
STUDIES ON SYNTHESIS OF -HYDROXY …shodhganga.inflibnet.ac.in/bitstream/10603/3442/10/10...STUDIES...
55
47 CHAPTER 2 (SECTION-A) STUDIES ON SYNTHESIS OF -HYDROXY CARBOXYLIC ACIDS 2.1 INTRODUCTION: The three most important classes of compounds in the pool of chiral building blocks [1] are carbohydrates, amino acids and hydroxy carboxylic acids. In contrast to the carbohydrates, many representatives of the other two groups are now available in both the enantiomeric forms, such as lactic acid [2, 3], mandelic acid, maleic acid [4], tartaric acid [5], and phenyl lactic acid [6]. The variety is further increased by the fact that some of the optically active amino acids can be converted to - hydroxycarboxylic acids with retention of configuration [7]. Thus, it is not surprising that compounds of this type are used frequently as starting materials for the synthesis of other enantiomerically pure products. Some of the -hydroxycarboxylic acids commonly encountered in natural products and synthetic work are shown in Table-2.1. Table 2.1 Some -hydroxycarboxylic acids S. No Name of the compound Structure 1 2-Hydroxyethanoic acid (Glycollic acid) H OH O OH H
Transcript of STUDIES ON SYNTHESIS OF -HYDROXY …shodhganga.inflibnet.ac.in/bitstream/10603/3442/10/10...STUDIES...

47
CHAPTER 2 (SECTION-A)
STUDIES ON SYNTHESIS OF -HYDROXY CARBOXYLIC ACIDS
2.1 INTRODUCTION:
The three most important classes of compounds in the pool of
chiral building blocks [1] are carbohydrates, amino acids and hydroxy
carboxylic acids. In contrast to the carbohydrates, many representatives
of the other two groups are now available in both the enantiomeric forms,
such as lactic acid [2, 3], mandelic acid, maleic acid [4], tartaric acid [5],
and phenyl lactic acid [6]. The variety is further increased by the fact that
some of the optically active amino acids can be converted to -
hydroxycarboxylic acids with retention of configuration [7]. Thus, it is not
surprising that compounds of this type are used frequently as starting
materials for the synthesis of other enantiomerically pure products.
Some of the -hydroxycarboxylic acids commonly encountered in natural
products and synthetic work are shown in Table-2.1.
Table 2.1 Some -hydroxycarboxylic acids
S. No Name of the compound Structure
1 2-Hydroxyethanoic acid
(Glycollic acid) H
OH
O
OHH

48
2 2-hydroxypropanoic acid
(Lactic acid) H3C
OH
O
OH
3 2-Methyl-2-hdroxypropanoic acid
(Methyl lactic acid) H3C
OH
O
OHH3C
4 2-Hydroxybutanoic acid H3C OH
O
OH
5 2-Hydroxypentanoic acid, OH
O
OH
H3C
6 2-Hydroxyhexanoic acid OH
O
H3C
OH
7 Maleic acid
COOH
COOH
HO
The synthesis of enantiomerically pure -hydroxy carboxylic acids
is of general interest and applicability to asymmetric organic synthesis as
it provides a direct route to the preparation of a large variety of
heterocyclic systems, as well as natural and pharmacologically
interesting products. In this regard, the preparation of -hydroxy
carboxylic acids is particularly important. Several different methods for
the synthesis of -hydroxycarboxylic acids have been reported. Some of
them are described below:

49
2.2 LITERATURE REPORTED METHODS:
Hulin et al., [8] have reported a racemic approach (Scheme-2.1) for
the synthesis of the title compound, where in trimethylsilyl cyanide was
added to phenyl acetaldehyde followed by acid hydrolysis of the silylated
cyanohydrin in alcoholic medium to afford racemic -hydroxy phenyl
propanoic ester, which was hydrolyzed to acid. The major disadvantage
of this method being the handling of raw materials, they are quite
unstable and difficult to store and use.
CO2R
OH
CHO CN
OSi(CH3)3
(CH3)3SiCNROH/HClHydrolysis
1 2 3
………Scheme-2.1
The approach adopted by Gerhard et al., (9) (Scheme-2.2) involves
substituted benzaldehyde, which is more stable, compared to aryl
acetaldehydes used in Hulin et al., method. The strategy involved in this
method is the condensation of aldehyde moiety with hydantoin to give
the corresponding hydantoin derivative. The later on hydrolysis under
basic conditions results in the formation of -ketoacid. Reduction of the
-ketoacid with sodium borohydride in alcoholic medium affords the
required racemic product in moderate yield. The disadvantages are being
the solubility of the intermediates and the instability of -keto acid.

50
CHO
HOHO
CO2H
O
HO
CO2H
OH
HO NH
HN
O
O
NH
HN
O
O
Piperidine Aq OH-
NaBH4 / IPA
45 6
7
………Scheme-2.2
The azalactone method developed by Herbest et al., (10) (Scheme-
2.3) for the synthesis racemic -hydroxycarboxylic acids is quite
interesting since the raw materials are quite easily available and
reactions are quite feasible. The major disadvantage is being the use of
mercury, for the reduction, which is quite poisonous and causes serious
effluent problem. The synthetic method consists of condensation of an
aromatic aldehyde with N-acetylglycine to furnish the corresponding
azalactones, which on further reductive hydrolysis using zinc amalgam &
conc. hydrochloric acid afforded the required racemic -hydroxy
carboxylic acid in moderate yield.
CHOCO2H
OHNO
O
CH2CO2H
NHCOCH3
AC2O/CH3CO2Na Zn-Hg/HCl
8 9 3
………Scheme-2.3

51
The method developed by Aston et al., (11) (Scheme-2.4), the title
compound was prepared by the oxidation of the methyl group and
subsequent reduction of the keto group of the acetophenone. The
oxidation of the methyl group was achieved by the halogenation of
acetophenone in acidic medium followed by base hydrolysis and
acidification to afford the -ketoacid, which was reduced to alcohol. The
major disadvantage is being the difficulty in controlling the halogenation
and the separation of the by- products. The yields are moderate and such
reactions are difficult to handle on the plant scale.
O O
Cl2 CO2H
OHNaOHHCl
2Cl2
10 11 12
………Scheme-2.4
The synthetic strategy adopted by Green et al., (12) (Scheme-2.5)
consists in the introduction of acetyl and the benzyl groups on to the
active methylene group of acetoacetic ester by acetyl chloride and the
benzyl bromide respectively in the presence of a strong base. The
resulting disubstituted acetoacetic ester was subjected to base hydrolysis
to obtain the required product in moderate yield. The drawbacks of this
method being the use of costly and sensitive reagents like LTA. The other
disadvantage is that the method has poor atom conservation and hence
it is not eco friendly.

52
H3COCO CO2C2H5
OH3COCO CO2C2H5
O
CH3COCH2COOC2H5
CO2H
OH
LTA
Benzene
350 C
NaHBnBrDioxane
NaethanolReflux
13 14 15
16
………Scheme-2.5
Kenji Koga et al., (13) have introduced a new method (Scheme-2.6)
for preparing chiral -hydroxycarboxylic acids by Chiron approach. In
this method the synthesis of required -hydroxy acid was achieved via
diazotizing the appropriate - amino acid with sodium nitrite and
mineral acid. The major disadvantage of the method is partial
racemisation.
CO2H
YOH
Y
HO
CO2H
YNH2
NaNO2
1NH2SO4
COOH
YOH
COOH
Y Y
COOH
/H2O+ +
+
16 17 18 19
20 21
………Scheme-2.6
Milton et al., (14) (Scheme-2.7) have modified the Kenji Koga
approach by introducing acetic acid in place of sulfuric acid. The author

53
has utilized acetic acid as one of the medium in the diazotization process
instead of aqueous sulfuric acid in the previous approach to obtain the
required product. The specific rotation data did not match with the
reported literature data, which is one of the drawbacks of the process.
CO2H
1N HCl
CH3CO2H
NaNO2CO2H
NH2 OH
22 23
………Scheme-2.7
Kenji Mori (15) (Scheme-2.8) approach is much more improvised in
obtaining relatively pure chiral form in spite of some racemisation. The
method is similar to the one demonstrated by Kenji Koga. The product
was recrystalized three times to attain the reported chiral purity. This
approach is similar to the earlier approach mentioned by Kenji Koga et
al., the difference being found in the workup procedure. The product
after three recrystallizations was found to be very close to the reported
value in terms of its optical rotation.
COOH COOH
NH2 OH
HNO2
24 25
………Scheme-2.8

54
Palomo et al., [16] used (Scheme 2.9) the different reaction
conditions were employed where in acid and nitrite solutions were added
simultaneously to the aqueous -amino acid solution to perform
diazotization for Valine. The product was isolated in 57% yield. The major
disadvantage of this method being the three recrystallizations required
for getting the pure product.
H2SO4/2N NaNO2
H2O,O0C-rtR COOH
NH2
R COOH
OHR= (26) CHMe2 (27)
(24) CH2CHMe2 (25)
(28) CH2Ph (3)
………Scheme- 2.9
The method developed by Junji Inanaga et al., (17) (Scheme-2.10)
is based on the regioselective ring opening of the,-epoxy esters. This
method consists of regioselective ring opening followed by methyl iodide
and subsequent reduction of the resulting iodohydrins to afford racemic
-hydroxycarboxylic acid. The disadvantage is being the possibility of the
by products and costly raw materials.
CO2H
OH
CO2H
OHY:77%
oCO2H
IMgI2 Bu3SnH
28 29 23
………Scheme-2.10

55
2.3. PRESENT WORK:
The literature review on the various methods of synthesis of -
hydroxycarboxylic acids revealed that the synthesis of this moiety was
achieved by using a racemic synthetic method followed by resolution to
its desired enantiomer. This method is traditional and no novelty is
involved. The other method followed is Chiron approach starting from
amino acids. This method is quite good in the sense that a single isomer
is expected to be the desired product. The mechanism involved in this
method is illustrated in the scheme-2.11.
Y
CH2
CH2N H
CO2H
Y
CH2
CH
OC
O
Y
CH2
CX H
CO2H
Y C
CH2X(30) (31)
(32)
(33)
CO2H
H
………Scheme-2.11
The amine group is subjected to diazotization and subsequently
eliminated. During the process,
the free hydroxy group of the carboxylic acid extends the anchimeric
assistance leading to the formation of oxirane derivative, which leads to
the inversion of the configuration. Subsequently hydroxyl group attacks
the ring from the rear side in SN2 type leading to the inversion of the

56
configuration for a second time. The two inversions amount to the
retention of the configuration. Hence the configuration of the amino acid
is retained in the -hydroxy carboxylic acid.
2.4. RESULTS AND DISCUSSION:
The reactions of nitrous acid on optically active -amino acids
having hydrogen and having an asymmetric -carbon atom are reported
to give exclusively -hydroxycarboxylic acids with retention of
configuration, due to the participation of the neighbouring carboxylate
group. From literature precedent it was known that the reaction is highly
dependent on the solvent.
In the present modified process, water miscible solvent namely
acetone was used along with water as a medium for diazotization
reaction and after completion of the reaction the pH of the solution was
increased by addition of conc. HCl followed by extraction. With these
modifications, the yields dramatically improved from 30% to 75%
subsequently this methodology was applied on different amino acid
substrates resulting in consistently higher yields.
The methods followed by various other authors have some
disadvantages like racemisation of the product, low yield, tedious work
ups and purification of the product by several recrystallisations. In view

57
of the present situation, a simple and efficient method has been
developed. The major advantages of this method are:
(i) The racemisation is arrested to a minimal level,
(ii) The work up has been simplified and
(iii) Yields and the purities have been improved.
This method has been utilized for conducting experiments on both
aromatic and aliphatic amino acids. The detailed experimental procedure
and the spectral data are given in the experimental section.
2.4.1. GENERAL PROCEDURE:
The reactions were carried out as follows: To a solution of optically
pure L-amino acids in 6% H2SO4 and acetone (1:1 ratio), were added 3
molar equivalents of sodium nitrite in water solution at 00c for 15 min.
After complete addition, the reaction was maintained at same temp for
additional 120 min and the whole was allowed to stand at room
temperature overnight. Reaction mass was poured in water (25ml) and
hydrochloric acid (5ml) was added. The product was extracted into ethyl
acetate (3x25ml), combined organic layers were washed with water
(1x25ml) and concentrated to a syrupy residue. Optical rotation of the
work up compound was checked and matched with reported values.

