
THE JOURNAL BIOLOGICAL CHEMISTRY Vol. April … JOURNAL OF BIOLOGICAL CHEMISTRY 0 1986 hy The...
9
THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1986 hy The American Society of Biological Chemists, Inc. Vol. 261, No. 10, Issue of April 5, pp. 4620-4628,1986 Printed in U.S.A. The Effect of Mild Diamide Oxidation on the Structure and Function of Human Erythrocyte Spectrin* (Received for publication, July 19, 1985) Pamela S. Becker, Carl M. CohenS, and Samuel E. Lux From the Division of Hematology-Oncology, The Children’s Hospital and the Dana-Farber Cancer Institute, the Department of Pediatrics, Harvard Medical School, the Program on Cell and Developmental Biology, Division of Medical Sciences, Harvard University, Boston, Massachusetts 02115, and the #Department of Biomedical Research, St. Elizabeth’s Hospital and Tufts University School of Medicine, Boston, Massachusetts 02135 Oxidants can alter erythrocyte membraneproperties and cause ultimate hemolysis, but the mechanisms re- sponsible for these changes are not understood. A pro- tein skeleton preservesthenormal integrity ofthe erythrocyte membrane.In this study, we investigated the effects of limited chemical oxidation on the struc- ture and function of the major skeletal protein, spec- trin. After mild treatment of spectrin with 2.5 PM diamide, with formationofanaverageofonlyone disulfide bond, we observed a 50% reduction in the ability of protein 4.1 to amplify spectrin-actin binding. The oxidized spectrin specifically lacked theability to bind protein 4.1, whereas all other spectrin functions remained intact. However, oxidation also produced a structural change in spectrin. A rapidly migrating spe- cies appeared on non-denaturing gels in a dose-depend- ent manner with increasing diamide concentrations. By electron microscopy, the oxidized spectrin appeared as single-stranded signet rings with irregular knob- like protrusions. Fifty per cent of spectrin was con- verted to the ring form after the formation of an av- erage of two disulfide bonds. Both the structural and functional defects were reversed by chemical reduc- tion. The loss of spectrin function or the structural transformation in spectrin may contribute to erythro- cyte membrane failure in theoxidative environment. The major function of the red blood cell is oxygen transport via hemoglobin. Hemoglobin is capable of generating oxidiz- ing radicals, since it is capable of mixed-function oxidase-like activity in vitro in a reconstituted hydroxylase system (1, 2) or in the intact erythrocyte in the presence of exogenous substrates (3). The erythrocyte has several intracellular modes of protection against such oxidation. For example, it carries enzymes such as superoxide dismutase, glutathione peroxidase, and catalase capable of disarming oxidizing radi- * This investigation was supported by Public Health Service Na- tional Research Service Award 2T 32 GM07753-06 from the National Institute of General Medical Sciences (to P. S. B.) and grants from the National Institute for Arthritis, Diabetes, Digestive and Kidney Diseases (5R01 AM34083) and theNational Heart, Lung, and Blood Institute (5P01 HL32262 and 5P60 HL15157 (to S. E. L.)). Portions of this article were presented at the American Society of Hematology meetings in San Francisco, CA, in December 1983 and in Miami Beach, FL, in December, 1984. The work has appeared in abstract form (Becker, P. S., Donner, S., and Lux, S. E. (1983) Blood 62, 43a and Becker, P. S., Cohen, C. M., and Lux, S. E. (1984) Blood 64, 23a). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. cals, and it contains antioxidants such as glutathione and vitamin E (4). However, in certain hereditary disorders, ex- cessive oxidant generation or altered cellular metabolism de- creases levels of glutathione or vitamin E and can lead to oxidant damage to membrane lipids and proteins. Such altered physiology and subsequent oxidant damage has been reported in sickle cell disease (5, 6, 7), glucose-6-phosphate dehydro- genase deficiency (8), and unstable hemoglobinopathies (9, lo), among others. The mechanism by whichoxidant damage leads to hemolytic anemia in such disorders is unknown. Erythrocytes may also be damaged by exogenous oxidants. For example, intracellular hemoglobin is oxidized and the red cells are lysed after suspension in a medium containing su- peroxide and hydrogen peroxide (ll), and intracellularhemo- globin and glutathione are oxidized when erythrocytes are suspended with activated neutrophils generating oxidants (12). Since hemoglobin is oxidized, it is possible that other red cell proteins may undergo similar damage that somehow leads to membrane failure and cell lysis. A number of studies have examined the effect of oxidants and sulfhydryl-active reagents on the integrity of the red cell. Sulfhydryl group modification of red blood cells with p-chlo- romercuribenzoate (pCMB1) or N-ethylmaleimide (NEM) in- duces spheroidal cell shape, increased osmotic fragility, de- creased cellular deformability, decreased survival, and splenic sequestration (13, 14). Oxidation of intact erythrocytes with diamide caused similar perturbations (15), as well as an alter- ation in membrane lipid asymmetry (16-19). The latter phe- nomenon is also observed after oxidation with sodium tetra- thionate (20). Bifunctional thiol reagents such as diamide cause a 50% reduction in cell deformability with modification of less than 5% of the membrane sulfhydryl groups. In con- trast, with monofunctional SH reagents such as NEM, 20% of the membrane sulfhydryl groups must be modified to achieve the same effect (21). The erythrocyte membrane skeleton stabilizes the cell shape and maintains the membrane structure (22). The skel- eton is composed of spectrin, actin, and proteins 4.1 and 4.9 (25): Spectrin dimer, consisting of one a chain (protein 1) and one /3 chain (protein 2), self-associates (26) at a specific site at the head end of the molecule (27) to form tetramers The abbreviations used are: pCMB, p-chloromercuribenzoic acid; DTNB, 5,5’-dithiobis-(2-nitrobenzoic acid); DTT, dithiothreitol; FMB, fast migratingband (on non-denaturing gels of diamide-treated spectrin); IAEDANS, 5-(iodoacetamidoethy1)aminonaphthalene-1- sulfonic acid; NEM, N-ethylmaleimide; PAGE, polyacrylamide gel electrophoresis; SDS, sodium dodecyl sulfate. The proteins are designated numerically according to thesystem of nomenclature devised by Fairbanks et al. (23) and modified by Steck (24). 4620
-
Upload
phungduong -
Category
Documents
-
view
215 -
download
0
Transcript of THE JOURNAL BIOLOGICAL CHEMISTRY Vol. April … JOURNAL OF BIOLOGICAL CHEMISTRY 0 1986 hy The...

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1986 hy The American Society of Biological Chemists, Inc.
Vol. 261, No. 10, Issue of April 5, pp. 4620-4628,1986 Printed in U.S.A.
The Effect of Mild Diamide Oxidation on the Structure and Function of Human Erythrocyte Spectrin*
(Received for publication, July 19, 1985)
Pamela S. Becker, Carl M. CohenS, and Samuel E. Lux From the Division of Hematology-Oncology, The Children’s Hospital and the Dana-Farber Cancer Institute, the Department of Pediatrics, Harvard Medical School, the Program on Cell and Developmental Biology, Division of Medical Sciences, Harvard University, Boston, Massachusetts 02115, and the #Department of Biomedical Research, St. Elizabeth’s Hospital and Tufts University School of Medicine, Boston, Massachusetts 02135
Oxidants can alter erythrocyte membrane properties and cause ultimate hemolysis, but the mechanisms re- sponsible for these changes are not understood. A pro- tein skeleton preserves the normal integrity of the erythrocyte membrane. In this study, we investigated the effects of limited chemical oxidation on the struc- ture and function of the major skeletal protein, spec- trin. After mild treatment of spectrin with 2.5 PM diamide, with formation of an average of only one disulfide bond, we observed a 50% reduction in the ability of protein 4.1 to amplify spectrin-actin binding. The oxidized spectrin specifically lacked the ability to bind protein 4.1, whereas all other spectrin functions remained intact. However, oxidation also produced a structural change in spectrin. A rapidly migrating spe- cies appeared on non-denaturing gels in a dose-depend- ent manner with increasing diamide concentrations. By electron microscopy, the oxidized spectrin appeared as single-stranded signet rings with irregular knob- like protrusions. Fifty per cent of spectrin was con- verted to the ring form after the formation of an av- erage of two disulfide bonds. Both the structural and functional defects were reversed by chemical reduc- tion. The loss of spectrin function or the structural transformation in spectrin may contribute to erythro- cyte membrane failure in the oxidative environment.
