
Theoretical Analysis of Calcium Phosphate Precipitation in...
34
1 Theoretical Analysis of Calcium Phosphate Precipitation in Simulated Body Fluid Xiong Lu and Yang Leng 1 Department of Mechanical Engineering Hong Kong University of Science and Technology Kowloon, Hong Kong, China Abstract The driving force and nucleation rate of calcium phosphate (Ca-P) precipitation in simulated body fluid (SBF) were analyzed based on the classical crystallization theory. SBF supersaturation with respect to hydroxyapatite (HA), octacalcium phosphate (OCP) and dicalcium phosphate (DCPD) was carefully calculated, considering all the association/dissociation reactions of related ion groups in SBF. The nucleation rates of Ca-P were calculated based on a kinetics model of heterogeneous nucleation. The analysis indicates that the nucleation rate of OCP is substantially higher than that of HA, while HA is most thermodynamically stable in SBF. The difference in nucleation rates between HA and OCP reduces with increasing pH in SBF. The HA nucleation rate is comparable with that of OCP when the pH value approaches 10. DCPD precipitation is thermodynamically impossible in normal SBF, unless calcium and phosphate ion concentrations of SBF increase. In such case, DCPD precipitation is the most likely because of its highest nucleation rates among Ca-P phases. We examined the influences of different SBF recipes, interfacial energies, contact angle and molecular volumes, and found that the parameter variations do not have significant impacts on analysis results. The effects of carbonate incorporation and calcium deficiency in HA were also estimated with available data. Generally, such apatite precipitations are more kinetically favorable than HA. 1 Corresponding author, email address: [email protected] ; fax: 852-23581543 This is the Pre-Published Version
Transcript of Theoretical Analysis of Calcium Phosphate Precipitation in...

1
Theoretical Analysis of Calcium Phosphate Precipitation in Simulated Body Fluid
Xiong Lu and Yang Leng1
Department of Mechanical Engineering
Hong Kong University of Science and Technology
Kowloon, Hong Kong, China
Abstract
The driving force and nucleation rate of calcium phosphate (Ca-P) precipitation in simulated
body fluid (SBF) were analyzed based on the classical crystallization theory. SBF
supersaturation with respect to hydroxyapatite (HA), octacalcium phosphate (OCP) and
dicalcium phosphate (DCPD) was carefully calculated, considering all the
association/dissociation reactions of related ion groups in SBF. The nucleation rates of Ca-P
were calculated based on a kinetics model of heterogeneous nucleation. The analysis indicates
that the nucleation rate of OCP is substantially higher than that of HA, while HA is most
thermodynamically stable in SBF. The difference in nucleation rates between HA and OCP
reduces with increasing pH in SBF. The HA nucleation rate is comparable with that of OCP
when the pH value approaches 10. DCPD precipitation is thermodynamically impossible in
normal SBF, unless calcium and phosphate ion concentrations of SBF increase. In such case,
DCPD precipitation is the most likely because of its highest nucleation rates among Ca-P phases.
We examined the influences of different SBF recipes, interfacial energies, contact angle and
molecular volumes, and found that the parameter variations do not have significant impacts on
analysis results. The effects of carbonate incorporation and calcium deficiency in HA were also
estimated with available data. Generally, such apatite precipitations are more kinetically
favorable than HA.
1 Corresponding author, email address: [email protected]; fax: 852-23581543
This is the Pre-Published Version

2
Keywords: Calcium Phosphate; Simulated Body Fluid; Thermodynamics; Kinetics.
Introduction
The precipitation of bioactive calcium phosphate (Ca-P) in simulated body fluid (SBF), a
solution with ion concentrations and a pH value similar to those of human blood plasma, or in
other solutions with supersaturated calcium and phosphate has attracted extensive research
interest [1-27]; because such Ca-P precipitation is similar to biological mineralization. Also, the
Ca-P precipitation in such biomimetic solutions provides an alternative method of creating Ca-P
coatings on titanium implants [15-27]. In addition, Ca-P precipitation in SBF has been widely
used to assess the bioactivity of bioactive glass [28, 29], A-W glass-ceramic [1], ceramic α-
CaSiO3 [30], silica gel [31], bioceramics/polymer composites [32-34] and surface treated
titanium ([2-4, 8-14].
It is well known that precipitated Ca-P phases in aqueous solutions mainly include dicalcium
phosphate [CaHPO4⋅2H2O, DCPD], octacalcium phosphate [Ca8(HPO4)2(PO4)4⋅5H2O, OCP] and
hydroxyapatite [Ca10(OH)2(PO4)6, HA]. HA is considered the most thermodynamically stable in
physiological environment. OCP and DCPD, however, have been regarded as precursors of HA
or the metastable phases of Ca-P because that they are kinetically favorable [35-39].
Experimental studies of Ca-P formation in simulated physiological environments have not
clearly indicated the conditions for forming specific Ca-P phases. Although HA or bone-like
apatite formation in SBF has frequently been reported, OCP formation in similar environments
has also been observed. Marques et al. reported both OCP and HA precipitation on HA/TCP

3
biphasic ceramics in a simulated inorganic plasma (CSIP) solution with the same ion
concentrations as conventional SBF except for HCO3− [40, 41]. Feng et al. claimed to have
found a double layer Ca-P (HA/OCP) deposition by immersing alkali-treated Ti in a
supersaturated HA solution [19-21, 24]. Barrere et al. found a pure OCP layer deposited on a
non-alkaline treated Ti surface through a two-step SBF immersion procedure [15-16].
Koutsopoulos et al. studied calcification of fibrin and elastin in a simple solution of calcium
chloride and potassium dihydrogen phosphate, and they found OCP formation in certain cases
even though the driving force of HA formation was larger than that of OCP [42, 43]. Leng et al.
examined the crystal structure of Ca-P precipitated in conventional and revised SBF using single
crystal electron diffraction and identified exclusive OCP formation [44-45]. We believe that the
inconsistencies in reported Ca-P formation in supersaturated solutions are partially attributed to
misidentification of the Ca-P crystal phases [44-46]. On the other hand, we feel that there is a
lack of theoretical guidelines for Ca-P precipitation in biomimetic solutions such as SBF. Thus,
we are inspired to analyze Ca-P formation in SBF through theories of thermodynamics and
kinetics.
Thermodynamic analyses of Ca-P precipitation have been reported in the following solution
systems: CaCl2 + KH2PO4 + KOH [36, 37], Ca(NO3)2 + KH2PO4 + NaOH [38, 39], CaCl2 +
KH2PO4 + NaCl + KOH [42-43], CaCl2 + NaH2PO4 + NaCl + NaOH [47], Ca(OH)2 + H3PO4 +
KOH + HNO3 + CO2 [48] and supersaturated HA solution [20]. Although analyses of the
kinetics of Ca-P precipitation were investigated experimentally [36-39, 45-46], little theoretical
analysis of the kinetics has been conducted, except in the work of Boistelle et al. [47]. They
proposed a method to calculate a kinetic factor of nucleation rates based on the probability that

