THE JOURNAL OF CHEMISTRY Vol. No. of June pp. by for · PDF fileFree and Albumin-bound...
8
THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1994 by The American Society for Biochemistry and Molecular Biology, Inc. Vol. 269, No. 24, Issue of June 17, pp. 16712-16719, 1994 Printed in U.S.A. Free and Albumin-bound Bilirubin Are Efficient Co-antioxidants for a-Tocopherol, Inhibiting Plasma and Low Density Lipoprotein Lipid Peroxidation* (Received forpublication, January 4, 1994, and in revised form, March 23, 1994) JiEi Neuiil and Roland Stocker$ From the Biochemistrv GrouD. The Heart Research Institute, 145 Missenden Road, Camperdown, Sydney, 2050 New South Wales, Australia Peroxidation of the lipid moieties of low density li- poproteins (LDL) is regarded as an early event in atherogenesis. Because bilirubin is a physiological re- ductant with antioxidant activities, we investigated its inhibitory action on the radical-mediated oxidation of LDL and plasma lipids. Exposing fresh human blood plasma to lipophilic peroxyl radicals generated from 2,2’-azobis(2,4-dimethylvaleronitrile) (AMVN) resulted in rapid oxidation of ubiquinol-10, followed by that of ascorbate and bilirubin. Plasma lipids were well pro- tected from peroxidation as long as these three antioxi- dants were present, as assessed by the amounts of cho- lesterylester hydroperoxides formed during this period. Following consumption of these antioxidants, and in the presence of a-tocopherol, the rate of hydroperoxide for- mation increased sharply with roughly 2 molecules of cholesterylester hydroperoxides being formed for each peroxidation initiating event. Supplementation of AMVN-oxidizingplasma with exogenous bilirubin at the onset of rapid lipid peroxidation, i.e. after depletion of endogenous ubiquinol-10, ascorbate, and bilirubin, led to a halt in both hydroperoxide formation and consump- tion of a-tocopherol. When isolated LDL was incubated with AMVN, 4 molecules of cholesterylester hydroper- oxides were formed per peroxidation initiating event and while a-tocopherol was consumed. Addition of free or albumin-bound bilirubin to isolated LDL at the onset of oxidation resulted in a strong inhibition of hydroper- oxide formation and a-tocopherol consumption, the ef- fect beingmore pronounced with the freepigment. Ad- dition of the corresponding amounts of albumin alone was without effect. In the presence of albumin-bound bilirubin, some 30% of the pigment was initially con- verted into biliverdin, whereas formation of this oxida- tion product was not observed with the free pigment. Also, the presence of bilirubin oxidase partially re- versed the inhibitory activity of bilirubin on AMVN-in- duced LDL oxidation in the absence but not presence of albumin. An attenuation of hydroperoxide formation and a temporary increasein LDL’s a-tocopherol concen- tration were observed when free- or albumin-bound bil- irubin were addedto AMVN-oxidizing, a-tocopherol- containing LDL. In contrast, hydroperoxide formation was not inhibited significantly when the albumin-bound pigment was added to oxidizing LDL after complete * This study was supported by Grants 910284 and 940915 (to R. S.) from the Australian National Health & Medical Research Council and Grant A09030140 from the Australian Research Council. The costs of publication of this article were defrayedin part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. The Heart Research Institute, 145 Missenden Rd., Camperdown, Syd- $ To whom correspondence should be addressed: Biochemistry Group, ney, N.S.W., Australia. Tel.: +61-2-550-3560; Fax: +61-2-550-3302. consumption of its a-tocopherol. Our results show that bilirubin inhibits oxidation of LDL lipids initiated within the lipoprotein coreand indicate that this activ- ity is mediated by interaction of the pigment with LDL’s a-tocopherol. Inadvertent modification of biological macromolecules due to free radical-mediated oxidation is implicated in the pathogen- esis of a number of human diseases, with ischemic heart dis- ease having received particular attention (1, 2). It has been proposed that oxidation of low density lipoprotein (LDL)’ con- tributes to the onset of atherosclerosis, as such modified LDL can accumulate in macrophages beneath the endothelium thereby forming fatty streaks which appear early in atherogen- esis (reviewed in Refs. 3-6). Oxidation of LDL’s lipids is thought to precede and may lead to oxidative modification of the lipoprotein’s protein moiety, apoprotein B-100 (7). The lat- ter modification can result in recognition of the lipoprotein by the scavenger (8) and/or oxidized LDL receptor(s) (91, giving rise to lipid-laden cells. Hence, a great deal of interest has focused on antioxidation of LDL, primarily the prevention of oxidation of LDL’s lipids by non-proteinaceous, low molecular weight antioxidants such as the vitamins E and C (10). This interest is fueled by recent epidemiological studies (11, 12). The efficacy of various compounds to prevent LDL (lipid) oxidation is commonly assessed in vitro by exposing the iso- lated lipoprotein to an oxidant, and the oxidizability of the lipoprotein is gauged by how rapidly oxidized lipids are formed or the LDL is converted into a high uptake form. With strong redox oxidants, such as copper, or with cells in a transition metal-containing cell culture medium, relatively little lipid hy- droperoxides are formed until LDL’s major antioxidant a-to- copherol (TOH, the biologically most active form of vitamin E) and the minor antioxidants (carotenoids) have been depleted (13,14). Following consumption of TOH, lipid peroxidation may enter a rapid, “uninhibited phase, toward the end of which the LDL becomes recognizable to the scavengerand/or oxidized LDL receptors (8, 9). These observations have led to the con- clusion that TOH is an efficient antioxidant for LDL in vitro and, together with the epidemiological evidence (11, 12, 151, that TOH protects the LDL from oxidation in uiuo. We have studied the earliest events in lipid peroxidation in human plasma and isolated LDL using a mild flux of organic peroxyl radicals or low concentrations of transition metals as The abbreviations used are: LDL, low density lipoprotein; m, 2,2’-azobis(2, 4-dimethylvaleronitrile); BR, bilirubin; CE-OOH, cho- lesterylester hydroperoxides; HSA, human serum albumin; HSA-BR, albumin-bound bilirubin; TOH, a-tocopherol; TMP, tocopherol-medi- ated peroxidation; TO‘, a-tocopheroxyl radical; HPLC, high perform- ance liquid chromatography. 16712
-
Upload
duongkhuong -
Category
Documents
-
view
215 -
download
2
Transcript of THE JOURNAL OF CHEMISTRY Vol. No. of June pp. by for · PDF fileFree and Albumin-bound...

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1994 by The American Society for Biochemistry and Molecular Biology, Inc.
Vol. 269, No. 24, Issue of June 17, pp. 16712-16719, 1994 Printed in U.S.A.
Free and Albumin-bound Bilirubin Are Efficient Co-antioxidants for a-Tocopherol, Inhibiting Plasma and Low Density Lipoprotein Lipid Peroxidation*
(Received for publication, January 4, 1994, and in revised form, March 23, 1994)
J i E i Neuiil and Roland Stocker$ From the Biochemistrv GrouD. The Heart Research Institute, 145 Missenden Road, Camperdown, Sydney, 2050 New South Wales, Australia
Peroxidation of the lipid moieties of low density li- poproteins (LDL) is regarded as an early event in atherogenesis. Because bilirubin is a physiological re- ductant with antioxidant activities, we investigated its inhibitory action on the radical-mediated oxidation of LDL and plasma lipids. Exposing fresh human blood plasma to lipophilic peroxyl radicals generated from 2,2’-azobis(2,4-dimethylvaleronitrile) ( A M V N ) resulted in rapid oxidation of ubiquinol-10, followed by that of ascorbate and bilirubin. Plasma lipids were well pro- tected from peroxidation as long as these three antioxi- dants were present, as assessed by the amounts of cho- lesterylester hydroperoxides formed during this period. Following consumption of these antioxidants, and in the presence of a-tocopherol, the rate of hydroperoxide for- mation increased sharply with roughly 2 molecules of cholesterylester hydroperoxides being formed for each peroxidation initiating event. Supplementation of AMVN-oxidizing plasma with exogenous bilirubin at the onset of rapid lipid peroxidation, i.e. after depletion of endogenous ubiquinol-10, ascorbate, and bilirubin, led to a halt in both hydroperoxide formation and consump- tion of a-tocopherol. When isolated LDL was incubated with AMVN, 4 molecules of cholesterylester hydroper- oxides were formed per peroxidation initiating event and while a-tocopherol was consumed. Addition of free or albumin-bound bilirubin to isolated LDL at the onset of oxidation resulted in a strong inhibition of hydroper- oxide formation and a-tocopherol consumption, the ef- fect being more pronounced with the free pigment. Ad- dition of the corresponding amounts of albumin alone was without effect. In the presence of albumin-bound bilirubin, some 30% of the pigment was initially con- verted into biliverdin, whereas formation of this oxida- tion product was not observed with the free pigment. Also, the presence of bilirubin oxidase partially re- versed the inhibitory activity of bilirubin on AMVN-in- duced LDL oxidation in the absence but not presence of albumin. An attenuation of hydroperoxide formation and a temporary increase in LDL’s a-tocopherol concen- tration were observed when free- or albumin-bound bil- irubin were added to AMVN-oxidizing, a-tocopherol- containing LDL. In contrast, hydroperoxide formation was not inhibited significantly when the albumin-bound pigment was added to oxidizing LDL after complete
* This study was supported by Grants 910284 and 940915 (to R. S.) from the Australian National Health & Medical Research Council and Grant A09030140 from the Australian Research Council. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The Heart Research Institute, 145 Missenden Rd., Camperdown, Syd- $ To whom correspondence should be addressed: Biochemistry Group,
ney, N.S.W., Australia. Tel.: +61-2-550-3560; Fax: +61-2-550-3302.
