Telomerasa Cancer
28
Telomerase Inhibition in Cancer Therapeutics: Molecular-Based Ap p r o ac hes A.P. Cunn in gh am a , W.K. Love a , R.W. Zhang b,c,d,e , L.G. Andr ews a , and T.O. Tollefsbol *,a,b,c a Department of Biology, Universit y of Alabama at Birmingham, AL 35294, USA bComprehensive Cancer Center, University of Alabama at Birmingham, AL 35294, USA cCenter for Aging, University of Alabama at Birmingham, AL 35294, USA dCenter for AIDS Research, University of Alabama at Birmingham, AL 35294, USA eGene Therapy Center, University of Alabama at Birmingham, AL 35294, USA Ab st rac t Current standard cancer therapies (chemotherapy and radiation) often cause serious adverse off-target effects. Drug design strategies are therefore being developed that will more precisely target cancer cells for destruction while leaving surrounding normal cells relatively unaffected. Telomerase, widely expressed in most human cancers but almost undetectable in normal somatic cells, provides an exciting drug target. This review focuses on recent pharmacogenomic approaches to telomerase inhibition. Antisense oligonucleotides, RNA interference, ribozymes, mutant expression, and the exploitation of differential telomerase expression as a strategy for targeted oncolysis are discussed here in the context of cancer therapeutics. Reports of synergism between telomerase inhibitors and traditional cancer therapeutic agents are also analyzed. Keywords Telomerase inhibition; hTR; hTERT; antisense; RNAi; ribozyme; dominant-negative hTERT; mutant-template telomerase RNA INTRODUCTION Drug design in the area of cancer therapeutics is developing a trend toward more precise mechanisms of cancer cell destruction thereby minimizing adverse off-target effects incurred during the course of standard cancer treatment (nausea, vomiting, hair loss, fatigue, organ toxicity, etc). The key to selectively targeting cancer cells is to exploit some basic difference these cells have developed compared to their normal precursors. One such difference is the activity of the enzyme telomerase. Telomeres, Telomerase, and Cancer Telomerase is a ribonucleoprotein that synthesizes telomeres. Telomeres consist of tandem oligonucleotide repeats (5'-TTAGGG-3' in humans) that cap the ends of eukaryotic chromosomes. Normal human somatic cells contain up to 10-15 kilobases of telomeric repeats [1,2]. Telomeres appear to have a dual function. First, telomeres may serve to protect the ends of chromosomes from damage and prevent the cell from recognizing the ends as double-strand * Address correspondence to this author at the Department of Biology, 175 Campbell Hall, 1300 University Boulevard, University of Alabama at Birmingham, Birmingham, AL 35294-1170, USA; Tel: 205-934-4573 ; Fax: 205-975-609 7; E-mail: [email protected] NIH Public Access Author Manuscript Curr Med Chem. Author manuscript; available in PMC 2008 June 9. Published in final edited form as: Curr Med Chem. 2006 ; 13(24): 2875–2888. I P A A u t h o r a u s c r i p t I - P A A u t h o r a u s c r i p t I - P A A u t h o r a u s c r i p t
-
Upload
edwarrivera -
Category
Documents
-
view
220 -
download
0
Transcript of Telomerasa Cancer

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 1/28
Telomerase Inhibit ion in Cancer Therapeutics: Molecular-Based
Approaches
A.P. Cunninghama, W.K. Lovea, R.W. Zhangb,c,d,e, L.G. Andrewsa, and T.O. Tollefsbol*,a,b,c
a Department of Biology, University of Alabama at Birmingham, AL 35294, USA
bComprehensive Cancer Center, University of Alabama at Birmingham, AL 35294, USA
cCenter for Aging, University of Alabama at Birmingham, AL 35294, USA
dCenter for AIDS Research, University of Alabama at Birmingham, AL 35294, USA
eGene Therapy Center, University of Alabama at Birmingham, AL 35294, USA
Abstract
Current standard cancer therapies (chemotherapy and radiation) often cause serious adverse off-targeteffects. Drug design strategies are therefore being developed that will more precisely target cancer
cells for destruction while leaving surrounding normal cells relatively unaffected. Telomerase,
widely expressed in most human cancers but almost undetectable in normal somatic cells, provides
an exciting drug target. This review focuses on recent pharmacogenomic approaches to telomerase
inhibition. Antisense oligonucleotides, RNA interference, ribozymes, mutant expression, and the
exploitation of differential telomerase expression as a strategy for targeted oncolysis are discussed
here in the context of cancer therapeutics. Reports of synergism between telomerase inhibitors and
traditional cancer therapeutic agents are also analyzed.
Keywords
Telomerase inhibition; hTR; hTERT; antisense; RNAi; ribozyme; dominant-negative hTERT;mutant-template telomerase RNA
INTRODUCTION
Drug design in the area of cancer therapeutics is developing a trend toward more precise
mechanisms of cancer cell destruction thereby minimizing adverse off-target effects incurred
during the course of standard cancer treatment (nausea, vomiting, hair loss, fatigue, organ
toxicity, etc). The key to selectively targeting cancer cells is to exploit some basic difference
these cells have developed compared to their normal precursors. One such difference is the
activity of the enzyme telomerase.
Telomeres, Telomerase, and Cancer Telomerase is a ribonucleoprotein that synthesizes telomeres. Telomeres consist of tandem
oligonucleotide repeats (5'-TTAGGG-3' in humans) that cap the ends of eukaryotic
chromosomes. Normal human somatic cells contain up to 10-15 kilobases of telomeric repeats
[1,2]. Telomeres appear to have a dual function. First, telomeres may serve to protect the ends
of chromosomes from damage and prevent the cell from recognizing the ends as double-strand
*Address correspondence to this author at the Department of Biology, 175 Campbell Hall, 1300 University Boulevard, University of Alabama at Birmingham, Birmingham, AL 35294-1170, USA; Tel: 205-934-4573; Fax: 205-975-6097; E-mail: [email protected]
NIH Public AccessAuthor ManuscriptCurr Med Chem. Author manuscript; available in PMC 2008 June 9.
Published in final edited form as:
Curr Med Chem. 2006 ; 13(24): 2875–2888.
NI H-P A A u
t h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or M
anus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 2/28
breaks that might lead to adverse recombination. Second, telomeres have been proposed to act
as a ‘mitotic clock’ that counts the number of divisions a cell has undergone and its capacity
for continued division [3,4]. Leonard Hayflick observed years ago that normal cells have a
finite replicative capacity in vitro after which they remain metabolically active but cease to
proliferate [5]. This period of growth arrest is referred to as M1 (mortality 1) or cellular
senescence. A direct relationship between telomere length and cellular senescence has been
established [6]. Because of the end-replication problem [7,8], 50-200 bp of telomeric DNA is
lost with every round of replication. The non-coding telomeric repeats provide a buffer that prevents the loss of genetic information during each cycle of replication. When the telomeres
have eroded to a critical minimum length (∼5 kb), cellular senescence is triggered. Cellular
senescence might be bypassed by repression of tumor suppressor genes, activation of
oncogenes, or other mutations [9]. By escaping senescence, rare cells continue to divide and
their telomeres continue to shorten until they reach a critical stage (M2 or crisis). At this point,
chromosomal instability arises due to end-to-end fusions and/or chromosome breakage. DNA
damage checkpoints are activated along with apoptosis. Unless the cell develops a mechanism
through which to stabilize telomere length, it will not survive. Cells that escape crisis and
become immortalized generally achieve telomeric stability through the reactivation of
telomerase (Fig. 1).
Telomerase is a ribonucleoprotein that acts to elongate telomeres in cells that possess its
activity. This enzyme is expressed during embryonic development, loses its expression duringdifferentiation of somatic cells, and is almost undetectable in most normal human somatic cells
[10]. By contrast, telomerase is expressed in ∼85% of human cancers [11]. There are a few
types of cells that normally express telomerase including germ line cells, stem cells,
hematopoietic cells, cells lining the intestine, and other rapidly proliferating cells. The
widespread expression of telomerase in a variety of human cancers, while being almost
undetectable in most normal cells, makes it a very attractive drug target. Normal somatic cells
are thought to harbor enough telomeric DNA reserve to withstand telomere-based therapeutics,
and the few normal cells which express telomerase should also have enough reserve to
withstand treatment with telomerase inhibitors. It has been shown that cancer cells often
maintain much shorter telomeres than normal cells (3-9 kb compared to 10-15 kb) [12-15].
Additionally, the rapid proliferative nature of cancer cells leads to steady telomere erosion in
the absence of telomerase. Telomerase-based therapeutics should therefore impact tumor cells
before having any appreciable effect on telomerase-positive normal cells. Potential outcomesof telomerase-based therapeutics are illustrated in Fig. 2.
Telomerase contains an RNA component, hTR, and a catalytic reverse transcriptase
component, hTERT. While hTR is ubiquitously expressed in normal cells, it is the presence
of hTERT that confers telomerase activity [6]. Without expression of hTERT there is no
telomerase activity and consequently elongation of telomeres does not occur. Several different
approaches to telomerase inhibition are currently in laboratory use with new ones continually
being sought as a result of the growing interest in selective telomerase inhibition as a strategy
for rational pharmaceutical design. Small molecules such as AZT (azidothymidine, a non-
specific reverse transcriptase inhibitor) [16], chemicals such as retinoids [17], tamoxifen
[18], or EGCG (epigallocatechin gallate) [19], and molecules which interfere with telomere
structure (i.e., G-quadruplex stabilizers) [20,21] have been shown to be effective in vitro
inhibitors of telomerase transcription or function. While these compounds may be effective in
vitro, there is legitimate concern surrounding their specificity. AZT has been shown to inhibit
cell growth and telomerase activity of breast cancer cells in vitro [16]. However, AZT is not
specific for telomerase, being most recognized for its use in managing HIV infection. Retinoids,
which are vitamin A analogues, are able to induce telomere shortening, cell growth arrest, and
cell death in acute promyelocytic leukemia (APL) cells [17]. High levels of some retinoids,
Cunningham et al. Page 2
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 3/28
however, can cause toxicity in vivo. The need is apparent, therefore, to develop specific
molecular-based therapies that are both effective and exhibit an acceptable side effect profile.
The RNA component hTR and the reverse transcriptase hTERT are not the only components
of telomerase required to maintain telomere length and structure. There are also a number of
proteins associated with telomeric DNA that have been explored as possible drug targets. While
their potential as drug targets will not be covered in this review, some basic understanding of
these proteins is helpful. Although quite a number of proteins play a role in maintainingtelomere structure and length, some of the more notable ones include the human telomeric
proteins TRF1 and TRF2 and POT1. (For an extensive review of these and other telomeric
proteins, see [22]). Briefly, TRF1 and TRF2 are telomeric repeat binding factors. TRF1 binds
to the telomeric DNA duplex and acts to regulate telomere length through a negative feedback
loop. The number of TRF1 proteins present on the end of a chromosome correlates with the
length of the telomere [22]. TRF2 helps to form the t-loop structure at the end of the telomere
[23], and likely helps to physically prevent telomerase from acting on telomere ends [22]. TRF2
plays a key role in the protection of chromosome ends (see review [24]). POT1 (protection of
telomeres) is a single-stranded telomeric DNA binding protein that can associate with TRF1
and may regulate the amount or the frequency of telomere elongation [22].
The focus of this review will be on inhibitors of either the hTR or hTERT components of
telomerase and therapies currently being developed that exploit the unique nature of telomeraseexpression.