58
ROH
NH2
O
R'OH
OH
O
34 (a-f) 35 (a-f)
………Eqn-2.1
Reaction conditions: H2SO4/NaNO2/ H2O/Acetone/ (-) 50C-RT
2.5. EXPERIMENTAL SECTION
Preparation of 35 a-f: (General procedure)
A mixture of optically pure L-amino acids 34 a-f (1.0 g) in acetone
(5.0 ml) and dilute sulfuric acid (6%, w/v 5.0ml) was placed in a three-
necked round bottomed flask, equipped with a magnetic stirrer and
cooled to (-) 50C.To this was added a solution of sodium nitrite (2.32 g,
0.033 mole) in water (5.0 ml) slowly below 00c for 15min. After complete
addition, the reaction was maintained at same temp for additional 120
min and brought to room temperature while stirring overnight.
Reaction mass was poured in water (25 ml) and hydrochloric acid
(5 ml) was added. The product was extracted into ethyl acetate (3X25 ml)
combined organic layers were washed with water (1X25 ml) and
concentrated to a syrupy residue. This was purified by column
chromatography, eluting the product with mixtures of pet-ether and ethyl
acetate to yield pale yellow syrup.

59
Representative Spectral data
35a: IR (Neat): 3448 (OH, broad), 2923 (m), 1731 (s), cm-1; 1HNMR
(DMSO-d6) δ 3.0-3.2 (dd, 2H, CH2), 4.5 (dd, 1H, CH), 5.0 (OH), 7.3 (m,
5H, Ar-H). Mass (CI method): (M++1) 167.
35b: IR (Neat): 3449 (s), 2928 (br), 1723 (s) cm-1; 1HNMR (in DMSO) δ 3.5
(brs, 1H, OH), 5.0 (s, 1H, CH), δ 7.4 (m, 5H, Ar-H). Mass (CI method):
(M++1) 153.
35c: IR (Neat): 3448 (OH), 2923 (m), 1731 (s), cm-1; 1HNMR (in CDCl3) δ
0.95 (d, 3H, CH3), 1.1 (d, 3H, CH3), 2.1 (M, 1H, CH), 4.15 (d, 1H, CH), 5.9
(OH). Mass (CI method): (M++1) 119.
35d: IR (CHCl3):3414 (br), 3018 (w), 2967 (s), 2928 (w), 2875 (w), 1719
(br) cm-1; 1HNMR (in CDCl3) δ 0.9 (t, 3H, CH3), 1.1 (d, 3H, CH3), 1.4 (m,
2H, CH2), 1.9 (m, 1H, CH), 4.1 (d, 1H, CH) 4.6 (OH). Mass (CI method):
(M++1) 133.
35e: IR (Neat): 3448 (OH), 2923 (m), 1731 (s), cm-1; 1HNMR (in CDCl3) δ
1.0 (d, 6H, 2XCH3), 1.66 (q, 2H, CH2), 2.0 (m, 1H, CH), 4.9 (s, 2H, ) 5.2
(dd, 2H). Mass (CI method): (M++1) 133.
35f: IR (Neat): 3398 (OH, br), 2007 (br), 1730 (br) cm-1; 1HNMR: (in
DMSO) δ1.1 (d, 3H, CH3), 3.5 (s, OH), 4.0 (q, 1H, CH). Mass (CI method):
(M++1) 91.

60
CHAPTER 2 (SECTION-B)
SYNTHESIS OF 3-ARYL-2-QUINAZOLINYL-PHENYLACETIC ACID
ESTER DERIVATIVES
2.6. INTRODUCTION TO QUINAZOLINONES:
Quinazolinones (fig-1) are a building block for several naturally
occurring compounds isolated till date from different families of the plant
kingdom, animals and microorganisms. The primary quinazolinone was
synthesized [18] in the late 1860s from anthranilic acid and cyanogens to
give 2-cyanoquinazolinone. Awareness in the medicinal chemistry of
quinazolinone derivatives was inspired in the early 1950s with the
clarification of a quinazolinones alkaloid named as Febrifugine from an
Asian plant Dichroa febrifuga, which is an element of a conventional
Chinese herbal solution useful against malaria.
N
N
O
R1
R2
Figure-1. Quinazolinone basic structure
In view of the significance of the above quinazolinone derivatives
are possible for several activities, it has been considered of interest to
intend and synthesize new quinazolinone moieties for linking them to
derivatives of α-hydroxy carboxylic acids through piperazine moiety.

61
2.7. LITERATURE REPORTED METHODS
Bhatta et al., [19] has introduced a new method for preparing
substituted quinazol-4(3H)-ones by condensation in presence of pyridine
& cyclization in presence of potassium carbonate. In this method the
synthesis from anthranilic acid with chloroacetyl chloride in pyridine
followed by cyclization with appropriate amine in presence of potassium
carbonate at reflux. The disadvantage of this method is usage of highly
carcinogenic solvent like benzene and lesser yields (Scheme 2.12)
NH2
R1
R2
CO2H
NH
R1
R2
CO2H
COCH2Cl
N
N
Cl
O
RR2
R1
RNH2/K2CO3
ethanolClCOCH2Cl
Pyridine/reflux
36 37 38
………Scheme 2.12
Pandey et al., [20] utilized the substituted anthranilic acids to
condense with benzoyl chloride in pyridine giving benzoxazinone addition
product, which on reaction with a substituted amine in pyridine resulted
in the substituted quinazolines. The disadvantage of this method is use
of a large excess of pyridine and removal of pyridine as solvent by
addition of excess of water leading to the loss of yields (Scheme 2.12)
NH2
R1
CO2H
N
O
O
R1
Ph N
N
O
R1
R
Ph
PHCOClRNH2
pyridine
Pyridine/reflux
39 40 41 ………Scheme 2.13

62
Ichizo Inoue et al., [21] have attempted the alternate reaction of
anthranilic acid with thionylchloride in boiling benzene, followed by
treatment with the amines afforded the anthranilamines in moderate
yields and cyclization of anthranilamines with excess chloroacetyl
chloride in acetic acid at 110°C gives substituted quinazolines. The
disadvantage of this method is using highly carcinogenic benzene and
fewer yields (Scheme 2.14)
NH2
R2
CO2H
R1
NH2
NH
O
R2
R
R1
N
N
O
R2
R
Cl
R1
42 43 44
SOCl2
R-NH2
ClCOCH2Cl
………Scheme 2.14
Substituted quinazolinones Ahmad et al., [22] have been
synthesized in high to excellent yields through the one-pot condensation
of anthranilic acid, trimethyl othoformate and primary amines in the
presence of 5 mol% of Bismuth (III) trifluoroacetate immobilized on n-
butyl pyridinium tetrachloro ferrate ionic liquid at room temperature.
The major disadvantage in this approach is using of costly reagents like
bismuth (III) and n-butyl pyridinium tetrachloro ferrate (Scheme 2.15)
NH2
CO2H
N
N
O
R
45 46
R-NH2+ + HC(OR)3
Bi(TFA)3-[nbp]FeCl4
………Scheme 2.15
63
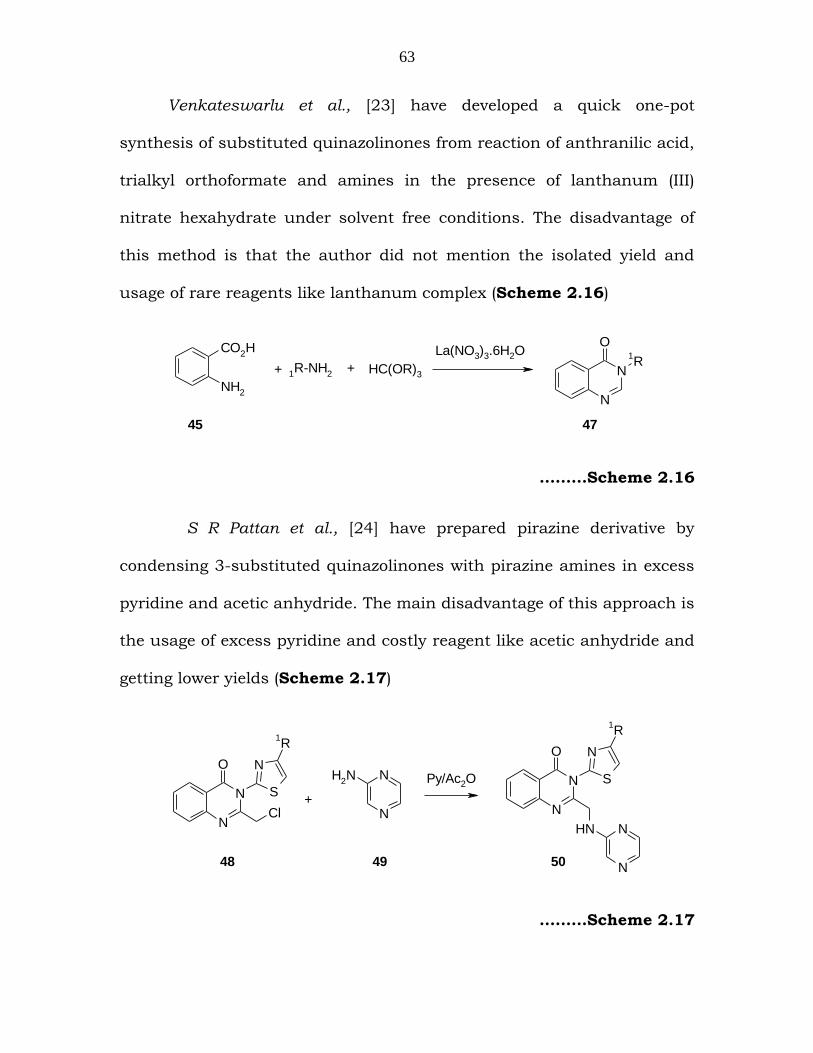
Venkateswarlu et al., [23] have developed a quick one-pot
synthesis of substituted quinazolinones from reaction of anthranilic acid,
trialkyl orthoformate and amines in the presence of lanthanum (III)
nitrate hexahydrate under solvent free conditions. The disadvantage of
this method is that the author did not mention the isolated yield and
usage of rare reagents like lanthanum complex (Scheme 2.16)
NH2
CO2H
N
N
O
R1
1R-NH
2
45 47
+ + HC(OR)3
La(NO3)3.6H2O
………Scheme 2.16
S R Pattan et al., [24] have prepared pirazine derivative by
condensing 3-substituted quinazolinones with pirazine amines in excess
pyridine and acetic anhydride. The main disadvantage of this approach is
the usage of excess pyridine and costly reagent like acetic anhydride and
getting lower yields (Scheme 2.17)
N
N
O
Cl
S
N
R1
N
NNH2
N
N
O
S
N
R1
NH
N
N
+
Py/Ac2O
48 49 50
………Scheme 2.17

