
The role of the scoring system for the diagnosis of Wilson's ...
21
Seoul National University Children’s Hospital Gastroenterology, hepatology and Nutrion F. Sang hee, Cho ATP7B mutation in patients with Wilson disease
Transcript of The role of the scoring system for the diagnosis of Wilson's ...

Seoul National University Children’s Hospital
Gastroenterology, hepatology and Nutrion
F. Sang hee, Cho
ATP7B mutation in patients with
Wilson disease

Wilson disease (WD) was first described in 1912 by
Kinnear Wilson as “progressive lenticular
degeneration,” a familial, lethal neurological disease
accompanied by chronic liver disease leading to
cirrhosis.
In 1993, the abnormal gene in WD was identified.
ATP7B encodes a metal-transporting P-type
adenosine triphosphatase (ATPase)
Introduction

Critical role in human metabolism as a cofactor of key metabolic enzymes, which are involved in respiration, neurotransmitter biosynthesis, radical detoxification, iron metabolism, and many other physiological processes
Average daily intake of copper : 1-3mg
When intake is less than 1mg/day, more than 50% of the copper is absorbed, when copper intake is more than 5mg/day, less than 20% is absorbed
Liver is the principal storage site for copper and regulates its excretion into the bile
Copper Homeostasis

High-affinity copper transporter (Ctr1) : polytopicmembrane protein involved in the copper uptake at the hepatocyte plasma membrane
Metallothioneins (MT) : a group of cysteine-rich intracellular proteins capable of binding metal ions
Metallochaperones (ATOX1/HAH1, CCS, Cox17) : involved in the transfer of copper to specific cellular targets
Cu-ATPase : ATP7B, ATP7A
Copper transporters
P. Ferenci et al.Pathophysiology and clinical features of Wilson disease 2004;19:229-39

