fevers and infections require promptevaluation and appropriate treatment. Ibuprofenshould be used as an antipyretic and aspirin shouldbe avoided. Acetaminophen is safe, although adher-ence to proper dosing is important because it canpose an oxidative stress. If an intercurrent illnessresults in poor oral intake of fluid or calories, earlyintravenous hydration with a dextrose-containingfluid is required and could avoid complications.
AnesthesiaMost anesthetic and surgical procedures are welltolerated in patients with documented or suspectedmitochondrial cytopathies. As part of a diagnosticevaluation, many patients undergo a muscle biopsy,in which general anesthesia has not been reported
to cause problems. However, anesthesia probablydoes pose a small additional risk to those withmitochondrial diseases.General anesthesia consists of induction with
intravenous agents (i.e., thiopental, propofol, etomi-date) followed by inhalation agents (i.e., nitrousoxide, halothane, enflurane, isoflurane, sevoflurane,and desflurane) for maintenance of anesthesia.Finally, muscle relaxants are occasionally used andinclude depolarizing (succinylcholine) and nonde-polarizing agents.There is intrinsically a greater risk of experienc-
ing medication side effects in the setting ofmitochondrial dysfunction. Despite the fact thatsome agents may interfere directly with mitochon-drial function, complications associated withanesthesia are more likely to be related to thepatient’s clinical status prior to surgery. Reportedadverse events include significant deteriorationof baseline neurologic status, seizures, stroke, car-diac rhythm disturbances, respiratory failure, coma,and death. Increased sensitivity to several agentshas been described, though reports are limited andusually anecdotal. Furthermore, a single anestheticagent can rarely be implicated as the etiology of thedecompensation. The absolute risk of an adverseanesthetic outcome in those with mitochondrialdysfunction is not known, although there is expand-ing literature on anesthetic associated problems.Despite this, the majority of patients with mito-chondrial cytopathies tolerate surgery andanesthesia without complications. The anesthesiolo-gist should be informed about the underlyingpathophysiology of these disorders and potentialcomplications related to general anesthesia.Additionally, preoperative assessment of patientsshould encompass a wide scope of clinical consid-erations given the multiple organ involvement fre-quently observed. Overall, the goal during anesthe-sia and surgery should be to maintain metabolicbalance, which may require monitoringbiochemical parameters throughout the procedureand sometimes for hours to days following surgery.This monitoring should include blood glucose,body temperature, and acid-base balance.There are reports that document tolerance to
many anesthetic medications. A 12-year-old boywith Kearns-Sayre syndrome tolerated anesthesiawith opioids and isoflurane.2 A 6-week-old infant

Think Mitochondria UMDF19
girl with mitochondrial encephalomyopathy due tofumarase deficiency tolerated induction with intra-venous thiopentone followed by isoflurane andnitrous oxide in oxygen. There were no intra- orpostoperative complications or deterioration fromher baseline status.3 A 23-year-old with Kearns-Sayre syndrome was studied for her response tosuccinylcholine (1 mg per kg) and pancuronium(three divided doses of 0.02 mg per kg per dose).There was no change in her neurologicexamination, and her response to succinylcholineand pancuronium was normal.4
Complications consisting of severe, diffuse whitematter degeneration and death following anesthesiawith thiopental, fentanyl, isoflurane, and pancuroni-um are described in a 13-month-old girl withmitochondrial myopathy.5 A 51-year-old patientwith Kearns-Sayre syndrome underwent emergentexploratory laparotomy for possible appendicitis.Anesthesia included thiopental (200 mg), vecuroni-um (0.1 mg per kg), nitrous oxide, oxygen,isoflurane, and supplemental fentanyl andvecuronium. Intraoperatively, the patient receivedlactated Ringer’s solution. Postoperatively, hedeveloped cyanosis and dyspnea, and subsequentlyrequired reintubation. EKG revealed left bundle-branch block and subsequent atrial fibrillation withST-segment depression. Following appropriatetreatment, the EKG reverted to his preoperativebaseline. It is likely that the volatile anestheticscontributed directly to myocardial depression. Therespiratory muscle weakness itself could have beendue to the effects of premedication or as residual ofinhaled anesthetics and/or muscle relaxants.6
The lack of uniformity from case reports make itimpossible to draw conclusions regarding the haz-ards of a specific anesthetic agent, and the safestregimen of anesthesia for patients with mitochondr-ial cytopathies remains unknown. Review of theliterature and personal experience, however, doesallow for the application of some general rules anddeductions, and global management considerationscan be inferred.An increased risk of perioperative pneumonia
exists in the setting of hypotonia, bulbar dysfunc-tion, and diminished respiratory capacity, a scenariocommon in patients with mitochondrial disease.Therefore, respiratory function should be strictlyattended to during the perioperative period, as
should heightened awareness for the possibility ofinfection. Chest physiotherapy should be includedas a standard postoperative measure for thosepatients with premorbid pulmonary dysfunction.Moreover, patients may not have adequate respons-es to hypoxemia and/or hypercarbia. If inhalationagents that are known to depress the ventilatoryresponse to CO2 are to be administered (isoflurane,desflurane), patients must be adequately monitoredin the perioperative and recuperative periods forhypoventilation and impending respiratory failure.7
Patients with an underlying seizure disorder mayexperience an increase in seizures immediately fol-lowing surgery, and these should be appropriatelymanaged. Dextrose-containing intravenous fluidsshould be provided if patients are required to fastpreoperatively. Lactated Ringer’s solution containslactic acid and should probably be avoided.Risk for malignant hyperthermia (MH) may be aconsideration in those with mitochondrial dysfunc-tion especially if myopathy is present. MH istriggered by inhalation anesthetics (i.e., halothane,enthrane) and/or depolarizing muscle relaxants (i.e.,succinylcholine). Those agents known to triggerMH should be avoided if there has been a prioradverse reaction involving either the patient or afamily member, but inhalation agents are routinelyused safely in patients known to have a mitochon-drial cytopathy. Regardless, dantrolene should beavailable and used at the first signs of malignanthyperthermia.In association with infectious illnesses and other
stressors, it is frequently noted that patients withmitochondrial cytopathies are at risk for respiratoryfailure and/or worsening of their underlying neuro-logic status. This deterioration is seen outside thesetting of surgery and anesthesia, but can also occurwith the stress of an illness requiring surgery andthe necessary anesthesia. This worsening isbelieved to be in part related to the increase ofcytokine production and subsequent formation ofnitric oxide, which, in high amounts, mayadversely affect energy production. In response tosurgery, cytokines, including tumor necrosis factor(TNF), are also released. Consequently, thesepatients are at increased risk of worsened neurolog-ic status, infections, and potential respiratory failureduring the perioperative period. Elective surgery forpatients with concurrent infection or other stressors

UMDF Think Mitochondria20
should be delayed in an effort to avoid an exacerba-tion or clinical worsening of the underlying diseaseprocess.For those patients at risk for cardiac conduction
block (i.e., Kearns-Sayre syndrome), isoflurane ispreferred over halothane because of the reduced riskof causing heart rhythm disturbance. Precautionarymeasures (i.e., readily available external cardiac pace-maker) and cardiac monitoring must be undertaken,given the risk for cardiac conduction abnormalities.Spinal anesthesia should be used with caution in
those patients with neuropathy or myopathy as partof their disease manifestations. However, it may per-mit monitoring of the patient’s neurologic statusintraoperatively and will allow for airway patency,and agents that may potentially trigger malignanthyperthermia can be avoided. For these reasons,spinal anesthesia (tetracaine) was administered to a40-year-old man with mitochondrial encephalopathywho underwent open reduction and internalfixation of an ankle fracture. There were noimmediate or delayed postoperative problems.8
It has been demonstrated in animal studies thatpropofol impairs mitochondrial function.Nonetheless, propofol has been safely utilizedduring anesthesia for many patients with mitochon-drial dysfunction. Observations have been made,however, that extended and high-dose use over aperiod of days in treatment of refractory seizuresleads to a syndrome analogous to mitochondrialfailure.9 Sodium nipride acts as an inhibitor of therespiratory chain and should be avoided as well.Despite precautions, clinical worsening may
occur following otherwise successful surgery andmay be due to either the natural course of the mito-chondrial disease process or the exacerbation due tothe stress of surgery and consequent cytokine-mediated changes.
Nonpharmacologic TreatmentsDietaryNutritional management of patients with disordersof energy production must be individualized,depending mainly on the specific underlying defect.Dietary management is likely to impact on theunderlying disease process by activation of alterna-tive pathways of energy production as well as play arole in decreasing endogenous formation of toxicmetabolites.10 Some key points with regards to
dietary therapy, though, are applicable to themajority of patients. Patients should avoid pro-longed periods without a meal; this may require fre-quent, small meals in an attempt to maintain normo-glycemia. There is a subset of patients who areunable to tolerate an overnight fast and maytherefore require a prebedtime snack consisting ofcomplex carbohydrates. A good source of complexcarbohydrate is uncooked cornstarch; however, it isnot very palatable. Patients with long-chain fattyacid oxidation disorders may need to avoid dietaryfats and ingest fats in the form of medium chaintriglycerides (MCT oil).
Aerobic ExerciseThe effects of aerobic training on exercise tolerance,fatigability, lactic acidosis, and muscle pain havebeen studied in patients with mitochondrialmyopathies. Ten patients with primary manifestingsymptoms of exercise intolerance and muscle weak-ness were enrolled in a training program consistingof aerobic exercise on a motorized treadmill three tofour times per week. Following the 8-week program,the mean estimated aerobic capacity was 30% high-er than at baseline (P <0.01). Aerobic trainingresulted in an increase of total exercise duration byapproximately 30% (P <0.02). Both resting lactateand that obtained following exercise were decreasedfollowing the 8-week training program. Finally,there was a demonstrable decrease in heart rate andin the half-time for ADP recovery after exercise asshown by phosphorous magnetic resonance spec-troscopy. In this group of patients those with definedmitochondrial DNA mutations (n = 7) showedslightly less of a response when compared to thosewith nuclear DNA mutations. The results of thisstudy lend support to the use of aerobic training aspart of the treatment regimen for patients with mito-chondrial myopathies. If this is to be undertaken, itmust be carried out in a wellsupervised and moni-tored setting, such that safety is not compromised.11
Pharmacologic TreatmentsSymptomatic Treatment
SeizuresManagement of seizures typically involves the useof common anticonvulsants including (but not limit-ed to) phenobarbital, phenytoin, carbamazepine,

Think Mitochondria UMDF21
gabapentin, lamotrigine, benzodiazepines, andzonisamide. Valproate has been identified as apotentially dangerous medication because of itshepatotoxic side effect in some patients withmetabolic diseases. There is considerabledebate as to whether this medication should everbe used, regardless of the situation, or whether, inmetabolic disease, it can be considered in the set-ting of seizures that have been refractory to othermedications. Valproate is known to inhibitcytochrome oxidase (COX) as well as cause mito-chondrial ultrastructural changes, but it is not knowif these are clinically relevant. Phenobarbital andthe benzodiazepines do interfere with mitochondri-al function in vitro but it is not clear if this is clini-cally relevant. There may be a theoretical advantageto using some of the newer neuroprotective drugssuch as gabapentin or lamotrigine. The ketogenicdiet has been used safely in many patients withoxidative phosphorylation disorders. It should beavoided in those with fatty acid oxidation diseaseand in those patients that either do not enter rapidketosis (indicating a primary or functional defect infatty acid oxidation) or those that becomeencephalopathic with the onset of fasting orinitiation of high-fat feeds. The use of levo-carnitine in all patients on the diet is controversial,but those with known metabolic disease shouldhave carnitine monitoring every 6 months.
Neuropathic PainTreatment of pain is beyond the scope of this man-uscript. However, gabapentin and/or carbamazepinecan be used to treat neuropathic pain in associationwith mitochondrial cytopathies. Effective dosagesmay be less than that needed for anticonvulsantactivity.
Cardiac DiseaseCardiac disease is common, especially in adultswith mitochondrial cytopathies. Cardiac rhythmshould be monitored by routine ECG frequently,probably on a yearly basis. In the setting of cardiacconduction defects or advanced heart block, pace-maker insertion may be used to reestablish normalcardiac rhythm. Cardiac failure should be managedwith medications by an experienced cardiologist.
Sodium Dichloroacetate
Dichloroacetate (DCA) is an investigational drugthat stimulates the activity of the pyruvate dehydro-genase multienzyme (PDH) complex. PDHcatalyzes the irreversible oxidation of pyruvate, theproduct of glycolysis, to acetyl coenzyme A andcarbon dioxide. Reducing equivalents in the formof NADH, which enter complex I of the ETC, arealso generated. Acetyl coenzyme A then is con-densed with oxaloacetate to form citrate, the firststep in the citric acid cycle. Regulation of theenzyme complex is mediated by phosphorylation ofone of its subunits, whereby in the phosphorylatedstate the PDH complex is rendered inactive. DCAstimulates PDH complex activity by inhibiting thePDH complex kinases that are responsible for phos-phorylation, thereby maintaining the PDH complexin its unphosphorylated, hence active, state. Theresult is improved oxidation of lactate and conse-quent increased supply of acetyl coenzyme A andNADH. This NADH is then utilized by complex I,but if there is a defect at or distal to complex I, it isnot known if lowering lactate concentrations orimproving the flux through PDH can improve ener-gy production. One property of DCA is that it mayinhibit its own metabolism. The major side effectof DCA is a reversible peripheral neuropathy thatmay have some relation to thiamine deficiency.12
A number of reports support the effectiveness ofDCA in treating congenital and acquired lactic aci-dosis. DCA has been associated with a lowering ofserum lactate in addition to clinical improvement.Stacpoole et al report on 53 patients with congeni-tal lactic acidosis who were treated over a 1- to 5-year period with oral DCA.13,14 Decreased serumlactate was demonstrated in 27 whereas decreasedserum and cerebrospinal fluid (CSF) lactate wasobserved in 11 patients. Some clinical improvement(vital signs, muscle tone, exercise endurance, cog-nition, stabilization of neurologic decline) wasobserved for 15% of the 39 patients whose subse-quent clinical course was known. For patients whorespond to DCA, there should be a 20% decrease ofserum lactate within 6 hours of the first dose. Forpatients who do not respond within 24 hours of oralor intravenous dosing, any response is not likelyand therefore treatment is probably unnecessary.13,14
In a controlled clinical trial involving adultpatients with varied etiologies of severe lactic aci-dosis, intravenous DCA was shown to significantly

UMDF Think Mitochondria22
reduce (P = 0.001) serum lactate concentrationswhen compared with placebo. The changes inserum lactate concentrations were not associatedwith clinical improvement or survival.15
More recently, DCA has been evaluated in thesetting of mitochondrial encephalomyopathies.Most reports are anecdotal and present conflictingclinical outcomes. Two siblings with MELAShaving clinical deterioration were treated with acombination of DCA (50 mg per kg) and multipleother medications, including vitamin B1. Theplasma lactate diminished within 2 days in bothpatients. The frequency and severity of myoclonicseizures (patient 1) were decreased within 1 month.The drug was maintained in this patient for 25months without apparent side effects. No additionalstrokelike episodes, headaches, or abdominal painwere observed in the second patient for the 22months of observation.16 A patient with MELASwas shown to improve after treatment with oralDCA on two separate occasions with regards toreduction in serum and CSF lactate levels.Additionally, this patient showed reduction of neu-rologic decline and cessation of auditory and visualhallucinations in conjunction with the normaliza-tion of the biochemical parameters.17 Improvementof magnetic resonance imaging (MRI) abnormali-ties occurred in two patients with Leigh syndromefollowing treatment with DCA (30 or 50 mg per kgper day). The improvement was mild and transitory(21.2 months) in one patient (PDH complex defi-ciency) and more significant and sustained over thefollow-up period of 9 months in the second patient(complex I deficiency). Both demonstratedreduction of serum and/or CSF lactate associatedwith initiation and continuation of therapy.18
Other than a lowering of serum lactate, Tuliniuset al19 did not find any significant difference clini-cally following treatment with DCA in a 6-month-old boy with myopathy and cardiomyopathy.DCA (50 mg per kg per day following a load of
50 mg per kg every 12 hours for three doses) wasused to treat a 1-year-old girl with Leigh’s syn-drome. She demonstrated gradual improvement inher clinical symptoms (respiratory status, physicalactivity, and muscle strength) and biochemical pro-file (lactate diminished in blood and CSF). Despitethis, 2 months after the start of therapy, MRIrevealed continued cerebral atrophy. At the same
time 1H magnetic resonance spectroscopy wasindicative of reduced neuronal function.The investigators conclude that DCA may lead tosome improvement of neurologic symptoms viareduction of serum lactate without truly affectingthe underlying disease process.20
In a double-blind, placebo-controlled study ofDCA in 11 patients with mitochondrial disease,DeStefano et al evaluated several measures ofoxidative metabolism following 1 week of treat-ment. A significant decrease (P <0.05) in serumlactate, pyruvate, and alanine occurred both at restand after exercise. In addition, proton magneticspectroscopy showed a decrease of brainlactate/creatine ratio by 42% (P <0.05) in additionto other changes indicative of improvements ofoxidative metabolism (NAA/creatine ratio increasedby 8%, P <0.05). Evaluation of the gastrocnemiusmuscle by phosphorous magnetic spectroscopyshowed no significant change following treatmentwith DCA. No significant clinical improvementwas noted following treatment despite the biochem-ical improvements, but may be due to the shorttreatment time of one week.21
In association with the administration of intra-venous DCA at a dose of 50 mg per kg per day for13 days followed by 25 mg per kg per day for 6days, arterial lactate decreased as did seizure activi-ty in a patient with MELAS. Serial proton magneticresonance spectroscopy revealed improvement interms of the magnitude of the lactate peak andNAA/Cr ratio in the region compatible with thispatient’s symptoms.22
Three children with mitochondrial encephalomy-opathy were administered DCA at a dose of 30 mgper kg per day. These children demonstrated radio-logic and clinical improvements following this oraltreatment regimen. MRS findings revealed amarked reduction of lactic acid peaks in two of thepatients. Both serum and CSF lactate levels dimin-ished. Serial MRI scans demonstratedgradual decrease of white matter lesions intwo patients, and the pontine and medullary lesionsin the third. Developmental progress was observedfollowing treatment in the two patients with Leigh’ssyndrome. These patients received oral DCA for 21or more months without any significant sideeffects.23
In summary, DCA will lower serum lactate and

Think Mitochondria UMDF23
improve other biochemical markers such as CSFlactate or serum alanine in some patients. Forpatients with a DCA-responsive PDH deficiency,the use of DCA can be helpful. It is not clear forthose with electron transport defects whether or notlowering lactate is helpful.By increasing the flux of pyruvate through PDH,the kinetics of the reaction pyruvate + NADH →lactate + NAD+ lowers the lactate, but also increas-es the ratio of NADH/NAD+. The NADH producedcannot be utilized by the (impaired) ETC.Furthermore, the putative role of the increasedconcentration of NAD+, produced by the conver-sion of pyruvate to lactate, is to allow glycolysisto proceed and generate ATP under anaerobicconditions. A reduced amount of available NAD+results in reduced production of anerobicallygenerated ATP.Unfortunately, despite lowering of serum and/or
CSF levels of lactate, DCA treatment does not uni-versally lead to overall clinical improvement.
Vitamins and SupplementsCoenzyme Q10Coenzyme Q10 (CoQ10), also known as ubiquinone,is a lipid soluble antioxidant that is synthesizedfrom tyrosine and mevalonic acid by animal cells.Multiple vitamins and trace elements are requiredfor its biosynthesis. Ubiquinones are componentsof all cell membranes, including mitochondrialmembranes.Normal CoQ10 levels are maintained by
endogenous synthesis and dietary sources, whichinclude primarily animal products. This compoundcan also be administered as an exogenous supple-ment. Normal muscle mitochondria, blood, andfibroblast levels have been established at 1811 ± 99ng/mg (n = 10), 637 ± 84 ng/mL (n = 8), and 48 ±1.3 ng/mg (n = 5) respectively. 24 Ubiquinone alsoexists in a partially reduced form (ubisemiquinone)and a fully reduced form (ubiquinol).The role of CoQ10 in energy metabolism is well
documented. Large amounts of CoQ10 are found inthe mitochondrial inner membrane where it acts asa mobile electron carrier. Specifically, CoQ10shuttles electrons from ETC complex I to complexIII and from complex II to complex III. In addition,CoQ10 absorbs free radicals, which are probablygenerated to the greatest extent at the level of
complex I, thereby acting as an antioxidant andpreventing propagation of lipid peroxidation.CoQ10 also assists in regenerating active vitaminE from the tocopheroxyl radical.Deficient CoQ10 occurs in a wide range of
human diseases and may occur due to insufficientdietary intake, impaired biosynthesis either due toendogenous causes or exogenous toxins, dispropor-tionate utilization, or any combination of these.Given its hydrophobic nature and large particle
size, oral administration results in inconsistentabsorption, requiring oil-based liquid preparationsor suspension in oil. Typical dosing begins at 4 mgper kg per day but may require dosages as high as15 mg per kg per day to achieve clinical efficacy.CoQ10 is the most widely recognized supplement
used in the treatment of mitochondrial cytopathies.Reported beneficial effects have included decreasein serum lactate, improved exercise tolerance,increased muscle strength, andmagnetic resonance spectroscopy improvements.As a cellular antioxidant, its role is theoreticallyimportant, as free radical production is known toincrease in mitochondrial disease.A number of primarily anecdotal reports suggest
a favorable effect of CoQ10 in disorders of energymetabolism. A 17-year-old girl with MELAS hadsymptoms unresponsive to intravenous betametha-sone, oral nicotinamide, oral CoQ10 (150 mg perday), and intravenous cytochrome c. Following ini-tiation of CoQ10 at a dose of 300 mg per day shedemonstrated improvement of her ophthalmologicsymptoms, increased exercise tolerance, anddecreased serum lactate (both before and afterexercise).25 A 19-year-old girl with Kearns- Sayresyndrome and low serum CoQ10 was treated with120 mg per day of CoQ10. Serum CoQ10 increasedto normal with concomitant lowering of fasting andpostexercise lactate and improved ocular move-ments.26 Seven patients with mitochondrial cytopa-thy and lactic acidosis were treated with 120 mgper day of CoQ10. Five of the seven patients hadlow serum CoQ10. Serum lactate following exercisewas significantly diminished (P <0.05) in four ofthe patients. Monthly neurologic exams revealedimproved muscle strength in all but one patient. Noechocardiographic improvements were noted. Therewere no reported harmful side effects related to thistreatment regimen.27 Abe et al28 reported on a

