
The MEK5-ERK5 kinase axis controls lipid metabolism in small … · Author Manuscript Published...
38
1 The MEK5-ERK5 kinase axis controls lipid metabolism in small cell lung cancer Sandra Cristea 1,2 , Garry L. Coles 1,2 , Daniel Hornburg 2 , Maya Gershkovitz 1,2 , Julia Arand 1,2 , Siqi Cao 1,2 , Triparna Sen 3& , Stuart C. Williamson 1,2,4 , Jun W. Kim 1,2 , Alexandros P. Drainas 1,2 , Andrew He 1,2 , Laurent Le Cam 5 , Lauren A. Byers 3 , Michael P. Snyder 2 , Kévin Contrepois 2 , Julien Sage 1,2 * 1 Department of Pediatrics, 2 Department of Genetics, Stanford University, Stanford, CA 94305, USA. 3 Department of Thoracic/Head and Neck Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA. 4 Clinical and Experimental Pharmacology Group, Cancer Research UK Manchester Institute, Manchester M20 4BX, UK. 5 IRCM, Institut de Recherche en Cancérologie de Montpellier, INSERM, Université de Montpellier, Institut Régional du Cancer de Montpellier, 34298 Montpellier, France. & Present address: Memorial Sloan Kettering Cancer Center, New York, NY, USA. Running title: Control of Lipid Metabolism by MEK5 and ERK5 in SCLC To whom correspondence should be addressed: Julien Sage, Stanford University School of Medicine, Mail code: 5149; 265 Campus Drive, SIM1, Stanford, CA 94305. Phone: (650) 724-9246. E-mail: [email protected]. Financial Support: This work was supported by the Department of Defense (grant W81XWH- 15-1-0250 to J.S.), the National Institute of Health (grants R01CA206540, R01CA201513, U01CA213273, and R35CA231997 to J.S., grant F31CA206346 to S.C., grant CA16672 to MD Anderson RPPA facility, grant 3P50HG00773505S1 to M.P.S.), the American Cancer Society (ACS) postdoctoral fellowship (to G.L.C.), the National Science Foundation Graduate Research Fellowship (to S.C.), a CRUK-Fulbright scholarship (to S.C.W.), the Emerson Collective (to on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
Transcript of The MEK5-ERK5 kinase axis controls lipid metabolism in small … · Author Manuscript Published...

1
The MEK5-ERK5 kinase axis controls lipid metabolism in small cell lung cancer
Sandra Cristea1,2, Garry L. Coles1,2, Daniel Hornburg2, Maya Gershkovitz1,2, Julia Arand1,2, Siqi
Cao1,2, Triparna Sen3&, Stuart C. Williamson1,2,4, Jun W. Kim1,2, Alexandros P. Drainas1,2,
Andrew He1,2, Laurent Le Cam5, Lauren A. Byers3, Michael P. Snyder2, Kévin Contrepois2,
Julien Sage1,2*
1Department of Pediatrics, 2Department of Genetics, Stanford University, Stanford, CA 94305,
USA. 3Department of Thoracic/Head and Neck Medical Oncology, The University of Texas MD
Anderson Cancer Center, Houston, TX 77030, USA. 4Clinical and Experimental Pharmacology
Group, Cancer Research UK Manchester Institute, Manchester M20 4BX, UK. 5IRCM, Institut de
Recherche en Cancérologie de Montpellier, INSERM, Université de Montpellier, Institut
Régional du Cancer de Montpellier, 34298 Montpellier, France.
&Present address: Memorial Sloan Kettering Cancer Center, New York, NY, USA.
Running title: Control of Lipid Metabolism by MEK5 and ERK5 in SCLC
To whom correspondence should be addressed:
Julien Sage, Stanford University School of Medicine, Mail code: 5149; 265 Campus Drive,
SIM1, Stanford, CA 94305. Phone: (650) 724-9246. E-mail: [email protected].
Financial Support: This work was supported by the Department of Defense (grant W81XWH-
15-1-0250 to J.S.), the National Institute of Health (grants R01CA206540, R01CA201513,
U01CA213273, and R35CA231997 to J.S., grant F31CA206346 to S.C., grant CA16672 to MD
Anderson RPPA facility, grant 3P50HG00773505S1 to M.P.S.), the American Cancer Society
(ACS) postdoctoral fellowship (to G.L.C.), the National Science Foundation Graduate Research
Fellowship (to S.C.), a CRUK-Fulbright scholarship (to S.C.W.), the Emerson Collective (to
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

2
J.S.), the Lung Cancer Research Foundation (LCRF) (T.S.), a CRUK-Fulbright scholarship
(S.C.W.), and the UICC (Union for International Cancer Control) Yamagiwa Yoshida Memorial
International study fund and Cancéropôle Grand Sud Ouest (to L.L.C.). J.S. is the Harriet and
Mary Zelencik Scientist in Children’s Cancer and Blood Diseases.
Disclosure of Potential Conflicts of Interests: J.S. receives research funding from
Stemcentrx/Abbvie and Pfizer, and owns stock from Forty Seven Inc. M.P.S. is a founder and
member of the science advisory board of Personalis, SensOmics, and Qbio and a science
advisory board member of Genapsys.
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

3
ABSTRACT
Small cell lung cancer (SCLC) is an aggressive form of lung cancer with dismal survival rates.
While kinases often play key roles driving tumorigenesis, there are strikingly few kinases known
to promote the development of SCLC. Here we investigated the contribution of the MAP kinase
module MEK5/ERK5 to SCLC growth. MEK5 and ERK5 were required for optimal survival and
expansion of SCLC cell lines in vitro and in vivo. Transcriptomics analyses identified a role for
the MEK5-ERK5 axis in the metabolism of SCLC cells, including lipid metabolism. In-depth
lipidomics analyses showed that loss of MEK5/ERK5 perturbs several lipid metabolism
pathways, including the mevalonate pathway that controls cholesterol synthesis. Notably,
depletion of MEK5/ERK5 sensitized SCLC cells to pharmacological inhibition of the mevalonate
pathway by statins. These data identify a new MEK5-ERK5-lipid metabolism axis that promotes
the growth of SCLC.
Significance
This study is the first to investigate MEK5 and ERK5 in SCLC, linking the activity of these two
kinases to the control of cell survival and lipid metabolism.
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

4
INTRODUCTION
Small cell lung cancer (SCLC) is a subtype of lung cancer characterized by features of
neuroendocrine differentiation, rapid growth, and a high metastatic potential. More than 200,000
patients die from SCLC every year worldwide. As smoking rates increase in several parts of the
world, the number of patients developing and succumbing to SCLC continues to grow. SCLC
patients are usually treated with a combination of radiation therapy and chemotherapy.
However, resistant tumors usually emerge within months; at this point, therapeutic options are
very limited, leading to the dismal survival rates of this disease (reviewed in (1,2)). Recent
observations indicate that immunotherapies may help treat subsets of SCLC patients (3).
Similarly, targeting DNA repair pathways may prove useful to induce cell death in SCLC cells
and inhibit the growth of SCLC tumors (4). Nonetheless, it is critical to identify and investigate
additional therapeutic options, requiring a deeper understanding of SCLC biology, and the
pathways underlying its tumorigenicity.
Resection of SCLC is rare, which, for many years, has limited the number of samples
available for analysis. More recently, however, a global effort among multiple groups resulted in
a more substantial collection of SCLC samples, and an investigation of the genetic and genomic
events that may drive the growth of SCLC (5-7). A notable genetic feature of SCLC is that the
recurrent mutations observed are often loss-of-function events that inactivate tumor
suppressors, including nearly ubiquitous inactivation of the RB1 and TP53 tumor suppressor
genes. A few oncogenic drivers have been identified, including transcription factors such as
MYC family members and NFIB. Some of these gain- and loss-of-function events have been
validated as drivers of SCLC growth in genetically engineered mouse models and human cells
and may represent new therapeutic opportunities, including c-Myc (8) or CREBBP (9). However,
the striking rarity of reoccurring oncogenic driving mutations points to the existence of
unexplored key vulnerabilities in SCLC (5-7).
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

5
The dysregulation of kinase signaling is an essential driver of oncogenic growth in multiple
contexts (10). SCLC tumors have very few activating events in genes coding for kinases
(reviewed in (11)). Nevertheless, work on kinases implicated in the response to DNA damage,
including WEE1 and CHK1 (12-14), shows that such kinases are promising targets in this
disease. There is little evidence for a role for canonical MAPK signaling (MEK1-ERK1/2) in
SCLC (11), but the less-studied MEK5-ERK5 kinase axis has not yet been investigated in SCLC
oncogenesis. In other cancers, the MEK5-ERK5 axis has been observed to play roles in many
different pathways, with multiple phenotypic results, and these two kinases have emerged as
possible therapeutic targets (reviewed in (15-17)). This dual kinase axis is responsible for
increased growth or metastasis, lower overall survival, or resistance to therapies in multiple
tumor types, including breast cancer (16,18-20), prostate cancer (21), colon cancer (18),
hepatocellular carcinomas (18,21), and high-grade osteosarcomas (18). Overall, however, the
molecular mechanisms and intracellular consequences of MEK5 and ERK5 actions leading to
these cancer phenotypes are not well understood. Here we sought to investigate the role of
these two kinases in SCLC. We found that MEK5 and ERK5 play a critical role for the survival of
SCLC cells. We also determined that MEK5 and ERK5 control lipid metabolism in SCLC cells,
including cholesterol metabolism, suggesting possible future therapeutic avenues for SCLC
treatment.
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