ATPase 7A (ATP7A)
In intestine, choroid plexus, vascular smooth
muscle, adrenal gland
ATPase 7B (ATP7B)
primarily expressed in the liver, and also in the
brain, kidney, placenta.
membrane protein located in the trans-Golgi involved in
incorporation of free copper into apoceruloplasmin and
in transporting the excess copper to secretory vesicles
for excretion into biliary canaliculi.
Copper transporters
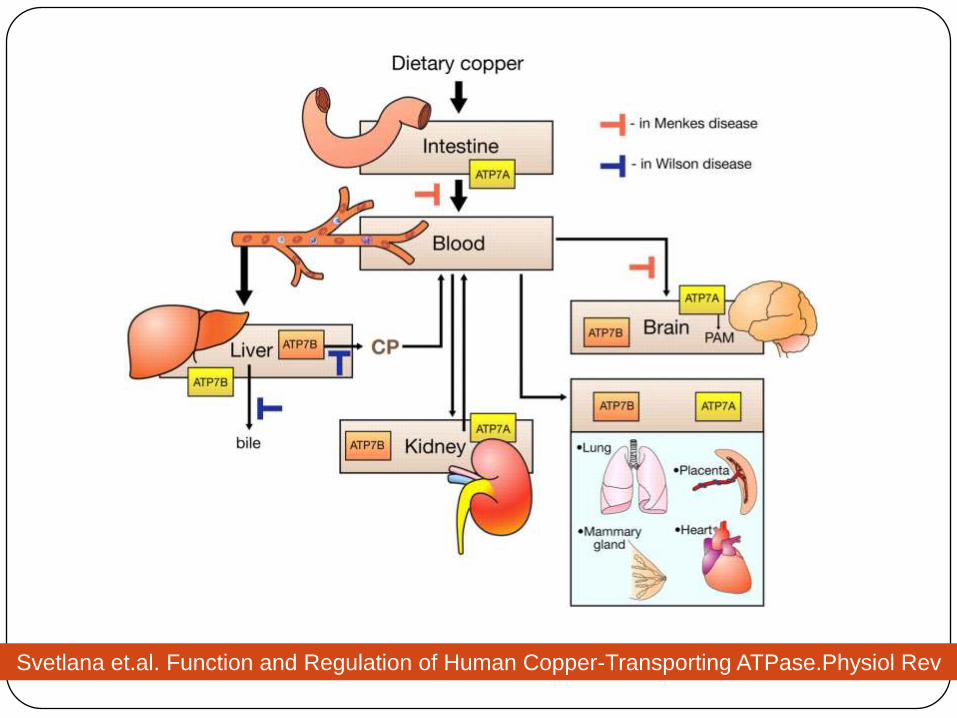
Svetlana et.al. Function and Regulation of Human Copper-Transporting ATPase.Physiol Rev
2007;87:1011-46

NML45
Intracellular pathways of copper distribution
Svetlana et.al. Function and Regulation of Human Copper-Transporting ATPase.Physiol Rev
2007;87:1011-46
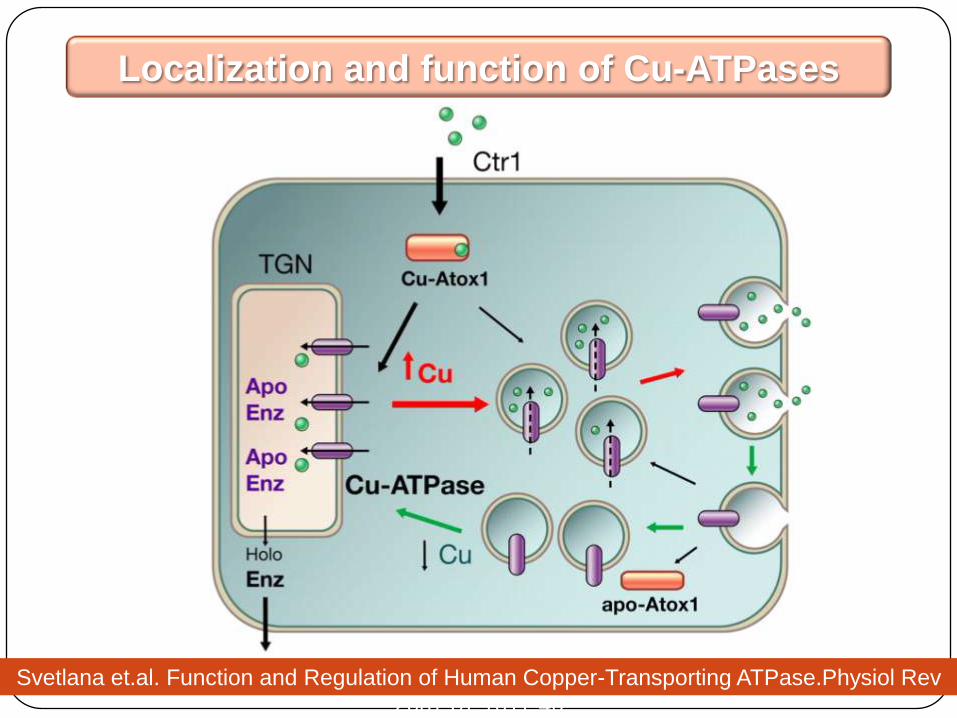
Localization and function of Cu-ATPases
Svetlana et.al. Function and Regulation of Human Copper-Transporting ATPase.Physiol Rev
2007;87:1011-46