4
the ions units of calcium phosphates encounter each other to form a nucleus. The theoretical
analysis of Ca-P precipitation in SBF is understandably scarce because of the complexity of its
chemical composition. The difficulties of analyzing precipitation in SBF arise from the
necessary considerations of all the association/dissociation balances between various ions and
ion groups.
In this report, we present our analysis of the free energy of Ca-P formation in SBF based on the
classic theory of crystallization with few assumptions about chemical activities of each ion in
SBF in the calculation. Our kinetics analysis of Ca-P heterogeneous nucleation in SBF is based
on the kinetic model of Boistelle et al. [47]. The analysis includes Ca-P precipitation in SBF
made from the various recipes listed in Table 1, which are conventional SBF (C-SBF), revised
SBF (R-SBF), ionized SBF (I-SBF), modified SBF (M-SBF) and synthetic body fluid (S-SBF).
Among them, C-SBF is the first SBF recipe being proposed. R-SBF has ion concentrations that
are equal to those of blood plasma. The Cl − and HCO3− concentration of C-SBF, M-SBF and S-
SBF are not exactly the same with those in blood plasma. Generally speaking, SBF does not
include proteins. I-SBF however simulates the influence of proteins by including only the free
ions which are not bound to the proteins. Of the magnesium ions in blood plasma (1.5 mmol
dm−3), 0.5 mmol dm−3 of the Mg2+ are bound to proteins. Of the calcium ions in blood plasma
(2.5 mmol dm−3), 0.9 mmol dm−3 of Ca2+ are bound to proteins [7]. The nominal Ca2+ and Mg2+
concentrations of I-SBF are equal to 1.6 and 1.0 mmol dm−3, respectively, after subtracting the
concentrations of protein-bound ions. We also analyzed the cases of SBF with excessive amounts
of calcium and phosphate ions, considering possible dissolution of calcium and phosphate from
the substrates containing calcium and phosphor.

5
Analytical Model
Driving forces of precipitation
The thermodynamic driving forces for Ca-P precipitation were calculated based on the classical
equation of free energy change in supersaturated solutions [49]:
ln( ) ln( / )p spRT RTG S A Kn n
∆ = − = − , (1)
where ∆G is the Gibbs energy per mole of ionic units that compose Ca-P in solution, R is the gas
constant (8.314 J K−1mol−1), T is the absolute temperature, n is the number of ion units in a Ca-P
molecule, and S is the supersaturation that is defined by the ratio of the activity product of ion
units composing precipitates (Ap) to the corresponding solubility product (Ksp).
The equations for precipitation in aqueous solutions given below define the ion units of HA,
OCP and DCPD:
2+ 24 4Ca HPO CaHPO−+ = ; (2)
2+ 2 3 2 34 4 4 4 4 24Ca HPO 2PO Ca (HPO )(PO )− − − −+ + = ; (3)
2+ 34 5 4 35Ca 3PO OH Ca (PO ) (OH)− −+ + = . (4)
Thus, the corresponding supersaturations (S) are defined as follows:
2 24(Ca ) (HPO )(DCPD)
(DCPD)sp
a aSK
+ −
= ; (5)
4 2 2 2 34 4(Ca ) (HPO ) (PO )(OCP)
(OCP)sp
a a aSK
+ − −
= ; (6)

6
5 2 3 34(Ca ) (PO ) (OH )(HA)
(HA)sp
a a aSK
+ − −
= , (7)
where Ksp(DCPD) = 10−6.622 [50], Ksp(OCP) = 10−36.48, Ksp(HA)= 2.35 × 10−59 [51].
To determine the activity of an individual ion unit, all possible association/dissociation reactions
in SBF are taken into account. In total, 17 association/dissociation reactions and 4 mass balance
equations are used to calculate the activities of all of the ion and ion units in SBF. The
association/dissociation reactions are listed in Table 2, while the 4 mass balance equations are
given as follows:
3
2 +2 3 3 3 3HCO
+3 3 3
= [H CO ] + [HCO ] + [CO ] + [Ca HCO ]
+ [CaCO ] + [MgHCO ] + [MgCO ]
C −− −
− ; ( 8 )
2+
2+ + +3 3Ca
+2 4 4 4
= [Ca ] + [CaOH ] + [Ca HCO ] + [CaCO ]
+ [CaH PO ] + [Ca HPO ] + [CaPO ]
C−
; ( 9 )
2+
2+ + +3 3Mg
+2 4 4 4
= [Mg ] + [MgOH ] + [MgHCO ] + [MgCO ]
+ [MgH PO ] + [MgHPO ] + [MgPO ]
C−
; ( 10 )
24
2 33 4 2 4 4 4HPO
+2 4 4 4
+2 4 4 4
= [H PO ] + [H PO ]+ [HPO ]+[PO ]
+ [MgH PO ] + [Mg HPO ] + [MgPO ]
+ [CaH PO ] + [CaHPO ] + [CaPO ]
C −− − −
−
−
, ( 11 )
where [ ] is the equilibrium concentration and 3HCO
C − , 2+Ca C , 2+Mg
C 24HPOC − are the nominal
concentrations in SBF listed in the recipes (Table 1).
The activity coefficients, γi, of each ion unit were obtained from the modified Debye-Hückel
equation proposed by Davies [55] : 1/ 2
21/ 2log 0.3
1i iIAz II
γ⎡ ⎤
= − −⎢ ⎥+⎣ ⎦ , ( 12 )
where A is the Debye-Hückel constant dependent on temperature; A = 0.5211 at 37°; zi is the

7
charge number of ions; and I is the total ionic strength of the solution and is defined as:
212 i i
iI c z= ∑ (13)
in which ci is the molar concentration of each ion unit.
A computer program based on the Newton-Raphson iteration method was written to solve
numerically the simultaneous equations, including the 17 equations in Table 2 and the mass
balance equations (Eqs. 8-11). The ionic strength was obtained by iterative computation, which
eliminated possible errors from estimation.
The equations for the activity calculations included those with magnesium in SBF, even though it
is not an element in the Ca-P composition. Magnesium has been considered the inhibitor of Ca-P
nucleation and growth in aqueous solution [15, 56]. This inhibitor role affecting activities of
calcium and phosphate ions was taken into account in the analysis. The possible function of
magnesium to block the active sites of Ca-P precipitation [56], however, could not be modeled
by the classic theories of thermodynamics and kinetics.
Nucleation rates
The kinetics analysis was focused on the Ca-P nucleation rate (J), which can be estimated based
on the classical model of heterogeneous nucleation [57]
( )
2 3
23 3
16 ( )exp( ) exp( )3 ln
G v fJ K KkT k T S
π γ θ∆= − = − (14)
in which k is the Boltzmann constant and T is the absolute temperature. The nucleation rate is
proportional to a kinetic factor (K), and is exponentially affected by the activation energy of
nucleation (∆G), which is determined by the interfacial tension (γ ) between Ca-P and the
solution, the supersaturation (S) and the contact angle function, f (θ ), for a nucleus on a substrate.

