
The chick embryo amnion as an in vitro culture system for ...
186
Louisiana State University LSU Digital Commons LSU Doctoral Dissertations Graduate School 2004 e chick embryo amnion as an in vitro culture system for IVF and NT embryos Tonya Renea Davidson Louisiana State University and Agricultural and Mechanical College Follow this and additional works at: hps://digitalcommons.lsu.edu/gradschool_dissertations Part of the Animal Sciences Commons is Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please contact[email protected]. Recommended Citation Davidson, Tonya Renea, "e chick embryo amnion as an in vitro culture system for IVF and NT embryos" (2004). LSU Doctoral Dissertations. 1280. hps://digitalcommons.lsu.edu/gradschool_dissertations/1280
Transcript of The chick embryo amnion as an in vitro culture system for ...

Louisiana State UniversityLSU Digital Commons
LSU Doctoral Dissertations Graduate School
2004
The chick embryo amnion as an in vitro culturesystem for IVF and NT embryosTonya Renea DavidsonLouisiana State University and Agricultural and Mechanical College
Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_dissertations
Part of the Animal Sciences Commons
This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion inLSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please [email protected].
Recommended CitationDavidson, Tonya Renea, "The chick embryo amnion as an in vitro culture system for IVF and NT embryos" (2004). LSU DoctoralDissertations. 1280.https://digitalcommons.lsu.edu/gradschool_dissertations/1280

THE CHICK EMBRYO AMNION AS AN IN VITRO CULTURE SYSTEM FOR IVF AND NT EMBRYOS
A Dissertation
Submitted to the Graduate Faculty of Louisiana State University and
Agricultural and Mechanical College In partial fulfillment of the
Requirements for the degree of Doctor of Philosophy
in
The Interdepartmental Program in Animal and Dairy Sciences
by Tonya Renea Davidson
B.S., Oklahoma State University, 1997 M.S., Oklahoma State University, 2000
December 2004

ACKNOWLEDGEMENTS
I would like to begin by dedicating this dissertation to my mom, Linda Millspaugh.
Mom, you have always been my role model, my support system and my foundation. You
taught me that I could be whatever I wanted to be, and you had faith in me even when I
didn’t have faith in myself. I am so proud to be your daughter. I would also like to
dedicate this dissertation to my brother, Casey Millspaugh. Thank you Casey, for always
believing in me and looking up to me. That has been more encouragement than you will
ever know.
I would like to express me sincere gratitude to my major professor, Dr. Robert
Godke. Thank you for allowing me to pursue my Ph.D. at LSU. You have provided me
with a firm scientific background, but more importantly, you have encouraged me to
stand up for that which I believe. You have been a wonderful mentor and I appreciate all
that you have done for me.
To Dr. Leibo, Dr. Lynn and Dr. Williams. Thank you so much for serving on my
committee, and for all of the assistance you have provided to make my time at LSU such
a success. It has been a great pleasure working with all of you. I have learned so much
from each of you and I am a better scientist for having known you.
To Trey Chapman, Kyle Hebert, Laurie Henderson, Andrea Huval, Angela
“Beanie” Klumpp, Luke Lenard, Alison Mosian, Kyle Hebert, Dr. Masao Murakami and
James “Par” Sterling. Thank you for all that you have done for me. You have each
touched my life in a very special way and I will always remember you.
To Kristy Bruce. Thank you for saying “Hello” that first day of class. Since that
time, you have been a wonderful source of entertainment and support, but more
importantly, you have been an exceptional friend. I am so much better for having known
you.
And finally, to Edward Ferguson. My words can not adequately express how I
truly feel for you. You are my best friend and the love of my life. You have never left my
side, no matter how hard things got, and for that I will always love you.
ii

TABLE OF CONTENTS
ACKNOWLEDGEMENTS ................................................................................................. ii LIST OF TABLES..............................................................................................................vi LIST OF FIGURES..........................................................................................................viii ABBREVIATIONS USED IN THE DISSERTATION..........................................................xi ABSTRACT......................................................................................................................xii CHAPTER I. INTRODUCTION .................................................................................... 1 CHAPTER II. LITERATURE REVIEW.......................................................................... 3
In Vitro Production of Embryos.............................................................................. 3 Production of Bovine Embryos by In Vitro Fertilization........................... 3 Production of Bovine Embryos by Nuclear Transfer............................... 4 In Vitro Embryo Culture .......................................................................... 6
Influence of In Vitro Production on Embryo Development................................... 11 Embryo Development Rates................................................................. 11 Embryo Morphology ............................................................................. 12 Pregnancy and Calving Rates .............................................................. 14 Large Offspring Syndrome and Associated Disorders.......................... 16
Apoptosis ............................................................................................. 18 Chick Embryo Co-Culture.................................................................................... 24
Biochemistry of the Avian Egg.............................................................. 24 Early Use of Chick Embryo Extracts in Mammalian Cell Culture ......... 25 In Vitro Culture of Chick Embryos ........................................................ 26 Chick Embryo Co-Culture..................................................................... 26
CHAPTER III. CULTURING IN VITRO PRODUCED BOVINE EMBRYOS IN THE
AMNIOTIC CAVITY OF THE DEVELOPING CHICKEN EMBYRO...... 28 Introduction.......................................................................................................... 28 Materials and Methods ........................................................................................ 29
Experimental Design ............................................................................ 29 Experimental Methods.......................................................................... 31 Statistical Analysis................................................................................ 36
Results ................................................................................................................ 36 Discussion........................................................................................................... 41 CHAPTER IV. USE OF THE AMNIOTIC CAVITY OF THE DEVELOPING CHICK EMBRYO FOR CULTURING BOVINE 8-CELL EMBRYOS DERIVED FROM SOMATIC CELL NUCLEAR TRANSFER ................................. 46 Introduction ......................................................................................................... 46 Materials and Methods........................................................................................ 47
Experimental Design ............................................................................ 47 Experimental Methods.......................................................................... 49
iii

Statistical Analysis................................................................................ 53 Results ................................................................................................................ 53
Discussion........................................................................................................... 54 CHAPTER V. THE EFFECT OF ALTERATIONS IN THE NORMAL ANGIOGENIC PATTERN OF THE CHICK CHORIOALLANTOIC MEMBRANE ON BOVINE EMBRYO DEVELOPMENT AFTER CHICK EMBRYO CO-CULTURE...................................................................................... 60
Introduction ..................................................................................................... 60 Materials and Methods .................................................................................... 61
Experimental Design ............................................................................ 61 Experimental Methods.......................................................................... 63 Statistical Analysis................................................................................ 69
Results ............................................................................................................ 69 Discussion ....................................................................................................... 78
CHAPTER VI. DEVELOPMENT OF 8-CELL BOVINE EMBRYOS IN CULTURE
MEDIUM SUPPLEMENTED WITH CHICK AMNIOTIC FLUID (CAF).. 80 Introduction...................................................................................................... 80
Materials and Methods.................................................................................... 81 Experimental Design ............................................................................ 81 Experimental Methods.......................................................................... 81 Statistical Analysis................................................................................ 85
Results ............................................................................................................ 86 Discussion ....................................................................................................... 86
CHAPTER VII. ESTABLISHMENT AND CULTURE OF CELLS DERIVED FROM DAY-6 CHICK EMBRYOS.................................................................... 93
Introduction.......................................................................................................... 93 Materials and Methods ........................................................................................ 95
Experimental Design ............................................................................ 95 Experimental Methods.......................................................................... 95 Statistical Analysis.............................................................................. 101
Results .............................................................................................................. 102 Discussion ......................................................................................................... 107
CHAPTER VIII. THE INCIDENCE OF APOPTOSIS IN BOVINE EMBRYOS AFTER IN VITRO CULTURE AND CHICK EMBRYO CO-CULTURE............. 112
Introduction........................................................................................................ 112 Materials and Methods ...................................................................................... 113
Experimental Design .......................................................................... 113 Experimental Methods........................................................................ 115 Statistical Analysis.............................................................................. 122
Results .............................................................................................................. 122 Discussion ......................................................................................................... 129
SUMMARY AND CONCLUSIONS............................................................................... 138
iv

LITERATURE CITED.................................................................................................... 140 APPENDIX A: BO-A STOCK SOLUTION..................................................................... 170 APPENDIX B: BO-B STOCK SOLUTION..................................................................... 171 APPENDIX C: CR1aa STOCK SOLUTION .................................................................. 172 VITA .............................................................................................................................. 173
v

LIST OF TABLES 3.1. Development of 1-cell bovine embryos cultured in CR1aa + 5% fetal bovine
serum (control) or co-cultured in the chick amnion from day 0 to day 3 (CEC-I) or from day 3 to day 7 (CEC-II) ........................................................................... 38 3.2. Development of 2-cell bovine embryos cultured in CR1aa + 5% fetal bovine serum (control) or co-cultured in the chick amnion from day 0 to day 3 (CEC-I) or from day 3 to day 7 (CEC-II) ........................................................................... 40
4.1. Day 6 development of somatic cell nuclear transfer bovine 8-cell embryos co- cultured in the chick embryo amnion................................................................... 55
5.1. Mortality of chick embryos after treatment application and subsequent 24 hour incubation............................................................................................................ 70
5.2. Effect of cytochalasin D, ethanol, PGE2 and PGF2α on capillary branching in the chorioallantoic membrane of the chick embryo. Treatment effects were quantified by counting the number of nodes in four random areas of each CAM ................................................................................................................... 71 5.3. Development of 8-cell bovine embryos cultured in CR1aa + 5% fetal bovine
serum (control), co-cultured in the chick embryo amnion co-cultured in the amnion of PGE2-treated chicks ........................................................................... 75
6.1. Development of 8-cell bovine embryos cultured in CR1aa supplemented with
5% fetal bovine serum (control) or 20% chick amniotic fluid (CAF) .................... 87 7.1. Experimental design for Experiment 7.1 for growth and proliferation of fetal
chick cells ............................................................................................................ 96
7.2. Experimental design for Experiment 7.2 for growth and proliferation of chick amnion cells ........................................................................................................ 97
7.3. Growth characteristics of fetal chick cells during in vitro culture in mTCM and mDMEM culture media...................................................................................... 103 7.4. Growth characteristics of chick amnion cells during in vitro culture in mTCM and mDMEM culture media............................................................................... 104 8.1. Response of cows to synchronization/superovulation protocol......................... 123 8.2. Number, stage and quality grade of embryos recovered from superovulated cows.................................................................................................................. 125 8.3. Effect of culture environment on mean number of apoptotic cells in morula, early blastocyst and blastocyst stage embryos ................................................. 126
vi

8.4. Effect of culture environment on mean number of apoptotic cells in morula, early blastocyst and blastocyst stage embryos................................................. 127 8.5. Effect of culture environment on apoptotic index in morula, early blastocyst, and blastocyst stage embryos........................................................................... 128 8.6. Effect of culture environment on mean number of apoptotic cells in grade 1,
grade 2, grade 3, grade 4 and degenerate embryos......................................... 130 8.7. Effect of culture environment on mean number of apoptotic cells in grade 1,
grade 2, grade 3, grade 4 and degenerate embryos......................................... 131
8.8. Effect of culture environment on apoptotic index in grade 1, grade 2, grade 3 and grade 4 embryos ........................................................................................ 132
vii

LIST OF FIGURES 3.1. Experimental design for the culture of 1-cell bovine embryos in the amnion of a developing chick embryo.............................................................................. 30 3.2. Experimental design for the culture of 2-cell bovine embryos in the amnion of a developing chick embryo.................................................................................. 32 3.3. Development of 1-cell bovine embryos cultured in CR1aa + 5% fetal bovine
serum (control) or co-cultured in the chick amnion from day 0 to day 3 (CEC-I) or from day 3 to day 7 (CEC-II) on day 3 of development (A) and on day 7 of development (B). ................................................................................................. 39
3.4. Development of 2-cell bovine embryos cultured in CR1aa + 5% fetal bovine
serum (control) or co-cultured in the chick amnion from day 0 to day 3 (CEC-I) or from day 3 to day 7 (CEC-II) on day 3 of development (A) and on day 7 of development (B). ................................................................................................. 42
3.5. Quality grade of bovine 2-cell embryos cultured in CR1aa + 5% fetal bovine
serum (control) or co-cultured in the chick amnion from day 0 to day 3 (CEC-I) or from day 3 to day 7 (CEC-II) on day 7 of development .................................. 43
4.1. Experimental design for the culture of bovine 8-cell embryos derived from somatic cell nuclear transfer. Culture was either in vitro in CR1aa
supplemented with 5% fetal bovine serum (control) or co-cultured in the chick embryo amnion.................................................................................................... 48
4.2. Bovine somatic cell nuclear transfer embryos developed into
morulae/blastocysts (day 6 of in vitro culture) after co-culture in the amniotic cavity of a developing chick embryo for 3 days. These embryos were agarose embedded (agar cylinders are indicated by arrows) at the 8-cell stage following in vitro culture in CR1aa medium from day 0 to day 3 of in vitro culture and then injected into the amnion of a 96-hour live chick embryo ............................. 56
4.3. Ultrasound image of a fetus (day 30) derived from bovine somatic cell nuclear
transfer embryos that were co-cultured in the amnion of a developing chick embryo from day 3 to day 6 following in vitro culture in CR1aa medium ........... 56
5.1. Experimental design to evaluate the effects of PGE2 and PGF2α on capillary development in the chick chorioallantoic membrane .......................................... 62 5.2. Experimental design for the culture of 8-cell bovine embryos cultured in
CR1aa + 5% fetal bovine serum (control), co-cultured in the chick embryo amnion (CEC) or co-cultured in the amnion of PGE2-treated chicks (PGE) ....................................................................................................... 64
5.3. Restructuring of vasculature networks in CAMs treated with (A) chick Ringer’s solution (control), (B) ethanol (vehicle), (C) PGE2 and (D) PGF2α. .................... 72
viii

5.4. Effect of cytochalasin D, ethanol (EtOH), PGE2 (PGE) and PGF2α (PGF) on
capillary branching in the chorioallantoic membrane of the chick embryo. The number on each bar indicates the total number of samples (four samples per CAM) in each treatment group ............................................................................ 73
5.5. Development of 8-cell bovine embryos cultured in CR1aa supplemented
with 5% fetal bovine serum (control), cultured in the chick embryo amnion (CEC) or cultured in the amnion of PGE2-treated chicks (PGE) on day 7 of
development (A), day 8 of development (B) and day 9 of development (C). ...... 76 5.6. Quality grade of bovine embryos cultured in CR1aa supplemented with 5%
fetal bovine serum (control), cultured in the chick embryo amnion (CEC) or cultured in the amnion of PGE2-treated chicks (PGE) on day 7 of develop- ment (A), day 8 of development (B) and day 9 of development (C). ................... 77
6.1. Experimental design for culturing 8-cell bovine embryos in culture medium
supplemented with chick amniotic fluid. In vitro culture was either in CR1aa supplemented with 5% fetal bovine serum (control group) or CR1aa supplemented with 20% chick amniotic fluid (CAF group) .................................. 82
6.2. Development of 8-cell bovine embryos cultured in CR1aa supplemented with 5% fetal bovine serum (control) or 20% chick amniotic fluid (CAF) on day 7 of development (A), day 8 of development (B) and day 9 of development (C). .. 88 6.3. Quality grade of 8-cell bovine embryos cultured in CR1aa supplemented with 5% fetal bovine serum (control) or 20% chick amniotic fluid (CAF) on day 7 of development (A), day 8 of development (B) and day 9 of development (C).................................................................................................. 89 7.1. Growth of chick amnion cells in vitro in mTCM culture medium........................ 105
7.2. Growth of chick amnion cells in vitro in mDMEM culture medium. ................... 106 7.3. Cell populations of chick amnion cells cultured in vitro in mTCM during the first passage...................................................................................................... 108 7.4. Cell populations of chick amnion cells cultured in vitro in mTCM during the second passage ................................................................................................ 109 7.5. Cell populations of chick amnion cells cultured in vitro in mTCM after freezing
and thawing ....................................................................................................... 110 8.1. Experimental design for the production and fluorescent labeling of in vivo- derived embryos................................................................................................ 114 8.2. Experimental design for the production and fluorescent labeling of embryos cultured in vitro ................................................................................................. 116
ix

8.3. Experimental design for the production and fluorescent labeling of embryos cultured in the chick embryo amnion................................................................. 117 8.4. Localization of apoptosis staining to the ICM of a grade 2 frozen in vivo- derived bovine blastocyst.................................................................................. 135 8.5. The presence of different degrees of fluorescence within an embryo.
Pinpoint areas of fluorescence are thought to represent cells in the early stages of apoptosis with only small amounts of caspase present in the cytoplasm (A), whereas extensive fluorescence are thought to represent cells in more advanced stages of apoptosis with much more caspase present in the cell (B) ......................................................................................................... 136
x

ABBREVIATIONS USED IN THE DISSERTATION
AI - artificial insemination BSA - bovine serum albumin BVDV - bovine viral diarrhea virus CAF - chick amniotic fluid CAM - chorioallantoic membane CEC - chick embryo co-culture CIDR - control internal drug release CL - corpus luteum DMEM - Dulbecco’s modified eagle’s medium DPBS - Dulbecco’s phosphate buffered saline ET - embryo transfer EtOH - ethanol FBS - fetal bovine serum FCS - fetal calf serum FSH - follicle stimulating hormone ICM - inner cell mass IGF-I - insulin-like growth factor-I IGF-II - insulin-like growth factor-II IGF2R - insulin-like growth factor-II receptor IVF - in vitro fertilization IVM - in vitro maturation IVP - in vitro produced M-II - metaphase-II mBMOC - modified Brinster medium for ovum culture MOET - multiple ovulation embryo transfer NT - nuclear transfer P4 - progesterone PGE2 - prostaglandin E2PGF2α - prostaglandin F2α PMSG - pregnant mare serum gonadotropin POEC - porcine oviduct epithelial cells SOFaa - synthetic oviduct fluid with amino acids TCM - tissue culture medium TE - trophoectoderm TGF-α - transforming growth factor-α TNF-α - tumor necrosis factor-α TNF-R1 - tumor necrosis factor receptor I TUNEL - TdT-mediated dUTP nick-end labeling WOW - well of the well
xi

ABSTRACT
Calving rates are significantly reduced following in vitro production of embryos.
Thus, if a technique could be developed that would increase calving rates by as little as
one viable offspring, significant research advances could be made. Therefore, in a series
of experiments, the efficiency and quality of culturing IVP bovine embryos in the amnion
of a domestic chicken egg was tested. In Experiment I, by culturing IVP bovine embryos
in the chick amnion (day 4 to 7 of incubation) it was discovered that there was no
significant difference in blastocyst rates compared with controls. In Experiment II, it was
shown that nuclear transfer bovine embryos cultured in the chick amnion reached the
blastocyst stage at rates equal to controls and were capable of producing pregnancies
following transplantation into recipient females. In a subsequent experiment, a method of
naturally improving the chick embryo co-culture (CEC) system was explored by treating
the developing chick embryo with prostaglandin E2 (PGE2) or prostaglandin F2α (PGF2α).
It was determined that treatment with PGE2 increased angiogenesis within the
developing chicken egg, while treatment with PGF2α decreased angiogenesis. When
bovine embryos were cultured in PGE2-treated chicks, developmental rates were not
increased. In Experiment IV, chick amniotic fluid (CAF) was evaluated as a media
supplement to the control culture system. Although replacing fetal bovine serum (FBS)
with CAF resulted in significantly fewer blastocysts on day 7 of culture, there were no
significant differences in the number of grade 1 embryos between the two treatments.
This finding was important because it demonstrated an ability to culture IVP bovine
embryos in the absence of FBS, a medium component that has been implicated in
numerous fetal and calf abnormalities. Another experiment was designed to develop a
method of culturing cells derived from the chick embryo and surrounding amniotic
membrane for later use as a co-culture system for bovine IVP embryos. In the final
experiment, using a novel method to detect apoptotic cells, it was determined that CEC
did not alter the number of apoptotic cells in the embryo when compared with in vivo-
derived or in vitro-cultured bovine embryos.
xii

CHAPTER I
INTRODUCTION
With the advent of in vitro fertilization technologies in the 1980s came the need
for methods of culturing embryos to later stages of development. As early as 1975, Dr.
R.A. Godke envisioned the use of avian eggs for the culture of mammalian embryos
(personal communication). Early attempts using unfertilized hen’s eggs proved largely
unsuccessful (Blakewood et al., unpublished data), but after several months of trial and
error a technique was developed that used shell-less chick embryos for the co-culture of
mammalian embryos. In the initial report, pronuclear stage murine embryos were
embedded in agarose and injected into the amniotic cavity of a 96-hour chick embryo.
After 72 to 96 hours of incubation in the chick amnion, significantly more embryos had
developed to the hatching blastocyst stage compared with those cultured in control
medium alone (Blakewood et al., 1989a). Further studies demonstrated that the chick
embryo co-culture system could support development of porcine (Ocampo et al., 1993),
caprine (Blakewood et al., 1989a) and bovine (Blakewood et al., 1989b) embryos at
rates equal to or better than controls.
In vitro culture techniques have progressed significantly since these early
experiments. Researchers have converted from culturing domestic animal embryos in
modified Tissue Culture Medium (mTCM) and Ham’s F-10 to culturing embryos in media
such as M2, CR1aa, CR2aa, NCSU-23, KSOM, SOFaa and even sequential mediums
such as G1/G2 and PPM1/PPM2. Pregnancy rates and calving rates do not appear to be
significantly different in sheep, pigs or cattle when embryos are cultured in these
different media. These observations suggest that the culture media may not be a major
factor in the increased abortion rates, increased placental problems and decreased
calving rates often observed after the transfer of embryos produced in vitro.
A more likely cause of the decreased calf production observed after transfer of in
vitro fertilized and cultured embryos is the use of fetal bovine serum (FBS) as a media
supplement. FBS is added to bovine embryo culture media (as with many other species)
to stimulate formation of the blastoceol and to increase the rate of blastocyst
development. Although there have been serum free media developed (known as defined
culture media), the blastocyst rates obtained with these media are not sufficient enough
1

to merit the use of in vitro fertilization and culture to produce offspring (Pinyopunnintr
and Bavister, 1991; Bavister, 1995).
Although serum is necessary for the efficient production of bovine blastocysts for
transfer, a number of studies have cited the use of serum as a cause for large offspring
syndrome, a collection of fetal and calf abnormalities often observed following the
transfer of in vitro-produced ovine and bovine embryos (Farin et al. 2000; Hasler et al.,
2000). Among these reports it was suggested that the presence of serum in the culture
medium resulted in significantly more embryo transfer recipients aborting during the later
stages of pregnancy and a higher rate of calf mortality. This effect has also been
reported when embryos were cultured in media supplemented with estrous cow serum
or serum derived from other species (Kruip and den Daas, 1997; Agca et al., 1998;
Hasler et al., 2000).
Large offspring syndrome has also been reported following transfer of embryos
co-cultured with different cell types, such as buffalo rat liver cells, oviduct epithial cells,
uterine epithelial cells and fibroblast cells (Sinclair et al., 1997). Although co-culture was
first used as a means to allow in vitro-produced embryos to develop beyond the
developmental block stage, neither its use nor supplementing media with serum has
improved calving rates compared with multiple ovulation and embryo transfer.
These problems do not seem to be inherent only to embryos produced by in vitro
fertilization and culture. Pregnancies produced following the transplantation of nuclear
transfer (NT)-derived embryos to recipient females have an even higher rate of large
offspring syndrome (Kruip and den Daas, 1997; Barnes, 2000). Although it has been
suggested that manipulation of the genetic material of the oocyte may be the primary
cause of large offspring syndrome with NT-derived embryos, the common link between
those embryos and in vitro-produced embryos is the in vitro culture medium and the use
of serum as a protein supplement.
Collectively, these findings indicate a need for an alternative in vitro culture
system that is capable of producing blastocyst rates at an acceptable level and that may
reduce or alleviate the occurrence of large offspring syndrome following in vitro embryo
production. Therefore, the objective of this dissertation is to re-evaluate the use of the
chick embryo co-culture system as an in vitro culture system for bovine embryos.
2

CHAPTER II
LITERATURE REVIEW
IN VITRO PRODUCTION OF EMBRYOS Production of Bovine Embryos by In Vitro Fertilization Prior to the 1980s early attempts to produce live offspring from in vitro-fertilized
bovine oocytes were largely unsuccessful. Initial success with bovine in vitro fertilization
(IVF) came in 1977 when Iritani and Niwa published the first report of IVF using in vitro-
matured (IVM) bovine oocytes. Much of the research in the following years was
dedicated to the study of the mechanics of fertilization. For example, Brackett et al.
(1980a) observed that sperm penetration (19 to 24 hours post-insemination) was favored
by in vivo-matured ova when compared with immature ova (recovered from 2- to 5-mm
follicles) matured in vitro for 18 to 25 hours prior to fertilization. In addition, Brackett et al.
(1980b) observed similarities, such as loss of cortical granules and the presence of
sperm remnants in oocytes and early embryos, between in vivo- and in vitro-fertilized
oocytes.
As with the development of IVF procedures in laboratory animals, preparation of
bovine sperm, particularly capacitation, proved to be a major challenge (Brackett, 2001).
Early methods of capacitating spermatozoa involved incubation of spermatozoa in the
oviduct or uterus of estrual females (Iritani and Niwa, 1977). However, by 1984 fresh
ejaculated spermatozoa could be capacitated in a chemically defined medium (Iritani et
al., 1984). By 1986, proteooglycans and glycosoaminoglycans (GAGs), capacitating
agents present at the site of fertilization in vivo (for review see Miller and Ax, 1990) were
determined to effectively induce the acrosome reaction and capacitate spermatozoa in
vitro (Parrish et al., 1985, 1986, 1988, 1989). Other agents used to capacitate
spermatozoa in vitro have included hyaluronic acid (Fukui, 1990), a combination of
heparin and caffeine (Niwa and Ohgoda, 1998), calcium ionophore A 23187 with or
without caffeine (Hanada et al., 1985; Goto et al., 1988) and calcium-free Tyrodes (Ijaz
and Hunter, 1989).
These studies culminated in the production of the first IVF calf in June, 1981 and
publication of the first repeatable protocol for bovine IVF in 1982 (Brackett et al., 1982).
These results were confirmed in 1984 with the birth of twin calves following in vitro
3

fertilization of in vivo-matured oocytes (Brackett et al., 1984). In these reports oocytes
were matured in vivo following hormonal stimulation of donor females with either
pregnant mare serum gonadotropin (PMSG) or follicle stimulating hormone (FSH) and
following IVF, presumptive zygotes were surgically transferred to the oviducts of
recipient females.
Because in vivo-matured oocytes were collected in the same manner (i.e.,
surgically) as early stage embryos, it became necessary to develop effective in vitro
maturation techniques for IVF to be considered a practical approach to the production of
bovine embryos. Two decades later, much research is still focused on the successful
maturation of oocytes in vitro.
Production of Bovine Embryos by Nuclear Transfer In 1997, Ian Wilmut et al. announced the birth of Dolly, the first live ‘clone’ of an
adult animal (Wilmut et al., 1997). The concept of cloning, however, originated long
before the birth of Dolly. As early as the 1880s, scientists sought the answer to one
fundamental question of biology – how does the nucleus control cell specialization
during embryogenesis (Di Berardina, 2001). To answer this question, scientists began to
examine the development of early stage embryos.
In 1892, Driesch (reviewed in Morgan, 1934; Speman, 1938; Di Berardina, 2001)
separated 2-cell sea urchin embryos in calcium-free seawater and found that in most
cases each separate cell could develop into a complete larva. Continued experiments in
other species, including amphibians, fish and mammals, yielded similar results.
In 1894, Loeb (reviewed in Di Berardina, 2001) conducted a primitive cloning
experiment using sea urchin eggs. In this study, the sea urchin eggs were placed in a
hypotonic solution, causing a break in the vitelline membrane and subsequent herniation
of a portion of the zygote through the break. In this particular study, the herniated portion
of the zygote lacked a nucleus. As cell division progressed in the nucleated portion of
the zygote, a daughter nucleus migrated into the non-nucleated potion of the zygote,
resulting in two whole and separate sea urchin embryos.
Hans Spemann (1914; reviewed by Di Berardina, 2001) extended Loeb’s
research into amphibians. In this study, Spemann (1914) bisected newt eggs with a baby
hair from his own son’s head. As in Loeb’s study, only one portion of the constricted egg
contained a nucleus. Development proceeded as normal in the nucleated side of the
4

egg, but the non-nucleated side failed to cleave. Once the nucleated portion had
developed to the 8- to 16-cell stage, he removed the ligature, allowing a nucleus to cross
over into the enucleated side and then severed the two halves. As development
continued, he found that each half could develop into a complete larva, thus proving that
an embryonic cell was capable of programming larval development.
Soon thereafter Robert Briggs set out to determine if the nuclei of somatic cells
could program development in the same manner that zygotic nuclei could. Many thought
this to be a “harebrained” idea, but in 1952 Briggs and King successfully produced
tadpoles from blastula nuclei injected into enucleated frog eggs, creating the prototype
for today’s nuclear transfer procedure (Briggs and King, 1952).
It was not for another 30 years after the initial successes of Briggs and King that
the first mammal was successfully cloned. The first report of live offspring after nuclear
transfer was that of Illmensee and Hoppe (1981) using inner cell mass (ICM) cells of
mouse blastocysts injected into an enucleated murine zygote. However, these results
could not be replicated for almost 17 years. Tsunoda and Kato (1998) were eventually
able to produce live offspring from both ICM and trophoblast cells. However, to
accomplish this they had to use different host cells and a serial nuclear transfer
procedure.
The first uncontested cloned offspring were produced by Willadsen in 1986. He
produced three lambs by fusing enucleated metaphase-II (M-II) oocytes to a single
blastomere derived from an 8- to 16-cell embryo. These results were verified by the
production of live calves from blastomeres of 4- to 16-cell embryos (Prather et al., 1987).
Following these initial successes, cloned offspring have been produced from embryonic
cells of many different species including rabbits (Collas and Robl, 1991), pigs (Prather et
al., 1989), sheep (Campbell et al., 1996; Wilmut et al., 1997), goats (Yong et al., 1991),
cattle (Sims and First, 1993; Collas and Barnes, 1994; Keefer et al., 1994) and rhesus
monkeys (Meng et al., 1997).
After the success of nuclear transfer with embryonic cells, researchers began to
question if cloned offspring could be produced from adult cells. Early attempts to clone
an adult animal using differentiated cells were completely unsuccessful (Di Berardina,
2001). However with the birth of Dolly, the belief that it was ‘biologically impossible’ to
clone a mammal by nuclear transfer was dispelled (McGrath and Solter, 1984).
5

Furthermore, the production of a live offspring from totipotent mammary tissue (as with
Dolly) suggested that other types of somatic cells could be used for cloning adult
animals (Edwards et al., 2003). These findings were confirmed in mice with cumulus
cells (Wakayama et al., 1998), pigs with granulosa cells (Polejaeva et al., 2000), goats
with cumulus granulosa cells (Zou et al., 2001) and cattle with cumulus and oviduct cells
(Kato et al., 1998). Cloned offspring have since been produced from many different cell
types including leukocytes (Galli et al., 1999), muscle (Shiga et al., 1999), mammary
tissue (Zakhartchenko et al., 1999; Kishi et al., 2000), ear tissue (Zakhartchenko et al.,
1999), skin cells (Hill et al., 2000a; Kubota et al., 2000) and sertoli cells (Ogura et al.,
2000) and from many different species including mice (Wakayama et al., 1998), rabbits
(Chesne et al., 2002), pigs (Polejaeva et al., 2000), sheep (Wilmut et al., 1997), goats
(Zou et al., 2001), cattle (Kato et al., 1998; Zakhartchenko et al., 1999; Hill et al., 2000a;
Kishi et al., 2000; Kubota et al., 2000; Kasinathan et al., 2001; Heyman et al., 2002) and
cats (Shin et al., 2002).
In Vitro Embryo Culture Attempts to culture embryos outside the protective environment of the uterus
date as far back as 1912 when Brachet attempted to culture 5- to 7-day rabbit embryos
in coagulated blood plasma (referenced in Kane, 2003) and in the later study of Lewis
and Gregory (1929) who reported developing rabbit embryos in vitro.
The first successful report of in vitro embryo development came when Hammond
(1949) reported that 8-cell mouse embryos could develop to the blastocyst stage in vitro
when cultured in a simple saline solution supplemented with chicken egg white and yolk.
During this same period, Chang (1949) reported that heat inactivated serum could be
used as a supplement in culture medium for 2-cell rabbit embryos.
The first culture medium specifically designed for mammalian embryos was
described by Whitten (1957). He observed that a simple Kreb’s Ringer’s bicarbonate
solution supplemented with serum albumin could promote development of 2-cell mouse
embryos to the blastocyst stage (Whitten, 1957). McLaren and Biggers (1958) later
reported that these embryos could result in live offspring after transfer to a surrogate
female. The most important outcome of this research, however, was the fundamental
discovery that early stage mouse embryos required lactate as an energy source for
continued development (Bavister, 1995; reviewed by Kane, 2003). This crucial
6

observation paved the way for future research to examine the precise substrate
requirements of pre-implantation mammalian embryos (Bavister, 1995).
Progress in in vitro embryo culture was relatively slow for the next 20 years as
attempts to support in vitro development of embryos from species other than mice and
rabbits met with limited success (Bavister, 1995). It became obvious that embryos of
most species suffered from an ‘in vitro developmental block’ that prevented further pre-
implantation development in vitro (Bavister, 1995). These blocks were observed to occur
at a characteristic stage of development in each species.
Early attempts at circumventing the ‘block’ initially involved culturing in vitro-
produced embryos in the ligated oviduct of a rabbit. This technique has been used
extensively for the culture of embryos of many different species including pigs (Polge et
al., 1972), sheep (Averill et al., 1955; Lawson et al., 1972), cattle (Sreenan et al., 1968;
Boland, 1984; Ectos et al., 1993) and horses (Allen et al., 1976). Similarly, hamster
embryos have been cultured in the mouse oviduct (Minami et al., 1988), pig embryos
have been cultured in the sheep oviduct (Prather et al., 1991) and cattle embryos have
been cultured in the oviducts of sheep (Eyestone et al., 1987; Galli and Lazzari, 1996;
Enright et al., 2000) and mice (Krisker et al., 1989; Sharif et al., 1991).
It is interesting to note that despite the advances of embryo culture in recent
years, current research indicates that modern culture media fail to produce embryos of
the same quality as those cultured in the oviduct of an intermediary recipient. Lonergan
et al. (2001) reported that IVM-IVF embryos cultured in the ewe oviduct survived
vitrification and warming much better than IVM-IVF embryos cultured in vitro in SOF
(88.0% and 5.6% hatched blastocysts, respectively). In a reciprocal experiment, in vitro
culture of in vivo-derived presumptive zygotes had a significantly lower post-warming
survival and hatching rate than did in vivo-derived presumptive zygotes cultured in the
ewe oviduct (Lonergan et al., 2001). These findings have been corroborated in sheep
(Tervit et al., 1994) and cattle (Holm et al., 1994; Galli and Lazari, 1996; Enright et al.,
2000; Jimeneze et al., 2001; Pugh et al., 2001; Rizos et al., 2002).
Although oviductal culture allowed development of in vitro-produced embryos
through the ‘block’ it was both impractical and expensive; thus, there remained a need
for a way to culture embryos completely in vitro. In 1965, Cole and Paul reported that a
high percentage of mouse embryos could develop and hatch in vitro when cultured on
7

HeLa cells (Cole and Paul, 1965). This classic study provided a more efficient method
for maintaining embryos in vitro for extended periods of time with minimal reductions in
viability (Rexroad, 1989; Thibodeaux and Godke, 1995) and laid the foundation for many
of the embryo co-culture systems still in use today (Thibodeaux and Godke, 1995).
Since the classic study of Cole and Paul (1965) many different cell types have
been found to support mammalian embryo development in vitro. One of the earliest
reports of embryo co-culture was that of Kuzan and Wright (1982) who reported that a
higher percentage of embryos developed to the hatched blastocyst stage when cultured
on either uterine or testicular fibroblasts when compared with culture medium alone.
Later, Weimer et al. found that fetal uterine fibroblasts could enhance development of
bovine (1987a,b) and equine (1989a,b) embryos. Rexroad and Powell (1986, 1988)
were among the first to report that oviduct epithelial cells could be used to culture early
stage ovine embryos and soon thereafter, Gandolfi and Moor (1987) reported that
culture of early stage ovine embryos on oviduct epithelial cells resulted in more
expanded blastocysts and a higher pregnancy rate than when embryos were cultured on
uterine fibroblasts. Similarly, Eyestone and First (1989) reported a higher percentage of
morula and blastocyst stage bovine embryos when IVF-derived 1-cell embryos were
cultured on oviduct cells than when cultured in medium alone (22% vs 3%, respectively).
In 1988, Goto et al. reported the establishment of viable pregnancies following in
vitro culture of IVF-derived bovine embryos on bovine granulosa cells. In a similar
experiment, Fukuda et al. (1989) reported live offspring from both fresh and frozen-
thawed bovine embryos cultured on cumulus cells. Interestingly, bovine cumulus cells
have been used successfully to culture embryos from other species including pigs
(Zhang et al., 1990) and horses (Rodriquez et al., 1991). These findings corroborate the
early findings of Baird et al. (1990), who reported that culturing mouse embryos on
hamster cumulus cells significantly increased in vitro development when compared with
culture medium alone.
Many other cells types have been evaluated for use in embryo co-culture in
addition to the ones described above. The work of Cole and Paul (1965) led others to
use different types of helper cells, such as L cells, liver cells, JLS-V11 cells and
teratocarcinoma cells (Glass et al., 1979). Others have used hamster hepatocytes
(Overskei and Cincotta, 1987), buffalo rat liver cells (Hu et al., 1989) bovine fetal spleen
8

cells (Kim et al., 1989; Kim et al., 1991), chick embryo fibroblasts (Kim et al., 1989; Kim
et al., 1991) and kidney (VERO and MDBK) cells (Ouhibi et al., 1990).
Despite the extensive research conducted in this area researchers have yet to
explain how co-culture cells benefit embryo development. Three hypotheses seem to
exist: 1) cells detoxify the culture medium by removing harmful contaminates, such as
heavy metal ions, 2) cells reduce the concentration of components in the medium that
inhibit embryo development (e.g., glucose) and 3) cells secrete embryotropic factors into
the culture medium, such as amino acids, pyruvate, proteins and growth factors
(Bavister, 1995). It seems more likely, however, that the benefits of co-culture are a
combination of all three hypotheses (Bavister, 1995).
In recent years, the use of co-culture systems has largely given way to culture in
semi-defined or defined culture media supplemented to varying degrees with amino
acids, vitamins, serum and/or other compounds. The first medium formulated specifically
for in vitro embryo culture (synthetic oviduct fluid, SOF) was a simple medium based on
the concentrations of ions and carbohydrates present in ovine oviduct fluid (Tervit et al,
1972). With this medium it was possible to develop 1-cell bovine embryos up to the 16-
cell stage and 8-cell bovine embryos up the blastocyst stage (Tervit, et al., 1972). This
approach to embryo culture was largely ignored by the research community, who
favored the more physiological environment of the rabbit or ewe oviduct for embryo
culture (Gardner, 1999).
It has not been the formulation of exceptional new media, but rather the small
observations along the way, that have led to the advances in embryo culture
technologies. One of the major, but largely unappreciated, milestones in embryo culture
was the realization that IVM and IVF should occur at a temperature equal to the core
body temperature of the animal (39°C in cattle; Lenz et al., 1983); this was later applied
to sheep (Gandolfi and Moor, 1987; Fukui et al., 1988) and cattle (Fukui and Ono, 1988;
Fukuda et al., 1990) embryo culture. Another very significant milestone in the
development of in vitro embryo culture was that of Thompson et al. (1990). These
researchers reported that development was significantly improved when embryos were
cultured in an O2 concentration between 5 and 10% as opposed to the 20% O2 content
of air.
9

During that same time period, Bavister and Arlotto (1990) first reported the
benefits of amino acids during in vitro culture. Soon thereafter, amino acids were
incororporated into mSOF (Takahasi and First, 1992), CR1 (Rosenkrans and First, 1994)
and KSOM (Liu and Foote, 1995) culture media. In all three of these reports, addition of
amino acids to the culture media resulted in significantly more embryos developing to
the blastocyst stage. In addition, transfer of embryos cultured in amino acid
supplemented SOF resulted in a 55% calving rate (Takahashi and First, 1992).
With the routine addition of amino acids to culture media, Gardner et al. (1994)
observed that embryos were particularly sensitive to ammonia produced by the
spontaneous deamination of the amino acids and/or amino acid metabolism. Based on
this observation, they proposed that media be removed and replaced with fresh medium
every 48 hours to optimize embryo development. In hindsight this was a critical finding.
Recent research indicates that chronic exposure of embryos to ammonium in culture
impairs development (Gardner and Lane, 1993; Gardner et al., 1994), retards fetal
growth (Lane and Gardner, 1994) and can lead to abnormal fetal development (Lane
and Gardner, 1994; McEvoy et al., 1997; Sinclair et al., 1998).
One could not thoroughly discuss in vitro embryo culture without mentioning the
use of serum in in vitro culture media. Serum has been used throughout the history of
embryo culture, both alone and as a supplement (reviewed in Wright and Bondioli,
1981). Early attempts at embryo culture utilized pure serum as a medium, however little
to no development was reported (Pincus, 1951; Brock and Rowson, 1952). Later studies
utilized serum as a supplement to the culture medium. In 1963, Hafez et al. cultured 1-
cell bovine embryos in serum supplemented saline, but again, failed to achieve high
rates of embryonic development. However, when Moore (1970) cultured 2- to 8-cell
ovine embryos in Brinster’s Salt Solution supplemented with 15% ovine serum, 38 of 57
(66%) of the embryos cleaved and 4 of 30 (13%) resulted in pregnancies.
In 1976, Wright et al. compared embryo development in several different media.
Of the media tested, Ham’s F-10 with 50% heat treated fetal calf serum (FCS) was the
only medium evaluated that was able to support development from the 8-cell stage to
the expanded blastocyst stage (Wright et al., 1976a,b,c). This was the first report of
bovine embryos expanding and hatching in vitro. Unbeknownst to them, this was likely
the first report of the biphasic effect of serum. In 1993, Kolver and MacMillan reported
10

that serum was inhibitory to embryos when added during early cleavage but was
stimulatory when present at the initiation of compaction. Many current in vitro culture
systems have taken advantage of this to maximize blastocyst yield by leaving serum out
of the culture medium during early development but adding it later in development
(Carolan et al., 1995; Massip et al., 1996; Pinyopmmintr and Bavister, 1996a,b; Van
Langendonkt et al., 1996).
In spite of the apparent benefits of serum supplementation, recent evidence
suggests that prolonged culture of embryos in the presence of serum alters embryo
morphology and biochemistry (Gardner et al., 1994; Shamsuddin and Rodriquez-
Martinez, 1994; Thompson et al., 1995). Serum has been implicated in precocious
blastoceol formation (Walker et al., 1992; Thompson et al., 1995), sequestration of lipids
(Thompson et al., 1995), abnormal mitochondrial ultrastructure (Thompson et al., 1995),
perturbations in metabolism (Gardner et al., 1995), altered embryo morphology
(Gardner, 1999), abnormally large offspring (Willadsen et al., 1991; Keefer et al, 1994;
Behboodi et al., 1995; Thompson et al., 1995), increased gestation lengths (Walker et
al., 1996) and increased perinatal (Behboodi et al., 1995; Wilson et al., 1995; Garry et
al., 1996; Schmidt et al., 1996; Kruip and den Daas, 1997; Hill et al., 1999) and neonatal
mortality (Behboodi et al., 1995; Wilson et al., 1995; Garry et al., 1996; Schmidt et al.,
1996; Kruip and den Daas, 1997; Hill et al., 1999). The effects of in vitro embryo
production on embryo development will be discussed in detail later in this review.
INFLUENCE OF IN VITRO PRODUCTION ON EMBRYO DEVELOPMENT Embryo Developmental Rates
Historically, the most widely used method of evaluating embryo quality has been
whether an embryo has attained an appropriate stage of development by a
predetermined time point (Elsden et al., 1978; Schneider et al., 1980; Shea, 1981; Lidner
and Wright, 1983). In a retrospective analysis of embryo collections at a commercial
bovine embryo transfer center, Lindner and Wright (1983) reported that the highest
proportion of morulae and compact morulae were recovered 5 to 6 days after estrus,
whereas early blastocysts and blastocysts were more prevalent on day 7. On days 8 and
9, blastocysts, expanded blastocysts and hatched blastocysts were most often
recovered. Similar findings have been reported with both superovulated females (Shea,
11

1981; Wright, 1981) and with single ovulating cows (Hamilton and Laing, 1946; Linares
and King, 1980).
In contrast, in vitro-produced bovine embryos exhibit delayed development when
compared to their in vivo-derived counterparts (Sirard and Lambert, 1985; Hytel et al.,
1989; Grisart et al., 1994). Early cleavage divisions (up to the 8- to 16-cell stage) of in
vitro-produced embryos have been reported to occur at a rate similar to those of in vivo-
recovered embryos (Hamilton and Laing, 1946; McGaugh et al., 1974; Christenson et
al., 1975; Moore, 1975; Betteridge and Flechon, 1988; Barnes and Eyestone, 1990).
However, Grisart et al. (1994) reported that the appearance of 8- to 16-cell embryos,
morulae and blastocysts in vitro is delayed by 30 to 40 hours when compared to
embryos produced in vivo. These findings are in agreement with those of McGaugh et al.
(1974), Prather and First (1988) and Funahashi et al. (1994). Van Soom et al. (1992)
suggested that this delay indicates that development past the 8- to 16-cell stage is
extremely sensitive to culture environment. Moreover, developmental arrest is very
common at this stage if culture conditions are inadequate (Van Soom et al., 1992),
lending additional support to Van Soom’s theory.
In addition, production of embryos in vitro is much less efficient than in vivo
embryo production, with less than 40% of immature oocytes reaching the blastocyst
stage (Wright and Ellington, 1995; Lonergan et al., 2001). It has been suggested that the
first embryos to cleave are more likely to (up to 70%) reach the morula to blastocyst
stage by day 8 (Plante and King, 1992; Grisart et al., 1994; Dinnyes et al., 1999;
Lonergan et al., 1999). Similar results have been reported in other species (buffalo –
Totey et al., 1996; rhesus monkey – Bavister et al., 1983; McKiernan and Bavister, 1994;
human – Sakkas et al., 1998; Shouker et al., 1998). In addition, Van Soom et al. (1992)
observed that embryos that cleaved at least once before 36 hours post-insemination
were more likely to form compacted morulae. Similarly, Vergos et al. (1989) reported
that embryos that had developed to at least the 4-cell stage by 44 to 48 hours post-
insemination were 23% more likely to form blastocysts than those that had only reached
the 1- to 3- cell stage by this time.
Embryo Morphology Morphological characteristics of early bovine embryos have been described by
several authors (Hamilton and Laing, 1946; Chang, 1952; Shea, 1981; Wright, 1981;
12