UMDF Think Mitochondria24
patient with MELAS whose CSF lactate and pyru-vate decreased, with noted improvement in seizuresand myopathy following treatment with CoQ10.In a double-blinded placebo-controlled crossover
trial of CoQ10 at 160 mg per day for 1 month,improved muscle strength and reduced fatigabilitywere observed for those patients whose CoQ10 lev-els were lower than controls prior to initiation oftreatment. Following 3 months of therapy, therewas a statistically significant increase in overallmuscle strength testing (P <0.05) but strength ofspecific proximal and distal musculature did notdemonstrate significant improvement. The meanserum CoQ10 levels increased to abovenormal range within 2 months of treatment with nosignificant change thereafter. These investigatorsdid not find any significant change in the metabo-lism of lactate while patients were on CoQ10. Theglobal MRC score was the only significantimprovement observed in this study.29
Chan et al30 sought to determine clinically valu-able metabolic parameters of patients with mito-chondrial encephalomyopathies treated with CoQ10(150 mg per day) under exercise conditions. Ninepatients were evaluated with bicycle ergometryprior to, during (3 months), and following treatmentfor 6 months. At rest, only two patients demonstrat-ed elevated serum lactate levels, whereas, followingexercise, seven patients had elevations of the same.In addition, the lactate-to-pyruvate ratio was abnor-mal at rest in eight patients and in all patientsfollowing exercise. At 3 months following onset oftherapy, no clear change was noted in thesebiochemical parameters. At 6 months, however,there was a decrease in the lactate-to-pyruvate ratioat rest and in association with exercise (P <0.05 formale patients, n = 4) in six of the patients.There are several additional reports of patients
who either showed equivocal changes or did notseem to demonstrate improvements while receivingCoQ10. In a multicenter trial, 44 patients with mito-chondrial myopathy were treated for 6 monthsexclusively with CoQ10 at a dose of 2 mg per kgper day. Sixteen of the 44 patients showed a 25%decrease of postexercise lactate levels. All patientsdemonstrated statistically significant increases inmuscle strength. These 16 patients were subse-quently studied an additional 3 months in a blindedstudy of CoQ10 and placebo. Serum lactate levels
were not further decreased in this time interval inthose treated with CoQ10, although thosereceiving placebo developed worsening ofpostexercise lactate. Muscle strength did notimprove during the second treatment period. Nochange was noted in terms of cardiac conductionabnormalities or ophthalmologic findings duringthe study period of 6 months.31
Two patients with mitochondrial myopathieswere treated for 1 year with either 100 mg or 50mg daily of CoQ10. Neither of these patients hadabnormal (low) CoQ10 levels. Following 1 year oftreatment, neither patient demonstrated improve-ment of their ophthalmologic symptoms,quantitative isometric strength testing revealed nosignificant improvement, and CoQ10 levelsremained essentially unchanged. It must be notedthat the dose of administered CoQ10 in this study isless than is generally used to treat most adults.32
Sixteen patients with varied mitochondrialcytopathies were treated with CoQ10 in addition tovitamins K3 and C, riboflavin, thiamine, andniacin. This open study, using 300 mg per day overa 2-month time period, did not show any benefit.The parameters evaluated included resting and pos-texercise serum lactate, phosphorous magneticresonance spectroscopy, and regular follow-up ofclinical symptoms.33
The reliance on clinical and biochemicalparameters exclusively in the evaluation of patientswith mitochondrial cytopathies who undergo anexperimental treatment may not provide an entirelyaccurate sense of effectiveness of therapy. 31Pmagnetic resonance spectroscopy can be utilized toevaluate energy parameters in the specific tissue ofinterest, providing a noninvasive, quantitative meas-ure of brain and/or muscle metabolism. In thepresence of disordered mitochondrial metabolismone might expect to see a low concentration ofphosphocreatine, a high concentration of inorganicphosphate, and a high calculated ADP.34 Muscle canbe evaluated at rest, during exercise, and duringimmediate postexercise recovery. In disorders ofmitochondrial respiration, a decrease in the phos-phocreatine/inorganic phosphate (PCr/Pi) ratio canbe observed at rest. There is delayed replenishmentof phosphocreatine following exercise. In somepatients with mitochondrial cytopathy there may beno abnormality on 31P MRS, likely indicating that

Think Mitochondria UMDF25
skeletal muscle mitochondria are not involved.35
Again, with utilization of this technique, conflictingreports exist as to the effectiveness of CoQ10therapy.Eight patients and 18 healthy controls were treat-
ed with 150 mg of CoQ10 per day for 6 monthsand evaluated by 31P MRS of the calf muscle at rest,during exercise, and during the postexercise recov-ery period. MRS was performed at the beginning oftreatment and following 3 and 6 months of therapy.The mean PCr/Pi was significantly higher for con-trols prior to treatment and did not significantlychange throughout the supplementationperiod. By 3 months of treatment, there was a non-significant repletion of phosphocreatine in thepatient population. One patient had a dramaticimprovement of the PCr/Pi at rest in addition toincreased repletion of phosphocreatine postexercisefollowing 3 months of treatment.36
Barbiroli et al37 utilized in vivo phosphorousmagnetic resonance spectroscopy to evaluate theeffectiveness of CoQ10 on improving brain andskeletal muscle mitochondrial respiration. Tenpatients with mitochondrial cytopathies were evalu-ated by 31P MRS prior to and 6 months followingtreatment with 150 mg per day of CoQ10. Therewere 36 age-matched, healthy controls. Prior totreatment all patients demonstrated low concentra-tions of phosphocreatine and high ADP, indicativeof mitochondrial dysfunction. There was a signifi-cant increased (P <0.02) brain concentration ofphosphocreatine following treatment with CoQ10,in addition to a significant decrease (P <0.01) ofbrain concentration of inorganic phosphorous. Withregards to the skeletal muscle evaluation, the 31PMRS spectra were not significantly different at resteither prior to or following treatment when com-pared with controls. Despite this, all patients diddemonstrate a faster recovery of phosphocreatinefollowing treatment and some (those with CPEO)reported increased strength.Two patients with mitochondrial encephalomy-
opathy were treated with 150 mg per day of CoQ10and evaluated by bicycle ergometry and 31P nuclearmagnetic resonance (NMR) spectroscopy prior toand 10 months after initiation of treatment. In bothpatients before treatment there was a low PCr/Pi atrest, in addition to a high resting serum lactate.Acidosis occurred during the exercise phase, fol-
lowed by a delay in recovery after exercise.Furthermore, the bicycle ergometer test revealed alowering of the ventilatory threshold aswell as reduction of the maximum oxygen uptake.Pretreatment 31P NMR spectroscopy demonstratedthe following abnormalities: twofold decrease ofATP concentration and abnormally low PCr/Piratio. Following 10 months of treatment, there wassignificant improvement during the exercise testalong with a decrease in resting lactate, increase inoxygen consumption, increase in maximal load,and ventilatory threshold reached normal range. 31PNMR spectroscopy (performed on flexor digitorumsuperficialis muscle) corroborated these findings inthat there was a significant increase from thebaseline PCr/Pi ratio at rest in addition to improvedrecovery of all measured parameters. The decreasedATP concentration was still present, thought to alesser degree. The postexercise PCr/Pi ratio wasessentially unchanged for one patient but demon-strated a fourfold increase for the second patient.These results lend support to the efficacy ofhigh-dose administration of CoQ10.
38
The literature suggests significant controversyregarding the efficacy of CoQ10 supplementation.Regardless, many patients report improvedfunction, and the side effects associated with its useare rare. The majority of treating clinicians willadminister a therapeutic trial in escalating doses (4to 15 mg per kg per day) to determine its efficacyin an individual patient.
IdebenoneIdebenone is an analog of CoQ10 and acts both as afree radical scavenger as well as stimulating ATPformation by functioning as a mobile electron carri-er. It is currently not available in the United States.A patient with LHON and myopathy was treated
with oral idebenone (45 mg three times per daywith increase by 135 mg per day every 2 days)following the onset of marked spasticity and weak-ness. By the sixth day of treatment (135 mg threetimes per day), the patient was able to walk, run,and climb stairs. On neurologic examination, therewas marked reduction of spasticity of the lowerextremities in addition to improved strength.Following observation of this improvement,idebenone was continued at a maximum of 405 mgper day for an additional 3 months, during which

UMDF Think Mitochondria26
the patient remained clinically stable. Followingwithdrawal of idebenone, the patient again demon-strated weakness of the lower extremities and spasticparaparesis. Idebenone was reinitiated and within 2weeks clinical improvement was again observed.This patient was additionally evaluated by brain andmuscle 31P MRS. Following approximately 3 monthsof treatment there was an increase in phosphocrea-tine concentration and a decrease from baseline ofinorganic phosphate toward reference values. Whenreimaged following withdrawal of idebenone, theseparameters were markedly worsened and did notmarkedly improve following reinstitution of medica-tion. In addition, 3 weeks following initiation oftreatment, muscle studies showed an increased rateof recovery of phosphocreatine and inorganic phos-phate. Though similarworsening of variables were observed following dis-continuation of this medication, once resumed therewas no return to the previously observed recoverylevels.39
A 10-year-old boy with LHON due to homoplas-mic 11778 mutation was treated with oral idebenone(90 mg per day) after presenting with early onsetsymptoms. After 7 months of treatment, visual acu-ity was improved slightly (6/90 bilaterally to 6/6).40
However, spontaneous improvement in visual acuityis frequently reported in LHON.A patient with MELAS was treated with CoQ10
augmented by the addition of idebenone. Following8 months of treatment with 210 mg per day ofCoQ10, the patient demonstrated some improvementin terms of amelioration of sensorydisturbance, ataxia, and muscle weakness. Serumlactate decreased slightly. Electroencephalogram(EEG) and Wechsler Adult Intelligence Scale(WAIS) testing remained unchanged when comparedto prior to treatment. Motor and sensory conductionvelocities normalized. Following the addition of 90mg of idebenone, muscle strength improved further.Her EEG revealed marked improvement from base-line and from during treatment with CoQ10 alone.Her WAIS scores increased by 14 points. CSF pro-tein decreased from 64 mg/dL to 45 mg/dL. Theidebenone dose was then increased to 180 mg perday for an additional 11 months. CSF lactatedecreased. The improvements were maintained for afollow-up time of 20 months.41
The experience with idebenone may be too limit-
ed to draw any definitive conclusions. Furtherinvestigations may elucidate a role for this agent ina subset of patients with mitochondrial disease.Levo-Carnitine Endogenous levo-carnitine (beta-hydroxy-gammatrimethylammonium butyrate),found in many human tissues, is an amino acidderivative. It is synthesized in the liver and kidneyfrom protein-bound lysine (supplemental orallysine cannot improve carnitine synthesis) andmethionine. It is a water-soluble compound thatexhibits biologic activity only when in the levo iso-form. Several enzymes and cofactors (iron, ascorbicacid, niacin, and pyridoxine) are involved in itsbiosynthesis, and only one matrix mitochondrialenzyme is involved in the pathway. Of note, skele-tal and heart muscle are unable to synthesize carni-tine, and these tissues are therefore dependent onuptake of carnitine from blood.Normal plasma and tissue levels are maintained
by both de novo synthesis and exogenous dietarysources. Meat and milk products contain the high-est concentrations of dietary carnitine whereasplant products are poor sources. Normal plasmacarnitine concentrations are about 25 umol/L ininfants and 54 umol/L in adults.42 The highest con-centration of carnitine is found in skeletal muscle(98%), although distribution is shared with heart,kidney, liver, and brain.43
Carnitine is present in tissues and physiologicfluids as either free carnitine or as the acylcarnitineester. In normal circumstances, approximately 85 to90% is present in the free state. The majority ofplasma acylcarnitine is represented by acetylcarni-tine, which is often nonpathologically elevated inthe fasting state. The ratio between acylcarnitine tofree carnitine varies with timing of the last meal,composition of that meal, nutritional status, exer-cise, and disease conditions and is quite sensitive tochanges in mitochondrial metabolism. A ratio of0.25 is considered to be normal, whereas greaterthan 0.4 is abnormal and is indicative of carnitineinsufficiency or insufficient carnitine in light of themetabolic demands.44
Carnitine is necessary for transporting long-chainfatty acids across the inner mitochondrial mem-brane for the process of beta-oxidation. This occursmainly in skeletal muscle, heart, and liver and iscarried out by carnitine palmitoyltransferase I (CPTI), acylcarnitine translocase, and CPT II. A second

Think Mitochondria UMDF27
major task of carnitine is to maintain intracellularhomeostasis of acyl-CoA. Carnitine transesterifiesthe acyl-CoA esters that arise during beta-oxidationthrough the action of carnitine-acyltransferases.The acylcarnitine can then cross the mitochondrialmembrane in exchange for free carnitine, thusallowing for restoration of free CoA within themitochondria. In addition to these major functions,carnitine may also play some role in altering thephysiologic properties of cell membranes, such asmembrane stabilization.45 In the setting of inbornerrors of metabolism, carnitine serves to detoxifythe poisonous metabolic intermediates by forming aless toxic ester.A number of pathologic conditions have been
associated with abnormal metabolism of carnitine,the most frequent of which is carnitine deficiency.Carnitine deficiency can be defined as a state wherethe concentration is not adequate to meet thebody’s normal carnitine requirement. Systemiccarnitine deficiency can be primary but may occurin many disease states, including disorders ofoxidative phosphorylation, betaoxidation, organicacidurias, malnutrition, valproate, and zidovudineuse and in those receiving total parenteralnutrition without adequate carnitine replacement.Many metabolic disorders lead to elevated levelsof acyl-CoA intermediates, which impair thefunction of adenine nucleotide translocase, theenzyme that exchanges ADP for ATP across theinner mitochondrial membrane. Carnitine forms anester linkage with the acyl-CoA, forming therelatively nontoxic acylcarnitine, which is excretedin the urine. Elevated levels of acyl-CoA intermedi-ates over time can lead to a secondary carnitinedeficiency. Likewise, a carnitine deficiency itselfcan result in increased toxicity of the accumulatedacyl-CoA compounds.Clinical manifestations of a carnitine-deficient
state are varied, including but not limited tocardiomyopathy, acute encephalopathy, myopathy,cognitive delay, central nervous system dysfunc-tion, gastrointestinal dysmotility, and recurrentincidences of metabolic decompensation.Treatment with levo-carnitine should be consid-
ered for any person with a primary or secondarycarnitine deficiency. The role of carnitine therapy inmitochondrial disease is threefold. As alreadydiscussed, carnitine plays a role in reestablishing
homeostasis of acyl groups, a process that is aber-rant when mitochondrial dysfunction exists, leadingto inhibition of respiratory enzymes. In addition,secondary carnitine deficiency exists in the settingof mitochondrial cytopathies; thus, carnitinereplacement is essential. Finally, carnitine mayprovide improved integrity of the mitochondrialmembrane, thus adding to membrane stabilization.46
The typical dose of levo-carnitine is 100 mg perkg per day for children and 2 to 4 grams per day foradults in three divided doses. In the nonacute setting,levo-carnitine is available as a liquid or tablet, but itis also available as an intravenous preparation. Theintravenous dose is the same as the oral dose. Prior toinitiation of carnitine therapy in any patient, plasmaand urine carnitine and acylcarnitine profiles shouldbe obtained. The primary adverse effects includediarrhea and nausea, though carnitine is usually welltolerated at typical doses. It should be noted that oralabsorption is variable, and as little as 15% of the oraldose may actually be absorbed.Of 48 patients studied by Campos et al,47 four had
both total and free plasma carnitine deficiency(both defined as <30 µmol/L with normals in the lowto mid 50s) with carnitine insufficiency (defined asratio of esterified to free carnitine >0.25 with normal0.13 ± 0.016), and 17 had isolated carnitine insuffi-ciency. All 21 patients with carnitine deficiency orinsufficiency were treated with 50 to 200 mg per kgper day of levocarnitine. The following improvementswere observed following initiation of treatment: 20 of21 patients with muscle weakness demonstrated sub-jective improvement in muscle tone, four of eightpatients with failure to thrive showed growth acceler-ation, and eight of eight patients with cardiomyopa-thy demonstrated improved echocardiographic find-ings and clinical improvement. The average treatmentduration was 11 months (range 1 to 24 months).Plasma carnitine levels 10 days after initiation oftreatment were normal or above normal. Many addi-tional reports have demonstrated the beneficial effectsof carnitine in the setting of cardiomyopathy due tounderlying metabolic etiologies, including mitochon-drial abnormalities.
CreatineCreatine is an amino acid produced endogenously inthe liver from arginine and glycine, and it is alsofound in meat products. Creatine phosphate is syn-

UMDF Think Mitochondria28
thesized from creatine and ATP, and it is catalyzedby creatine kinase (CK). Unlike ATP, which the bodyis unable to store, creatine phosphate can be storedto a limited degree in tissues, allowing for a supplyof the high-energy phosphate bond, which can be uti-lized when needed. The hydration of phosphocrea-tine to creatine and ATP thereby allows the ATP tobe utilized by the tissue. Creatine is found in highestconcentrations in skeletal muscle and to lesserdegrees in cardiac muscle, smooth muscle, brain,sperm, and kidney.Intramuscular phosphocreatine may be reduced
in patients with mitochondrial cytopathies.Supplemental creatine seems to be most effective atincreasing phosphocreatine and creatine in this set-ting. Harris et al demonstrated that administration ofcreatine to healthy subjects resulted in a greater effectfor those patients whose initial total creatine concen-tration was low. There were no side effects from sup-plementation with doses ranging from 70 g to 330 gwith maximum treatment time of 21 days.48
The rationale for using creatine is to increase thetissue concentrations and possibly increase theability of muscle (or other organs) to accumulate cre-atine phosphate. Tarnopolsky et al have shown thatcreatine monohydrate (at doses of 5 g twice per dayfor 2 weeks followed by 2 g twice per day for 1week) improved strength for high-intensityanaerobic and aerobic activities and lean musclemass in patients with neuromuscular diseases,including those with mitochondrial myopathy. Bothtrials were based on short-term results, and the long-term beneficial effects of creatine remain to beproven. Regardless, the use of creatine in critical sit-uations seems to be reasonable.49,50
In one study of nine healthy men provided witheither oral creatine (as creatine monohydrate, 20 gdose) or placebo demonstrated no effect onperformance for maximum exercise or on phospho-creatine levels. However, the supplement was onlyadministered over a 3-day period and was in asetting of likely normal muscle creatine levels; there-fore, its relevancy to those with mitochondrial dis-ease is not known.51
AntioxidantsNumerous pharmacologic agents have been used inthe treatment of mitochondrial cytopathies, includingantioxidants. Antioxidants may improve enzyme
function or slow the process of oxidative damage,although the benefit over time is not possible tomeasure. The more commonly employed antioxi-dants (and the typical daily dosages) for patientswith mitochondrial disease includeselenium (50 to 100 mcg), vitamin C (250 to 4000mg in divided dosages), vitamin E (400 to 1200 IUin divided dosages), and lipoic acid (200 to 600 mgin divided dosages). Given the vast clinical presen-tation of the mitochondrial cytopathies and the factthat these agents have not systematically been stud-ied in this setting, it is not possible to state thatthere are proven benefits. Despite this, they are rou-tinely used in patients with these diseases.
RiboflavinRiboflavin (vitamin B2) is a precursor to flavinmononucleotide (FMN) and flavin adenine dinu-cleotide (FAD), which are cofactors of ETC com-plexes I and II, respectively. Riboflavin has beenproposed to act therapeutically by one of severalpotential mechanisms including inhibition of thebreakdown of complex I by providing more resist-ance to proteolysis or stabilizing the mitochondrialmembrane. It has been used with some success insome patients with mitochondrial disease withoutany apparent side effects.A 33-year-old patient with MELAS and complex
I deficiency was administered a combination ofriboflavin (100 mg three times per day) andnicotinamide (1 g four times per day) in a double-blinded, randomized fashion. In conjunction withthis treatment, there was cessation of this patient’sencephalopathic and myopathic symptoms. Thespectroscopic findings were equivocal: restingPCr/Pi did not show any treatment effect, whereashigh-energy phosphate recovery deteriorated uponwithdrawal of nicotinamide (but not riboflavin).Sural nerve sensory testing revealed a drop inamplitude once therapy was withdrawn with recov-ery following the reinitiation of treatment (P =0.00007 right, 0.017 left).52
A 10-month-old infant girl with a partial defectof complex I was treated with increasing doses ofriboflavin (beginning at 3 mg per kg). It wasobserved that lactate levels normalized and muscleweakness improved at the maximal dose of 13 mgper kg.53 Riboflavin was given to five patientswith mitochondrial myopathy due to complex I