8
The Ca-P molecular volume (v) is defined by the Ca-P crystal structure. The v values of HA,
OCP and DCPD are 263.24, 310.59, 126.54 Å3, respectively [47, 60]. The geometrical factor
(16π/3) represents spherical nuclei, which should be changed for non-spherical nuclei (e.g., 32
for a cube). In this analysis, 16π/3 was used because our SEM examinations found the nuclei of
Ca-P in SBF were in a hemispherical shape.
The kinetic factor (K) should be proportional to the probability (P) that the appropriate ion units
of Ca-P meet to compose a nucleus in the solution, i.e., K = K’P in which K’ is a constant and the
probability depends upon the concentrations of the ion units in the solutions [47]. The P values
were calculated using the concentrations of ion units for DCPD, OCP and HA according to the
method of Boiselle et al. [47]:
2 24
2 2 24
2![Ca ][HPO ]([Ca ] [HPO ])
P+ −
+ −=+
for DCPD ; (15)
2 4 2 3 24 4
2 2 3 74 4
7![Ca ] [HPO ][PO ]4!2!([Ca ] [HPO ] [PO ])
P+ − −
+ − −=+ +
for OCP; (16)
2 5 3 34
2 3 94
9![Ca ] [PO ] [OH ]5!3!([Ca ] [PO ] [OH ])
P+ − −
+ − −=+ +
for HA . (17)
Boiselle et al. determined the K’ values from the experimentally measured J with given
supersaturation and interfacial energies. According to their report, K’ should be 13.64×10−24
cm−3s−1 and this value was adopted in this analysis [47]. Accuracy of the K’ value does not
affect the comparison of nucleation rates of DCPD, OCP and HA.
The interfacial energy (γ) is also an uncertain parameter in calculating J in Eq. 14. In our

9
calculations, γ values were chosen as 10.4 mJm-2 for HA, 4.3 mJm-2 for OCP, 0.4 mJm-2 for
DCPD based on the studies by Wu et al. [58-60]. Possible effects of the γ variations on the J
calculations are addressed in the discussion section. The contact angle (θ) between the nucleus
and the substrate surface can be determined experimentally. The globule-like Ca-P nucleus is
widely reported [3, 13, 14, 45] regardless of the crystalline phase. We estimated the contact
angle (θ) of Ca-P globule-like nuclei on surfaces of the alkali-treated titanium and calcium
phosphate using SEM images and found that θ = 90° is a good estimation. The effects of the
contact angle variations on the Ca-P nucleation rates are also addressed the discussion section.
Results and Discussion
The analysis results of the free energy changes of Ca-P precipitation (∆G) and the Ca-P
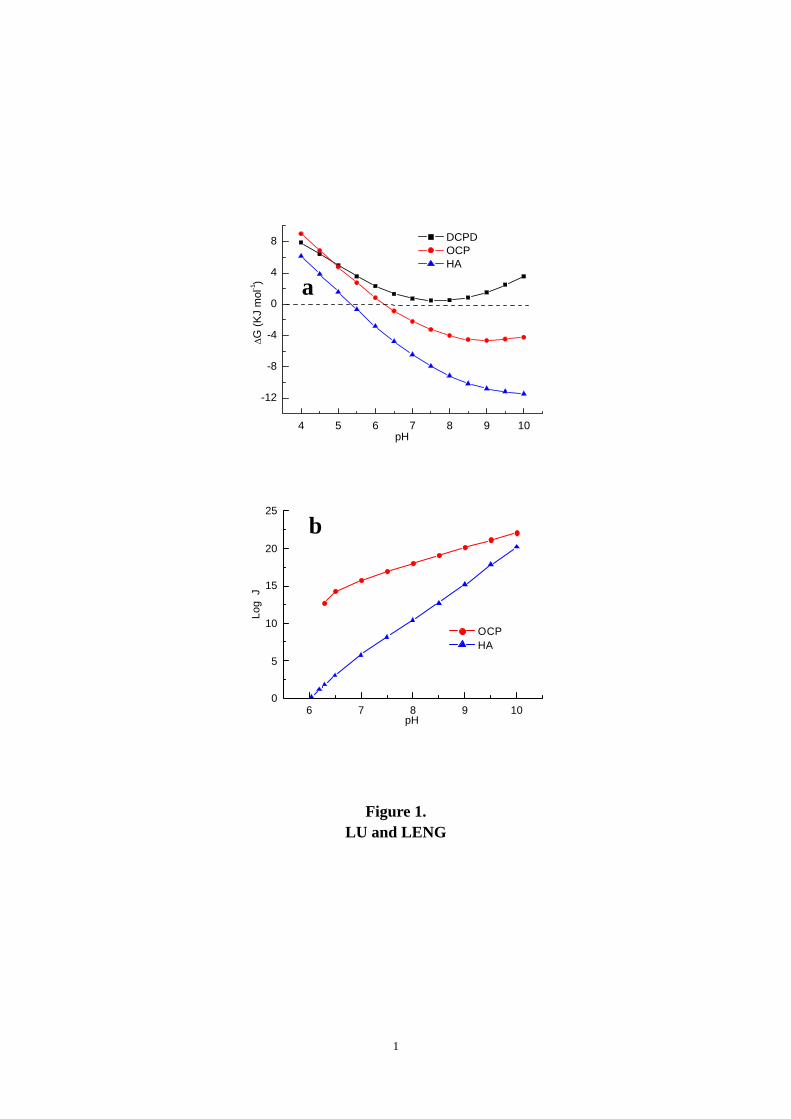
nucleation rates (J) in SBF are presented for a range of pH values. Figure 1 shows the
comparison of ∆G and J for the HA, OCP and DCPD precipitations in R-SBF, which is the
primary SBF recipe in this analysis. The HA precipitation is thermodynamically favorable
(∆GHA becomes negative) when pH ≥ 5.4, while the OCP precipitation obtains its
thermodynamic driving force when pH ≥ 6.3. However, there is no thermodynamic driving force
for the DCPD precipitation (∆GDCPD > 0) over the entire pH range. The driving force of HA is
always larger than that of OCP, although both increase with increasing pH values. On the other
hand, the nucleation rate of OCP (JOCP) is higher than that of HA (JHA) by 10 orders of
magnitude under physiological conditions (pH = 7.4) as shown in Fig. 1b. This difference in
nucleation rates reduces with increasing pH values, and JHA approaches JOCP when the pH value
approaches 10. The results suggest that the pH value is the critical factor that affects Ca-P
nucleation. Note that the DCPD nucleation rate (JCDPD) is not shown in Fig. 1b because

10
nucleation rates can only be calculated when ∆G is less than zero, according to the classical
nucleation theory (Eq. 14). We found that Fig. 1 represents the general characteristics of ∆G
and J of Ca-P precipitation in SBF. The parameters that possibly affect the ∆G and J calculations
are discussed in the following sections.
Solution composition
Different SBF recipes
Variation in the SBF composition results in changes in supersaturation and therefore affects ∆G
and J of Ca-P formation. The effects of different SBF recipes on ∆G and J are shown in Fig. 2.
The differences in ∆G and J for different SBF recipes are insignificant, particularly when pH
values are less than 9. In other words, variations in supersaturation in different SBF recipes are
not sufficient to alter the general characteristics of Ca-P precipitation that are shown in Fig. 1.
Note that there is no driving force for DCPD precipitation in SBF because ∆GDCPD is always
larger than zero (Fig 2e). This might be the reason that no DCPD precipitation has been
previously reported in SBF, although DCPD is commonly believed to be a precursor to HA
formation in aqueous solutions [37, 61]. According to Fig 2, the SBF recipes however can be
divided into three groups: C-SBF and M-SBF (Group 1), R-SBF and S-SBF (Group 2), I-SBF
(Group 3). The driving force of precipitation in Group 3 is lower than that of the others because
the Ca2+ concentration in the Group 3 is the lowest (Table 1). Note that the higher
concentration of HCO3− in Group 2 than in Group 1 can also reduce the driving force. Since
there are only small differences in the ∆G and J of the different SBF recipes, we discuss the
cases with R-SBF in the following sections.

