
THE J BIOLOGICAL C © 2004 by The American Society for ... · Two Periplasmic Disulfide...
13
Two Periplasmic Disulfide Oxidoreductases, DsbA and SrgA, Target Outer Membrane Protein SpiA, a Component of the Salmonella Pathogenicity Island 2 Type III Secretion System* Received for publication, March 11, 2004, and in revised form, May 28, 2004 Published, JBC Papers in Press, May 28, 2004, DOI 10.1074/jbc.M402760200 Tsuyoshi Miki, Nobuhiko Okada‡, and Hirofumi Danbara From the Department of Microbiology, School of Pharmaceutical Sciences, Kitasato University, Tokyo 108-8641, Japan The formation of disulfide is essential for the folding, activity, and stability of many proteins secreted by Gram-negative bacteria. The disulfide oxidoreductase, DsbA, introduces disulfide bonds into proteins exported from the cytoplasm to periplasm. In pathogenic bacte- ria, DsbA is required to process virulence determinants for their folding and assembly. In this study, we exam- ined the role of the Dsb enzymes in Salmonella patho- genesis, and we demonstrated that DsbA, but not DsbC, is required for the full expression of virulence in a mouse infection model of Salmonella enterica serovar Typhimurium. Salmonella strains carrying a dsbA mu- tation showed reduced function mediated by type III secretion systems (TTSSs) encoded on Salmonella path- ogenicity islands 1 and 2 (SPI-1 and SPI-2). To obtain a more detailed understanding of the contribution of DsbA to both SPI-1 and SPI-2 TTSS function, we identi- fied a protein component of the SPI-2 TTSS apparatus affected by DsbA. Although we found no substrate pro- tein for DsbA in the SPI-1 TTSS apparatus, we identified SpiA (SsaC), an outer membrane protein of SPI-2 TTSS, as a DsbA substrate. Site-directed mutagenesis of the two cysteine residues present in the SpiA protein re- sulted in the loss of SPI-2 function in vitro and in vivo. Furthermore, we provided evidence that a second disul- fide oxidoreductase, SrgA, also oxidizes SpiA. Analysis of in vivo mixed infections demonstrated that a Salmo- nella dsbA srgA double mutant strain was more attenu- ated than either single mutant, suggesting that DsbA acts in concert with SrgA in vivo. The secretion of proteins is a prerequisite for interactions between pathogenic bacteria and their hosts. In Gram-negative bacteria, several types of secretion pathways for proteins that are important for such bacterial interactions with host cells have been identified. The type I secretion mechanism requires three accessory proteins to form a transmembrane structure, which includes a channel spanning the inner and outer mem- branes. Proteins secreted by this pathway have an uncleaved C-terminal secretion signal responsible for directing the secre- tion of protein (1–3). The secretion of proteins by the type II secretion pathway (also known as the “general secretion path- way”) involves two different steps. Proteins are first translo- cated through the inner membrane via signal peptides that interact with the Sec-dependent pathway, and the proteins are then transported across the outer membrane by either of sev- eral different terminal branches of the process (4, 5). The type III secretion system (TTSS) 1 functions as a pathway for secre- tion across bacterial membranes and for the translocation of secreted proteins across the plasma membrane of eukaryotic cells (6). The type IV secretion system also translocates pro- teins in a single step from the cytoplasm to the cytosol of a host cell (7). Proteins secreted via the type V secretion system are autotransporter proteins, which are characterized by a unify- ing structure possessing an N-terminal signal sequence and a pore-forming C-terminal domain (8, 9). In all of these secretion pathways, many of the proteins residing in or transiting through the periplasmic space acquire disulfide bonds after their translocation across the inner membrane. The formation of disulfide bonds is a key step in the folding of many secreted and membrane proteins. In Escherichia coli, disulfide bond formation is catalyzed by the Dsb proteins (10). DsbA is a 21-kDa periplasmic protein with a CXXC motif in its active site, and it interacts with reduced substrate proteins, catalyzing the oxidation of their cysteine residues to disulfide bonds (11). The inner membrane protein DsbB oxidizes DsbA (12, 13) and is re-oxidized directly by membrane-bound ubiqui- nones (14 –16). DsbC and DsbG are the periplasmic compo- nents of the isomerization pathway. These proteins reshuffle misfolded multiple disulfide bonds (17, 18). The active sites of DsbC and DsbG are maintained in the reduced form by the inner membrane protein DsbD, which transfers electrons from the cytoplasmic protein thioredoxin onto DsbC and DsbG (17, 19 –21). DsbA plays a central role in periplasmic protein folding. A dsbA mutant of an E. coli strain exhibits numerous in vivo phenotypes, including a loss of motility, an absence of alkaline phosphatase activity, sensitivity to benzylpenicillin and dithio- threitol, and resistance to phage M13, because of severe defects in disulfide bond formation in proteins requiring these processes (11, 22). In addition, dsbA-null mutants have a decreased growth rate in minimal media, as compared with the wild-type strain, and show mucoid colonies when grown on plates in minimal media (11). Although DsbC mutants are defective at disulfide bond formation, a mutation in dsbC does not display any obvious phenotype, except for a defect in the expression of dsbC sub- strates that contain multiple disulfide bonds (17). * This work was supported in part by a Grant-in-aid for Exploratory Research 15659105 and by a 21st Century Center of Excellence pro- gram grant from the Japanese Ministry of Education, Culture, Sports, Sciences, and Technology. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. ‡ To whom correspondence should be addressed: Dept. of Microbiol- ogy, School of Pharmaceutical Sciences, Kitasato University, 5-9-1 Shi- rokane, Minato-ku, Tokyo 108-8641, Japan. Tel.: 81-3-5791-6256; Fax: 81-3-3444-4831; E-mail: [email protected]. 1 The abbreviations used are: TTSS, type III secretion system; CI, competitive index; HA, hemagglutinin; cfu, colony-forming units; PBS, phosphate-buffered saline; FBS, fetal bovine serum; DTT, dithiothre- itol; SIF, Salmonella-induced filament. THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 279, No. 33, Issue of August 13, pp. 34631–34642, 2004 © 2004 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A. This paper is available on line at http://www.jbc.org 34631 by guest on April 5, 2019 http://www.jbc.org/ Downloaded from
Transcript of THE J BIOLOGICAL C © 2004 by The American Society for ... · Two Periplasmic Disulfide...

Two Periplasmic Disulfide Oxidoreductases, DsbA and SrgA, TargetOuter Membrane Protein SpiA, a Component of the SalmonellaPathogenicity Island 2 Type III Secretion System*
Received for publication, March 11, 2004, and in revised form, May 28, 2004Published, JBC Papers in Press, May 28, 2004, DOI 10.1074/jbc.M402760200
Tsuyoshi Miki, Nobuhiko Okada‡, and Hirofumi Danbara
From the Department of Microbiology, School of Pharmaceutical Sciences, Kitasato University, Tokyo 108-8641, Japan
The formation of disulfide is essential for the folding,activity, and stability of many proteins secreted byGram-negative bacteria. The disulfide oxidoreductase,DsbA, introduces disulfide bonds into proteins exportedfrom the cytoplasm to periplasm. In pathogenic bacte-ria, DsbA is required to process virulence determinantsfor their folding and assembly. In this study, we exam-ined the role of the Dsb enzymes in Salmonella patho-genesis, and we demonstrated that DsbA, but not DsbC,is required for the full expression of virulence in amouse infection model of Salmonella enterica serovarTyphimurium. Salmonella strains carrying a dsbA mu-tation showed reduced function mediated by type IIIsecretion systems (TTSSs) encoded on Salmonella path-ogenicity islands 1 and 2 (SPI-1 and SPI-2). To obtain amore detailed understanding of the contribution ofDsbA to both SPI-1 and SPI-2 TTSS function, we identi-fied a protein component of the SPI-2 TTSS apparatusaffected by DsbA. Although we found no substrate pro-tein for DsbA in the SPI-1 TTSS apparatus, we identifiedSpiA (SsaC), an outer membrane protein of SPI-2 TTSS,as a DsbA substrate. Site-directed mutagenesis of thetwo cysteine residues present in the SpiA protein re-sulted in the loss of SPI-2 function in vitro and in vivo.Furthermore, we provided evidence that a second disul-fide oxidoreductase, SrgA, also oxidizes SpiA. Analysisof in vivo mixed infections demonstrated that a Salmo-nella dsbA srgA double mutant strain was more attenu-ated than either single mutant, suggesting that DsbAacts in concert with SrgA in vivo.
The secretion of proteins is a prerequisite for interactionsbetween pathogenic bacteria and their hosts. In Gram-negativebacteria, several types of secretion pathways for proteins thatare important for such bacterial interactions with host cellshave been identified. The type I secretion mechanism requiresthree accessory proteins to form a transmembrane structure,which includes a channel spanning the inner and outer mem-branes. Proteins secreted by this pathway have an uncleavedC-terminal secretion signal responsible for directing the secre-tion of protein (1–3). The secretion of proteins by the type II
secretion pathway (also known as the “general secretion path-way”) involves two different steps. Proteins are first translo-cated through the inner membrane via signal peptides thatinteract with the Sec-dependent pathway, and the proteins arethen transported across the outer membrane by either of sev-eral different terminal branches of the process (4, 5). The typeIII secretion system (TTSS)1 functions as a pathway for secre-tion across bacterial membranes and for the translocation ofsecreted proteins across the plasma membrane of eukaryoticcells (6). The type IV secretion system also translocates pro-teins in a single step from the cytoplasm to the cytosol of a hostcell (7). Proteins secreted via the type V secretion system areautotransporter proteins, which are characterized by a unify-ing structure possessing an N-terminal signal sequence and apore-forming C-terminal domain (8, 9). In all of these secretionpathways, many of the proteins residing in or transitingthrough the periplasmic space acquire disulfide bonds aftertheir translocation across the inner membrane.
The formation of disulfide bonds is a key step in the foldingof many secreted and membrane proteins. In Escherichia coli,disulfide bond formation is catalyzed by the Dsb proteins (10).DsbA is a 21-kDa periplasmic protein with a CXXC motif in itsactive site, and it interacts with reduced substrate proteins,catalyzing the oxidation of their cysteine residues to disulfidebonds (11). The inner membrane protein DsbB oxidizes DsbA(12, 13) and is re-oxidized directly by membrane-bound ubiqui-nones (14–16). DsbC and DsbG are the periplasmic compo-nents of the isomerization pathway. These proteins reshufflemisfolded multiple disulfide bonds (17, 18). The active sites ofDsbC and DsbG are maintained in the reduced form by theinner membrane protein DsbD, which transfers electrons fromthe cytoplasmic protein thioredoxin onto DsbC and DsbG(17, 19–21).
DsbA plays a central role in periplasmic protein folding. AdsbA mutant of an E. coli strain exhibits numerous in vivophenotypes, including a loss of motility, an absence of alkalinephosphatase activity, sensitivity to benzylpenicillin and dithio-threitol, and resistance to phage M13, because of severe defectsin disulfide bond formation in proteins requiring these processes(11, 22). In addition, dsbA-null mutants have a decreased growthrate in minimal media, as compared with the wild-type strain,and show mucoid colonies when grown on plates in minimalmedia (11). Although DsbC mutants are defective at disulfidebond formation, a mutation in dsbC does not display any obviousphenotype, except for a defect in the expression of dsbC sub-strates that contain multiple disulfide bonds (17).
* This work was supported in part by a Grant-in-aid for ExploratoryResearch 15659105 and by a 21st Century Center of Excellence pro-gram grant from the Japanese Ministry of Education, Culture, Sports,Sciences, and Technology. The costs of publication of this article weredefrayed in part by the payment of page charges. This article musttherefore be hereby marked “advertisement” in accordance with 18U.S.C. Section 1734 solely to indicate this fact.