Think Mitochondria UMDF29
deficiency in dosages ranging from 9 to 60 mg perday. Patients presented with either pure myopathicsymptoms or encephalomyopathy. One patient hadstabilization of the previously regressive disease andthree showed clinical improvement, especiallyin the myopathic component. There was normaliza-tion of complex I activity in three of the patientsin addition to improved lactate levels or musclehistopathology.54 A 13-year-old girl with progressiveexercise intolerance, severe lactic acidosis, and com-plex I deficiency was treated with 100 mg of oralriboflavin daily. There was remarkable and persistentclinical improvement with an increase in exercisetolerance. Exercise parameters (maximal workcapacity, O2 uptake, and base excess) alsoimproved.55 A 6-year-old boy with a defect of com-plex I and myopathy presenting as slowlyprogressive weakness was successfully treated withriboflavin and carnitine. Clinically, muscle strengthand motor conduction velocities were improved. Inaddition, the complex I activity and carnitine levelsnormalized 7 months after the start of treatment.56
More recently, Ogle et al57 report on a 21⁄2-year-oldpatient with complex I deficiency, mitochondrialDNA mutation, and myopathy and who had persist-ent response to riboflavin therapy (20 to 25 mg twicedaily) over a 3-year period. Despite persistent lacticacidosis, this patient demonstrated improvements interms of overall muscle strength and endurance.Riboflavin is not always effective, as demonstrat-
ed by the case of a 4-month-old infant with severe-congenital lactic acidosis and complex I deficiency,who underwent a trial of riboflavin at a dose of 100mg daily. The child’s serum lactate and ratio to pyru-vate remained significantly elevated, and the patientcontinued to demonstrate features of severe myopa-thy and cardiomyopathy until his death.58 Fourpatients with MELAS in association with complex Ideficiency treated with riboflavin, other supple-ments, and the ketogenic diet did not show improve-ment.59 Another large trial of riboflavin in combina-tion with other supplements found no significanttherapeutic success.33 The results of treatment withriboflavin, with a large variation in doses and treat-ment duration in this diverse population, are not uni-form but demonstrate that those with complex I defi-ciency and pure myopathy may benefit fromsupplemental riboflavin, with or without other sup-plements. Of course, the clinical course of these
diseases remains variable such that any improve-ment observed may not be due to therapeuticinterventions.
ThiamineThe PDH complex catalyzes the thiamine-dependent decarboxylation of pyruvate. Thiaminepyrophosphate, the physiologically active form ofthiamine, acts as a coenzyme for this decarboxyla-tion. The use of thiamine has been established inthe treatment of some forms of PDH deficiency,although its use in disorders involving more distalcomponents of energy metabolism, such as electrontransport chain disorders, is not established.Thereare no reported side effects with administration.Following treatment over a several week period
with thiamine at 300 mg three times per day, threepatients with Kearns-Sayre syndrome were found tohave normalization of previously abnormal lactateand pyruvate levels. There was some change inoverall level of fatigue, but clinical improvement ingeneral was trivial.60 Another patient, a 23-year-oldwith mitochondrial myopathy, cardiomyopathy, andlactic acidosis, was treated with thiamine (100 mgtwo times per day) in combination with prednisone(60 mg per day). Over a 3-week period, the patientdemonstrated progressive improvement in overallstrength. The previously life-threatening episodesof lactic acidosis also ceased, a finding that persist-ed over a 7-year follow-up period.61 One largerstudy of patients with mitochondrial cytopathiesfailed to demonstrate efficacy of treatment at dosesof 100 mg per day for 2 months.33
Vitamin K3In vitro studies show that in the presence of com-plex I inhibitors, vitamin K3 (menadione) stimu-lates oxygen utilization in mitochondria, resultingin an increase of NADH oxidation.62 One group ofinvestigators utilized 31P nuclear magneticresonance to assess the response to treatment withmenadione (40 to 80 mg per day) and ascorbate(4 g per day) of a 19-year-old patient with mito-chondrial myopathy associated with complex IIIdeficiency. This treatment approach resulted in bothclinical and metabolic improvement that persistedat 1-year follow-up. She was no longer wheelchair-bound though overall her weakness remainedunchanged. The clinical improvements were sup-

UMDF Think Mitochondria30
ported by 31P NMR data, which revealed anincrease in phosphocreatine concentration at restand postexercise. A doubling of the initial dose ofK3 improved the phosphorous NMR spectra evenfurther without apparent side effects. Symptomsdeteriorated with withdrawal of treatment withrecovery of function once reinstated.63,64
Toscano et al65 treated a 16-year-old girl withataxia, myoclonus, lactic acidosis, and complex IIIdeficiency with vitamins K3 (40 mg per day) and C(4 g per day). There was no significant change inlactic acidosis but there was mild improvement ofher ataxia. After 5 months of therapy, the brain31P-MRS indices were restored to normal. Skeletalmuscle evaluation, though, revealed only slightimprovement of mitochondrial bioenergetics.One larger trial in which patients were treated
with 60 mg per day of menadione and 2 g per dayof vitamin C (in addition to multiple other supple-ments) did not substantiate the therapeutic benefitseen by these other investigators.33
K3 is known to cause hemolytic anemia, hyper-bilirubinemia, and kernicterus and is therefore con-traindicated for neonates, pregnant females, orthose being treated with coumadin. It is also diffi-cult to find, and K1, the common form of vitaminK, may have no benefit. As a practical alternative,CoQ10 may provide the same benefit.
FolateA 23-year-old woman with Kearns-Sayre syndromeon phenytoin for a seizure disorder was found tohave diminished CSF and plasma folate levels inaddition to low free carnitine. She was treated withfolate (40 µg per kg of body weight or 15 mg perday), D, levocarnitine (10 g per day), and methion-ine (500 mg per day). Plasma folate increased tonormal with supplementation but CSF folateremained unchanged. She also demonstrated clini-cal improvement to the point of being able toambulate after having been bedridden.66
UridineUridine will soon be under investigation as a treat-ment for mitochondrial disorders. Uridine is apyrimidine nucleotide, required for synthesis ofRNA and DNA. Normal cell and organ functionrely on adequate synthesis, transport, and intercon-versions of pyrimidines. The synthetic pathway for
uridine synthesis involves the mitochondrial dehy-drogenation of dihydrooroate to oroate, which isintimately linked with CoQ10 recycling and normalelectron transport chain function. Any process thatinterferes with CoQ10 recycling or electron trans-port chain function can impair oroate formation.The process of uridine synthesis concludes withcondensing orotic acid with phosphoribosylpyrophosphate (PRPP) to form uridine monophos-phate. Disordered oxidative phosphorylation willimpede the de novo synthesis of pyrimidines andfurther exacerbate cellular dysfunction. The theo-retical argument for treating with exogenousuridine is to overcome the relative deficit due toimpaired synthesis leading to improved cellularhealth.Hereditary orotic aciduria produces a primary
uridine deficiency and results in a syndrome of fail-ure to thrive, megaloblastic anemia, orotic aciduria,congenital malformations, transient immunoglobu-lin deficiency, immune deficiency, and developmen-tal delays. Lifelong supplemental uridine isrequired and may reverse some of the manifesta-tions of this deficient state. Postulated beneficialeffects of uridine are based primarily on animalresearch and include: appetite stimulation, anticon-vulsant effects, prevention of lactic acid overpro-duction, antidepressant, cardioprotective, andprevention of cerebral edema, among others.Human trials are planned. Side effects have beenreported and more commonly include a transientincrease in seizure activity upon initiation oftherapy, nausea, vomiting, and/or diarrhea (RobertNaviaux, personal communication, 2001).
CombinationTherapy/Miscellaneous1. Sixteen patients with mitochondrial myopathy,Kearns-Sayre syndrome, MELAS, or MERRF weretreated with multiple supplements including vita-min K3 (20 to 60 mg per day), vitamin C (1 gtwice a day), α-tocopherol (200 IU twice a day),CoQ10 (30 to 120 mg per day), and methylpred-nisolone (2 to 16 mg every other day). All patientsunderwent a baseline 31P-NMR spectroscopy,although only five were evaluated during treatmentwith this method. Ten patients were evaluated usingnear-infrared spectroscopy. Neither the 31P-NMRspectroscopy nor near-infrared spectroscopy

Think Mitochondria UMDF31
demonstrated any acute changes in association withtherapy. Follow-up range was 0.5 months to 15years. Ten of the patients died. Despite the lack ofobjective data to demonstrate efficacy with thisintervention, the authors felt there was a subset ofpatients who appeared to benefit from this treat-ment with prolonged survival, less functionaldisability, and/or fewer medical complications.67
2. Thirteen patients treated with daily doses oflevo-carnitine (100 mg to 200 mg), ubiquinone (80to 300 mg), tocopherol (50 to 100 mg per kg),riboflavin (50 to 100 mg), vitamin K3 (80 to 160mg), and vitamin C (2 g) underwent monitoring ofserum carnitine, ubiquinone, and erythrocyte toco-pherol levels. Total and free carnitine levelsincreased with treatment (P <0.05, total carnitine)but the acylcarnitine/free carnitine index demon-strated sustained elevation above that of controls(implying an increase of acylcarnitine generation).Serum ubiquinone and erythrocyte tocopherollevels were higher after treatment, though this didnot reach statistical significance. Additionally,blood lactate levels were significantly decreased infour patients who clinically either stabilized orimproved (P <0.05).68
3. Lipoic acid functions as a coenzyme of thepyruvate dehydrogenase complex. In one study,treatment of a 33-year-old woman with CPEO with600 mg per day of lipoic acid for 7 months wasassociated with a subjective improvement insymptoms and objective changes in 31P NMRspectroscopy of brain and muscle. At baseline thispatient demonstrated reduced phosphocreatine con-centration in conjunction with increased ADPconcentration on brain MRS. Following treatmentwith lipoic acid there was an increased phosphocre-atine content and decreased ADP, indicating thatthe brain was functioning under more stableconditions.Additionally, some improvements wereseen with muscle 31P NMR spectroscopy though notas impressive (rate of phosphocreatineresynthesis postexercise did not improve afterinitiation of therapy).69
SummarEvaluating the efficacy of treatments for mitochon-drial cytopathies is influenced by the relative rarityof these conditions and their variable andunpredictable natural course, as well as their
extensive clinical and biochemical heterogeneity.The lack of conclusive evidence in favor of any onetreatment, or combinations of treatments, makes itimpossible to determine conclusively which meta-bolic therapies might be effective. It is important tonote that there is no evidence suggesting therapyalters the ultimate course of these otherwise poten-tially progressive diseases. There is evidence tosuggest some patients may have an improvement insymptoms and an improved quality of life. Themajority of data available with regards to specifictherapies is anecdotal, though more recently therehas been an emphasis on controlled trials.Unfortunately, even in this setting, the patientpopulation varies significantly and statistical con-straints make it impossible to evaluate all potentialresponses.Because mitochondrial diseases have so many
potential symptoms, identifying which symptom orsign to evaluate as part of any study is problematic.If a study relies on one endpoint, improvements inother functions may be ignored and miss a potentialbenefit of a treatment. Attempting to measure everysymptom and sign of the disease would be soburdensome that a study would be prohibitivelyexpensive. As an example, relying on serum or CSFmeasurements of lactate alone may not fullydemonstrate the full therapeutic benefit of a partic-ular medication. It has also been demonstrated inmany of the studies discussed that even withdiminishment of lactate levels, there is not neces-sarily concurrent symptomatic improvement.Therefore, the use of other objective measures andpossibly many objective measurements (i.e.,nuclear magnetic resonance spectroscopy of brainand/or muscle and bicycle ergometry) to evaluatethe efficacy of these interventions remains crucialto proving or disproving their benefit.Determination of clinical improvements should alsobe included in an effort to prove a link between thebiochemical and functional improvements.Despite the lack of consistent data, providing
supplements as part of an individual trial in whichthe patient serves as their own control seems to bea reasonable approach. The clinician and patientwill need to use their best judgment as to the issuesof efficacy and cost. The use of medications suchas dichloroacetate is still under investigation andwill likely remain reserved for those patients with

UMDF Think Mitochondria32
life-threatening lactic acidosis that is not responsiveto conventional treatment. Uridine remains underinvestigation with regards to its utility in the treat-ment of mitochondrial cytopathies.
References
1. Chinnery PF, Howell N, Lightowlers RN, TurnbullDM. Molecular pathology of MELAS andMERRF: the relationship between mutation loadand clinical phenotypes. Brain 1997;120:1713–1721
2. Lauwers MH, Van Lersberghe C, Camu F.Inhalation anaesthesia and the Kearns-Sayre syn-drome. Anaesthesia 1994;49:876–878
3. Burns AM, Shelly MP. Anaesthesia for patientswith mitochondrial myopathy. Anaesthesia1989;44:975–977
4. D’Ambra MN, Dedrick D, Savarese JJ. Kearns-Sayre syndrome and pancuronium-succinyl-choline-induced neuromuscular blockade.Anesthesiology 1979;51:343–345
5. Casta A, Quackenbush EJ, Houck CS, et al.Perioperative white matter degeneration and deathin a patient with a defect in mitochondrial oxida-tive phosphorylation. Anesthesiology1997;87:420–425
6. Kitoh T, Mizuno K, Otagiri T, Ichinose A, Sasao J,Goto H. Anesthetic management for a patientwith Kearns-Sayre syndrome. Anesth Analg1995;80:1240–1242
7. Barohn RJ, Clanton T, Sahenk Z, Mendell JR.Recurrent respiratory insufficiency and depressedventilatory drive complicating mitochondrialmyopathies. Neurology 1990;40:103–106
8. Maslow A, Lisbon A. Anesthetic considerations inpatients with mitochondrial dysfunction. AnesthAnalg 1993;76:884–886
9. Schenkman KA, Yan S. Propofol impairment ofmitochondrial respiration in isolated perfusedguinea pig hearts determined by reflectant spec-troscopy. Crit Care Med 2000;28:172–177
10. Przyrembel H. Therapy of mitochondrial diseases.J InheritMetab Dis 1987;10(suppl 1):129–146
11. Taivassalo T, De Stefano N, Argov Z, et al.
Effects of aerobic training in patients withmitochondrial myopathies. Neurology1998;50:1055–1060
12. Stacpoole PW, Henderson GN, Yan Z, CornettR, James MO. Pharmacokinetics, metabolism,and toxicology of dichloroacetate. Drug MetabRev 1998;30:499–539
13. Stacpoole PW, Barnes CL, Hurbanis MD,Cannon SL, Kerr DS. Treatment of congenitallactic acidosis with dichloroacetate. Arch DisChild 1997;77:535–541
14. Stacpoole PW, Lorenz AC, Thomas RG,Harman EM. Dichloroacetate in the treatmentof lactic acidosis. Ann Inter Med1988;108:58–63
15. Stacpoole PW, Wright EC, Baumgartner TG, etal. A controlled clinical trial of dichloroacetatefor treatment of lactic acidosis in adults. N EnglJ Med 1992;327:1564–1569
16. Kuroda Y, Ito M, Naito E, et al. Concomitantadministration of sodium dichloroacetate andvitamin B1 for lactic acidemia in children withMELAS syndrome. J Pediatr1997;131:450–452
17. Saijo T, Naito E, Ito M, Takeda E, HashimotoT, Kuroda Y. Therapeutic effect of sodiumdichloroacetate on visual and auditory halluci-nations in a patient with MELAS.Neuropediatrics 1991;22:166–167
18. Kimura S, Osaka H, Saitou K, Ohtuki N,Kobayashi,Nezu A. Improvement of lesionsshown on MRI and CT scan by administrationof dichloroacetate in patients with Leigh syn-drome. J Neurol Sci 1995;134:103–107
19. Tulinius MH, Eriksson BO, Hjalmarson O,Holme E, Oldfors A. Mitochondrial myopathyand cardiomyopathy in siblings. Pediatr Neurol1989;5:182–188
20. Takanashi J, Sugita K, Tanabe Y, Maemoto T,Niimi H. Dichloroacetate treatment in Leighsyndrome caused by mitochondrial DNA muta-tion. J Neurol Sci 1997;145:83–86
21. DeStefano N, Matthews PM, Ford B, Genge A,

Think Mitochondria UMDF33
Karpati G, Arnold DL. Short-term dichloroac-etate treatment improves indices of cerebralmetabolism in patients with mitochondrial dis-orders. Neurology 1995;45:1193–1198
22. Pavlakis SG, Kingsley PB, Kaplan GP,Stacpoole PW, O’Shea M, Lustbader D.Magnetic resonance spectroscopy: use in moni-toring MELAS treatment. Arch Neurol1998;55:849–852
23. Kimura S, Ohtuki N, Nezu A, Tanaka M,Takeshita S. Clinical and radiologic improve-ments in mitochondrial encephalomyopathy fol-lowing sodium dichloroacetate therapy. BrainDev 1997;19:535–540
24. Ogasahara S, Engel A, Frens D, Mack D.Muscle coenzyme Q deficiency in familialmitochondrial encephalomyopathy. Proc NatlAcad Sci U S A 1989;86:2379–2382
25. Goda S, Hamada T, Ishimoto S, Kobayashi T,Goto I, Kuroiwa Y. Clinical improvement afteradministration of coenzyme Q10 in a patientwith mitochondrial encephalomyopathy. JNeurol 1987;234:62–63
26. Ogasahara S, Yorifuji S, NishikawaY, et al.Improvement of abnormal pyruvate metabolismand cardiac conduction defect with coenzymeQ10 in Kearns-Sayre syndrome. Neurology1985;35:372–377
27. Bresolin N, Bet L, Binda A, et al. Clinical andbiochemical correlations in mitochondrialmyopathies treated with coenzyme Q10.Neurology 1988;38:892–899
28. Abe K, Fujimura H, NishikawaY, et al. Markedreduction in CSF lactate and pyruvate levelsafter CoQ therapy in a patient with mitochondr-ial myopathy, encephalopathy, lactic acidosisand stroke-like episodes (MELAS). ActaNeurol Scand 1991;83:356–359
29. Chen RS, Huang CC, Chu NS. Coenzyme Q10treatment in mitochondrial encephalomy-opathies: short-term doubleblind, crossoverstudy. Eur Neurol 1997;37:212–218
30. Chan A, Reichmann H, Koegel A, Beck A,
Gold R. Metabolic changes in patients withmitochondrial myopathies and effects of coen-zyme Q10 therapy. J Neurol 1998;245:681–685
31. Bresolin N, Doriguzzi C, Ponzetto C, et al.Ubidecarenone in the treatment of mitochondri-al myopathies: a multi-center double-blind trial.J Neurol Sci 1990;100:70–78
32. Zierz S, von Wersebe O, Bleistein J, JerusalemF. Exogenous coenzyme Q (CoQ) fails toincrease CoQ in skeletal muscle of two patientswith mitochondrial myopathies. J Neurol Sci1990;95:283–290
33. Matthews PM, Ford B, Dandurand RJ, et al.Coenzyme Q10 with multiple vitamins is gener-ally ineffective in treatment of mitochondrialdisease. Neurology 1993;43:884–890
34. Eleff SM, Barker PB, Blackband SJ, et al.Phosphorous magnetic resonance spectroscopyof patients with mitochondrial cytopathiesdemonstrates decreased levels of brain phos-phocreatine. Ann Neurol 1990;27:626–630
35. Argov Z, Bank WJ. Phosphorous magnetic res-onance spectroscopy (31P MRS) in neuromus-cular disorders. Ann Neurol 1991;30:90–92
36. Gold R, Seibel P, Reinelt G, et al. Phosphorousmagnetic resonance spectroscopy in the evalua-tion of mitochondrial myopathies: results of a6-month therapy study with coenzyme Q. EurNeurol 1996;36:191–196
37. Barbiroli B, Iotti S, Lodi R. Improved brain andmuscle mitochondrial respiration with CoQ: anin vivo study by 31P-MR spectroscopy inpatients with mitochondrial cytopathies. Bio-Factors 1999;9:253–260
38. Bendahan D, Desnuelle C, Vanuxen D, et al. 31PNMR spectroscopy and ergometer exercise testas evidence for muscle oxidative performanceimprovement with coenzyme Q in mitochondrialmyopathies. Neurology 1992;42:1203–1209
39. Cortelli P, Montagna P, Pierangeli G, et al.Clinical and brain bioenergetics improvementwith idebenone in a patient with Leber’s heredi-tary optic neuropathy: a clinical and 31P-MRSstudy. J Neurol Sci 1997;148:25–31
40. MashimaY, Hiida Y, Oguchi Y. Remission of