6
METHODS
Ethics statement
Mice were maintained according to practices prescribed by the NIH at Stanford’s Research
Animal Facility accredited by the American Association for Accreditation of Laboratory Animal
Care (AAALAC). All animal studies were conducted following approval from the Stanford Animal
Care and Use Committee (IACUC).
In Vivo Growth Assays
Cells to be injected were stained for viability with Trypan Blue solution (Sigma-Aldrich cat #
T8154) and counted using a Countess II FL Automated Cell Counter. 1 million cells were
injected subcutaneously per flank of each NSG mouse, in 100 µL RPMI media without any
serum or antibiotics, and 100 µL Corning Matrigel Matrix (Phenol-Red-Free). Tumors were then
monitored for growth, and mice were sacrificed at 21 days post-injection. Tumors were
measured by caliper, and tumor volume was calculated using the formula
(4π/3)((length+width)/4))3. Each cell line was injected into both flanks for 2 different mice. When
graphing, the volumes of tumors on different flanks of the same mouse were averaged.
Cell culture
SCLC cell lines were maintained and passaged as described before (22). All cell lines were
passaged and grown in RPMI-1640 media supplemented with 10% bovine growth serum (BGS)
(Fisher Scientific) (unless stated as 2% serum), and penicillin-streptomycin-glutamine (Gibco).
These cells grow as suspension spheres or aggregates in culture. All cell lines were maintained
at 37°C in a humidified chamber with 5% CO2. KP1 murine SCLC cells and NJH29 human
SCLC cells have previously been described (5). All cell lines were routinely testing for
mycoplasma (MycoAlertTM Mycoplasma detection Kit, Lonza, LT07-418) and all the cells used in
this study were negative. Mouse cell lines were derived from mouse tumors and genotyped for
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

7
genetic loss of Rb and p53. Human cell lines were purchased from ATCC (except for NJH29,
which was developed at Stanford University from a SCLC patient) and not further authenticated.
Proliferation assays, cell cycle and cell death assays, gene knock-down and gene
expression were performed largely as before (22). See Supplementary Methods file for details.
Atorvastatin Treatment and IC50 Assays
Atorvastatin Calcium (Selleckchem, #S2077) was dissolved in DMSO as per manufacturer’s
instructions. 2 x 104 cells of each cell line treated were plated per well of a 96-well plate, in 90
µL of reduced (2% BGS) serum media, in triplicate for each treatment condition. Cells were
allowed to re-form their spheroids (in the case of mSCLC KP1 cells) or clumps (in the case of
many hSCLC cells) or to adhere (in the case of some hSCLC cell lines, such as SBC5) for 24
hours, after which a 10x concentration of atorvastatin or vehicle control (DMOS) in 2% BGS
media was added to each well, in a 10 µL volume for a total volume of 100µL per well. 48 hours
or 5 days later, plates were read using the alamarBlue reagent (ThermoScientific, DAL1100), as
described above. Atorvastatin was used at final concentrations of 0 (DMSO only), 2, 5, 10 and
20 µM. The fraction of remaining viable cells was calculated by dividing the averaged
fluorescence signal of each concentration replicate set for each cell line, by the vehicle control
averaged values for that same cell line.
For atorvastatin IC50 assays on MEK5/ERK5-knockdown cells, hSCLC NJH29 cells were
plated similarly, at 2 x 104 cells per well of a 96-well plate, for each shRNA-expressing sample,
in 90 µL of reduced (2% BGS) serum media, in triplicate for each treatment condition. Plates
were read using alamarBlue after 48 hours of atorvastatin treatment, and. IC50 values were
calculated using GraphPad Prism 7.
Immunoassays
Protein levels were determined by immunoblot or using the Simple WesternTM quantitative
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

8
immunoassay and the Compass software, according to the manufacturer’s protocol. Cells were
lysed in RIPA lysis buffer from ThermoFisher Scientific (cat # 89900), supplemented with
proteasome and phosphatase inhibitors, and lysates were cleared by centrifugation at maximum
speed for 10 minutes, and sonicated for 30 seconds each. Total protein was quantified using the
Pierce BCA Protein Assay Kit (Thermo Fisher, Cat. #23277). For Simple WesternTM, whole-cell
lysates were diluted to a final concentration of 0.2 µL/mL. For Simple WesternTM immunoassays
the antibodies and dilutions used were as follows: ERK5 (D23E9) rabbit mAb (Cell Signaling
Technology, CST #3552, 1:100), ERK5 (D3I5V) rabbit mAb (CST, #12950, 1:50) phospho-
ERK5 (Thr218/Tyr220) rabbit polyclonal antibody (CST #3371, 1:50), PCNA (PC10) mouse
mAb (CST #2586, 1:100), cleaved PARP (D124) rabbit mAb (CST #9664, 1:100), Tubulin
mouse mAb, (Sigma-Aldrich T9026, 1:500), HSP90 (C45G5) rabbit mAb (CST #4877,
1:10,000), NEUROD1 (D35G2) rabbit mAb (CST #4373, 1:2000), and MASH1/ASCL1 mouse
mAb (BD Biosciences, # 556604, 1:1000). All other conditions and reagents were as suggested
by the manufacturer. A representative example of quantification with raw data is shown in
Supplementary Table S16. For immunoblot, washing was done in Tris-buffered saline washing
buffer with 0.1% Tween-20, blocking was done with 10% milk in washing buffer, and antibodies
were diluted in 5% milk in washing buffer. Antibodies used in immunoassays were as follows:
MEK5 mouse mAb antibody (sc-135986, 1:1000), HMGCR rabbit mAb (Abcam, ab174830,
1:500), CC3 (D175) rabbit polyclonal antibody (CST #9541, 1:100) HSP90 rabbit mAb (CST
#4877, 1:10,000), and peroxidase-AffiniPure goat anti-Rabbit IgG antibody (Jackson Immuno-
Research, # 111-035-144, 1:10,000).
Reverse-Phase Protein Array
Human SCLC NJH29 cells were infected with lentiviral constructs expressing an shRNA
against MEK5 and two independent shRNAs against ERK5, as well as two control shRNAs
(shGFP and shSCR). These samples were infected, selected, expanded, and grown in reduced
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

9
serum media (2% BGS) for 3-4 days in independent triplicates, Lysates were prepared as
required by the MD Anderson RPPA facility as previously reported (23), and submitted to the
facility, where RPPA processing and statistical analysis was performed as before (23). Fold
change values are shown unlogged, and p-values are from simple student’s t test calculations,
with all replicates of shRNAs against MEK5 in one group, against ERK5 in another group, and
against “controls” (shGFP and shSCR) in an “shCTRL” group.
RNA-seq library preparation, RNA-seq analysis, and Gene Set Enrichment Analysis
mSCLC KP1 cells and hSCLC NJH29 cells were grown in RPMI-1640 media supplemented
with 10% bovine growth serum (BGS) (Fisher Scientific) and penicillin-streptomycin-glutamine
(Gibco). After infection with respective lentiviral shRNA constructs in triplicate, the cells were
subjected to 4 days of puromycin selection (ThermoFisher cat # A1113803, 2 µg/mL for mSCLC
cells and 2.5 µg/mL for hSCLC cells), then allowed to recover for 1-2 days without puromycin,
and expanded for 3-4 days in 2% serum RPMI media. The details of RNA extraction, library
preparation, sequencing and analysis can be found in the Supplementary Methods file.
Lipid staining and Lipidomics
Lipid staining was performed with BODIPY™ 493/503 dye (4,4-Difluoro-1,3,5,7,8-Pentamethyl-
4-Bora-3a,4a-Diaza-s-Indacene, ThermoFisher, #D3922) according to the manufacturer’s
instructions. Details can be found in the Supplementary Methods file.
Sample Preparation. mSCLC KP1 cells were infected with two hairpins per gene (MEK5 and
ERK5), as well as two control hairpins (shGFP and shSCR), in completely independent
triplicates. These samples were subjected to selection and recovery, and allowed to expand in
2% BGS media. Cells were trypsinized and counted, and 107 live cells per sample were washed
three times with 10 mL phosphate-buffered saline (PBS), and snap-frozen in Eppendorf Safe-
Lock 2 mL tubes (Eppendorf, # 022363344) by dropping in liquid nitrogen, and stored at -80ºC.
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

10
Cell pellets were then stored at -80ºC until further processing. The details of lipid extraction,
measurements, and analysis can be found in the Supplementary Methods file.
DepMap Analysis
Dependency scores for all assayed genes in all 25 hSCLC cell lines were extracted from the
complete Combined RNAi (Broad, Novartis, Marcotte) dataset available on the Cancer
Dependency Map website, downloaded from the Data tab (depmap.org/portal/download/) in
August of 2018. Pearson correlations between the dependency scores of MEK5 and ERK5,
respectively, and those of all other genes across the hSCLC cell lines, were calculated using the
corrplot R package. These sets of genes were then compared to find genes with dependency
scores that correlated “highly” (r>0.5) with both MEK5 and ERK5 dependencies in hSCLC cell
lines. The resulting 63 genes were then analysed with Enrichr (amp.pharm.mssm.edu/Enrichr/)
to find GO Molecular Functions, GO Biological Processes, KEGG Pathways, and WikiPathways
lists, as well as statistical significance for each term.
REVIGO Analysis
MEK5 and ERK5 dependency-correlated genes were analysed by Enrichr as stated above; GO
Biological Processes terms associated with the 63 genes overlapping for MEK5 and ERK5
(Table S10) were then analysed and visualized using REVIGO (revigo.irb.hr/). GO ID numbers
and adjusted p values were entered into the REVIGO field, and analysis was performed with
“Medium(0.7)” allowed similarity, the Homo sapiens GO database, and the SimRel (default)
semantic similarity measure. The resulting scatterplot is shown with slight aesthetic changes,
and only GO terms with dispensability scores < 0.05 are labelled.
Statistics
Statistical significance was assayed with the GraphPad Prism 7 software. *: p-value<0.05; **: p-
value<0.01; ***: p-value<0.001; ****: p-value<0.0001; ns: not significant. The tests used are
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