‡ To whom correspondence should be addressed: Dept. of Microbiol-ogy, School of Pharmaceutical Sciences, Kitasato University, 5-9-1 Shi-rokane, Minato-ku, Tokyo 108-8641, Japan. Tel.: 81-3-5791-6256; Fax:81-3-3444-4831; E-mail: [email protected].
1 The abbreviations used are: TTSS, type III secretion system; CI,competitive index; HA, hemagglutinin; cfu, colony-forming units; PBS,phosphate-buffered saline; FBS, fetal bovine serum; DTT, dithiothre-itol; SIF, Salmonella-induced filament.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 279, No. 33, Issue of August 13, pp. 34631–34642, 2004© 2004 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org 34631
by guest on April 5, 2019
http://ww
w.jbc.org/
Dow
nloaded from

In many pathogenic bacteria, DsbA is involved in pathogenic-ity through the catalysis of oxidative protein folding in virulencedeterminants. These virulence factors include the following:the cholera toxin of Vibrio cholerae (23, 24); the heat-stabletoxin of enterotoxigenic E. coli (25, 26); a molecular chaperone,PapD, of P pili of uropathogenic E. coli (27); bundle-forming piliand Intimin of enteropathogenic E. coli (28, 29); and Invasin ofYersinia pseudotuberculosis (30). DsbA is also required for theproper function of the TTSS in Yersinia pestis (31), Shigellaflexneri (32), and Pseudomonas aeruginosa (33). In Y. pestis, amutation in dsbA results in the unstable expression of an outermembrane protein, YscC, that constitutes the TTSS apparatus,which leads to the decreased translocation of Yop proteins.Substitution of cysteine residues in YscC reproduced all of thephenotypes seen in a dsbA mutation, suggesting that DsbAcatalyzes the YscC as a substrate (31).
Salmonella enterica is a Gram-negative and facultative in-tracellular bacterium that is pathogenic to humans and ani-mals; this pathogen is known to cause a broad spectrum ofdiseases such as gastroenteritis and bacteremia, as well astyphoid fever. The nature and severity of infection by Salmo-nella is generally dependent upon both the serovar and the hostspecies. Typhoid and paratyphoid fevers result from systemicinfection with human-adapted serovars such as S. entericaserovar Typhi and S. enterica serovar Paratyphi A. In contrast,infection with the broad host range-adapted serovar S. entericaserovar Typhimurium usually causes gastroenteritis in hu-mans but produces a systemic infection similar to typhoid feverin susceptible mice. S. enterica utilize two different virulence-associated TTSS, encoded in Salmonella pathogenicity islands1 and 2 (SPI-1 and SPI-2, respectively), for different stages ofpathogenesis. The SPI-1 TTSS is required for the invasion ofintestinal epithelial cells and the induction of the inflammatoryresponse in the intestinal mucosa (6, 34, 35). In contrast, SPI-2TTSS is required for intracellular survival in macrophages andfor systemic infection in the mouse model (36–38).
The complete genome sequence of S. enterica serovar Typhi-murium strain LT2 has revealed the presence of Dsb proteinhomologues (39). Southern hybridization analysis has alsodemonstrated the wide distribution of a dsbA gene amongSalmonella serovars (40). A dsbA gene cloned from theS. enterica serovar Typhimurium can restore the dsbA� phe-notype in an E. coli strain (40), demonstrating that Salmonelladisulfide oxidoreductase DsbA is functional, although the en-zymatic activity of Salmonella DsbA seems to be different fromthat of E. coli (40). Recently, a number of proteins affected bythe dsbA mutation have been identified using two-dimensionalgel electrophoresis with comparison of periplasmic proteinsexpressed in the wild-type and dsbA mutant strains inS. enterica serovar Typhi (41). However, the presence of disul-fide bonds in these proteins has not been determined. Thus, themembrane and secreted proteins, the folding of which is af-fected by DsbA in Salmonella, remain still obscure.
In the S. enterica serovar Typhimurium, the SalmonellaDsbA paralogue SrgA, encoded on the 94-kb virulence plasmid,functions as a disulfide oxidoreductase, whereas the enzymaticactivity of SrgA is less efficient than that of DsbA when E. colialkaline phosphatase is used as a substrate (42). SrgA specifi-cally oxidizes the disulfide bond of PefA, the major structuralsubunit of the plasmid-encoded fimbriae Pef, and thus thedisulfide oxidoreductase activity of SrgA is required for theassembly of Pef on the bacterial surface (42). Moreover, SrgAactivity is dependent upon the presence of functional DsbB(42), suggesting that, similar to DsbA, SrgA is recycled to anactive oxidized form by DsbB.
Recently, it has been shown that a Salmonella strain that
contains a mutation in dsbA is highly attenuated in a systemicinfection of mice (43). A Salmonella dsbA mutant strain hasalso shown decreased SPI-1 and SPI-2 TTSS function in termsof the translocation of effector proteins into mouse macro-phage-like RAW264.7 cells (43). However, the interaction be-tween DsbA and the component proteins of SPI-1 and SPI-2TTSS has not yet been determined. Thus, in order to obtain amore detailed understanding of the role of DsbA in Salmonellapathogenesis, we characterized the effect of DsbA on the activ-ity of both SPI-1 and SPI-2 TTSS, and we identified a proteincomponent of the SPI-2 TTSS apparatus affected by DsbA.Although we found no substrate for DsbA in the SPI-1 TTSSapparatus, we identified SpiA (also referred as SsaC), an outermembrane protein of SPI-2 TTSS, as a DsbA substrate. Fur-thermore, we demonstrated that a second disulfide oxidoreduc-tase, SrgA, also oxidizes SpiA in vitro. An analysis of in vivomixed infections showed that a dsbA srgA double mutant strainwas more attenuated than either single mutant strain, sug-gesting that DsbA acts in concert with SrgA in vivo.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, Primers, and Growth Conditions—Thebacterial strains and plasmids used in this study are listed in Table I.The oligonucleotide primers used in this study are listed in Table II.E. coli and Salmonella strains were grown in Luria-Bertani (LB) brothor on LB agar under selection for resistance to ampicillin (100 �g/ml),chloramphenicol (25 �g/ml for plasmid-containing strains, 5 �g/ml forchromosomal integrants), kanamycin (25 �g/ml), nalidixic acid (50 �g/ml), or streptomycin (50 �g/ml), as required. A previously describedintracellular salts-based minimal medium with limiting magnesium(MgM, pH 5.8) was used to induce SPI-2 gene expression (44). PhageP22-mediated transductions for Salmonella have been described previ-ously (45). The dsbA::Tn5 mutation of E. coli strain SK101 (46) wasintroduced into strain MC1061 by using P1 phages.
Construction of Plasmids—The dsbA and dsbC genes were amplifiedby PCR with the following primers: dsbAST-1 and dsbAST-2 for dsbA,and dsbC-FW and dsbC-RV for dsbC; strain SL1344 genomic DNA wasused as the template. The PCR products were cloned into TA cloningvector pGEM-T Easy (Promega) in order to produce pGEM-dsbA andpGEM-dsbC, respectively. Plasmids pMW-dsbA and pAC-dsbA, used tocomplement the dsbA mutant strain, were constructed by inserting theBamHI-XhoI fragment containing the dsbA gene from pGEM-dsbA intothe BamHI-SalI sites of pMW118 (Nippon Gene) and pACYC184 (NewEngland Biolabs), respectively. The srgA gene was amplified from the94-kb virulence plasmid of strain SL1344 using the primers srgA-FWand srgA-RV, and the PCR product was cloned into pGEM-T Easy. Thefragment was then subcloned into pMW119 (Nippon Gene) at the SacIand SphI sites in order to generate pMW-srgA. The spiA gene wasamplified by PCR with the primers spiA-FW1 and spiA-RV1, and strainSL1344 genomic DNA was used as the template. The PCR fragmentwas cloned into pGEM-T Easy, and the fragments were subcloned intopMW118 at the SacI and SphI sites in order to generate pMW-spiA.
The SpiA point mutants were constructed by site-directed mutagen-esis using the plasmid pGEM-sipA as a template and the respectiveoligonucleotides using the Quikchange XL site-directed mutagenesis kit(Stratagene) according to the manufacturer’s instructions. The oligonu-cleotides C133S-1 and C133S-2, and C152S-1 and C152S-2 were used toreplace cysteine residues with serine residues at positions 133 and 152in the SpiA protein, respectively. The mutated plasmids were trans-formed into E. coli XL10-Gold supercompetent cells (Stratagene), andthe presence of a respective mutation was confirmed by DNA sequenc-ing. The resulting plasmids obtained were designated as pGEM-spiAC133S and pGEM-spiAC152S, respectively.
To construct FLAG-tagged fusion proteins, the target genes were am-plified by PCR using the following primers: sipB-FW and sipB-RV forsipB; sipC-FW and sipC-RV for sipC; sseB-FW and sseB-RV for sseB;invG-FW and invG-RV for invG; and spiA-FW2 and spiA-RV2 for spiA,spiAC133S, and spiAC152S. The PCR products were digested with XhoI andBamHI and were cloned into the XhoI-BglII site of pFLAG-CTC (Sigma).
To construct the hemagglutinin (HA) epitope-tagged SseJ fusionprotein, DNA fragment containing sseJ and its promoter region wasamplified by PCR using sseJ-Pro and sseJ-RV. The PCR product wasdigested with XhoI and BamHI and was ligated to the same site ofpMW118, yielding pTM21. To insert the HA epitope into the C-terminalSseJ, pTM21 was amplified by reverse PCR using the primers HA-R1
Disulfide Bond Formation of SPI-2 Outer Membrane Protein34632
by guest on April 5, 2019
http://ww
w.jbc.org/
Dow
nloaded from

and HA-R2. The PCR product was digested with StuI and then self-ligated, yielding plasmid pTM22, which encodes the SseJ-HA fusionprotein.
Construction of Mutant Strains—Nonpolar mutants of dsbA anddsbC were constructed by allele exchange using the temperature- andsucrose-sensitive suicide vector pCACTUS (47). A disruption mutationwas created by the insertion of the SmaI-digested Kmr-encoding gene(kan) cassette from pUC18K (48) or promoterless cat gene into a uniqueEcoRV site in the coding region of dsbA and dsbC on pGEM-dsbA andpGEM-dsbC, respectively. The disrupted gene was then subcloned us-ing SalI and SphI into similarly digested pCACTUS, and the resultingplasmid was introduced into strain SL1344 by electroporation for alleleexchange mutagenesis, which was carried out as described previously(47). Chromosomal mutations were verified by PCR and DNA sequenc-ing analyses.
For the construction of mutant strains expressing point-mutatedSpiA proteins, the SacI-SphI fragments from pGEM-spiAC133S andpGEM-spiAC152S were subcloned into similarly digested pCACTUS, andeach resulting plasmid was introduced into strain SL1344 by electro-poration. The gene replacement of spiA to a point-mutated spiA wasconfirmed by DNA sequencing and by restriction enzyme digestion ofthe PCR-amplified segments with BamHI for spiAC135S and PvuIIfor spiAC152S.
The ssaV mutant was constructed by the Red disruption system (49).The primers used for this series were ssaV-red-FW and ssaV-red-RV.Strain SH100 carrying pKD46 was used for gene disruption, as de-scribed previously (49). The disrupted genes were transferred by phageP22 transduction into strain SL1344.
To construct lacZ transcriptional fusions, the DNA fragments con-taining the hilA, invF, and ssrA promoter regions were amplified byPCR using the primers hilA-Pro and hilA-RV, invF-Pro and invF-RV,and ssrA-Pro and ssrA-RV. The PCR products digested with SalI andBamHI were ligated into the same sites of pLD-lacZ�, a derivative ofsuicide vector pGP704 (50) containing a promoterless lacZ gene andreplacing the bla gene with the � fragment, producing pLD-hilAZ,pLD-invFZ, and pLD-ssrAZ. The resulting plasmids were transferredfrom E. coli SM10�pir to S. enterica serovar Typhimurium strainSH100 by conjugation. All fusion genes were introduced into the wild-type SL1344 and dsbA mutant TM100 strains by phage P22-mediatedtransduction. �-Galactosidase activities of reporter gene fusions weredetermined according to the standard procedures (51) with the sub-strate o-nitrophenyl-�-D-galactoside.