UMDF Think Mitochondria34
Leber’s hereditary optic neuropathy withidebenone. Lancet 1992;340:368–369
41. Ihara Y, Namba R, Kuroda S, Sato T, Shirabe T.Mitochondrial encephalomyopathy (MELAS):pathological study and successful therapy withcoenzyme Q10 and idebenone. J Neurol Sci1989;90:263–271
42. Schmidt-Sommerfield E, Werner D, Penn D.Carnitine plasma concentrations in 353 meta-bolically healthy children. Eur J Pediatr1988;147:356–360
43. Angelini C, Vergani L, Martinuzzi A. Clinicaland biochemical aspects of carnitine deficiencyand insufficiency: transport defects and inbornerrors of beta-oxidation. Crit Rev Clin Lab Sci1992;29:217–242
44. Pons R, DeVivo DC. Primary and secondarycarnitine deficiency syndromes. J ChildNeurology 1995;10(suppl 2):9–24
45. Carter AL, Abney TO, Lapp DF. Biosynthesisand metabolism of carnitine. J Child Neurol1995;10(suppl 2):3–7
46. Bellei M, Battelli D, Guarriero DM, et al.Changes in mitochondrial activity caused byammonium salts and the protective effect ofcarnitine. Biochem Biophys Res Commun1989; 158:181–188
47. Campos Y, Huertas R, Lorenzo G, et al. Plasmacarnitine insufficiency and effectiveness of L-carnitine therapy in patients with mitochondrialmyopathy. Muscle Nerve 1993;16:150–153
48. Harris RC, Soderlund K, Hultman E. Elevationof creatine in resting and exercised muscle ofnormal subjects by creatine supplementation.Clin Sci 1992;83:367–374
49. Tarnopolsky MA, Roy BD, MacDonald JR. Arandomized, controlled trial of creatine mono-hydrate in patients with mitochondrialcytopathies. Muscle Nerve 1997;20:1502–1509
50. Tarnopolsky MA, Martin J. Creatine monohy-drate increases strength in patients with neuro-muscular disease. Neurology 1999;52:854–857
51. Odland LM, MacDougall JD,Tarnopolsky MA,Elorriaga A, Borgmann A. The effect of oral Crsupplementation on muscle [PCr] and short-term maximal power output. Med Sci Sports
Exerc 1997;29:216–219
52. Penn AMW, Lee JWK, Thuillier P, et al.MELAS syndrome with mitochondrialtRNALeu(UUR) mutation: correlation of clini-cal state, nerve conduction, and muscle 31Pmagnetic resonance spectroscopy during treat-ment with nicotinamide and riboflavin.Neurology 1992;42:2147–2152
53. Griebel V, Kraegeloh-Mann I, Ruitenbeek W,Trijbels JMF, Paulus W. A mitochondrialmyopathy in an infant with lactic acidosis. DevMed Child Neurol 1990;32:528–531
54. Bernsen PLJA, Gabreels FJM, Ruitenbeek W,Hamburger HL. Treatment of complex I defi-ciency with riboflavin. J Neurol Sci1993;118:181–187
55. Arts WFM, Scholte HR, Bogaard JM, KerrebijnKF, Luyt-Houwen IEM. NADH-CoQ reductasedeficient myopathy: successful treatment withriboflavin. Lancet 1983;2:581–582
56. Bernsen PLJA, Gabreels FJM, Ruitenbeek W,Sengers RCA, Stadhouders AM, Renier WO.Successful treatment of pure myopathy, associ-ated with complex I deficiency, with riboflavinand carnitine. Arch Neurol 1991;48:334–338
57. Ogle RF, Christodoulou J, Fagan E, et al.Mitochondrial myopathy with tRNALeu(URR)mutation and complex I deficiency responsiveto riboflavin. J Pediatr 1997;130:138–145
58. Hoppel CL, Kerr DS, Dahms B, Roessmann U.Deficiency of the reduced nicotinamide adeninedinucleotide dehydrogenase component of com-plex I of mitochondrial electron transport. JClin Invest 1987;80:71–77
59. Ichiki T, Tanaka M, Nishikimi M, et al.Deficiency of subunits of complex I and mito-chondrial encephalomyopathy. Ann Neurol1988;23:287–294
60. Lou HC. Correction of increased plasma pyru-vate and plasma lactate levels using large dosesof thiamine in patients with Kearns-Sayre syn-drome. Arch Neurol 1981;38:469
61. Mastaglia FL, Thompson PL, Papadimitriou JM.Mitochondrial myopathy with cardiomyopathy,lactic acidosis: response to prednisone andthiamine. Aust N Z J Med 1980;10:660–664

Think Mitochondria UMDF35
62. Cooper JM, Hayes DJ, Challiss RAJ, Morgan-Hughes JA, Clark JB. Treatment of experimen-tal NADH ubiquinone reductase deficiency withmenadione. Brain 1992;115:991–1000
63. Eleff S, Kennaway NG, Buist NRM, et al. 31PNMR study of improvement in oxidative phos-phorylation by vitamins K3 and C in a patientwith a defect in electron transport at complexIII in skeletal muscle. Proc Natl Acad Sci U SA 1984;81:3529–3533
64. Argov Z, Bank WJ, Maris J, et al.Treatment ofmitochondrial myopathy due to complex IIIdeficiency with vitamins K3 and C: a 31P-NMR follow-up study. Ann Neurol1986;19:598–602
65. Toscano A, Fazio MC, Vita G, et al. Early-onsetcerebellar ataxia, myoclonus and hypogonadismin a case of mitochondrial complex III deficien-cy treated with vitamins K3 and C. J Neurol1995;242:203–209
66. Allen RJ, DiMauro S, Coulter DC,Papadimitriou A, Rothenberg SP. Kearns-Sayresyndrome with reduced plasma and cere-brospinal fluid folate. Ann Neurol1983;13:679–682
67. Peterson PL. The treatment of mitochondrialmyopathies and encephalomyopathies.Biochimica Biophysica Acta1995;1271:275–280
68. Artuch R, Vilaseca MA, Pineda M. Biochemicalmonitoring of the treatment in paediatricpatients with mitochondrial disease. J InheritMetab Dis 1998;21:837–845
69. Barbiroli B, Medori R, Tritschler HJ, et al.Lipoic (thioctic) acid increases brain energyavailability and skeletal muscle performance asshown by in vivo 31P-MRS in a patient withmitochondrial cytopathy. J Neurol1995;242:472–477

UMDF Think Mitochondria36
Mitochondrial medicine has become one of thefastest growing new disciplines in medicine (Luft1994; 1995, Graff 1999). New mitochondrialdiseases are being described every year. Nearlyone hundred different mutations in mitochondrialDNA have been described, and nearly 500 nucleargene defects are associated with mitochondrialdysfunction. Not all of these genetic defects causemeasurable declines in oxidative phosphorylation –the process by which food and oxygen arecombined to make energy (ATP). Nevertheless,even the mitochondrial diseases that do not causemeasurable energy failures can be catastrophic anddifficult to diagnose. Our expanding knowledge ofthe molecular, biochemical, and clinical features ofmitochondrial disorders has forced a change in howscientists understand these complex diseases. Thisarticle will review some of the hallmarks of thesedisorders in adults, and outline some of the teststhat are required for diagnosis.
“Any disease. Any organ. Any age.” Thisis perhaps the best general summary of thespectrum of mitochondrial disease available(Christodoulou 1999). Mitochondrial diseases arenotorious masqueraders (Kerr 1998). They cancause symptoms that are indistinguishable fromthose caused by common disorders. Only thebehavior of the mitochondrial disease over timesets it apart from its more common cousins.Mitochondrial dysfunction has now been linked tocommon maladies as diverse as infertility (Jansen1998), cancer (Susin 1998), migraine headaches(Welch 1995), diabetes (Damore 1999), heartdisease (Hatch 1998, DiMauro 1998), blindness(Latkany 1999), deafness (Fischel 1999), kidneydisease (Niaudet 1996), liver disease (Treem 1998),stroke (Heales 1999), the toxicity of AIDS drugs(Barile 1998), Parkinson disease (Kosel 1999),Alzheimer dementia (Fiskum 1999), and the agingprocess itself (Wallace 1997). Epidemiologicstudies have established beyond doubt that whenthese chronic disorders are studied as groups, they
are complex and have multiple causes – bothgenetic and environmental. Mitochondrial diseasedoes not cause a majority of any one of thedisorders listed. However, it is important toremember that mitochondrial disease can be acause of any of these disorders, because mitochon-drial disease tends to extend to other organ systems,progress with age, and respond poorly to currenttherapy. A comprehensive listing of signs andsymptoms of mitochondrial disease is beyond thescope of this article. This is true in part becausethe general statement quoted at the beginning ofthis paragraph is a clinical fact. The more patientswe evaluate with proven mitochondrial disease, thebroader the spectrum of signs and symptomsbecomes that we normally associate with anyparticular disease. A few examples will bereviewed below to clarify this point.Even a single point mutation in mitochondrial
DNA can produce many different diseases.Perhaps the best studied example of this is theA3243G mutation first linked to the disease calledMELAS (mitochondrial encephalomyopathy, lacticacidemia, stroke-like episodes). In our experienceat the Mitochondrial and Metabolic Disease Center,adults bearing this mutation in mitochondrial DNAfrequently presented with diabetes years beforeonset of brain disease. Some patients first sufferedpsychiatric disease and hearing loss for decadesbefore the onset of recurrent stroke-like episodesand diabetes led to the correct diagnosis. Otherpatients had early onset dementia in their 30s thatwas undiagnosed until the occurrence of a seizureand stroke-like episode. In still other patients, thesingle manifestation of this mutation was an unex-plained cardiomyopathy and mildly elevated lac-tate. Nearly a third of the patients who carry theA3243G mutation had near normal blood lacticacid levels. In these patients, only the cere-brospinal fluid lactic acid was elevated. In 10-15%,lactic acid was elevated neither in the blood norspinal fluid. The message is that not all patients
Adult Presentations of Mitochondrial Diseaseby Robert K. Naviaux, M.D., Ph.D.
The Mitochondrial and Metabolic Disease CenterUniversity of California, San Diego
© The Mitochondrial News, United Mitochondrial Disease Foundation, 2000

Think Mitochondria UMDF37
with the A3243G mutation have MELAS.Similarly, most adult patients who carry theT8993G mutation, often referred to as the NARPmutation, do not have the “neuropathy orneurogenic muscular weakness, ataxia, or retinitispigmentosa”, for which the acronym was coined.These facts illustrate that the name of a mitochon-drial disease can be misleading. Physicians andpatients who rely on an acronym like MELAS orNARP to guide them in making a diagnosis will bewrong more often than not. Even classical mito-chondrial diseases like Kearns-Sayre syndrome canbe difficult to diagnose when the first symptomsappear. Neither the onset nor the rate ofprogression to other organ systems is stereotyped.When ptosis, ophthalmoplegia, retinopathy, ataxia,weakness, exertional fatigue, cardiac conductionblock, elevated cerebrospinal fluid protein, andragged red fibers are all present together, thediagnosis is simple. However, it may take decadesof slow progression before all these symptoms arepresent together. Most often in adults, thesymptoms will appear one by one over severalyears. When progression is relatively slow, adultpatients will typically be referred to a new medicalspecialist every few years, to take care of each newsymptom as it appears. Often it is a medicalstudent trying to make sense of the multisystemdisease who prompts a referral to a mitochondrialand metabolic disease center where the finaldiagnosis is made.If mitochondrial disorders are so complex and
protean, how can general physicians make thediagnosis? A systematic approach is essential. Itwould be wrong to say that everyone with diabetesor heart disease should be checked for mitochondri-al disease. Table 1 lists three rules of thumb thatcan help guide anyone who suspects mitochondrialdisease. If the answer to any two of the three rulesof thumb is yes, and the reasons for these affirma-tive answers are not explained by the patient’s pres-ent diagnosis, then a mitochondrial work-up is jus-tified. Table 2 lists the standard tests that arerequired in the evaluation of suspected mitochondr-ial disease. I call this the “5 + 2” evaluation.Frequently, the results of these tests will suggestother studies that may need to be performed, butthese 7 tests will provide a solid database uponwhich the rational selection of additional studies
Table 1. Rules of ThumbThink mitochondria when:
1. A “common disease” has atypical featuresthat set it apart from the pack.
2. Three or more organ systems are involved.3. Recurrent setbacks or flare-ups in a chronic
disease occur with infections.
Table 2. Diagnostic Testing for Mitochondrial Disease
1. Blood for mtDNA (PCR and Southern)2. Blood and CSF for Lactate and Pyruvate, or Brain
MR Spectroscopy3. Urine Organic Acids (by GC/MS)4. Plasma and Urine Amino Acids5. Blood and Urine Carnitine6. Brain MRI7. Muscle Biopsy
Neuropathology and Electron MicroscopyMitochondrial Electron Transport StudiesFresh (coupled) mitochondrial PolarographyMuscle mtDNA (PCR and Southern)
can be based. The first five tests listed (omittingbrain spectroscopy for the moment) are relativelynon-invasive and can usually be performed forabout $1500. The last two tests are more costly,but are considered by many specialists to be themost informative. The brain MRI and musclebiopsy, along with the associated respiratory chainassays, and mtDNA testing may cost about $5000.When the results of these seven studies are com-bined with careful medical history, family history,and serial physical examinations, and assembled bya metabolic specialist with expertise in neurometa-bolic disease and mitochondrial medicine, anaccurate diagnosis can be reached in about 50% ofthe adult patients referred for suspicion of mito-chondrial disease. Among children, the yield ishigher. These studies permit an accurate diagnosisin about 75% of the children referred for evaluationof suspected mitochondrial disease.The figures quoted in the paragraph above are
influenced by the current state of the art, and by thenature of the patients referred to a specialty centerfor diagnosis. Research is being conducted todaythat will change these figures significantly. Newdiseases are being discovered. New methods ofdiagnosis are being developed. New experimentalsystems and animal models of mitochondrial dis-ease are being constructed, and new treatments are

9. Heales, SJ; Bola–os, JP; Stewart, VC; Brookes,PS; Land, JM; Clark, JB. Nitric oxide,mitochondria and neurological disease.Biochimica et Biophysica Acta, 1999 Feb 9,1410(2):215-28.
10. Jansen, RP; de Boer, K. The bottleneck:mitochondrial imperatives in oogenesis andovarian follicular fate. Molecular and CellularEndocrinology, 1998 Oct 25, 145(1-2):81-8.
11. Kerr, DS. Protean manifestations ofmitochondrial diseases: a mini review. Journalof Pediatric Hematology/Oncology, 1997 Jul-Aug, 19(4):279-86.
12. Kösel, S; Hofhaus, G; Maassen, A; Vieregge, P;Graeber, MB. Role of mitochondria inParkinson disease. Biological Chemistry, 1999Jul-Aug, 380(7-8):865-70.
13. Latkany P, Ciulla TA, Cacchillo PF, MalkoffMD. Mitochondrial maculopathy: geographicatrophy of the macula in MELAS associated Qto G 3243 mitochondrial DNA point mutation.Am J Ophthalmol 128:112-114, 1999.
14. Luft, R. The development of mitochondrialmedicine. Proceedings of the NationalAcademy of Sciences of the United States ofAmerica, 1994 Sep 13, 91(19):8731-8.
15. Luft, R; Landau, BR. Mitochondrialmedicine. Journal of Internal Medicine, 1995Nov, 238(5):405-21.
16. Niaudet, P; Rötig, A. Renal involvement inmitochondrial cytopathies. PediatricNephrology, 1996 Jun, 10(3):368-73.
17. Susin, SA; Zamzami, N; Kroemer, G.Mitochondria as regulators of apoptosis: doubtno more. Biochimica et Biophysica Acta, 1998Aug 10, 1366(1-2):151-65.
18. Treem, WR; Sokol, RJ. Disorders of themitochondria. Seminars in Liver Disease, 1998,18(3):237-53.
19. Wallace D. Mitochondrial DNA in aging anddisease. Sci Am 8(Aug):40-47, 1997.
20. Welch, KM; Ramadan, NM. Mitochondria,magnesium and migraine. Journal of theNeurological Sciences, 1995.
UMDF Think Mitochondria38
being tested. Both clinical and basic research areabsolutely essential for this progress. Thehistory of scientific progress has taught us that themost monumental discoveries are not predictable.We cannot foresee what the next five years will belike for mitochondrial medicine, but based on thecurrent rate of growth, it is safe to say that there arestill a number of surprises in store, that manycherished beliefs will fall, and new ideas about therole and function of mitochondria in human diseasewill be expanded dramatically.
References1. Barile, M; Valenti, D; Quagliariello, E;
Passarella, S. Mitochondria as cell targets ofAZT (zidovudine). General Pharmacology,1998 Oct, 31(4):531-8.
2. Christodoulou J. Somatic cell mitochondrialmutations and respiratory chain disorders.Presented at The Bottleneck SeronoSymposium, Sydney Australia, 7-8 May 1999.
3. Damore, ME; Speiser, PW; Slonim, AE; New,MI; Shanske, S; Xia, W; Santorelli, FM;DiMauro, S. Early onset of diabetes mellitusassociated with the mitochondrial DNAT14709C point mutation: patient report andliterature review. Journal of PediatricEndocrinology and Metabolism, 1999 Mar-Apr,12(2):207-13.
4. DiMauro, S; Hirano, M. Mitochondria andheart disease. Current Opinion in Cardiology,1998 May, 13(3):190-7.
5. Fischel-Ghodsian, N. Mitochondrial deafnessmutations reviewed. Human Mutation, 1999,13(4):261-70.
6. Fiskum, G; Murphy, AN; Beal, MF.Mitochondria in neurodegeneration: acuteischemia and chronic neurodegenerativediseases. Journal of Cerebral Blood Flow andMetabolism, 1999 Apr, 19(4):351-69.
7. Graff C, Clayton DA, Larsson N-G.Mitochondrial medicine—recent advances. JInternal Med 1999; 246:11-23.
8. Hatch, GM. Cardiolipin: biosynthesis,remodeling and trafficking in the heart andmammalian cells (Review). InternationalJournal of Molecular Medicine,1998 Jan, 1(1):33-41.

Think Mitochondria UMDF39
acceleration. Under these circumstances, the carcan stop working completely. In everyday life, thepatient with a mitochondrial cytopathy mayfunction well enough to get by. But when thedemands of an illness or other stressful situationrequire a higher performance state, the ability ofthe body to manufacture the needed energy to meetthat demand is not optimal. It takes longer for amitochondrial patient to recover from an illness,and sometimes the illness is far more severe than ifthe same illness happened to someone with normalmitochondrial function. So when illness occurs,the mitochondrial cytopathy patient is faced with asituation of having both less energy reserves tofight off the illness and the inability to maximizeenergy production, to help overcome and recoverfrom the illness’ effects on the body.Little is known how to prevent an energy
IntroductionThe precarious health of a patient with a mito-
chondrial cytopathy represents the fine linebetween little energy reserve and potential energydeficiency. When demands of added energyrequirements occur, as they do in an acute illness,the decreased reservoir of stored energy in a patientwith a mitochondrial cytopathy often cannotcompensate for the new energy demand. Whencombined with the decreased inherit capacity tomanufacture energy, the patient’s bioenergetichealth is altered and a bioenergetic crisis can occur.This is especially true in young children, who havelittle energy reserves to begin with, and in thosewith a severe mitochondrial cytopathy. Althoughthe number of things that can cause excessivebioenergetic stress is large, we mostly seecompromised bioenergetic health in the context ofanother illness. Viral illnesses and fevers forexample, can have mitochondrial consequences.A quick and simple review of mitochondrial
function is important to understand this article. Weconsume food to make the energy our body needsto function. The energy in food is contained in thecleavable bonds between the atoms in molecules ofsugars (carbohydrate), fats, and proteins. Healthymitochondria will generate 36 molecules of ATP(adenine triphosphate, each ATP represents a unitof energy) for each molecule of glucose that themitochondria can burn or oxidize. If the mitochon-dria do not function (which is not compatible witheven a brief life of any person), glucose is not fullyburned and only 2 ATP molecules will be produced.In this situation, there is also the production of twomolecules of lactic acid. Studies of mitochondrialfunction in some of our sicker patients show thatunder ideal laboratory conditions, only about 40 –60% of the maximal energy can be produced (14 -21 ATP molecules for each glucose burned). Thisis an estimate of theoretical ATP production, whichwould decrease if laboratory conditions mimicked
what occurs in the body during severe viralillnesses, dehydration, and high fever.A useful analogy is to think of an eight-cylinder
car that is running on only 6 or 7 cylinders. Aslong as the car is on horizontal ground the car’sperformance can appear acceptable. However,when the demands are increased, such as when thecar is loaded with passengers, attempts to climb ahill, or accelerates to enter the freeway, there is notsufficient energy performance to accomplish thetask. The car knocks, sputters, and lags in
Management Strategy for Acute Illness in Patients withMitochondrial Cytopathy
by Russell P. Saneto, DO, PhD, Pediatric Epilepsyand Bruce H. Cohen, MD Pediatric Neurology
The Cleveland Clinic Foundation, Cleveland, OH© The Mitochondrial News, United Mitochondrial Disease Foundation, 2000
Table 1. Some Worrisome SignsUnexplained or excessive feverAlteration of usual level cognitive functionConfusion, excessive sleepiness, excessive cryingVomitingLoss of appetiteRapid breathingAbdominal pain