11
indicated in the figure legends. To compare growth curves, we used the 2-way ANOVA followed
by t-tests. When comparing more than 2 groups, we first performed one-way ANOVA, followed
by t-tests. If F-test for variance showed a significantly different distribution between two groups
being compared (F-test p<0.05), the nonparametric Mann-Whitney p-value is reported instead
of the student’s t-test p-value, with significance symbols as described above. Data are
represented as mean+/-SD unless otherwise stated. To calculate the significance of the overlap
between two groups of genes, the hypergeometric test was used
(systems.crump.ucla.edu/hypergeometric/index.php), with the “population size” being the sum of
all genes identified after filtering, regardless of p-value “number of successes in population”
being the size of one list being considered (list 1), “sample size” being the size of the second list
being overlapped (list 2), and “number of successes” being the overlap between list 1 and list 2.
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

12
RESULTS
Depletion of MEK5 or ERK5 inhibits the expansion of SCLC cell populations
Surveying the data from the RNAi Cancer Dependency Map (24), we found that the majority
of the 25 human SCLC cell lines tested show some dependency on MAP2K5, the gene coding
for MEK5 (Supplementary Fig. S1A). The Cancer Dependency Map analysis in SCLC cell lines
for MAPK7, coding for ERK5, showed little to no dependency (see depmap.org), suggesting that
MEK5 may have additional targets in SCLC cells or that ERK5 loss is easier to compensate for
in SCLC cells. Both MAP2K5 and MAPK7 are expressed at intermediate levels in human SCLC
tumors (Supplementary Table S1). Data from the cBioPortal show occasional genetic events
implicating the MAP2K5 and MAPK7 genes, with no reported recurrent events in SCLC but a
missense mutation in the MAPK7 gene (A501D) in 2/88 patients with adrenocortical carcinoma,
suggestive of a possible oncogenic role for this kinase in neuroendocrine cancers (see
cbioportal.org).
These observations and the absence of published data on MEK5-ERK5 in SCLC prompted
us to further investigate the role of MEK5 and ERK5 in this form of lung cancer. To this end, we
first knocked down these two kinases in mouse and human SCLC cells with independent sets of
shRNA molecules (Supplementary Fig. S1B-C). Upon knock-down of the MEK5-ERK5 axis,
human SCLC (hSCLC) NJH29 cells and murine SCLC (mSCLC) KP1 cell populations grew
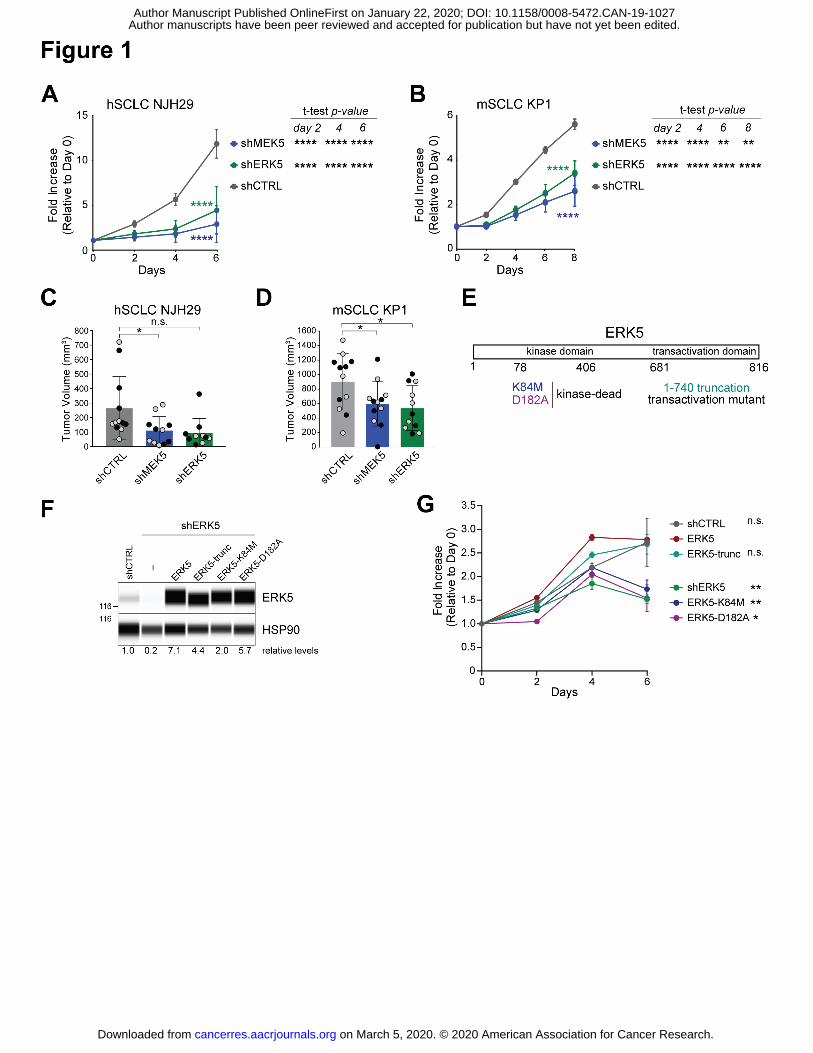
slower compared to cells expressing control shRNAs (Fig. 1A-B and Supplementary Fig. S1D).
As expected, phosphorylated ERK5 was downregulated when MEK5 was knocked down
(Supplementary Fig. S1E). We also performed subcutaneous tumor growth assays in
immunocompromised NOD-Scid-Gamma (NSG) mice and found that injection of MEK5- or
ERK5-depleted cells resulted in lower tumor volumes compared to control knock-down cells
(Fig. 1C-D). There was no evidence of counter-selection for the MEK5 knock-down in the
context of these experiments (Supplementary Fig. S1F). Thus, the MEK5 and ERK5 kinases
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

13
contribute to the optimal expansion of SCLC cell populations in culture and in vivo.
Depletion of MEK5 or ERK5 induces cell death in SCLC cell populations
The long-term growth of SCLC cells is driven by lineage transcription factors such as ASCL1
or NEUROD1 implicated in neuroendocrine identity (reviewed in (25)). We investigated whether
MEK5 and ERK5 regulate the levels of these proteins. However, protein levels of ASCL1 were
not affected by the loss of MEK5 in the ASCL1-high KP1 cells, and levels of NEUROD1
increased upon MEK5 loss in NEUROD1-high NJH29 cells (Supplementary Fig. S2A-B). These
results suggested that the inhibition of SCLC growth upon reduction of the MEK5-ERK5 axis
was not directly connected to these transcription factors and neuroendocrine cell identity.
ERK5 is a kinase that can also function as a transcription activator (26,27). To determine the
function(s) of ERK5 that are important for the expansion of SCLC cells, we re-introduced into
ERK5 knockdown NJH29 human SCLC cells either wild-type ERK5 or mutant forms of ERK5
impaired for its transcriptional or kinase activities (Figure 1E-F). The inhibition of growth
observed upon ERK5 knockdown was rescued by wild-type ERK5 as well as by a truncation
mutant that abolishes the transcriptional activity; in contrast, two separate kinase-dead mutant
failed to rescue the growth defects (Figure 1G), indicating that ERK5 kinase activity is important
for the optimal growth of SCLC cell populations in this context.
Based on these observations, to further investigate the role of the MEK5-ERK5 axis in
SCLC, we next queried a number of signaling pathways, many mediated by phosphocascades,
using a Reverse-Phase Protein Array (RPPA) approach (28) in MEK5 and ERK5 knock-down
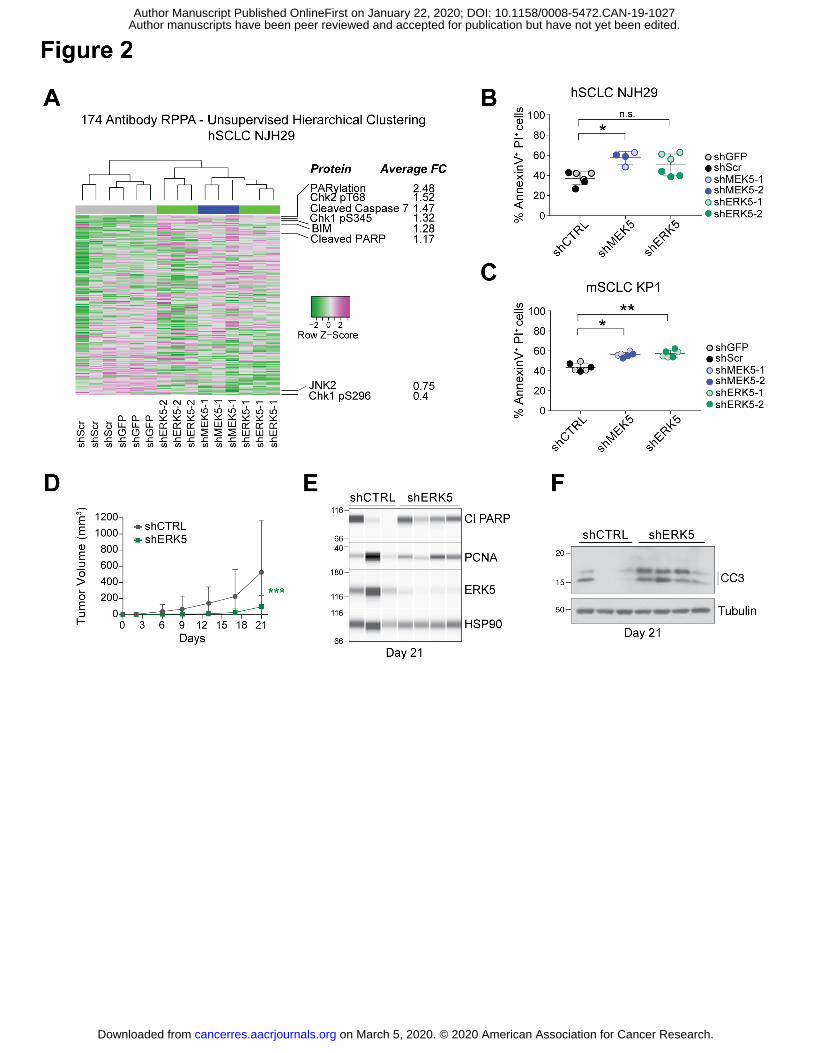
cells. Following this antibody‐based functional proteomic analysis, unsupervised clustering
grouped MEK5 and ERK5 knock-down cells together, while control cells had distinct profiles,
indicating that MEK5 and ERK5 belong to the same phosphocascade in SCLC cells (Fig. 2A,
Supplementary Fig. S2C-D, and Supplementary Table S2). However, few major concerted
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