Mouse Virulence Assays—Female BALB/c mice (5–6 weeks old) wereused for the mouse infection studies and were housed at KitasatoUniversity according to the standard Laboratory Animal Care AdvisoryCommittee guidelines. To prepare the inocula, bacteria were grownovernight at 37 °C in LB broth under conditions of shaking, and thenthe bacteria were used to inoculate fresh medium (1:100). The bacteriawere grown to an A600 of 0.5 to 0.6 under the same conditions. For oralinfection, 2 � 106 bacteria diluted in PBS were inoculated at a volumeof 20 �l into groups of five mice. For intraperitoneal infection, 1 � 105
bacteria were injected at a volume of 100 �l into groups of five mice. Thecfu were determined by plating serial dilutions of the inoculum. Thesurvival of infected mice was observed daily for 4 weeks. In the casesinvolving mixed infections, the wild-type and mutant strains weregrown separately and then were mixed prior to inoculation. The precise
TABLE IBacterial strains and plasmids used in this study
Name Relevant characteristics Source/Ref.
Salmonella strainsSL1344 Serovar Typhimurium, wild-type 70SB136 SL1344 invA::kan, Km 71TM100 SL1344 dsbA::kan, Kmr This studyTM202 SL1344 dsbC::kan, Kmr This studyTM133 SL1344 spiAC133S, Cys-133 to Ser substitution This studyTM152 SL1344 spiAC152S, Cys-152 to Ser substitution This studyTM114 SL1344 �ssaV::cat, Cmr This studyTM233 SL1344 ssrA::kan, transductant, Kmr This studyTM171 SL1344 �srgA::kan, Kmr This studyTM101 TM100 containing pMW-dsbA This studyTM502 SL1344 dsbA::cat, Cmr This studyTM505 SL1344 dsbA::cat, �srgA::kan This study
E. coli strainsDH5� K-12 recA1 endA1 gyrA96 thi-1 hsdR17 Invitrogen
supE44 �(lacXYA-argR)U169 deoR (�80 dlac�(lacZ)M15)SM10�pir thi-1 thr leu tonA lacY supE recA::RP4–2-Tc::Mu �pirSK101 K-12 dsbA::Tn5, Kmr 46MC1061 K-12 araD139 �(ara, leu)7697 �lac(IZY)X74 galE15 galK16 rpsL hsdR2
(rK� mK
�) mcrA mcrB172
TM161 MC1061 dsbA::Tn5, Kmr This studyXL10-Gold K-12 �(mcrA)183 �(mcrCB-hsdSMR-mrr)173 endA1supE44 thi-1 recA1
gyrA96 relA1 lac HteStratagene
�F� proAB lacIqZ�M15 Tn10(Tetr) Amy Cmr�Plasmids
pGEM-T Easy TA cloning vector, Ampr PromegapMW118 pSC101-based low copy number plasmid, Ampr Nippon GenepMW119 pSC101-based low copy number plasmid, Ampr New England BiolabspACYC184 p15A-based low copy number plasmid, Cmr, Tetr New England BiolabspFLAG-CTC FLAG tag expression vector, Ampr SigmapUC18K Plasmid coding Kmr-encoding gene cassette 48pCACTUS Suicide vector mobilized by RP4 and sucrose-sensitive, repts, Cmr 47pKD46 Plasmid expressing � Red recombinase, Ampr 49pLD-lacZ� Integrational plasmid with promoterless lacZ gene, Sper This studypMW-dsbA dsbA in pMW118, Ampr This studypAC-dsbA dsbA in pACYC184, Cmr This studypMW-srgA srgA in pMW119, Ampr This studypAC-srgA srgA in pACYC184, Cmr This studypMW-spiA spiA in pMW118, Ampr This studypTM9 invG in pFLAG-CTC, Ampr This studypTM10 spiA in pFLAG-CTC, Ampr This studypTM11 spiAC133S in pFLAG-CTC, Ampr This studypTM12 spiAC152S in pFLAG-CTC, Ampr This studypTM22 sseJ-HA under control of native promoter in pMW118, Ampr This studypLD-hilAZ hilA::lacZ operon fusion in pLD-lacZ� This studypLD-invFZ invF::lacZ operon fusion in pLD-lacZ� This studypLD-ssrAZ ssrA::lacZ operon fusion in pLD-lacZ� This study
Disulfide Bond Formation of SPI-2 Outer Membrane Protein 34633
by guest on April 5, 2019
http://ww
w.jbc.org/
Dow
nloaded from

ratio of the two strains was determined retrospectively by a comparisonof colony counts obtained on LB agar and LB agar with the appropriateantibiotics. Mice were sacrificed at 48 h after inoculation by carbondioxide inhalation. The spleens were removed, placed in PBS, andhomogenized by mechanical disruption. The number of wild-type andmutant bacteria in the spleen was then determined by plating a dilutionseries of the lysate onto LB agar alone and LB agar containing theappropriate antibiotics. Each competitive index (CI) value indicates themean of at least three independent infections S.E. CI data wereanalyzed by Student’s t test for statistical significance. p values of 0.05or less were considered as significant.
Antibodies—Anti-SipB, anti-SipC, and anti-SseB antisera were gen-erated by the immunization of mice with the SipB-, SipC-, and SseB-FLAG fusion proteins overexpressed in E. coli DH5� and purified onanti-FLAG M2 affinity gel (Sigma). Anti-�-lactamase antiserum (dilu-tion of 1:2,000) was kindly provided by Dr. Matsuhisa Inoue (KitasatoUniversity, Kanagawa, Japan). The following antibodies were obtainedfrom commercial sources: anti-CD107a H4A3 (LAMP-1, dilution of1:1000, BD Pharmingen), anti-HA epitope tag HA.11 (dilution of1:1000, Covance), anti-Salmonella lipopolysaccharide O4 group antigen(dilution of 1:1000, Denka Seiken), anti-FLAG M2 (dilution of 1:20,000,Sigma), and anti-DnaK (dilution of 1:1000, Calbiochem). Alexa 488-conjugated goat anti-mouse IgG and Alexa 594 goat anti rabbit IgGsecondary antibodies (dilution of 1:500) were obtained from MolecularProbes. Alkaline phosphatase-conjugated goat anti-mouse IgG antibodywas purchased from Sigma and was used at dilution of 1:10,000.
Cell Culture—HeLa cells (ATCC CCL-2) were grown in minimalessential medium (Sigma) supplemented with 10% fetal bovine serum(FBS), and HEp-2 cells (ATCC CCL-23) were grown in Dulbecco’s mod-ified Eagle’s medium (Sigma) supplemented with 10% FBS. All of thecell lines used here were cultured in the presence of gentamicin andkanamycin and were maintained in a humidified atmosphere contain-ing 5% CO2 at 37 °C.
Gentamicin Protection Assay—Bacteria were grown overnight at37 °C in LB broth with aeration, diluted at 1:33 into fresh LB brothcontaining 0.3 M NaCl, and grown for another 3 h to obtain an A600 of 0.6to 0.8. The bacteria were added to HEp-2 cells (2 � 105 cells/well) in24-well plates at a multiplicity of infection of 100. The plates werecentrifuged for 5 min at 500 � g to contact bacteria and HEp-2 cells, andthen the plates were incubated for 1 h at 37 °C in the presence of 5%
CO2. To remove extracellular bacteria, the cells were washed threetimes with Hanks’ balanced salt solution, and were incubated in Dul-becco’s modified Eagle’s medium containing 10% FBS and gentamicin(100 �g/ml) for 1 h at 37 °C in the presence of 5% CO2. The cells werewashed three times with Hanks’ balanced salt solution and were lysedwith 0.2 ml of 1% Triton X-100. After incubation for 15 min at 4 °C, thesamples were vigorously mixed with 0.8 ml of PBS. The number ofintracellular bacteria was determined by plating the cells on LB agar.
Bacterial Infection of HeLa Cells—HeLa cells were seeded onto glasscoverslips (12-mm diameter) in 24-well plates at a density of 1 � 105
cells/well. Bacteria were grown at 37 °C overnight with aeration andthen were subcultured in LB broth for 3 h. The cultures were diluted inminimal essential medium and added to the HeLa cells at a multiplicityof infection of 100. The cells were incubated for 10 min at 37 °C in a 5%CO2. For the dsbA mutant strain, infection was carried out for 1 h tocompensate for the invasion deficiency of the strain. Monolayers werewashed three times with Hanks’ balanced salt solution, and then thesamples were incubated for 2 h in minimal essential medium containing10% FBS and gentamicin (100 �g/ml) in order to kill the extracellularbacteria, after which the concentration of gentamicin was decreased to5 �g/ml.
Immunofluorescence Microscopy—For the immunofluorescence anal-ysis, the cells were fixed in 4% formaldehyde in PBS for 10 min at 4 °C.After being washed three times in PBS, the fixed cells were permeabi-lized in 0.1% Triton X-100 in PBS for 5 min. The samples were thenprobed with various primary and secondary antibodies, mounted usingVectashield solution (Vector Laboratories, Inc.), and viewed at �63magnification on a Zeiss confocal laser scanning microscope (LSM510META).
