
Temporal Changes in Tissue Repair Permit Survival of Diet ...
9
TOXJCOLOGICAL SCTENCES 45, 233-241 (1998) ARTICLE NO. T X 9 8 2 5 1 1 Temporal Changes in Tissue Repair Permit Survival of Diet-Restricted Rats from an Acute Lethal Dose of Thioacetamide Shashi K. Ramaiah, Thomas J. Bucci,* Alan Warbritton,* Madhusudan G. Soni, and Harihara M. Mehendale 1 Division of Toxicology and Louisiana Institute of Toxicology, College of Pharmacy and Health Sciences, Northeast Louisiana University, Monroe, Louisiana 71209-0470; and *Pathology Associates, Inc., National Center for Toxicological Research, Jefferson, Arkansas 72079 Received January 20, 1998; accepted May 29, 1998 Temporal Changes in Tissue Repair Permit Survival of Diet- Restricted Rats from an Acute Lethal Dose of Thioacetamide. Ramaiah, S. K., Bucci, T. J., Warbritton A., Soni, M. G., and Mehendale, H. M. (1998). Toxicol. Sci. 45, 233-241. Although, diet restriction (DR) has been shown to substantially increase longevity while reducing or delaying the onset of age- related diseases, little is known about the mechanisms underlying the beneficial effects of DR on acute toxic outcomes. An earlier study (S. K. Ramaiah et al., 1998, Toxicol. Appl. Pharmacol. 150, 12-21) revealed that a 35% DR compared to ad libitum (AL) feeding leads to a substantial increase in liver injury of thioacet- amide (TA) at a low dose (50 mg/kg, ip). Higher liver injury was accompanied by enhanced survival. A prompt and enhanced tis- sue repair response in DR rats at the low dose (sixfold higher liver injury) occurred, whereas at equitoxic doses (50 mg/kg in DR and 600 mg/kg in AL rats) tissue repair in AL rats was substantially diminished and delayed. The extent of liver injury did not appear to be closely related to the extent of stimulated tissue repair response. The purpose of the present study was to investigate the time course (0-120 h) of liver injury and liver tissue repair at the high dose (600 mg TA/kg, ip, lethal in AL rats) in AL and DR rats. Male Sprague-Dawley rats (225-275 g) were 35% diet restricted compared to their AL cohorts for 21 days and on day 22 they received a single dose of TA (600 mg/kg, ip). Liver injury was assessed by plasma ALT and by histopathological examination of liver sections. Tissue repair was assessed by [ 3 H]thymidine incor- poration into hepatonuclear DNA and proliferating cell nuclear antigen (PCNA) immunohistochemistry during 0-120 h after TA injection. In AL-fed rats hepatic necrosis was evident at 12 h, peaked at 60 h, and persisted thereafter until mortality (3 to 6 days). Peak liver injury was approximately twofold higher in DR rats compared to that seen in AL rats. Hepatic necrosis was evident at 36 h, peaked at 48 h, persisted until 96 h, and returned to normal by 120 h. Light microscopy of liver sections revealed progression of hepatic injury in AL rats whereas injury regressed completely leading to recovery of DR rats by 120 h. Progression of injury led to 90% mortality in AL rats vs 30% mortality in DR group. In the surviving AL rats, S-phase DNA synthesis was evident at 60 h, peaked at 72 h, and declined to base level by 120 h, whereas in DR rats S-phase DNA synthesis was evident at 36 h and was consistently higher until 96 h reaching control levels by 1 To whom correspondence should be addressed. Fax: (318) 342-1686. E-mail: [email protected]. 120 h. PCNA studies showed a corresponding increase in cells in S and M phase in the AL and DR groups. DR resulted in abolition of the delay in tissue repair associated with the lethal dose of TA in ad libitum rats. Temporal changes and higher tissue repair response in DR rats (earlier and prolonged) are the conduits that allow a significant number of diet restricted rats to escape lethal Consequence. O 1998 Society of Toxicology. Diet is one of the most important components of our envi- ronment that impacts public health. Diet restriction (DR) 2 alters a number of key biological processes in such a way as to promote good health, decrease disease, and increase longevity including cancer (Allaben et al, 1990; Masaro, 1988; Wein- druch and Walford, 1992; Hass et al, 1996). A variety of mechanisms such as DNA repair, drug metabolism, immune function, hormonal regulation, and cytoprotection against free radical injury have been invoked for the beneficial effects of DR (Turturro et al, 1993; Hart et al, 1992, 1995). Increased incidence of apoptosis as a mechanism for protection from DNA damage-inducing agents and disease by eliminating mu- tated cells (James and Mushkhelishvili, 1994), increased fidel- ity of DNA replication (Srivastava et al., 1993), lowered on- cogene expression and preserved inducible cellular responses (Himeno et al, 1992), and increased repair of various forms of DNA damage are examples of mechanisms invoked for the beneficial effects of DR (Busbee et al, 1995). Toxicity of chemotherapeutic agents such as ganciclovir (Berg et al, 1994) and isoproterenol (Duffy et al, 1995) has been shown to be decreased by diet restriction. In light of the potential exposure to hepatotoxicants understanding the mechanisms and the im- pact of these changes induced by diet restriction are important to better extrapolate to man the results of toxicity tests in animals, thereby allowing better assessment of risk from ex- posure to environmental toxicants, especially under the condi- tions of human dietary ecology. However, the mechanisms by which restricted human diet may modulate the toxicity of 2 Abbreviations used: AL, ad libitum; ALT, alanine aminotransferase; DR, diet restriction; GSH, glutathione; 3 H-T, [ 3 H-me7/i>'/]thymidine; PCNA, pro- liferating cell nuclear antigen; TA, thioacetamide; EGFR, epidermal growth factor receptor; STAT, signal transducers and activators of transcription. 233 1096-6080/98 $25.00 Copyright O 1998 by the Society of Toxicology. All rights of reproduction in any form reserved. Downloaded from https://academic.oup.com/toxsci/article-abstract/45/2/233/1653896 by guest on 08 April 2018
Transcript of Temporal Changes in Tissue Repair Permit Survival of Diet ...

TOXJCOLOGICAL SCTENCES 45, 233-241 (1998)
ARTICLE NO. TX982511
Temporal Changes in Tissue Repair Permit Survival of Diet-RestrictedRats from an Acute Lethal Dose of Thioacetamide
Shashi K. Ramaiah, Thomas J. Bucci,* Alan Warbritton,* Madhusudan G. Soni, and Harihara M. Mehendale1
Division of Toxicology and Louisiana Institute of Toxicology, College of Pharmacy and Health Sciences, Northeast Louisiana University, Monroe,Louisiana 71209-0470; and *Pathology Associates, Inc., National Center for Toxicological Research, Jefferson, Arkansas 72079
Received January 20, 1998; accepted May 29, 1998
Temporal Changes in Tissue Repair Permit Survival of Diet-Restricted Rats from an Acute Lethal Dose of Thioacetamide.Ramaiah, S. K., Bucci, T. J., Warbritton A., Soni, M. G., andMehendale, H. M. (1998). Toxicol. Sci. 45, 233-241.
