The Nature of Membrane Curvature-Induction by Amphipathic α-Helices Relies upon Protein Length:...
1
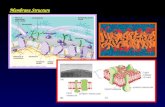
1194-Plat The Nature of Membrane Curvature-Induction by Amphipathic a-Helices Relies upon Protein Length: Simulations of a-Synuclein and H0 Anthony R. Braun 1 , Eva Sevcsik 2 , Elizabeth Rhoades 2 , Stephanie Tristram-Nagle 3 , Jonathan N. Sachs 1 . 1 University of Minnesota, Minneapolis, MN, USA, 2 Yale University, New Haven, CT, USA, 3 Carnegie Mellon University, Pittsburgh, PA, USA. Remodeling of membranes, for example the induction of membrane curvature, is a necessary step in vesicle fusion and fission. One mechanism for curvature induction relies upon the interfacial binding of amphipathic proteins, which act as a wedge in one leaflet of the membrane. In this study, we explore differences between the membrane remodeling effects of two biologically important am- phipathic proteins, specifically a-Synuclein (aS) and the N-BAR helix H0. aS is an intrinsically disordered protein that adopts an amphipathic a-helical structure upon binding a membrane. Recently it has been shown that aS can induce tubulation and vesiculation of vesicles (Varkey et al. 2010, J Biol Chem, 285(42):32486). In contrast, the amphipathic helix H0 is unable, on its own, to induce tubulation, as N-BAR relies instead upon its large scaffolding domain (Fernandes et al. 2008, Biophys J, 94(8):3065). Using a combination of x-ray scattering and coarse-grained molecular dynamics simulations, we ex- plored the membrane remodeling effects of aS. Our findings suggest that aS 1) causes a significant thinning of the bilayer; and 2) stabilizes an anisotropic curvature-field of both positive mean curvature and negative Gaussian curva- ture. Simulations of H0 show a similar magnitude of the local curvature as with aS, however owing to its molecular length the curvature-field around H0 is isotropic. We propose that the difference in curvature-field anisotropy ex- plains the difference in the two proteins’ abilities to induce macroscopic curva- ture (i.e. tubulation) of lipid vesicles. 1195-Plat Computer Simulation of Membrane Tubulation by EFC F-BAR Domain Lattices Hang Yu, Klaus Schulten, Ying Yin, Anton Arkhipov. University of Illinois, Urbana Champaign, Urbana, IL, USA. Cells are dynamically sculpted into many types of compartments by cellular membranes, in some cases with the help of BAR domain proteins. BAR domain proteins act under in vitro conditions are found to induce formation of tubules. We have seen in coarse-grained molecular dynamics simulation stretching over 100 microseconds how a flat membrane is curved into a tube when F-BAR do- main proteins are arranged on the membrane surface as a regular lattice of par- allel rows. The simulations could also characterize the membrane bending properties of F-BAR domains in different lattice arrangements, showing mem- brane curvatures with radii ranging from 25 to 100 nm. Lastly, the simulations reveal two key structural features of F-BAR domain that facilitate efficient binding to membranes and membrane curving: (1) Curving is promoted by close contact between phosphoserine lipid head groups and clus- ters of cationic residues along the membrane facing surface of F-BAR domains, namely lysine and arginine residues 30, 33, 110, 113, 114, and 139, 140, 146, 150, respectively. (2) Within the 100 ns of contact, the F-BAR domain hinge region, through a 20 degree rotation of the helix moment of inertia, establishes a close contact between protein and membrane. (1) and (2) result in membrane bending on a microsecond-to-millisecond time scale. 1196-Plat Invisible Binding: DnaA, the Initiator of Chromosome Replication in Bacteria, Associates with the Escherichia Coli Inner Membrane In Vivo Tomer Regev, Nadav Myers, Raz Zarivach, Itzhak Fishov. Ben Gurion University of the Negev, Beer-Sheva, Israel. DnaA initiates chromosome replication in most known bacteria and its activity is controlled to execute only once every cell division cycle. ATP in the active ATP-DnaA is hydrolyzed after initiation and ADP is replaced back to ATP on the verge of next initiation. Thus DnaA acts as a molecular switch, in which the nucleotide recycling couples key processes in the cell. Two putative recycling mechanisms presume binding of DnaA either to the membrane or to specific chromosomal sites, promoting nucleotide dissociation. While there is no doubt that DnaA interacts with artificial membranes in vitro, it is still controversial as to whether it binds the cytoplasmic membrane in vivo. We sought after DnaA- membrane interaction in E. coli cells employing fluorescent microscopy and cell fractionation with both native and fluorescent DnaA