
Radical mechanisms of methyl vinyl ketone oligomerization through ...

Theoretical study on reaction mechanism of the vinyl
radical with nitrogen atom
Yang Suna,*, Qi-yuan Zhanga, Xi-cheng Aia, Jian-ping Zhanga, Chia-chung Sunb
aThe State Key Laboratory for Structural Chemistry of Unstable and Stable Species, The Center for Molecular Science, Institute of Chemistry,
Chinese Academy of Sciences, Beijing 100080, People’s Republic of ChinabInstitute of Theoretical Chemistry, Jilin University, Changchun 130023, People’s Republic of China
Received 3 March 2004; accepted 5 August 2004
Abstract
The complex triplet potential energy surface of the C2H3N system is investigated at the UB3LYP and CCSD(T) (single-point) levels in
order to explore the possible reaction mechanism of C2H3 radical with N(4S). Eleven minimum isomers and 18 transition states are located.
Possible energetically allowed reaction pathways leading to various low-lying dissociation products are obtained. Starting from the energy-
rich reactant C2H3CN(4S), the first step is the attack of the N atom on the C atom having one H atom attached in C2H3 radical and form the
intermediate C2H3N(1). The associated intermediate 1 can lead to product P1 CH2CNCH and P23CH2C3HCN by the cleavage of C–H bond
and C–C bond, respectively. The most favorable pathway for the C2H3CN(4S) reaction is the channel leading to P1, which is preferred to that
of P2 due to the comparative lower energy barrier. The formation of P33C2H2C3NH through hydrogen-abstraction mechanism is also
feasible, especially at high temperature. The other pathways are less competitive comparatively.
q 2004 Elsevier B.V. All rights reserved.
Keywords: Potential energy surface; Vinyl radical; Isomers
1. Introduction
The vinyl radical, C2H3, is a reactive intermediate
formed during oxidation of hydrocarbons and played an
important role in the atmospheric and combustion
chemistry. The formation enthalpy of vinyl radical is
300.0G3.3 kJ/mol [1], which is much more active than its
saturated analog C2H5. The kinetics of small C2 radicals
such as vinyl has implicit importance in the atmospheric
chemistry of combustion processes and in ultralow-
temperature chemistry of dense interstellar clouds. It is of
great insignificance to learn the chemical behavior of the
C2H3 radical for environmental protection. So far, the
reactions of the C2H3 with other radicals, atoms and
molecules (H2, C2H2, CH3, CO, O2, NO, O, F, Cl) have
been studied extensively both experimentally and theoreti-
cally [2–12]. N(4S) is the ground state of nitrogen atom,
0166-1280/$ - see front matter q 2004 Elsevier B.V. All rights reserved.
doi:10.1016/j.theochem.2004.08.019
* Corresponding author. Tel.: C86 1062563101; fax: C86 1062588930.
E-mail address: [email protected] (Y. Sun).
and nitrogen atom reactions have been a subject of interest
among experimental and theoretical chemists for the past
decades. Nitrogen-containing compounds also play inter-
esting and important roles in atmospheric chemistry,
combustion of nitrogen-fuels, and explosion processes.
The reactions of the ground state atomic nitrogen with the
saturated or unsaturated hydrocarbon radicals are of
importance in a series of systems such as the chemistry of
the atmosphere in some planet [13,14]; the chemistry of
interstellar clouds [15–17]; nitrogen chemistry in hydro-
carbon combustion and the ‘active nitrogen’ hydrocarbon
reactions [18,19]. The reactions of the atomic nitrogen with
CH3 and C2H5 have been studied in detail before [20,21].
For the title reaction, the rate constant is determinate as
(7.7G2.9)!10K11 cm3 moleculeK1 sK1 by Payne et al.
[22] at TZ298 K and pressure of 1 Torr He. They also give
the branching ratio for the reaction and indicate that the
CH2CNCH is the primary product. Thorn et al. [23] also
investigate the reaction of C2H3CN(4S), whereas they
observed that CH3CN is the main product. For the C2H3N
system, there are some theoretical investigations, but mostly
Journal of Molecular Structure (Theochem) 686 (2004) 123–130
www.elsevier.com/locate/theochem

Y. Sun et al. / Journal of Molecular Structure (Theochem) 686 (2004) 123–130124
concentrating on the physical chemistry properties such as
formation heat and electron affinity [24–26]. Doughty et al.
