
The Genetics and Prevention of Sudden Cardiac...
12
The Genetics and Prevention of Sudden Cardiac Death
Transcript of The Genetics and Prevention of Sudden Cardiac...

The Genetics and Prevention of Sudden Cardiac Death

Sudden cardiac death (SCD), a serious public health problem
Every day, between 1,600 and 2,000 people die worldwide from genetically caused SCD.1
SCD is a complex disease. Its pathogenesis involves different genetic variants.2
Some heart diseases, particularly cardiomyopathies and channelopathies, play a fundamental role in SCD.3
SCD can be preventedThe identification of at-risk patients helps to prevent a fatal outcome. 4

Genetics, key for a comprehensive diagnosis
Genes involved in channelopathies. Adapted from text ref. 5.
Clinical diagnosis
Comprehensive diagnosis
Molecular diagnosis
Based on signs and symptoms. It can be determined before the disease is expressed.
• Medical record• Electrocardiogram• Echocardiography• Magnetic resonance imaging• Pharmacological tests• Electrophysiological studies
Genetics
Genes involved in cardiomyopathies. Adapted from text ref. 3.
subunit α
subunit β
Ca2+
K+
K+
K+
Ca2+
Ca2+
Na+
Na+
Cay1.2
CASQ2
SERCA2a
RyR2SR
Nay1.5
HCN4
Kv11.1
Kir2.1
Kv7.1
mink
/Na
/f
/Cal/
Ks
/K1
/Kr
PLNK+
subunit γ subunit δ
subunit α2
