
The Role of NF-kB in TNF-Related Apoptosis-Inducing … · The Role of NF-kB in TNF-Related...
10
of June 7, 2018. This information is current as Apoptosis of Melanoma Cells (TRAIL)-Induced Apoptosis-Inducing Ligand B in TNF-Related κ The Role of NF- Nguyen and Peter Hersey Jayne E. Sanders, Xi Yi Zhang, Wayne D. Thomas, Tam Agustin V. Franco, Xu Dong Zhang, Elisabeth Van Berkel, http://www.jimmunol.org/content/166/9/5337 doi: 10.4049/jimmunol.166.9.5337 2001; 166:5337-5345; ; J Immunol References http://www.jimmunol.org/content/166/9/5337.full#ref-list-1 , 26 of which you can access for free at: cites 48 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved. Copyright © 2001 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on June 7, 2018 http://www.jimmunol.org/ Downloaded from by guest on June 7, 2018 http://www.jimmunol.org/ Downloaded from
Transcript of The Role of NF-kB in TNF-Related Apoptosis-Inducing … · The Role of NF-kB in TNF-Related...

of June 7, 2018.This information is current as
Apoptosis of Melanoma Cells(TRAIL)-InducedApoptosis-Inducing Ligand
B in TNF-RelatedκThe Role of NF-
Nguyen and Peter HerseyJayne E. Sanders, Xi Yi Zhang, Wayne D. Thomas, Tam Agustin V. Franco, Xu Dong Zhang, Elisabeth Van Berkel,
http://www.jimmunol.org/content/166/9/5337doi: 10.4049/jimmunol.166.9.5337
2001; 166:5337-5345; ;J Immunol
Referenceshttp://www.jimmunol.org/content/166/9/5337.full#ref-list-1
, 26 of which you can access for free at: cites 48 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2001 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 7, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on June 7, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Role of NF-kB in TNF-Related Apoptosis-Inducing Ligand(TRAIL)-Induced Apoptosis of Melanoma Cells1
Agustin V. Franco, Xu Dong Zhang, Elisabeth Van Berkel, Jayne E. Sanders, Xi Yi Zhang,Wayne D. Thomas, Tam Nguyen, and Peter Hersey2
Previous studies have shown that activation of NF-kB can inhibit apoptosis induced by a number of stimuli. It is also known thatTNF-related apoptosis-inducing ligand (TRAIL) can activate NF-kB through the death receptors TRAIL-R1 and TRAIL-R2, and decoyreceptor TRAIL-R4. In view of these findings, we have investigated the extent to which activation of NF-kB may account for the variableresponses of melanoma lines to apoptosis induced by TRAIL and other TNF family members. Pretreatment of the melanoma lines withthe proteasome inhibitorN-acetyl-L-leucinyl-L-leucinyl-L-norleucinal (LLnL), which is known to inhibit activation of NF- kB, was shownto markedly increase apoptosis in 10 of 12 melanoma lines with death receptors for TRAIL. The specificity of results for inhibition ofNF-kB activation was supported by an increase of TRAIL-induced apoptosis in melanoma cells transfected with a degradation-resistantIkBa. Furthermore, studies with NF-kB reporter constructs revealed that the resistance of melanoma lines to TRAIL-induced apoptosiswas correlated to activation of NF-kB in response to TRAIL. TRAIL-resistant sublines that were generated by intermittent exposureto TRAIL were shown to have high levels of activated NF-kB, and resistance to TRAIL could be reversed by LLnL and by thesuperrepressor form of IkBa. Therefore, these results suggest that activation of NF-kB by TRAIL plays an important role in resistanceof melanoma cells to TRAIL-induced apoptosis and further suggest that inhibitors of NF-kB may be useful adjuncts in clinical use ofTRAIL against melanoma. The Journal of Immunology,2001, 166: 5337–5345.
Several members of the TNF family have been shown toinduce apoptosis in susceptible cells by activation of thecaspase pathways (1–3). Induction of apoptosis appears to
be restricted to receptors that contain “death domains” such asthose for Fas (CD95) ligand (FasL).3 TNF-a, TNF-related apop-tosis-inducing ligand (TRAIL)/apo-2 (4, 5), and apo-3 ligand (apo-3L; Ref. 6). TRAIL appears to be particularly important, as itappears to be able to induce apoptosis in a wide range of neoplasticbut not normal cells (4). TRAIL can induce apoptosis by interac-tion with two receptors referred to as DR4 (TRAIL-R1) (7, 8) andDR5/TRAIL-R2 (9–11). These receptors were found to be widelyexpressed on normal tissues, but the latter are believed to be pro-tected from apoptosis by two additional receptors, TRAIL-R3/DcR1 (12–14) and TRAIL-R4/DcR2 (15, 16). The latter are be-lieved to inhibit apoptosis either by competition for binding ofTRAIL (e.g., acting as decoys) or by providing signals that inhibitapoptosis (3, 17). Activation of the transcription factor NF-kB maybe one of the main pathways involved in the latter, as it is knownto be activated by TRAIL-R4 (17) and, paradoxically, also bydeath receptors for TRAIL (8). NF-kB is known to regulate theexpression of several proteins that inhibit apoptosis, such as in-
hibitors of apoptosis (XIAP, IAP1, and IAP2) that block apoptosisby inhibition of caspase-8 (18) and caspase-3 and -9 (19) and theprotein A1, which is a Bcl-2 homologue (20).
Evidence for the importance of activation of NF-kB in resis-tance to apoptosis came from studies on leukemia cells in whichinhibition of NF-kB was associated with increased sensitivity toapoptosis induced by TNF-a and activation of CD95 and TRAIL(21). TRAIL-induced apoptosis of keratinocyte lines was inhibitedby IL-1, which was shown to activate NF-kB (22). Some mela-noma cells were shown to become sensitive to TNF-a when theywere transfected with a dominant-negative form of IkB, whichinhibited NF-kB activation (23). Similarly, inhibition of NF-kBwas found to sensitize chemoresistant tumors to TNF-a and che-motherapy (24) and to reduce the in vivo survival of human headand neck squamous cell carcinomas (25).
We have shown previously that TRAIL (26–28) but not othermembers of the TNF family (29, 30) can induce apoptosis in ap-proximately two-thirds of melanoma cell lines. There was markedvariability in response to TRAIL, which in some lines was relatedto loss of TRAIL receptor expression (27) but in other lines, thebasis for resistance to TRAIL was not clear. In the present study,we have assessed the role of NF-kB in susceptibility of melanomacells to apoptosis induced by TRAIL and other members of theTNF family by use of agents which inhibit NF-kB activity. Wealso have generated TRAIL-resistant melanoma cells by culture inTRAIL and show that activation of NF-kB appears to be a keytranscription factor involved in resistance to TRAIL but not otherTNF family members.
Materials and MethodsCell lines
Melanoma cell lines with the prefix Mel were isolated from fresh surgicalbiopsies from patients attending the Sydney and Newcastle MelanomaUnits and established in the laboratory. FH, CV, AT, and GL were fromlymph nodes. MC was from skin. RM, JG, and SP were from bowel. Thecell lines had been in culture for 2–6 mo at the time of these studies.
Department of Oncology and Immunology Unit, David Maddison Clinical SciencesBuilding, Newcastle, New South Wales, Australia
Received for publication April 12, 2000. Accepted for publication February 16, 2001.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby markedadvertisementin accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work supported by the Melanoma and Skin Cancer Research Institute, Sydney,Australia, and New South Wales State Cancer Council, New South Wales, Australia.2 Address correspondence and reprint requests to Dr. Peter Hersey, Department ofOncology and Immunology, Room 443, David Maddison Building, Corner of Kingand Watt Streets, Newcastle, NSW 2300, Australia. E-mail address: [email protected] Abbreviations used in this paper: FasL, Fas ligand; TRAIL, TNF-related apoptosis-inducing ligand; TRAIL-R, receptors for TRAIL; hu, human; DcR, decoy receptor forTRAIL; DR, death receptor for TRAIL; GFP, green fluorescent protein; LLnL,N-acetyl-Leu-Leu-norleucinal; IAP, inhibitors of apoptosis.
Copyright © 2001 by The American Association of Immunologists 0022-1767/01/$02.00
by guest on June 7, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
MM200, Me1007, Me10538, Me4405, and IgR3 were from primary mel-anoma. The derivation of MM200, Me1007, Me10538, and Me4405, aredescribed elsewhere (27, 30). SK-MEL-110 and SK-MEL-28 were a kindgift from Dr. A. Albino (American Health Foundation, Valhalla, NY) andS. Ralph (Department of Biochemistry and Molecular Biology, MonashUniversity, Victoria, Australia), respectively. All melanoma cell lines werepositive for tyrosinase and MART-1 mRNA by RT-PCR tests describedelsewhere (31), except for Mel-SP, which was positive for Tyrosinase butnot MART-1. All cell lines were cultured in DMEM containing 5% FCS(Commonwealth Serum Laboratories, Melbourne, Victoria, Australia).