[27] also got some isomers of C2H3N system, but these
information is obviously inadequate for learning the detail
mechanism for the C2H3CN(4S) reaction. Thus, a high-
level and detail potential energy surface (PES) is desirable
for the full understanding of the title reaction.
Therefore, due to the importance of the title reaction and
the rather limited knowledge about its reaction mechanism,
we decide to carry out a systematic theoretical study. A
detailed triplet PES is explored by means of density
functional theory (DFT-B3LYP) and coupled cluster
[CCSD(T)] (single-point) methods. The aim in this
investigation is to identify the product distribution and the
exact channels for each of the products and thereby to gain
insight into the reaction kinetics.
2. Computational methods
All computations are carried out using the GAUSSIAN98
program package [28]. The optimized geometries and
harmonic frequencies of the reactant, products, local
minima and transition states are obtained at the UB3LYP/
6-311G(d,p) theory level.
Vibrational frequencies are calculated at the UB3LYP/
6-311G(d,p) level to check whether the obtained stationary
point is an isomer or a first-order transition state. In order to
confirm whether the obtained transition states connect the
right reactants and products, the intrinsic reaction coordinate
calculations are performed at the UB3LYP/ 6-311G(d,p)
level. Single-point calculations are performed at the
CCSD(T)/6-311G(2df,p) theory level at the UB3LYP/6-
311G(d,p) optimized geometries. The UB3LYP/6-311G(d,p)
zero-point vibration energy (ZPVE) is also included. Unless
otherwise specified, the CCSD(T) single-point energies with
Fig. 1. Potential energy curve for the initial attack of the nitrogen atom to C2H3 ra
pointwise optimization of the remaining varied bond lengths or bond angles with e
is constrained as Cs.
inclusion of UB3LYP/ 6-311G(d,p) zero-point energies are
used in the following discussions.
3. Results and discussions
For the title reaction, various dissociation products,
including P1 CH2CNCH, P23CH2C3HCN, P3
3C2H2C3NH,
P43CH2C3HNC, P5 CH3CCN, P6 C2HCNH2 and
P7 CH2NCCH (in Fig. 1), are considered. In Table 1,
total and relative energies including ZPVE of all the seven
products as well as the reactants are listed. Note that the
energy of reactant R is set zero for reference. Eleven
intermediate isomers and 18 transition states are located.
The optimized structures of isomers and the transition
states are given in Figs. 2 and 3, respectively. The energetic
of the isomers and the transition states are listed in Tables 2
and 3, respectively. The symbol TSm/n is used to denote
the transition state connecting the isomers m and n. By
means of the products, intermediate isomers, transition
states and their corresponding relative energies, a sche-
matic PES of C2H3N in triplet is plotted in Fig. 4.
3.1. Initial association
Both singlet and triplet PES are possible for the C2H3N
isomers. And the ground state atomic nitrogen can attack the
C2H3 radicals by two ways: the direct attack on the carbon
atom with only one hydrogen atom attached or the side CaC
p bond attack. Two association adduct will be formed, i.e.
the vinyl nitrene (isomer 1 in our triplet PES) and the cyclic
isomer 2H-azirine (isomer 4 in our triplet PES). The singlet
vinyl nitrene isomer cannot be optimized at the RB3LYP/
6-311G(d,p) level of theory, and all initial configuration
lead to the 2H-azirine isomer at last. In triplet PES, The
direct N attack on the carbon atom can barrierlessly lead to
dical at the UB3LYP/6-311G(d,p) level. The calculations are performed by
very fixed internal C–N bond length. During the optimization, the symmetry

Table 1
Total (a.u.) and relative energies in parentheses (kcal/mol) as well as those including zero-point vibration energies (kcal/mol) of the reactant products for the
C2H3CN reaction
Species UB3LYP/6-311G(d,p) CCSD(T)/6-311G(2df,p) CCSD(T)/6-311G(2df,p)CZPVE
R C2H3CN K132.5255749(0.0) K132.2489698(0.0) 0.0
