
The first example of an unusual rearrangement - …10.1007/s10593-015-1801...The first example of an...
16
SI - 1 Chemistry of Heterocyclic Compounds 2015, 51(10), 940–943 The first example of an unusual rearrangement in the van Leusen imidazole synthesis Mokhtar Fodili 1, *, Bellara Nedjar-Kolli 2 , Marc Vedrenne 3 , Nathalie Saffon-Merceron 3 , Christian Lherbet 4 , Pascal Hoffmann 4, * SUPPLEMENTARY INFORMATION 1 Université Ziane Achour, Laboratoire de Chimie Organique & Substances Naturelles, Djelfa, Algerie; e-mail: [email protected] 2 Université des Sciences et de la Technologie Houari Boumediene, Laboratoire de Chimie Organique, Alger, Algérie; e-mail:[email protected] 3 Université de Toulouse, UPS, Institut de Chimie de Toulouse, ICT FR 2599, 118 route de Narbonne, 31062 Toulouse cedex 9, France; e-mail:[email protected]; [email protected] 4 Université de Toulouse, UPS, LSPCMIB (Laboratoire de Synthèse et Physico-Chimie de Molécules d’Intérêt Biologique), UMR CNRS 5068, 118 route de Narbonne, 31062 Toulouse cedex 9, France; e-mail: [email protected]; [email protected]
-
Upload
phungduong -
Category
Documents
-
view
215 -
download
1
Transcript of The first example of an unusual rearrangement - …10.1007/s10593-015-1801...The first example of an...

SI - 1
Chemistry of Heterocyclic Compounds 2015, 51(10), 940–943
The first example of an unusual rearrangement in the van Leusen imidazole synthesis
Mokhtar Fodili1,*, Bellara Nedjar-Kolli2, Marc Vedrenne3, Nathalie Saffon-Merceron3, Christian Lherbet4, Pascal Hoffmann4,*
SUPPLEMENTARY INFORMATION
1 Université Ziane Achour, Laboratoire de Chimie Organique & Substances Naturelles,
Djelfa, Algerie; e-mail: [email protected] 2 Université des Sciences et de la Technologie Houari Boumediene, Laboratoire de Chimie Organique,
Alger, Algérie; e-mail:[email protected] 3 Université de Toulouse, UPS, Institut de Chimie de Toulouse,
ICT FR 2599, 118 route de Narbonne, 31062 Toulouse cedex 9, France; e-mail:[email protected]; [email protected]
4 Université de Toulouse, UPS, LSPCMIB (Laboratoire de Synthèse et Physico-Chimie de Molécules d’Intérêt Biologique), UMR CNRS 5068, 118 route de Narbonne, 31062 Toulouse cedex 9, France; e-mail: [email protected]; [email protected]
Dzintra
Typewritten Text
Dzintra
Typewritten Text
Dzintra
Typewritten Text
Dzintra
Typewritten Text
Dzintra
Typewritten Text
Dzintra
Typewritten Text

SI - 2
Synthesis of (3E)-6-Methyl-3-[1-(thiophen-2-ylmethylamino)ethylidene]-2H-pyran-2,4(3H)-dione (1).
A solution of dehydroacetic acid (4) (1.68 g, 0.01 mol) and 2-thienylmethanamine (1.13 g, 0.01 mol) in ethanol (40 ml) was stirred for 6 h at room temperature. The reaction mixture was then concentrated under reduced pressure to a volume of about 10 ml, and the pale-yellow precipitated enamine 1 was collected by filtration and washed with cold ethanol. Yield 68%. Yellow powder. Mp 112–114°C. IR spectrum, ν, cm–1: 3105, 3082, 1691, 1659, 1575, 1468, 1390, 1361, 1324, 1062, 998, 928, 728. 1H NMR spectrum (300 MHz, CDCl3), δ, ppm (J, Hz): 2.10 (3H, s, CH3); 2.69 (3H, s, CH3); 4.83 (2H, d, J = 5.4, CH2); 5.66 (1H, s, 5-CH pyran); 6.95–7.02 (2H, m, H-3,4 thiophene); 7.27 (1H, dd, J = 1.3, J = 5.0, H-5 thiophene); 14.55 (1H, br. s, NH). 13C NMR spectrum (75 MHz, CDCl3), δ, ppm: 18.3 (C-8); 19.8 (C-1); 42.8 (C-9); 96.9 (C-5); 107.3 (C-3); 126.0 (C-13); 126.6 (C-11); 127.3 (C-12); 137.2 (C-10); 162.9 (C-6); 163.6 (C-2); 176.0 (C-4); 184.8 (C-7). Found m/z: 264.0698 [M+H]+. C13H14NO3S. Calculated, m/z: 264.0689.