Plasmid vectors and transfectants
A superrepressor form of human IkBa protein S32A/36A resistant to deg-radation (in the vector PRC/CMV) was kindly provided by SteveGerondakis (Walter and Eliza Hall Institute, Melbourne Victoria, Austra-lia) (32). Stable and transient transfectants were conducted by electropo-ration by a Gene Pulser (Bio-Rad, Hercules, CA) as follows: MM200, 0.36kV and 960mF; Mel-RM, 0.33 kV and 500mF; Mel-FH, 0.36 kV and 500mF; SK-MEL-28, 0.32 kV and 500mF; Mel-CV, 0.36 kV and 500mF; andMe4405, 0.30 kV and 500mF. Twenty-four hours after electroporation,G418 was added to a final concentration of 400mg/ml for 21 days or untilcolonies appeared on the plate. For the analysis of NF-kB activation intransient transfectants, the cells were cotransfected with 30mg of pNF-kB-d2EGFP, a reporter vector encoding the green fluorescent protein(GFP) under the control of thek enhancer element (Clontech, Palo Alto,
CA) for 24 h. Fluorescence was measured by flow cytometry, as mentionedin Materials and Methods. To determine the efficiency of transfection, thecells were cotransfected with 30mg of a vector encoding the LacZ cDNA,and percentage of blue cells were estimated 24 h after transfection.
In some experiments, the mutant IkBa-containing vector at 30mgDNA/2,000,000 cells in 400ml was cotransfected with pEG FP-N1GFPvector (Clontech) at 1.5mg of DNA. Control cultures were transfected withthe GFP vector at 30mg of DNA/2,000,000 cells.
Monoclonal Abs, recombinant proteins, and peptides
Recombinant human (hu) TRAIL (lot 6321-19) prepared as described else-where (4) and huCD40L (lot 5753-56) were supplied by Immunex (Seattle,WA). Each preparation was supplied as a leucine zipper fusion protein,which required no further cross-linking for maximal activity. RecombinanthuFasL, produced from isolated cDNA (GenBank accession no. U08137)in vector pDC409 and transfected into COS cells, was kindly supplied assterile supernatants by Immunex. It produced 50% lysis of Jurkat T cells atdilutions of 2–4 (30). rTNF-a cytokines and control mAb antitrinitrophe-nyl (anti-TNP; IgG1) were purchased from BD PharMingen (Bioclone,Marrickville, Australia). Control mAb 1D4.5 (Ig2a) was against an Ag onSalmonellae (kindly supplied by L. Ashman, Hansen Cancer ResearchCentre, Adelaide, Australia). The MAbs against TRAIL-R1 (IgG2ahuTR1-M271; lot: 7136-07), TRAIL-R2 (IgG1 huTRAIL-R2-M413; lot:5274-96), TRAIL-R3 (IgG1 huTR3-M430; lot: 7313-217), and TRAIL-R4(IgG1 huTR4-M444; lot: 7136-15) were supplied by Immunex and aredescribed elsewhere (33). The rabbit Abs against p65 were raised againstthe C-terminal end peptide SGDEDFSSIADMDFSALLSQIS and anti-p75against the C-terminal end peptide MMTTSSDSMGETDNPRLLSM, andwere purchased from Stressgen (Victoria, British Columbia, Canada). Therabbit Ab against p50 was purchased from Fitzgerald (Concord, MA). Thecontrol purified rabbit IgG (prod. no. 15006) was purchased from Sigma(St. Louis, MO). The proteasome inhibitor (calpain inhibitor 1)N-acetyl-Leu-Leu-norleucinal (LLnL) (34) was purchased from Sigma (cat. no.Ab2185). Lactacystin (35) was purchased from ICN Biochemicals (Bio-medicals Australasia, Seven Hills, New South Wales, Australia).
Flow cytometry
Analysis was conducted with a Facscan flow cytometer (Becton Dickinson,Mountain View, CA) . In studies on relocalization of TRAIL receptors,melanoma cells were grown overnight in 24-well plates and exposed toTRAIL 100 ng/ml for 30 min. Adherent cells were removed by trypsiniza-tion in 0.25% trypsin at 37°C for 5 min, washed twice in cold DMEM andonce in PBS at 4°C, and fixed in 4% paraformaldehyde. Appropriate con-centrations of mAbs were added to the cells in 100ml of PBS containing20% human A serum and incubated for 7 min at room temperature. Cellswere washed either twice with PBS and analyzed if directly labeled, or ifindirectly labeled, cells the were incubated with F(ab9)2 affinity-isolatedFITC-conjugated sheep anti-mouse Ig (Silenus, cat. no. 985051020; AmradBiotech, Boronia, Victoria, Australia) plus 20ml of 100% human serum to
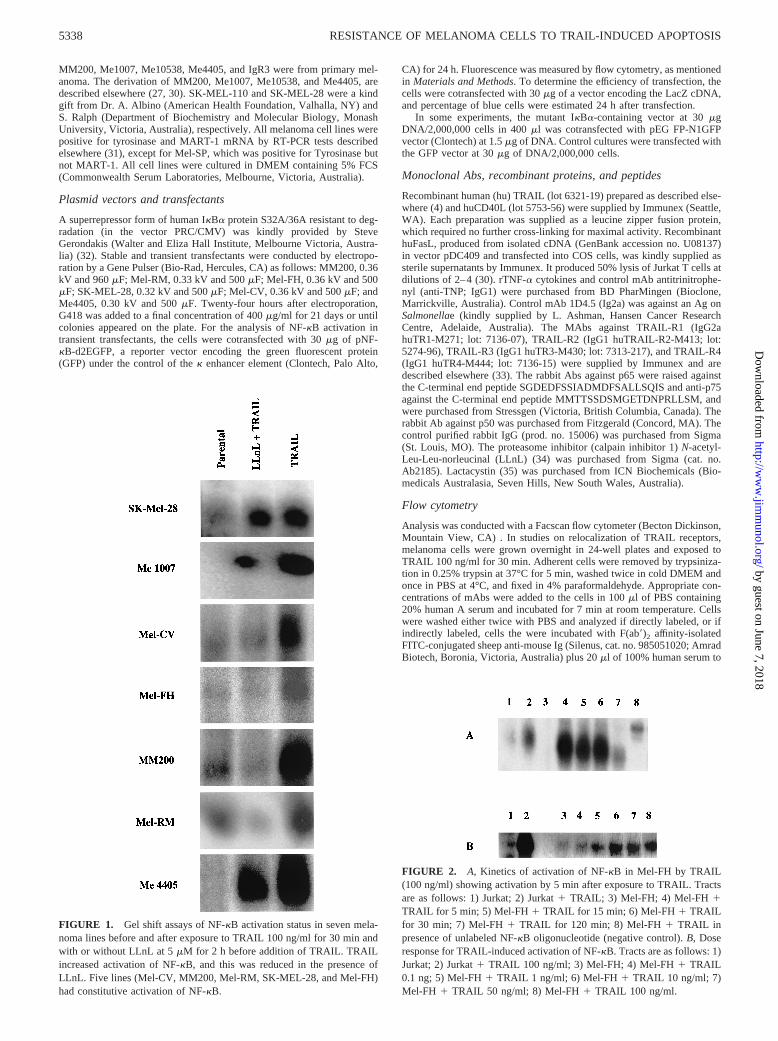
FIGURE 1. Gel shift assays of NF-kB activation status in seven mela-noma lines before and after exposure to TRAIL 100 ng/ml for 30 min andwith or without LLnL at 5mM for 2 h before addition of TRAIL. TRAILincreased activation of NF-kB, and this was reduced in the presence ofLLnL. Five lines (Mel-CV, MM200, Mel-RM, SK-MEL-28, and Mel-FH)had constitutive activation of NF-kB.