P1 CH2CNCH K132.6340804(K68.0) K132.3503811(K63.6) K66.9
P23CH2CHCN K132.6164891(K57.0) K132.336505(K54.9) K59.1
P3 C2H2C3NH K132.5917562(K41.5) K132.310237(K38.4) K40.7
P4 CH2CHNC K132.5928601(K42.2) K132.3118656(K39.5) K42.4
P5 CH3CCN K132.5906249(K40.8) K132.3094711(K37.9) K39.1
P6 C2HCNH2 K132.5245526(0.6) K132.2414626(4.7) 1.6
P7 CH2NCCH K132.50126(15.3) K132.2149145(21.4) 18.9
Y. Sun et al. / Journal of Molecular Structure (Theochem) 686 (2004) 123–130 125
adduct C2H3N(1), which can be confirmed by the calculated
pointwise potential curves at the B3LYP/6-311G(d,p) (in
Fig. 5) level of theory. The nitrogen atom can also attack the
CaC double bond of the C2H3 radical, but the energy of
such triplet three-membered isomer (4) is much higher than
that of C2H3N(1). This attacking process involves the
formation of the C–N bond and the activation of CaC
double bond, and it is commonly an energy-consuming
process, so the formation of triplet 2H-azirine is unfavorable
compared to the former attack ways. The energy of the
singlet 2H-azirine adduct is 15.7 kcal/mol lower than that of
the triplet vinyl nitrene at the B3LYP/6-311G(d,p) level of
theory, but no similar barrierless scan potential curves as
Fig. 5 are located for this p bond attacking ways. In
addition, the spin density is dominantly on the carbon atom
(1.027), which is much bigger than the other atoms in C2H3
radical, indicating that this carbon atom is the active site for
the C2H3 radical in the intermolecular reactions. Thus the
most reasonable initial association mechanism is the direct
attack on the carbon atom with one hydrogen atom attached
and form the triplet associated adduct 1.
3.2. Reaction pathways
Except for one pathway of the product P3, all the other
products undertake the addition–elimination mechanism, and
Fig. 2. B3LYP/6-311G(d,p) optimized geometries for reactant and
products. Bond lengths are in angstroms and angles in degrees.
they can be isomerized and dissociated from the initial
associated intermediate 1, so we can discuss the seven products
and their pathways in detail starting from the isomer 1.
Two pathways can be obtained for the formation of
product P1 CH2CNCH as follows:
Path P1ð1Þ : R C2H3 CN/C2H3Nð1Þ/P1
Path P1ð2Þ : R C2H3 CN/C2H3Nð1Þ/CH3CNð2Þ/P1
In the first pathway, P1 can be obtained by the dissociation
of one H atom via the transition state TS1/P1, and the barrier
for this process is 37.4 kcal/mol. The bond length for the
Fig. 3. B3LYP/6-311G(d,p) optimized geometries for C2H3N isomers.
Bond lengths are in angstroms and angles in degrees.

Table 2
Total (a.u.) and relative energies in parentheses (kcal/mol) as well as those including zero-point vibration energies (kcal/mol) of the isomers for the C2H3CN
reaction
Species UB3LYP/6-311G(d,p) CCSD(T)/6-311G(2df,p) CCSD(T)/6-
311G(2df,p)CZPVE
1 K132.6899679(K103.2) K132.3987072(K93.9) K90.5
2 K132.6246255(K62.2) K132.3367528(K55.1) K51.5
3 K132.6750637(93.8) K132.3804684 (K82.5) K78.9
4 K132.607076(K51.1) K132.3170104(K42.7) K39.8
5 K132.5401598(K9.2) K132.2467748(1.4) 2.2
6 K132.5844607(K36.9) K132.2966579(K29.9) K26.2
7 K132.6507256(K78.5) K132.360171(K69.8) K66.4
7 0 K132.6513557(K78.9) K132.3611808(K70.4) K66.8
7 00 K132.653024(K79.9) K132.3623581(K71.1) K67.7
8 K132.6158663(K56.6) K132.3212132(K45.3) K40.8
9 K132.6278225(K64.2) K132.3309935(K51.5) K47.1
Y. Sun et al. / Journal of Molecular Structure (Theochem) 686 (2004) 123–130126
C–H is elongated from 1.1 to 1.889 A. P1 can also be
obtained by the dissociation of one H atom in CH3CN via
TS2/P1, and the barrier is 27.0 kcal/mol, which is lower than
that of Path P1(1), whereas the formation of CH3CN(2) must
overcome a high isomerization barrier of TS1/2, which
involves a 1,3 H-shift process. The formation of such three-
membered transition state is energy-consuming, so this
pathway is unfavorable kinetically. The two pathways are
both energetically feasible, while it is obvious that Path
P1(1) will dominate over the other due to its less reaction
steps and lower barrier.