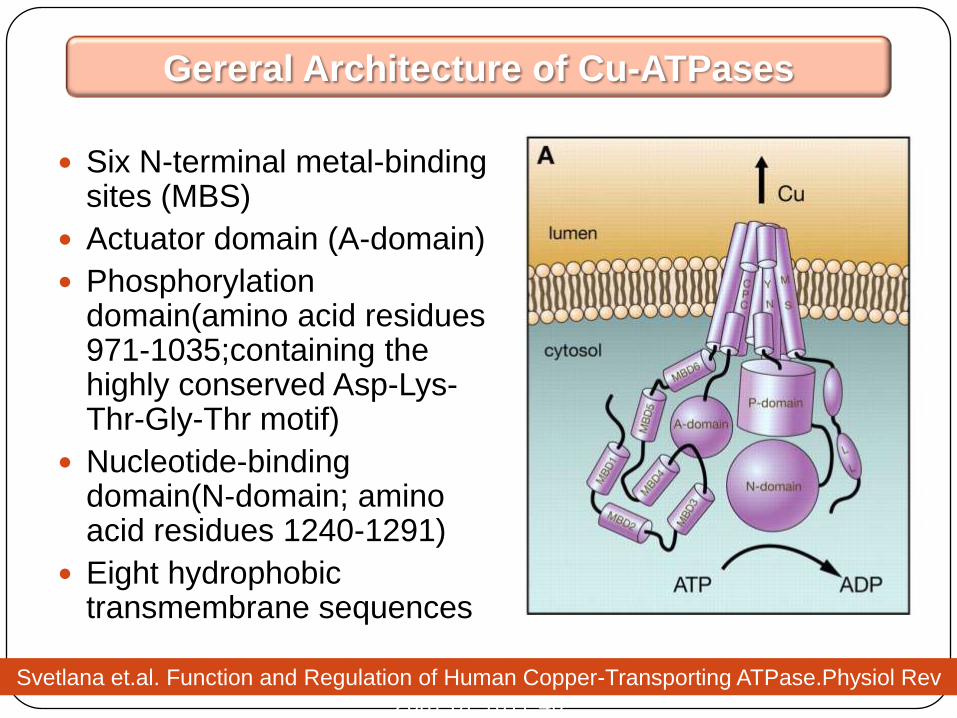
Six N-terminal metal-binding sites (MBS)
Actuator domain (A-domain)
Phosphorylationdomain(amino acid residues 971-1035;containing the highly conserved Asp-Lys-Thr-Gly-Thr motif)
Nucleotide-binding domain(N-domain; amino acid residues 1240-1291)
Eight hydrophobic transmembrane sequences
Gereral Architecture of Cu-ATPases
Svetlana et.al. Function and Regulation of Human Copper-Transporting ATPase.Physiol Rev
2007;87:1011-46
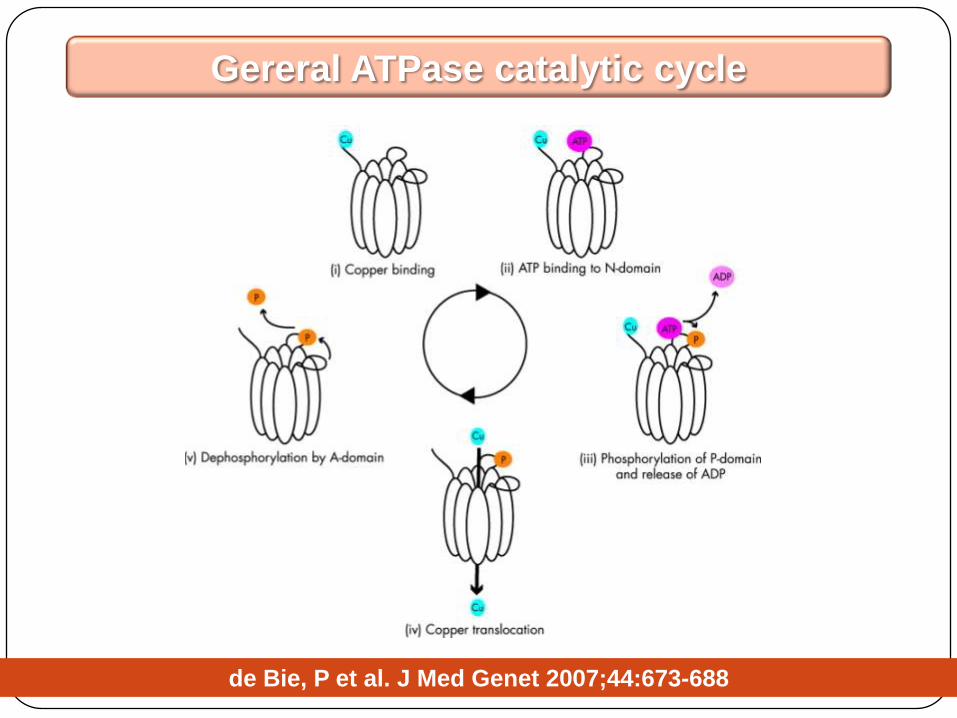
Gereral ATPase catalytic cycle
de Bie, P et al. J Med Genet 2007;44:673-688
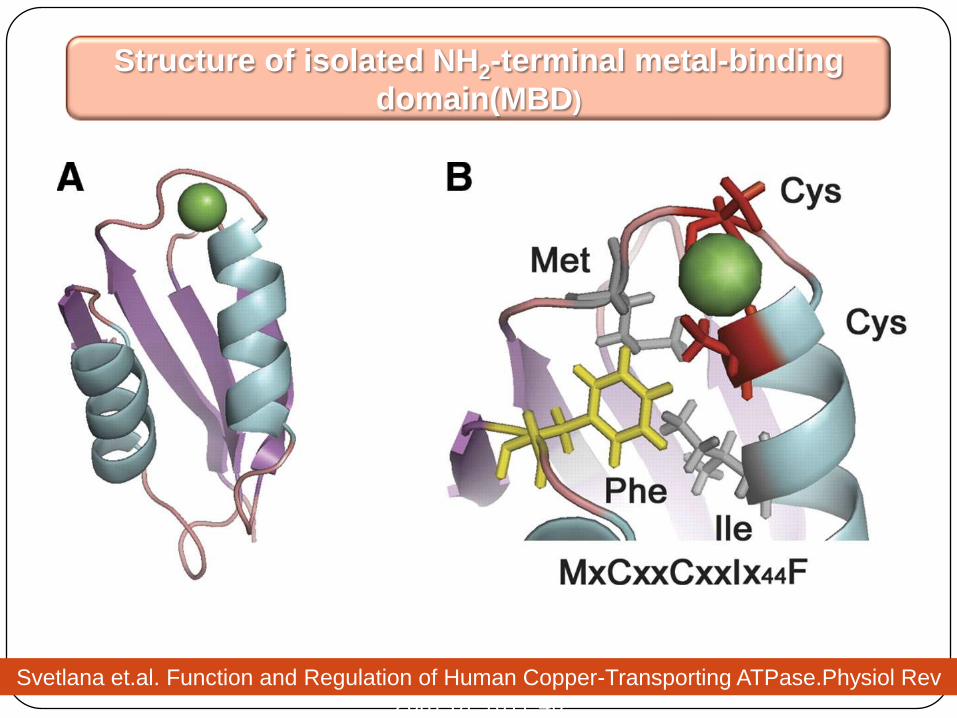
Structure of isolated NH2-terminal metal-binding
domain(MBD)
Svetlana et.al. Function and Regulation of Human Copper-Transporting ATPase.Physiol Rev
2007;87:1011-46
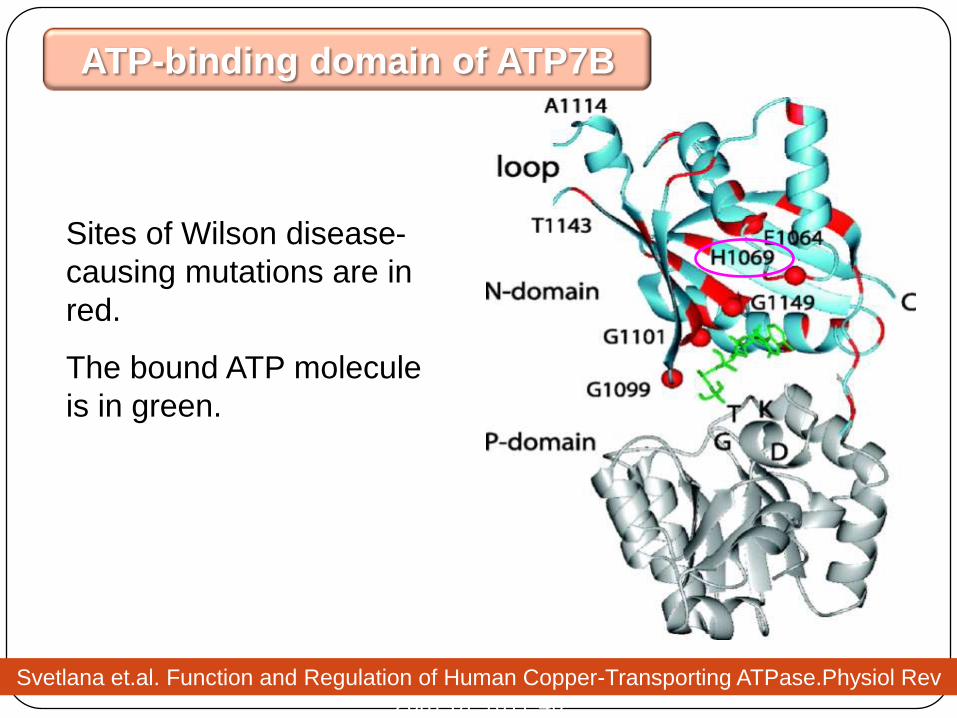
Sites of Wilson disease-
causing mutations are in
red.