The major function of the red blood cell is oxygen transport via hemoglobin. Hemoglobin is capable of generating oxidiz- ing radicals, since it is capable of mixed-function oxidase-like activity in vitro in a reconstituted hydroxylase system (1, 2) or in the intact erythrocyte in the presence of exogenous substrates (3). The erythrocyte has several intracellular modes of protection against such oxidation. For example, it carries enzymes such as superoxide dismutase, glutathione peroxidase, and catalase capable of disarming oxidizing radi-
* This investigation was supported by Public Health Service Na- tional Research Service Award 2T 32 GM07753-06 from the National Institute of General Medical Sciences (to P. S. B.) and grants from the National Institute for Arthritis, Diabetes, Digestive and Kidney Diseases (5R01 AM34083) and the National Heart, Lung, and Blood Institute (5P01 HL32262 and 5P60 HL15157 (to S. E. L.)). Portions of this article were presented at the American Society of Hematology meetings in San Francisco, CA, in December 1983 and in Miami Beach, FL, in December, 1984. The work has appeared in abstract form (Becker, P. S., Donner, S., and Lux, S. E. (1983) Blood 62, 43a and Becker, P. S., Cohen, C. M., and Lux, S. E. (1984) Blood 64, 23a). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
cals, and it contains antioxidants such as glutathione and vitamin E (4). However, in certain hereditary disorders, ex- cessive oxidant generation or altered cellular metabolism de- creases levels of glutathione or vitamin E and can lead to oxidant damage to membrane lipids and proteins. Such altered physiology and subsequent oxidant damage has been reported in sickle cell disease (5, 6, 7), glucose-6-phosphate dehydro- genase deficiency (8), and unstable hemoglobinopathies (9, lo), among others. The mechanism by which oxidant damage leads to hemolytic anemia in such disorders is unknown. Erythrocytes may also be damaged by exogenous oxidants. For example, intracellular hemoglobin is oxidized and the red cells are lysed after suspension in a medium containing su- peroxide and hydrogen peroxide (ll), and intracellular hemo- globin and glutathione are oxidized when erythrocytes are suspended with activated neutrophils generating oxidants (12). Since hemoglobin is oxidized, it is possible that other red cell proteins may undergo similar damage that somehow leads to membrane failure and cell lysis.
A number of studies have examined the effect of oxidants and sulfhydryl-active reagents on the integrity of the red cell. Sulfhydryl group modification of red blood cells with p-chlo- romercuribenzoate (pCMB1) or N-ethylmaleimide (NEM) in- duces spheroidal cell shape, increased osmotic fragility, de- creased cellular deformability, decreased survival, and splenic sequestration (13, 14). Oxidation of intact erythrocytes with diamide caused similar perturbations (15), as well as an alter- ation in membrane lipid asymmetry (16-19). The latter phe- nomenon is also observed after oxidation with sodium tetra- thionate (20). Bifunctional thiol reagents such as diamide cause a 50% reduction in cell deformability with modification of less than 5% of the membrane sulfhydryl groups. In con- trast, with monofunctional SH reagents such as NEM, 20% of the membrane sulfhydryl groups must be modified to achieve the same effect (21).
The erythrocyte membrane skeleton stabilizes the cell shape and maintains the membrane structure (22). The skel- eton is composed of spectrin, actin, and proteins 4.1 and 4.9 (25): Spectrin dimer, consisting of one a chain (protein 1) and one /3 chain (protein 2), self-associates (26) at a specific site at the head end of the molecule (27) to form tetramers
The abbreviations used are: pCMB, p-chloromercuribenzoic acid; DTNB, 5,5’-dithiobis-(2-nitrobenzoic acid); DTT, dithiothreitol; FMB, fast migratingband (on non-denaturing gels of diamide-treated spectrin); IAEDANS, 5-(iodoacetamidoethy1)aminonaphthalene-1- sulfonic acid; NEM, N-ethylmaleimide; PAGE, polyacrylamide gel electrophoresis; SDS, sodium dodecyl sulfate.
The proteins are designated numerically according to the system of nomenclature devised by Fairbanks et al. (23) and modified by Steck (24).
4620

Diamide Oxidation of Erythrocyte Spectrin 4621
(28) and oligomers (29). Tetramers and oligomers are believed to be the form of spectrin on the intact membrane (30). Spectrin binds to short filaments of F-actin (protein 5) at its tail end (31). Protein 4.1 binds to spectrin nearby (32) and greatly amplifies the spectrin-actin interaction (33-37). In order to investigate how oxidants might disrupt the red cell membrane, we studied the effects of limited intramolecular oxidation on the structure and function of spectrin, the major membrane skeletal protein.
EXPERIMENTAL PROCEDURES
Materials
5,5'-Dithiobis(2-nitrobenzoic acid) (DTNB) and NEM were ob- tained from Pierce. pCMB and diazenedicarboxylic. acid (diamide) were obtained from Sigma. 5-(Iodoacetamidoethyl)aminona~hth~- lene-1-sulfonic acid (IAEDANS) was obtained from Molecular Probes.
Protein Purification
Spectrin was isolated by a low ionic strength extraction of fresh human erythrocyte membranes with 0.1 mM sodium phosphate, pH 8, at 37 "C as described (38). Spectrin dimer was purified by gel filtration chromatography on Bio-Gel A15m, 200-400 mesh (Bio- Rad) in 10 mM Tris, 0.15 M NaC1, 0.1 mM EDTA, pH 8 (buffer A), as described (38). G-actin was prepared from rabbit muscle as de- scribed (39). G-actin (2.5 mg/ml) was polymerized to F-actin by adding KC1 to 50 mM and magnesium chloride to 2 mM in 2 mM Tris, 1 mM DTT, 0.2 mM ATP, 0.2 mM calcium chloride. Protein 4.1 was purified from human erythrocyte membranes using Tween 20 as described (40). Extraction of membranes with Tween 20 yields gly- cophorin in addition to protein 4.1 (41), but the two proteins resolve to some degree by DEAE chromatography. The early column fractions on the ascending limb of the protein 4.1 peak are free of both spectrin and glycophorin. The purified proteins were concentrated as needed by ultrafiltration. Protein concentrations were determined by use of 1% Am values of 10, 8, and 6.7 for spectrin, protein 4.1, and actin, respectively.
Spectrin Modification Spectrin was modified with NEM, pCMB, and DTNB in order to
determine the number of sulfhydryl groups. NEM modification was performed in 0.1 M sodium phosphate, pH 7, and the decrease in absorbance at 305 nm was monitored. A molar extinction coefficient of 620 was utilized in order to calculate the number of sulfhydryl groups (42). pCMB titration was performed with additions of pCMB from a stock 4 X M solution previously defined by titration to equivalence with a stock solution of glutathione (43). Absorbance at 250 nm was monitored with 20-p1 additions of pCMB to 1 mg of spectrin in 1 ml of 0.05 M sodium phosphate, pH 7, until there was no further change. Two methods were used for the calculation of SH groups from the pCMB modification: 1) calculation of moles of pCMB added at the point of equivalence, and 2) dividing the change in absorbance at 250 nm by the molar extinction coefficient for the pCMB-glutathione adduct, 7,600. Titrations of spectrin with DTNB were performed in buffer A and the change in absorbance was moni- tored at 412 nm. In order to calculate thiol concentration, the molar extinction coefficient of 13,600 for the nitrobenzoate ion was utilized (44). Spectrin was also assayed for sulfhydryl groups with DTNB after denaturation with 1% sodium dodecyl sulfate (SDS).
Spectrin (0.1-1 mg/ml) was modified with various concentrations of diamide from 1 to 1000 p~ for 1 h at 0 "C in buffer A, then dialyzed against buffer A.
Spectrin (0.3-1 mg/ml) was modified with 3 mM IAEDANS in 0.1 M sodium phosphate, pH 7, for 1 h on ice and then dials zed into the appropriate buffer.