Although, diet restriction (DR) has been shown to substantiallyincrease longevity while reducing or delaying the onset of age-related diseases, little is known about the mechanisms underlyingthe beneficial effects of DR on acute toxic outcomes. An earlierstudy (S. K. Ramaiah et al., 1998, Toxicol. Appl. Pharmacol. 150,12-21) revealed that a 35% DR compared to ad libitum (AL)feeding leads to a substantial increase in liver injury of thioacet-amide (TA) at a low dose (50 mg/kg, ip). Higher liver injury wasaccompanied by enhanced survival. A prompt and enhanced tis-sue repair response in DR rats at the low dose (sixfold higher liverinjury) occurred, whereas at equitoxic doses (50 mg/kg in DR and600 mg/kg in AL rats) tissue repair in AL rats was substantiallydiminished and delayed. The extent of liver injury did not appearto be closely related to the extent of stimulated tissue repairresponse. The purpose of the present study was to investigate thetime course (0-120 h) of liver injury and liver tissue repair at thehigh dose (600 mg TA/kg, ip, lethal in AL rats) in AL and DR rats.Male Sprague-Dawley rats (225-275 g) were 35% diet restrictedcompared to their AL cohorts for 21 days and on day 22 theyreceived a single dose of TA (600 mg/kg, ip). Liver injury wasassessed by plasma ALT and by histopathological examination ofliver sections. Tissue repair was assessed by [3H]thymidine incor-poration into hepatonuclear DNA and proliferating cell nuclearantigen (PCNA) immunohistochemistry during 0-120 h after TAinjection. In AL-fed rats hepatic necrosis was evident at 12 h,peaked at 60 h, and persisted thereafter until mortality (3 to 6days). Peak liver injury was approximately twofold higher in DRrats compared to that seen in AL rats. Hepatic necrosis wasevident at 36 h, peaked at 48 h, persisted until 96 h, and returnedto normal by 120 h. Light microscopy of liver sections revealedprogression of hepatic injury in AL rats whereas injury regressedcompletely leading to recovery of DR rats by 120 h. Progression ofinjury led to 90% mortality in AL rats vs 30% mortality in DRgroup. In the surviving AL rats, S-phase DNA synthesis wasevident at 60 h, peaked at 72 h, and declined to base level by 120 h,whereas in DR rats S-phase DNA synthesis was evident at 36 hand was consistently higher until 96 h reaching control levels by
1 To whom correspondence should be addressed. Fax: (318) 342-1686.E-mail: [email protected].
120 h. PCNA studies showed a corresponding increase in cells inS and M phase in the AL and DR groups. DR resulted in abolitionof the delay in tissue repair associated with the lethal dose of TAin ad libitum rats. Temporal changes and higher tissue repairresponse in DR rats (earlier and prolonged) are the conduits thatallow a significant number of diet restricted rats to escape lethalConsequence . O 1998 Society of Toxicology.
Diet is one of the most important components of our envi-ronment that impacts public health. Diet restriction (DR)2
alters a number of key biological processes in such a way as topromote good health, decrease disease, and increase longevityincluding cancer (Allaben et al, 1990; Masaro, 1988; Wein-druch and Walford, 1992; Hass et al, 1996). A variety ofmechanisms such as DNA repair, drug metabolism, immunefunction, hormonal regulation, and cytoprotection against freeradical injury have been invoked for the beneficial effects ofDR (Turturro et al, 1993; Hart et al, 1992, 1995). Increasedincidence of apoptosis as a mechanism for protection fromDNA damage-inducing agents and disease by eliminating mu-tated cells (James and Mushkhelishvili, 1994), increased fidel-ity of DNA replication (Srivastava et al., 1993), lowered on-cogene expression and preserved inducible cellular responses(Himeno et al, 1992), and increased repair of various forms ofDNA damage are examples of mechanisms invoked for thebeneficial effects of DR (Busbee et al, 1995). Toxicity ofchemotherapeutic agents such as ganciclovir (Berg et al, 1994)and isoproterenol (Duffy et al, 1995) has been shown to bedecreased by diet restriction. In light of the potential exposureto hepatotoxicants understanding the mechanisms and the im-pact of these changes induced by diet restriction are importantto better extrapolate to man the results of toxicity tests inanimals, thereby allowing better assessment of risk from ex-posure to environmental toxicants, especially under the condi-tions of human dietary ecology. However, the mechanisms bywhich restricted human diet may modulate the toxicity of
2 Abbreviations used: AL, ad libitum; ALT, alanine aminotransferase; DR,diet restriction; GSH, glutathione; 3H-T, [3H-me7/i>'/]thymidine; PCNA, pro-liferating cell nuclear antigen; TA, thioacetamide; EGFR, epidermal growthfactor receptor; STAT, signal transducers and activators of transcription.
2 3 3 1096-6080/98 $25.00Copyright O 1998 by the Society of Toxicology.All rights of reproduction in any form reserved.
Downloaded from https://academic.oup.com/toxsci/article-abstract/45/2/233/1653896by gueston 08 April 2018

234 RAMAIAH ET AL.
environmental toxicants are not understood. While studieshave shown that nutritional status is an important factor indetermining susceptibility to toxic chemicals (Chanda and Me-hendale, 1996a), it is also known that survival after hepatocel-lular injury and necrosis may depend on the ability of theremaining hepatocytes to divide and restore an adequate pop-ulation of functioning cells (Chanda and Mehendale, 1994,1996b). Several studies have established the critical role ofliver cell division and compensatory tissue repair (Calabreseand Mehendale, 1996; Mehendale, 1991; Mehendale et al,1994). Thioacetamide, originally used as a fungicide, is apotent model hepatotoxicant and exposure to environmentalagents like this is known to cause liver injury leading tohepatitis and fulminant hepatic failure (Mehendale et al,1997).
Our earlier studies showed that low-dose thioacetamide (50mg/kg ip) results in an enormous increase in liver injury in DRrats (Ramaiah et al, 1998). However, there was a higher and atimely enhancement of tissue repair response in DR group.Equitoxic dose3 (50 mg TA/kg in DR and 600 mg TA/kg in ALrats) studies revealed that despite equal liver injury, DR ratswere able to mount an adequate tissue repair response allowingthem to escape death. The purpose of this study was to inves-tigate if the mechanism of increased cell proliferation responseseen at the low dose of TA was also operating at the lethal doseto account for increased survival in DR rats. Since the low doseof TA caused a 6-fold higher liver injury in DR rats, it was ofinterest to investigate the extent of liver injury at the lethal dose(which is 12-fold higher) in DR and AL rats and to investigatethe accompanying tissue repair. We report here that enhancedresponsiveness of the liver to regenerate and replace dead cellsin DR rats allows animal survival despite substantially highermechanism-driven infliction of liver injury. Since DR is con-sidered as a lifestyle option to prolong disease-free life, thesefindings may substantially impact the outcome-based risk as-sessment of human exposure to environmental hepatotoxicants,under varied diet regimens such as voluntary diet restriction.