14
changes were identified in specific signaling pathways in MEK5 and ERK5 knock-down cells
compared to controls. One exception was changes in proteins implicated in apoptotic cell death,
such as increased detection of cleaved caspase 7, cleaved PARP, and the pro-apoptotic factor
BIM, upon MEK5-ERK5 depletion. Consistent with these findings, MEK5 and ERK5 knock-down
cells showed a higher propensity toward apoptotic cell death in culture (Fig. 2B-C). No
significant changes were observed in cell cycle profiles under the same conditions in culture
(Supplementary Fig. S2E-F). When we examined markers of cell cycle progression and cell
death in vivo in xenografts from NJH29 cells upon ERK5 knock-down, we found no obvious
changes in PCNA, a marker of DNA replication, and a trend towards increased cleaved PARP
and cleaved Caspase 3, two markers of apoptotic cell death (Fig. 2D-F and Fig. S2G).
Together, these observations indicate that the MEK5-ERK5 axis controls the expansion of
SCLC cells mainly by promoting their survival.
Depletion of MEK5 or ERK5 perturbs gene programs associated with metabolic pathways
To gain further insights into the mechanisms by which the MEK5 and ERK5 proteins
promote the survival and the expansion of SCLC cell populations, we performed transcriptional
RNA-seq analyses of SCLC cells with an impaired MEK5-ERK5 axis (Supplementary Fig. S3A
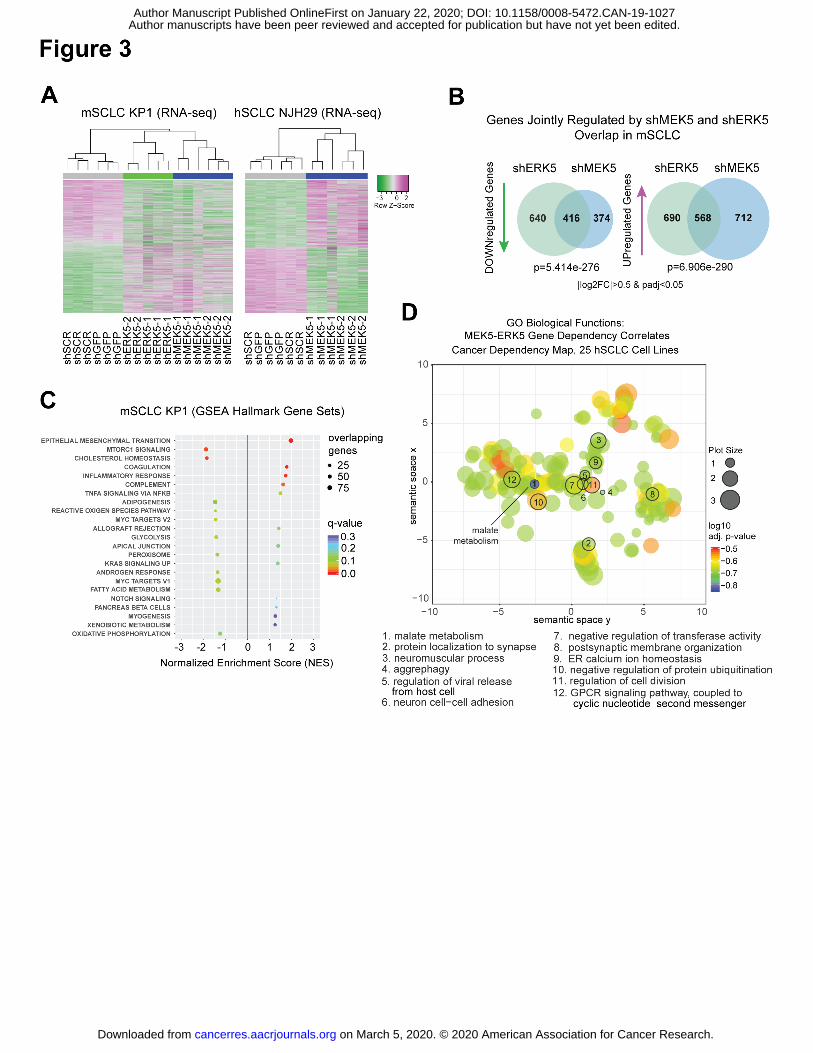
and Supplementary Tables S3-4). In unsupervised hierarchical clustering analyses (Fig. 3A) and
principal component analysis (PCA) of the gene expression data (Supplementary Fig. S3B),
mSCLC KP1 cells with MEK5 and ERK5 knock-down mSCLC KP1 cells clustered separately
from controls; similarly, MEK5 knock-down hSCLC NJH29 cells clustered separately from
control cells (Fig. 3A and Supplementary Fig. S3A-B). ERK5 knock-down NJH29 cells were not
investigated using this assay. A significant overlap was found between the genes
downregulated upon MEK5 knock-down in KP1 and NJH29 cells, suggesting that MEK5 has a
similar role in promoting gene expression programs in these two contexts
(Supplementary Fig. S3C). Additionally, the programs affected by MEK5 and ERK5 knock-down
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

15
overlapped significantly in KP1 cells, confirming that the two kinases, indeed, function in the
same phosphocascade (Fig. 3B). GSEA (Gene Set Enrichment Analysis) of averaged log2 fold
change values in murine MEK5 and ERK5 knock-down SCLC cells pointed at the
downregulation of metabolic pathways upon reduction in MEK5 and ERK5 levels (Fig. 3C and
Supplementary Table S5). Similar metabolic pathways were found downregulated at the
transcriptional levels in NJH29 cells upon MEK5 knock-down (Supplementary Figure S3D and
Supplementary Table S6). Neither mSCLC nor hSCLC cells with MEK5-ERK5 axis knock-down
showed an enrichment in cell cycle or neuroendocrine genes (Supplementary Figure S4A-B),
further supporting that the loss of viability of SCLC cells after MEK5-ERK5 depletion is not due
to a change in neuroendocrine status or their proliferation rate.
We also investigated the genes whose dependency scores were most correlated with those
for MEK5 or ERK5 in the 25 human SCLC cell lines of the Cancer Dependency Map project.
298 and 293 genes had a dependency score Pearson correlation coefficient greater than 0.5 for
MEK5 and ERK5, respectively, with an overlap of 63 genes (Supplementary Table S7). Enrichr
analysis of these 63 genes confirmed a link between MEK5 and ERK5 and metabolic pathways,
with the most significant GO Molecular Functions terms suggested links to malate metabolism
and phosphofructokinase activity (Supplementary Table S8); GO Biological Process analysis
further highlighted malate metabolism, NADH metabolism, oxaloacetate metabolism, and
Vitamin D biosynthesis (Supplementary Table S9 and summarized by REVIGO analysis in
Fig. 3D and Supplementary Table S10). KEGG pathway analysis and WikiPathways highlighted
connections between MEK5-ERK5 signaling and pyruvate metabolism and glyoxylate and
dicarboxylate metabolism, as well as the citrate cycle and glycolysis
(Supplementary Tables S11-S12).
Together, this analysis of transcriptional networks and dependency links loss of MEK5 or
ERK5 in SCLC cells to perturbations in metabolism. In particular, a number of genes and
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