hybrids. A small (5-10%) but reliable portion of DnaA is indeed membrane associated, though invisible in fluorescent cell images. This small fraction is physiologically sig- nificant as representing the free DnaA available for initiation. Using combina- tion of mCherry with variety of DnaA fragments, we demonstrate that the membrane binding function is delocalized on the protein structure. Analysis of E. coli DnaA structure model reveals a hydrophobic continuity on the protein surface, supporting a concerted interaction of distant residues with the membrane, rather than by an individual amphiphilic helix. A binding- bending mechanism is suggested, explaining the membrane-induced nucleo- tide release from DnaA. We have suggested previously that the enigmatic ‘initiation mass’ phenomenon may result from a highly cooperative inter- conversion between two functional states of DnaA driven by its membrane surface occupancy (Aranovich et al., 2006, Aranovich et al., 2007). Our present results provide a strong basis for extrapolation of this phenomenon to in vivo situation. 1197-Plat Curvature as a Mechanism for Biomolecule Localization in Bacterial Cells Lars D. Renner 1 , Prahathees Eswaramoorthy 2 , Kumaran S. Ramamurthi 2 , Douglas B. Weibel 1 . 1 University of Wisconsin Madison, Madison, WI, USA, 2 National Cancer Institute, Bethesda, MD, USA. One of the central questions in cell biology is how the temporal and spatial organization of the cell machinery within the cell is established, maintained, and replicated. In Eubacteria, an understanding of the cellular organization of proteins is just beginning to take shape. Numerous, functionally unrelated proteins have been found that localize to regions of rod-shaped bacterial cells that are characterized by a high intrinsic curvature (e.g. the poles and the divi- sion septum). Recent data suggests that there are geometric cues for the local- ization of proteins and lipids in bacteria. We have recently tested the hypothesis that membrane anisotropy occurs by mechanisms governed by physical and geometrical constrains. We found that microdomains of cardiolipin (CL) pref- erentially localize to regions of large, negative curvature. In this presentation we explore whether these domains or curvature are responsible for protein localization. We present data for the localization of two functionally bacterial division proteins, MinD (from Escherichia coli) and DivIVA (from Bacillus subtilis) that localize to regions of large curvature in vivo. We use a top- down approach that combines in vivo and in vitro experiments with E. coli and B. subtilis cells. We find that a critical difference in the radius of curvature DC (curvature difference between cell poles and midcell) of ~0.5 mm 1 is re- quired to drive the polar localization of MinD and DivIVA. Our data provides support for curvature as a general mechanism for regulating the spatial organi- zation in bacterial membranes. This research expands our understanding of Eubacterial cell biology and provides insight into the spatial and temporal dynamics of membranes and their role in cell biology. Platform: Actin & Actin-binding Proteins 1198-Plat Coarse-Grained Analysis and Modeling of Nucleotide-Dependent Changes in F-Actin Marissa G. Saunders, Gregory A. Voth. Department of Chemistry, Institute for Biophysical Dynamics, James Franck Institute, and Computation Institute, University of Chicago, Chicago, IL, USA. Actin filaments represent a truly multiscale physical system in which changes at the chemical reaction level (ATP hydrolysis) affect the conformation of actin subunits which in turn affect the binding affinity of actin-binding proteins and the stability of actin filaments. Connecting information between these scales and understanding the physical basis of these changes is a significant and challenging problem. We present a coarse-grained (CG) model and analysis of molecular dynamics simulations of actin filaments in the ATP and ADP-bound states to understand the large scale changes in structure and dy- namics based on nucleotide state. Because the CG model is based on the underlying protein structure, changes in the distribution and stability of CG sites highlight specific areas in the filament that may respond to the nucleotide state, and which should be analyzed further at the atomistic scale. These CG models are essential to allow simulation of actin filaments at a network level and to characterize how nucleotide state affects the emergent properties of the network. Monday, February 27, 2012 237a
-
Upload
jonathan-n -
Category
Documents
-
view
212 -
download
0
Transcript of The Nature of Membrane Curvature-Induction by Amphipathic α-Helices Relies upon Protein Length:...