Only one pathway is located for the formation of product
P23CH2C3HCN.
Path P2 R C2H3 CN/C2H3Nð1Þ/P23CH2 C 3HCN
The C–C bond in the associated intermediate 1 has been
weakened compared with that in the C2H3, which is
elongated from 1.304 to 1.398 A, so the energy-rich
Table 3
Total (a.u.) and relative energies in parentheses (kcal/mol) as well as those includ
C2H3CN reaction
Species B3LYP/6-311G(d,p) C
TSR/P3 K132.5252074(0.2) K
TS1/2 K132.5755756(K31.4) K
TS1/4 K132.5864719(K38.2) K
TS1/7 K132.6044337(K49.5) KTS1/3 K132.6102725(K53.1) K
TS1/P1 K132.6274947(K63.9) K
TS1/P2 K132.6110059(K53.6) K
TS2/6 K132.4974926(17.6) KTS4/5 K132.5367757(K7.0) K
TS3/8 K132.559616(K21.3) K
TS3/7 0 K132.5770249(K32.3) K
TS2/P1 K132.5767272(K32.1) K
TS2/P5 K132.5780971(K32.9) K
TS7 00/8 K132.5454029(K13.5) K
TS6/P5 K132.5668462(K27.8) K
TS7 0/9 K132.570672(K28.3) K
TS7 0/7 00 K132.6110059(53.6) K
TS7/7 0 K132.6218133(60.4) K
intermediate 1 can undergo further dissociation and give
rise to the product P2 through TS1/P2. The C–C bond in
TS1/P2 is 2.291 A, whereas the other bonds such as C–H
and C–N bonds or angles between them is close to those in
CH2 and HCN, so TS1/P2 is a ‘loose’ transition state and can
be described as approximately separate CH2 and HCN. The
barrier for TS1/P2 is 42.6 kcal/mol.
Product P33C2H2C3NH can be formed by the following
two pathways through different mechanism:
Path P3ð1Þ R/P3
Path P3ð2Þ R C2H3 CN/C2H3N1/C2H2NH7/P3
The first pathway is the H-abstraction mechanism, and the
second one is the addition–elimination mechanism. In the
H-abstraction transition state TSR/P3, the angle of :CHN
is nearly 1808, and the CH bond which is to be broken is
only elongated from 1.094 to 1.168 A, while the NH bond to
be formed is 1.629 A. The conformation of TSR/P3 does not
ing zero-point vibration energies (kcal/mol) of the transition states for the
CSD(T)/6-311G(2df,p) CCSD(T)/6-311G(2df,p)CZPVE
132.2375445(7.6) 6.0
132.281498(K20.4) K20.1
132.2953092(K29.1) K23.5
132.3135871(K40.5) K39.9
132.3184876(K43.6) K43.5
132.3393152(K50.7) K53.1
132.3267737(K48.8) K47.9
132.2045336(27.8) 28.8
132.2429143(3.8) 4.6
132.2646945(K9.8) K10.2
132.2809001(K20.0) K20.9
132.2852885(K22.8) K24.5
132.2923947(K27.2) K26.7
132.247048(1.2) K0.7
132.2783942(K18.5) K17.0
132.2773797(K17.8) K16.8
132.3267737(K48.8) K49.3
132.3302044(K51.0) K49.2

Fig. 4. B3LYP/6-311G(d,p) optimized geometries for C2H3N transition
states. Bond lengths are in angstroms and angles in degrees.
Y. Sun et al. / Journal of Molecular Structure (Theochem) 686 (2004) 123–130 127
change much compared with the reactant, and the barrier for
TSR/P3 is 6.0 kcal/mol. In the addition–elimination mech-
anism, intermediate C2H3N(1) can isomerize to isomer 7 via
TS1/7, which is a four-membered transition state with a
barrier of 50.6 kcal/mol. In this energy-consuming process,
one hydrogen atom undergo 1,3 shift to the nitrogen atom on
the same side in C–C bond, then 7 can lead to P3 by the
fission of C–N bond with the dissociation energy of
25.7 kcal/mol.
There is only one pathway located for product P43CH2C3HNC
Path P4 R C2H3 CN/C2H3Nð1Þ/CH2CNHð3Þ/P4
P4 can be formed through the break of the C–N bond in
intermediate CH2CNH(3), the barrier for the isomerization
from 1 to 3 is 47.0 kcal/mol. TS1/3 is a three-membered
transition state involving the shift of hydrogen atom
between the two adjacent atoms. In TS1/3, the bond to be
breached and the bond to be formed are 1.266 and 1.249 A,
respectively, and the energy for the dissociation of isomer 3
to the product P4 is 36.5 kcal/mol.