64
Shanthan Rao et al., [25] have developed one-pot synthesis of
substituted quinazolinones has been carried out by the three-component
coupling of anthranilic acid and amines in presence of Nafion-H (a
perfluorinated resin-supported sulfonic acid) as a heterogeneous catalyst
under solvent free microwave irradiation condition. The major
disadvantage of this method is that the reactions cannot be handled on
larger scales (>50-100 g) & industrially not feasible method (Scheme
2.18)
NH2
OH
O
NH2
N
N
O
+
Nafion-HMW
45 51 52
………Scheme 2.18
Kurosh Rad et al., [26] have developed a convenient method for the
synthesis of 3-substituted quinazolin-4(3H)-ones using the convergent
reactions of formic acid a primary amine and isotoic anhydride under
solvent free conditions and with microwave irradiation (Scheme 2.19)
NH2
NH
O
R
N
N
O
R
53 54
HCO2H,MW
………Scheme 2.19

65
Besson et al., [27] have reported 3H-quinazolin- 4-one by using
microwave irradiation and improved the yields and reduced the reaction
time (Scheme 2.20)
NH2
OH
O
N
NH
O
N
OH
O
NH2
+ H2N-CHO
45 55 56
reflux130-150°C
………Scheme 2.20
2.8 PRESENT WORK:
The present work describes the design and synthesis of novel
quinazolinone derivatives of 2-(2-(4-((3,4-dihydro-4-oxo-3-arylquina
zolin-2-yl)methyl)piperazin-1-yl)acetoyloxy)-2-phenyl acetic acid esters
(57) incorporating three biologically active moieties such as
quinazolinones, piperazine and L-mandelic acid derivatives in a single
molecule. (Scheme 2.21)
OR
O
O
N
O
N
N
N
O
R1
57

66
The present work also describes the synthesis of several novel
derivatives of quinazolinones by incorporating different substituted aryl
amines at 3rd position and cyclic rings containing nitrogen nucleophiles
such as ethyl 4-piperidinecarboxylate, morpholine and piperidone at 2nd
position of quinazolinone nucleus in solution phase and also under
solvent-free conditions using Poly ethylene glycol (PEG-400) by simple
physical grinding with mortar and pestle.
The present work illustrate the synthesis of various sulfones of 2-
(4-methanesulfonylpiperazin-1-yl-methyl)-3-phenyl-quinazolin-4(3H)-one
& 2-(4-benzenesulfonyl-piperazin-1-yl-methyl)-3-phenyl-quinazolin-4(3H)
-one from anthranilic acid.
2.10. RESULTS AND DISCUSSION:
The retro synthetic analysis of present route (Scheme-2.21)
revealed that the molecule could be dissected into two intermediates
namely 2-chloroacetoxyphenyl acetic acid esters (58) and 3-aryl-2-
(piperazin-1-yl)quinazolin-4(3H)-one (59), which can be condensed
conveniently to afford the final product. Accordingly, the present work
describes here a more efficient method wherein the two key intermediates
58 & 59 are made simultaneously and upon condensation produce 2-(2-
(4-((3,4-dihydro-4-oxo-3-arylquinazolin-2-yl) methyl)piperazin-1-yl)
acetoyloxy)-2-phenyl acetic acid esters (57).

67
O
O
OR
O
N
N
N
N
O
Ar
O
O
OR
O
Cl
NH
N
N
N
O
Ar+
57 58 59
………Scheme-2.21
The synthetic strategy described above was implemented by
making the two key intermediates 58 & 59 simultaneously. The crucial
α-hydroxy compound process was developed by same group & prepared
by a stereo selective diazotization of L-phenyl glycine (34b) in aqueous
acetone, using sodium nitrite in presence of dil. H2SO4, to afford (L)-2-
hydroxy-2-phenyl acetic acid (35b) in 70% yield with more than 99%ee.
These results are further supported by comparison with known
compound of L-Mandelic acid by optical rotation and chiral HPLC. When
compare the previous literature reports, this method is very
advantageous because product is obtained in good yields and no side
products are formed. 35b on treatment with isopropyl alcohol in
presence of catalytic amount of Conc. H2SO4 under refluxing conditions,
gave, the previously reported [28] isopropyl (2S)-2-hydroxy-2-
phenylethanoate (60). The latter on treatment with chloroacetyl chloride
in dichloromethane using triethylamine as a base at room temperature to
obtain desired isopropyl (2S)-2-[(2-chloroacetyl) oxy)-2-phenylethanoate
(58) and further it was characterized by IR, 1H NMR & Mass. (Scheme-

68
2.22) In IR spectra showed two strong absorptions at 1768.4 & 1747.19
cm-1 for carbonyl group. The main resonances in 1HNMR spectra in
CDCl3 are, i) two resonances at 1.05-1.1 and 1.15-1.20 ppm, usually
two three proton doublets which corresponds to two methyl groups on
isopropyl, ii) a doublet of doublet at 4.2-4.3 ppm, usually two proton
adjacent to chlorine group iii) a multiplet single proton at 5.1 ppm of
isopropyl group iv) a sharp singlet at 5.9 ppm of chiral proton and v)
five proton multiplet at 7.4-7.5ppm for aromatic protons. The (M++1)
peak 217.9 showed in mass spectra.
O
O
OR
O
ClOH
O
OH
OH
O
OR
35b 60 58
i ii
R = Me, Et, Isopropyl
………Scheme-2.22
Reagents and Conditions: i) R-OH, Conc.H2SO4 (R= Me, Et, Isopropyl)/
Δ; ii) Chloro acetyl chloride, TEA, DCM, RT.
In another sequence of reactions anthranilic acid (45) was treated
with chloroacetyl chloride in presence of triethylamine in
dichloromethane at room temperature to obtain previously reported [20]
2-chloromethyl benzo [d][1,3]oxazin-4-one (61). The latter on treatment
with aniline in refluxing pyridine for 5-6 h, followed by simple processing

69
resulted in the formation of 2-(chloromethyl)-3-phenyl-3,4-dihydro-4-
quinazolinone (62) [21]. 62 reacted with N-Boc-piperazine in acetonitrile
and K2CO3 as a base in presence of catalytic amount of KI under
refluxing conditions for 1-2 h, yielded a neat product which has been
characterized as tert-butyl-4-[(4-oxo-3-phenyl-3,4-dihydro-2-quinazolin
yl)methyl]-1-piperazinecarboxylate (63), on the basis of IR, 1H NMR &
Mass spectral data (Scheme-2.23). In IR spectra a strong absorption
showed at 1687.4 cm-1 for carbonyl group. The major resonances in
1HNMR spectra of the 63 in CDCl3 are i) a sharp singlet at 1.4 ppm
usually nine proton of tert-butyl group, ii) a broad singlet at 2.25 ppm
usually four protons of piperazine, iii) a sharp singlet corresponds to CH2
protons between quinazolinone and piperazine and four piperazine
protons together appeared at 3.2-3.25 ppm, iv) nine aromatic proton
multiplet appeared at 7.3-8.3. The mass peak showed at 421.1 (M+1
peak) with 100% abundance, a 40% abundance showed at 365 with loss
of tert-butyl group. Treatment of 63 with methanolic HCl (10% w/w)
solution at room temperature for Boc deprotection gave 3-phenyl-2-
(piperazinomethyl)-3,4-dihydro-4-quinazolinone hydrochloride salt (59).
The compound 59 further characterized by IR, 1H, 13C NMR & Mass
spectral data. In IR spectra a strong absorption showed at 1676.8 cm-1
for quinazolinone carbonyl group. The major resonances in the 1HNMR
spectra in CDCl3 are i) a two broad singlets at 2.2 and 2.8 ppm for
piperazine CH2 protons, ii) a sharp singlet at 3.25 usually a CH2

70
protons between quinazolinone and piperazine and iii) a multiplet at
7.35-8.35 for nine aromatic protons. The mass peak showed at 321.4
(M++1) with 100% abundance.
NH2
CO2H
N
O
O
Cl N
N
O
Cl
Ar
N
N
O
N
ArN
O
O
N
N
O
N
ArNH. HCl
45 61 62 63
59
i ii iii
iv
………Scheme-2.23
Reagents and Conditions: i) chloroacetyl chloride, TEA, DCM; ii) Ar-
NH2, (Ar = a=-C6H4-2-CF3; b = -C6H4-3-OCH3; c=-C6H4-2-CH3; d=-C6H5),
pyridine, Δ; iii) N-BOC-piperazine, K2CO3, KI, CH3CN, Δ; iv) 10%
methanolic HCl.
Having made two key intermediate 58 & 59 simultaneously, the
two units were condensed (Scheme-2.24) in presence of potassium
carbonate and catalytic amount of KI in refluxing acetonitrile to afford
desired compound isopropyl (2S)-2-[(2-{4-[(4-oxo-3-phenyl-3,4-dihydro-2-
quinazolinyl)methyl]piperazine} oxy]-2-phenylethanoate (57) in good
yields. The obtained product was characterized by IR, 1H, 13C NMR, &
Mass spectral analysis. The major IR absorptions showed at 1747, 1695,
1602 and 1585 cm-1. The main resonances in the 1HNMR in CDCl3 are i)
a two three proton doublets at 1.3 & 1.5 ppm corresponds to methyl

71
groups on isopropyl, ii) a two broad singlet’s at 2.55-2.8 corresponds to
CH2 piperazine protons, iii) a singlet at 3.45 ppm corresponds to CH2 in
between quinazolinone & piperazine ring, iv) a doublet of doublet at
3.5-3.65 ppm which corresponds for CH2 protons in between piperazine
and carbonyl group, v) a multiplet showed at 5.2 ppm for CH proton on
isopropyl group, vi) a sharp singlet showed at 6.1 ppm usually
corresponds to chiral proton of mandalate and vii) a 14 proton multiplet
at 7.5-8.5 ppm correspond to aromatic protons. The major resonances
in the 13CNMR in CDCl3 are 21.28, 21.55 (two CH3), 52.48 (four
piperazine CH2), 58.7 (one CH2 between quinazolinone & piperazine),
61.0 (one CH2 between piperazine and carbonyl), 69.4 (CH of isopropyl),
74.6 (CH of Chiral carbon on mandalate), 121.1 (quaternary Carbon on
quinazolinone ring attached to keto), 146.9 (quaternary Carbon on
quinazolinone adjacent to N), 162 (carbonyl of quinazolinone), 167.9 &
169.5 ( two carbonyls of adjacent to chiral carbon). The mass spectra
showed a major 100% abundance at 555 (M++1 peak), this is strongly
supports the M++1 ion peak (compound mol weight is 554), another
molecular ion showed with 10% abundance at 379 which corresponds to
cleavage of mandalate ester. During this, reaction racemisation takes
place which is confirmed by checking of isolated compounds for optical
purity by Polarimeter and value showed is 0°. The explanation for the
racemisation is α- hydrogen is labile and thus causing racemisation
under the given conditions.