METHODS
Chemicals. Thioacetamide and [3H-methyl]thymidinc (3H-T, sp act 1.7Ci/mmol) were obtained from Sigma Chemical Company (St. Louis, MO). Thescintillation fluid (Scintiverse SX 16-4) was purchased from Fisher ScientificCo (Pittsburgh, PA). All other biochemicals and chemicals were obtained fromSigma.
Animals and treatment. Male Sprague-Dawley rats (Harlan Sprague-Dawley, Inc. Indianapolis, IN; 225-275 g, 8-9 weeks of age) were maintainedin our central animal facility. They were housed individually over sawdustbedding known to be free of any chemical contamination. The ad libitum grouphad free access to water and normal rodent chow (Harlan Teklad Rat Chow No.7001, Madison, WI; protein 25%, fat 4.25%, fiber 4.67%, vitamins andminerals supplemented, calories 3.94 kcal/g ) at all times. The rats in thediet-restricted group were allowed 65% (17.7 g/day) of the ad libitum dailyfood consumption (27.2 ± 5.4 g/day) for a period of 21 days and maintained
3 Doses that induce equivalent acute hepatic injury (enzyme elevations andrelative degrees of necrosis).
on the same regimen during and after TA treatment (Ramaiah et aL, 1998).Food consumption of the DR group was adjusted relative to that observed inAL group twice weekly. Forty rats were diet restricted individually in twobatches of 20 in our central animal facility for time course studies (0-120 h,n = 4 for each time point). The first batch of 20 rats was used for 0-, 12-, 24-,36-, and 48-h time course studies. Sixteen rats from the second batch of 20 ratswere used for the remaining time points (60, 72, 96, and 120 h). On day 22 ofthe diet restriction regimen they were used for TA treatment. The AL (n = 32)and DR (n = 32) rats received a single ip injection of TA (600 mg/kg)dissolved in normal saline (0.9% NaCl). The respective controls received onlythe saline (1 ml/kg) vehicle. The plasma and liver tissue from these rats wasused for further studies. Since administration of 600 mg TA/kg resulted inmortality in the AL group at 72 h and after, the surviving rats were used toobtain plasma and liver tissues for various parameter estimations.
Plasma enzymes. Blood was collected in heparinized tubes from the dorsalaorta of rats under diethyl ether anesthesia at 0, 12, 24, 36, 48, 60, 72, 96, and120 h after TA or vehicle administration, and the plasma was separated bycentrifugation to estimate alanine aminotransferase (ALT; EC 2.6.1.2.) asmarker of liver injury using Sigma kit No. 59 UV (ALT, Sigma,).
Histopathology. Livers were surgically removed from rats under diethylether anesthesia. Portions of liver from each rat collected at various periodsafter TA treatment were washed with ice-cold normal saline (0.9% NaCl), cutinto small slices, and fixed in 10% phosphate-buffered formaldehyde fixativefor 48 h. The tissues were then transferred to 70% ethyl alcohol, processed, andembedded in paraffin. Liver sections (5>im thick) were stained with hema-toxylin-eosin (H & E) for histological examination under a light microscope.Unstained liver sections were prepared for proliferating cell nuclear antigenimmunohistochemistry on these slides.
In vivo incorporation of [3 HJthymidine into rat liver nuclear DNA. Stim-ulation of S-phase synthesis was quantified by 3H-T incorporation into hepa-tonuclear DNA using the procedure of Chang and Looney (1965) and Chau-veau et al., (1956). The DNA content was estimated according to the methodby Burton (1956). Since 3H-T incorporation study may indicate DNA repairinstead of DNA synthesis, proliferating cell nuclear antigen assay was used toconfirm S-phase stimulation and cell cycle progression.
Proliferating cell nuclear antigen (PCNA) assay. The proliferating cellnuclear immunohistochcmical analysis was conducted as described by Green-well et al. (1991). Briefly, the liver sections mounted on glass slides were firstblocked with casein and then reacted with monoclonal antibody to PCNA(Dako Corporation, Carpinteria, CA). The antibody was then linked withbiotinylated goat anti-mouse IgG antibody (Boehringer/Mannheim, Indianap-olis, IN), which was then labeled with streptavidin-conjugated peroxidase(Jackson Immunoresearch, West Grove, PA). Color was developed by expos-ing the peroxidase-labeled streptavidin to diaminobenzidine, which forms abrown reaction product. The sections were then counterstained with Gill'shematoxylin. Go cells were blue and did not take the PCNA stain, G, cells werelight brown in color, S-phase cell nuclei stained dark brown, G2 cells hadcytoplasmic staining with or without a speckled nuclear appearance, and Mcells were visualized as mitotic figures. PCNA studies were conducted in orderto confirm stimulated cell division and cell cycle progression suggested by3H-T incorporation studies. Each liver section was quantified for different cellsin the cell cycle by counting 1000 cells in 10 high-power fields (Kulkarni etal., 1996, 1997; Rao et aL, 1996).
liver glycogen levels. Total hepatic glycogen was estimated as marker ofhepatic energy status as total nonprotein anthrone-positive sugars (Seifer et aL,1950) and expressed as milligrams of glycogen equivalents per gram of liverweight.
Statistics. Means ± SEM were calculated for all the values. Comparisonbetween values was made using general linear models followed by least squaremeans or by Duncan's statistical analysis. Data were analyzed using StatisticalAnalysis System (SAS Institute, Cary, NC). The acceptable level of statisticalsignificance was established at the criterion of P £ 0.05.
Downloaded from https://academic.oup.com/toxsci/article-abstract/45/2/233/1653896by gueston 08 April 2018

DIET RESTRICTION AND TISSUE REPAIR 235
3500 1
0 12 24 36 48 60 72 96 120
Hours after thioacetamide Admin. (600 mg/kg)
FIG. 1. Plasma alanine aminotransferase (ALT) activity in ad libitum anddiet-restricted rats treated with a single dose of thioacetamide (600 mg/kg, ip).Plasma ALT activity was determined at various times after the administrationof thioacetamide. ALT was measured as a marker of liver injury over a timecourse (0-120 h) after TA administration in the DR and AL group. Results areexpressed as means ± SE (n = 4). *Values significantly different from the ALgroup at the corresponding time point. #Values significantly different fromcontrol AL group. 'Values significantly different from control DR group.
RESULTS
Lethality study. In the AL group 90% mortality occurredwhereas 30% mortality was recorded in the DR rats. Deathsoccurred between 3 and 6 days (average time of death, 4.5days) in AL-fed rats whereas they occurred between 5 and 6days in DR rats (average time of death, 5.5 days; Ramaiah etal, 1998).
Plasma enzyme elevations. ALT was estimated as amarker of liver injury over a time course (0, 12, 24, 36, 48, 60,72, 96, and 120 h) following administration of TA at 600mg/kg (Fig. 1). An approximately twofold increase in peakALT levels was evident in DR rats compared to the AL rats.Maximal liver injury was evident between 48 and 60 h in DRrats and rapidly regressed thereafter whereas in AL rats thepeak of liver injury occurred at 60 h (Fig. 1) and persistedthereafter culminating in mortality (one rat experienced mor-tality at 72 h, one at 96 h, two at 120 h, and five after 120 h andbefore 5 days). The temporal persistence and progression ofliver injury is concordant with the time of mortality as seenwith lethality studies (Ramaiah et al., 1998).