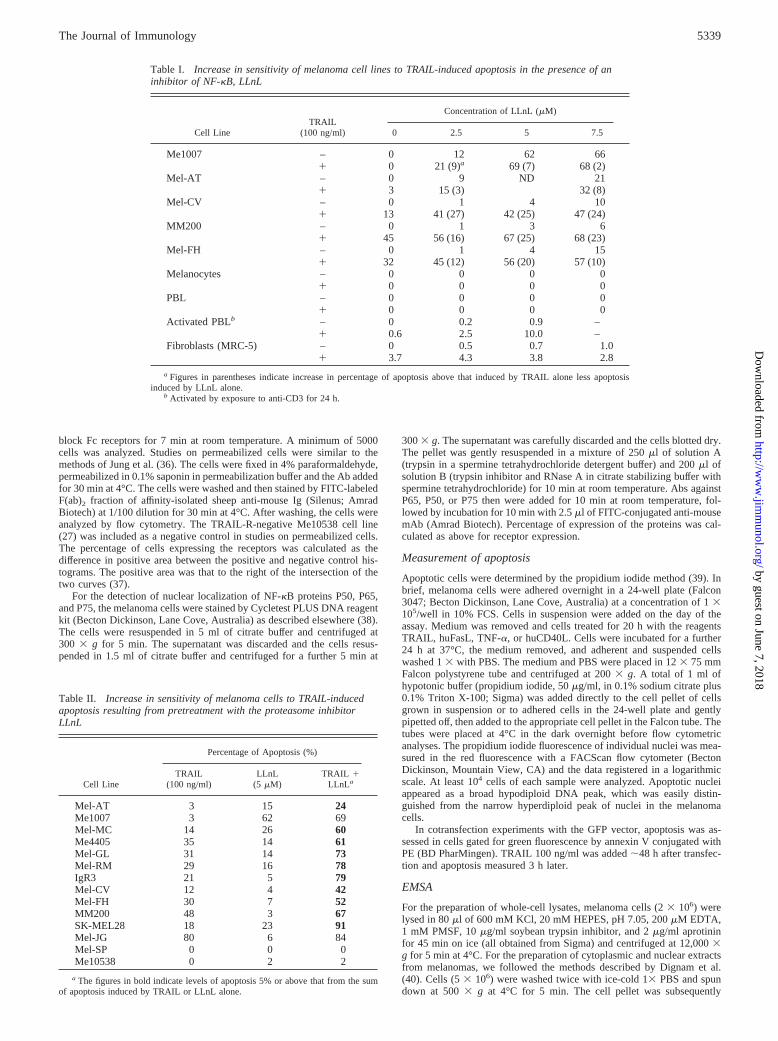
FIGURE 2. A, Kinetics of activation of NF-kB in Mel-FH by TRAIL(100 ng/ml) showing activation by 5 min after exposure to TRAIL. Tractsare as follows: 1) Jurkat; 2) Jurkat1 TRAIL; 3) Mel-FH; 4) Mel-FH 1TRAIL for 5 min; 5) Mel-FH 1 TRAIL for 15 min; 6) Mel-FH1 TRAILfor 30 min; 7) Mel-FH1 TRAIL for 120 min; 8) Mel-FH1 TRAIL inpresence of unlabeled NF-kB oligonucleotide (negative control).B, Doseresponse for TRAIL-induced activation of NF-kB. Tracts are as follows: 1)Jurkat; 2) Jurkat1 TRAIL 100 ng/ml; 3) Mel-FH; 4) Mel-FH1 TRAIL0.1 ng; 5) Mel-FH1 TRAIL 1 ng/ml; 6) Mel-FH1 TRAIL 10 ng/ml; 7)Mel-FH 1 TRAIL 50 ng/ml; 8) Mel-FH1 TRAIL 100 ng/ml.
5338 RESISTANCE OF MELANOMA CELLS TO TRAIL-INDUCED APOPTOSIS
by guest on June 7, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
block Fc receptors for 7 min at room temperature. A minimum of 5000cells was analyzed. Studies on permeabilized cells were similar to themethods of Jung et al. (36). The cells were fixed in 4% paraformaldehyde,permeabilized in 0.1% saponin in permeabilization buffer and the Ab addedfor 30 min at 4°C. The cells were washed and then stained by FITC-labeledF(ab)2 fraction of affinity-isolated sheep anti-mouse Ig (Silenus; AmradBiotech) at 1/100 dilution for 30 min at 4°C. After washing, the cells wereanalyzed by flow cytometry. The TRAIL-R-negative Me10538 cell line(27) was included as a negative control in studies on permeabilized cells.The percentage of cells expressing the receptors was calculated as thedifference in positive area between the positive and negative control his-tograms. The positive area was that to the right of the intersection of thetwo curves (37).
For the detection of nuclear localization of NF-kB proteins P50, P65,and P75, the melanoma cells were stained by Cycletest PLUS DNA reagentkit (Becton Dickinson, Lane Cove, Australia) as described elsewhere (38).The cells were resuspended in 5 ml of citrate buffer and centrifuged at300 3 g for 5 min. The supernatant was discarded and the cells resus-pended in 1.5 ml of citrate buffer and centrifuged for a further 5 min at
3003 g. The supernatant was carefully discarded and the cells blotted dry.The pellet was gently resuspended in a mixture of 250ml of solution A(trypsin in a spermine tetrahydrochloride detergent buffer) and 200ml ofsolution B (trypsin inhibitor and RNase A in citrate stabilizing buffer withspermine tetrahydrochloride) for 10 min at room temperature. Abs againstP65, P50, or P75 then were added for 10 min at room temperature, fol-lowed by incubation for 10 min with 2.5ml of FITC-conjugated anti-mousemAb (Amrad Biotech). Percentage of expression of the proteins was cal-culated as above for receptor expression.
Measurement of apoptosis
Apoptotic cells were determined by the propidium iodide method (39). Inbrief, melanoma cells were adhered overnight in a 24-well plate (Falcon3047; Becton Dickinson, Lane Cove, Australia) at a concentration of 13105/well in 10% FCS. Cells in suspension were added on the day of theassay. Medium was removed and cells treated for 20 h with the reagentsTRAIL, huFasL, TNF-a, or huCD40L. Cells were incubated for a further24 h at 37°C, the medium removed, and adherent and suspended cellswashed 13 with PBS. The medium and PBS were placed in 123 75 mmFalcon polystyrene tube and centrifuged at 2003 g. A total of 1 ml ofhypotonic buffer (propidium iodide, 50mg/ml, in 0.1% sodium citrate plus0.1% Triton X-100; Sigma) was added directly to the cell pellet of cellsgrown in suspension or to adhered cells in the 24-well plate and gentlypipetted off, then added to the appropriate cell pellet in the Falcon tube. Thetubes were placed at 4°C in the dark overnight before flow cytometricanalyses. The propidium iodide fluorescence of individual nuclei was mea-sured in the red fluorescence with a FACScan flow cytometer (BectonDickinson, Mountain View, CA) and the data registered in a logarithmicscale. At least 104 cells of each sample were analyzed. Apoptotic nucleiappeared as a broad hypodiploid DNA peak, which was easily distin-guished from the narrow hyperdiploid peak of nuclei in the melanomacells.
In cotransfection experiments with the GFP vector, apoptosis was as-sessed in cells gated for green fluorescence by annexin V conjugated withPE (BD PharMingen). TRAIL 100 ng/ml was added;48 h after transfec-tion and apoptosis measured 3 h later.
EMSA
For the preparation of whole-cell lysates, melanoma cells (23 106) werelysed in 80ml of 600 mM KCl, 20 mM HEPES, pH 7.05, 200mM EDTA,1 mM PMSF, 10mg/ml soybean trypsin inhibitor, and 2mg/ml aprotininfor 45 min on ice (all obtained from Sigma) and centrifuged at 12,0003g for 5 min at 4°C. For the preparation of cytoplasmic and nuclear extractsfrom melanomas, we followed the methods described by Dignam et al.(40). Cells (53 106) were washed twice with ice-cold 13PBS and spundown at 5003 g at 4°C for 5 min. The cell pellet was subsequently
Table I. Increase in sensitivity of melanoma cell lines to TRAIL-induced apoptosis in the presence of aninhibitor of NF-kB, LLnL
Cell LineTRAIL
(100 ng/ml)
Concentration of LLnL (mM)
0 2.5 5 7.5
Me1007 – 0 12 62 661 0 21 (9)a 69 (7) 68 (2)
Mel-AT – 0 9 ND 211 3 15 (3) 32 (8)
Mel-CV – 0 1 4 101 13 41 (27) 42 (25) 47 (24)
MM200 – 0 1 3 61 45 56 (16) 67 (25) 68 (23)
Mel-FH – 0 1 4 151 32 45 (12) 56 (20) 57 (10)
Melanocytes – 0 0 0 01 0 0 0 0
PBL – 0 0 0 01 0 0 0 0
Activated PBLb – 0 0.2 0.9 –1 0.6 2.5 10.0 –
Fibroblasts (MRC-5) – 0 0.5 0.7 1.01 3.7 4.3 3.8 2.8
a Figures in parentheses indicate increase in percentage of apoptosis above that induced by TRAIL alone less apoptosisinduced by LLnL alone.
b Activated by exposure to anti-CD3 for 24 h.
