
Subunit Heterogeneity Human Serum Lipoprotein* - PNAS · an ethanol-ether mixture yields a protein...
8
Proceedings of the National Academy of Sciences Vol. 66, No. 4, pp. 1075-1082, August 1970 Subunit Heterogeneity in Human Serum Beta Lipoprotein* J. P. Kane,t E. G. Richards, and R. J. Havel CARDIOVASCULAR RESEARCH INSTITUTE AND DEPARTMENT OF MEDICINE, UNIVERSITY OF CALI- FORNIA (SAN FRANCISCO); AND INSTITUTE OF MARINE RESOURCES, UNIVERSITY OF CALIFORNIA, BERKELEY Communicated by Julius H. Comroe, Jr., May 21, 1970 Abstract. The apoprotein of human serum beta lipoprotein has been prepared in a lipid-free and soluble form by a technique of interfacial extraction in the presence of guanidinium hydrochloride and nonionic amphiphile. Gel permea- tion chromatography of maleylated apobeta lipoprotein separates two protein fractions with different amino acid compositions. The smaller particle has a molecular weight of 2.6 X 104. Efforts to study the subunit composition of the protein moiety of human serum beta lipoprotein have been impeded by the extreme difficulty of obtaining a molecular dispersion of apoprotein following removal of the lipid moiety. While virtually all of the lipid is bound noncovalently to protein, the apoprotein ex- hibits a marked affinity for phosphatide. Quantitative delipidation is achieved only by the use of mixed solvent systems containing one solvent of low polarity and another of intermediate polarity, usually an alcohol or ketone. In contrast to the high-density lipoproteins,I delipidation of native serum beta lipoprotein by such solvent extraction yields a protein product virtually insoluble in neutral aqueous buffer systems. The presence of 0.2 -M sodium dodecyl sulfate during and after extraction by an ethanol-ether mixture yields a protein soluble in aqueous buffers and con- taining only 2-3% phospholipid.2 Studies of sedimentation behavior, how- ever, show this material to be aggregated. Gel permeation chromatography of a similar apoprotein preparation using dodecyl sulfate in the eluting buffer gave a single peak of protein with an elution volume corresponding to a molecular weight of 8.0 X 104 daltons.3 Asymmetry of the peak and the presence of a second, minor protein band on cellulose acetate electrophoresis were noted, how- ever. Shore and Shore applied the technique of sedimentation equilibrium ultracentrifugation to carboxymethylated apobeta lipoprotein in the presence of 8 M\I urea and dodecyl sulfate.4 Protein distribution in the cell at equilibrium indicated homogeneity with respect to particle size. Using a value for the partial specific volume of the protein moiety, in water, calculated from its amino acid composition (0.725), they computed a particle weight of 4.2 X 104 daltons. However, determination of partial specific volume (v) by the method of Casassa and Eisenberg5 yielded a value of 0.82, indicating marked preferential interaction between the apoprotein and water in the multicomponent solvent system. 1075
Transcript of Subunit Heterogeneity Human Serum Lipoprotein* - PNAS · an ethanol-ether mixture yields a protein...

Proceedings of the National Academy of SciencesVol. 66, No. 4, pp. 1075-1082, August 1970
Subunit Heterogeneity in Human SerumBeta Lipoprotein*
J. P. Kane,t E. G. Richards, and R. J. HavelCARDIOVASCULAR RESEARCH INSTITUTE AND DEPARTMENT OF MEDICINE, UNIVERSITY OF CALI-
FORNIA (SAN FRANCISCO); AND INSTITUTE OF MARINE RESOURCES, UNIVERSITY OF
CALIFORNIA, BERKELEY
Communicated by Julius H. Comroe, Jr., May 21, 1970
Abstract. The apoprotein of human serum beta lipoprotein has been preparedin a lipid-free and soluble form by a technique of interfacial extraction in thepresence of guanidinium hydrochloride and nonionic amphiphile. Gel permea-tion chromatography of maleylated apobeta lipoprotein separates two proteinfractions with different amino acid compositions. The smaller particle has amolecular weight of 2.6 X 104.