72
O
O
OR
O
N
N
N
N
O
Ar
O
O
OR
O
Cl
NH
N
N
N
O
Ar+
58 59 57
i
………Scheme-2.24
Reagents and Conditions: i) K2CO3, KI, CH3CN, Δ.
2.10.1 NITROGEN NUCLEOPHILES
The reaction of 2-(chloromethyl)-3-(2-methylphenyl)-3,4-dihydro-4-
quinazolinone 62 (Ar = C6H4-3-OCH3) with 4-Piperidone, K2CO3 and KI,
under refluxing acetonitrile for 90-120 min. resulted in the formation of
3-(2-methylphenyl)-2-[(4-oxopiperidino)methyl]-3,4-dihydro-4-quinazolin-
one (64). Structure of the latter compound was established by spectral
and analytical data. (Scheme-2.25) This reaction found general one and
has been extended to other nitrogen nucleophilic substrates such as
ethyl-4-Piperidinecarboxylate, morpholine and products thus obtained
were assigned structures 65 and 66 respectively on the basis of their
spectral data.

73
N
N
O
Cl
Ar
N
N
O
Ar
N
O
N
N
O
Ar
N
O
N
N
O
Ar
N
CO2Et
i ii iii
62c
64 65 66
(Ar = a = -C6H4-3-OCH3; b=-C6H5; c=-C6H4-2-CH3; d=-C6H4-2-CF3)
………Scheme 2.25
Reagents and Conditions: i) 4-Piperidone, K2CO3, KI, CH3CN, Δ; ii)
morpholine, K2CO3, KI, CH3CN, Δ; iii) 4-ethyl piperdinecarboxylate,
K2CO3, KI, CH3CN, Δ; Under Solvent-Free conditions: i/ii/iii) 4-
Piperidone / morpholine/ ethyl 4-Piperdinecarboxylate, K2CO3, KI, PEG-
400, mortar and pestle.
The IR spectrum of 64 showed the main absorption showed at
region 1712.48 & 1684.52 cm-1 corresponds to carbonyl groups present
in quinazolinone and piperidine. The main resonances in the 1HNMR in
CDCl3 are at 2.15 a sharp singlet corresponds to CH3 group on

74
benzene, ii) a two four proton multiplet at 2.3-2.4 & 2.5-2.66 ppm
usually corresponds to piperidone iii) a two proton doublet of doublet at
3.25-3.44 corresponds to CH2 protons between quinazolinone and
piperidone iv) a series of multiplet at 7.2-8.3 corresponds to eight
aromatic protons. The mass spectrum showed the major peak at 348.3
(M++1) with 100% abundance and
Conversion of 62 to corresponding 64, 65, and 66 derivatives is
favoured in presence of KI. This is probably due to the fact that in
presence of KI, the chlorine of 62 is initially replaced by iodine and
subsequent reaction of iodo derivative of 62 with the nitrogen
nucleophile is facile.
R-Cl + KI R-I+ KCl
………Scheme-2.26
In recent years considerable attention has been paid to reactions
done under solvent-free conditions [29]. One of the areas of central
attention in this field includes reactions between solids. These reactions
are not only of interest from an economical point of view in many cases
they also offer considerable synthetic advantages in terms of yield,
selectivity and simplicity of the reaction procedure.
PEG-400 [30] has been applied here as an efficient reaction
medium for the preparation of quinazolinone derivatives containing

75
nitrogen cyclic ring systems. It is a biologically acceptable inexpensive
polymer and an eco-friendly reagent. The PEG-400 is widely used in
many organic reactions for several conversions
Alternatively, reaction of 62 with 4-Piperidone in presence of PEG-
400 is carried out in solid phase by physical grinding in a mortar and
pestle for ~8 min. and subsequent work-up yielded product identical with
64 obtained in solution phase in all respects by comparison with mp, IR
data.
It was found that above reactions between 62 and 4-Piperidone did
not occur in the absence of PEG-400 even after grinding mixture of solids
for 3-4 hrs (scheme 2.27). Thus it appears that PEG-400 acts like a
Crown ether and that is why the addition of KI makes the reaction much
faster because PEG-400 enhances the nucleophilicity of the iodide ion
and facilitating reaction between 62 and 4-Piperidone.
N
N
O
Cl
Ar
N
N
O
R
ArR, K2CO3, KI, Solvent-free
Ar= -C6H5-2-CH3; R= 4-Piperidone
………Scheme-2.27
Above reactions are of significant in nature when compare to the
solution phase method in terms of time, yield and eco-friendly nature of
the reaction.

76
Reaction between 62 and 4-Piperidone in solution phase and also
under solvent-free conditions using PEG-400 as catalyst has been found
to be a general one and has been extended to other nitrogen nucleophilic
substrates such as ethyl-4-Piperidinecarboxylate, morpholine and
products thus obtained were assigned structures 65 and 66 respectively
on the basis of their spectral data. Furthermore, reactions of ethyl
piperdine-4-carboxylate, morpholine and 4-Pieridone are very general
and have been found to occur with other quinazolinone derivatives
resulting in the formation of 64, 65, 66 (whose structures were assigned
based on spectral data
2.10.2 SULFONES:
The compound 59 was treated with methanesulfonyl chloride
(MsCl) in dichloromethane containing TEA as base to obtain 2-(4-
methanesulfonyl-piperazin-1-yl-methyl)-3-phenyl-quinazolin-4(3H)-one
(67, R=-CH3). The structure of 67 has been established on the basis of
their spectral data. The above reaction of 59 to 67 has been found to be
general and has been extended to other aryl groups. The conversion of
59 with benzenesulfonyl chloride give 2-(4-benzenesulfonyl-piperazin-1-
yl-methyl)-3-phenyl-quinazolin-4(3H)-one (67, i.e. R=-C6H5) (Scheme-
2.28). All the products obtained in the above work have been
characterized by spectral data.

77
N
N
O
N
ArNH
N
N
O
N
ArN
SR
O
O
59 67
R-SO2-Cl
TEA, DCM
(R= Me, Ph)
………Scheme 2.28
Alternatively the title compounds 67 could also be prepared by the
following sequence of reactions. N-BOC Piperazine was treated with
methanesulfonyl chloride in acetone containing pyridine at ambient
temperature to yield 4-methanesulfonyl piperazine-1-carboxylic acid tert-
butyl ester 68, which was treated with isopropyl alcohol-hydrochloric
acid (IPA-HCl, 5-10%) at rt gave the hydrochloride salt of 1-
methanesulfonyl-Piperazine 69, (i.e. R=-CH3) by the de protection of the
BOC group. Reaction of 62 with 69 in presence of K2CO3 as a base and
catalytic amount of KI in refluxing acetonitrile for 2h followed by simple
processing resulted in the formation of 67 which was found to be
identical with the product obtained from 59 in all respects i.e. TLC, mp,
NMR and IR (Scheme-2.29)
N
N
O
N
ArN
SR
O
O
N NH
O
O
N N
O
O
S
O
O
R HN N S
O
O
R
67
68 69
i ii
62, iii
………Scheme 2.29

78
Reagents and conditions: i) methanesulfonyl chloride/benzenesulfonyl
chloride, pyridine, acetone, RT; ii) IPA-HCl (5-10%), rt; iii) 62, K2CO3, KI,
CH3CN, Δ
2.11. CONCLUSION:
In conclusion, we have achieved the synthesis of {2-(2-(4-((3,4-
dihydro-4-oxo-3-arylquinazolin-2-yl)methyl)piperazin-1-yl)acetoyloxy)-2-
phenyl acetic acid esters, through the preparation of two key
intermediates, by incorporating three moieties such as quinazolinones,
piperazine and L-mandelic acid in a single molecule. We have developed
a simple and efficient method for preparation of new 4(3H)-Quinazolinone
derivatives in solution phase and also under solvent-free conditions
using PEG-400 by simple physical grinding in mortar and pestle at room
temperature. A simple, efficient, novel method for synthesis of various
sulfones has been developed by incorporating three biologically active
moieties such as quinazolinone, piperazine and a sulfones group in a
single molecule.

79
2.12. EXPERIMENTAL SECTION
Preparation of 60:
To a solution of 35 (1.52 g, 10 mmol) in isopropanol (20 mL) were
added few drops of conc. H2SO4 and the mixture refluxed on a water bath
for 3 h. At the end of this period, the excess solvent was removed by
distillation. The residue was dissolved in water, cooled to 0oC and
gradually neutralized with solid NaHCO3. Reaction mass was extracted
into dichloromethane (3x20 mL). Combined organic layers were washed
with water (25 mL) and dried over anhyd.Na2SO4. Finally the solvent was
distilled off under reduced pressure to dryness to give a desired product
60 as light yellow liquid (1.66g, 100% yield). (Scheme-2.22)
Preparation of 58:
A mixture of 60 (10 mmol), triethylamine (4.17 ml, 30 mmol) in
dichloromethane (20 ml) was cooled to 0-5°C. Chloro acetyl chloride
(0.956 ml, 12 mmol) was added slowly at 0-5°C during the period of 15-
20 min. After complete addition reaction mass was brought to rt and
maintained for 5-6 h. The progress of the reaction was monitored by TLC
for disappearance of 60. After completion of reaction, water (20 ml) was
added and the layers separated. Aq. layer further extracted in to
dichloromethane (2x10 ml). Combined organic layers were washed with
water (20 ml) and dried over anhyd.Na2SO4. The organic layer was
evaporated to dryness to give a pure product 58. (Scheme-2.22)

80
Representative Spectral Data
58a: R= -CH3; Yield= 82%; Mp. Colourless liquid; IR (KBr): 1754.9 (COO)
cm-1; 1H NMR (CDCl3, 200 MHz): δ 3.8 (s, 3H, -OCH3), 4.18-4.22 (d, 2H, -
CH2), 6.20 (S, 1H, -CH), 7.40-7.50 (m, 5H, Ar-H); m/z (M++1): 243. Anal.
Calcd. For (C11H11ClO4); require: C, 54.45, H, 4.57; Found: C, 54.41, H,
4.51%.
58b: R= -C2H5; Yield= 76%; Mp Colourless liquid; IR (KBr): 1747.19
(COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 1.09-1.30 (t, 3H, -CH3), 4.05-
4.25 (q, 2H, -CH2), 5.90-6.0 (s, 1H, -CH), 7.30-7.50 (m, 5H, Ar-H); m/z
(M++1): 257. Anal. Calcd. For (C12H13ClO4); require: C, 56.15, H, 5.10;
Found: C, 56.09, H, 5.02%.
58c: R= CH(CH3)2; Yield= 82%; Mp Colourless liquid; IR (KBr): 1768.5
(COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 1.10-1.15 (d, 3H, -CH3), 1.22-
1.30 (dd, 3H, -CH3), 4.07-4.25 (q, 2H, -CH2), 5.00-5.10 (m, 1H, -CH),
5.90-5.95 (s, 1H, -CH), 7.38-7.50 (m, 5H, Ar-H); m/z (M++1): 271. Anal.
Calcd. For (C13H15ClO4); require: C, 57.68, H, 5.59; Found: 57.62, H,
5.54%.
Preparation of 63:
A mixture of 62 (10 mmol), N-BOC-piperazine (2.66 g, 15 mmol),
K2CO3 (2.76 g, 20 mmol), KI (0.016 g, 0.01 mmol) and CH3CN (20 mL)
was heated at 80 oC for 90-120 min. The progress of reaction was