subunit α2
Cytosol
Cytoskeleton
Caveolin 3
subunit β
Cell membrane
Syntrophin α1
KCNE1, KCNE2, KCNH2, KCNJ2, KCNQ1,KCND3, KCNE3, KCNE1L (KCNE5),
KCNJ5, KCNJ8, KCNA5, KCNE4
CACNA1C, CACNB2, CACNA2D1, CACNA1G, CACNA1H, CACNA1I
SCN4B, SCN5A, SCN1B, SCN2B, SCN3B
Cytoplasm
Nuclear membrane
BAG3, CSRP3, FHL2, LDB3,
MYOZ2, NEBL, NEXN, TCAP, VCL
TMPO
CRYAB
Extracellular matrix
Laminin
Desmosome
Sarcoplasmic reticulum
Nucleus
ACTC1, MYBPC3, MYH6, MYH7, MYL2, MYL3,
TNNC1, TNNI3, TNNT2, TPM1,
TTN
EMD
LMNA
LAMA4
a
a
b
b
DSP, DSG2, DSC2, JUP,
PKP2
CASQ2, PLN, RYR2
DES
CAV3
Protein complex associated with dystrophin
Z disk
Sarcomere
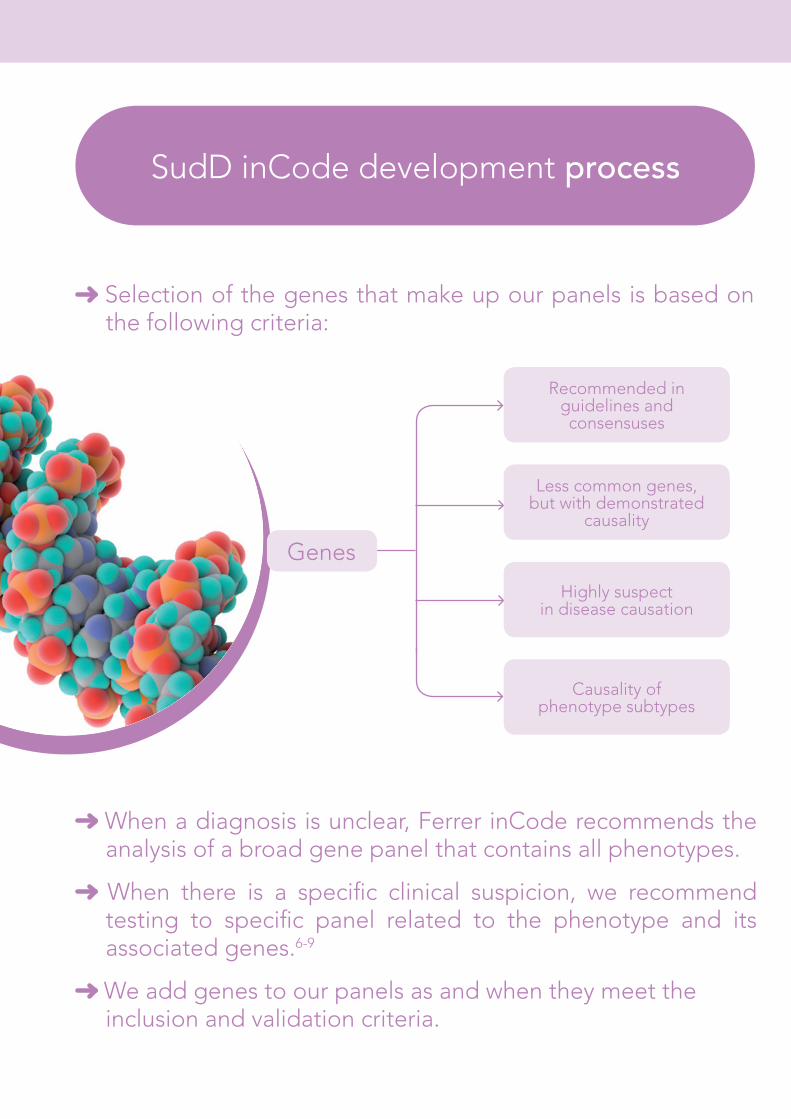
Selection of the genes that make up our panels is based on the following criteria:
When a diagnosis is unclear, Ferrer inCode recommends the analysis of a broad gene panel that contains all phenotypes.
When there is a specific clinical suspicion, we recommend testing to specific panel related to the phenotype and its associated genes.6-9
We add genes to our panels as and when they meet the inclusion and validation criteria.
SudD inCode development process
Less common genes, but with demonstrated
causality
Recommended in guidelines and consensuses
Highly suspect in disease causation
Causality of phenotype subtypes
Genes