Table II. Increase in sensitivity of melanoma cells to TRAIL-inducedapoptosis resulting from pretreatment with the proteasome inhibitorLLnL
Cell Line
Percentage of Apoptosis (%)
TRAIL(100 ng/ml)
LLnL(5 mM)
TRAIL 1LLnLa
Mel-AT 3 15 24Me1007 3 62 69Mel-MC 14 26 60Me4405 35 14 61Mel-GL 31 14 73Mel-RM 29 16 78IgR3 21 5 79Mel-CV 12 4 42Mel-FH 30 7 52MM200 48 3 67SK-MEL28 18 23 91Mel-JG 80 6 84Mel-SP 0 0 0Me10538 0 2 2
a The figures in bold indicate levels of apoptosis 5% or above that from the sumof apoptosis induced by TRAIL or LLnL alone.
5339The Journal of Immunology
by guest on June 7, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

homogenized in 200ml of Dignam buffer A containing protease inhibitorsas described above. The homogenate was spun down, and the nuclear pelletwas washed twice with buffer A. The nuclear pellet then was resuspendedin 30 ml of Dignam buffer C. A total of 10mgrams of supernatant wasincubated with 4ml of 53 gel shift binding buffer (Promega, Madison, WI)for 30 min at room temperature to double-stranded oligonucleotides en-coding the consensus binding sites of NF-kB (59-AGTTGAGGGGACTTTCCCAGGC-39) that had been labeled with [32P]ATP (GeneWorks, Ad-elaide, Australia) by polynucleotide kinase (Promega). For supershiftassays, Abs against P50 and P65 were incubated at room temperature for30 min with the melanoma lysates in the presence of the radiolabeled oligo.Protein-oligo complexes were separated on a nondenaturing 6% PAGE in0.53 Tris-borate-EDTA buffer. Gels were dried and exposed overnight toan x-ray film. Included in each assay was a positive control of extracts fromthe Jurkat T cell line exposed to TRAIL and a negative control of unlabeledNF-kB and random AP2 oligonucleotide included in the Promega NF-kBgel shift assay system (cat. no. E3300).
ResultsInhibition of TRAIL-induced activation of NF-kB by LLnL
The proteasome inhibitor LLnL was shown in previous studies toinhibit the degradation of IkB and thereby to prevent activation ofNF-kB (21, 34). To determine whether LLnL inhibited activationof NF-kB in melanoma cells, we used EMSA to measure NF-kBactivation in the melanoma lines before and after activation byTRAIL and the effect of LLnL on this activation. As shown in Fig.1, enhanced NF-kB binding activity induced by TRAIL was shownin seven of the melanoma lines selected for study. ConstitutiveNF-kB activation was found in five cell lines, SK-MEL-28, Mel-CV, Mel-FH, MM200, and Mel-RM, before addition of TRAIL(Fig. 1). LLnL (5 mM) added 2 h before TRAIL (and throughoutculture with TRAIL) reduced the binding to barely detectable lev-els, except in extracts from Me4405 that still had relatively highlevels of activated NF-kB after pretreatment with LLnL. The levelof NF-kB activation shown in the gel shift assays were consistentwith the levels shown by NF-kB reporter assays shown below. Asshown in Fig. 2A, NF-kB activation occurred within 5 min and was
maximal from 15 to 60 min after exposure to TRAIL. NF-kBactivation was maximal at a concentration of 10 ng/ml (Fig. 2B).This is less than the optimal concentration of 100 ng/ml shown forTRAIL-induced apoptosis (26). Similar results were found in stud-ies on the Mel-CV line.
The proteasome inhibitor LLnL that inhibits activation of NF-kBcan reverse resistance to TRAIL-induced apoptosis
We then examined whether LLnL would inhibit TRAIL-inducedapoptosis of melanoma cells in studies on a large number of mel-anoma lines. The dose titration studies shown in Table I indicatethat two of the lines, Me1007 and Mel-AT, showed marked sen-sitivity to LLnL alone, and there was only a small increase inapoptosis induced by TRAIL in the presence of LLnL. Three mel-anoma lines that were partially sensitive to TRAIL-induced apo-ptosis had more marked increases in sensitivity to TRAIL in thepresence of LLnL that approached maximum levels in the assay. Italso was noted that treatment of melanocytes, fibroblasts, and rest-ing PBL with even high concentrations of LLnL did not makethem sensitive to TRAIL-induced apoptosis. However, there was asmall increase in sensitivity of PBL activated by anti-CD3 toTRAIL-induced apoptosis.
These studies were repeated at a fixed concentration of LLnL (5mM) in a wider panel of melanoma cells, as shown in Table II.Again, there was wide variation in toxicity of LLnL between linesbut in most lines there was a marked increase in TRAIL-inducedapoptosis in the presence of LLnL, the exceptions being the controlcell lines Me10538 and Mel-SP, which did not have death recep-tors for TRAIL (27). The TRAIL-sensitive line Mel-JG was max-imally killed by TRAIL alone, and no further increase was inducedby LLnL. Studies with a second proteasome inhibitor, lactacystin(35), at 10mM on the cell lines Me1007, Mel-CV, Mel-FH, andMM200 gave a similar increase in TRAIL-induced apoptosis (datanot shown).
Inhibition of NF-kB with LLnL has minimal effects on the levelof apoptosis induced by other TNF family members
In view of the increased sensitivity of melanoma cells to TRAIL-induced apoptosis in melanoma cells exposed to LLnL, we alsoexamined whether activation of NF-kB may be involved in resis-tance of melanoma cells to TNF-a, FasL, and CD40L. Six celllines that showed a wide range of sensitivity to TRAIL-inducedapoptosis were pretreated with LLnL 3 h before addition ofTRAIL, FasL, TNF-a, and CD40L in optimum concentrations es-tablished in previous studies (26, 29, 30). The results shown inTable III indicate that LLnL did not sensitize the melanoma cells toapoptosis induced by TNF-a or CD40L but did potentiate low levelsof apoptosis induced by FasL in the cell line Mel-CV. In contrast,LLnL increased apoptosis induced by TRAIL in all but the controlTRAIL-R-negative line Me10538. It was of interest that FasL induced
FIGURE 3. Gel shift assay of NF-kB activation status in two melanomalines showing activation of NF-kB in SK-MEL-28 cells by CD40L, FasL,and TRAIL, and in Mel-FH cells by FasL and TRAIL. TRAIL 100 ng/mlwas added 30 min before the assay.
Table III. The proteasome inhibitor LLnL does not increase apoptosis in melanoma cells induced by other TNF family membersa
Cell Line LLnL TRAILLLnL 1TRAIL FasL
LLnL 1FasL % Fas TNF-a
LLnL 1TNF-a
% TNF-R
CD40LLLnL 1CD40L % CD4021 22
Mel-CV 2 11 66b 3 10 83 0 8 21 18 0 5 63Mel-FH 7 20 46 1 7 34 1 6 17 0 0 8 9MM200 3 48 70 2 9 20 0 4 12 11 0 3 12SK-MEL-28 3 23 47 0 2 8 0 3 9 0 0 0 13Me10538 2 0 2 19 21 6 4 6 9 11 0 3 8Mel-JG 2 73 86 2 5 0 0 2 2 0 0 2 0
a LLnL was used at 2.5mM, TRAIL and TNF-a at 100 ng/ml, and CD40L at 300 ng/ml. FasL was used at a dilution of 1:100.b The figures in bold indicate levels of apoptosis 5% or above the sum of apoptosis due to LLnL or TRAIL alone.
5340 RESISTANCE OF MELANOMA CELLS TO TRAIL-INDUCED APOPTOSIS
by guest on June 7, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

low levels of apoptosis in the latter and this was not increased byLLnL. All of the cell lines expressed receptors for the other membersof the TNF family except the TRAIL-sensitive line Mel-JG. The re-sults shown are representative of two experiments.
To determine whether NF-kB was activated by other membersof the TNF family, NF-kB activation was measured in gel-shiftassays on two of the cell lines (SK-MEL-28 and Mel-FH) that didnot undergo apoptosis after exposure to the TNF family members.As shown in Fig. 3, NF-kB was activated by FasL and TRAIL inboth melanoma lines and by CD40L in the SK-MEL-28 line, butNF-kB activation was not associated with apoptosis. However, theresults show that the receptors were expressed at sufficient levelsto be functionally active.