O
O
Me
HN
S
Me O
1 1H-NMR (recorded on a Bruker spectrometer at 300 MHz using CDCl3 as the solvent. For 1H NMR the residual proton signal of the deuterated solvent was used as an internal reference: CDCl3 7.26 ppm).
ppm (t1)0.05.010.015.0
-1000
0
10000
20000
30000
40000
50000
60000
70000
80000
14
.54
91
4.5
47
7.2
81
7.2
77
7.2
64
7.2
60
7.0
20
7.0
16
7.0
08
7.0
05
6.9
85
6.9
74
6.9
69
6.9
57
5.6
57
4.8
29
4.8
10
2.6
90
2.0
98
2.0
21
.00
1.0
1
2.0
6
3.0
7
2.8
5
0.9
1

SI - 3
ppm (t1)2.03.04.05.06.07.0
-1000
0
10000
20000
30000
40000
50000
60000
70000
80000
72
77
7.2
64
7.2
60
7.0
20
7.0
16
7.0
08
7.0
05
6.9
85
6.9
74
6.9
69
6.9
57
5.6
57
4.8
29
4.8
10
2.6
90
2.0
98
2.0
2
1.0
0
1.0
1
2.0
6
3.0
7
2.8
5
J-MOD 13C- NMR (recorded on a Bruker spectrometer at 75 MHz. The carbon signal of the deuterated solvent was used as an internal reference: CDCl3 = 77.0 ppm. Quaternary and CH2 carbons are positive and CH3 / CH carbons are negative).
ppm (t1)50100150
-4000
-3000
-2000
-1000
0
10000
20000
30000
4000018
4.7
49
17
6.0
36
16
3.6
26
16
2.9
13
13
7.1
67
12
7.3
03
12
6.5
53
12
6.0
04
10
7.3
08
96
.89
4
42
.82
7
19
.80
0
18
.25
4
TOF MS ES+ (high-resolution mass spectra (HRMS) recorded on a Xevo® G2 Q-TOF-Waters, using electrospray ionization methods).

SI - 4
FT-IR (recorded on a Thermo Scientific Nicolet Nexus 670 FT-IR Spectrometer Thermo Nicolet Corporation, equipped with a Smart iTR with a diamond window Thermo Fisher Scientific Inc).

SI - 5
Synthesis of 4-Hydroxy-6-methyl-3-[5-methyl-1-(thiophen-2-ylmethyl)imidazol-4-yl]-2H-pyran-2-one (2).
A solution of compound 1 (2.63 g, 0.01 mol), toluenesulfonylmethyl isocyanide (1.95 g, 0.01 mol), bismuth(III) trifluoromethanesulfonate (66 mg, 0.1 mmol) and tert-butylamine (3.65 g, 0.05 mol) in methanol (40 ml) was stirred at room temperature for 48 h. The solution was then evaporated to dryness under reduced pressure and the crude product was purified by column chromatography on silica gel using a mixture of ethyl acetate–methanol, 9:1, as eluent to afford compound 2. Yield 2.11 g (70%). Pale-yellow powder. Mp 227–228°C. IR spectrum, ν, cm–1: 3101, 3087, 1666, 1655, 1626, 1537, 1479, 1434, 1414, 1294, 1270, 1226, 1207, 994, 904, 778, 737. 1H NMR spectrum (500 MHz, DMSO-d6), δ, ppm (J, Hz): 2.09 (3H, d, J = 0.9, CH3C–O); 2.24 (3H, s, CH3C=N); 5.53 (2H, s, CH2); 5.78 (1H, q, J = 0.9, 5-CH pyran); 7.05 (1H, dd, J = 3.5, J = 5.1, H-4 thiophene); 7.16–7.18 (1H, m, H-3 thiophene); 7.55 (1H, dd, J = 1.3, J = 5.1, H-5 thiophene); 8.52 (1H, s, H imidazole). 13C NMR spectrum (125 MHz, DMSO-d6), δ, ppm: 10.5 (C 10); 19.7 (C-6); 44.0 (C-11); 90.1 (C-2); 105.0 (C-4); 124.5 (C-7); 127.4 (C-15); 127.8 (C-13); 128.0 (C-14); 129.4 (C-9); 133.2* (C-8); 138.6 (C-12); 160.3 (C-1); 162.8 (C5); 173.5 (C-3). Found, m/z: 303.0799 [M+H]+. C15H15N2O3S. Calculated, m/z: 303.0798.
1H-NMR (recorded on a Bruker spectrometer at 300 MHz using DMSO-d6 as the solvent. For 1H NMR the residual proton signal of the deuterated solvent was used as an internal reference: DMSO = 2.508 ppm).
2D NOESY 1H-1H (2D NOESY NMR spectrum of imidazole 2 in dmso-d6 recorded on a Bruker Avance 500 MHz equipped with a 5 mm TCI cryoprobe with mixing time of 0.5 s; Cross-peaks, circled in green, indicate the spatial proximity of C(10)H3 and C(11)H2.)