consumption of its a-tocopherol. Our results show that bilirubin inhibits oxidation of LDL lipids initiated within the lipoprotein core and indicate that this activ- ity is mediated by interaction of the pigment with LDL’s a-tocopherol.
Inadvertent modification of biological macromolecules due to free radical-mediated oxidation is implicated in the pathogen- esis of a number of human diseases, with ischemic heart dis- ease having received particular attention (1, 2). It has been proposed that oxidation of low density lipoprotein (LDL)’ con- tributes to the onset of atherosclerosis, as such modified LDL can accumulate in macrophages beneath the endothelium thereby forming fatty streaks which appear early in atherogen- esis (reviewed in Refs. 3-6). Oxidation of LDL’s lipids is thought to precede and may lead to oxidative modification of the lipoprotein’s protein moiety, apoprotein B-100 (7). The lat- ter modification can result in recognition of the lipoprotein by the scavenger (8) and/or oxidized LDL receptor(s) (91, giving rise to lipid-laden cells. Hence, a great deal of interest has focused on antioxidation of LDL, primarily the prevention of oxidation of LDL’s lipids by non-proteinaceous, low molecular weight antioxidants such as the vitamins E and C (10). This interest is fueled by recent epidemiological studies (11, 12).
The efficacy of various compounds to prevent LDL (lipid) oxidation is commonly assessed in vitro by exposing the iso- lated lipoprotein to an oxidant, and the oxidizability of the lipoprotein is gauged by how rapidly oxidized lipids are formed or the LDL is converted into a high uptake form. With strong redox oxidants, such as copper, or with cells in a transition metal-containing cell culture medium, relatively little lipid hy- droperoxides are formed until LDL’s major antioxidant a-to- copherol (TOH, the biologically most active form of vitamin E) and the minor antioxidants (carotenoids) have been depleted (13,14). Following consumption of TOH, lipid peroxidation may enter a rapid, “uninhibited phase, toward the end of which the LDL becomes recognizable to the scavenger and/or oxidized LDL receptors (8, 9). These observations have led to the con- clusion that TOH is an efficient antioxidant for LDL in vitro and, together with the epidemiological evidence (11, 12, 151, that TOH protects the LDL from oxidation in uiuo.
We have studied the earliest events in lipid peroxidation in human plasma and isolated LDL using a mild flux of organic peroxyl radicals or low concentrations of transition metals as
The abbreviations used are: LDL, low density lipoprotein; m, 2,2’-azobis(2, 4-dimethylvaleronitrile); BR, bilirubin; CE-OOH, cho- lesterylester hydroperoxides; HSA, human serum albumin; HSA-BR, albumin-bound bilirubin; TOH, a-tocopherol; TMP, tocopherol-medi- ated peroxidation; TO‘, a-tocopheroxyl radical; HPLC, high perform- ance liquid chromatography.
16712

Bilirubin and Lipoprotein Lipid Antioxidation 16713
the oxidants (16-20). Several observations made in these stud- ies were paradoxical, z.e. not compat ible with the conventional view of TOHs molecular action as a chain-breaking, lipid- soluble antioxidant (21). The findings led us to conclude that lipid peroxidation in isolated LDL is actually propagated by the a-tocopheroxyl radical (TO') rather than the lipid peroxyl radi- cal. To be an efficient antioxidant, TOH requires a suitable "co-antioxidant" which prevents the adverse effect of TO' by scavenging and "exporting" this radical from the LDL par t ic le (for a detailed repor t see Ref. 20). Ascorbate, the reduced form of vitamin C, is t h o u g h t t o a c t as such a co-antioxidant in biological systems. It readily reacts with (22) and thereby con- ver t s the lipophilic TO' in to the charged, aqueous ascorbyl radical. LDL-associated ubiquinol-10 (the reduced form of co- enzyme Q) has been proposed to act in a similar fashion (19, 20). It reacts with TO' (23) as well, producing a semiquinone radical that itself readily combines with molecular oxygen to give ubiquinone-10 and superoxide anion radical. The l a t t e r may diffuse into the aqueous medium surrounding LDL (19). Indeed, both ascorbate and ubiquinol-10 are most efficient in- hibitors of radical-mediated oxidation of LDL lipids (17, 18,20, 24).
A question arising from the aforementioned is whether o ther biological co-antioxidants exist for TOH in LDL and other bio- logical lipids. Recent observations by Packer and colleagues (25) suggest that cer ta in membrane-associated proteins in- volved in electron transfer reactions can reduce TO' in lipid bilayers. Among the sulfhydryl compounds, cysteine (26) ap- pea r s t o be more active than reduced glutathione (27, 28) and dehydrolipoic acid (25, 291, a l though the low levels of cysteine in extracellular f luids make its role in reducing LDL's TO' doubtful. Urate (20, 30) and p-carotene (29) are no t able t o reduce TO'. In contrast, a potential candidate for a physiologi- cal reductant of TO' is bilirubin (BR). The pigment, in its free form and when bound to its physiological carrier protein of extracellular f luids, albumin, efficiently scavenges radicals (31, 32), and a water-soluble form of BR has been shown to syner- gize wi th TOH in liposomal lipid antioxidation (28). We there- fore tested whether free- and albumin-bound BR inhibited lipid oxidation in human b lood plasma and isolated LDL exposed to lipophilic peroxyl radicals. The results obtained show that both forms of the pigment efficiently inhibit lipid oxidation and that this antioxidant activity is likely due to an interaction of BR wi th TOH incorporated within lipoproteins.
EXPERIMENTAL PROCEDURES MateriaZs-2,2'-Azobis(2,4-dimethylvaleronitrile) ( A M V N ) was ob-
tained from Polysciences (Warrington, PA), urate, ascorbate, BR (con- taining less than 10% of the non-physiological IIIa and XIIIa isomers), BR oxidase (bi1ilubin:oxygen oxidoreductase, EC 1.3.3.51, and human serum albumin (HSA, essentially fatty acid free) were from Sigma, and biliverdin was from Porphyrin Products (Logan, UT). Solvents were from Malinckrodt and of HPLC quality. All other chemicals were of the highest punty available. Nanopure water was used for preparation of all aqueous buffers that were subsequently treated with Chelexa 100 resin (Bio-Rad). This treatment resulted in elimination of contaminat- ing, redox-active amounts of transition metals, as verified by the ascor- bate autoxidation method (33).
Preparation of Plasma and LDL-Blood was obtained from a non- fasted healthy male donor (35 years old) and drawn into heparinized vacutainers. Plasma was prepared by centrifuging blood a t 1000 x g at 4 "C for 15 min and used undiluted. Freshly prepared plasma con- tained, as a rule, -7 PM BR, 55 p~ ascorbate, 400 urate, 22 pv TOH, 0.45 p~ p-carotene, 0.7 p~ lycopene, and 0.6 PM ubiquinol-10 as non- proteinaceous antioxidants and -0.7 mM unesterified cholesterol, 0.18 mM cholesteryl arachidonate, 1.54 mM cholesteryl linoleate, and 1.03 mM cholesteryl oleate as the major lipids.
LDL, essentially free of other plasma proteins, was isolated from freshly prepared plasma by a rapid density ultracentrifugation using a Beckman (Palo Alto, CA) TL-100 table-top centrifuge equipped with a
TL-100.4 rotor (2 h, 15 "C, 100,000 rpm) (34) and used within 48 h of preparation. No special care was taken to preserve the small amounts of ubiquinol-10 in LDL. One LDL particle contained 550 molecules of unesterified cholesterol, -735 molecules of cholesteryl oleate, =1135 cholesteryl linoleate, -123 cholesteryl arachidonate, -12 TOH, and 1 molecule of apoprotein B-100. Prior to oxidation, potassium bromide and other low molar mass contaminants (including small amounts of hydrophilic antioxidants) were removed from LDL by passage of the lipoprotein solution through a PD-10 gel-filtration column (Pharmacia, Uppsala, Sweden).