Lidner and Wright, 1983; Betteridge and Flechon, 1988), and numerous reports have
demonstrated that embryos produced under in vitro conditions display distinct
morphological differences when compared with in vivo-derived embryos (Plante and
King, 1994; Thompson, 1997; Holm and Callesen, 1998; Abe et al., 1999). For example,
embryos produced under in vitro conditions have been found to have darker cytoplasm,
a grainy appearance, less perivitelline space and swollen blastomeres when compared
with in vivo-derived embryos of similar degrees of development. Other observable
differences in ultrastructural features of in vitro-produced embryos include the amount of
lipid droplets contained in the cytoplasm, the development of junctional complexes
between trophoblast cells and/or cells of the ICM, development of microvilli on the apical
surface of trophoblast cells and alterations in morphological features of the zona
pellucida (Abe et al., 1999).
Interestingly, Abe et al. (1999) reported that morulae and blastocysts cultured in
vitro in the presence of serum contained a higher number of lipid droplets, particularly
within trophoblastic cells, than in vivo-derived morulae and blastocysts. These findings
are in agreement with other reports (Brackett et al., 1980b; Mohr and Trounson, 1981;
Shamsuddin et al., 1993; Plante and King, 1994; Crosier et al., 2000). Moreover, Abd El
Razek et al. (2000) determined that the lipids observed in embryos produced in vitro
were predominately triglycerides, with less lipids from other classes. Crosier et al. (2000)
observed an increase in lipids in compact morulae produced in vitro compared with in
vivo-derived morulae, regardless of the type of medium in which the embryos were
cultured [TCM-199 with 10% estrous cow serum (ECS), TCM-199 with BSA or TCM-199
with BSA and ECS from 72 hours post-insemination (hpi) to 144 hpi or mSOF)]. Taken
together, these findings suggest that the accumulation of lipid droplets within the
cytoplasm is characteristic of in vitro-produced embryos (Abe et al., 1999). The reason
for this lipid accumulation remains unknown, but it has been suggested that it is a result
of membrane breakdown in response to an artificial culture environment (Crosier et al.,
2000).
Alternatively, it has been suggested that insufficient mitochondrial metabolism
may cause the elevated lipid levels noted in in vitro-produced embryos (Dorland et al.,
1994). Mitochondria, specifically the cristae, are the primary site of lipid metabolism.
Immature mitochondria, lacking sufficient cristae, have been found in early cleavage and
13

morula stage bovine and primate embryos (Enders and Schlafke, 1981; Betteridge and
Flechon, 1988). These embryos may have a decreased ability to metabolize lipids into
ATP, thus causing a buildup of lipids within the embryo (Crosier et al., 2000).
Pregnancy and Calving Rates In addition to producing lower quality embryos, in vitro maturation and fertilization
of oocytes and/or in vitro culture of embryos results in other developmental problems.
Generally, a bovine embryo will develop for ~12 to 14 days in vitro, but will fail to
elongate as seen in vivo at this time. Therefore, in order to produce live offspring from in
vitro-produced embryos, the embryos must be transferred to recipient females.
Unfortunately, pregnancy rates achieved from the transfer of in vitro-produced embryos
are lower than those achieved from in vivo-derived embryos. One of the first studies
illustrating this difference was reported by Hasler et al. (1995). In this study 1884 fresh in
vitro-produced embryos were transferred to recipient females resulting in a 56%
pregnancy rate. In comparison, 320 in vivo-derived embryos were transferred, resulting
in a 66% pregnancy rate. The pattern of decreased pregnancy rates to embryo transfer
(ET) was consistent across embryo quality grades (grade 1, grade 2 and grade 3
embryos) and day of transfer (day 7 or day 8, post onset of estrus) when pregnancy
rates for in vitro-produced embryos were compared with pregnancy rates for in vivo-
derived embryos.
However, as in vitro embryo culture systems have improved over time so have
pregnancy rates resulting from the transfer of in vitro-produced embryos. Farin et al.
(2001) reported no difference between pregnancy rates from the transfer of in vitro-
produced (63%) and in vivo-derived embryos (65%). However, when serum-restricted in
vitro-produced embryos were transferred, pregnancy rates were lower than those
achieved from the transfer of serum supplemented in vitro-produced or in vivo-derived
embryos. There was also a higher incidence of degenerate conceptuses resulting from
the transfer of in vitro-produced embryos compared with their in vivo-derived
counterparts. Taken together, these reports indicate that improvements to in vitro culture
conditions have improved pregnancy rates achieved from the transfer of in vitro-
produced embryos; however, many critical problems remain. This is predominantly a two
part problem consisting of: (1) a higher incidence of abortion and (2) a higher incidence
of post-natal loss. Hasler et al. (1995) reported that abortions occurring from the transfer
14

of in vitro-produced embryos were not randomly distributed. In a two year study, abortion
occurred in 11% (range = 8% to 16% ) of 542 pregnancies resulting from the transfer of
in vitro-produced embryos. In comparison, an abortion rate of only 5% has been
reported for in vivo-derived ET pregnancies (Hasler et al., 1987). The timing of these
abortions in regard to stage of gestation is also different from that of normally mated
cattle. Ayalon (1978) and Diskin and Sreenan (1980) reported that the majority of
pregnancy loss occurs during the embryonic period of gestation (day 1 to day 42).
However, in pregnancies resulting from the transfer of in vitro-produced embryos, an
abortion rate of 13 to 20% has been reported to occur during the last 2 trimesters of
gestation (Hasler et al., 1995; Hasler et al., 2000; Agca et al., 1998).
It has been suggested that an increase in placental abnormalities is a
contributing factor in the increased incidence of abortion observed among ET
pregnancies resulting from in vitro-produced embryos. Farin et al. (2000) reported that
placental weight of fetuses resulting from in vitro-produced embryos were significantly
heavier than those from fetuses resulting from in vivo-derived embryos. Although there
were no differences in the number of placentomes and placental fluid volume, placentas
of in vitro-produced embryos had a significantly smaller caruncular surface area
compared with placentas from in vivo-derived embryos. Furthermore the fetal binucleate
cell volume was significantly less for placentas resulting from in vitro-produced embryos
compared with in vivo-derived pregnancies. In addition, Hasler et al. (1995) reported that
hydrallantois occurred in 1 of every 200 pregnancies. This was significantly higher than
the rate reported for normal pregnancies (1 of every 7500 pregnancies). Similar findings
have been reported by Hill et al. (1999) and Mello et al. (2003).
In addition to problems associated with placental malformations and increased
abortion rates, in vitro-produced ovine and bovine fetuses that survive to term often
experience significantly longer gestation periods (Walker et al., 1992; Sinclair et al.,
1995; Kruip and den Daas, 1997). The mean increase in gestation length for cattle has
been reported to be 3.6 days longer for in vitro-produced embryos compared with
gestation lengths from normal mating, artificial insemination (AI) or ET (Sinclair et al.,
1995; Kruip and den Daas, 1997).
Because of the increased incidence of abortion, the calving rate for cows
transplanted with in vitro-produced embryos is significantly lower than for cows receiving
15

in vivo-derived embryos (Behboodi et al., 1995; Hasler et al., 1995). Furthermore, of the
cows that calve, there is an increase in dystocia in pregnancies achieved from in vitro-
produced embryos (Behboodi et al., 1995; Schmidt et al., 1996). Increases in dystocia
for in vitro-produced embryo established pregnancies have been reported to range from
20% (Kruip and den Daas, 1997) to 62% (Behboodi et al., 1995). This increase is a six-
fold increase over that observed with natural mating, AI or ET.
In addition to lower calving rates a significant increase in perinatal morality has
been reported for sheep and cattle (Walker et al., 1992; Behboodi et al., 1995; Hasler et
al., 1995). Kruip and den Daas (1997) reported a two-fold increase in perinatal death
(5.6% to 6.7% occurrence) from in vitro-produced embryos compared with in vivo-
derived embryos. However, one of the original reports showed a 50% calf mortality
following parturition in calves resulting from in vitro-produced embryos (Behboodi et al.,
1995). In respect to the perinatal death rate increase there has been a report of an
increase in congenital abnormalities as a result of the transplantation of in vitro-produced
embryos (Schmidt et al., 1996).
Large Offspring Syndrome and Associated Disorders One of the first reported incidences of ‘large offspring syndrome’ was described
by Walker et al. (1992) as possibly resulting from transplantation of in vitro-produced
ovine embryos. Lambs resulting from these in vitro-produced embryos were significantly
heavier at birth (mean = 5.6±0.31 kg) compared with lambs produced from in vivo-
derived embryos (mean = 4.8±0.27 kg). Since this time there have been many
descriptions of large offspring syndrome occurring in sheep and cattle after transfer of in
vitro-produced embryos (Behboodi et al., 1995; Hasler et al., 1995; Schmidt et al., 1996).
In cattle, birth weights from in vitro-produced embryos have been reported to deviate
from that of naturally produced calves by as little as 1.9 kg to as much as 15.9 kg (Kruip
and den Daas, 1997). These adverse affects were attributed to factors within the culture
environment.
Since this time there have been many articles describing and attempting to
explain the causes of large offspring syndrome. It has been hypothesized that in vitro
culture is an abnormal environment and may be a leading cause of this problem. Others
have attributed the increased birth weight to the use of co-culture systems, and still
16

others believe that the addition of bovine serum albumin (BSA) or serum to the culture
medium is the primary cause.
In the absence of in vitro culture there are two reported methods to increase
embryo and/or conceptus size in utero. One of these methods is to transfer an embryo
into a more advanced stage uterus. Wilmut and Sales (1981) demonstrated this by
transferring early stage sheep embryos (4- to 8-cell and blastocyst stage) into uteri that
were -72 to +72 hours in synchrony with the transferred embryos. They concluded that
transfer of embryos into a uterus that is developmentally behind that of the embryo
resulted in rapid degeneration of the embryo. However, when an embryo was transferred
into a uterus that is developmentally advanced compared with the embryo the embryo
experienced a rapid increase in development, so that it matched the stage of the uterus
within a few days.
A second method to increase the rate of development and size of the fetus is
progesterone (P4) treatment to early pregnant females. Kleemann et al. (1994) reported
that if a pregnant ewe was administered P4 during the 3 days following ovulation fetal
crown-rump size and weight would be significantly larger compared with nontreated
controls. Later, Kleemann et al. (2001) found that the increase in fetal length and weight
were a response to increased heart, skeletal and muscle growth of the conceptus.
These findings are similar to reported increases in fetal size following in vitro culture.
Farin and Farin (1995) reported that following the transfer of in vitro-produced embryos
the resulting fetuses had significant increases in body weight, heart girth, long-bone
lengths and heart weight compared with fetuses produced from the transfer of in vivo-
derived embryos.
There are conflicting reports regarding the incidence of large offspring syndrome
following culture of in vitro-produced embryos with co-culture cells. The first incidence of
large offspring syndrome was reported to occur in sheep as a result of in vitro culture;
this was coincidentally one of the first reports of successful in vitro production of
embryos in the absence of co-culture (Walker et al., 1992). But one of the first reports of
large offspring syndrome occurring in in vitro-produced embryo pregnancies used an
oviduct epithelium co-culture system (Behboodi et al., 1995). Even more puzzling are the
reports of a very low incidence of large offspring syndrome as a result of the
transplantation of in vitro-produced embryos (Massip et al., 1996; Breukelman et al.,
17

2002). However, these two reports have two things in common, low numbers and low
pregnancy rates that may be the cause of these contradictory results.
When ovine pregnancies were established from in vitro-produced embryos
cultured in SOF supplemented with human serum or BSA there was a significant
reduction in birth weight from embryos cultured in SOF supplemented with BSA
compared with human serum (Thompson et al., 1995). Furthermore, when in vivo-
derived embryos were cultured in SOF supplemented with BSA the lambing weights
were not different from the in vitro-produced embryos cultured in the same medium
(Thompson et al., 1995). This finding demonstrated early that regardless of the origin of
the embryo, in vitro culture could result in large offspring syndrome at parturition, but the
addition of serum significantly magnified this effect.
Later, however, Thompson et al. (1998) reported that calves resulting from
embryos cultured in SOF supplemented with FCS where lighter at birth compared with
calves produced from embryos cultured in SOF supplemented with BSA or charcoal
striped FCS, although the differences were not significant. However, culturing embryos
in the presence of serum still results in significantly higher birthweights in sheep (Sinclair
et al., 1998; Brown and Radziewic, 1996) and in cattle (Farin and Farin, 1995; Kruip and
den Daas, 1997) compared with naturally mated, AI or MOET produced control calves.
In addition, pregnancies resulting from the transfer of embryos cultured in the presence
of serum or co-culture systems resulted in significantly greater fetal liver and heart sizes
compared with those of fetuses derived from embryos cultured in vitro without serum or
co-culture (Sinclair et al. 1997).
Apoptosis There are two distinct forms of cell death, necrosis and apoptosis, which can be
distinguished by specific morphological criteria (Wyllie et al., 1980; Majno and Joris,
1995). Necrosis is generally considered to be caused by accidental injury to the cell
initiated by environmental insults such as severe hypoxia, ischemia, reactive oxygen
species, heat shock or exposure to metabolic toxins or infections (Wyllie et al., 1980;
Betts and King, 2001). It affects large groups of cells or tissues, causing cellular swelling
and membrane rupture (Wyllie et al., 1980). Necrosis will not be discussed in detail in
this dissertation.
18

In contrast, apoptosis is a self-directed form of cell suicide that characteristically
affects single cells (Hardy, 1997; Betts and King, 2001). Initially, the cell shrinks, and the
chromatin condenses and migrates against the inner nuclear membrane, causing gross
indentions in the membrane (Hardy, 1997; Betts and King, 2001). The nucleus
fragments, and the DNA is cleaved between nucleosomes forming oligonucleosomal-
sized fragments (~185 bp; Hardy, 1997; Betts and King, 2001). During the final phases
of apoptosis, the cytoplasm condenses and the nucleus breaks up (karyohexis) into
pyknotic nuclear fragments (Betts and King, 2001). The fragmenting cellular components
can be found in budding membrane processes (called blebs) that break off into apoptotic
bodies, which are then either extruded into intracellular tissue spaces or phagocytized by
macrophages or neighboring tissue cells (Wyllie et al., 1980; Majno and Joris, 1995).
Cell death during early embryo development
Characteristic events of apoptosis have been documented in blastocysts of
several species including mice (El-Shershaby and Hinchliffe, 1974; Copp, 1978;
Handyside and Hunter, 1986), cattle (Mohr and Trouson, 1982; Plante and King; 1994),
baboons (Enders et al., 1990), rhesus monkeys (Hurst et al., 1978; Enders and
Schlafke, 1981; Enders et al., 1982) and humans (Mohr and Trouson, 1982; Hardy,
1999; Hardy et al., 1989; Hardy et al., 1996; Sathananthan et al., 1982). There is little
evidence, however, that apoptosis occurs prior to compaction in developing embryos of
good quality (Hardy et al., 1989). For example, nuclear DNA fragmentation has been
observed in situ using the TdT-mediated dUTP nick-end labeling (TUNEL) method in in
vitro-produced bovine oocytes, 8-cell embryos, morulae and blastocysts, but not in
zygotes or early cleavage-stage embryos (Byrne et al., 1999; Matwee et al., 2000), nor
in in vivo-derived embryos recovered from superovulated cows (referenced in Betts and
King, 2001). Similar results have been observed in pigs in which DNA fragmentation was
detected in in vitro-produced oocytes (Tatemoto et al., 2000) and blastocysts (Long et
al., 1998), but not 4- to 8-cell embryos. Normal developing human embryos have also
failed to show signs of apoptosis from the 2-cell to uncompacted morula stage
(Jurisocova et al., 1996).
It must be kept in mind, however, that it is difficult to estimate the full extent of
apoptosis in the embryo, as current techniques provide only a ‘snapshot’ of the embryo
at a specific point in time (Hardy, 1997). The initial stages of apoptosis appear to occur
19

very rapidly, with cytoplasmic fragmentation occurring within minutes (Wyllie et al.,
1980). In addition, phagocytosis of cell remnants can occur within as little as 12 to 18
hours (Wyllie et al., 1980). These findings suggest that observation of only a moderate
number of dead cells by current techniques may in all actuality represent extensive cell
death (Hardy, 1997).
Apoptosis is a critical process during pre-implantation development for the
disposal of excess cells, cells that are in the way, abnormal or potentially dangerous
(Gjorret et al., 2003). Moreover, elevated levels of apoptotic cells have been correlated
with abnormal embryo morphology (Hardy et al., 1989) and it has been suggested that
when apoptosis surpasses a certain ‘threshold level’ further embryonic development is
compromised (Tam, 1988; Juriscova et al., 1998; Hardy, 1999) and nonviable offspring
are eliminated (Byrne et al., 1999). Apoptosis appears to be a normal part of embryonic
development as in vivo-derived embryos from mice (El-Shershaby and Hinchliffe, 1974;
Copp, 1978; Handyside and Hunter, 1984; Brison and Schultz, 1997; Devreker and
Hardy, 1997), rats (Pampfer et al., 1990a,b), baboons (Enders et al., 1990), rhesus
monkeys (Enders and Schlafke, 1981) and humans (Hertig et al., 1954) have exhibited
characteristics of apoptosis immediately upon recovery from the reproductive tract.
Apoptosis appears to predominate in the ICM (El-Shershaby and Hinchliffe, 1974; Copp,
1978; Mohr and Trouson, 1982; Handyside and Hunter, 1986; Hardy and Handyside,
1996), with much lower levels detected in the trophoectoderm (Copp, 1978; Hardy and
Handyside, 1996; Brison and Schultz, 1997) and it has been suggested that apoptosis
occurs naturally in vivo as a means to help eliminate ICM cells that still have the
potential to form trophoectoderm, thus decreasing the chance of inappropriate
trophoectoderm expression during germ layer differentiation (Handyside and Hunter,
1986; Pierce et al., 1989). In support of this theory, a wave of cell death is apparent in
mouse embryos recovered from the reproductive tract 96 hours post-hCG administration
coincident with the time that ICM cells loose the ability to differentiate into
trophoectoderm cells (Handyside and Hunter, 1986). In contrast, in vivo-derived rabbit
(Giles and Foote, 1995) and porcine (Papaioannou and Ebert, 1988) embryos show no
evidence of apoptotic activity upon recovery from the reproductive tract. Although, the
technique (differential labeling) used in these reports may not be adequate to detect
apoptotic activity in these species.
20

Apoptosis may also function to remove abnormal cells from the embryo (Hardy,
1997; Hardy, 1999). Nuclear and chromosomal abnormalities have been identified in
both human and bovine embryos. Specifically, ~ 20% of human embryos exhibit gross
chromosomal abnormalities and more than 40% of embryonic cells are mosaic (i.e.,
some cells within the same embryo are diploid while others are aneuploid, haploid or
triploid; Harper et al., 1995). Similarly, 25% of in vivo- and 72% of in vitro-produced
bovine embryos were identified as mosaic (Viuff et al., 1999). These data are in
agreement with those of Jacobs (1990) and King (1991) in that the in vitro production
environment appears to increase nuclear and chromosomal abnormalities and
consequently reduce their developmental potential.
Effect of in vitro culture in the incidence of apoptosis
In vitro embryo production has been correlated with alterations of normal
apoptotic patterns; however, conflicting data does exist. In one study, the incidence of
apoptotic cells in murine blastocysts was similar to those seen in in vivo-derived
embryos (Devreker and Hardy, 1997). In a separate study, the percentage of apoptotic
cells was significantly higher in murine embryos produced in vitro as compared to their in
vivo-derived counterparts (Brison and Schultz, 1997). Similarly, in vitro-produced bovine
blastocysts had a significantly higher number of apoptotic cells than those that
developed completely in vivo (Byrne et al., 1999; Gjorret et al., 2001).
It is generally accepted that maternal- and embryonic-derived growth factors
significantly influence pre-implantation development (Kaye, 1997; Watson et al., 2000)
and it has been suggested that the effect of in vitro culture may be a result of the
presence or absence of these factors in the culture medium (Brison and Schultz, 1997;
Hardy, 1997; Moley et al., 1998; Gjorret et al., 2001). Both transforming growth factor α
(TGF-α) and insulin-like growth factor I (IGF-I) increase blastocyst formation and
blastocyst cell number, presumably by reducing blastomere cell death (Brison and
Schultz, 1997; Lighten et al., 1998). Similarly, the incidence of apoptosis is elevated in
blastocysts from diabetic rats (Pampfer et al., 1990a), but can be reduced by maternal
treatment with insulin (de Hertogh et al., 1992). In contrast, both in vitro- and in vivo-
derived murine blastocysts had a significant increase in nuclear fragmentation in ICM
cells with a concomitant reduction in the number of ICM cells and embryo viability after
treatment with tumor necrosis factor-α (TNF-α; Wuu et al., 1999). Byrne et al. (1999)
21

suggested that the addition of TBS to embryo culture media, while beneficial, may also
be detrimental in that it may contain ‘death’ factors such as TNF-α that could trigger
apoptosis within the embryo.
Regulation of apoptosis and expression of apoptotic genes during early embryo development
The Bcl-2 family of genes are key regulators of apoptosis in mammalian cells
(Betts and King, 2001). The Bcl-2 family of proteins can be subdivided into two distinct
groups: (1) the anti-apoptotic proteins or survival proteins, including Bcl-2, Bcl-XL and
Bcl-W and (2) the pro-apoptotic proteins or death proteins, including Bax, Bcl-XS, Bak,
Bas, Bik and Bid (Krajewski et al., 1993). It is believed that the ratio of pro- and anti-
apoptotic proteins, rather than the expression levels of individual Bcl-2 genes, determine
a cell’s susceptibility to apoptosis (Rheostat hypothesis; Oltvai et al., 1993).
There are several mechanisms through which apoptosis can be induced in cells;
however, there are thought to be two general pathways: the receptor or extrinsic
pathway and the mitochondrial or intrinsic pathway (Lawen, 2003). In the receptor
mediated or extrinsic pathway, extracellular ligands bind to specific death receptors
bound to the plasma membrane of the targeted cell. The most common death receptors
are Fas (also called Apo-1 or CD95) and tumor-necrosis factor receptor-1 (TNF-R1;
Lawen, 2003). Activation of a death receptor results in activation of an initiator caspase,
caspase-8 or -10 in particular. The initiator caspase then activates other caspases in the
cascade eventually leading to the activation of effector caspases, such as caspase-3 or -
6 (Lawen, 2003).
In contrast to the receptor-mediated induction pathway, the mitochondrial or
intrinsic pathway is activated by intracellular stresses, such as withdrawal from growth
factors (Betts and King, 2001), oxidative stress (Lawen, 2003) or exposure to DNA
damaging agents (Betts and King, 2001; Lawen, 2003). In this pathway, pro-apoptotic
members of the Bcl-2 family, usually Bax, undergo a conformational change that allows
them to integrate themselves into the outer mitochondrial membrane. Once incorporated
into the membrane they interact with voltage-dependent anion channels, causing the
formation of pores in the membrane and allowing release of cytochrome c into the
cytosol (Shimizu et al., 1999). Within the cytosol, cytochrome c binds to Apoptotic
Protease Factor-I (APF-I) to form an apoptosome. The apoptosome contains the initiator
22

caspase procaspase-9 (Waterhouse et al., 2002) that, when activated, initiates a
cascade of events leading to activation of an effector caspase and cellular breakdown
(Lawen, 2003). It has been suggested the fate of the cell via the intrinsic pathway is
influenced by the ratio of death factors to survival factors (Wyllie, 1995). Specifically,
survival factors, such as Bax and Bcl-XL, may act to prevent the release of cytochrome c
from the mitochondria by binding to Bax or by closing the pores in the mitochondrial
membrane (Shimizu et al., 1999).
As indicated, regardless of the type of induction, both signaling pathways
converge at the level of the caspases (Lawen, 2003), a family of cysteine-dependent
aspartate-specific proteases that cleave key cellular proteins and cause disassembly of
the cell (Barinaga, 1998; Mehmet, 2000). To date, 14 mammalian caspases have been
identified (Strasser et al., 2000) and divided into three groups based on function (Grutter,
2000): (1) the initiator caspses (caspase-2, -8, -9 and -10) recognize specific receptors
in the cell and ‘declare the death sentence’ (Betts and King, 2001), (2) the executioner or
effector caspases (caspase-3, -6 and -7) which ‘carry out the death sentence’ by
breaking down cellular proteins (Betts and King, 2001) and (3) the caspases involved in
cytokine maturation rather than apoptosis (caspase-1, -4, -5, -11, -12, -13 and -14).
Briefly, initiator caspases cleave and activate effector caspases, which then cleave
cellular proteins causing the typical morphological changes observed in cells undergoing
apoptosis (i.e., membrane blebbing, disassembly of the cell structure and DNA
fragmentation; Lawen, 2003).
Transcripts for apoptosis related genes have been detected in early stage
embryos of mice (Exley et al., 1999), cattle (Kolle et al., 2002; Yang and
Rajamahendran, 2002; Rizos et al., 2003) and humans (Warner et al., 1998).
Specifically, transcripts for both Bax and Bcl-2 have been identified in 2- to 12-cell
human embryos (Warner et al., 1998). Moreover, levels of Bcl-2 were higher in
blastocysts of good morphology; whereas, Bax transcript levels were higher in
blastocysts with extensive fragmentation (Hardy, 1999). Similar results have been
observed in mice in that fragmented blastocysts contained lower levels of Bcl-2 (Exley et
al., 1999) adding support to the hypothesis that alteration in the ratio of Bcl-2 to Bax
determines susceptibility to apoptosis (Oltvai et al., 1993). In cattle, Bcl-2 expression has
been detected from the 4-cell stage throughout development and in both the TE and ICM
23

of blastocysts and hatched blastocysts (Kolle et al., 2002; Neuber et al., 2002), with
apoptosis being proportionally higher in the ICM than the TE (Neuber et al., 2002).
Similar to results reported in mice, Bcl-2 expression varied between embryos and was
suggested to be related to degree of fragmentation of the embryo (Kolle et al., 2002).
Similarly, Bax expression has been detected from the 8-cell stage throughout
development (Byrne et al., 1999; Matwee et al., 2000; Paula-Lopes and Hansen, 2002;
Lonergan et al., 2003). In addition, Yang and Rajamahendran (2002) found that Grade I
bovine embryos had a much higher expression of Bcl-2 compared with Grade IV
embryos; whereas Bax expression was much more prominent in Grade IV embryos.
These authors suggest that Grade IV embryos are in an advanced stage of apoptosis,
as indicated by the elevated levels of Bax expression (Yang and Rajamanhendran,
2002). Furthermore, these authors concur with the concept proposed by Oltvai et al.
(1993) that the ratio of Bcl-2 to Bax determined cell fate (Yang and Rajamanhendran,
1993). Similar results have been observed with bovine embryos produced in the
presence of serum (Rizos et al., 2003).
CHICK EMBRYO CO-CULTURE Biochemistry of the Avian Embryo The avian egg is a complete environment capable of sustaining embryonic
development from the earliest stages of blastulation through hatching. At the time of
oviposition, the chicken egg is composed of ~ 56% albumen, 32% yolk and 12% shell
(Romanoff and Romanoff, 1949). At this time, the albumen contains ~ 88% water, 10%
protein, 0.05% lipid and 0.8% ash (Runge, 1982). In contrast, the yolk is composed of ~
49% water, 17% protein, 32% lipid and 2% ash (Runge, 1982).
The yolk is the main source of nutrients for the developing chick during early
development. The nutrients within the yolk are quite concentrated and because fat is
much more concentrated calorically than sugar, the egg is rich in fat rather than
carbohydrates (Romanoff, 1967). High energy lipids make up almost 64% of the dry
weight of the egg, including lecithin, cephalin, cholesterol, carotenoids and ergosterol
(Romanoff, 1967). In addition, egg yolk contains glucose, glycogen, lactic acid and high
energy adenosine triphosphate, as well as many amino acids, vitamin A, ascorbic acid,
vitamin B12, thiamine and minerals such as chloride, calcium, sodium, potassium and
iron (Romanoff, 1967).
24

During early embryo development, the embryo receives nutrients from the yolk
via the vitelline circulation (Runge, 1982). It is believed that the amniotic fluid that bathes
the developing embryo is a result of the metabolism of these nutrients (Runge, 1982).
However, the regulation of the volume and contents of the amniotic fluid are poorly
understood (Pierce, 1933; Romanoff, 1960). Recent reports indicate that there are three
barriers (the blood/amnion barrier, the blood/allantois barrier and the allantois/amnion
barrier) that specifically control amino acid concentration within these compartments (ten
Busch et al., 1997b). In birds, the allantois and amnion lack innervation (Pierce, 1933;
Romanoff, 1960), so it is believed that the amniotic and allantoic fluids are controlled
largely by hormones (Schmidek et al., 2001).
The components of avian amniotic fluid still remain largely undefined; however,
ions of chloride, sodium, potassium, phosphorus, magnesium, calcium, iron and sulfur
are present as well as low levels of proteins and carbohydrates (Smoczkiewiczowa,
1959). In addition, 47 free amino acids and related compounds have been identified in
the plasma, amniotic fluid and/or allantoic fluid of the chick embryo (Epple et al., 1992;
Gill et al., 1994; ten Busch et al., 1997a,b; Epple et al., 1997; Piechotta et al., 1998) and
IGF-I has also been reported to be widespread in the tissues of the chick (McFarland et
al., 1992; De Pablo et al., 1993; Yang et al., 1993; Tanaka et al., 1996; McMurtry et al.,
1997).
Early Use of Chick Embryo Extracts in Mammalian Cell Culture Extracts obtained from developing chick embryos have been used throughout the
history of mammalian cell culture. In 1913, Alexis Carrel reported that when extracts
from 20-day old chick embryos were added to in vitro cultures of canine connective
tissue, there was a dramatic increase in growth rate. In a second experiment he noted
that a 30-fold increase in growth rate could be obtained if the chick embryos were stored
for 20 days in cold storage prior to extraction (Carrel, 1913). Later, Willmer and Jacoby
(1936) reported that extracts from day 7 chick embryos could stimulate the development
of quiescent avian cells. Interestingly, the rate of cell proliferation was proportional to the
amount of extract in the medium.
Since these early experiments, chick embryo extract has been used as a
supplement for many different cell types including HeLa cells (Takano, 1957), heart
endothelial cells (Namikawa, 1969), retinal pigmented epithelium cells (Chader et al.,
25

1975), rat pancrease cells (McEvoy and Hegre, 1976), chick epidermis (Matsuhashi and
Sugihara, 1984), dorsal root ganglion (Xue et al., 1992) and hematopoietic cells
(Nicolas-Bolnet et al., 1995) and it is commonly used for modern neural crest (Hou and
Takeuchi, 1992; Wu et al., 2001) and muscle (Slater, 1976; Scarpini et al., 1982; Ii et al.,
1982; Bischoff, 1986; Smith and Schroedl, 1992; Nicolas-Bolnet and Dieterlen-Lievre,
1995) cell cultures.
In Vitro Culture of Chick Embryos The earliest report of the culture of whole egg contents outside of the egg shell
were those of Preyer (1885; referenced in Rowlett, 1990). With this system, embryo
survival was limited to only 2 days. Since that time, many researchers have attempted to
improve embryo viability following shell-less chick culture. The most successful method
has been that of Dunn (1974) in which he suspended the egg contents in commercial
plastic wrap placed in culture vessels made of modified Playtex® nurser holders. More
importantly, he determined that incubation in a 5% CO2 humidified environment
improved embryo viability with chicks surviving up to 22 to 23 days, but were unable to
hatch. This technique has since been modified (Dunn and Boone, 1976, 1977, 1978;
Dunn et al., 1981; Dunn et al., 1987, Blakewood and Godke, 1989).
The advantages to using this method of chick culture is that development of the
chick embryo is easily observed and reasonable survival rates can be obtained. More
important to this dissertation, this culture system allows manipulations of the chick
embryo and access to the amnion for injection of mammalian embryos. The main
drawback to culturing chick embryos in the absence of a shell is that the shell provides
70 to 80% of the calcium required by the developing chick (Johnston and Comar, 1955;
Romanoff, 1960, Crooks and Simnkiss, 1974). This is a problem in that it appears to
prevent the chick from “hatching” from this shell-less culture system. In addition,
because the embryo is maintained on plastic film, the egg contents assume abnormal
spatial relationships due to the shape of the plastic film sac and excessive evaporation
may occur from the surface of the cultures. All of these factors have significant effects on
the growth and development of chick embryos in shell-less culture (Rowlett, 1990).
Chick Embryo Co-Culture The use of avian eggs for the culture of mammalian embryos was envisioned by
Dr. R.A. Godke in 1975 (personal communication). However, it took more than 15 years
26

for this idea to come to fruition. Early attempts using unfertilized hen’s eggs proved
largely unsuccessful (Blakewood et al., unpublished), but after much trial and error a
technique using shell-less chick embryos for the co-culture of mammalian embryos was
developed (Blakewood et al., 1989a,b). With this technique, mammalian embryos were
embedded in agar and injected into the amnion of a live chick embryo maintained in
shell-less culture. After 72 to 96 hours of culture, the mammalian embryos could be
recovered from the amnion and transferred to recipient females.
In the initial report, pronuclear stage murine embryos from two separate strains,
one of which characteristically showed a 2-cell developmental block in vitro, were placed
in the amniotic cavity of 96-hour chick embryos and cultured for 72 hours. Upon
recovery, there were significantly more hatched blastocysts resulting from culture in the
chick embryo in both strains of mice (Blakewood et al., 1989a) when compared with
Whitten’s control medium.
Later studies were conducted to determine if the chick embryo amnion could
support development of caprine embryos. Blakewood et al. (1990) reported that culture
of 2- to 8-cell caprine embryos in the chick amnion allowed development past the in vitro
developmental block. Furthermore, these embryos developed to blastocysts at rates
equal to co-culture of similar stage embryos on uterine fibroblasts. When culture was
extended to 96 hours, significantly more embryos developed to the blastocyst stage after
culture in the chick amnion. Furthermore, 12 of the resulting embryos were surgically
transferred to recipient females and resulted in 6 live offspring.
Further studies indicated that the chick amnion could promote development of
pre-compaction stage bovine morulae to the expanded blastocyst stage at rates
significantly greater than embryos cultured on uterine fibroblasts or in medium alone. In
addition, chick embryo co-culture prior to freezing improved post-thaw viability when
compared with embryos cultured in medium alone prior to freezing.
27

CHAPTER III
CULTURING IN VITRO-PRODUCED BOVINE EMBRYOS IN THE AMNIOTIC CAVITY OF THE DEVELOPING CHICKEN EMBRYO
INTRODUCTION
With the advent of in vitro fertilization technologies in the 1980s came the need
for methods of culturing embryos to later stages of development. Early methods included
in vitro co-culture with feeder cells (Cole and Paul, 1965), fibroblast monolayers (Kuzan
and Wright, 1982; Voelkel et al., 1985) or trophoblastic vesicles (Camous et al., 1984;
Heyman et al., 1987; Pool et al., 1988) and in vivo culture in the ligated oviducts of
sheep (Willadsen, 1979).
As early as 1975, Dr. R.A. Godke envisioned the use of avian eggs for the
culture of mammalian embryos (personal communication). Early attempts using
unfertilized hen’s eggs proved largely unsuccessful (Blakewood et al., unpublished
data), but after several months of trial and error a technique was developed that used
shell-less chick embryos for the co-culture of mammalian embryos. Chick embryo co-
culture improved development of murine (Blakewood et al., 1989a), caprine (Blakewood
et al., 1989b) and bovine (Blakewood et al., 1989b) embryos compared with the culture
media of the time, with embryos consistently passing thru the in vitro developmental
block and a greater percentage reaching the blastocyst stage compared to control
methods.
Since that time, in vitro embryo culture techniques have progressed and there is
no longer a need for in vitro co-culture or culture in the oviducts of sheep. However,
many problems still remain. With current embryo culture methods, only 30 to 40% of in
vitro-matured and fertilized ova routinely reach the blastocyst stage (Krisher and
Bavister, 1998; Lonergan et al., 2003). In addition, in vitro-produced embryos show
marked differences in morphology (Greve et al., 1995), timing of development (Van
Soom et al., 1997), embryonic metabolism (Khurana and Niemann, 2000) and
pregnancy rates following transfer (Enright et al., 2000) when compared with embryos
derived in vivo.
It has been hypothesized that exposure of embryos to in vitro culture conditions,
particularly exposure to fetal bovine serum (FBS), may result in abnormal fetal and
neonatal development and growth, known collectively as large calf or large offspring
28

syndrome (reviewed by Kane, 2003). Specifically, offspring produced from embryos
cultured in vitro have exhibited increased birth weights (Willadsen et al., 1991; Keefer et
al, 1994; Behboodi et al., 1995), longer gestation lengths (Walker et al., 1996), increased
placental anomalies (Hasler, et al., 1995; Hill et al., 1999, 2001, 2002) and increased
incidences of abortion (Walker et al., 1996; Hill et al., 1999) and perinatal (Behboodi et
al., 1995; Wilson et al., 1995; Garry et al., 1996; Schmidt et al., 1996; Kruip and den
Daas, 1997; Hill et al., 1999) and neonatal mortality (Schmidt et al., 1996; Kruip and den
Daas, 1997; Hill et al., 1999). Breathing difficulties (Young et al., 1998) and disruptions
in the growth and development of body organs are also common (Kane, 2003).
The problems associated with modern in vitro embryo culture systems have led
us to re-visit the chick embryo co-culture system. In the present study, the development
of bovine 1-cell and 2-cell embryos produced by in vitro fertilization was assessed after
culture in the chick embryo amnion. This study was conducted as a preliminary
evaluation of the use of this novel culture technique as a means to improve in vitro
development of bovine embryos.
MATERIALS AND METHODS Experimental Design Experiment 3.1
The design for Experiment 3.1 is illustrated in Figure 3.1. This experiment was
conducted to determine if IVF-derived 1-cell embryos could develop to morulae and
blastocysts at rates similar to IVF-derived embryos cultured in CR1aa. Upon removal
from in vitro insemination, 1-cell embryos were randomly allotted to one of four treatment
groups for in vitro culture. Bovine 1-cell embryos allotted to Treatment A (control) were
cultured in CR1aa from day 0 to day 3 post-insemination and CR1aa containing 5% FBS
from day 3 to day 7. Bovine 1-cell embryos allotted to Treatment B (agarose control)
were embedded in agarose immediately upon removal from insemination and cultured in
CR1aa from day 0 to day 3 post-insemination and CR1aa containing 5% FBS from day 3
to day 7. Bovine 1-cell embryos allotted to Treatment C (CEC-I) were embedded in
agarose immediately upon removal from insemination and then injected into the amniotic
cavity of 96-hour chick embryos. After 72 hours of co-culture, the bovine embryos were
recovered from the amnion and placed in CR1aa containing 5% FBS for the remaining
29

Control Groups Media Change
Onset of Egg Incubation
Preparation of Shell-less Cultures
48 h
~24 h
Injection of CEC-I
IVF IVC
72 h
Recovery of CEC-I Injection of CEC-II
96 h
Recovery of CEC-IIEvaluation
Evaluation Addition of FBS
5 h Chick Culture Treatment Groups Figure 3.1. Experimental design for the culture of 1-cell bovine embryos in the amnion of a developing chick embryo.
30

culture period. Bovine 1-cell embryos allotted to Treatment D (CEC-II) were embedded
in agarose immediately upon removal from insemination and cultured in CR1aa from day
0 to day 3 post-insemination. On day 3, agar-embedded embryos were injected into the
amniotic cavity of 96-hour chick embryos. After 96 hours of culture, the bovine embryos
were recovered from the amnion and evaluated.
Experiment 3.2
The design for Experiment 3.2 is illustrated in Figure 3.2. This experiment was
conducted to determine if IVF-derived 2-cell embryos could develop to morulae and
blastocysts at rates similar to IVF-derived embryos cultured in CR1aa. Presumptive
zygotes were purchased from a commercial source and upon arrival were randomly
allotted to one of four treatment groups for in vitro culture. Bovine 2-cell embryos allotted
to Treatment A (control) were cultured in CR1aa from day 0 to day 3 post-insemination
and CR1aa containing 5% FBS from day 3 to day 7. Bovine 2-cell embryos allotted to
Treatment B (agar control) were embedded in agarose immediately upon arrival and
cultured in CR1aa from day 0 to day 3 post-insemination and CR1aa containing 5% FBS
from day 3 to day 7. Bovine 2-cell embryos allotted to Treatment C (CEC-I) were
embedded in agarose immediately upon arrival and then injected into the amniotic cavity
of 96-hour chick embryos. After 72 hours of co-culture, the bovine embryos were
recovered from the amnion and placed in CR1aa containing 5% FBS for the remaining
culture period. Bovine 2-cell embryos allotted to Treatment D (CEC-II) were embedded
in agarose immediately upon arrival and cultured in CR1aa from day 0 to day 3 post-
insemination. On day 3, agar-embedded embryos were injected into the amniotic cavity
of 96-hour chick embryos. After 96 hours of culture, the bovine embryos were recovered
from the amnion and evaluated.
Experimental Methods Production of In Vitro Embryos
Media Preparation
Brackett-Oliphant (BO) media were prepared as previously described by Brackett
and Oliphant (1975). Briefly, BO-A stock solutions contain 150.2 mM NaCL (S-5886,
MO), 3.0 mM CaCl2·2H2O (C-7902, Sigma-Aldrich Inc., St. Louis, MO), 1.0 mM
NaH2PO4·H2O (S-9638, Sigma-Aldrich Inc., St. Louis, MO), 0.6857 mM MgCl2·6H2O
31

Control Groups Shipment of
Presumptive Media Change
Onset of Egg Incubation
48 h ~24 h
Injection of CEC-I
IVF IVC
72 h
Recovery of CEC-I Injection of CEC-II
96 h
Recovery of CEC-I Evaluation
Evaluation Addition of FBS
Chick Culture Treatment Groups
Zygotes 18 h
Preparation of Shell-less Cultures
Figure 3.2. Experimental design for the culture of 2-cell bovine embryos in the amnion of a developing chick embryo.
32

(M-2393, Sigma-Aldrich Inc., St. Louis, MO) and 0.1 ml of 0.5% phenol red (P-0290,
Sigma-Aldrich Inc., St. Louis, MO) per 500 ml of sterile water. BO-B stock solutions
contain 154.0 mM NaHCO3 (S-8875, Sigma-Aldrich Inc., St. Louis, MO) and 0.04 ml of
0.5% phenol red per 200 ml of sterile water.
CR1aa medium was prepared as previously described by Rosenkrans and First
(1994). Briefly, CR1aa stock solution was prepared by adding 114.7 mM NaCl, 1.6 mM
KCl, 26.2 mM NaHCO3, 0.3999 mM pyruvic acid (P-4562, Sigma-Aldrich Inc., St. Louis,
MO), 2.5 mM L(+) lactic acid (L-4388, Sigma-Aldrich Inc., St. Louis, MO), 0.5195 mM
glycine (G-8790, Sigma-Aldrich Inc., St. Louis, MO), 0.5051 mM L-alanine (A-7469,
Sigma-Aldrich Inc., St. Louis, MO) and 0.2 ml of 0.5% phenol red per 100 ml of sterile
water.
Fertilization stock medium (BO-AB) was prepared by adding 1.2 mM pyruvic
acid, 0.05 ml of gentamicin (50 mg/ml; 15750-060, Gibco, Grand Island, NY) and 0.018
ml of heparin sodium (10,000 IU/ml; Elkins-Sinn, Cherry Hill, NJ) per 38 ml of BO-A
stock solution and 12 ml of BO-B stock solution. Fertilization medium (BO-caffeine) was
prepared by adding 0.0243 g of caffeine sodium (C-4144, Sigma-Aldrich Inc., St. Louis,
MO) per 25 ml of BO-AB. Sperm washing medium (0.6% BSA-BO) was prepared by
adding 0.03 g of bovine serum albumin (BSA; Fraction V, A-4503, Sigma-Aldrich Inc., St.
Louis, MO) to 5 ml of BO-AB. Oocyte washing medium (0.3% BSA-BO) was prepared by
adding 0.03 g of BSA (Fraction V) to 10 ml of BO-AB. TCM oocyte washing medium was
prepared by adding 0.03 g of BSA (Fraction V) to 10 ml of Tissue Culture Medium-199
(Eagle’s salt) with 25 mM HEPES Buffer (TCM-199; Gibco Laboratories, Grand Island,
NY).
CR1aa culture medium for day 0 to day 3 was prepared by adding 0.2 ml of
Basal Medium Eagle (BME) amino acid solution (50X, B-6766, Sigma-Aldrich Inc., St.
Louis, MO), 0.1 ml of Modified Eagle Medium (MEM) amino acid solution (10X, 11140-
050, Gibco Laboratories, Grand Island, NY), 0.01 ml of gentamicin, 1.1 mM L-glutamine
(G-5763, Sigma-Aldrich Inc., St. Louis, MO) and 0.03 g of BSA (fatty acid free, A-7511,
Sigma-Aldrich Inc., St. Louis, MO) to 9.5 ml of CR1aa stock solution. CR1aa culture
medium for day 3 to day 7 was prepared by adding 0.2 ml of BME amino acid solution,
0.1 ml of MEM amino acid solution, 0.5 ml of FBS (SH30070.02; HyClone, Logan, UT),
33