11
High Ca and P concentrations
Although variation in SBF recipe is not sufficient to cause significant ∆G and J changes, it is of
interest to calculate ∆G and J of the Ca-P precipitation in SBF with higher than normal
concentrations of calcium and phosphate ions. Such solutions with high calcium and phosphate
concentrations have been used for biomimetic mineralization [16-21]. A SBF recipe with high
calcium and phosphate ion concentrations might represent a local physiological environment in
which a calcium phosphate implant is partially dissolved and releases calcium and phosphate
ions to the surrounding body fluid. Figure 3 shows the effects of high Ca2+ and HPO42−
concentrations in R-SBF on ∆G and J at pH levels, 5, 7.4, and 10. The x-axis indicates that the
Ca2+ and HPO42− concentrations range from 1 to 5 times higher than those of normal R-SBF.
The other ion concentrations remain the same as in normal R-SBF during the analysis. The
results show that ∆GHA, ∆GOCP and ∆GDCPD decrease with increasing Ca2+ and HPO42−
concentrations at the same rate at pH levels of 5, 7.4 and 10. Note that ∆GDCPD become negative
in solutions when the Ca2+ and HPO42−
concentrations increase slightly at pH = 7.4. The
significance is that JDCPD is extremely high, even higher than JOCP, when ∆GDCPD becomes
negative. Thus, DCPD becomes the most kinetically favorable phase in SBF when the
concentrations of calcium and phosphate are higher than the normal level at pH = 7.4.
Interfacial energy
The interfacial energy (γ) is one of the most difficult parameters in Eq. 14 to determine. Note
that interfacial energy and interfacial tension are identical and often are used interchangeably in
the literature. The interfacial energy data used in our analysis actually are interfacial tension data
that were determined experimentally. Note that these data of HA, OCP and DCPD are far from

12
certain and were determined by several means including the crystal growth kinetics, dissolution
kinetics and contact angle measurements [58-60, 62-63]. Wu and Nancollas reviewed the
measurements of the Ca-P interfacial tension and summarized the measured γHA, γOCP and γDCPD
as shown in Table 3 [60].
The kinetics methods employ the classical nucleation theory (Eq. 14) to determine the interfacial
tension by measuring the nucleation or dissolution rate. Unfortunately, the measurements from
different studies based on the kinetics method vary to a large degree. As shown in Table 3, from
the kinetics method, γHA varies from 9.3 to 87 mJm−2, while γDCPD varies form 0.4 to 70 mJm−2.
The kinetics method involves a number of variables that are difficult to control or measure
experimentally. On the other hand, the contact angle method measures the rate of the rise of a
solution in a thin-layer wicking capillary [58-50, 64]. The rising rate in a capillary of a solution
relates to the interfacial tension according to the Washburn equation. The contact angle method
generates more consistent results than does the kinetic method [60]. Thus, the interfacial tension
values obtained with this method were chosen for analysis as listed in Table 3, except for DCPD
because γDCPD is negative from the contact angle method. The negative interfacial energy cannot
be used in the calculation of nucleation rates in the classical nucleation model. As an alternative,
we chose the minimum value of γDCPD = 0.4 mJm−2 from the kinetics method.
Considering the uncertainty of the interfacial tension data, we evaluated the effects of γHA, γOCP
and γDCPD on the J calculations. The curves in Fig. 4 represent the J dependence on choosing
the γ values listed in Table 3. Fig. 4a shows the comparison of the γHA and γOCP effects on JHA,
JOCP in R-SBF, and Fig. 4b shows the comparison of the γHA, γOCP and γDCPD effects in solutions

13
with the Ca2+ and HPO42−
concentrations that are five-fold higher than those of normal R-SBF.
Although γHA, γOCP and γDCPD vary to a large degree, JOCP is always larger than JHA as shown in
Fig. 4a. The solution with high Ca2+ and HPO42−
concentrations follow the same trend (Fig 4b).
Thus, we conclude that variations in interfacial energy data from different measurement methods
do not change the general trends shown in Figs. 1-3; that is, JOCP is always larger than JHA in
SBF, and JDCPD is the highest when DCPD precipitation is thermodynamically possible.
Molecular volume
Note that the molecular volume (v) used in the nucleation rate calculations (Eq. 14) is defined as
the Ca-P crystal volume per molecule. Wu et al. argued that the volume of a crystal unit cell (vc)
should be used in Eq. 14, because the interfacial tension of calcium phosphate in solution is from
the crystal/solution interface [60]. The vc values of HA, OCP and DCPD are 582.7, 1242.4, 506.2
Å3, respectively. This argument was supported by Wu et al.’s experimental interfacial tension
measurements by substituting vc for v in the kinetics method. When vc is adopted in the kinetics
method, Wu et al. were able to obtain γ values comparable with those values obtained by the
contact angle method [60]. Thus, we feel that it is necessary to evaluate the effect of replacing v
with vc on the Ca-P nucleation rates. We compared the JHA and JOCP calculated with vc with the
those calculated with v in Fig. 5. The comparison clearly indicates that we can ignore the
variations of nucleation rates by using either v or vc in Eq. 14.
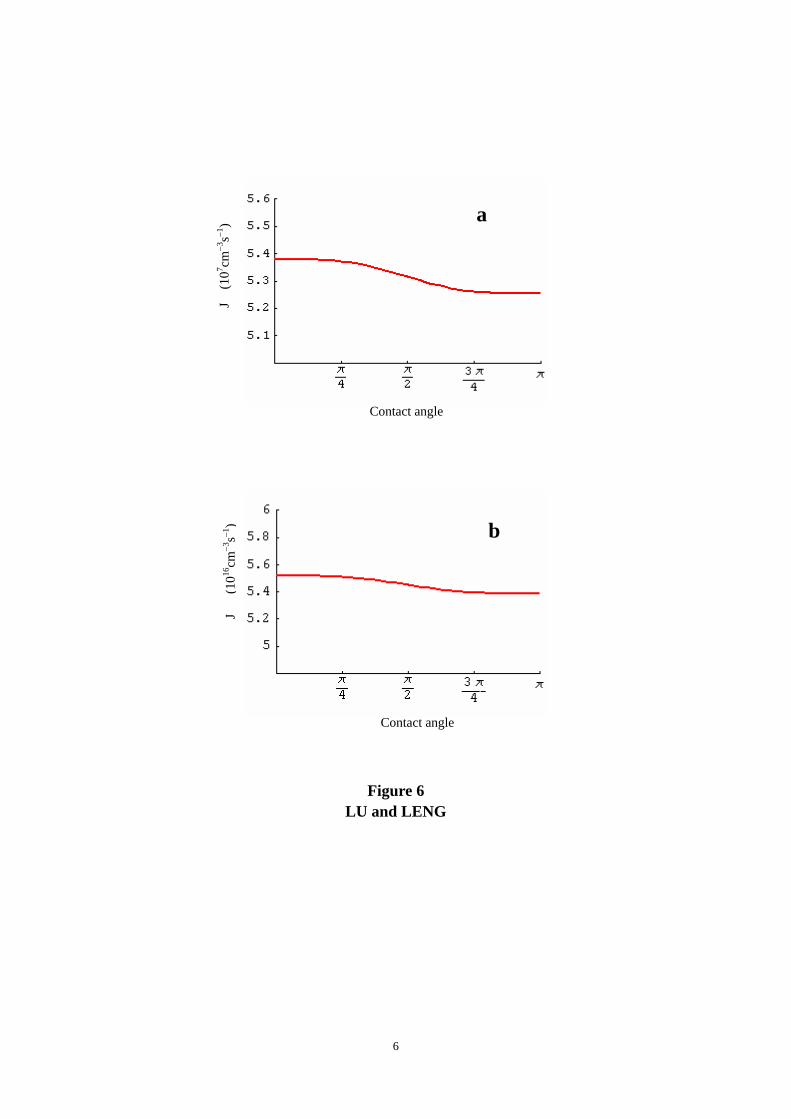
Contact angle
The contact angle function, f(θ), in Eq. 14 is given by the classical theory of heterogeneous
nucleation as [57] :