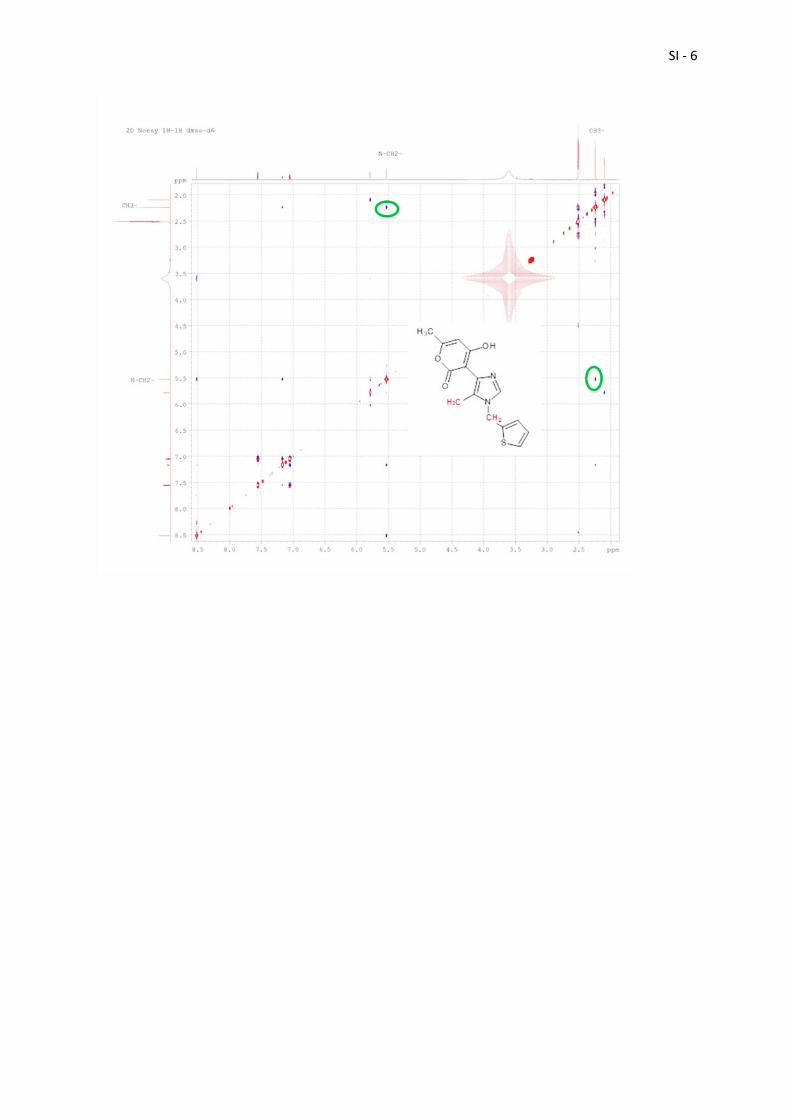
SI - 6

SI - 7
J-MOD 13C- NMR (recorded on a Bruker spectrometer at 125 MHz. The carbon signal of the deuterated solvent was used as an internal reference: DMSO = 40.10 ppm. Quaternary and CH2 carbons are positive and CH3 / CH carbons are negative).
TOF MS ES+
(high-resolution mass spectra (HRMS) recorded on a Xevo® G2 Q-TOF-Waters, using electrospray ionization methods)

SI - 8
FT-IR ( recorded on a Thermo Scientific Nicolet Nexus 670 FT-IR Spectrometer Thermo Nicolet Corporation, equipped with a Smart iTR with a diamond window Thermo Fisher Scientific Inc)

SI - 9
Crystallographic data
Intensity data for compounds 1 and 2 were collected using MoK radiation (wavelength =
0.71073 Å) on a Bruker-AXS APEX II Quazar diffractometer using a 30 W air-cooled
microfocus source (ImS) with focusing multilayer optics (2) and on a Bruker-AXS D8-
Venture diffractometer equipped with a CMOS Area detector (1). Phi- and omega-scans were
used. Crystals were mounted in inert oil and crystal structure determinations were effected at
193K. The data were integrated with SAINT[1] and an empirical absorption correction with
SADABS was applied (SADABS, Program for data correction, Bruker−AXS). The structures
were solved by direct methods, using SHELXTL Software Package and refined using the
least-squares method on F2.[2] All non-H atoms were treated anisotropically. The NH
hydrogen atoms were located on difference Fourier maps and isotropically refined without
any restraint. CCDC-1407101 (1) and CCDC-1407102 (2) contain the supplementary
crystallographic data. These data can be obtained free of charge from The Cambridge
Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Data for compound 1.