0.01 ml of gentamicin, 1.1 mM L-glutamine and 0.03 g of BSA (fatty acid free) to 9.5 ml
of CR1aa stock solution.
In Vitro Fertilization
Oocytes were purchased weekly from a commercial supplier (BoMed, Inc.,
Madison, WI) and matured in a portable incubator (~38.5°C) during the overnight travel
interval (20 to 22 hours). Upon arrival at the laboratory, shipping vials were removed
from the portable incubator and stored in a humidified atmosphere of 5% CO2 in air at
39°C until insemination.
Semen was prepared by thawing a straw of frozen semen from a Holstein bull
(CSS 7H5188, Genex Cooperative Inc., Shawano, WI) of proven fertility in a 39° to 40°C
water bath for ~45 seconds. After thawing, the contents of the straw were deposited into
a 15-ml plastic conical tube (Corning, New York, NY). Then, 9 ml of BO-caffeine was
added to the tube and the suspension was centrifuged for 6 minutes at 200 x g. After
centrifugation, the supernatant was removed and the remaining pellet was resuspended
in an additional 9 ml of BO-caffeine. The resulting suspension was centrifuged for 6
additional minutes at 200 x g. The supernatant was removed again and the pellet
resuspended in 4 ml of BO-caffeine and 4 ml of 0.6% BSA-BO and placed in a 38°C
incubator for ~10 minutes to equilibrate. After equilibration, four 80-µl insemination drops
were created in a preheated 35 x 10-mm Falcon® plastic petri dish (Becton Dickinson,
Lincoln Park, NJ) and covered with warmed mineral oil (M-8140, Sigma-Aldrich Inc., St.
Louis, MO).
After the 20 to 22 hour maturation period, cumulus oocyte complexes were
removed from the shipping vials and washed through two 35 x 10 mm plastic petri
dishes containing 2 to 3 ml of 0.3% BSA-BO. Then, 10 to 20 oocytes were placed in
each of the 80-µl insemination drops previously prepared and were co-incubated for 5
hours at 38°C and 5% CO2 in air.
After fertilization, oocytes were washed through a single 35 x 10-mm plastic petri
dish of TCM oocyte washing medium. Cumulus cells were removed by vortexing for 3
minutes in 1 ml of TCM-199 containing 0.0010 g of hyaluronidase (H-3506, Sigma-
Aldrich Inc., St. Louis, MO). After vortexing, oocytes were washed through two 35 x 10-
mm plastic petri dishes containing TCM oocyte washing medium and one 35 x 10-mm
plastic petri dish of CR1aa. Then, 10 to 15 oocytes per drop were placed in 50-µl drops
34

of CR1aa covered with warmed mineral oil and incubated in a modular incubator
containing 5% CO2, 5% O2 and 90% N2 at 39°C.
Chick Embryo Co-Culture
The basic procedure for the culture of bovine 8-cell IVF embryos using the chick
embryo amnion was modified from that previously described by Blakewood and Godke
(1989). Briefly, fertilized domestic chicken eggs were collected on the day of oviposition
and stored at 10°C until the onset of incubation. Four days prior to the introduction of
bovine embryos, the eggs were removed from cold storage and allowed to come to room
temperature (25°C) before placing them in a 37°C commercial egg incubator. After 72
hours of incubation, the eggs were rinsed with a solution of 70% ethanol (Aaper Alcohol
and Chemical Company, Shelbyville, KY) and then coated with 7.5% providone iodine
solution (Betadine®, Purdue Fredrick Company, Stanford, CT) and allowed to air dry in a
horizontal position to ensure proper positioning of the chick embryo.
To remove the egg contents, a crack was made in the shell by striking the
center of the egg against the rim of a sterile beaker. The egg contents were then
released into a 100-ml plastic embryo collection dish (Veterinary Concepts, Spring
Valley, WI) covered by a piece of cellophane kitchen wrap (Saran®, Dow Chemical,
Midland, MI). The dish, including the egg contents, was loosely capped with a plastic lid
(Veterinary Concepts, Spring Valley, WI) and maintained in an incubator at 38°C for an
additional 24 hours before introducing the bovine embryos into the amniotic cavity of the
chick embryo.
Prior to embedding and injection of bovine embryos, suitable chick embryos
were selected for injection based on the following criteria: 1) round intact yolk, 2) good
vascularity in the chorioallantoic membrane, 3) vigorous heartbeat and 4) no apparent
abnormalities.
The procedure for embedding embryos in agarose was modified from that of
Willadsen (1979) for the culture of ovine embryos in the ligated oviducts of sheep. On
day 0 of culture, two or three 8-cell bovine IVF embryos were placed in a 50-µl drop of
low-melting agarose solution [1.2% agarose (A-9799, Sigma-Aldrich Inc., St. Louis, MO)
dissolved in Dulbecco’s Phosphate-Buffered Saline (DPBS; D1408, Sigma-Aldrich Inc.,
St. Louis, MO)] at ~37oC and then aspirated into a beveled glass pipette (200 µm outer
diameter). After holding in air for ~30 seconds, the agar (containing the embryos) within
35

the pipette was expelled as a gel cylinder into room temperature TL-HEPES
(BioWittaker, Walkersville, MD). The agarose cylinder, including the bovine embryos,
was then aspirated back into the beveled pipette and carefully expelled into the amniotic
cavity of a 96-hour chick embryo.
After introducing the embedded embryos into the amniotic cavity, the shell-less
culture systems were returned to incubation for an additional 72 hours at 39oC. Thus, a
single agarose cylinder containing two or three 8-cell bovine IVF embryos was co-
cultured in the amnion of each developing chick embryo for 3 to 4 days.
On day 3 or day 7 of in vitro culture, the amnion containing the agarose cylinder
was dissected away from the rest of the egg components, placed in a 35 x 10-mm
plastic petri dish and rinsed several times with DPBS containing 1% FBS. After rinsing,
the petri dish was placed under a stereomicroscope (Nikon SMZ-2B, Tokyo, Japan)
equipped with an overhead light source and the agarose cylinder was removed from the
amniotic cavity by using two 22-gauge, 37.5-mm needles (Kendall, Mansfield, MA) to
make a small opening in the amniotic membrane. The agarose cylinder was carried from
the amnion with the escaping amniotic fluid, settling on the bottom of the dish. The
cylinder was then washed twice in CR1aa medium and dissected using two 22-gauge
needles to isolate and remove the developing embryos.
Statistical Analysis For both experiments, data were analyzed using PROC GLM of the SAS
Statistical Package (v. 8e, 1999, SAS Institute, Inc., Gary, NC). Significant differences
between embryo developmental rates (Experiment 3.1 and Experiment 3.2) and embryo
quality grade (Experiment 3.2) in each group were tested using a post hoc least
significant difference (LSD) test. Differences with a probability level (P) of 0.05 or less
were considered significant.
RESULTS In Experiment 3.1, 60 chicken eggs were initially placed in incubation. After 72
hours of incubation, the eggs were removed from their shell and placed in shell-less
culture. At this time, six (10%) of the eggs were found to be unfertilized, four (7%)
contained embryos that failed to develop and one (2%) contained an embryo that was
nonviable. The remaining 49 eggs were placed in shell-less culture and returned to
36

incubation. After 24 hours of incubation in shell-less culture, 21 eggs were selected for
injection of bovine embryos.
In the CEC-I treatment group, 29 of 36 (81%) embryos were recovered from the
amniotic cavity of nine live chicks after 72 hours of culture. Of the 29 embryos
recovered, 16 (55%) had successfully cleaved with seven (24%) of those appearing to
be viable 6- to 8-cell embryos (Table 3.1). Overall cleavage rate of the CEC-I treatment
group was significantly lower (P<0.05) than the control (34/36), agar (33/36) and CEC-II
treatment groups (29/36). Similarly, there were significantly fewer (P<0.05) 6- to 8-cell
embryos in the CEC-I treatment group compared with the control (25/36), agar (23/36) or
CEC-II (21/36) treatment groups. At this time, the CEC-II embryos (n=36) were injected
into the amniotic cavity of nine chick embryos (four embryos per chick amnion). These
embryos were recovered after 96 hours of culture.
On day 7 of culture, 32 of the 36 (89%) embryos originally injected on day 3 were
successfully recovered from the chick amnion. Of these, six (20%) had developed to the
blastocyst stage. This was not significantly different (P>0.05) from the total number of
blastocysts in the other treatment groups [control = 16/36 (44%); agar = 17/36 (47%);
CEC-I = 2/29 (7%)]. However, there were significantly fewer (P<0.05) total blastocysts in
the CEC-I treatment group when compared with the control or agar treatment groups
(Table 3.1). There was no significant difference (P>0.05) in the number of morulae, early
blastocysts, blastocysts or expanded blastocysts between the four treatment groups
(Figure 3.3).
In Experiment 3.2, 50 chicken eggs were initially placed in incubation. When the
eggs were prepared for shell-less culture, two (4%) eggs were unfertile, six (12%)
contained embryos that failed to develop and three (6%) contained embryos that were
nonviable. The remaining 36 eggs were placed in shell-less culture and returned to
incubation. After 24 hours of incubation in shell-less culture, 13 eggs were selected for
injection of bovine embryos.
In the CEC-I treatment group, 28 of 40 (70%) embryos were recovered from the
amniotic cavity of nine live chicks after 72 hours of culture. Of the 28 embryos
recovered, 17 (61%) appeared to be viable 6- to 8-cell embryos and seven (25%) had
developed past the 8-cell stage (Table 3.2). There were no significant differences
(P>0.05) in development between the four treatment groups. At this time, the CEC-II
37

Table 3.1. Development of 1-cell bovine embryos cultured in CR1aa + 5% fetal bovine serum (control) or co-cultured in the chick amnion from day 0 to day 3 (CEC-I) or from day 3 to day 7 (CEC-II)
a Number of 1-cell embryos allotted to each treatment. The number in parenthesis indicates the number of embryos successfully recovered from live chick embryos on day 3 (CEC-I) or day 7 (CEC-II). b The percentage of embryos developing in the chick co-culture groups (CEC-I and CEC-II) were based on the number of embryos successfully recovered from live chick embryos. c MORL = morulae. d BLST = blastocysts. e,f Within a column, numbers without a common superscript are significantly different (P<0.05).
38

0102030405060708090
100
1-cell 2-4 cell 6-8 cell cleavage
% o
f Em
bryo
s
Control Agar CEC-I CEC-IIA
a
b
a a
a
b
aa
aaaa
a
b
a a
0
10
20
30
40
50
MORL Early BLST BLST ExpandedBLST
% o
f Em
bryo
s
Control Agar CEC-I CEC-II
a
a
a
a
a
a
a
aa
a
a
a a aaa
B
Figure 3.3. Development of 1-cell bovine embryos cultured in CR1aa + 5% fetal bovine serum (control) or co-cultured in the chick amnion from day 0 to day 3 (CEC-I) or from day 3 to day 7 (CEC-II) on day 3 of development (A) and on day 7 of development (B). Within a developmental stage, columns without a common superscript are significantly different (P<0.05).
39

Table 3.2. Development of 2-cell bovine embryos cultured in CR1aa + 5% fetal bovine serum (control) or co-cultured in the chick amnion from day 0 to day 3 (CEC-I) or from day 3 to day 7 (CEC-II)
Treatment Total Grade 1 group na % 2-4 cellb % 6-8 cellb % >8 cellb % MORLbc % BLSTbd % BLSTbd
Control 39 21 ± 7e 54 ± 8e 3 ± 5e 5 ± 4e 64 ± 8e 26 ± 7e
Agar 39 21 ± 7e 62 ± 8e 15 ± 6e 5 ± 4e 38 ± 8e 23 ± 7e
CEC-I 40 (28) 7 ± 5e 61 ± 9e 25 ± 8e 7 ± 5e 42 ± 10e 31 ± 12e
CEC-II 39 (19) 23 ± 7e 46 ± 8e 21 ± 7e 11 ± 7e 37 ± 11e 16 ± 9e
Day 3 Day 7
a Number of 2-cell embryos allotted to each treatment. The number in parenthesis indicates the number of embryos successfully recovered from live chick embryos on day 3 (CEC-I) or day 7 (CEC-II). b The percentage of embryos developing in the chick co-culture groups (CEC-I and CEC-II) were based on the number of embryos successfully recovered from live chick embryos. c MORL = morulae. d BLST = blastocysts. e Within a column, numbers without a common superscript are significantly different (P<0.05).
40

embryos (n=39) were injected into the amniotic cavity of 13 chick embryos (three
embryos per chick). These embryos were recovered after 96 hours of culture.
On day 7 of culture, 19 of the 39 (49%) embryos originally injected on day 3 were
successfully recovered from the chick amnion. Of these, seven (37%) had developed to
at least the early blastocyst stage. This was not significantly different (P>0.05) from the
total number of blastocysts in the other treatment groups [control = 25/39 (64%); agar =
15/39 (38%); CEC-I = 12/28 (25%)]. There was no significant difference (P>0.05) in the
number of embryos developing to the morula or early blastocyst stage of development
between the four treatment groups (Figure 3.4). More embryos developed to the
blastocyst stage in the CEC-I group than in the CEC-II group (P<0.05). However, there
were more expanded blastocysts in the CEC-II group than in either the agar control
group or the CEC-I group (P<0.05). The number of embryos developing to the hatching
or hatched blastocyst stage did not differ (P>0.05) between treatments.
There was no significant difference (P>0.05) in the number of grade1, grade 2 or
grade 4 embryos when embryos in the four treatments were evaluated for quality (Figure
3.5). There were significantly more (P<0.05) grade 3 embryos in the control group when
compared with the agar group; however, neither group was significantly different
(P>0.05) from either chick treatment.
DISCUSSION In the early 1990s, the preferred method of culturing bovine embryos in vitro
involved the inclusion of co-culture cells in the embryo production system (Gandolfi and
Moor, 1987; Eyestone and First, 1989). In vitro embryo production systems have
progressed, and co-culture is no longer required for embryos to progress through the in
vitro developmental block and develop to the blastocyst stage. However, despite the
progress that has been made, current in vitro embryo culture conditions are unable to
mimic the in vivo conditions of the oviducts and uterus. Moreover, in vitro embryo culture
has been implicated in problems such as delayed embryo development (Van Soom et
al., 1997), reduced embryo quality (Duby et al., 1997; Boni et al., 1999; Viuff et al.,
1999), reduced pregnancy rates (Enright et al., 2000) and numerous fetal and/or calf abnormalities (Thompson et al., 1995; Walker et al., 1996; Sinclair et al., 1999). These
problems have led researchers to develop and test alternative culture methods such as perifusion culture (Lim et al., 1996; McGowan and Thompson, 1997), microfluidic
41

0102030405060708090
100
2-4 cell 6-8 cell >8-cell
% o
f Em
bryo
s
Control Agar CEC-I CEC-IIA
a a aa
a
aaa
a
a
a a
0
10
20
30
40
50
MORL Early BLST BLST ExpandedBLST
Hatching orHatched
BLST
% o
f Em
bryo
s
Control Ag a r CEC-I CEC-II
B
ab
b
a
b
a
a
aa
a a a b
ab ab
aa a
a a a
Figure 3.4. Development of 2-cell bovine embryos cultured in CR1aa + 5% fetal bovine serum (control) or co-cultured in the chick amnion from day 0 to day 3 (CEC-I) or from day 3 to day 7 (CEC-II) on day 3 of development (A) and on day 7 of development (B). Within a developmental stage, columns without a common superscript are significantly different (P<0.05).
42
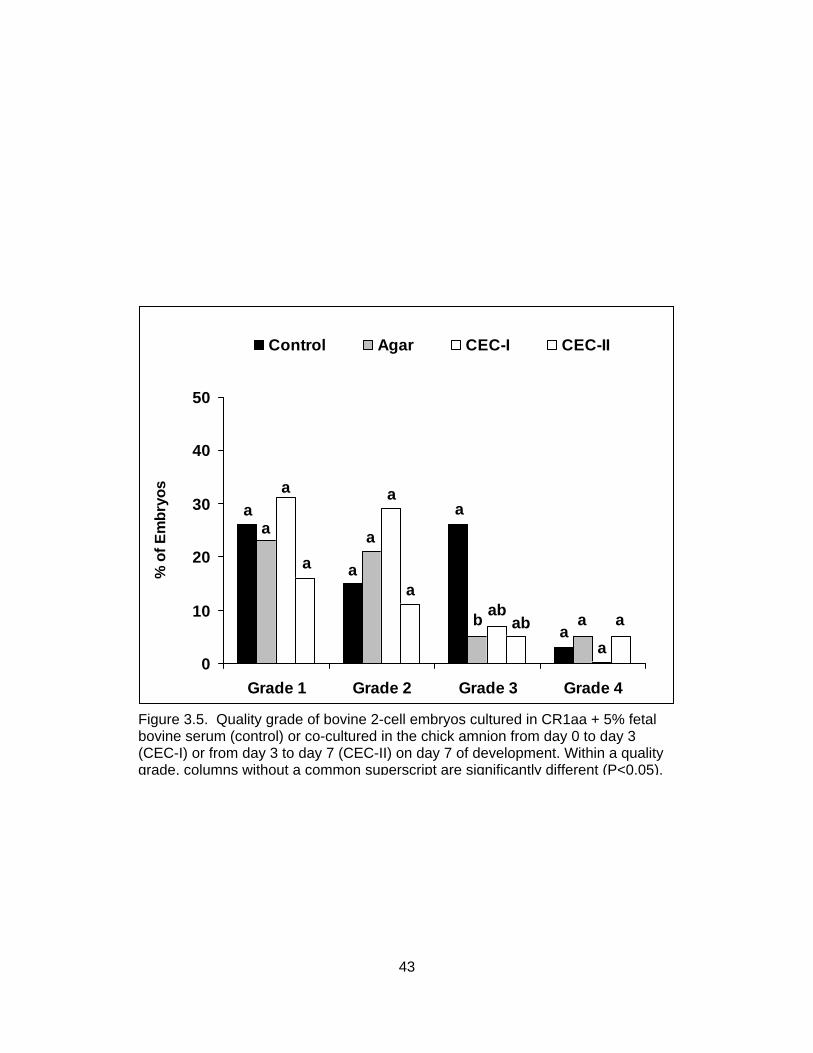
0
10
20
30
40
50
Grade 1 Grade 2 Grade 3 Grade 4
% o
f Em
bryo
s
Control Agar CEC-I CEC-II
a
a
a aab
abb
a
a
a
a
aa
a
a a
Figure 3.5. Quality grade of bovine 2-cell embryos cultured in CR1aa + 5% fetal bovine serum (control) or co-cultured in the chick amnion from day 0 to day 3 (CEC-I) or from day 3 to day 7 (CEC-II) on day 7 of development. Within a quality grade, columns without a common superscript are significantly different (P<0.05).
43

“microchannel” systems (Glasgow et al., 1998) and the “well-of-well” (WOW) method
(Vajta et al., 2000) for in vitro embryo culture.
In the early 1990s, one such alternative culture system was developed in which
mammalian embryos were cultured in the amnion of a living chicken embryo (Blakewood
and Godke, 1989). The trials and tribulations of current in vitro embryo culture and the
need to develop alternative culture environments have led us back to the chick embryo
co-culture system. Results of the present study indicate that bovine embryos can
develop in the environment of the chick amnion at rates similar to those of embryos
cultured in a semi-defined culture medium (CR1aa in the present study).
Culture of bovine embryos in the chick amnion from the 1-cell stage resulted in
significantly fewer 6- to 8-cell embryos on day 3 of development and a significantly lower
total blastocyst (early blastocyst, blastocyst and expanded blastocyst) rate on day 7. In
contrast, there was no significant difference in development on day 3 or in total
blastocysts on day 7 when chick embryo co-culture was initiated at the 2-cell stage.
These findings are interesting and may indicate that embryos are particularly sensitive to
culture environment during the first 24 hours of development. If this is in fact the case,
inadequate conditions during this time could have profound impacts on embryo
developmental potential. The low number of observations in the present study must be
taken into account. However, further investigation into the effects of culture environment
on the first 24 hours of embryo development is warranted.
In theory, the chick embryo co-culture system should provide a more
physiological “normal” environment for embryo development. Because the live chick
embryo actively regulates the pH, osmolarity and O2 content of its own environment, the
amnion should provide a more stable environment for mammalian embryo development
than current “static” in vitro culture systems (Blakewood et al., 1989a). In addition, the
chick embryo continuously synthesizes growth factors needed for its own development.
These growth factors could provide a stimulus to mammalian embryo development that
is currently unavailable in current embryo culture systems without the addition of serum
or other exogenous growth factors (Blakewood et al., 1989a).
In Experiment 3.2, there was no difference in the number of grade 1, grade 2 or
grade 4 embryos when embryos in the four treatments were evaluated for quality.
Numerically, the number of grade 3 embryos was greater in controls than in either chick
44

treatment. The lack of statistical differences is likely due to the low number of
observations in each chick treatment group. These results are promising and suggest
that chick embryo culture may in fact increase embryo quality over current culture
conditions. As stated above, the low number of observations must be taken into account,
but it is apparent that further research may be warranted.
One major drawback of the chick embryo co-culture system, however, is the
inherent mortality rate of shell-less chick embryos. Dunn and Boone (1976) reported that
chick survival rates of 90 to 95% appear to be as high as can be expected with shell-less
culture and a rapid degeneration of mammalian embryos has been reported following
chick embryo death (Blakewood and Godke, 1989). The exact cause of the decline in
mammalian embryo viability following chick embryo death is not known, but it can be
speculated that with the death of the chick comes alterations in amniotic fluid pH,
osmolarity and temperature, as well as build-up of waste products.
In conclusion, although bovine embryos can develop in the embryotropic
environment of the chick embryo amnion, the use of this novel culture system does not
appear to be advantageous for embryo development when compared with the semi-
defined culture medium, CR1aa. This approach, however, should not be ruled out.
Further research should be conducted to determine the effects of the avian amniotic fluid
on embryo quality, embryo metabolism and pregnancy and calving rates.
45

CHAPTER IV
USE OF THE AMNIOTIC CAVITY OF THE DEVELOPING CHICK EMBRYO FOR CULTURING BOVINE 8-CELL EMBRYOS DERIVED FROM SOMATIC
CELL NUCLEAR TRANSFER (NT)*
INTRODUCTION Somatic cell NT has been utilized in cattle for production of transgenic animals as
well as to produce copies of elite animals, but success rates are low (Cibelli et al., 1998;
Hill et al., 1999; Shiga et al., 1999; Wells et al., 1999; Hill et al., 2000b). It remains to be
determined whether this inefficiency is a result of the NT procedure itself or conditions in
which the embryos are cultured following the NT procedure.
Despite sustained efforts in laboratories throughout the world, present in vitro
culture conditions are unable to fully replicate in vivo conditions. In vitro culture
conditions are routinely enhanced by the addition of serum to the culture medium
(Bavister, 1995). It has been suggested that serum may contain components
advantageous for blastoceol development, such as growth factors or extracellular matrix
components like fibronectin (Pinyopummintr and Bavister, 1994). However, it is
hypothesized that the components of serum may also contribute to fetal and calf
abnormalities (Thompson et al., 1995; Walker et al., 1996; Sinclair et al., 1999).
Calves produced from embryos cultured in vitro often exhibit increased birth
weight (Willadsen et al., 1991; Keefer et al., 1994; Behboodi et al., 1995), longer
gestation lengths (Walker et al., 1996), increased placental anomalies (Hasler et al.,
1995; Hill et al., 1999, 2001, 2002) and increased incidences of abortion (Walker et al.,
1996; Hill et al., 1999) and perinatal (Behboodi et al., 1995; Wilson et al., 1995; Garry et
al., 1996; Schmidt et al., 1996; Kruip and den Daas, 1997; Hill et al., 1999) and neonatal
mortality (Schmidt et al., 1996; Kruip and den Daas, 1997; Hill et al., 1999).
Furthermore, bovine viral diarrhea virus (BVDV) may be introduced into in vitro
production systems via the addition of serum, extracted components of serum or somatic
cell lines (Stringfellow et al., 2000). BVDV is a ubiquitous pathogen that can cause
abortion resulting in severe economic losses, and undetected association of BVDV in
* Used with permission of Molecular Reproduction and Development, Wiley-Liss,Inc.,
Hoboken, NJ
46

transferred in vitro-produced embryos may result in early embryonic death or abortion.
Shin et al. (2000) have reported that the rate of pregnancy loss between 30 and
40 days of gestation was significantly greater in bovine fetuses derived from BVDV-
infected somatic cells than in those from noninfected somatic cell lines. Total elimination
of materials of bovine origin, including serum and serum albumin, from the standard
oocyte maturation and embryo or somatic cell culture systems could reduce the risk of
virus introduction.
Previous reports indicate that factors within the amniotic cavity of developing
chick embryos are capable of supporting the maturation of porcine oocytes (Ocampo et
al., 1994) and development of embryos in several mammalian species. In these studies,
researchers found significantly more expanded and hatched blastocysts compared with
controls in mice (control = Whitten's medium), goats (control = Ham's F-10), cattle
(control = Ham's F-10; Blakewood et al., 1989a, 1989b, 1990) and pigs (control =
mBMOC + POEC) (Ocampo et al., 1993). Unfortunately, most of these studies used a
relatively low number of observations per treatment group (mean of 40 observations per
treatment across studies). In addition, the media used in these studies are now
considered somewhat obsolete by today’s standards. Even with these drawbacks, this
technique warrants further investigation as an alternative in vitro embryo culture system
that is free from materials of bovine origin for the culture of in vitro-produced mammalian
embryos.
In the present study, the development of bovine 8-cell embryos produced by
somatic cell NT was assessed after culture in the chick embryo amnion. The viability of
bovine embryos developing into morulae or blastocysts after culture in the chick amnion
was further investigated by transferring a representative sample of these embryos into
recipient females. This study was conducted as a preliminary trial in evaluating the use
of this unique co-culture system as an alternative to present in vitro embryo culture
systems used for the production of somatic cell NT calves.
MATERIALS AND METHODS Experimental Design
The design for this experiment is illustrated in Figure 4.1. After reconstruction
(day=0), all embryos were cultured in vitro in CR1aa medium for 3 days. Viable 8-cell
embryos were then randomly allotted to one of two treatment groups for in vitro culture.
47

Onset of Egg Incubation
72 hours Nuclear Transfer
In Vitro Culture in CR1aa
72 hours Preparation of Shell-less Cultures
Selection of
8-cell Embryos
24 hours
Injection of 8-cell Embryos into Chick Amnion
Culture in CR1aa + 5% FBS
72 hours
72 hours
Evaluation Recovery Evaluation
Embryo Transfer
Figure 4.1. Experimental design for the culture of bovine 8-cell embryos derived from somatic cell nuclear transfer. Culture was either in vitro in CR1aa supplemented with 5% fetal bovine serum (control) or co-cultured in the chick embryo amnion.
48

Embryos allotted to Treatment A (control) were cultured in CR1aa supplemented with 5%
fetal bovine serum (FBS) from day 3 to day 6 post-reconstruction. Embryos allotted to
Treatment B were cultured in the amniotic cavity of 96-hour chick embryos from day 3 to
day 6 post-reconstruction. Embryos that had reached the morula and blastocyst stages
after culture in the chick embryo amnion on day 6 of culture were transferred to recipient
females.
Experimental Methods Media Preparation
CR1aa medium was prepared according to Rosenkrans and First (1994). First,
CR1aa stock solution was prepared by adding 114.7 mM NaCl (S-5886, Sigma-Aldrich
Inc., St. Louis, MO), 1.6 mM KCl (P-5405, Sigma-Aldrich Inc. St. Louis, MO), 26.2 mM
NaHCO3 (S-8875, Sigma-Aldrich Inc., St. Louis, MO), 0.3999 mM pyruvic acid (P-4562,
Sigma-Aldrich Inc., St. Louis, MO), 2.5 mM L(+) lactic acid (L-4388, Sigma-Aldrich Inc.,
St. Louis, MO), 0.5195 mM glycine (G-8790, Sigma-Aldrich Inc., St. Louis, MO), 0.5051
mM L-alanine (A-7469, Sigma-Aldrich Inc., St. Louis, MO) and 0.2 ml of 0.5% phenol red
per 100 ml of sterile water.
For culture from day 0 to day 3, CR1aa culture medium was prepared by adding
0.2 ml of Basal Medium Eagle (BME) amino acid solution (50X, B-6766, Sigma-Aldrich
Inc., St. Louis, MO), 0.1 ml of Modified Eagle Medium (MEM) amino acid solution (10X,
11140-050, Gibco Laboratories, Grand Island, NY), 0.01 ml of gentamicin, 1.1 mM L-
glutamine (G-5763, Sigma-Aldrich Inc., St. Louis, MO) and 0.03 g of bovine serum
albumin (BSA; fatty acid free, A-7511, Sigma-Aldrich Inc., St. Louis, MO) to 9.5 ml of
CR1aa stock solution. For culture from day 3 to day 7, CR1aa culture medium was
prepared by adding 0.2 ml of BME amino acid solution, 0.1 ml of MEM amino acid
solution, 0.5 ml of FBS (SH30072.02; HyClone, Logan, UT), 0.01 ml of gentamicin, 1.1
mM L-glutamine and 0.03 g of BSA (fatty acid free) to 9.5 ml of CR1aa stock solution.
Preparation of Adult Bovine Donor Cells
The donor cells used for the NT procedure were established from skin biopsy
samples of a 3-year-old Brahman cow. Briefly, skin biopsies were obtained from the
dorsal aspect of the shoulder directly over the supraspinatus muscle using a 6-mm
diameter biopsy punch (Miltex Instrument Co, Betpage, NY). Using heat sterilized iris
scissors, the dermal portion of the biopsy was finely minced into pieces measuring
49

~1 mm2. Culture medium (mDMEM) (Cibelli et al., 1998; Shiga et al., 1999) consisting of
Dulbeco’s Modified Eagle Medium (DMEM; Gibco Laboratories, Grand Island, NY), 10%
FBS and 1 µl/ml of gentamicin was added and the tissue suspension transferred to a 15-
ml conical tube (Corning, New York, NY). The tissue suspension was washed by swirling
and the pieces of tissue were allowed to settle to the bottom of the conical tube. The
supernatant was discarded and 3 ml of fresh mDMEM was added. This washing process
was repeated two additional times. After the last wash, the tissue was resuspended in 3
ml of mDMEM and transferred to a 35 x 10-mm Falcon® plastic petri dish (Becton and
Dickinson, Lincoln Park, NJ). All cultures were maintained at 39°C and 5% CO2 in a
humidified atmosphere.
Once a fibroblast cell layer was established, the cells were maintained in 25-cm2
tissue culture flasks (CostarTM, Cambridge, MA) until they reached ~95% confluency.
Cells were then trypsinized (Trypsin EDTA, 1x; T3924, Sigma-Aldrich Inc., St. Louis,
MO), counted on a hemocytometer and reseeded at a density of 100,000 cells per 25-
cm2 tissue culture flask. The cells were passaged twice prior to cryopreservation in
DMEM supplemented with 20% FBS and 5% dimethylsulfoxide (DMSO; D128-500,
Fischer Scientific, Fairlawn NJ) and then frozen in aliquots of 500,000 cells per cryovial.
To prepare adult fibroblasts as donors for NT, cells were thawed and cultured in mDMEM
in Nunc® 4-well plates (Apogent Technologies, Rochester, NY).
Nuclear Transfer
Oocytes were obtained on a weekly basis from a commercial source (BoMed,
Madison, WI). Oocytes were matured in a portable incubator (~38.5oC) during the
overnight travel interval for 20 to 22 hours. After arrival, the cumulus cells were removed
from the oocytes by vortexing in 1 ml of TCM-199 (Eagle’s salt) with 25 mM HEPES
buffer (TCM-199, Gibco, Grand Island, NY) containing 0.001 g of hyaluronidase (H-3506,
Sigma-Aldrich Inc., St. Lois, MO) for 3 minutes. The denuded oocytes were washed
twice in modified TCM (mTCM: 19 ml of TCM-199 supplemented with 1 ml of FBS and
0.02 ml of gentamicin) and were selected for the presence of a first polar body.
Micromanipulation was performed in a 200-µl drop of manipulation medium [4.0
ml of Dulbecco’s Phosphate Buffered Saline (DPBS; D-1408, Sigma-Aldrich Inc., St.
Louis, MO) containing 1 ml of FBS, 0.005 ml of gentamicin and 0.005 ml of cytochalasin
D (C-8273, Sigma-Aldrich Inc., St. Louis, MO)] overlaid with warmed mineral oil (M-8140,
50

Sigma-Aldrich Inc., St. Louis, Mo). All manipulations were conducted on an inverted
microscope (Nikon Diphot, Tokyo Japan) equipped with a pair of Lietz
micromanipulators.
The NT procedure was modified from that previously described by Shiga et al.
(1999). Briefly, oocytes were held in place by negative pressure applied to a holding
pipette (120 µm outside diameter). After brief exposure (<10 seconds) to ultraviolet light,
the first polar body and adjacent cytoplasm containing the nuclear material, as visualized
by Hoechst-33342 (5 µg/ml; bisBenzimide; B-2261; Sigma-Aldrich Inc., St. Lois, MO)
staining, were manipulated out of a rent in the zona pellucida using a flexible glass
needle. A pipette (25 µm outer diameter) containing the donor cell (passages 2 to 8) was
introduced through the rent in the zona pellucida made during enucleation, and the cell
was wedged between the zona and vitelline membrane to ensure close membrane
contact required for subsequent fusion.
The reconstructed cytoplast-karyoplast couplets were induced to fuse in a
microslide fusion chamber with stainless steel electrodes separated by a 1-mm gap. The
chamber was filled with fusion buffer consisting of 0.3 M mannitol (D-3651, Sigma-
Aldrich Inc., St. Louis, MO), 0.05 mM CaCl2·2H2O (C-7902, Sigma-Aldrich Inc., St. Louis,
MO) and 0.1 mM MgSO4 (M-7774, Sigma-Aldrich Inc., St. Louis, MO) and the couplets
were fused with two direct current pulses of 2.25 kV/cm for 15 microsecconds, delivered
by a BTX Electrocell Manipulator 200 unit (Genetronics, San Diego, CA). The fused
couplets were further activated with 10 µg/ml of cyclohexamide (C-7698, Sigma-Aldrich
Inc., St. Louis, MO) in CR1aa medium for 4 hours.
After activation, embryos were cultured in 50-µl drops of CR1aa medium in a
modular incubator containing 5% CO2, 5% O2 and 90% N2 (activation = day 0). On day 3
of culture, good quality (as indicated by equal size, well defined blastomeres), 8-cell NT
embryos were randomly selected for in vitro culture in CR1aa supplemented with 5%
FBS (control group) or co-culture in the amniotic cavity of a developing chick embryo as
described below.
Chick Embryo Co-Culture
A technique modified from that originally described by Blakewood and Godke
(1989) was used for the shell-less culture of chick embryos. Fertilized domestic chicken
eggs were collected on the day of oviposition and stored at 10°C until the onset of
51

incubation. Four days prior to introduction of bovine embryos into the chick embryo
amnion the eggs were removed from cold storage and allowed to come to room
temperature (25°C). They were then placed in a 37°C commercial egg incubator. After
72 hours of incubation, the eggs were rinsed with a solution of 70% ethanol (Aaper
Alcohol and Chemical Company, Shelbyville, KY) and then coated with 7.5% providone
iodine (Betadine®, Purdue Fredrick Company, Stanford, CT) and allowed to air dry in a
horizontal position to ensure proper positioning of the chick embryo.
To remove the egg contents, the egg shell was cracked against the rim of a
sterile beaker and the egg contents were released into a 100-ml plastic embryo
collection dish (Veterinary Concepts, Spring Valley, WI) covered by a piece of cellophane
kitchen wrap (Saran®, Dow Chemical, Midland, MI). The dish, including the egg contents,
was loosely capped with a plastic lid (Veterinary Concepts, Spring Valley, WI) and
maintained in an incubator at 38°C for an additional 24 hours before introducing the
bovine embryos into the amniotic cavity of the chick embryo. Prior to embedding and
injection of bovine embryos, suitable chick embryos were selected for injection based on
the following criteria: 1) round intact yolk, 2) good vascularity in the chorioallantoic
membrane, 3) vigorous heartbeat and 4) no apparent abnormalities.
A technique modified from that originally described by Willadsen (1979) was used
for embedding embryos in agarose. On day 3 of culture, two or three 8-cell bovine NT
embryos were placed in a 50-µl drop of low-melting agarose solution [1.2% agarose (A-
9799; Sigma-Aldrich Inc., St. Louis, MO) dissolved in DPBS] at ~37oC. The embryos
were then aspirated into a beveled glass pipette (200 µm outer diameter). After holding
in air for ~30 seconds, the agar (containing the embryos) within the pipette was expelled
as a gel cylinder into room temperature TL-HEPES (BioWittaker, Walkersville, MD). The
agarose cylinder, including the bovine embryos, was then aspirated back into the
beveled pipette, and carefully expelled into the amniotic cavity of a 96-hour chick
embryo.
After introducing the embedded embryos into the amniotic cavity the shell-less
culture systems were returned to incubation for an additional 72 hours at 39oC. Thus, a
single agarose cylinder containing two or three 8-cell bovine NT embryos was co-
cultured in the amnion of each developing chick embryo for 3 days.
52

On day 6 of in vitro culture, the amnion containing the agarose cylinder was
dissected away from the rest of the egg components, placed in a 35 x 10-mm plastic
petri dish and rinsed several times with DPBS containing 1% FBS. Under a
stereomicroscope (Nikon SMZ-2B, Tokyo, Japan), the agarose cylinder was removed
from the amniotic cavity by using two 22-gauge, 37.5 mm needles (Kendall, Mansfield,
MA) to make a small opening in the amniotic membrane. The agarose cylinder was
carried from the amnion with the escaping amniotic fluid and allowed to settle on the
bottom of the dish. The cylinder was then washed twice in CR1aa medium and the
developing embryos were dissected out of the agar using two 22-gauge needles.
Embryos that had reached the morula and blastocyst stages after culture in the
chick embryo amnion (day 6) were nonsurgically transferred to naturally cycling beef
females (age = 2 to 10 years; BCS = 5 to 7) on days 6 to 7 of their estrous cycle (estrus
= day 0) when recipients were available. Pregnancy status of recipient females was
diagnosed by ultrasonography (Aloka, SSD-500V, Japan) on day 30 of gestation.
Statistical Analysis Data are expressed as means and were analyzed using PROC GLM of the SAS
Statistical Package (v. 8e, 1999, SAS Institute, Inc., Gary, NC). Significant differences
between means for embryo development rates in each group were tested using a post
hoc, Fisher’s protected least significant difference test (PLSD test). Differences with a
probability (P) value of 0.05 or less were considered significant in this study.
RESULTS For preparation of shell-less chick embryos, 210 chicken eggs were initially
placed in incubation. After 72 hours of incubation, the eggs were removed from their
shell and placed in shell-less culture. At this time, 25 (12%) of the eggs were found to be
unfertilized, 21 (10%) contained embryos that failed to fully develop and 11 (5%)
contained an embryo that was nonviable. The remaining 153 eggs were returned to
incubation. After 24 hours of incubation in shell-less culture, 39 eggs were selected for
injection of bovine embryos.
Of 485 cytoplast-karyoplast couplets reconstructed, 387 (79.8%) successfully
fused during the NT procedure. Of the 372 viable fused embryos, 232 (62.4%)
developed to the 8-cell stage by day 3 of culture in CR1aa; 12 of these embryos were
discarded due to poor quality. Of the 220 8-cell embryos remaining, 113 embryos were
53

randomly allotted for culture in CR1aa + 5% FBS (control). The remaining 107 8-cell
embryos were embedded in agar and injected into the amniotic cavity of chick embryos.
Of the embryos injected, 54 (50.5%) were successfully recovered for evaluation. Embryo
loss occurred either during the manual recovery procedure or was due to chick embryo
death. Ten of the 39 (25.6%) chick embryos originally injected did not survive shell-less
incubation, accounting for the majority of loss of the bovine embryos.
Developmental rates for bovine 8-cell embryos co-cultured in the chick-embryo
amnion or cultured in CR1aa medium containing 5% FBS (control) are summarized in
Table 4.1. There was no effect (P>0.05) of culture system on the percentage of 8-cell
embryos that developed to the morula (control = 54.2% and chick embryo co-culture =
42.6%) or blastocyst (control = 19.8% and chick embryo co-culture = 17.2%) stage on
day 6 of culture. A total of 21 morula to blastocyst stage embryos from the chick embryo
co-culture group (Figure 4.2) were transferred nonsurgically to 7 synchronized recipient
females (2 to 4 embryos per female) on day 6 or day 7 of their estrous cycle (estrus =
day 0) corresponding to day 6 of culture. Of those recipients, three females (42.9%)
were diagnosed (via ultrasonography and visualization of fetal heartbeats) pregnant with
singletons on day 30 of gestation (Figure 4.3). By day 120 of gestation, however, the
recipients had lost their pregnancies.
DISCUSSION Previous reports have shown that an embryo co-culture system using domestic chicken
egg shell-less culture is capable of supporting the development of pre-hatching
mammalian embryos (Blakewood and Godke, 1989; Blakewood et al., 1989a, 1989b),
and live transplant offspring have been produced following chick embryo co-culture of 2-
to 8-cell caprine embryos. (Blakewood et al.,1990). Results of the present study indicate
that the chick embryo amnion is also capable of supporting development of bovine 8-cell
embryos derived from somatic cell NT at rates comparable with embryos cultured in vitro
in CR1aa medium containing 5% FBS.
Supplementation of bovine embryo culture medium with serum from the initiation
of compaction, as performed here in the control group, is widely practiced to stimulate
blastoceol development, despite the undefined and variable nature of its components
(Pinyopummintr and Bavister, 1994; Thompson et al., 1998). However, addition of serum
has been implicated in several fetal and/or calf abnormalities including increased birth
54
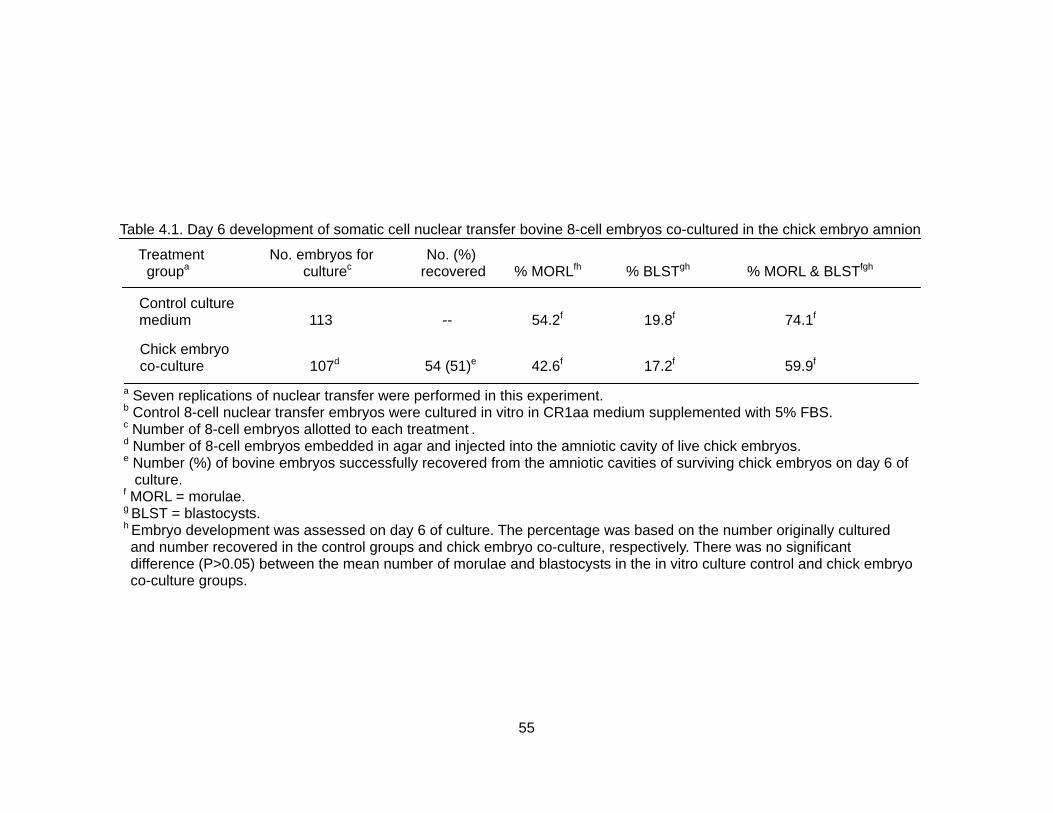
Table 4.1. Day 6 development of somatic cell nuclear transfer bovine 8-cell embryos co-cultured in the chick embryo amnion
Treatment No. embryos for No. (%) groupa culturec recovered % MORLfh % BLSTgh % MORL & BLSTfgh
a Seven replications of nuclear transfer were performed in this experiment. b Control 8-cell nuclear transfer embryos were cultured in vitro in CR1aa medium supplemented with 5% FBS. c Number of 8-cell embryos allotted to each treatment . d Number of 8-cell embryos embedded in agar and injected into the amniotic cavity of live chick embryos. e Number (%) of bovine embryos successfully recovered from the amniotic cavities of surviving chick embryos on day 6 of culture. f MORL = morulae. g BLST = blastocysts. h Embryo development was assessed on day 6 of culture. The percentage was based on the number originally cultured and number recovered in the control groups and chick embryo co-culture, respectively. There was no significant difference (P>0.05) between the mean number of morulae and blastocysts in the in vitro culture control and chick embryo
Chick embryo co-culture 107d 54 (51)e 42.6f 17.2f 59.9f
Control culture medium 113 -- 54.2f 19.8f 74.1f
co-culture groups.
55

Figure 4.2. Bovine somatic cell nuclear transfer embryos developed into morulae/blastocysts (day 6 of in vitro culture) after co-culture in the amniotic cavity of a developing chick embryo for 3 days. These embryos were agarose embedded (agar cylinders are indicated by arrows) at the 8-cell stage following in vitro culture in CR1aa medium from day 0 to day 3 of in vitro culture and then injected into the amnion of a 96-hour live chick embryo. Figure 4.3. Ultrasound image of a fetus (day 30) derived from bovine somatic cell nuclear transfer embryos that were co-cultured in the amnion of a developing chick embryo from day 3 to day 6 following in vitro culture in CR1aa medium.
56

weight and abortion of NT-derived conceptuses (Walker et al., 1996). Also, Stringfellow
et al. (2000) indicated that materials of bovine origin, including serum and extracted
components of serum, used in bovine in vitro embryo production systems could possibly
originate from cattle that were exposed to BVDV. The BVDV infection has been
implicated as a cause of early embryonic death or abortion in cattle (Shin et al., 2000).
Although the embryotropic properties of avian amniotic fluid remain largely
undefined, amniotic fluid may serve as a beneficial embryo culture medium. Because the
living chick embryo actively regulates the pH, osmolarity and O2 content of its own
environment, it may serve as a more stable physiological system for mammalian embryo
development than current in vitro culture systems (Blakewood et al., 1989a). In addition,
the chick embryo actively synthesizes growth factors and other materials that may
stimulate growth and development of mammalian embryos, a benefit unavailable in
current embryo culture systems without supplementation of serum or bovine serum
albumin (Blakewood et al., 1989a).
The use of the chick embryo amnion for culturing bovine NT embryos may also
be a way of circumventing potential risks associated with present bovine in vitro embryo
culture conditions. In particular, the risk of transmitting BVDV or other pathogens into
reconstructed embryos may be lowered when chick amniotic fluid is used for donor cell
culture, as well as for both oocyte maturation and embryo culture.
In the present study, 42.9% of the recipient females that received NT embryos
co-cultured in the chick embryo amnion were pregnant on day 30 post estrus. This
percentage was similar to contemporary day-30 pregnancy data for NT embryos cultured
in vitro (in CR1aa medium for the first 72 hours and in CR1aa medium plus 5% FBS for
the second 72 hours) using the same methods described herein (8/21 for 38.1%,
unpublished data). All three recipient females lost their pregnancies prior to 120 days of
gestation in the present study, while 37.5% (3/8) of the recipients carrying NT-derived
fetuses remained pregnant in a corresponding study at this laboratory.
It is widely acknowledged that NT in cattle results in increased rates of abortion
throughout pregnancy, particularly during the first trimester. Since the timing of early fetal
loss coincides with the timing of placentome formation in normal conceptuses, failure of
placental development has been considered a contributing factor in pregnancy loss for
NT embryos derived from embryonic cell lines (Stice et al., 1996). The effects appear
57

more extreme with somatic cell NT (Hill et al., 2000b) and may relate to a deficiency or a
combination of deficiencies either in the NT procedure itself, leading to incomplete
nuclear reprogramming of the cultured donor cells, or in the in vitro maturation and/or
embryo culture systems used (Wells et al., 1999). The lack of placentome development
could be the result of diminished or aberrant production of key regulatory hormones,
such as insulin-like growth factors (IGFs), or abnormalities in maternal-placental cell
communication (Hill et al., 2000a).
It has been postulated that these anomalies, including fetal overgrowth, may
reflect epigenetic changes, especially in the patterns of methylation, resulting from
cloning and/or culture conditions (Niemann and Wrenzycki, 2000; Niemann et al., 2002;
Young et al., 2001). Niemann and Wrenzycki (2000) have shown, in their studies of
mRNA expression in viable in vitro-produced bovine embryos, that extended exposure to
in vitro conditions leads to significant divergence from normal gene expression patterns.
They suggested that alterations of gene expression during the pre-implantation period
may affect fetal development of bovine embryos and could be relevant to the large
offspring phenomenon. These alterations may be an attempt by the embryo to
compensate for suboptimal conditions and further investigation of current in vitro embryo
production systems is needed to determine the cause(s).
One drawback of the chick embryo co-culture system is the embryo loss that
occurs when the chick embryo dies. This death might contribute to a decrease in overall
viable embryo yield, unless this co-culture system would produce embryos that were
more viable at transfer and subsequently produce more live offspring than a standard
culture system.
In conclusion, results in the present study suggest that somatic cell NT bovine
8-cell embryos co-cultured in the chick embryo amnion are capable of developing into
transferable stage embryos with a pregnancy rate comparable with embryos cultured in
vitro in serum-supplemented medium. However, the problem of early pregnancy loss has
not been ameliorated at this time. Several factors must be considered when determining
the efficacy of this procedure for production of live offspring. First, embryos were only
transferred when recipients were available, resulting in a low number of embryos
transferred in this study. In addition, these transfers occurred at the end of May through
July, 2002, a suboptimal time of year due to climate at this location. Further studies,
58

such as the use of the chick embryo co-culture system for both oocyte maturation and
embryo culture, as well as transfer of additional embryos, must be conducted to
determine the potential benefits of this culture system in bovine NT.
59