TARGETING TELOMERASE ENZYME COMPONENTS
Antisense and Related Oligonucleot ides
One of the oldest and most commonly used classes of telomerase inhibitory agents is antisense
DNA oligonucleotides. The use of antisense molecules to block the translation of mRNA into
a functional protein has been commonly used since the 1990s. In the classical sense, antisense
technology utilizes nucleotides with sequence complementarity to sense RNAs. These
oligonucleotides may be designed to occupy the ribosome binding site, preventing the ribosome
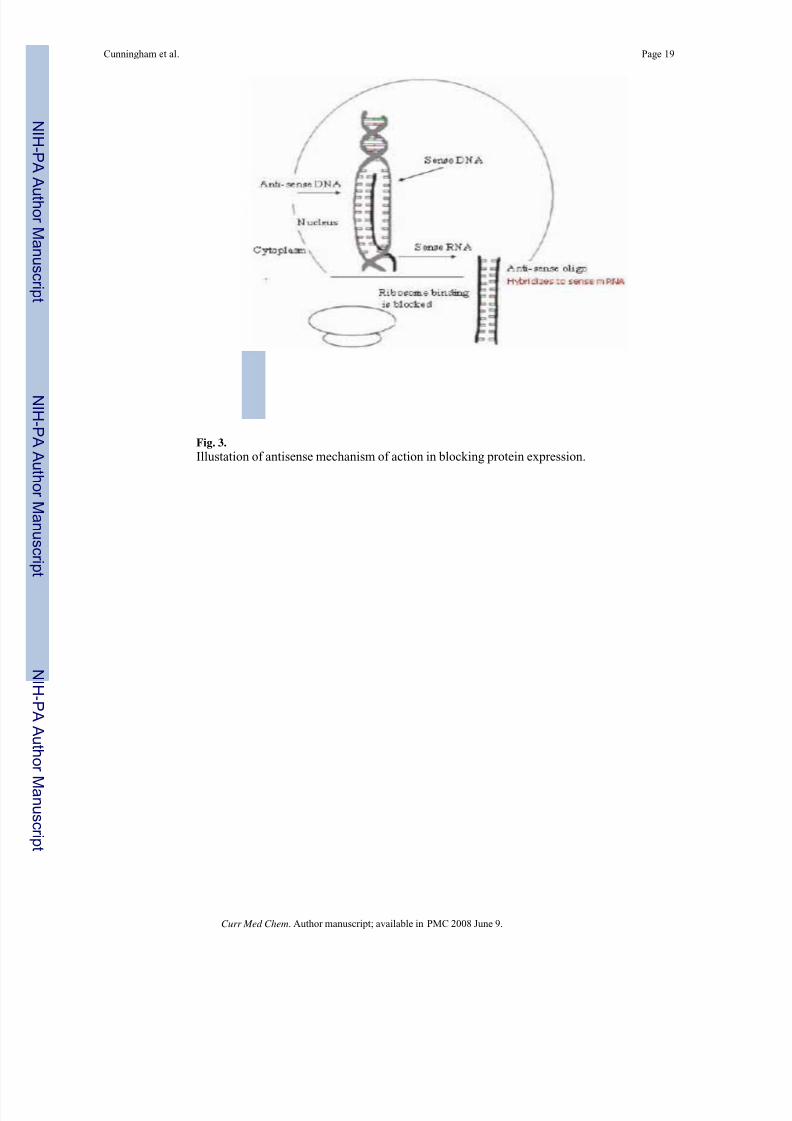
from binding the target mRNA (Fig. 3). Antisense oligos targeted to sequences downstream
of the ribosome binding site anywhere in the coding sequence will prevent ribosomal
translocation, halting translation and producing a non-functional or truncated protein [25].There have been numerous modifications of antisense molecules reflecting efforts to enhance
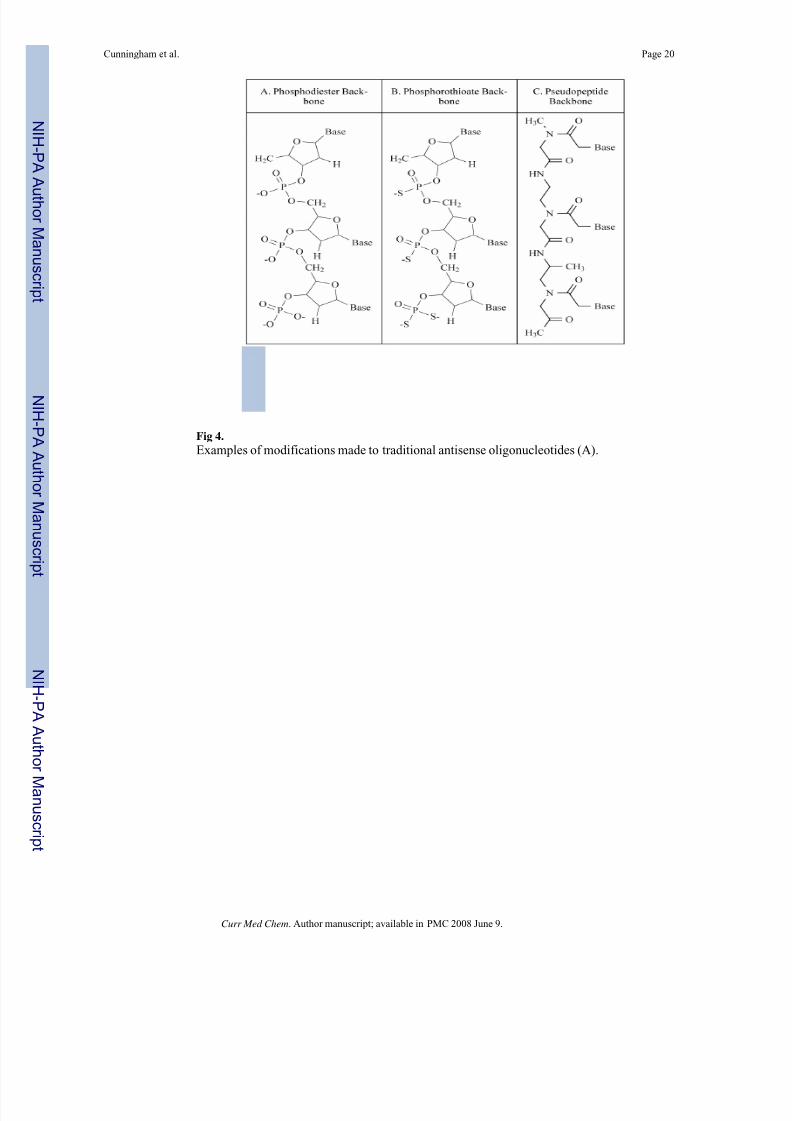
their cellular up-take, potency, and half-life (Fig. 4). Some modifications help to recruit the
activity of RNase H, which degrades the RNA strand of an RNA-DNA duplex. After the
degradation of the mRNA component of the duplex, the antisense DNA molecule is released
and becomes free to bind other target mRNA molecules. Phosphorothioate (PS) linkages slow
the degradation of the antisense molecules in the cells and enhance their half-life [26]. Peptide
nucleic acids (PNA) are molecules in which the antisense bases are connected to various peptide
backbones (Fig. 5). These modifications have been found to improve the half-life of antisense
oligomers and enhance hybridization properties [27]. 2'-O-Methyl- and 2'-methoxyethyl-
modified RNAs increase the affinity of antisense molecules for their specific targets [25].
Several of the various types of antisense molecules have made their way into clinical trials.
One, Vitravene, has gained FDA approval for the treatment of CMV-induced retinitis [28].
The use of antisense technology in telomerase inhibition is not new. In fact the first report of
successful inhibition of telomerase activity involved the use of antisense oligonucleotides
against hTR 10 years ago [29]. Feng et al. reported in 1995 that antisense oligonucleotides
complementary to sequences within or near the human telomeric template RNA resulted in
suppression of telomerase activity while antisense oligonucleotides against targets that were
more distant from the telomeric template failed to inhibit the action of the ribonucleoprotein.
Cunningham et al. Page 3
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 4/28
As a result, HeLa cells that were transfected with the antisense-hTR expression construct
underwent crisis after 23 to 26 population doublings and exhibited a loss of telomeric DNA
repeats. In contrast, telomerase-negative foreskin cells transfected with the construct
expressing antisense-hTR did not enter crisis during the same time period. This study was the
first to demonstrate the therapeutic potential of antisense oligonucleotide expression as a
treatment for human cancers. Building on the findings of Feng et al., others have used the anti-
hTR expression vector to inhibit telomerase activity in other cancer cell lines. Human gastric
cancer cells transfected with a vector expressing antisense-hTR demonstrated shortening of telomere length and an elevated level of apoptosis, suggesting that the antisense-mediated
telomere shortening in gastric cancer cells acts to induce apoptosis [30]. Even if apoptosis is
not induced by introduction of the antisense-hTR expression vector, the construct may still be
effective in reducing the aggressiveness of cancer cells (lowering invasive capacity and
tumorigenicity). This was found to be the case with various malignant glioma cell lines [31].
Upon introduction of the antisense-hTR construct, apoptosis occurred in only some of the cell
populations after 30 population doublings. Other cell populations avoided apoptosis, but
appeared to differentiate and diverge in morphology from their parental cells, demonstrating
that telomerase inhibition can trigger distinctly different desirable results: apoptosis or
differentiation. Although not a direct effecter of tumor cell death, differentiation may be a
viable therapeutic outcome as it tempers the effect of the malignant phenotype.
Delivery of telomerase-inhibiting agents as a component of cancer therapeutics presents theneed for stable expression of the inhibitor in vivo. An increasingly common approach to stable
expression of specific genomic therapeutics is the use of modified retroviral, lentiviral, or
adenoviral delivery systems. Successful delivery and expression of antisense RNA by
replication-deficient retrovirus in HeLa cells and human kidney carcinoma cells has been
shown to be effective in inhibiting telomerase activity by at least 75% in vitro [32].
Additionally, a hybrid adenovirus/adeno-associated virus has been used to express antisense-
hTR in MCF-7 breast cancer cells [33]. The stable expression of antisense hTR in these cells
resulted in significant suppression of telomerase activity and progressive telomere shortening
for 30 population doublings along with induction of apoptosis, reduction of cell proliferation,
and reduction of colony formation as demonstrated by soft agar assay.
While the preponderance of literature appears to report the use of antisense molecules targeting
the RNA component of telomerase (hTR), antisense-mediated hTERT inhibition has also beensuccessfully achieved. Antitumor action was observed when antisense oligodeoxynucleotides
(mostly 20-mers) against various regions of the hTERT mRNA were introduced into human
bladder cancer cells by transfection [34]. The amount of hTERT transcript was reduced, cell
viability was significantly impaired, and G1 arrest was induced. Interestingly, when an
antisense expression vector coding for antisense RNA against hTERT was transfected into
human breast cancer cells, decreased telomerase activity was observed as well as significant
apoptosis [35]. However, these phenomena were observed 24 hours post-transfection and were
not accompanied by significant shortening of telomeric DNA. These findings support other
observations of telomerase inhibition rapidly inducing apoptosis independent of telomere
erosion [36,37], and are significant because they address the traditional concern that telomerase
inhibition would incur a substantial lag time between the onset of telomerase inhibition and
the sufficient erosion of telomeres as to cause growth arrest of cancer cells. The simultaneous
inhibition of hTR and hTERT has been found to inhibit telomerase activity synergistically[38], suggesting another strategy for antisense oligonucleotide therapy.
Improvements in antisense technology have led to improvements in introduction of the
molecules into cells, stability, lengthening of half-life, and specificity of target binding.
Modifications of traditional antisense oligonucleotides used in telomerase inhibition include
2'-5'-oligoadenylate (2-5A) linkages [39,40], 2'-O-methyl-RNA [41], phosphorothioate-
Cunningham et al. Page 4
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 5/28
modified oligodeoxynucleotides (PS-ODN) [34,42,43], peptide nucleic acids (PNA) [44-46],
and locked nucleic acids (LNA) [47].