Efforts to study the subunit composition of the protein moiety of human serumbeta lipoprotein have been impeded by the extreme difficulty of obtaining amolecular dispersion of apoprotein following removal of the lipid moiety. Whilevirtually all of the lipid is bound noncovalently to protein, the apoprotein ex-hibits a marked affinity for phosphatide. Quantitative delipidation is achievedonly by the use of mixed solvent systems containing one solvent of low polarityand another of intermediate polarity, usually an alcohol or ketone. In contrastto the high-density lipoproteins,I delipidation of native serum beta lipoproteinby such solvent extraction yields a protein product virtually insoluble in neutralaqueous buffer systems.The presence of 0.2 -M sodium dodecyl sulfate during and after extraction by
an ethanol-ether mixture yields a protein soluble in aqueous buffers and con-taining only 2-3% phospholipid.2 Studies of sedimentation behavior, how-ever, show this material to be aggregated. Gel permeation chromatography ofa similar apoprotein preparation using dodecyl sulfate in the eluting buffer gavea single peak of protein with an elution volume corresponding to a molecularweight of 8.0 X 104 daltons.3 Asymmetry of the peak and the presence of asecond, minor protein band on cellulose acetate electrophoresis were noted, how-ever. Shore and Shore applied the technique of sedimentation equilibriumultracentrifugation to carboxymethylated apobeta lipoprotein in the presenceof 8 M\I urea and dodecyl sulfate.4 Protein distribution in the cell at equilibriumindicated homogeneity with respect to particle size. Using a value for the partialspecific volume of the protein moiety, in water, calculated from its amino acidcomposition (0.725), they computed a particle weight of 4.2 X 104 daltons.However, determination of partial specific volume (v) by the method of Casassaand Eisenberg5 yielded a value of 0.82, indicating marked preferential interactionbetween the apoprotein and water in the multicomponent solvent system.
1075

BIOCHEMISTRY: KANE, RICHARDS, AND HAVEL PROC. N. A. S.
Calculation of particle weight using the experimentally determined value for vyielded a value of 6.4 X 104 daltons.Scanu et al.6 demonstrated that succinylation of beta lipoprotein followed by
two-solvent extraction of the lipid moiety yields a protein product soluble inaqueous buffer systems without amphiphilic agents. Agarose gel filtration ofthis material indicated the presence of an associating system and sedimentationequilibrium ultracentrifugation revealed a nonlinear relationship of the logarithmof protein concentration to the square of radial distance. Minimum particleweight calculated from the linear portion of the plot from the upper levels of thecell was 3.6-3.8 X 104 daltons. A subsequent sedimentation equilibrium studyin 8 M urea, of beta apoprotein solubilized at high pH after extraction, demon-strated heterogeneity of particle size with a minimum molecular weight of ap-proximately 2.7 X 104 daltons, which was assumed to represent the only mono-meric unit in the protein moiety. This finding provided the basis for formulationof a structural model for beta lipoprotein involving an icosahedral arrange-ment of 20 subunits of identical dimensions around a lipid core.7The presence of subunit heterogeneity in this class of lipoprotein is supported
by the recent finding of two antigenic determinants in a maleylated apoproteinpreparation8 and the separation of two proteins, differing in amino acid composi-tion, by ion exchange chromatography of protein from the Sf 4-8 lipoproteinfraction.9