Monday, February 27, 2012 237a
1194-PlatThe Nature of Membrane Curvature-Induction by Amphipathic a-HelicesRelies upon Protein Length: Simulations of a-Synuclein and H0Anthony R. Braun1, Eva Sevcsik2, Elizabeth Rhoades2,Stephanie Tristram-Nagle3, Jonathan N. Sachs1.1University of Minnesota, Minneapolis, MN, USA, 2Yale University,New Haven, CT, USA, 3Carnegie Mellon University, Pittsburgh, PA, USA.Remodeling of membranes, for example the induction of membrane curvature,is a necessary step in vesicle fusion and fission. One mechanism for curvatureinduction relies upon the interfacial binding of amphipathic proteins, which actas a wedge in one leaflet of the membrane. In this study, we explore differencesbetween the membrane remodeling effects of two biologically important am-phipathic proteins, specifically a-Synuclein (aS) and the N-BAR helix H0.aS is an intrinsically disordered protein that adopts an amphipathic a-helicalstructure upon binding a membrane. Recently it has been shown that aS caninduce tubulation and vesiculation of vesicles (Varkey et al. 2010, J BiolChem, 285(42):32486). In contrast, the amphipathic helix H0 is unable, onits own, to induce tubulation, as N-BAR relies instead upon its large scaffoldingdomain (Fernandes et al. 2008, Biophys J, 94(8):3065). Using a combination ofx-ray scattering and coarse-grained molecular dynamics simulations, we ex-plored the membrane remodeling effects of aS. Our findings suggest that aS1) causes a significant thinning of the bilayer; and 2) stabilizes an anisotropiccurvature-field of both positive mean curvature and negative Gaussian curva-ture. Simulations of H0 show a similar magnitude of the local curvature aswith aS, however owing to its molecular length the curvature-field aroundH0 is isotropic. We propose that the difference in curvature-field anisotropy ex-plains the difference in the two proteins’ abilities to induce macroscopic curva-ture (i.e. tubulation) of lipid vesicles.
1195-PlatComputer Simulation of Membrane Tubulation by EFC F-BAR DomainLatticesHang Yu, Klaus Schulten, Ying Yin, Anton Arkhipov.University of Illinois, Urbana Champaign, Urbana, IL, USA.Cells are dynamically sculpted into many types of compartments by cellularmembranes, in some cases with the help of BAR domain proteins. BAR domainproteins act under in vitro conditions are found to induce formation of tubules.We have seen in coarse-grained molecular dynamics simulation stretching over100 microseconds how a flat membrane is curved into a tube when F-BAR do-main proteins are arranged on the membrane surface as a regular lattice of par-allel rows. The simulations could also characterize the membrane bendingproperties of F-BAR domains in different lattice arrangements, showing mem-brane curvatures with radii ranging from 25 to 100 nm.Lastly, the simulations reveal two key structural features of F-BAR domain thatfacilitate efficient binding to membranes and membrane curving: (1) Curving ispromoted by close contact between phosphoserine lipid head groups and clus-ters of cationic residues along the membrane facing surface of F-BAR domains,namely lysine and arginine residues 30, 33, 110, 113, 114, and 139, 140, 146,150, respectively. (2) Within the 100 ns of contact, the F-BAR domain hingeregion, through a 20 degree rotation of the helix moment of inertia, establishesa close contact between protein and membrane. (1) and (2) result in membranebending on a microsecond-to-millisecond time scale.