Cytoplasmic and nuclear location of activated NF-kB
To further substantiate the gel shift assays of NF-kB activation weexamined the site of the activated NF-kB proteins in the melanomacells by studies on extracts from the nucleus and cytoplasm of thecells that were prepared as described elsewhere (40). As shown inFig. 4A, TRAIL induced a typical increase in NF-kB activation inextracts from the nuclei of Mel-RM cells and there were onlyrelatively small amounts in the cytoplasm. The specificity of theprobe for the NF-kB proteins also was also shown by the super-shift electromobility assays with Abs to p50 and p65 NF-kB pro-teins, illustrated in Fig. 4B.
The levels of TRAIL-induced activation of NF-kB measured inreporter assays are higher in TRAIL-resistant compared withTRAIL-sensitive cell lines
In view of the variable levels of NF-kB activation in the mel-anoma lines before and after exposure to TRAIL shown in Fig.1, we used more quantitative NF-kB reporter assays to examinethe relation between NF-kB activation and TRAIL-induced ap-optosis. Transient transfection with the NF-kB reporter vectorpNF-kB-d2EGFP was possible in eight of nine melanoma linesbut was not possible in the Me4405 line. The results shown inTable IV havebeen arranged in order of the sensitivity of the celllines to TRAIL and %GFP levels have been adjusted to take intoaccount the variable transfection efficiency. The relatively resistantcell lines Mel-FH and Me1007 had low basal NF-kB levels, but aftertreatment with TRAIL, the levels of NF-kB increased by 73 and166% respectively (Table IV). IgR3, Mel-RM, and SK-MEL-28 hadmoderate sensitivity to TRAIL and moderately high basal levels ofNF-kB, which was increased further by TRAIL. In contrast, theTRAIL-sensitive lines, Mel-JG and MM200, had high basal levels ofNF-kB that were relatively unchanged after treatment with TRAIL.Mel-CV also had high basal levels and relatively low levels ofTRAIL-induced activation of NF-kB but remained relatively resistantto TRAIL-induced apoptosis. Essentially similar results were found ina repeat of the study, with the exception of Mel-CV. These results
FIGURE 4. A, Gel shift assay of acti-vated NF-kB protein binding in extracts ofthe cytoplasm and nuclei of Mel-RM. Af-ter exposure to TRAIL some degradationand increased mobility of NF-kB proteinswas evident in cytoplasmic extracts but therewas, as expected, a marked increase inNF-kB protein binding in the nucleus afterexposure to TRAIL.B,Supershift gel assaysof extracts from Mel-RM with Abs againstp50 and p65. This confirms that the proteinsbound to the probe are NF-kB proteins.
Table IV. Sensitivity of melanoma cells to TRAIL-induced apoptosis appears inversely related to the level of TRAIL-induced NF-kB activationmeasured by GFP reporter assays
Cell LineTRAIL
(100 ng/ml) % Apoptosis
NF-kBActivation %Expression of
GFPa MFIb
CorrectedValues %
Expression ofGFPc
% ChangeInduced by
TRAILd % Transfection
Mel-JG – 2.1 10.2 1.61 93 111 85 10.7 1.65 97 4 11
MM200 – 3.2 54.0 1.39 81 671 61.6 57.8 1.46 86 6 67
IgR3 – 1.6 37 2.03 69 531 42 47 2.29 88 28 53
Mel-RM – 2.2 33 1.16 75 441 31.4 41 1.29 93 24 44
Mel-FH – 3.1 17.7 1.1 50.6 351 23.0 30.6 1.31 87.4 73 35
SK-MEL28 – 1.6 16.5 1.12 71.7 231 17.9 22.3 1.21 96.9 35 23
Mel-CV – 2.6 57.1 1.12 84.0 681 11.8 66.0 1.25 97.0 16 68
Me1007 – 0 3 1.1 33 91 4 8 1.28 88 166 9
a Percentage of cells expressing GFP by flow cytometry.b MFI, Median fluorescent intensity.c Values corrected for percentage of transfection by dividing by percentage of transfection. The latter was assessed by percentage of blue/blue1 white colonies.d Percentage of change induced by TRAIL was calculated by dividing the increase in GFP by the basal GFP level.
5341The Journal of Immunology
by guest on June 7, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
suggest a reciprocal relation between the TRAIL-induced activationof NF-kB and TRAIL-induced apoptosis of melanoma. Regressionanalysis of percentage of apoptosis vs percentage of increase in GFPexpression, including all the results in Table IV, showed a significantinverse relationship (y5 54.02 0.393 r2 5 0.55;p 5 0.03.).
A superrepressor form of IkBa increases TRAIL-inducedapoptosis in melanoma cells
The findings above that TRAIL-induced apoptosis was increasedby treatment with LLnL did not prove that the increase was at-tributable to inhibition of NF-kB activation, as proteasome inhib-itors also inhibit the breakdown of other proteins that may be in-volved in apoptosis, such as p53 (41). In view of this, wetransfected a mutated degradation-resistant form of IkBa into themelanoma cells that specifically inhibits activation of NF-kB bybinding the proteins in the cytoplasm. Transient transfection of thedegradation-resistant IkBa was conducted in two cell lines,
Mel-FH and Mel-RM. The levels of IkBa in the transfectants wereconfirmed by FACS with anti-IkBa (data not shown). As shown inTable V, TRAIL-induced apoptosis was increased in each of themelanoma lines to levels comparable to that seen with the protea-some inhibitor LLnL. This was accompanied by decreased bindingof p50, p65, and p75 to the nuclei of Mel-FH and Mel-RM, shownby flow cytometry on isolated nuclei. Inhibition of NF-kB activa-tion also was shown in NF-kB GFP reporter assays on Mel-RMand Mel-FH in that there was a decrease in %GFP expression afterexposure to TRAIL in the IkBa-transfected melanoma cells butnot in the control cells transfected with the vector alone.
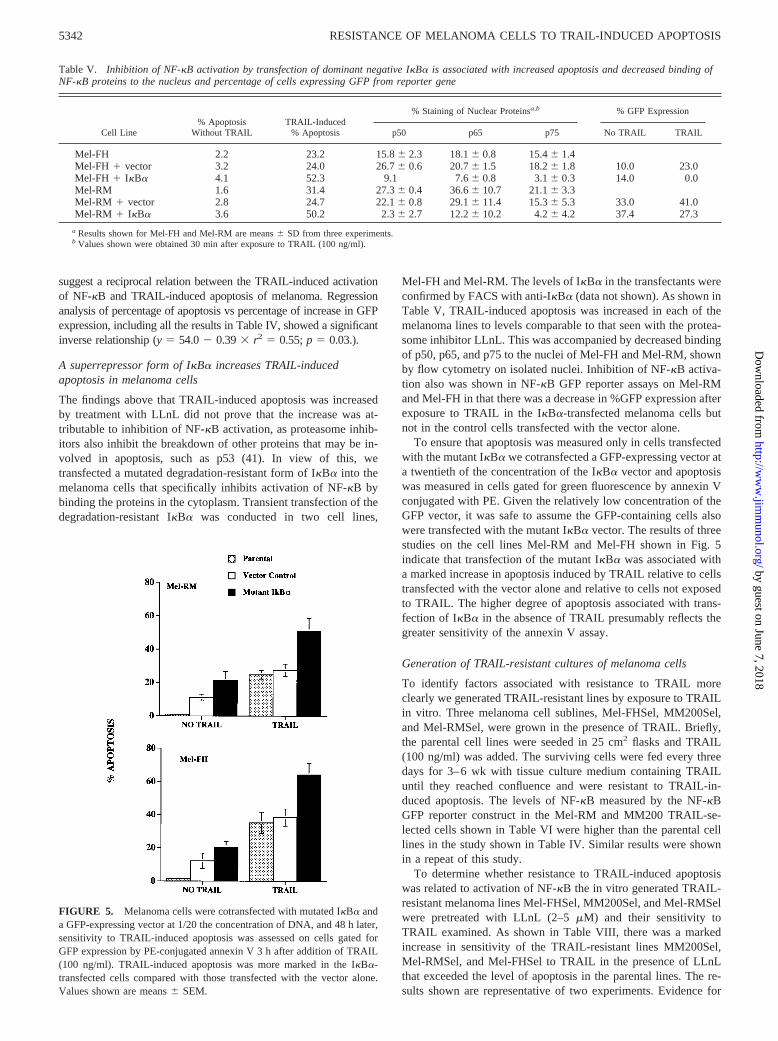
To ensure that apoptosis was measured only in cells transfectedwith the mutant IkBa we cotransfected a GFP-expressing vector ata twentieth of the concentration of the IkBa vector and apoptosiswas measured in cells gated for green fluorescence by annexin Vconjugated with PE. Given the relatively low concentration of theGFP vector, it was safe to assume the GFP-containing cells alsowere transfected with the mutant IkBa vector. The results of threestudies on the cell lines Mel-RM and Mel-FH shown in Fig. 5indicate that transfection of the mutant IkBa was associated witha marked increase in apoptosis induced by TRAIL relative to cellstransfected with the vector alone and relative to cells not exposedto TRAIL. The higher degree of apoptosis associated with trans-fection of IkBa in the absence of TRAIL presumably reflects thegreater sensitivity of the annexin V assay.