CHAPTER V
THE EFFECT OF ALTERATIONS IN THE NORMAL ANGIOGENIC PATTERN OF THE CHICK CHORIOALLANTOIC MEMBRANE ON BOVINE EMBRYO
DEVELOPMENT AFTER CHICK EMBRYO CO-CULTURE
INTRODUCTION In vitro embryo production techniques are continuously being reinvestigated and
reinvented (Wheeler et al., 2004). Despite much progress, however, in vitro-produced
embryos still lag behind their in vivo-derived counterparts with respect to morphology
(Greve et al., 1995), timing of development (Van Soom et al., 1997), embryonic
metabolism (Khurana and Niemann, 2000) and pregnancy rates (Enright et al., 2000). In
addition, a significant percentage of offspring resulting from embryos produced in vitro
have been reported to exhibit distinct developmental abnormalities, known collectively as
large calf or large offspring syndrome (Behboodi et al., 1995; Farin and Farin, 1995;
Sinclair et al., 1995; Walker et al., 1996; Kruip and den Daas, 1997; McEvoy et al., 1998;
van Wagtendonk-de Leeuw et al., 1998, 2000). Abnormalities associated with large
offspring syndrome include increased birth weight (Behboodi et al., 1995; Sinclair et al.,
1995; van Wagtendonk-de Leeuw et al., 1998, Jacobsen et al., 2000; van Wagtendonk-
de Leeuw et al., 2000), hydroallantois (Hasler et al., 1995; van Wagtendonk-de Leeuw et
al., 1998, 2000), placental abnormalities (Hasler et al., 1995; Farin and Farin, 1995) and
perinatal and neonatal mortality (Behboodi et al., 1995; Schmidt et al., 1996).
The inadequacies of current in vitro culture conditions have led researchers to
develop alternative culture systems such as perifusion culture (Lim et al., 1996;
McGowan and Thompson, 1997), microfluidic “microchannel” systems (Glasgow et al.,
1998) and the “well-of-well” (WOW) method (Vajta et al., 2000) for culturing embryos in
vitro. Previously, a culture system was developed in which mammalian embryos could
be cultured in the amnion of a developing chick embryo. In early studies, researchers
found that chick embryo co-culture resulted in significantly more expanded and hatched
blastocysts compared with controls in mice (Blakewood et al., 1989a), goats (Blakewood
et al., 1989a) and pigs (Ocampo et al., 1993). More recent research indicates that
bovine embryos cultured in the chick amnion develop into blastocysts at rates similar to
those of embryos cultured in a semi-defined medium (Davidson et al., unpublished data).
It is believed that the chick amnion may serve as a more physiologically “normal” culture
60

environment compared with current “static” in vitro culture systems. Because of this the
chick embryo culture system should not be abandoned without further investigation.
Therefore the present study was conducted in an effort to improve the chick
embryo co-culture system. It is hypothesized that by increasing blood flow to the chick
embryo, chick embryo viability and growth factor production may be improved, thus
potentially improving development of the bovine embryos contained within the amnion.
Thus, the purpose of the first experiment was to determine if normal blood vessel
patterns could be altered with angiogenic compounds. To accomplish this, angiogenesis
was examined in chick chorioallantoic membranes (CAM) treated with prostaglandin E2
(PGE2) and prostaglandin F2α (PGF2α). Although many angiogenic compounds exist,
PGE2 and PGF2α were chosen for this experiment because they are similar in chemical
composition, but have very opposite physiological actions. Specifically, PGE2 is a
vasodialator involved in maintenance of the corpus luteum (CL) and implantation. PGF2α,
in contrast, is a vasoconstrictor involved in regression of the CL and parturition. Based
on the findings of the first experiment, a second experiment was conducted to determine
if PGE2 treatment prior to or during bovine embryo co-culture could improve bovine
embryo development over traditional culture techniques.
MATERIALS AND METHODS Experimental Design The design for Experiment 5.1 is illustrated in Figure 5.1. Fertilized chicken eggs
were obtained from a commercial supplier and incubated at 37°C and 85 to 90% relative
humidity. On day 4 of incubation, eggs were prepared for treatment by creating a
window in the egg shell. The window was sealed with transparent tape, and the eggs
were returned to incubation for 24 hours. On day 5, eggs allotted to Treatment A
(control) were treated with chick ringers solution, eggs allotted to Treatment B (EtOH)
were treated with ethanol (vehicle), eggs allotted to Treatment C (cytochalasin) were
treated with cytochalasin-D (negative control), eggs allotted to Treatment D (PGE) were
treated with PGE2 and eggs allotted to Treatment E (PGF) were treated with PGF2α. All
eggs were then returned to incubation. On day 6, CAMs were evaluated for treatment
effects.
The design for Experiment 5.2 is illustrated in Figure 5.2. Presumptive zygotes
were purchased from a commercial supplier and upon arrival were placed in CR1aa for
61

EXPERIMENTAL DESIGN
Prepare eggs for assay
by removing albumin and creating a window
Return to incubation
Apply treatments and return to incubation
Incubate eggs at 37°C and 85 to 90% relative humidity for 96 hours
Quantify results
0 1 2 3 4 5 6
Time (Days) Figure 5.1. Experimental design to evaluate the effects of PGE2 and PGF2α on capillary development in the chick chorioallantoic membrane.
62

48 hours. On day 3 post-insemination, good quality 8-cell embryos were randomly
allotted to one of three treatment groups. Embryos allotted to Treatment A (control) were
cultured in CR1aa supplemented with 5% fetal bovine serum (FBS) from day 3 to day 7
post-insemination. Embryos allotted to Treatment B (CEC) were embedded in agar and
injected into the amnion cavity of 96-hour chick embryos. After 96 hours of culture, the
bovine embryos were recovered from the chick amnion and evaluated. Embryos allotted
to Treatment C (PGE) were embedded in agar and injected into the amniotic cavity of
96-hour chick embryos that had been treated with PGE2. After 96 hours of culture, the
bovine embryos were recovered from the chick amnion and evaluated.
Experimental Methods Evaluation of the Effects of Prostaglandins on Angiogenesis
Media Preparation
Chick Lactated Ringers solution was prepared by addition of 123.2 mM NaCl (S-
5886, Sigma-Aldrich Inc., St. Louis, MO), 1.6 mM CaCl2·2H2O (C-7902 Sigma-Aldrich
Inc., St. Louis, MO), 2.5 mM KCl (P-5404, Sigma-Aldrich Inc., St. Louis, MO), 10 mM
HEPES (49329, Sigma-Aldrich Inc., St. Louis, MO) per 1 L of distilled water. The pH was
then adjusted to 7.4.
CAM Assay
Angiogenesis was evaluated using the CAM assay modified from that previously
described by Martins-Green and Feugate (1998). Fertilized chicken eggs were collected
on the day of oviposition and stored at 10°C until incubation. Incubations were
conducted at 37°C and 85 to 90% relative humidity throughout the duration of the
experiment. On the fourth day of incubation, 3 to 4 ml of albumin was aspirated from the
pole adjacent to the air pocket using an 18-gauge, 37.5-mm needle (Kendall, Mansfield,
MA) and 3-ml disposable syringe (Sherwood, Davis and Geck, St. Louis, MO) and
sealed with transparent tape (Gould QUIKSTIK; Gould Packaging, Inc. Vancouver, WA).
This was done to allow the embryo to drop away from the egg shell and to allow the
CAM to develop in a way that is accessible to treatments. A window (~25 mm in
diameter) was opened in each egg to allow subsequent access to the CAM and then
sealed with transparent tape. On day 5, 0.2 ml of chick ringers (control), 100% EtOH
(vehicle), cytochalasin D (negative control; 0.99 µM; C-8273, Sigma-Aldrich Inc., St.
Louis, MO), PGE2 (2 µM; P-4172, Sigma-Aldrich Inc., St. Louis, MO) or PGF2α (2 µM;
63

Control Group
Shipment of Presumptive Media Change
Zygotes
64
Onset of Egg Incubation
IVF IVC
Injection of Bovine Embryos
Recovery Evaluation
Injection of Bovine Embryos
Recovery Evaluation
PGE Treatment Group
CEC Treatment Group Shipment of Presumptive
Zygotes
Preparation of Shell-less Cultures Application of PGE2
Preparation of Shell-less Cultures
Onset of E g Incubation g
~24 h 96 h18 h 24 h 24 h
Shipment of Presumptive
Zygotes
IVF IVC
~24 h 96 h18 h 48 h
IVC Addition of FBS Evaluation IVF
Figure 5.2. Experimental design for the culture of 8-cell bovine embryos cultured in CR1aa + 5% fetal bovine serum (control), co-cultured in the chick embryo amnion (CEC) or co-cultured in the amnion of PGE2-treated chicks (PGE).

P-0424, Sigma-Aldrich Inc., St. Louis, MO) were applied to the surface of each CAM by
inserting a 22-gauge, 37.5-mm needle (Kendall, Mansfield, MO) through the transparent
tape and delivering treatment solutions in a back and forth motion to ensure coverage of
the entire CAM. On day 6, CAMs were prepared for evaluation as described below.
Quantification of CAM Results
Control and experimental CAMs were prepared for quantification by removing an
additional 3 to 4 ml of albumin as described above and enlarging the window to expose
as much of the CAM as possible. Digital images of each CAM were taken with an
Olympus C-2100 digital camera from a distance of 31.3 cm with the addition of close-up
lenses with diopters of +1, +2 and +4 for macro-imaging.
All digital images were processed using Adobe Photoshop (v 6.0). Degree of
capillary branching was quantified by placing a 256 pixel x 256 pixel box on each CAM
image at random. The chosen area was then pasted into a new blank image and
processed using the brightness/contrast option to contrast enhance the blood vessels.
The nodes or branch points were then counted. This process was repeated three
additional times to obtain node counts for a total of four random areas of each CAM.
Data from the four boxes were combined to produce a mean number of nodes for each
CAM.
Production of In Vitro Embryos
Media Preparation
Brackett-Oliphant (BO) media were prepared according to Brackett and Oliphant
(1975). Briefly, BO-A stock solutions were prepared by addition of 150.2 mM NaCL (S-
5886, Sigma-Aldrich Inc., St. Louis, MO), 2.7 mM KCl (P-5405, Sigma-Aldrich Inc., St.
Louis, MO), 3.0 mM CaCl2·2H2O (C-7902, Sigma-Aldrich Inc., St. Louis, MO), 1.0 mM
NaH2PO4·H2O (S-9638, Sigma-Aldrich Inc., St. Louis, MO), 0.6857 mM MgCl2·6H2O (M-
2393, Sigma-Aldrich Inc., St. Louis, MO) and 0.1 ml of 0.5% phenol red (P-0290, Sigma-
Aldrich Inc., St. Louis, MO) per 500 ml of sterile water. BO-B stock solutions were
prepared by addition of 154.0 mM NaHCO3 (S-8875, Sigma-Aldrich Inc., St. Louis, MO)
and 0.04 ml of 0.5% phenol red per 200 ml of sterile water.
Fertilization stock medium (BO-AB) was prepared from the stock solutions by
adding 1.2 mM pyruvic acid, 0.05 ml of gentamicin (50 mg/ml; 15750-060, Gibco, Grand
Island, NY) and 0.018 ml of heparin sodium (10,000 IU/ml; Elkins-Sinn, Cherry Hill, NJ)
65

per 38 ml of BO-A stock solution and 12 ml of BO-B stock solution. Fertilization medium
(BO-caffeine) was prepared by adding 0.0243 g of caffeine sodium (C-4144, Sigma-
Aldrich Inc., St. Louis, MO) per 25 ml of BO-AB. Sperm washing medium (0.6% BSA-
BO) was prepared by adding 0.03 g of bovine serum albumin (BSA; Fraction V, A-4503,
Sigma-Aldrich Inc., St. Louis, MO) to 5 ml of BO-AB. Oocyte washing medium (0.3%
BSA-BO) was prepared by adding 0.03 g of BSA (Fraction V) to 10 ml of BO-AB. TCM
oocyte washing medium was prepared by adding 0.03 g of BSA (Fraction V) to 10 ml of
Tissue Culture Medium-199 (Eagle’s salt) with 25 mM HEPES Buffer (TCM-199; Gibco
Laboratories, Grand Island, NY).
CR1aa medium was prepared according to Rosenkrans and First (1994). CR1aa
stock solution was first prepared by adding 114.7 mM NaCl, 1.6 mM KCl, 26.2 mM
NaHCO3, 0.3999 mM pyruvic acid (P-4562, Sigma-Aldrich Inc., St. Louis, MO), 2.5 mM
L(+) lactic acid (L-4388, Sigma-Aldrich Inc., St. Louis, MO), 0.5195 mM glycine (G-8790,
Sigma-Aldrich Inc., St. Louis, MO), 0.5051 mM L-alanine (A-7469, Sigma-Aldrich Inc.,
St. Louis, MO) and 0.2 ml of 0.5% phenol red per 100 ml of sterile water.
For embryo culture from day 0 to day 3, CR1aa culture medium was prepared by
adding 0.2 ml of Basal Medium Eagle (BME) amino acid solution (50X, B-6766, Sigma-
Aldrich Inc., St. Louis, MO), 0.1 ml of Modified Eagle Medium (MEM) amino acid solution
(10X, 11140-050, Gibco Laboratories, Grand Island, NY), 0.01 ml of gentamicin, 0.00146
g of L-glutamine (G-5763, Sigma-Aldrich Inc., St. Louis, MO) and 0.03 g of BSA (fatty
acid free, A-7511, Sigma-Aldrich Inc., St. Louis, MO) to 9.5 ml of CR1aa stock solution.
For embryo culture from day 3 to day 7, CR1aa culture medium was prepared by adding
0.2 ml of BME amino acid solution, 0.1 ml of MEM amino acid solution, 0.5 ml of fetal
bovine serum (FBS; SH30070.02, HyClone, Logan, UT), 0.01 ml of gentamicin, 0.00146
g of L-glutamine and 0.03 g of BSA (fatty acid free) to 9.5 ml of CR1aa stock solution.
In Vitro Fertilization
Oocytes were purchased weekly from a commercial supplier (BoMed, Inc,
Madison, WI) and shipped in a portable incubator (~39°C). Upon arrival at the
laboratory, shipping vials were removed from the portable incubator and stored in a
humidified atmosphere of 5% CO2 in air at 39°C for completion of maturation (maturation
time = 20 to 22 hours).
66

To prepare the semen for in vitro fertilization, a straw of frozen semen from a
Holstein bull (CSS 7H5188, Genex Cooperative Inc., Shawano, WI) of proven fertility
was thawed in a 39° to 40°C water bath for ~45 seconds. After thawing, the contents of
the straw were deposited into a 15-ml plastic conical tube (Corning, New York, NY).
Then, 9 ml of BO-caffeine was added to the tube and the suspension was centrifuged for
6 minutes at 200 x g. After centrifugation the supernatant was removed and the
remaining pellet was resuspended in an additional 9 ml of BO-caffeine. The resulting
suspension was centrifuged for 6 additional minutes at 200 x g. The supernatant was
removed again and the pellet resuspended in 4 ml of BO-caffeine and 4 ml of 0.6% BSA-
BO. The semen preparation was then allowed to equilibrate in a 39°C incubator for ~10
minutes. After equilibration, four 80-µl insemination drops were created in a preheated
35 x 10-mm Falcon® plastic Petri dish (Becton and Dickinson, Lincoln Park, NJ) and
covered with warmed mineral oil (M-8140, Sigma-Aldrich Inc., St. Louis, MO).
After 20 to 22 hours of maturation, cumulus oocyte complexes were removed
from the shipping vials and washed through two 35 x 10-mm plastic petri dishes
containing 2 to 3 ml of 0.3% BSA-BO. Then, 10 to 20 oocytes were placed in each of the
80-µl insemination drops previously prepared and were co-incubated for 5 hours at 39°C
and 5% CO2 in air.
After the 5-hour fertilization period, oocytes were removed from insemination
drops and washed through a single 35 x 10-mm plastic petri dish of TCM oocyte
washing medium. Cumulus cells were then removed by vortexing for 3 minutes in 1 ml of
TCM-199 containing 0.001 g of hyaluronidase (H-3506, Sigma-Aldrich Inc., St. Louis,
MO). After vortexing, oocytes were washed through two 35 x 10 mm plastic petri dishes
containing TCM oocyte washing medium and one 35 x 10-mm plastic petri dish of
CR1aa. Then, 10 to 15 oocytes per drop were placed in 50-µl drops of CR1aa covered
with warmed mineral oil and incubated in a modular incubator containing 5% CO2, 5%
O2 and 90% N2 at 39°C.
Chick Embryo Co-Culture
The procedure for the culture of bovine 8-cell IVF embryos using the chick
embryo amnion was modified from that of Blakewood and Godke (1989). Briefly,
fertilized domestic chicken eggs were collected on the day of oviposition and stored at
10°C until the onset of incubation. Four days prior to the introduction of bovine embryos,
67

the eggs were removed from cold storage and allowed to come to room temperature
(25°C) before placing them in a 37°C commercial egg incubator. After 72 hours of
incubation, the eggs were sterilized with 70% ethanol (Aaper Alcohol and Chemical
Company, Shelbyville, KY) and 7.5% providone iodine (Betadine®, Purdue Fredrick
Company, Stanford, CT). The eggs were then allowed to air dry in a horizontal position
to ensure proper positioning of the chick embryo.
To remove the egg contents, the egg was broken against the rim of a sterile
beaker and the contents released into a 100-ml plastic embryo collection dish
(Veterinary Concepts, Spring Valley, WI) covered by a piece of cellophane kitchen wrap
(Saran®, Dow Chemical, Midland, MI). The dish, including the egg contents, was then
loosely capped with a plastic lid (Veterinary Concepts, Spring Valley, WI) and placed in a
38°C incubator for an additional 24 hours before introducing the 8-cell bovine embryos
into the amniotic cavity of the chick embryo. Prior to embedding and injection of bovine
embryos, suitable chick embryos were selected for injection based on the following
criteria: 1) round intact yolk, 2) good vascularity in the chorioallantoic membrane, 3)
vigorous heartbeats and 4) no apparent abnormalities.
Bovine embryos were embedded in agarose according to the procedure set
forth by Willadsen (1979) for the culture of ovine embryos in the ligated oviducts of
sheep. On day 3 of culture, two or three 8-cell bovine IVF embryos were placed in a 50-
µl drop of low-melting agarose solution [1.2% agarose (A-9799, Sigma-Aldrich Inc., St.
Louis, MO) dissolved in Dulbecco’s Phosphate-Buffered Saline (DPBS; D1408, Sigma-
Aldrich Inc., St. Louis, MO)] at ~37oC. The embryos were then aspirated into a beveled
glass pipette (200 µm outer diameter) and held in air for ~30 seconds to allow the
agarose to harden. Then, the agar (containing the embryos) within the pipette was
expelled as a gel cylinder into room temperature TL-HEPES (BioWittaker, Walkersville,
MD). The agarose cylinder, including the bovine embryos, was then aspirated back into
the beveled pipette, and carefully expelled into the amniotic cavity of a 96-hour chick
embryo.
After introducing the embedded embryos into the amniotic cavity the shell-less
culture systems were returned to incubation for an additional 72 hours at 39oC. Thus, a
single agarose cylinder containing two or three 8-cell bovine IVF embryos was co-
cultured in the amnion of each developing chick embryo for 4 days.
68

On day 7 of in vitro culture, the amnion containing the agarose cylinder was
dissected away from the rest of the egg components and placed in a 35 x 10-mm plastic
petri dish. The amnion was then rinsed several times with DPBS containing 1% FBS.
After rinsing, the petri dish was placed under a stereomicroscope (Nikon SMZ-2B,
Tokyo, Japan) equipped with an overhead light source and the agarose cylinder was
removed from the amniotic cavity using two 22-gauge, 37.5-mm needles (Kendall,
Mansfield, MA) to make a small opening in the amniotic membrane. The agarose
cylinder was carried from the amnion with the escaping amniotic fluid, settling on the
bottom of the dish. The cylinder was then washed twice in CR1aa medium and dissected
using two 22-gauge needles to isolate and remove the developing embryos.
Statistical Analysis For Experiment 5.1, data were analyzed using PROC GLM in the SAS Statistical
Package (v. 8e, 1999, SAS Institute, Inc., Gary, NC). When significance was detected
(P<0.05), post-hoc comparisons were made using Duncan’s Multiple Range test. For
Experiment 5.2, data were analyzed using PROC GLM of SAS. Significant differences
between embryo development rates in each group were tested using a post hoc least
significant difference test (LSD test). Differences with a probability (P) value of 0.05 or
less were considered significant in this study
RESULTS In Experiment 5.1, chick embryo mortality was 41%, with the highest mortality
rate (86%) occurring in CAMs treated with cytochalasin D (Table 5.1). To determine the
affect of prostaglandins on blood vessel development, CAMs were exposed to
treatments at an early stage of blood vessel development (day 5) and then evaluated 24
hours later. Average node counts per CAM are provided in Table 5.2. Capillary
branching was not affected (P>0.05) when CAMs were treated with cytochalasin D
(negative control), but was significantly greater (P<0.05) when CAMs were treated with
EtOH (carrier), PGE2 or PGF2α (Figure 5.3). Both PGE2 and PGF2α stimulated (P<0.05)
capillary branching when compared with control and cytochalasin-treated CAMs, and
PGE2 increased (P<0.05) capillary branching over that of EtOH. PGF2α did not differ
(P>0.05) from either EtOH or PGE2 (Figure 5.4).
In Experiment 5.2, 60 chicken eggs were initially placed in incubation. After 72
hours of incubation, the eggs were removed from their shell and placed in shell-less
69
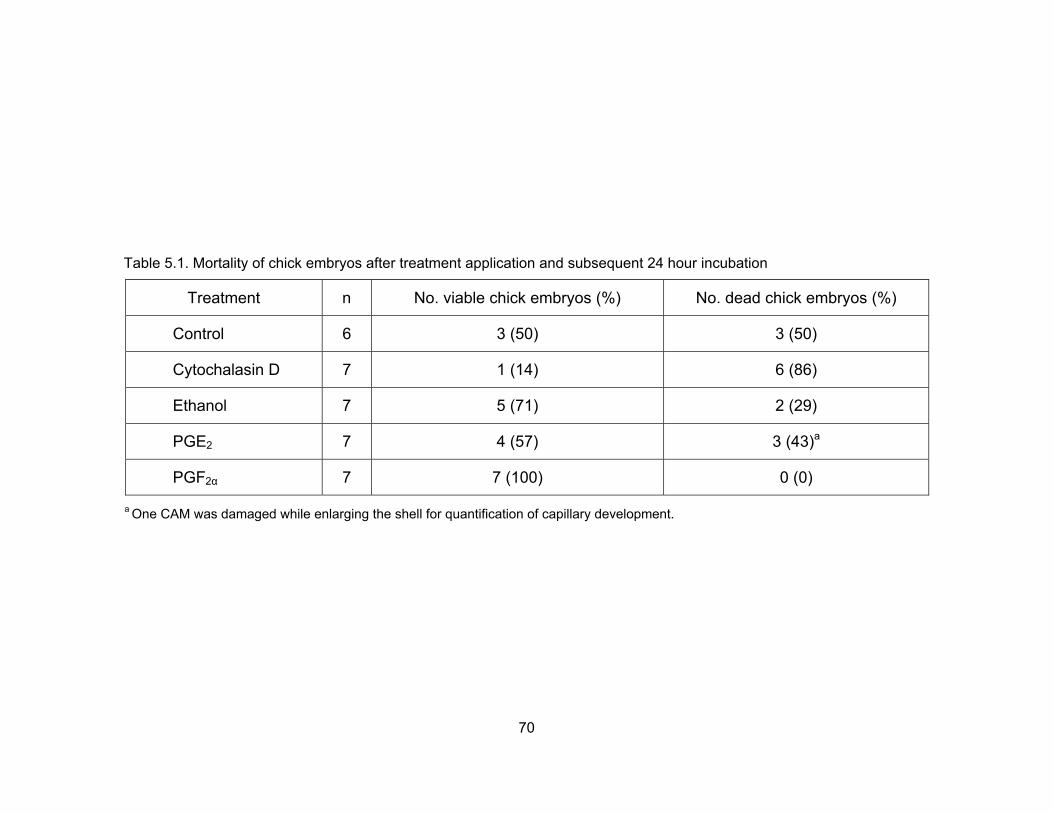
Table 5.1. Mortality of chick embryos after treatment application and subsequent 24 hour incubation
Treatment n No. viable chick embryos (%) No. dead chick embryos (%)
Control 6 3 (50) 3 (50)
Cytochalasin D 7 1 (14) 6 (86)
Ethanol 7 5 (71) 2 (29)
PGE2 7 4 (57) 3 (43)a
PGF2α 7 7 (100) 0 (0) a One CAM was damaged while enlarging the shell for quantification of capillary development.
70
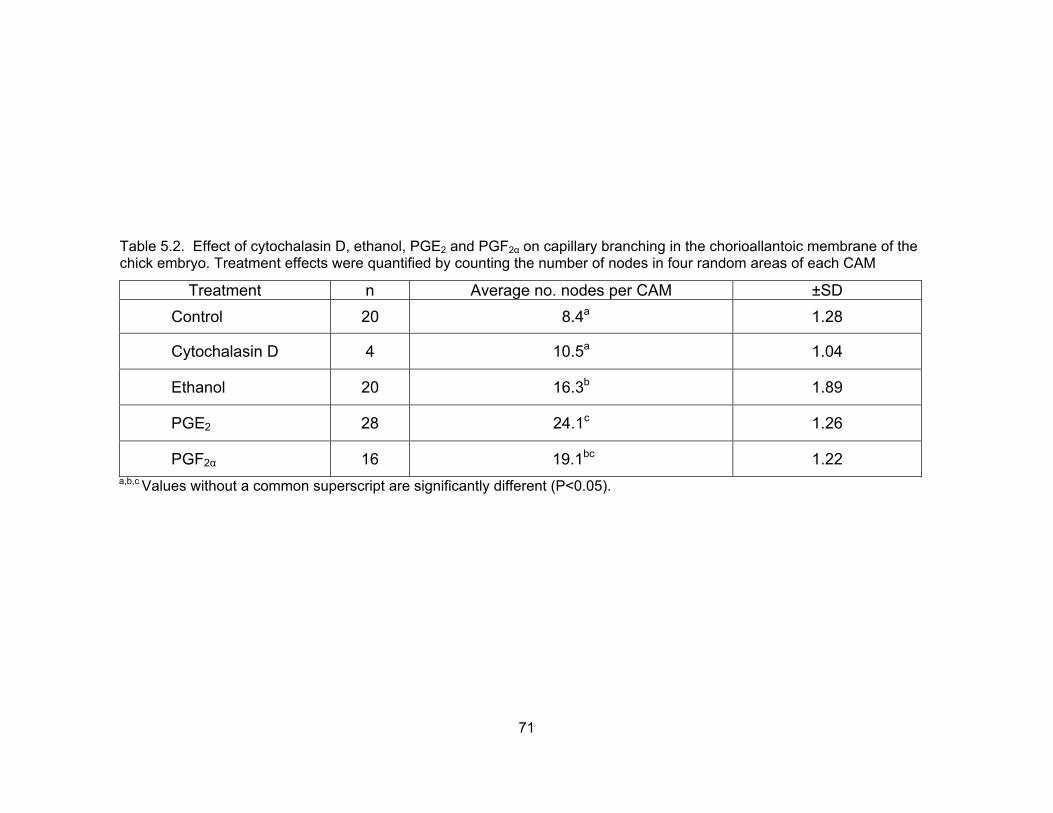
Table 5.2. Effect of cytochalasin D, ethanol, PGE2 and PGF2α on capillary branching in the chorioallantoic membrane of the chick embryo. Treatment effects were quantified by counting the number of nodes in four random areas of each CAM
a,b,c Values without a common superscript are significantly different (P<0.05).
Treatment n Average no. nodes per CAM ±SD Control 20 8.4a 1.28
Cytochalasin D 4 10.5a 1.04
Ethanol 20 16.3b 1.89
PGE2 28 24.1c 1.26
PGF2α 16 19.1bc 1.22
71

A B
C D
Figure 5.3. Restructuring of vasculature networks in CAMs treated with (A) chick Ringer’s solution (control), (B) ethanol (vehicle), (C) PGE2 and (D) PGF2α.
72
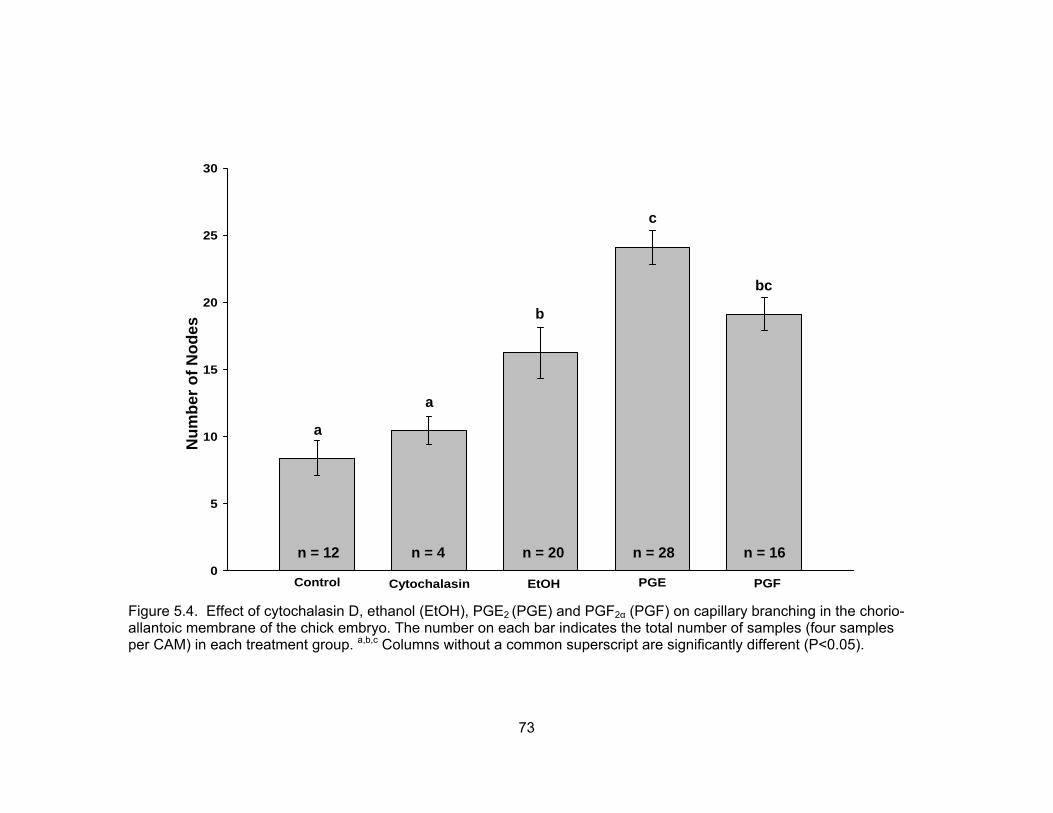
Control Cytochalasin EtOH PGE PGF0
5
10
15
20
25
30
c
bc
b
Num
ber o
f Nod
es
a a
n = 12 n = 4 n = 20 n = 28 n = 16
Figure 5.4. Effect of cytochalasin D, ethanol (EtOH), PGE2 (PGE) and PGF2α (PGF) on capillary branching in the chorio-allantoic membrane of the chick embryo. The number on each bar indicates the total number of samples (four samples per CAM) in each treatment group. a,b,c Columns without a common superscript are significantly different (P<0.05).
73

culture. At this time, four (6%) of the eggs were found to be unfertilized, four (6%)
contained embryos that failed to fully develop and one (2%) contained an embryo that
was nonviable. The remaining 51 eggs were returned to incubation. After 24 hours of
incubation in shell-less culture, 34 eggs were selected for injection of bovine embryos.
For chick embryo co-culture, 2-cell bovine embryos (n=102) were embedded and placed
into the amniotic cavities of chick embryos, and 51 (50%) were successfully recovered
on day 7 for evaluation. Loss of embryos occurred during recovery or was due to chick
embryo mortality. Death of chick embryos was the primary reason for the loss, as six of
the 17 (35%) chick embryos in the CEC treatment group, and four of the 17 (24%) chick
embryos in the PGE2 treatment group did not survive injection and subsequent shell-less
culture.
On day 7 of culture, a significantly greater (P<0.05) percentage of morulae were
noted following culture in PGE2-treated chick embryos than in untreated chicks. There
was no significant difference (P<0.05) between the CEC treatment group and the control
treatment group (Table 5.3). Although there was no difference detected in the number of
early blastocysts or blastocysts among the three treatments, there were significantly more expanded blastocysts after culture in control medium alone than in either chick
embryo co-culture treatment group (Figure 5.5). The number of hatching blastocysts did
not differ (P>0.05) across treatments (Figure 5.5). Although there were no differences in
the percentage of embryos developing to the early blastocyst, hatching blastocysts or
hatched blastocyst stage on day 8 of development, there were significantly more
expanded blastocysts in the control treatment group than the CEC treatment group.
Embryo development to the expanded blastocyst stage in the PGE2 treatment group was
not significantly different (P>0.05) from the other two groups. On day 9 of development,
there were more blastocysts in the CEC and PGE2 treatment groups compared with
controls (P<0.05). However, embryo development to the expanded blastocyst, hatching
blastocyst or hatched blastocyst stage did not differ with treatment (P>0.05). Embryo
quality grade did not differ (P>0.05) between the three treatments on day 7 or day 8 of
culture, but there were significantly more (P<0.05) grade 1 blastocysts in the CEC
treatment group than in the PGE2 or control treatment groups on day 9 of development
(Figure 5.6).
74

Table 5.3. Development of 8-cell bovine embryos cultured in CR1aa + 5% fetal bovine serum (control), co-cultured in the chick embryo amnion or co-cultured in the amnion of PGE2-treated chicks
a Embryos allotted to the control group were cultured in CR1aa + 5% FBS from day 3 to day 7 post-insemination. Embryos allotted to the CEC group were culture in the amnion of a developing chick embryo from day 3 to day 7 post insemination. Embryos cultured in the PGE2 group were cultured in the amnion of PGE2 treated chicks from day 3 to day 7 post insemination. b The number of 8-cell embryos allotted to each treatment. The number in parenthesis indicates the number of embryos successfully recovered from live chick embryos. c The percentage of embryos developing in the chick co-culture group and the PGE2 treated chick co-culture group were based on the number of embryos successfully recovered from live chick embryos. d MOR = morulae.
e BLST = blastocysts. f HT/HD BLST = hatching and/or hatched blastocysts. g,h Within a column, values without a common superscript are significantly different (P<0.05).
75

0
10
20
30
40
MORL EarlyBLST
BLST ExpandBLST
HatchingBLST
% o
f Em
bryo
s
ControlCECPGE
A
aa
a
bb
a
a
aa
a a
a
b
ab
a
0
10
20
30
40
MORL Early BLST Expanded Hatching Hatched
% o
f Em
bryo
BLST BLST BLST BLST
s
ControlCECPGE
a
a a a
a
aa
a
a
b
ab
aaa
aa
B
a
a
0
5
10
15
20
BLST ExpandBLST
HatchingBLST
HatchedBLST
% o
f Em
bryo
s
ControlCECPGE
C
a
aa
aa
a
a
a
a
bb
a
Figure 5.5. Development of 8-cell bovine embryos cultured in CR1aa supplemented with 5% fetal bovine serum (control), cultured in the chick embryo amnion (CEC) or cultured in the amnion of PGE2-treated chicks (PGE) on day 7 of development (A), day 8 of development (B) and day 9 of development (C). Within a developmental stage, columns without a common superscript are significantly different (P<0.05).
76
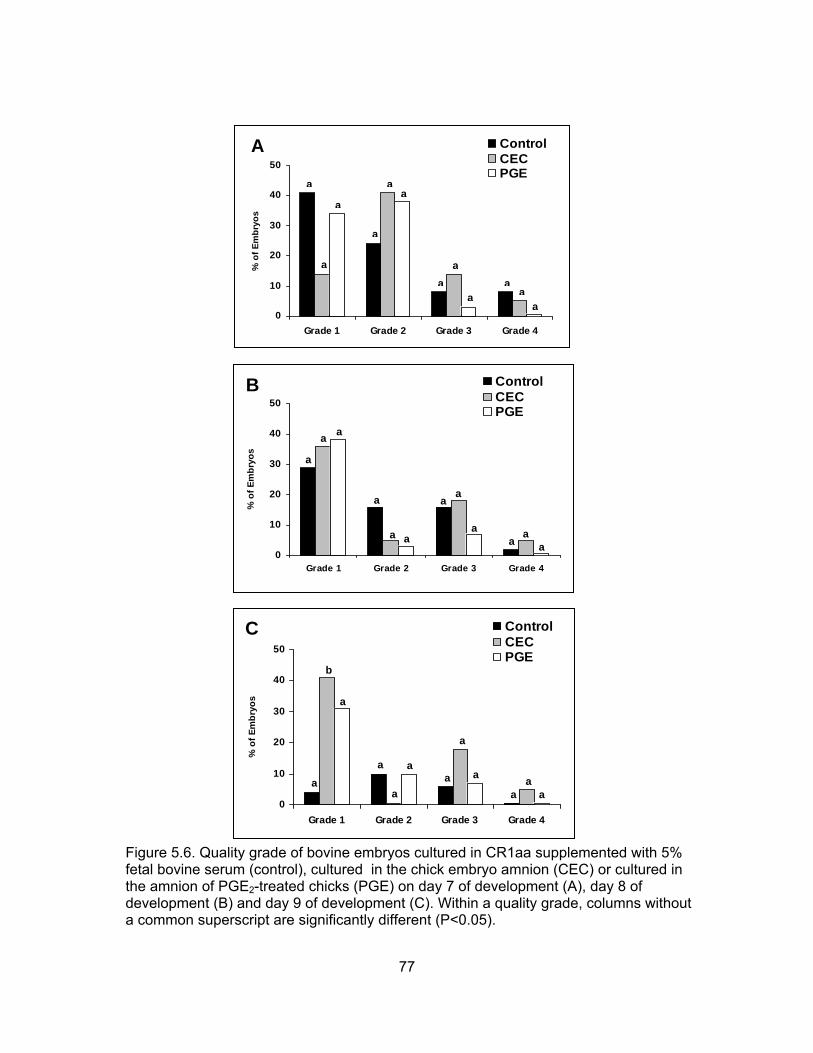
0
10
20
30
40
50
Grade 1 Grade 2 Grade 3 Grade 4
% o
f Em
bryo
s
ControlCECPGE
A
aa
aa
aa
aa
a
a
a
a
0
10
20
30
40
50
Grade 1 Grade 2 Grade 3 Grade 4
% o
f Em
bryo
s
ControlCECPGE
B
aa
aa
aa
aa
a
aa
a
0
10
20
30
40
50
Grade 1 Grade 2 Grade 3 Grade 4
% o
f Em
bryo
s
ControlCECPGE
C
aa
a
a
a
aa
a
a
a
b
a
Figure 5.6. Quality grade of bovine embryos cultured in CR1aa supplemented with 5% fetal bovine serum (control), cultured in the chick embryo amnion (CEC) or cultured in the amnion of PGE2-treated chicks (PGE) on day 7 of development (A), day 8 of development (B) and day 9 of development (C). Within a quality grade, columns without a common superscript are significantly different (P<0.05).
77

DISCUSSION Angiogenesis is the formation of new blood vessels by branching and growth,
and migration of pre-existing blood vessels (Rissau, 1995; Rissau and Flamme, 1995;
Rissau, 1997). This capillary proliferation is vital during embryogenesis (Wilting et al.,
1995; Flamme et al., 1997), wound healing (Arbiser, 1996; Martins-Green and Hanafusa,
1997) and bone fracture repair (Klagsburn and D’Amore, 1991). Rapid or persistent
capillary growth also has been associated with tumors, retinopathies, haemangiomas,
fibrosis and rheumatoid arthritis (for review see Folkman and Klagsburn, 1987;
Klagsburn and D’Amore, 1991). In most adult tissues, angiogenesis is rare (Denekamp,
1984; Klagsbrun and D’Amore, 1991). Exceptions can be found in the reproductive
organs of adult females, where angiogenesis occurs cyclicly in the ovary and the uterus,
as well as in the uterine endometrium and placenta during pregnancy (Augustin et al.,
1995; Reynolds and Redmer, 1995; Redmer and Reynolds, 1996).
The purpose of the first experiment of this series was to determine if normal
blood flow patterns to the chick embryo could be altered by the application of prostaglandins. To investigate this hypothesis, angiogenesis was examined in chick
chorioallantoic membranes (CAM) treated with either PGE2 or PGF2α. The CAM forms between day 4 and day 5 of development by fusion of the allantois and chorion. The
resulting membrane consists of the chorion (ectoderm), mesoderm with blood vessels
and allantois (endoderm) (DeFouw et al., 1989). The CAM is of physiologic importance
to the chick because it serves as the major respiratory organ for gaseous exchange until
hatching, and it provides a bladder into which waste products can be delivered
(Romanoff, 1967). Because the chorioallantoic membrane is easily accessible for in vivo work and is highly vascularized, it has proven to be an excellent model for the study of
the angiogenic process (Ribatti et al., 1996).
Findings of the first experiment suggest that PGF2α may directly inhibit
angiogenesis. Based on visual observation, blood vessels in CAMs treated with PGF2α
appeared thicker and larger with fewer main vessels than those in controls or vehicle-
treated CAMs. The response of CAMs to PGF2α was not dramatic, however, and
quantification of branch nodes was not significantly different than vehicle. It is interesting
to note that although blood vessel development was visually impaired in PGF2α -treated
chicks, 100% of the treated chicks survived shell-less culture. These findings are
78

counterintuitive to our original hypothesis, but the small number of observations should
be taken into account. It is possible that these findings would not hold true with
additional observations.
In contrast, CAMs treated with PGE2 showed a significant increase in capillary
development compared with controls and vehicle-treated CAMs, suggesting that PGE2
directly influences angiogenesis. It is well known that increased blood flow to a tissue
results in an increase of function (Guyton, 1971). Should PGE2 in fact increase
angiogenesis, as indicated here, then it is possible to infer that the increase in blood
vessel formation would increase blood flow to the chick embryo, resulting in increased
chick viability.
The results of the first study were encouraging because the inherent mortality
rate of chick embryos maintained in shell-less culture poses a major drawback to the
efficacy of this procedure for use as an alternative embryo culture technique. Dunn and
Boone (1976) reported that chick survival rates of 90 to 95% appear as high as can be
expected with shell-less culture. More importantly, Rowlett and Simkiss (1987) have
reported losses as high as 27% between day 4 and day 7 of shell-less culture, the time
period in which bovine embryos are maintained in the chick amnion. If chick viability
could be improved with the application of prostaglandins, loss of valuable bovine
embryos upon chick death could be minimized.
In the second experiment, CAMs were treated with PGE2 24 hours before
introduction of bovine embryos into the amniotic cavity. In this study, there was only a
slight improvement in chick embryo viability in PGE2 treated chicks (65% in CEC and
76% in PGE2); although, as in the first study, an obvious increase in capillary
development in PGE2-treated chicks compared with non treated chicks was observed.
Unfortunately, increasing blood flow to the chick embryo did not appear to improve
development or quality of bovine embryos cultured in untreated chicks or in semi-defined
medium (control). These findings suggest that the benefits of the chick embryo culture
system may not be due to growth factors and other embryotropic properties within the
amniotic fluid, but rather in the ability of the chick embryo to actively regulate the pH,
osmolarity and O2 content of its own environment.
79

CHAPTER VI
DEVELOPMENT OF 8-CELL BOVINE EMBRYOS IN CULTURE MEDIUM SUPPLEMENTED WITH CHICK AMNIOTIC FLUID (CAF)
INTRODUCTION
Even with continual efforts worldwide, researchers have been unable to fully
replicate the conditions within the oviduct and uterus in vitro. The development of
immature bovine oocytes to the blastocyst stage appear to have plateaued at 30 to 40%
following in vitro maturation, fertilization and culture (Krisher and Bavister, 1998;
Lonergan et al., 2003). In addition, the quality of blastocysts produced in vitro rarely, if
ever, reach that of in vivo-derived blastocysts (Duby et al., 1997; Boni et al., 1999; Viuff
et al., 1999) and marked differences have been reported to exist between in vivo- and in
vitro-derived embryos with regard to morphology (Greve et al., 1995), timing of
development (Van Soom et al., 1997), embryonic metabolism (Khurana and Niemann,
2000) and pregnancy rates (Enright et al. 2000).
Current culture conditions rely heavily on the addition of fetal bovine serum (FBS)
and/or bovine serum albumin (BSA) to the culture medium (Bavister, 1995), despite the
undefined and variable nature of serum composition (Rizos et al., 2003). It has been
suggested that serum may contain components advantageous to blastoceol
development, such as growth factors or extracellular matrix components, like fibronectin
(Pinyopunnintr and Bavister, 1994). However, it has also been suggested that the
components of serum may contribute to fetal and calf abnormalities (Thompson et al.,
1995; Walker et al., 1996; Sinclair et al., 1999). Specifically, offspring produced from
embryos cultured in vitro often exhibit increased birth weight (Willadsen et al., 1991;
Keefer et al, 1994; Behboodi et al., 1995), longer gestation lengths (Walker et al., 1996),
increased placental anomalies, (Hasler, et al., 1995; Hill et al., 1999, 2001, 2002) and
increased incidences of abortion (Walker et al., 1996; Hill et al., 1999) and perinatal
(Behboodi et al., 1995; Wilson et al., 1995; Garry et al., 1996; Schmidt et al., 1996; Kruip
and den Daas, 1997; Hill et al., 1999) and neonatal mortality (Schmidt et al., 1996; Kruip
and den Daas, 1997; Hill et al., 1999).
Attempts to circumvent potential risks associated with the addition of serum to
embryo culture media have included culturing embryos in the amniotic cavity of a
developing chicken embryo. Previous reports have indicated that the chick amnion is
80

capable of supporting the development of murine (Blakewood et al., 1989a), caprine
(Blakewood et al., 1989a), bovine (Blakewood et al., 1989b; Murakami et al., in press)
and porcine (Ocampo et al., 1993) embryos, and live transplant offspring have been
produced following chick embryo co-culture of 2- to 8-cell caprine embryos (Blakewood
et al., 1990). However, the inherent mortality rate of shell-less chick embryos poses a
major drawback to the efficacy of this procedure. Dunn and Boone (1976) reported that
chick survival rates of 90 to 95% appear to be as high as can be expected with shell-less
culture, and a rapid degeneration of mammalian embryos has been reported following
chick embryo death (Blakewood and Godke, 1990). Therefore, the objective of this study
was to determine if amniotic fluid extracted from a developing chick embryo could be
used as a supplement to standard in vitro culture media and support development of
bovine embryos.
MATERIALS AND METHODS Experimental Design The design for this experiment is illustrated in Figure 6.1. Upon removal from in
vitro insemination, all presumptive zygotes were placed in 50-µl drops of CR1aa (10 to
15 per 50-µl drop) for in vitro culture (Rosenkrans and First, 1994). After 72 hours of
culture, good quality (as indicated by equal size, well defined blastomeres) 6- to 8-cell
embryos were randomly allotted to one of two treatment groups. Embryos allotted to
Treatment A (control group) were cultured in CR1aa medium supplemented with 5%
FBS. Embryos allotted to Treatment B (CAF group) were cultured in CR1aa medium
supplemented with 20% CAF obtained from a day 7 chick embryo. All embryos were
cultured in a modular incubator containing 5% CO2, 5% O2 and 90% N2 at 39°C. Media
were changed on day 7 of culture and embryonic development was assessed on days 7,
8 and 9. At each evaluation time point, each embryo was assigned a quality grade score
from 1 to 4 (grade 1 = excellent quality to grade 4 = poor quality or degenerate).
Experimental Methods Media Preparation
For the preparation of Brackett-Oliphant (BO) media, BO-A stock solutions of
150.2 mM NaCL (S-5886, Sigma-Aldrich Inc., St. Louis, MO), 2.7 mM KCl (P-5405,
Sigma-Aldrich Inc. St. Louis, MO), 3.0 mM CaCl2·2H2O (C-7902, Sigma-Aldrich Inc., St.
81
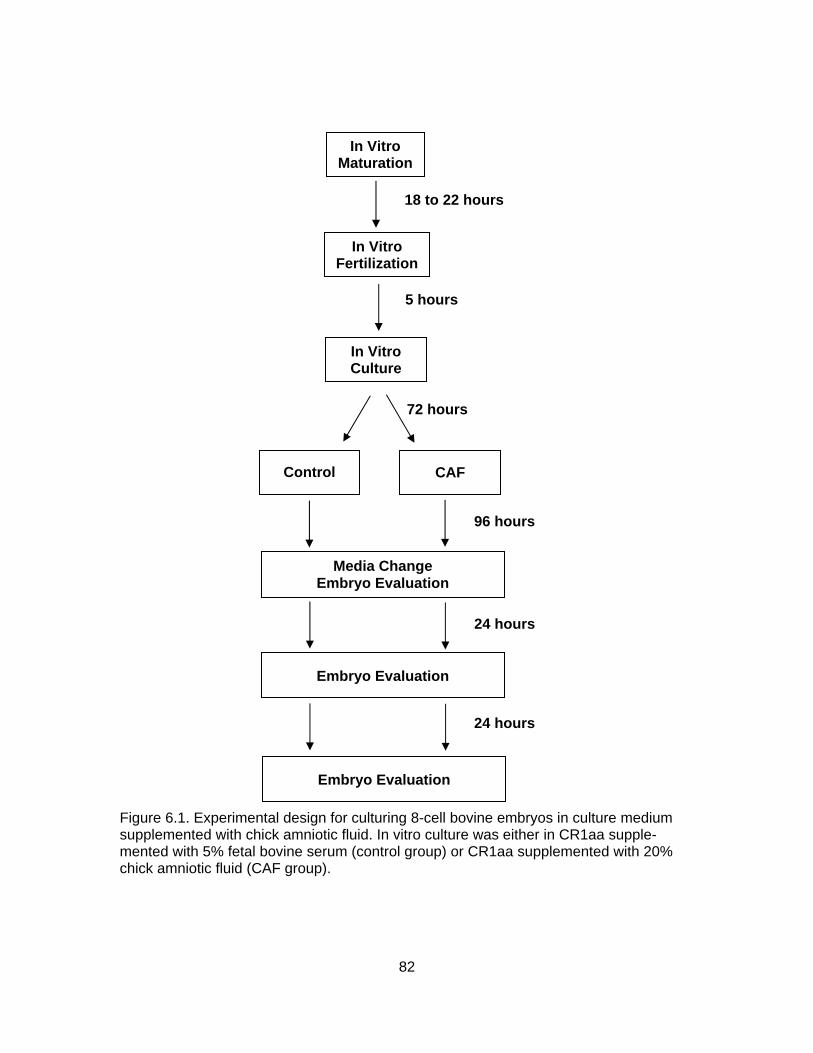
In Vitro
Maturation
CAF
Control
In Vitro Culture
In Vitro Fertilization
96 hours
72 hours
5 hours
18 to 22 hours
Media Change Embryo Evaluation
Embryo Evaluation
24 hours
24 hours
Embryo Evaluation
Figure 6.1. Experimental design for culturing 8-cell bovine embryos in culture medium supplemented with chick amniotic fluid. In vitro culture was either in CR1aa supple-mented with 5% fetal bovine serum (control group) or CR1aa supplemented with 20% chick amniotic fluid (CAF group).
82