Preparation of BR-containing Samples-HSA-BR was prepared by addition of freshly dissolved BR (in 50 mM NaOH; concentration of BR, 1 mM) to a solution of HSA (in 0.1 M phosphate buffer, pH 7.41, at a final molar ratio of BR to HSA of up to 1:5. Previous studies by others have shown that addition of BR to HSA in such way results in binding of the pigment to its primary, physiological binding site (35). For some experi- ments BR was added to plasma or isolated LDL as an aliquot of a freshly prepared pigment solution in 50 mM NaOH. In these cases the volume of NaOH added was <0.15% (viv), and the corresponding control samples (no BR) were supplemented with the appropriate amount of 50 mM NaOH. To assess whether BR was incorporated into LDL, the com- plex was passed through a gel-filtration column (PD-10, Pharmacia), and the pigment concentration was determined in the resulting LDL.
Oxidation of Plasma and LDL-Oxidations were carried out for the indicated period of time in aerated reaction tubes protected from light and placed in a shaking water bath set a t 37 "C. AMVN (2 mM, final concentration) was used to initiate all oxidation experiments. This ther- molabile, lipophilic azo-compound decomposes within hydrophobic sites to an initial (single) pair of alkyl radicals which under aerobic condi- tions gives rise to lipophilic peroxyl radicals formed at a constant rate (36, 37). The radical generator was added to plasma or isolated LDL (1-2 mg proteidml) as an ethanolic solution and dropwise, as otherwise precipitation was observed. Attempts to incorporate more than 2 mM AMVN also led to precipitation, likely due to destabilization of the lipoprotein emulsion at higher molar AMVN-to-LDL ratios.
The distribution of AMVN in whole plasma was assessed by density fractionation of plasma. Briefly, the density of AMVN-containing plasma was adjusted to 1.06 g/ml by addition and dissolving of the appropriate amount of solid KBr. Density-adjusted plasma samples were then placed into 5.1-ml tubes, centrifuged for 2 h at 15 "C and 100,000 rpm (TL-100 centrifuge, TL100.4 rotor) before individual frac- tions (550 pl) were removed by aspiration (from top to bottom) and examined for their concentration of A M V N . For this, the various frac- tions (500 pl) were extracted with hexane and acidified methanol (den- sity fraction/MeOWhexane = 1:4:20, v/v/v), the organic phase removed, dried, and redissolved in EtOH. AMVN was determined by reversed- phase HPLC (C18 column, 250 x 4.6 mm inner diameter, 5-pm particles, Supelco, Bellefonte, PA) using MeOWH,O, 95:5 (v/v) as the mobile phase at 1 ml min" and monitoring the eluant a t 345 nm. The distri- bution of AMVN in human plasma is shown in Fig. 1. The extent of incorporation of AMVN into LDL was verified by following the rate of oxidation at 37 "C in the supplemented (2 mM A M V N ) lipoprotein with or without prior passing through a PD-10 column. For experiments with HSA-BR or BR, the AMVN-supplemented LDL was preincubated for 5 min at 37 "C before addition of the pigment. Where indicated, BR oxi- dase was added to the sample at the beginning of the oxidation at the final enzyme activity of 0.98 nkat ml".
Analysis of Antioxidants, Lipids, and Lipid Hydroperoxides- Aliquots (200 pl) of the reaction mixtures were withdrawn at the time points indicated and extracted with 2 ml of cold methanol (containing 0.1% (v/v) acetic acid) and 10 ml of hexane, vortexed vigorously for about 30 s, and centrifuged at 4 "C and 1,000 x g for 5 min. The resulting hexane layer (5 ml) was removed, dried in uacuo, the residue dissolved in 200 pl of ethanol, and used for subsequent HPLC analyses of lipophilic antioxidants, lipids, and cholesterylester hydroperoxides (CE-OOH) (see below). The aqueous methanol phase was removed, fil- tered (0.2 pm), and used immediately for HPLC analysis of ascorbate and urate by electrochemical detection (38). For analysis of bile pig- ments, an aliquot of the reaction mixture was mixed with cold ethanol (1:4, v/v), vortexed, centrifuged at 16,000 x g for 15 min a t room tem- perature, filtered (0.2-pm), and separated on a C,, column (50 x 4.6 mm inner diameter, 5-pm particles; Supelco) eluted with 0.1 M methanolic dioctylamine/H,O (95:5 (v/v), pH 7.9) a t 0.5 ml min" (39). Biliverdin (eluting at 2.6 m i d and BR (7.7 min) were monitored at 440 and 370 nm, respectively.
Lipid-soluble antioxidants (TOH, ubiquinol-10, p-carotene, and lyco- pene), unoxidized (unesterified cholesterol and cholesterylesters), and oxidized lipids (CE-OOH) present in the hexane extract were analyzed
167 14 Bilirubin and Lipoprotein Lipid Antioxidation
30 A Plasma
proteins LDL T
1 2 3 4 5 6 7 8 9
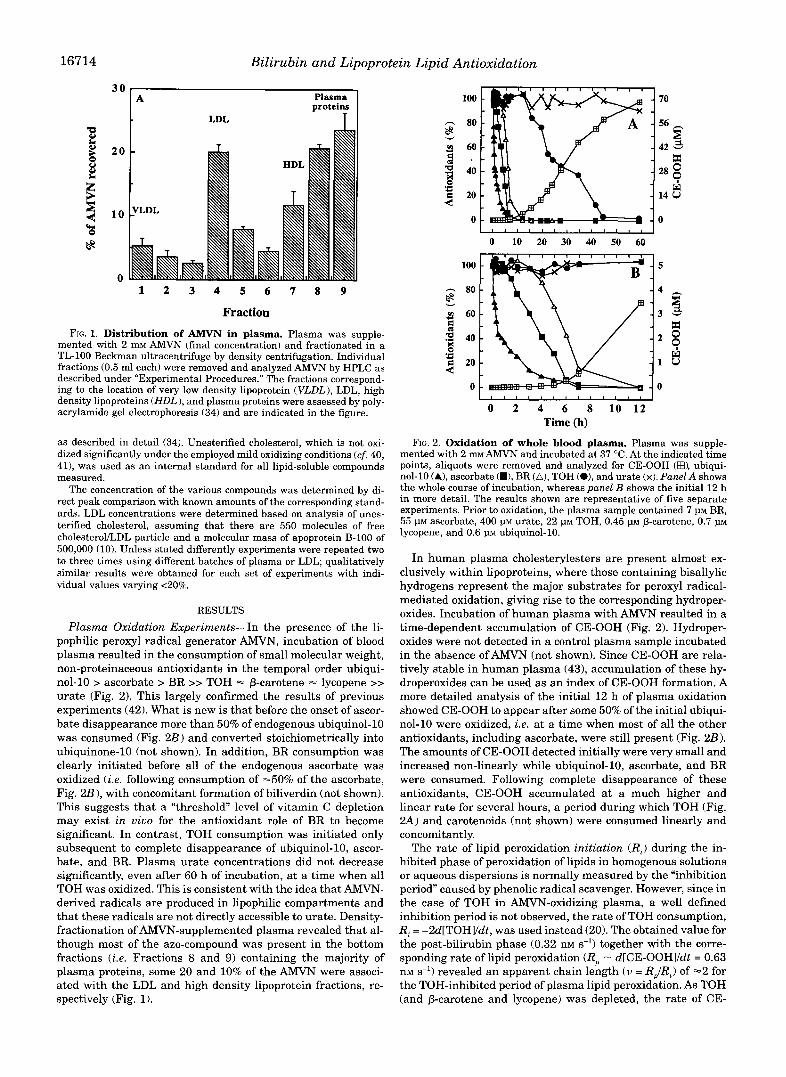
Fraction FIG. 1. Distribution of AMVN in plasma. Plasma was supple-
mented with 2 mM AMVN (final concentration) and fractionated in a TL-100 Beckman ultracentrifuge by density centrifugation. Individual fractions (0.5 ml each) were removed and analyzed AMVN by HPLC as described under “Experimental Procedures.” The fractions correspond- ing to the location of very low density lipoprotein (VLDL), LDL, high density lipoproteins (HDL), and plasma proteins were assessed by poly- acrylamide gel electrophoresis (34) and are indicated in the figure.
as described in detail (34). Unesterified cholesterol, which is not oxi- dized significantly under the employed mild oxidizing conditions (cf. 40, 411, was used as an internal standard for all lipid-soluble compounds measured.