Generation of TRAIL-resistant cultures of melanoma cells
To identify factors associated with resistance to TRAIL moreclearly we generated TRAIL-resistant lines by exposure to TRAILin vitro. Three melanoma cell sublines, Mel-FHSel, MM200Sel,and Mel-RMSel, were grown in the presence of TRAIL. Briefly,the parental cell lines were seeded in 25 cm2 flasks and TRAIL(100 ng/ml) was added. The surviving cells were fed every threedays for 3–6 wk with tissue culture medium containing TRAILuntil they reached confluence and were resistant to TRAIL-in-duced apoptosis. The levels of NF-kB measured by the NF-kBGFP reporter construct in the Mel-RM and MM200 TRAIL-se-lected cells shown in Table VI were higher than the parental celllines in the study shown in Table IV. Similar results were shownin a repeat of this study.
To determine whether resistance to TRAIL-induced apoptosiswas related to activation of NF-kB the in vitro generated TRAIL-resistant melanoma lines Mel-FHSel, MM200Sel, and Mel-RMSelwere pretreated with LLnL (2–5mM) and their sensitivity toTRAIL examined. As shown in Table VIII, there was a markedincrease in sensitivity of the TRAIL-resistant lines MM200Sel,Mel-RMSel, and Mel-FHSel to TRAIL in the presence of LLnLthat exceeded the level of apoptosis in the parental lines. The re-sults shown are representative of two experiments. Evidence for
FIGURE 5. Melanoma cells were cotransfected with mutated IkBa anda GFP-expressing vector at 1/20 the concentration of DNA, and 48 h later,sensitivity to TRAIL-induced apoptosis was assessed on cells gated forGFP expression by PE-conjugated annexin V 3 h after addition of TRAIL(100 ng/ml). TRAIL-induced apoptosis was more marked in the IkBa-transfected cells compared with those transfected with the vector alone.Values shown are means6 SEM.
Table V. Inhibition of NF-kB activation by transfection of dominant negative IkBa is associated with increased apoptosis and decreased binding ofNF-kB proteins to the nucleus and percentage of cells expressing GFP from reporter gene
Cell Line% Apoptosis
Without TRAILTRAIL-Induced
% Apoptosis
% Staining of Nuclear Proteinsa,b % GFP Expression
p50 p65 p75 No TRAIL TRAIL
Mel-FH 2.2 23.2 15.86 2.3 18.16 0.8 15.46 1.4Mel-FH 1 vector 3.2 24.0 26.76 0.6 20.76 1.5 18.26 1.8 10.0 23.0Mel-FH 1 IkBa 4.1 52.3 9.1 7.66 0.8 3.16 0.3 14.0 0.0Mel-RM 1.6 31.4 27.36 0.4 36.66 10.7 21.16 3.3Mel-RM 1 vector 2.8 24.7 22.16 0.8 29.16 11.4 15.36 5.3 33.0 41.0Mel-RM 1 IkBa 3.6 50.2 2.36 2.7 12.26 10.2 4.26 4.2 37.4 27.3
a Results shown for Mel-FH and Mel-RM are means6 SD from three experiments.b Values shown were obtained 30 min after exposure to TRAIL (100 ng/ml).
5342 RESISTANCE OF MELANOMA CELLS TO TRAIL-INDUCED APOPTOSIS
by guest on June 7, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
the specificity of these results for inhibition of NF-kB was ob-tained by transient transfection of the TRAIL-resistant MM200Seland Mel-RMSel lines with the degradation-resistant IkBa (66 and48% transfection efficiency, respectively). As shown in Table VII,the cells transfected with the latter had increased sensitivity toTRAIL-induced apoptosis that was similar to that seen in mela-noma cells treated with LLnL. When the transfection efficiency isallowed for, the percentage of apoptosis levels induced by TRAILapproximates that in the lines pretreated with LLnL. The resultswere similar in a repeat of the study and confirm that activation ofNF-kB was responsible in part for the resistance of the cell lines toTRAIL-induced apoptosis.
DiscussionOur previous studies (26–28) and those by others (33, 42) showthat TRAIL appears to be particularly active in induction of apo-ptosis in melanoma cells whereas other members of the TNF fam-ily had little or no such activity (26, 29, 30). Nevertheless, wefound previously that there was a large variation in response ofdifferent melanoma cell lines to TRAIL. This was accounted for inpart by the level of expression of the TRAIL death receptors, R1and R2, on melanoma cells but some cell lines were resistant toTRAIL despite relatively high levels of TRAIL-R expression (27).A number of studies have shown that TRAIL-R not only activatesthe caspase pathway leading to apoptosis but also transmits signalsthat activate the transcription factor NF-kB (8, 17). This was con-firmed in the present studies by gel shift assays, GFP reporterconstructs for NF-kB, and nuclear staining with Abs specific forp50, p65, and p75. NF-kB is known to activate a number of genescoding for proteins that inhibit apoptosis by the transmembranesignaling pathway (18, 43) and mitochondrial stress pathway (20,
44). To investigate the possible role of NF-kB in resistance toTRAIL-induced apoptosis, we used an inhibitor, LLnL, that blocksthe ability of proteasomes to degrade IkB. Therefore, IkB remainsbound to NF-kB in the cytoplasm and prevents its relocation to thenucleus. A mutant form of IkBa that was not degraded by protea-somes also was used to confirm the specificity of the effects forinhibition of the activation of NF-kB.
The results indicated that LLnL reversed the resistance toTRAIL-induced apoptosis in 10 of 12 melanoma lines with deathreceptors for TRAIL. Two lines did not show an increase in sen-sitivity to TRAIL in the presence of LLnL. One (Me10538) did notexpress TRAIL-R (27) and another line, Me1007, was difficult tointerpret because it underwent cell death in the presence of LLnLalone. The latter result was similar to studies on Hodgkin’s lym-phoma cells that found that proliferation and viability was depen-dent on activation of NF-kB in the cells (45). Another line, Mel-JG, was very sensitive to TRAIL-induced apoptosis alone, andLLnL could not further increase this sensitivity. These results sug-gest that NF-kB activation is a significant factor in protectionagainst TRAIL-induced apoptosis of melanoma, as reported byothers in studies on lymphoid cells (21) and other cancers (24, 25).In contrast to the increased sensitivity to TRAIL induced by LLnL,the latter did not increase the sensitivity of the melanoma lines toapoptosis induced by CD40L, TNF-a, or FasL, the exception beingsmall increases in sensitivity of one line to FasL.
However, proteasome inhibitors are not specific for NF-kB andare responsible for degradation of proteins involved in cell cycleregulation (41), which might have indirect effects on induction ofapoptosis. Therefore, we transfected melanoma cells with a deg-radation-resistant form of IkBa that binds NF-kB proteins in thecytoplasm and thereby specifically inhibits activation of NF-kB. In
Table VI. TRAIL-selected resistant melanoma cells express high levels of NF-kB activation measured inGFP reporter assays
Cell LineTRAIL
(100 ng/ml) % Apoptosis
NF-kBActivation %Expression of
GFPCorrectedValuesa
% ChangeInduced by
TRAIL % Transfection
MM200 – 3.2 54 81 6 671 61.6 57.8 86 67
MM200Sel – 2.8 67.2 97 691 6.5 69.0 100.0 2.5 69
Mel-RM – 2.2 33 75 441 31.4 41 93 24 44
Mel-RMSel – 3.0 39.7 84.5 471 4.5 43.6 89.0 7 49
a Values corrected for percentage of transfection by dividing by percentage of transfection. The latter was assessed bypercentage of blue/blue1 white colonies.
Table VII. Inhibition of NF-kB activation by proteasome inhibitors and by transfection of mutated IkBaincreases TRAIL-induced apoptosis of parental and TRAIL-selected resistant melanoma sublines
Cell Line
Percentage of TRAIL-Induced Apoptosis
TRAIL Alone TRAIL 1 LLnLa
TRAIL 1 cellstransfected with
vector alone
TRAIL 1 cellstransfected withmutated IkBa
MM200 49.0 86.5 48.6 74.2MM200Sel 1.0 53.7 3.8 21Mel-RM 24.3 42.9 24.7 50.2Mel-RMSel 3.4 55.8 8.0 19Mel-FH 32 53 24.2 52.3Mel-FHSel 6 62 16.4 ND
a LLnL was used at 2.5mM and added 2 h prior to TRAIL. Apoptosis levels induced by LLnL alone were,6%.b Apoptosis levels were,5% in the transfected cells in the absence of TRAIL.