Spectrin was radioiodinated as needed for protein binding assays with lZ5I-labeled Bolton-Hunter reagent (New England Nuclear) as described (45). Unbound Bolton-Hunter reagent was removed by dialysis or gel filtration on Sephadex G-25 (Pharmacia). The radio- labeled spectrin was then divided into a portion for diamide treatment and a portion for the untreated control.
Binding Assays
Spectrin to Inside-Out Membrane Vesicles-Spectrin-depleted membrane vesicles (known as inside-out vesicles) were prepared as described (46). The binding of '%I-labeled spectrin dimer to mem- brane vesicles was performed as described (47), but without DTT.
Spectrin Self-association: Kinetics of Dimer to Oligomer Conver- sion-Spectrin dimer (0.4 to 1 mg/ml) in buffer A was incubated at 30 "C for various times from 0 to 6 h. Thirty-pg aliquots were withdrawn at 30-60-min intervals and placed on ice prior to electro- phoresis. In order to assess the state of oligomerization, electropho- resis was performed on 3% polyacrylamide disc gels, as described (23), and as modified (24), but without sodium dodecyl sulfate (SDS) and without reducing agents in the sample buffer. 'These gels were run at 2 mA/gel a t 4 "C for 12-16 h.
Spectrin-Actin-Protein 4.1 "Various amounts of '%I-labeled spec- trin (0-20 pg) were incubated with a fixed amount of F-actin (36-42 pg) with or without a fixed amount of protein 4.1 (6 pg) in a final volume of 80 pl at 21 "C for 1.5 h and the binding was assayed as described (40), but without DTT.
Spectrin-Protein 4.1 Binding-Spectrin's ability to associate with protein 4.1 was first assessed by immunoprecipitation of spectrin and '9-labeled protein 4.1 with rabbit anti-human spectrin antibody and formaldehyde-fixed, protein A-bearing Staphylococcus aureus (The Enzyme Center) as described (32), but without DTT. However, we encountered high nonspecific binding (70-90%) with this assay, pos- sibly related to the absence of DTT, so we developed a competition assay utilizing spectrin-Sepharose 4B to assay the binding of protein 4.1 to spectrin. Dry CNBr-Sepharose 4B (Pharmacia) was pre-treated with 1 mM HCl(200 ml/g of dry beads) for 15 min immediately before use, and then washed with 100 ml of 0.1 M sodium bicarbonate, pH 8. Unmodified spectrin dimer was attached to Sepharose 4B by adding spectrin (5 mg of spectrin/g of dry beads) at 2 mg/ml in 0.1 M NaHC03, pH 8, to the swollen beads and mixing end-over-end at 4 "C for 16 h. The coupling efficiency was generally greater than 90%. The same amount of bovine serum albumin was attached to an equal amount of Sepharose 4B to assess nonspecific binding. A fixed amount of spectrin-Sepharose (40-50 pg of spectrin) or a similar quantity of albumin-Sepharose was incubated with various amounts of lZ5I-labeled protein 4.1 (2-20 pg) in 2.5 mM Tris, 2.5 mM sodium phosphate, 130 mM KCl, 37.5 mM NaC1,l mM EDTA, 2 mM sodium azide, 0.1% Nonidet P-40 (Calbiochem-Behring), 1 mg/ml bovine serum albumin (Sigma) for 90 min at 0 "C. One hundred and fifty microliters of the spedrin-Sepharose or albumin-Sepharose and pro- tein 4.1 mixture were layered over 200 pl of 20% sucrose in the same buffer and centrifuged at 5000 rpm for 15 min. The tubes were then frozen and cut 7 mm from the bottom and the pellets assayed for radioactivity. A graph of micrograms of protein 4.1 bound versus 4.1 concentration was then constructed for both the spectrin-Sepharose and the albumin-Sepharose, with the latter representing the nonspe- cific binding curve. A 4.1 concentration was chosen from the graph just prior to saturation for the subsequent competition assays. Using the same fixed amount of spectrin-Sepharose (e.g. 50 pg), this chosen amount of lZ5I-labeled protein 4.1 (e.g. 8 pg) was incubated with increasing amounts of unlabeled test spectrin (0-100 pg) to compete for binding. The test spectrin samples were either 10 p~ diamide- treated spectrin or an untreated control.
In order to ensure that the failure of the diamide-treated spectrin to compete for protein 4.1 binding was not a result of aberrant interaction with the spectrin-Sepharose, the ability of diamide- treated spectrin-to bind to spectrin-Sepharose was compared to that of untreated spectrin in the buffer system used for the competition assay. Two to 23 pg of untreated of 10 p~ diamide-treated '%I-labeled spectrin were incubated with a fixed amount of spectrin-Sepharose (50 pg of spectrin) in a total volume of 200 pl for 90 min at 0 "C. The amount of bound spectrin was determined after centrifugation of the mixtures over 20% sucrose, as described for the competition assay. No difference was observed between the binding of the untreated and that of the diamide-treated spectrin to the spectrin-Sepharose.
Gel Electrophoresis Denaturing SDS-polyacrylamide gels were prepared as described
(23) and as modified (24), but without DTT in the sample buffer.
Electrophoretic Elution Elution of protein from non-denaturing gels was performed by a
modification of the method of Hunkapillar et al. (48). A portion of
4622 Diamide Oxidation of Erythrocyte Spectrin
the protein (10%) to be eluted was modified with IAEDANS prior to electrophoresis so that the band could be visualized under ultraviolet light and cut out of the gel. The gel fragment was macerated with a razor blade and the gel pieces were loaded into the gel chamber of the elution apparatus. The elution buffer was 20 mM Tris, pH 8.3. A constant voltage of 150 V was applied to the chamber for 16 h and the buffers in the anode and cathode chambers were mixed and circulated by a pump.
Electron Microscopy Low-angle rotary shadowing electron microscopy was performed
as described (49). The stock spectrin solutions were 0.3-0.4 mg/ml in buffer A. The final protein concentration was 15 pg/ml in 70% glycerol.
Proteolytic Digestions of Spectrin Spectrin (0.5-1 mg/ml) in buffer A was digested with trypsin in a
1:20 ratio (w/w) at 0 "C for 1.5 -2 h, similar to the method described (50). Two types of two-dimensional electrophoresis were performed (1) isoelectric focusing versus SDS-polyacrylamide (PAGE), known as a spectrin domain map, and (2) unreduced versus reduced SDS- PAGE. The isoelectric focusing versus SDS-PAGE gel was prepared by a modification (50) of the method of O'Farrell (51). The second gel system consisted of a 5%/10% (stacking/separating) discontin- uous first dimensional SDS disc gel as described (52) , but without 2- mercaptoethanol. The proteins in the first dimensional gel were then reduced by soaking the gel in 100 mM DTT, 3% SDS, 2 mM EDTA, 10% glycerol for 30 min at 21 "C. The gel was then run in a second dimension 5%/10% (stacking/separating) discontinuous SDS slab gel (52). In order to visualize free sulfhydryl groups within peptides on a domain map, the peptides were treated with 3 mM IAEDANS after tryptic digestion for 1 h on ice. The peptides were then dialyzed against 50 mM ammonium bicarbonate and lyophilized prior to two dimensional electrophoresis as described (50).
Disulfide Bonds The spectrin used for these studies was purified using buffers
degassed under nitrogen immediately after the preparation of eryth- rocyte ghosts. To determine the domains containing the disulfide bonds, spectrin was first cleaved into domains with trypsin, then treated with 2 mM NEM for 2 h on ice in 0.1 M Na phosphate, pH 7, with 6 M urea to block free sulfhydryl groups and then dialyzed against the same buffer without urea. The peptides were then treated with 10 mM DTT for 1 h on ice and then again dialyzed. Finally, they were modified with 3 mM IAEDANS for 1 h on ice, dialyzed against 0.1 M ammonium bicarbonate, and lyophilized. The two-dimensional peptide maps were photographed on a shortwave ultraviolet lightbox.