Histopathology. H & E-stained liver sections were scoredfor necrosis using a graded scoring pattern for multifocalnecrosis and hepatitis (Table 1). The liver sections were ex-amined for necrotic cells, extent of inflammation, polymorpho-nuclear infiltration, lipid laden, and swollen hepatocytes (Figs.2A and 2B). Swollen cells were identified by enlargement, andthe pale staining of the cytoplasm and necrotic cells wasidentified by pyknotic nuclei. Cells containing lipid were iden-tified by round vacuoles within the cytoplasm. Liver necrosisindicated that the injury regressed to restore normal appearanceby 120 h in the DR rats while in AL rats injury progressed. At
earlier time points (24 and 36 h) centrilobular cell swelling wasevident in both AL and DR rats. Also, vacuolations and pyk-notic nuclei were visible. Beyond 36 h, further progression ofinjury and massive lesions of necrosis were evident. Multifocalcytologic alterations like lipid deposition in the cytoplasm,swelling, and enlargement of cells were noted in the DR ratswhile in AL group multifocal necrosis was noted. These ne-crosed cells were concentrated around the central vein. Exten-sive multifocal necrosis appeared by 48 h in both groups. InAL rats massive multifocal necrotic lesions appeared as brokenup liver cell columns with cells separated from each otherindicating degeneration (Fig. 2A). There were signs of damageto the periportal tracts indicating massive spread of necrosis.These lesions further persisted and progressed (Fig. 2A) ac-counting for deaths in these rats. Although liver histomor-phometry in DR rats indicated multifocal cytologic alterations,there were no indications of lesions spreading presumably dueto opposing tissue repair response via mitotic activity in DRrats (Fig. 2B). Clear morphological differences were notedbetween the two groups; mitotic cells were abundant after 96 hin DR rats while they were absent in the AL rats. Oval cellproliferation was also noted only in DR rats after 72 h. Thus,liver injury declined in DR rats giving normal appearance by120 h. The histological findings agreed with ALT elevationswith respect to time of occurrence of liver injury and progres-sion. Mortality and its time of occurrence were consistent withhistopathological findings.
3H-T incorporation study. Figure 3 illustrates 3H-T incor-poration into the hepatonuclear DNA over a time course afterthe administration of lethal dose of TA in DR and AL rats.Liver tissue repair indicated by S-phase DNA synthesis was
TABLE 1Effects of Diet Restriction on Hepatic Injury
of Thioacetamide (600 mg/kg)
Time after TA ~injection (h)
012243648607296
120
AL
Within normal1, 1, 1, 11, 1, 2, 12, 3, 2, 33, 4, 4, 54, 3, 4, 33, 5, 5, D5, 5, 5, D5, 5, D, D
Necrosis
X
limits11.252.54.03.54.35.05.0
score"
DR
Within normal1, 1, 1, 12, 2, 2, 23, 3, 3, 33, 3, 3, 44, 4, 3, -4, 5, 4, 45, 5, 4, 43, 3, 1, 1
X
limits1233.253.64.254.52.0
Note. A single dose of thioacetamide was administered ip to male Sprague-Dawley rats (225-275 g) in saline (1 ml/kg).
" Lesions scored as multifocal necrosis and hepatitis. Scoring: 0, no necro-sis; + 1, minimal, defined as only occasional necrotic cells in any lobule; +2,defined as less than one-third of the lobular structure affected; +3, moderate,defined as between one-third and two-thirds of the lobular structure affected;+4, severe, defined as greater than two-thirds of the lobular structure affected;+5, more severe, defined as damage to most of the parenchyma of the liver.
Downloaded from https://academic.oup.com/toxsci/article-abstract/45/2/233/1653896by gueston 08 April 2018

236 RAMAIAH ET AL.
DR 12h
: „ -| D R 60h • c
- . Y
- ^
DR 120h c
p
FIG. 2. Representative liver histopathology during a time course afteradministration of thioacetamide (600 mg/kg, ip) in diet-restricted and adlibitum rats. (A) Photomicrographs of liver sections from ad libitum rats at 12,36, 48, 60, 96, and 120 h after 600 mg/kg TA treatment. (B) Photomicrographsof liver sections from diet restricted rats at 12, 36, 48, 60, 96, and 120 h after600 mg TA/kg treatment, c, central vein; p, pyknotic nuclei; v, vacuolization;n, areas of necrosis.
delayed and was evident at 60 h, peaked at 72 h, and returnedto control levels by 120 h (two of four deaths at this time point)in the AL rats. Although the peak of S-phase DNA synthesiswas higher than DR rats, the peak was delayed leading toprogression of injury characteristic of fulminant hepatic fail-ure. In contrast, DR rats showed an enhanced S-phase DNAsynthesis which occurred at 36 h and persisted until 96 h beforeit returned to control levels by 120 h. The delay in S-phase
DNA synthesis observed in AL rats was abolished in DR rats.The deficiency of tissue repair response corresponds with themortality pattern in ad libitum rats whereas prompt and con-sistent tissue repair in DR rats is concordant with substantialsurvival (70%) in DR rats.
Proliferating cell nuclear antigen assay. 3H-T incorpora-tion studies (Fig. 3) were further corroborated by PCNA im-munohistochemical staining procedure (Figs. 4 and 5 ). Nor-mally most cells are in resting phase and a relatively smallproportion of cells are in other phases of cell cycle with about3-4% in the G2 phase. Lethal dose of TA administration to DRrats resulted in a prompt and continuous cell cycle progressionresulting in a large number of cells in G, at 24 h and propor-tional increase in S-phase cells starting from 36 h and there-after until 96 h, finally resulting in mitosis as evidenced bymitotic figures at and beyond 72 h. An increased number ofcells in S-phase was evident in AL rats at 60 h and peaked at72 h. However, at later time points number of S-phase cells didnot increase. The maximum number of cells in S-phase wasseen at 72 h in both AL and DR rats by this method, inagreement with the wave of S-phase stimulation in 3H-T in-corporation observed at 72 h, after TA administration in DRand AL rats. Most of the cells had progressed to the M phaseby 120 h in DR rats while in AL rats this was minimal to none.This was evidenced by predominant number of Go cells in ALrats at 120 h. It should be noted that wave of S-phase stimu-lation was apparent at 36 h in the DR rats while there was asignificant delay in the appearance of these cells in the AL ratsuntil 60 h. Thus, diet restriction caused the abolition ofdelay in tissue repair response induced by lethal dose of TAin ad libitum rats, as evidenced by 3H-T incorporation andPCNA data.
Liver glycogen levels. Liver injury results in breakdown ofglycogen causing a significant reduction in glycogen stores. As
0 12 24 36 48 60 72 96 120
Hr after thioacetamide admin.(600 mg/kg)
FIG. 3. [3H]Triymidine incorporation into hepatonuclear DNA after TAtreatment in AL and DR rats. 3H-T (50 jiCi/300 g body wt) was administered2 h prior to termination at each time point. Results are expressed as means ±SE (n = 4). 'Values significantly different from the AL group at the corre-sponding time point. #Values significantly different from control AL group.IValues significantly different from control DR group.