Louis, MO), 1.0 mM NaH2PO4·H2O (S-9638, Sigma-Aldrich Inc., St. Louis, MO), 0.6857
mM MgCl2·6H2O (M-2393, Sigma-Aldrich Inc., St. Louis, MO) and 0.1 ml of 0.5% phenol
red (P-0290, Sigma-Aldrich Inc., St. Louis, MO) per 500 ml of sterile water were
prepared. BO-B stock solutions of 154.0 mM NaHCO3 (S-8875, Sigma-Aldrich Inc., St.
Louis, MO) and 0.04 ml of 0.5% phenol red per 200 ml of sterile water were also
prepared (Brackett and Oliphant, 1975).
For the preparation of CR1aa culture medium, a CR1aa stock solution of 114.7
mM NaCl, 1.6 mM KCl, 26.2 mM NaHCO3, 0.3999 mM pyruvic acid (P-4562, Sigma-
Aldrich Inc., St. Louis, MO), 2.5 mM L(+) lactic acid (L-4388, Sigma-Aldrich Inc., St.
Louis, MO), 0.5195 mM glycine (G-8790, Sigma-Aldrich Inc., St. Louis, MO), 0.5051 mM
L-alanine (A-7469, Sigma-Aldrich Inc., St. Louis, MO) and 0.2 ml of 0.5% phenol red per
100 ml of sterile water was prepared (Rosenkrans and First, 1994).
From the BO-A and BO-B stock solutions, fertilization stock medium (BO-AB)
was prepared by adding 1.2 mM pyruvic acid, 0.05 ml of gentamicin (50 mg/ml; 15750-
060, Gibco, Grand Island, NY) and 0.018 ml of heparin sodium (10,000 IU/ml; Elkins-
Sinn, Cherry Hill, NJ) per 38 ml of BO-A stock solution and 12 ml of BO-B stock solution.
Fertilization medium (BO-caffeine) was prepared by adding 0.0243 g of caffeine sodium
(C-4144, Sigma-Aldrich Inc., St. Louis, MO) per 25 ml of BO-AB. Sperm washing
medium (0.6% BSA-BO) was prepared by adding 0.03 g of BSA (Fraction V, A-4503,
Sigma-Aldrich Inc., St. Louis, MO) to 5 ml of BO-AB. Oocyte washing medium (0.3%
BSA-BO) was prepared by adding 0.03 g of BSA (Fraction V) to 10 ml of BO-AB. TCM
oocyte washing medium was prepared by adding 0.03 g of BSA (Fraction V) to 10 ml of
Tissue Culture Medium-199 (Eagle’s salt) with 25 mM HEPES Buffer (TCM-199; Gibco
Laboratories, Grand Island, NY).
From the CR1aa stock solutions, CR1aa culture medium for day 0 to day 3 was
prepared by adding 0.2 ml of Basal Medium Eagle (BME) amino acid solution (50X, B-
6766, Sigma-Aldrich Inc., St. Louis, MO), 0.1 ml of Modified Eagle Medium (MEM) amino
acid solution (10X, 11140-050, Gibco Laboratories, Grand Island, NY), 0.01 ml of
gentamicin, 1.1 mM L-glutamine (G-5763, Sigma-Aldrich Inc., St. Louis, MO) and 0.03 g
of BSA (fatty acid free, A-7511, Sigma-Aldrich Inc., St. Louis, MO) to 9.5 ml of CR1aa
stock solution. CR1aa culture medium for day 3 to day 7 was prepared by adding 0.2 ml
of BME amino acid solution, 0.1 ml of MEM amino acid solution, 0.5 ml of FBS
83

(SH30070.02; HyClone, Logan, UT), 0.01 ml of gentamicin, 1.1 mM L-glutamine and
0.03 g of BSA (fatty acid free) to 9.5 ml of CR1aa stock solution.
In Vitro Fertilization
Oocytes were purchased weekly from a commercial supplier (BoMed, Inc,
Madison, WI) and matured in a portable incubator (~39°C) during the overnight travel
interval (20 to 22 hours). Upon arrival at the laboratory, shipping vials were removed
from the portable incubator and stored in a humidified atmosphere of 5% CO2 in air at
39°C until insemination.
To prepare semen for fertilization, a straw of frozen semen from a Holstein bull
(CSS 7H5188, Genex Cooperative Inc., Shawano, WI) of proven fertility was thawed in a
39° to 40°C water bath for ~45 seconds. After thawing, the contents of the straw were
deposited into a 15-ml plastic conical tube (Corning, New York, NY) and 9 ml of BO-
caffeine was added to the tube. The suspension was then centrifuged for 6 minutes at
200 x g. After centrifugation, the washing step was repeated by resuspending the semen
pellet in an additional 9 ml of BO-caffeine and centrifuging for 6 additional minutes at
200 x g. The supernatant was removed and the pellet resuspended in 4 ml of BO-
caffeine and 4 ml of 0.6% BSA-BO. The semen preparation was then placed in a 39°C
incubator for ~10 minutes to equilibrate. After equilibration, four 80-µl insemination drops
were created in a preheated 35 x 10-mm Falcon® plastic petri dish (Becton and
Dickinson, Lincoln Park, NJ) and covered with warmed mineral oil (M-8140, Sigma-
Aldrich Inc., St. Louis, MO).
Following 20 to 22 hours of maturation, cumulus oocyte complexes were
removed from the shipping vials and washed through two 35 x 10-mm plastic petri
dishes containing 2 to 3 ml of 0.3% BSA-BO. Then, 10 to 20 oocytes were placed in
each of the 80-µl insemination drops previously prepared and were co-incubated for 5
hours at 39°C and 5% CO2 in air.
After the 5-hour fertilization period, oocytes were washed through a single 35 x
10-mm plastic petri dish of TCM oocyte washing medium and the cumulus cells were
removed by vortexing for 3 minutes in 1 ml of TCM-199 containing 0.001 g of
hyaluronidase (H-3506, Sigma-Aldrich Inc., St. Louis, MO). The oocytes were then
washed through two 35 x 10-mm plastic petri dishes containing TCM oocyte washing
medium and one 35 x 10-mm plastic petri dish of CR1aa. Then, 10 to 15 oocytes per
84

drop were placed in 50-µl drops of CR1aa covered with warmed mineral oil and
incubated in a modular incubator containing 5% CO2, 5% O2 and 90% N2 at 39°C.
Collection of Chick Amniotic Fluid (CAF)
The basic procedure used for the shell-less culture of chick embryos was
modified from that previously described by Blakewood and Godke (1989). Briefly,
fertilized domestic chicken eggs were collected on the day of oviposition and stored at
10°C until the onset of incubation. Seven days prior to the day of amniotic fluid collection
the eggs were removed from cold storage and allowed to come to room temperature
(25°C) before placing them in a 37°C commercial egg incubator. After 72 hours of
incubation, the eggs were rinsed with a solution of 70% ethanol (Aaper Alcohol and
Chemical Company, Shelbyville, KY) and then coated with 7.5% providone iodine
(Betadine®, Purdue Fredrick Company, Stanford, CT) and allowed to air dry in a
horizontal position to ensure proper positioning of the chick embryo.
To remove the egg contents, a crack was made in the shell by striking the center
of the egg against the rim of a sterile beaker. The egg contents were then released into
a 100-ml plastic embryo collection dish (Veterinary Concepts, Spring Valley, WI) covered
by a piece of cellophane kitchen wrap (Saran®, Dow Chemical, Midland, MI). The dish,
including the egg contents, was loosely capped with a plastic lid (Veterinary Concepts,
Spring Valley, WI) and maintained in an incubator at 39°C for an additional 96 hours
before collection of the amniotic fluid.
On day 7 of shell-less culture, fluid was aspirated from the amnion using a 22-
gauge, 37.5-mm needle (Kendall, Mansfield, MA) and a 3-ml disposable syringe
(Sherwood, Davis and Geck, St. Louise, MO). Amniotic fluid obtained from three chick
embryos was pooled in a 15-ml plastic conical tube and stored at -20°C until use.
Statistical Analysis Data were analyzed using PROC GLM of the SAS Statistical Package (v. 8e,
1999, SAS Institute, Inc., Gary, NC). Significant differences between embryo
developmental rates in each group were tested using a post hoc least significant
difference (LSD) test. Differences with a probability level (P) of 0.05 or less were
considered significant in this study.
85

RESULTS Developmental rates for bovine 8-cell embryos cultured in CR1aa medium
supplemented with 5% FBS (control) or in CR1aa supplemented with 20% CAF are
summarized in Table 6.1. On day 7 of development a significantly greater (P<0.05)
percentage of morula stage embryos resulted from bovine 8-cell embryos cultured in
medium supplemented with 20% CAF than in the control culture medium (33% and 14%,
respectively). In contrast, culture in FBS-supplemented medium resulted in significantly
more (P<0.05) blastocysts and expanded blastocysts compared with CAF-supplemented
medium (blastocysts = 31% and 17%, expanded blastocysts = 30% and 18%,
respectively; Figure 6.2). On day 8 post-insemination there was no effect (P>0.05) of
culture medium on the percentage of embryos that developed to the morula, early
blastocyst, blastocyst, expanded blastocyst or hatched blastocyst stage. However, there
were significantly more (P<0.05) hatching blastocysts in FBS-supplemented medium
than in CAF-supplemented medium (11% and 3%, respectively; Figure 6.2). Although
there was no difference (P>0.05) in the total number of blastocysts present in each
treatment group on day 9 post-insemination, there were significantly more (P<0.05)
hatched blastocysts in the FBS-supplemented medium compared with the CAF-
supplemented medium (18% and 9%, respectively; Figure 6.2).
Embryo quality grades are presented in Figure 6.3. Although culture in FBS-
supplemented medium resulted in a significantly higher (P<0.05) blastocyst yield on day
7 post-insemination, there was no significant difference (P>0.05) in the percentage of
grade 1 blastocysts between the two treatment groups (control = 16% and CAF = 15%).
On day 8 post-insemination, there was no effect of culture medium on grade 1 or grade
2 embryos, but fewer grade 3 embryos were observed in the CAF-supplemented
medium compared with the control medium (9% and 20%, respectively). Similar results
were noted on day 9 of in vitro culture.
DISCUSSION Bovine embryo culture media often require the addition of serum at the initiation of
compaction to stimulate development of the blastocoel (Pinyopummintr and Bavister,
1994; Thompson et al., 1998). Addition of serum to in vitro culture systems, however,
has been implicated in several fetal and/or calf abnormalities (Willadsen et al., 1991;
Keefer et al., 1994; Behboodi et al., 1995; Hasler et al., 1995; Wilson et al., 1995; Garry
86

Table 6.1. Development of 8-cell bovine embryos cultured in CR1aa supplemented with 5% fetal bovine serum (control)
or 20% chick amniotic fluid (CAF)
a Embryos allotted to the control group were cultured in CR1aa + 5% FBS from day 3 to day 7 post-insemination. Embryos allotted to the CAF group were cultured in CR1aa + 20% FBS from day 3 to day 7 post-insemination. b The number of 8-cell embryos allotted to each treatment. c MOR = morulae. d BLST = blastocysts. e HT/HD BLST = hatching and/or hatched blastocysts. f,g Within a column, numbers without a common superscript are significantly different (P<0.05).
87

0
10
20
30
40
50
MORL Early BLST BLST Expand BLST
% o
f Em
bryo
s
ControlCAF
a
b
a a
a
b
a
b
A
0
10
20
30
40
50
MORL Early BLST BLST ExpandedBLST
HatchingBLST
HatchedBLST
% o
f Em
bryo
s
B ControlCAF
a a a
aaa a a
a baa
0
10
20
30
40
50
BLST ExpandBLST
HatchingBLST
HatchedBLST
% o
f Em
bryo
s
ControlCAF
b
a
aa
aa
a a
C
Figure 6.2. Development of 8-cell bovine embryos cultured in CR1aa supplemented with 5% fetal bovine serum (control) or 20% chick amniotic fluid (CAF) on day 7 of development (A), day 8 of development (B) and day 9 of development (C). Within a developmental stage, columns without a common superscript are significantly different (P<0.05).
88
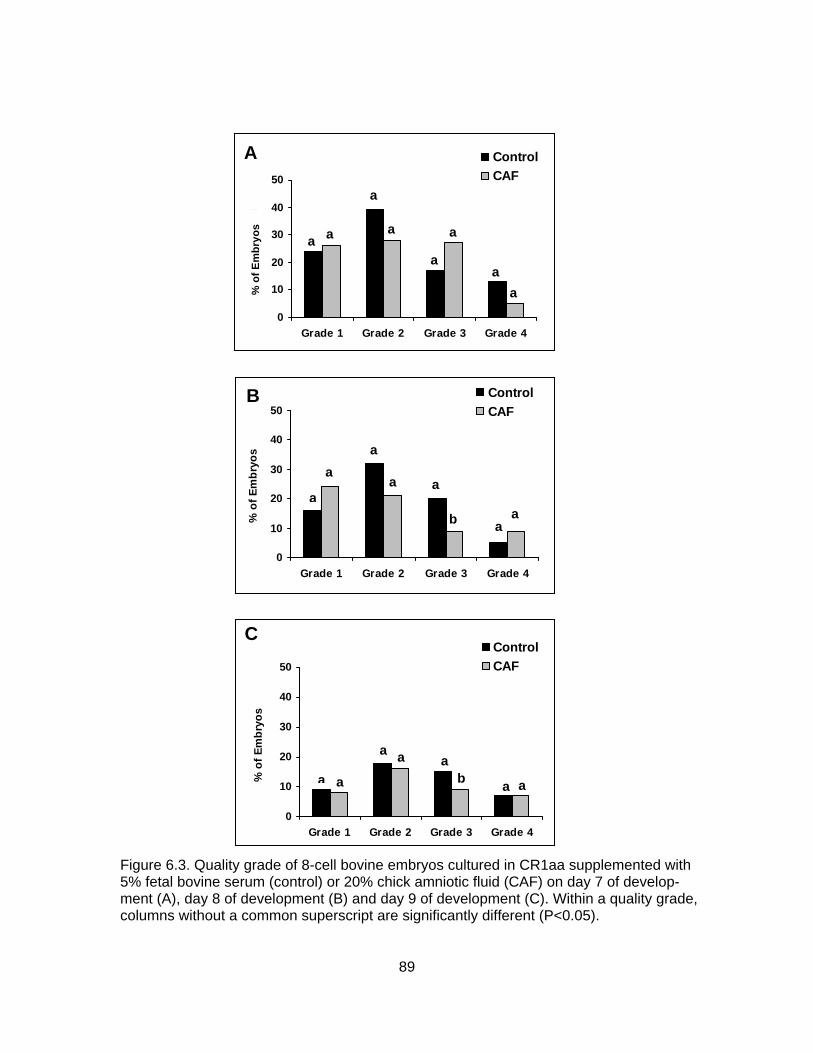
0
10
20
30
40
50
Grade 1 Grade 2 Grade 3 Grade 4
No.
of E
mbr
yos
ControlCAF
% o
f Em
bryo
s
aa
a
a
a
a
aa
A
0
10
20
30
40
50
Grade 1 Grade 2 Grade 3 Grade 4
% o
f Em
bryo
s
ControlCAF
a
aa
a a
b aa
B
0
10
20
30
40
50
Grade 1 Grade 2 Grade 3 Grade 4
% o
f Em
bryo
s
ControlCAF
a a
a a ab a a
C
Figure 6.3. Quality grade of 8-cell bovine embryos cultured in CR1aa supplemented with 5% fetal bovine serum (control) or 20% chick amniotic fluid (CAF) on day 7 of develop-ment (A), day 8 of development (B) and day 9 of development (C). Within a quality grade, columns without a common superscript are significantly different (P<0.05).
89

et al., 1996; Schmidt et al., 1996; Walker et al., 1996; Kruip and den Daas, 1997; Hill et al., 1999, 2001, 2002). In addition, despite extensive quality control measures, the
growth promoting properties of FBS and BSA have been observed to vary from batch to
batch. Early studies by Sirard and Lambert (1985) found that batches of serum from
different animals prepared by identical methods had very different effects on embryo
development.
Although the embryotropic properties of avian amniotic fluid remain largely
undefined, results of the present study suggest that it may serve as a potential
alternative to serum supplementation of in vitro embryo culture media, thus possibly
circumventing some of the potential risks associated with the use of FBS and other
materials of bovine origin. Amniotic fluid is an ultrafiltrate produced in close proximity to
the developing fetus and has been reported to be less variable in chemical composition
than serum or plasma (Shirley et al., 1985; Gianaroli et al., 1986) and is likely to contain
growth factors, hormones and other proteins beneficial to embryo development
(Mohamed and Noakes, 1985; Wintour et al., 1986). In addition, amniotic fluid, unlike
serum, is not likely to contain unexpected contaminates that would adversely affect
embryo development (Gianaroli et al., 1986).
Amniotic fluid has been used for the maturation of porcine (Nagi et al., 1990) and
bovine (Ocana Quero et al., 1995) oocytes as well as for the culture of murine (Gianaroli
et al., 1986; Sueldo et al., 1988; Coetzee et al., 1989; Dorfman et al., 1989; Oettle and
Wiswedel, 1989), bovine (Javed and Wright, 1990) and human (Gianaroli et al., 1986;
Dorfman et al., 1989) embryos. The results of these studies are somewhat conflicting,
however. For example, Gianaroli et al. (1986) and Coetzee et al. (1989) found that
culturing murine embryos in undiluted human amniotic fluid did not improve blastocyst
rates over controls. In contrast, Dorfman et al. (1989) and Oettle and Wiswedel (1989)
reported that culturing murine embryos in undiluted human amniotic fluid significantly
increased blastocyst development over controls. Similar discrepancies were noted when
culturing human embryos in amniotic fluid. Both Gianaroli et al (1986) and Dorfman et al.
(1989) indicated no significant difference in cleavage rates. In contrast, Dorfman et al.
(1989) noted a marked decrease in pregnancy rates after culture in human amniotic
fluid, whereas, Gianaroli et al. (1986) found no significant difference. One explanation for
these discrepancies may be the use of amniotic fluid from women of advanced maternal
90

age by Dorfman et al. (1989). It must be assumed that these women also served as
embryo recipients, as it is not stated otherwise in these publications. All in all, these
results must be evaluated with caution as each experiment utilized different control
media and amniotic fluid samples from varying stages of gestation.
Although the use of CAF in the present experiment resulted in significantly fewer
total blastocysts on day of 7 of development, there was no significant difference (P<0.05)
in the number of embryos classified as quality grade 1 when compared with embryos
cultured in CR1aa supplemented with 5% FBS. In addition, there were fewer grade 3
embryos in the CAF treatment group on both day 8 and day 9 of development. These
results suggest that the significant increase in blastocyst yield in the control group was
composed mostly of embryos of quality grade 3 or grade 4.
Results of the present study do indicate, however, that embryos cultured in the
presence of CAF undergo a delay in development of ~24 hours. It has been previously
reported that development of in vitro-produced bovine embryos is delayed by ~30 to 40
hours when compared with embryos produced in vivo (McGaugh et al, 1974; Prather
and First, 1988; Funahashi et al, 1994; Grisart et al., 1994). Van Soom et al. (1992)
suggested that this delay indicates that development past the 8- to 16-cell stage is
extremely sensitive to culture environment. The fact that embryos cultured in the
presence of CAF develop slower than the controls is somewhat concerning. There are
reports that indicate that morula and blastocyst stage embryos are able to increase their
rate of development to match that of the uterus after transplantation, however (Wilmut
and Sales, 1981). This finding would theoretically allow for the normal development of a
transferred embryo from the CAF treatment group even though it may be
developmentally slower than embryos in the control group.
In conclusion, results of the present study indicate that CAF, when added as a
supplement to bovine culture medium, provides proteins and growth factors which are
able to support in vitro development of bovine embryos. Thus, supplementation of
bovine embryo culture medium with chick amniotic fluid may be a feasible alternative to
serum supplemented medium for the production of quality bovine embryos, particularly
in situations where FBS is not readily available. The fact that CAF-supplemented
medium resulted in more grade 1 embryos from fewer total blastocysts is promising and
warrants further investigation. More thorough examination of such factors as CAF
91

concentration in the medium, and more importantly, evaluation of pregnancy and calving
rates following transfer of CAF-cultured embryos to recipient females, is warranted and
must be conducted to determine the potential benefits of this supplement to bovine in
vitro embryo production.
92

CHAPTER VII
ESTABLISHMENT AND CULTURE OF CELLS DERIVED FROM DAY-6 CHICK EMBRYOS
INTRODUCTION
In vitro embryo culture has been an active area of research for many years;
however, few researchers have completely identified the developmental needs of the
resulting embryos (Liebfried-Rutledge, 1999). With current embryo culture methods, only
30 to 40% of in vitro-matured and fertilized ova routinely reach the blastocyst stage
(Krisher and Bavister, 1998; Lonergan et al., 2003). Moreover, in vitro-produced (IVP)
embryos have marked differences in morphology (Greve et al., 1995), timing of
development (Van Soom et al., 1997), embryonic metabolism (Khurana and Niemann,
2000) and subsequent pregnancy rates (Enright et al., 2000) when compared with in
vivo-derived embryos.
In vitro embryo production has also been associated with abnormal fetal and
neonatal development and growth, known collectively as large calf or large offspring
syndrome (reviewed by Kane, 2003). Specifically, offspring produced from embryos
cultured in vitro often exhibit increased birth weights (Willadsen et al., 1991; Keefer et al,
1994; Behboodi et al., 1995), longer gestation lengths (Walker et al, 1996), increased
placental anomalies, (Hasler, et al., 1995; Hill et al., 1999, 2001, 2002) and increased
incidences of abortion (Walker et al., 1996; Hill et al., 1999) and perinatal (Behboodi et
al., 1995; Wilson et al., 1995; Garry et al., 1996; Schmidt et al., 1996; Kruip and den
Daas, 1997; Hill et al., 1999) and neonatal mortality (Schmidt et al., 1996; Kruip and den
Daas, 1997; Hill et al., 1999). Breathing difficulties (Young et al., 1998) and perturbations
in the growth and development of body organs are also common following culture of IVP
embryos (Kane, 2003). It has been hypothesized that exposure of pre-elongation
embryos to unusual environmental conditions may increase the incidence of large
offspring syndrome (reviewed by Kane, 2003); however, the exact cause of this
condition still remains unclear.
The inadequacies associated with in vitro embryo production have led to the
development of several alternative culture systems. One such system involves culturing
mammalian embryos in the amniotic cavity of a live chick embryo. Previous reports
93

indicate that factors within the amniotic cavity of developing chick embryos are capable
of supporting the maturation of porcine oocytes (Ocampo et al., 1994) and development
of embryos in various mammalian species (Blakewood et al., 1989a, 1989b, 1990). In
early experiments, chick embryo co-culture consistently resulted in significantly more
expanded and hatched blastocysts compared with controls in mice (Blakewood et al.,
1989a), pigs (Ocampo et al., 1993), goats (Blakewood et al., 1989a, 1990) and cattle
(Blakewood et al., 1989b). In addition, live offspring were produced following transfer of
caprine embryos that had been cultured in the chick amnion (Blakewood et al., 1990).
Experiments presented in this dissertation indicate that the chick embryo co-culture
system is capable of supporting development of bovine embryos at rates similar to more
modern embryo culture systems.
It is unclear, however, exactly where the benefits of this system lie. One theory is
that the chick embryo is a “live” system compared with the static embryo culture systems
currently in use. Because the living chick embryo actively regulates the pH, osmolarity
and O2 content of its own environment, it may serve as a more stable physiological
system for mammalian embryo development than current in vitro culture systems
(Blakewood et al., 1989a). An alternative hypothesis is that the chick embryo and/or
amnion actively synthesize growth factors and other factors that may stimulate growth
and development of mammalian embryos, a benefit unavailable in current embryo
culture systems without supplementation of fetal bovine serum (FBS) or bovine serum
albumin (BSA) (Blakewood et al., 1989a). It is also possible that the embryotropic
properties of the chick embryo amnion are a combination of both aforementioned
hypotheses.
Therefore the objective of the current research is to develop a protocol for the in
vitro culture of fetal chick cells and chick amnion cells as a preliminary experiment to
study the contributions of these cell types to bovine embryo development. More
specifically, we wanted to: 1) determine which of two media support cell growth and
proliferation most efficiently, 2) evaluate the growth characteristics of these cells in vitro
and 3) evaluate the post-thaw viability of these cells.
94

MATERIALS AND METHODS Experimental Design Experimental designs for Experiment 7.1 and 7.2 are given in Table 7.1 and
Table 7.2, respectively. Treatment groups for Experiment 7.1 consisted of bovine
fibroblasts cultured in modified Tissue Culture Medium-199 (mTCM; Treatment A;
control), bovine fibroblasts cultured in modified Dulbecco’s Modified Eagle’s Medium
(mDMEM; Treatment B; control), fetal chick cells cultured in mTCM (Treatment C) and
fetal chick cells cultured in mDMEM. Treatment groups for Experiment 7.2 consisted of
bovine fibroblasts cultured in mTCM (Treatment A; control), bovine fibroblasts cultured in
mDMEM (Treatment B, control), chick amnion cells cultured in mTCM (Treatment C) and
chick amnion cells cultured in mDMEM (Treatment D). In both experiments the
parameters evaluated for each treatment included number of days to 50% confluence,
number of days before each of two passages, evaluation of morphology and cell type,
population doublings, plating efficiency and post-thaw viability.
Primary cultures of bovine fibroblasts, fetal chick cells or chick amnion cells were
taken and processed. All cell types were maintained in both mTCM and mDMEM to
determine the effect of culture media on cell growth and proliferation. Media were
changed every 48 hours and cells were passaged at ~ 95% confluence. Cells were
passaged two times before being frozen and stored in liquid nitrogen. Plating efficiency
was determined 24 hours after passage and growth curves were performed every 24
hours for 5 days or until cells reached confluence. Cell viability was determined at each
passage.
To determine post-thaw viability, cells were maintained in liquid nitrogen for a
minimum of 48 hours before being thawed and replated. Plating efficiency was
determined 24 hours after thawing, and viability was determined when cells reached
~ 95% confluence.
Experimental Methods Media Preparation
Modified TCM was prepared by adding 5 ml of FBS (SH30070.02 HyClone,
Logan, UT) and 0.05 ml of gentamicin (50 mg/ml; 15750-060, Gibco, Grand Island, NY)
to 45 ml of TCM-199 (Gibco Laboratories, Grand Island, NY). Modified DMEM was
95
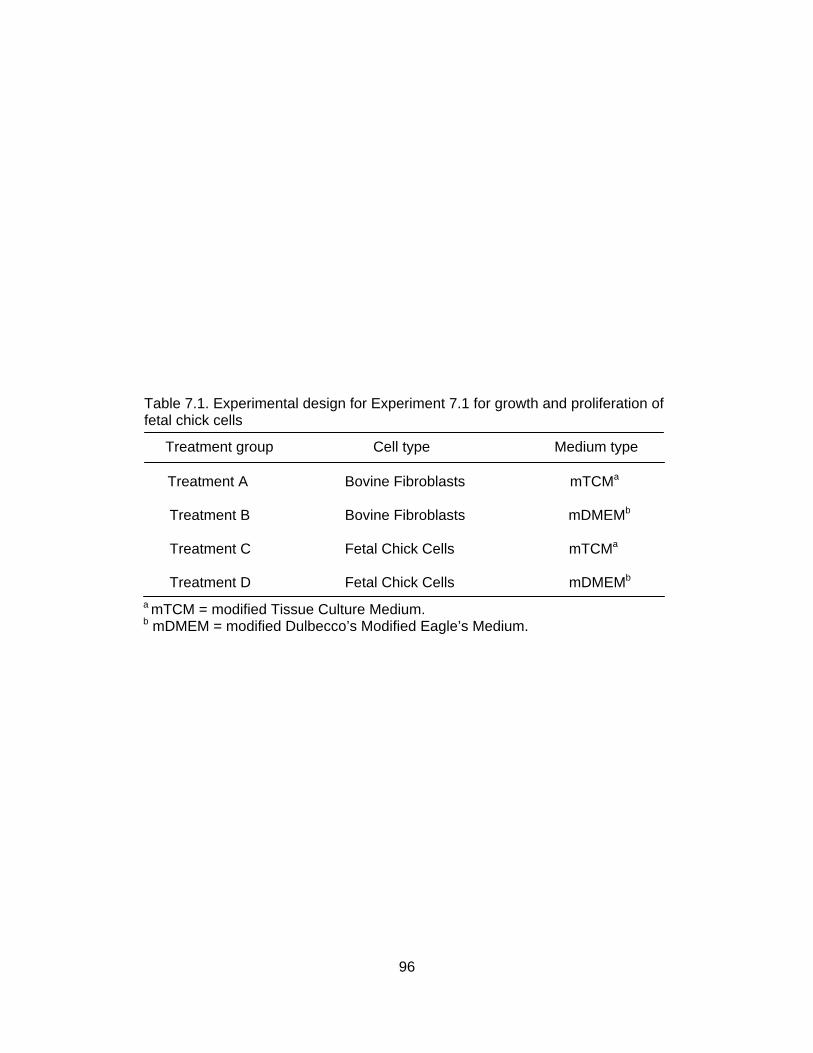
Table 7.1. Experimental design for Experiment 7.1 for growth and proliferation of fetal chick cells Treatment group Cell type Medium type
a mTCM = modified Tissue Culture Medium. b mDMEM = modified Dulbecco’s Modified Eagle’s Medium.
Treatment A Bovine Fibroblasts mTCMa
Treatment B Bovine Fibroblasts mDMEMb
Treatment C Fetal Chick Cells mTCMa
Treatment D Fetal Chick Cells mDMEMb
96

Table 7.2. Experimental design for Experiment 7.2 for growth and proliferation of chick amnion cells Treatment group Cell type Medium type
a mTCM = modified Tissue Culture Medium. b mDMEM = modified Dulbecco’s Modified Eagle’s Medium.
Treatment A Bovine Fibroblasts mTCMa
Treatment B Bovine Fibroblasts mDMEMb
Treatment C Chick Amnion Cells mTCMa
Treatment D Chick Amnion Cells mDMEMb
97

prepared by adding 7.5 ml of FBS, 0.5 ml of Modified Eagle Medium (MEM) amino acid
solution (10X, 11140-050, Gibco Laboratories, Grand Island, NY) and 0.05 ml of gentamicin to 41.5 ml of DMEM (10313-021, Gibco, Grand Island, NY). All media was
filtered through 0.2 µm Acrodisc filters (Pall Corporation, East Hills, NY) and allowed to
equilibrate in a 39°C and 5% CO2 incubator for a minimum of 30 minutes. Sterile, pre-
warmed TCM-199 was used to wash the serum residue from cells prior to trypsinization.
Shell-less Chick Culture for Chick Embryo Retrieval
The basis for the shell-less chick embryo culture procedure was modified from
that previously described by Blakewood and Godke (1989). Briefly, fertilized domestic
chicken eggs were obtained on the day of oviposition and stored at 10°C until the onset
of incubation. Six days prior to the day of embryo retrieval, eggs were removed from cold
storage and allowed to come to room temperature (25°C) before placing them in a 37°C
commercial egg incubator. After 72 hours of incubation, the eggs were rinsed with a 70%
ethanol solution (Aaper Alcohol and Chemical, CO, Shelbyville, KY) and then coated
with 7.5% providone iodine solution (Betadine®, Purdue Fredrick Company, Stanford,
CT) and allowed to air dry in a horizontal position to ensure proper positioning of the
chick embryo.
Egg contents were removed by making a crack in the shell by striking the center
of the egg against the rim of a sterile beaker. The egg contents were then released into
a 100-ml plastic embryo collection dish (Veterinary Concepts, Spring Valley, WI) that
had been covered with a piece of cellophane kitchen wrap (Saran®, Dow Chemical,
Midland, MI). The dish, including the egg contents, was loosely capped with a plastic lid
(Veterinary Concepts, Spring Valley, WI) and maintained in an incubator at 38°C for an
additional 72 hours before retrieval of the chick embryo and surrounding amnion.
On day 6 of shell-less culture the chick embryo and surrounding amnion were
dissected away from the rest of the egg components and then placed in a 35 x 10-mm
Falcon® plastic Petri dish (Becton and Dickerson, Lincoln Park, NJ).They were then
rinsed three times with sterile Dulbecco’s Phosphate Buffered Saline (DPBS; D1408,
Sigma-Aldrich Inc., St. Louis, MO) containing 1% FBS. The amniotic membrane was
gently dissected away from the chick embryo and placed in a 35 x 10-mm plastic petri
dish of mDPBS (mDPBS: 10 ml DPBS containing 0.01 ml of gentamicin). Any remaining
extraembryonic membranes were trimmed away from the chick embryo and the embryo
98

was placed in a 15-ml plastic conical tube (Corning, New York, NY) of mDPBS until
processing.
Establishment of Bovine Fibroblast Cell Lines.
Skin biopsies were taken from three adult cows of similar age (3 to 5 years of
age), body weights (408 to 498 kg) and body condition scores (4.5 to 5.5) using a 6-mm
diameter biopsy punch (Miltex Instrument Co, Bethphage, NY). Biopsies were obtained
from the dorsal aspect of the shoulder directly over the supraspinatus muscle. The tissue
was maintained in mDPBS until processing.
Using heat sterilized iris scissors, the dermal portion of the biopsy was finely
minced into pieces measuring ~1 mm2 in mDPBS. The tissue suspension was
transferred to a 15-ml plastic conical tube and washed in DPBS by swirling. The tissue
pieces were allowed to settle to the bottom of the tube, the supernatant discarded and 3
ml of fresh mDPBS was added. The process was repeated two additional times. After
the last wash the tissue was resuspended in 3 ml of culture medium (either mTCM or
mDMEM) and transferred to a 35 x 10-mm plastic petri dish. All cultures were
maintained at 39°C and 5% CO2 in a humidified atmosphere.
Fibroblasts began to grow from the tissue explants by about the fifth day at which
time the tissue explants were removed from the culture. Once a fibroblast cell layer was
established, the cells were maintained in the 35 x 10-mm plastic petri dish until they
reached ~95% confluency. Cells were then trypsinized by cold trypsinization. Briefly,
cells were rinsed two times with pre-warmed TCM and then maintained in cold (4°C)
trypsin (Trypsin EDTA, 1X, T3924, Sigma-Aldrich Inc., St. Louis, MO) at room
temperature for 5 minutes and then incubated at 39°C for an additional 5 to 10 minutes.
The resulting cell suspension was transferred to a 15-ml plastic conical tube and the dish
rinsed twice with culture medium. The cells were then washed by centrifugation at 500 x
g for 10 minutes and the resulting pellet resupsended in 1 ml of culture medium for
counting. Small tissue culture flasks (25 cm2; Costar, Cambridge, MA) were seeded with
1x106 cells/ml and filled with 2 to 3 ml of culture medium before being placed in
incubation. The cells were passaged twice prior to cryopreservation in TCM or DMEM
supplemented with 20% FBS and 5% dimethylsulfoxide (DMSO; D128-500, Fischer
Scientific, Fairlawn, NJ) and then frozen in aliquots of 1x106 cells per cryovial.
99

Establishment of Fetal Chick Cell Lines
Primary cell cultures were prepared from day 6 chicken embryos. After removal
of the head and limbs, the chick embryos were finely minced into pieces measuring
~1 mm2 in mDBPS using heat-sterilized iris scissors. Tissue pieces were washed three
times in mDPBS by allowing the tissue to settle to the bottom of the petri dish and
removing the supernatant. To further disassociate the cells, the tissue pieces were
subjected to cold trypsinization as described above. Any undigested tissue pieces were
allowed to settle to the bottom of the dish and the supernatant was removed and placed
in a 15-ml plastic conical tube. The cells were then washed by centrifugation at 500 x g
for 10 minutes in 4 ml of mTCM. The resulting pellet was subjected to cold trypsinization
one additional time.
Prior to centrifugation, 0.5 ml of the resulting cell suspension was diluted in 3 ml
of mTCM and 0.5 ml in 3 ml of mDMEM. Each cell suspension was then centrifuged at
500 x g for 10 minutes and counted using a hemocytometer. Small tissue culture flasks
(25 cm2) were seeded at a density of 1x106 cells per ml and cultured at 39°C and 5%
CO2 in a humidified atmosphere. The cells were passaged twice prior to cryopreservation
in either TCM or DMEM supplemented with 20% FBS and 5% DMSO and then frozen in
aliquots of 1x106 cells per cryovial.
Establishment of Chick Amnion Cell Lines
The basic procedure for the establishment of chick amnion cell lines was adapted
from that previously reported for use in humans (Neeper et al., 1990). Using heat
sterilized forceps, pieces of the amnion were washed through three 35 x 10-mm plastic
petri dishes of mDPBS before being subjected to cold trypsinization as described above.
Any tissue pieces remaining after cold trypsinization were manually disassociated by
pipetting. The resulting cell suspension was aspirated from the dish and placed in a 15-
ml plastic conical tube and the dish washed twice with 2 ml of mTCM. The rinse medium
was added to the conical tube and the suspension mixed well. A 2.0 ml aliquot of cell
suspension was added to each of two 15-ml plastic conical tubes and the cells in each
tube were counted using a hemocytometer. Each cell suspension was then centrifuged
at 500 x g for 6 minutes and resuspended in the appropriate culture media (mTCM or
mDMEM). The cells were subpassaged twice prior to cryopreservation in either TCM or
100

DMEM supplemented with 20% FBS and 5% DMSO and then frozen in aliquots of 1x106
cells per cryovial.
Determination of Cell Viability
Cell viability was determined by a dye exclusion test with trypan blue (0.05%;
Sigma-Aldrich Inc., St. Louis, MO) as previously described by Phillips (1973). Briefly,
cells were trypsinized by cold trypsinization. After centrifugation, the pellet was
resupsended in 1 ml of culture medium. The cell suspension was diluted with trypan blue
at a 1:1 (v/v) ratio and cells were counted using a hemocytometer. Exclusion of trypan
blue from the interior of the cell was considered an indicator of viability. Viability was
determined by the formula [(number of live cells/total number of cells) x 100] and
reported as a percentage.
Determination of Plating Efficiency
To determine plating efficiency, each well of a Nunc® 4-well plate (Apogent
Technologies, Rochester, NY) was seeded with ~15,000 cells and incubated at 39°C
and 5% CO2. After 24 hours, medium was removed from each well and pooled for
counting. The pooled cell suspension was centrifuged at 500 x g for 6 minutes and the
pellet resuspended in 1 ml of culture medium. The cell suspension was diluted with
culture medium at a 1:1 (v/v) ratio and cells were counted using a hemocytometer.
Plating efficiency was determined by the formula [1 – (number of unplated cells/total
number of cells seeded)] x 100 and was reported as a percentage.
Determination of Population Doublings
For determination of amnion cell population doublings, five 35 x 10-mm plastic
petri dishes were seeded at 300,000 cells/ml and incubated at 39°C and 5% CO2 in a
humidified atmosphere. Every 24 hours, one petri dish from each treatment was
removed from incubation, trypsinized and counted to determine total cell number.
Population doublings were determined by the formula [log (final concentration/initial
concentration) x 3.33].
Statistical Analysis Data were analyzed using PROC GLM of the SAS Statistical Package (v. 8e,
1999, SAS Institute, Inc., Gary, NC). To determine statistical differences for time to first,
second and post-thaw passages, cell viability, plating efficiency and population
doublings means were compared using a post hoc least significant difference (LSD) test.
101

Differences with a probability (P) value of 0.05 or less were considered significant in this
study.
RESULTS The growth characteristics of in vitro-cultured fetal chick cells are summarized in
Table 7.3. There was no effect of culture media (P>0.05) on the time required for fetal
chick cells to become confluent. With the exception of time required for primary culture,
these cells displayed a similar growth pattern to bovine fibroblasts (P>0.05). Plating
efficiency was not affected (P>0.05) by culture media in either the first (84.2% and
70.4% for mTCM and mDMEM, respectively) or the second (84.7% and 93.2% for
mTCM and mDMEM, respectively) passage (data not shown). Similar results were
observed for viability [first passage: mTCM = 98.3% and mDMEM = 90.4%; second
passage: mTCM = 85.0% and mDMEM = 91.9% (data not shown)]. Plating efficiency
and viability were not evalutated for primary culture or post-thaw passages.
The growth characteristics of in vitro-cultured chick amnion cells are summarized
in Table 7.4. There was no effect of culture media (P>0.05) on the time required for chick
amnion cells to become confluent (Table 7.4). Plating efficiency was not affected
(P>0.05) by culture media in primary culture (93.4% and 94.0% for mTCM and mDMEM,
respectively), first passage (73.9% and 59.0% for mTCM and mDMEM, respectively),
second passage (72.7% and 69.4% for mTCM and mDMEM, respectively) or after
freezing and thawing (47.0% and 39.5% for mTCM and mDMEM, respectively; data not
shown). Similar results were observed for viability (first passage: mTCM = 74.0% and
mDMEM = 72.1%; second passage: mTCM = 66.0% and mDMEM = 58.0%; post-thaw:
mTCM = 77.0% and mDMEM = 63.4%; data not shown).
Population doublings were evaluated for each passage to determine if chick
amnion cells were actively proliferating in vitro. When chick amnion cells were cultured in
mTCM, population doubling rate increased over the five day evaluation period for the
first passage, but remained relatively constant for the second passage and after freezing
and thawing (Figure 7.1). There was no significant difference (P>0.05) between
passages, however. In contrast, when chick amnion cells were cultured in mDMEM, the
population doubling rate remained fairly constant for all three passages evaluated
(Figure 7.2). As with the cells grown in mTCM, there was no significant difference
between passages (P>0.05).
102
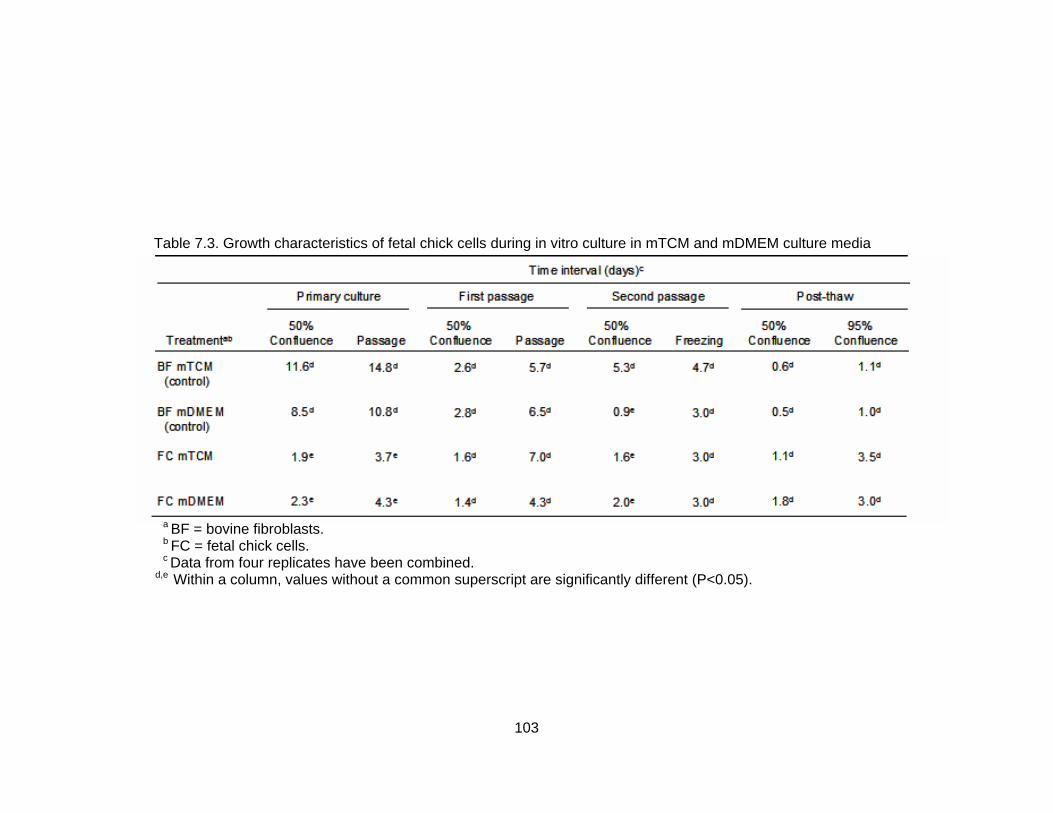
Table 7.3. Growth characteristics of fetal chick cells during in vitro culture in mTCM and mDMEM culture media
a BF = bovine fibroblasts. b FC = fetal chick cells. c Data from four replicates have been combined. d,e Within a column, values without a common superscript are significantly different (P<0.05).
103
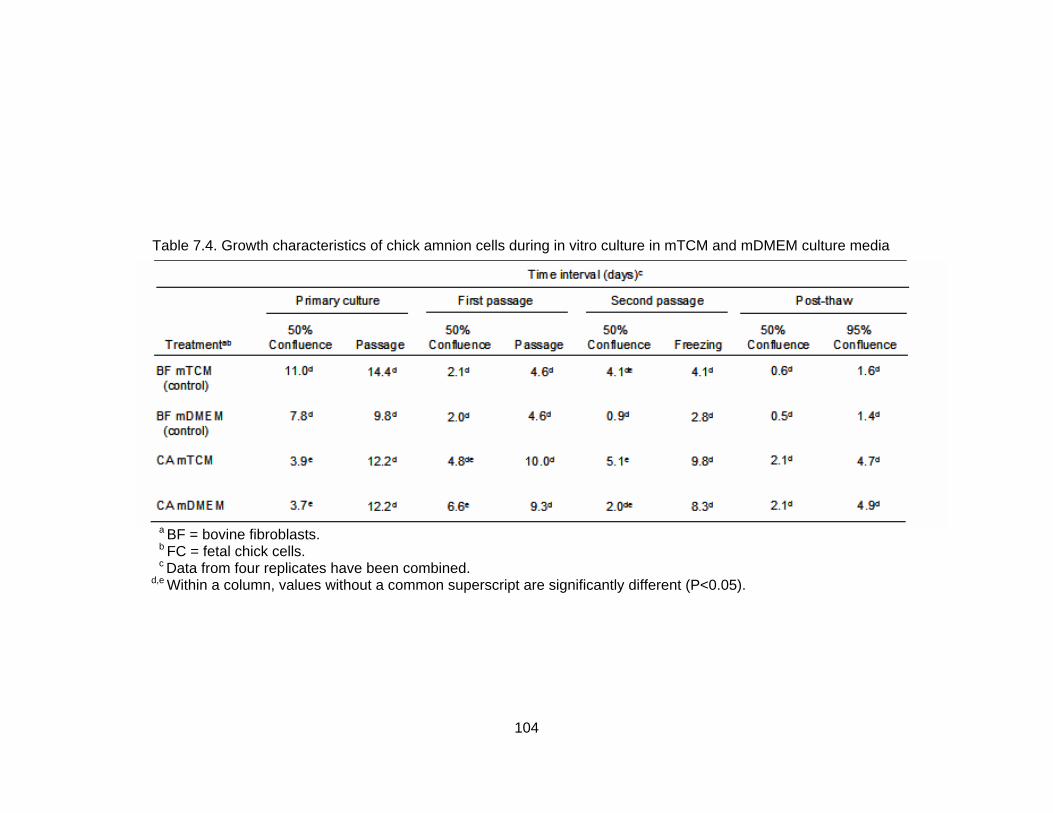
Table 7.4. Growth characteristics of chick amnion cells during in vitro culture in mTCM and mDMEM culture media
a BF = bovine fibroblasts. b FC = fetal chick cells. c Data from four replicates have been combined. d,e Within a column, values without a common superscript are significantly different (P<0.05).
104
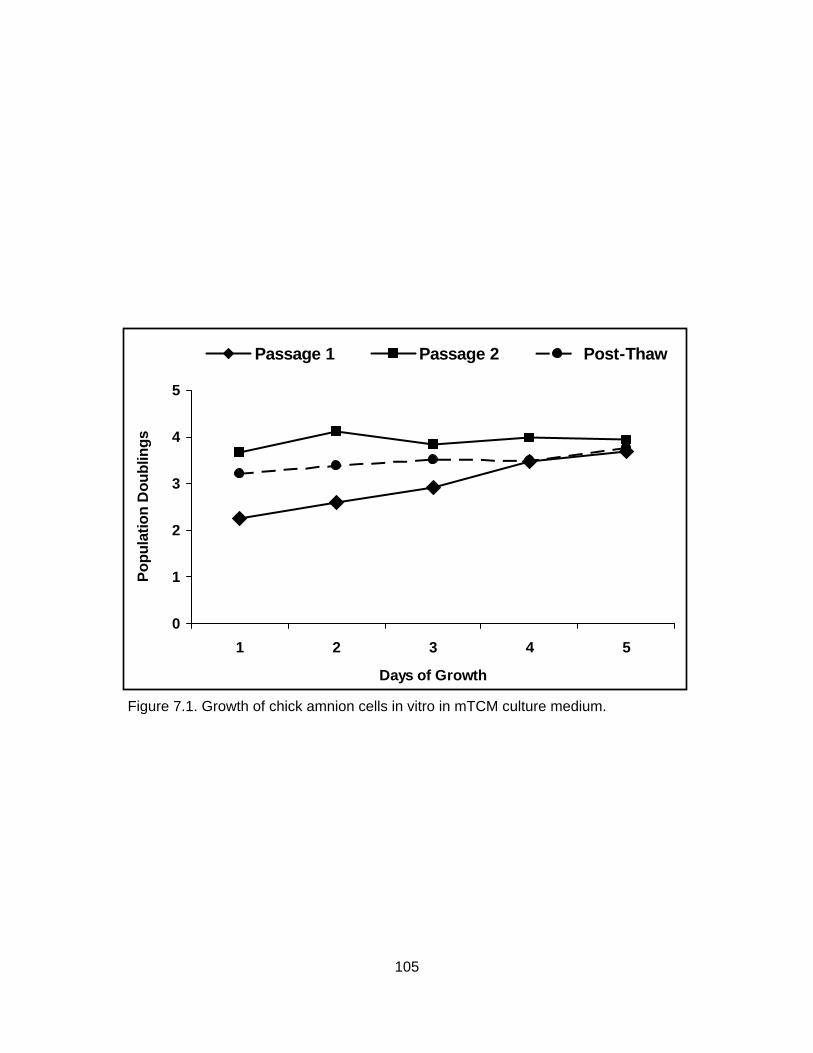
0
1
2
3
4
5
1 2 3 4 5
Days of Growth
Popu
latio
n D
oubl
ings
Passage 1 Passage 2 Post-Thaw
Figure 7.1. Growth of chick amnion cells in vitro in mTCM culture medium.
105