One remaining group of compounds that warrants discussion includes the oligonucleotide N3'-
P5' phosphoramidates and N3'-P5' thio-phosphoramidates. This class of oligonucleotides has
attracted attention lately as one of its derivatives, GRN163L, is well on its way to becoming
the first telomerase inhibitor to be available for cancer treatment. In the last five years,
oligonucleotide phosphoramidates were designed with sequence complementarity to either thetemplate region of hTR or a region ∼100 nucleotides downstream of the template region
[48-50]. The N3'-P5' phosphoramidates (NP oligonucleotides) and the N3'-P5' thio-
phosphoramidates (NPS oligonucleotides) form sequence-specific duplexes with target RNA.
N3'-P5' thio-phosphoramidates were designed to combine the RNA binding affinity and
sequence specificity of phosphoramidates with the abilitiy of the phosphorothioate
oligonucleotides to interact with proteins (i.e., hTERT) [51]. Both NP and NPS
oligonucleotides inhibited telomerase activity, causing telomere shortening, senescence, and
eventual apoptosis; however, NP oligonucleotides were inefficient without the use of a lipid
carrier to facilitate their introduction into the cells. NPS oligonucleotides were significantly
more potent telomerase inhibitors than their parent molecules, even without the use of lipid
carriers. NP deoxyoligonucleotides exhibited IC50 values in the 0.5-1 μM range compared to
NPS deoxyoligonucleotides, which had IC50 values in the 0.5-5 nM range (both with cellular
uptake enhancers) [49].
The GRN163 13-mer has emerged as an exciting N3'-P5' thio-phosphoramidate therapeutic
candidate, resulting from the optimization of NP and NPS inhibitory strategies [51]. Early
testing in cell-free assays demonstrated IC50 values as low as 26-44 pM [51]. Significantly,
GRN163 was shown to inhibit telomerase activity in a variety of cancer cell lines in vitro 24-72
hours after treatment (using the cell-based TRAP assay Telomeric Repeat Amplification
Protocol); the survival of normal human cells, WI-38 and BJ fibroblasts, was not affected, even
with treatments of up to 100 μM GRN163 for 72 hours [51]. Treatment with GRN163 has been
shown to induce telomere shortening, growth arrest, and cell death in human multiple myeloma
[52,53] and non-Hodgkin lymphoma [53] cell lines, as well as tumor growth suppression in a
prostate cancer xenograft model [54].
A second generation oligonucleotide, GRN163L, has recently been shown to be more potentthan its GRN163 predecessor, causing more rapid shortening of telomeres and cell growth
inhibition and having an average of sevenfold lower IC50 values in various cell lines tested
[55]. The difference in GRN163L is a lipid modification of the first generation oligonucleotide.
Challenges in therapeutic oligonucleotide delivery have often made it necessary to use lipid-
based transfection reagents or carriers for in vitro experimentation. Lipid modification of the
oligonucleotide in this case has apparently eliminated the requirement for an extraneous lipid
carrier. Manufacturing of GRN163L has been ongoing in order to supply enough of the drug
for toxicity and pharmacokinetic studies in animals and Phase I clinical studies, making it
potentially the first telomerase inhibitor for cancer therapeutics1. The uses of antisense
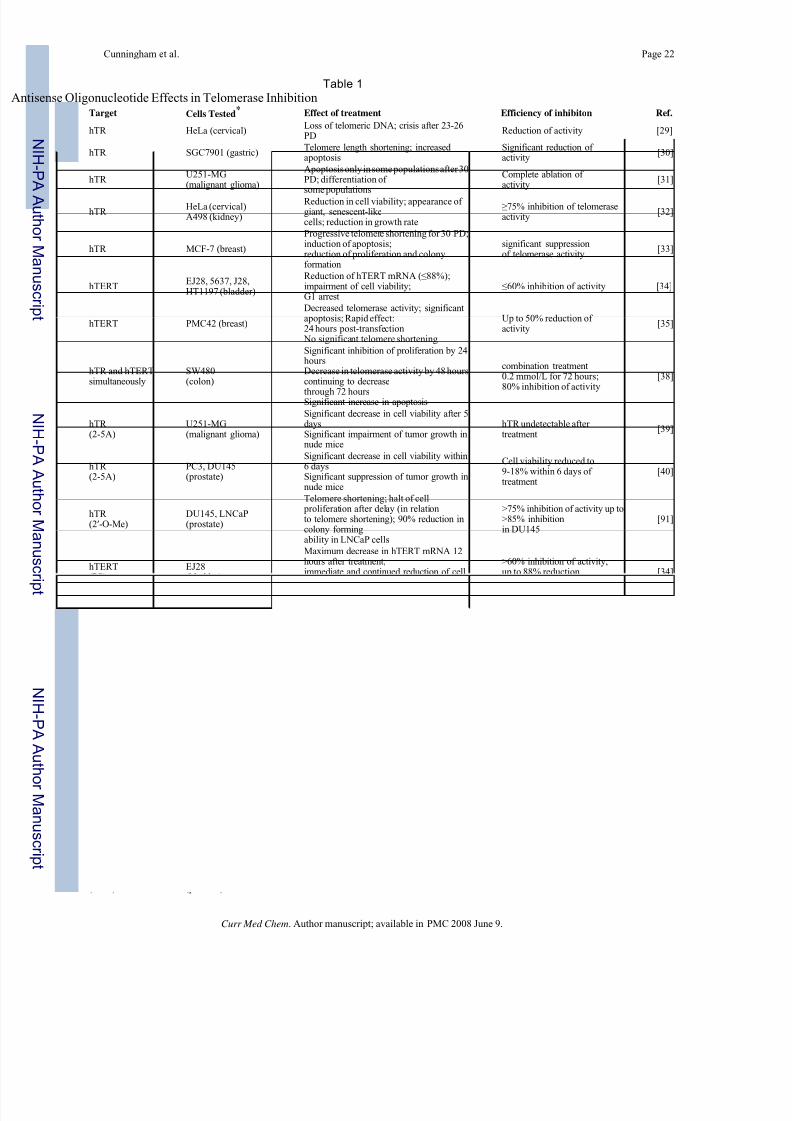
technology in the inhibition of telomerase are summarized in Table 1.
RNA Interference
Since the RNA interference (RNAi) phenomenon was first described in 1998 [56], RNAi-
mediated gene knockdown has become the latest must-have tool for analysis of gene function
and is emerging as a potential treatment strategy for a variety of human diseases, including
cancer. Briefly, RNAi involves the use of short double-stranded RNA molecules to activate a
natural pathway which results in the degradation of the homologous target mRNA.
Cunningham et al. Page 5
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 6/28
Clearly the essential components of telomerase, hTR and hTERT, present themselves as
attractive targets for RNAi therapeutics. There have been attempts made to knock down
expression of hTR and hTERT by transfection of small interfering RNAs (siRNAs) and stable
expression of siRNA constructs in vitro using plasmids or viral expression vectors. Kosciolek
et al. first inhibited telomerase using RNAi in 2003 [57]. This group used 21-nucleotide
siRNAs against either hTR or hTERT. The region of hTERT that was targeted by this group
was the same one used by Hahn et al. in their dominant negative mutation [58]. Human colon
carcinoma cells were transfected with siRNA against either hTR or hTERT. Telomeraseactivity was inhibited in a dose-dependent manner, with the siRNA against hTR proving more
effective than the siRNA against hTERT. The maximum effect observed in that cell line was
a 75% decrease in telomerase activity in cells transfected with siRNA against hTR compared
to a 65% decrease in activity in those cells treated with siRNA against hTERT. Perhaps this
difference may be attributed to target selection for each component. It is also possible that
direct RNAi of the RNA subunit of telomerase is more easily achieved than indirect RNAi of
the protein component, hTERT. Similar results were observed when the siRNAs were used in
HeLa cervical carcinoma cells as well as other types of carcinoma cells and sarcoma cells of
mesodermal origin. Simultaneous treatment of HeLa cells with siRNAs against both hTR and
hTERT was not found to result in greater inhibition of telomerase activity than each siRNA
separately. As expected, the effects of direct siRNA transfection were transient in nature. A
DNA construct expressing a hairpin structure targeting hTR was used to transfect HeLa cells
to assess long-term effects [57]. At 75 days after transfection, 4 out of 5 transfected clonesexhibited reduced telomeric DNA content (45%) compared to control cells. The apparent
effectiveness of RNA interference directed against telomerase came as somewhat of a surprise,
as RNAi is usually considered as being restricted to the cytoplasm while telomerase is regarded
as being generally confined to the nucleus. While telomerase may be manufactured in the
cytoplasm, its site of function is the nucleus. However, it does appear that telomerase remains
in the cytoplasm at least long enough for RNA interference to cause hTR or hTERT mRNA
degradation with respectable efficiency.
Soon after the initial publication of results demonstrating the ability of RNAi to diminish
telomerase activity, results were published that proved the usefulness of retroviral-mediated
RNA interference against hTERT [10]. The retroviral vector pMKO.1-puro [59] was used to
express a short hairpin RNA (shRNA) in HeLa cells, causing suppression of hTERT mRNA
and protein expression and abrogation of telomerase enzymatic function. Normal humanfibroblasts (BJ and WI-38) were shown by this group to have transient telomerase activity
during S phase of the cell cycle; introduction of shRNA against hTERT in these cells caused
complete disruption of this transient activity, slowed their proliferation, and delayed their entry
into S phase (cells accumulated in the G2/M phase). These cells continued to proliferate,
although more slowly than control cells. After extended culture, morphological changes and
senescence-associatedβ-galactosidase staining indicated that fibroblasts infected with hTERT
retroviral shRNA constructs entered replicative senescence more quickly than uninfected
control cells. Members of the same group recently expanded their findings on retroviral
delivery of hTERT shRNA in cervical cancer cells [60]. Cells with compromised hTERT
expression had impaired telomerase activity, shortened telomeres and shortened telomeric 3'
overhangs (necessary for formation of the t-loop) with increasing population doublings. Even
after a relatively short time (5 population doublings), infected cells demonstrated a decreased
proliferative rate. Low-passage cells also had impaired colony-forming ability and
tumorigenicity in mice. Finally, treatment with shRNA against hTERT appeared to
significantly enhance sensitivity of cells to treatment with conventional therapeutic agents that
are known to induce DNA double-strand breaks (topoisomerase inhibitors, bleomycin, and
radiation).
Cunningham et al. Page 6
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 7/28
Another example of RNA interference against hTERT has been demonstrated in Barrett's
adenocarcinoma cells [61]. Two regions of hTERT were targeted using two siRNA duplexes;
a mixture of these siRNAs was used to transfect the esophageal adenocarcinoma cell line
SEG-1. The siRNA treatment was extremely efficient, causing a reduction in telomerase
protein expression to only 5% of cells treated with control siRNAs. Telomerase activity was
observed to be inhibited by day 1 and was essentially completely ablated by day 3. Greater
than 80% of cells treated with the siRNAs for 3 weeks underwent apoptosis. Treatment also
led to significant telomeric attrition, with more than 50% of treated cells having a completeloss of detectable telomeres. Gene expression profiling of treated cells revealed increased levels
of p73, p63, and E2F1 as well as cell cycle arrest genes p21, p16, and GADD45. There was
also elevated expression of several genes involved in apoptosis, including FasL, Fas, caspases
8, 7, and 3, and CARD 9.
Interesting results have been obtained using lentiviral delivery of shRNA against hTR [62].