It is evident that the intense tendency toward aggregation exhibited by theseapoprotein preparations, coupled with the difficulty of determining partialspecific volume accurately in the multicomponent solvent systems necessary fortheir solubilization, has limited the applicability of sedimentation techniques todetermination of the size of monomeric units. The presence of heterogeneoussubunits would be expected to complicate these determinations further, espe-cially if hybrid oligomers exist. A search for heterogeneity of subunits in betalipoprotein must recognize that contamination by other lipoproteins, especiallythe HDL1 fraction, is to be expected within the classical density interval of1.019 < d < 1.063. Hence, a narrower density interval must be chosen forseparation by preparative ultracentrifugation. In this study, human serumbeta lipoprotein, prepared by ultracentrifugation between such narrowed densitylimits and subsequently maleylated, has been delipidized and solubilized by anew technique of two-phase extraction in the presence of guanidine hydrochlorideand nonionic amphiphile. Gel permeation chromatography of the resultingapoprotein in 6 M guanidine hydrochloride permits the separation of two proteinfractions and an estimation of their molecular weights. Distinct differences inthe total amino acid composition of the two fractions indicate that at least twospecies of subunit exist in human serum beta lipoprotein.Materials and Methods. Preparation of beta lipoprotein: Serum from healthy
male Caucasian donors is adjusted to d = 1.024, and layered under an NaCl-KBrsolution of d = 1.024 with EDTA,1 X 10- M, pH 8.6, and centrifuged at 40,000 rpmfor >20 hr in the 40.3 rotor of a Beckman model L ultracentrifuge at 12'C. The tubesare sliced just above the beta lipoprotein band and the infranatant yellow lipoproteinlayer adjusted to a density of 1.055 or 1.050 with a concentrated KBr-NaCl solution,layered under a salt solution of similar density with EDTA as above, and centrifuged
1076

VOL. 66, 1970 BIOCHEMISTRY: KANE, PICIARDS, AND IAVEL
at 40,000 rpm for more than 24 hr. The lipoprotein in the top centimeter of the tube is re-moved, layered under salt solution of the same density, and recentrifuged as above for24-72 hr. The material in the upper centimeter is removed and dialyzed against 1 X10- M EDTA, pH 8.6.
Criteria of purity: Immunodiffusion by the technique of Ouchterlony'0 reveals asingle precipitin band with both goat anti-beta lipoprotein antibody (Hyland Labora-tories) and with rabbit antiwhole human serum antibody. Electrophoresis on paper"and on agarose gel'2 reveals a single narrow band when stained with Lipid Crimson (Ed.Gurr Ltd., London), and agarose gel electrophoretograms run with protein-free buffer,reveal a single band when stained with Amido Schwartz 10-B (National Aniline). Elec-tron microscopy of beta lipoprotein prepared by negative staining with 2% phospho-tungstic acid'3 and by low angle platinum shadowing after osmium fixation'4 revealed ahomogeneous population of spheres, with very few particles compatible in size withhigh-density or very low-density lipoproteins.
Maleylation: Beta lipoprotein was maleylated by the technique of Butler et al. 'using maleic anhydride (Eastman Organic Chemicals) in 100-fold molar excess overlysine residues. Residual maleate was removed by dialysis against 1 X 10-3 M EDTA,pH 8.6. The maleylated beta lipoprotein appeared as a single narrow band of markedlyincreased mobility in agarose gel electrophoresis, whether stained with Lipid Crimson orAmido Schwartz. Appearance in electron photomicrographs was that of unmodifiedbeta lipoprotein.