The bound ATP molecule
is in green.
ATP-binding domain of ATP7B
Svetlana et.al. Function and Regulation of Human Copper-Transporting ATPase.Physiol Rev
2007;87:1011-46
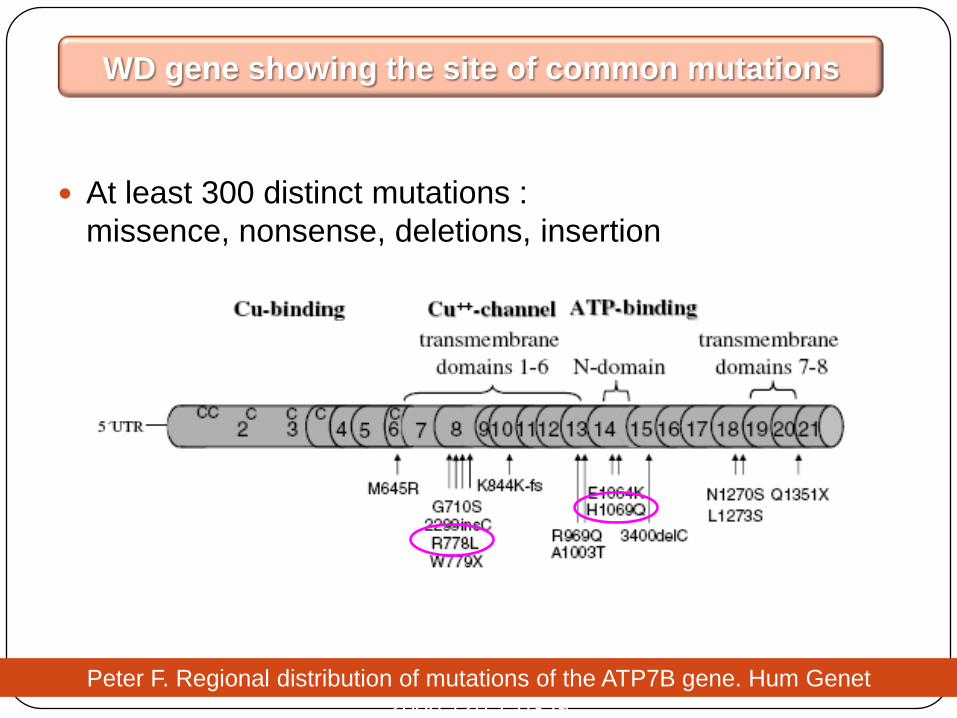
At least 300 distinct mutations :
missence, nonsense, deletions, insertion
WD gene showing the site of common mutations
Peter F. Regional distribution of mutations of the ATP7B gene. Hum Genet
2006;120:151-59

Genotype-to-phenotype correlations in WD are hampered by the high prevalence of compound heterozygotes and the relative paucity of homozygotes.