The concentration of the various compounds was determined by di- rect peak comparison with known amounts of the corresponding stand- ards. LDL concentrations were determined based on analysis of unes- terified cholesterol, assuming that there are 550 molecules of free cholesteroYLDL particle and a molecular mass of apoprotein B-100 of 500,000 (10). Unless stated differently experiments were repeated two to three times using different batches of plasma or LDL; qualitatively similar results were obtained for each set of experiments with indi- vidual values varying <20%.
RESULTS
Plasma Oxidation Experiments-In the presence of the li- pophilic peroxyl radical generator AMVN, incubation of blood plasma resulted in the consumption of small molecular weight, non-proteinaceous antioxidants in the temporal order ubiqui- nol-10 > ascorbate > BR >> TOH - p-carotene - lycopene >> urate (Fig. 2). This largely confirmed the results of previous experiments (42). What is new is that before the onset of ascor- bate disappearance more than 50% of endogenous ubiquinol-10 was consumed (Fig. 2B) and converted stoichiometrically into ubiquinone-10 (not shown). In addition, BR consumption was clearly initiated before all of the endogenous ascorbate was oxidized (i.e. following consumption of -50% of the ascorbate, Fig. 2B), with concomitant formation of biliverdin (not shown). This suggests that a “threshold” level of vitamin C depletion may exist in vivo for the antioxidant role of BR to become significant. In contrast, TOH consumption was initiated only subsequent to complete disappearance of ubiquinol-10, ascor- bate, and BR. Plasma urate concentrations did not decrease significantly, even after 60 h of incubation, at a time when all TOH was oxidized. This is consistent with the idea that AMVN- derived radicals are produced in lipophilic compartments and that these radicals are not directly accessible to urate. Density- fractionation of AMVN-supplemented plasma revealed that al- though most of the azo-compound was present in the bottom fractions (i.e. Fractions 8 and 9) containing the majority of plasma proteins, some 20 and 10% of the AMVN were associ- ated with the LDL and high density lipoprotein fractions, re- spectively (Fig. 1).
- 100 -
80 -
60 - 40 -
20 -
0 -
70
56 h
42 - X 3
28 9 14 U
W
0
100 B i 5
- 4 - E - 3 2
- 2 0 - x
- s - 1 U
- , o 0 2 4 6 8 1 0 1 2
Time (h)
mented with 2 m~ AMVN and incubated at 37 “C. At the indicated time FIG. 2. Oxidation of whole blood plasma. Plasma was supple-
points, aliquots were removed and analyzed for CE-OOH (EEL ubiqui- nol-10 (A), ascorbate (W), BR (A), TOH (O), and urate (x). PaneZA shows the whole course of incubation, whereas panel B shows the initial 12 h in more detail. The results shown are representative of five separate experiments. Prior to oxidation, the plasma sample contained 7 p~ BR, 55 p~ ascorbate, 400 PM urate, 22 p~ TOH, 0.45 PM p-carotene, 0.7 PM lycopene, and 0.6 PM ubiquinol-10.
In human plasma cholesterylesters are present almost ex- clusively within lipoproteins, where those containing bisallylic hydrogens represent the major substrates for peroxyl radical- mediated oxidation, giving rise to the corresponding hydroper- oxides. Incubation of human plasma with AMVN resulted in a time-dependent accumulation of CE-OOH (Fig. 2). Hydroper- oxides were not detected in a control plasma sample incubated in the absence of AMVN (not shown). Since CE-OOH are rela- tively stable in human plasma (431, accumulation of these hy- droperoxides can be used as an index of CE-OOH formation. A more detailed analysis of the initial 12 h of plasma oxidation showed CE-OOH to appear after some 50% of the initial ubiqui- nol-10 were oxidized, i.e. at a time when most of all the other antioxidants, including ascorbate, were still present (Fig. 2 B ) . The amounts of CE-OOH detected initially were very small and increased non-linearly while ubiquinol-10, ascorbate, and BR were consumed. Following complete disappearance of these antioxidants, CE-OOH accumulated at a much higher and linear rate for several hours, a period during which TOH (Fig. 2 A ) and carotenoids (not shown) were consumed linearly and concomitantly.
The rate of lipid peroxidation initiation (R,) during the in- hibited phase of peroxidation of lipids in homogenous solutions or aqueous dispersions is normally measured by the “inhibition period caused by phenolic radical scavenger. However, since in the case of TOH in AMVN-oxidizing plasma, a well defined inhibition period is not observed, the rate of TOH consumption, Ri = -2d[TOH]/dt, was used instead (20). The obtained value for the post-bilirubin phase (0.32 nM s-’) together with the corre- sponding rate of lipid peroxidation (R, - d[CE-OOHYdt = 0.63 nM s-’) revealed an apparent chain length (v = R,/R,) of -2 for the TOH-inhibited period of plasma lipid peroxidation. As TOH (and p-carotene and lycopene) was depleted, the rate of CE-

Bilirubin and Lipoprotein Lipid Antioxidation 16715
c
100
- 20
- 40
60-
- 80
-
0 - 4 .,,,,. 40
32 E 5.
24- X
16 8 c j
8 U
0
0 10 20 30 40 50
Time (h) FIG. 3. Oxidation of plasma and its inhibition by HSA-BR in the
absence of ubiquinol-10 and ascorbate. Plasma was supplemented with 2 mMAMVN and incubated at 37 “C. Following complete consump- tion of endogenous ubiquinol-10, ascorbate, and BR (i.e. after 20.3 h of incubation as indicated by arrow), the mixture was supplemented with
and incubated for further 24 h. At the indicated time points, aliquots 10 VM BR (solid lines) or the appropriate amount of NaOH (broken lines)
were removed and analyzed for CE-OOH (0, H), ubiquinol-10 (A), and TOH (0,O) as described under “Experimental Procedures.”
OOH formation decreased for at least a further 10 h. Oxidation experiments were not extended beyond 60 h of incubation. Thus, the overall pattern of CE-OOH accumulation during the examined oxidation period of plasma was complex, with the maximum rate achieved during the inhibited phase of peroxi- dation (i.e. when more than 40% of TOH were still present) after consumption of ubiquinol-10, ascorbate, and BR.
Addition of physiological amounts of BR (which would bind to albumin) to AMVN-oxidizing human plasma subsequent to complete consumption of all endogenous ubiquinol-10, ascor- bate, and BR but in the presence of TOH, slowed down the rate of TOH consumption (Fig. 3). More importantly, the rate of CE-OOH formation also decreased (by 70%) thereby producing a rather well defined period of inhibited lipid peroxidation. The length of this inhibition period corresponded to the time re- quired to consume the added BR and was directly proportional to the amounts of BR added (not shown).
Oxidation of Isolated LDL-Addition of an ethanolic solution of AMVN to isolated LDL resulted in “incorporation” of the radical generator into the lipoprotein. Incorporation is defined here based on the observation that virtually all of the AMVN added to an LDL suspension was recovered with the lipoprotein following its gel filtration through a PD-10 column (data not shown). Additionally, the rate of CE-OOH accumulation in “containing LDL was indistinguishable whether or not the lipoprotein was gel filtered subsequent to addition of the azo-compound and prior to incubation at 37 “C (not shown).
Fig. 4 shows the oxidation and antioxidation of isolated LDL incubated with AMVN in the absence and presence of HSA (75 VM) containing increasing amounts of BR. In the absence of HSA-BR, LDL’s cholesterylesters were peroxidized in a radical chain reaction with an apparent chain length of -9. This is consistent with recent reports (17,20,24). The presence of HSA alone a t 75 p~ had no significant effect on either CE-OOH formation or TOH consumption in “oxidizing LDL (not shown). However, when used at much higher concentrations (e.g. 500 p~), significant inhibition of both parameters was observed (data not shown). LDL oxidation was inhibited strongly by 75 VI HSA containing BR, with the extent of inhi- bition of CE-OOH accumulation and TOH consumption directly proportional to the initial concentration of the pigment added (Fig. 4). A sparing action of BR present in the aqueous phase as
100 - 80 -
- 60- c 4 0 -
20 - 0 t
- 40
- 30
- 20
- 10
- 0
E 5. v
0 5 0 100 150 200 250 Time (min)
FIG. 4. Peroxidation of isolated LDL supplemented with HSA-
AMVN (final concentration), preincubated for 5 min at 37 “C, and then BR. LDL isolated from fresh plasma was supplemented with 2 mM
incubated at 37 “C after addition of HSA-BR at the BR concentration of 15 (rhombs), 4.5 (triangles), or 1.5 PM (squares). The concentration of HSA was 75 PM in all cases. The control sample (no HSA or BR, circles) was supplemented with a corresponding volume of 50 mM NaOH. At the time points indicated, aliquots were taken, extracted, and analyzed for TOH (empty symbols in A), BR (empty symbols in B ) , and CE-OOH (filled symbols in A and B ) as described under “Experimental Proce- dures.’’ The numbers refer to the concentrations of BR used.