81
monitored by TLC for complete disappearance of 62. On completion of
reaction, mixture was diluted with water and extracted with ethyl acetate
(2x 25 mL). The combined organic layer was washed with water,
saturated solution of NH4Cl (25 mL) (to remove un reacted BOC-
piperazine), brine and then dried with anhydrous Na2SO4. The organic
layer was distilled under reduced pressure, gave 63 (Scheme-2.23)
Representative Spectral Data
63a: R1= -C6H4-2-CF3; Yield= 86%; Mp 166-68; IR (KBr): 1684 (C=O),
1695 (COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 1.4 (t, 9H, BOC -CH3),
2.1-2.4 (m, 4H, Piperazine), 3.1-3.2 (dd, 2H, -CH2), 3.2-3.4 (m, 4H,
Piperazine), 7.45 -7.85 (m, 7H, Ar-H), 8.30 (m, 1H, Ar-H); m/z (M++1):
489. Anal. Calcd. For (C25H27 F3N4O3); require: C, 61.47, H, 5.57; N,
11.47; Found: 61.38, H, 5.49, N, 11.41%.
63b: R1= -C6H4-3-OCH3; Yield= 78%; Mp 152-54 °C; IR (KBr): 1680
(C=O), 1697 (COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 1.4 (s, 9H, BOC -
CH3), 2.0-2.4 (m, 4H, Piperazine), 3.1-3.2 (dd, 2H, -CH2), 3.3-3.4 (m, 4H,
Piperazine), 3.7 (s, 3H, Ar-OCH3), 7.1-8.3 (m, 7H, Ar-H); m/z (M+.+1):
451.5. Anal. Calcd. For (C25H30N4O4); require: C, 66.65, H, 6.71; N,
12.44; Found: 66.58, H, 6.65, N, 12.38 %.
63c: R1= -C6H4-2-CH3; Yield= 74%; Mp 132-34 °C ; IR (KBr): 1680 (C=O),
1694 (COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 1.4 (s, 9H, BOC CH3),
2.10-2.35 (m, 4H, Piperazine), 2.2 (s, 3H, Ar-CH3), 3.1-3.2 (dd, 2H, -CH2),

82
3.2-3.4 (m, 4H, Piperazine), 7.18-7.20 (d, 1H, Ar-H),7.3-7.4 (m, 3H, Ar-
H), 7.45-7.70 (t, 1H, Ar-H), 7.75-7.81 (m, 2H, Ar-H), 8.3 (d, 1H, Ar-H);
m/z (M+.+1): 435. Anal. Calcd. For (C25H30N4O3); require: C, 69.10, H,
6.96; N, 12.89; Found: C, 69.00, H, 6.92, N, 12.83 %.
63d: R1= -C6H5; Yield= 84%; Mp. 126-30 °C; IR (KBr): 1687 (C=O), 1696
(COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 1.4 (s, 9H, BOC CH3), 2.2-2.3
(m, 4H, Piperazine), 3.2 (d, 2H, -CH2), 3.2-3.3 (m, 4H, Piperazine), 7.30-
7.35 (d, 2H, Ar-H), 7.42-7.55 (m, 4H, Ar-H), 7.75-7.80 (m, 2H, Ar-H),
8.28 (d, 1H, Ar-H); M/Z (M+.+1): 421. Anal. Calcd. For (C24H28N4O3);
require: C, 68.55, H, 6.71; N, 13.32; Found: C, 68.48, H, 6.65, N,
13.28%.
Preparation of 59: (deprotection of-BOC)
Compound 63 (10 mmol) was suspended in methanolic HCl (15
mL, 10-12% w/v) at ambient temperature. The mixture was stirred at
same temp for 1-2 h. After completion of reaction, MtBE (15 mL) was
added and stirred the mixture for another 30 min at RT. The separated
solid was filtered and washed with MtBE (5 mL) (to remove excess of
HCl). The solid was dissolved in water (5 mL) and pH was adjusted to ~
8-9 by adding sufficient amount of saturated aqueous Na2CO3 solution.
The aq. layer was extracted in to DCM (3x20 mL). The organic layers were
combined, washed successively with water (2x 10 mL, until aq layer pH
is neutral), brine and then dried with anhydrous. Na2SO4. The organic

83
layer was distilled under reduced pressure, to obtain a residue of 59
(Scheme-2.23)
Representative Spectral Data
59a: R1= -C6H4-2-CF3; Yield= 80%; Mp 142-145°C; IR (KBr): 1754.9
(COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 2.2-2.4 (m, 4H, Piperazine), 2.7-
2.8 (m, 4H, Piperazine), 3.15-3.25 (dd, 2H, -CH2), 7.5-7.9 (m, 7H, Ar-H),
8.3 (m, 1H, Ar-H); M/Z (M+.+1): 389. Anal. Calcd. For (C20H19F3N4O);
require: C, 61.85, H, 4.93; N, 14.43; Found: 61.81, H, 4.89, N, 14.38%
59b: R1= -C6H4-3-OCH3; Yield= 78%; Mp 112-114°C; IR (KBr): 1674
(C=O) cm-1; 1H NMR (CDCl3, 200 MHz): δ 2.0-2.4 (m, 4H, Piperazine), 2.6-
2.8 (m, 4H, Piperazine), 3.1-3.4 (dd, 2H, -CH2), 3.7 (s, 3H, Ar-OCH3), 7.1-
7.85 (m, 7H, Ar-H), 8.3 (m, 1H, Ar-H); M/Z (M+.+1): 351.5. Anal. Calcd.
For (C20H22N4O2); require: C, 68.55, H, 6.33; N, 15.99; Found: C, 68.51,
H, 6.28, N, 15.93%.
59c: R1= -C6H4-2-CH3; Yield= 72%; Mp 170-74°C; IR (KBr): 1684 (C=O)
cm-1; 1H NMR (CDCl3, 200 MHz): δ 2.1 (s, 3H, Ar-CH3), 2.2-2.4 (m, 4H,
Piperazine), 2.7-2.9 (m, 4H, Piperazine), 3.05-3.4 (dd, 2H, -CH2), 7.1-7.4
(m, 4H, Ar-H), 7.4-7.6 (m, 1H, Ar-H), 7.7-7.8 (m, 2H, Ar-H), 8.3 (d, 1H,
Ar-H); M/Z (M+.+1): 335.5. Anal. Calcd. For (C20H22N4O); require: C,
71.83, H, 6.63; N, 16.75; Found: C, 71.78, H, 6.58, N, 16. 68%.

84
59d: R1= -C6H5; Yield= 76%; Mp 152-54°C ; IR (KBr): 1676 (C=O) cm-1;
1H NMR (CDCl3, 200 MHz): δ 2.2-2.45 (m, 4H, Piperazine), 2.7-2.8 (m, 4H,
Piperazine), 3.2 (s, 2H, -CH2), 7.3-7.4 (d, 2H, Ar-H),7.42-7.60 (m, 4H, Ar-
H), 7.7-7.80 (m, 2H, Ar-H), 8.3 (d, 1H, Ar-H); M/Z (M+.+1): 321.5. Anal.
Calcd. For (C19H20N4O); require: C, 71.23, H, 6.29; N, 17.49; Found: C,
71.18, H, 6.25, N, 17.45 %.
Preparation of 57:
A mixture of 58 (10 mmol, 1.0 equiv.), 59 (10 mmol, 1.0 equiv)
were dissolved in CH3CN (20 mL), K2CO3 (2.76 g, 20 mmol) and KI (0.016
g, 0.1 mmol) were added in respective order. The mixture (a suspension)
was heated at 80 °C for 90-120 min. The progress of the reaction was
monitored by TLC. On completion of reaction, the mixture was diluted
with water and extracted with ethyl acetate (2x 25 mL). The combined
organic layer was washed with water, brine and then dried with
anhydrous Na2SO4. The organic layer was distilled under reduced
pressure, to obtain the final compound 57 (Scheme-2.24).
Representative Spectral Data
57a: R= -CH3; R1= -C6H4-2-CF3; Yield= 78%; Mp liquid; IR (KBr): 1670
(C=O), 1754.9 (COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 2.2-2.4 (m, 4H,
Piperazine), 2.4-2.6 (m, 4H, Piperazine), 3.10-3.20 (d, 2H, N-CH2), 3.30-
3.50 (d, 2H, N-CH2-CO), 3.75 (s, 3H, -OCH3), 5.95-6.00 (s, 1H, -CH),
7.40-7.9 (m, 12H, Ar-H), 8.3 (m, 1H, Ar-H); M/Z (M+.+1): 595.4. Anal.

85
Calcd. For (C31H29F3N4O5); require: C, 62.62, H, 4.92; N, 9.42; Found: C,
62.58, H, 4.89, N, 9.36 %.
57b: R= -CH3; R1= -C6H4-3-OCH3; Yield= 80%; Mp liquid; IR (KBr):
1688.4 (C=O), 1749.1 (COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 2.4-2.5
(m, 4H, Piperazine), 2.5-2.6 (m, 4H, Piperazine), 3.20-3.30 (d, 2H, N-
CH2), 3.30-3.40 (d, 2H, N-CH2-CO), 3.75 (s, 3H, -OCH3), 3.82 (s, 3H, -
OCH3), 5.95-6.00 (s, 1H, -CH), 6.8-7.0 (m, 3H, Ar-H),7.4-7.5 (m, 7H, Ar-
H), 7.75-7.80 (m, 2H, Ar-H), 8.3 (m, 1H, Ar-H); M/Z (M+.+1): 557.4. Anal.
Calcd. For (C31H32N4O6); require: C, 66.89, H, 5.79; N, 10.07; Found: C,
66.85, H, 5.74, H, 10.01 %.
57c: R= -CH3; R1= -C6H4-2-CH3; Yield= 82%; Mp liquid; IR (KBr): 1698
(C=O), 1750 (COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 2.2 (s, 3H, -CH3),
2.4-2.5 (m, 4H, Piperazine), 2.5-2.6 (m, 4H, Piperazine), 3.10-3.20 (dd,
2H, N-CH2), 3.30-3.40 (d, 2H, N-CH2-CO), 3.75 (s, 3H, -OCH3), 5.95 (s,
1H, -CH), 7.1-7.2 (m, 1H, Ar-H),7.3-7.4 (m, 8H, Ar-H), 7.4-7.5 (m, 1H,
Ar-H),7.7-7.8 (m, 2H, Ar-H), 8.3 (m, 1H, Ar-H); M/Z (M+.+1): 541. Anal.
Calcd. For (C31H32N4O5); require: C, 68.87, H, 5.97; N, 10.36; Found: C,
68.81, H, 5.95, N, 10.32%.
57d: R= -CH3; R1= -C6H5; Yield= 89%; Mp liquid; IR (KBr): 1683 (C=O),
1753 (COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 2.3-2.4 (m, 4H,
Piperazine), 2.4-2.5 (m, 4H, Piperazine), 3.20-3.25 (d, 2H, N-CH2), 3.30-
3.40 (d, 2H, N-CH2-CO), 3.75 (s, 3H, OCH3), 5.95 (s, 1H, -CH), 7.3-7.6