With an infection efficiency of >95%, eliminating the need for antibiotic selection and
consequential delay in assessing results, the effects of hTR inhibition were able to be analyzed
almost immediately after infection. The hairpin RNA expressed by the lentiviral vector
efficiently knocked down endogenous levels of hTR in vitro and caused rapid cell growth
inhibition and apoptosis independent of cellular p53 status and telomere length, and without
bulk telomere shortening. The same group found similar results when employing lentiviral
delivery of mutant-template telomerase RNA (to be discussed in more detail later in thisreview). When co-expressed in vitro, the shRNA and mutant-template hTR synergistically
killed cancer cells. Further experimentation with the lentiviral expression of shRNA against
hTR has brought forth evidence that the aforementioned hTR knockdown in cancer cells
actually induces global changes in gene expression through a novel response pathway, causing
the suppression of genes likely involved in other facets of cancer progression, including
angiogenesis and metastasis [63]. This exciting finding has implications for expanding not only
the current knowledge base concerning the nature of telomerase inhibition and its resulting
phenotypes (telomere uncapping, changes in gene expression, senescence, and apoptosis), but
also potential therapeutic insights. Table 2 presents a summary of the reports of RNA
interference against telomerase.
Although RNAi technology is relatively new, advances are emerging rapidly. Two siRNA
drugs intended for the treatment of age-related macular degeneration (AMD) are undergoingPhase I clinical trials, the first human clinical trials for pharmaceutical siRNA [64].
Ribozymes
Ribozymes are catalytic RNA molecules which have specific endoribonuclease activity. The
ability of these molecules to cleave specific sites of target RNAs has generated considerable
interest in using them as human gene therapeutic agents. The first description of a ribozyme
serving as a telomerase inhibitor came in 1996. In that report [65], a hammerhead ribozyme
designed to cleave at the downstream end of the template region of hTR RNA was mixed with
extracts of human hepatocellular carcinoma cells. The result was inhibition of telomerase
activity by up to 90%, even under reaction conditions which were sub-optimal for ribozyme
activity. Later efforts to target hTR RNA with ribozymes resulted in reduction of telomerase
activity in both studies [66,67] but with slightly different outcomes. While human melanomacells transfected with an expression vector delivering a ribozyme against hTR RNA displayed
a significant increase in doubling time, a decrease in DNA synthesis, and some increased
apoptosis compared to control cells, telomere shortening was not observed, even after 30
population doublings [66]. Ribozyme expression by a vector introduced into the human breast
cancer cell line MCF-7 caused significant decreases in hTR expression among cell clones and
dramatic decreases in telomerase activity (complete ablation in some clones, weak activity in
Cunningham et al. Page 7
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 8/28
others correlating with hTR expression) [67]. Cells bearing the ribozyme expression vector
coding the ribozyme R1 (which targeted the template region of the hTR RNA) showed
markedly decreased cell proliferation, morphologic changes, and signs of apoptosis. In contrast
to the results caused by the ribozyme-mediated telomerase inhibition in melanoma cells [66],
ribozyme-mediated telomerase inhibition in the MCF-7 cell line caused a significant shortening
of telomere lengths in treated MCF-7 cells compared to control MCF-7 cells [67].
Ribozyme inhibition has also been successfully attempted against various targets on the hTERTtranscript. Early attempts targeted the extreme ends of the transcript outside of the open reading
frame [68]. Selecting sites at a number of locations distributed throughout the mRNA transcript,
Yokoyama et al. found that 13 nucleotides downstream from the 5'-end and 59 nucleotides
upstream from the poly(A) tail made the best targets for their ribozymes. In their experiments,
endometrial carcinoma cells stably transfected with ribozyme-expressing plasmids maintained
cell growth rates for 2 months after transfection, then slowed gradually. One clone died out
completely within 4 months, and some growth-arrested cells displayed signs of apoptosis. An
interesting finding was reported by a group delivering the same ribozyme into cells by two
different methods [69]. Stable transfection of a plasmid encoding a ribozyme against hTERT
mRNA in the region encoding the T-motif caused a reduction in mRNA levels, attenuation of
telomerase activity, significantly decreased growth and evidence of apoptosis with concomitant
telomere shortening in MCF-7 cells and the immortal human breast epithelial cell line
HBL-100. However, upon expressing the same ribozyme via recombinant adenoviral infectionin the HBL-100 cells, the group observed massive cell death through apoptosis in a matter of
days before any telomere shortening could take place. These findings were expanded later by
employing ovarian cancer cell lines having widely different telomere lengths [37]. Cells were
transduced with adenovirus expressing the same ribozyme described previously [69]. Rapid
effects were observed, with marked decreases in telomerase activity between 2 and 7 days after
transduction; massive apoptosis also occurred without lag time in ovarian cancer cell lines with
either long or short telomeres and without telomere shortening. Ribozyme-mediated telomerase
inhibition studies are summarized in Table 3.
DISRUPTING NORMAL FUNCTION BY EXPRESSION OF MUTANTS
So far this review has focused on either ways to actively destroy hTR and hTERT transcripts
or ways to prevent these normal components of the telomerase enzyme from carrying out their intended functions. Expression of mutant forms of either hTR or hTERT may be equally
effective in disrupting telomerase activity by overwhelming cells with faulty versions of the
primary enzyme components.
Dominant Negative hTERT
Two reports utilizing dominant negative mutations in the catalytic subunit of human telomerase
made their way into the literature in 1999 [58,70]. The first report in the literature [70] of the
use of dominant negative hTERT protein mutants (DN-hTERT) for telomerase inhibition
studies describes four such mutants at the following amino acid residues: 712, 868, 869, and
a double mutant at residues 868 and 869. In epidermoid tumor cells with short telomeres (A431,
1-4 kb), expression of the DN-hTERT led to morphological changes and massive apoptosis
within 5-10 days after induction of expression; cell division continued until death of the
populations, which indicated that further telomere shortening led to eventual chromosomal
damage and induction of an apoptotic response. Actual telomere length measurements were
not possible due to the rapid loss of the clones. The group turned to embryonic kidney cells
(HEK 293 cells) with longer telomeres in order to measure the effect of DN-hTERT expression.
In HEK 293 clones expressing DN-hTERT protein, reduced telomerase activity was observed
along with pronounced telomere shortening. Initial telomere lengths of the HEK 293 clones
used were 10-12 kb; cell proliferation continued unabated for >109 population doublings with
Cunningham et al. Page 8
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 9/28
telomere lengths approaching 4 kb. Unfortunately, around this time the DN-hTERT expression
was lost, preventing the observation of a possible apoptotic outcome. In short, the study showed
that cells having short telomeres were more rapidly susceptible to the effects of telomerase
inhibition than were cells with longer telomeres, perhaps because those cells with very short
telomeres might require telomerase for telomere maintenance at each cell division while cells
with longer telomeres do not. The senescent phenotype was not induced in cells with short
telomeres, only apoptosis. These observations are important when considering the implications
of telomerase inhibition as a therapeutic strategy in human cancer treatment. The averagetelomere length characterizing a tumor might be a factor when determining whether to use a
telomerase inhibitor as a treatment either alone or in conjunction with another therapy.
Additionally, the initial telomere lengths within a tumor might roughly predict the amount of
time required to produce a therapeutic outcome.
A critical step in the development of telomerase inhibition as a therapeutic strategy was the
description of a DN-hTERT [58] used by others [71-73] in years to follow. In this report,
aspartic acid and valine at amino acid residues 710 and 711 were substituted with alanine and
isoleucine, respectively, resulting in the creation of a catalytically inactive form of hTERT
[58]. The expression of this DN-hTERT in several telomerase-positive human cancer cell lines
produced multiple cell clones lacking detectable telomerase activity. Expression of the mutant
in ovarian and breast cancer cell lines caused gradual telomere shortening as would be predicted
by the rate of telomere erosion in normal, telomerase-negative cells [4]. Chromosomalmetaphase spread analysis revealed a number of dicentric chromosomes and chromosomal
fusions indicative of DN-hTERT -induced telomere dysfunction. Cell proliferation results in
cells expressing the mutant at levels sufficient to inhibit telomerase activity showed impaired
growth and eventually growth arrest and induction of apoptosis at intervals that correlated with
initial telomere length. Tumorigenicity was decreased in vivo by expression of the catalytic
mutant. Of therapeutic interest is the finding that the apoptotic response induced by the
expression of the DN-hTERT is p53-independent [58], which is in harmony with previous
findings [70]. As p53 mutations are found in at least 50% of human cancers [74], it is
encouraging that there is evidence that DN-hTERT as a therapeutic telomerase inhibitor may
be useful in a wide range of human malignancies, not simply those which retain p53 function.
Dominant-negative hTERT studies are summarized in Table 4.
Mutant-Template Telomerase RNA
Early theory behind this type of telomerase inhibition rested upon the highly conserved nature
of the human telomeric DNA sequence. Experiments with Tetrahymena and yeast [75,76]
demonstrated that mutations in the template region of telomerase RNA caused the synthesis
of mutant telomeric repeats which in turn impaired cell growth and survival. It is possible that
proteins that bind telomeric DNA may not be able to bind mutant telomeric sequences and
consequently may be unable to fulfill their roles in the regulation of telomere length [77].
Accordingly, early experiments in this area showed that expression of mutant-template
telomerase RNA (MT-hTR) caused mutant telomerase activity and the synthesis of mutant
telomeres [78]. Use of MT-hTR constructs as a possible cancer therapeutic strategy was
supported by a study expressng MT-hTR in human prostate and breast cancer cell lines, in
which expression decreased cellular viability and increased apoptosis despite the retention of
normal telomerase RNA and stable telomere lengths [79]. Similar results were obtained invivo with breast cancer xenografts expressing MT-hTR [79].
In the aforementioned uses of MT-hTR, only low expression of mutant template RNA was
observed along with relatively slight phenotypic changes. In an effort to make delivery and
expression of MT-hTR more efficient, and therefore to enable more rapid analysis of the short-
term effects of expression on cancer cells, Li et al. developed a lentiviral delivery method
Cunningham et al. Page 9
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 10/28
which resulted in >95% target cell infection efficiency [62]. Rapid results were observed in
human melanoma cells (LOX) and human bladder transitional epithelial carcinoma cells (UM-
UC-3) infected with lentivirus expressed MT-hTR. LOX cells exhibited both rapid growth
inhibition and the onset of apoptosis by day 5 post-infection, while UM-UC-3 cells were growth
inhibited by day 8 and showed signs of apoptosis by day 9. Interestingly, bulk telomere
shortening was not required for the onset of visible phenotypic effects, as was proven by the
rapid onset of growth inhibition in LOX cells, whose telomeres are >40 kb long. In addition,
the phenotypic effects were not dependent upon p53 status of the cells (see also DN-hTERT[58,70]). These findings are summarized in Table 5.
UNIQUENESS OF TELOMERASE EXPRESSION AS A TARGETING
STRATEGY FOR ONCOLYSIS
In addition to actively destroying telomerase enzyme components and interfering with their
function, we are increasingly able to use the very nature of telomerase itself as a therapeutic
weapon. The widespread expression of telomerase in human cancers [11] has already been
discussed. It would be reasonable then to attempt directed cancer cell death using the hTR and
hTERT enzyme components as targets for delivery.
Virus-Mediated Specific Lysis of Tumor Cells
The prospect of specifically targeting cancer cells for death while leaving surrounding healthy
cells unharmed is the Holy Grail of therapeutic drug design, yet that is exactly what
conditionally-replicating viruses are being designed to do in vitro and in vivo. The adenovirus
TRAD (tumor- or telomerase-specific replication-competent adenovirus) has been designed to
express E1A and E1B genes under the control of the hTERT promoter [80]. The result was a
selective cell lysis of cancer cells (several non-small cell lung cancer and colorectal carcinoma
cell lines) and no apparent cytopathic effects in normal cells (WI38 and NHLF) 7 days after
adenoviral infection. Additionally, TRAD infection of human tumor xenografts in mice caused
marked suppression of tumor growth as well as the induction of tumor cell necrosis. Analysis
showed systemic distribution of TRAD by blood and lymph circulation in mice without adverse
effect. In fact, experiments successfully demonstrated that intratumoral injection of TRAD
could result in viral replication in other, distant tumors. This study has exciting implications
not only for primary tumor therapy but also for treating distant metastases which may goundetected clinically for extended periods, dramatically affecting the patient's prognosis and
outcome. However, although no apparent ill effects were observed in normal cells in this study,
one must carefully weigh the consequences of expressing E1A and E1B (a potent combination
of oncogenes) in normal human cells. Additionally, the relatively new concept of using
telomerase-specific oncolytic adenoviruses seems promising but requires a deeper
understanding of viral replication and associated tumor specificity (see [81]).