16
pathways found to be altered in the RNA-seq and Cancer Dependency Map analyses of MEK5-
ERK5 deficient SCLC cells pointed to lipid metabolism-related pathways, including those
implicated in cholesterol homeostasis and de novo fatty acid (FA) synthesis. These data
suggest that altered metabolism, including lipid metabolism, may contribute to decreased
survival and growth inhibition upon inactivation of the MEK5-ERK5 module.
MEK5 and ERK5 knockdown affects cholesterol synthesis pathways
Little to nothing is known about lipid metabolism in SCLC and the links between the MEK5-
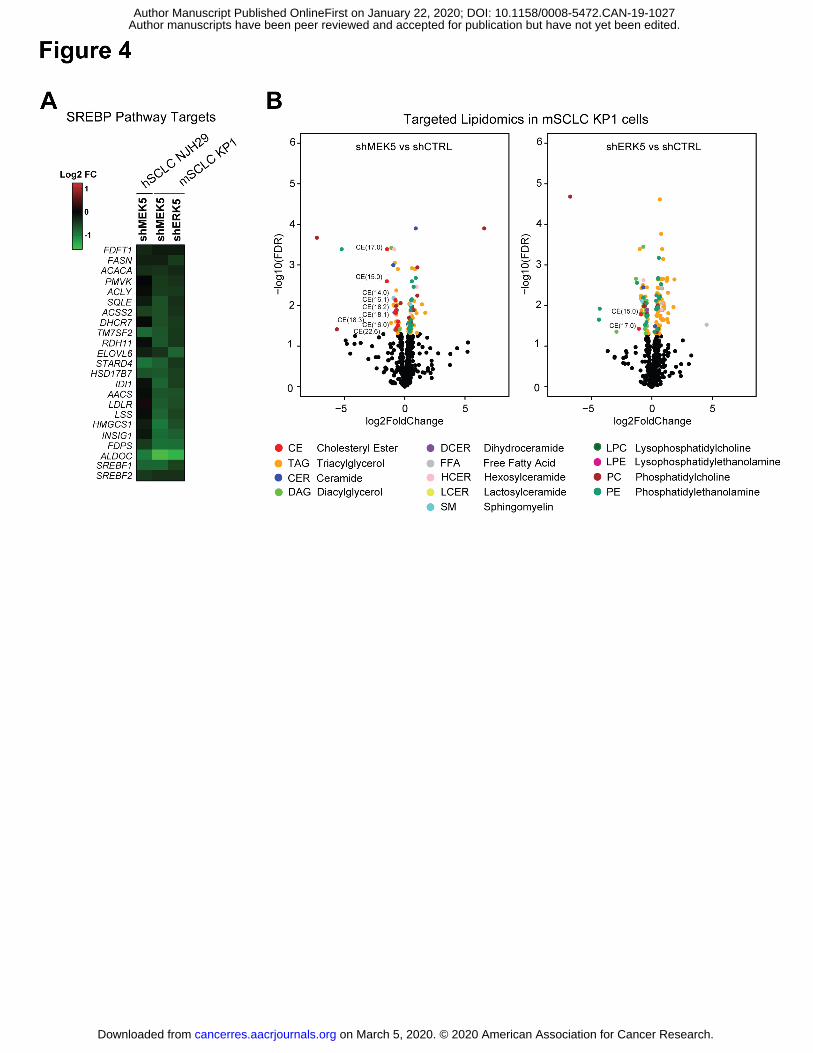
ERK5 axis and lipid homeostasis (29,30). Consistent with our findings, one of the most
significant transcription factor target gene sets to be identified by GSEA as disenriched in MEK5
and ERK5-deficient cells, was SREBP (Sterol-Regulatory Element Binding Protein) target genes
(Supplementary Figure S5A and Supplementary Table S13). Indeed, SREBP targets (31) were
downregulated in MEK5-deficient human and murine cells and ERK5-deficient murine cells
(Fig. 4A and Supplementary Figure S5B). Briefly, the SREBP pathway has two arms, including
the mevalonate pathway regulated by SREBF2, which results in cholesterol synthesis, and the
fatty acid synthesis pathway regulated by SREBF1 (32). Staining of MEK5 and ERK5 knock-
down cells with BODIPY showed no significant decrease in total neutral lipid content compared
to controls (Supplementary Fig. S5C). To more specifically ascertain which components of lipid
synthesis were dysregulated following MEK5 and ERK5 knock-down, we subjected murine
SCLC cells (KP1) to targeted lipidomics analyses (Fig. 4B, Supplementary Fig. S5D,
Supplementary Tables S14-S15). Unsupervised hierarchical clustering and PCA clustered
MEK5 and ERK5 knock-down SCLC cells separately from controls
(Supplementary Figure S6A). Additionally, though multiple lipid species were changing
significantly, the only lipid classes to show all significantly changing species as reduced in
abundance in both MEK5 and ERK5 knock-down cells were cholesteryl esters (CE),
diacylglycerols (DAG) and dihydroceramides (DCER) (Fig. 4B and Supplementary Figure S6B).
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

17
Out of the classes with relative decreased abundance as a consequence of MEK5-ERK5
knockdown, the DCER lipid class was represented by a single specific DCER significantly
downregulated in both MEK5 and ERK5 knock-down samples (Supplementary Figure S6B).
Among the remaining two classes, decreased CE abundance was predominant in MEK5 knock-
down cells, while DAG was the lipid class predominantly downregulated in ERK5 knock-down
cells. Free fatty acids (FFAs) were not significantly altered in MEK5-ERK5 cells; in addition,
although the relative abundance of a diverse range of triacylglycerol (TAG) species, into which
FAs are incorporated for energy storage, changed, they did so in both directions (Fig. 4B and
Supplementary Tables S14-S15). In contrast, the relative abundance of all detected CEs was
significantly decreased in MEK5-knockdown mSCLC KP1 cells, and several were also reduced
in ERK5-knockdown cells (Supplementary Fig. S6C – see also Methods and
Supplementary Fig. S6D). Together, these data point to cholesterol biosynthesis pathways
downstream of MEK5-ERK5 and suggest that these metabolic defects could contribute to the
loss of viability of SCLC cells upon MEK5 or ERK5 depletion.
Inhibition of the MEK5-ERK5 axis and the mevalonate pathway can both limit the
expansion of SCLC cells
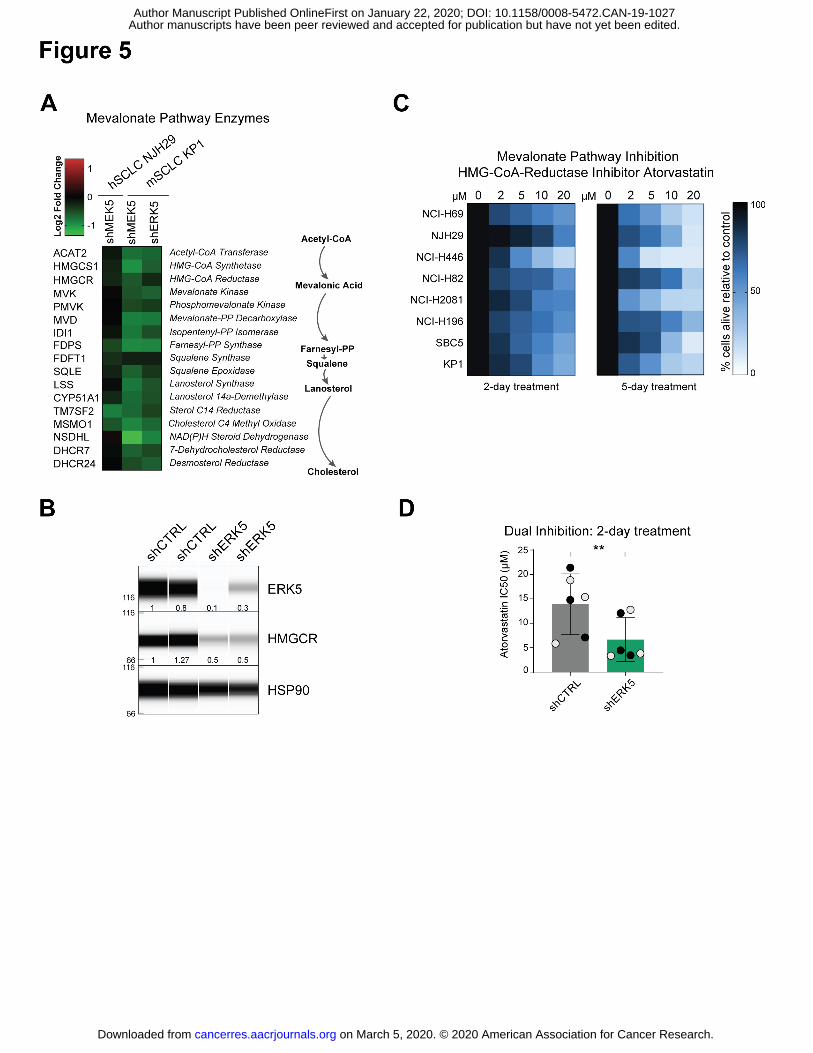
The mevalonate pathway is composed of a sequence of enzymatic steps that convert
Acetyl-CoEnzymeA into cholesterol and isoprenoids (33). Consistent with our lipidomic
analyses, genes encoding enzymes involved in different steps of this pathway were
downregulated by shMEK5 in human and murine SCLC cells, and by shERK5 in murine SCLC
cells (Fig. 5A and Supplementary Fig. S7A). One of the most clinically-relevant inhibitors of this
pathway is atorvastatin calcium (known commercially as Lipitor) (34), an inhibitor of the rate-
limiting enzyme HMG-CoA-Reductase (HMGCR). HMGCR levels were lower in MEK5 and
ERK5 knock-down cells at the RNA level (Fig. 5A). HMGCR protein levels were also lower in
ERK5 knock-down cells (Fig. 5B). To evaluate the importance of the mevalonate pathway in
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

18
SCLC, we first treated 8 cell lines (7 hSCLC and 1 mSCLC cell line (KP1)) with low micromolar
doses of atorvastatin for 2 or 5 days. Atorvastatin decreased viability by an average of 50% cells
across the human SCLC cell lines and ~60% in mouse KP1 cells alive after 2 days of treatment,
with a decreased viability of ~80% by day 5 (Fig. 5C). These results are in line with the inhibitory
activity of two related compounds, simvastatin and fluvastatin on hSCLC cell lines in a recent
large-scale screening effort (35) (Supplementary Fig. S7B).
These data identifying a previously unknown connection between the MEK5-ERK5 axis and
the mevalonate pathway in SCLC cells led us to test the possibility that inhibiting both pathways
simultaneously may have a greater effect on the expansion of SCLC cells than the inhibition of
each single pathway. We focused on the NJH29 cell line - one of the most resistant to
atorvastatin treatment in our study. Strikingly, ERK5 depletion in NJH29 cells sensitized these
cells to further inhibition of the mevalonate pathway by low doses of atorvastatin (Fig. 5D and
(Supplementary Fig. S7C). These experiments further the functional link between the MEK5-
ERK5 axis and the mevalonate pathway in SCLC.
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