** Familial segregation studies are essential to analyse pathogenicity. 10, 11
Graphs adapted from text ref. 10.
Clinical utility of SudD inCode
Benefits
Familial
Index case
• Confirmation of diagnosis*• Establishment of preventive
measures• Reproductive counselling
• Presymptomatic diagnosis • Establishment of preventive
measures• Reproductive counselling• Adjustment of frequency of clinical
monitoring• Provides the basis for the
pathogenicity study**
* when the causal mutation is found

Current guidelines12, 13 recommend genetic analysis of the genes associated with the clinical suspicion.
There are more than 50 different genes associated with channelopathies and cardiomyopathies14, so next-generation sequencing (NGS) is the solution of choice for identifying them.15
Sequencing the complete gene is extremely important.16
Clinical guidelines
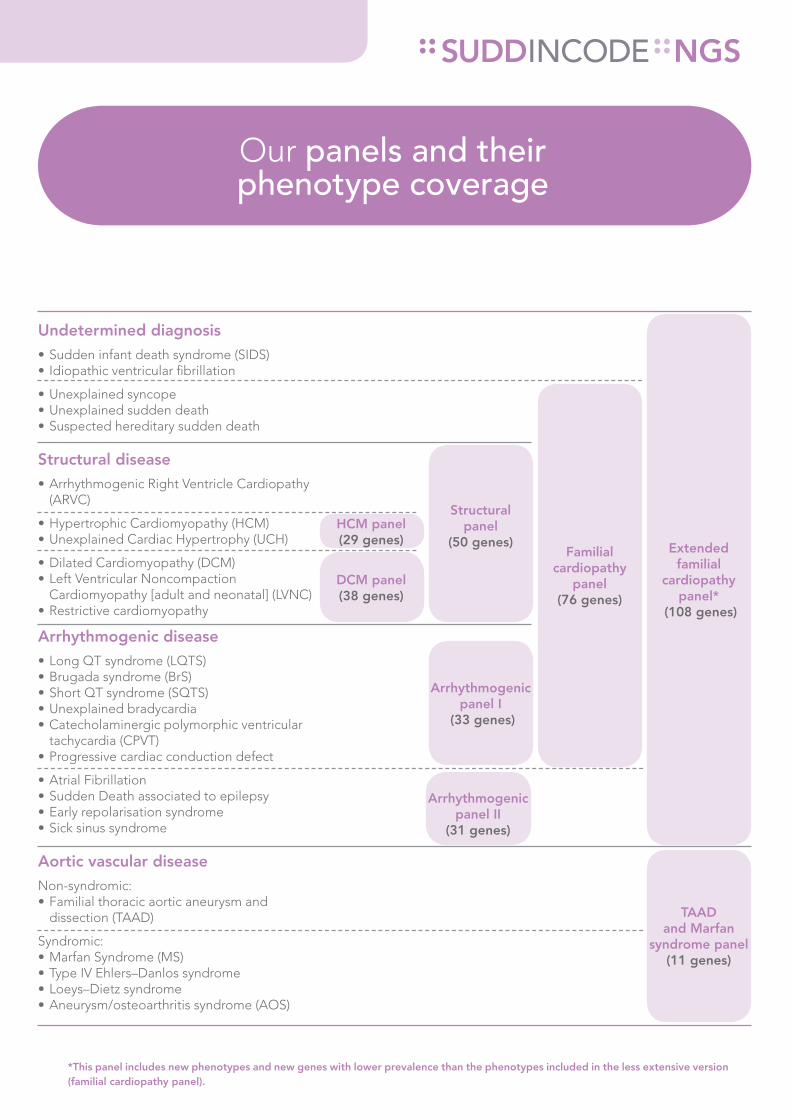
Extended familial
cardiopathy panel*
(108 genes)
Familial cardiopathy
panel (76 genes)
Structural panel
(50 genes)
Arrhythmogenic panel II
(31 genes)
Arrhythmogenic panel I
(33 genes)
HCM panel (29 genes)
DCM panel (38 genes)
TAAD and Marfan
syndrome panel (11 genes)
Our panels and their phenotype coverage
*This panel includes new phenotypes and new genes with lower prevalence than the phenotypes included in the less extensive version (familial cardiopathy panel).
Undetermined diagnosis• Sudden infant death syndrome (SIDS)• Idiopathic ventricular fibrillation
• Unexplained syncope• Unexplained sudden death • Suspected hereditary sudden death
Structural disease• Arrhythmogenic Right Ventricle Cardiopathy
(ARVC)
• Hypertrophic Cardiomyopathy (HCM)• Unexplained Cardiac Hypertrophy (UCH)
• Dilated Cardiomyopathy (DCM)• Left Ventricular Noncompaction
Cardiomyopathy [adult and neonatal] (LVNC)• Restrictive cardiomyopathy
Arrhythmogenic disease• Long QT syndrome (LQTS)• Brugada syndrome (BrS)• Short QT syndrome (SQTS)• Unexplained bradycardia• Catecholaminergic polymorphic ventricular
tachycardia (CPVT)• Progressive cardiac conduction defect
• Atrial Fibrillation • Sudden Death associated to epilepsy• Early repolarisation syndrome• Sick sinus syndrome
Aortic vascular diseaseNon-syndromic:• Familial thoracic aortic aneurysm and
dissection (TAAD)
Syndromic:• Marfan Syndrome (MS)• Type IV Ehlers–Danlos syndrome• Loeys–Dietz syndrome• Aneurysm/osteoarthritis syndrome (AOS)

NGS, next-generation sequencing
NGS, the present and the future of genetic analysis for clinical diagnosis
Multiple studies have been published that have led to genetic diagnosis programmes based on the use of NGS.17, 18
NGS sequencing is around 20 times cheaper than Sanger sequencing as well as being much faster, without losing precision.13
NGS, technological evolution.15*
• More genes analysed• Lower cost • Less time• With the same precision
*Compared to the Sanger technique, used to date.