Downloaded from https://academic.oup.com/toxsci/article-abstract/45/2/233/1653896by gueston 08 April 2018
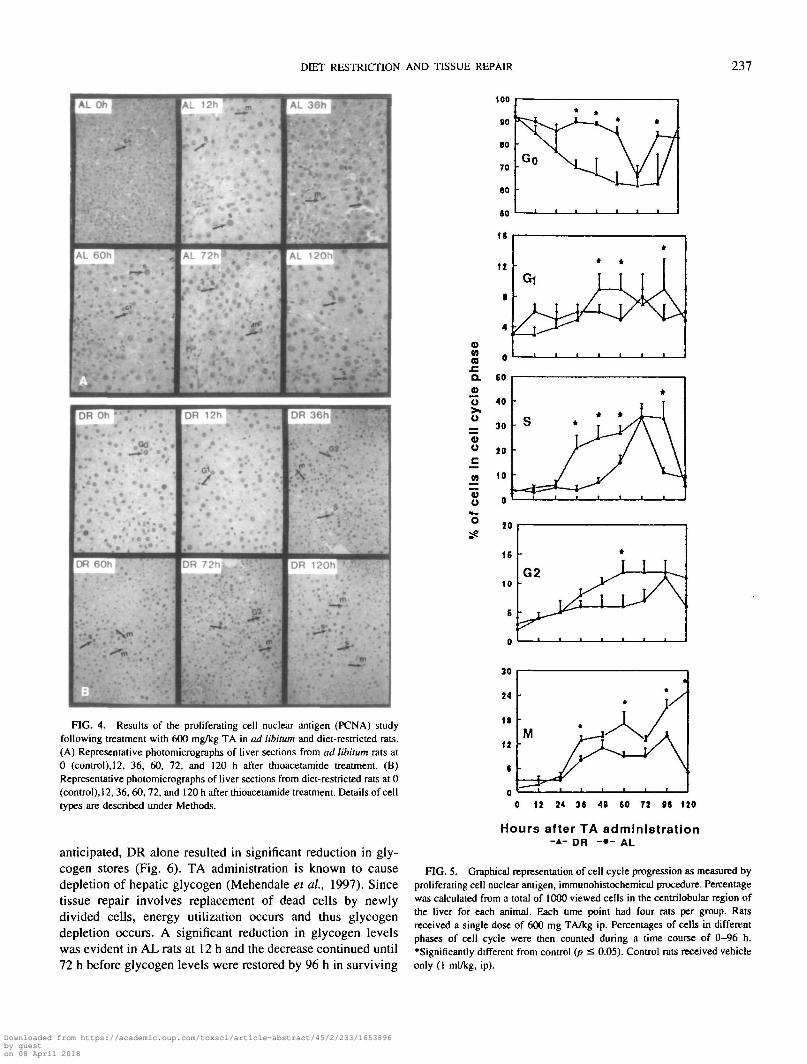
DIET RESTRICTION AND TISSUE REPAIR 237
DR
y
DR
Oh • ' . '
V *
•
1 •
• -
DR 12h *
•
• • * » '•>DR
A
DR 120h "'
oMacQ.
u
u
FIG. 4. Results of the proliferating cell nuclear antigen (PCNA) studyfollowing treatment with 600 mg/kg TA in ad libitum and diet-restricted rats.(A) Representative photomicrographs of liver sections from ad libitum rats at0 (control), 12, 36, 60, 72, and 120 h after thioacetamide treatment. (B)Representative photomicrographs of liver sections from diet-restricted rats at 0(control), 12, 36, 60, 72, and 120 h after thioacetamide treatment. Details of celltypes are described under Methods.
anticipated, DR alone resulted in significant reduction in gly-cogen stores (Fig. 6). TA administration is known to causedepletion of hepatic glycogen (Mehendale et al, 1997). Sincetissue repair involves replacement of dead cells by newlydivided cells, energy utilization occurs and thus glycogendepletion occurs. A significant reduction in glycogen levelswas evident in AL rats at 12 h and the decrease continued until72 h before glycogen levels were restored by 96 h in surviving
20
is
10 •
G2
T / \ I
*I I I
y \
0 12 24 36 At 60 72 16 120
Hours after TA administration-A- DR - • - AL
FIG. 5. Graphical representation of cell cycle progression as measured byproliferating cell nuclear antigen, immunohistochemical procedure. Percentagewas calculated from a total of 1000 viewed cells in the centrilobular region ofthe liver for each animal. Each time point had four rats per group. Ratsreceived a single dose of 600 mg TA/kg ip. Percentages of cells in differentphases of cell cycle were then counted during a time course of 0-96 h.•Significantly different from control (p =S 0.05). Control rats received vehicleonly (1 ml/kg, ip).
Downloaded from https://academic.oup.com/toxsci/article-abstract/45/2/233/1653896by gueston 08 April 2018

238 RAMAIAH ET AL.
70 1
>_l
en3 .
C
oO)ou
5
60
50
40
30
20
10
0
l #
* T . T . I.; II
S3 i f
0 12 24 36 48 60 72 96 120
Hr after TA admin. ( 600 mg/kg )
FIG. 6. Glycogen levels in the liver over a time course of 0-120 h afterthioacetamide administration in diet-restricted and ad libitum fed rats. Glyco-gen is expressed as equivalents of micrograms of glucose per gram of liver.Results are expressed as means ± SE (n = 4). *Values are significantlydifferent from the AL group at the corresponding time point. 'Values signif-icantly different from the AL group at the corresponding time point. #Valuessignificantly different from control AL group. (Values significantly differentfrom control DR group.
rats. Glycogen levels decreased at 36 h in DR rats and after awavy recovery and decline at 72 and 96 h, respectively, recov-ered to normal levels by 120 h.
DISCUSSION
Highly stimulated compensatory tissue repair is a biologicalresponse that follows partial hepatectomy or chemical-inducedinjury (Michalopoulus, 1990; Chanda et al, 1995; Kulkarni etal, 1997). In chemical hepatectomy, the regeneration responseis a dose-related event in which cell division and tissue repairoccur in a dose-dependent fashion until a threshold is reached,beyond which injury results in lethality due to unrestrainedprogression of injury (Mangipudy et al., 1995a; Mehendale,1994; Rao et al., 1997). These studies suggest that prompt andexacting tissue repair plays a critical role in the final outcomeof injury and that this biological event is an opposing andparallel response to tissue injury.
Modulation of food intake can influence the development ofchronic disease, cancer, and longevity (Hart et al, 1992). Dietrestriction enhances liver regeneration in aged animals follow-ing partial hepatectomy (Chou et al, 1995), significantly de-creases the cardiotoxicity of isoproterenol (Duffy et al, 1995),and decreases the mortality in mice treated with an antiviraldrug, ganciclovir (Berg et al, 1994). A number of mechanismssuch as increased apoptosis, increase in repair of DNA damage,and increased fidelity of DNA replication have been proposedas the benefits of diet restriction. However, the exact mecha-nism for the decreased mortality was not investigated (Berg etal, 1994).