Samples of purified proteins for calibration of gel columns were maleylated under thesame conditions. Included were bovine serum albumin (Armour Pharmaceutical Co.),ribonuclease, muramidase and ovalbumin (Worthington Biochemical Co.), cytochrome c(Sigma), chymotrypsinogen (Calbiochem), and rabbit muscle myosin prepared by amodification of the method of Richards et al.'6
Delipidation by interfacial extraction: Maleylated beta lipoprotein was concen-trated by dialysis against solid polyethylene glycol 20 M (Union Carbide) to a con-centration of 5 mg/ml. In a 50-ml stoppered centrifuge tube, 1.0 ml of lipoproteinsolution is added to 1.0 ml of 6 M guanidine hydrochloride (Eastman), pH 8.6, andchilled to 0°C in an ice bath. The solution is then made 10- M with respect toEDTAand 3 X 10-2 M with respect to a nonionic amphiphile, polyethyleneoxy(20)sorbitan mono-laurate (Tween 20, J. T. Baker Chemical Co.). Ten volumes of 3: 1 diethyl ether; 95%ethanol (ether from Mallinkrodt Chemical Co.), precooled to 0°C, is added and the tubeis shaken at 0°C for 1 hr, just vigorously enough to maintain an emulsion. The tube isthen centrifuged briefly in a refrigerated centrifuge to split phases, and the supernatant,organic solvent phase is removed by suction. Dissolved ether is carefully removed fromthe guanidine-water phase in vacuo. The volume of the original aqueous phase is re-stored using 3 M guanidinium hydrochloride, pH 8.6, the tube is chilled to 0°C and thesolution made 6 X 10-s M with respect to the amphiphile. A second extraction at 0°Cis performed with 10 volumes of solvent for 4 hr, as previously. After removal of the organicphase and dissolved ether as above, the clear aqueous phase is dialyzed against guanidinesolutions for ultracentrifugation or for gel permeation studies.For evaluation of the extent of lipid removal, portions of the final aqueous phase con-
taining the apolipoprotein were extracted by the technique of Carlson.17 Aliquots of thisextract were used for determination of cholesterol'8 and phospholipid.19 To measurerecovery of protein, portions of the apoprotein-containing aqueous phase were dialyzedagainst distilled water to remove guanidine. The contents of the dialysis tubing werethen removed quantitatively and protein was determined by the technique of Lowryet al.20 For comparison, portions of the lipoprotein solutions were removed before inter-facial extraction. The protein moiety was precipitated by repeated extraction of lipidswith 25 volumes of ethanol: diethyl ether, 3:1. After centrifugation, the solvent wasremoved and the precipitates were dissolved in 0.1 N NaOH for estimation of their pro-tein content.2YGel permeation chromatography: Two gels were employed, Sephadex G-150 and
1077

BIOCHEMISTRY: KANE, RICHARDS, AND HAVEL PROC. N. A. S.
Agarose (Sepharose 4B) (both from Pharmacia Fine Chemicals), in columns 2.5 by55 cm. The solvent employed for preparation and operation of the columns was 6 Mguanidine hydrochloride (Mann Research Laboratories) with 2 X 10-' M Tween 20,and 10-' M EDTA at pH 8.6. Just before each use, the column was washed with thissolvent containing 1 X 10-2 M 2-mercaptoethanol (Eastman) delivered from a vesselwith a nitrogen atmosphere. Solid sucrose was dissolved in the samples, which were thenapplied by layering under the solvent at the top of the columns. The Sephadex G-150column was run at 0.5 ml/min and the agarose column at 0.12 ml/min at 250C. Calibra-tion curves were determined for both columns2' using assorted purified proteins bothunmodified and maleylated, to determine the effect of side chain modification upon gelpermeation behavior. Protein content of serial fractions was determined by absorbanceat 280 nm for all proteins and by absorbance at 450 nm for cytochrome c. Distributionof protein was verified by the Lowry technique.20 Calculations of molecular weights arebased on the mean of the elution volumes of three separate apolipoprotein preparationsfrom different donor pools, each representing beta lipoprotein from three individuals.Amino acid analysis: Samples for amino acid analysis on fractions from gel
permeation chromatography were taken only from narrow central portions of thetwo peaks. After removal of guanidine from these fractions by dialysis against dis-tilled water, they were lyophilized. The lyophilisate was extracted with 3:1 absoluteethanol: diethyl ether to remove any residual lipids and amphiphile and then hydrolyzed,after careful deaeration, in 6 N HCI at 110'C for periods varying up to 24 hr. Apoproteinfrom samples of native beta lipoprotein prepared by repetitive ultracentrifugation be-tween d = 1.024 and d = 1.055 was delipidated by repeated extractions with 3:1 ethanol:diethyl ether and the dried protein residue was hydrolyzed as above. After hydrolysis,the hydrolysates were lyophilized and redissolved in citrate buffer pH 2.2 for amino acidanalysis, which was performed on a Beckman model 120B amino acid analyzer22 induplicate. Recoveries of serine, threonine, and methionine after various periods ofhydrolysis were plotted and extrapolation was made to zero time to correct for lossesduring hydrolysis. No correction was necessary for tyrosine. Cysteine content of frac-tions obtained by gel permeation and of protein derived from native beta lipoprotein wasestimated by determination of cysteic acid after performic acid oxidation.23 The aver-age difference of duplicate determinations for all amino acids was 2.5%. Tryptophancontent of the maleylated derivatives could not be estimated by ultraviolet spectroscopybecause of absorbancy of the maleyl functions.