1196-PlatInvisible Binding: DnaA, the Initiator of Chromosome Replication inBacteria, Associates with the Escherichia Coli Inner Membrane In VivoTomer Regev, Nadav Myers, Raz Zarivach, Itzhak Fishov.Ben Gurion University of the Negev, Beer-Sheva, Israel.DnaA initiates chromosome replication in most known bacteria and its activityis controlled to execute only once every cell division cycle. ATP in the activeATP-DnaA is hydrolyzed after initiation and ADP is replaced back to ATP onthe verge of next initiation. Thus DnaA acts as a molecular switch, in which thenucleotide recycling couples key processes in the cell. Two putative recyclingmechanisms presume binding of DnaA either to the membrane or to specificchromosomal sites, promoting nucleotide dissociation. While there is no doubtthat DnaA interacts with artificial membranes in vitro, it is still controversial asto whether it binds the cytoplasmic membrane in vivo. We sought after DnaA-membrane interaction in E. coli cells employing fluorescent microscopy andcell fractionation with both native and fluorescent DnaA hybrids. A small(5-10%) but reliable portion of DnaA is indeed membrane associated, thoughinvisible in fluorescent cell images. This small fraction is physiologically sig-nificant as representing the free DnaA available for initiation. Using combina-tion of mCherry with variety of DnaA fragments, we demonstrate that the
membrane binding function is delocalized on the protein structure. Analysisof E. coli DnaA structure model reveals a hydrophobic continuity on theprotein surface, supporting a concerted interaction of distant residues withthe membrane, rather than by an individual amphiphilic helix. A binding-bending mechanism is suggested, explaining the membrane-induced nucleo-tide release from DnaA. We have suggested previously that the enigmatic‘initiation mass’ phenomenon may result from a highly cooperative inter-conversion between two functional states of DnaA driven by its membranesurface occupancy (Aranovich et al., 2006, Aranovich et al., 2007). Ourpresent results provide a strong basis for extrapolation of this phenomenonto in vivo situation.
1197-PlatCurvature as a Mechanism for Biomolecule Localization in Bacterial CellsLars D. Renner1, Prahathees Eswaramoorthy2, Kumaran S. Ramamurthi2,Douglas B. Weibel1.1University of Wisconsin Madison, Madison, WI, USA, 2National CancerInstitute, Bethesda, MD, USA.One of the central questions in cell biology is how the temporal and spatialorganization of the cell machinery within the cell is established, maintained,and replicated. In Eubacteria, an understanding of the cellular organizationof proteins is just beginning to take shape. Numerous, functionally unrelatedproteins have been found that localize to regions of rod-shaped bacterial cellsthat are characterized by a high intrinsic curvature (e.g. the poles and the divi-sion septum). Recent data suggests that there are geometric cues for the local-ization of proteins and lipids in bacteria. We have recently tested the hypothesisthat membrane anisotropy occurs by mechanisms governed by physical andgeometrical constrains. We found that microdomains of cardiolipin (CL) pref-erentially localize to regions of large, negative curvature. In this presentationwe explore whether these domains or curvature are responsible for proteinlocalization. We present data for the localization of two functionally bacterialdivision proteins, MinD (from Escherichia coli) and DivIVA (from Bacillussubtilis) that localize to regions of large curvature in vivo. We use a top-down approach that combines in vivo and in vitro experiments with E. coliand B. subtilis cells. We find that a critical difference in the radius of curvatureDC (curvature difference between cell poles and midcell) of ~0.5 mm�1 is re-quired to drive the polar localization of MinD and DivIVA. Our data providessupport for curvature as a general mechanism for regulating the spatial organi-zation in bacterial membranes. This research expands our understanding ofEubacterial cell biology and provides insight into the spatial and temporaldynamics of membranes and their role in cell biology.
Platform: Actin & Actin-binding Proteins
1198-PlatCoarse-Grained Analysis and Modeling of Nucleotide-Dependent Changesin F-ActinMarissa G. Saunders, Gregory A. Voth.Department of Chemistry, Institute for Biophysical Dynamics, James FranckInstitute, and Computation Institute, University of Chicago, Chicago,IL, USA.Actin filaments represent a truly multiscale physical system in which changes atthe chemical reaction level (ATP hydrolysis) affect the conformation of actinsubunits which in turn affect the binding affinity of actin-binding proteinsand the stability of actin filaments. Connecting information between thesescales and understanding the physical basis of these changes is a significantand challenging problem.We present a coarse-grained (CG) model and analysis of molecular dynamicssimulations of actin filaments in the ATP and ADP-bound states to understand
the large scale changes in structure and dy-namics based on nucleotide state. Becausethe CG model is based on the underlyingprotein structure, changes in the distributionand stability of CG sites highlight specificareas in the filament that may respondto the nucleotide state, and which shouldbe analyzed further at the atomistic scale.These CG models are essential to allowsimulation of actin filaments at a networklevel and to characterize how nucleotidestate affects the emergent properties of thenetwork.