19
DISCUSSION
Small cell lung cancer (SCLC) is a disease with a dire survival rate reflective of late
diagnosis, incredibly quick metastasis, few and ineffective treatment choices, and an underlying
lack of knowledge about its basic biology. Here we focused on the relatively unexplored MEK5-
ERK5 axis, a pathway that is not recurrently altered at the genetic level but that we
hypothesized could be a driving force in SCLC. Accumulating evidence supports an important
role for MEK5 and ERK5 in various phenotypes associated with cancer, even though the pro-
tumorigenic effects of these two enzymes have not been associated with genetic events leading
to their activation (15-17). We identified a pro-survival role for these kinases in SCLC cells and
show that they are implicated in the control of cholesterol synthesis and other lipid metabolism
pathways in SCLC cells.
We identified a pro-survival role for these kinases in SCLC cells. A similar role in the control
of survival has been described for MEK5 and ERK5 in multiple normal and cancerous cell types
(21,36,37), suggesting that this function is broadly conserved for the MEK5-ERK5 axis. The
upstream signals that activate MEK5 and ERK5 remain poorly understood, especially in the
context of pro-survival signals in cancer cells. The downstream mechanisms by which this
kinase axis promotes survival are also poorly understood but likely to be diverse. Our work and
recent work by others suggest that it may include the regulation of metabolic pathways. A recent
study identified a link between MEK5/ERK5 and the stability of MYC, a regulator of cell
metabolism and growth (38); this link is corroborated by our RNA-seq analysis (Fig. 3C).
Emerging evidence also links MEK5 and ERK5 to the control of oxidative phosphorylation (39).
Notably, one study makes a connection between forced oxidative phosphorylation and
downstream effects on cholesterol levels, via an increase in LDLR expression and intracellular
LDL-cholesterol intake – a process dependent on the MEK5-ERK5 axis (29). To our knowledge,
however, there is no prior direct description of a control of de novo cholesterol synthesis by the
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

20
MEK5-ERK5 axis. Further studies are required to determine if our observation is specific to
SCLC or if it applies to other cell types. Importantly, our data show that the lipid metabolism
phenotypes of MEK5 loss and ERK5 loss are very similar, but not identical, in SCLC cells, and it
is thus also possible that some of the mechanisms of survival control are different for the two
kinases. Notably, our results suggest that the kinase activity of ERK5 contributes to its pro-
tumorigenic role in SCLC, and inhibitors of both kinases in the pathway may provide therapeutic
strategies in the future, especially in combination with other therapeutic agents.
Our work also highlights the current general lack of knowledge of SCLC metabolism. SCLC,
like many tumors, is thought to be highly glycolytic. However, unlike most cancers, which
predominantly express the PKM2 isoform of pyruvate kinase M, a glycolytic enzyme that
specifies the fate of glucose-derived carbons, SCLC cells express a higher ratio of PKM1 to
PKM2 (40). This results in SCLC cells having higher glucose flux into lactate conversion and the
TCA cycle, attenuated glutamine metabolism, and likely are better at performing mitophagy, and
attaining lower ROS (reactive oxygen species) levels (41). Another recent study shows that the
ASCL1-low subtype of SCLC tumors is specifically dependent on de novo purine synthesis in
vivo (42).
While large amounts of lipids have been demonstrated to be necessary to support the rapid
proliferation of cancer cells, the implication of lipid metabolism in SCLC remains poorly
understood. A single study utilizing metabolomic profiling of one SCLC cell line (NCI-H446)
found elevated carnitine palmitoyltransferase 1A (CPT1A) and 2 (CPT2), key enzymes in fatty
acid oxidation, compared to NSCLC and normal epithelial cell controls (43). The only SCLC
study to date related to cholesterol metabolism, to our knowledge, is a correlative study that
found that low serum LDL (low-density lipoprotein), and low protein expression of LDLR (low-
density lipoprotein receptor), both independently correlate with better overall survival (44). A
previous study using simvastatin, a pharmacological inhibitor of HMCGR, the rate-limiting
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

21
enzyme of the mevalonate pathway, in SCLC cell lines observed a decrease in proliferation but
linked these effects to possible changes in Ras signaling and not to general cholesterol
metabolism (45). Our studies provide novel insights into lipid biology in SCLC cells and
comparisons with future analyses in other cancer types will identify unique aspects of lipid
metabolism in SCLC cells. For example, SCLC cells have been observed to require lipid-raft
mediated SRC-PI3K/AKT activation for sustained growth in culture (46). Additionally, the
isoprenoid byproducts of the mevalonate pathway are crucial for the prenylation of multiple
proteins (including RAS superfamily members), which is critical for their correct tethering,
localization, and protein-protein binding signaling functions (47). Furthermore, cholesterol itself
can be attached directly to proteins, including Smoothened (SMO) in the Hedgehog pathway, a
pathway that has been shown to control SCLC tumor initiation and progression (48,49). These
cholesterol-mediated functions regulate a wide range of cellular processes, including cell
polarity and cell body dynamics, cell proliferation and survival, protein and intracellular vesicular
trafficking, cell cycle, and nuclear transport dynamics – all of which may affect the survival and
the expansion of SCLC cells.
Targeting the mevalonate pathway as a therapeutic intervention is being investigated in
multiple tumor types (50). Our experiments exposing SCLC cell lines to atorvastatin suggest
that at least a subset of SCLC cells is sensitive to HMGCR inhibition. Interestingly, a few clinical
trials have included SCLC patients treated with statins (45,51,52), with no visible benefit for the
survival of these patients. However, given the evidence building up that HMGCR inhibitors such
as statins have multiple effects, systemically and tumor-specifically, the failure of these trials
may say more about the complexity of cholesterol inhibition in tumors and in human tissues than
it does about the degree of dependence of SCLC on cholesterol synthesis. Consistent with our
findings, a recent study identified sensitivity to inhibition of the cholesterol biosynthetic pathway
enzyme squalene epoxidase (SQLE) in SCLC cells (53). A large meta-analysis of small cell
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

22
neuroendocrine tumors also suggests susceptibility to disruption of lipid and sterol metabolism
(54). Additional investigation into the latter will need to be performed in order to ascertain which
of the many roles of the mevalonate pathway and its byproducts are truly critical in SCLC.
Our work suggests that the anti-cancer effects of inhibiting the mevalonate pathway may be
enhanced by inhibition of the MEK5/ERK5 axis, which may be achieved when potent and
specific inhibitors of these kinases have been developed (55). Additionally, mevalonate pathway
inhibitors were found to have vaccine-adjuvant activities and to synergize with anti-PD-1
antibodies to kill tumor cells, by enhancing the functions of antigen-presenting cells (56).
Therefore, mevalonate pathway inhibition may also have the ability to increase antigen
presentation to the immune system, perhaps serving as a future adjuvant for an SCLC vaccine
in conjunction with FDA-approved immunotherapies.
In this study, we present the first experiments suggesting that a less-studied arm of the MAP
Kinase pathway – the MEK5/ERK5 dual kinase axis – is crucial for sustained SCLC cell viability.
Furthermore, we have connected this axis to downstream cholesterol biology in SCLC,
especially the mevalonate pathway. Additional experiments need to be performed to identify the
detailed molecular mechanism of these connections, but our studies already present multiple
single and combinatorial therapeutic strategies which can be further tested and validated
preclinically, in hopes of clinical success for the hundreds of thousands of patients who die
yearly from small cell lung cancer.
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

23
ACKNOWLEDGMENTS
We thank the staff of the Stanford Functional Genomics Facility, the FACS Core at the Institute
for Stem Cell Biology and Regenerative Medicine, and the Stanford Veterinary Service Center,
as well as Pauline Chu, for their technical support and expertise. We acknowledge members of
the Sweet-Cordero and Sage laboratories for technical and materials support, including Andrea
Chaikovsky for her help with immunoblots. This work was supported by the Department of
Defense (grant W81XWH-15-1-0250 to J.S.), the National Institute of Health (grants
R01CA206540, R01CA201513, U01CA213273 and R35CA231997 to J.S., grant F31CA206346
to S.C., grant CA16672 to MD Anderson RPPA facility, grant P50HG007735 to M.P.S.), the
American Cancer Society (ACS) postdoctoral fellowship (to G.L.C.), the National Science
Foundation Graduate Research Fellowship (to S.C.), a CRUK-Fulbright scholarship (to S.C.W.),
the Emerson Collective (to J.S.), the Lung Cancer Research Foundation (LCRF) (T.S.), a
CRUK-Fulbright scholarship (S.C.W.), and the UICC (Union for International Cancer Control)
Yamagiwa Yoshida Memorial International study fund (to L.L.C.). J.S. is the Harriet and Mary
Zelencik Scientist in Children’s Cancer and Blood Diseases.
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