RESULTS
Number of Sulfhydryl Groups-By modification with DTNB, native spectrin was found to have 20.1 f 2.4 (S.D.) free sulfhydryl groups whereas SDS-denatured spectrin had 28.4. Twenty-seven sulfhydryl groups were accessible to pCMB in native spectrin as determined by titration to equiv- alence or 24 f 3 (S.D.) as determined by assuming that the extinction coefficient for the reaction with the cysteine of glutathione was equal to that for reaction with a cysteine of spectrin. NEM modified 60.8 f 2.4 (S.D.) residues of native spectrin, suggesting that its actin was not limited to sulfhydryl groups, but included other nucleophilic residues. Two-dimen- sinal domain maps of IAEDANS-modified spectrin peptides exhibited fluorescence in all domains, indicating that all spec- trin domains have free sulfhydryl groups.
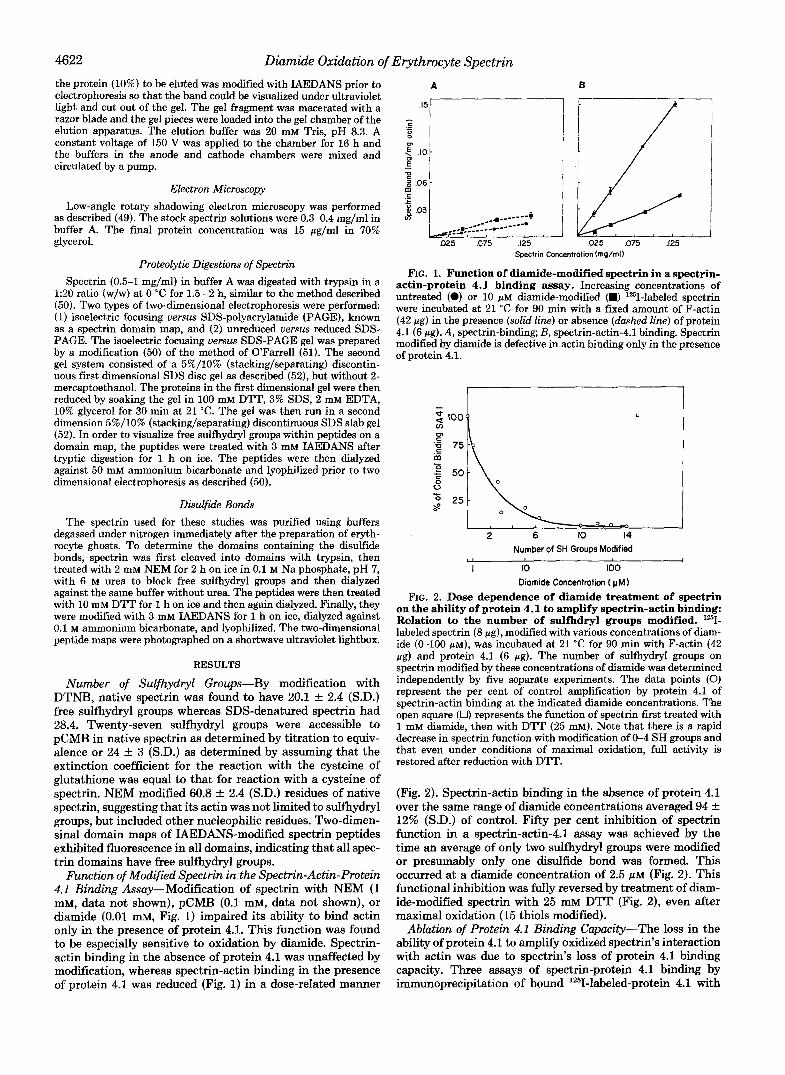
Function of Modified Spectrin in the Spectrin-Actin-Protein 4.1 Binding Assay-Modification of spectrin with NEM (1 mM, data not shown), pCMB (0.1 mM, data not shown), or diamide (0.01 mM, Fig. 1) impaired its ability to bind actin only in the presence of protein 4.1. This function was found to be especially sensitive to oxidation by diamide. Spectrin- actin binding in the absence of protein 4.1 was unaffected by modification, whereas spectrin-actin binding in the presence of protein 4.1 was reduced (Fig. 1) in a dose-related manner
A B
Spectrin Concentration (rng/rnl)
FIG. 1. Function of diamide-modified spectrin in a spectrin- actin-protein 4.1 binding assay. Increasing concentrations of untreated (0) or 10 PM diamide-modified (W) 1251-labeled spectrin were incubated at 21 "C for 90 min with a fixed amount of F-actin (42 pg) in the presence (solid line) or absence (dashed line) of protein 4.1 (6 pg). A, spectrin-binding; B, spectrin-actin-4.1 binding. Spectrin modified by diamide is defective in actin binding only in the presence of protein 4.1.
0
0 " "
2 6 10 14 Number of SH Groups Modified
I f I
I IO 100 Diamide Concentration (JIM 1
FIG. 2. Dose dependence of diamide treatment of spectrin on the ability of protein 4.1 to amplify spectrin-actin binding: Relation to the number of sulfhdryl groups modified. lz5I- labeled spectrin (8 rg), modified with various concentrations of diam- ide (0-100 p ~ ) , was incubated at 21 "C for 90 min with F-actin (42 pg) and protein 4.1 (6 pg). The number of sulfhydryl groups on spectrin modified by these concentrations of diamide was determined independently by five separate experiments. The data points (0) represent the per cent of control amplification by protein 4.1 of spectrin-actin binding at the indicated diamide concentrations. The open square (0) represents the function of spectrin first treated with 1 mM diamide, then with DTT (25 mM). Note that there is a rapid decrease in spectrin function with modification of 0-4 SH groups and that even under conditions of maximal oxidation, full activity is restored after reduction with DTT.
(Fig. 2). Spectrin-actin binding in the absence of protein 4.1 over the same range of diamide concentrations averaged 94 f 12% (S.D.) of control. Fifty per cent inhibition of spectrin function in a spectrin-actin-4.1 assay was achieved by the time an average of only two sulfhydryl groups were modified or presumably only one disulfide bond was formed. This occurred at a diamide concentration of 2.5 p~ (Fig. 2). This functional inhibition was fully reversed by treatment of diam- ide-modified spectrin with 25 mM DTT (Fig. 2), even after maximal oxidation (15 thiols modified).
Ablation of Protein 4.1 Binding Capacity-The loss in the ability of protein 4.1 to amplify oxidized spectrin's interaction with actin was due to spectrin's loss of protein 4.1 binding capacity. Three assays of spectrin-protein 4.1 binding by immunoprecipitation of bound 1251-labeled-protein 4.1 with

Diamide Oxidation of Erythrocyte Spectrin 4623
anti-spectrin antibody showed a decrease in the ability of diamide-treated spectrin to bind protein 4.1 (data not shown). By using a competition assay, we found that 10 p~ diamide- treated spectrin was unable to compete with bead-bound spectrin as well as untreated spectrin for protein 4.1 binding (Fig. 3).
Intact Spectrin-Ankyrin Binding-Modification of spectrin with diamide (50 p ~ ) or NEM (1 mM) (data not shown) had no effect on its ability to associate with spectrin-depleted inside-out vesicles, presumably via ankyrin (53,54).
Intact Dimer-Dimer Self-association-Oxidation of spectrin dimer with 2.5 or 5 p~ diamide did not impair its ability to associate to higher-order oligomers (see Fig. 7). The initial amount of spectrin present as dimer in the oxidized sample was reduced (by 8-22%) in the diamide-treated samples, es- pecially at higher diamide concentrations, due to inter-dimer cross-linking by disulfide bonds. Despite the differences in the initial amounts of free dimer, the kinetics of association to oligomeric species proceeded in parallel with that of un- treated spectrin (data not shown). However, the number of oligomeric species were more numerous in the diamide-treated spectrin than in the untreated spectrin, as will be described later in the text.
Tryptic Digestions of Diamide-treated Spectrin-Treatment of spectrin with 2.5-5 p~ diamide did not produce any marked alterations of the two-dimensional tryptic domain map (data not shown). Similarly, two-dimensional SDS-PAGE in the absence (first dimension) or presence (second dimension) of DTT did not reveal any significant alterations in the peptide pattern (data not shown). This suggests that interdomain disulfide bonds were not formed during mild diamide oxida- tion.