5343The Journal of Immunology
by guest on June 7, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

some experiments, cotransfection of a GFP expression vector wasused to identify the transfected cells. These studies showed that thetransfected melanoma cells had increased sensitivity to TRAIL-induced apoptosis and decreased binding of NF-kB proteins in thenucleus.
NF-kB reporter assays indicated that some melanoma had highconstitutive levels of NF-kB activation but this was not necessarilyrelated to resistance to TRAIL-induced apoptosis. Instead, it ap-peared from the NF-kB reporter assays that resistance to apoptosiscorrelated more with the increase in the level of NF-kB activationafter exposure to TRAIL. This was shown particularly in studieson Mel-FH and Me1007 and in studies on melanoma lines that hadbeen selected for resistance to TRAIL, such as Mel-RMSel.
Melanoma cells with high constitutive levels of NF-kB activa-tion (Mel-JG and MM200) underwent a small increase in NF-kBactivation after exposure to TRAIL and were relatively sensitive toTRAIL-induced apoptosis. High constitutive levels of activatedNF-kB in melanoma were reported previously to be associatedwith the level of oxidative radicals in the cells (46) or autocrineproduction of IL-1, which is known to activate NF-kB (47). Athird possible mechanism was accelerated decay of IkB in mela-noma cells allowing NF-kB proteins to enter the nucleus (48).Reasons for the sensitivity of melanoma cells to TRAIL-inducedapoptosis despite the presence of high constitutive levels of acti-vated NF-kB remain the subject of ongoing studies.
Studies on melanoma lines made resistant to TRAIL by pro-longed culture in TRAIL indicated that NF-kB appeared to beactivated to a greater extent in the TRAIL-selected resistant linescompared with the parental line. This was consistent with themarked increases in sensitivity to TRAIL-induced apoptosis in theTRAIL-resistant sublines after treatment with LLnL to inhibit NF-kB. Transfection of the degradation-resistant IkB into the cellsalso resulted in an increase in TRAIL-induced apoptosis but not tothe same level as seen with the proteasome inhibitor. The latterresults may reflect the transfection efficiency or indicate that fac-tors other than those involved in activation of NF-kB are involvedin resistance of the cells to TRAIL-induced apoptosis.
Some insights into how activation of NF-kB may inhibit apo-ptosis resulting from signals triggered from the same receptor wasderived from kinetic studies, which suggested that NF-kB wasactivated much earlier than the caspase pathway and at lower con-centrations of TRAIL; e.g., activation of NF-kB was evident by 5min and at concentrations of 1 ng/ml. This compares with reportedactivation of caspase-8 30 min after exposure to TRAIL (42) andpeak activation of the main effector, caspase-3, by 2–4 h (49) andat concentrations of;100 ng (26). These possible differences inkinetics of the two pathways needs to be confirmed by studies onthe induction of NF-kB proteins involved in inhibition of apoptosissuch as the Bcl-2 homologue A1 and members of the IAP family.If confirmed, these findings might suggest that TRAIL may onlyinduce apoptosis when present at high concentrations and that lowconcentrations may act to increase resistance to apoptosis.
In summary, these studies on melanoma lines and TRAIL-se-lected lines support the view that mechanisms dependent on acti-vation of NF-kB play a key role in resistance of melanoma cells toTRAIL-induced apoptosis and suggest that inhibitors of NF-kBmay be a valuable adjunct to treatment of patients with TRAIL.Further studies on fresh melanoma tissue and studies in vivo areneeded to support these conclusions.
References1. Nagata, S. 1997. Apoptosis by death factor.Cell 88:355.2. Lincz, L. F. 1998. Deciphering the apoptotic pathway: all roads lead to death.
Immunol. Cell Biol. 76:1.
3. Griffith, T. S., and D. H. Lynch. 1998. TRAIL: a molecule with multiple receptorsand control mechanisms.Curr. Opin. Immunol. 10:559.
4. Wiley, S. R., K. Schooley, P. J. Smolak, W. S. Din, C.-P. Huang, J. K. Nicholl,G. R. Sutherland, T. D. Smith, C. Rauch, C. A. Smith, and R. G. Goodwin. 1995.Identification and characterization of a new member of the TNF family thatinduces apoptosis.Immunity 3:673.
5. Pitti, R. M., S. A. Marsters, S. Ruppert, C. J. Donahue, A. Moore, andA. Ashkenazi. 1996. Induction of apoptosis by Apo-2 ligand, a new member ofthe tumor necrosis factor cytokine family.J. Biol. Chem. 271:12687.
6. Marsters, S. A., J. P. Sheridan, R. M. Pitti, J. Brush, A. Goddard, andA. Ashkenazi. 1998. Identification of a ligand for the death-domain-containingreceptor Apo3.Curr. Biol. 8:525.
7. Pan, G., K. O’Rourke, A. M. Chinnaiyan, R. Gentz, R. Ebner, J. Ni, andV. M. Dixit. 1997. The receptor for the cytotoxic ligand TRAIL.Science 276:111.
8. Schneider, P., M. Thome, K. Burns, J. L. Bodmer, K. Hofmann, T. Kataoka,N. Holler, and J. Tschopp. 1997. TRAIL receptors 1 (DR4) and 2 (DR5) signalFADD-dependent apoptosis and activate NF-kB. Immunity 7:831.
9. Pan, G., J. Ni, Y.-F. Wei, G. Yu, R. Gentz, and V. M. Dixit. 1997. An antagonistdecoy receptor and a death domain-containing receptor for TRAIL.Science 277:815.
10. Walczak, H., M. A. Degli-Esposti, R. S. Johnson, P. J. Smolak, J. Y. Waugh,N. Boiani, M. S. Timour, M. J. Gerhart, K. A. Schooley, C. A. Smith,R. G. Goodwin, and C. T. Rauch. 1997. TRAIL-R2: a novel apoptosis-mediatingreceptor for TRAIL.EMBO J. 16:5386.
11. Screaton, G. R., J. Mongkolsapaya, X.-N. Xu, A. E. Cowper, A. J. McMichael,and J. I. Bell. 1997. TRICK2, a new alternatively spliced receptor that transducesthe cytotoxic signal from TRAIL.Curr. Biol. 7:693.
12. Degli-Esposti, M. A., P. J. Smolak, H. Walczak, J. Waugh, C.-P. Huang,R. F. DuBose, R. G. Goodwin, and C. A. Smith. 1997. Cloning and character-ization of TRAIL-R3, a novel member of the emerging TRAIL receptor family.J. Exp. Med. 186:1165.
13. Sheridan, J. P., S. A. Marsters, R. M. Pitti, A. Gurney, M. Skubatch, D. Baldwin,L. Ramakrishnan, C. L. Gray, K. Baker, W. I. Wood, et al. 1997. Control ofTRAIL-induced apoptosis by a family of signaling and decoy receptors.Science277:818.
14. Schneider, P., J. L. Bodmer, M. Thome, K. Hofmann, N. Holler, and J. Tschopp.1997. Characterization of two receptors for TRAIL.FEBS Lett. 416:329.
15. Pan, G., J. Ni, G. Yu, Y. F. Wei, and V. M. Dixit. 1998. TRUNDD, a newmember of the TRAIL receptor family that antagonizes TRAIL signalling.FEBSLett. 424:41.
16. Marsters, S. A., J. P. Sheridan, R. M. Pitti, A. Huang, M. Skubatch, D. Baldwin,J. Yuan, A. Gurney, A. D. Goddard, P. Godowski, and A. Ashkenazi. 1997. Anovel receptor for Apo2L/TRAIL contains a truncated death domain.Curr. Biol.7:1003.
17. Degli-Esposti, M. A., W. C. Dougall, P. J. Smolak, J. Y. Waugh, C. A. Smith, andR. G. Goodwin. 1997. The novel receptor TRAIL-R4 induces NF-kB and protectsagainst TRAIL-mediated apoptosis, yet retains an incomplete death domain.Im-munity 7:813.
18. Wang, C.-Y., M. W. Mayo, R. G. Korneluk, D. V. Goeddel, and A. S. Baldwin,Jr. 1998. NF-kB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 andc-IAP2 to suppress caspase-8 activation.Science 281:1680.