24
REFERENCES
1. Bunn PA, Jr., Minna JD, Augustyn A, Gazdar AF, Ouadah Y, Krasnow MA, et al. Small Cell Lung Cancer: Can Recent Advances in Biology and Molecular Biology Be Translated into Improved Outcomes? J Thorac Oncol 2016;11:453-74
2. Sabari JK, Lok BH, Laird JH, Poirier JT, Rudin CM. Unravelling the biology of SCLC: implications for therapy. Nature reviews Clinical oncology 2017
3. Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med 2018;379:2220-9
4. Pietanza MC, Waqar SN, Krug LM, Dowlati A, Hann CL, Chiappori A, et al. Randomized, Double-Blind, Phase II Study of Temozolomide in Combination With Either Veliparib or Placebo in Patients With Relapsed-Sensitive or Refractory Small-Cell Lung Cancer. J Clin Oncol 2018:JCO2018777672
5. George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015;524:47-53
6. Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 2012;44:1104-10
7. Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet 2012;44:1111-6
8. Mollaoglu G, Guthrie MR, Bohm S, Bragelmann J, Can I, Ballieu PM, et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 2017;31:270-85
9. Jia D, Augert A, Kim DW, Eastwood E, Wu N, Ibrahim AH, et al. Crebbp Loss Drives Small Cell Lung Cancer and Increases Sensitivity to HDAC Inhibition. Cancer Discov 2018;8:1422-37
10. Knapp S, Sundstrom M. Recently targeted kinases and their inhibitors-the path to clinical trials. Curr Opin Pharmacol 2014;17C:58-63
11. Cristea S, Sage J. Is the Canonical RAF/MEK/ERK Signaling Pathway a Therapeutic Target in SCLC? J Thorac Oncol 2016
12. Sen T, Tong P, Stewart CA, Cristea S, Valliani A, Shames DS, et al. CHK1 inhibition in small cell lung cancer produces single-agent activity in biomarker-defined disease subsets and combination activity with cisplatin or olaparib. Cancer Res 2017
13. Sen T, Tong P, Diao L, Li L, Fan Y, Hoff J, et al. Targeting AXL and mTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clin Cancer Res 2017
14. Lallo A, Frese KK, Morrow C, Szczepaniak Sloane R, Gulati S, Schenk MW, et al. The combination of the PARP inhibitor olaparib and the Wee1 inhibitor AZD1775 as a new therapeutic option for small cell lung cancer. Clin Cancer Res 2018
15. Wang X, Tournier C. Regulation of cellular functions by the ERK5 signalling pathway. Cell Signal 2006;18:753-60
16. Hoang VT, Yan TJ, Cavanaugh JE, Flaherty PT, Beckman BS, Burow ME. Oncogenic signaling of MEK5-ERK5. Cancer Lett 2017;392:51-9
17. Stecca B, Rovida E. Impact of ERK5 on the Hallmarks of Cancer. Int J Mol Sci 2019;20 18. Simoes AE, Rodrigues CM, Borralho PM. The MEK5/ERK5 signalling pathway in cancer:
a promising novel therapeutic target. Drug Discov Today 2016;21:1654-63 19. Liu F, Zhang H, Song H. Upregulation of MEK5 by Stat3 promotes breast cancer cell
invasion and metastasis. Oncol Rep 2017;37:83-90
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

25
20. Antoon JW, Martin EC, Lai R, Salvo VA, Tang Y, Nitzchke AM, et al. MEK5/ERK5 signaling suppresses estrogen receptor expression and promotes hormone-independent tumorigenesis. PloS one 2013;8:e69291
21. Lochhead PA, Gilley R, Cook SJ. ERK5 and its role in tumour development. Biochemical Society transactions 2012;40:251-6
22. Denny SK, Yang D, Chuang CH, Brady JJ, Lim JS, Gruner BM, et al. Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell 2016;166:328-42
23. Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov 2012;2:798-811
24. Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a Cancer Dependency Map. Cell 2017;170:564-76 e16
25. Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer 2019
26. Kasler HG, Victoria J, Duramad O, Winoto A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol Cell Biol 2000;20:8382-9
27. Nithianandarajah-Jones GN, Wilm B, Goldring CE, Muller J, Cross MJ. ERK5: structure, regulation and function. Cell Signal 2012;24:2187-96
28. Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Molecular cancer therapeutics 2006;5:2512-21
29. Khan AUH, Allende-Vega N, Gitenay D, Gerbal-Chaloin S, Gondeau C, Vo DN, et al. The PDK1 Inhibitor Dichloroacetate Controls Cholesterol Homeostasis Through the ERK5/MEF2 Pathway. Sci Rep 2017;7:10654
30. Belkahla S, Haq Khan AU, Gitenay D, Alexia C, Gondeau C, Vo DN, et al. Changes in metabolism affect expression of ABC transporters through ERK5 and depending on p53 status. Oncotarget 2018;9:1114-29
31. Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proceedings of the National Academy of Sciences of the United States of America 2003;100:12027-32
32. Guo D, Bell EH, Mischel P, Chakravarti A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr Pharm Des 2014;20:2619-26
33. Buhaescu I, Izzedine H. Mevalonate pathway: a review of clinical and therapeutical implications. Clin Biochem 2007;40:575-84
34. Jones P, Kafonek S, Laurora I, Hunninghake D. Comparative dose efficacy study of atorvastatin versus simvastatin, pravastatin, lovastatin, and fluvastatin in patients with hypercholesterolemia (the CURVES study). Am J Cardiol 1998;81:582-7
35. Polley E, Kunkel M, Evans D, Silvers T, Delosh R, Laudeman J, et al. Small Cell Lung Cancer Screen of Oncology Drugs, Investigational Agents, and Gene and microRNA Expression. J Natl Cancer Inst 2016;108
36. Hayashi M, Kim SW, Imanaka-Yoshida K, Yoshida T, Abel ED, Eliceiri B, et al. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. The Journal of clinical investigation 2004;113:1138-48
37. Watson FL, Heerssen HM, Bhattacharyya A, Klesse L, Lin MZ, Segal RA. Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat Neurosci 2001;4:981-8
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

26
38. Vaseva AV, Blake DR, Gilbert TSK, Ng S, Hostetter G, Azam SH, et al. KRAS Suppression-Induced Degradation of MYC Is Antagonized by a MEK5-ERK5 Compensatory Mechanism. Cancer Cell 2018;34:807-22 e7
39. Charni S, de Bettignies G, Rathore MG, Aguilo JI, van den Elsen PJ, Haouzi D, et al. Oxidative phosphorylation induces de novo expression of the MHC class I in tumor cells through the ERK5 pathway. Journal of immunology 2010;185:3498-503
40. Morita M, Sato T, Nomura M, Sakamoto Y, Inoue Y, Tanaka R, et al. PKM1 Confers Metabolic Advantages and Promotes Cell-Autonomous Tumor Cell Growth. Cancer Cell 2018;33:355-67 e7
41. Nomura M, Morita M, Tanuma N. A metabolic vulnerability of small-cell lung cancer. Oncotarget 2018;9:32278-9
42. Huang F, Ni M, Chalishazar MD, Huffman KE, Kim J, Cai L, et al. Inosine Monophosphate Dehydrogenase Dependence in a Subset of Small Cell Lung Cancers. Cell Metab 2018;28:369-82 e5
43. Yu L, Li K, Xu Z, Cui G, Zhang X. Integrated omics and gene expression analysis identifies the loss of metabolite-metabolite correlations in small cell lung cancer. Onco Targets Ther 2018;11:3919-29
44. Zhou T, Zhan J, Fang W, Zhao Y, Yang Y, Hou X, et al. Serum low-density lipoprotein and low-density lipoprotein expression level at diagnosis are favorable prognostic factors in patients with small-cell lung cancer (SCLC). BMC Cancer 2017;17:269
45. Khanzada UK, Pardo OE, Meier C, Downward J, Seckl MJ, Arcaro A. Potent inhibition of small-cell lung cancer cell growth by simvastatin reveals selective functions of Ras isoforms in growth factor signalling. Oncogene 2006;25:877-87
46. Arcaro A, Aubert M, Espinosa del Hierro ME, Khanzada UK, Angelidou S, Tetley TD, et al. Critical role for lipid raft-associated Src kinases in activation of PI3K-Akt signalling. Cell Signal 2007;19:1081-92
47. Ahmadi Y, Ghorbanihaghjo A, Argani H. The balance between induction and inhibition of mevalonate pathway regulates cancer suppression by statins: A review of molecular mechanisms. Chem Biol Interact 2017;273:273-85
48. Park KS, Martelotto LG, Peifer M, Sos ML, Karnezis AN, Mahjoub MR, et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat Med 2011;17:1504-8
49. Chen C, Breslin MB, Lan MS. Sonic hedgehog signaling pathway promotes INSM1 transcription factor in neuroendocrine lung cancer. Cell Signal 2018;46:83-91
50. Clendening JW, Penn LZ. Targeting tumor cell metabolism with statins. Oncogene 2012;31:4967-78
51. Seckl MJ, Ottensmeier CH, Cullen M, Schmid P, Ngai Y, Muthukumar D, et al. Multicenter, Phase III, Randomized, Double-Blind, Placebo-Controlled Trial of Pravastatin Added to First-Line Standard Chemotherapy in Small-Cell Lung Cancer (LUNGSTAR). J Clin Oncol 2017;35:1506-14
52. Ung MH, MacKenzie TA, Onega TL, Amos CI, Cheng C. Statins associate with improved mortality among patients with certain histological subtypes of lung cancer. Lung Cancer 2018;126:89-96
53. Mahoney CE, Pirman D, Chubukov V, Sleger T, Hayes S, Fan ZP, et al. A chemical biology screen identifies a vulnerability of neuroendocrine cancer cells to SQLE inhibition. Nat Commun 2019;10:96
54. Balanis NG, Sheu KM, Esedebe FN, Patel SJ, Smith BA, Park JW, et al. Pan-cancer Convergence to a Small-Cell Neuroendocrine Phenotype that Shares Susceptibilities with Hematological Malignancies. Cancer Cell 2019;36:17-34 e7
55. Nguyen D, Lemos C, Wortmann L, Eis K, Holton SJ, Boemer U, et al. Discovery and Characterization of the Potent and Highly Selective (Piperidin-4-yl)pyrido[3,2-
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