Localization of Disulfide Bonds Induced by Diamide-After modification of spectrin domain-sized peptides by NEM, DTT, and IAEDANS sequentially in order to limit fluorescent labeling to sulfhydryl groups that had been in disulfide bonds, more intense fluorescence was observed in the /3 IV and a IV domains of 2.5 p~ diamide-modified spectrin compared to control untreated spectrin (Fig. 4). In order to check that the difference in fluorescence intensity was not due to a difference
2i, 40 eb 80 100
FREE SPECTRIN ADDEDWg)
FIG. 3. Spectrin-protein 4.1 binding aesay. Spectrin (50 pg) linked to Sepharose 4B was incubated with '=I-labeled protein 4.1 (4 pg) and increasing concentrations of free untreated spectrin (0) or diamide-modified spectrin (W). Soluble, untreated spectrin competes effectively for the binding of '?-labeled protein 4.1 with Sepharose- bound spectrin, whereas spectrin oxidized with 10 p~ diamide is unable to compete.
A 0
5 5 5.0 5.5 5 0 "-pH -
FIG. 4. Location of disulfide bonda in spectrin domain maps. After tryptic digestion of untreated spectrin ( p a n e l A ) or 2.5 p~ diamide-treated spectrin ( p a n e l R). free SH groups were blocked with NEM, then disulfide bonds were reduced with 10 mM DTT, and the newly exposed SH groups were fluorescently labeled with 3 mM IAEDANS. Each gel contains 250 pg of spectrin peptides. The hori- zontal dimension is isoelectric focusing and the vertical dimension is SDS-PAGE. The f l IV ( l o n g arrow) and n IV (short arrow) were the only domains to change appreciably in fluorescence intensity with diamide modification.
0 1 2 5 5 1 0 S D T T DIAMIDE CONCENTRATION fpM I
hc. 5. Dose dependence of the formation of the fast mi- grating band (FMB) with increasing diamide concentration. Each 3% non-denaturing gel contains 30 pg of spectrin treated for 1 h at 0 "C with 0, 1, 2.5,5, or 10 p~ diamide as indicated. The last gel contains a sample first treated with 5 p M diamide, then with 10 mM D T T for 10 h at 0 "C. Higher diamide concentrations lead to a higher proportion of FMB. Reduction causes the FMR to disappear.
in the quantity of the peptide, the amount of peptide was quantified by dye elution (55). Quantitation confirmed that there was not a significantly greater quantity of the particular peptide in the oxidized sample than in the untreated sample, since there was 1.2 times the amount of protein in the /3 IV domain and 0.6 times the amount in the a IV domain in the oxidized than the untreated spectrin. With higher concentra- tions of diamide (5-50 p ~ ) , disulfide bonds formed in numer- ous peptides, so that it was difficult to localize a particular site of disulfide bond formation. The background fluorescence of peptides after NEM modification and IAEDANS treatment varied, from essentially no fluorescence to scattered low level labeling of a few domains, including the a I, a IV, and B IV domains. This background labeling may represent free sulfhy- dryl groups not adequately blocked by NEM, or disulfide bonds that form during isolation and storage of the spectrin. In contrast, virtually all spectrin peptides become intenaely fluorescent after direct labeling with IAEDANS without prior blocking ofthe sulfhydryl groups with NEM (data not shown).
Appearance of Diamide-treated Spectrin on Non-denaturing Gels-Oxidation of spectrin with 1-10 p~ diamide resulted in increasing amounts ( 5 4 % of total protein) of a band that migrated more rapidly than epectrin dimer on non-denaturing gels (Fig. 5). This band contained the a and B spectrin chains in a 1:l ratio (Fig. 6). I t was capable of associating with

4624 Diamide Oxidation of Erythrocyte Spectrin
I I 0 D F
A
FIG. 6. Composition of the fast migrating band. The first dimension gel (run left to right) is a non-denaturing 3% polyacryl- amide gel containing 30 pgof 5 p~ diamide-treated spectrin incubated for 6 h at 30 "C to promote oligomer formation. The second dimension (run top to bottom) is a 3%/6% (stacking/separating) gel run after soaking the first dimensional gel in sample buffer containing DTT. F, fast migrating band; D, dimer; 0, oligomeric species. The FMB and all oligomeric species contain a and B chains in a one-to-one ratio.
B.
1
0 . 5 1 2 6 TIMEIHRsI
FIG. 7. Dimer to oligomer conversion. Each gel contains 30 pg of spectrin treated with 0 (panel A) or 2.5 p~ (panel E ) diamide and then incubated in buffer A at 30 'C for 0-6 h as indicated. F, fast migrating band; D, dimer; T, tetramer; 0, oligomeric species. Note that untreated dimer converta to tetramer and hexamer, whereas the FMB associates to multiple oligomeric species, including a band below tetramer and two or three bands in the region of hexamer.
spectrin dimer under conditions that promoted self-associa- tion to produce multiple oligomeric species (Fig. 7). The fast migrating species appears to have more types of oligomeric species than untreated spectrin (Fig. 7). For example, there was a band below tetramer, and there were two or three species in the hexamer region. When the fast migrating pro- tein from the non-denaturing gels was electrophoretically eluted and then run on an SDS-polyacrylamide gel in the absence of Dl", only the CY chain was observed in the spectrin
B
FIG. 8. Composition of the fast migrating band after elec- trophoretic elution. A 4% SDS-polyacrylamide gel without reduc- ing agents of 20 pg spectrin dimer ( l a n e A ) or spectrin from the fast migrating band of diamide (IO pM)-treated spectrin after electropho- retic elution ( l a n e E ) . The sample in lane B was prepared by treating spectrin dimer (400 pg at 0.4 mg/ml) with 10 p~ diamide, running the sample on 10 non-denaturing gels, cutting the FMR from the gels, and eluting the protein in an electrophoretic elution apparatus (48) without SDS at 4 ' C . Note that on the gel run on the eluted FMB, only the a chain is visible in the region of apectrin.
region (Fig. 8). Two higher molecular weight species were also present, which migrated to positions consistent with their identification aa a /3 chain dimer and a heterodimer of a and /3 chains (Fig. 8). It is likely that the two spectrin chains became separated at the time of oxidant modification and that during the elution of the fast migrating band, at the high concentrations present on the dialysis membrane, aggregated species containing /3 chain formed. In fact, others have ob- served aggregation of the isolated /3 chain (56). Since the elution was performed in the absence of reducing agents, the aggregated spectrin species may be linked by intermolecular disulfide bonds.
Interchain Disulfide Bond Formation-On an SDS-polyac- rylamide gel without DTT, most of a sample of 10 p~ diamide- treated spectrin (0.4 mg/ml) migrated as individual a and 6 subunits. Only a small percentage (approximately 10%) formed a higher molecular weight species (data not shown). This may represent the proportion of interchain disulfide bond cross-linking. Alternatively, it may represent intermo- lecular (interdimer) disulfide bond cross-linking (as previ- ously described on non-denaturing gels) that results in a species migrating with the size of tetramer on denaturing gels (e.g. p a n e l B, 0 h in Fig. 7).
Electron Microscopy of Diamide-treated Spectrin-Rotary shadowed platinum replicas of diamide-treated spectrin dimer showed ringlet forms with irregular and occasional knob-like protrusions (Fig. 9). Fewer of these forms were observed in samples of unmodified spectrin prepared and stored in the absence of DTT. Treatment with DTT eliminated all circular forms from the electron micrographs of both untreated and diamide-treated spectrin. The circular forms appeared to be single stranded, as compared to dimeric species on the same

Diamide Oxidution of Erythrocyte Spectrin 4625
FIG. 9. Low angle rotary shadowed electron micrographs of untreated, diamide-treated, and DTT- treated spectrin. Spectrin (0.4 mg/ml) was treated with 0 ( p a n e l A ) or 10 p~ ( p a n e l C) diamide without reducing agents or with 0 ( p a n e l B ) or 10 p~ (panel D) diamide followed by 12-h treatment with 20 mM DTT. The samples were shadowed at a final concentration of 15 pg/ml in 70% glycerol. The s m f l arrows indicate circular forma and the large arrows indicate linear forms. Circular forms are the major form observed in diamide-treated spectrin. Untreated spectrin in the absence of reducing agents also has some circular forms. Chemical reduction causes the disappearance of all circular forms from both untreated and diamide-treated spectrin.
shadowed replica. Their circumference was equivalent to the contour length of one spectrin dimer. Electron micrographs of diamide-treated spectrin after incubation under conditions that promoted oligomer formation showed a wide variety of forms, including attached rings, rings associated with ex- tended forms (lollipop configuration), and commonly, figure- eight shapes.