Selective Expression of Cytotoxic or Pro-Apoptotic Genes
The differences in hTR and hTERT expression in malignant and normal tissues also lends itself
to the possibility of expressing either toxic genes (suicide genes) or genes which are important
in the induction of the apoptotic process selectively in cancer cells. Several such studies have
demonstrated the usefulness of the hTR or hTERT promoters in driving the expression of the
diphtheria toxin A-chain gene [82], the pro-apoptotic gene Bax [83], the Fas associated protein
with death domain [84], and the apoptotic signal transducers caspase-6 [85] and caspase-8
[86]. Additionally, selective expression of the herpes simplex virus thymidine kinase gene
under the control of the hTERT promoter has been shown to confer sensitivity to ganciclovir
treatment on cancer cells [87,88] and the expression of bacterial nitroreductase under control
of the hTR or hTERT promoters sensitizes cancer cells in vitro and in vivo to the pro-drug
CB1954 [89,90].
Cunningham et al. Page 10
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 11/28
TELOMERASE INHIBITORS IN COMBINATION THERAPY
As reviewed here, telomerase inhibitors have been shown to be effective by themselves as
potentially valuable therapeutic agents. However, their greatest use may come not as stand-
alone pharmaceuticals but as part of a coordinated treatment strategy in conjunction with
standard treatments including various chemotherapeutics and radiation therapy. Several of the
studies cited in this review demonstrated success with their telomerase inhibitors both alone
and with synergistic effects when combined with other chemotherapeutics. The method of action of the various chemotherapeutic agents include topoisomerase inhibitors [60,69,72],
inducers of DNA damage and double-strand breaks [60,71,91,92], mitotic inhibitors [71], and
one agent shown to cause telomere erosion (paclitaxel) [93], [94]. Additionally, one study
showed the ability of gene expression (noradrenaline transporter gene) under the control of the
hTR promoter to induce the uptake of a radiopharmaceutical ([131I] MIBG) by cancer cells
[95]. These combination approaches are summarized in Table 6.
POTENTIAL DRAWBACKS OF TELOMERASE INHIBITORS
Despite the obvious benefits that could be garnered from the development of this class of novel
therapeutic agents, there are a few risks that warrant mentioning. While telomerase is not
significantly expressed in normal cells, some, such as hematopoietic stem cells, intestinal crypt
cells, and cells lining the endometrium do express telomerase and could theoretically be
adversely affected by treatment with telomerase inhibitors. It is thought, however, that adverse
effects would be minimal, as these normal cells express telomerase infrequently and at lower
levels as compared to cancer cells. The telomeres are also generally of greater length in the
rare telomerase-positive normal cells which makes these cells less susceptible to telomerase
inhibition. These cells also proliferate at a slower rate than most cancer cells and should
therefore incur less telomeric attrition. Another concern is that treatment with telomerase
inhibitors might incur a clinically significant lag time before telomeric erosion initiates cell
senescence or apoptosis. This appears to be less of a concern with the recent appearance of
numerous reports of rapid apoptotic responses upon telomerase inhibition [37,62,63] resulting
from possible telomere uncapping or the induction of a novel gene expression pathway [63].
Also, standard chemotherapeutic agents such as paclitaxel, a known telomere-shortening agent
[93], could be used to offset any lag time to therapeutic effect.
As previously mentioned, telomerase inhibitors may exert a delayed effect on cells dependent
upon telomeric attrition or they can act rapidly without bulk telomere shortening [35-37,62,
63,69]. Interestingly, this phenomenon cannot be explained solely by initial telomere length
or p53 status (see [62]). Perhaps these alternate outcomes are in some way related to the putative
extratelomeric functions of telomerase. These functions may include cell protection [96,97],
anti-apoptotic function [98], and support of the tumorigenic phenotype in a manner beyond
that of simple telomere maintenance [99] (see [100] for review). It may be the case that targeting
either hTR or hTERT in various ways affects the telomerase enzyme at different levels,
resulting in slightly different outcomes. Not all telomerase inhibitors cause telomeric attrition
(and consequently a delayed effect on the cell) by abrogating telomerase activity. Perhaps some
of the rapid responses to telomerase inhibition may be the result of telomere uncapping as a
consequence of inhibition, since telomerase is known to play a role in the capping of telomeres
as well as their elongation [101]. It is desirable for these different outcomes to be further studied and the mechanisms of action of the inhibitors to be better understood before advancing into
clinical trials, as the extratelomeric effects of telomerase are not yet well understood. Better
understanding of the mechanisms of action will allow better targeting of drug therapies, design
of more effective combination therapies, and minimize undesirable side effects of treatment.
Cunningham et al. Page 11
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 12/28
A significant consideration when contemplating using telomerase inhibitors is the potential for
selecting for the alternative lengthening of telomeres (ALT) pathway [102]. Cells that maintain
their telomere lengths while proliferating indefinitely must use either telomerase or the ALT
pathway. Eliminating telomerase activity may promote the development of the ALT pathway
in cancer cells. Such a phenomenon was observed after telomerase inhibition in a colon cancer
cell line having a mismatch repair defect [103]. Efforts should therefore be made to further
examine the relationship between telomerase inhibition and induction of ALT and to develop
mechanisms to block the ALT pathway.
The studies reviewed here, in addition to others outside the scope of this article, demonstrate
the considerable therapeutic potential of molecular-based methods of telomerase inhibition.
Either alone or in conjunction with existing cancer therapies, telomerase inhibition promises
to be a targeted, specific means of treatment with minimal side effects.
ACKNOWLEDGEMENTS
This work was supported in part by grants from the National Cancer Institute, the University of Alabama at Birmingham
Ovarian SPORE, and the American Institute for Cancer Research.
ABBREVIATIONS
hTR, Human telomerase RNAhTERT, Human telomerase reverse transcriptase
PS, Phosphorothioatecd
PNA, Peptide nucleic acid
2-5A, 2'-5'-oligoadenylate
LNA, Locked nucleic acid
NP, N3'-P5' phosphoramidates
NPS, N3'-P5' thio-phosphoramidates
TRAP, Telomeric repeat amplification protocol
RNAi, RNA interference
siRNA, Small interfering RNA
PD, Population doubling
nt, Nucleotide
shRNA, Short hairpin RNADN-hTERT, Dominant negative human telomerase re verse transcriptase
MT-hTR, Mutant template human telomerase RNA
TRAD, Tumor- or telomerase-specific replication competent adenovirus
ALT, Alternative lengthening of telomeres
REFERENCES
1. Blackburn EH. Nature 1991;350:569. [PubMed: 1708110]
2. Greider CW. Curr. Opin. Cell Biol 1991;3:444. [PubMed: 1892656]
3. Harley CB. Mutat. Res 1991;256:271. [PubMed: 1722017]
4. Harley CB, Futcher AB, Greider CW. Nature 1990;345:458. [PubMed: 2342578]
5. Hayflick L. Exp. Cell Res 1965;37:614. [PubMed: 14315085]
6. Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner
S, Wright WE. Science 1998;279:349. [PubMed: 9454332]
7. Olovnikov AM. Dokl. Akad. Nauk. SSSR 1971;201:1496. [PubMed: 5158754]
8. Watson JD. Nat. New Biol 1972;239:197. [PubMed: 4507727]
9. Shay JW, Pereira-Smith OM, Wright WE. Exp. Cell Res 1991;196:33. [PubMed: 1652450]
10. Masutomi K, Yu EY, Khurts S, Ben-Porath I, Currier JL, Metz GB, Brooks MW, Kaneko S, Murakami
S, DeCaprio JA, Weinberg RA, Stewart SA, Hahn WC. Cell 2003;114:241. [PubMed: 12887925]
Cunningham et al. Page 12
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 13/28
11. Shay JW, Bacchetti S. Eur. J. Cancer 1997;33:787. [PubMed: 9282118]
12. de Lange T, Shiue L, Myers RM, Cox DR, Naylor SL, Killery AM, Varmus HE. Mol. Cell Biol
1990;10:518. [PubMed: 2300052]
13. Engelhardt M, Drullinsky P, Guillem J, Moore MA. Clin. Cancer Res 1997;3:1931. [PubMed:
9815582]
14. Engelhardt M, Ozkaynak MF, Drullinsky P, Sandoval C, Tugal O, Jayabose S, Moore MA. Leukemia
1998;12:13. [PubMed: 9436916]
15. Hastie ND, Dempster M, Dunlop MG, Thompson AM, Green DK, Allshire RC. Nature 1990;346:866.[PubMed: 2392154]
16. Melana SM, Holland JF, Pogo BG. Clin. Cancer Res 1998;4:693. [PubMed: 9533539]
17. Pendino F, Flexor M, Delhommeau F, Buet D, Lanotte M, Segal-Bendirdjian E. Proc. Natl. Acad.
Sci. USA 2001;98:6662. [PubMed: 11371621]
18. Aldous WK, Marean AJ, DeHart MJ, Matej LA, Moore KH. Cancer 1999;85:1523. [PubMed:
10193942]
19. Naasani I, Seimiya H, Tsuruo T. Biochem. Biophys. Res. Commun 1998;249:391. [PubMed:
9712707]
20. Gomez D, Lemarteleur T, Lacroix L, Mailliet P, Mergny JL, Riou JF. Nucleic Acids Res 2004;32:371.
[PubMed: 14729921]
21. Fu W, Begley JG, Killen MW, Mattson MP. J. Biol. Chem 1999;274:7264. [PubMed: 10066788]
22. Smogorzewska A, de Lange T. Annu. Rev. Biochem 2004;73:177. [PubMed: 15189140]
23. Stansel RM, de Lange T, Griffith JD. EMBO J 2001;20:5532. [PubMed: 11574485]
24. de Lange T. Oncogene 2002;21:532. [PubMed: 11850778]
25. Braasch DA, Corey DR. Biochemistry 2002;41:4503. [PubMed: 11926811]
26. Geary RS, Yu RZ, Levin AA. Curr. Opin. Investig. Drugs 2001;2:562.
27. Egholm M, Buchardt O, Christensen L, Behrens C, Freier SM, Driver DA, Berg RH, Kim SK, Norden
B, Nielsen PE. Nature 1993;365:566. [PubMed: 7692304]
28. Orr RM. Curr. Opin. Mol. Ther 2001;3:288. [PubMed: 11497353]
29. Feng J, Funk WD, Wang SS, Weinrich SL, Avilion AA, Chiu CP, Adams RR, Chang E, Allsopp RC,
Yu J, et al. Science 1995;269:1236. [PubMed: 7544491]