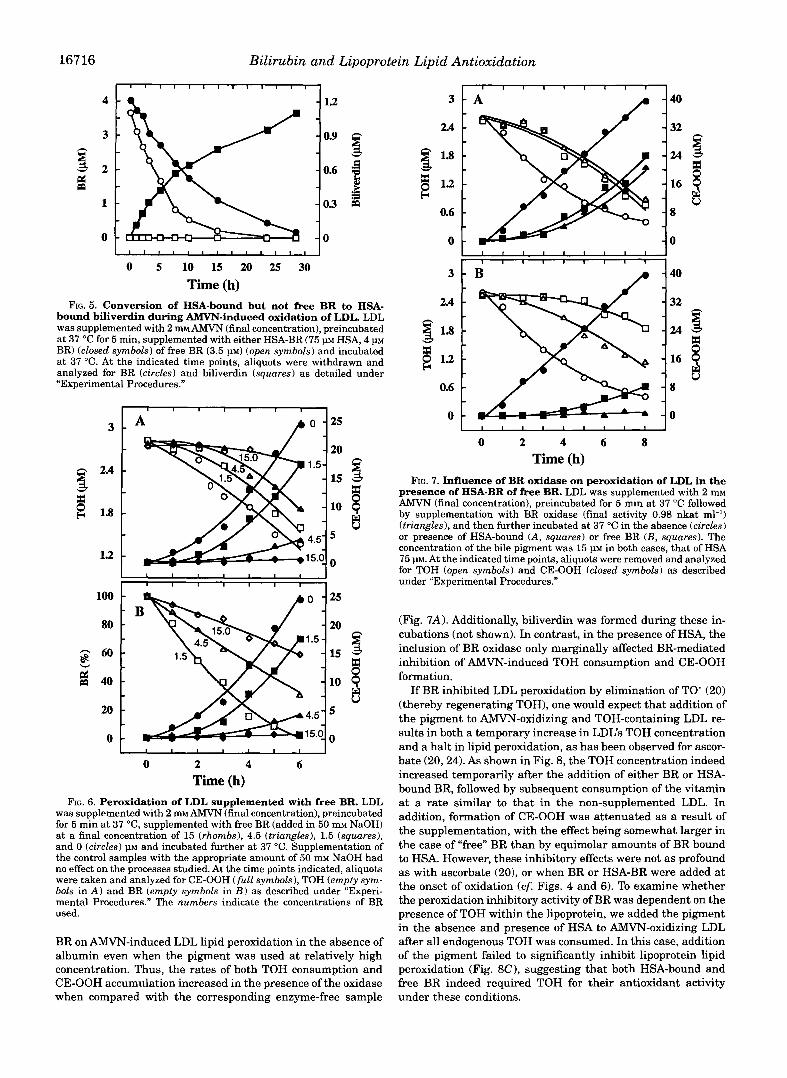
an albumin complex on AMVN-induced TOH consumption is indicative of regeneration of TOH from TO’ by the pigment. The rate of CE-OOH accumulation increased markedly after an initial inhibition period of 3-4 h corresponding to the loss of 50-60% of the BR (not shown), reminiscent of the situation in plasma where BR was also bound to HSA (cf. Fig. 2B). As judged by their initial rates of consumption and formation, some 30% of the BR were initially converted into biliverdin, its two-electron oxidation product (Fig. 5).
An even stronger inhibition of AMVN-induced LDL lipid oxi- dation was observed with free BR (Fig. 6). Thus, low micromo- lar amounts of the pigment in the absence of HSA initially almost completely prevented CE-OOH formation and TOH con- sumption. The duration of the resulting clear inhibition period of lipid peroxidation was proportional to the initial BR concen- tration, and BR was consumed during this phase. Following the inhibition period, CE-OOH accumulated a t higher rates, simi- lar to that of oxidizing LDL in the absence of the pigment. Consumption of some TOH was observed before the first traces of CE-OOH were detected. In contrast to the situation observed with HSA-bound BR, oxidation of the pigment in the absence of HSA did not result in formation of biliverdin (Fig. 5). Separate gel filtration experiments with LDL-BR mixtures showed that the water-insoluble BR was “associated with rather than “in- corporated” in the lipoproteins, as the pigment was retained by the Sephadex matrix (not shown). In contrast, endogenous TOH, ubiquinol-10, and carotenoids remained within LDL when the lipoprotein solution was treated in the same way (not shown).
Fig. 7 shows that the presence of BR oxidase, an enzyme that oxidizes free BR much more efficiently than albumin-bound BR to biliverdin (44), partially reversed the inhibitory activity of
16716 Bilirubin and Lipoprotein Lipid Antioxidation
3 3
3 -
2 -
1 -
0 -
- 0.9
- 0.6
- 0 3
- 0 I ~ I I I I I I I I I I ~ I J
0 5 10 15 20 25 30 Time (h)
bound biliverdin during AMVN-induced oxidation of LDL. LDL FIG. 5. Conversion of HSA-bound but not free BR to HSA-
was supplemented with 2 mMAMVN (final concentration), preincubated at 37 “C for 5 min, supplemented with either HSA-BR (75 p HSA, 4 PM BR) (closed symbols) of free BR (3.5 PM) (open symbols) and incubated at 37 “C. At the indicated time points, aliquots were withdrawn and analyzed for BR (circles) and biliverdin (squares) as detailed under “Experimental Procedures.”
- 20
3 2A I 1.5: 15 3 8 18- 110 8 X
M
6 4.5-
1.2 - 15.0
100 - 80 -
- 60- 3 40 -
20 - 0 -
E
0 - 2 5
20
- 15 - 10
- 1.5 -
4.5-
15.Q I I I I t I I I
0 2 4 6 Time (h)
FIG. 6. Peroxidation of LDL supplemented with free BR. LDL was supplemented with 2 mM AMVN (final concentration), preincubated for 5 min a t 37 “C, supplemented with free BR (added in 50 rn NaOH) at a final concentration of 15 (rhombs), 4.5 (triangles), 1.5 (squares), and 0 (circles) p~ and incubated further at 37 “C. Supplementation of the control samples with the appropriate amount of 50 mM NaOH had no effect on the processes studied. At the time points indicated, aliquots were taken and analyzed for CE-OOH (full symbols), TOH (empty sym- bols in A ) and BR (empty symbols in E ) as described under “Experi- mental Procedures.” The numbers indicate the concentrations of BR used.
BR on “induced LDL lipid peroxidation in the absence of albumin even when the pigment was used at relatively high concentration. Thus, the rates of both TOH consumption and CE-OOH accumulation increased in the presence of the oxidase when compared with the corresponding enzyme-free sample
2.4 -
18 - 1.2 - 0.6 -
0 -
- 32
-24
116 1 - 8
2.4 -
3 lJ3 : X 13 - 0.6 -
0 -
- 40
- 32
-24
I16
- 8
- 0 I I I I I I I I I I l
0 2 4 6 8 Time (h)
FIG. 7. Influence of BR oxidase on peroxidation of LDL in the presence of HSA-BR of free BR. LDL was supplemented with 2 mM AhWN (final concentration), preincubated for 5 min at 37 “C followed by supplementation with BR oxidase (final activity 0.98 nkat ml-I) (triangles), and then further incubated at 37 “C in the absence (circles) or presence of HSA-bound (A, squares) or free BR ( E , squares). The concentration of the bile pigment was 15 PM in both cases, that of HSA
for TOH (open symbols) and CE-OOH (closed symbols) as described 75 PM. At the indicated time points, aliquots were removed and analyzed
under “Experimental Procedures.”
(Fig. 7A). Additionally, biliverdin was formed during these in- cubations (not shown). In contrast, in the presence of HSA, the inclusion of BR oxidase only marginally affected BR-mediated inhibition of AMVN-induced TOH consumption and CE-OOH formation.