Two pathways are located for the product P5 CH3CCN:
Path P5ð1Þ R C2H3 CN/C2H3Nð1Þ/CH3CNð2Þ/P5
Path P5ð2Þ R C2H3 CN/C2H3Nð1Þ/CH3CNð2Þ
/CH3NCð6Þ/P5
P5 can be obtained by the breach of the C–C bond and C–N
bond in CH3CN and CH3NC, respectively. The correspond-
ing transition states are TS2/P5 and TS6/P5. Intermediate
CH3CN and CH3NC are resembled to each other in
geometry, and the difference is that the carbon and nitrogen
atoms are in opposite position. The two isomers both
possess Cs symmetry, and CH3CN is more stable than
CH3NC thermodynamically. The barrier for TS2/P5 is
24.8 kcal/mol, whereas the barrier for TS6/P5 is much
lower, which is 9.2 kcal/mol, but the formation of isomer 6
must through the isomerization from isomer CH3CN, and
the barrier for this isomerization is rather high. In TS2/6, the
distance between the carbon atom in methyl with another
carbon atom and nitrogen atom is 1.723 and 1.749 A,
respectively. Such methyl migration process is very energy-
consuming due to the steric hindrance. The barrier for TS2/6
is 80.3 kcal/mol, and it is also 28.8 kcal/mol higher than the
reactant, thus the pathway Path P5(2) is unfeasible both
kinetically and thermodynamically.
There are three pathways for the formation of product P6
C2HCNH2 including:
Path P6ð1Þ R C2H3 CN/C2H3Nð1Þ/CH2CNHð3Þ
/HC2NH2ð8Þ/P6
Path P6ð2Þ R C2H3 CN/C2H3Nð1Þ/CH2CNHð3Þ
/C2H2NHð70Þ/C2HNH2ð9Þ/P6
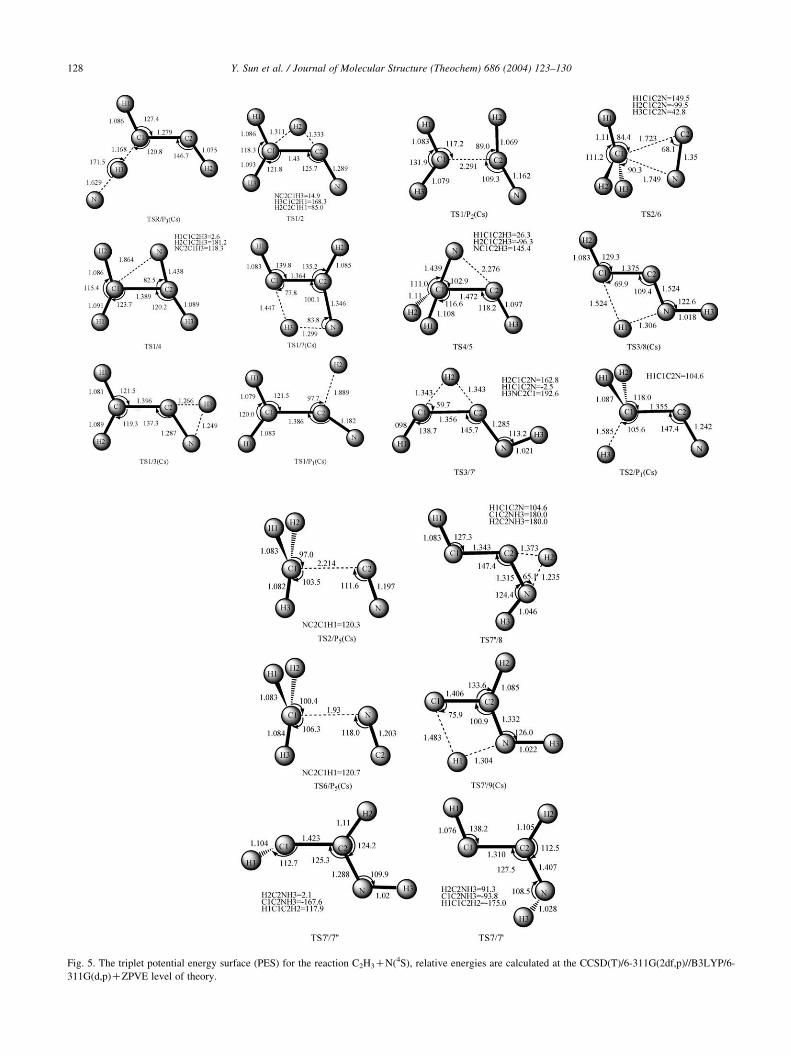
Fig. 5. The triplet potential energy surface (PES) for the reaction C2H3CN(4S), relative energies are calculated at the CCSD(T)/6-311G(2df,p)//B3LYP/6-
311G(d,p)CZPVE level of theory.