86
(m, 11H, Ar-H), 7.75-7.8 (m, 2H, Ar-H), 8.35 (m, 1H, Ar-H); M/Z
(M+.+1): 527.5. Anal. Calcd. For (C30H30N4O5); require: C, 68.43, H, 5.74;
N, 10.64; Found: C, 68.39, H, 5.70, N, 10.58 %.
57e: R= -C2H5; R1= -C6H4-2-CF3; Yield= 80%; Mp liquid; IR (KBr): 1692.2
(C=O), 1747.2 (COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 1.19-1.20 (t, 3H,
-CH3), 2.2-2.3 (m, 4H, Piperazine), 2.4-2.6 (m, 4H, Piperazine), 3.05-3.20
(dd, 2H, -CH2), 3.2-3.4 (dd, 2H, N-CH2-CO), 4.10 (q, 2H, -CH2), 5.95 (s,
1H, -CH), 7.2-7.8 (m, 12H, Ar-H), 8.3 (m, 1H, Ar-H); M/Z (M+.+1): 609.0.
Anal. Calcd. For (C32H31F3 N4O5); require: C, 63.15, H, 5.13; N, 9.21;
Found: C, 63.15, H, 5.08, N, 9.17%.
57f: R= -C2H5; R1= -C6H4-3-OCH3; Yield= 84%; Mp liquid; IR (KBr):
1688.4 (C=O), 1746.2 (COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 1.19-1.20
(t, 3H, -CH3), 2.4-2.5 (m, 4H, Piperazine), 2.5-2.6 (m, 4H, Piperazine),
3.25-3.30 (dd, 2H, -CH2), 3.3-3.4 (dd, 2H, N-CH2-CO), 3.85 (s, 3H, -
OCH3), 4.15 (q, 2H, -CH2), 5.95 (s, 1H, -CH), 6.8-7.0 (m, 3H, Ar-H), 7.3-
7.5 (m, 7H, Ar-H), 7.75-7.80 (m, 2H, Ar-H), 8.3 (m, 1H, Ar-H); M/Z
(M+.+1): 571.5. Anal. Calcd. For (C32H34N4O6); require: C, 67.35, H, 6.01;
N, 9.82; Found: C, 67.29, H, 5.98, N, 9.78%.
57g: R= -C2H5; R1= -C6H5; Yield= 86%; Mp liquid; IR (KBr): 1686.4 (C=O),
1748.2 (COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 1.19-1.20 (t, 3H, -CH3),
2.4-2.5 (m, 4H, Piperazine), 2.5-2.6 (m, 4H, Piperazine), 3.25-3.30 (dd,
2H, -CH2), 3.3-3.4 (dd, 2H, N-CH2-CO), 4.15 (q, 2H, -CH2), 5.95 (s, 1H, -

87
CH), 7.2-7.5 (m, 11H, Ar-H), 7.75-7.8 (m, 2H, Ar-H), 8.3 (m, 1H, Ar-H);
M/Z (M+.+1): 541.5. Anal. Calcd. For (C31H32N4O5); require: C, 68.87, H,
5.97; N, 10.36; Found: C, 68.82, H, 5.92, 10.30%.
57h: R= -CH(CH3)2; R1= -C6H4-2-CF3; Yield= 76%; Mp liquid; IR (KBr):
1691.2 (C=O), 1746.2 (COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 1.10-1.15
(d, 3H, -CH3), 1.22-1.30 (d, 3H, CH3), 2.2-2.4 (m, 4H, Piperazine), 2.4-2.6
(m, 4H, Piperazine), 3.1-3.2 (d, 2H, N-CH2), 3.3-3.4 (d, 2H, N-CH2-CO),
4.90-5.1 (m, 1H, -CH), 5.9 (s, 1H, -CH), 7.35-7.8 (m, 10H, Ar-H), 7.75-
7.80 (m, 2H, Ar-H), 8.3 (m, 1H, Ar-H); M/Z (M+.+1): 623.5. Anal. Calcd.
For (C33H33 F3N4O5); require: C, 63.66, H, 5.34; N, 9.00; Found: C, 63.63,
H, 5.29, N, 8.95%.
57i: R= -CH(CH3)2; R1= -C6H4-3-OCH3; Yield= 80%; Mp liquid; IR (KBr):
1693.2 (C=O), 1747.2 (COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 1.10-1.15
(d, 3H, -CH3), 1.22-1.30 (d, 3H, -CH3), 2.4-2.5 (m, 4H, Piperazine), 2.5-
2.6 (m, 4H, Piperazine), 3.2-3.3 (d, 2H, N-CH2), 3.3-3.4 (d, 2H, N-CH2-
CO), 3.85 (s, 3H, -OCH3), 5.00-5.10 (m, 1H, -CH), 5.9 (s, 1H, -CH), 6.8-
7.0 (m, 3H, Ar-H), 7.35-7.45 (m, 7H, Ar-H), 7.75-7.80 (m, 2H, Ar-H),
8.35 (m, 1H, Ar-H); M/Z (M+.+1): 585.0. Anal. Calcd. For (C33H36N4O6);
require: C, 67.79, H, 6.21; N, 9.58; Found: C, 67.75, H, 6.15, 9.54%.
57j: R= -CH(CH3)2; R1= -C6H4-2-CH3; Yield= 80%; Mp liquid; IR (KBr):
1698 (C=O), 1750 (COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 1.10-1.15 (d,
3H, -CH3), 1.22-1.30 (d, 3H, -CH3), 2.15 (s, 3H, -CH3), 2.2-2.3 (m, 4H,

88
Piperazine), 2.4-2.6 (m, 4H, Piperazine), 3.15-3.2 (d, 2H, N-CH2), 3.25-3.4
(dd, 2H, N-CH2-CO), 5.0-5.10 (m, 1H, -CH), 5.9 (s, 1H, -CH), 7.15-7.2
(m, 2H, Ar-H), 7.3-7.55 (m, 8H, Ar-H), 7.75-7.80 (m, 2H, Ar-H), 8.35
(m, 1H, Ar-H); M/Z (M+.+1): 569.0. Anal. Calcd. For (C33H36N4O5); require:
C, 69.70, H, 6.38; N, 9.85; Found: C, 69.66, H, 6.34, N, 9.82%.
57k: R= -CH(CH3)2; R1= -C6H5; Yield= 88%; Mp liquid; IR (KBr): 1695.1
(C=O), 1747.2 (COO) cm-1; 1H NMR (CDCl3, 200 MHz): δ 1.25-1.20 (d, 3H,
-CH3), 1.4-1.50 (d, 3H, -CH3), 2.15 (s, 3H, -CH3), 2.5-2.6 (m, 4H,
Piperazine), 2.6-2.8 (m, 4H, Piperazine), 3.40-3.50 (d, 2H, N-CH2), 3.5-3.6
(d, 2H, N -CH2-CO), 5.15-5.25 (m, 1H, -CH), 6.05 (s, 1H, CH), 7.5-7.75
(m, 11H, Ar-H), 7.9-8.0 (m, 2H, Ar-H), 8.4 (m, 1H, Ar-H); M/Z (M+.+1):
555.0. Anal. Calcd. For (C32H34N4O5); require: C, 69.30, H, 6.18; N,
10.10; Found: C, 69.26, H, 6.14, N, 10.04%.
Preparation of compounds 64-66 (General procedure in solution)
A mixture of 62 (10 mmol, 1.0 equiv), ethyl piperdine-4-
carboxylate/morpholine/ piperidine -4-one (10 mmol, 1.0 equiv), K2CO3
(2.76 g, 20 mmol, 2.0 equiv.), KI (0.57 g, 3 mmol, 0.3 equiv.) and CH3CN
(20 mL) was heated at 80 oC for 90-120 min. The progress of reaction
was monitored by TLC for complete disappearance of 62. On completion
of reaction, mixture was diluted with water and extracted with ethyl
acetate (2x 25 mL). The combined organic layer was washed with water,
brine and then dried with anhydrous Na2SO4. The organic layer was

89
distilled under reduced pressure, gave respectively 64/65/66. (Scheme-
2.25)
Preparation of compounds 64-66 (General procedure under solvent-
free)
A mixture of powdered anhydrous K2CO3 (4.14 g, 30 mmol, 3.0 eq),
PEG-400 (10mol%), KI (0. 57 g, 3 mmol, 0.3 equiv) and ethyl piperdine-4-
carboxylate/morpholine/piperidine (10 mmol, 1.0equiv) were taken in a
mortar and ground with a pestle for few minutes. To this mixture,
starting material 62 (10mmol) was added and the whole mixture was
ground with pestle in the same mortar at room temperature for 7-15 min.
The progress of reaction was monitored by TLC. After complete
disappearance of starting material, mixture was treated with ice-cold
water (50ml). Product separated was filtered, washed with water, and
dried to obtain 65/66 (Table-1). Product 64 separated out as oil, which
was extracted in to DCM, washed with water, and organic layer on
evaporation gave pure 64 as residue.
Representative Spectral data for 64-66 compounds:
64a: IR (KBr) cm-1:1670, 1711. 1H NMR (200MHz, CDCl3): δ 2.3. (m, 4H,
piperidone), 2.6-2.8 (m, 4H, piperidone), 3.4 (s, 2H, -CH2), 3.8 (s, 3H, -
OCH3), 6.9-8.3 (m, 8H, Ar-H). M/z (M +. +1): 364. Anal. Calcd. for
(C21H21N3O3) requires: C, 69.41, H, 5.82; N, 11.56; Found: C, 69.38; H,
5.80; N, 11.54%.