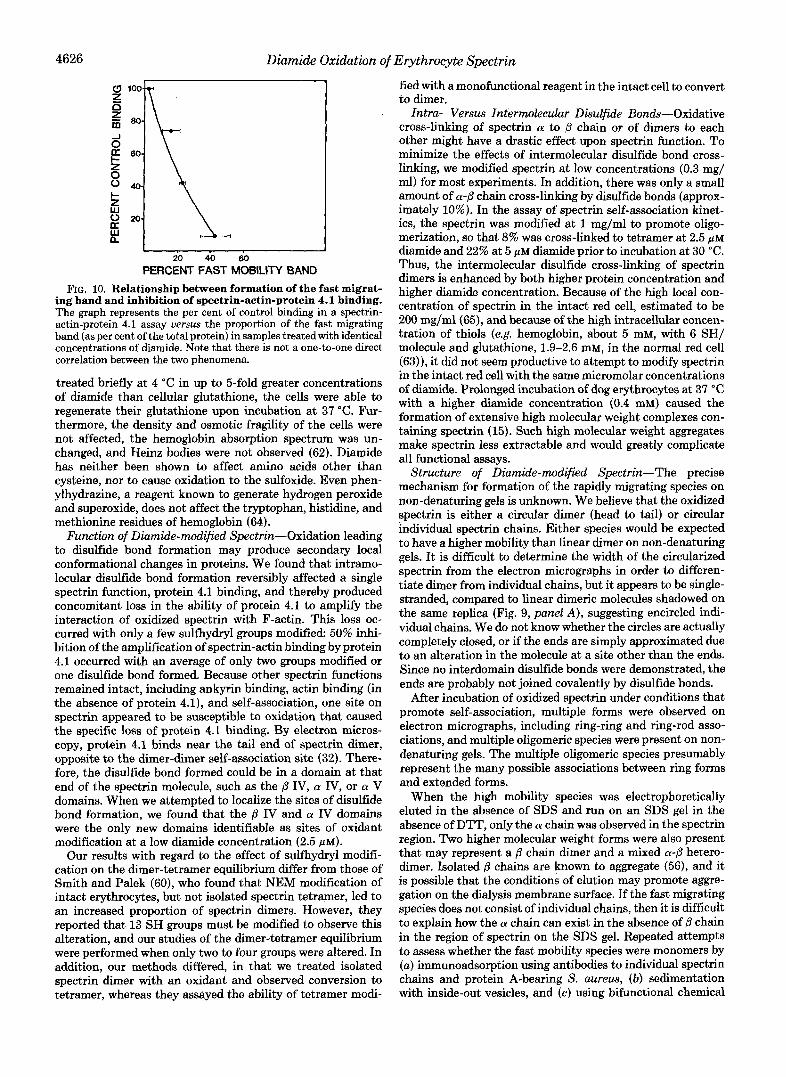
Relationship of Formation of the Rapidly Migrating Species to Inhibition of Spectrin-Actin-Protein 4.1 Binding-A plot of the per cent inhibition of spectrin-actin-protein 4.1 binding by diamide versus the per cent fast migrating band formed under identical conditions of modification did not reveal a direct one-to-one correlation of these two phenomena (Fig. 10). Instead, a 50% loss in the ability of protein 4.1 to amplify spectrin-actin binding occurs when an average of only one disulfide bond exists in spectrin, whereas 50% of spectrin is in a rapidly migrating species when there are two disulfide bonds.
DISCUSSION
Sulfhydryl Groups of Spectrin-The number of cysteines per spectrin dimer has been estimated in several studies. Values of 21 (57), 24.4 (58), and 38 (59) were calculated from amino acid analysis data. By DTNB titration, 21 were found in native spectrin and 28.5 in denatured spectrin (60). Our values correlate well with these latter numbers by the same method.
Disulfide bonds have not been obeerved between domains (50) or within the sequenced a 1-80 kDa domain (61). We have no evidence to suggest that any exist within the native molecule, although intradomain disulfide bonds do appear to form during storage in the absence of reducing agents. even at 0 "C.
Properties of Diamide-Diamide has been shown to stoi- chiometrically oxidize the sulfhydryl group of cysteine in glutathione (62) and proteins (63) to the disulfide without generating radicals in aqueous solutions at neutral pH (62). The oxidation was mild enough that, after red cells were
4626 Diamide Oxidation of Erythrocyte Spectrin
0 n z z rn
PERCENT FAST MOBILITY BAND FIG. 10. Relationship between formation of the fast migrat-
ing band and inhibition of spectrin-actin-protein 4.1 binding. The graph represents the per cent of control binding in a spectrin- actin-protein 4.1 assay versus the proportion of the fast migrating band (as per cent of the total protein) in samples treated with identical concentrations of diamide. Note that there is not a one-to-one direct correlation between the two phenomena.
treated briefly a t 4 "C in up to &fold greater concentrations of diamide than cellular glutathione, the cells were able to regenerate their glutathione upon incubation at 37 "C. Fur- thermore, the density and osmotic fragility of the cells were not affected, the hemoglobin absorption spectrum was un- changed, and Heinz bodies were not observed (62). Diamide has neither been shown to affect amino acids other than cysteine, nor to cause oxidation to the sulfoxide. Even phen- ylhydrazine, a reagent known to generate hydrogen peroxide and superoxide, does not affect the tryptophan, histidine, and methionine residues of hemoglobin (64).
Function of Diamide-modified Spectrin-Oxidation leading to disulfide bond formation may produce secondary local conformational changes in proteins. We found that intramo- lecular disulfide bond formation reversibly affected a single spectrin function, protein 4.1 binding, and thereby produced concomitant loss in the ability of protein 4.1 to amplify the interaction of oxidized spectrin with F-actin. This loss oc- curred with only a few sulfhydryl groups modified 50% inhi- bition of the amplification of spectrin-actin binding by protein 4.1 occurred with an average of only two groups modified or one disulfide bond formed. Because other spectrin functions remained intact, including ankyrin binding, actin binding (in the absence of protein 4.1), and self-association, one site on spectrin appeared to be susceptible to oxidation that caused the specific loss of protein 4.1 binding. By electron micros- copy, protein 4.1 binds near the tail end of spectrin dimer, opposite to the dimer-dimer self-association site (32). There- fore, the disulfide bond formed could be in a domain at that end of the spectrin molecule, such as the ,8 IV, a IV, or a V domains. When we attempted to localize the sites of disulfide bond formation, we found that the ,8 IV and a IV domains were the only new domains identifiable as sites of oxidant modification at a low diamide concentration (2.5 pM).
Our results with regard to the effect of sulfhydryl modifi- cation on the dimer-tetramer equilibrium differ from those of Smith and Palek (60), who found that NEM modification of intact erythrocytes, but not isolated spectrin tetramer, led to an increased proportion of spectrin dimers. However, they reported that 13 SH groups must be modified to observe this alteration, and our studies of the dimer-tetramer equilibrium were performed when only two to four groups were altered. In addition, our methods differed, in that we treated isolated spectrin dimer with an oxidant and observed conversion to tetramer, whereas they assayed the ability of tetramer modi-
fied with a monofunctional reagent in the intact cell to convert to dimer.
