
THALASSEMIA Hope Y. Agbemenyah. The hemoglobin is critical for gaseous exchange and transportation...
17
THALASSEMI A Hope Y. Agbemenyah
-
Upload
tamsyn-underwood -
Category
Documents
-
view
214 -
download
0
Transcript of THALASSEMIA Hope Y. Agbemenyah. The hemoglobin is critical for gaseous exchange and transportation...

THALASSEMIAHope Y. Agbemenyah

• The hemoglobin is critical for gaseous exchange and transportation between tissues.
• Hemoglobins are encoded in two tightly linked gene clusters; • the α-like globin genes are clustered on chromosome 16, and the β-like genes
on chromosome 11. • The α-like cluster consists of two α-globin genes and a single copy of the ζ-
gene. • The non-α gene cluster consists of a single ε gene, the Gγ and Aγ fetal globin
genes, and the adult δ and β genes.
• Thalassemias are heterogeneous group of inherited haemoglobinopathies due to mutations leading impaired α- or β- globin biosynthesis.
-diminished production of adult haemoglobin tetramers HbA (α2β2): hypochromic and microcytic anemia.
-increased cell membrane damage or ineffective erythropoiesis or both
Thalassemia syndromes

Demographics and Epidemiology
Certain ancestry: endemic in the Mediterranean basin, Middle East, Asian and tropical Africa and the Indian subcontinent ancestry. Distribution parallels that of Plasmodium falciparum

Molecular Pathogenesis• Mutations
• Trancriptional: Promoter region mutations – Inability to recruit transcription factors eg GATA-1, NFE-2, the SWI/SNF-like protein
called ATRX
• Posttranscriptional: mRNA processing – Splicing mutations some normal beta globin– β+- Thalassemia (reduced but detectable β-globin synthesis)
• Translational: Chain termination mutation– de-novo stop codon and indel blocking translation– βo- Thalassemia (absent β-globin)– Deletion: α-Thalassemia
• Deletion: α-Thalassemia
• Other causes • α-protein stabilizing protein (AHSP)

Classifications• α-Thalassemias
• due to deletions that cause reduced or lack of synthesis of alpha globin chain.
• hence unpaired gamma globins (fetal) or beta (adults)
• these are soluble so hemolysis and ineffective erythropoiesis is less severe than beta thalassemia.
Thalassemia Genetic mutation Remarks
Silent carrier -α/αα No effect
α-Thalassemia traitMinor
-α/-α--/αα
Slight anemia
Hb H, β4 --/-α Severely anemic and susceptible to oxidation
Barts hydrops fetalis, γ4 --/-- Lethal

Classificationsβ-Thalassemias
variable mutations leading to increased levels of alpha globin proteins
Thalassemia Genetic mutation Remarks
β-Thalassemia trait β/βo or β/β+ Mild anemia and small RBCs due to lack of Hb
β-Thalassemia intermedia Milder variant of β+/β+ or βo/β+
Significant lack of Hb, Moderate anemia
β-Thalassemia major βo/βo, βo/β+ or β+/β+
Severe anemia
• accumulation of highly insoluble unpaired α chains. They form toxic inclusion bodies that kill developing erythroblasts in the marrow, few escape with membrane damaging insoluble α chains that undergo extravascular lyses
• Provoking compensatory erythroid hyperplasia but the marrow is sabotaged by ineffective erythropoiesis.
Persistence of anemia causes exuberant extramedullary erythropoietic tissues recruitment

Clinical presentationsDespite differences in the molecular mechanisms underlying the cause of the disease
patients generally present common symptoms of
• severe tissue hypoxia, hemolytic anemia, marked hepatomegaly, marked
erythroid hyperplasia, iron overload
Other symptoms include fatigue, weakness, pale appearance, excessive hemolysis
leading to yellowish discoloration of skin (jaundice), facial bone deformities, dark
urine. The signs and symptoms the patient experiences depend on the type and
severity of thalassemia.
Hyperplasia of bone marrow and extramedullary erythropoiesis