Preparation of Secreted Proteins—For the preparation of secretedproteins that are dependent on SPI-1 TTSS, the bacteria were grown inLB broth containing 0.3 M NaCl overnight at 37 °C without aeration.For the preparation of secreted proteins that are dependent on SPI-2TTSS, the bacteria were grown in MgM minimal medium containing0.1% casamino acids overnight at 37 °C with aeration. For the isolationof proteins released into the culture supernatants, the supernatantsfrom bacterial cultures were filtered, and trichloroacetic acid was addedto the samples at a final concentration of 10%. After incubation on icefor 3 h, the samples were centrifuged at 16,000 � g for 45 min, and theresulting precipitated proteins were dissolved in SDS-PAGE sample
TABLE IIPrimers used in this study
Primer Sequence (restriction sites underlined)
dsbAST-1 5�-GATTACTGGCCTCGAGAGACAACG-3� (XhoI)dsbAST-2 5�-CTGTGGGATCCGAAAGATATACAG-3� (BamHI)dsbC-FW 5�-CAACTTCATCAACTGCACCATCCGC-3�dsbC-RV 5�-ATGGAGGAAGATCGGCGGGGAGTTC-3�srgA-FW 5�-CTGGTCAGCAGCAGGAGAATCAGTG-3�srgA-RV 5�-ACGCATAACCGGAATATTCCGGCTG-3�spiA-FW1 5�-CACAAGTAGTAGCTCTGAGCTTATTGC-3�spiA-RV1 5�-GGTTACCTTCATTCAGCCATACTTCCC-3�C133S-1 5�-CCTTTCATCACCGGGATCCGAGGTTAAAGAAATTACC-3�C133S-2 5�-GGTAATTTCTTTAACCTCGGATCCCGGTGATGAAAGG-3�C152S-1 5�-GTGAGCGGTGTTCCCAGCTCCCTGACTCGTATTAGTC-3�C152S-2 5�-GACTAATACGAGTCAGGGAGCTGGGAACACCGCTCAC-3�sipB-FW 5�-AGGCTCGAGAATGACGCAAGTAGCATTAGC-3� (XhoI)sipB-RV 5�-AAAGGATCCTGCGCGACTCTGGCGCAGAAT-3� (BamHI)sipC-FW 5�-GGGCTCGAGATTAGTAATGTGGGAATAAAAT-3� (XhoI)sipC-RV 5�-AAAGGATCCAGCGCGAATATTGCCTGCGAT-3� (BamHI)sseB-FW 5�-GGGAAGCTTTCAGGAAACATCTTATGGGGA-3� (HindIII)sseB-RV 5�- CTCGAGGTACGTTTTCTGCGCTATCATA-3� (XhoI)invG-FW 5�-ACCGGACTCGAGACACATATTCTTTTGGCC-3� (XhoI)invG-RV 5�-CAAGGATCCTTTAATTGCCTCCTGACCTCT-3� (BamHI)spiA-FW2 5�-GGGCTCGAGGTAAATAAACGTTTAATCTTA-3� (XhoI)spiA-RV2 5�-CCCGGATCCACCATGAGATATGCCATTATT-3� (BamHI)sseJ-Pro 5�-GGGCTCGAGTCACATAAAACACTAGCACTT-3� (XhoI)sseJ-RV 5�-CCCGGATCCTTCAGTGGAATAATGATGAGC-3� (BamHI)HA-R1 5�-GGGAGGCCTTATCCGTATGATGTGCCGGATTATGCGT AGGACTACAAGGACGACGATGACAAG-3� (StuI)HA-R2 5�-GGGAGGCCTGACAGATCCTTCAGTGGAATA-3� (StuI)ssaV-red-FW 5�-ATTATATCGTTTGTCACTCACAATCAGCACATCACG GCTGGTGTAGGCTGGAGCTGCTTC-3�ssaV-red-RV 5�-TCTGCGTCTTATGACGAGACGACAGCGCAGTATAGG TCCCCATATGAATATCCTCCTTAG-3�hilA-Pro 5�-GTAAGTCGACGTCCAGATGACACTA-3� (SalI)hilA-Rv 5�-AACCGGATCCTGCATCTGAAAAGGA-3� (BamHI)invF-Pro 5�-ACCGGTCGACTCACGGAGGAAGGGA-3� (SalI)invF-RV 5�-TCCTGGATCCGGCAATTTTCATTGT-3� (BamHI)ssrA-Pro 5�-GGGGTCGACCTGTGAAATTCGCTCACAACC-3� (SalI)ssrA-RV 5�-GGGGGATCCTCAGTCGCTAATGAGCATTGA-3� (BamHI)
Disulfide Bond Formation of SPI-2 Outer Membrane Protein34634
by guest on April 5, 2019
http://ww
w.jbc.org/
Dow
nloaded from

buffer. Proteins detached from the bacterial surface (detached fraction)were prepared as described previously (52). Briefly, bacteria grown in100 ml of MgM minimal medium containing 0.1% casamino acids werepelleted by centrifugation at 6,000 � g for 10 min and resuspended in10 ml of PBS. The bacterial suspension was then agitated on a Vortexmixer (Vortex Genie 2, Scientific Industries, Inc) at maximum speed for1 min. Bacterial cells were pelleted by centrifugation at 10,000 � g for10 min, and the supernatant was pass through a filter (0.2 �m-poresize) to remove residual bacteria. Protein in the detached fraction wasrecovered by trichloroacetic acid precipitation. After incubation on icefor 3 h, the samples were centrifuged at 16,000 � g for 45 min. Theresulting precipitated proteins were air-dried and dissolved in SDS-PAGE sample buffer.
SDS-PAGE and Western Blot Analysis—The protein samples werenormalized according to bacterial cfu, separated by SDS-PAGE, andtransferred to polyvinylidene difluoride membranes (Immobilon, Milli-pore) for immunoblotting. Western blot analysis was carried out asdescribed previously (53). Oxidized and reduced states of the proteinswere examined by comparing the gel mobilities of the proteins in sam-ples with and without DTT.
RESULTS
Characterization of Salmonella dsbA Mutation—A nonpolardsbA mutant strain of S. enterica serovar Typhimurium,TM100, was examined for its pleiotropic phenotype, as reportedpreviously (11, 40). The mutant strain exhibited mucoidy whengrown on agar plates, as well as reduced motility and a highsensitivity to DTT; the mutant strain was unable to grow in LBmedium in the presence of 8 mM DTT, as compared with thewild-type strain, which did grow in the same concentration ofDTT. The growth rate of the dsbA mutant was similar to thatof the wild-type strain when grown in rich media. To furtherconfirm the effect of the dsbA mutation on the disulfide bondformation of �-lactamase, a periplasmic protein with a singledisulfide bond, we determined the mobility of �-lactamase bySDS-PAGE under nonreducing conditions. Bacterial cell ex-tracts from the wild-type strain SL1344 carrying plasmidpBlueScriptII SK(�), which expresses �-lactamase, and a dsbAmutant strain TM100 carrying the same plasmid, were sub-jected to SDS-PAGE in the absence of DTT. �-Lactamase wasdetected by immunoblotting with antiserum specific for �-lac-tamase. As expected, the oxidized form of �-lactamase wasfound in the wild-type strain but not in the dsbA mutant strain,confirming the defects in periplasmic disulfide bond formation(Fig. 1). These results were consistent with the phenotype of adsbA mutant in E. coli (11). The dsbA mutant phenotype wascompletely restored by introducing the wild-type dsbA allele onthe plasmid into S. enterica serovar Typhimurium strainTM100 (data not shown).
Virulence of the dsbA::kan, but Not dsbC::kan, Mutant IsAttenuated in Vivo—To elucidate the role of dsbA in virulence,we examined lethality in mice after oral administration. A
mutant strain, TM100 (dsbA::kan), did not lead to lethal infec-tion when BALB/c were inoculated orally with 2 � 106 cfu/mouse, the dose at which the wild-type strain was 100% lethal(50-fold higher than the reported LD50 for SL1344) (Fig. 2).Complementation with pMW-dsbA resulted in restoration ofthe virulence defect of the mutant strain TM100 in mice. Incontrast, a strain with a mutation in dsbC was shown to haveno changes in virulence when the bacteria were administeredorally at 3 � 106 cfu/mouse (Fig. 2). These results demon-strated that DsbA, but not DsbC, is required for the full ex-pression of pathogenesis in mice by infection with Salmonella.
Effects of dsbA Mutation on SPI-1 TTSS Function—It hasbeen demonstrated that DsbA is essential for TTSS-mediatedprotein secretion in several pathogenic bacteria, includingY. pestis (31), S. flexneri (32), and P. aeruginosa (33). Therefore,to investigate the effects of dsbA mutation on SPI-1 TTSSfunction, secretion of Sip proteins by the wild-type and theisogenic dsbA mutant strain was determined by SDS-PAGEand immunoblotting analysis. The dsbA mutant strain TM100secreted SipA, SipB, and SipC proteins at lower levels into theculture medium as compared with the corresponding wild-typelevels (Fig. 3A). In addition, Western blot analysis using anti-SipB antiserum showed that the amount of these proteins inthe whole-cell extracts was similar to that of the wild-type level(Fig. 3B). We next examined the interaction of the S. entericaserovar Typhimurium dsbA mutant strain with culturedHEp-2 cells using an assay for the examination of bacterialinternalization and actin cytoskeleton reorganization. The in-activation of the dsbA gene in strain SL1344 resulted in an 85%reduction in the invasive capacity into HEp-2 cells comparedwith that of the wild-type strain (Fig. 3C). Consistent withthese results, the mutant strain was also found to be lesseffective than the wild-type strain at inducing actin cytoskele-ton rearrangement (Fig. 3D). Complementation of a mutantstrain TM100 with plasmid pMW-dsbA completely restoredthese SPI-1 TTSS-dependent phenotypes, indicating that thedisruption of the dsbA gene in S. enterica serovar Typhi-murium was responsible for the observed reduced SPI-1TTSS function.
Effects of dsbA Mutation on SPI-2 TTSS Function—S. en-terica serovar Typhimurium contains two distinct TTSS en-coded by SPI-1 and SPI-2. Thus, we next determined the effectsof dsbA mutation on the second TTSS encoded by SPI-2. First,we compared the levels of the major SPI-2 TTSS-secreted pro-teins, SseB, SseC, and SseD, in fractions of the bacterial cellsurface obtained from dsbA mutant and the wild-type strains.Bacteria were grown under conditions that induced the secre-tion of SPI-2 TTSS, and the cultures were subjected to vigorousmixing. The bacterial surface proteins (i.e. the detached frac-tion) were then analyzed by SDS-PAGE. Proteins with appar-ent molecular masses of about 20 (SseD), 21 (SseB), and 52 kDa(SseC) secreted by the wild-type strain were reduced in thedetached fraction of the dsbA mutant strain (Fig. 4A). Theseproteins were absent in the detached fraction obtained from amutant strain, TM114, defective in ssaV, a structural compo-nent of SPI-2 TTSS. Furthermore, immunoblot analysis usingmouse anti-SseB antiserum showed that the secretion of SseBin the detached fraction of the dsbA mutant strain was de-creased to 20% of the wild-type level, whereas the totalamount of SseB obtained from the whole-cell fraction of themutant strain amounted to more than 90% of that obtainedfrom the wild-type strain (Fig. 4B).
Because SPI-2 TTSS activates and functions when bacteriareside within host cells, we next examined the ability of thedsbA mutant to form Salmonella-induced filaments (SIFs). SIFformation is an intracellular phenotype that is dependent on
FIG. 1. Disulfide bond formation is defective in the S. entericaserovar Typhimurium dsbA::kan mutant strain. S. enterica sero-var Typhimurium SL1344 (wild type) and TM100 (dsbA::kan) mutantstrains were transformed with pBlueScript II SK(�) (pSK) carrying blaand were grown at 37 °C. Proteins of the whole-cell lysate were sepa-rated by SDS-PAGE under nonreducing conditions, and �-lactamasewas detected by Western blot analysis with antibody to �-lactamase.The positions of the oxidized (ox.) and reduced (red.) forms of the proteinare indicated.
Disulfide Bond Formation of SPI-2 Outer Membrane Protein 34635
by guest on April 5, 2019
http://ww
w.jbc.org/
Dow
nloaded from

the presence of functional SPI-2 TTSS (54). HeLa cells wereinfected with the wild-type strain SL1344 and the dsbA mutantstrain TM100. Twenty-two hours after infection, infected cellswere fixed, permeabilized, and stained for SIFs and bacteriausing antibodies to LAMP-1 and Salmonella, respectively. SIFswere clearly detected in HeLa cells infected with the wild-typestrain, whereas no SIFs were observed in cells infected with thedsbA mutant. This result was similar to that obtained with thephenotype of the SPI-2 TTSS mutant strain TM114 (ssaV::cat)(Fig. 4C). These findings confirmed that intracellular SPI-2TTSS function is dependent on the activity of DsbA.
A dsbA Mutation Does Not Affect the Expression of EitherSPI-1 or SPI-2—In the above experiments, the defect in DsbAfunction appeared to render the phenotype that resembled thatof strains deficient in the SPI-1 and SPI-2 secretion apparatus;however, there was no effect on the expression of the effectormolecules secreted by SPI-1 and SPI-2 TTSS. Therefore, tofurther determine the role of dsbA in the expression of SPI-1and SPI-2 TTSS, we constructed mutant strains containinghilA::lacZ and invF::lacZ transcriptional fusions for SPI-1 ex-pression and ssrA::lacZ transcriptional fusion for SPI-2 expres-sion on the chromosome both with a dsbA� and a dsbA� back-ground. Salmonella strains were grown under SPI-1- or SPI-2-inducing conditions, and �-galactosidase activity wasmeasured in the wild-type and dsbA mutant strains. As shownin Table III, transcription of hilA and invF, which are regula-tory genes for SPI-1 (55), was not affected by the loss of DsbA,whereas the transcription of ssrA, a regulatory gene for SPI-2(38), was slightly increased in the dsbA mutant strain forreasons that remain unclear. Thus, disulfide bond formationmay be essential for SPI-1 and SPI-2 TTSS apparatus assem-bly. These data were consistent with results showing that theamount of effector proteins of SPI-1 and SPI-2 in the total cellfractions was at the same level in the dsbA mutant strain as inthe wild-type strain (see Figs. 3B and 4B).