Intra- Versus Intermolecular Disulfide Bonds-Oxidative cross-linking of spectrin a to ,i3 chain or of dimers to each other might have a drastic effect upon spectrin function. To minimize the effects of intermolecular disulfide bond cross- linking, we modified spectrin at low concentrations (0.3 mg/ ml) for most experiments. In addition, there was only a small amount of a-p chain cross-linking by disulfide bonds (approx- imately 10%). In the assay of spectrin self-association kinet- ics, the spectrin was modified at 1 mg/ml to promote oligo- merization, so that 8% was cross-linked to tetramer at 2.5 p~ diamide and 22% at 5 p~ diamide prior to incubation at 30 "C. Thus, the intermolecular disulfide cross-linking of spectrin dimers is enhanced by both higher protein concentration and higher diamide concentration. Because of the high local con- centration of spectrin in the intact red cell, estimated to be 200 mg/ml(65), and because of the high intracellular concen- tration of thiols (e.g. hemoglobin, about 5 mM, with 6 SH/ molecule and glutathione, 1.9-2.6 mM, in the normal red cell (63)), it did not seem productive to attempt to modify spectrin in the intact red cell with the same micromolar concentrations of diamide. Prolonged incubation of dog erythrocytes at 37 "C with a higher diamide concentration (0.4 mM) caused the formation of extensive high molecular weight complexes con- taining spectrin (15). Such high molecular weight aggregates make spectrin less extractable and would greatly complicate all functional assays.
Structure of Diamide-modified Spectrin-The precise mechanism for formation of the rapidly migrating species on non-denaturing gels is unknown. We believe that the oxidized spectrin is either a circular dimer (head to tail) or circular individual spectrin chains. Either species would be expected to have a higher mobility than linear dimer on non-denaturing gels. It is difficult to determine the width of the circularized spectrin from the electron micrographs in order to differen- tiate dimer from individual chains, but it appears to be single- stranded, compared to linear dimeric molecules shadowed on the same replica (Fig. 9, panel A), suggesting encircled indi- vidual chains. We do not know whether the circles are actually completely closed, or if the ends are simply approximated due to an alteration in the molecule a t a site other than the ends. Since no interdomain disulfide bonds were demonstrated, the ends are probably not joined covalently by disulfide bonds.
After incubation of oxidized spectrin under conditions that promote self-association, multiple forms were observed on electron micrographs, including ring-ring and ring-rod asso- ciations, and multiple oligomeric species were present on non- denaturing gels. The multiple oligomeric species presumably represent the many possible associations between ring forms and extended forms.
When the high mobility species was electrophoretically eluted in the absence of SDS and run on an SDS gel in the absence of DTT, only the a chain was observed in the spectrin region. Two higher molecular weight forms were also present that may represent a /3 chain dimer and a mixed a-,8 hetero- dimer. Isolated ,8 chains are known to aggregate (561, and it is possible that the conditions of elution may promote aggre- gation on the dialysis membrane surface. If the fast migrating species does not consist of individual chains, then it is difficult to explain how the a chain can exist in the absence of ,i3 chain in the region of spectrin on the SDS gel. Repeated attempts to assess whether the fast mobility species were monomers by (a) immunoadsorption using antibodies to individual spectrin chains and protein A-bearing S. aureus, (b) sedimentation with inside-out vesicles, and (c ) using bifunctional chemical

Diamide Oxidation of Erythrocyte Spectrin 4627
cross-linkers were not successful. Despite the alterations of spectrin structure observed on
non-denaturing gels and by electron microscopy, we did not find a difference in the limited tryptic digestion pattern. This finding is in contrast to the results of Smith and co-workers (66), who observed multiple changes in the tryptic digestion pattern of NEM-treated spectrin. For example, they demon- strated an increase in the (Y 1-74 kDa peptide relative to the (Y 1-80 kDa domain. Again, however, this result was obtained after extensive sulfhydryl blocking by a monofunctional re- agent, N-ethylmaleimide.
Relationship of Altered Structure and Function-If oxida- tion changes spectrin's morphology to that of ring-like ShC- tures, then the consequence may be loss of protein 4.1 binding simply due to loss of the free tail end. However, we believe that the loss of protein 4.1 binding occurs prior to formation of the high mobility species. A 50% loss in the ability of protein 4.1 to amplify spectrin-actin binding occurred at a diamide concentration that caused formation of only one disulfide bond (on average), whereas 50% of the spectrin is present as the rapidly migrating species at a diamide concen- tration that resulted in the formation of an average of two disulfide bonds. This lack of direct correlation suggests that the two phenomena are separate events. Perhaps the most oxidant-sensitive site leads to loss of protein 4.1 binding and the second most sensitive site leads to formation of the rapidly migrating species.
Possible Sources for in Vivo Oxidant Damage-We hypoth- esize that intramolecular oxidation of membrane proteins might occur in the intact cell due to local generation of oxidizing radicals in certain red cell disorders or during nor- mal cellular aging. For example, others. have observed lipid peroxidation in sickle erythrocytes (5) and aged red cells (67), and oxidized protein complexes in glucose-6-phosphate de- hydrogenase deficient red cells (8, 63, 68), in glucose phos- phate isomerase deficiency (69), and in aged red cells (70). If oxidation of spectrin similar to that observed in the present studies also occurs in vivo, it would greatly weaken spectrin- actin-protein 4.1 interactions. This could lead to fragile mem- branes and hemolysis in conditions such as the unstable hemoglobinopathies, sickle cell disease, and glucose-6-phos- phate dehydrogenase deficiency. In support of this argument, recent studies of red cells from a patient with the unstable hemoglobin, Hb Nottingham, disclosed a defect in the func- tion of spectrin in the spectrin-actin-4.1 assay (73)? In addi- tion, mechanically fragile members (71) have been described in one family with hereditary spherocytosis in which the mutant spectrin does not bind protein 4.1 and thereby spec- trin-actin binding is impaired (72).
In summary, a number of physiological or pathological processes can potentially cause oxidant damage to the red cell. Such damage may ultimately result in hemolysis. We have investigated the effects of in vitro oxidation of the structure and function of the major erythrocyte skeletal pro- tein spectrin in an effort to determine the mechanism for membrane failure in the oxidative environment, and have found two major consequences to such modification. First, the ability of spectrin to bind protein 4.1 and thereby allow amplification of actin binding is abolished. Second, there is a structural alteration such that spectrin migrates more rapidly than native dimer in non-denaturing gels and acquires a circular configuration when examined by electron microscopy. Either or both of these changes might lead to failure of the red cell membrane skeleton.
0. S. Platt, J. F. Falcone, and S. E. Lux, manuscript in prepara- tion.
1.
2.
3.
4.
5. 6.
7.
8.
9.
10.
11.
12.
13. 14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24. 25. 26.
27.
28.
29.
30.
31.
32.
33. 34.
35. 36.
37.
REFERENCES
Acknowledgments-We thankfully acknowledge the technical ad- vice of Kathryn John and helpful discussions of oxidation with Dr. Michael Marletta. We are grateful to Susan Donner for her invaluable contributions to these experiments.
Mieyal, J. J., Ackerman, R. S., Blumer, J. L., and Freeman, L. S.
Ferraiolo, B. L., Onady, G. M., and Mieyal, J. J. (1984) Biochern-
Blisard, K. S., and Mieyal, J. J. (1979) J. Biol. Chem. 254,5104-
Carrell, R. W., Winterbourn, C. C., and Rachmilewitz, E. A.
Das, S. K., and Nair, R. C. (1980) Br. J. Haernatol. 4 4 , 87-92 Hebbel, R. P., Eaton, J. W., Balasingam, M., and Steinberg, M.
Rank, B. H., Carlsson, J., and Hebbel, R. P. (1985) J. Clin. Invest.
Johnson, G. J., Allen, D. W., Cadman, S., Fairbanks, V. F., White, J. G., Lampkin, B. C., and Kaplan, M. E. (1979) N . Engl. J. Med. 301,522-527
Alloisio, N., Michelson, D., Bannier, E., Revol, A., Beuzard, Y., and Delaunay, J. (1982) Biochim. Biophys. Acta 691,300-308
Flynn, T. P., Allen, D. W., Johnson, G. J., and White, J. G. (1983) J. Clin. Invest. 71, 1215-1223
Kellogg, E. W., 111, and Fridovich, 1. (1977) J. Biol. Chem. 252 , 6721-6728
Grisham, M. B., Jefferson, M. M., and Thomas, E. L. (1984) J. Biol. Chem. 259,6757-6765
Jacob, H. S., and Jandl, J. H. (1962) J. Clin. Invest. 41,779-792 Jacob, H. S., and Jandl, J. H. (1962) J. Clin. Inuest. 4 1 , 1514-
1523 Johnson, G. J., Allen, D. W., Flynn, T. P., Finkel, B., and White,
J. G. (1980) J. Clin. Inuest. 66,955-961 Haest, M. B., Jefferson, M. M., and Thomas, E. L. (1978)
Biochim. Biophys. Acta 5 0 9 , 21-32 Mohandas, N., Wyatt, J., Mel, S. F., Rossi, M. E., and Shohet, S.