SI - 10
Table 1. Crystal data and structure refinement for compound 1. _____________________________________________________________________________________ Empirical formula C13 H13 N O3 S Formula weight 263.30 Temperature 193(2) K Wavelength 0.71073 Å Crystal system Monoclinic Space group P21/c Unit cell dimensions a = 11.2522(6) Å = 90°. b = 6.5289(3) Å = 107.002(2)°. c = 17.7516(9) Å = 90°. Volume 1247.11(11) Å3 Z 4 Density (calculated) 1.402 Mg/m3 Absorption coefficient 0.259 mm-1 F(000) 552 Crystal size 0.400 x 0.200 x 0.080 mm3 Theta range for data collection 3.343 to 32.186°. Index ranges -16<=h<=16, -9<=k<=9, -26<=l<=26 Reflections collected 55736 Independent reflections 4395 [R(int) = 0.0445] Completeness to theta = 25.242° 99.8 % Absorption correction Semi-empirical from equivalents Max. and min. transmission 0.7464 and 0.6931 Refinement method Full-matrix least-squares on F2 Data / restraints / parameters 4395 / 0 / 169 Goodness-of-fit on F2 1.048 Final R indices [I>2sigma(I)] R1 = 0.0441, wR2 = 0.1152 R indices (all data) R1 = 0.0701, wR2 = 0.1345 Extinction coefficient n/a Largest diff. peak and hole 0.461 and -0.274 e.Å-3
_____________________________________________________________________________________ Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2x 103) for 1. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. ________________________________________________________________________________ x y z U(eq) ________________________________________________________________________________ S(1) 3657(1) 3336(1) 4492(1) 38(1) O(1) 9088(1) 7801(2) 3268(1) 28(1) O(2) 9818(1) 5003(2) 3911(1) 36(1) O(3) 5499(1) 6513(2) 3124(1) 30(1) N(1) 6211(1) 3214(2) 3905(1) 24(1) C(1) 8611(2) 10883(2) 2549(1) 37(1) C(2) 8144(1) 9088(2) 2892(1) 25(1) C(3) 6964(1) 8697(2) 2854(1) 25(1) C(4) 6624(1) 6856(2) 3194(1) 22(1) C(5) 7623(1) 5515(2) 3608(1) 21(1) C(6) 8880(1) 5996(2) 3628(1) 24(1) C(7) 7373(1) 3711(2) 3996(1) 22(1) C(8) 8370(1) 2400(3) 4523(1) 33(1) C(9) 5783(1) 1466(2) 4272(1) 29(1) C(10) 4402(1) 1497(2) 4108(1) 25(1) C(11) 3578(1) 73(2) 3689(1) 29(1) C(12) 2336(1) 501(2) 3686(1) 34(1) C(13) 2244(1) 2210(3) 4091(1) 36(1) ________________________________________________________________________________

SI - 11
Table 3. Bond lengths [Å] and angles [°] for 1.
_____________________________________________________ S(1)-C(13) 1.7069(17) S(1)-C(10) 1.7155(14) O(1)-C(2) 1.3652(16) O(1)-C(6) 1.3933(16) O(2)-C(6) 1.2147(16) O(3)-C(4) 1.2559(15) N(1)-C(7) 1.3106(16) N(1)-C(9) 1.4634(17) N(1)-H(1) 0.90(3) C(1)-C(2) 1.486(2) C(1)-H(1A) 0.9800 C(1)-H(1B) 0.9800 C(1)-H(1C) 0.9800 C(2)-C(3) 1.3336(18) C(3)-C(4) 1.4453(18) C(3)-H(3) 0.9500 C(4)-C(5) 1.4446(17) C(5)-C(7) 1.4330(18) C(5)-C(6) 1.4390(17) C(7)-C(8) 1.5009(18) C(8)-H(8A) 0.9800 C(8)-H(8B) 0.9800 C(8)-H(8C) 0.9800 C(9)-C(10) 