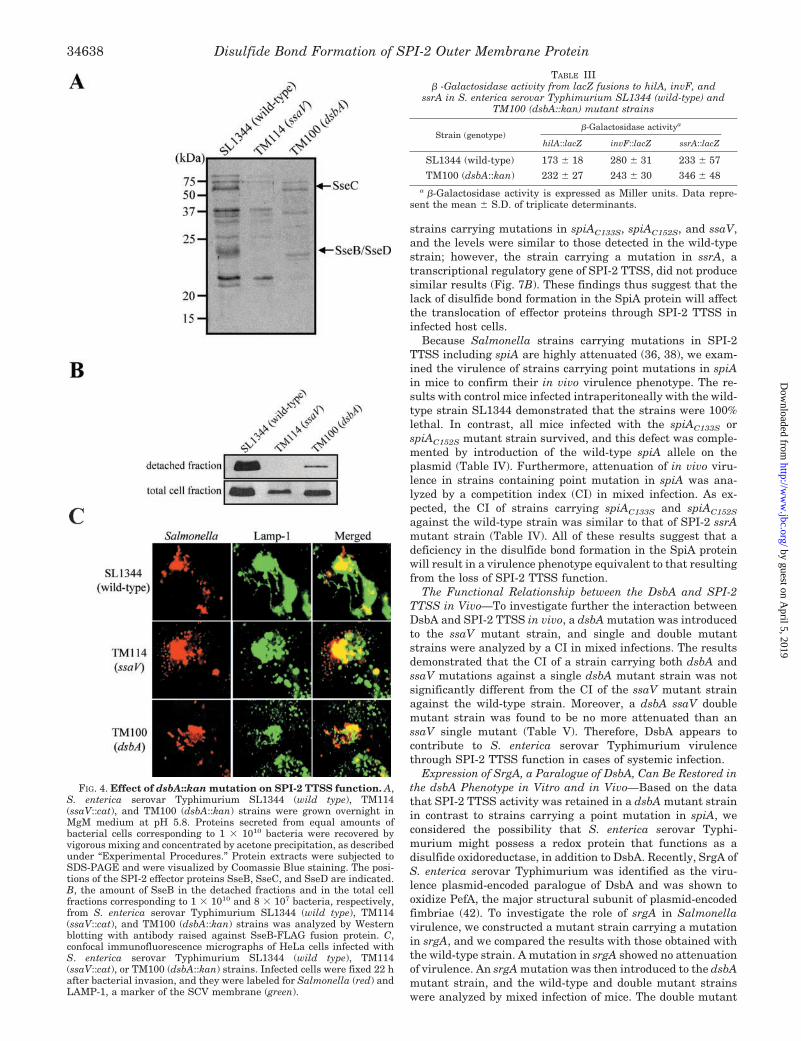
SPI-2 TTSS Outer Membrane Protein SpiA Requires DsbA toFunction—It has been reported that the outer membrane pro-tein YscC, a component of the Y. pestis TTSS apparatus, isoxidized by DsbA (31). Therefore, Salmonella DsbA may targetYscC homologues, InvG and SpiA, which are major outer mem-brane proteins of SPI-1 and SPI-2 TTSS, as substrates. Thus,the ability of Salmonella DsbA to catalyze the outer membraneproteins, InvG of SPI-1 TTSS and SpiA of SPI-2 TTSS, wasexamined. The plasmids pTM9 and pTM10 expressing C-ter-minal FLAG-tagged InvG fusion protein (InvG-FLAG) and C-terminal FLAG-tagged SpiA fusion protein (SpiA-FLAG), re-spectively, were introduced into an S. enterica serovarTyphimurium strain that carried either the dsbA� or dsbA�
allele. The folding of these fusion proteins was investigated bySDS-PAGE in the presence or absence of 1 mM DTT, and byimmunoblotting with anti-FLAG-M2 monoclonal antibody forthe detection of the proteins. The mobility of the InvG-FLAGfusion protein was not affected by the absence or presence ofDTT in the wild-type and dsbA mutant strains (Fig. 5A). Incontrast, SpiA-FLAG fusion protein was found as a single bandin all strains tested under reducing conditions, whereas differ-ent forms of the SpiA-FLAG fusion protein corresponding to thereduced and oxidized forms were observed in the wild-type andTM101 (dsbA�/dsbA�) strains, but not in the TM100 strain(dsbA::kan) under nonreducing conditions (Fig. 5B). These re-sults indicated that an intramolecular disulfide bond wasformed in SpiA. Consistent with these results, examination ofthe predicted amino acid sequences of InvG and SpiA revealedthat SpiA contained two cysteine residues at positions 133 and152, but no cysteine was present in the InvG protein.
To further examine whether the folding of SpiA could bemediated directly by the two cysteines in SpiA, we carried outsite-directed mutagenesis by using a plasmid pTM10 contain-ing spiA-FLAG fusion gene with Cys-133 or Cys-152 replacedwith serine residues. The plasmids pTM11 and pTM12 express-ing point-mutated SpiA fusion proteins, SpiAC133S-FLAG andSpiAC152S-FLAG, respectively, were introduced into S. entericaserovar Typhimurium wild-type strain SL1344. SDS-PAGEand immunoblotting analysis of the point-mutated SpiAC133S
and SpiAC152S proteins expressed by the wild-type strainshowed that the mobility of these mutant proteins was notaffected by either the presence or the absence of a reducingagent (Fig. 5C) in a manner that was consistent with the redoxstatus of the SpiA protein. These results demonstrated thatCys-133 and Cys-152 in SpiA are involved in the formation ofan intramolecular disulfide bond.
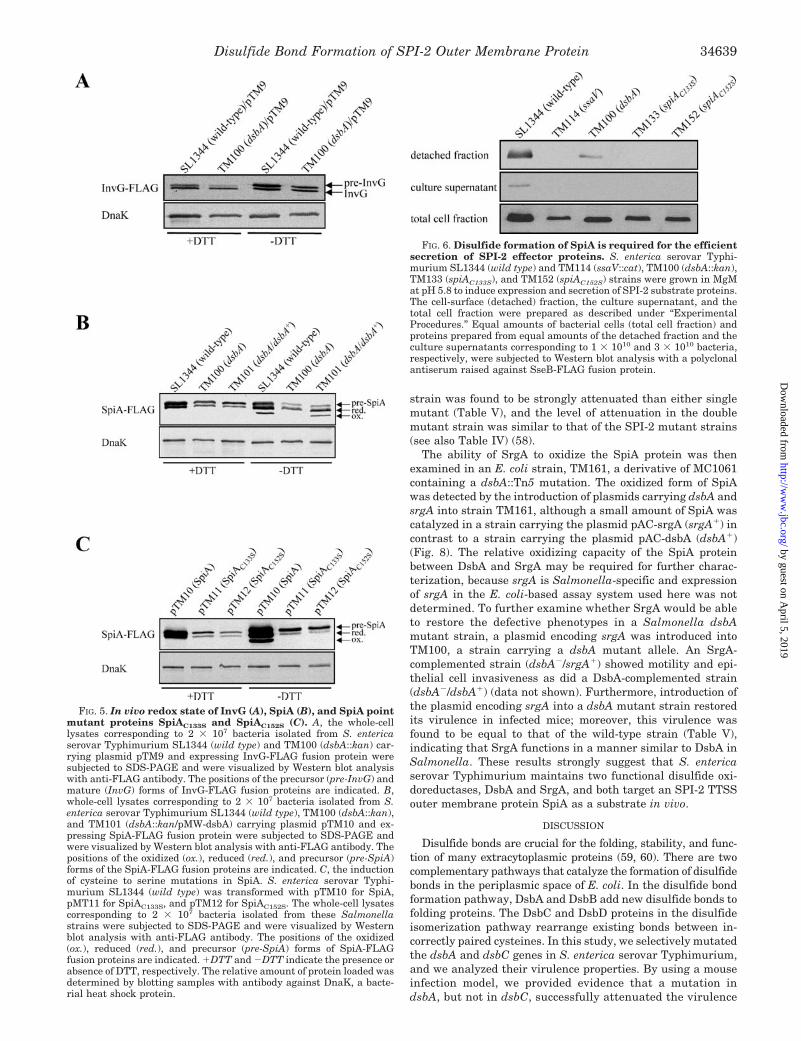
Two Cysteine Residues in SpiA Are Essential for SPI-2 TTSSFunction—To investigate SPI-2 TTSS function in the SpiApoint-mutated strains, TM133 (spiAC133S) and TM152(spiAC152S), we examined the level of SseB protein expressed onthe cell surface as well as the level of that released into theextracellular medium. The wild-type strain SL1344 was able totranslocate SseB protein on the cell surface and release it intothe culture supernatant. However, less SseB was released fromthe fractions of both mutant strains TM133 and TM152, as wasthe case with the SPI-2 TTSS mutant strain TM114 (ssaV::cat)(Fig. 6). In the dsbA mutant strain TM100, a reduced amountof SseB was expressed on the cell surface, and no SseB wasfound in the supernatant fraction (Fig. 6).
To analyze whether these Salmonella mutant strains couldassemble functional SPI-2 TTSS in an intracellular manner, we
FIG. 2. Virulence of S. enterica serovar Typhimurium dsbA::kan and dsbC::kan mutant strains. S. enterica serovar TyphimuriumSL1344 (wild type) and TM100 (dsbA::kan) and TM202 (dsbC::kan) mutant strains were administered orally with 106 cfu to female BALB/c mice.The virulence of S. enterica serovar Typhimurium carrying a mutation in dsbC was not altered, but the loss of dsbA had a marked effect onvirulence. Introduction of a cloned dsbA gene on the low copy number plasmid into the dsbA::kan mutant strain was able to restore virulence tothe wild-type level.
Disulfide Bond Formation of SPI-2 Outer Membrane Protein34636
by guest on April 5, 2019
http://ww
w.jbc.org/
Dow
nloaded from

examined the SPI-2 TTSS-dependent translocation of an effec-tor protein, SseJ, a protein that is translocated by intracellularSalmonella, and is associated with Salmonella-containingvacuoles (SCVs) and SIFs after translocation (56, 57). HeLacells were infected with Salmonella strains carrying the plas-mid pTM22 that expresses the HA-tagged sseJ under the con-trol of its native promoter. The cells were fixed at 22 h afterinfection and then were stained for immunofluorescence usinganti-HA and anti-Salmonella lipopolysaccharide antibodies.SseJ-HA fusion protein was detected in the HeLa cells infectedwith the wild-type strain but was not found in the cells infectedwith the SpiA point-mutated strains equivalent to the SPI-2TTSS mutant strain (Fig. 7A). Although a dsbA mutant strain
failed to induce LAMP-1-positive SIFs in infected cells (see Fig.4C), the same mutant strain harboring pTM22 was able totranslocate the SseJ-HA fusion protein into the infected cellcytoplasm and form SIF structures (Fig. 7A). This phenotypicdifference was probably due to limitations in the sensitivity ofthe antibodies used for immunostaining and/or the differencemay have been due to the induction of SIF formation by theplasmid-based expression of sseJ.