90
64b: IR (KBr) cm-1: 1675, 1714. 1H NMR (200MHz, CDCl3): δ 2.3 (t, 4H,
piperidone), 2.6 (t, 4H, piperidone), 3.4 (s, 2H, -CH2), 7.2-8.3 (m, 9H, Ar-
H). M/z (M +. +1): 334. Anal. Calcd. for (C20H19N3 O2) requires: C, 72.05; H,
5.74; N, 12.60; Found: C, 72.01; H, 5.78; N, 12.58%.
64c: IR (KBr) cm-1: 1678, 1714: 1H NMR (200MHz, CDCl3): δ 2.2 (s, 3H, -
CH3), 2.3 (dd, 4H, piperidone), 2.6 (dd, 4H, piperidone), 3.2-3.4 (dd, 2H, -
CH2), 7.2-8.3 (m, 8H, Ar-H). M/z (M +. +1): 348. Anal. Calcd. for
(C21H21N3O2) requires: C, 72.60, H, 6.09, N, 12.10; Found: C, 72.62; H,
6.01; N, 12.04%.
64d: IR (KBr) cm-1: 1686, 1717. 1H NMR (200MHz, CDCl3): δ 2.3 (m, 4H,
piperidone), 2.7-2.9 (m, 4H, piperidone) 3.3 (dd, 2H, CH2), 7.5-8.3 (m,
8H, Ar-H). M/z (M++1): 402. Anal. Calcd. for (C21H18F3N3O2) requires: C,
62.84; H, 4.52, N, 10.47; Found: C, 62.80; H, 4.46; N, 10.42%.
65a: IR (KBr) cm-1: 1672. 1H NMR (200MHz, CDCl3): δ 2.3-2.4 (m, 4H,
morpholine), 3.2 (s, 2H,-CH2), 3.6 (m, 4H, morpholine), 3.9 (s, 3H, OCH3),
6.9-8.3 (m, 8H, Ar-H). M/z (M +. +1): 352. Anal. Calcd. for (C20H21N3O3)
requires: C, 68.36; H, 6.02, N, 11.96; Found: C, 68.32; H, 6.08; N,
11.98%.
65b: IR (KBr) cm-1: 1676. 1H NMR (200MHz, CDCl3): δ 2.4 (m, 4H,
morpholine), 3.3 (s, 2H, -CH2), 3.5-3.6 (m, 4H, morpholine), 7.3-8.3 (m,

91
9H, Ar-H). M/z (M +. +1): 322. Anal. Calcd. for (C19H19N3O2) requires: C,
71.01; H, 5.96; N, 13.08; Found: C, 71.05; H, 5.92; N, 13.02%.
65c: IR (KBr) cm-1: 1684. 1H NMR (200MHz, CDCl3): δ 2.15 (s, 3H, -
CH3), 2.2-2.4 (m, 4H, morpholine), 3.1-3.3 (dd, 2H, -CH2), 3.5-3.6 (m,
4H, morpholine), 7.2-8.3 (m, 8H, Ar-H). M/z (M +. +1): 336. Anal. Calcd.
for (C20H21N3O2) requires: C, 71.62; H, 6.31; N, 12.53; Found: C, 71.58;
H, 6.26; N, 12.55%.
65d: IR (KBr) cm-1: 1678. 1H NMR (200MHz, CDCl3): δ 2.2-2.4 (dd, 4H,
morpholine), 3.1-3.2 (dd, 2H, -CH2), 3.6 (q, 4H, morpholine), 7.5-8.3 (m,
8H, Ar-H). M/z (M +. +1): 390. Anal. Calcd. for (C20H18F3N3O2) requires: C,
61.69; H, 4.66; N, 10.79 ; Found: C, 61.65; H, 4.62; N, 10.72%.
66a: IR (KBr) cm-1:1670, 1730. 1H NMR (200MHz, CDCl3): δ 1.2 (t, 3H, -
OCH2-CH3), 1.5-2.6 (m, 9H, -piperidine), 3.1-3.3 (dd, 2H, -CH2), 3.8 (s,
3H, -OCH3), 4.1 (q, 2H, -OCH2-CH3), 6.85-8.3 (8H, Ar-H). M/z (M+. +1):
422. Anal. Calcd. for (C24H27N3O4) requires: C, 68.39; H, 6.46; N, 9.97;
Found: C, 68.34; H, 6.40; N, 9.92%.
66b: IR (KBr) cm-1:1685, 1728. 1H NMR (200MHz, CDCl3): δ 1.2 (t, 3H, -
OCH2-CH3), 1.6-2.7 (m, 9H, piperidine), 3.2 (dd, 2H, -CH2), 4.1 (q, 2H, -
OCH2-CH3), 7.3-8.3 (m, 9H, Ar-H). M/z (M +. +1): 392. Anal. Calcd. for
(C23H25N3O3) requires: C, 70.57; H, 6.44; N, 10.73; Found: C, 70.51; H,
6.38; N, 10.75%.

92
66c: IR (KBr) cm-1:1678, 1730. 1H NMR (200MHz, CDCl3): δ 1.2 (t, 3H, -
OCH2-CH3), 1.6-2.6 (m, 9H, piperidine), 2.2 (s, 3H, -CH3), 3.1-3.3 (dd,
2H, -CH2), 4.1 (q, 2H, -OCH2-CH3), 7.2-8.3 (m, 8H, Ar-H). M/z (M +. +1):
406. Anal. Calcd. for (C24H27N3O3) requires: C, 71.09; H, 6.71; N, 10.36;
Found: C, 71.00; H, 6.65; N, 10.28%.
66d: IR (KBr) cm-1: 1680, 1730. 1H NMR (200MHz, CDCl3): δ 1.2 (t, 3H, -
OCH2-CH3), 1.6-2.6 (m, 9H, piperidine), 3.15 (dd, 2H, -CH2), 4.1 (q, 2H, -
OCH2-CH3), 7.5-8.3 (m, 8H, Ar-H). M/z (M +. +1): 460. Anal. Calcd. for
(C24H24F3N3O3) requires: C, 62.74; H, 5.26; N, 9.15; Found: C, 62.68; H,
5.20; N, 9.11%.
Preparation of 67:
A mixture of 59 (10 mmol), TEA (4.17 ml, 30 mmol) and DCM (20
ml), was cooled to 0-5°C. Methanesulfonyl chloride (11 mmol) was added
slowly at the same temp during a period of 5-10 min. After complete
addition, reaction mass was brought to rt and maintained at RT with
stirring for 5-6 hours. The progress of reaction was monitored by TLC till
disappearance of 59. After completion of reaction, water (20 ml) was
added and the layers separated. Aq. layer was further extracted in to
DCM (2x10 ml). Combined organic layers were washed with water (20ml)
and dried over anhydrous Na2SO4. The organic layer was evaporated to
dryness to yield a crude product 67. This was purified by column

93
chromatography, eluting the product with a mixture of hexane and ethyl
acetate to obtain pure as off-white crystalline solids (Scheme-2.28).
67a: 7a: Ar =-C6H5; R=-CH3; Yield 76%; m.p. 80-84°C; IR (KBr): 1682
(C=O), 1325 cm-1 (S=O); 1H NMR (CDCl3, 200 MHz): δ 2.4-2.5 (m, 4H,
piperazine), 2.7 (s, 3H, SO2-CH3), 3.1-3.2 (m, 4H, piperazine), 3.3 (s, 2H,
-CH2), 7.3-8.3 (m, 9H, Ar-H); 13C NMR (CDCl3, 50 MHz): δ 162.30,
152.37, 146.76, 136.61, 134.50, 129.17, 129.13, 128.76, 127.41,
127.25, 126.92, 121.10, 60.61, 52.00, 45.56, 34.41; MS: m/z (M+.+1)
399. Anal. Calcd for (C20H22N4O3S) requires: C, 60.28; H, 5.56; N, 14.06.
Found: C, 60.22; H, 5.53; N, 14.02%.
67b: Ar =-C6H4-3-OCH3; R=-CH3; Yield 80%; m.p. 76-78°C; IR (KBr):
1684 (C=O), 1327 cm-1 (S=O); 1H NMR (CDCl3, 200 MHz): δ 2.2-2.5 (m,
4H, piperazine), 2.2 (s, 3H, Ar-CH3), 2.7 (s, 3H, SO2-CH3), 3.1-3.2 (m, 4H,
piperazine), 3.2-3.4 (dd, 2H, -CH2), 7.1-8.3 (m, 8H, Ar-H); 13C NMR
(CDCl3, 50 MHz): δ 161.44, 151.99, 146.81, 137.28, 135.5, 135.35,
134.74, 130.13, 127.53, 127.41, 127.07, 127.05, 126.78, 120.88, 60.9,
52.15, 45.42, 34.10, 15.83; MS: m/z (M+.+1) 429.5. Anal. Calcd for
(C21H24N4O4S) requires: C, 58.86; H, 5.65; N, 13.07. Found: C, 58.84; H,
5.61; N, 13.02%.
67c: Ar =-C6H4-2-CH3; R=-CH3; Yield 78%; m.p. 73-75°C; IR (KBr): 1682
(C=O), 1325 cm-1 (S=O); 1H NMR (CDCl3, 200 MHz): δ 2.2 (s, 3H, Ar-CH3),
2.3-2.5 (m, 4H, piperazine), 2.7 (s, 3H, SO2-CH3), 3.1-3.2 (m, 4H,

94
piperazine), 3.2-3.4 (dd, 2H, -CH2), 7.2-8.3 (m, 8H, Ar-H); 13C NMR
(CDCl3, 50 MHz): δ 161.43, 152.36, 146.78, 136.02, 135.68, 134.39,
130.75, 129.30, 128.33, 127.28, 127.08, 126.82, 126.68, 120.88, 60.51,
52.02, 45.30, 34.22, 17.82; MS: m/z (M+.+1) 413. Anal. Calcd for
(C21H24N4O3S) requires: C, 61.14; H, 5.86; N, 13.58. Found: C, 61.10; H,
5.82; N, 13.56%.
67d: Ar =-C6H4-3-Cl; R=-CH3; Yield 82%; m.p. 168-72°C; IR (KBr): 1671
(C=O), 1327 cm-1 (S=O); 1H NMR (CDCl3, 200 MHz): δ 2.3 (m, 2H,
piperazine), 2.5 (m, 2H, piperazine), 2.8 (s, 3H, SO2 -CH3), 3.2 (m, 4H,
piperazine), 3.4 (q, 2H, -CH2), 7.3-8.3 (m, 8H, Ar-H); 13C NMR (CDCl3, 50
MHz): δ 162.10, 151.77, 146.62, 137.72, 134.72, 134.28, 130.15,
130.11, 129.28, 127.51, 126.92, 126.70, 120.97, 60.79, 51.85, 45.62,
34.02; MS: m/z (M+.+1) 434. Anal. Calcd for (C20H21ClN4O3S) requires: C,
55.49; H, 4.89; N, 12.94. Found: C, 55.46; H, 4.82; N, 12.92%.
67e: Ar =-C6H4-3-F; R=-CH3; Yield 75%; m.p. 140-44°C; IR (KBr): 1685
(C=O), 1326 cm-1 (S=O); 1H NMR (CDCl3, 200 MHz): δ 2.4 (m, 2H,
piperazine), 2.5-2.6 (m, 2H, piperazine), 2.8 (s, 3H, SO2-CH3), 3.1 (m, 4H,
piperazine), 3.4 (q, 2H, -CH2), 7.1-8.3 (m, 8H, Ar-H); 13C NMR (CDCl3, 50
MHz): δ 162.10, 151.8, 146.63, 134.71, 130.37, 130.30, 127.51, 127.47,
126.93, 124.49, 120.97, 117.15, 116.96, 116.23, 60.68, 51.99, 45.58,
34.26; MS: m/z (M+.+1) 417. Anal. Calcd for (C20H21FN4O3S) requires: C,
57.68; H, 5.08; N, 13.45. Found: C, 57.64; H, 5.07; N, 13.41%.