In an earlier study we found diet restriction to enhance tissuerepair response upon hepatotoxic injury with thioacetamide(Ramaiah et al, 1998). A lethal dose of thioacetamide admin-istration resulted in 30 vs 90% mortality in diet-restricted vs ad
libitum fed rats, respectively. Paradoxically, diet restrictionsubstantially increased liver injury of thioacetamide. Tissuerepair was also enhanced in the DR rats. Equitoxic dose studyrevealed that a dose of 600 mg TA/kg in AL rats yields liverinjury similar to that observed in DR rats receiving 50 mgTA/kg. However, tissue repair response was quite dissimilarsuggesting that higher tissue repair in DR rats was not merelydue to higher liver injury. The objective of the present workwas to investigate the dynamic relationship between liver in-jury and tissue repair in rats maintained on DR and AL regi-mens and challenged with 600 mg TA/kg dose.
Plasma enzyme elevations of ALT revealed an approxi-mately twofold higher peak liver injury in DR compared to theAL rats. Injury progressed in die AL group while it regressedtoward normal in the DR rats. Liver necrosis scoring andhistomorphometric analysis of H & E-stained liver sectionsrevealed liver necrosis concordant with ALT elevations. ALTlevels and histopathology indicated progression of liver injuryin AL rats as opposed to regression and recovery in DR rats.The time of mortality in AL rats was concordant with themagnitude of ALT elevation and liver histopathology.
Liver injury as evidenced by ALT and necrosis scoring ofH & E-stained liver sections revealed approximately 2-foldhigher liver injury in DR rats receiving 600 mg TA/kg. A6-fold higher liver injury was noted in DR rats receiving a lowdose of 50 mg TA/kg (Ramaiah et al, 1998). Interestingly, a12-fold higher dose did not produce the corresponding increasein liver injury in DR rats. The present study revealed that thepeak liver injury in both DR and AL rats was delayed by 12 to24 h at the higher dose. The peak of liver injury in DR rats atthe low dose (Ramaiah et al, 1998) was evident at 24 h whileit appeared at 48 h after the high dose, whereas in AL rats itwas evident at 36 h at the low dose while it was delayed until60 h after the high dose. The mechanism underlying this delayin the causation of injury is worthy of experimental scrutiny. Aplausible explanation based on substrate inhibition of bioacti-vation enzymes merits consideration (Scott et al., 1991;Schlosser et al, 1993; Mangipudy et al, 1995a). The mecha-nism of thioacetamide hepatotoxicity is by the formation of asulfoxide which is metabolized further to thioacetamide sul-fone by cytochrome P450 and flavine-containing monooxygen-ases (Chieli and Mavaldi, 1984; Hunter et al, 1977). It ispossible that 600 mg TA/kg may exert inhibition of the me-tabolism of sulfoxide to reactive sulfone. Since the enzymeswhich catalyze the conversion of thioacetamide to sulfoxidealso catalyze sulfoxide to sulfone, high thioacetamide concen-tration reached at 600 mg TA/kg dose may inhibit the forma-tion of reactive species. Lower liver injury at earlier points wasalso observed in a dose-response study with TA, where liverinjury at 600 mg/kg dose was significantly lower at 12 and 24 hcompared to that observed after 50, 150, or 300 mg/kg doses(Mangipudy et al, 1995a). Kulkarni et al. (1996) reportedsimilar delay in maximum liver injury in Sprague-Dawley andFischer 344 rats treated with higher doses of 1,2-dichloroben-zene.
Downloaded from https://academic.oup.com/toxsci/article-abstract/45/2/233/1653896by gueston 08 April 2018

DIET RESTRICTION AND TISSUE REPAIR 239
Liver tissue repair measured by S-phase DNA synthesis andcell cycle progression by proliferating cell nuclear antigenimmunohistochemistry reveal substantial differences betweenDR and AL rats. Normally most cells are in the resting phaseand a relatively small proportion of cells is in the other phasesof cell cycle with about 3-4% in the G2 phase. Followinghigh-dose TA administration, there was a peak of S-phaseDNA synthesis at 72 h in the AL rats. However, stimulation ofS-phase occurred significantly earlier in DR rats receiving thesame dose: the S-phase started at 36 h and persisted until 96 hbefore declining to control levels by 120 h; PCNA studiesrevealed corresponding increases in mitotic cells in bothgroups. A substantial number of mitotic figures was present at120 h in DR rats while in AL rats the mitotic figures haddisappeared by 120 h. The most striking feature from S-phaseDNA synthesis and PCNA study was the abolition of delay intissue repair response in rats maintained on the DR regimen.Thus, recovery from liver injury results from the prompt andsustained liver regeneration response in DR rats. These find-ings are consistent with other reports of enhanced animalsurvival if tissue repair is stimulated (Chanda et ai, 1995;Mehendale, 1991, 1994; Rao et ai, 1997; Mangipudy et ai,1995a,b) and failure to survive even a nonlethal challenge iftissue repair is inhibited (Mangipudy et ai, 1996; Rao et ai,1997). Thus, the lethal dose of TA in AL rats was converted toan infralethal dose not because of decreased liver injury butdue to enhanced tissue repair response in DR rats. Otherpossible mechanisms of protection such as higher hepatic GSHmay be considered. It is known that hepatotoxicity of TA isenhanced after fasting but not after pretreatment with diethylmaleate, an agent that causes GSH depletion with minimaltoxicity (Reed and Fariss, 1984). Moreover, Chanda et al.(1995) have shown that following thioacetamide (50 mg/kg)treatment the levels of GSH and oxidized glutathione were notaffected. Since 50 mg/kg TA in DR rats caused the same levelof injury as 600 mg/kg in AL rats (equitoxic doses), it isreasonable to suggest that changes in GSH levels after thelethal dose are unlikely to explain the differences between thetoxic outcomes in DR and AL rats. Therefore, it is unlikely thatincreased GSH may play a role in the ability of DR rats toescape mortality. Survival or death thus is a function of timelyamplitude (efficiency) of repair response in these animals.