UMDF Think Mitochondria40
imbalance and possible subsequent physiologicaldamage. There is not much in the medicalliterature that is instructive in medically managingthe crises of illness or other stressful event in amitochondrial patient. What follows is based onour experience and understanding of some of thepractical and theoretical implications of how thebody’s biochemistry affects the bioenergetic healthof a mitochondrial patient.
Preparation is the Key to Energetic HealthThere is a wide spectrum of mitochondrial
cytopathies. Each one is expressed in a unique waythat is particular to a specific person. Therefore,there is no “one” best treatment for those with amitochondrial cytopathy. Each patient has to becared for on an individual basis. Furthermore, ourcurrent understanding of mitochondrial diseases islimited and hence, a best treatment protocol doesnot exist. At present, there are many moreunknowns than proven treatments in the medicalcare of a patient with a mitochondrial cytopathy.The fragility of someone with a mitochondrial
cytopathy requires a working knowledge of whatsigns to look for during an acute illness. For thepurposes of this discussion, we will be discussingthe management of fevers, inability to consumeenough liquid and food, and dehydration. Somepatients vomit excessively, others have increasedtremulousness, while still others stop drinking andeating, and some have cognitive changes. It isimportant for the parent or caregiver to know thesesigns (Table 1 on page 39) for their loved one andcall their doctor if these signs develop. In addition,there should be a good working relationship withthe primary care physician, so when the parent orcaregiver begins to see the signs of rapid bioener-getic decline, the physician can make arrangementsfor hospitalization. A plan for such events shouldbe well developed by the physician prior to a crisis.Past experiences will dictate the need for hospital-ization and the immediate treatment once thepatient has arrived at the hospital. Unnecessaryhospitalization may occur on occasion, when thepatient is not as sick as originally believed to be,but both physician and caregivers quickly learnwhen and when not hospitalization is needed.At the first signs of an illness, the parent or
caregiver should be quick to implement treatment.
This generally includes fluid and sugar, and we findthat some common sports drinks such as Gatoradehelp. High carbohydrate meals, given by frequentfeedings, also may help replenish and sustain theneeded levels of glucose for metabolism. Forexample, we have used added uncooked corn starchin a meal to increase the level of glucose in themeal.The use of medication to reduce fever, such as
ibuprofen or acetaminophen should be used. Thedoctor should calculate the proper dose of thesemedications (10 – 15 mg/kg/dose given every 4 – 6hours), so that there are no fevers.
Treatment for Worsening Clinical StatusIt is difficult to pinpoint the time when the
patient needs hospitalization. Experience andcommunication with the physician/health care teamare needed. Decisions can be made when theparent/caregiver notices that, despite the addedmeasures of increased fluids and extra carbohy-drate-containing meals/drinks, the patient has notresponded appropriately. This would be moreurgent if the patient continues to worsen. For eachpatient, the dictating symptoms are different,however, through consultation with the health careteam the appropriate decision can be made.Once the decision is made, the patient and
parent/caregiver should go to the nearest hospital.We tend to have our patients admitted directly tothe hospital, but this will vary according to thepatient’s doctor. Our decisions on what to do nextare based on our knowledge of what deficit ourpatient may have. Once the patient has beenchecked into the hospital, we have a general planfor proceeding ahead. Most patients will needblood and urine tests, placement of an IV, and flu-ids initiated.
Laboratory Tests: The underlying reason for thechange in bioenergetic status needs to be addressed.For example, if there is an infection, this may needto be treated. If it is asthma, then the proper respi-ratory medications need to be given. Laboratorytests may need to be obtained, including lactate,pyruvate, ammonia, electrolytes and urine analysis.These values may assist in understanding the depthof bioenergetic compromise. For instance, if thelactate level is high, then the amount of dextrose

Think Mitochondria UMDF41
added to the balanced salt solution used for hydra-tion can be determined. The level of BUN willhelp determine the level of dehydration and the rateof fluid administration. If the urine analysis indi-cates that ketone bodies are being excreted in theurine, this is an indication that fats are being mobi-lized and maybe carnitine needs to be added to thefluids.
Dehydration: The degree of dehydration is veryimportant. This is because dehydration mayadversely affect the brain, muscle, heart, and kid-ney. Even mild degrees of dehydration, caused byvomiting, diarrhea, or fever may greatly limit thekidney’s ability to get rid of a toxic metabolite, setthe conditions for rising metabolite levels andinduce further injury. This is the likely mechanismfor the evolution of basal ganglia injury in cases ofmethylmalonic aciduria and type I glutaric aciduria.We usually begin dextrose containing a balanced
salt solution, usually D5 or D10 with 1⁄4 or 1⁄2 normalsaline (a salt mixture containing 5% or 10% dex-trose) and added carnitine. Given a particular situa-tion, the amount of salt or sugar could be higher orlower in the IV solution. The percentage of dex-trose containing fluid depends on the abnormalityof the patient. The rate at which fluid is given isindividualized depending on the degree of dehydra-tion, and is the same regardless of whether or notsomeone has a mitochondrial disease. The normalcriteria used to decide whether to administer IVfluids should be abandoned in those with acute ill-ness and dehydration, as oral rehydration therapydoes not offer the same degree of control and thereis not as much room for error in someone with amitochondrial disease.
Glucose:Why use added dextrose (glucose) in amitochondrial cytopathy patient that is dehydratedand/or has lactic acidosis? Let’s use the automo-bile engine analogy again. In a mitochondrialcytopathy patient, the need for fuel is more pro-nounced than in a normal patient. Since the enginedoes not function optimally, we need to eitherincrease the octane of the fuel so the engine getsmore output from the fuel or give the engine morefuel to burn. By giving the patient more glucose inintravenous fluids we are accomplishing both, moreglucose or fuel to burn and a higher octane by
enhancing the purity of the fuel to burn, and there-fore produce more immediate energy (instead of thefatty acids from the breakdown of fats). By treat-ing dehydration, we are also producing an environ-ment for the engine, which is better for energy effi-ciency.In more scientific terms, what we are trying to
do is decrease the lactic acidosis while expandingthe volume of fluid in the body. Lactate, but alsoother toxins, can be poisons to the brain and as pre-viously mentioned dehydration can concentratetoxic metabolites and decrease the kidney’s abilityto get rid of these metabolites. Lactate is the by-product of inefficient glucose metabolism due tomitochondrial dysfunction. When lactate builds up,it causes the blood to become acidotic. The liver,in a non-mitochondrial patient, can utilize much ofthe lactate produced to remake glucose for storageand also burn it for fuel. However, when the pHfalls below a certain point, below 7.1, the liverceases using lactate and instead produces lactate.By giving fluid, we are expanding the volume ofthe blood and allowing the kidneys to help removesome of the toxins. In addition, the added fluid ishelping the kidneys reverse the acidosis. Underconditions of severe illness, it is easier for the bodyto burn glucose, rather than fat, for energy. Thehopeful result of IV fluids with added glucose andcarnitine is the resolution of lactic acidosis, correc-tion of electrolyte balance, and the resolution ofsymptoms. It is critical to note that excess of glu-cose can be highly toxic to a person with pyruvatedehydrogenase (PDH) deficiency. In some situa-tions of severe mitochondrial failure, excess glu-cose can result in worsening lactic acidosis as well.In certain emergent cases, we have had to add an
insulin drip (0.03 units/kg/hr – 0.1 units/kg/hr) tohelp improve mitochondrial function by makingglucose more available to the mitochondria andlowering free fatty acid levels, which can improvethe function of sick mitochondria. These are veryselect cases, and consultation with a mitochondriaexpert is needed to assess and implement insulin inthese special cases.
Levo-Carnitine: In some patients, we will give abolus of levo-carnitine (usually 50 mg/kg, followedby 100 mg/kg/day in 3 divided doses) as thesepatients usually present with a lactate acidosis and

would only recommend these types of treatment inconsultation with a mitochondria expert.
ConclusionThere are a few important points to remember
when dealing with an intervening illness in a per-son with a mitochondrial cytopathy.1. The patient or caregiver, along with the primarycare physician, should develop a plan of howthese illnesses will be approached ahead of time.
2. In many patients, there is little ability to com-pensate during an acute illness, so early interven-tion and the use of IV fluids are often warranted.
3. Once IV hydration has started, it may be neces-sary to stop all attempts at feeding, allowing thebowel to rest, until the patient begins to requestfluids or food.
4. Improvement can be slower than would beexpected in otherwise healthy persons, but mostpatients restart oral hydration and feeding within12 – 24 hours of the IV fluids having begun.
5. The source of infection should be sought, and ifthere is a bacterial infection, it should be treatedwith appropriate antibiotics. Viral infectionsshould not be treated with antibiotics, as these donot work against viruses and many antibioticscan further limit mitochondrial function.
Glossary of TermsSugars: a general term used to define a simple car-
bohydrate.Glucose: a common sugar contained in sucrose
(table sugar) or lactose (milk sugar).Dextrose is the pharmaceutical term forglucose.
Bioenergetic Health: a descriptive term used todefine the ability to produce an adequatesupply of energy that will meet the body’senergy demands.
UMDF Think Mitochondria42
have begun to break down fats into fatty acids. Inthe analogy of the car engine, when the fuel isimproper for the engine there are by-products pro-duced that can decrease the performance of theengine. One can think of carnitine as a fuel addi-tive to help prevent the build up of toxic by-prod-ucts created by inefficient fuel utilization.Carnitine binds toxic-free fatty acids and organic
acids. In addition, it acts as a mitochondrial mem-brane stabilizer (seals the “leaky” gasket). Oftenpatients with mitochondrial cytopathies have a sec-ondary carnitine deficiency as a result of overpro-duction of free fatty acids. The carnitine deficiencywould be worsened by an acute insult to the mito-chondria and the metabolic machinery. Added car-nitine during the acute stage of an illness wouldhelp in removing toxins and improving the carni-tine deficiency. However, there are situations whenadded carnitine may not be needed or may poten-tially worsen the situation.
Other Supplements: There are occasions whenother supplements, in addition to those above, maybe needed. We have patients who have severe mus-cle and peripheral nerve impairment when illnessalso induces changes in bioenergetic homeostasis,very similar to chronic inflammatory demyelinatingpolyneuropathy. Other patients have severe move-ment disorders, such as dystonia. We have foundthat intravenous gamma globulin (1 – 2 gm/kg in 1or 2 doses) temporarily improves these conditions.Although not FDA approved for these diseases, wehave seen this treatment help reverse neuropathicweakness and dystonic movements. There is someexperience with the use of creatine in patients hav-ing mitochondrial myopathies. We have used crea-tine as only a short-term treatment when trying toprevent a patient from being placed on a breathingmachine. The body adapts to long term use of cre-atine and presumably its effectiveness lessens. We

Think Mitochondria UMDF43
IntroductionThis article will outline some basic aspects of
anesthesia and address the issue of the special risksof anesthesia in patients with mitochondrialcytopathies. The applicability of these recommen-dations to a particular patient is complex andshould be individualized by your physician.Considerations include whether the patient isundiagnosed (i.e. receiving their first evaluation todetermine whether a mitochondrial disease ispresent), whether the patient carries the diagnosisof a mitochondrial disease, and what criteria wereused in making the diagnosis. The clinicalcondition of the patient is probably the mostimportant aspect of the pre-operative evaluation.Some patients have minimal disease manifestationsand are at low risk for complications, whereas,other patients have significant disease manifesta-tions such as respiratory muscle weakness,swallowing difficulties, liver disease, and heartdisease, and are at high risk for complications.Anesthesia, often referred to as general
anesthesia, is the medical procedure that renderspatients unconscious, insensible to pain andprovides muscle relaxation. With local anesthesia,or regional anesthesia, patients are awake or sedat-ed, but do not feel pain because the pain pathwaysare “blocked” by the local anesthetics. Spinalanesthesia involves the use of local anestheticsor narcotics injected around the spinal cord,causing loss of sensation below the level that themedication is injected.
Intravenous AnestheticsIn ancient times orally administered extracts of
poppy seeds (opium and morphine), extracts of thedeadly night shade (hyoscene and belladonna), andextracts of fermentations (alcohol) were used for“anesthesia”. All of these drugs decreaseconsciousness and the awareness of pain. Atpresent, most of these drugs or their moderncounterparts are administered intravenously.Thiopental is a rapidly acting barbiturate used forinduction (the first part of anesthesia when the
awake patient is put to sleep) of anesthesia. Propofoland etomidate are new rapidly acting inductionagents. Diazepam (Valium) andmidazolam (Versed) are drugs in the benzodiazepinecategory, which are potent hypnotics (induce asleep-like state). Morphine, meperidine (Demerol)and fentanyl are examples of potent narcotic painrelievers (analgesics) that are used as part of someanesthetics.
Inhalation AnestheticsThe earliest modern inhalation anesthetics were
the gas nitrous oxide (also referred to as laughinggas), and ether, a potent inhalational anesthetic.Potent inhalational anesthetics are vapors producedfrom a liquid that evaporates easily. Nitrous oxide isstill a basic in modern anesthesia; but other modernpotent inhalational anesthetics include halothane,enflurane, isoflurane, sevoflurane, and desflurane.The potent inhalational anesthetic agents provide allthe modalities of general anesthesia, which includeunconsciousness, analgesia and mild muscle relax-ation.
Muscle RelaxantsIn addition to the drugs that induce the sleep-like
state, a group of drugs that provides musclerelaxation are often part of a modern anesthetic.These drugs interfere with the communicationbetween nerves and muscles, and induce aparalyzed state so that the patient does notunconsciously move during surgery. There are oftwo types of muscle relaxants. The depolarizingmuscle relaxants (succinylcholine), cause thepatient’s muscles to move before paralysis occursand are relatively short acting, while thenon-depolarizing muscle relaxants do not cause suchmovements. When patients are paralyzedwith these drugs, the anesthesiologist or anesthetistmust breathe for the patient by either hookingthe breathing tube to a machine or manually squeez-ing a “bag” often containing a mixture of oxygen,laughing gas and a potent inhalational anesthetic.
Anesthesia and Mitochondrial CytopathiesBy Bruce H. Cohen, MD, John Shoffner, MD, Glenn DeBoer, MD
© The Mitochondrial News, United Mitochondrial Disease Foundation, 1998

UMDF Think Mitochondria44
Issues of Anesthesia in Mitochondrial CytopathiesModern general anesthesia consists of induction
with intravenous agent and maintenance with inhala-tional agents and/or with intravenous agents. Musclerelaxants may or may not be used. Concerns about theside effects and possiblecomplications associated with surgery andanesthesia are shared by patients with mitochondrialcytopathies, their families and their physicians.The vast majority of patients with mitochondrial
cytopathies have an uneventful surgery andanesthesia.Patients rarely experience a complication with a
simple elective surgical procedure such as a musclebiopsy or gastrostomy tube placement. Patients withpreoperative respiratory problems are at greater riskfor worse problems after surgery. Similarly, those withseizures may experience post-operative seizures.There are a limited number of reports describing
adverse events and outcomes in patients with mito-chondrial diseases following surgery and anesthesia.Our knowledge about these potentialcomplications are based on these anecdotal reports. Itis not possible to draw conclusions about the safety ofa particular anesthetic agent based on the outcome ofthese cases. Although it is possible to test a particularanesthetic in a laboratory setting to see how it affectsmitochondrial function, this work is based on animalexperiments. How these animal studies relate tohumans in a clinical (not laboratory) setting is impos-sible to determine. In reviewing these reports, a num-ber of inferences can be made:• Patients with mitochondrial cytopathies, on
average, are “sicker” than the unaffected patientundergoing surgery and are having an operative proce-dure for potentially more serious reasons than theunaffected patient.• As a general rule, patients with mitochondrial
cytopathies are at greater risk than unaffectedpeople for side effects of some medications. Althoughsome medications may interfere with energy metabo-lism to some degree, complications are usually relatedto the clinical condition of the patient prior to surgery.• The adverse events reported include new neuro-
logic problems such a strokes, worsening of the over-all neurologic status, respiratory difficulties, seizures,cardiac arrhythmias, prolonged coma and death.• Hypotonia (low muscle tone), bulbar dysfunction
(weakness of the muscles that protect the airway) and
relatively poor ventilatory function (decreased abilityto breathe deeply and cough) are common in patientswith mitochondrial diseases and pose an increasedrisk for perioperative pneumonia. In one study ofpatients with typical Leigh syndrome (which general-ly represents one of the more severe forms of mito-chondrial cytopathies), respiratory difficulties prior toanesthesia and surgery were a predictor of postopera-tive respiratory failure and death. In the cases report-ed, the patients awakened from anesthesia but deteri-orated within a day. There did not seem to be anyspecific anesthetic agent or technique that triggeredthese adverse events.It is not clear from this study whether the deterio-
ration was a direct result of the surgical procedure,the anesthetic drugs, or mitochondrial failure due toinadequate oxygen, resulting from an unrecognizedpneumonia or respiratory failure. Understandingthese factors may make anesthesia safer, but will notavoid all risks.(J Child Neurol 1990;5:137-41)• Malignant Hyperthermia (MH) is a life threaten-
ing, inherited syndrome triggered by potentinhalational anesthetic agents and/or depolarizingmuscle relaxants. It is caused by abnormalincreases in muscle calcium concentrations leading touncontrolled muscle metabolism, subsequent meta-bolic acidosis, muscle damage, and elevated potassi-um levels. Without treatment, MH will often result indeath. Patients at risk for MH may develop the disor-der with their first anesthetic, or may have a dozen ormore anesthetics without a problem, only to developMH with the next anesthetic. There are specific treat-ments available for MH if it should develop, but thebest approach is to identify patients at risk and useanesthetics that do not trigger MH. Risk factors forMH include 1) prior MH episode in the patient, 2) afamily history of MH and 3) muscle disease.Many patients with mitochondrial cytopathies
sometimes have an associated myopathy (muscle dis-ease), which places them at potential risk for MH.There are anesthetics that are “safe” for those with orat risk for MH, but these anesthetics may adverselyaffect mitochondrial function in some patients.• The risk of respiratory failure and worsening of
neurologic function is often noted in patients withmitochondrial cytopathies after “stressful” illnesses,including infections such as routine viral illnesses orpneumonia. Infections may result from a complica-

Think Mitochondria UMDF45
tion of surgery or from the need for surgery, as in thecase of a ruptured appendix. Infections, such as thecommon cold, can also occur randomly around thetime of surgery. Certainly surgery itself, even if for anon-emergency condition, is a major stress. The fol-lowing discussion is quite complicated butnecessary in order to understand that anesthetic drugsalone should not be considered the onlyelement in leading to these adverse outcomes. Duringinfections, the body responds by making chemicalsknown as cytokines. Cytokines help the body fightinfection, and are also responsible for the fever, aches,chills and the overall “rotten” feeling we get when weare ill. Cytokines induce the formation of nitric oxide.Nitric oxide (the chemical formula is NO) is a power-ful oxidant with many useful purposes in our bodies,some of which seem quite unrelated, such as formingnew memories and killing bacteria. However, nitricoxide inhibits cis-acotinase (a citric acid cycleenzyme) and the iron-containing cytochromes of therespiratory chain. Therefore, NO in high amounts maydecrease energy production, which is ill-afforded inpatients who already have an impaired ability to gen-erate energy. Nitric oxide can also interact with otherchemicals in the body that result in damage to themitochondrial DNA and mitochondrial structure itself.TNF (tumor necrosis factor) is one cytokine that isknown to be released by the body during surgery, andis also known to be a potent inhibitor of complex III.TNF has many essential functions, and serves as anatural defense against infections and cancer. In other-wise healthy people, the inhibitory effect on complexIII is obviously not harmful, but may play some rolein people with mitochondrial diseases, who are notable to tolerate any small decrement in mitochondrialfunction. Therefore, anesthetic agents may not beresponsible, at least without additional factors, forcausing neurologic deterioration. Both the stress ofsurgery as well as any associated infections may trig-ger the events leading to a deterioration in susceptiblepatients. (Anesthesiology 1997;87:420-5)• Patients with heart rhythm problems, such as
those with Kearn-Sayres, are at risk for severe heartelectrical conduction blocks, which can lead to death.Isoflurane may be a preferred inhalational agent asopposed to Halothane, because Isoflurane causes lessdisturbances in heart rhythms. (Anesthesiology1994;49:876-878)• Although spinal anesthesia is safe, it should be
used with extra caution in patients with neuropathiesor myopathies, because of the possible deleteriouseffects on blood pressure andrespiratory function.
RecommendationsThere is no doubt that patients with mitochondrial
disease can undergo general anesthesia safely, asdemonstrated by untold thousands of uneventful sur-geries and anesthetic exposures. The question thatpatients and their physicians wish to know is how tofurther decrease the risk. The followingrecommendations are made with the understandingthat there is little data suggesting that any specificprecautions can lower the risk of neurologic events.However, these recommendations seem to beprudent given what is known about the effects of sur-gical stress, infections and anesthetic agents inpatients with mitochondrial cytopathies:1. Strict attention should be made to respiratory func-tion before, during and after surgery, especially inpatients with abnormal preoperativerespiratory signs and symptoms. Vigorous respira-tory physiotherapy should be standard postopera-tive care in any patient with pulmonarydifficulties. Early use of artificial ventilation, main-taining normal oxygenation, CO2 elimination, andvigorous respiratory physiotherapy are recommend-ed at the first sign of respiratory deterioration.
2. There should be a heightened level ofsuspicion for infections such as pneumonia, whichshould be promptly treated.
3. Lactated Ringer’s solution (also known as Ringer’sLactate) should be avoided as an intravenous fluid,as it contains lactic acid.
4. Normal blood glucose, body temperature, andacid-base balance should be maintained duringsurgery. Low blood glucose should be avoided.However, a high blood glucose may indicate anacute disturbance in pyruvate metabolism oroxidative phosphorylation. In this situation, thelactic acid levels may also be elevated.
5. Avoid depolarizing muscle relaxants, althoughthese have been used safely in many patients withmitochondrial diseases. (Anesthesiology1979;51:343-345.)
6. Delay elective surgery if there is any evidence ofinfection.
7. Potent inhalational anesthetic agents appear to be

UMDF Think Mitochondria46
safe in the majority of people with mitochondrialdiseases. In patients at risk for MH, such as thosepatients with myopathies that are often associatedwith their mitochondrial disease, the risks andalternative methods of anesthesia must be consid-ered by the physician. Certainly if there has beena previous adverse reaction in the patient or familymember, these agents should be avoided. (Table 1)
8. Anesthesia with combinations of barbiturates, nar-cotics, benzodiazepines, and nitrous oxide alsopose a theoretical risk for patients with disordersof oxidative phosphorylation. (Table 2) This riskshould be considered only as a potential riskunless a patient has experienced a bad reaction toany of the medications. This apparent paradoxbetween the two methods of general anesthesiamust be addressed with each patient, and the anes-thesiologist must determine what is the safestroute.
9. Animal studies indicate that propofol, a new intra-venous anesthetic, impairs mitochondrialfunction to a greater degree than other anesthetics.However, this drug has been used safely as an
Table 1: Malignant Hyperthermia (MH) PrecautionsFactor Treatment
History Acknowledge the potential for problems inpatients with muscle disease, those with a pasthistory or a family history of MH.
Muscle Relaxants Avoid depolarizing drugs such assuccinylcholine; and use non-depolarizingagents such as pancuronium instead.
Anesthetic Agent Avoid the potent inhalational agents such ashalothane and enthrane. Use agents such asnitrous oxide, barbiturates, benzodiazepines,and narcotics.
Preparation for MH Have adequate amounts of Dantrolene®
available and use it as soon as the first signs ofMH occur.
Table 2: Effects of Anesthetic Agents of Mitochondrial FunctionMedication Biochemical and Clinical Effects on
Mitochondrial FunctionBarbiturates Inhibits Complex I activity at high levels.Benzodiazepines Inhibits adenosine nucleotide translocasePropofol and/or lipid carrier Inhibits mitochondrial function.Halothane Increased risk for heart rhythm disturbance.Nitrous Oxide (chemical formula is N2O) Neurotoxic, possibly by increasing nitric oxide
production, which inhibits cis-acotinase andiron-containing electron transport enzymes;affecting energy production.
Non-depolarizing Agents Increasing sensitivity to the paralytic effectsand prolonged responses reported.
Local Anesthetics Bupivacaine uncouples oxidation andphosphorylation.
anesthetic in many patients with mitochondrialcytopathies. There have been observations thatprolonged continuous use (days) at high dosages totreat frequent seizures causes a syndrome similarto mitochondrial failure, and therefore prolongeduse in a patient with a mitochondrial cytopathymay not be safe.
ConclusionAn increased awareness is needed whenever a per-
son with a mitochondrial cytopathy is contemplatingor undergoing a surgical procedure. By virtue of theillness itself, there are greater risks involved withevery medical intervention. The safest anesthetic isnot known and the choice of anesthetic must be indi-vidualized to the patient’s particular needs. Althoughanesthetic agents may play a contributing factor incausing an adverse event associated with surgery, theillness, the stress of that illness, the surgical proce-dure and concurrent infections may play a larger rolein causing neurologic deterioration. With additionalresearch, more will be learned about these problems.