Our NGS service
NGS panels are performed using the oligonucleotide-based target-capture technique (enrichment array designed and validated for exclusive use by Ferrer inCode).
Ultrasequencing on Illuminainc platform
(HiSeq2000 or MiSeq).
Obtaining genetic variants combines SAMTOOLS and our own program (GendiCall) to identify possible variations.
Data are expressed based on the hg19 version of the genome.
NGS is supplemented with Sanger technology (ABI 3710) to obtain > 99.9% coverage of encoding areas and exon-intron boundaries.
More than 99.9% of substitution variants (SNPs) are detected, as well as small insertions and deletions of 1 to 6 nucleotides.
All variants with possible pathogenic classification are confirmed by Sanger sequencing.
NGS, next-generation sequencing
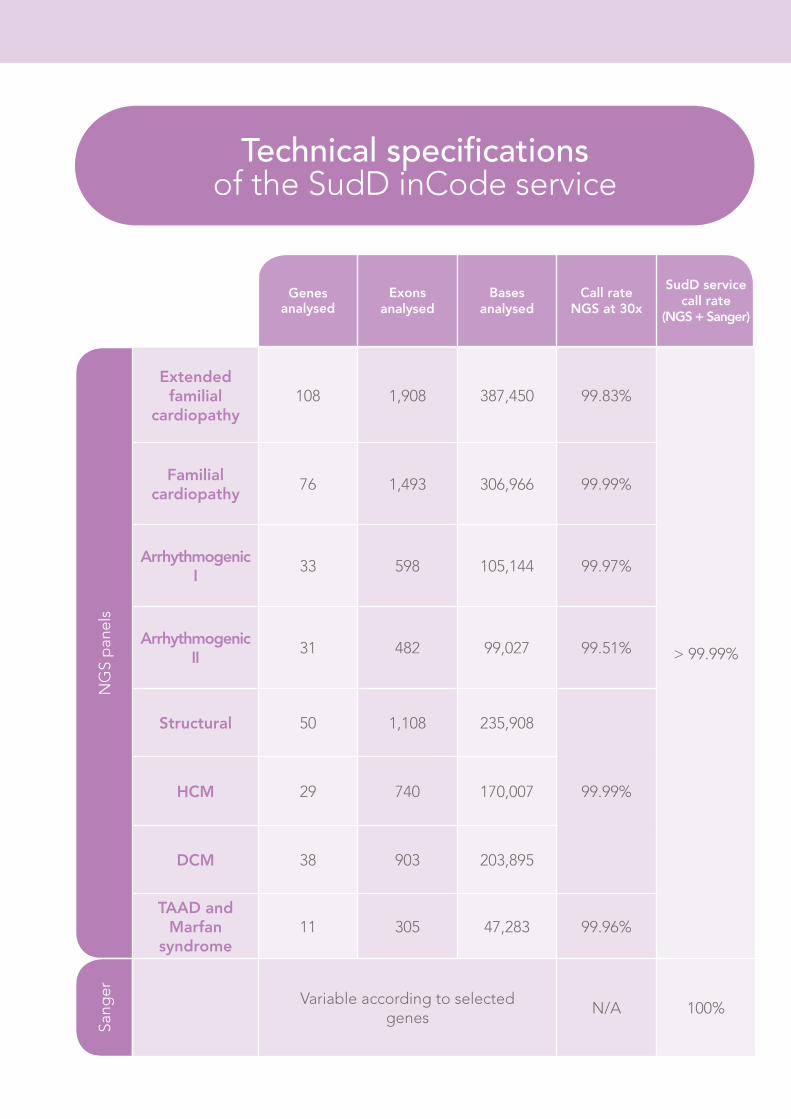
Genes analysed
Exons analysed
Bases analysed
Call rate NGS at 30x
SudD service call rate
(NGS + Sanger)
NG
S p
anel
s
Extended familial
cardiopathy108 1,908 387,450 99.83%
> 99.99%
Familial cardiopathy
76 1,493 306,966 99.99%
Arrhythmogenic I
33 598 105,144 99.97%
Arrhythmogenic II
31 482 99,027 99.51%
Structural 50 1,108 235,908
99.99%HCM 29 740 170,007
DCM 38 903 203,895
TAAD and Marfan
syndrome11 305 47,283 99.96%
Sang
er Variable according to selected genes
N/A 100%
Technical specifications of the SudD inCode service