To investigate whether the reduced secretion of these mu-tant strains resulted from the decreased expression of the sseJgene, the level of SseJ-HA fusion protein present in the whole-cell lysate was analyzed by Western blotting using anti-HAmonoclonal antibody. The fusion protein was detected in the
FIG. 3. Effect of the dsbA::kan mutation on SPI-1 TTSS function. A, SDS-PAGE profiles of supernatant proteins from S. enterica serovarTyphimurium SL1344 (wild type), SB136 (invA::kan), TM100 (dsbA::kan), and TM101 (TM100 carrying pMW-dsbA) cultures. Proteins from theculture supernatants corresponding to 1 � 109 bacteria were precipitated as described under “Experimental Procedures,” and the proteins wereseparated by SDS-PAGE and were visualized by Coomassie Blue staining. The arrows indicate previously identified proteins that were present inthe culture supernatant from the wild-type strain but that were reduced in the case of the dsbA mutant strain (69). B, the amount of SipB in theculture supernatants and in the total cell fractions corresponding to 1 � 109 and 8 � 107 cells, respectively, from S. enterica serovar TyphimuriumSL1344 (wild type), SB136 (invA::kan), TM100 (dsbA::kan), and TM101 (TM100 with pMW-dsbA) strains was analyzed by Western blotting withantibody raised against SipB-FLAG fusion protein. The same results were obtained when mouse anti-SipC antiserum was used against the samepreparations. C, the invasiveness of S. enterica serovar Typhimurium SL1344 (wild type), SB136 (invA::kan), TM100 (dsbA::kan), and TM101(TM100 with pMW-dsbA) into HEp-2 cells was determined by gentamicin protection assays. The data represent the percentage of the wild-typeintracellular cfu and are presented as the means S.E. from triplicate experiments. D, the induction of localized actin cytoskeleton rearrange-ments in HeLa cells infected with SL1344 (wild type) (a), SB136 (invA::kan) (b), TM100 (dsbA::kan) (c), or TM101 (TM100 complemented withpMW-dsbA) (d). The actin cytoskeleton was visualized by phalloidin staining (green). Bacteria (red) were stained with anti-Salmonella antiserum.The exposure times for all of the images are identical.
Disulfide Bond Formation of SPI-2 Outer Membrane Protein 34637
by guest on April 5, 2019
http://ww
w.jbc.org/
Dow
nloaded from
strains carrying mutations in spiAC133S, spiAC152S, and ssaV,and the levels were similar to those detected in the wild-typestrain; however, the strain carrying a mutation in ssrA, atranscriptional regulatory gene of SPI-2 TTSS, did not producesimilar results (Fig. 7B). These findings thus suggest that thelack of disulfide bond formation in the SpiA protein will affectthe translocation of effector proteins through SPI-2 TTSS ininfected host cells.
Because Salmonella strains carrying mutations in SPI-2TTSS including spiA are highly attenuated (36, 38), we exam-ined the virulence of strains carrying point mutations in spiAin mice to confirm their in vivo virulence phenotype. The re-sults with control mice infected intraperitoneally with the wild-type strain SL1344 demonstrated that the strains were 100%lethal. In contrast, all mice infected with the spiAC133S orspiAC152S mutant strain survived, and this defect was comple-mented by introduction of the wild-type spiA allele on theplasmid (Table IV). Furthermore, attenuation of in vivo viru-lence in strains containing point mutation in spiA was ana-lyzed by a competition index (CI) in mixed infection. As ex-pected, the CI of strains carrying spiAC133S and spiAC152S
against the wild-type strain was similar to that of SPI-2 ssrAmutant strain (Table IV). All of these results suggest that adeficiency in the disulfide bond formation in the SpiA proteinwill result in a virulence phenotype equivalent to that resultingfrom the loss of SPI-2 TTSS function.
The Functional Relationship between the DsbA and SPI-2TTSS in Vivo—To investigate further the interaction betweenDsbA and SPI-2 TTSS in vivo, a dsbA mutation was introducedto the ssaV mutant strain, and single and double mutantstrains were analyzed by a CI in mixed infections. The resultsdemonstrated that the CI of a strain carrying both dsbA andssaV mutations against a single dsbA mutant strain was notsignificantly different from the CI of the ssaV mutant strainagainst the wild-type strain. Moreover, a dsbA ssaV doublemutant strain was found to be no more attenuated than anssaV single mutant (Table V). Therefore, DsbA appears tocontribute to S. enterica serovar Typhimurium virulencethrough SPI-2 TTSS function in cases of systemic infection.
Expression of SrgA, a Paralogue of DsbA, Can Be Restored inthe dsbA Phenotype in Vitro and in Vivo—Based on the datathat SPI-2 TTSS activity was retained in a dsbA mutant strainin contrast to strains carrying a point mutation in spiA, weconsidered the possibility that S. enterica serovar Typhi-murium might possess a redox protein that functions as adisulfide oxidoreductase, in addition to DsbA. Recently, SrgA ofS. enterica serovar Typhimurium was identified as the viru-lence plasmid-encoded paralogue of DsbA and was shown tooxidize PefA, the major structural subunit of plasmid-encodedfimbriae (42). To investigate the role of srgA in Salmonellavirulence, we constructed a mutant strain carrying a mutationin srgA, and we compared the results with those obtained withthe wild-type strain. A mutation in srgA showed no attenuationof virulence. An srgA mutation was then introduced to the dsbAmutant strain, and the wild-type and double mutant strainswere analyzed by mixed infection of mice. The double mutant
FIG. 4. Effect of dsbA::kan mutation on SPI-2 TTSS function. A,S. enterica serovar Typhimurium SL1344 (wild type), TM114(ssaV::cat), and TM100 (dsbA::kan) strains were grown overnight inMgM medium at pH 5.8. Proteins secreted from equal amounts ofbacterial cells corresponding to 1 � 1010 bacteria were recovered byvigorous mixing and concentrated by acetone precipitation, as describedunder “Experimental Procedures.” Protein extracts were subjected toSDS-PAGE and were visualized by Coomassie Blue staining. The posi-tions of the SPI-2 effector proteins SseB, SseC, and SseD are indicated.B, the amount of SseB in the detached fractions and in the total cellfractions corresponding to 1 � 1010 and 8 � 107 bacteria, respectively,from S. enterica serovar Typhimurium SL1344 (wild type), TM114(ssaV::cat), and TM100 (dsbA::kan) strains was analyzed by Westernblotting with antibody raised against SseB-FLAG fusion protein. C,confocal immunofluorescence micrographs of HeLa cells infected withS. enterica serovar Typhimurium SL1344 (wild type), TM114(ssaV::cat), or TM100 (dsbA::kan) strains. Infected cells were fixed 22 hafter bacterial invasion, and they were labeled for Salmonella (red) andLAMP-1, a marker of the SCV membrane (green).
TABLE III� -Galactosidase activity from lacZ fusions to hilA, invF, and
ssrA in S. enterica serovar Typhimurium SL1344 (wild-type) andTM100 (dsbA::kan) mutant strains
Strain (genotype)�-Galactosidase activitya
hilA::lacZ invF::lacZ ssrA::lacZ
SL1344 (wild-type) 173 18 280 31 233 57
TM100 (dsbA::kan) 232 27 243 30 346 48a �-Galactosidase activity is expressed as Miller units. Data repre-
sent the mean S.D. of triplicate determinants.
Disulfide Bond Formation of SPI-2 Outer Membrane Protein34638
by guest on April 5, 2019
http://ww
w.jbc.org/
Dow
nloaded from
strain was found to be strongly attenuated than either singlemutant (Table V), and the level of attenuation in the doublemutant strain was similar to that of the SPI-2 mutant strains(see also Table IV) (58).
The ability of SrgA to oxidize the SpiA protein was thenexamined in an E. coli strain, TM161, a derivative of MC1061containing a dsbA::Tn5 mutation. The oxidized form of SpiAwas detected by the introduction of plasmids carrying dsbA andsrgA into strain TM161, although a small amount of SpiA wascatalyzed in a strain carrying the plasmid pAC-srgA (srgA�) incontrast to a strain carrying the plasmid pAC-dsbA (dsbA�)(Fig. 8). The relative oxidizing capacity of the SpiA proteinbetween DsbA and SrgA may be required for further charac-terization, because srgA is Salmonella-specific and expressionof srgA in the E. coli-based assay system used here was notdetermined. To further examine whether SrgA would be ableto restore the defective phenotypes in a Salmonella dsbAmutant strain, a plasmid encoding srgA was introduced intoTM100, a strain carrying a dsbA mutant allele. An SrgA-complemented strain (dsbA�/srgA�) showed motility and epi-thelial cell invasiveness as did a DsbA-complemented strain(dsbA�/dsbA�) (data not shown). Furthermore, introduction ofthe plasmid encoding srgA into a dsbA mutant strain restoredits virulence in infected mice; moreover, this virulence wasfound to be equal to that of the wild-type strain (Table V),indicating that SrgA functions in a manner similar to DsbA inSalmonella. These results strongly suggest that S. entericaserovar Typhimurium maintains two functional disulfide oxi-doreductases, DsbA and SrgA, and both target an SPI-2 TTSSouter membrane protein SpiA as a substrate in vivo.
DISCUSSION
Disulfide bonds are crucial for the folding, stability, and func-tion of many extracytoplasmic proteins (59, 60). There are twocomplementary pathways that catalyze the formation of disulfidebonds in the periplasmic space of E. coli. In the disulfide bondformation pathway, DsbA and DsbB add new disulfide bonds tofolding proteins. The DsbC and DsbD proteins in the disulfideisomerization pathway rearrange existing bonds between in-correctly paired cysteines. In this study, we selectively mutatedthe dsbA and dsbC genes in S. enterica serovar Typhimurium,and we analyzed their virulence properties. By using a mouseinfection model, we provided evidence that a mutation indsbA, but not in dsbC, successfully attenuated the virulence
FIG. 5. In vivo redox state of InvG (A), SpiA (B), and SpiA pointmutant proteins SpiAC133S and SpiAC152S (C). A, the whole-celllysates corresponding to 2 � 107 bacteria isolated from S. entericaserovar Typhimurium SL1344 (wild type) and TM100 (dsbA::kan) car-rying plasmid pTM9 and expressing InvG-FLAG fusion protein weresubjected to SDS-PAGE and were visualized by Western blot analysiswith anti-FLAG antibody. The positions of the precursor (pre-InvG) andmature (InvG) forms of InvG-FLAG fusion proteins are indicated. B,whole-cell lysates corresponding to 2 � 107 bacteria isolated from S.enterica serovar Typhimurium SL1344 (wild type), TM100 (dsbA::kan),and TM101 (dsbA::kan/pMW-dsbA) carrying plasmid pTM10 and ex-pressing SpiA-FLAG fusion protein were subjected to SDS-PAGE andwere visualized by Western blot analysis with anti-FLAG antibody. Thepositions of the oxidized (ox.), reduced (red.), and precursor (pre-SpiA)forms of the SpiA-FLAG fusion proteins are indicated. C, the inductionof cysteine to serine mutations in SpiA. S. enterica serovar Typhi-murium SL1344 (wild type) was transformed with pTM10 for SpiA,pMT11 for SpiAC133S, and pTM12 for SpiAC152S. The whole-cell lysatescorresponding to 2 � 107 bacteria isolated from these Salmonellastrains were subjected to SDS-PAGE and were visualized by Westernblot analysis with anti-FLAG antibody. The positions of the oxidized(ox.), reduced (red.), and precursor (pre-SpiA) forms of SpiA-FLAGfusion proteins are indicated. �DTT and �DTT indicate the presence orabsence of DTT, respectively. The relative amount of protein loaded wasdetermined by blotting samples with antibody against DnaK, a bacte-rial heat shock protein.
FIG. 6. Disulfide formation of SpiA is required for the efficientsecretion of SPI-2 effector proteins. S. enterica serovar Typhi-murium SL1344 (wild type) and TM114 (ssaV::cat), TM100 (dsbA::kan),TM133 (spiAC133S), and TM152 (spiAC152S) strains were grown in MgMat pH 5.8 to induce expression and secretion of SPI-2 substrate proteins.The cell-surface (detached) fraction, the culture supernatant, and thetotal cell fraction were prepared as described under “ExperimentalProcedures.” Equal amounts of bacterial cells (total cell fraction) andproteins prepared from equal amounts of the detached fraction and theculture supernatants corresponding to 1 � 1010 and 3 � 1010 bacteria,respectively, were subjected to Western blot analysis with a polyclonalantiserum raised against SseB-FLAG fusion protein.
Disulfide Bond Formation of SPI-2 Outer Membrane Protein 34639
by guest on April 5, 2019
http://ww
w.jbc.org/
Dow
nloaded from

of Salmonella. Because DsbA is a key protein-folding catalyst,inactivation of dsbA might block the folding of various secretedproteins, which include important virulence determinants ofsystemic infection by Salmonella, and such inactivation maythus have conferred a reduction of virulence in this mouseinfection model.