30. Zhang FX, Zhang XY, Fan DM, Deng ZY, Yan Y, Wu HP, Fan JJ. World J. Gastroenterol 2000;6:430.
[PubMed: 11819619]
31. Kondo S, Tanaka Y, Kondo Y, Hitomi M, Barnett GH, Ishizaka Y, Liu J, Haqqi T, Nishiyama A,
Villeponteau B, Cowell JK, Barna BP. FASEB J 1998;12:801. [PubMed: 9657520]32. Bisoffi M, Chakerian AE, Fore ML, Bryant JE, Hernandez JP, Moyzis RK, Griffith JK. Eur. J. Cancer
1998;34:1242. [PubMed: 9849487]
33. Zhang X, Chen Z, Chen Y, Tong T. Oncogene 2003;22:2405. [PubMed: 12717417]
34. Kraemer K, Fuessel S, Schmidt U, Kotzsch M, Schwenzer B, Wirth MP, Meye A. Clin. Cancer Res
2003;9:3794. [PubMed: 14506173]
35. Cao Y, Li H, Deb S, Liu JP. Oncogene 2002;21:3130. [PubMed: 12082628]
36. Cao Y, Li H, Mu F-T, Ebisui O, Funder JW, Liu J-P. FASEB J 2001;01
37. Saretzki G, Ludwig A, von Zglinicki T, Runnebaum IB. Cancer Gene Ther 2001;8:827. [PubMed:
11687906]
38. Fu XH, Zhang JS, Zhang N, Zhang YD. World J. Gastroenterol 2005;11:785. [PubMed: 15682468]
39. Kondo S, Kondo Y, Li G, Silverman RH, Cowell JK. Oncogene 1998;16:3323. [PubMed: 9681832]
40. Kondo Y, Koga S, Komata T, Kondo S. Oncogene 2000;19:2205. [PubMed: 10822370]41. Pitts AE, Corey DR. Proc. Natl. Acad. Sci. USA 1998;95:11549. [PubMed: 9751703]
42. Matthes E, Lehmann C. Nucleic Acids Res 1999;27:1152. [PubMed: 9927750]
43. Ye J, Wu YL, Zhang S, Chen Z, Guo LX, Zhou RY, Xie H. World J. Gastroenterol 2005;11:2230.
[PubMed: 15818731]
44. Shammas MA, Liu X, Gavory G, Raney KD, Balasubramanian S, Shmookler Reis RJ. Exp. Cell Res
2004;295:204. [PubMed: 15051503]
Cunningham et al. Page 13
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 14/28
45. Shammas MA, Simmons CG, Corey DR, Shmookler Reis RJ. Oncogene 1999;18:6191. [PubMed:
10597217]
46. Folini M, Berg K, Millo E, Villa R, Prasmickaite L, Daidone MG, Benatti U, Zaffaroni N. Cancer
Res 2003;63:3490. [PubMed: 12839932]
47. Elayadi AN, Braasch DA, Corey DR. Biochemistry 2002;41:9973. [PubMed: 12146961]
48. Gryaznov S, Pongracz K, Matray T, Schultz R, Pruzan R, Aimi J, Chin A, Harley C, Shea-Herbert
B, Shay J, Oshima Y, Asai A, Yamashita Y. Nucleosides Nucleotides Nucleic Acids 2001;20:401.
[PubMed: 11563055]49. Herbert BS, Pongracz K, Shay JW, Gryaznov SM. Oncogene 2002;21:638. [PubMed: 11850790]
50. Pruzan R, Pongracz K, Gietzen K, Wallweber G, Gryaznov S. Nucleic Acids Res 2002;30:559.
[PubMed: 11788719]
51. Gryaznov S, Asai A, Oshima Y, Yamamoto Y, Pongracz K, Pruzan R, Wunder E, Piatyszek M, Li
S, Chin A, Harley C, Akinaga S, Yamashita Y. Nucleosides Nucleotides Nucleic Acids 2003;22:577.
[PubMed: 14565232]
52. Akiyama M, Hideshima T, Shammas MA, Hayashi T, Hamasaki M, Tai YT, Richardson P, Gryaznov
S, Munshi NC, Anderson KC. Cancer Res 2003;63:6187. [PubMed: 14559802]
53. Wang ES, Wu K, Chin AC, Chen-Kiang S, Pongracz K, Gryaznov S, Moore MA. Blood
2004;103:258. [PubMed: 12969977]
54. Asai A, Oshima Y, Yamamoto Y, Uochi TA, Kusaka H, Akinaga S, Yamashita Y, Pongracz K, Pruzan
R, Wunder E, Piatyszek M, Li S, Chin AC, Harley CB, Gryaznov S. Cancer Res 2003;63:3931.
[PubMed: 12873987]55. Herbert BS, Gellert GC, Hochreiter A, Pongracz K, Wright WE, Zielinska D, Chin AC, Harley CB,
Shay JW, Gryaznov SM. Oncogene 2005;24:5262. [PubMed: 15940257]
56. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Nature 1998;391:806. [PubMed:
9486653]
57. Kosciolek BA, Kalantidis K, Tabler M, Rowley PT. Mol. Cancer Ther 2003;2:209. [PubMed:
12657713]
58. Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A, Beijersbergen RL, Knoll JH,
Meyerson M, Weinberg RA. Nat. Med 1999;5:1164. [PubMed: 10502820]
59. Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn
WC, Sharp PA, Weinberg RA, Novina CD. RNA 2003;9:493. [PubMed: 12649500]
60. Nakamura M, Masutomi K, Kyo S, Hashimoto M, Maida Y, Kanaya T, Tanaka M, Hahn WC, Inoue
M. Hum. Gene Ther 2005;16:859. [PubMed: 16000067]
61. Shammas MA, Koley H, Batchu RB, Bertheau RC, Protopopov A, Munshi NC, Goyal RK. Mol.Cancer 2005;4:24. [PubMed: 16022731]
62. Li S, Rosenberg JE, Donjacour AA, Botchkina IL, Hom YK, Cunha GR, Blackburn EH. Cancer Res
2004;64:4833. [PubMed: 15256453]
63. Li S, Crothers J, Haqq CM, Blackburn EH. J. Biol. Chem 2005;280:23709. [PubMed: 15831499]
64. Weil, N. Running Interference. 2004.
http://www.benitec.com/PRDownloads/Running%20Interference% 20RNAi%20021805.pdf
65. Kanazawa Y, Ohkawa K, Ueda K, Mita E, Takehara T, Sasaki Y, Kasahara A, Hayashi N. Biochem.
Biophys. Res. Commun 1996;225:570. [PubMed: 8753802]
66. Folini M, Colella G, Villa R, Lualdi S, Daidone MG, Zaffaroni N. J. Invest. Dermatol 2000;114:259.
[PubMed: 10651984]
67. Yeo M, Rha SY, Jeung HC, Hu SX, Yang SH, Kim YS, An SW, Chung HC. Int. J. Cancer
2005;114:484. [PubMed: 15551309]
68. Yokoyama Y, Takahashi Y, Shinohara A, Wan X, Takahashi S, Niwa K, Tamaya T. Biochem.
Biophys. Res. Commun 2000;273:316. [PubMed: 10873604]
69. Ludwig A, Saretzki G, Holm PS, Tiemann F, Lorenz M, Emrich T, Harley CB, von Zglinicki T.
Cancer Res 2001;61:3053. [PubMed: 11306487]
70. Zhang X, Mar V, Zhou W, Harrington L, Robinson MO. Genes Dev 1999;13:2388. [PubMed:
10500096]
Cunningham et al. Page 14
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 15/28
71. Misawa M, Tauchi T, Sashida G, Nakajima A, Abe K, Ohyashiki JH, Ohyashiki K. Int. J. Oncol
2002;21:1087. [PubMed: 12370759]
72. Nakajima A, Tauchi T, Sashida G, Sumi M, Abe K, Yamamoto K, Ohyashiki JH, Ohyashiki K.
Leukemia 2003;17:560. [PubMed: 12646945]
73. Preto A, Singhrao SK, Haughton MF, Kipling D, Wynford-Thomas D, Jones CJ. Oncogene
2004;23:4136. [PubMed: 15064743]
74. Levine AJ. Cell 1997;88:323. [PubMed: 9039259]
75. Yu GL, Bradley JD, Attardi LD, Blackburn EH. Nature 1990;344:126. [PubMed: 1689810]76. McEachern MJ, Blackburn EH. Nature 1995;376:403. [PubMed: 7630414]
77. Fang, G.; Cech, TR. Telomere proteins. In: Greider, E.H.B.a.C.W., editor. Telomeres. Cold Spring
Harbor Laboratory Press; Cold Spring Harbor, NY: 1995. p. 69-105.
78. Marusic L, Anton M, Tidy A, Wang P, Villeponteau B, Bacchetti S. Mol. Cell. Biol, 1997;17:6394.
[PubMed: 9343401]
79. Kim MM, Rivera MA, Botchkina IL, Shalaby R, Thor AD, Blackburn EH. Proc. Natl. Acad. Sci.
USA 2001;98:7982. [PubMed: 11438744]
80. Kawashima T, Kagawa S, Kobayashi N, Shirakiya Y, Umeoka T, Teraishi F, Taki M, Kyo S, Tanaka
N, Fujiwara T. Clin. Cancer Res 2004;10:285. [PubMed: 14734481]
81. O'Shea CC. Oncogene 2005;24:7640. [PubMed: 16299526]
82. Abdul-Ghani R, Ohana P, Matouk I, Ayesh S, Ayesh B, Laster M, Bibi O, Giladi H, Molnar-Kimber
K, Sughayer MA, de Groot N, Hochberg A. Mol. Ther 2000;2:539. [PubMed: 11124054]
83. Gu J, Kagawa S, Takakura M, Kyo S, Inoue M, Roth JA, Fang B. Cancer Res 2000;60:5359. [PubMed:
11034071]
84. Komata T, Koga S, Hirohata S, Takakura M, Germano IM, Inoue M, Kyo S, Kondo S, Kondo Y. Int.
J. Oncol 2001;19:1015. [PubMed: 11605003]
85. Komata T, Kondo Y, Kanzawa T, Hirohata S, Koga S, Sumiyoshi H, Srinivasula SM, Barna BP,
Germano IM, Takakura M, Inoue M, Alnemri ES, Shay JW, Kyo S, Kondo S. Cancer Res
2001;61:5796. [PubMed: 11479218]
86. Koga S, Hirohata S, Kondo Y, Komata T, Takakura M, Inoue M, Kyo S, Kondo S. Hum. Gene Ther
2000;11:1397. [PubMed: 10910137]
87. Majumdar AS, Hughes DE, Lichtsteiner SP, Wang Z, Lebkowski JS, Vasserot AP. Gene Ther
2001;8:568. [PubMed: 11319624]
88. Song JS, Kim HP, Yoon WS, Lee KW, Kim MH, Kim KT, Kim HS, Kim YT. Biosci. Biotechnol.
Biochem 2003;67:2344. [PubMed: 14646192]
89. Plumb JA, Bilsland A, Kakani R, Zhao J, Glasspool RM, Knox RJ, Evans TR, Keith WN. Oncogene
2001;20:7797. [PubMed: 11753658]
90. Bilsland AE, Anderson CJ, Fletcher-Monaghan AJ, McGregor F, Evans TR, Ganly I, Knox RJ, Plumb
JA, Keith WN. Oncogene 2003;22:370. [PubMed: 12545158]
91. Chen Z, Koeneman KS, Corey DR. Cancer Res 2003;63:5917. [PubMed: 14522918]
92. Kondo Y, Kondo S, Tanaka Y, Haqqi T, Barna BP, Cowell JK. Oncogene 1998;16:2243. [PubMed:
9619833]
93. Multani AS, Li C, Ozen M, Imam AS, Wallace S, Pathak S. Oncol. Rep 1999;6:39. [PubMed:
9864398]
94. Mo Y, Gan Y, Song S, Johnston J, Xiao X, Wientjes MG, Au JL. Cancer Res 2003;63:579. [PubMed:
12566299]
95. Boyd M, Mairs RJ, Mairs SC, Wilson L, Livingstone A, Cunningham SH, Brown MM, Quigg M,
Keith WN. Oncogene 2001;20:7804. [PubMed: 11753659]96. Lu C, Fu W, Mattson MP. Brain Res. Dev. Brain Res 2001;131:167.