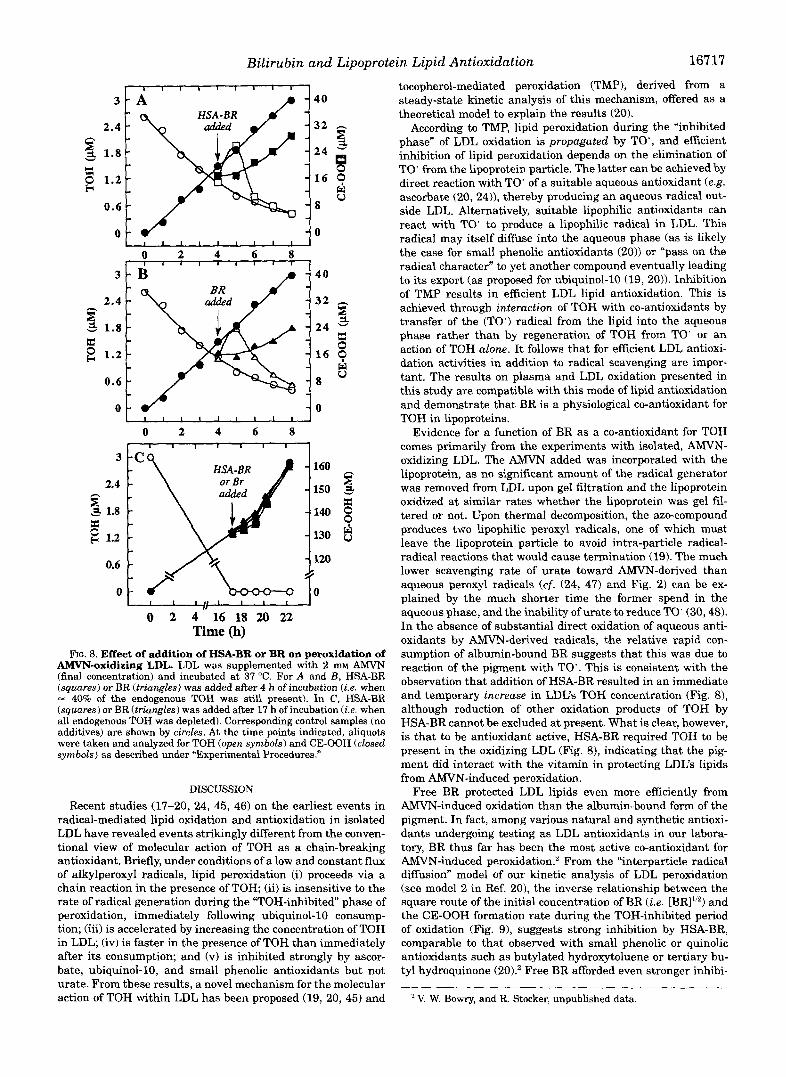
If BR inhibited LDL peroxidation by elimination of TO’ (20) (thereby regenerating TOH), one would expect that addition of the pigment to “oxidizing and TOH-containing LDL re- sults in both a temporary increase in LDL’s TOH concentration and a halt in lipid peroxidation, as has been observed for ascor- bate (20,24). As shown in Fig. 8, the TOH concentration indeed increased temporarily after the addition of either BR or HSA- bound BR, followed by subsequent consumption of the vitamin at a rate similar to that in the non-supplemented LDL. In addition, formation of CE-OOH was attenuated as a result of the supplementation, with the effect being somewhat larger in the case of “free” BR than by equimolar amounts of BR bound to HSA. However, these inhibitory effects were not as profound as with ascorbate (20), or when BR or HSA-BR were added at the onset of oxidation (cf. Figs. 4 and 6). To examine whether the peroxidation inhibitory activity of BR was dependent on the presence of TOH within the lipoprotein, we added the pigment in the absence and presence of HSA to AMVN-oxidizing LDL after all endogenous TOH was consumed. In this case, addition of the pigment failed to significantly inhibit lipoprotein lipid peroxidation (Fig. 8C), suggesting that both HSA-bound and free BR indeed required TOH for their antioxidant activity under these conditions.
Bilirubin and Lipoprotein Lipid Antioxidation 16717
3 40
2.4 32 h E 3 1.8
8 1.2
=L 24 -
Y
0 16 0
CI w 0.6 8
u
0 0
0 2 4 6 8
3 F B 1 l 1 1 1 1 1 1 1
E
8 1.2
2.4
s 1.8 EE
0.6
0
3 2
24 - 16
8
0
E 3.
3 w u
3 -c -
- -
160 2.4 -
B
g 1.2 - 130 8 3 1.8 X
-
0.6 - 0 -
120 -
0 2 4 16 18 20 22 Time (h)
FIG. 8. Effect of addition of HSA-BR or BR on peroxidation of AMVN-oxidizing LDL. LDL was supplemented with 2 mM AMVN (final concentration) and incubated at 37 “C. For A and B, HSA-BR (squares) or BR (triangles) was added after 4 h of incubation (Le. when - 40% of the endogenous TOH was still present). In C , HSA-BR (squares) or BR (triangles) was added after 17 h of incubation ( i . e . when all endogenous TOH was depleted). Corresponding control samples (no additives) are shown by circles. At the time points indicated, aliquots were taken and analyzed for TOH (open symbols) and CE-OOH (closed symbols) as described under “Experimental Procedures.”
DISCUSSION
Recent studies (17-20, 24, 45, 46) on the earliest events in radical-mediated lipid oxidation and antioxidation in isolated LDL have revealed events strikingly different from the conven- tional view of molecular action of TOH as a chain-breaking antioxidant. Briefly, under conditions of a low and constant flux of alkylperoxyl radicals, lipid peroxidation (i) proceeds via a chain reaction in the presence of TOH; (ii) is insensitive to the rate of radical generation during the “TOH-inhibited phase of peroxidation, immediately following ubiquinol-10 consump- tion; (iii) is accelerated by increasing the concentration of TOH in LDL; (iv) is faster in the presence of TOH than immediately after its consumption; and (v) is inhibited strongly by ascor- bate, ubiquinol-10, and small phenolic antioxidants but not urate. From these results, a novel mechanism for the molecular action of TOH within LDL has been proposed (19, 20, 45) and
tocopherol-mediated peroxidation (TMP), derived from a steady-state kinetic analysis of this mechanism, offered as a theoretical model to explain the results (20).
According to TMP, lipid peroxidation during the “inhibited phase“ of LDL oxidation is propagated by TO’, and effkient inhibition of lipid peroxidation depends on the elimination of TO’ from the lipoprotein particle. The latter can be achieved by direct reaction with TO’ of a suitable aqueous antioxidant (e.g. ascorbate (20,24)), thereby producing an aqueous radical out- side LDL. Alternatively, suitable lipophilic antioxidants can react with TO’ to produce a lipophilic radical in LDL. This radical may itself diffuse into the aqueous phase (as is likely the case for small phenolic antioxidants (20)) or “pass on the radical character” to yet another compound eventually leading to its export (as proposed for ubiquinol-10 (19, 20)). Inhibition of TMP results in efficient LDL lipid antioxidation. This is achieved through interaction of TOH with co-antioxidants by transfer of the (TO‘) radical from the lipid into the aqueous phase rather than by regeneration of TOH from TO‘ or an action of TOH alone. It follows that for efficient LDL antioxi- dation activities in addition to radical scavenging are impor- tant. The results on plasma and LDL oxidation presented in this study are compatible with this mode of lipid antioxidation and demonstrate that BR is a physiological co-antioxidant for TOH in lipoproteins.
Evidence for a function of BR as a co-antioxidant for TOH comes primarily from the experiments with isolated, AMVN- oxidizing LDL. The AMVN added was incorporated with the lipoprotein, as no significant amount of the radical generator was removed from LDL upon gel filtration and the lipoprotein oxidized at similar rates whether the lipoprotein was gel fil- tered or not. Upon thermal decomposition, the azo-compound produces two lipophilic peroxyl radicals, one of which must leave the lipoprotein particle to avoid intra-particle radical- radical reactions that would cause termination (19). The much lower scavenging rate of urate toward AMVN-derived than aqueous peroxyl radicals (cf. (24, 47) and Fig. 2) can be ex- plained by the much shorter time the former spend in the aqueous phase, and the inability of urate to reduce TO‘ (30,481. In the absence of substantial direct oxidation of aqueous anti- oxidants by AMVN-derived radicals, the relative rapid con- sumption of albumin-bound BR suggests that this was due to reaction of the pigment with TO‘. This is consistent with the observation that addition of HSA-BR resulted in an immediate and temporary increase in LDL‘s TOH concentration (Fig. 8), although reduction of other oxidation products of TOH by HSA-BR cannot be excluded at present. What is clear, however, is that to be antioxidant active, HSA-BR required TOH to be present in the oxidizing LDL (Fig. 8), indicating that the pig- ment did interact with the vitamin in protecting LDL’s lipids from AMVN-induced peroxidation.