0
1
2
3
4
5
1 2 3 4 5
Days of Growth
Popu
latio
n D
oubl
ings
Passage 1 Passage 2 Post-Thaw
Figure 7.2. Growth of chick amnion cells in vitro in mDMEM culture medium.
106

Both epithelial cells and fibroblast cells were present in in vitro chick amnion cell
cultures. During the first passage, epithelial cell populations increased through day 3 but
then declined. In contrast, fibroblast cell populations increased progressively throughout
the five day evaluation period (Figure 7.3). Similar results were observed during the
second passage and after freezing and thawing (Figures 7.4 and 7.5, respectively).
DISCUSSION Results of the present study indicate that cultures of fetal chick cells and chick
amnion cells can be maintained in vitro. Furthermore, these culture systems appear to
be able to promote cell growth and proliferation through multiple passages. In general,
cultures of both cell types maintained under the conditions described herein survive well
in vitro and do not appear to be adversely affected by freezing and thawing.
The two media (mTCM and mDMEM) were selected for this experiment because
one (mTCM-199) is a traditional medium developed for chick embryonic cells and the
other (mDMEM) was developed for the culture of amniocytes. TCM-199, the major
component of mTCM, was originally reported for the culture of chick embryo fibroblasts
(Morgan et al., 1950) and is now commonly used for the culture of many mammalian cell
types. It is the traditional medium used in our laboratory for the cultivation and
maintenance of bovine and caprine fibroblast cell lines. DMEM was chosen as an
alternative medium as it has been successfully used for the propagation of human
amnion cells (Neeper et al., 1990; Wang et al., 1992) and is the base medium for several
commercial human amnniocyte cell culture media.
Plating efficiency has been suggested to be a highly sensitive test for the
detection of noxious substances in the culture environment (Seemayer et al., 1987).
Since plating efficiency was not significantly different between the two media, it can be
concluded that there were no immediate adverse affects of either culture medium on
fetal chick cells. It must be pointed out, however, that although plating efficiency can be used as a measure of the cell’s response to its surroundings, it does not indicate if those
surroundings are capable of supporting sustained cell growth. The results of the trypan
blue exclusion test indicate that there was no significant difference in cell viability
between the two media. Viability was determined at the end of each passage and when
taken together with the outcome of the plating efficiency test, it can be concluded that
both media are capable of sustaining fetal chick cells in vitro.
107
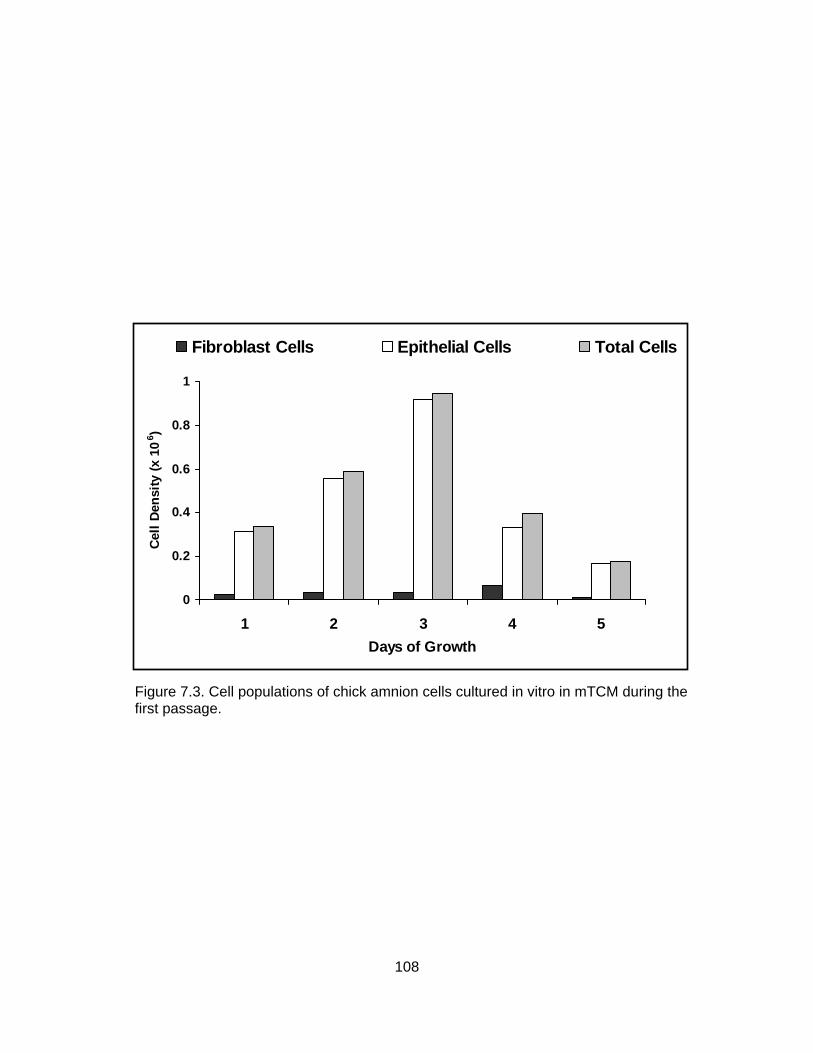
0
0.2
0.4
0.6
0.8
1
1 2 3 4 5Days of Growth
Cell
Dens
ity (x
106 )
Fibroblast Cells Epithelial Cells Total Cells
Figure 7.3. Cell populations of chick amnion cells cultured in vitro in mTCM during the first passage.
108

0
1
2
3
4
5
6
1 2 3 4 5Days of Growth
Cell
Dens
ity (x
106 )
Fibroblast Cells Epithelial Cells Total Cells
Figure 7.4. Cell populations of chick amnion cells cultured in vitro in mTCM during the second passage.
109

0
0.5
1
1.5
2
2.5
3
3.5
1 2 3 4 5Days of Growth
Cell
Dens
ity (x
106 )
Fibroblast Cells Epithelial Cells Total Cells
Figure 7.5. Cell populations of chick amnion cells cultured in vitro in mTCM after freezing and thawing. .
110

As with the fetal cells, both media appear to support the growth of chick amnion
cells in vitro. Neither plating efficiency nor viability were significantly different between
the two media. These findings were somewhat unexpected since DMEM has been used
quite successfully for the culture of human amnion cells (Neeper et al., 1990; Wang et
al., 1992). However, although both media are capable of sustaining growth of chick
amnion cells in vitro, neither appear to promote adequate cell proliferation. With the
exception of the primary culture passage of mTCM, evaluation of population doublings
indicate that the rate of cell growth was approximately equal to the rate of cell death.
These findings suggest that while capable of sustaining cell growth, neither medium is
suitable for the proliferation of chick amnion cells in vitro at an optimal level.
General morphometic features of both the fetal chick cells and chick amnion cells
indicate a mixed culture of epithelial cells and fibroblast cells. Epithelial cells appeared
generally round and tended to form dense colonies. In contrast, fibroblasts were spindle
shaped with long, branching pseudopodia and tended to form monolayers rather than
colonies. It is generally believed that trypsinization of cell cultures, as performed in this
experiment, selects against epithelial cells, resulting in cell populations predominated by
fibroblast cells (Kenneth Bondioli, personal communication). This does not appear to be
the case when culturing bovine amnion cells as epithelial cells were present in all three
passages evaluated. Further passages may be required in order to observe this
phenomenon. When population doublings were evaluated within a single passage,
epithelial cell numbers increased progressively through day 3 of culture before declining.
This suggests that neither media type are conducive to epithelial cell proliferation. It may
be necessary to use a medium capable of supporting growth and proliferation of both
epithelial and fibroblast cells if the integrity of the amnion is to be maintained.
In conclusion, we have successfully developed a repeatable protocol for culturing
both fetal chick cells and chick amnion cells. However, both systems may require some
optimization. This is believed to be the first report of culture of cells derived from the
chick embryo amnion. The current research provides a tool for in vitro studies of the
amnion and the chick embryo and provides an in vitro model for the study of the effects
of chick embryo co-culture on mammalian embryo development.
111

CHAPTER VIII
THE INCIDENCE OF APOPTOSIS IN BOVINE EMBRYOS AFTER IN VITRO CULTURE AND CHICK EMBRYO CO-CULTURE
INTRODUCTION
Although much research has been devoted to the development and improvement
of in vitro embryo production techniques, differences still remain between in vitro- and in
vivo-derived embryos. The development of immature bovine oocytes to the blastocyst
stage appear to have plateaued at about 30 to 40% following in vitro maturation,
fertilization and culture (Krisher and Bavister, 1998; Lonergan et al., 2003). In addition,
the quality of blastocysts produced in vitro rarely, if ever, reach that of in vivo-derived
blastocysts (Duby et al., 1997; Boni et al., 1999; Viuff et al., 1999), and marked
differences have been reported to exist between in vivo- and in vitro-derived embryos
with regard to morphology (Greve et al., 1995), timing of development (Van Soom et al.,
1997), embryonic cell numbers (Van Soom et al., 1997), embryonic metabolism
(Khurana and Niemann, 2000) and gene expression (Wrenzycki et al., 1996; Knijn et al.,
2002) and incidences of chromosomal abnormalities (Viuff et al., 2001).
With many of the current embryo culture systems, the addition of fetal bovine
serum (FBS) is necessary for successful development of the embryonic blastoceol and
continued embryo development (Pinyopummintr and Bavister, 1994; Thompson et al.,
1998). However, serum and other compounds of bovine origin have been implicated in
delayed embryo development (Van Soom et al., 1997), reduced embryo quality (Duby et
al., 1997; Boni et al., 1999; Viuff et al., 1999), reduced pregnancy rates (Enright et al.,
2000) and numerous fetal and/or calf abnormalities (Thompson et al., 1995; Walker et
al., 1996; Sinclair et al., 1999).
Apoptosis is a critical process during pre-implantation development for the
disposal of excess cells, cells that are in the way, abnormal or potentially dangerous
(Gjorret et al., 2003). However, elevated levels of apoptotic cells have been correlated
with abnormal embryo morphology (Hardy et al., 1989), and it has been hypothesized
that when apoptosis surpasses a certain “threshold level” further embryonic
development is compromised (Tam, 1988; Juriscova et al., 1998; Hardy, 1999) and
nonviable offspring are eliminated (Byrne et al., 1999). In addition, apoptosis may be a
cellular response to stress and suboptimal culture conditions (Gjorret et al., 2003) and
112

evidence suggests that the fetal anomalies and increased incidences of abortion
associated with in vitro culture may be the result of distortions to normal apoptotic
processes (Sadler and Hanter, 1994).
As an alternative to traditional in vitro culture systems utilizing serum
supplementation, Blakewood and Godke (1989) developed an alternative culture system
in which pre-implantation mammalian embryos were cultured in the amniotic cavity of
live chicken embryos. This culture system has been successful in supporting the
development of pre-implantation embryos from several mammalian species including
mice (Blakewood et al., 1989a), goats (Blakewood et al., 1989a), cattle (Blakewood et
al., 1989b; Murakami et al., in press) and pigs (Ocampo et al., 1993). Moreover, live
transplant offspring have been produced following chick embryo co-culture of 2- to 8-cell
caprine embryos (Blakewood et al., 1990).
In this study, we propose to compare the occurrence of apoptosis in fresh and
frozen-thawed in vivo-derived embryos and in embryos cultured in vitro and in the chick
embryo co-culture system in order to evaluate the efficacy of the use of the chick embryo
co-culture system as a potential embryo culture environment. Traditional analysis of
apoptosis in pre-implantation mammalian embryos have utilized the TUNEL reaction to
label fragmented DNA in apoptotic cells. However, in this study we evaluated the
incidence of apoptosis using a fluorescent labeled inhibitor to caspases-3 and -7.
MATERIALS AND METHODS Experimental Design
The synchronization and superovulation scheme for in vivo embryo production is
illustrated in Figure 8.1. In vivo embryos were collected from beef females which were
synchronized and superstimulated. Briefly, females were synchronized with CIDRs for 8
days. On day 11 of the synchronized cycle, females were superstimulated with follicle
stimulating hormone (FSH) in a decreasing dose schedule and inseminated 12 and 24
hours after visual observation of estrus. Embryos were nonsurgically recovered either
6.5 or 7 days after the onset of estrus (estrus = day 0). Embryos were fluorescently
labeled and evaluated for caspase activity immediately upon recovery.
The process for production of in vitro embryos is illustrated in Figure 8.2.
In vitro embryos were produced using the standard IVF protocol for our laboratory.
113

EXPERIMENTAL DESIGN
Figure 8.1. Experimental design for the production and fluorescent labeling of in vivo-derived embryos.
114

Briefly, oocytes were matured for 20 to 22 hours before being exposed to motile
spermatozoa from a bull of proven fertility for IVF. After 5 hours of co-incubation with
spermatazoa, oocytes were removed from fertilization and placed into culture in CR1aa.
On day 3 post-insemination medium was removed and replaced with CR1aa containing
5% FBS. Embryos were fluorescently labeled and evaluated for caspase activity on day
7 post-insemination. Because it was not possible to perform all treatments concurrently,
two replications of IVF were performed (IVF-I and IVF-II).
The process for production of chick co-cultured embryos is illustrated in Figure
8.3. Chick co-cultured embryos were produced as described for in vitro embryos;
however on day 3 post-insemination, bovine embryos were embedded in agar and
injected into the amniotic cavity of 96-hour chick embryos. After 96 hours of culture,
embryos were recovered from the chick amnion and fluorescently labeled for caspase
activity.
Experimental Methods Production of In Vivo Embryos
To obtain in vivo embryos, clinically healthy, non-lactating, beef females (age = 4
to 7 years of age; body weights = 386 to 612 kg; BCS = 4 to 6) were first synchronized
with a progesterone device (CIDR; Agtech, Inc, Manhattan, KS) for 8 days. Two days
prior to CIDR withdrawl, prostaglandin F2α (25 mg Lutalyse®; Pharmacia and Upjohn
Company, Kalamazoo, MI) was administered to ensure complete regression of the
corpus luteum (CL). At the time of CIDR withdrawl, GnRH (100 µg; Factrel®; Fort Dodge
Animal Health, Fort Dodge, IA) was administered to induce an LH surge. Beginning on
day 11 of the synchronized cycle (CIDR removal = Day 0), females received porcine
FSH (Sioux Biochemical, Inc., Sioux City, IA) twice daily, in decreasing doses, for 4
consecutive days: 5 units on the first day; 4 units on the second day; 4 units on the third
day; and 3 units on the fourth day. Prostaglandin-F2α (25 mg) was administered
concomitant with the sixth and seventh doses of FSH. Females were inseminated 12
and 24 hours after visual observation of standing heat with two straws of semen from a
bull of proven fertility. Embryos were collected nonsurgically from donors 6.5 or 7 days
after the first insemination. All embryos were evaluated for morphological stage and
quality grade by three technicians and a mean quality grade was determined.
115

EXPERIMENTAL DESIGN
Figure 8.2. Experimental design for the production and fluorescent labeling of embryos cultured in vitro.
116

EXPERIMENTAL DESIGN
Figure 8.3. Experimental design for the production and fluorescent labeling of embryos cultured in the chick embryo amnion.
117

Frozen in vivo-derived embryos were graciously provided by Dr. Neil Schrick (University
of Tennessee, Knoxville, TN). All embryos were obtained from beef females (Charolais,
Angus or Senepol) and were frozen in glycerol. All embryos were thawed for 2 minutes
in air and cryoprotectant removed using a 3-step protocol.
Production of In Vitro Embryos
Media Preparation
Brackett-Oliphant (BO) media were prepared as previously described by Brackett
and Oliphant (1975). Briefly, BO-A stock solutions contain 150.2 mM NaCL (S-5886,
Sigma-Aldrich Inc., St. Louis, MO), 2.7 mM KCl (P-5405, Sigma-Aldrich Inc. St. Louis,
MO), 3.0 mM CaCl2·2H2O (C-7902, Sigma-Aldrich Inc., St. Louis, MO), 1.0 mM
NaH2PO4·H2O (S-9638, Sigma-Aldrich Inc., St. Louis, MO), 0.6857 mM MgCl2·6H2O (M-
2393, Sigma-Aldrich Inc., St. Louis, MO) and 0.1 ml of 0.5% phenol red (P-0290, Sigma-
Aldrich Inc., St. Louis, MO) per 500 ml of sterile water. BO-B stock solutions contain
154.0 mM NaHCO3 (S-8875, Sigma-Aldrich Inc., St. Louis, MO) and 0.04 ml of 0.5%
phenol red per 200 ml of sterile water.
CR1aa medium was prepared as previously described by Rosenkrans and First
(1994). Briefly, CR1aa stock solution was prepared by adding 114.7 mM NaCl, 1.6 mM
KCl, 26.2 mM NaHCO3, 0.3999 mM pyruvic acid (P-4562, Sigma-Aldrich Inc., St. Louis,
MO), 2.5 mM L(+) lactic acid (L-4388, Sigma-Aldrich Inc., St. Louis, MO), 0.5195 mM
glycine (G-8790, Sigma-Aldrich Inc., St. Louis, MO), 0.5051 mM L-alanine (A-7469,
Sigma-Aldrich Incorporated, St. Louis, MO) and 0.2 ml of 0.5% phenol red per 100 ml of
sterile water.
Fertilization stock medium (BO-AB) was prepared by adding 1.2 mM pyruvic
acid, 0.05 ml of gentamicin (50 mg/ml; 15750-060, Gibco, Grand Island, NY) and 0.018
ml of heparin sodium (10,000 IU/ml; Elkins-Sinn, Cherry Hill, NJ) per 38 ml of BO-A
stock solution and 12 ml of BO-B stock solution. Fertilization medium (BO-caffeine) was
prepared by adding 0.0243 g of caffeine sodium (C-4144, Sigma-Aldrich Inc., St. Louis,
MO) per 25 ml of BO-AB. Sperm washing medium (0.6% BSA-BO) was prepared by
adding 0.03 g of bovine serum albumin (BSA; Fraction V, A-4503, Sigma-Aldrich Inc., St.
Louis, MO) to 5 ml of BO-AB. Oocyte washing medium (0.3% BSA-BO) was prepared by
adding 0.03 g of BSA (Fraction V) to 10 ml of BO-AB. TCM oocyte washing medium was
prepared by adding 0.03 g of BSA (Fraction V) to 10 ml of Tissue Culture Medium-199
118

(Eagle’s salt) with 25 mM HEPES Buffer (TCM-199; Gibco Laboratories, Grand Island,
NY).
CR1aa culture medium for day 0 to day 3 was prepared by adding 0.2 ml of
Basal Medium Eagle (BME) amino acid solution (50X, B-6766, Sigma-Aldrich Inc., St.
Louis, MO), 0.1 ml of Modified Eagle Medium (MEM) amino acid solution (10X, 11140-
050, Gibco Laboratories, Grand Island, NY), 0.01 ml of gentamicin, 1.1 mM L-glutamine
(G-5763, Sigma-Aldrich Inc., St. Louis, MO) and 0.03 g of BSA (fatty acid free, A-7511,
Sigma-Aldrich Inc., St. Louis, MO) to 9.5 ml of CR1aa stock solution. CR1aa culture
medium for day 3 to day 7 was prepared by adding 0.2 ml of BME amino acid solution,
0.1 ml of MEM amino acid solution, 0.5 ml of FBS (SH30070.02; HyClone, Logan, UT),
0.01 ml of gentamicin, 1.1 mM L-glutamine and 0.03 g of BSA (fatty acid free) to 9.5 ml
of CR1aa stock solution.
In Vitro Fertilization
Oocytes were purchased weekly from BoMed, Inc. (Madison, WI). The oocytes
were shipped in a portable incubator (~39°C) and allowed to mature during travel (20 to
22 hours). Upon arrival at the laboratory, shipping vials were removed from the portable
incubator and stored in a humidified atmosphere of 5% CO2 in air at 39°C until
insemination.
Semen was prepared by thawing a straw of frozen semen from a Holstein bull
(CSS 7H5188, Genex Cooperative Inc., Shawano, WI) of proven fertility in a 39° to 40°C
water bath for ~45 seconds. The contents of the straw were then deposited into a 15-ml
plastic conical tube (Corning, New York, NY) and 9 ml of BO-caffeine was added to the
tube. The suspension was then centrifuged for 6 minutes at 200 x g. After centrifugation
the supernatant was removed and the remaining pellet was resuspended in an additional
9 ml of BO-caffeine. The resulting suspension was centrifuged for 6 additional minutes at
200 x g. The supernatant was removed again and the pellet resuspended in 4 ml of BO-
caffeine and 4 ml of 0.6% BSA-BO and placed in a 39°C incubator for ~10 minutes to
equilibrate. After equilibration, four 80-µl insemination drops were created in a preheated
35 x 10-mm Falcon® plastic Petri dish (Becton and Dickinson, Lincoln Park, NJ) and
covered with warmed mineral oil (M-8140, Sigma-Aldrich Inc., St. Louis, MO).
After the 20 to 22 hour maturation period, cumulus oocyte complexes were
washed through two 35 x 10-mm plastic petri dishes containing 2 to 3 ml of 0.3% BSA-
119

BO. Then, 10 to 20 oocytes were placed in each of the 80-µl insemination drops
previously prepared and were co-incubated for 5 hours at 39°C and 5% CO2 in air.
After fertilization, oocytes were washed through a single 35 x 10-mm plastic petri
dish of TCM oocyte wash media. Cumulus cells were removed by vortexing for 3
minutes in 1 ml of TCM-199 containing 0.001 g of hyaluronidase (H-3506, Sigma-Aldrich
Inc., St. Louis, MO). After vortexing, oocytes were washed through two 35 x 10 mm
plastic petri dishes containing TCM oocyte wash and one 35 x 10-mm plastic petri dish
of CR1aa. Then, 10 to 15 oocytes per drop were placed in 50-µl drops of CR1aa
covered with warmed mineral oil and incubated in a modular incubator containing 5%
CO2, 5% O2, and 90% N2 at 39°C.
Chick Embryo Co-Culture
The basic procedure for chick embryo co-culture was modified from that
previously described by Blakewood and Godke (1989). Briefly, fertilized domestic
chicken eggs were collected on the day of oviposition and stored at 10°C until the onset
of incubation. Four days prior to the introduction of bovine embryos, the eggs were
removed from cold storage and allowed to come to room temperature (25°C). They were
then placed in a commercial egg incubator maintained at 37°C and 80 to 90% relative
humidity. After 72 hours of incubation, the eggs were rinsed with a solution of 70%
ethanol (Aaper Alcohol and Chemical Company, Shelbyville, KY) and then coated with
7.5% providone iodine solution (Betadine®, Purdue Fredrick Company, Stanford, CT)
and allowed to air dry in a horizontal position to ensure proper positioning of the chick
embryo.
To remove the egg contents, a crack was made in the shell by striking the
center of the egg against the rim of a sterile beaker. The egg contents were then
released into a 100-ml plastic embryo collection dish (Veterinary Concepts, Spring
Valley, WI) covered by a piece of cellophane kitchen wrap (Saran®, Dow Chemical,
Midland, MI). The dish, including the egg contents, was loosely capped with a plastic lid
(Veterinary Concepts, Spring Valley, WI) and maintained in an incubator at 38°C for an
additional 24 hours before introducing the reconstructed bovine embryos into the
amniotic cavity of the chick embryo.
The procedure for embedding embryos in agarose was modified from that of
Willadsen (1979) for the culture of ovine embryos in the ligated oviducts of sheep. On
120

day 3 of culture, two or three 8-cell bovine IVF embryos were placed in a 50-µl drop of
low-melting agarose solution [1.2% agarose (A-9799, Sigma-Aldrich Inc., St. Louis, MO)
dissolved in Dulbecco’s Phosphate-Buffered Saline (DPBS; D-1408, Sigma-Aldrich Inc.,
St. Louis, MO)] at ~37oC and then aspirated into a beveled glass pipette (200 µm outer
diameter). After holding in air for ~30 seconds the agar (containing the embryos) within
the pipette was expelled as a gel cylinder into room temperature TL-HEPES
(BioWittaker, Walkersville, MD). The agarose cylinder, including the bovine embryos,
was then aspirated back into the beveled pipette, and carefully expelled into the amniotic
cavity of a 96-hour chick embryo.
After introducing the embedded embryos into the amniotic cavity the shell-less
culture systems were returned to incubation for an additional 72 hours at 39oC. Thus, a
single agarose cylinder containing two or three 8-cell bovine IVF embryos was co-
cultured in the amnion of each developing chick embryo for 4 days.
On day 7 of in vitro culture, the amnion containing the agarose cylinder was
carefully dissected away from the rest of the egg components and placed in a 35 x 10-
mm plastic petri dish. The amnion was then rinsed several times with DPBS containing
1% FBS. After rinsing, the petri dish was placed under a stereomicroscope (Nikon SMZ-
2B; Tokyo, Japan) equipped with an overhead light source and the agarose cylinder was
removed from the amniotic cavity by using two 22-guage, 37.5-mm needles (Kendall,
Mansfield, MA) to make a small opening in the amniotic membrane. The agarose
cylinder was carried from the amnion with the escaping amniotic fluid, settling on the
bottom of the dish. The cylinder was then washed twice in CR1aa medium and dissected
using two 22-gauge needles to isolate and remove the developing embryos.
Caspase Assay
Embryos were evaluated for the presence of cells undergoing apoptosis using a
fluorescent based assay for the detection of active caspases-3 and -7 (Caspatag®
Caspase 3,7 In Situ Assay Kit, Chemicon International, Inc.). Briefly, a 150X stock
solution of Caspatag® reagent [a carboxyflouroscein-labeled flouromethyl ketone peptide
inhibitor of caspases-3 and -7 (SR-DEVD-FMK)] was prepared by reconstituting 1 vial of
Caspatag® reagent in 0.05 ml of dimethyl sulfoxide (DMSO, D128-500, Fischer
Scientific, Fair Lawn, NJ). Embryos were then incubated in a 1500x Caspatag® working
solution (1 µl 150x Caspatag® stock solution diluted in 1.5 ml of phosphate buffered
121

saline, protected from light, for 1 hour at 39°C under 5% CO2 in air). After the 1 hour
incubation period, embryos were washed in 1x Caspatag® wash buffer (1ml of
Caspatag® wash buffer diluted in 10 ml of deionized water) for 5 minutes and then
stained in Hoerscht 33342 (1 mg/ml; bisBenzimide, B-2261, Sigma-Aldrich Inc., St.
Louis, MO).
After Hoerscht staining, embyos were placed on slides and the total number of
apoptotic cells were counted using an inverted microscope (Nikon Diphot; Tokyo, Japan)
equipped with a ultraviolet light and a rhodamine filter. Each embryo was counted three
times and the mean number of apoptotic cells was determined. Embryos in which
individual fluorescent points were not apparent were categorized as having profuse
staining patterns and were assigned an arbitrary apoptotic cell value of 20. Apoptotic
indices were calculated by dividing the mean number of apoptotic cells by the total
number of cells. The total number of cells for in vivo- and in vitro-derived embryos were
previously reported by Knijn et al. (2003; in vivo morulae and early blastocysts = 128
cells; in vivo blastocysts = 166; in vitro morulae and early blastocysts = 113; in vitro
blastocysts = 114). These number of cells per embryo were in agreement with those
reported by Van De Velde et al. (1999) and Koo et al. (2002). Positive controls were
prepared by incubating in vitro-produced embryos in cyclohexamide (1 mg/ml; C-7698;
Sigma-Aldrich Inc., St. Louis, MO) for 3 to 4 hours prior to staining.
Statistical Analysis Data are expressed as means and were analyzed using PROC GLM of the SAS
Statistical Package (v. 8e, 1999 SAS Institute, Inc., Gary, NC). Significant differences
among means for number of apoptotic cells and apoptotic index were tested using a post
hoc least significant difference test (LSD test). Differences with a probability (P) value of
0.05 or less were considered significant in this study.
RESULTS Estrous synchronization and superovulation response is summarized in Table
8.1. Of the 18 females receiving CIDRs, 17 (94%) were administered GnRH at the time
of CIDR removal. One female was removed from the study at this time due to a lost
CIDR. Of the females remaining, 17 (100%) were determined to have a CL, as
122

Table 8.1. Response of cows to synchronization/superovulation protocol
No. cows receiving CIDRs 18
No. cows receiving GnRH 17
No. cows with a CL at initiation of FSH 17
No. cows observed in heat after FSH and PGF2α 13
No. cows inseminated 17
No. cows nonsurgically collected 13
123

determined by ultrasound (Aloka, SSD 500V, Japan) evaluation, at the initiation of FSH
treatment. Following PGF2α administration, 13 of these females were detected in estrus
and were inseminated 12 and 24 hours following observation of standing estrus. Four
females did not display any sign of behavioral estrus and were inseminated at 60 and 72
hours after the last PGF2α injection. Following insemination, five females were collected
nonsurgically 6.5 days post-insemination and 8 females were collected nonsurgically 7
days post-insemination. These time points were selected to ensure both morula and
blastocyst stage embryos were harvested. Of the four females not collected, two were
found to have uterine infections, one was diagnosed with a cystic follicle without a CL
present, and one did not have a CL present on either ovary. There were 62 embryos
recovered from 146 detectable CL (42% recovery rate) in 13 females (Table 8.2). Of the
62 embryos recovered, 28 were morulae, 13 were blastocysts and 21 were degenerate.
Of the 62 total embryos recovered, 38 (61%) were classified as quality grade 1 or grade
2 embryos.
The mean number of apoptotic cells in morula, early blastocyst and blastocyst
stage embryos are presented in Table 8.3. Overall, there was no effect (P>0.05) of
treatment on the mean number of apoptotic cells in morulae and early blastocysts. When
blastocyst stage embryos were evaluated there was a significant increase in the number
of apoptotic cells in frozen-thawed in vivo embryos when compared with fresh in vivo
embryos. There was no difference (P>0.05) detected between fresh in vivo embryos, in
vitro-cultured embryos and chick-cultured embryos. When only those embryos exhibiting
specific staining patterns were evaluated (Table 8.4), there were fewer apoptotic cells in
fresh in vivo morulae than in frozen-thawed in vivo morulae (P<0.05); morulae resulting
from in vitro culture did not differ from fresh in vivo morulae. There was no effect of
treatment on mean number of apoptotic cells in early blastocyst and blastocyst stage
embryos (P>0.05).
The apoptotic indices of embryos are presented in Table 8.5. There was no effect
(P>0.05) of treatment on apoptotic index in morula and blastocyst stage embryos. The
apoptotic indices of in vitro-cultured and chick-cultured early blastocysts were similar to
those of fresh in vivo embryos. No differences were detected (P>0.05), however,
between in vitro culture, chick culture and positive controls.
124

Table 8.2. Number, stage and quality grade of embryos recovered from superovulated cows
a MORL = morulae. b BLST = blastocysts. c G1 = quality grade 1. d G2 = quality grade 2. e G3-G4 = quality grade 3 and quality grade 4.
125

Table 8.3. Effect of culture environment on mean number of apoptotic cells in morula, early blastocyst and blastocyst stage embryos
Mean no. of apoptotic cellsab
In vivo embryos Frozen-thawed in vivo embryos In vitro-cultured embryos Chick-cultured embryos Positive control
32
32
64c
21
21
10.1d
8.9d
10.3d
13.3de
20.0e
No. of Early Treatment group embryos Morulae blastocysts Blastocysts Degenerate
1.0d
na
8.9de
0.0de
15.0e
0.9d
8.2e
3.8de
6.7de
18.0f
9.5d
na
13.6de
19.0d
na
a Includes both specific and profuse patterned embryos. b na, not applicable. No embryos were available for evaluation in these categories. c Both in vitro culture groups were combined for statistical evaluation. d,e, f Within a column, values with different superscripts are significantly different (P<0.05).
126

Table 8.4. Effect of culture environment on mean number of apoptotic cells in morula, early blastocyst and blastocyst stage embryos
Mean no. of apoptotic cellsab
No. of Early Treatment group embryos Morulae blastocysts Blastocysts Degenerate
In vivo embryos Specific Profuse Frozen-thawed in vivo embryos Specific Profuse In vitro-cultured embryos Specific Profuse Chick-cultured embryos Specific Profuse Positive control Specific Profuse
23 9
2111
43c
21
516
219
1.0d
na
na
na
0.6d
20.0
0.0d
na
0.0d
20.0
1.7d
20.0
na
na
3.1de
20.0
5.7e
20.0
na na
1.5d
20.0
3.4e
20.0
0.5d
20.0
0.0de
20.0
na 20.0
0.9d
na
1.5d
20.0
1.2d
20.0
0.0d
20.0
0.0d
20.0
a Embryos were categorized as having either specific or profuse staining patterns. In embryos exhibiting specific staining patterns, individual fluorescent points were apparent and could be quantified. Individual fluorescent points were not apparent in embryos exhibiting profuse staining patterns. These embryos were assigned an arbitrary apoptotic cell value of 20. b na, not applicable. No embryos were available for evaluation in these categories c Both in vitro culture groups were combined for statistical evaluation.
d,e Within a column, values with different superscripts are significantly different (P<0.05).
127

Table 8.5. Effect of culture environment on apoptotic index in morula, early blastocyst, and blastocyst stage embryos
Apoptotic indexabcde
Treatment group No. Morulae Early blastocysts Blastocysts
a Apoptotic index = mean number of apoptotic cells/total number of cells in the embryo. Total number of cells for in vivo- derived morulae and early blastocysts = 128; for in vivo-derived blastocysts = 166; for in vitro-produced morulae and early blastocysts = 113; for in vitro-produced blastocysts = 114 (Knijn et al., 2003).
In vivo embryos Frozen-thawed in vivo embryos In vitro-cultured embryos Chick-cultured embryos Positive control
25
32
56f
7
21
7.9g
7.0g
9.1g
11.8gh
17.7h
0.8g
na
7.8gh
0.0gh
13.3h
0.5g
5.0g
3.8g
5.8g
15.8h
b Embryos were categorized as having either specific or profuse staining patterns. In embryos exhibiting specific staining patterns, individual fluorescent points were apparent and could be quantified. Individual fluorescent points were not apparent in embryos exhibiting profuse staining patterns. These embryos were assigned an arbitrary apoptotic cell value of 20. c Includes both specific and profuse patterned embryos. d Apoptotic index could not be calculated for degenerate embryos because embryo stage could not always be determined. e na, not applicable. No embryos were available for evaluation in these categories
f Both in vitro culture groups were combined for statistical evaluation. g,h Within a column, values with different superscripts are significantly different (P<0.05).
128

The mean number of apoptotic cells per embryos in relation to quality grade are
presented in Table 8.6. There was no effect (P>0.05) of treatment on the mean number
of apoptotic cells in grade 1 or grade 2 embryos (morulae, early blastocysts and
blastocysts). For grade 3 embryos, there were significantly more (P<0.05) apoptotic cells
in chick-cultured embryos than in in vitro-cultured embryos; however, when only those
embryos exhibiting specific staining patterns were evaluated there was no treatment
effect (P>0.05) (Figure 8.7). For grade 4 embryos, frozen-thawed in vivo embryos had
significantly more (P<0.05) apoptotic cells than fresh in vivo embryos; in vitro-cultured
grade 4 embryos did not differ (P>0.05) from either treatment group. As with the mean
number of apoptotic cells, there was no effect (P>0.05) of treatment on apoptotic indices
of grade 1 or grade 2 embryos; similar results were observed for grade 4 embryos
(Table 8.8). For grade 3 embryos, chick cultured-embryos had a greater apoptotic index
than in vitro-cultured embryos; however, neither treatment was significantly different
(P>0.05) from either fresh or frozen-thawed in vivo-derived embryos.
DISCUSSION Dead cells were first observed in mammalian embryos in 1967 by Potts and Wilson.
Soon thereafter it was determined that cells die by apoptosis (Kerr et al., 1972), a self
directed form of cell suicide (Hardy, 1997; Betts and King, 2001). Since that time,
apoptosis has been determined to be a critical process during pre-implantation
development for the disposal of excess cells, cells that are in the way, abnormal or
potentially dangerous (Gjorret et al., 2003). A normal pattern of apoptosis is critical for
continued embryonic development (Brill et al., 1999), and any deviation in this pattern
can compromise future development, often resulting in early embryonic death or fetal
abnormalities (Knijn et al., 2003).
There are several mechanisms by which apoptosis can be induced in cells, but
regardless of the type of induction, receptor binding initiates a cascade of events that
results in activation of an effector caspase (generally caspase-3 or -7) (Lawen, 2003).
Effector caspases cleave cellular proteins and are responsible for the typical
morphological changes observed in cells undergoing apoptosis, such as DNA
fragmentation (Lawen, 2003).
To our knowledge, all studies to date evaluating apoptosis in pre-implantation
mammalian embryos utilize the TUNEL (terminal deoxynucleotidyl transferase mediated
129
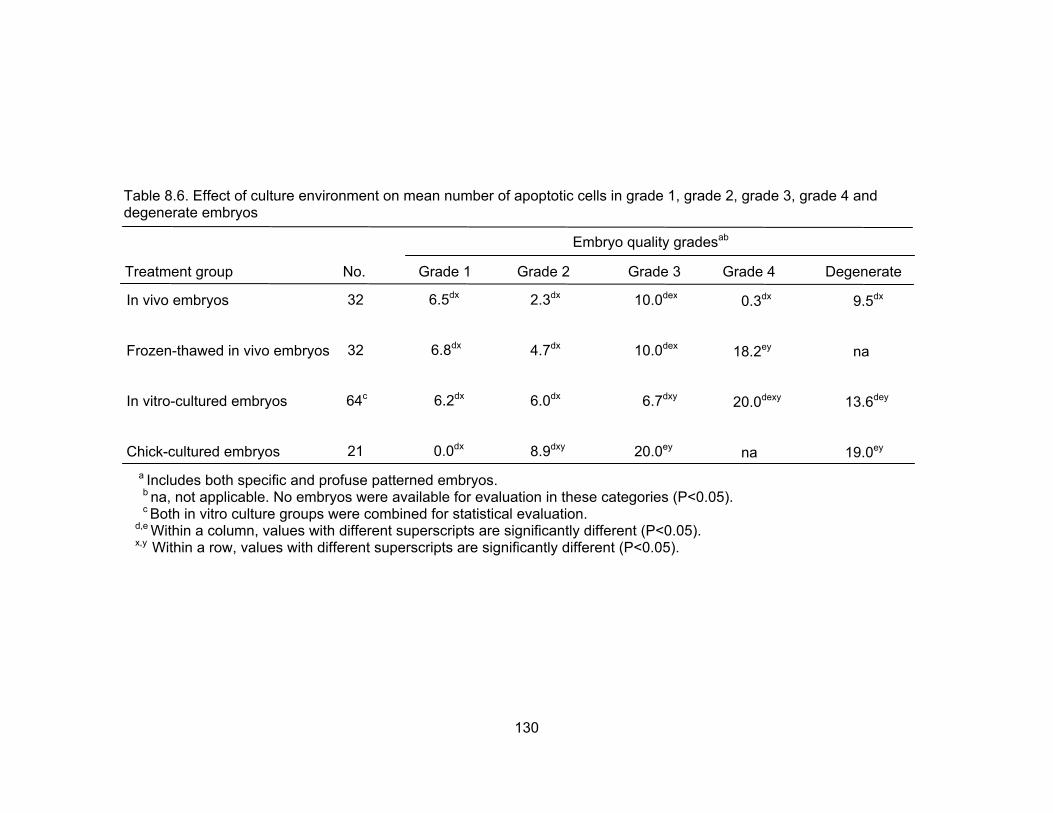
Table 8.6. Effect of culture environment on mean number of apoptotic cells in grade 1, grade 2, grade 3, grade 4 and degenerate embryos
a Includes both specific and profuse patterned embryos.
Embryo quality gradesab
Treatment group No. Grade 1 Grade 2 Grade 3 Grade 4 Degenerate
In vivo embryos Frozen-thawed in vivo embryos In vitro-cultured embryos Chick-cultured embryos
32
32
64c
21
6.5dx
6.8dx
6.2dx
0.0dx
9.5dx
na
13.6dey
19.0ey
0.3dx
18.2ey
20.0dexy
na
10.0dex
10.0dex
6.7dxy
20.0ey
2.3dx
4.7dx
6.0dx
8.9dxy
b na, not applicable. No embryos were available for evaluation in these categories (P<0.05). c Both in vitro culture groups were combined for statistical evaluation. d,e Within a column, values with different superscripts are significantly different (P<0.05). x,y Within a row, values with different superscripts are significantly different (P<0.05).
130

Table 8.7. Effect of culture environment on mean number of apoptotic cells in grade 1, grade 2, grade 3, grade 4 and degenerate embryos
Embryo quality gradesab
Treatment group No. Grade 1 Grade 2 Grade 3 Grade 4 Degenerate
In vivo embryos Specific Profuse Frozen-thawed in vivo embryos Specific Profuse In vitro-cultured embryos Specific Profuse Chick-cultured embryos Specific Profuse
239
2111
43c
21
5 16
0.8dx
20.0
1.8dx
20.0
1.2dx
20.0
0.0dx
na
2.3dfxy
na
3.3dy
20.0
0.7efx
20.0
0.0fx
20.0
0.0dxy
na
0.0dx
20.0
1.0dx
20.0
na 20.0
0.3dx
na
7.3ez
20.0
na 20.0
na na
1.7dy
20.0
na na
3.1dey
20.0
5.7ey
20.0
a Embryos were categorized as having either specific or profuse staining patterns. In embryos exhibiting specific staining patterns, individual fluorescent points were apparent and could be quantified. Individual fluorescent points were not apparent in embryos exhibiting profuse staining patterns. These embryos were assigned an arbitrary apoptotic cell value of 20. b na, not applicable. No embryos were available for evaluation in these categories. c Both in vitro culture groups were combined for statistical evaluation.
d,e,f Within a column, values with different superscripts are significantly different (P<0.05). x,y,z Within a row, values with different superscripts are significantly different (P<0.05).
131