References: 1. De Asmundis C et al. Epidemiology of Sudden Cardiac Death. Rev Esp Cardiol Supl. 2013;13(A):2-6 - Vol. 13 Núm.Supl.A. 2. Teekakirikul P et al. Review: Inherited Cardiomyopathies. Molecular Genetics and Clinical Genetic Testing in the Postgenomic Era. J Mol Diagn. 2013 Mar;15(2):158-70. 3. Tang Y et al. Molecular diagnostics of cardiovascular diseases in sudden unexplained death. Cardiovasc Pathol. 2014 Jan-Feb;23(1):1-4. 4. Brugada J et al. Sudden Cardiac Death. The Need for a Comprhensive Disease Managment Strategy. Rev Esp Cardiol Supl. 2013;13(A):1. 5. Arthur A et al. Genetic testing for inherited cardiac disease. Nat Rev Cardiol. 2013 Oct;10(10):571-83. 6. Voelkerding KV et al. Next-generation sequencing: from basic research to diagnostics. Clin Chem. 2009 Apr;55(4):641-58. 7. Voelkerding KV et al. Next generation sequencing for clinical diagnostics-principles and application to targeted resequencing for hypertrophic cardiomyopathy: a paper from the 2009 William Beaumont Hospital Symposium on Molecular Pathology. J Mol Diagn. 2010 Sep;12(5):539-51. 8. Dames S et al. Comparison of the Illumina Genome Analyzer and Roche 454 GS FLX for resequencing of hypertrophic cardiomyopathy-associated genes. J Biomol Tech. 2010 Jul;21(2):73-80. 9. Gowrisankar S et al. Evaluation of second-generation sequencing of 19 dilated cardiomyopathy genes for clinical applications. J Mol Diagn. 2010 Nov;12(6):818-27. 10. Riuró H et al. Genetic analysis, in silico prediction, and family segregation in long QT syndrome. Eur J Hum Genet. 2015 Jan;23(1):79-85. 11. Pérez-Serra A et al. A novel mutation in lamin a/c causing familial dilated cardiomyopathy associated with sudden cardiac death. J Card Fail. 2015 Mar;21(3):217-25. 12. Ackerman MJ et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011 Aug;8(8):1308-39. 13. Elliott PM et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014 Oct 14;35(39):2733-79. 14. Tester DJ et al. Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice. Circulation. 2011 Mar 8;123(9):1021-37. 15. Allegue C. New Genetic Diagnostic Tests for Sudden Cardiac Death. Rev Esp Cardiol Supl. 2013;13(A):24-29. 16. Ashley EA et al. Genetics and cardiovascular disease: a policy statement from the American Heart Association. Circulation. 2012 Jul 3;126(1):142-57. 17. Brion M et al. New technologies in the genetic approach to sudden cardiac death in the young. Forensic Sci Int. 2010 Dec 15;203 (1-3):15-24. 18. Meder B et al. Targeted next-generation sequencing for the molecular genetic diagnostics of cardiomyopathies. Circ Cardiovasc Genet. 2011 Apr;4(2):110-22.

Customer [email protected]
Tel. +34 936 003 883 SIC
1500
2 - e
d. 0
4/15
• F
OR
THE
EXC
LUSI
VE U
SE O
F H
EALT
HC
ARE
PRO
FESS
ION
ALS
Ability to analyse related genetic variants in a single process.
NGS, a cost-effective technological evolution in the genetic analysis of SCD.
Analytical quality, analysis of more than 99.99% in gene sequencing.
Genetic and clinical counselling from specialists with extensive international experience.
Results in 10 weeks


