Studies in homozygotes suggest that mutations affecting critical portions of the protein, including copper-binding domains or the ATPase loop, may lead to early onset of hepatic disease, but strict concordance is difficult to prove.
In general, convincing genotype-phenotype correlations remain elusive.
Genotype-to-phenotype correlations in WD

In the genetically confirmed 92 WD patients,
Common mutations (allele frequency)
R778L (40%),
A874V and N1270S (8.7%)
L1083F and V872X (6.0%)
Fourteen (15%) WD patients were homozygous
and 46(50%) heterozygous for the R778L.
Mutation analysis of ATP7B in 114 WD patients
(SNUCH 2009)
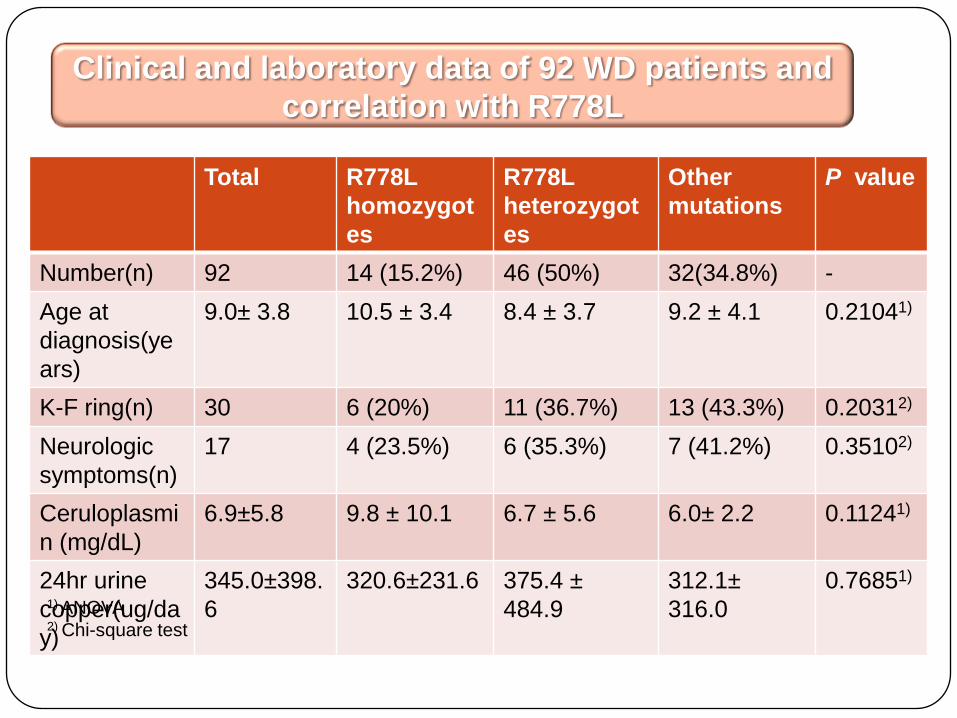
Total R778L
homozygot
es
R778L
heterozygot
es
Other
mutations
P value
Number(n) 92 14 (15.2%) 46 (50%) 32(34.8%) -
Age at
diagnosis(ye
ars)
9.0± 3.8 10.5 ± 3.4 8.4 ± 3.7 9.2 ± 4.1 0.21041)
K-F ring(n) 30 6 (20%) 11 (36.7%) 13 (43.3%) 0.20312)
Neurologic
symptoms(n)
17 4 (23.5%) 6 (35.3%) 7 (41.2%) 0.35102)
Ceruloplasmi
n (mg/dL)
6.9±5.8 9.8 ± 10.1 6.7 ± 5.6 6.0± 2.2 0.11241)
24hr urine
copper(ug/da
y)
345.0±398.
6
320.6±231.6 375.4 ±
484.9
312.1±
316.0
0.76851)
Clinical and laboratory data of 92 WD patients and
correlation with R778L
1) ANOVA2) Chi-square test

THANK YOU~~!!!




THANK YOU~~!!!


