Diagnosis• β-Thalassemia major: basis of severe microcytic anemia, ineffective erythropoiesis-
hepatosplenomegaly, and elevated levels of HbF, HbA2 or both
• Intermedia exhibit similar stigmata but can survive without chronic hypertransfusion
• Minor: profound microcytosis and hypochromia with target cells, but only minimal or mild anemia. Mean corpuscular volume is rarely >75fL; hematocrit is rarely <30-33%. Hb electrophoresis reveals normal/elevated HbA2 or normal/elevated HbF
• α-thalassemia trait- mild hypochromia and microcytosis usually without anemia • Hb electrophoresis• Microcytosis and hypochromia• Reduction in HbA and increase in Hb F• Increased RBC count for such level of anemia
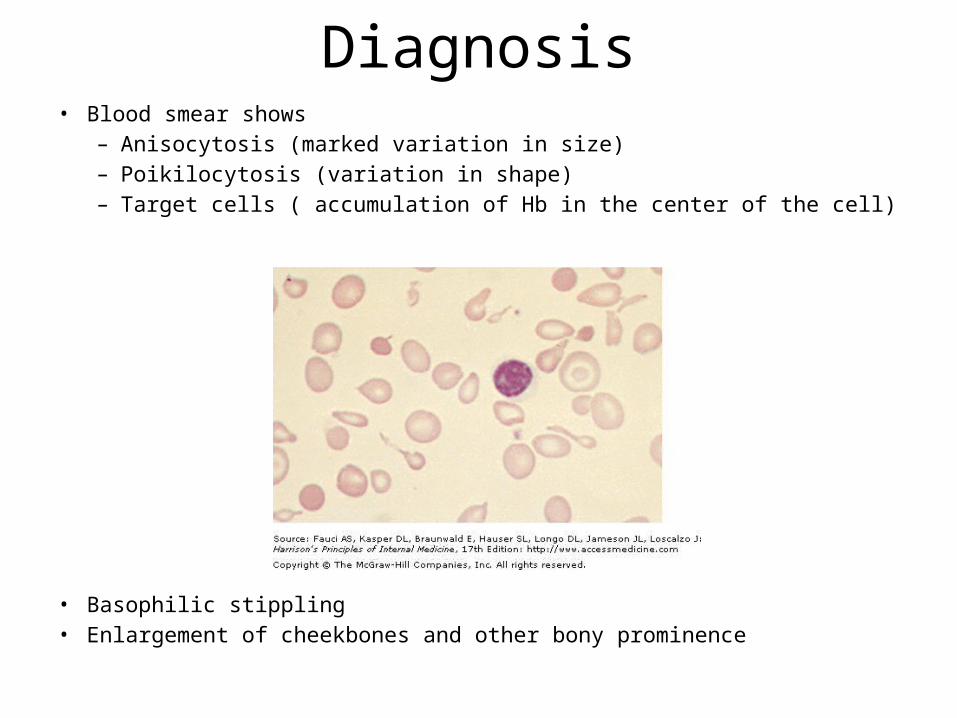
Diagnosis• Blood smear shows
– Anisocytosis (marked variation in size)– Poikilocytosis (variation in shape)– Target cells ( accumulation of Hb in the center of the cell)
• Basophilic stippling • Enlargement of cheekbones and other bony prominence

Treatment• Blood transfusion and as complication you treat iron overload iron chelator-
deferoxamine or deferasirox– Cardiac disease resulting from progressive iron overload and secondary hemochromatosis- an
important cause of death
• Regular monitoring of extramedullary hematopoiesis and spleen size (Splenectomy)
• Hydroxyurea (established) and HDACi (worth testing experimentally) indicated in patients with extramedullary hematopoiesis and leg ulcers
• Allogenic stem cells transplantation is the only curative therapy
• Vitamin D supplementation
• Gene therapy, lentiviral constructs expressing corresponding correct gene
• Premature Termination Codon treatment by the use of aminoglycosides

PreventionAntenatal diagnosisDNA diagnosis by PCR amplification of fetal DNA obtained by amniocentesis or chorionic villus biopsy which is then hybridized to allele-specific oligonucleotide probes
This examination is only appropriate if both parents are known to be carriers (β-Thalassemia minor) and will accept a termination
Seek genetic counseling if a carrier of α-Thalassemia gene
Assisted reproductive therapy

Differential Diagnoses
Anemia of Chronic Disease and Renal Failure
Lead Nephropathy
Lead poisoning
Sideroblastic Anemias

Case
• A 10-month-old boy of Arabic extraction is brought to the physician by his parents who complain that their child is failing to thrive. Physical examination reveals splenomegaly and jaundice. A CBC shows a microcytic, hypochromic anemia (hemoglobin = 7.4 g/dL). Fetal hemoglobin accounts for most of the hemoglobin. A peripheral blood smear is shown in the image. Which of the following is the appropriate diagnosis?
• What is the pathogenesis of splenomegaly seen in the patient described above

• Adriana is a 7-year-old who lives with her parents in a surburban community. Her parents brought her to the united states from their homeland in Greece when she was 1 year old. At the of 3, she was in the 10th percentile for her height and weight, pale and her hemoglobin was 5.8 g/dL. Following further diagnostic studies, she was diagnosed with beta-thalassemia major. Over the course of the next 4 years, Adriana was hospitalized every 1-2 months so she could be transfused with packed red blood cells.
• Case study• During a routine follow-up visit at the hematology clinic, Adriana’s laboratory
results were as follows:• Hemoglobin: 10 g/dL• Total serum iron: 150 g/L
• As hematologist, you discuss planned treatment with Adriana and her parents

• What are the clinical manifestations of b-Thalassemia?
• What are the priorities of care for Adriana?
• Which options are available for Adriana to prevent life-long blood transfusion?

References
• http://www.ncbi.nlm.nih.gov/pubmed/7290983• http://www.sciencedirect.com/science/article/pii/
S0891584914001907• http://www.thalassemia.com• http://www.thalassemia.org • http://www.thalassemia.org.cy• Pathology by Kumar and Robbins• Harrison’s principles of internal medicine 17th ed McGraw-
Hill • Davidson’s principles and practice of internal medicine 20th
ed McGraw-Hill

Vielen Dank fuer eure Aufmerksamkeit


