Free BR protected LDL lipids even more efficiently from AMVN-induced oxidation than the albumin-bound form of the pigment. In fact, among various natural and synthetic antioxi- dants undergoing testing as LDL antioxidants in our labora- tory, BR thus far has been the most active co-antioxidant for AMVN-induced peroxidation? From the “interparticle radical diffusion” model of our kinetic analysis of LDL peroxidation (see model 2 in Ref. 201, the inverse relationship between the square route of the initial concentration of BR (i.e. [BR]”2) and the CE-OOH formation rate during the TOH-inhibited period of oxidation (Fig. 91, suggests strong inhibition by HSA-BR, comparable to that observed with small phenolic or quinolic antioxidants such as butylated hydroxytoluene or tertiary bu- tyl hydroquinone (20)? Free BR afforded even stronger inhibi-
V. W. Bowry, and R. Stocker, unpublished data.
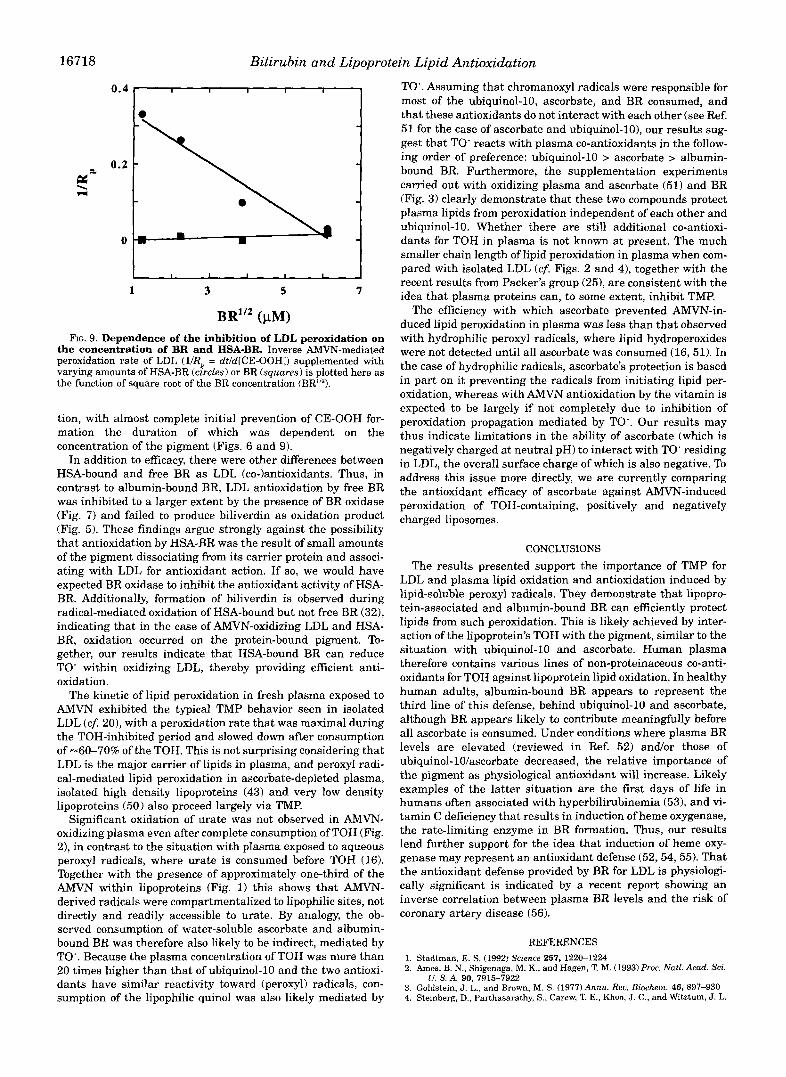
16718
0.4
0.2 \ wp rl
0
Bilirubin and Lipoprotein Lipid Antioxidation
I- 1 I I I I I I 1 3 5 7
BR”’ (pM)
the concentration of BR and HSA-BR. Inverse AMVN-mediated FIG. 9. Dependence of the inhibition of LDL peroxidation on
peroxidation rate of LDL (1/R = dt/d[CE-OOHl) supplemented with varying amounts of HSA-BR (c!rdes) or BR (squares) is plotted here as the function of square root of the BR concentration (BR’”).
tion, with almost complete initial prevention of CE-OOH for- mation the duration of which was dependent on the concentration of the pigment (Figs. 6 and 9).
In addition to efficacy, there were other differences between HSA-bound and free BR as LDL (co-)antioxidants. Thus, in contrast to albumin-bound BR, LDL antioxidation by free BR was inhibited to a larger extent by the presence of BR oxidase (Fig. 7) and failed to produce biliverdin as oxidation product (Fig. 5). These findings argue strongly against the possibility that antioxidation by HSA-BR was the result of small amounts of the pigment dissociating from its carrier protein and associ- ating with LDL for antioxidant action. If so, we would have expected BR oxidase to inhibit the antioxidant activity of HSA- BR. Additionally, formation of biliverdin is observed during radical-mediated oxidation of HSA-bound but not free BR (321, indicating that in the case of AMVN-oxidizing LDL and HSA- BR, oxidation occurred on the protein-bound pigment. To- gether, our results indicate that HSA-bound BR can reduce TO‘ within oxidizing LDL, thereby providing efficient anti- oxidation.
The kinetic of lipid peroxidation in fresh plasma exposed to AMVN exhibited the typical TMP behavior seen in isolated LDL (cf: 20), with a peroxidation rate that was maximal during the TOH-inhibited period and slowed down after consumption of -60-70% of the TOH. This is not surprising considering that LDL is the major carrier of lipids in plasma, and peroxyl radi- cal-mediated lipid peroxidation in ascorbate-depleted plasma, isolated high density lipoproteins (43) and very low density lipoproteins (50) also proceed largely via TMP.
Significant oxidation of urate was not observed in AMVN- oxidizing plasma even after complete consumption of TOH (Fig. 2), in contrast to the situation with plasma exposed to aqueous peroxyl radicals, where urate is consumed before TOH (16). Together with the presence of approximately one-third of the AMVN within lipoproteins (Fig. 1) this shows that AMVN- derived radicals were compartmentalized to lipophilic sites, not directly and readily accessible to urate. By analogy, the ob- served consumption of water-soluble ascorbate and albumin- bound BR was therefore also likely to be indirect, mediated by TO’. Because the plasma concentration of TOH was more than 20 times higher than that of ubiquinol-10 and the two antioxi- dants have similar reactivity toward (peroxyl) radicals, con- sumption of the lipophilic quinol was also likely mediated by
TO’. Assuming that chromanoxyl radicals were responsible for most of the ubiquinol-10, ascorbate, and BR consumed, and that these antioxidants do not interact with each other (see Ref. 51 for the case of ascorbate and ubiquinol-lo), our results sug- gest that TO’ reacts with plasma co-antioxidants in the follow- ing order of preference: ubiquinol-10 > ascorbate > albumin- bound BR. Furthermore, the supplementation experiments carried out with oxidizing plasma and ascorbate (51) and BR (Fig. 3) clearly demonstrate that these two compounds protect plasma lipids from peroxidation independent of each other and ubiquinol-lo. Whether there are still additional co-antioxi- dants for TOH in plasma is not known at present. The much smaller chain length of lipid peroxidation in plasma when com- pared with isolated LDL (cf, Figs. 2 and 4), together with the recent results from Packer’s group (251, are consistent with the idea that plasma proteins can, to some extent, inhibit TMP.
The efficiency with which ascorbate prevented AMVN-in- duced lipid peroxidation in plasma was less than that observed with hydrophilic peroxyl radicals, where lipid hydroperoxides were not detected until all ascorbate was consumed (16,511. In the case of hydrophilic radicals, ascorbate’s protection is based in part on it preventing the radicals from initiating lipid per- oxidation, whereas with AMVN antioxidation by the vitamin is expected to be largely if not completely due to inhibition of peroxidation propagation mediated by TO’. Our results may thus indicate limitations in the ability of ascorbate (which is negatively charged at neutral pH) to interact with TO’ residing in LDL, the overall surface charge of which is also negative. To address this issue more directly, we are currently comparing the antioxidant efficacy of ascorbate against AMVN-induced peroxidation of TOH-containing, positively and negatively charged liposomes.