Table 8.8. Effect of culture environment on apoptotic index in grade 1, grade 2, grade 3 and grade 4 embryos
a Apoptotic index = mean number of apoptotic cells/total number of cells in the embryo. Total number of cells for in vivo-
Treatment group No. Grade 1 Grade 2 Grade 3 Grade 4
Apoptotic indexabcde
In vivo embryos Frozen-thawed in vivo embryos In vitro-cultured embryos Chick-cultured embryos
25
29
28f
7
5.0gx
4.3gx
5.4gx
0.0gx
0.3gx
13.2gy
17.7gx
na
7.8fgx
6.0fgxy
6.7fx
17.6gy
1.7gx
3.5gx
5.2gx
8.9xxy
derived morulae and early blastocysts = 128; for in vivo-derived blastocysts = 166; for in vitro-produced morulae and early blastocysts = 113; for in vitro-produced blastocysts = 114 Knijn et al., 2003). b Embryos were categorized as having either specific or profuse staining patterns. In embryos exhibiting specific staining patterns, individual fluorescent points were apparent and could be quantified. Individual fluorescent points were not apparent in embryos exhibiting profuse staining patterns. These embryos were assigned an arbitrary apoptotic cell value of 20.
c Includes both specific and profuse patterned embryos. d Apoptotic index could not be calculated for degenerate embryos because embryo stage could not always be determined. e na, not applicable. No embryos were available for evaluation in these categories.
f Both in vitro culture groups were combined for statistical evaluation. g,h Within a column, values with different superscripts are significantly different (P<0.05). x,y Within a row, values with different superscripts are significantly different (P<0.05).
132

dUTP nick end labeling) technique to label fragmented DNA (mice - Wuu et al., 1999;
Brison and Schultz, 1997; rats - Pampfer et al., 1997; Hinck et al., 2001; pigs – Long et
al., 1998; cattle - Byrne et al., 1999; Matwee et al., 2000; Makarevich and Markkula,
2002; Kolle et al., 2002; Neuber et al., 2002; Paula-Lopes and Hansen, 2002; humans –
Yang et al., 1998; Hardy, 1999). However, DNA fragmentation is not an event unique to
apoptosis as cells undergoing necrosis (a second form of cell death) also exhibit
substantial DNA fragmentation (Grasl-Kraupp et al., 1995). In addition, it has been
reported that improper tissue handling is sufficient to induce enough DNA fragmentation
for TUNEL positive staining of nuclei (Negoescu et al., 1996).Thus there is a critical
need for evaluation of different markers of apoptotic cell death for adequate
determination of apoptosis in pre-implantation embryos.
The objective of the present study was to evaluate the presence of caspases-3
and -7 as indicators of apoptosis in bovine embryos fertilized and cultured under
different conditions. Caspase-3 mRNA and protein have been detected in mouse and rat
blastocysts (Hinck et al., 2001). However, this is believed to be the first report of
caspase activity in bovine embryos.
In the present study, more than 65% of all embryos displayed at least one
fluorescent cell, regardless of treatment (data not shown). These findings are consistent
with those of previous studies (Byrne et al., 1999; Matwee et al., 2000; Neuber et al.,
2002; Gjorret et al., 2003) and suggest that apoptosis is a normal process during early
embryo development in cattle.
Overall, culture conditions did not dramatically affect the incidence of apoptosis
in bovine embryos in the current research. Frozen-thawed in vivo embryos did have
significantly more apoptotic cells than did their fresh counterparts; however, when only
those embryos exhibiting specific staining patterns were evaluated, this statistical
difference was no longer present. It is somewhat unclear why some embryos exhibit
specific staining patterns while others exhibit profuse staining patterns, but we
hypothesize that profuse staining embryos are likely degenerate or degenerating.
There is conflicting information in the literature regarding the effect of in vitro
culture on apoptosis. Byrne et al. (1999) reported that the extent of apoptosis in embryos
was affected by in vitro production systems; however, they did not evaluate in vivo-
derived embryos in this study. Based on TUNEL analysis, Knijn et al. (2003) reported
133

that apoptotic index tended to be higher in in vitro-produced embryos than in embryos
produced in vivo. Similarly, Gjorret et al. (2003) reported that the incidence of apoptotic
cells was higher in blastocysts cultured in vitro than their in vivo-derived counterparts
when analyzed with TUNEL. They contributed this difference specifically to higher levels
of apoptosis in the inner cell mass (ICM) of in vitro-produced embryos.
Although we were not able to specifically localize apoptotic cells to the ICM or
trophoectoderm, there were consistently more caspase-positive (flourescent) cells in the
region of the ICM, regardless of culture system (Figure 8.4). These findings are in
agreement with previous reports indicating that apoptosis predominates in the ICM (El-
Shershaby and Hinchliffe, 1974; Copp, 1978; Mohr and Trouson, 1982; Handyside and
Hunter, 1986; Hardy and Handyside, 1996), with much lower levels detected in the
trophoectoderm (Copp, 1978; Hardy and Handyside, 1996; Brison and Schultz, 1997). It
has been hypothesized that apoptosis occurs naturally in vivo as a means to help
eliminate ICM cells that still have the potential to form trophoectoderm, thus decreasing
the chance of inappropriate trophoectoderm expression during germ layer differentiation
(Handyside and Hunter, 1986; Pierce et al., 1989). In addition, Gjorret et al. (2003)
suggested that the aberrations in apoptosis in the ICM could result in morphologically
normal appearing blastocysts carrying subcellular deviations that could affect the
developmental competence of the embryo.
One interesting observation was the presence of different degrees of fluorescence within
an embryo. Often, small, pinpoint areas of fluorescence were detected while other times
the fluorescence appeared to consume the entire cytoplasm of a cell (Figure 8.5). The
capase protein has been localized to the cytoplasm of the cell, therefore we hypothesize
that the different degrees of fluorescence represent different degrees of apoptosis.
Those cells with small pinpoint areas of fluorescence are likely in the very early stages of
apoptosis with only small amounts of caspase present in the cytoplasm. In contrast, the
instances in which the entire cell appears to fluoresce likely represent a more advanced
stage of apoptosis with greater amounts of caspase present.
In the present study, there was no major effect of in vitro culture environment on
the incidence of apoptosis when embryos were categorized by quality grade. Moreover,
within a treatment, there was no significant difference in apoptosis across quality grades. This is believed to be the first report of apoptosis evaluated in relation to embryo quality
134
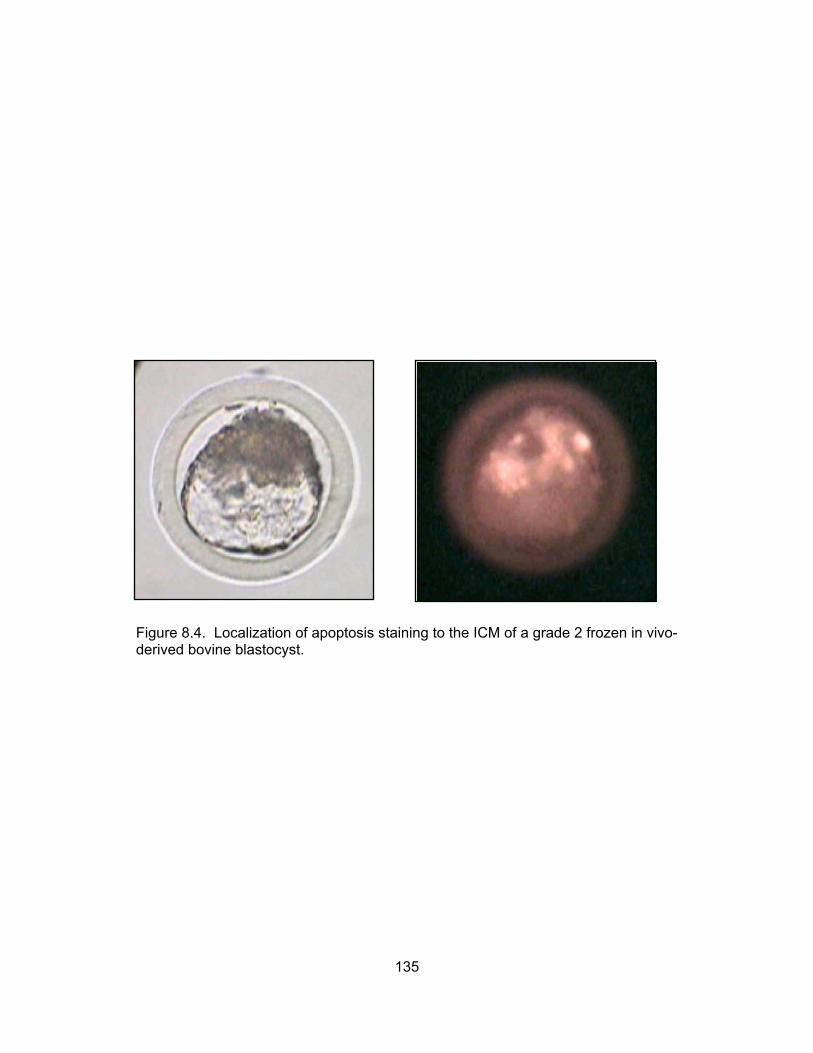
Figure 8.4. Localization of apoptosis staining to the ICM of a grade 2 frozen in vivo- derived bovine blastocyst.
135

B
A
Figure 8.5. The presence of different degrees of fluorescence within an embryo. Pinpoint areas of fluorescence are thought to represent cells in the early stages of apoptosis with only small amounts of caspase present in the cytoplasm (A), whereas extensive fluorescence are thought to represent cells in more advanced stages of apoptosis with much more caspase present in the cell (B).
136

grade. Based on observations in the present study, it is likely that apoptosis is a naturally
occurring event in embryo development and is not necessarily a reliable indicator of
embryo viability or quality grade.
The value of morphological evaluation as an indicator of embryo viability and
quality has been contested for many years. In the early 1980s, Shea (1981) reported
that embryos showing obvious signs of degeneration resulted in lower pregnancy rates
than embryos with a normal morphology; however, these embryos were still able to
produce pregnancies in many instances. It was further speculated that the extent of
embryo development was a better indicator of embryo quality than morphological
assessments. In the present study, some morphologically high quality embryos were
observed to have multiple apoptotic cells while some presumptive degenerate embryos
were observed to have no apoptotic cells. These observations add support to Shea’s
hypothesis that morphological evaluation is not an optimal indicator of embryo viability
and quality.
In summary, the majority of embryos evaluated had more than one apoptotic cell,
suggesting that apoptosis is a normal event in bovine embryo development both in vivo
and in vitro. There was no apparent effect of culture system on the incidence of
apoptosis when evaluated by embryo stage or quality grade. Based on the findings
presented in this report, apoptosis does not appear to be heavily involved in the reduced
developmental potential of in vitro-derived bovine embryos.
137

CHAPTER IX
SUMMARY AND CONCLUSIONS The primary purpose of this series of experiments was to evaluate the use of the
chick embryo co-culture system in bovine embryo culture. In the early 1990s, this system
was shown to improve development of pre-implantation embryos from various
mammalian species when compared with the medium used at that time. Since that time,
in vitro embryo culture media have progressed, but many problems still exist (e.g., poor
developmental rates, poor embryo quality, fetal and neonatal abnormalities). These
problems led us to revisit the use of the chick embryo co-culture system as an
alternative to in vitro embryo culture.
In the initial experiment, development of bovine 1-cell and 2-cell embryos
produced by IVF was assessed after culture in the chick embryo amnion. Results
indicated that bovine embryos could develop in the embryotropic environment of the
chick embryo amnion; however, development was not significantly different when
compared with the semi-defined culture medium (CR1aa). It was concluded that
although not significantly better, the chick embryo co-culture system could serve as a
viable alternative to more traditional methods of in vitro culture.
In the second experiment of this series, development of bovine 8-cell embryos
derived from somatic cell nuclear transfer was assessed after culture in the chick
embryo amnion. Results indicated that these embryos are capable of developing into
transferable embryos and implanting at a rate comparable with embryos culture in vitro
in a semi-defined medium (CR1aa).
The third experiment was conducted in an effort to improve the chick embryo co-
culture system. To accomplish this, angiogenesis was examined in chick chorioallantoic
membranes treated with a vasodilator (PGE2) and a vasoconstrictor (PGF2α). Based on
the findings of the first part of this experiment, the development of bovine embryos after
co-culture in PGE2-treated chicks was evaluated. It was concluded that increasing blood
flow to the chick choriallantoic membrane did not appear to improve development or
quality of bovine embryos cultured in untreated chicks over embryos cultured in semi-
defined medium (control). These findings suggest that the benefits of the chick co-
culture system lie in the ability of the chick to actively regulate its own environment
rather than in embryotropic properties within the amniotic environment.
138

Because of the inherent mortality rate of shell-less chick embryos a fourth
experiment was conducted to determine if amniotic fluid extracted from a developing
chick embryo could be used as a supplement to in vitro culture medium and support
development of bovine embryos. It was concluded that chick amniotic fluid could provide
the proteins and growth factors needed to support in vitro development of bovine
embryos, thus providing a feasible alternative to the use of fetal bovine serum in bovine
embryo culture.
In the fifth experiment of this series, protocols for culturing fetal chick cells and
chick amnion cells were developed. This research provides an in vitro model for future
studies into the effects of chick embryo co-culture on mammalian embryo development.
In the last experiment, apoptosis was evaluated in in vivo-derived embryos, in
vitro-produced embryos and embryos cultured in the chick amnion. This experiment
utilized a novel approach to evaluate the incidence of apoptosis in embryos. Findings of
this study indicate that apoptosis is a normal event in bovine embryo development both
in vivo and in vitro and does not appear to be affected by the in vitro culture
environment. Based on these findings, it was concluded that apoptosis is not extensively
involved in the reduced developmental potential of in vitro derived bovine embryos.
In conclusion, it was demonstrated that bovine embryos could develop in the
amniotic cavity of a developing chick embryo at rates similar to those of embryos
cultured in current culture media. Although the chick embryo co-culture system does not
surpass current in vitro culture systems, it should not be completely dismissed.
Pregnancy and calving rates resulting from current in vitro embryo production systems
are significantly lower than those achieved with artificial insemination and embryo
transfer. Moreover, numerous fetal and calf abnormalities have been associated with in
vitro embryo production, particularly with the use of serum. If the chick embryo culture
system could produce even one more viable offspring, free from developmental
abnormalities, then its use may be warranted and significant research gains could be
obtained.
139

LITERATURE CITED Abd El Razek, I.M., G. Charpigny, S. Kodja, B. Marquant-Leguienne, P. Mermillod, C.
Guyader-Joly, P. Humblot. 2000. Differences in lipid composition between in vivo- and in vitro-produced bovine embryos. Theriogenology 53:346 (abstr.).
Abe, H., T. Otoi, S. Tachikawa, S. Yamashita, T. Satoh, H. Hoshi. 1999. Fine structure of
bovine morulae and blastocysts in vivo and in vitro. Anat. Embryol. 199:519-527. Agca, Y. R.L. Monson, D.L. Northey, O.A. Mazni, D.M. Schaefer, J.J. Rutledge. 1998.
Transfer of fresh and cryopreserved IVP bovine embryos: normal calving, birth weight and gestation lengths. Theriogenology 50:147-162.
Allen, W.R., F. Steward, A.O. Trounson, M. Tischner, W. Bielanski. 1976. Viability of
horse embryos after storage and long-distance transport in the rabbit. J. Reprod. Fertil. 47:387-390
Arbiser, J.L. 1996. Angiogenesis and the skin: a primer. J. Am. Acad. Dermatol. 34:486-
497. Ashworth, C. J., F.W. Bazer. 1989. Changes in ovine conceptus and endometrial
function following asynchronous embryo transfer or administration of progesterone. Biol. Reprod. 40:425-33.
Augustin, H.G., K. Braun, I. Telemenakis, U. Modlich, W. Kuhn. 1995. Ovarian angiogenesis. Phenotypic characterization of endothelial cells in a physiological model of blood vessel growth and regression. Am. J. Pathol. 147:339-351.
Ayalon, N. 1978. A review of embryonic mortality in cattle. J. Reprod. Fertil. 54:483-493. Averill, R.L.W., C. E. Adams, L.E.A. Rowson. 1955. Transfer of mammalian ova
between species. Nature 176:167-170. Baird, J.W.C., C.A. Johnson, S.R. Williams, R.A. Godke, C.L. Jenkins, G. Schmidt.
1990. Increased blastocyst formation in the mouse following culture of hamster cumulus cell monolayers. Proc. 46th Amer. Fertil. Soc. Meeting., Birmingham, AL S9:0-020 (abstr.).
Barinaga, M. 1998. Death by dozens of cuts. Science 280:32-34. Barnes, F.L., W.H. Eyestone. 1990. Early cleavage and the maternal zygotic transition in
bovine embryos. Theriogenology 33:141-152. Bavister, B.D. 1995. Culture of preimplantation embryos: Facts and artifacts. Human
Reprod. Update. 1:91-148. Bavister, B.D., T. Arlotto. 1990. Influence of single amino acids on the development of
hamster one-cell embryos in vitro. Mol. Reprod. Dev. 25:45-51.
140

Bavister, B.D., D.E. Boatman, M.L. Leibfried, M. Loose, M.W. Vernon. 1983. Fertilization and cleavage of rhesus monkey oocytes in vitro. Biol. Reprod. 28:983-999.
Behboodi, E., G.B. Anderson, R.H. Bon Durant, S.L. Cargill, B.R. Kreuscher, J.F.
Medrano, J.D. Murray. 1995. Birth of large calves that developed from in vitro-produced bovine embryos. Theriogenology 44:227-232.
Betteridge, K.J., J.E. Flechon. 1988. The anatomy and physiology of the pre-
implantation bovine embryo. Theriogenology 29:216-222. Betts, D.H., W.A. King. 2001. Genetic regulation of embryo death and senescence.
Theriogenology 55:171-191. Bischoff, R. 1986. Proliferation of muscle satellite cells on intact myofibers in culture.
Dev. Biol. 115:129-139. Blakewood, E.G., R.A. Godke. 1989. A method using the chick embryo amnion for
mammalian embryo culture. J. Tiss. Cult. Meth. 12:73-76. Blakewood, E.G., J.M. Jaynes, W.A. Johnson, R.A. Godke. 1989a. Using the amniotic
cavity of the developing chick embryo for the in vivo culture of early-stage mammalian embryos. Poult. Sci. 68:1695-1702.
Blakewood, E.G., S.H. Pool, J.F. Prichard, R.A. Godke. 1990. Culturing two- to eight-cell
caprine embryos using domestic chicken eggs. Mol. Reprod. Dev. 27:288-294. Blakewood, E.G., S.H. Pool, K.E. Wiemer, R.A. Godke. 1989b. Culturing early-stage
bovine morulae using domestic chicken eggs. Theriogenology 31:176 (abstr.). Boland, M.P. 1984. Use of the rabbit oviduct as a screening tool for the viability of
mammalian eggs. Theriogenology 126-137. Boni, R., E. Tosti, S. Roviello, B. Dale. 1999. Intercellular communication in in
vivo- and in vitro-produced bovine embryos. Biol. Reprod. 61:1050-1055. Brackett, B.G. 2001. Advances in animal in vitro fertilization. In: Contemporary
Endocrinology: Assisted Fertilization and Nuclear Transfer in Mammals. Humana Press Inc.: Totowa, NJ. pp 21-51.
Brackett, B.G., D. Bousquet, M.L. Boice, W.J. Donawick, J.F. Evans, M.A. Dressel.
1982. Normal development following in vitro fertilization in the cow. Biol. Reprod. 27:147-158.
Brackett, B.G., J.F. Evans, W.J. Donawick, M.L. Boice, M.A. Cofone. 1980a. In vitro penetration of cow oocytes by bull sperm. Arch Androl 5:69-71. Brackett, B.G., C.L. Keefer, C.G. Troop, W.A. Donawick, K.A. Bennet. 1984. Bovine twins resulting from in vitro fertilization. Theriogenology 21:224 (abstr.).
141

Brackett, B.G., Y.K. Oh, J.F. Evans, W.J. Donawick. 1980b. Fertilization and early development of cow ova. Biol. Reprod. 23:189-205. Brackett, B.G., G. Oliphant. 1975. Capacitation of rabbit spermatozoa in vitro. Biol.
Reprod.12:260-274. Breukelman, S.P., J.M.C. Reinders, F.H. Jonkers, L. de Ruigh, L.M.T.E. Kaal, A.M. van
Wagtendonk-de Leeuw, P.L.A.M. Vos, S.J. Dieleman, J.F. Beckers, Z. Perenyi, M.A.M. Taverne. 2004. Fetometry and fetal heart rates between day 35 and 108 in bovine pregnancies resulting from transfer of either MOET, IVP-co-culture or IVP-SOF embryos. Theriogenology 61:867-882.
Briggs, R., T.J. King.1952. Transplantation of living nuclei from blastula cells into
enucleated frog’s eggs. Proc. Natl. Acad. Sci. USA 38:455-463. Brill, A., A. Torchinsky, H. Carp, V. Toder. 1999. The role of apoptosis in normal and
abnormal embryonic development. J. Assist. Reprod. Genetics 26:512-519. Brison, D.R., R.M. Schultz. 1997. Apoptosis during mouse blastocyst formation:
evidence for a role for survival factors including transforming growth factor alpha. Biol. Reprod. 56:1088-1096.
Brock, H., L.E. Rowson. 1952. The production of viable bovine ova. J. Agric. Sci. (Camb.):42:47. Brown, B.W., T. Radziewic. 1996. In vitro production of sheep embryos and growth rates
of resultant lambs. Proc. 13th Int. Congr. Anim. Reprod. 3:16-22. Byrne, A.T., J. Southgate, D.R. Brison, H.J. Leese. 1999. Analysis of apoptosis in the
preimplantation bovine embryo using TUNEL. J. Reprod. Fert. 117:97-105. Campbell, K.H.S., J. McWhir, W.A. Ritchie, I. Wilmut. 1996. Sheep cloned by nuclear
transfer from a cultured cell line. Nature 380:64-66. Camous, S., Y., Heyman, W. Meziou, Y. Menezo. 1984. Cleavage beyond the block
stage and survival after transfer of early bovine embryos cultured with trophoblastic vesicles. J. Reprod. Fert. 72:479-485.
Carolan, C., P. Lonergan, A. Van Langendonckt, P. Mermillod. 1995. Factors affecting
bovine embryo development in synthetic oviduct fluid following oocyte maturation and fertilization in vitro. Theriogenology 43:1115-1128.
Chader, G.J., D.A. Newsome, R.E. Bensinger, R.T. Fletcher. 1975. Studies on
differentiation of retinal pigmented epithelium cells in culture. Invest. Ophthalmol. 14:108-113.
Chang, M.C. 1949. Effects of heterologous sera on fertilized rabbit ova. J. Gen. Physiol.
32:29.
142

Chang, M.C. 1952. Development of bovine blastocysts with a note on implantation. Anat. Rec. 113:143-161.
Chesne, P., P.G. Adenot, C. Viglietta, M. Baratte, L. Boulanger, J.P. Renard. 2002.
Cloned rabbits produced by nuclear transfer from adult somatic cells. Nat. Biotechnol. 20:366-369.
Christenson, R.K., S.E. Echternkamp, P.B. Laster. 1975. Oestrus, LH, ovulation and
fertility in beef heifers. J. Reprod. Fertil. 43:543-547. Cibelli, J.B., S.L. Stice, P.J. Golueke, J.J. Kane, J. Jerry, C. Blackwell, F.A. Ponce de
Leon, J.M. Robl. 1998. Cloned transgenic calves produced from nonquiescent fetal fibroblasts. Science 280:1256-1258.
Coetzee, J.C., C.B.V.O. Sevenster, F. Le, R. Fourie, J.V. Van Der Merwe, L.A. Helberg.
1989. In vitro culture of mouse embryos in human amniotic fluid. South African Med. J. 76:62-63.
Cole, R.J., J. Paul. 1965. Properties of cultured preimplantation mouse and rabbit
embryos and cell strains developed from them. In: Preimplantation Stages of Pregnancy. Little, Brown & Co. Boston, MA. pp. 82-155.
Collas, P., F.L. Barnes. 1994. Nuclear transplantation by microinjection of inner cell
mass and granulosa cell nuclei. Mol. Reprod. Dev. 38:264-267. Collas, P., J.M. Robl. 1991. Development of rabbit nuclear transplant embryos from
morula and blastocyst stage donor nuclei. Theriogenology 35:190 (abstr.). Copp, A.J. 1978. Interaction between inner cell mass and trophoectoderm of the mouse
blastocyst I. A study of cellular proliferation. J. Embryol. Exp. Morphol. 48:109- 125.
Crooks, R.J., K. Simkiss. 1974. Respiratory acidosis and eggshell resorption by the
chick embryo. J. Exp. Biol. 61:197-202. Crosier, A.E., P.W. Farin, M.J. Dykstra, J.E. Alexander, C.E. Farin. 2000. Ultrastructural
morphometry of bovine compact morulae produced in vivo or in vitro. Biol. Reprod. 62:1459-1465.
Defouw, D.O., V.J. Rizzo, R. Steinfeld, R.N. Feinberg. 1989. Mapping of the
microcirculation in the chick chorioallantoic membrane during normal angiogenesis. Microvasc. Res. 38:136-147.
De Hertogh, R., I. Vanderheyden, S. Pampfer, D. Robin, J. Delcourt. 1992. Maternal
insulin treatment improves preimplantation embryo development in diabetic rats. Diabetologia 35:406-408.
143

Denekamp, J. 1984. Vasculature as a target for tumour therapy. Prog. Applied Microcirculation 4:28-38.
De Pablo, F., B. Perez-Villamil, J. Serva, P. Gonzalez-Guerrero, A. Lopez-Carranza, E.J.
De la Rosa, J. Alemany, T. Caldez. 1993. IGF-I and the IGF-I receptor in development of nonmammalian vertebrates. Mol. Reprod. Dev. 35:427-433.
Devreker, F., K. Hardy. 1997. Effects of glutamine and taurine on preimplantation
development and cleavage of mouse embryos in vitro. Biol. Reprod. 57:921-928. Di Berardino, M.A. 2001. Animal cloning – the route to new genomics in agriculture and
medicine. Differentiation 68:67-83. Dinnyes, A., P. Lonergan, T. Fair, M.P. Boland, X. Yang. 1999. Timing of the first
cleavage post-insemination affects cryosurvival of in vitro produced bovine blastocysts. Mol. Reprod. Dev. 53:318-324.
Diskin, M.G. and J.M. Sreenan. 1980. Fertilization and embryonic mortality rates in beef
heifers after artificial insemination. J. Reprod. Fertil. 59:463-468. Dorfmann, A.D., S.D. Bender, P. Robinson, H.F. Fugger, M. Bustillo, G. Reed, J.S.
Schulman. 1989. Cell-free human amniotic fluid as culture medium for mouse and human embryos. Fertil. Steril. 51:671-674.
Dorland, M., D.K. Gardner, A.O. Trounson. 1994. Serum in SOF causes mitochondrial
degeneration in ovine embryos. J. Reprod. Fert. 13:70 (abstr.). Driesch, H. 1892. Entwicklungsmechanische Studien. I. Der Wert der beiden ersten
Furchungszellen in der Echinodermentwick-lung. Experimentelle Erzeugung von Teil und Doppelbildungen. Zeirchr. Wiss. Zool. 53:160-178, 183-184.
Duby, R.T., J.L. Hill, D. O’Callaghan, E.W. Overstrom, M.P. Boland. 1997. Changes
induced in the bovine zona pellucida by ovine and bovine oviducts. Theriogenology 47:332 (abstr.).
Dunn, B.E. 1974. Technique for “shell-less” culture of the 72 hour avian embryo. Poult.
Sci. 53:409-412. Dunn, B.E., M.A. Boone. 1976. Growth of the chick embryo in vitro. Poult. Sci. 55:1067-
1071. Dunn, B.E., M.A. Boone. 1977. Growth and mineral content of cultured chick embryos.
Poult. Sci. 56:662-672. Dunn, B.E., M.A. Boone. 1978. Photographic study of chick embryo development in
vitro. Poult. Sci. 57:370-377.
144

Dunn, B.E., T.P. Fitzharris, B.D. Barnett. 1981. Effects of varying chamber construction and embryo pre-incubation age on survival and growth of chick embryos in “shell-less” culture. Anatomical Rec. 199:33-43.
Dunn, B.E., N.B. Clark, K.E. Scharf. 1987. Effect of calcium supplementation on growth
of shell-less cultured embryos. J. Exp. Zool. Supp. 1:33-37. Ectors F.J., F. Thonon, A. Delval, R.S. Fontes, K. Touati, J.F. Beckers, F. Ectors.
1993. Comparison between culture of bovine embryos in vitro versus development in rabbit oviducts and in vivo. Livestock Prod. Sci. 36:29–34.
Edwards, J.L., F.N. Schrick, M.D. McCracken, S.R. van Amstel, F.M. Hopkins, M.G.
Welbron, C.J. Davies. 2003. Cloning adult farm animals: a review of the possibilities and problems associated with somatic cell nuclear transfer. Am. J. Reprod. Immunology 50:113-123.
Elsden, R.P., L.D. Nelson, G.E. Seidel. 1978. Superovulating cows with follicle
stimulating hormone and pregnant mare’s serum gonadotrophin. Theriogenology 9:17-26.
El-Shershaby, A.M., J.R. Hinchliffe. 1974. Cell redundancy in the zona-intact
preimplantation mouse blastocyst: a light and electron microscopic study of dead cells and their fate. J. Embryol. Exp. Morph. 31:643-654.
Enders, A.C., A.G. Hendricks, P.E. Binkard. 1982. Abnormal development of blastocysts
and blastomeres in the Rhesus monkey. Biol. Reprod. 26:353-366. Enders, A.C., K.C. Lantz, S. Schlafke. 1990. Differentiation of the inner cell mass of the
baboon blastocyst. Anat. Rec. 226:237-248. Enders, A.C., S. Schlafke. 1981. Differentiation of the blastocyst of the Rhesus monkey.
Am. J. Anat. 162:1-21. Enright, B.P., P. Lonergan, A. Dinnyes, T. Fair, F.A. Ward, X. Yang, M.P. Boland. 2000.
Culture of in vitro produced bovine zygotes in vitro vs in vivo: implications for early embryo development and quality. Theriogenology 54:659-673.
Epple, A., T.S. Gill, B. Nibbio. 1992. The avian allantois: a depot for stress-released
catecholamines. Gen. Comp. Endocrinol. 85:462-476. Epple, A., B. Gower, M. ten Busch, T. Gill, L. Milakofsky, R. Piechotta, B. Nibbio, T.
Hare, M. Stetson. 1997. Stress in avian embryos. Am. Zool. 37:536-545. Exley, G.E., C. Tang, A.S. McElhinny, C.M. Warner. 1999. Expression of caspase and
bcl-2 apoptotic family members in mouse preimplantation embryos. Biol. Reprod. 61:231-239.
145

Eyestone, W.H., N.L. First. 1989. Co-culture of early cattle embryos to the blastocyst stage with oviductal tissue or in conditioned media. J. Reprod. Fertil. 85:715-720.
Eyestone, W.H., M.L. Leibfried-Rutledge, D.L. Northey, B.G. Gilligan, N.L. First. 1987.
Culture of one- and two-cell bovine embryos to the blastocyst stage in the ovine oviduct. Theriogenology 28:1–7.
Farin, P.W., C.E. Farin. 1995. Transfer of bovine embryos produced in vivo or in vitro:
survival and fetal development. Biol. Reprod. 52:697-703. Farin, P.W., A.E. Crosier, C.E. Farin. 2001. Influence of in vitro systems on embryo
survival and fetal development in cattle. Theriogenology 55:151-170. Flamme, I., T. Frolich, W. Rissau. 1997. Molecular mechanisms of vasculogenesis and
embryonic angiogenesis. J. Cell. Physiol. 173:206-210. Folkman, J., M. Klagsburn. 1987. Angiogenic factors. Science 235:442-447. Fukuda, Y., M. Ichikawa, K. Naito, Y. Toyoda. 1989. Birth of normal calves resulting from
bovine oocytes matured, fertilized and cultured with cumulus cells in vitro up to the blastocyst stage. Biol. Reprod. 42:114-119.
Fukui, Y. 1990. Effect of follicle cells on the acrosome reaction, fertilization and
developmental competence of bovine oocytes matured in vitro. Mol. Reprod. Dev. 26:40-46.
Fukui, Y., A.M. Glew, F. Gandolfi, R.M. Moor. 1988. In vitro culture of sheep oocytes
matured and fertilized in vitro. Theriogenology 29:883-891. Fukui, Y., H. Ono. 1988. In vitro development to blastocyst of in vitro matured and
fertilized bovine oocytes. Vet. Rec. 122:282 (abstr.). Galli, C., R. Dushi, R.M. Moor, G. Lazzari. 1999. Mammalian leukocytes contain all the
genetic information necessary for the development of a new individual. Cloning 1:161-170.
Galli, C., G. Lazzari. 1996. Practical aspects of IVM/IVF in cattle. Anim. Reprod. Sci. 42:
371–379. Gandolfi, F., R.M. Moor. 1987. Stimulation of early embryonic development in the sheep
by co-culture with oviduct epithelial cells. J. Reprod. Fertil. 81:23-28. Gardner, D.K. 1999. Development of serum-free culture systems for the ruminant
embryo and subsequent assessment of embryo viability. J. Reprod. Fert. Supp. 54:461-475.
Gardner, D.K., M. Lane. 1993. Amino acids and ammonium regulate mouse embryos
development in culture. Biol. Reprod. 48:377-385.
146

Gardner, D.K., M. Lane, A. Spitzer, P. Batt. 1994. Enhanced rates of cleavage and
development for sheep zygotes cultured to the blastocyst stage in vitro in the absence of serum and somatic cells: amino acids, vitamins, and culturing embryos in groups stimulate development. Biol. Reprod. 50:390-400.
Garry, F.B., R. Adams, J.P. McCann, K.G. Odde. 1996. Postnatal characteristics of
calves produced by nuclear transfer cloning. Theriogenology 45:141-152. Gianaroli, L., R. Seracchioli, A.P. Ferraretti, A. Trouson, C. Flamigni, L. Bovicelli. 1986.
The successful use of human amniotic fluid for mouse embryos culture and human in vitro fertilization, embryo culture and transfer. Fertil. Seril. 46:907-913.
Giles, J.R., R.H. Foote. 1995. Rabbit blastocyst: allocation of cells to the inner cell mass
and trophoectoderm. Mol. Reprod. Dev. 41:204-211. Gill, T.S., B. Porta, B. Nibbio, A. Epple. 1994. Sulfate conjugates of catecholamines in
the allantoic fluid of the chicken embryo. Gen. Comp. Endocrinol. 96:244-258. Gjorret, J.O., B. Avery, L.I. Larsson, K. Schellander, P. Hyttel. 2001. Apoptosis in bovine
blastocysts produced in vivo and in vitro. Theriogenology 55:321 (abstr.). Gjorret, J.O., H.M. Knijn, S.J. Dieleman, B. Avery, L.I. Larson, P. Maddox-Hyttel. 2003.
Chronology of apoptosis in bovine embryos produced in vivo and in vitro. Biol. Reprod. 69:1193-1200.
Glascow, I.K., H.C. Zeringue, D.J. Beebe, S.J. Choi, J. Lyman, M.B. Wheeler. 1998.
Individual embryo transport on a chip for a total analysis system. In: Third International Symposium on Micro Total Analysis System. Kluwer Academic Publishers: Boston, MA. pp 199-202.
Glass, R.H., A.I. Spindle, R.A. Pederson. 1979. Mouse embryo attachment to
substratum and interaction of trophoblast with cultured cells. J. Exp. Zool. 208:327-335.
Goto, K., Y. Kajihara, S. Kosaka, Y. Kobam, Y. Nakashi, K. Ogawa. 1988. Pregnancies
after co-culture of cumulus cells with bovine embryos derived from in vitro fertilization of in vitro matured follicular oocytes. J. Reprod. Fert. 83:753-758.
Grasl-Kraupp, B., B. Ruttkay-Nedecky, H. Koudelka, K. Bukowska, W. Bursch, R.
Schulte-Hermann. 1995. In situ detection of fragmented DNA (TUNEL) assay fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology 21:1465-1468.
Greve, T., H. Callesen, P. Hyttel, B. Avery. 1995. From oocytes to calf: In vivo and in
vitro. In: Animal Production and Biotechnology. Biofutur, Paris. pp 71-97.
147

Grisart, B., A. Massip, F. Dessy. 1994. Cinematographic analysis of bovine embryo development in serum-free oviduct-conditioned medium. J. Reprod. Fert. 101:257-264.
Grutter, M.G. 2000. Caspases: key players in programmed cell death. Curr. Opin. Struct.
Biol. 10:649-655. Guyton, A.C. 1971. Intrinsic regulations of the circulation. 1971. In: Textbook
of Medical Physiology. WB Saunders Co: Philadelphia, PA. pp 270. Hafez, E.S.E., T. Sugie, I. Gordon. 1963. Superovulation and related phenomena in the
beef cow. I. Superovulatory response following PMSG and HCG injections. J. Reprod. Fert. 5:359-379.
Hamilton, W.J., J.A. Laing. 1946. Development of the egg of the cow up to the stage of
blastocyst formation. J. Anat. 80:194-204. Hammond, J. 1949. Recovery and culture of tubal mouse ova. Nature 163:28-29. Hanada, A. 1985. In vitro fertilization in cattle with particular reference to sperm capacitation by ionophore A23187. Jpn. J. Anim. Reprod. 31:56-61. Handyside, A.H., S. Hunter. 1984. A rapid procedure for visualizing the inner cell mass
and trophectoderm nuclei of mouse blastocysts in situ using polynucleotide-specific fluorochromes. J. Exp. Zool. 31:429-434.
Handyside, A.H., S. Hunter. 1986. Cell division and death in the mouse blastocyst before
implantation. Roux’s Arch. Dev. Biol. 195:519-526. Hardy, K. 1997. Cell death in the mammalian blastocyst. Mol. Hum. Reprod. 3:919-925. Hardy, K. 1999. Apoptosis in the human embryo. Rev. Reprod. 4:125-134. Hardy, K., A.H. Handyside, R.M.L. Winston. 1989. The human blastocyst: cell number,
death and allocation during late preimplantation development in vitro. Development 107:597-604.
Hardy, K., A.H. Handyside. 1996. Metabolism and cell allocation during parthenogenetic
preimplantation mouse development. Mol. Reprod. Dev. 43:313-322. Hardy, K., A. Warner, R.M.L. Winston, D.L. Becker. 1996. Expression of intercellular
junctions during preimplantation development of the human embryo. Mol. Human Reprod. 2:621-632.
Harper, J.C., E. Coonen, A.H. Handyside, R.M. Winston, A.H. Hopman, J.D. Delhanty.
1995. Mosaicism of autosomes and sex chromosomes in morphologically normal, monospermic preimplantation human embryos. Prenat. Diagn. 15:41-49.
148

Hasler, J.F. 2000. In vitro culture of bovine embryos in Menezo’s B2 medium with and without coculture and serum: the normalcy of pregnancies and calves resulting from transferred embryos. Anim. Reprod. Sci. 60-61:81-91.
Hasler, J.F., W.B. Henderson, P.J. Hurtgen, Z.Q. Jin, A.D. McCauley, S.A. Mower, B.
Neely, L.S. Shuey, J.E. Stokes, S.A. Trimmer. 1995. Production, freezing and transfer of bovine IVF embryos and subsequent calving results. Theriogenology 43:141-152.
Hasler, J.F., A.D. McCauley, W.F. Lathrop, R.H. Foote. 1987. Effect of donor-embryo-
recipient interactions on pregnancy rate in a large-scale bovine embryo transfer program. Theriogenology 27:139-168.
Hertig, A.T., J. Rock, E.C. Adams, W.J. Mulligan. 1954. On the preimplantation stages of
the human ovum: a description of four normal and four abnormal specimens ranging from the second to fifth day of development. Contrib. Embryol. 35:199-220.
Heyman, Y., P. Chavatte-Palmer, D. LeBourhis, S. Camous, X. Vignon, J.P. Renard.
2002. Frequency and occurrence of late-gestation losses from cattle cloned embryos. Biol. Reprod. 66:6-13.
Heyman, Y., Y. Menezo, P. Chesne, S. Camous, V. Garnier. 1987. In vitro cleavage of
bovine and ovine early embryos: Improved development using co-culture with trophoblastoc vesicles. Theriogenology 27:59-68.
Hill, J.R., R.C. Burghardt, K. Jones, C.R. Long, T. Shin, T.E. Spence, J.A. Thompson,
Q.A. Winger, L. Bovicelli. 2000a. Evidence for placental abnormality as the major cause of mortality in first-trimester somatic cell cloned bovine fetuses. Biol. Reprod. 63:1787-1794.
Hill, J.R., J.F. Edwards, N. Sawyer, C. Blackwell, J.B. Cibelli. 2001. Placental anomalies
in a viable cloned calf. Cloning 3:83-88. Hill, J.R., A.J. Roussel, J.B. Cibellli, J.F. Edwards, N.L. Hooper, M.W. Miller, J.A.
Thompson, C.R. Looney, M.W. Westhusin, J.M. Robl, S.L. Stice. 1999. Clinical and pathological features of cloned transgenic calves and fetuses (13 case studies). Theriogenology 51:1451-1465.
Hill J.R., D.H. Schlafer, P.J. Fisher, C.J. Davies. 2002. Abnormal expression of
trophoblast major histocompatability complex class I antigens in cloned bovine pregnancies is associated with a pronounced endometrial lymphocytic response. Biol. Reprod. 67:55-63.
Hill, J.R., Q.A. Winger, C.R. Long, C.R. Looney, J.A. Thompson, M.E. Westhusin.
2000b. Developmental rates of male bovine nuclear transfer embryos derived from adult and fetal cells. Biol. Reprod. 62:1135-1140.
149

Hinck, L., P. Van Der Smissen, M. Heusteroreute, I. Donnay, R. De Hertogh, S. Pampfer. 2001. Identification of caspase-3 and caspase-activated deoxyribonuclease in rat blastocysts and their implication in the induction of chromatin degradation (but not nuclear fragmentation) by high glucose. Biol. Reprod. 64:555-562.
Holm, P., H. Callesen. 1998. In vivo versus in vitro produced bovine ova: similarities and
differences relevant for practical application. Reprod. Nutr. Dev. 38:579-594. Holm, P., S.K. Walker, B.A. Petersen, R.J. Ashman, R.F. Seamark. 1994. In vitro vs in
vivo culture of ovine IVM-IVF ova: effect on lambing. Theriogenology 41:217 (abstr.).
Hou, L., T. Takeuchi. 1992. Differentiation of reptilian neural crest cells in vitro. In Vitro
Cell Dev. Biol. 28A:387-390. Hurst, P.R., K. Jefferies, P. Eckstein, A.G. Wheeler. 1978. An ultrastructural study of
preimplantation uterine embryos of the Rhesus monkey. J. Anat. 126:209-220. Hyttel, P.H., H. Callesen, T. Greve. 1989. A comparative ultrastructural study of in vivo
versus in vitro fertilization of bovine oocytes. Anat. Embryol. 179:435-442. Ii, I., I. Kimura, E. Ozawa. 1982. A myotrophic protein from chick embryo extract: its
purification, identity to transferring, and indispensability for avian myogenesis. Dev. Biol. 94:366-377.
Ijaz, A., A.G. Hunter. 1989. Evaluation of calcium-free Tyrode’s sperm capacitation medium for use in bovine in vitro fertilization. J. Dairy Sci. 72:3280-3285. Illmensee, K., P.C. Hoppe. 1981. Nuclear transplantation in Mus musculus:
developmental potential of nuclei from perimplantation embryos. Cell 23:9-18. Iritani, A., K. Niwa. 1977. Capacitation of bull spermatozoa and fertilization in vitro of cattle follicular oocytes maturated in culture. J. Reprod. Fert. 50:119-121. Iritani, A., M. Kasai, K. Niwa, H.B. Song. 1984. Fertilization in vitro of cattle follicular
oocytes with ejaculated spermatozoa capacitated in a chemically defined medium. J. Reprod. Fert. 70:487-492.
Jacobs, P.A. 1990. The role of chromosomal abnormalities in reproductive failure.
Reprod. Nutr. Dev. Supp. 1:63s-74s. Jacobsen, H., M. Schmidt, P. Holm, P.T. Sangild, G. Vajta, T. Greve, H. Callesen. 2000.
Body dimensions and birth and organ weights of calves derived from in vitro produced embryos cultured with or without serum and oviduct epithelium cells. Therigenology 53:1761-1769.
150

Javed, M.H., R.W. Wright, Jr. 1990. Bovine amniotic and allantoic fluids for the culture of murine embryos. Theriogenology 33:257 (abstr.).
Jimenez C., N. Rumph, H. Moore, S. Walmsely, B. Buckrell, J.W. Pollard. 2001. Sheep
intermediate recipient culture of in vitro produced bovine embryos: culture in ligated oviducts versus combined oviductal-uterine culture. Theriogenology 55: 336 (abstr.).
Johnston, P.M., C.L. Comar. 1955. Distribution and contribution of calcium from the
albumen, yolk and shell to the developing chick embryo. Am. J. Physiol. 183:365-370.
Jurisicova, A., K.E. Latham, R.F. Casper, S.L. Varmuza. 1998. Expression and
regulation of genes associated with cell death during murine preimplantation embryo development. Mol. Reprod. Dev. 51:243-253.
Juriscova, A., S. Varmuza, R.F. Casper. 1996. Programmed cell death and human
embryo fragmentation. Mol. Hum. Reprod. 2:93-98. Kane, M.T. 2003. A review of in vitro gamete maturation and embryo culture and
potential impact on future animal biotechnology. Anim. Reprod. Science 79:171-190.
Kasinathan, P., J.G. Knott, Z. Wang, D.J. Jerry, J.M. Robl. 2001. Production of calves
from G1 fibroblasts. Nat. Biotechnol. 19:1176-1178. Kato, Y., T. Tani, Y. Sotomura, K. Kurokawa, J. Kato, H. Doguchi, H. Yasue, Y.
Tsunoda. 1998. Eight calves cloned from somatic cells of a single adult. Science 282:2095-2098.
Keefer, C.L., S.L. Stice, D.L. Matthews. 1994. Bovine inner cell mass cells as donor
nuclei in the production of nuclear transfer embryos and calves. Biol. Reprod. 50:935-939.
Kerr, J.F., A.H. Wylie, A.R. Currie. 1972. Apoptosis: a basic biological phenomenon with
wide ranging implications in tissue kinetics. British J. Cancer. 26:239-257. Khurana, N.K., H. Niemann. 2000. Energy metabolism in preimplantation bovine
embryos derived in vitro or in vivo. Biol. Reprod. 62:847-856. Kim, H.N., Y.X. Hu, J.D. Roussel, R.A. Godke. 1989. Culturing murine embryos on
bovine fetal spleen cell fibroblast and chick embryo fibroblast monolayers. Theriogenology 21:211 (abstr.).
Kim, H.N., J.D. Roussel, G.F. Amborski, Y.X. Hu, R.A. Godke. 1989. Monolayers of
bovine fetal spleen cells and chick embryo fibroblasts for co-culture of bovine embryos. Theriogenology 31:212 (abstr.).
151

King, W.A. 1991. Embryo-mediated pregnancy failure in cattle. Can. Vet. J. 32:99-103. Kishi, M., Y. Itagaki, R. Takakura, M. Imamura, T. Sudo, M. Yoshinari, M. Tanitmoto, H.
Yasue, N. Kashima. 2000. Nuclear transfer in cattle using colostrum-derived mammary gland epithelial cells and ear-derived fibroblast cells. Theriogenology 54:675-684.
Klagsbrun, M.A., P.A. D’Amore. 1991. Regulators of angiogenesis. Annual Rev. Physiol.
53:217-239.
Kleemann, D.O., S.K. Walker, K.M. Hartwich, L. Fong, R.F. Seamark, J.S. Robinson, J.A. Owens. 2001. Fetoplacental growth in sheep administered progesterone during the first three days of pregnancy. Placenta 22:14-23.
Kleemann, D.O., S.K. Walker, R.F. Seamark. 1994. Enhanced fetal growth in sheep administered progesterone during the first three days of pregnancy. J. Reprod. Fertil. 102:411-417.
Knijn, H.M., J.O. Gjorret, P.L.A.M. Vos, P.J.M. Hendriksen, B.C. van der Weijden, P.
Maddox-Hyttel, S.J. Dieleman. 2003. Consequences of in vivo development and subsequent culture on apoptosis, cell number, and blastocyst formation in bovine embryos. Biol. Reprod. 69:1371-1378.
Knijn, H.M., C. Wrenzycki, P.J.M. Hendriksen, P.L.A.M. Vos, D. Herrmann, G.C. van der
Weijden, H. Niemann, S.J. Dieleman. 2002. Effects of oocyte maturation regimen on the relative abundance of gene transcripts in bovine blastocysts derived in vitro or in vivo. Reproduction 124:365-375.
Kolle, S., M. Stojkovic, G. Boie, E. Wolf, F. Sinowatz. 2002. Growth hormone inhibits
apoptosis in in vitro produced bovine embryos. Mol. Reprod. Dev. 61:180-186. Kolver, E.S., K.L. MacMillian. 1993. Short term changes in selected metabolites in
pasture fed dairy cows during peak lactation. Proc. NZ Soc. Anim. Prod. 53:77- 81.
Koo, D.B., Y.K. Kang, Y.H. Choi, J.S. Park, H.N. Kim, K.B. Oh, D.S. Son, H. Park, K.K.
Lee, Y.M. Han. 2002. Aberrant allocations of inner cell mass and trophoectoderm cells in bovine nuclear transfer blastocysts. Biol. Reprod. 67:487-492.
Krajewski, S., S. Tanaka, S. Takayama, M.J, Schibler, W. Fenton, J.C. Reed. 1993.
Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and the outer mitochondrial membranes. Cancer Res. 53:4701-4714.
Krisher, R.L., B.D. Bavister. 1998. Reponses of oocytes and embryos to the culture
environment. Theriogenology 49:103-114.
152