Results. The interfacial extraction technique applied to either native ormaleylated beta lipoprotein produces a clear solution of apoprotein containingguanidine hydrochloride and amphiphile. Recovery of protein is quantitative.Residual sterol is less than 0.05% and residual phospholipid is less than 0.2%of the original content. Apoprotein prepared from both maleylated and un-modified beta lipoprotein remains soluble when guanidine is removed by dialysisagainst 1 X 10-2 M EDTA but marked broadening of the schlieren peak andincreased sedimentation velocity upon ultracentrifugation indicate that extensiveaggregation occurs as the guanidine is removed. Immunodiffusion studies usingantibody to native beta lipoprotein show complete loss of recognition of apopro-tein prepared from native or maleylated beta lipoprotein.
Gel permeation chromatography on Sephadex G-150 of maleylated apobetalipoprotein prepared by the technique of interfacial extraction resulted in theelution of two protein peaks (Fig. 1). The first (fraction I) emerged at the voidvolume of the column as determined with a high molecular weight dextranmarker. The mean of the elution volumes of the second peaks (fraction II)from three separate preparations corresponded to a random coil protein having amolecular weight of 2.6 X 104 daltons (+ 0.18 SEM). Figure 2 is a calibration
1078
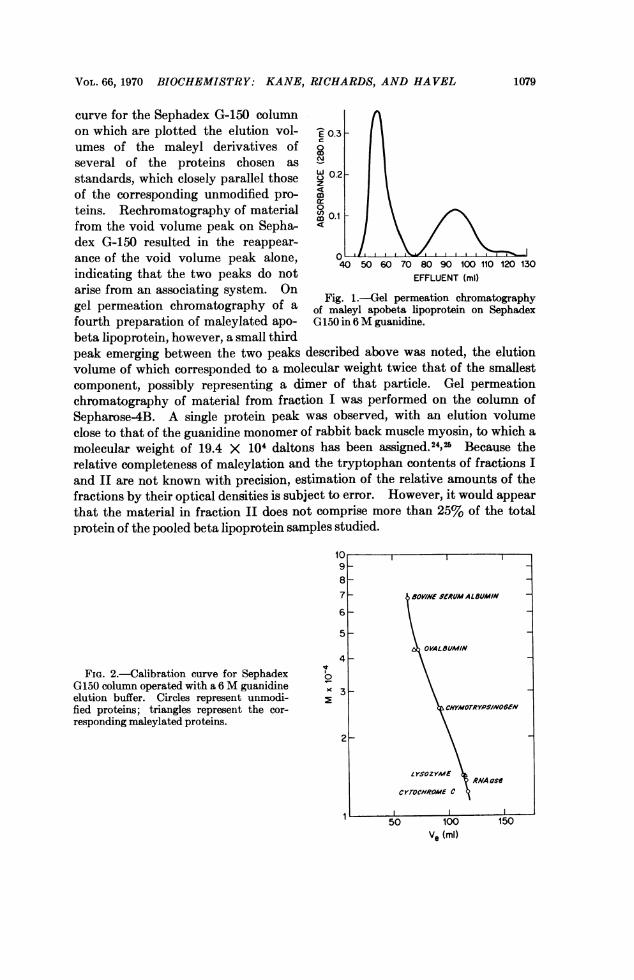
VOL. 66, 1970 BIOCHEMISTRY: KANE, RICHARDS, AND HAVEL
curve for the Sephadex G-150 columnon which are plotted the elution vol- C- 0.3umes of the maleyl derivatives of lseveral of the proteins chosen as Ncostandards, which closely parallel those w 0.2of the corresponding unmodified pro-
crteins. Rechromatography of material 00 0.1from the void volume peak on Sepha- I \dex G-150 resulted in the reappear-ance of the void volume peak alone, C 0 70 8 9 110 120 130indicating that the two peaks do not EFFLUENT (ml)arise from an associating system. Ongelermetiochrmatorapy ofa~ Fig. 1.-el permeation chromatographygel permeation chromatography of a of nmaleyl apobeta lipoprotein on Sephadex
fourth preparation of maleylated apo- G150 in 6 M guanidine.beta lipoprotein, however, a small thirdpeak emerging between the two peaks described above was noted, the elutionvolume of which corresponded to a molecular weight twice that of the smallestcomponent, possibly representing a dimer of that particle. Gel permeationchromatography of material from fraction I was performed on the column ofSepharose4B. A single protein peak was observed, with