1.4948(18) C(9)-H(9A) 0.9900 C(9)-H(9B) 0.9900 C(10)-C(11) 1.369(2) C(11)-C(12) 1.4249(19) C(11)-H(11) 0.9500 C(12)-C(13) 1.348(2) C(12)-H(12) 0.9500 C(13)-H(13) 0.9500 C(13)-S(1)-C(10) 92.15(7) C(2)-O(1)-C(6) 122.28(10) C(7)-N(1)-C(9) 125.77(11) C(7)-N(1)-H(1) 112.0(16) C(9)-N(1)-H(1) 122.1(16) C(2)-C(1)-H(1A) 109.5 C(2)-C(1)-H(1B) 109.5 H(1A)-C(1)-H(1B) 109.5 C(2)-C(1)-H(1C) 109.5 H(1A)-C(1)-H(1C) 109.5 H(1B)-C(1)-H(1C) 109.5 C(3)-C(2)-O(1) 121.83(12)
C(3)-C(2)-C(1) 126.60(13) O(1)-C(2)-C(1) 111.57(12) C(2)-C(3)-C(4) 121.26(12) C(2)-C(3)-H(3) 119.4 C(4)-C(3)-H(3) 119.4 O(3)-C(4)-C(5) 123.65(12) O(3)-C(4)-C(3) 119.29(11) C(5)-C(4)-C(3) 117.06(11) C(7)-C(5)-C(6) 119.79(11) C(7)-C(5)-C(4) 120.65(11) C(6)-C(5)-C(4) 119.55(11) O(2)-C(6)-O(1) 113.54(11) O(2)-C(6)-C(5) 128.51(13) O(1)-C(6)-C(5) 117.93(11) N(1)-C(7)-C(5) 118.25(11) N(1)-C(7)-C(8) 118.18(12) C(5)-C(7)-C(8) 123.53(11) C(7)-C(8)-H(8A) 109.5 C(7)-C(8)-H(8B) 109.5 H(8A)-C(8)-H(8B) 109.5 C(7)-C(8)-H(8C) 109.5 H(8A)-C(8)-H(8C) 109.5 H(8B)-C(8)-H(8C) 109.5 N(1)-C(9)-C(10) 111.21(11) N(1)-C(9)-H(9A) 109.4 C(10)-C(9)-H(9A) 109.4 N(1)-C(9)-H(9B) 109.4 C(10)-C(9)-H(9B) 109.4 H(9A)-C(9)-H(9B) 108.0 C(11)-C(10)-C(9) 126.65(12) C(11)-C(10)-S(1) 111.05(10) C(9)-C(10)-S(1) 122.21(11) C(10)-C(11)-C(12) 112.14(13) C(10)-C(11)-H(11) 123.9 C(12)-C(11)-H(11) 123.9 C(13)-C(12)-C(11) 112.89(14) C(13)-C(12)-H(12) 123.6 C(11)-C(12)-H(12) 123.6 C(12)-C(13)-S(1) 111.78(11) C(12)-C(13)-H(13) 124.1 S(1)-C(13)-H(13) 124.1
_________________________________________
____________________
Symmetry transformations used to generate equivalent atoms:

SI - 12
Table 4. Anisotropic displacement parameters (Å2x 103) for 1. The anisotropic
displacement factor exponent takes the form: -22[ h2 a*2U11 + ... + 2 h k a* b* U12 ]
______________________________________________________________________________
U11 U22 U33 U23 U13 U12
______________________________________________________________________________ S(1) 33(1) 33(1) 52(1) -11(1) 18(1) 0(1) O(1) 20(1) 28(1) 37(1) 2(1) 10(1) -1(1) O(2) 18(1) 38(1) 50(1) 7(1) 10(1) 6(1) O(3) 15(1) 30(1) 44(1) 5(1) 6(1) 2(1) N(1) 19(1) 26(1) 28(1) 4(1) 8(1) 1(1) C(1) 37(1) 32(1) 44(1) 5(1) 16(1) -5(1) C(2) 25(1) 24(1) 27(1) -1(1) 9(1) 0(1) C(3) 23(1) 24(1) 28(1) 1(1) 6(1) 2(1) C(4) 17(1) 24(1) 24(1) -3(1) 5(1) 0(1) C(5) 15(1) 24(1) 23(1) -2(1) 5(1) 0(1) C(6) 18(1) 25(1) 28(1) -3(1) 7(1) 0(1) C(7) 19(1) 25(1) 22(1) -2(1) 6(1) 1(1) C(8) 22(1) 38(1) 34(1) 10(1) 3(1) 2(1) C(9) 23(1) 31(1) 36(1) 10(1) 11(1) 3(1) C(10) 23(1) 25(1) 28(1) 3(1) 11(1) 2(1) C(11) 26(1) 31(1) 31(1) 2(1) 10(1) 3(1) C(12) 23(1) 37(1) 40(1) 0(1) 5(1) 0(1) C(13) 25(1) 38(1) 46(1) 3(1) 15(1) 6(1) ______________________________________________________________________________ Table 5. Hydrogen coordinates ( x 104) and isotropic displacement parameters (Å2x 10 3) for 1.