27
d]pyrimidine Based in Vitro Probe BAY-885 for the Kinase ERK5. J Med Chem 2019;62:928-40
56. Xia Y, Xie Y, Yu Z, Xiao H, Jiang G, Zhou X, et al. The Mevalonate Pathway Is a Druggable Target for Vaccine Adjuvant Discovery. Cell 2018;175:1059-73 e21
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

28
FIGURE LEGENDS
Figure 1: MEK5 and ERK5 knock-down inhibits the expansion of SCLC cells
(A-B) Quantification of populations growth in reduced serum (2%) of human NJH29 SCLC cells
(A) and murine KP1 SCLC cells (B) with shRNA-mediated knockdown of MEK5 and ERK5
compared to shCTRLs by alamarBlue assay. 2-way ANOVA interaction p-values comparing the
knock-down curves to the control are shown in colors; t-test p-values shown at the right of each
graph specify comparison of each knock down controls, with ** signifying p<0.01, ***, p<0.001,
and ****, p<0.0001; n = 2 independent shRNAs per group (for hSCLC NJH29, n=5-6, and for
mSCLC KP1, n=3 independent experiments per individual hairpin).
(C-D) Volume of tumors resulting from subcutaneous injections of hSCLC NJH29 cells (C) or
mSCLC KP1 cells (D) expressing shMEK5, shERK5, or shCTRL (shGFP and shSCR), after 3
weeks of growth in the flanks of NSG recipient mice; grey and black dots represent two
independent shRNAs per group (n=4-6 independent experiments per individual hairpin, *,
p<0.05, n.s., p>0.05 by t-test following one-way ANOVA p=0.0192 for (C) and p=0.0318 for (D)).
(E) Schematic representation of the ERK5 protein with the mutants used in (F-G).
(F) Immuno-assays for ERK5 and HSP90 (loading control) in hSCLC NJH29 cells with ERK5
knockdown and re-expression of wild-type or mutant forms of ERK5, as indicated. Levels of
ERK5 relative to HSP90 are indicated below. The 116 kDa molecular weight marker is shown
on the left.
(G) Quantification of populations growth in reduced serum (2%) of human NJH29 SCLC cells as
in (B) by alamarBlue assay (n=3). 2-way ANOVA p-values comparing the kinase-dead mutants
to the wild-type ERK5 rescue are significant (p<0.0001 for both K84M and D182A); t-test p-
values shown on the right are day 6 values compared to ERK5 rescue, with * signifying p<0.05,
** signifying p<0.01 and n.s. for non significant.
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

29
Figure 2: MEK5 and ERK5 knock-down induces cell death in SCLC cell populations
(A) Unsupervised hierarchical clustering of all protein quantities measured by Reverse Phase
Protein Array (RPPA) separates hSCLC NJH29 cells with MEK5 and ERK5 knockdown, from
those with shCTRL knockdowns (shGFP and shSCR) (top); for proteins with average fold
change (FC) across all ERK5 and MEK5 hairpins larger than 1.15 or smaller than 0.75, protein
names and average FCs are noted.
(B-C) Knockdown of MEK5 and ERK5 in hSCLC NJH29 (B) and mSCLC KP1 (C) SCLC cells
results in a higher rate of cell death by apoptosis as measured by AnnexinV/PI staining and flow
cytometry after 2 days of growth in 2% serum conditions; n.s., p>0.05, *, p<0.05, **, p<0.01 by t-
test following one-way ANOVA (p=0.0115 for (B) and p=0.004 for (D)); grey and colored dots
represent 2 different shRNAs per group, and n=2-3 independent experiments per individual
hairpin.
(D) Volume of tumors resulting from subcutaneous injections of hSCLC NJH29 cells expressing
shERK5 (one shRNA) or shCTRL (shGFP) during 3 weeks of growth in the flanks of NSG
recipient mice (n=12 tumors per group, error bars represent S.E.M.). 2-way ANOVA interaction
p=0.0002, *** shown in graph.
(E) Immunoassays for ERK5, the cell death marker cleaved PARP (Cl PARP), and the cell cycle
marker PCNA on extracts from NJH29 tumors in (D) at day 21 of growth. Tumors were selected
to minimize differences due to tumor size (n=3 shCTRL and n=4 shERK5). Protein levels are
relative to the loading control HSP90.
(F) Immunoblot as in (E) for the cell death marker cleaved caspase 3 (CC3). Tubulin is a loading
control.
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

30
Figure 3: MEK5 and ERK5 knock-down perturbs metabolic pathways in SCLC cells
(A) Unsupervised clustering from RNA-seq data upon MEK5-ERK5 knockdown in mSCLC KP1
cells, and MEK5 knockdown in hSCLC NJH29 cells; allgenes with |log2FoldChange|>0.5 and
adjusted p-values <0.05 were included in the analysis from any comparison); n=2-3
independent replicates per hairpin.
(B) MEK5 and ERK5 knockdown in mSCLC KP1 cells change the transcriptome in similar ways,
with significantly overlapping gene sets being downregulated and upregulated; hypergeometric
test used to obtain p-value of overlap; only genes changing by a |log2 fold change|>0.5 and an
adjusted p-value<0.05 were considered.
(C) Hallmarks Gene Set Enrichment Analysis (GSEA) gene sets significantly enriched or
disenriched when the MEK5-ERK5 axis is downregulated in mSCLC KP1 cells compared to
controls; log2 fold change values averaged for shMEK5 and shERK5, respectively, each
compared to shCTRLs (shGFP and shSCR), to focus analysis on genes controlled by both
kinases; only enriched sets with q-values <0.3 are shown.
(D) Gene Ontology Biological Function term results from Enrichr, for the set of 63 genes with a
Pearson correlation coefficient of over 0.5 between their dependency scores and those of both
MEK5 and ERK5 in 25 hSCLC cell lines from the Cancer Dependency Map project, analyzed by
ReviGO, and mapped based on their semantic similarity; GO IDs with a dispensability score
<0.15 are numbered and stated in the legend below; bubble or plot size is proportional to
frequency of Homo sapiens UniProt entries associated with that GO ID, color specifies log10
adjusted p-value for that GO ID from Enrichr.
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

31
Figure 4: MEK5 and ERK5 loss changes the lipidomic profile of SCLC cells
(A) MEK5 and ERK5 knockdown cells downregulate SREBP pathway targets compared to cells
infected with shCTRLs (from RNA-seq FPM values).
(B) Significantly changing lipid species (p<0.05) between shMEK5 and shCTRL cells (left) or
between shERK5 and shCTRL cells (right) are shown in volcano plots with -log10(FDR) on the
Y axis and log2 fold change on the X axis; significantly changing lipid species (FDR<0.05) are
shown as colored dots according to their lipid classes (colors corresponding to different lipid
classes are shown in the legend at bottom); significantly changing cholesteryl esters (CE) are
labeled with their number of carbons and unsaturations contained on the fatty acid moeity; CE,
cholesteryl esters, TAG, triacylglycerols, CER, ceramides, DAG, diacylglycerols, DCER,
dihydrocermides, FFA, free fatty acids, HCER, hexosylceramides, LCER, lactosylceramindes,
SM, sphingomyelins, LPC, lysophosphatidylcholines, LPE, lysophosphatidylethanolamines, PC,
phosphatidylcholines, PE, phosphatidylethanolamines; n=2-3 independent replicates per
hairpin.
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

32
Figure 5: MEK5 and ERK5 knockdown results in inhibition of the mevalonate pathway
and increased sensitivity to mevalonate pathway inhibitors
(A) MEK5 and ERK5 knockdown cells downregulate Mevalonate pathway enzymes
(biosynthesis schematic on right) compared to cells infected with shCTRL hairpins (from
RNAseq FPM values).
(B) Immunoassay for ERK5, HMGCR, and HSP90 expression in control and ERK5 knock-down
NHJ29 hSCLC cells. The molecular weights are indicated on the left (in kDa). The amount of
ERK5 and HMGCR relative to the first control and to HSP90 are indicated below the signal
corresponding to each protein.
(C) Treatment of 8 SCLC cell lines (7 human cell lines and the mouse cell line KP1) with
increasing doses of atorvastatin in reduced (2%) serum media; color corresponds to % cells
alive compared to vehicle-treated controls after 2 or 5 days at each concentration; n=3
independent experiments per treatment.
(D) Concentrations of atorvastatin necessary to kill 50% of NHJ29 cells (IC50) infected with
shCTRLs are higher than concentrations needed to kill cells with ERK5 knockdown; grey and
black dots represent two independent hairpins per group; n=3 experiments, **, p≤0.005 (t-test).
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027

Published OnlineFirst January 22, 2020.Cancer Res Sandra Cristea, Garry L. Coles, Daniel Hornburg, et al. cell lung cancerThe MEK5-ERK5 kinase axis controls lipid metabolism in small
Updated version
10.1158/0008-5472.CAN-19-1027doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2020/01/22/0008-5472.CAN-19-1027.DC1
Access the most recent supplemental material at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/early/2020/01/22/0008-5472.CAN-19-1027To request permission to re-use all or part of this article, use this link
on March 5, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 22, 2020; DOI: 10.1158/0008-5472.CAN-19-1027


