97. Zhu H, Fu W, Mattson MP. J. Neurochem 2000;75:117. [PubMed: 10854254]
98. Luiten RM, Pene J, Yssel H, Spits H. Blood 2003;101:4512. [PubMed: 12586632]
99. Stewart SA, Hahn WC, O'Connor BF, Banner EN, Lundberg AS, Modha P, Mizuno H, Brooks MW,
Fleming M, Zimonjic DB, Popescu NC, Weinberg RA. Pro.c Natl. Acad. Sci. USA 2002;99:12606.
100. Chung HK, Cheong C, Song J, Lee HW. Curr. Mol. Med 2005;5:233. [PubMed: 15974878]
Cunningham et al. Page 15
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 16/28
101. Chan SW, Blackburn EH. Oncogene 2002;21:553. [PubMed: 11850780]
102. Henson JD, Neumann AA, Yeager TR, Reddel RR. Oncogene 2002;21:598. [PubMed: 11850785]
103. Bechter OE, Zou Y, Walker W, Wright WE, Shay JW. Cancer Res 2004;64:3444. [PubMed:
15150096]
Cunningham et al. Page 16
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 17/28
Fig. 1.
Telomeres erode in normal somatic cells with every population doubling due to the virtual
absence of telomerase. Reactivation of telomerase appears to play a key role in the development
of cancer.
Cunningham et al. Page 17
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 18/28
Fig. 2.
Diagram illustrating the possible outcomes of telomerase inhibition. Inhibition of telomerase
prevents the maintenance of telomere length in telomerase-positive cells. As a result,
telomerase may shorten, leading to eventual replicative senescence or apoptosis. Telomerase
inhibition may also cause rapid cell death without telomere shortening and the induction of a
novel gene expression pathway (discussed later in review).
Cunningham et al. Page 18
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt
7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 19/28
Fig. 3.
Illustation of antisense mechanism of action in blocking protein expression.
Cunningham et al. Page 19
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt
7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 20/28
Fig 4.
Examples of modifications made to traditional antisense oligonucleotides (A).
Cunningham et al. Page 20
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 21/28
Fig 5.
Three variations of peptide nucleic acids (PNAs).
Cunningham et al. Page 21
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
NI H-P A A
ut h or Manus c r i pt
NI H-P A A ut h or Manus c r i pt
NI H-P A A ut h or
Manus c r i pt
7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 22/28
NI H-P A
A ut h or Manus c r i pt
NI H-P A A ut h or Manus c r
i pt
NI H-P A A ut h
or Manus c r i pt
Cunningham et al. Page 22
Table 1
isense Oligonucleotide Effects in Telomerase InhibitionTarget Cells Tested
* Effect of treatment Efficiency of inhibiton Ref.
hTR HeLa (cervical)Loss of telomeric DNA; crisis after 23-26PD
Reduction of activity [29]
hTR SGC7901 (gastric)Telomere length shortening; increased apoptosis
Significant reduction of activity
[30]
hTR U251-MG(malignant glioma)
Apoptosis only in some populations after 30PD; differentiation of some populations
Complete ablation of activity
[31]
hTR HeLa (cervical)A498 (kidney)
Reduction in cell viability; appearance of giant, senescent-likecells; reduction in growth rate
≥75% inhibition of telomeraseactivity
[32]
hTR MCF-7 (breast)
Progressive telomere shortening for 30 PD;induction of apoptosis;reduction of proliferation and colonyformation
significant suppressionof telomerase activity
[33]
hTERTEJ28, 5637, J28,HT1197 (bladder)
Reduction of hTERT mRNA (≤88%);impairment of cell viability;G1 arrest
≤60% inhibition of activity [34]
hTERT PMC42 (breast)
Decreased telomerase activity; significantapoptosis; Rapid effect:24 hours post-transfection
No significant telomere shortening
Up to 50% reduction of activity
[35]
hTR and hTERTsimultaneously
SW480(colon)
Significant inhibition of proliferation by 24hoursDecrease in telomerase activity by 48 hourscontinuing to decrease
through 72 hoursSignificant increase in apoptosis
combination treatment0.2 mmol/L for 72 hours;80% inhibition of activity
[38]
hTR (2-5A)
U251-MG(malignant glioma)
Significant decrease in cell viability after 5daysSignificant impairment of tumor growth innude mice
hTR undetectable after treatment
[39]
hTR (2-5A)
PC3, DU145(prostate)
Significant decrease in cell viability within6 daysSignificant suppression of tumor growth innude mice
Cell viability reduced to9-18% within 6 days of treatment
[40]
hTR (2′-O-Me)
DU145, LNCaP(prostate)
Telomere shortening; halt of cell proliferation after delay (in relationto telomere shortening); 90% reduction incolony formingability in LNCaP cells
>75% inhibition of activity up to>85% inhibitionin DU145
[91]
hTERT(PS)
EJ28(bladder)
Maximum decrease in hTERT mRNA 12hours after treatment;immediate and continued reduction of cellviability with successivetransfections of constructs
>60% inhibition of activity;up to 88% reductionin hTERT mRNA
[34]
hTR (PS)
HL-60(leukemia)
Efficient but not selective; non-complementary constructs had nearly same effect as complementary; moreefficient than PNA
IC50 of 0.5 and 0.6 nM [42]
hTR (PS)
MKN-28, SGC7901,MKN-45(gastric)
After 96 hours, significant growthinhibition in poor and moderatelydifferentiated cell lines but not in well-differentiated Apoptosis in poor and moderatelydifferentiated lines
Inhibition of activity at 5mmol/L; complete inhibition at10 mmol/L
[43]
hTERT(PNA)
DU145 (prostate)U2OS(osteogenic sarcoma)
Efficient introduction of naked PNAagainst hTERT using photochemicalinternalization approach; effect most
prominent 6 hoursafter treatment becoming less pronounced 24-48 hours after treatment
Telomerase activityreduced to 8.4 ± 0.79%of control
[46]
single stranded G-rich overhang(PNA)
AT-SV1†(Ataxiatelangiectasia)
No inhibition of telomerase activity;
decrease in colony sizes;slight decrease in median telomere lengthSynergistic effect with PNA blockingtelomerase activity
Virtual elimination of
colony formation whencombined with telomeraseinhibitor
[44]
hTR (PNA)
AT-SV1†(Ataxiatelangiectasia)
Inhibition of telomerase activity; proliferation arrest after 5 to 30generations; median telomere lengthshortened by 377 bp; reduction incolony size
62% reduction of telomeraseactivity with 10mM treatment
[45]
hTR (LNA)
DU145(prostate)
Inhibition of activity up to 40 hours post-transfection
Some LNAs resulted in [47]
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 23/28
NI H-P A
A ut h or Manus c r i pt
NI H-P A A ut h or Manus c r
i pt
NI H-P A A ut h
or Manus c r i pt
Cunningham et al. Page 23
Target Cells Tested* Effect of treatment Efficiency of inhibiton Ref.
High-affinity binding and selectivity>80% inhibition of telomeraseactivity
hTR (NP and NPS)
various
Telomerase inhibition by both NP and NPSresulting in reductionof telomere length
NP: most effective targets within templateregion
NPS: may use PS group interacting withhTERT to stabilize secondary structure
NP: sub-nM IC50 NPS: ∼50 pM IC50
[48]
hTR (NP and NPS)
HME50-5E†(spontaneouslyimmortalized
breast epithelialcells)
NP inefficient without lipid carrier; NPSefficient with or withoutcarrier; 0.5 mM NPS caused telomereshortening, senescence byday 100, massive apoptosis by day 115Addition of -thio group significantlyincreased potency
NP: IC50∼0.5-1 mMwith lipid
NPS: IC50 0.5-5 nMwith lipid
[49]
hTR (GRN163)
various
Inhibition of telomerase activity within24-72 hours after treatmentWI-38 and BJ fibroblast survival notaffected by up to 100 mMtreatment for 72 hoursInduction of telomere shortening, growtharrest, cell death, and tumor growth suppression in nude mice
IC50 of 26-44 pM[51-54]
(GRN163L) various
Sevenfold lower IC50 in tested cell lines;more rapid telomericattrition and growth inhibition; lipid modification eliminates need
for carrier In early clinical trials
[55]
Antisense modifications: 2-5A = 2′-5′-oligoadenylate; 2′-O-Me = 2′-O-methyl-RNA; PS = phosphorothioate; PNA = peptide nucleic acid; LNA = locked
nucleic acid; NP = oligonucleotide N3′-P5′ phosphoramidates; NPS = N3′-P5′ thio-phosphoramidates.
*All listed are cancer cell lines unless otherwise specified (†)
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
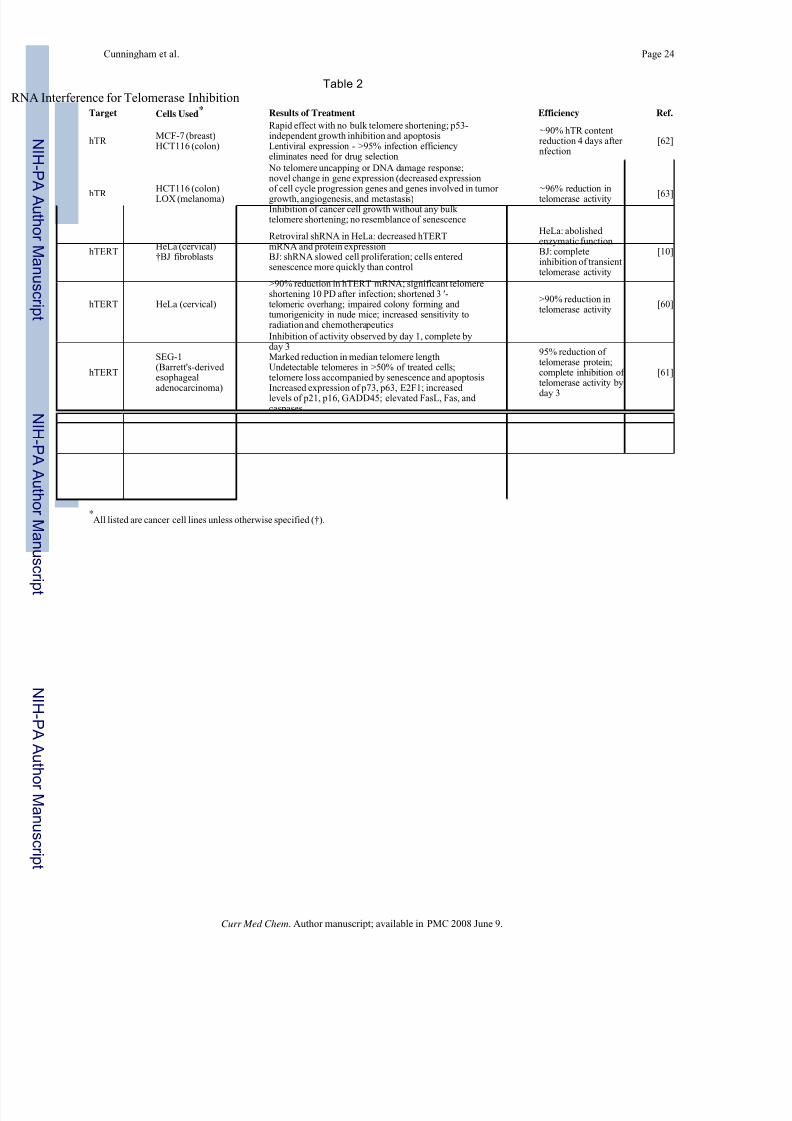
7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 24/28
NI H-P A
A ut h or Manus c r i pt
NI H-P A A ut h or Manus c r
i pt
NI H-P A A ut h
or Manus c r i pt
Cunningham et al. Page 24
Table 2
A Interference for Telomerase InhibitionTarget Cells Used
* Results of Treatment Efficiency Ref.