19. Deveraux, Q. L., and J. C. Reed. 1999. IAP family proteins: suppressors ofapoptosis.Genes Dev. 13:239.
20. Zong, W. X., L. C. Edelstein, C. Chen, J. Bash, and C. Gelinas. 1999. Theprosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-kBthat blocks TNFa-induced apoptosis.Genes Dev. 13:382.
21. Jeremias, I., C. Kupatt, B. Baumann, I. Herr, T. Wirth, and K. M. Debatin. 1998.Inhibition of nuclear factorkB activation attenuates apoptosis resistance in lym-phoid cells.Blood 91:4624.
22. Kothny-Wilkes, G., D. Kulms, B. Poppelmann, T. A. Luger, M. Kubin, andT. Schwarz. 1998. Interleukin-1 protects transformed keratinocytes from tumornecrosis factor-related apoptosis-inducing ligand.J. Biol. Chem. 273:29247.
23. Bakker, T. R., D. Reed, T. Renno, and C. V. Jongeneel. 1999. Efficient adenoviraltransfer of NF-kB inhibitor sensitizes melanoma to tumor necrosis factor-medi-ated apoptosis.Int. J. Cancer 80:320.
24. Wang, C.-Y., J. C. Cusack, R. Liu, and A. S. Baldwin. 1999. Control of induciblechemoresistance: enhanced anti-tumor therapy through increased apoptosis byinhibition of NF-kB. Nat. Med. 5:412.
25. Duffey, D. C., Z. Chen, G. Dong, F. G. Ondrey, J. S. Wolf, K. Brown,U. Siebenlist, and C. Van Waes. 1999. Expression of a dominant-negative mutantinhibitor-kB in human head and neck squamous cell carcinoma inhibits survival,proinflammatory cytokine expression, and tumor growth in vivo.Cancer Res.59:3468.
26. Thomas, W. D., and P. Hersey. 1998. TNF-related apoptosis-inducing ligand(TRAIL) induces apoptosis in Fas Ligand-resistant melanoma cells and mediatesCD4 T cell killing of target cells.J. Immunol. 161:2195.
27. Zhang, X., A. Franco, K. Myers, C. Gray, T. Nguyen, and P. Hersey. 1999.Relation of TRAIL receptor and FLIP expression to TRAIL induced apoptosis ofmelanoma.Cancer Res. 59:2747.
28. Zhang, X. D., A. V. Franco, T. Nguyen, and P. Hersey. 2000. Differential local-ization and regulation of death and decoy receptors for TNF-related apoptosis-inducing ligand (TRAIL) in human melanoma cells.J. Immunol. 164:3961.
29. Thomas, W. D., M. J. Smith, Z. Si, and P. Hersey. 1996. Expression of theco-stimulatory molecule CD40 on melanoma cells.Int. J. Cancer 68:795.
30. Thomas, W. D., and P. Hersey. 1998. CD4 T cells kill melanoma cells by mech-anisms that are independent of Fas (CD95).Int. J. Cancer 75:1.
5344 RESISTANCE OF MELANOMA CELLS TO TRAIL-INDUCED APOPTOSIS
by guest on June 7, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

31. Curry, B. J., K. Myers, and P. Hersey. 1998. Polymerase chain reaction detectionof melanoma cells in the circulation: relation to clinical stage, surgical treatment,and recurrence from melanoma.J. Clin. Oncol. 16:1760.
32. Whiteside, S. T., M. K. Ernst, O. LeBail, C. Laurent-Winter, N. Rise, andA. Israel. 1995. N- and C-terminal sequences control degradation of MAD3/IkBa in response to inducers of NF-kB activity. Mol. Cell Biol. 15:5339.
33. Griffith, T. S., C. Rauch, P. J. Smolak, J. Y. Waugh, N. Boiani, D. H. Lynch,C. A. Smith, R. G. Goodwin, and M. Z. Kubin. 1999. Functional analysis ofTRAIL receptors using monoclonal antibodies.J. Immunol. 162:2597.
34. Palombella, V. J., O. J. Rando, A. L. Goldberg, and T. Maniatis. 1994. Theubiquitin-proteasome pathway is required for processing the NF-kB1 precursorprotein and the activation of NF-kB. Cell 78:773.
35. Fenteany, G., R. F. Standaert, W. S. Lane, S. Choi, E. J. Corey, andS. L. Schreiber. 1995. Inhibition of proteasome activities and subunit-specificamino-terminal threonine modification by lactacystin.Science 268:726.
36. Jung, T., U. Schauer, C. Heusser, C. Neumann, and C. Rieger. 1993. Detectionof intracellular cytokines by flow cytometry.J. Immunol. Methods 159:197.
37. Sharrow, C. O. 1996. Analysis of flow cytometry data. InCurrent Protocols inImmunology, Section 5.2.2. J. E. Coligan, A. M. Kruisbeek, D. H. Margulies,E. M. Shevach, and W. Strober, eds. John Wiley & Sons, New York.
38. Foulds, S. 1997. Novel flow cytometric method for quantifying nuclear bindingof the transcription factor nuclear factorkB in unseparated human monocytes andpolymorphonuclear cells.Cytometry 29:182.
39. Nicoletti, I., G. Migliorati, M. C. Pagliacci, F. Grignani, and C. Riccardi. 1991.A rapid and simple method for measuring thymocyte apoptosis by propidiumiodide staining and flow cytometry.J. Immunol. Methods 139:271.
40. Dignam, J. D., R. M. Lebovitz, and R. G. Roeder. 1983. Accurate transcriptioninitiation by RNA polymerase II in a soluble extract from isolated mammaliannuclei.Nucleic Acids Res. 11:1475.
41. Adams, J., V. J. Palombella, E. A. Sausville, J. Johnson, A. Destree,D. D. Lazarus, J. Maas, C. S. Pien, S. Prakash, and P. J. Elliott. 1999. Proteasome
inhibitors; a novel class of potent and effective antitumor agents.Cancer Res.59:2615.
42. Griffith, T. S., W. A. Chin, G. C. Jackson, D. H. Lynch, and M. Z. Kubin. 1998.Intracellular regulation of TRAIL-induced apoptosis in human melanoma cells.J. Immunol. 161:2833.
43. Opipari, A. W. Jr., H. M. Hu, R. Yabkowitz, and V. M. Dixit. 1992. The A20 zincfinger protein protects cells from tumor necrosis factor cytotoxicity.J. Biol.Chem. 267:12424.
44. Deveraux, Q. L., N. Roy, H. R. Stennicke, T. Van Arsdale, Q. Zhou,S. M. Srinivasula, E. S. Alnemri, G. S. Salvesen, and J. C. Reed. 1998. IAPsblock apoptotic events induced by caspase-8 and cytochromec by direct inhibi-tion of distinct caspases.EMBO J. 17:2215.
45. Bargou, R. C., F. Emmerich, D. Krappmann, K. Bommert, M. Y. Mapara,W. Arnold, H. D. Royer, E. Grinstein, A. Greiner, C. Scheidereit, and B. Dorken.1997. Constitutive nuclear factor-kB-RelA activation is required for proliferationand survival of Hodgkin’s disease tumor cells.J. Clin. Invest. 100:2961.
46. Meyskens, F. L. Jr., J. A. Buckmeier, S. E. McNulty, and N. B. Tohidian. 1999.Activation of nuclear factor-kB in human metastatic melanoma cells and theeffect of oxidative stress.Clin. Cancer Res. 5:1197.
47. Kobayashi, H., T. Kokubo, K. Sato, S. Kimura, K. Asano, H. Takahashi,H. Iizuka, N. Miyokawa, and M. Katagiri. 1998. CD41 T cells from peripheralblood of a melanoma patient recognize peptides derived from nonmutated ty-rosinase.Cancer Res. 58:296.
48. Shattuck-Brandt, R. L., and A. Richmond. 1997. Enhanced degradation of I-kBacontributes to endogenous activation of NF-kB in Hs294T melanoma cells.Can-cer Res.57:3032.
49. Zhang, X. D., T. Nguyen, W. D. Thomas, J. E. Sanders, and P. Hersey. 2000.Mechanisms of resistance of normal cells to TRAIL-induced apoptosis vary be-tween different cell types.FEBS Lett. 482:193.
5345The Journal of Immunology
by guest on June 7, 2018http://w
ww
.jimm
unol.org/D
ownloaded from


















![Antiproliferative and Apoptosis-inducing Effects of Abrus … · 2019-01-22 · of apoptosis in the development of therapeutic agents for treating cancer[8]. The homeostasis in eukaryotic](https://static.fdocuments.net/doc/165x107/5e7531a7e0b7db69cc34d0b0/antiproliferative-and-apoptosis-inducing-effects-of-abrus-2019-01-22-of-apoptosis.jpg)