The mechanisms leading to the efficient tissue repair in DRrats are of investigational interest. The ability of the liver toregenerate is regulated by complex signaling and transducingevents that depend on the metabolic activity of the liver and theendocrine, autocrine, paracrine, and neural regulatory circuits(Diehl and Rai, 1996; Steer, 1995). Cell cycle check points arerecognized as positions of control that ensure the order ofevents in the cell cycle and that integrate DNA repair with cellcycle progression (Hartwell and Kastan, 1994). Although TA isnot known to cause DNA damage, the presence of early sig-naling mechanisms following DR can be envisaged to enhancecell cycle transition. It is well established that the Go to G,transition begins almost immediately after partial hepatectomy
(Steer, 1995) and chemical hepatectomy (Mehendale 1991,1994), triggered by growth factors already present in blood orliver. Growth factors regulate hepatic regeneration by provid-ing both stimulatory and inhibitory signals for cell prolifera-tion. TGF-a and EGF exert their effects on cells throughbinding to EGF-receptor (EGF-R), EGF-R dimerization, andautophosphorylation of tyrosine residues (Kumar et ai, 1995).Mitogenic growth factors promote G, phase cell cycle progres-sion by stimulating the formation or activation of cyclin D andcyclin E (Assoian, 1997). Other cellular proteins that transducecell proliferation signals are signal transducers and activatorsof transcription (STAT), nuclear factor-xB, and certainCCAAT/enhancer binding proteins which are under regulatorycontrol of proinflammatory cytokines such as tumor necrosisfactor-a, interleukin-1 (IL-1), and IL-6 (Diehl and Rai, 1996).Studies have shown that administration of EGF to mice resultsin the phosphorylation of tyrosine residues of STATla andSTAT1/3 which can be detected within 6 and 20 min aftertreatment (Zhong et ai, 1994). Metallothionein (Waalkes andGoering, 1990; Moffatt et ai, 1995) and heat shock proteinslike hsp 70 (Heydari et ai, 1995) are other proteins implicatedin cell growth and hepatic regeneration. The later events of cellproliferation like the expression of protooncogenes are alsolikely candidates for regulation. Early protooncogenes likec-fos and c-myc may be expressed early in DR rats and may beresponsible for early and increased tissue repair response.Stimulation of TA-induced cell division by fatty acid supple-ment has been shown to be associated with protooncogeneexpression (Chanda and Mehendale, 1994, 1996b). Dissectingthese signal-transducing events responsible for transforminglethal dose to an infralethal dose in DR rats should help usunderstand the molecular players for increased tissue repairresponse in DR rats.
ACKNOWLEDGMENTS
These investigations were supported by NIEHS Grant ES-08604. This effortwas supported by the Louisiana Board of Regents Support Fund throughNortheast Louisiana University the Kitty DeGree Endowed Chair in Pharmacy(Toxicology). The authors also acknowledge Dr. Richard Brown, DVM, Mis-sissippi Board of Animal Health, for his help on histopathology slides.
REFERENCES
Allaben, W. T., Chou, M. W., Pegram, R. A., Leakey, J., Feuers, R. J., Duffy,P. H., Turturro., and Hart, R. W. (1990). Modulation of toxicity andcarcinogenesis by caloric restriction. Korean J. Toxicol. 6, 167-182
Assoian, R. K. (1997). Anchorage-dependent cell cycle progression. J. Cell
Biol. 136, 1-4.
Berg, T. F., Breen, P J., Feuers, R J., Oriaku, E. T., Chen, F. X., and Hart,R. W. (1994). Acute toxicity of ganciclovir: Effect of dietary restriction andchronobiology. Food Chem. Toxicol. 32, 45-50.
Burton, K. (1956). A study of the conditions and mechanisms of the diphe-nylamine reaction for positive colorimetric estimation of DNA. Biochem. J.62,315-323
Busbee, D., Miller, S., Schroeder, M., Srivastava, V., Guntupalli, B., Merriam,E., Holt, S., Wilson, V., and Hart, R. W. (1995). DNA polymerase n
Downloaded from https://academic.oup.com/toxsci/article-abstract/45/2/233/1653896by gueston 08 April 2018

240 RAMAIAH ET AL.
function and fidelity: Dietary restriction as it affects age related enzymes InDietary Restriction: Implications for the Design and Interpretation ofTox-icity and Carcinogenicity Studies (R. W. Hart, D. A Neumann, and R. TRobertson Eds.), pp. 245-269. ILSI Press, Washington, DC.
Calabrese, E. J., and Mehendale, H. M. (1996). A review of the role of tissuerepair as an adaptive strategy: Why low dose are often non-toxic and whyhigh doses can be fatal. Food Chem. Toxicol. 34, 301—311.
Chanda, S., and Mehendale, H. M. (1994). Role of nutritional fatty acid andL-camitine in the final outcome of thioacetamide hepatotoxicity. FASEB J.8, 1061-1068.
Chanda, S., Mangipudy, R. S., Warbritton, A., Bucci, T. J., and Mehendale,H. M. (1995). Stimulated hepatic tissue repair underlies heteroprotection bythioacetamide against acetaminophen-induced lethality. Hepatology 21,477-486.
Chanda, S., and Mehendale., H. M. (1996a). Role of nutrition in the survivalafter hepatotoxic injury. Toxicology 111, 163-178.
Chanda, S., and Mehendale, H. M. (1996b). Hepatic cell division and tissuerepair: A key to survival after liver injury. Mol. Med. Today 2, 82—89.
Chang, L. O., and Looncy, W. B. (1965). A biochemical and autoradiographystudy of the in vivo utilization of tritiated thymidine in regenerating rat liver.Cancer Res. 25, 1817-1822.
Chauveau, J., Mouley, Y., and Roullier, C. H. (1956). Isolation of pure andunaltered liver nuclei morphology and biochemical composition. Exp. CellRes. 11, 317-321.
Chieli, E., and Mavaldi, G. (1984). Role of microsomal FAD-containingmonooxygenases in liver toxicity of thioacetamide s-oxide. Toxicology 31,41-52.
Chou, M. W., Shaddock, J. G., Hart, R. W., and Casciano, D. A. (1995). Effectof dietary restriction on partial hepatectomy induced liver regeneration ofaged F344 rats. Cancer Lett. 91, 191-197.
Diehl, A. M., and Rai, R M. (19%). Regulation of signal transduction duringliver regeneration. FASEB J. 10, 215-227.
Duffy, P. H., Feuers, R, J., Pipkin, J. L., Berg, T. F., Leakey, J. E. A., Turturro,A., and Hart R. W. (1995). The effect of dietary restriction and aging on thephysiological response of rodents to drugs. In Dietary Restriction: Implica-tions for the Design and Interpretation of Toxicity and CarcinogenicityStudies (R. W. Hart, D. A Neumann, and R. T Robertson Eds.), pp. 127-140.ILSI Press, Washington, DC.
Greenwell, A., Foley, J. F., and Maronpot, R. R. (1991). An enhancementmethod for immunohistochemical staining of proliferating cell nuclear an-tigen in archival rodent tissues. Cancer Lett. 54, 251-256.
Hart, R. W., Chou, R., Feuers, J., Leakey, J., Duffy, P., Lyn-Cook, B.,Turturro, A., and Allaben, W. (1992). Caloric restriction and chemicaltoxicity/carcinogenesis. Quality Assur.: Good Practice Regul. Law 1, 120-131.
Hart, R. W., Keenan, K., Turturro, A., Abdo, K. M., Leakey, J., and Lyn-Cook,B. (1995). Caloric restriction and toxicity. Fundam. Appl. Toxicol. 25,184-195.
Hartwell, L. H, and Kastan, M. B. (1994). Cell cycle control and cancer.Science 266, 1821-1828.
Hass, B. S., Lewis, S. M., Duffy, P. H., Ershler, W., Feuers, R. J., Good, R. A.,Ingram, D. K., Lane, M. A., Leakey J. E. A., Lipschitz, D., Poehlman, E. T.,Roth, G. S., Sprott, R. L., Sullivan, D. H., Turturro, A., Verdery, R. B., Walford,R. L., Weindruch, R., Yu, B. P., and Hart, R. W. (1996). Dietary restriction inhumans: report on the little rock conference on the value, feasibility andparameters of a proposed study. Mech. Ageing Dev. 91, 79-94.