hTR MCF-7 (breast)HCT116 (colon)
Rapid effect with no bulk telomere shortening; p53-independent growth inhibition and apoptosisLentiviral expression - >95% infection efficiencyeliminates need for drug selection
∼90% hTR contentreduction 4 days after nfection
[62]
hTR
HCT116 (colon)
LOX (melanoma)
No telomere uncapping or DNA damage response;novel change in gene expression (decreased expressionof cell cycle progression genes and genes involved in tumor
growth, angiogenesis, and metastasis)Inhibition of cancer cell growth without any bulk telomere shortening; no resemblance of senescence
∼96% reduction in
telomerase activity [63]
hTERTHeLa (cervical)†BJ fibroblasts
Retroviral shRNA in HeLa: decreased hTERTmRNA and protein expressionBJ: shRNA slowed cell proliferation; cells entered senescence more quickly than control
HeLa: abolished enzymatic functionBJ: completeinhibition of transienttelomerase activity
[10]
hTERT HeLa (cervical)
>90% reduction in hTERT mRNA; significant telomereshortening 10 PD after infection; shortened 3 ′-telomeric overhang; impaired colony forming and tumorigenicity in nude mice; increased sensitivity toradiation and chemotherapeutics
>90% reduction intelomerase activity
[60]
hTERT
SEG-1(Barrett's-derived esophageal
adenocarcinoma)
Inhibition of activity observed by day 1, complete byday 3Marked reduction in median telomere lengthUndetectable telomeres in >50% of treated cells;telomere loss accompanied by senescence and apoptosis
Increased expression of p73, p63, E2F1; increased levels of p21, p16, GADD45; elevated FasL, Fas, and caspases
95% reduction of telomerase protein;complete inhibition of telomerase activity by
day 3
[61]
hTR/hTERT
various
siRNA against hTR more effective than siRNAagainst hTERT;increased siRNA concentrations did not increaseeffectiveness of telomerase inhibition; successivesiRNA treatments more effective than single treatment Hairpinconstruct against hTR effective at least 75daysCombination treatment of siRNA against hTR and hTERT not better than either alone
hTR siRNA: 75%inhibition of activityhTR hairpin: up to87% reduction of activityhTERT siRNA: 65%decrease in activity
[57]
*All listed are cancer cell lines unless otherwise specified (†).
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 25/28
NI H-P A
A ut h or Manus c r i pt
NI H-P A A ut h or Manus c r
i pt
NI H-P A A ut h
or Manus c r i pt
Cunningham et al. Page 25
Table 3
ozyme-Mediated Inhibition of TelomeraseTarget Cells Used
* Results of Treatment Efficiency Ref.
hTR HepG2, Huh-7 (liver)First report of ribozyme inhibition of telomerase;dose-dependent telomerase inhibition
Up to 90% inhibitionof telomerase activity
[65]
hTR JR8, M14 (melanoma)
Significantly increased cell doubling time; decreasein DNA sythesis;morphological changes; No telomere shortening (30PD)
≥75% inhibition of telomeraseactivity
[66]
hTR MCF-7 (breast)
>50% reduction of growth rate of some clones;telomere shortening;morphological changes; visible reduction intelomerase activity by 72hours
Complete inhibition of activity in some clones
[67]
hTERT Ishikawa, AN3CA (endometrial)
Targets outside of hTERT open reading frame (13nt downstream from5′-end, 59 nt upstream from poly(A) tail); cellgrowth rate maintained 2months followed by slowed proliferation then deathwithin 4 months
Stronger inhibitionusing 5′-end targetthan 3′-end target
[68]
hTERTMCF-7 (breast) HBL-100†(immortalized breastepithelial cells)
First use of ribozymes against hTERT; plasmid expression - decrease inhTERT mRNA and telomerase activity, decrease ingrowth rate, telomere shortening and apoptosis;viral expression - massive apoptosiswithout lag time or telomere shortening (HBL-100)
HBL-100: 62%reduction intelomerase activityMCF-7:46% inhibition of telomeraseactivity
[69]
hTERTOV-2774, OVCAR-3, OV-MZ-2A, OV-MZ-38 (ovarian)
Adenoviral delivery of ribozyme; marked decreasein telomerase activity 2-7 days after transduction;massive apoptosis without delay (regardless of initial telomere length); no telomere shortening
>90% inhibition of
telomeraseactivity in OV-2774and OV-MZ-38; 75%inhibition inOVCAR-3 and OV-MZ-2A
[37]
*All cells are cancer cell lines unless otherwise noted (†).
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.
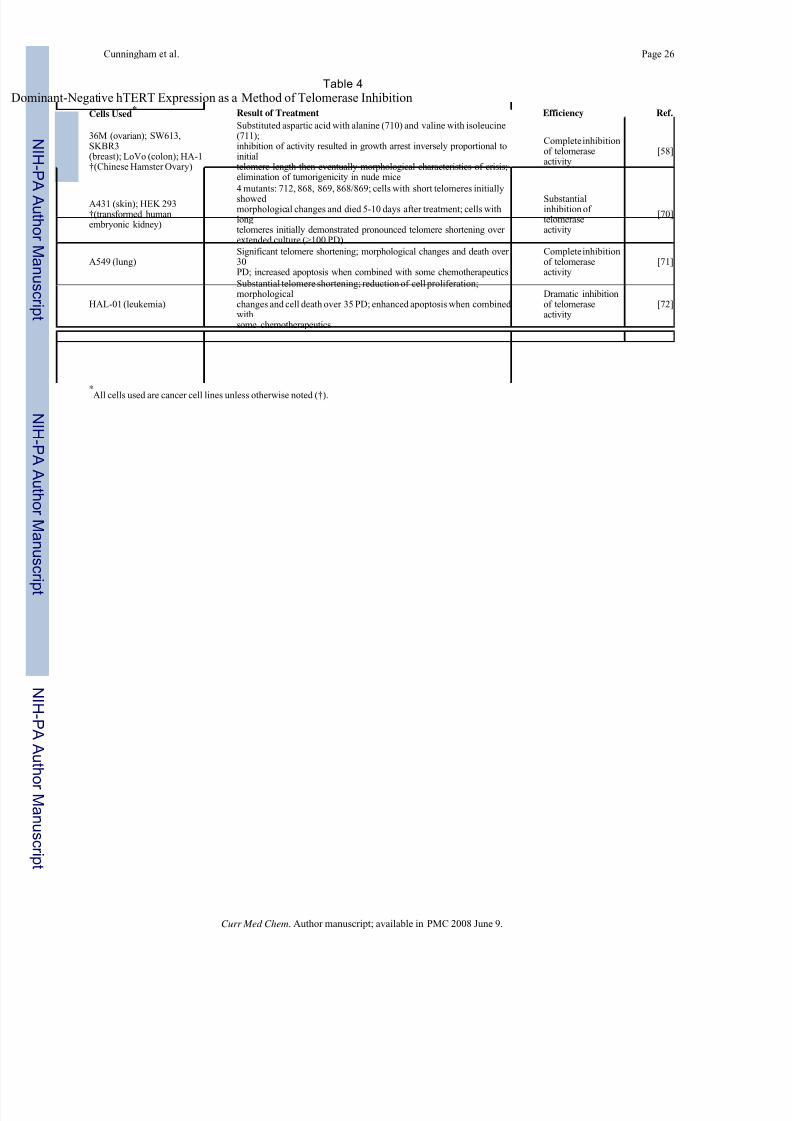
7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 26/28
NI H-P A
A ut h or Manus c r i pt
NI H-P A A ut h or Manus c r
i pt
NI H-P A A ut h
or Manus c r i pt
Cunningham et al. Page 26
Table 4
minant-Negative hTERT Expression as a Method of Telomerase Inhibition
Cells Used* Result of Treatment Efficiency Ref.
36M (ovarian); SW613,SKBR3(breast); LoVo (colon); HA-1†(Chinese Hamster Ovary)
Substituted aspartic acid with alanine (710) and valine with isoleucine(711);inhibition of activity resulted in growth arrest inversely proportional toinitialtelomere length then eventually morphological characteristics of crisis;elimination of tumorigenicity in nude mice
Complete inhibitionof telomeraseactivity
[58]
A431 (skin); HEK 293†(transformed humanembryonic kidney)
4 mutants: 712, 868, 869, 868/869; cells with short telomeres initially
showed morphological changes and died 5-10 days after treatment; cells withlongtelomeres initially demonstrated pronounced telomere shortening over extended culture (>100 PD)
Substantialinhibition of telomeraseactivity
[70]
A549 (lung)Significant telomere shortening; morphological changes and death over 30PD; increased apoptosis when combined with some chemotherapeutics
Complete inhibitionof telomeraseactivity
[71]
HAL-01 (leukemia)
Substantial telomere shortening; reduction of cell proliferation;morphologicalchanges and cell death over 35 PD; enhanced apoptosis when combined withsome chemotherapeutics
Dramatic inhibitionof telomeraseactivity
[72]
K1, K2 (thyroid)
Delayed growth arrest with characteristics of senescence; elevated expressionof p21WAF1 and senescence-associatedβ-galactosidase expression;obvioustelomere erosion
Complete inhibitionof telomeraseactivity
[73]
*All cells used are cancer cell lines unless otherwise noted (†).
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 27/28
NI H-P A
A ut h or Manus c r i pt
NI H-P A A ut h or Manus c r
i pt
NI H-P A A ut h
or Manus c r i pt
Cunningham et al. Page 27
Table 5
ant-Template Telomerase RNA Expression Exerts Effects Without Telomere Shortening
Cells Used* Results of Treatment Ref.
HT-1080 (fibrosarcoma);HEK 293 †(transformed human embryonic kidney)
Synthesis of mutant telomeric repeats; expression of mutant for >90 PD after transfection; impaired growth; accumulation of senescent cellsLow expression of MT-hTR
[78]
LNCaP (prostate); MCF-7(breast)
Decreased viability and increased apoptosis; stable telomere lengths; reduced tumor growth rate in nudemice; evenlow expression of MT-hTR produced results despite retention of wild-type hTR and telomerase activity
[79]
LOX (melanoma); UM-UC-3 (bladder)
Rapid growth inhibition and apoptosis within 9 days; coexpression with siRNA sensitized cells to MT-hTR; smaller,less vascular tumors in nude mice; no bulk telomere shortening; effect independent of cellular p53 status;inductionof sustained DNA damage response in telomerase-positive cancer cells >95% infection of target cells withMT-hTR construct
[63]
Treatment with these agents may be particularly advantageous since telomere erosion is not necessary for a phenotypic result and the time to therapeutic
effect is significantly decreased compared to telomerase inhibitors that cause telomeres to gradually shorten.
*All cells used are cancer cell lines unless otherwise noted (†).
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.

7/23/2019 Telomerasa Cancer
http://slidepdf.com/reader/full/telomerasa-cancer 28/28
NI H-P A
A ut h or Manus c r i pt
NI H-P A A ut h or Manus c r
i pt
NI H-P A A ut h
or Manus c r i pt
Cunningham et al. Page 28
Table 6
omerase Inhibitors may Sensitize Cancer Cells to Standard Therapeutic AgentsInhibitor/Sensitizer Standard Therapeutic Agent Outcome
2'-O-methoxyethyl RNA Cisplatin/carboplatin Synergistic effect
Antisense-hTR Paclitaxel Significant increase in sensitivity
Antisense-hTR Cisplatin Increase in sensitivity
DN-hTERT Cisplatin/taxanes/etoposide Increase in induction of apoptosis
DN-hTERT Daunorubicin Increase in apoptosis
Ribozyme-hTERT Doxorubicin Increase in sensitivity
RNAi-hTERT Topoisomerase inhibitors/bleomycin/radiation Increase in sensitivityhTR-NAT [131I]MIBG Induced uptake
AZT Paclitaxel Increased activity and effect
NAT = noradrenaline transporter gene; MIBG = [131I]meta-iodobenzylguanidine; AZT = azidothymidine.
Curr Med Chem. Author manuscript; available in PMC 2008 June 9.