Y. Sun et al. / Journal of Molecular Structure (Theochem) 686 (2004) 123–130128

Y. Sun et al. / Journal of Molecular Structure (Theochem) 686 (2004) 123–130 129
Path P6ð3Þ R C2H3 CN/C2H3Nð1Þ/CH2CNHð3Þ
/C2H2NHð70Þ/ ð700Þ/HC2NH2ð8Þ/P6
The energy of product P6 is a bit higher than the reactant.
Intermediate C2H2NH has three isomers, i.e. 7, 7 0 and 7 00.
The energy of the three isomers is similar, and they can
convert to each other easily with low barrier. Isomer 7 00 is
the lowest energetic isomer. Intermediate 3 can also
isomerize to 8. Similar to TS7 0/9, the hydrogen atom
connected to carbon atom can undergo 1,3 migration to
nitrogen atom on the same side. TS3/8 is also a four-
membered planar transition state, and the corresponding
barrier is 68.7 kcal/mol. TS7 00/8 is a three-membered
structure, the formation of such transition state need high
energy, and this can be proved by the high energy barrier of
67.0 kcal/mol. The energy for isomer 8 and 9 is close, and
the difference between them is that the hydrogen atoms are
connected to different carbon atoms, and they can both give
rise to the product P6 by the rupture of C–N bond, the
dissociation energy is 42.4 and 48.6 kcal/mol, respectively.
We can notice that the pathways leading to the product P6 all
need to overcome high barrier and involve complex steps,
thus the formation of P6 is less favorable kinetically.
The product P7 CH2NCCH can be formed through the
ring open process and the subsequent C–C bond dissociation
of the three-membered intermediate 4, but P7 lies
18.9 kcal/mol higher than the reactants, so P7 can be
neglected for it is unfavorable thermodynamically.
3.3. Reaction mechanism
Through the analysis in Section 3.2, we can notice that
that the product P6 and P7 lies energetically higher than the
reactants, so the pathways leading to them can be ruled out
for the discussion. The pathway Path P5(2) is also excluded
due to the factors mentioned above. The other pathways
such as follows:
Path P1ð1Þ R C2H3 CN/C2H3Nð1Þ/P1CH2CN CH
Path P1ð2Þ R C2H3 CN/C2H3Nð1Þ/CH3CNð2Þ
/P1CH2CN CH
Path P2R C2H3 CN/C2H3Nð1Þ/P23CH2 C 3HCN
Path P3ð1ÞR/P33CH2 C 3NH
Path P3ð2Þ R C2H3 CN/C2H3Nð1Þ/C2H2NHð7Þ
/P33CH2 C 3NH
Path P4 R C2H3 CN/C2H3Nð1Þ/CH2CNHð3Þ
/P43CH2 C 3HNC
Path P5ð1Þ R C2H3 CN/C2H3Nð1Þ/CH3CNð2Þ
/P5CH3 CCN
are below the reactants energetically, so they all can
contribute to the final products to some extent. By means of
these feasible pathways, let us discuss the possible
mechanism of the title reaction. Noticing that except for
Path P3(1), the common intermediate 1 are involved in the
other pathways, so some qualitative conclusions can be
drawn about the distributions of these products. As shown in
Fig. 4, the lowest dissociation or isomerization barrier
starting from isomer 1 is TS1/P1, which is 37.4 kcal/mol. In
addition, the product P1 obtained by this pathway is also the
most energetic low-lying product, so P1 is the most
favorable product both kinetically and thermodynamically.