It is known that DsbA is involved in the biogenesis of bacte-rial toxins and organelles on the bacterial surface, e.g. fimbriaeand other adhesive molecules (27–30). In addition to these
virulence factors, the dsbA gene is required for the transloca-tion of effector proteins by TTSS in Y. pestis (31), S. flexneri(32), and P. aeruginosa (33). In Y. pestis, DsbA contributes tothe stable expression of YscC, an outer membrane protein ofthe Yersinia TTSS apparatus (31). Therefore, a mutation indsbA results in unstable YscC, leading to the reduced secretionof Yop proteins. An additional effect on the expression of theTTSS apparatus has also been reported in P. aeruginosa (33).To elucidate the mechanism of virulence attenuation achievedby dsbA mutation in S. enterica serovar Typhimurium, wecharacterized various virulence phenotypes dependent on bothSPI-1 and SPI-2 TTSS in a dsbA mutant strain. Our datademonstrated that the translocation of effector proteins by bothSPI-1 and SPI-2 TTSS was deficient in the dsbA mutant strain,whereas in contrast to P. aeruginosa, the expression of SPI-1and SPI-2 TTSS was not affected by the loss of DsbA. Becauseeffector proteins are directly translocated from the cytoplasmto the extracellular space of the bacterium via the TTSS appa-ratus, the DsbA protein of S. enterica serovar Typhimurium isrequired for the formation of the TTSS apparatus.
Assembly of the TTSS apparatus is thought to proceed in amanner similar to that of a flagella basal body-hook assembly(61). For example, the first step in the assembly process of theSPI-1 TTSS is the sec-dependent export of the proteins PrgH,PrgK, and InvG, which form the stable base substructure of theTTSS machinery. Therefore, the secretion of these proteinsdoes not require a functional TTSS apparatus. This incompletebase structure is required for the transport of the main subunitof the needle substructure, PrgI, and other exported secretioncomponents including InvJ. The integration of inner membranecomponents into base substructure is completed prior to secre-tion of PrgI and needle assembly. Thus, it is likely that DsbAcould catalyze Salmonella TTSS component proteins that areexported to the periplasmic space in a sec-dependent manner.In the case of the SPI-1 TTSS apparatus, the base structurecomponent proteins PrgH, PrgK, and InvG lack multiple cys-teine residues after the cleavage of the putative signal se-quence, and they are therefore not directly affected by muta-tions of DsbA. However, InvG forms a ring-like pore structureessential for the secretion of SPI-1 effector proteins from bac-terial cells and requires an accessory lipoprotein, InvH, for itsproper location (61). Although not essential for needle complexassembly, InvH facilitates the efficient outer membrane inser-tion of InvG (62, 63). Additional evidence has shown reducedvirulence associated with invH mutation, and InvH is known tohave two cysteine residues (64). Such evidence suggests thepossibility that a mutation in dsbA causes a failure to formintramolecular disulfide bonds in InvH, resulting in the re-duced stability of InvH, which leads to the less efficient secre-tion of SPI-1 effectors. The molecular characterization of therole played by DsbA in InvH is now in progress.
To provide a possible explanation for the role of DsbA in effi-cient SPI-1 TTSS function, the pleiotropic effect caused by thedsbA mutation should also be considered. Many proteins that failto form proper disulfide bonds are degraded rapidly in bacterialcells (59). Additionally, it is known that abnormal states on thebacterial surface due to an aberrant redox balance in theperiplasm occur as a result of dsbA mutation (11). Therefore,dsbA mutant strains are hypersensitive to the reductant DTT,benzylpenicillin, and some metal ions including Hg2� and Cd2�
(22, 65). These structural changes occurring in bacterial mem-branes may affect the proper localization of SPI-1 TTSS secretorycomponents such as the InvG outer membrane protein.
In contrast to the InvG protein of the SPI-1 TTSS apparatus,SpiA, an outer membrane component of SPI-2 TTSS, bears thesec-dependent signal sequence for secretion in its N-terminal
FIG. 7. Disulfide formation of SpiA is required for the efficientSPI-2-dependent translocation of SseJ into host cells. A, confocalimmunofluorescence micrographs of HeLa cells infected with S. entericaserovar Typhimurium SL1344 (wild type), TM114 (ssaV::cat), TM133(spiAC133S), TM152 (spiAC152S), or TM100 (dsbA::kan) strains, all car-rying plasmid pTM22 expressing an HA-tagged SseJ fusion protein.The cells were fixed 22 h after bacterial entry and were examined byconfocal immunofluorescence microscopy. HA-tagged SseJ was detectedby anti-HA monoclonal antibody (green), and whole bacteria were de-tected with anti-Salmonella antiserum (red). B, the expression of HA-tagged SseJ was dependent on the SPI-2 transcriptional activator SsrA.S. enterica serovar Typhimurium SL1344 (wild type), TM114(ssaV::cat), TM133 (spiAC133S), TM152 (spiAC152S), TM100 (dsbA::kan),or TM233 (ssrA::kan) strains, all carrying plasmid pTM22, were grownin MgM at pH 5.8, and equal amounts of bacterial lysates correspondingto 8 � 107 cfu and were analyzed by Western blotting with anti-HAantibody. The relative amount of protein loaded was determined byblotting the samples with antibody against DnaK.
Disulfide Bond Formation of SPI-2 Outer Membrane Protein34640
by guest on April 5, 2019
http://ww
w.jbc.org/
Dow
nloaded from

region and possesses two cysteine residues after the cleavage ofa putative signal sequence. Thus, we investigated whetherSpiA is a disulfide bond-containing protein, the folded proteinstructure of which is required for SPI-2 TTSS function. Ourresults demonstrated that the SpiA protein produced by thewild-type strain, but not that produced by a dsbA mutantstrain, exhibited different structures corresponding to reducedand oxidized forms of the protein, as shown by SDS-PAGE
conducted under both reducing and nonreducing conditions.The replacement of the cysteine with a serine at position 133 or152 in SpiA by site-directed mutagenesis has also revealed thatthese mutated proteins produced by the wild-type strain couldnot change their oxidized structure under nonreducing condi-tions. In addition, similar to the SPI-2 TTSS-deficient mutantstrains, a Salmonella strain expressing the mutated SpiAC133S
or SpiAC152S protein had less efficient SPI-2 TTSS function, asshown by both in vitro and in vivo studies. These results thussuggested that DsbA plays an important role in the assembly ofthe SPI-2 TTSS apparatus through the disulfide bond forma-tion in the outer membrane protein SpiA.
A mutation of dsbA usually leads to phenotypic effects thatresemble those of a mutation in the gene encoding the virulencefactor per se. Our data showed that S. enterica serovar Typhi-murium strains carrying a mutation in spiA completely lostSPI-2 TTSS function, but a dsbA mutant strain was shown tohave retained reduced activity of SPI-2 TTSS, indicating theexistence of a DsbA-like protein that was able to catalyze thefolding of SpiA. In this study, we demonstrated that a virulenceplasmid-encoded SrgA efficiently oxidized the disulfide bond ofSpiA in an E. coli dsbA� background. Furthermore, because acombination of mutations in both dsbA and srgA genes con-ferred a greater reduction in virulence in mice than did singlemutations in either the dsbA or srgA alone, both the DsbA andSrgA proteins appear to be functionally important for the ac-tivity of SPI-2 TTSS in Salmonella.
The distribution of the srgA allele among Salmonella sero-vars revealed that srgA is only carried by limited strains (42).An SrgA homologue, Dlp, encoded on the virulence plasmid ofS. enterica serovar Enteritidis, and another homologue, Dlt,encoded on the chromosome of S. enterica serovar Typhi, werealso shown to have disulfide oxidoreductase enzymatic activity(66). These data suggest that a second gene encoding disulfideoxidoreductase may be an additional horizontally acquired de-terminant of some of the Salmonella serovars. Most interest-ing, the enzymatic activity of SrgA has been shown to besubstrate-specific (42). Thus, the maintenance of two differenttypes of disulfide oxidoreductase may play an important role inthe enhanced virulence of these Salmonella serovars.
Based on our data and previous studies showing that severalenvironmental factors are required to induce the functionalassembly of the SPI-2 TTSS apparatus (67), we propose here amodel for the role of Salmonella periplasmic disulfide oxi-doreductases in SPI-2 TTSS function. The expression of spiA isinduced when Salmonella reside in phagosomes, where bacte-ria impose nutrient limitations. The product of the spiA gene istransported across the inner membrane via a sec-dependentpathway; this product is in the form of a precursor protein with
TABLE IVMouse virulence for S. enterica serovar Typhimurium strains after peritoneal infection
Strain (genotype) Plasmid (genotype) Dose Survival/total micea CIb
cfu/mouse
SL1344 (wild type) 1.9 � 105 0/5 0.97 0.15c
TM133 (spiAC133S) 2.0 � 105 5/5 0.0039 0.001d
TM133 (spiAC133S) pMW-spiA (spiA�) 2.0 � 105 0/5 NDe
TM152 (spiAC152S) 1.8 � 105 5/5 0.0047 0.002d
TM152 (spiAC152S) pMW-spiA (spiA�) 1.6 � 105 0/5 NDTM232 (ssrA::kan) 2.0 � 105 5/5 0.0039 0.001d
a Survival of BALB/c mice infected with S. enterica serovar Typhimurium strains was determined at least 28 days postinoculation.b The CI was calculated as the ratio of the number of mutant strain to the wild-type strain carrying plasmid pMW118 (Ampr) bacteria recovered
from infected spleens removed 48 h after intraperitoneal injection of 105 bacteria into mice. The CI shown is the mean of the CIs obtained fromthe infection of at least three mice with corresponding standard deviations.
c The CI obtained for mixed infections with the wild-type strain and the wild-type strain carrying plasmid pMW118 was not significantlydifferent from 1 (p � 0.05).
d Significantly different (p � 0.001) from the CI obtained for mixed infections with the wild-type strain and the wild-type strain carrying plasmidpMW118.
e Not determined.
TABLE VCI value for mixed infection
GenotypeCIa
Strain A Strain B
Mixed infection 1Wild type dsbA 0.13 0.05b
Wild type ssaV 0.012 0.002b
dsbA dsbA ssaV 0.024 0.003b
ssaV dsbA ssaV 0.67 0.30c
Mixed infection 2Wild type srgA 0.89 0.13c
Wild type dsbA srgA 0.002 0.001d
Wild type dsbA/dsbA� 1.41 0.58c
Wild type dsbA/srgA� 1.15 0.35c
a The CI was calculated as the ratio of the number of strain B to thatof strain A bacteria recovered from infected spleens removed 48 h afterintraperitoneal injection of 105 bacteria into mice. The CI shown is themean of the CI values obtained from the infection of at least five mice,together with the corresponding standard deviations.
b Significantly different from 1.0 (p � 0.001).c Not significantly different from 1.0 (p � 0.05)d Significantly different from the CI of the corresponding single dsbA
mutant strain versus that of the wild-type strain (p � 0.05).
FIG. 8. Two distinct periplasmic disulfide oxidoreductases,DsbA and SrgA, target SpiA. E. coli strains TM161 (dsbA::Tn5)carrying pTM10 expressing SpiA-FLAG fusion protein were trans-formed with pACYC184 (vector only), pAC-dsbA (cloned dsbA), or pAC-srgA (cloned srgA), and protein extracts corresponding 2 � 107 cellswere subjected to SDS-PAGE under nonreduced conditions and visual-ized by Western blot analysis with anti-FLAG antibody. The positionsof the oxidized (ox.), reduced (red.), and precursor (pre-SpiA) forms ofSpiA-FLAG fusion proteins are indicated.