an elution volumeclose to that of the guanidine monomer of rabbit back muscle myosin, to which amolecular weight of 19.4 X 104 daltons has been assigned.24,25 Because therelative completeness of maleylation and the tryptophan contents of fractions Iand II are not known with precision, estimation of the relative amounts of thefractions by their optical densities is subject to error. However, it would appearthat the material in fraction II does not comprise more than 25% of the totalprotein of the pooled beta lipoprotein samples studied.
1098-
7 - 8 VNE SERUM ALBUMIN
6
5-VAOALBUM N
4-
FIG. 2.-Calibration curve for Sephadex gG150 column operated with a 6 M guanidine , 3-elution buffer. Circles represent unmodi-fied proteins; triangles represent the cor- CrNOTRYPS/NWOENresponding maleylated proteins.
2~
50 100Ve (ml)
150
L YSOZYME
CrTOCHROME CRN/A OSe
1079

BIOCHEMISTRY: KANE, RICHARDS, AND HAVEL PROC. N. A. S.
The amino acid compositions of the two apoprotein fractions separated bygel permeation chromatography of maleylated apobeta lipoprotein on SephadexG-150 are compared in Table 1. A subsequent set of analyses of neutral and
TABLE 1. Amino acid composition of fractions I, Il*Fraction I Fraction II
Lys 68.8 72.0His 22.2 19.5Arg 35.6 39.0Asp 104.9 82.2Thr 73.0 67.2Ser 86.7 104.1Glu 116.7 137.6Pro 42.7 37.8Gly 45.0 103.6Ala 56.8 74.71/2 Cys 12.4 29.4Val 53.8 48.4Met 14.9 0.9Ile 60.2 37.1Leu 121.3 89.9Tyr 32.2 30.1Phe 53.5 26.3
* Mol/103 mol.
acidic amino acids from the two fractions prepared from a different beta lipopro-tein pool was in excellent agreement with the molar ratios of those amino acids inTable 1. The glycine content of fraction II exceeds that of fraction I by a factorof more than 2 while the phenylalanine content of fraction I is more than doublethat of the smaller particle. Fraction I is significantly richer in isoleucine andleucine as well. It is possible that the principal peptide species represented infraction II is devoid of methionine, as the amount of that amino acid detected inthat fraction could have arisen from contamination to the extent of only 6%7with a protein as rich in methionine as fraction I. Differences between the totalamino acid composition of the native beta lipoprotein prepared by ultracentrif-ugation between the density limits of 1.024 and 1.055 and that of the 1.019 <d < 1.063 fraction were found to be small. The principal differences noted werea relative deficiency of glycine of 10% and glutamic acid of 7% in the materialprepared within the narrower interval.