14
2(2 cos )(1 cos )( )4
f θ θθ⎧ ⎫+ −
= ⎨ ⎬⎩ ⎭
. (18)
The f(θ) range corresponding to 0° < θ < 180° is from 0 to 1, while θ = 180° corresponds to
homogeneous nucleation. Thus, the effect of the contact angle change on the nucleation rates is
minor. Figure 6 shows that JHA and JOCP variations with θ are less than one order of magnitude
and therefore can be ignored. This implies that there is little difference in the analysis results
using either the homogeneous or the heterogeneous nucleation model. The selection of θ = 90°
for the J calculations shown in Figs. 1-3 was simply based on the shape of Ca-P globular nuclei
on titanium surfaces.
Ion units of OCP formation
Notice that the ion units that compose OCP nuclei can be expressed alternatively as in the
following equation.
2+ 3 +4 4 4 34Ca 3PO H Ca (PO ) H−+ + = . (19)
Thus, the corresponding S and P should be
4 2 3 34(Ca ) (PO ) (H )(OCP)
(OCP)sp
a a aSK
+ − +
= ; (20)
2 4 3 34
2 + 3 84
8![Ca ] [H ][PO ]4!3!([Ca ] [H ] [PO ])
P+ + −
+ −=+ +
, (21)
where Ksp(OCP) = 1.00 × 10−48.7 [65]. The ∆GOCP and JOCP calculations based on Eqs. 19 and
21 are different from those based on Eqs. 3 and 16. Figure 7a shows that the ∆GOCP calculated
based on Eqs. 19 and 21 is only slightly different from the previous calculation. The ∆G
difference results from the difference in the number of ion units (n = 7 in Eq. 3 and n = 8 in Eq.

15
25) during calculation. The n∆G is the same in both cases because the value of supersaturation
does not change with the expressions. However, the difference in calculating JOCP cannot be
ignored because the kinetic factor in Eq. 14 significantly changes when the ion units of PO43−
and H+ are used for the probability calculation. The probability (P) based on ion units in Eq. 21
is 1010 times lower than that in Eq. 16. As a consequence, the HA nucleation becomes more
kinetically favorable than OCP in SBF as shown in Fig 7b. We do not adopt Eq. 19 in the
calculation because it implies that the hydrogen is a separate ion existing in OCP. This is not
consistent with the OCP crystal structure in which one hydrogen ion is always associated with
one PO43− to form HPO4
2− [66]. In addition, the probability calculation indicates that the chance
of ion units in Eq. 19 combining together is too low compared with the chance of the ion units in
Eq. 3. Thus, we believe that Eq. 19 should not be used for the OCP nucleation rate analysis.
Apatite with carbonate or calcium-deficiency
Carbonate can incorporate into apatite and substitute for PO4 or OH in the apatite crystal
structure and subsequently change its properties [67]. Helebrant et al. found that the
supersaturation of slightly carbonated apatite (SCHA) was even higher than that of apatite [68] in
SBF. Also, calcium-deficient apatite (DOHA) is an important form of Ca-P in biological systems
[56]. Comparing the driving force and nucleation rates of such irregular apatites with those of
the stoichiometric HA, OCP and DCPD is important. The difficulties in analyzing such apatites
come from uncertainties in chemical compositions and the lack of thermodynamic and kinetic
data.
Based on the available data, we managed to analyze two special cases: 1) Ca10(PO4)6(CO3)0.5(OH)
(SCHA) and 2) Ca9 (HPO4) (PO4)5OH (DOHA) . The ∆GSCHA and ∆GDOHA were evaluated from

16
their supersaturation in SBF:
10 2 6 3 0.5 24 3(Ca ) (PO ) (CO ) (OH )(SCHA)(SCHA)sp
a a a aSK
+ − − −
= ; (22)
9 2 5 34 4(Ca ) (HPO ) (PO ) (OH )(DOHA)
(DOHA)sp
a a a aSK
+ − − −
= , (23)
in which Ksp(SCHA) = 10−115.6 [68], and Ksp(SCHA) = 10−85.1 [56].
There was lack of experimental data, particularly the kinetic factor used in Eq. 14, to calculate
nucleation rates for SCHA and DOHA. We however note that the kinetic factor is mainly
determined by the probability (P) that the appropriate ion units meet to compose a nucleus in the
solution. The probabilities (P) of SCHA and DOHA can be calculated and compared with the
stoichiometric HA. Actually, P plays a dominant role in determining the nucleation rates
according to the Boistelle’s theory [47]. The calculation of PSCHA was based on the concentration
equivalency of OH− and CO32− :
2 10 3 6 24
2 3 184
18![Ca ] [PO ] [OH ]10!6!([Ca ] [PO ] [OH ] )
eq
eq
P+ − −
+ − −=+ +
(24)
in which 23OH OH 2COeq
− − −= + because one CO32− can substitute for two OH− in the apatite
crystal structure. The calculation of P DOHA is rather straightforward:
2 9 2 1 3 5 14 4
2 2 3 164 4
16![Ca ] [HPO ] [PO ] [OH ]9!5!([Ca ] [HPO ] [PO ] [OH ])
P+ − − −
+ − − −=+ + +
. (25)
Fig 8 compares the analysis results of SCHA and DOHA with the other Ca-P. The results
indicate that the SCHA precipitation obtains a similar level of thermodynamic driving force as
the stoichiometric HA. However, the DOHA precipitation is less thermodynamically favorable
and only comparable with OCP (Fig. 8a). The nucleation probabilities of SCHA and DOHA are

17
considerably higher than that of stoichiometric HA, but still less than those of OCP and DCPD
(Fig. 8b). Thus, higher nucleation rates of SCHA and DOHA than that of stoichiometric HA are
expected based on the analysis. We believe that analysis of these special cases indicates a
general trend of carbonate-containing and calcium-deficient apatites. The general trend should
be that precipitation of carbonate-containing and calcium-deficient apatites in SBF exhibit their
kinetic advantages, compared with the stoichiometric HA. The analysis of DOHA indicates that
the calcium-deficient apatite exhibits a lower thermodynamic driving force than does
stoichiometric HA.
Concluding remarks Ca-P formations in SBF were analyzed based on classical crystallization theories of
thermodynamics and kinetics. We analyzed the possible effects of data variations and chemistry
changes on the nucleation driving force and nucleation rates with best available thermodynamic
and kinetic data. The analysis indicates that HA precipitation exhibits a higher thermodynamic
driving force than does OCP and DCPD in SBF. OCP precipitation is kinetically favorable in
SBF. The HA nucleation rate is significantly affected by the pH value. High pH environment is
favorable for HA nucleation and the HA nucleation rate approaches the nucleations rates of OCP
when the pH value approaches 10. DCPD does not have a thermodynamic driving force of
precipitation in SBF, even though it has kinetic advantages in nucleation. DCPD precipitation
becomes possible when the concentrations of calcium and phosphate ions increase to a higher
than normal level in SBF. Possible variation of parameter values in the analysis model, such as
discrepancies in the interfacial energy data or uncertainty of the contact angle of nuclei on
surfaces, does not change these conclusions. However, HA precipitation can be considerably

18
affected by containing carbonate or being deficient in calcium. Generally, precipitation of
carbonate-containing HA is more kinetically favorable than that of stoichiometric HA and has a
same level of thermodynamic driving force; precipitation of calcium-deficient HA is also more
kinetically favorable, but its thermodynamic driving force is lower than that of stoichiometric
HA.
Acknowledgements
This project was financially sponsored by the Research Grants Council of Hong Kong (HKUST
6037/02E) and the Funds for High Impact Areas at Hong Kong University of Science &
Technology. The authors wish to acknowledge the valuable discussion about chemical
equilibrium with Mr. Hu Quanyuan in the Department of Chemistry, Hong Kong University of
Science and Technology.