CONCLUSIONS
The results presented support the importance of TMP for LDL and plasma lipid oxidation and antioxidation induced by lipid-soluble peroxyl radicals. They demonstrate that lipopro- tein-associated and albumin-bound BR can efficiently protect lipids from such peroxidation. This is likely achieved by inter- action of the lipoprotein’s TOH with the pigment, similar to the situation with ubiquinol-10 and ascorbate. Human plasma therefore contains various lines of non-proteinaceous co-anti- oxidants for TOH against lipoprotein lipid oxidation. In healthy human adults, albumin-bound BR appears to represent the third line of this defense, behind ubiquinol-10 and ascorbate, although BR appears likely to contribute meaningfully before all ascorbate is consumed. Under conditions where plasma BR levels are elevated (reviewed in Ref. 52) and/or those of ubiquinol-lO/ascorbate decreased, the relative importance of the pigment as physiological antioxidant will increase. Likely examples of the latter situation are the first days of life in humans often associated with hyperbilirubinemia (531, and vi- tamin C deficiency that results in induction of heme oxygenase, the rate-limiting enzyme in BR formation. Thus, our results lend further support for the idea that induction of heme oxy- genase may represent an antioxidant defense (52,54,55). That the antioxidant defense provided by BR for LDL is physiologi- cally significant is indicated by a recent report showing an inverse correlation between plasma BR levels and the risk of coronary artery disease (56).
REFERENCES 1. Stadtman, E. S. (1992) Science 257, 122&1224 2. Ames, B. N., Shigenaga, M. K., and Hagen, T. M. (1993) Proc. Natl. Acad. Sci.
3. Goldstein, J. L., and Brown, M. S. (1977) Annu. Reu. Biochem. 46, 897-930 4. Steinberg, D., Parthasarathy, S.. Carew, T. E., Khoo, J. C., and Witztum, J. L.
U. S. A. SO, 7915-7922

Bilirubin and Lipoprotein Lipid Antioxidation 16719
5. Steinbrecher, U. P., Zhang, H., and Lougheed, M. (1990) Free Rad. Biol. Med. (1989) N . Engl. J. Med. 320,915-924
6. Ross, R. (1993) Nature 382,801409 7. Steinbrecher, U. P., Lougheed, M., Kwan, W.-C., and Dirks, M. (1989) J. Biol.
8. Goldstein, J. L., Ho, Y. K., Basu, S. K., and Brown, M. S. (1979) h o c . Natl.
9. Sparrow, C. P., Parthasarathy, S., and Steinberg, D. (1989) J. Biol. Chem. 264,
10. Esterbauer, H., Gebicki, J., Puhl, H., and Jiirgens, G. (1992) Free Rad. Bid.
11. Rimm, E. B., Stampfer, M. J., Ascherio, A., Giovannucci, E., Colditz, G. A., and
12. Stampfer, M. J., Hennekens, C. H., Manson, J. E., Colditz, G. A., Rosner, B.,
13. Steinbrecher, U. P., Parthasarathy, S., Leake, D. S., Witztum, J. L., and Stein-
14. Esterbauer, H., Jiirgens, G., Quehenberger, O., and Koller, E. (1987) J. Lipid
15. Gey, K. E , Puska, P., Jordan, P., and Moser, U. K. (1991)Am. J. Clin. Nutl: 63,
16. Frei, B., Stocker, R., and Ames, B. N. (1988)Proc. Natl. Acad. Sci. U. S. A. 66,
17. Stocker, R., Bowry, V. W., and Frei, B. (1991)Proc. Natl. Acad. Sci. U. S. A. 88,
18. Mohr, D., Bowry, V. W., and Stocker, R. (1992) Biochim. Biophys. Acta 1128,
19. Ingold, K. U., Bowry, V. W., Stocker, R., and Walling, C. (1993) Proc. Natl. Acad.
20. Bowry, V. W., and Stocker, R. (1993) J. Am. Chem. Soc. 116,6029-6040
22. Packer, J. E., Slater, T. F., and Willson, R. L. (1979) Nature 278, 737-738 21. Burton, G. W., and Ingold, K. U. (1986)Acc. Chem. Res. 19, 194-201
23. Mukai, I C , Kikuchi, S., and Urano, S. (1990) Biochim. Biophys. Acta 1036,
24. Sato, K., Niki, E., and Shimasaki, H. (1990) Arch. Biochem. Biophys. 279,
25. Constantinescu, A,, Han, D., and Packer, L. (1993) J. Biol. Chem. 288,10906-
26. Motoyama, T., Miki, M., Mino, M., Takahashi, M., and Niki, E. (1989)Arch.
27. Tsuchiya, J . , Yamada, T., Niki, E., and Kamiya. Y (1985) Bull. Chem. SOC. Jvn.
9, 155-168
Chem. 264,15216-15223
Acad. Sci. U. S. A. 78, 333437
2599-2604
Med. 13,341-390
Willett, W. C. (1993) N . Engl. J. Med. 328, 1450-1456
and Willett, W. C. (1993) New Engl. J. Med. 328, 1444-1449
berg, D. (1984) Proc. Natl. Acad. Sci. U. S. A. 81,38834887
Res. 28,495-509
3268-3348
974EL9752
1646-1650
247-254
Sci. U. S. A. 90, 45-49
77-82
402-405
10913
Biochem. Biophys. 270,655-661
68,326-330 28. Stocker, R., and Peterhans, E. (1989) Biochim. Biophys. Acta 1002, 238-244
29. Kagan, V. E., Serbinova, E. A,, Forte, T., Scita, G., and Packer, L. (1992) J.
30. Niki, E., Saito, M., Yoshikawa, Y., Yamamoto, Y., and Kamiya, Y (1986) Bull.
31. Stocker, R., Yamamoto, Y., McDonagh, A. F., Glazer, A. N., and Ames, B. N.
32. Stocker, R., Glazer, A. N., and Ames, B. N. (1987) Proc. Natl. Acad. Sci. U. 5'. A.
33. Buettner, G. R. (1988) J . Biochem. Biophys. Methods 18,27-40
35. Brodersen, R. (1979) CRC Crit. Rev. Clin. Lab. Inuest. 11, 305-399 34. Sattler, W., Mohr, D., and Stocker, R. (1994) Methods Enzymol. 233,46%489
36. Yamamoto, Y., Haga, S., Niki, E., and Kamiya, Y. (1984) Bull. Chem. SOC. Jpn.
37. Barclay, L. R. C., Locke, S. J., MacNeil, J. M., and VanKessel, J . (1984) J. Am.
38. Behrens, W. A., and Madere, R. (1987) Anal. Biochem. 186, 102-107 39. Stocker, R., McDonagh, A. E , Glazer, A. N., and Ames, B. N. (1990) Methods
40. Barclay, L. R. C., Baskin, K.A., Kong, S., and Locke, S. J. (1987) Can. J . Chem.
41. Barclay, L. R., Cameron, R. C., Forrest, B. J., Locke, S. J., Nigam, R., and
42. Frei, B., Stocker, R., England, L., and Ames, B. N. (1990) Adu. Exp. Med. B i d .
43. Bowry, V. W., Stanley, K. K., and Stocker, R. (1992) Proc. Natl. Acad. Sci.
44. Kimura, M., Matsumura, Y., Konno, T., Miyauchi, Y., and Maeda, H. (1990)
45. Bowry, V. W., Ingold, K. U., and Stocker, R. (1992) Biochem. J . 288, 341-344 46. Holley, A. E., and Slater, T. F. (1991) Free. Radical Res. Commun. 16, 51-63 47. Rubin, B. Y., Anderson, S. L., Lunn, R. M., Richardson, N. I C , Hellermann, G .
R., Smith, L. J., and Old, L. J. (1988) J. Immunol. 141, 1180-1184 48. Davies, M. J. , Forni, L. G., and Willson, R. L. (1988) Biochem. J . 266,513-522 49. Frei, B., Kim, M., and Ames, B. N. (1990) Proc. Natl. Acad. Sci. U. S. A. 87,
50. Mohr, D., and Stocker, R. (1994)Arteriosclel: Thromb., in press 51. Frei, B., England, L., and Ames, B. N. (1989) Proc. Natl. Acad. Sci. U. S. A. 88,
52. Stocker, R. (1990) Free Radical Res. Comms 9, 101-112 53. Newman, T., and Maisels, M. J. (1992) Pediatrics 89,809-818 54. Applegate, L. E., Liischer, P., and Tyrrell, R. M. (1991) CancerRes. 61,974-978 55. Nath, K. A,, Balla, G., Varcellotti, G. M., Jacob, H. S., Levitt, M. D., and
56. Schwertner, H. A,, Jackson, W. G., and Tolan, G. (1994) Clin. Chem. 40,18-23
Lipid. Res. 33, 385-397
Chem. Soc. Jpn. 69,471477
(1967) Science 235, 1043-1046
~ , 5 9 1 8 - 5 9 2 2
67,1260-1264
Chen. Soc. 106,2479-2481
Enzymol. 188,301409
86,2541-2550
Wnqvist, M. R. (1990) Biochim. Biophys. Acta 1047,255-263
264, 155-163
U. S. A. 89, 1031€-10320
Proc. SOC. Exp. B i d . Med. 196, 64-69
4879-4883
6377-6381
Rosenberg, M. E. (1993) J. Clin. Inuest. 90,267-270