________________________________________________________________________________
x y z U(eq)
________________________________________________________________________________
H(1) 5680(20) 4140(40) 3617(15) 63(7) H(1A) 7922 11827 2321 55 H(1B) 8966 10418 2136 55 H(1C) 9252 11587 2962 55 H(3) 6336 9646 2599 30 H(8A) 8895 3241 4949 49 H(8B) 8879 1800 4216 49 H(8C) 7986 1303 4748 49 H(9A) 6182 1504 4848 35 H(9B) 6034 178 4066 35 H(11) 3809 -1067 3430 35 H(12) 1645 -331 3426 41 H(13) 1484 2725 4147 43
________________________________________________________________________________
Table 6. Hydrogen bonds for 1 [Å and °]. ____________________________________________________________________________ D-H...A d(D-H) d(H...A) d(D...A) <(DHA) ____________________________________________________________________________ N(1)-H(1)...O(3) 0.90(3) 1.76(3) 2.5601(15) 147(2) ____________________________________________________________________________
Symmetry transformations used to generate equivalent atoms:
SI - 13
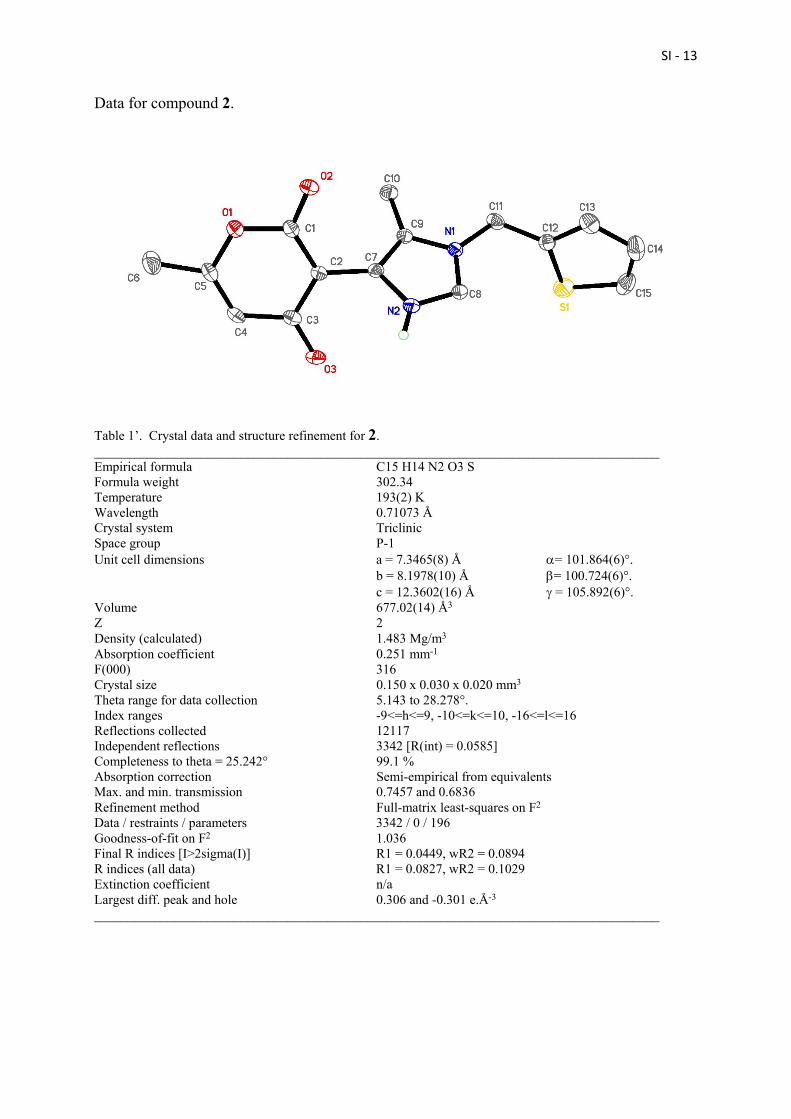
Data for compound 2.
Table 1’. Crystal data and structure refinement for 2. _____________________________________________________________________________________ Empirical formula C15 H14 N2 O3 S Formula weight 302.34 Temperature 193(2) K Wavelength 0.71073 Å Crystal system Triclinic Space group P-1 Unit cell dimensions a = 7.3465(8) Å = 101.864(6)°. b = 8.1978(10) Å = 100.724(6)°. c = 12.3602(16) Å = 105.892(6)°. Volume 677.02(14) Å3 Z 2 Density (calculated) 1.483 Mg/m3 Absorption coefficient 0.251 mm-1 F(000) 316 Crystal size 0.150 x 0.030 x 0.020 mm3 Theta range for data collection 5.143 to 28.278°. Index ranges -9<=h<=9, -10<=k<=10, -16<=l<=16 Reflections collected 12117 Independent reflections 3342 [R(int) = 0.0585] Completeness to theta = 25.242° 99.1 % Absorption correction Semi-empirical from equivalents Max. and min. transmission 0.7457 and 0.6836 Refinement method Full-matrix least-squares on F2 Data / restraints / parameters 3342 / 0 / 196 Goodness-of-fit on F2 1.036 Final R indices [I>2sigma(I)] R1 = 0.0449, wR2 = 0.0894 R indices (all data) R1 = 0.0827, wR2 = 0.1029 Extinction coefficient n/a Largest diff. peak and hole 0.306 and -0.301 e.Å-3
_____________________________________________________________________________________

SI - 14
Table 2’. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2x 103)
for 2. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________________________
x y z U(eq)