19
References
1. Kokubo T, Kushitani H, Sakka S, Kitsugi T, Yamamuro T. Solutions able to reproduce in
vivo surface-structure changes in bioactive glass-ceramic A-W. J Biomed Mater Res
1990;24:721-734.
2. Kokubo T, Miyaji F, Kim HM, Nakamura T. Spontaneous formation of bonelike apatite layer
on chemically treated titanium metals. J Am Ceram Soc 1996; 79:1127-1129.
3. Kokubo T, Kim HM, Kawashita M. Novel bioactive materials with different mechanical
properties. Biomaterials 2003;24: 2161-2175.
4. Kim HM, Miyaji F, Kokubo T, Nakamura T. Preparation of bioactive Ti and its alloys via
simple chemical surface treatment. J Biomed Mater Res 1996;32:409-417.
5. Kim HM, Miyaji F, Kokubo T, Nakamura T. Effect of heat treatment on apatite-forming
ability of Ti metal induced by alkali treatment. J Mater Sci: Mater Med 1997;8:341-347.
6. Kim HM, Miyazaki T, Kukubo T, Nakamura T. Revised simulated body fluid. Key Eng
Mater 2001;192-195: 47-50.
7. Oyane A, Kim HM, Furuya T, Kokubo T, Miyazaki T, Nakamura T. Preparation and
assessment of revised simulated body fluids. J Biomed Mater Res 2003;65A:188-195.
8. Nishiguchi S, Nakamura T, Kobayashi M, Kim HM, Miyaji F, Kokubo T. The effect of heat
treatment on bone-bonding ability of alkali-treated titanium. Biomaterials 1999; 20: 491-500.
9. Takadama H, Kim HM, Kokubo T, Nakamura T. An X-ray photoelectron spectroscopy
study of the process of apatite formation on bioactive titanium metal. J Biomed Mater Res
2001; 55: 185-193.
10. Takadama H, Kim HM, Kokubo T, Nakamura T. TEM-EDX study of mechanism of
bonelike apatite formation on bioactive titanium metal in simulated body fluid. J Biomed
Mater Res 2001; 57: 441-448.
11. Lee BH, Kim YD, Shin JH, Lee KH. Surface modification by alkali and heat treatments in
titanium alloys. J Biomed Mater Res 2002; 61: 466-473.
12. Kaneko H, Uchida M, Kim HM, Kokubo T, Nakamura T. Process of apatite formation
induced by anatase on titanium metal in simulated body fluid. Key Eng Mater 2002; 218-220:
649-652.

20
13. Uchida M, Kim KM, Kokubo T, Fujibayashi S, Nakamura T. Effect of water treatment on
the apatite-forming ability of NaOH-treated titanium metal. J Biomed Mater Res Appl
Biomater 2002;63:522-530.
14. Wei M, Kim HM, Kokubo T, Evans JH. Opitmising the bioactivity of alkaline-treated
titanium alloy. Mat Sci Eng C 2002;20:125-134.
15. Barrère F, Layrolle P, van Blitterswijk CA, de Groot K. Biomimetic calcium phosphate
coatings on Ti6Al4V: a crystal growth study of octacalcium phosphate and inhibition by
Mg2+ and HCO3−. Bone 1999;25:107S-111S.
16. Barrère F, Layrolle P, van Blitterswijk CA, de Groot K. Biomimetic coatings on titanium: a
crystal growth study of calcium phosphate. J Mater Sci: Mater Med 2001;12:529-34.
17. Barrère F, van Blitterswijk CA, de Groot K, Layrolle P. Nucleation of biomimetic Ca-P
coatings on Ti6Al4V from a SBF×5 solution: influence of magnesium. Biomaterials 2002;
23: 2211-2220.
18. Barrère F, van Blitterswijk CA, de Groot K, Layrolle P. Influence of ionic strength and
carbonate on the Ca-P coating formation from SBF×5 solution. Biomaterials 2002;23: 1921-
1930.
19. Feng QL, Wang H, Cui FZ, Kim TN. Controlled crystal growth of calcium phosphate on
titanium by NaOH-treatment. J Crystal Growth 1999;200:550-557.
20. Feng QL, Cui FZ, Wang H, Kim TN, Kim JO. Influence of solutions conditions on
deposition of calcium phosphate on titanium by NaOH-treatment. J Crystal Growth 2000;
210: 735-740.
21. Mao CB, Li HD, Cui FZ, Ma CL, Feng QL. Oriented growth of phosphates on
polycrystalline titanium in a process mimicking biomineralization. J Crystal Growth
1999;206:308-321.
22. Tas AC. Synthesis of biomimetic Ca-hydroxyapatite powder at 37°C in synthetic body
fluids. Biomaterials 2000;21:1429-1438.
23. Cui FZ, Zhou LF, Cui H, Ma CL, Lu HB, Li HD. Phase diagram for controlled
crystallization of calcium phosphate under acidic organic monolayers. J Crystal Growth
1996;169:557-562.
24. Li F, Feng QL, Cui FZ, Li HD, Schubert H. A simple biomimetic method for calcium
phosphate coating. Surf Coat Tech 2002;154:88-93.

21
25. Liu Y, Layrolle P, de Bruijn J, van Blitterswijk C, de Groot K. Biomimetic coprecipiation of
calcium phosphate and bovine serum albumin on titanium alloy. J Biomed Mater Res 2001;
57: 327-335.
26. Liu Y, Hunzikerc EB, Randalld NX, de Groot K, Layrolle P. Proteins incorporated into
biomimetically prepared calcium phosphate coatings modulate their mechanical strength and
dissolution rate. Biomaterials 2003;24:65-70.
27. Stigter M, de Groot K, Layrolle P. Incorporation of tobramycin into biomimetic
hydroxyapatite coating on titanium. Biomaterials 2002;23: 4143-4153.
28. Rámila A, Vallet-Regí M. Static and dynamic in vitro study of a sol-gel glass bioactivity,
Biomaterials 2001; 22: 2301-2306.
29. Fujibayashi S, Neo M, Kim HM, Kokubo T, Nakamura T. A comparative study between in
vivo bone ingrowth and in vitro apatite formation on Na2O-CaO-SiO2 glasses. Biomaterials
2003;24:1349-1356.
30. Siriphannon P, Kameshima Y, Yasumori A, Okada K, Hayashi S. Comparative study of the
formation of hydroxyapatite in simulate body fluid under static and flowing system. J
Biomed Mater Res 2002;60:175-185.
31. Li P, Ohtsuki C, Kokubo T, Nakanishi K, Soga N, Nakamura T, Yamamuro T. Process of
formation of bone-like apatite layer on silica gel. J Mater Sci: Mater Med 1993; 4:127-131.
32. Ni J, Wang M. In vitro evaluation of hydroxyapatite reinforced polyhydroxybutyrate
composite. Mat Sci Eng C 2002;20:101–109.
33. Leonor IB, Ito A, Onuma K, Kanzaki N, Reis RL. In vitro bioactivity of starch
thermoplastic/hydroxyapatite composite biomaterials: an in situ study using atomic force
microscopy. Biomaterials 2003;24:579–585.
34. Rich J, Jaakkola T, Tirri T, Närhi T, Yli-Urpo A, Seppälä J. In vitro evaluation of poly(ε-
caprolactone-co-DL-lactide)/bioactive glass composite. Biomaterials 2002;23: 2143-2150.
35. LeGeros RZ. Apatites in biological systems. Prog Crystal Growth Charact 1981;4:1-45.
36. Koutsoukos PG, Amjad Z, Tomson MB, Nancollas GH. Crystallization of calcium
phosphates-a constant composition study. J Am Chem Soc 1980;102:1553-1557.
37. Koutsoukos PG, Nancollas GH. Crystal growth of calcium phosphates-Epitaxial
Considerations. J Crystal Growth 1981;53:10-19.