Heydari, A. R., Gutsmann, A., You, S., Takahashi, R., and Richardson, A.(1995). Effect of dietary restriction on the genome function of cells: Alter-ations in the transcriptional apparatus of cells In Dietary Restriction: Im-plications for the Design and Interpretation of Toxicity and CarcinogenicityStudies (R. W. Hart, D. A Neumann, and R. T Robertson, Eds.), pp.213-228. ILSI Press, Washington, DC.
Himeno, Y., Engelman, R. W., and Good, R. A. (1992). Influence of caloricrestriction on oncogene expression and DNA synthesis during liver regen-eration. Proc. Natl. Acad. Sci. USA 89, 5497-5501.
Hunter, A. L, Hosscher, M. A., and Neal, R. A. (1977). Thioacetamide-induced hepatic necrosis. I. Involvement of the mixed function oxidaseenzyme system. J. Phamacol. Exp. Ther. 200, 439-448.
James, S. J., and Muskhelishvili, L. (1994). Rates of apoptosis and prolifera-tion vary with caloric intake and may influence incidence of spontaneoushepatoma in B6C3F1 mice. Cancer Res. 54, 5508-5510.
Kulkarni, S. G., Duong, H., Gomila, R., and Mehendale, H. M. (1996). Straindifferences in tissue repair response to 1,2-dichlorobenzene. Arch. Toxicol.70,714-723.
Kulkarni, S. G., Warbritton, A., Bucci, T. J, and Mehendale, H. M. (1997).Antimitotic intervention with colchicine alters the outcome of o-DCB-induced hepatotoxicity in Fischer 344 rats. Toxicology 120, 79-88.
Kumar, V., Bustin, S. A., and McKay, I. A. (1995). Transforming growthfactor alpha. Cell Biol. Internal. 19, 373-388.
Mangipudy, R. S., Chanda, S., and Mehendale, H. M. (1995a). Tissue repairresponse as a function of dose in thioacetamide hepatotoxicity. Environ.Health Perspecl. 103, 260-267
Mangipudy, R. S., Chanda, S., and Mehendale, H. M. (1995 b) Stimulatedhepatocellular regeneration: Key to thioacetamide autoprotection. Pharma-col. Toxicol. 77, 182-188.
Mangipudy, R. S., Rao, P. S., and Mehendale, H. M. (1996). Effect ofcolchicine antimitosis on thioacetamide hepatotoxicity. Environ. HealthPerspect. 104, 744-749.
Masaro, E. F. (1988). Mini-review—Food restriction in rodents: An evaluationof its role in the study of aging. J. Gerontol. 43, B59-B64.
Mehendale, H. M.(1991). Role of hepatocellular regeneration and hepatolobu-lar healing in the final outcome of liver injury: A two stage model ofhepatotoxicity. Biochem. Pharmacol. 42, 1155—1162.
Mehendale, H. M. (1994). Amplified interactive toxicity of chemicals atnontoxic levels: Mechanistic considerations and implications to publichealth. Environ. Health Perspect. 10, 139-149.
Mehendale, H. M., Mangipudy, R. S., and Chanda, S. (1997). Thioacetamide.In Comprehensive Toxicology (I. G. Sipes, C. A. McQueen, and A. J.Gandolfi, Eds.), Vol. 9, pp. 483-492. Elsevier, New York.
Mehendale, H. M., Thakore, K. N., and Rao, V. C. (1994). Autoprotection:Stimulated tissue repair permits recovery from injury. J. Biochem. Toxicol.9, 131-139.
Michalopoulos, G. K. (1990). Liver regeneration: Molecular mechanisms ofgrowth control. FASEB J. 4, 176-187.
Moffatt, P., Plaa, G. L., and Denizeau, F. (1995). Induction of metallothioneingene expression by epidermal growth factor and its inhibition by transform-ing growth factor-beta and dexamethasone in rat hepatocytes. Hepatology21, 1038-1044.
Ramaiah, S. K., Soni, M. G., Bucci, T. J., and Mehendale, H. M. (1998). Dietrestriction enhances compensatory liver tissue repair and survival followingadministration of lethal dose of thioacetamide. Toxicol. Appl. Pharmacol.150, 12-21.
Rao, P. S., Dalu, A., Kulkarni, S. G., and Mehendale, H. M. (1996).Stimulated tissue repair prevents lethality in isopropanol-induced poten-tiation of carbon tetrachloride hepatotoxicity. Toxicol. Appl. Pharmacol.140, 235-244.
Rao, P. S., Mangipudy, R. S., and Mehendale, H. M. (1997). Tissue injury andrepair as parallel and opposing responses to CC14 hepatotoxicity: a noveldose response. Toxicology 118, 181-193.
Reed, D. J., and Fariss, M. W. (1984). Glutathione depletion and susceptibility.Pharmacol. Rev. 36, 25S-33S
Schlosser, P. M., Bond. J. A., and Medinsky, M. A. (1993). Benzene and
Downloaded from https://academic.oup.com/toxsci/article-abstract/45/2/233/1653896by gueston 08 April 2018

DIET RESTRICTION AND TISSUE REPAIR 241
phenol metabolism by mouse and rat liver microsomes. Carcinogenesis 14,2477-2486.
Scott, J. G., Darryl, H. P., Douglas, K., Peter, F. S., Eugene, G. H., John, G. D.,and Anthony, G. Z. (1991). Thiobenzamide-induced hepatotoxicity: Effectsof substifuents and route of administration on the nature and extent of liver
injury. ToxicoL Appl. Pharmacol. I l l , 388-408.
Seifer, S., Dayton, S., Novic, B., and Muntwyler, E. (1950). Estimation of
glycogen with anthrone reagent. Arch Biochem. 25, 191-200.
Srivastava, V. K., Miller, S., Schroeder, M. D., Hart, R. W., and Busbee,D. (1993). Age-related changes in cellular levels and functions ofDNA polymerase a: Effects of caloric restriction. Mulat. Res. 295,265-280.
Steer, C. J. (1995). Liver regeneration. FASEB J. 9, 1396-1400.
Turturro, A., Duffy, P., and Hart, R. W. (1993). Modulation of toxicity by dietand dietary macro nutrient restriction. Mulat. Res. 295, 15-24.
Waalkes, M., and Goering, P. (1990). Metallothionein and other cadmium-binding proteins: Recent developments. Chetn. Res. Toxicol. 3, 281-288.
Weindruch, R., and Walford, R. L. (1992). Dietary restriction in mice begin-ning at one year of age: Effect on life span and spontaneous cancerincidence. Science 215, 1415-1418.
Zhong, Z., Wen, Z., and Darnell, J. E., Jr. (1994). Stat3: A STAT familymember activated by tyrosine phosphorylation in response to epidermalgrowth factor and interleukin-6. Science 264, 95—98.
Downloaded from https://academic.oup.com/toxsci/article-abstract/45/2/233/1653896by gueston 08 April 2018


