Disulfide Bond Formation of SPI-2 Outer Membrane Protein 34641
by guest on April 5, 2019
http://ww
w.jbc.org/
Dow
nloaded from

an N-terminal signal peptide. The signal peptide is then re-moved, and the mature SpiA protein undergoes folding that isassisted by two distinct periplasmic disulfide oxidoreductases,DsbA and SrgA, and is released into the periplasmic compart-ment. During maturation of the SCV, the pH of the phagosomallumen may change to acidic levels by fusion with vacuolarATPase (68). This acidification could induce the oligomeriza-tion of SpiA subunits, resulting in the formation of functionalpores in the outer membrane that are required for the secretionand translocation of effector proteins by SPI-2 TTSS (67). Thetranslocation of effector proteins into the host cytosol allowsthe bacteria to form SIFs and to maintain SCV where Salmo-nella can then replicate and escape from intracellular killingcaused by host defense mechanisms.
Acknowledgments—We thank Koreaki Ito for providing the E. colistrain SK101 and Matsuhisa Inoue for the gift of rabbit anti-�-lacta-mase antibody.
REFERENCES
1. Koronakis, V., Sharff, A., Koronakis, E., Luisi, B., and Hughes, C. (2000)Nature 405, 914–919
2. Koronakis, V., Andersen, C., and Hughes, C. (2001) Curr. Opin. Struct. Biol.11, 403–407
3. Delepelaire, P., and Wandersman, C. (1990) J. Biol. Chem. 265, 17118–171254. Sandkvist, M. (2001) Mol. Microbiol. 40, 271–2835. Pugsley, A. P. (1993) Microbiol. Rev. 57, 50–1086. Hueck, C. J. (1998) Microbiol. Mol. Biol. Rev. 62, 379–4337. Christie, P. J., and Vogel, J. P. (2000) Trends Microbiol. 8, 354–3608. Henderson, I. R., Cappello, R., and Nataro, J. P. (2000) Trends Microbiol. 8,
529–5329. Henderson, I. R., and Nataro, J. P. (2001) Infect. Immun. 69, 1231–1243
10. Kadokura, H., Katzen, F., and Beckwith, J. (2003) Annu. Rev. Biochem. 72,111–135
11. Bardwell, J. C., McGovern, K., and Beckwith, J. (1991) Cell 67, 581–58912. Bardwell, J. C., Lee, J. O., Jander, G., Martin, N., Belin, D., and Beckwith, J.
(1993) Proc. Natl. Acad. Sci. U. S. A. 90, 1038–104213. Guilhot, C., Jander, G., Martin, N. L., and Beckwith, J. (1995) Proc. Natl.
Acad. Sci. U. S. A. 92, 9895–989914. Kobayashi, T., Kishigami, S., Sone, M., Inokuchi, H., Mogi, T., and Ito, K.
(1997) Proc. Natl. Acad. Sci. U. S. A. 94, 11857–1186215. Bader, M., Muse, W., Ballou, D. P., Gassner, C., and Bardwell, J. C. (1999) Cell
98, 217–22716. Bader, M. W., Xie, T., Yu, C. A., and Bardwell, J. C. (2000) J. Biol. Chem. 275,
26082–2608817. Rietsch, A., Belin, D., Martin, N., and Beckwith, J. (1996) Proc. Natl. Acad. Sci.
U. S. A. 93, 13048–1305318. Bessette, P. H., Cotto, J. J., Gilbert, H. F., and Georgiou, G. (1999) J. Biol.
Chem. 274, 7784–779219. Missiakas, D., Schwager, F., and Raina, S. (1995) EMBO J. 14, 3415–342420. Rietsch, A., Bessette, P., Georgiou, G., and Beckwith, J. (1997) J. Bacteriol.
179, 6602–660821. Katzen, F., and Beckwith, J. (2000) Cell 103, 769–77922. Missiakas, D., Georgopoulos, C., and Raina, S. (1993) Proc. Natl. Acad. Sci.
U. S. A. 90, 7084–708823. Yu, J., Webb, H., and Hirst, T. R. (1992) Mol. Microbiol. 6, 1949–195824. Peek, J. A., and Taylor, R. K. (1992) Proc. Natl. Acad. Sci. U. S. A. 89,
6210–621425. Yamanaka, H., Kameyama, M., Baba, T., Fujii, Y., and Okamoto, K. (1994) J.
Bacteriol. 176, 2906–291326. Okamoto, K., Baba, T., Yamanaka, H., Akashi, N., and Fujii, Y. (1995) J.
Bacteriol. 177, 4579–458627. Jacob-Dubuisson, F., Pinkner, J., Xu, Z., Striker, R., Padmanhaban, A., and
Hultgren, S. J. (1994) Proc. Natl. Acad. Sci. U. S. A. 91, 11552–1155628. Zhang, H. Z., and Donnenberg, M. S. (1996) Mol. Microbiol. 21, 787–79729. Hicks, S., Frankel, G., Kaper, J. B., Dougan, G., and Phillips, A. D. (1998)
Infect. Immun. 66, 1570–1578
30. Leong, J. M., Morrissey, P. E., and Isberg, R. R. (1993) J. Biol. Chem. 268,20524–20532
31. Jackson, M. W., and Plano, G. V. (1999) J. Bacteriol. 181, 5126–513032. Watarai, M., Tobe, T., Yoshikawa, M., and Sasakawa, C. (1995) Proc. Natl.
Acad. Sci. U. S. A. 92, 4927–493133. Ha, U. H., Wang, Y., and Jin, S. (2003) Infect. Immun. 71, 1590–159534. Galan, J. E. (2001) Annu. Rev. Cell Dev. Biol. 17, 53–8635. Kaniga, K., Tucker, S., Trollinger, D., and Galan, J. E. (1995) J. Bacteriol. 177,
3965–397136. Ochman, H., Soncini, F. C., Solomon, F., and Groisman, E. A. (1996) Proc. Natl.
Acad. Sci. U. S. A. 93, 7800–780437. Shea, J. E., Hensel, M., Gleeson, C., and Holden, D. W. (1996) Proc. Natl. Acad.
Sci. U. S. A. 93, 2593–259738. Hensel, M., Shea, J. E., Waterman, S. R., Mundy, R., Nikolaus, T., Banks, G.,
Vazquez-Torres, A., Gleeson, C., Fang, F. C., and Holden, D. W. (1998) Mol.Microbiol. 30, 163–174
39. McClelland, M., Sanderson, K. E., Spieth, J., Clifton, S. W., Latreille, P.,Courtney, L., Porwollik, S., Ali, J., Dante, M., Du, F., Hou, S., Layman, D.,Leonard, S., Nguyen, C., Scott, K., Holmes, A., Grewal, N., Mulvaney, E.,Ryan, E., Sun, H., Florea, L., Miller, W., Stoneking, T., Nhan, M., Water-ston, R., and Wilson, R. K. (2001) Nature 413, 852–856
40. Turcot, I., Ponnampalam, T. V., Bouwman, C. W., and Martin, N. L. (2001)Can. J. Microbiol. 47, 711–721
41. Agudo, D., Mendoza, M. T., Castanares, C., Nombela, C., and Rotger, R. (2004)Proteomics 4, 355–363
42. Bouwman, C. W., Kohli, M., Killoran, A., Touchie, G. A., Kadner, R. J., andMartin, N. L. (2003) J. Bacteriol. 185, 991–1000
43. Ellermeier, C. D., and Slauch, J. M. (2004) J. Bacteriol. 186, 68–7944. Beuzon, C. R., Banks, G., Deiwick, J., Hensel, M., and Holden, D. W. (1999)
Mol. Microbiol. 33, 806–81645. Sternberg, N. L., and Maurer, R. (1991) Methods Enzymol. 204, 18–4346. Kamitani, S., Akiyama, Y., and Ito, K. (1992) EMBO J. 11, 57–6247. Morona, R., van den Bosch, L., and Manning, P. A. (1995) J. Bacteriol. 177,
1059–106848. Menard, R., Sansonetti, P. J., and Parsot, C. (1993) J. Bacteriol. 175,
5899–590649. Datsenko, K. A., and Wanner, B. L. (2000) Proc. Natl. Acad. Sci. U. S. A. 97,
6640–664550. Miller, V. L., and Mekalanos, J. J. (1988) J. Bacteriol. 170, 2575–258351. Miller, J. H. (1992) A Short Course in Bacterial Genetics, pp. 72–74, Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, NY52. Nikolaus, T., Deiwick, J., Rappl, C., Freeman, J. A., Schroder, W., Miller, S. I.,
and Hensel, M. (2001) J. Bacteriol. 183, 6036–604553. Gotoh, H., Okada, N., Kim, Y. G., Shiraishi, K., Hirami, N., Haneda, T., Kurita,
A., Kikuchi, Y., and Danbara, H. (2003) Microb. Pathog. 34, 227–23854. Garcia-del Portillo, F., Zwick, M. B., Leung, K. Y., and Finlay, B. B. (1993)
Proc. Natl. Acad. Sci. U. S. A. 90, 10544–1054855. Bajaj, V., Hwang, C., and Lee, C. A. (1995) Mol. Microbiol. 18, 715–72756. Ruiz-Albert, J., Yu, X. J., Beuzon, C. R., Blakey, A. N., Galyov, E. E., and
Holden, D. W. (2002) Mol. Microbiol. 44, 645–66157. Freeman, J. A., Ohl, M. E., and Miller, S. I. (2003) Infect. Immun. 71, 418–42758. Beuzon, C. R., and Holden, D. W. (2001) Microbes Infect. 3, 1345–135259. Gilbert, H. F. (1997) J. Biol. Chem. 272, 29399–2940260. Ritz, D., and Beckwith, J. (2001) Annu. Rev. Microbiol. 55, 21–4861. Sukhan, A., Kubori, T., Wilson, J., and Galan, J. E. (2001) J. Bacteriol. 183,
1159–116762. Daefler, S., and Russel, M. (1998) Mol. Microbiol. 28, 1367–138063. Crago, A. M., and Koronakis, V. (1998) Mol. Microbiol. 30, 47–5664. Altmeyer, R. M., McNern, J. K., Bossio, J. C., Rosenshine, I., Finlay, B. B., and
Galan, J. E. (1993) Mol. Microbiol. 7, 89–9865. Stafford, S. J., Humphreys, D. P., and Lund, P. A. (1999) FEMS Microbiol.
Lett. 174, 179–18466. Rodriguez-Pena, J. M., Alvarez, I., Ibanez, M., and Rotger, R. (1997) Microbi-
ology 143, 1405–141367. Rappl, C., Deiwick, J., and Hensel, M. (2003) FEMS Microbiol. Lett. 226,
363–37268. Rathman, M., Sjaastad, M. D., and Falkow, S. (1996) Infect. Immun. 64,
2765–277369. Komoriya, K., Shibano, N., Higano, T., Azuma, N., Yamaguchi, S., and Aizawa,
S. I. (1999) Mol. Microbiol. 34, 767–77970. Hoiseth, S. K., and Stocker, B. A. (1981) Nature 291, 238–23971. Galan, J. E., Ginocchio, C., and Costeas, P. (1992) J. Bacteriol. 174, 4338–434972. Casadaban, M. J., and Cohen, S. N. (1980) J. Mol. Biol. 138, 179–207
Disulfide Bond Formation of SPI-2 Outer Membrane Protein34642
by guest on April 5, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Tsuyoshi Miki, Nobuhiko Okada and Hirofumi DanbaraType III Secretion System
Pathogenicity Island 2SalmonellaMembrane Protein SpiA, a Component of the Two Periplasmic Disulfide Oxidoreductases, DsbA and SrgA, Target Outer
doi: 10.1074/jbc.M402760200 originally published online May 28, 20042004, 279:34631-34642.J. Biol. Chem.
10.1074/jbc.M402760200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/279/33/34631.full.html#ref-list-1
This article cites 71 references, 37 of which can be accessed free at
by guest on April 5, 2019
http://ww
w.jbc.org/
Dow
nloaded from


