22
38. Heughebaert JC, Nancollas GH. Kinetics of crystallization of octacalcium phosphate. J Phys
Chem 1984;88:2478-2481.
39. Heughebaert JC, Nancollas GH. Mineralization kinetics-the role of octacalcium phosphate in
the precipitation of calcium phosphates. Colloid Surf 1984;9:89-93.
40. Marques PAAP, Magalhães MCF, Dorozhkin SV, Correia RN. Hydrogencarbonate as a
biological buffer in simulated plasma. Key Eng Mater 2001;192-195:27-30.
41. Marques PAAP, Magalhães MCF, Correia RN. Inorganic plasma with physiological
CO2/HCO3- buffer. Biomaterials 2003;24:1541-1548.
42. Koutsopoulos S, Paschalakis PC, Dalas E. The calcification of elastin in vitro. Langmuir
1994;10:2423-2428.
43. Koutsopoulos S, Dalas E. The calcification of fibrin in vitro. J Crystal Growth 2000;216:
450-458.
44. Leng Y, Chen JY, Qu SX. TEM examinations of calcium phosphate precipitation on HA/TCP.
Biomaterials 2003; 24: 2125-2131.
45. Lu X, Leng Y. TEM study of calcium phosphate precipitation on bioactive titanium surfaces.
Biomaterials 2004; 25: 1779-1786.
46. Brown WE, Smith JP, Lehr FR, Frazier AW. Octacalcium phosphate and hydroxyapatite.
Nature 1962; 196: 1048-1055.
47. Boistelle R, Lopez-Valero I. Growth units and nucleation: the case of calcium phosphates. J
Crystal Growth 1990; 102: 609-617.
48. Vereecke G, Lemaitre J. Calculation of the solubility diagrams in the system Ca(OH)2-
H3PO4-KOH-HNO3-CO2-H2O. J Crystal Growth 1990; 104: 820-832.
49. Nielsen AE. Electrolyte crystal growth mechanisms. J Crystal Growth 1984;67:289-310.
50. Gregory TM, Moreno EC, Brown WE, Solubility of CaHPO4⋅2H2O in the system Ca(OH)2-
H3PO4-H2O at 5, 15, 25, and 37°C. J Res Natl Bur Std (US) 1970;74A: 461-475.
51. McDowell H, Gregory TM, Brown WE. Solubility of Ca5(PO4)3OH in the system Ca(OH)2-
H3PO4-H2O at 5, 15, 25, and 37°C. J Res Natl Bur Std (US) 1977;81A: 273-281.
52. Chughtai A, Marshall R, Nancollas GH. Complexes in calcium phosphate solutions. J Phys
Chem 1968; 72: 208-211.
53. Butler JN, Cogley DR. Ionic equilibrium: solubility and pH calculations. New York: Wiley,
1998. p.410.

23
54. Childs CW. A potentiometric study of equilibria in aqueous divalent metal orthophosphate
solutions. Inorg Chem 1970; 9: 2465-2469.
55. Davies CW. Ion association. London: Butterworths, 1962. p41.
56. Driessens FCM, Verbeeck RMH. Biominerals. Boca Raton, Fla: CRC Press, 1990. p38.
57. Mullin JW. Crystallization, 3rd ed. Oxford: Butterworth-Heinemann,1993. p172-201.
58. Wu W, Nancollas GH. The relationship between surface free-energy and kinetics in the
mineralization and demineralization of dental hard tissue. Adv Dent Res 1997;11: 566-575.
59. Wu W, Nancollas GH. The dissolution and growth of sparingly soluble in organic salts: A
kinetics and surface energy approach. Pure Appl Chem 1998;70:1867-1872.
60. Wu W, Nancollas GH. Determination of interfacial tension from crystallization and
dissolution data: a comparison with other methods. Adv Colloid Interface Sci 1999;79: 229-
279.
61. Wong ATC, Czernuszka JT. Transformation behavior of calcium phosphate I. Theory and
modeling. Colloid Surface A 1993;78:245-253.
62. Liu Y, Wu W, Sethuraman G, Nancollas GH. Intergrowth of calcium phosphates: an
interfacial energy approach. J Crystal Growth 1997;174: 386-392
63. Liu Y, Sethuraman G, Wu W, Nancollas GH, Grynpas M. The crystallization of fluorapatite
in the presence of hydroxyapatite seeds and of hydroxyapatite in the presence of fluorapatite
seeds. J Colloid Interface Sci 1997; 186: 102-109.
64. Liu Y, Nancollas GH. Fluorapatite growth kinetics and the influence of solution composition.
J Crystal Growth 1996;165:116-123.
65. Tung MS, Eidelman N, Sieck B, Brown WE. Octacalcium phosphate solubility product from
4 to 37°C. J Res Natl Bur Std (US) 1988;93:613-624.
66. Mathew M, Brown WE, Schroeder LW. Crystal structure of octacalcium
bis(hydrogenphosphate) tetrakis(phosphate)pentahydrate Ca8(HPO4)2(PO4)4⋅5H2O. J Crystal
Spectr Res 1988; 18: 235-249.
67. Elliott JC. Structure and chemistry of the apatites and other calcium orthophosphates.
Amsterdam: Elsevier, 1994. p191-301.
68. Helebrant A, Jonášová L, Šanda L. The influence of simulated body fluid composition on
carbonated hydroxyapatite formation. Ceramics − Silikáty 2002; 46: 9-14.

24
Table 1 Nominal Concentrations of Different SBF
Concentration (mmol dm−3) Ion Blood
Plasma [6,7]
C-SBF [6,7]
R-SBF [6,7]
i-SBF [7]
m-SBF [7]
Syn-SBF [22]
Na+ 142.0 142.0 142.0 142.0 157.0d 142.0 K+ 5.0 5.0 5.0 5.0 5.0 5.0
Mg2+ 1.5 1.5 1.5 1.0 1.5 1.5 Ca2+ 2.5 2.5 2.5 1.6 2.5 2.5
Cl− 103.0 187.8 a 103.0 103.0 103.0 165.0a HCO3
− 27.0 4.2 27.0 27.0 4.2 27.0 HPO4
2− 1.0 1.0 1.0 1.0 1.0 1.0 SO4
2− 0.5 0.5 0.5 0.5 0.5 0.5 buffer − 6.063b 11.928c 11.928c 17.892c 6.063b
a. Considering 40 mmol dm−3 of extra chloride ion added with the Tris-HCl buffer. b. Tris (hydroxylmethyl) aminomethane (g/L). c. HEPES 2-(4-(2-hydroxyethyl)-1-piperazinyl) ethane surfonic acid (g/L) d. Considering 15 mmol dm−3 of extra Na+ added with the HEPES-NaOH buffer