Think Mitochondria UMDF47
IntroductionLeigh syndrome or Leigh disease is a
degenerative disease of childhood, and is one of themore common and best known of the mitochondrialdiseases. Initially the child appears to be growingnormally and may develop the usual early skillssuch as smiling interactively, rolling over, pullingthemselves up, talking and walking. Suddenly(often following a viral infection) or sometimesgradually, the child begins to lose these skills (iedevelopmental regression). This loss of skills typi-cally becomes apparent between 4 and 12 monthsof age, but it can be much earlier or later, andoccasional patients apparently don’t show symp-toms until adulthood. The clinical symptoms varybetween children, but common features include anabnormal breathing rhythm (panting or sighing),clumsy or jerky movements and either wobbley orfrozen eyeballs. The disease typically progressesbut at a variable rate, often with sharp declinesfollowed by plateaus in which the child may remainrelatively well and even recover some lost skills,but usually followed by another decline. We haveno cure for Leigh disease, although some patientsdo show a partial response to drug or diet therapy.The UMDF asked me recently for permission to
use a scientific article (Rahman and colleagues,1996, Annals of Neurology (1996) volume 39,pages 343-351) about Leigh disease on which I wasthe senior author. Unfortunately, as with mostpublications, the copyright belongs to the publish-ing company rather than to the original authors.Since the permission was not mine to give, Ioffered to write a summary, and attempt to removemost of the scientific jargon so it would be a littlemore easily understood by the non-specialist. I havehad to leave in quite a lot of detail though, and Iwould suggest reading the Introduction of the leadarticle by Dr. Bruce Cohen in the Fall 1997 issue ofUMDF Mitochondrial News for more backgroundon mitochondrial disease. Our 1996 article wasbasically a summary of my laboratory’s experience
of Leigh disease, and an attempt to answerquestions such as: In how many patients can weidentify a basic biochemical defect? How commonis each defect? Do patients with different basicdefects have different clinical features? Howcommon is Leigh disease? What does all this tell usabout the genetics of Leigh disease? We reasonedthat pulling together what we knew of the basicbiochemistry and genetics would improve ourability to choose rational therapy for patients and togive accurate genetic counselling and prenataldiagnosis.
A Brief History of Leigh DiseaseThe characteristic feature of Leigh disease is the
presence of distinctive changes or lesions in certainparts of the brain. Thus until recently, the diagnosisof Leigh disease could only be made after death.This neuropathology was first described as“Subacute Necrotizing Encephalomyelopathy” by aBritish pediatrician, Dr. Denis Leigh in 1951, andSNE is another name sometimes used for Leighdisease. Since Dr. Leigh pronounced his name inthe English way, the correct pronunciation of Leighdisease is to rhyme with “Lee” rather than “Lay”.The brain lesions in Leigh disease were noticed tobe similar to those found in a disease calledWernicke’s encephalopathy. Dietary deficiency ofvitamin B1 (thiamine) contributes to Wernicke’sencephalopathy, and so early studies of Leighdisease concentrated on thiamine metabolism, andit was suggested that urine from patients containeda chemical that blocked activation of thiamine.Other studies around this time (the 1960s)suggested two other abnormalities in Leigh disease.Blood from patients was found to have largeamounts of lactic acid, a chemical that tends toaccumulate when the body cannot generate energynormally from fuels such as sugar. Secondly, it wassuggested that a particular enzyme (ie a proteincatalyst needed to convert one chemical into a dif-ferent form) named pyruvate carboxylase was
Leigh Syndrome: Clinical Features andBiochemical and DNA Abnormalities
by David R. Thorburn, PhDRoyal Children’s Hospital, Melbourne, Australia
© The Mitochondrial News, United Mitochondrial Disease Foundation, 1998

UMDF Think Mitochondria48
missing in some patients.In hindsight, some of the early reports on the
roles of a thiamine inhibitor and pyruvate carboxy-lase deficiency in Leigh disease were probablymistaken, due to problems with the methods oftesting available at that time. During the 1970showever, it became clear that some cases of Leighdisease were caused by inherited defects in twoother enzymes involved in energy metabolism,namely the pyruvate dehydrogenase complex(PDHC) and cytochrome c oxidase (COX). Figure1 shows how these enzymes are involved ingenerating a chemical called ATP from sugars likeglucose. ATP is the key energy carrier that all ourcells need, whether it is used for our body to grow,our muscles to contract, our heart to pump, ourbrain cells to communicate, or our pancreas tomake and excrete insulin. COX is the fourthcomponent of our central energy generating path-way, the mitochondrial respiratory chain, and isalso known as complex IV. The terms “electrontransport chain” and “oxidative phosphorylation”or “OXPHOS” mean much the same thing asrespiratory chain.
During the 1980s, occasional patients wererecognized with defects of respiratory chaincomplex I (also known as NADH dehydrogenase).We also began to realize the amazing complexity ofthe respiratory chain, the normal function of whichrequires more than 100 different genes to beworking properly. It became apparent that somedisorders of the respiratory chain were not due tomutations (or changes) in the genes of the cellnucleus, but to mutations in the mitochondrial DNA(mtDNA). The mtDNA is present in thousands ofcopies in each cell inside our cellular power plants,the mitochondria, which we inherit from ourmother. In the early 1990s, mtDNA mutationswere shown to be responsible for some cases ofLeigh disease. When one considers how long it hastaken to work out the genetics of disorders likecystic fibrosis and Huntington disease, which bothinvolve just a single gene, it is not surprising thatprogress seems slow with disorders of the mito-chondrial respiratory chain.
Our Survey of Leigh DiseaseIn 1992, I had the good fortune to take over a
laboratory which had been started by Dr. GarryBrown (now at Oxford University). It had been themain Australian referral laboratory for childrensuspected of disorders of energy generation, andhad a collection of skin cell samples (or fibroblasts)dating back to the mid-1970s. Given the recentfindings of mtDNA mutations in Leigh disease, Iwas keen to determine how many of our patientscarried such mutations. This was possible thanks toa British pediatrician, Dr. Shamima Rahman, whospent a year in Melbourne on an exchangeprogram. She and I decided to look for mtDNAmutations in all the patients we had been referredfor investigation of Leigh disease. She alsoreviewed all the medical records and brain imagingstudies of each child. Another important aspect ofour study was that prior to 1994, we (and mostother laboratories) had not been able to measurerespiratory chain complex I in skin cells. Thanks toa conversation at a scientific conference, webecame able to do this by modifying a method usedin Dr. Doug Turnbull’s laboratory in Newcastle,UK, enabling us to diagnose complex I deficiencyin an additional 9 patients.A definite diagnosis of Leigh disease is only
NADH AcetylCoA
TCAcycleSuccinate
ATP
Lactic acid
Pyruvate
Glucose
Glycolysis
PDHC
OXPHOSComplexes:
II
III IV VI
Q c
Figure 1. The enzymes involved inconverting sugars to the energy carrier
ATP. Enzyme names are shown in gray highlight, Q is coenzyme Q, and
c is cytochrome c.

Think Mitochondria UMDF49
possible if the child has had brain imaging (CATscan or MRI) or autopsy studies showing thetypical brain changes. The group of 67 children westudied included 35 who had a definite diagnosis,and 32 Leigh-like children who had clinicalsymptoms suggesting Leigh disease but who had notbeen shown to have the typical brain changes. Someof these 32 almost certainly had Leighdisease, but had not had imaging or autopsyperformed. On average, these Leigh-like childrenwere milder than those with Leigh disease ie, theylived longer. The following sections describe the dif-ferent basic defects we found in our patients.
How Common Is Each Basic Defect?mtDNA ATPase6 MutationsOur mtDNA is a circular genome consisting of
16,569 base pairs or nucleotides, which we numberfrom 1 to 16,569. If an mtDNA mutation affects justone nucleotide (ie a point mutation), we refer to itby its number in the mtDNA sequence. Ten childrenhad mutations at nucleotide 8993, in which the nor-mal T base had been changed to either a G or a C.The TG mutation was first described in an mtDNAsyndrome known as NARP, that involves Neuropathy(muscle weakness), Ataxia (a movement disorder)and Retinitis Pigmentosa (a form of blindness), and astudy from NewYork had found previously that itwas a relatively common cause of Leigh disease. The8993 mutations are in the ATPase6 gene of mtDNAwhich encodes part of the ATP synthase (complex V)that forms the final part of the energy generatingpathway. It is difficult to measure complex V activityby enzyme assays, and so none of our 10 patients hadhad an enzyme diagnosis made previously. We there-fore sequenced the ATPase 6 gene in 4 patients (andsubsequently in a further 10) but found no othermutations in this gene.
Other mtDNA MutationsOne child had a large deletion that removed about
a quarter of their mtDNA genome, and asecond had a mutation at nucleotide 8344, which isalso sometimes found in another mtDNAsyndrome, known as MERRF, a form of epilepsy.This mutation is in the tRNA-Lysine gene, whichalong with the 21 other tRNA genes is needed formitochondrial protein synthesis ie to make theproteins encoded by the other mtDNA genes such as
ATPase6. Thus most patients with tRNAmutations also have an enzyme defect of one ormore of the respiratory chain enzymes.
Respiratory Chain Enzyme DefectsThe most common basic defect in our group of
patients was complex I deficiency, affecting 13 ofour 67 children. This enzyme defect has beenunderdiagnosed in the past because the enzymeassay was problematic, particularly in frozenmuscle and in skin cells. Until recently, perhapsonly one laboratory (Dr. Brian Robinson inToronto) had been able to measure complex I reli-ably in skin cells, and he had been the only one todescribe more than one or two cases of Leigh dis-ease with complex I deficiency. The availability ofnew synthetic substrates for the assay, and new pro-tocols means that it can now be diagnosed reliably.Nine other children had deficiency of another respi-ratory chain enzyme, namely complex IV (COX).
Pyruvate Deyhdrogenase DefectsSeven children had PDHC deficiency. Seven
different genes are necessary for normal PDHCfunction, but one of these is a “hotspot” formutations, namely the E1alpha subunit gene, whichis found on the X chromosome. The other PDHCgenes are “autosomal”, ie they are not present onthe X or Y chromosomes that determine our gender,but on some of the other 22 chromosomes. Six ofour PDHC patients were boys, and all six hadmutations in the E1alpha subunit gene. This geneappeared normal in the only girl with PDHCdeficiency, suggesting she had a mutation in one ofthe six autosomal genes for PDHC.
In How Many Patients Can We IdentifyA Basic Defect?In our study, 80% of the patients with definite
Leigh disease, and 41% of the Leigh-like childrenhad a basic defect identified. Shortly after ourstudy, Morris and colleagues (Annals of Neurology,1996, volume 40, pages 25-30) reported a similarsurvey of British Leigh disease patients. Theyclassified patients as definite (pathologicallyproven) and probable, and identified a defect in74% of the definite families, and 39% of theprobable families. They also confirmed thatcomplex I deficiency was a common diagnosis. In
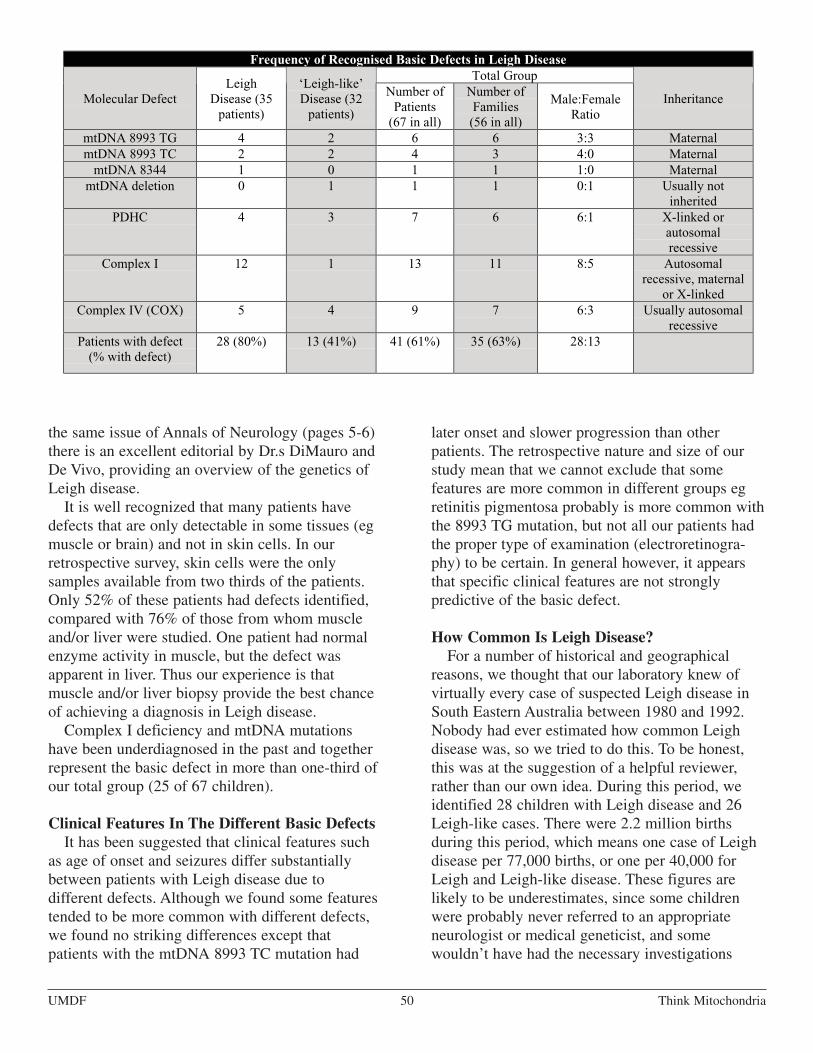
UMDF Think Mitochondria50
Frequency of Recognised Basic Defects in Leigh DiseaseTotal Group
Molecular DefectLeigh
Disease (35patients)
‘Leigh-like’Disease (32patients)
Number ofPatients(67 in all)
Number ofFamilies(56 in all)
Male:FemaleRatio
Inheritance
mtDNA 8993 TG 4 2 6 6 3:3 MaternalmtDNA 8993 TC 2 2 4 3 4:0 MaternalmtDNA 8344 1 0 1 1 1:0 MaternalmtDNA deletion 0 1 1 1 0:1 Usually not
inheritedPDHC 4 3 7 6 6:1 X-linked or
autosomalrecessive
Complex I 12 1 13 11 8:5 Autosomalrecessive, maternalor X-linked
Complex IV (COX) 5 4 9 7 6:3 Usually autosomalrecessive
Patients with defect(% with defect)
28 (80%) 13 (41%) 41 (61%) 35 (63%) 28:13
the same issue of Annals of Neurology (pages 5-6)there is an excellent editorial by Dr.s DiMauro andDe Vivo, providing an overview of the genetics ofLeigh disease.It is well recognized that many patients have
defects that are only detectable in some tissues (egmuscle or brain) and not in skin cells. In ourretrospective survey, skin cells were the onlysamples available from two thirds of the patients.Only 52% of these patients had defects identified,compared with 76% of those from whom muscleand/or liver were studied. One patient had normalenzyme activity in muscle, but the defect wasapparent in liver. Thus our experience is thatmuscle and/or liver biopsy provide the best chanceof achieving a diagnosis in Leigh disease.Complex I deficiency and mtDNA mutations
have been underdiagnosed in the past and togetherrepresent the basic defect in more than one-third ofour total group (25 of 67 children).
Clinical Features In The Different Basic DefectsIt has been suggested that clinical features such
as age of onset and seizures differ substantiallybetween patients with Leigh disease due todifferent defects. Although we found some featurestended to be more common with different defects,we found no striking differences except thatpatients with the mtDNA 8993 TC mutation had
later onset and slower progression than otherpatients. The retrospective nature and size of ourstudy mean that we cannot exclude that somefeatures are more common in different groups egretinitis pigmentosa probably is more common withthe 8993 TG mutation, but not all our patients hadthe proper type of examination (electroretinogra-phy) to be certain. In general however, it appearsthat specific clinical features are not stronglypredictive of the basic defect.
How Common Is Leigh Disease?For a number of historical and geographical
reasons, we thought that our laboratory knew ofvirtually every case of suspected Leigh disease inSouth Eastern Australia between 1980 and 1992.Nobody had ever estimated how common Leighdisease was, so we tried to do this. To be honest,this was at the suggestion of a helpful reviewer,rather than our own idea. During this period, weidentified 28 children with Leigh disease and 26Leigh-like cases. There were 2.2 million birthsduring this period, which means one case of Leighdisease per 77,000 births, or one per 40,000 forLeigh and Leigh-like disease. These figures arelikely to be underestimates, since some childrenwere probably never referred to an appropriateneurologist or medical geneticist, and somewouldn’t have had the necessary investigations

Think Mitochondria UMDF51
(such as MRI) for a definite diagnosis. We felt thatthe figure of 1 per 40,000 births was a reasonableestimate for the incidence of Leigh disease. Thisfigure would imply that about 100 new casesshould be diagnosed each year in the USA.
The Genetics Of Leigh DiseaseThe genetics of Leigh disease are very complex
but the usual modes of inheritance of each basicdefect are summarized in the Table. Up until theearly 1990s, it had been assumed that Leigh diseasewas nearly always an autosomal recessivecondition. Autosomal recessive inheritance impliesthat the child has inherited a faulty copy of anuclear gene from each parent. All of us carry 5 to10 such faulty gene copies that are harmless, unlessour partner happens to also carry a faulty copy ofthe same gene. If that is the case, then eachpregnancy is at a 1 in 4 risk of the two faulty genecopies coming together, resulting in an affectedchild. Perhaps the major finding of our study wasthat the assumption of autosomal recessiveinheritance would have been wrong in nearly onehalf of the families in which a basic defect wasidentified.It has been known for many years that for every
two girls with Leigh disease, about three boys areaffected, and our study confirmed this. Mutations ingenes on the X chromosome tend to affect moreboys than girls, since boys have only one Xchromosome, and the second copy in girls providessome protection. PDHC deficiency causes some ofthis excess of affected boys, but not all of it. Acomplex I gene has also recently been shown to beon the X chromosome, but other factors areprobably also involved, which could be hormonal,environmental or genetic.It is clear that mutations in various parts of the
energy production system can cause Leigh disease.Other patients with defects in the same componentsmay die soon after birth or develop a different rangeof symptoms in childhood or adulthood. This sug-gests that Leigh disease is due more to the degree ofimpairment of energy production in certain brainregions than to the particular gene involved. Thus
we view Leigh disease as part of a spectrum ofenergy generation disorders (see Figure 2). The fac-tors involved in determining where each patient fitsin this spectrum may include genetic factors such asthe gene involved, the particular mutation, themutant load (for mtDNA mutations), the X-chromo-some inactivation pattern (in girls) and environmen-tal factors including infections and diet.
An Update on the Causes of Leigh or Leigh-likeDiseaseSince our article was submitted, a number of
other types of basic defect have been reported inLeigh disease. These include: other mutations inthe ATPase6 gene; other mtDNA mutations in thetRNA genes for valine and tryptophan; mtDNAdepletion (which means not enough copies ofmtDNA, in contrast to mtDNA deletion whichmeans normal numbers of mtDNAs but somecopies having a large chunk missing) ; otherrespiratory chain enzyme defects such as complexII deficiency and combined complex I and IVdeficiency.
Summary Of Major FindingsWe were able to find the basic defect in about
80% of children with Leigh disease, and 40% ofchildren with Leigh-like disease. All the maincauses affect energy generation, and we confirmedthat complex I deficiency and mtDNA mutations(together with complex IV and PDHC deficiency)are common abnormalities. At least three differenttypes of inheritance are common, so accurategenetic counselling can only be offered to a familyif the basic defect is identified. Research advancesduring the last decade mean that major specialistcentres are now able to identify the basic problemin most children with Leigh disease. We have alsoimproved our ability to offer prenatal diagnosis insome families to avoid having further affectedchildren. Clearly, the challenge is to build on thisresearch base, to offer improved diet and drugtherapy and to develop new treatments such asgene therapy.