Krisher R.L., R.M. Petters, B.H. Johnson, B.D. Bavister, A.E. Archibong. 1989. Development of porcine embryos from the one-cell stage to blastocyst in mouse oviducts maintained in organ culture. J. Exp. Zool. 249: 235-239.
Kruip, T.A.M., J.H.G. den Daas. 1997. In vitro produced and cloned embryos: effects on
pregnancy, parturition and offspring. Theriogenology 47:43-52. Kubota, C., H. Yamakuchi, J. Todoroki, K. Mizoshita, N. Tabara, M. Barber, X. Yang.
2000. Six cloned calves produced from adult fibroblast cells after long term culture. Proc. Natl. Acad. Sci. USA 97:990-995.
Kuzan, F.B., R.W. Wright, Jr. 1982. Observations on the development of bovine
morulae on various cellular and noncellular substrata. J. Anim. Sci. 54:811-816. Lane, M., D.K. Gardner. 1994. Amino acids increase post-implantation development of
cultured mouse embryos while ammonium induces exencephaly and fetal retardation. J. Reprod. Fert. 192:305-312.
Lawen, A. 2003. Apoptosis – an introduction. BioEssays 25:888-896. Lawson, R.A., L.E. Rowson, C.E. Adams. 1972. The development of cow eggs in the
rabbit oviduct and their viability after re-transfer to heifers. J. Reprod. Fertil. 28:313-315.
Leibfried-Rutledge, M.L. 1999. Factors determining competence of in vitro produced
cattle embryos. Theriogenology 51:473-485. Lenz, R.W., G.D. Ball, M.L. Leibfried, R.L. Ax, N.L. First. 1983. In vitro maturation and
fertilization of bovine oocytes are temperature-dependent processes. Biol. Reprod. 29:173-179.
Lewis, W.H., P.W. Gregory. 1929. Cinematographs of living developing rabbit-eggs.
Science 69: 226-229. Lidner, G.M., R.W. Wright, Jr. 1983. Bovine embryo morphology and evaluation.
Theriogenology 20:407-416. Lighten, A.D., G.M. Moore, R.M.L. Winston, K. Hardy. 1998. Routine addition of IGF-I
could benefit clinical IVF culture. Human Reprod. 13:3144-3150. Lim, J.M., B.C. Reggio, R.A. Godke, W.A. Hansel. 1996. A continuous flow, perifusion
culture system for 8- to 16-cell bovine embryos developed from in vitro culture. Theriogenology 51:1565-1576.
Linares, T., W.A. King. 1980. Morphological study of the bovine blastocyst with phase
contrast microscopy. Theriogenology 14:123-129.
153

Liu, A., R.H. Foote. 1995. Effects of amino acids on the development of in-vitro matured/in-vitro fertilized bovine embryos in a simple protein-free medium. Human Reprod. 11:2985-2991.
Loeb, J. 1894. Über eine einfache Methode, zwei oder mehr zusammengewachsene
Embryonen aus einem Ei hervorzubringer. Pflügers. Arch. 55:525-530. Lonergan, P., H. Khatir, F. Piumi, D. Rieger, P. Humblot, M.P. Boland. 1999. Effect of
time interval from insemination to first cleavage on the developmental characteristics, sex, and pregnancy rates following transfer of bovine preimplantation embryos. J. Reprod. Fert. 117:159-167.
Lonergan, P., D. Rizos, A. Gutierrez-Adan, T. Fair, M.P. Boland. 2003. Oocyte and
embryo quality: effect of origin, culture conditions and gene expression patterns. Reprod. Dom. Anim. 38:259-267.
Lonergan, P., D. Rizos, F. Ward, M.P. Boland. 2001. Factors influencing oocyte and
embryo quality in cattle. Reprod. Nutr. Dev. 41:427-437. Long, C.R., J.R. Dobrinsky, W.M. Garrett, L.A. Johnson. 1998. Dual labeling of the
cytoskeleton and DNA strand breaks in porcine embryos produced in vivo and in vitro. Mol. Reprod. Dev. 51:59-65.
Makarevich, A.V., M. Markkula. 2002. Apoptosis and cell proliferation potential of bovine
embryos stimulated with insulin-like growth factor I during in vitro maturation and culture. Biol. Reprod. 66:386-392.
Majno, G., I. Joris. 1995. Apoptosis, oncosis, and necrosis. Amer. J. Path. 146:3-15. Martins-Green, M., J.E. Feugate. 1998. The 9E3/CEF4 gene product is a chemotactic
and angiogenic factor that can initiate the wound-healing cascade in vivo. Cytokine 10:522-535.
Martins-Green, M., H. Hanafusa. 1997. The chicken chemotactic and angiogenic factor
(cCAF): potential roles in would healing and tumor development. Cytokine Growth Factor Rev. 8:221-232.
Massip, A., P. Mermillod, A. Van Langendonckt, H.D. Reichenback, P. Lonergan, U.
Berg, C. Carolan, R. De Roover, G. Brem. 1996. Calving outcome following transfer of embryos produced in vitro in different conditions. Anim. Reprod. Sci. 44:1-10.
Matsuhashi, S., H. Sugihara. 1984. Development of chicken epidermis cultured with
embryo extract. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 46:53-64. Matwee, C., D.H. Betts, W.A. King. 2000. Apoptosis in the early bovine embryo. Zygote
8:57-68.
154

McEvoy, R.C., O.D. Hegre. 1976. Foetal rat pancreas in organ culture: effects of media supplementation with various steroid hormones on the acinar and islet components. Differentitation 6:105-111.
McEvoy, T.G., J.J. Robinson, R.P. Aitken, P.A. Findlay, I.S. Robertson. 1997. Dietary
excess of urea influence the viability and metabolism of preimplantation sheep embryos and may affect fetal growth among survivors. Anim. Reprod. Sci. 47:71-90.
McEvoy, T.G., K.D. Sinclair, P.J. Broadbent, K.L. Goodhand, J.J. Robinson. 1998. Post-
natal growth and development of Simmental calves derived from in vivo or in vitro embryos. Reprod. Fertil. Dev. 10:459-464.
McFarland, D.C., N.H. Ferrin, K.K. Gilkerson, J.E. Pesall. 1992. Tissue distribution of
insulin-like growth factor in the turkey. Comp. Biochem. Physiol. 103B:601-607. McGaugh, J.W., D. Oids, D.D. Kratzer. 1974. Ovum recovery in superovulated cows and
cleavage rates in the fertilized ova. Theriogenology 6:213-217. McGowan, L.T., J.G. Thompson. 1997. Perfusion culture of bovine in vitro produced
embryos. Proc Aust Soc Reprod Biol 29:24 (abstr.). McGrath, J., D. Solter. 1984. Completion of mouse embryogenesis requires both
maternal and paternal genomes. Cell 37:179-183. McKiernan, S.H., B.D. Bavister. 1994. Timing of development is a critical parameter for
predicting successful embryogenesis. Human Reprod. 9:2123-2129. McLaren, A., J.D. Biggers. 1958. Successful development and birth of mice cultivated in
vitro as early embryos. Nature 182:877-888. McMurtry, J.P., G.L. Francis, Z. Upton. 1997. Insulin-like growth factors in poultry. Dom.
Anim. Endocrinol. 14:199-229. Mehmet, L.R. 2000. Caspases find a new place to hide. Nature 403:29-30. Mello, M.R., H.V. Caetano, M.G. Marques, M.S. Padilha, J.F. Garcia, M.P. Milazzotto,
M.E. Assumpcao, A.S. Lima, A.C. Nicacio, C.M. Mendes, V.P. Oliveira, J.A. Visintin. 2003. Production of a cloned calf from a fetal fibroblast cell line. Braz. J. Med. Biol. Res. 36:1485-1489.
Meng, L., J.J. Ely, R.L. Stouffer, D.P. Wolf. 1997. Rhesus monkeys produced by nuclear
transfer. Biol. Reprod. 57:454-459. Miller, D.J., R.L. Ax. 1990. Carbohydrates and fertilization in animals. Mol. Reprod. Dev. 26:184-198.
155

Minami, N., B.D. Bavister, A. Iritani. 1988. Development of hamster two-cell embryos in the isolated mouse oviduct in organ culture system. Gamete Res. 9:235-240.
Mohamed, A.R., D.E. Noakes. 1985. Enzymatic activities in amniotic fluid and maternal
blood in cattle before and after induced fetal death and abortion. British Vet. J. 141:49-59.
Mohr, L.R., A.O. Trounson. 1982. Comparative ultrastructure of hatched human, mouse
and bovine blastocysts. J. Reprod. Fert. 66:499-504. Moley, K.H., M.M.Y. Chi, C.M. Knudson, S.J. Korsmeyer, M.M. Mueckler. 1998.
Hyperglycemia induces apoptosis in pre-implantation embryos through cell death effector pathways. Nat. Med. 4:1421-1424.
Moore, N.W. 1970. Preliminary studies on in vitro culture of fertilized sheep ova.
Aust. J. Biol. Sci. 23:721-724. Moore, N.W. 1975. The control of time of oestrus and ovulation and the induction of
superovulation in cattle. Aust. J. Res. 26:295-304. Morgan, T.H. 1934. Embryology and Genetics. Columbia University Press: New York,
NY. Morgan, J.F., H.J. Morgan, R.C. Parker. 1950. Nutrition of animal cells in tissue culture.
I. Initial studies on a synthetic medium. Proc. Soc. Exp. Biol. Med. 73:1-8. Murakami, M., T.R. Davidson, C.E. Ferguson, D. Hylan, Y. Echelard, R.A. Godke. 2004.
Use of the amniotic cavity of the developing chick embryo for culturing bovine 8-cell embryos derived from somatic cell nuclear transfer. Mol. Reprod. Dev. (in press).
Nagai, T., T. Takahashi, Y. Chioya, N. Oguri. 1990. Maturation and fertilization of pig
follicular oocytes cultured in pig amniotic fluid. Theriogenology 34:195-203. Namikawa, S. 1969. Effects of Sephadex fractions of chick embryo extract on growth
and differentiation of endothelial cells from chick embryo hearts in culture. Mie. Med. J. 19:29-40.
Neeper, I.D., D.L. Patton, C.C. Kuo. 1990. Cinematographic observations of growth
cycles of Chlamydia trachomatis in primary cultures of human amniotic cells. Infect. Immunity 58:2042-2047.
Negoescu, A., P. Lorimier, F. Labat-Moleur, C. Drouet, C. Robert, C. Guillermet, C.
Brambilla, E. Brambilla. 1996. In situ apoptotic cell labeling by the TUNEL method: improvement and evaluation on cell preparations. J. Histochem. Cytochem. 44:959-968.
156

Neuber, E., C.M. Luetjens, A.W. Chan, G.P. Schatten. 2002. Analysis of DNA fragmentation of in vitro cultured bovine blastocysts using TUNEL. Theriogenology 57:2193-2202.
Nicolas-Bolnet, C., F. Dieterlen-Lievre. 1995. In vitro survival and multiplication of
chicken myeloblasts promoted for several weeks by chick embryo extract. Poult. Sci. 74:942-950.
Niemann, H., C. Wrenzycki. 2000. Alterations of expression of developmentally
important genes in preimplantation bovine embryos by in vitro culture conditions: implications for subsequent development. Theriogenology 53:21-34.
Niemann, H., C. Wrenzycki, A. Lucas-Hahn, T. Brambrink, W.A. Kues, J.W. Carnwath.
2002. Gene expression patterns in bovine in vitro-produced and nuclear transfer-derived embryos and their implications for early development. Cloning Stem Cells 4:29-38.
Niwa, K., O. Ohgoda. 1998. Synergistic effect of caffeine and heparin on in vitro fertilization of cattle oocytes matured in culture. Theriogenology 30:733-741. Ocampo, M.B., L.C. Ocampo, T. Mori, J. Ueda, H. Kanagawa. 1994. Nuclear and
cytoplasmic maturation of pig oocytes cultured in the amniotic fluid of developing chick embryos. J. Vet. Med. Sci. 56:173-176.
Ocampo, M.B., L.C. Ocampo, K. Suzuki, T. Mori, J. Ueda, H. Shimizu, H. Kanagawa.
1993. Development to the blastocyst stage of pig embryos cultured in the amniotic fluid of developing chick embryos. J. Vet. Med. Sci. 55:889-891.
Ocana Quero, J.M., M. Moreno Millan, M. Valera Cordoba, M. Pinedo Merlin, A. Rodero
Franganillo. 1995. The effect of bovine amniotic fluid on in vitro maturation of bovine oocytes. British Vet. J. 151:547-554.
Oettle, E.E., K. Wiswedel. 1989. Mouse embryos cultured in amniotic fluid. South African
Med. J. 76:63-64. Ogura, A., K. Inoue, N. Ogonuki, A. Noguchi, K. Takano, R. Nagano, O. Suzuki, J. Lee,
F. Ishino, J. Matsuda. 2000. Production of male cloned mice from fresh, cultured, and cryopreserved immature Sertoli cells. Biol. Reprod. 62:1579-1584.
Oltvai, Z.N., C.L. Milliman, S.J. Korsmeyer. 1993. Bcl-2 heterodimerizes in vitro with a
conserved homology, Bax, that accelerates programmed cell death. Cell 74:609-619.
Ouhibi, N., J. Hamidi, J. Guillaud, Y. Menezo. 1990. Co-culture of 1-cell mouse embryos
on different cell supports. Human Reprod. 5:737-743. Overskei, T.L., A.H. Cincotta. 1987. A new approach to embryo co-culture.
Theriogenology 27:266 (abstr.).
157

Pampfer, S., R. de Hertogh, I. Vanderheyden, B. Michiels, M. Vercheval. 1990a. Decreased inner cell mass proportion in blastocysts from diabetic rats. Diabetes 39:471-476.
Pampfer, S., I. Vanderheyden, B. Michiels, R. de Hertogh. 1990b. Cell allocation to the
inner cell mass and the trophoectoderm in rat embryos during in vivo preimplantation development. Roux’s Arch Dev Biol 198:257-263.
Pampfer, S., I. Vanderheyden, J.E. McCracken, J. Vesela, H.R. De. 1997. Increased cell
death in rat blastocysts exposed to maternal diabetes in utero and to high glucose or tumor necrosis factor-alpha in vitro. Development 124:4827-4836.
Papaionnaou, V.E., K.M. Ebert. 1988. The preimplantation pig embryo cell number and
allocation to trophoectoderm and inner cell mass of the blastocyst in vivo and in vitro. Development 102:793-803.
Parrishh, J.J., J.L. Susko-Parrish, N.L. First. 1985. Effect of heparin and chondroitin
sulfate on the acrosome reaction and fertility of bovine sperm in vitro. Theriogenology 24:537-549.
Parrish, J.J., J.L. Susko-Parrish, R.R. Handrow, M.M. Sims, N.L. First. 1989.
Capacitation of bovine spermatozoa by oviduct fluid. Biol. Reprod. 40:1020-1025. Parrish, J.J., J.L. Susko-Parrish, M.L. Leibfried-Rutledge, E.S. Critser, W.H. Eyestone,
N.L. First. 1986. Bovine in vitro fertilization with frozen-thawed semen. Theriogenology 25:591-600.
Parrish, J.J., J.L. Susko-Parrish, M.A. Winer, N.L. First. 1988. Capacitation of bovine sperm by heparin. Biol. Reprod. 38:1171-1180. Paula-Lopes, F.F., P.J.Hansen. 2002. Heat shock-induced apoptosis in preimplantation
bovine embryos is a developmentally regulated phenomenon. Biol. Reprod. 66:1169-1177.
Phillips, H.J. 1973. Dye exclusion test for cell viability. In: Tissue Culture. Methods and
Application. Academic Press: New York, NY. pp. 406-408. Piechotta, R., L. Milakofsky, B. Nibbio, T. Hare, A. Epple. 1998. Impact of exogenous
amino acids on endogenous amino compounds in the fluid compartments of the chicken embryo. Comp. Biochem. Physiol. A 120:325-337.
Pierce, G.B., A.L. Lewellyn, R.E. Parchment. 1989. Mechanism of programmed cell
death in the blastocyst. Proc. Natl. Acad. Sci. USA 86:3654-3658. Pierce, M.E. 1933. The amnion of the chick as an independent effector. J. Exp. Zool.
65:443-473.
158

Pincus, G. 1951. Observations on the development of cow ova in vivo and in vitro. In: Proc First Natl Egg Transfer Breeding Conf. San Antonia, TX. p 18.
Pinyopummintr, T., B.D. Bavister. 1994. Development of bovine embryos in a cell-free
culture medium: effects of type of serum, timing of its inclusion and heat inactivation. Theriogenology 41:1241-1249.
Pinyopummintr, T., B.D. Bavister. 1996a. Effects of amino acids on development in vitro
of cleavage stage bovine embryos into blastocysts. Reprod. Fert. Dev. 8:835- 841.
Pinyopummintr, T., B.D. Bavister. 1996b. Energy substrate requirements for in vitro
development of early cleavage stage bovine embryos. Mol. Reprod. Dev. 44:193-199.
Plante, L., W.A. King. 1992. Effect of time to first cleavage on hatching rate of bovine
embryos in vitro. Theriogenology 37:274 (abstr.). Plante, L., W.A. King. 1994. Light and electron microscopic analysis of bovine embryos
derived by in vitro and in vivo fertilization. J. Assist. Reprod. Genet. 11:515-529. Polejaeva, I.A., S.H. Chen, T.D. Vaught, R.L. Page, J. Mullins, S. Ball, Y. Dai, J. Boone,
S. Walker, D.L. Ayares, A. Coleman, K.S. Campbell. 2000. Cloned pigs produced by nuclear transfer from adult somatic cells. Nature 407:86-90.
Polge, C.,C.E. Adams, R.D. Baker. 1972. Development and survival of pig embryos in
the rabbit oviduct. Proc. 7th Int. Congr. Anim. Reprod. 5:513–517. Pool, S.H., R.W. Rorie, R.J. Pendleton, A.R. Menino, R.A. Godke. 1988. Culture of
early-stage bovine embryos inside day-13 and day-14 precultured trophoblastic vesicles. Ann. NY Acad. Sci. 541:407-418.
Potts, D.M., I.B. Wilson. 1967. The preimplantation conceptus of the mouse at 90 hours
post-coitum. J. Anat. 102:1-11. Prather, R.S., F.L. Barnes, M.M. Sims, J.M. Robl, W.H. Eyestone, N.L. First. 1987.
Nuclear transplantation in the bovine embryo: assessment of donor nuclei and recipient oocyte. Biol. Reprod. 37:859-866.
Prather, R.S., N.L. First. 1988. A review of early mouse embryogenesis and it’s
applications to domestic species. J. Anim. Sci. 66:2626-2635. Prather, R.S., M.M. Simms, N.L. First. 1989. Nuclear transplantation in early pig
embryos. Biol. Reprod. 41:414-418. Prather R.S., M.M. Sims, N.L. First. 1991. Culture of porcine embryos from the one- and
two-cell stage to the blastocyst stage in sheep oviducts. Theriogenology 35: 1147–1151.
159

Pugh A., S. Cox, J. Peterson, A. Ledgard, J. Forsyth, K. Cockrem, R. Tervit. 2001. Culture of bovine embryos in sheep oviducts improves frozen but not fresh embryo survival. Theriogenology 55:314 (abstr.).
Redmer, D.A., L.P. Reynolds. 1996. Angiogenesis in the ovary. Rev. Reprod.
1:182-192. Rexroad, C.E. 1989. Co-culture of domestic animal embryos. Theriogenology 31:105-
114. Rexroad, C.E., A.M. Powell. 1986. Co-culture of sheep ova and cells from sheep
oviduct. Theriogenology 37:859-866. Rexroad, C.E., A.M. Powell. 1989. Co-culture of ovine eggs with oviductal cells in
medium 199. J. Anim. Sci. 66:647-953. Reynolds, L.P., D.A. Redmer. 1995. Utero-placental vascular development and placental
function. J. Anim. Sci. 73:1839-1851. Ribatti, D., A. Vacca, L. Roncali, F. Dammacco. 1996. The chick chorioallantoic
membrane as a model for in vivo research on angiogenesis. Int. J. Dev. Biol. 40: 1189-1197.
Rissau, W. 1995. Differentiation of endothelium. FASEB J. 9:926-933. Rissau, W. 1997. Mechanisms of angiogenesis. Nature 386:671-674. Rissau, W., I. Flamme. 1995. Vasculogenesis. Ann. Rev. Cell Dev. Biol. 11:73-91. Rizos, D., A. Gutierrez-Adan, S. Perez-Garnelo, J. de la Fuente, M.P. Boland, P.
Lonergan. 2003. Bovine embryo culture in the presence or absence of serum: Implications for blastocyst development, cryotolerance, and messenger RNA expression. Biol. Reprod. 68:236-243.
Rizos D., F. Ward, P. Duffy, M.P. Boland, P. Lonergan. 2002. Consequences of bovine
oocyte maturation, fertilization or early embryo development in vitro versus in vivo: implications for blastocyst yield and blastocyst quality. Mol. Reprod. Dev. 61: 234-248.
Romanoff, A.L. 1960. The avian embryo. MacMillian: New York, NY. Romanoff, A.L. 1967. Biochemistry of the avian embryo. Wiley: New York, NY. pp 143-
176. Romanoff, A.L., A.J. Romanoff. 1949. The avian egg. John Wiley and Sons: New York,
NY.
160

Rosenkrans, C.F., Jr., N.L. First. 1994. Effect of free amino acids and vitamins on cleavage and developmental rate of bovine zygotes in vitro. J Anim Sci 72:434-437.
Rowlett, K., K. Simkiss. 1987. Explanted embryo culture: in vitro and in ovo techniques
for domestic fowl. Br. Poult. Sci. 28:91-101. Runge, G. 1982. Incubation and hatching for poultry. Queensland Agric. J. 6:305-316. Sadler, T.W., E.S. Hanter. 1994. Principles of abnormal development. Past, present and
future. In: Development Toxicology. Raven Press: New York, NY. pp 53-63. Sakkas, D., Y. Shoukir, D. Chardonnens, P.G. Bianchi, A. Campana. 1998. Early
cleavage of human embryos to the two-cell stage after intracytoplasmic sperm injection as an indicator of embryo viability. Human Reprod. 13:182-187.
Sathananthan, A.H., C. Wood, J.F. Leeton. 1982. Ultrastructural evaluation of 8-16 cell
human embryos cultured in vitro. Micron 13:193-203. Scarpini, E., G. Meola, P. Baron, M. Moggio, V. Silani, G. Scarlato. 1982. Morphological
studies and quantitative assessment of the effect of chick embryo extract on monolayer cultures of normal human muscle. Acta Neurol. (Napoli) 4:403-410.
Schmidek, A., T. Hare. L. Milakofsky, B. Nibbio, A. Epple. 2001. Insulin-like growth
factor-I affects amnion compounds in the fluids of the chicken embryo. Gen. Comp. Endocrin. 123:235-243.
Schmidt, M., T. Greve, B. Avery, J.F. Beckers, J. Sulon, H.B. Hansen. 1996.
Pregnancies, calves and calf viability after transfer of in vitro produced bovine embryos. Theriogenology 46:527-539.
Schneider, H.J., Jr., R.S. Castleberry, J.L. Griffin. 1980. Commercial aspects of bovine
embryo transfer. Theriogenology 13:73-85. Seemayer, N.H., W. Hadnagy, R. Tomingas. 1987. The effect of automobile
exhaust particulates on cell viability, plating efficiency and cell division of mammalian tissue culture cells. Sci. Total Environ. 61:107-115.
Shamsuddin, M., B. Larson, H. Rodriguez-Martinez. 1993. Culture of bovine IVM/IVF
embryos up to blastocyst stage in defined medium using insulin, transferin and selenium or growth factors. Reprod. Dom. Anim. 28:209-210.
Shamsuddin, M., H. Rodriguez-Martinez. 1994. Fine structure of bovine blastocysts
developed either in serum-free medium or in conventional co-culture with oviduct epithelial cells. J. Vet. Med. 41:307-316.
161

Sharif H., E. Vergos, P. Lonergan, M. Gallagher, A. Kinis, I. Gordon. 1991. Development of early bovine embryos in the isolated mouse oviduct maintained in organ culture. Theriogenology 35:270 (abstr.).
Shea, B.F. 1981. Evaluating the bovine embryo. Theriogenology 15:13-42. Shiga, K., T. Fujita, K. Hirose, Y. Sasae, T. Nagai. 1999. Production of calves by transfer
of nuclei from culture somatic cells obtained from Japanese black bulls. Theriogenology 52:527-535.
Shimizu, S., M. Narita, Y. Tsujimoto. 1999. Bcl-2 family proteins regulate the release of
apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399:483-487.
Shin, T., D. Kraemer, J. Pryor, L. Liu, J. Rigila, L. Howe, S. Buck, K. Murphy, L. Lyons,
M. Westhusin. 2002. A cat cloned by nuclear transplantation. Nature 415:859. Shin, T., L. Sneed, J.R. Hill, M.E. Westhusin. 2000. High incidence of developmental
failure in bovine fetuses derived by cloning bovine viral diarrhea virus-infected cells. Theriogenology 53:243 (abstr.).
Shirley, B., J.W.E. Wortham, Jr., J. Witmyer, M. Condon-Mahony, G. Fort. 1985. Effects
of human serum and plasma on development of mouse embryos in culture media. Fertil. Steril. 43:129-134.
Shoukier, Y., D. Chardonnens, A. Campana, P. Bischof, D. Sakkas. 1998. The rate of
development and time of transfer play different roles in influencing the viability of human blastocysts. Human Reprod. 13:676-681.
Sims, M., N.L. First. 1993. Production of calves by nuclear transfer of nuclei from
cultured inner cell mass cells. Proc. Natl. Acad. Sci. USA 90:6143-6147. Sinclair, K.D., P.J. Broadbent, D.F. Dolman. 1995. In vitro produced embryos as a
means of achieving pregnancy and improving productivity in beef cows. Anim. Sci. 60:55-64.
Sinclair, K.D., E.K. Maxfield, J.J. Robinson, C.A. Maltin, T.G. McEvoy, L.D. Dunne, L.E.
Young, P.J. Broadbent. 1997. Culture of sheep zygotes can alter fetal growth and development. Theriogenology 47:380 (abstr.).
Sinclair, K.D., T.G. McEvoy, C. Carolan, E.K. Maxfield, C.A. Maltin, L.E. Young, I.
Wilmut, J.J. Robinson, P.J. Broadbent. 1998. Conceptus growth and development following in vitro culture of ovine embryos in media supplemented with bovine sera. Theriogenology 49:218 (abstr.).
Sinclair, K.D., T.G. McEvoy, E.K. Maxfield, C.A. Maltin, L.E. Young, I. Wilmut, P.J.
Broadbent, J.J. Robinson. 1999. Aberrant fetal growth and development after in vitro culture of sheep zygotes. J. Reprod. Fertil. 60:674-682.
162

Sirard, M.A., R.D. Lambert. 1985. In vitro fertilization of bovine follicular oocytes obtained by laproscopy. Biol. Reprod. 33:487-494.
Smith, S.M., N.A. Schroedl. 1992. Heme-containing compounds replace chick embryo
extract and enhance differentiation in avian muscle cell culture. In Vitro Cell Dev. Biol. 28A:387-390.
Smoczkiewiczowa, A. 1959. Sodium, potassium, calcium and chloride ion contents and
protein fractions in the fluids of chick embryos. Nature 183:1260-1261. Spemann, H. 1914. Über verzögerte Kernversorung von Keimteilen. Verh. Deutsch.
Zool. Ges. Leipzig. (Freiburg) 24:216-221. Spemann, H. 1938. Embryonic development and induction. Yale University Press: New
Haven, CN. Sreenan, J.M., P. Scanlon, I. Gordon. 1968. Culture of fertilized cattle eggs. J. Agri. Sci.
70:183–185. Stice, S.L., N.S. Strelchenko, C.L Keefer, L. Matthews. 1996. Pluripotent bovine
embryonic cell lines direct embryonic development following nuclear transfer. Biol. Reprod. 54:100-110.
Strasser, A., L. O’Conner, V.M. Dixit. 2000. Apoptosis signaling. Annu. Rev. Biochem.
69:217-245. Stringfellow, D.A., K.P. Riddell, P.K. Galik, P. Damiani, M.D. Bishop, J.C. Wright. 2000.
Quality controls for bovine viral diarrhea virus-free IVF embryos. Theriogenology 53:827-839.
Sueldo, C., L. Montoro, P. Young, E. Kelly, E. Subias, M. Baccaro, J. Swanson, H.
Lambert. 1988. The use of amniotic fluid in in-vitro fertilization (IVF). Fertil. Steril. 50:Suppl s70-71 (abstr.).
Takahashi, Y., N.L. First. 1992. In vitro development of bovine one-cell embryos:
influences of glucose, lactate, pyruvate, amino acids and vitamins. Theriogenology 37:963-978.
Takano, K. 1957. Role of chick embryo extract for growth mode in vitro of human
carcinon HeLa. Jpn. J. Med. Sci. Biol. 10:35-45. Tam, P.P. 1988. Postimplantation development of mitomycin C-treated mouse
blastocysts. Teratology 37:205-212.
Tanaka, M., Y. Hayashida, K. Sakaguchi, T. Ohkubo, M. Wakita, S. Hoshino, K. Nakashima. 1996. Growth hormone-independent expression of insulin-like growth factor I messenger ribonucleic acid in extrahepatic tissues of the chicken. Endocrinology 137:30-34.
163

Tatemoto, H., N. Sakurai, N. Muto. 2000. Protection of porcine oocytes against apoptotic cell death caused by oxidative stress during in vitro maturation: role of cumulus cells. Biol. Reprod. 63:805-810.
Taverne, M.A.M., S.P. Breukelman, Z. Perenyi, S.J. Dieleman, P.L.A.M. Vos, H.H.
Jonker, L. de Ruigh, J.M. van Wagtengonk-de Leeiuw, J.F. Beckers. 2002. The monitoring of bovine pregnancies derived from transfer of in vitro produced embryos. Reprod. Nutr. Develop. 42:613-642.
ten Busch, M., L. Milakofsky, T. Hare, B. Nibbio, A. Epple. 1997a. Impact of ethanol
stress on components of the allantoic fluid of the chicken embyo. Comp. Biochem. Physiol. 116A:125-129.
ten Busch, M., L. Milakofsky, T. Hare, B. Nibbio, A. Epple. 1997b. Regulation of
substances in allantoic and amniotic fluid of the chicken embryo. Comp. Biochem. Physiol. 116A:131-136.
Tervit, H.R., P.A. Pugh, L.T. McGowan, A.C.S. Bell, R.W. Wells. 1994. The freezability
of sheep embryos is affected by culture system and source (in vivo or in vitro derived). Theriogenology 41:315 (abstr.).
Tervit, H.R., D.G. Whittingham, L.E.A. Rowson. 1972. Successful culture in vitro of
sheep and cattle ova. J. Reprod. Fert. 30:495-497. Thibodeaux, J.K., R.A. Godke. 1992. In vitro enhancement of early-stage embryos with
co-culture. Arch. Pathol. Lab. Med. 116:364-372. Thompson, J.G. 1997. Comparison between in vivo-derived and in vitro-produced pre-
elongation embryos from domestic ruminants. Reprod. Fert. Dev. 9:341-354.
Thompson, J.G., 2000. In vitro culture and embryo metabolism of cattle and sheep
embryos - a decade of achievement. Anim. Reprod. Sci. 60-61:263-75.
Thompson, J.G., N.W. Allen, L.T. McGowan, A.C. Bell, M.G. Lambert, H.R. Tervit. 1998.
Effect of delayed supplementation of fetal calf serum to culture medium on bovine embryo development in vitro and following transfer. Theriogenology 49:1239-1249.
Thompson, J.T., D.K. Gardner, P.A. Pugh, W.H. Macmillan, H.R. Tervit. 1995. Lamb
birth weight is affected by culture system utilized during in vitro pre-elongation development of ovine embryos. Biol. Reprod. 53:1385-1391.
Thompson, J.G., A.C. Simpson, P.A. Pugh, P.E. Donnelly, H.R. Tervit. 1990. Effect of
oxygen concentration on in-vitro development of preimplantation sheep and cattle embryos. J. Reprod. Fert. 89:573-578.
164

Totey, S.M., M. Daliri, K.B.C.A. Rao, C.H. Pawshe, M. Taneja, R.S. Chillar. 1996. Differential cleavage and developmental rates and their correlation with cell numbers and sex ratios in buffalo embryos generated in vitro. Theriogenology 45:521-533.
Tsunoda, Y., Y. Kato. 1998. Not only inner cell mass cell nuclei but also trophoectoderm
nuclei of mouse blastocysts have a developmental totipotency. J. Reprod. Fert. 113:181-184
Vajta, G.T., T. Puera, P. Holm, A. Paldi, T. Greve, A.O. Trousan, H. Callesen. 2000.
New method for the culture of zona-included or zona-free embryos: the well of the well (WOW) system. Mol. Reprod. Dev. 55:256-264.
Van De Velde, A., L. Liu, P.E.J. Bols, M.T. Ysebaert, X. Yang. 1999. Cell allocation and
chromosomal complement of parthenogentic and IVF derived bovine embryos. Mol. Reprod. Dev. 54:57-62.
Van Langendonkt, A., P. Auquier, I. Donnay, A. Massip, F. Dessy. 1996. Acceleration of
in vitro bovine embryo development in the presence of fetal calf serum. Theriogenology 45:194 (abstr.).
Van Soom, A., M.L. Boerjan, P.E.J. Bols, G. Vanrose, A. Lein, M. Coryn, A. de Kruif.
1997. Timing of compaction and inner cell allocation in bovine embryos produced in vivo after superovualtion. Biol. Reprod. 57:1041-1049.
Van Soom, A., I. Van Vlaenderen, A.R. Mahmoudzadeh, H. Deluyker, A. de Kruif. 1992.
Compaction rates of in vitro fertilized bovine embryos related to the interval from insemination to first cleavage. Theriogenology 38:905-919.
van Wagtendonk-de Leeuw, A.M., B.J.G Aerts, J.H.G. den Daas. 1998. Abnormal
offspring following in vitro production of bovine preimplantation embryos: a field study. Therigenology 49:883-894.
van Wagtendonk-de Leeuw, A.M., E. Mullaart, A.P.W. de Roos, J.S. Merton, J.H.G. den
Daas, B. Kemp, L. de Ruigh. 2000. Effects of different reproduction techniques: AI, MOET, or IVP on health and welfare of bovine offspring. Theriogenology 53:575-597.
Vergos, E., A. Kinis, M. Gallagher, A. Gordon. 1989. Development of bovine embryos in
an oviductal monolayer in relation to cell stage reached at 48 hours after in vitro fertilization. J. Reprod. Fert. 4:37 (abstr.).
Viuff, D., P.J.M. Hendriksen, P.L.A.M. Vos, S.J. Dielemann, B.M. Bibby, T. Greve, P.
Hyttel, P.D. Thompsen. 2001. Chromosomal abnormalities and developmental kinetics in in vivo-derived cattle embryos at days 2 to 5 after ovulation. Biol. Reprod. 2001 65:204-208.
165

Viuff, D., L. Rickords, H. Offenberg, P. Hyttel, B. Avery, T. Greve, I. Olsaker, J.L. Williams, H. Callesen, P.D. Thomsen. 1999. A high proportion of bovine blastocysts produced in vitro are mixoploid. Biol. Reprod. 60:1273-1278.
Voelkel, S.A., G.F. Amborski, K.G. Hill, R.A. Godke. 1985. Use of uterine-cell monolayer
culture system for micromanipulated bovine embryos. Theriogenology 24:271-281.
Wakayama, T., A.C.F. Perry, M. Zuccotti, K.R. Johnson, R. Yanagimachi. 1998. Full
term development of mice from enucleated oocytes injected with cumulus cell nuclei. Nature 394:369-374.
Walker, S.K., T.M. Heard, R.F. Seamark. 1992. In vitro culture of sheep embryos
without co-culture: successes and perspectives. Theriogenology 37:111-127. Walker, S.K., K.M. Hartwich, R.F. Seamark. 1996. The production of unusually large
offspring following embryo manipulation: concepts and challenges. Theriogenology 45:111-120.
Wang, S.K., D.L. Patton, C.C. Kou. 1992. Effects of ascorbic acid on Chlamydia
trachmomatis infection and on erythromycin treatment in primary cultures of human amniotic cells. J. Clinical. Micro. 30:2551-2554.
Warner, C.M., W. Cao, G.E. Exley, A.S. McElhinny, M. Alikani, J. Cohen, R.T. Scott,
C.A. Brenner. 1998. Genetic regulation of egg and embryo survival. Human Reprod. 13:178-190.
Waterhouse, N.J., J.E. Ricci, D.R. Green. 2002. And all of a sudden it’s over:
mitochondrial outer-membrane permeabilization in apoptosis. Biochemie 84:113-121.
Watson, A.J., P. De Sousa, A. Caveney, L.C. Barcroft, D. Natale, J. Urquhart, M.E.
Westhusin. 2000. Impact of bovine oocyte maturation medium on oocyte transcript levels, blastocyst development, cell number, and apoptosis. Biol. Reprod. 62:355-364.
Weimer, K.E., G.F. Amborski, R.S. Denniston, K.L. White, R.A. Godke. 1987a. Use of
hormone-treated fetal uterine fibroblast monolayer system for in vitro culture of bovine embryos. Theriogenology 27:294 (abstr.).
Weimer, K.E., P.L. Casey, P.S. Mitchell, R.A. Godke. 1989a. Pregnancies following 24-
hour co-culture of equine embryos on feotal bovine uterine monolayer cells. Equine Vet. J. 8(suppl):117-122.
Weimer, K.E., J. Cohen, G.F. Amborski, G. Wright, S. Wiker, L. Munyakazi, R.A. Godke.
1989b. In vitro development and implantation of human embryos following culture on fetal bovine uterine fibroblast cells. Human Reprod. 45:595-600.
166

Weimer, K.E., R.S. Denniston, G.F. Amborski, K.L. White, R.A. Godke. 1987b. A fetal fibroblast monolayer system of in vitro culture of bovine embryos. J. Anim. Sci. 65(suppl 1):122 (abstr.).
Wells, D.N., P.M. Misica, H.R. Tervit. 1999. Production of cloned calves following
nuclear transfer with cultured adult mural granulosa cells. Biol. Reprod. 60:996-1005.
Wheeler, M.B., S.G. Clark, D.J. Beebe. 2004. Developments in in vitro technologies for
swine embryo production. Reprod. Fert. Dev. 16:1-11. Whitten, W.K. 1957. Culture of tubal ova. Nature 179:1081-1082. Willadsen, S.M. 1979. A method for culture of micromanipulated sheep embryos and its
use to produce monozygotic twins. Nature 227:229-300. Willadsen, S.M. 1986. Nuclear transplantation in sheep embryos. Nature 320:63-65. Willadsen, S.M., R.E. Janzen, R.J. McAlister, B.F. Shea, G. Hamilton, D. McDermand.
1991. The viability of late morulae and blastocysts produced by nuclear transplantation in cattle. Theriogenology 35:161-170.
Willmer, E.N., F. Jacoby. 1936. Studies on the growth of tissues in vitro. J. Exp. Biol.
13:237-248. Wilmut, I., D.I. Sales. 1981. Effect of an asynchronous environment on embryonic
development in the sheep. J. Reprod. Fertil. 61:179-184. Wilmut, I., A.E. Schnieke, J. McWhir, A.J. Kind, K.H.S. Campbell. 1997. Viable offspring
derived from fetal and adult mammalian cells. Nature 385:810-813. Wilson, J.M., J.D. Williams, K.R. Bondioli, C.R. Looney, M.E. Westhusin, D.F. McCalla.
1995. Comparison of birth weight and growth characteristics of bovine claves produced by nuclear transfer (cloning), embryo transfer, and natural mating. Anim. Reprod. Sci. 38:73-83.
Wilting, J., B. Brand-Saberi, H. Kurz, B. Christ. 1995. Development of the embryonic
vascular system. Cell. Mol. Biol. Res. 41:219-232. Wintour, E.M., B.M. Laurence, B.E. Lingwood. 1986. Anatomy, physiology and pathology
of the amniotic and allantoic compartments in the sheep and cow. Australian Vet. J. 63:216-221.
Wrenzycki, C., D. Herrmann, J.W. Carnwath, H. Niemann. 1996. Expression of the gap
junction gene connexin43 (Cx43) in preimplantation bovine embryos derived in vitro or in vivo. J. Reprod. Fertil. 108:17-24.
167

Wright, J.M. 1981. Non-surgical embryo transfer in cattle: embryo-recipient interactions. Theriogenology 15:43-56.
Wright, R.Q., Jr., G.B. Anderson, P.T. Cupps. M. Drost. 1976a. Successful culture in
vitro of bovine embryos to the blastocyst stage. Biol. Reprod. 14:157. Wright, R.Q., Jr., G.B. Anderson, P.T. Cupps. M. Drost. 1976b. Blastocyst expansion
and hatching of bovine ova cultured in vitro. J. Anim. Sci. 43:170 (abstr.). Wright, R.Q., Jr., G.B. Anderson, P.T. Cupps. M. Drost. 1976c. In vitro culture of
embryos from adult and prepubertal ewes. J. Anim. Sci. 43:170 (abstr.). Wright, R.W., Jr., K.R. Bondioli. 1981. Aspects of in vitro fertilization and embryonic
culture in domestic animals. J. Anim. Sci. 53:702-729. Wright, R.W., Jr., J. Ellington. 1995. Morphological and physiological differences
between in vivo- and in vitro-produced preimplantation embryos from livestock species. Theriogenology 44:1167-1189.
Wu, X., M.J. Howard. 2001. Two signal transduction pathways involved in the
catecholaminergic differentiation of avian neural crest-derived cells in vitro. Mol. Cell. Neurosci. 18:394-406.
Wuu, Y.D., S. Pampfer, P. Becquet, I. Vanderheyden, K.H. Lee, R. De Hertogh. 1999.
Tumor necrosis factor alpha decreases the viability of mouse blastocysts in vitro and in vivo. Biol. Reprod. 60:479-483.
Wyllie, A.H. 1995. The genetic regulation of apoptosis. Curr. Opin. Genet. Dev. 5:97-
104. Wyllie, A.H., J.F.R. Kerr, A.R. Curie. 1980. Cell death: the significance of apoptosis. Int.
Rev. Cyt. 68:251-306. Xue, Z.G., X.J. Xue, M. Fauquet, J. Smith, N. Le Douarin. 1992. Expression of the gene
encoding tyrosine hydroxylase in a subpoplation to quail dorsal root ganglion cells cultured in the presence of insulin or chick embryo extract. Brain Res. Dev. Brain Res. 69:23-30.
Yang, H.W., K.J. Hwang, H.C. Kwon, H.S. Kim, K.W. Choi, K.S. Oh. 1998. Detection of
reactive oxygen species (ROS) and apoptosis in human fragmented embryos. Human Reprod. 13:998-1002.
Yang, Y.W., D.R. Brown, H.L. Robcis, M.M. Rechler, F. de Pablo. 1993. Developmental
regulation of insulin-like growth factor binding protein-2 in chick embryo serum and vitrious humor. Reg. Pept. 48:145-155.
168

Yang, M.Y., R. Rajamahendran. 2002. Expression of Bcl-2 and Bax proteins in relation to quality of bovine oocytes and embryos produced in vitro. Anim. Reprod. Sci. 70:159-169.
Yong, Z., W. Jianchen, Q. Jufen, H. Zhiming. 1991. Nuclear transplantation in goats.
Theriogenology 35:299 (abstr.). Young, L.E., K. Fernandes, T.G. McEvoy, S.C. Butterwith, C.G. Gutierrez, C. Carolan,
P.J. Broadbent, J.J. Robinson, I. Wilmut, K.D. Sinclair. 2001. Epigenetic change in IGF2R is associated with fetal overgrowth after sheep embryo culture. Nat. Genet. 27:153-154.
Young, L.E., K.D. Sinclair, I. Wilmut. 1998. Large offspring syndrome in cattle and
sheep. Rev. Reprod. 3:155-163. Zakhartchenko, V., R. Alberio, M. Stojkovic, K. Prelle. W. Schernthaner, P. Stojkovic, H.
Wenigerkind, R. Wanke, M. Duchler, R. Steinborn, M. Mueller, G. Brem, E. Wolf. 1999. Adult cloning in cattle: potential for nuclei from a permanent cell line and from primary cultures. Mol. Reprod. Dev. 54:264-272.
Zou, X., Y. Chen, Y. Wang, J. Lou, Q. Zhang, X. Zhang, Y. Yang, H. Ju, Y. Shen, W.
Lao, S. Xu, M. Du. 2001. Production of cloned goats from enucleated oocytes injected with cumulus cell nuclei or fused with cumulus cells. Cloning 3:31-37.
169

APPENDIX A
BO-A STOCK SOLUTION1
Component Product number Company Amount
NaCl S-5886 Sigma 4.3092 g
KCl P-5405 Sigma 0.1974 g
CaCl2•2H2O C-7902 Sigma 0.2171 g
NaH2PO4•H2O S-9638 Sigma 0.0743 g
MgCl2•6H2O M-2393 Sigma 0.0697 g
Milli Q water – Milli Q 500 ml
Phenol red 0.5% P-0290 Sigma 0.1 ml
1 Prepare solution in a 500-ml bottle and store in a refrigerator for up to 3 months.
170

APPENDIX B
BO-B STOCK SOLUTION1
Component Product number Company Amount
NaHCO3 S-8875 Sigma 2.5873 g
Milli Q water – Milli Q 200 ml
Phenol red 0.5% P-0290 Sigma 0.04 ml
1 Prepare solution in a 250-ml bottle. Inject CO2 gas into the bottle for 1 to 2 minutes until a color change occurs. Seal the bottle with parafilm to maintain proper pH and store in a refrigerator for up to 3 months.
171

APPENDIX C
CR1aa STOCK SOLUTION1
Component Product number Company Amount
NaCl S-5886 Sigma 0.6703 g
KCl P-5405 Sigma 0.0231 g
NaHCO3 S-8875 Sigma 0.2201 g
Pyruvic acid P-4562 Sigma 0.0044 g
L (+) Lactic acid L-4388 Sigma 0.0546 g
Glycine G-8790 Sigma 0.0039 g
L-Alanine A-7469 Sigma 0.0045 g
Milli Q water – Milli Q 500 ml
Phenol red 0.5% P-0290 Sigma 0.2 ml
1 Prepare solution in a 100-ml bottle and stored in a refrigerator for up to 3 months.
172

VITA
Tonya Renea Davidson was born on September 24, 1972 in Lufkin, Texas to
James and Linda Davidson. Tonya has one younger brother, Casey Millspaugh, who
resides in Fayetteville, Arkansas. Tonya grew up in Fort Smith, Arkansas where she
attended Woods Elementary School and later attended Southside High School.
Following graduation from high school in 1991, Tonya enrolled in Carl Albert State
University, in Poteau, Oklahoma, completing an Associate of Science degree in May of
1993. She then transferred to Oklahoma State University in Stillwater, Oklahoma to
pursue a Bachelor of Science degree in Animal Science. After receiving her Bachelor’s
degree in December of 1997, Tonya entered the graduate program at Oklahoma State
University under Dr. Leon J. Spicer. After completing her class work and research,
Tonya received a Master’s of Science in December of 2000. She was then accepted to
begin a doctoral degree at Louisiana State University under the guidance of Dr. Robert
Godke, Boyd Professor of Reproductive Physiology. She is a candidate for the Doctor of
Philosophy degree in reproductive physiology in the Department of Animal Sciences at
Louisiana State University, Baton Rouge, Louisiana.
173


