Discussion. The distinguishing feature of the technique of interfacial ex-traction introduced here is the removal of lipid while the lipoprotein is exposedto a denaturing aqueous solvent system. The absence of either guanidine oramphiphile from the system results in insolubility of apoprotein. While non-ionic detergents, in contrast to the alkyl sulfates, are incapable of solubilizingbeta apoprotein previously delipidated by solvent extraction,2 several such agentseffect apoprotein solubilization in the interfacial extraction procedure, where thepresence of guanidine interdicts the use of anionic amphiphiles. Presumablysolubilization is effected by interaction of guanidine and amphiphile with themore hydrophobic areas of the protein which become exposed as removal of lipidprogresses, thereby preventing protein-protein interaction of an irreversible
1080

VOL. 66, 1970 BIOCHEMISTRY: KANE, RICHARDS, AND HAVEL
nature. The guanidine-amphiphile solution used in the interfacial extractionsystem does not solubilize lipoprotein previously delipidated by solvent extrac-tion.
Modification of the apolipoprotein by maleylation permits the separation ofthe smaller component, fraction II, by gel filtration. The finding that similarmodification of several proteins selected for calibration of gel columns had littleeffect on their elution volumes is in keeping with the evidence that the effectivemolecular radius is relatively independent of amino acid composition of poly-peptides in the random coil conformation.2" The assumption may be made thatlittle error is introduced by such modification in determination of molecularweight by gel permeation chromatography in 6 M guanidine solutions. It maythus be concluded that human serum beta lipoprotein contains a protein subunitwhich has a molecular weight at least as small as 2.6 X 104 daltons. The ap-parent absence of a particle of this size in sedimentation equilibrium studiesby Shore and Shore4 of apoprotein from Sf 4-8 lipoprotein may be attributableto the rotor speeds employed. It may be calculated that a 26,000 molecularweight subunit representing up to 25% of the protein in solution would not haveproduced sufficient fringe displacement at the rotational velocity employed to berecognized as a separate component.The substantial differences in amino acid composition between fractions I and
II indicate that at least two species of protein subunit must be present in the betalipoprotein of human serum. The fact that the composition of fraction I re-sembles that of whole beta lipoprotein much more closely than does that offraction II suggests that fraction I represents the predominant peptide or pep-tides of beta lipoprotein and that the protein of fraction II cannot be representedto any appreciable extent in some hybrid oligomeric form in fraction I. Thelarge particle size of fraction I, demonstrable on agarose gel permeation chro-matography, suggests that it represents some form of oligomeric protein.
Since several peptide species have recently been identified in both the high-density and very low-density classes of human serum lipoproteins, the possibilitythat the protein of fraction II may be identical with one of these entities mustbe considered. Of the peptides so far identified in the high-density lipopro-tein,26 all fractions have amino acid compositions which are distinctly differentfrom that of the protein in fraction II and, specifically, both the dominant specieswith C-terminal glutamine and threonine exhibit categorical differences, as theR-glutamine peptide contains no histidine or arginine and the R-threoninepeptide, no isoleucine. The amino acid composition of a hybrid of these twosubunits, which has been identified in delipidated preparations of high-densitylipoprotein,26 is also markedly dissimilar from fraction II protein. Additionally,neither threonine nor glutamine can be demonstrated as carboxyl terminal aminoacids in beta lipoprotein, by application of methods which readily indicate theirpresence in high-density lipoprotein.9 Similarly, all of the soluble proteinsisolated to date from very low-density lipoprotein of human serum show distinctdifferences in amino acid composition from this protein.9 27 Since both of theprotein fractions which can be eluted from DEAE-cellulose after application ofapobeta lipoprotein9 contain substantially greater amounts of methionine than
1081

BIOCHEMISTRY: KANE, RICHARDS, AND HAVEL PROC. N. A. S.
fraction II protein, it is likely that this fraction represents a heretofore un-recognized protein constituent of human serum beta lipoprotein.