The second pathway Path P1(2) is much less competitive
compared to Path P1(1) and would not play an important
role in the formation of P1. P2 can also be obtained from
intermediate 1 via transition state TS1/P2, and the
corresponding barrier is 42.6 kcal/mol, only 5.2 kcal/mol
higher than TS1/P1, so Path P2 and Path P1(1) can compete
with each other. The pathway in addition–elimination
mechanism for formation of product P3 is unfavorable
kinetically due to the high barrier as discussed above,
whereas the barrier for the pathway in hydrogen-abstraction
mechanism is only 6.0 kcal/mol, which is feasible for the
intermolecular hydrogen transition, and this channel will
play an important role for the title reaction at high
temperature and vice versa, can be neglected at low
temperature. The barrier for TS1/3 in the Path P4 is
9.6 kcal/mol higher than that of TS1/P1, for such three-
membered transition state is not very easy to be attained, so
product P4 will be less competitive compared to P1, P2 and
P3. The formation of P5 is also by the isomerization of
CH3CN, although CH3CN is stable energetically, but the
formation of it must undergo a 1,2 H-shift migration
process, and this is a very energy-consuming mechanism,
the barrier for it is 70.4 kcal/mol, so P5 is also a less
competitive product kinetically.
In the study of Payne et al. [22], the branching ratio for
the product CH2CNCH is 0.8, which is in agreement with
our calculation result that the P1 is the mostly kinetically
and thermodynamically preferable product. The branching
ratio for the product C2H2CNH is 0.16, which is in
coincidence with the hydrogen-abstraction mechanism of
our calculation. They also verify the branching ratio for the
isomer CH3CN is 0.04. Through the analysis above, the
formation for CH3CN is difficult kinetically in triplet PES.
The energy of singlet CH3CN is about 100 kcal/mol lower
than the triplets CH3CN, athough it has high energetic
stability but the direct formation of the singlet CH3CN is
unfeasible by the triplet mechanism, so we infer maybe a
part of CH3CN is obtained by the secondary reaction of
CH2CN with the H atom. The heat formation by our
calculation is also in good agreement with the experiment
data given by their paper. Notice that the product P23CH2C3HCN is not observed in the experiment, and this
perhaps due to the triplet carbene radical is so active that it
cannot keep stable and undergo further reactions rapidly.

Y. Sun et al. / Journal of Molecular Structure (Theochem) 686 (2004) 123–130130
We expect that the product CH2CHCN can be detected in
future experimental detection.
It is also worthy to compare the mechanism of the similar
reaction of CH3C4N with the title reaction. In the quantum
chemistry, investigation carried out by Hadjebar et al. [29],
they also identify that the reaction go through the triplet
PES, and the most favorable pathway is also the first
formation of the adduct 3NCH3 with subsequently hydrogen
dissociation and at last give rise to the product H2CNCH.
The barrier for the Path P1(1) (37.4 kcal/mol) is very close
to the activation energy of 3NCH3, which is 37 kcal/mol
obtained at high-level of theory (MPW1PW91). All this
indicated that the PES calculated by us is reliable and
reasonable.
4. Conclusions
A detailed triplet PES of the C2H3CN(4S) reaction
system is built up at the B3LYP and CCSD(T) (single-point)
level of theory. For the title reaction, the initial step is the
attack of N(4S) on the carbon atom having one H atom
attached in C2H3 radical and form the energy-rich triplet
intermediate C2H3N. The intermediate C2H3N can undergo
the dissociation of the hydrogen atom and give rise to the
primary product P1 CH2CNCH. This pathway is the most
favorable channel for the title reaction both thermodynami-
cally and kinetically. The intermediate C2H3N can also lead
to product P23CH2C3HCN by the cleavage of C–C bond,
this pathway is less competitive compared to that of P1. The
formation of P33C2H2C3NH through hydrogen-abstraction
mechanism is a feasible pathway and would be an important
channel at high temperature. The possibility for the
formation of CH3CN is small, and the pathways leading to
other products can be neglected at normal conditions. We
hope our calculated results may provide some useful
information for understanding the reaction mechanism of
the unsaturated hydrocarbon with the nitrogen atom.
Acknowledgements
This project was supported by The National Natural
Science Foundation of China (20133020, 39890390, and
20073014) and The State Key Basic Research and
Development Plan (G1998010100).
References
[1] D.R. Lide, CRC Handbook of Chemistry and Physics, 74th ed.,
Chemical Rubber, Boca Raton, 1993.
[2] J.Y. Park, M.C. Heaven, D. Gutman, Chem. Phys. Lett. 104 (1984)
469.