UMDF Think Mitochondria52
IntroductionTo the extent that physicians understand
mitochondrial disease, the brain is viewed as themost vulnerable organ, both because it is oftenaffected and also because any brain injury can becatastrophic. Most would agree that braindysfunction occurring in mitochondrial diseasesaffects the quality of life more than the dysfunc-tion of other organs systems. Whether it be mentalretardation, seizures or strokes, brain dysfunctionis obvious. One of the most common neurologicevents commonly associated with mitochondrialcytopathies are strokes. Despite this association,strokes are seemingly less common in those withmitochondrial cytopathies than other neurologicproblems such as seizures, weakness or cognitivedelays. The true incidence of strokes is notknown.From a historical perspective, strokes were
known to be part of mitochondrial diseases. Oneof the earliest mitochondrial diseases went by theacronym of MELAS (Mitochondrial Myopathy,Encephalopathy, Lactic Acidosis and Stroke-Likeepisodes), which is still one of the most commonlydescribed combination of symptoms. Almost bydefinition, those with MELAS suffer strokes or“stroke-like” events that seem like a stroke at first,but where the patient returns to normal (or theirneurologic ‘baseline’) quickly. In addition tostroke(s), those with MELAS also have varyingdegrees of lactic acidosis, as well as cognitiveimpairment, weakness, seizures and otherneurologic problems. MELAS will be discussedin greater detail later in this article.The terminology of mitochondrial diseases is
not easy to understand. In the past, the entiregroup of disorders were often referred to as mito-chondrial encephalomyopathies, referring to dis-eases (pathy) affecting the brain (encephalo) andmuscle (myo). However, we now understand thatmany organs other than the brain and muscle wereaffected (Table 1) and therefore the term ‘mito-chondrial cytopathy’ is now considered the accept-ed designation.
Brief Historical Review And Basics OfMitochondrial GeneticsIt is important to understand terms ‘genotype’
and ‘phenotype’ when discussing mitochondrialdiseases. This concept is important to understandbecause it is not always possible, using currenttechnology, to find the genetic mutation that isresponsible for the signs and symptoms expressedas part of the mitochondrial disease. The ‘geno-type’ refers to the specific genetic abnormality thatcauses the disease. In the case of a mitochondrialdisease, this gene mutation can occur either in themitochondrial DNA or in the nuclear DNA. Overthe last two decades, the major effort has been toidentify the mutations on the mitochondrial DNAthat are responsible for the disease. However,it is becoming increasingly evident that nucleargene mutations must also be responsible for some,and possibly the majority, of all mitochondrialdisorders.The ‘phenotype’ refers to how the person
expresses the genetic abnormality, specifically thesigns and symptoms of the disease. It is somewhatunique to mitochondrial diseases that individualmembers of a family with the exact gene mutation,or genotype, can have different clinical expres-sions, or phenotypes. Conversely, people with thesimilar clinical manifestations can have a varietyof different gene mutations. Advances in molecu-lar genetics and mitochondrial medicine occurringin the 1980s and 1990s resulted in identifying anumber of mitochondrial DNA point mutations(the genotype) specifically associated with thesyndrome that historically is referred to asMELAS (the phenotype).It is important to remember that the acronym
‘MELAS’ is used to describe those with mitochon-drial cytopathies who have lactic acidosis andstrokes or stroke-like events as part of their clinicalexpression. Although some point mutations forMELAS have been identified and are easy to testfor, not all patients with clinical MELAS harboredthe known MELAS point mutations.
Strokes and Transient Events in Mitochondrial CytopathiesBy: Bruce H. Cohen, MD
© The Mitochondrial News, United Mitochondrial Disease Foundation, 1998

Think Mitochondria UMDF53
StrokesStrokes are common neurologic events that result
in permanent brain injury and are usually caused bya lack of oxygen or blood flow to a region of thebrain. Strokes are now often referred to as “brainattacks”, as one would refer to heart attacks.Outside the setting of a mitochondrial disease, theblockage occurs as part of the process of athero-sclerosis, where a waxy substance builds up on theinside of the arteries and finally causes thecomplete blockage of that artery. In other circum-stances, a blood clot or other substance can travelthrough the bloodstream and block the artery,which is called an embolus. This interruption inblood flow or oxygen causes the affected area ofthe brain to be deprived of energy. As the energystores are depleted the affected brain cells die. Theextent of the injury is determined by the amount ofbrain involved. Strokes can also be caused bybleeding into the brain, which may occur if aperson has very high blood pressure or a braintumor. When bleeding occurs, brain tissue isinjured and causes loss of neurologic function.Modern technology allows the doctor to confirmthe clinical suspicion of a stroke by performing anMRI scan of the brain. Symptoms of a stroke mayinclude visual loss, weakness, sensory loss andintellectual problems. Although a stroke defines apermanent injury, the symptoms may improve overtime. The process of neurologic recovery is notwell understood, and it is not well known whysome patients improve and others do not. In somecases, the neurologic event is quickly reversible,with the weakness or visual loss disappearing inminutes to hours, and this event is commonlyreferred to as a TIA or transient ischemic attack.One common cause of TIAs are thought to be dueto blockages in blood flow that quickly reversethemselves.We tend to think that strokes mainly occur in
older people, but they can occur at any age.Strokes were described in the early reports ofpatients with mitochondrial diseases, and we willexplore those associated with mitochondrialcytopathies. Strokes that occur in the setting of amitochondrial disease are usually not due to any ofthe factors listed above, but are most likely due to adeficiency of energy production in a particularregion of the brain. Brain cells, like other cells,
need energy to function and survive. This energy isdelivered to the blood stream as glucose (or othersubstances like ketones). In patients with disordersof oxidative phosphorylation, the blood carrying theglucose, ketones and oxygen is delivered properly,but the ability to produce energy in the form ofATP is diminished. This lack of energy can resultin brain injury, just as if the blood carrying the glu-cose and oxygen never got to the brain because of ablockage in the artery. In summary, the stroke isdue to a critical energy crisis, and the extent of theenergy crisis will determine the size and severity ofthe stroke. In some patients with disorders of ener-gy metabolism, there may be a systemic inability togenerate proper amounts of either glucose or ketonebodies, usually in the setting of relative starvationor illness; this lack of energy substrate can alsoresult in a stroke. As with more common types ofstrokes, those with mitochondrial diseases can havebrief events, with mild or severe symptoms thatquickly reverse.Sometimes transient neurologic events are
accompanied by a headache. These headaches aresimilar to the common migraine headaches thataffect millions of people. The headaches tend toinvolve only one side of the head at a time.Patients usually describe the pain as “pounding.”The term complicated migraine is used to describethe situation when neurologic events such as visualloss or muscle weakness accompanies a headache.In patients with mitochondrial diseases, transientneurologic events with or without headaches mayoccur and can be viewed as a reversible “stroke-likeevents.” The gradation between the transientneurologic symptoms associated with complicatedmigraines, reversible stroke-like events, and strokesis probably continuous, with the irreversible strokerepresenting the most severe end of the spectrum.Symptoms of a stroke or stroke-like event can
include:• Various patterns of visual disturbance or visualloss• Weakness of varying degrees, generally affectingonly one side of the body• Clumsiness, generally affecting only one side ofthe body• Inability to speak properly or understand speech• Mental confusion, loss of perceptual abilities, orin severe cases, loss of consciousness

UMDF Think Mitochondria54
Given the large energy needs of the brain, it is nowonder why strokes could occur in those withOXPHOS disorders. However, there are still morequestions than answers with respect to strokes. Itis not fully understood:• What can trigger a stroke after years of normalfunctioning• Why strokes tend to involve rather specific partsof the brain that are not usually affected outsidethe setting of mitochondrial cytopathies• Why strokes occur in some patients withotherwise minimal or no evidence of prior braininvolvement, while other patients with severeepilepsy and mental retardation never have astroke• Why a patient with a severe impairment ofOXPHOS function as measured by muscle analy-sis would never have a stroke, while a patient witha mild impairment may have a stroke
MelasAs mentioned earlier, the most well-described
syndrome in which strokes are known to occur isMELAS, or Mitochondrial Myopathy,Encephalopathy, Lactic Acidosis and Stroke-Likeepisodes. The syndrome of MELAS was proposedin 1984, and was used to describe patients withnormal early development, growth delay, seizures,lactic acidosis, ragged-red fibers on muscle biopsy(signifying overproduction of abnormal mitochon-dria) and episodes that resembled strokes. The
strokes do not always follow the pattern that areseen in strokes due to blocked blood vessels. Thepatient can fully recover from the stroke or thepatient may develop a progressive neurologicsyndrome consisting of multiple strokes with orwithout seizures. Some patients with MELAS mayalso have other neurologic problems that are due toan insufficient energy supply in the brain and else-where in the body. These include ataxia (clumsymovements), optic atrophy and retinopathy (with orwithout visual loss), dystonia/myoclonus/chorea(abnormal twisting or shaking movements), andneuropathy (nerve injury). Neuro-psychiatricsyndromes may be a transient or progressiveproblem in MELAS and other mitochondrialcytopthies. The strokes in MELAS usually occurbetween the ages of 5 to 15 years, but can occur atany time. The stroke generally involve posteriorbrain regions, especially the occipital lobes (affect-ing vision) or posterior parietal or temporal lobes.The initial stroke in MELAS can be severe, or thepatient may have a migraine-like event where thepatient develops weakness, visual loss, or mentalchanges associated with a sever headache. Theneurologic event may reverse (as would be with amigraine) or may not.All patients with MELAS have similar
symptoms, but like all mitochondrial diseases, theseverity of symptoms vary from patient to patient,even in those patients that are members of the samefamily. This variability is due to a number of
Table 1: Problems That May Be Associated with Mitochondrial CytopathiesOrgan System Possible ProblemsBrain developmental delays, mental retardation, dementia, seizures, neuro-
psychiatric disturbances, atypical cerebral palsy, migraines, strokesNerves weakness (which may be intermittent), neuropathic pain, absent reflexes,
gastrointestinal problem (ge reflux, constipation, pseudo-obstruction),fainting, absent or excessive sweating resulting in temperature regulationproblems
Muscles weakness, hypotonia, cramping, muscle painKidneys proximal renal tubular wasting resulting in loss of protein, magnesium,
phosphorous, calcium and other electrolytesHeart cardiac conduction defects (heart blocks), cardiomyopathyLiver hypoglycemia (low blood sugar), liver failureEyes visual loss and blindnessEars hearing loss and deafnessPancreas and OtherGlands
diabetes and exocrine pancreatic failure (inability to make digestiveenzymes), parathyroid failure (low calcium)
Systemic failure to gain weight, short statue, fatigue, respiratory problemsincluding intermittent air hunger, vomiting

Think Mitochondria UMDF55
factors. It is critical to note that the syndrome ofMELAS is not due to a single genetic mutation.About 80% of patients with MELAS have an A toG point mutation at base pair 3243, with otherpatients having a T to C point mutation at bp 3271or a mutation of G to A at bp 3252. All of thesemutations affect the transfer RNA that encodes forleucine (leucine UUR tRNA). Secondly , as withall other mitochondrial cytopathies due to amutation in the mtDNA, the percentage of abnor-mal mtDNA varies randomly among the differenttissues in the body. In other words, the brain ofone patient may have more abnormal mtDNA thanthe muscle of that same patient; consequently thestroke manifestations may be more or less evidentthan the other features of the condition. Finally,patients with the same mutation in the same familymay inherit variable quantities of abnormalmtDNA, which means that the brain of one familymember may be more abnormal mtDNA, and there-fore more severe neurologic disease, than the brainof another family member.
Strokes Outside the Setting of MelasIt is not uncommon for a person with mitochon-
drial cytopathy to have many of the same featuresof MELAS, including the myopathy, encephalopa-thy, lactic acidosis and strokes, but test negative forthe point mutations associated with MELAS.As we learn more about the point mutations,biochemistry, respiratory chain associatedstructures and ultra-structure of the mitochondria,we may understand more about what causesstrokes.
Prevention, Diagnosis and TreatmentThere are no proven preventative measures that
will reduce the risk of strokes in patients withmitochondrial diseases. Furthermore, in patientswith MELAS, there probably is no treatment todaythat will prevent a stroke. Illnesses such as the flumay increase the risk of a stroke or stroke-likeevent, but most strokes occur outside the setting ofsuch an illness. Stroke-like events and neurologicdeterioration have been described after viralinfections (with subsequent dehydration andtemporary starvation), vaccinations, alcoholingestion, hypoxia (as would occur at very highaltitudes or with cigarette smoking), ingestion of
monosodium glutamate (MSG) and exposure toextreme temperatures. It is impossible to elimi-nate exposure to viruses, and as a general rule,children with mitochondrial cytopathies shouldhave routine vaccinations. It is a matter of per-sonal choice to avoid alcohol, cigarette smoke andMSG. Aside from locations such as SanFrancisco, it is not possible to avoid temperatureextremes, and if a person shows a particular sensi-tivity to extreme heat or cold, these should beavoided. Regardless, proper bundling is importantin the winter, and over-dressing can be as harmfulas under-dressing. In the summer, overheatingcan be particularly dangerous to the child with amitochondrial disease, so loose fitting clothes,proper shading (a cool hat), plenty of water, andavoidance of prolonged direct sunlight is impor-tant. Regardless, strict adherence to all of theabove will not prevent a stroke from occurring,nor is there any evidence that one can lower therisk of strokes in the setting of MELAS, even withavoidance of all precipitating factors and use ofmedication and vitamins.The evaluation of a person with a stroke of the
first stroke-like event would include a physicalexamination and neuroimaging, preferably withmagnetic resonance imaging (MRI). In thesituation of the first event when the person has yetto be diagnosed with a mitochondrial cytopathy,the MRI can aid in the diagnosis if the pattern ofbrain injury fits with the typical patterns seen inthese diseases. Once the clinical patterns ofstroke-like events has been established, repeatedimaging is not necessary with each event.There is no specific treatment for strokes that
occur in people with mitochondrial disease. Thenew treatments for strokes that occur outside thesetting of mitochondrial diseases, such as tPA, arenot beneficial for the strokes that are due to mito-chondrial dysfunction. Supportive care is essen-tial and should consist of bed rest, and in severecases intravenous fluids may be needed.Metabolic parameters such as electrolytes, glu-cose, lactate and ammonia should be evaluatedimmediately and as needed during recovery. Lowblood glucose should be treated immediately. Ifthe patient is not alert enough to eat, nutritionalsupport is necessary.

UMDF Think Mitochondria56
ConclusionAside from MELAS, in which, by definition,
stroke is part of the syndrome, it is not possible todetermine the risk of stroke in a person with mito-chondrial cytopathy. Even in those with MELAS,the timing of the stroke and stroke-like events arenot predictable and occur at random. It seems to bethe experience of those that care for a lot ofpatients, that testing positive for the known pointmutations associated with MELAS generallycarries with it a worse overall prognosis, in termsof ability to recover from the stroke. Whether ornot most people with mitochondrial diseases are at
increased risk of strokes over their lifetime is notknown, although less severe stroke-like events areknown to occur rather frequently in some.Disorders of brain function that occur in peoplewith mitochondrial cytopathies may includeseizures, developmental delays and severe cognitiveproblems that include mental retardation andautism, dementia (loss of acquired cognitive func-tions), stokes and stroke-like events. Again, notall people with mitochondrial cytopathies havedisorders of brain function, and those that do mayhave mild, moderate or severe symptoms.

Think Mitochondria UMDF57
IntroductionThe disorders of energy metabolism comprise a
group of sometimes devastating disorders. Theyusually have their onset in infancy and can be fataldue to their rapid progression in a matter of weeks,months or years. A smaller subset of patients haveslower developing diseases with onset in theteenage or adult years, but may be at risk forhaving a child who may be much more severelyaffected. How do we deal with this from the pointof view of prenatal diagnosis? How do we counselparents about the chances of having another affect-ed child, and how do we counsel them about theprenatal detection of the mitochondrial disorders asa group?A search of the literature will not find very much
written about any of these questions. The lack ofliterature stems from two major problems. The firstproblem is that very little is known about the pat-tern of inheritability of many of these disorders. Insome we assume autosomal recessive the mode ofinheritance by default. In others we just do notknow whether the defect is nuclear or mitochondri-ally encoded in any one family. The secondproblem is the lack of a collective database ofinformation for those physicians, scientists andparents brave enough to go through prenatal diag-nosis procedures with no guaranteed result. Anyparents reading this, who then ask the question “isprenatal diagnosis available for us?” need to haveat their disposal as much information as possibleabout the disorder that has affected their family.
The major information you need is:1) What is the name attached to this disorder?2) Is there an enzyme diagnostic test for thisdisorder?
3) Has the disorder been narrowed down to thelevel of the gene?
4) Can the disorder be detected in skin fibrolasts?
Why These Questions?The name attached to the disorder tells you
either about the symptomatology of the diseaseprocess, or about the name of the principle enzymethat (by interrupting biochemical pathways) isresponsible for the symptomatology of the diseaseprocess. It may be an acronym such as MELAS,MERRF or NARP, or it may be a name likecytochrome oxidase deficiency or PDH complexdeficiency. This information in some cases isenough for you to discern whether prenatal diagno-sis is possible. Very often you also need to knowthe subtype of the disorder. Your physician can helpyou with this if you don’t know the answer to thequestion right now. It is beyond the scope of thisarticle to fill in the differential diagnostic featuresof all mitochondrial diseases so in the table below Ihave set out each of the disorders with known sub-types and some information which could be helpfulabout prenatal diagnosis.
What Are You Likely To Find?The simple truth at the moment is that the
minority of these diseases is amenable to prenataldiagnosis. That is the bad news. The good news isthat the situation is improving all the time. Theimprovement comes from two sources, the firstbeing improvements in cellular detection tech-niques. Better testing procedures are beingdeveloped all the time in laboratories around theworld. The second source of discovery is thedescription of the genes and genetic mutationsresponsible for this group of disorders. This yearthe gene defect forPDH-X deficiency, one form of Complex I defi-
ciency (AQDQ gene defect), and one form of COXdeficiency (classical Leigh disease due to theSURF1 gene) have been described. More are on theway.(The following more detailed information is
divided into discussion of nuclear DNA defects andmtDNA encoded defects.)
Prenatal Diagnosis of Mitochondrial Diseaseby Brian H. Robinson, Ph.D.
Hospital for Sick Children/University of Toronto© The Mitochondrial News, United Mitochondrial Disease Foundation, 1999

UMDF Think Mitochondria58
Nuclear-Encoded Defects:There are a lot of these and most are inherited in
an autosomal recessive pattern. That means thatalthough the parents are unaffected they both con-tribute a mutation in their genes to the affected child.
Pyruvate Dehydrogenase Deficiency:Most patients having this defect have a defect in
their E1a gene which is carried on the X-chromo-some. Thus the affected subtype is E1. However,this is an unusual X-linked disease because bothboys and girls can have it. When it occurs, most ofthe time the defective gene cannot be detected inthe mother or the father. This is because the muta-tion happened in the formation of the egg or spermbut is not present in the other tissues of the parent.This is good news for those seeking prenatal diag-nosis because the recurrence risk drops from 1 in 2for boys to a very low number. To my knowledge,32 prenatal diagnoses have been done for thisdefect, and 30 have been unaffected, quite normalchildren.Since molecular genetic diagnosis for this dis-
ease is feasible, this is the method of choice.Measurement of enzyme activity can be risky,especially if the expected child is female. The otherdefects - E2, E3 and X ñ are all autosomalrecessive and could be detected either by moleculartechniques or by measurement of enzyme activity.
Pyruvate Carboxylase Deficiency:This defect has been detected prenatally by
enzyme measurement. Molecular techniques maysupercede this, but are much more expensive.
Respiratory Chain DefectsComplex I (NADH-UbiquinoneOxidoreductase):This is a very difficult enzyme to measure in cul-
tured cells, so trying to detect it in amniocytes orchorionic villous cells is next to impossible.However, the more severe defects in this enzymecan be detected by the high lactate to pyruvate ratiopresent in the cells. Eighteen prenatal tests for thisdefect have been done by this technique. Fifteenwere predicted unaffected, three were affected. Oneof these predictions was incorrect by this method.Better methods for this defect will come as moremolecular defects are defined.
Complex II (Succinate-CoQ Oxidoreductase):Defects in the subunits of succinate dehydroge-
nase have been defined in molecular terms. Thisshould be amenable to prenatal diagnosis in thefuture.
Complex III (Nuclear):No Complex III defect has been defined in
molecular terms. These defects are poorlyexpressed in fibroblasts, and therefore probably notamenable to prenatal diagnosis in amniocytes.
Complex IV (Cytochrome Oxidase Deficiency):The gene for the most common form of
cytochrome oxidase deficiency has recently beenshown by Dr. Eric Shoubridge to be the SURF1gene, a gene responsible in some way for theassembly of the cytochrome oxidase complex. Thisdefect was already diagnosable in amniocytes byenzyme techniques. The less common Quebec-Saguenay Lac St Jean COX deficiency is caused bya different gene located on Chromosome 2 (BHR).It is difficult to diagnose prenatally, but a new tech-nique based on Chromosome 2 markers is beingdeveloped. The other forms of COX deficiency arenot well expressed in fibroblasts, and little informa-tion on prenatal diagnosis has been published.
mtDNA Depletion Syndrome:While this syndrome entails gradual loss of
mtDNA, the gene responsible is a nuclear one andprobably is responsible for some critical function inmitochondrial maintenance. The defect is onlysometimes expressed in cultured cell systems,which means that looking at amniocytes or chorion-ic villous cells may not be reliable. The majorenzyme deficit is cytochrome oxidase deficiencyand the disease is sometimes mistaken for a pri-mary defect in this enzyme. Prenatal diagnosis hasnot been attempted in this disease to my knowl-edge.
mtDNA Deletion Syndrome:In this defect, multiple deletions of mtDNA
appear in tissues, usually skeletal muscle and heart.The disease is often autosomal dominant in inheri-tance, which means you can inherit the diseasefrom one parent and it is again a nuclear-encodedgene which is responsible. Several chromosomal