The authors wish to thank Dr. R. D. Cole, Professor of Biochemistry, University of Cali-fornia, Berkeley, for valuable suggestions and for the assistance of his laboratory in perform-ing amino acid analyses.
* This work was conducted during Dr. Kane's tenure as an advanced postdoctoral fellow ofthe American Heart Association and a research and development scholar of the AmericanDiabetes Association and was supported by USPHS grants HE-06285 and GM 09899.
t Requests for reprints may be addressed to Dr. J. P. Kane, Cardiovascular Research Insti-tute and Department of Medicine, University of California, San Francisco, Calif. 94122.
1 Scanu, A., L. A. Lewis, and F. M. Bumpus, Arch. Biochem. Biophys., 74, 390 (1958).2 Granda, J. L., and A. Scanu, Biochemistry, 5, 3301 (1966).8 Day, C. E., and R. S. Levy, J. Lipid Res., 9, 789 (1968).4Shore, B., and V. Shore, Biochem. Biophys. Res. Commun., 28, 1003 (1967).5 Casassa, E. F., and H. Eisenberg, Advan. Protein Chem., 19, 287 (1964).6 Scanu, A., H. Pollard, and W. Reader, J. Lipid Res., 9, 342 (1968).7Pollard, H., A. M. Scanu, and E. W. Taylor, these PROCEEDINGS, 64, 304 (1969).8 Simons, K., and A. Helenius, Ann. Med. Exp. Biol. Fenn., 47, 48 (1969).' Shore, B., and V. Shore, Biochemistry, 8, 4510 (1969).
10 Ouchterlony, O., in Immunological Methods (Oxford: Blackwell, 1964), pp. 55-78.11Lees, R. S., and F. T. Hatch, J. Lab. Clin. Med., 61, 518 (1963).12 Noble, R. P., J. Lipid Res., 9, 693 (1968).13 Hamilton, R. L., D. M. Regen, M. E. Gray, and V. S. LeQuire, Lab. Invest., 16, 305 (1967).14 Jones, A. L., and J. M. Price, J. Histochem. Cytochem., 16, 366 (1968).15 Butler, P. J. G., J. J. Morris, B. S. Hartley, and R. Leberman, Biochem. J., 103, 78P
(1967).16 Richards, E. G., C-S. Chung, D. E. Menzel, and H. S. Olcott, Biochemistry, 6, 528 (1967).17 Carlson, L. A., J. Atheroscler. Res., 3, 334 (1963).18 Sperry, W. M., and M. Webb, J. Biol. Chem., 187, 97 (1950).19 Stewart, C. P., and E. B. Hendry, Biochem. J., 29, 1683 (1935).20 Lowry, 0. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall, J. Biol. Chem., 193, 265
(1951).21 Fish, N. H., K. G. Mann, and C. Tanford, J. Biol. Chem., 244, 4989 (1969).22 Spackman, D. H., N. H. Stein, and S. Moore, Anal. Chem., 30, 1190 (1958).28 Hirs, C. H. W., in Methods in Enzymology (New York: Academic Press, 1967), vol. 11,
p. 59.24 Gazith, J., S. Himmelfarb, and W. F. Harrington, J. Biol. Chem., 245, 15 (1970).25 Richards, E. G., unpublished data.26 Shore, V., and B. Shore, Biochemistry, 7, 3396 (1968).27 Brown, W. V., R. I. Levy, and D. S. Fredrickson, J. Biol. Chem., 244, 5687 (1969).
1082


