[3] P.R. Westmoreland, Combust. Sci. Technol. 82 (1992) 151.
[4] J.W. Bozzelli, A.M. Dean, J. Phys. Chem. 97 (1993) 4427.
[5] J.G. Moeblmann, J.T. Gleaves, J.W. Hudgens, J.D. McDonald,
J. Chem. Phys. 60 (1974) 4790.
[6] D.J. Norfolk, R.F. Skinner, W.J. Williams, Radiat. Phys. Chem. 21
(1983) 307.
[7] A.B. Callear, G.B. Smith, Chem. Phys. Lett. 105 (1984) 119.
[8] A.B. Callear, G.B. Smith, J. Phys. Chem. 90 (1986) 3229.
[9] B.K. Carpenter, J. Phys. Chem. 99 (1995) 9801.
[10] A.M. Mebel, K. Morokuma, M.C. Lin, J. Chem. Phys. 103 (1995)
3440.
[11] A.M. Mebel, E.W.G. Diau, M.C. Lin, K. Morokuma, J. Am. Chem.
Soc. 118 (1996) 9759.
[12] R. Sumathi, H.M.T. Nhuyen, M.T. Nhuyen, J. Peeters, J. Phys. Chem.
104 (2000) 1905.
[13] Y.L. Yung, M.A. Allen, J.P. Pinto, Astrophys. J. Suppl. Ser. 55 (1984)
465.
[14] E. Lellouch, P.N. Romani, J. Rosenqvist, Icarus 108 (1994) 112.
[15] L.A.M. Nejad, T.J. Miller, Mon. Not. R. Astr. Soc. 230 (1988) 79.
[16] W.D. Langer, T.E. Graedel, Astrophys. J. Suppl. Ser. 69 (1989) 241.
[17] T.J. Miller, A. Freeman, Mon. Not. R. Astr. Soc. 207 (1984) 405.
[18] J.A. Miller, C.T. Bowman, Prog. Energy Combust. Sci. 15 (1989) 287.
[19] J.V. Machael, Chem. Phys. Lett. 76 (1980) 129.
[20] L.J. Stief, G. Marston, D.F. Nava, W.A. Payne, F.L. Nesbitt, Chem.
Phys. Lett. 147 (1989) 570.
[21] L.J. Stief, F.L. Nesbitt, W.A. Payne, W. Tao, S.C. Kao, R.B. Klemm,
J. Chem. Phys. 102 (1995) 5309.
[22] W.A. Payne, P.S. Monks, F.L. Nesbitt, L.J. Stief, J. Chem. Phys. 104
(1996) 9308.
[23] R. Peyton Thorn Jr., P.S. Monks, L.J. Stief, S. Kuo, Z. Zhang,
S.K. Ross, R.B. Klemm, J. Phys. Chem. A 102 (1998) 846.
[24] P.M. Mayer, M.S. Taylor, M.W. Wong, L. Radom, J. Phys. Chem. A
102 (1998) 7074.
[25] M.T. Nguyen, T.K. Ha, J. Chem. Soc., Perkin Trans. 8 (1984) 1401.
[26] E.U. Wuerthwein, J. Org. Chem. 49 (1984) 2971.
[27] A. Doughty, G.B. Bacskay, J.C. Mackie, J. Phys. Chem. 98 (1994)
13546.
[28] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb,
J.R. Cheeseman, V.G. Zakrzewski, J.A. Montgomery, Jr., R.E.
Stratmann, J.C. Burant, S. Dapprich, J.M. Millam, A.D. Daniels,
K.N. Kudin, M.C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi,
R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J.
Ochterski, G.A. Petersson, P.Y. Ayala, Q. Cui, K. Morokuma, D.K.
Malick, A.D. Rabuck, K. Raghavachari, J.B. Foresman, J. Cioslowski,
J.V. Ortiz, B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I.
Komaromi, R. Gomperts, R.L. Martin, D.J. Fox, T. Keith, M.A. Al-
Laham, C.Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe,
P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong, J.L. Andres, C.
Gonzalez, M. Head-Gordon, E.S. Replogle, and J.A. Pople, G98W
A.7, Gaussian, Inc., Pittsburgh PA, 1998.
[29] I. Hadjebar, M.N. Achour, A. Boucekkine, G. Berthier, Chem. Phys.
264 (2001) 153.


