________________________________________________________________________________ S(1) 5892(1) 2922(1) 1678(1) 31(1) O(1) 3744(2) 2916(2) 8758(1) 23(1) O(2) 6110(2) 2454(2) 7995(1) 26(1) O(3) 20(2) 1916(2) 5630(1) 22(1) N(1) 5817(2) 2599(2) 4490(1) 16(1) N(2) 3204(2) 853(2) 4708(1) 18(1) C(1) 4440(2) 2541(2) 7785(2) 19(1) C(2) 3203(2) 2328(2) 6713(2) 17(1) C(3) 1215(2) 2225(2) 6596(2) 18(1) C(4) 604(2) 2528(2) 7640(2) 21(1) C(5) 1855(3) 2914(3) 8658(2) 21(1) C(6) 1469(3) 3411(3) 9801(2) 33(1) C(7) 4015(2) 2194(2) 5719(2) 16(1) C(8) 4307(2) 1121(2) 3981(2) 18(1) C(9) 5670(2) 3292(2) 5575(2) 17(1) C(10) 7144(3) 4927(2) 6373(2) 23(1) C(11) 7450(3) 3435(3) 4028(2) 22(1) C(12) 7325(2) 2579(2) 2825(2) 20(1) C(13) 8469(3) 1638(3) 2467(2) 27(1) C(14) 8184(3) 1200(3) 1267(2) 35(1) C(15) 6837(3) 1802(3) 730(2) 34(1)
________________________________________________________________________________

SI - 15
Table 3’. Bond lengths [Å] and angles [°] for 2.
_____________________________________________________ S(1)-C(15) 1.705(2) S(1)-C(12) 1.722(2) O(1)-C(5) 1.370(2) O(1)-C(1) 1.402(2) O(2)-C(1) 1.230(2) O(3)-C(3) 1.275(2) N(1)-C(8) 1.336(2) N(1)-C(9) 1.381(2) N(1)-C(11) 1.484(2) N(2)-C(8) 1.329(2) N(2)-C(7) 1.388(2) N(2)-H(2) 0.90(2) C(1)-C(2) 1.408(3) C(2)-C(3) 1.418(2) C(2)-C(7) 1.459(3) C(3)-C(4) 1.440(3) C(4)-C(5) 1.332(3) C(4)-H(4) 0.9500 C(5)-C(6) 1.489(3) C(6)-H(6A) 0.9800 C(6)-H(6B) 0.9800 C(6)-H(6C) 0.9800 C(7)-C(9) 1.367(2) C(8)-H(8) 0.9500 C(9)-C(10) 1.485(3) C(10)-H(10A) 0.9800 C(10)-H(10B) 0.9800 C(10)-H(10C) 0.9800 C(11)-C(12) 1.482(3) C(11)-H(11A) 0.9900 C(11)-H(11B) 0.9900 C(12)-C(13) 1.360(3) C(13)-C(14) 1.413(3) C(13)-H(13) 0.9500 C(14)-C(15) 1.349(3) C(14)-H(14) 0.9500 C(15)-H(15) 0.9500 C(15)-S(1)-C(12) 92.12(11) C(5)-O(1)-C(1) 120.87(15) C(8)-N(1)-C(9) 109.13(15) C(8)-N(1)-C(11) 128.24(17) C(9)-N(1)-C(11) 122.64(15) C(8)-N(2)-C(7) 109.68(16) C(8)-N(2)-H(2) 124.2(16) C(7)-N(2)-H(2) 126.1(16) O(2)-C(1)-O(1) 113.93(17) O(2)-C(1)-C(2) 128.10(18) O(1)-C(1)-C(2) 117.96(15) C(1)-C(2)-C(3) 121.06(18) C(1)-C(2)-C(7) 118.19(15)
C(3)-C(2)-C(7) 120.75(16) O(3)-C(3)-C(2) 122.76(18) O(3)-C(3)-C(4) 120.89(16) C(2)-C(3)-C(4) 116.33(17) C(5)-C(4)-C(3) 121.53(16) C(5)-C(4)-H(4) 119.2 C(3)-C(4)-H(4) 119.2 C(4)-C(5)-O(1) 121.58(18) C(4)-C(5)-C(6) 127.05(17) O(1)-C(5)-C(6) 111.37(17) C(5)-C(6)-H(6A) 109.5 C(5)-C(6)-H(6B) 109.5 H(6A)-C(6)-H(6B) 109.5 C(5)-C(6)-H(6C) 109.5 H(6A)-C(6)-H(6C) 109.5 H(6B)-C(6)-H(6C) 109.5 C(9)-C(7)-N(2) 106.01(17) C(9)-C(7)-C(2) 129.06(17) N(2)-C(7)-C(2) 124.93(16) N(2)-C(8)-N(1) 108.05(17) N(2)-C(8)-H(8) 126.0 N(1)-C(8)-H(8) 126.0 C(7)-C(9)-N(1) 107.13(16) C(7)-C(9)-C(10) 130.31(18) N(1)-C(9)-C(10) 122.55(16) C(9)-C(10)-H(10A) 109.5 C(9)-C(10)-H(10B) 109.5 H(10A)-C(10)-H(10B) 109.5 C(9)-C(10)-H(10C) 109.5 H(10A)-C(10)-H(10C) 109.5 H(10B)-C(10)-H(10C) 109.5 