25
Table 2 Reactions in SBF
Reaction K Ref.
2 3 3H CO (aq.) H HCO+ −⇔ + 10−6.31 [7] 2
3 3HCO H CO− + −⇔ + 10−10.25 [7] +
3 4 2 4H PO (aq.) H + H PO −⇔ 10−2.196 [7]
+ 22 4 4H PO H + HPO − −⇔ 10−7.185 [7]
2 + 34 4HPO H + PO − −⇔ 10−12.19 [7]
2+ +3 3Ca + HCO Ca HCO− ⇔ 101.16 [7]
2+ 23 3Ca + CO CaCO (aq.)− ⇔ 103.38 [7]
2+ +Ca + OH CaOH − ⇔ 25.12 [37] 2+ +
2 4 2 4Ca + H PO Ca H PO− ⇔ 31.9 [52] 2+
4 4Ca + HPO Ca HPO (aq.)− ⇔ 6.81 × 102 [52] 2+ 3
4 4Ca + PO Ca PO− −⇔ 3.46 × 106 [52]
2+3 3Mg + HCO MgHCO − +⇔ 100.62 [53]
2+ 23 3Mg + CO MgCO (aq.)− ⇔ 101.87 [53]
2+ +Mg + OH MgOH − ⇔ 102.19 [53] 2+ +
2 4 2 4Mg + H PO Mg H PO− ⇔ K = 100.4 [54] 2+
4 4Mg + HPO Mg HPO (aq.)− ⇔ K = 101.8 [54]
2+ 34 4Mg + PO Mg PO− −⇔ K = 103.3 [54]
Table 3 Summary of Reported Interfacial Tensions[60]
Researcher Year HA OCP DCPD Measurement method Nancollas et al 99 10.0 4.3 −4.2 Contact angle 98 17 7 0.4 Crystal growth kinetics 98 9.3 3 0.4 Dissolution kinetics Christoffersen et al 88 − − 70 Crystal growth kinetics 87-92 80-87 − − Crystal growth kinetics 96 46 − − Dissolution kinetics Lundager et al 84 − 45 22 Crystal growth kinetics
The unit of interfacial tension is mJm−2.

26
Figure Captions
Figure 1. a) Free energy change (∆G) of Ca-P precipitation in R-SBF as a function of pH
value; b) Nucleation rates (J) of HA and OCP precipitation in R-SBF.
Figure 2. Effects of SBF recipes on Ca-P precipitation: a) ∆GHA; b) JHA; c) ∆GOCP; d) JOCP;
and e) ∆GDCPD.
Figure 3. Effects of Ca2+ and HPO42− concentrations in SBF on Ca-P precipitation. The
[Ca2+] and [HPO42−] vary from 1 to 5 times of those in normal SBF. a) ∆G at
pH = 5; b) J at pH = 5; c ) ∆G at pH = 7.4; d ) J at pH = 7.4; e ) ∆G at pH = 10;
and f ) J at pH = 10.
Figure 4. Effects of interfacial energy (γ) variations on the calculations of nucleation rates
at pH = 7.4. The solid line represents JHA; the dashed line represents JOCP; and
the dotted line represents JDCPD. The triangles mark the data obtained from the
contact angle method; the circles mark the data from the crystal growth kinetics
method; and the rhombohedra mark the data from the dissolution kinetics
method. a) in R-SBF; and b) in R-SBF with high [Ca2+] (12.5 mmol dm−3) and
[HPO42−] (5.0 mmol dm−3).
Figure 5. Comparison of the nucleation rates calculated from the molecular volume (v) and
the volume of the crystal unit cell (vc) in R-SBF.
Figure 6. Effects of the contact angle (θ) on the nucleation rates in R-SBF at pH = 7.4: a)
JHA; and b) JOCP.
Figure 7. Comparison of using different ion units for OCP formation: a) Small differences
in ∆GOCP values using different ion unit; and b) questionable JOCP based on Eqs
19 and 21 compared with JHA.
Figure 8. Comparison of precipitation of SCHA and DOHA with other Ca-P: a) ∆G; b)
kinetic probability (P).
1
4 5 6 7 8 9 10
-12
-8
-4
0
4
8∆G
(KJ
mol
-1)
pH
DCPD OCP HA
Figure 1. LU and LENG
a
b
6 7 8 9 100
5
10
15
20
25
Log
J
pH
OCP HA

2
Figure 2 LU and LENG
6 7 8 9 100
5
10
15
20
25Lo
g J
pH
C-SBF R-SBF I-SBF M-SBF S-SBF
6 7 8 9 100
5
10
15
20
25
Log
J
pH
C-SBF R-SBF I-SBF M-SBF S-SBF
4 5 6 7 8 9 10
-12
-8
-4
0
4
8
∆G (K
J m
ol-1)
pH
C-SBF R-SBF I-SBF M-SBF S-SBF
dc
a
HA
b
HA
OCP OCP
DCPD
4 5 6 7 8 9 10
-12
-8
-4
0
4
8
∆G (K
J m
ol-1)
pH
C-SBF R-SBF I-SBF M-SBF S-SBF
4 5 6 7 8 9 10
-12
-8
-4
0
4
8
∆G (K
J m
ol-1)
pH
C-SBF R-SBF I-SBF M-SBF S-SBF
e

3
Figure 3
LU and LENG
0 1 2 3 4 50
5
10
15
20
25
Log
J
Ca_P
DCPD OCP HA
0 1 2 3 4 5
-12
-8
-4
0
4
∆G (K
J m
ol-1
)
Ca_P
DCPD OCP HA
0 1 2 3 4 50
5
10
15
20
25
Log
J
Ca_P
DCPD OCP HA
0 1 2 3 4 5
-12
-8
-4
0
4
∆G (K
J m
ol-1
)
Ca_P
DCPD OCP HA
0 1 2 3 4 5
-12
-8
-4
0
4
∆G (K
J m
ol-1
)
Ca_P
DCPD OCP HA
0 1 2 3 4 50
5
10
15
20
25Lo
g J
Ca_P
DCPD OCP HA
c
e f
b
d
a
pH = 5 pH = 5
pH = 7.4 pH = 7.4
pH = 10 pH = 10

4
.
Figure 4 LU and LENG
a
b
Log
J
γ ( mJm−2 )
Log
J
γ ( mJm−2 )

5
6 7 8 9 100
5
10
15
20
25Lo
g J
pH
OCP v HA v OCP vc HA vc
Figure 5 LU and LENG
6
Figure 6 LU and LENG
Contact angle
J (
107 cm
−3s−
1 ) J
(1
016cm
−3s−
1 ) a
b
Contact angle

7
4 5 6 7 8 9 10
-12
-8
-4
0
4
8
∆G (K
J m
ol-1)
pH
OCP Eqs 3 & 16 OCP Eqs 19 & 21
6 7 8 9 100
5
10
15
20
25
Log
J
pH
OCP HA
Figure 7
LU and LENG
a
b

8
4 5 6 7 8 9 10
-12
-8
-4
0
4
8
12
∆G (K
J m
ol-1)
pH
DCPD OCP HA SCHA DOHA
4 5 6 7 8 9 10-80
-70
-60
-50
-40
-30
-20
-10
0
Log
P
pH
DCPD OCP HA SCHA DOHA
Figure 8 LU and LENG
a
b


