B. (1982) J. Biol. Chem. 257,6537-6543 Bergmann, W. L., Dressler, V., Haest, C. W. M., and Deuticke,
B. (1984) Biochim. Biophys. Acta 7 6 9 , 390-398 Dressler, V., Haest, C. W. M., Plasa, G., Deuticke, B., and
Erusalimsky, J. D. (1984) Biochirn. Biophys. Acta 7 7 5 , 189- 196
Williamson, P., Bateman, J., Kozarsky, K., Mattocks, K., Her- manowicz, N., Choe, H. R., and Schlegel, R. A. (1982) Cell 30 , 125-7.13
(1976) J. Biol. Chem. 251,3436-3441
istry 23,5528-5534
5110
(1975) Br. J. Haematol. 30,259-264
H. (1982) J. Clin. Invest. 70, 1253-1259
75,1531-1537
Fischer, T. M., Haest, C. W. M., Stohr, M., Kamp, D., and
Lange, Y. , Hadesman, R. A., and Steck, T. L. (1982) J. Cell Biol.
Fairbanks, G., Steck, T. L., and Wallach, D. F. H. (1971) Bio-
Steck, T. L. (1972) J. Mol. Biol. 66, 295-305 Sheetz, M. P. (1979) Biochim. Biophys. Acta 557,122-134 Ungewickell, E., and Gratzer, W. (1978) Eur. J . Biochem. 8 8 ,
Morrow, J. S., Speicher, D. W., Knowles, W. J., Hsu, C. J., and Marchesi, V. T. (1980) Proc. Natl. Acad. Sci. U. S. A. 77,6592- 6596
Shotton, D. M., Burke, B. E., and Branton, D. (1979) J. Mol.
Morrow, J. S., and Marchesi, V. T. (1981) J. Cell Biol. 8 8 , 463-
Liu, S. C., Windisch, P., Kim, S., and Palek, J. (1984) Cell 373 ,
Cohen, C. M., Tyler, J. M., and Branton, D. (1980) Cell 2 1 , 691-
." ."
Deuticke, B. (1978) Biochim. Biophys. Acta 510,270-282
92,714-721
chemistry 10,2606-2617
379-385
Biol. 131,303-309
468
587-594
70 1 TyI&, J. M., Reinhardt, B. N., and Branton, D. (1980) J. Biol.
Chem. 255, 7034-7039 Cohen, C. M., and Foley, S. F. (1980) J. Cell Biol. 86,694-698 Cohen, C. M., and Korsgren, C. (1980) Biochern. Biophys. Res.
Fowler, V., and Taylor, D. L. (1980) J. Cell Biol. 85,361-376 Fowler, V. M., Luna, E. J., Hargreaves, W. R., Taylor, D. L., and
Ohanian, V., Wolfe, L. C., John, K. M., Pinder, J. C., Lux, S. E.,
Commun. 97,1429-1435
Branton, D. (1981) J. Cell Biol. 88,388-395

4628 Diamide Oxidation of Erythrocyte Spectrin
38.
39.
40.
41.
42.
43.
44. 45.
46.
47.
48.
49.
50.
51. 52. 53.
and Gratzer, W. B. (1984) Biochemistry 23, 4416-4420 Harris, H. W., Jr., and Lux, S. E. (1980) J. Bid, Chem. 255,
Spudich, J. A., and Watt, S. J. (1971) J, Biol. Chem. 246, 4866-
Becker, P. S., Spiegel, J. E., Wolfe, L. C., and Lux, S. E. (1983)
Elliot, C., and Ralston, G. (1984) Biochim. Biophys. Acta 775,
Glazer, A. N., Delange, R. J., and Sigman, D. S. (1975) in Laboratory Techniques in Biochemistry and Molecular Biology, Vol 4, Part I (Work, T. S., and Work, E., eds) pp. 110-111, Elsevier/North-Holland Biomedical Press, Amsterdam
Benesch, R., and Benesch, R. E. (1962) Methods Biochem. Anal.
Habeeb, A. F. S. A. (1972) Methods Enzymol. 25,457-464 Bolton, A. E., and Hunter, W.M. (1973) Bwchem. J. 133, 529-
Bennett, V., and Branton, D. (1977) J. Biol. Chem. 252, 2753-
Goodman, S. R., and Weidner, S. A. (1980) J. Biol. Chem. 255,
Hunkapillar, M. W., Lujan, E., Ostrander, F., and Hood, L. E.
Tyler, J. M., and Branton, D. (1980) J. Ultrastruct. Res. 71, 95-
Speicher, D. W., Morrow, J. S., Knowles; W. J., and Marchesi,
O’Farrell, P. H. (1975) J. Bwl. Chem. 250,4007-4021 Laemmli, U. K. (1970) Nature 227,680-685 Bennett, V., and Stenbuck, P. J. (1980) J. Biol. Chem. 255,2540-
11512-11520
4871
Anal. Bwchem. 132,195-201
313-319
10,43-70
539
2763
8082-8086
(1983) Methods Enzymol. 91,227-236
102
V. T. (1982) J. Biol. Chem. 257,9093-9101
2548
54.
55.
56.
57.
58.
59. 60. 61.
62.
63.
64.
65. 66.
67.
68. 69. 70.
71.
72.
73.
Yu, J., and Goodman, S. R. (1979) Proc. Natl. Acud. Sei. U. S. A.
Fenner, C., Traut, R. R., Mason, D. T., and Wikman-Coffelt, J.
Calvert, R., Bennett, P., and Gratzer, W. (1980) Eur. J . Biochem.
Fuller, G. M., Boughter, J. M., and Momzzani, M. (1974) Bio-
Eshdat, Y., and Lemay, A. (1979) Biochim. Bwphys. Acta 577,
Anderson, J. M. (1979) J. Biol. Chem. 254, 939-944 Smith, D. K., and Palek, J. (1983) Blood 62, 1190-1196 Speicher, D. W., Davis, G., and Marchesi, V. T. (1983) J. Biol.
Kosower, N. S., Kosower, E. M., and Werthelm, B. (1969)
Kosower, N. S., Zipser, Y., and Faltin, Z. (1982) Biochim. Biophys.
Vilsen, B., and Nielson, H. (1984) Biochem. Pharmacol. 33,2739-
Lux, S. E. (1979) Semin. Hematol. 16,21-51 Smith, D. K., Lawler, J., and Palek, J. (1982) J. Cell Biol. 95,
76,2340-2344
(1975) Anal. Biochem. 63,595-602
107,355-361
chemistry 13,3036-3041
360-370
Chem. 258,14938-14947
Bwchem. Bwphys. Res. Commun. 37,593-596
Acta 691,345-352
2748
253a
Commun. 92,247-254 Jain, S. K., and Hochstein, P. (1980) Biochem. Biophys. Res.
Coetzer, T.. and Zail. S. (1980) Blood 56. 159-167 Coetzer; T.; and Zail; S. (1979) J. Clin. Inuest. 63, 552-561 Snyder, L. M., Leb, L., Piotrowski, J., Sauberman, N., Liu, S. C.,
and Fortier, N. L. (1983) Br. J. Huematol. 53,379-384 Mohandas, N., Clark, M. R., Health, B. P., Rossi, M., Wolfe, L.
C., Lux, S. E., and Shohet, S. B. (1982) Blood 59, 768-774 Wolfe, L. C., John, K. M., Falcone, J. C., Byme, A. M., and Lux,
S. E. (1982) N. Engl. J. Med. 307, 1367-1374 Platt, 0. S., Falcone, J. F., and Lux, S. E. (1984) Blood 64, Suppl.
1,30a


