C(12)-C(11)-N(1) 115.73(15) C(12)-C(11)-H(11A) 108.3 N(1)-C(11)-H(11A) 108.3 C(12)-C(11)-H(11B) 108.3 N(1)-C(11)-H(11B) 108.3 H(11A)-C(11)-H(11B) 107.4 C(13)-C(12)-C(11) 126.12(18) C(13)-C(12)-S(1) 110.64(16) C(11)-C(12)-S(1) 122.69(14) C(12)-C(13)-C(14) 112.70(19) C(12)-C(13)-H(13) 123.7 C(14)-C(13)-H(13) 123.7 C(15)-C(14)-C(13) 113.1(2) C(15)-C(14)-H(14) 123.4 C(13)-C(14)-H(14) 123.4 C(14)-C(15)-S(1) 111.43(19) C(14)-C(15)-H(15) 124.3 S(1)-C(15)-H(15) 124.3
_____________________________________________________________
Symmetry transformations used to generate equivalent atoms:

SI - 16
Table 4’. Anisotropic displacement parameters (Å2x 103) for 2. The anisotropic
displacement factor exponent takes the form: -22[ h2 a*2U11 + ... + 2 h k a* b* U12 ]
______________________________________________________________________________
U11 U22 U33 U23 U13 U12
______________________________________________________________________________ S(1) 32(1) 39(1) 25(1) 13(1) 4(1) 14(1) O(1) 19(1) 32(1) 21(1) 7(1) 7(1) 10(1) O(2) 16(1) 39(1) 23(1) 8(1) 4(1) 12(1) O(3) 15(1) 24(1) 25(1) 8(1) 1(1) 4(1) N(1) 14(1) 17(1) 16(1) 5(1) 4(1) 4(1) N(2) 14(1) 18(1) 19(1) 4(1) 3(1) 2(1) C(1) 18(1) 19(1) 22(1) 5(1) 8(1) 6(1) C(2) 14(1) 17(1) 19(1) 5(1) 4(1) 4(1) C(3) 16(1) 11(1) 26(1) 6(1) 5(1) 2(1) C(4) 13(1) 23(1) 29(1) 9(1) 8(1) 6(1) C(5) 20(1) 22(1) 27(1) 9(1) 13(1) 9(1) C(6) 34(1) 40(1) 33(1) 11(1) 18(1) 17(1) C(7) 15(1) 18(1) 16(1) 4(1) 2(1) 7(1) C(8) 16(1) 22(1) 16(1) 5(1) 3(1) 7(1) C(9) 17(1) 19(1) 18(1) 6(1) 4(1) 8(1) C(10) 21(1) 19(1) 24(1) 4(1) 4(1) 3(1) C(11) 18(1) 24(1) 25(1) 11(1) 8(1) 3(1) C(12) 20(1) 22(1) 20(1) 9(1) 6(1) 4(1) C(13) 26(1) 22(1) 35(1) 11(1) 12(1) 6(1) C(14) 40(1) 26(1) 38(2) 5(1) 23(1) 6(1) C(15) 43(1) 33(1) 21(1) 6(1) 11(1) 3(1)
_____________________________________________________________________________
Table 5’. Hydrogen coordinates ( x 104) and isotropic displacement parameters (Å2x 10 3) for 2.
________________________________________________________________________________
x y z U(eq)
________________________________________________________________________________
H(2) 2100(30) -70(30) 4560(20) 48(7) H(4) -721 2452 7602 25 H(6A) 83 3304 9703 50 H(6B) 2280 4631 10201 50 H(6C) 1790 2626 10251 50 H(8) 4064 390 3230 22 H(10A) 6684 5272 7052 34 H(10B) 7331 5874 5991 34 H(10C) 8391 4714 6602 34 H(11A) 7515 4674 4091 26 H(11B) 8691 3453 4516 26 H(13) 9363 1311 2970 32 H(14) 8870 549 878 42 H(15) 6462 1624 -75 41
________________________________________________________________________________ 1. SAINT-NT; Bruker AXS Inc.: Madison, Wisconsin, 2000.
2. Sheldrick, G. M. Acta Cryst. 2008, A64, 112–122


