Think Mitochondria UMDF59
Expressed in
Defect InheritanceMode
MolecularTest
Available
Muscle Fibroblasts Amniocytes CVS Prenatal Test available
PDHE1a
E2E3X
X-L; AD; AR
ARARAR
Yes
YesYesYes
Yes*
YesYesYes
Yes
YesYesYes
Yes
YesYesYes
Yes
YesYesYes
Activity (Males)Molecular (Females)
--Yes
PC Nuclear AR Yes X Yes Yes ? YesComplex I Nuclear AR X Yes Yes � ? � Under Investigation
Complex II SDH Nuclear AR Yes(some)
Yes ? ? ? ?
Complex III AR Yes Yes ? ? ? ?Complex IV NuclearClassic Leigh DiseaseSLSJ Leigh DiseaseFatal InfantileCardiomyopathy
ARARARAR
YesXXX
YesXYesYes
Yes�
XYes
Yes�
X?
Yes???
Enzyme Analysis� Under Investigation
??
mtDNA Depletion SyndromeNuclear
AR X � � ? ? ?
mtDNA Deletion (Familial)Nuclear
AR; AD X Yes X ? ? ?
mtDNA Deletion (Sporadic) S or M Yes Yes X ? ? ?mtDNA tRNA (MELAS,MERRF)
M Yes Yes � ? ? ?
mtDNA rRNA M Yes Yes Yes ? ? ?mtDNA Protein (NARP) M Yes Yes Yes ? ? � Under InvestigationLegend AR = autosomal recessive AD = autosomal dominant S = sporadic M = maternal (through mtDNA) X-L = X-linked
loci have been located in different families, andprenatal diagnosis might be possible but for what isoften an adult onset disease, there is understandablynot a lot of interest in this procedure. Deletions arenot often seen in cultured cells.
mtDNA Deletion (sporadic, Kearn SayreSyndrome, CPEO, Pearsons Syndrome):Usually the deletions are not seen in cultured
cells and the affected cases are sporadic, i.e. notlinked to other family members, and rarely present
How to Read the TableFirst of all, locate the name of the defect, then look for the subtype. For instance, if you know your child hasPDH complex deficiency, the information is different depending on whether the defect is located in the E1, E2,or X components. If you are looking up cytochrome oxidase deficiency you need to know if you are dealingwith the classical Leigh form, the Saguenay Lac St Jean Leigh form, the fatal infantile form, the cardiomyo-pathic form, or a non-classified variety. For each of them the information is different.
Under each subtype is listed:Nuclear/Mitochondrial: Is this disorder determined by the nuclear DNA or by mitochondrial DNA (mtDNA)?Molecular: This says whether the gene responsible has been identified, yes or no.Muscle, Fibroblasts: These headings tell you whether the enzyme defect is measurable in fibroblasts (culturedskin cells).Amniocytes: This tells you whether fetal cells in the amniotic fluid express the defect.CVS: This tells you if it is known whether chorionic villous cells from the placenta express the defect.Prenatal Diagnosis: This tells you whether prenatal diagnosis for this disorder has been performed successfully.
continued on page 60

UMDF Think Mitochondria60
in siblings. This puts the chances of having twoaffected children at a low probability. It is doubtfulthat looking for deleted mtDNA in amniocytes orchorionic villous tissue would be useful.
Primary Mitochondrial DNADisordersThese mtDNA defects are inherited maternally.
For those mothers that the gene defect, there is lit-tle to be gained by testing the fetus, as the fetuswill also have the gene defect, and because thepercentage of abnormal mtDNA in the tested cellsmay not reflect the percentage of abnormal mtDNAin the muscles or brain, there is no method to pre-dict how sick the baby would be. There has beenlittle interest in prenatal diagnosis for those mtDNAdefects that involve transfer RNA genes. Theseinclude the most common diseases, 3243 MELASand 8344 MERRF. The amount of mutated mtDNAin any one tissue determines whether the tissue isaffected. The amount or extent of mutated mtDNAin muscle, heart and brain of a developing fetusmay bear no relationship at all to the amount thatcan be measured in amniocytes or chorionic villoustissue. This has precluded any attempt to diagnosethese defects in utero. For several diseases affectingprotein coding genes of mtDNA, especially 8993NARP, the situation may be different. While experi-ence is limited it seems that amniocyte levels of themutation may reflect at least partially the levels inthe tissues of the fetus. Obviously more data isneeded in this area, but at best the predictability ofsuch testing is only likely to provide an approxi-mate guide to the percentage of the mutation. For
other conditions such as the pigmentary retinopathyassociated with the11778 LHON mutation, noinformation is available, but again, for obviousreasons there is little interest in prenatal determina-tion of status.
Facilities For Prenatal DiagnosisHaving read the above guide, you may be inter-
ested in pursuing the subject further. The onecourse of action that is not advisable is to begin apregnancy on the basis of what you know, and thenseek out prenatal diagnosis. The two major prob-lems that any center embarking on a prenatal diag-nosis have to deal with are:1) Being satisfied that the criteria needed for prena-tal diagnosis i.e.: expression of the defect in cul-tured cells, have been met.
2) Fear of litigation, should a prediction be incor-rect. The first problem is one that often needs tobe looked at very carefully and itself may takeseveral months to resolve. The second problemis more difficult and prevents many institutionsfrom attempting prenatal diagnosis for mito-chondrial disorders. In either case, lengthy dis-cussions of the facts surrounding your case and arealistic exploration of expectations with yourgenetic counselor or physician are mandatorybefore you do anything.
ReferencePoulton, J. and Marchington, D.R. (1996) Prospectsfor DNA-based prenatal diagnosis of mitochondrialdisorders. Prenatal Diagnosis 16:1247-1256.

Think Mitochondria UMDF61
Gastrointestinal (GI) symptoms are commonlyencountered in patients with mitochondrial disease.Most often, symptoms are episodic in that theycome and go, and are related to ‘functional’problems of the bowel. In particular, vomiting iscommon among sufferers of many different mito-chondrial disorders, and among these individuals,vomiting itself has many different causes.Occasional bouts of vomiting is common,especially in infants, and is often found to becaused by gastroesophageal reflux. This articlediscusses one particular type of vomiting disorder,called ‘cyclic vomiting’.Cyclic vomiting is not new as it was first
described in the eighteenth century, although eventoday very few physicians or other clinical careproviders have heard of it. Cyclic vomiting refersto discrete and severe episodes of vomiting, nauseaand lethargy (severe tiredness). Episodes arediscrete in that the sufferer is free of nausea andvomiting between episodes. Episodes are severe inthat the sufferer almost always feels quite ill,preferring to lie down in a dark and/or quiet place,and not interested in any of the activities of life.Vomiting and loss of appetite can be severe enoughto necessitate hospitalization for intravenous (IV)fluids with each episode. In other cases, nauseaand lethargy may be much more troublesome thanvomiting. Episodes can occur on a routineschedule (such as once a week or once a month), betriggered by physical or psychological stress, orappear to come at random. Each episode can lastfor hours to many days, but usually there is acharacteristic duration in each individual patient.In some cases, an episode may be stopped if thechild sleeps. Some sufferers have additional symp-toms during episodes such as loose stools, droolingor headache. There may or may not be an ‘aura’,or symptoms which occur before vomiting begins.In most cases, cyclic vomiting starts in childrenranging from about age 3 to 8 years, although thedisorder can start at any age including in infantsand adults. Cyclic vomiting can run its course andresolve, continue indefinitely, or change into
migraine headaches. Most sufferers have normalintelligence and are generally healthy betweenepisodes, however, many of them have variousdegrees of developmental delay and/or additionalproblems such as epilepsy.Cyclic vomiting has many known causes,
including intestinal blockage, brain disorders,kidney disease, and several different metabolicdisorders. Many of these causes are treatable, anda careful diagnostic work-up is important.However, in the vast majority of cases, none of theabove causes can be found, and these individualsare given the diagnosis of ‘cyclic vomitingsyndrome’ or ‘CVS’. Migraine headaches andepisodic severe abdominal pain (abdominalmigraine) are very common in CVS sufferers andtheir family members alike (usually in the maternalrelatives!). At present, migraine headaches,abdominal migraine and CVS are considered to berelated, and possibly are different manifestations ofthe same disorder. Cyclic vomiting has beenreported in individuals with the A3243G ‘MELAS’mutation in the mitochondrial DNA (mtDNA). Inaddition, one child with CVS, developmental delay,seizures, growth delay and additional problems wasreported to have a large mtDNA deletion andduplication. However, in my experience as ametabolic geneticist, CVS with or withoutadditional problems is not rare in children withmitochondrial disorders, and among this group,‘routine’ mtDNA analysis fails to identify previous-ly known mtDNA mutations in most of them.To date, I have personally evaluated about 15
children with CVS and suspected mitochondrialdisease. These and an additional 50 cases collectedworldwide with CVS and additional neuromuscularproblems (a group at risk for possible mitochondri-al disease) have been entered into an ongoingresearch study. Many of these children have aspecific pattern of additional clinical and laboratoryfindings including GI dysmotility (reflux, delayedgastric emptying, constipation), dysautonomia(unexplained fevers, high heart rate, etc.), muscleweakness, chronic fatigue, seizures and pain (head,
Cyclic Vomiting in Mitochondrial Diseaseby Richard G. Boles, M.D., Division of Medical Genetics, Childrens Hospital Los Angeles
© The Mitochondrial News, United Mitochondrial Disease Foundation, 2001

UMDF Think Mitochondria62
abdomen and/or extremities). The latter, referred tosometimes as “muscle cramps”, is occasionallyassociated with swelling and skin discoloration in amanner suggestive of neuro-vascular dystrophy.No single child suffers from all of these problems,and when present in a given child the symptomstend to be episodic and variable. In some of thesechildren, cyclic vomiting itself is a minor part ofthe child’s problems, and may disappear or neverhave been present. Intelligence ranges from giftedto severe mental retardation.Laboratory analysis in children with CVS and
mitochondrial disease demonstrates elevated lacticacid and abnormal urine organic acids (ketones,Kreb cycle intermediates, and/or ethylmalonate)early in vomiting episodes, but biochemical testsare rarely abnormal at other times. A few childrenhave received muscle biopsies which revealedfindings suggestive of mitochondrial dysfunction,including increased variation in fiber size, mito-chondrial proliferation, and/or complex 1deficiency. However, in my opinion, the moststriking finding is maternal inheritance of the samekind of episodic problems seen in the affectedchildren themselves, but usually to a lesser degree,including migraine, cyclic vomiting, GI dysmotility,dysautonomia, muscle weakness or pain, chronicfatigue, and/or seizures. At the time of this writing,at least 5 unrelated cases were found to haveheteroplasmic (two different mtDNA sequencespresent in the same individual) nucleotide changesin the mtDNA control region. These molecularvariants are maternally inherited (present in motherand siblings, even if they themselves are withoutsymptoms) and were not found in over 100 childrenwithout mitochondrial disease. The same controlregion variants were found in children with mito-chondrial disease but without CVS, and thesignificance of our recent findings are not yet clearand are the subject of ongoing investigation.However, our data does demonstrate that mitochon-drial disease with cyclic vomiting is oftenmaternally inherited.Unlike most published cases with mitochondrial
disorders, disease progression appears to be rare inchildren with maternally-inherited cyclic vomiting.One exception to the general benign disease courseis that a few families have had infants under age 2years who died suddenly and were labeled as
“SIDS”. Most children, and especially their affect-ed relatives, attend normal schools or havejobs/careers, and their lives are fairly normalbetween disease episodes. In many school-agedaffected children, severe fatigue and muscleweakness has necessitated the occasional usage ofwheelchairs and/or half day or home schooling. Alltoo often, clinical care providers and/or schoolpersonnel have down-played the disease process,even to the extent of labeling the child/family asexaggerating symptoms, being psychologicallydisturbed, or having caused the illness(Münchausen By Proxy).The good news is that treatments are available
for cyclic vomiting in individuals with mitochondr-ial disease. In mitochondrial disease, symptomsare believed to occur when energy supply cannotmeet energy demand. Since often little can be doneto increase energy supply, decreasing energydemand is a major part of therapy. In practicalterms, this means the reduction of stress, includingthe avoidance of fasting, limiting exposure to envi-ronmental temperature extremes, and the prompttreatment of infections and dehydration. Cyclicvomiting and some other symptoms often improvewith frequent feedings of complex carbohydrate,including between meals and at bedtime. Otherchildren improve if awakened during sleep for asnack and/or placed on a low fat diet. In additionto physical stress, the reduction of psychologicalstress is important: not because this is the cause ofthe disease, but because stress increases energydemand and can trigger an episode. In cases inwhich the response to these simple measures is notadequate, anti-migraine medication includingamitriptyline (Elavil), cyproheptadine (Periactin) orpropranolol (Inderal) taken at bedtime or moreoften can reduce the number of vomiting episodesin most cases, sometimes dramatically. When theydo occur, vomiting episodes are treated with IVfluids (10% Dextrose with standard electrolytes at arate of 1.5 to 2 times maintenance) in a dark andquiet room in order to facilitate sleep. In somecases, ondansetron (Zofran) and/or medications toinduce sleep (i.e. lorazepam/Ativan) are helpful.Diagnostic work-up (testing) must be tailor-fit to
each individual child. Of course, confirming thediagnosis of mitochondrial disease and ruling outother treatable metabolic disorders (urea cycle dis-

Think Mitochondria UMDF63
orders, organic acidemias) should be pursued. Isuggest that a minimum work-up should includeserum electrolytes, routine urinalysis, plasma lac-tate, quantitative plasma amino acids and quantita-tive urine organic acids (including full quantitationof Kreb cycle intermediates and other potential‘mitochondrial markers’), with samples obtainedearly in a severe or typical vomiting episode.Mitochondrial DNA analysis should include at aminimum PCR for A3243G and Southern blotting.Unless the diagnosis of mitochondrial disease isfirm and CVS symptoms respond to treatment,work-up for other potential causes of cyclic vomit-ing should be performed, possibly including but notnecessarily limited to: upper GI series, abdominalultrasound, brain CT scan, and testing for sinusitis,porphyria and pregnancy. Probably no singleindividual requires, or should receive, all of thestudies listed, and it is important to discuss thework-up with your child’s physician.This is a very new and rapidly evolving field,
and not nearly half of the answers are in yet. Muchof our understanding of, and hopefully our abilityto treat, this disorder will improve over the nextseveral months to years. I am writing this article atthis early stage in the hope that some children willbe steered towards treatments now which may besomewhat helpful to them. For more information,the Cyclic Vomiting Syndrome AssociationUSA/Canada, an organization much like theUMDF, may be helpful. I suggest browsing theirweb-site at: www.beaker.iupui.edu/cvsahttp://www.beaker.iupui.edu/cvsa . In addition,information on any available studies in CVS (withor without mitochondrial disease) and their
entrance criteria and procedures are listed there.Alternatively, you can reach the association bycontacting:Debra Waites CVSA Administrator3585 Cedar Hill Rd NWCanal Winchester, Ohio 43110614-837-2586cvsadwaites @msn.comhttp://www.beaker.iupui.edu/cvsahttp://www.beaker.iupui.edu/cvsa>
References1. Fleisher DR, The cyclic vomiting syndromedescribed. J Pediatr Gastroenterol Nutr 1995;21(Suppl. 1):S1-S5.
2. Li BUK, Cyclic vomiting: The pattern and syn-drome paradigm. J Pediatr Gastroenterol Nutr1995;21(Suppl. 1):S6-S10.
3. Boles RG, Chun N, Senadheera D, WongL-JC (1997): Cyclic vomiting syndrome andmitochondrial DNA mutations. Lancet350:1299-1300.
4. Boles RG, Williams JC. Mitochondrial Diseaseand Cyclic Vomiting Syndrome. Dig Dis Sci1999;44(Suppl.):103S-107S.
5. Li, B U.K. & Balint, J. Cyclic vomiting syn-drome, the evolution of understanding of a brain-gut disorder. In Louis A. Barness (Ed.) Advancesin Pediatrics. St. Louis: Mosby Inc. 2000;47,117-160.
6. Li, B U.K., Sarna, S . Issenman, R. (Guest Eds.).Proceedings of the 2nd Scientific Symposium onCVS, Held at the Medical College of Wisconsin,USA, April 17-18, 1998. Digestive Diseases andSciences. 1999;44(Suppl.):1S-120S.

www.geneclinics.org
Funded by
National Institutes of Health (NIH)
Health Resources and Services Adminstration (HRSA)
U.S. Department of Energy (DOE)
Administrative Support:
University of Washington School of Medicine
Children’s Hospital Regional Medical Center,
Seattle, Washington
�������������������������������� ����������������������������������������������������������� ��������������������������������
• ��������������������������������������������������� ����
�������������������� ��������������������������������������������������������������������������������������������������������������������������
• !���������������������������� ��� �������������
• !� ���������������������������������������������������
• ���� ��� �� ��� ������ ���������������������������������
��������
������"������������������� ��� ��������������������������������������
����� ���������������������������� ��������������������!��������������������������������������#�����$���������%��������������������������
������"�����������&�������������'����������(�����)���� ��������*�
����������$�� ��������+��������������������,��-�����.������.������/001�233/�
$�������4����! ���� ������������ !"�������!��"��������� !"��������!��"�
As of June 2002, this NIH-funded website is the most comprehensive list we’ve found of testing facilitiesfor mitochondrial disease. Membership is free. Log on and select the “Laboratory Directory” tab.

Benefits of UMDF Membership
Tri-annual Newsletter
Symposia Meeting Notices
Notices of Research Grant Opportunities (Goal of
$5.25 Million to be awarded in next 5 years)
Educational Materials - for staff and patients
UMDF Patient Registry - A dynamic resource forfamilies and specialists to facilitate the exchange of
information (with written consent of participants)
Web Site - www.umdf.org - Access to MembersOnly Areas
Membership Application/Contribution Form
� Enclosed are my annual dues - within the U.S.: $40
� Enclosed are my annual dues - outside of the U.S.: $50 (drawn on U.S. bank)
� Enclosed is my gift to help sustain research and family support: $
� (Please make checks payable to UMDF)
For more information about UMDF membership, please contact the UMDF
office at 412-793-8077, [email protected] or visit www.umdf.org.
To Join NOW, please mail the form below to:
The United Mitochondrial Disease Foundation
8085 Saltsburg Road, Suite 201
Pittsburgh, PA 15239
Please supply us with your information to send a membership packet and/or to acknowledge your gift:
Name: ____________________________ Address: _____________________________________________________
City: ______________________________ State: ____________________________________ Zip: _______________
Office Phone: ______________________ Fax: _________________________ Email __________________________
The above gift is made: � In Memory of _______________________ � In Honor of ______________________
Name _________________________________________________________________________________
On the occasion of: _____________________________________________ (Memorial, birthdate, deathdate, other)
Please send an acknowledgement of my gift to:
Name: ____________________________ Address: _____________________________________________________
City: ______________________________ State: ____________________________________ Zip: _______________
UMDF also accepts � Visa or � Mastercard _______________________________________________________
(card number)
Name as listed on card (print): __________________________ Signature of Cardholder: _________________________
Visit the UMDF Web Site for Upcoming Conferences at www.umdf.org
Mission Statement
To promote research for cures and
treatments of Mitochondrial
Disorders and to provide support
to affected individuals and
families.

Could it be Mitochondrial Disease?
developmental delays
visual and hearing problems
lactic acidosis
cardiac disease
liver disease
seizures
susceptibility to infections
muscle weakness
diabetes
respiratory complications
loss of motor control
gastro-intestinal disorders and swallowing difficulties
poor growth
Think MitochondriaVolume 1, June 2002 This publication serves to
increase awareness and informmedical professionals, families
and individuals aboutMitochondrial Disease.
UMDF MissionTo promote research for curesand treatments of MitochondrialDisorders and to provide support
to affected families.
UNITED MITOCHONDRIAL DISEASE FOUNDATION
8085 Saltsburg Road, Suite 201Pittsburgh, PA 15239
Voice: 412-793-8077Fax: 412-793-6477
Email: [email protected] Site: www.umdf.org
UNITED MITOCHONDRIAL DISEASE FOUNDATION
ThinkMitochondria


