95
67f: Ar =-C6H3-3-F-4-F; R=-CH3; Yield 73%; m.p. 156-58°C; IR (KBr): 1685
(C=O), 1324 cm-1 (S=O); 1H NMR (CDCl3, 200 MHz): δ 2.4 (m, 2H,
piperazine), 2.5-2.6 (m, 2H, piperazine), 2.8 (s, 3H, SO2 -CH3), 3.1 (m, 4H,
piperazine), 3.3 (s, 2H, -CH2), 7.1-8.3 (m, 7H, Ar-H); 13C NMR (CDCl3, 50
MHz): δ 162.15, 151.58, 146.54, 134.82, 127.57, 126.91, 125.25, 120.84,
119.22, 119.07, 117.65, 117.50, 60.77, 52.05, 45.56, 34.34; MS: m/z
(M+.+1) 435. Anal. Calcd for (C20H20F2N4O3S) requires: C, 55.29; H, 4.64;
N, 12.90. Found: C, 55.28; H, 4.62; N, 12.88%.
67g: Ar =-C6H3-2-F-3-Cl; R=-CH3; Yield 74%; m.p. 186-90°C; IR (KBr):
1686 (C=O), 1328 cm-1 (S=O); 1H NMR (CDCl3, 200 MHz): δ 2.1-2.2 (m,
2H, piperazine), 2.5-2.6 (m, 2H, piperazine), 2.7 (s, 3H, SO2 -CH3), 2.9-
3.1 (m, 4H, piperazine), 3.3-3.5 (dd, 2H, -CH2), 7.2-8.3 (m, 7H, Ar-H); 13C
NMR (CDCl3, 50 MHz): δ 161.31, 151.66, 146.56, 134.91, 131.33,
127.65, 127.48, 127.24, 126.98, 124.52, 124.48, 121.45, 120.70, 61.23,
51.59, 45.36, 33.47; MS: m/z (M+.+1) 452. Anal. Calcd for
(C20H20ClFN4O3S) requires: C, 53.27; H, 4.47; N, 12.43. Found: C, 53. 21;
H, 4.45; N, 12.40%.
67h: Ar =-C6H4-3-OCH3; R=-C6H5; Yield 72%; m.p. 168-70°C; IR (KBr):
1685 (C=O), 1329 cm-1 (S=O); 1H NMR (CDCl3, 200 MHz): δ 2.2-2.4 (m,
4H, piperazine), 2.2 (s, 3H, Ar-CH3), 3.1-3.2 (m, 4H, piperazine), 3.2-3.4
(dd, 2H, -CH2), 7.1-8.3 (m, 13H, Ar-H); 13C NMR (CDCl3, 50 MHz): δ
161.26, 151.55, 147.65, 140.89, 137.23, 135.61, 134.82, 133.78,
132.26, 129.65, 128.97, 127.45, 127.33, 127.16, 124.87, 122.54,

96
120.88, 119.50, 61.55, 51.68, 45.88, 17.36; MS: m/z (M+.+1) 495.5.
Anal. Calcd for (C26H26N4O4S) requires: C, 63.66; H, 5.34; N, 11.42.
Found: C, 63.62; H, 5.30; N, 11.40%.
67i: Ar =-C6H4-2-CH3; R=-C6H5; Yield 77%; m.p. 174-76°C; IR (KBr):
1687 (C=O), 1326 cm-1 (S=O); 1H NMR (CDCl3, 200 MHz): δ 2.2-2.4 (m,
4H, piperazine), 3.1 (m, 4H, piperazine), 3.3 (s, 2H, -CH2), 7.2-8.3 (m,
13H, Ar-H); 13C NMR (CDCl3, 50 MHz): δ 162.16, 152.11, 146.35, 141.19,
136.83, 136.21, 135.12, 134.18, 133.06, 129.45, 128.27, 127.54,
127.13, 127.10, 124.27, 122.14, 120.48, 119.22, 61.35, 51.68, 44.65;
MS: m/z (M+.+1) 475.5. Anal. Calcd for (C26H26N4O3S): requires: C, 65.80;
H, 5.52; N, 11.81. Found: C, 65.82; H, 5.54; N, 11.79%.
Preparation of 68:
A mixture of N-Boc Piperazine (10 mmol), pyridine (2.42 ml, 30
mmol) and acetone (20 ml) was cooled to 0-5°C. Methanesulfonyl chloride
(11 mmol) was added slowly at same temp during a period of 5-10 min.
After the completion of addition, reaction was maintained at the same
temp for 1h and then brought to rt and maintained for 2h. TLC was used
to monitor the progress of the reaction till the disappearance of N-Boc
Piperazine. After completion of reaction, the excess solvent was removed
by distillation, and the residue treated with chloroform (25 ml). The
chloroform layer was washed with water (3x25 ml) and dried (anhyd.
Na2SO4). The organic layer was evaporated to dryness to obtain crude 68

97
which was used as such without further purification in the next step
(Scheme-2.29).
68a: R =-CH3; Yield 85%; m.p. 86-88°C; IR (KBr): 1720 (COO), 1315 cm-1
(S=O); 1H NMR (CDCl3, 200 MHz): δ 1.4 (s, 9H, BOC -CH3), 2.8 (s, 3H, -
SO2-CH3), 3.2 (m, 4H, piperazine), 3.5 (m, 4H, piperazine); MS: m/z
(M+.+1) 265. Anal. Calcd for (C10H20N2O4S) requires: C, 45.44; H, 7.63; N,
10.60. Found: C, 45.40; H, 7.61; N, 10.56%.
68b: R =-C6H5; Yield 87%; m.p. 124-26°C; IR (KBr): 1710 (COO), 1325
cm-1 (S=O); 1H NMR (CDCl3, 200 MHz): δ 1.4 (s, 9H, BOC CH3), 3.2 (m,
4H, piperazine), 3.5 (m, 4H, piperazine), 7.3-7.9 (m, 5H, Ar-H); MS: m/z
(M+.+1) 326. Anal. Calcd for (C15H22N2O4S) requires: C, 55.19; H, 6.79; N,
8.58. Found: C, 55.17; H, 6.77; N, 8.56%.
Preparation of 69: (deprotection of BOC)
Compound 68 (10 mmole) in IPA-HCl (10-12% w/v, 30 mmole) was
mixed at ambient temperature. The reaction mixture was stirred for 1-2
hr at the same temperature. The progress of the reaction was monitored
by TLC till the disappearance of 68. After the disappearance of starting
material, MTBE (15 mL) was added and the mixture stirred for another
30 min at RT. The separated solid was filtered and washed with MTBE (5
mL) (to remove excess of HCl), yielding hydrochloride salt of 69 (Scheme
2.29)

98
69a: R =-CH3; Yield 90%; m.p. 142-46°C; IR (KBr): 1320 cm-1 (S=O); 1H
NMR (CDCl3, 200 MHz): δ 3.0 (s, 3H, -SO2-CH3), 3.4 (m, 4H, piperazine),
3.6 (m, 4H, piperazine); MS: m/z (M+.+1) 164. Anal. =-C6H5; Yield 88%;
m.p. 108-10°C; IR (KBr): 1330 cm-1 (S=O); 1H NMR (CDCl3, 200 MHz): δ
3.2 (m, 4H, piperazine), 3.5 (m, 4H, piperazine), 7.3-7.9 (m, 5H, Ar-H);
MS: m/z (M+.+1) 226. Anal. Calcd for (C11H15NO2S) requires: C, 58.64; H,
6.71; N, 6.22. Found: C, 58.60; H, 6.75; N, 6.18%.Found: C, 44.12; H,
8.01; N, 8.56%.
Preparation of 67 (from 69):
A mixture of 69 (10 mmole), 62 (10 mmole), solid K2CO3 (2.76 g, 20
mmole) and KI (0.016 g, 0.1 mmole) in CH3CN (20 mL) was refluxed for 2
hr. The progress of the reaction was monitored on TLC, till the
disappearance of 62. On completion of reaction (~2 hr), the reaction
mixture was filtered and the insoluble material washed with CH3CN (2 ×
5 mL); the combined filtrate was distilled to dryness under reduced
pressure and the residue treated with chloroform (25 mL). The organic
layer was washed with water, brine and dried (anhyd. Na2SO4). The
chloroform layer was distilled under reduced pressure to obtain a residue
of 67. This was purified by column chromatography, eluting the product
with a mixture of hexane and ethyl acetate to yield pure 67 as off-white
crystalline solids (Scheme 2.29).
Yield (%): a= 75, b= 76, c= 78, d= 75, e= 72, f=75, g= 69, h= 70, i = 75.

99
2.13 REFERENCES:
1. Seebach D. and Kalinowski H.O., Nachr.chem.Tech.24, 415,1976
2. Haltr C.H., Laticacid Verlag chemie., Weinheim 1971
3. Patterson M.A.K, Szajewski R.P and Whutesides G.M., J.Org.Chem,
46, 4682, 1981
4. Hunger Buhler E., Seebach D and Wasmuth D., Helv.chim. Acta.,
64, 1467, 1981. B) Wynberg H and Staring E.G., J.Am.Chem
Soc.104, 166, 1982
5. Barre P., Ann. Techol. Arg.,.15, 203, 1966
6. a) Neuenberer, Advances in protein chemistry and 333. Acadmic
Press, New York (1948). b) Brewster, Hiron F., et al., Nature 166,
179, 1950
7. Seebach D and Hungerbubler E., Modern Synthetic Methods, 2,
91, 1980
8. David, H; Rami, Kantilal H., Chem. Abst., 125, 58491, 1996
9. Gerhard Billek, Organic Synthesis coll. vol, 5, 627, 1976
10. Herbest R.M and Shemin. D, Organic Synthesis coll. vol., 2, 1,
1943
11. Aston J.G., Newkirk J.D., Organic Synthesis Coll Vol., 3, 538 1955
12. Nathan G, and Laforge. F.B, J.Am.Chem. Soc., 70, 2812 (1948)
13. Kenji Koga, chin c.wu and shun-ichi yamada., Tetrahedron
Letters., 25, 2287 (1971)
14. Miltonwinitz et al., J.Am.Chem Soc., 78, 2423 (1956)

100
15. Mori K., Tetrahedron. 32, 1101 (1976)
16. Palomo C, et al., J.Am.Chem Soc., 112, 7659 (1990)
17. Otsubo K, Inanaga J, et al., Tetrahedron Letters, 28, 4435 (1987)
18. Griess, P, Ber. 1869, 2, 415; & 1878, 11, 1985
19. Pattanaik et al., Indian Journal of Chemistry, 37B, 1998, 1304-
1306
20. VK Pandey, et al., Indian Journal of Chemistry, 44B, 2005, 1940-
1943
21. Junichi Tani, et al., Journal of Medicinal Chemistry, 1979, 22, 95-
99
22. Ahmad R. Khosropour et al., Tetrahedron Letters 47, 2006, 3561-
3564
23. Venkateswarlu et al., Tetrahedron Letters, 47, 2006, 4381-4383
24. Shashikant R Pattan et al., Indian Journal of Chemistry, sec B,
2006, 1778-1781
25. Shanthan Rao et al., Synlett, 2006, 2507-2509
26. Kurosh Rad et al., Journal of Heterocyclic Chemistry 2006,
Volume 43(4), 913–91
27. Besson et al., Tetrahedron Letters, 2002, 43, 3911-3913.
28. Yongzheng, C.; Jinggang, Xu et al., Tetrahedron Asymmetry,
2007,18, 2537-2540
29. Tanaka, T.; Toda, F. Chem. Rev. 2000, 100, 1025

101
30. Dickerson, T. J.; Reed, N. N.; Janda, K. D. Chem. Rev. 2002, 102,
3325







![Synthesis of the New 2-[6-Nitro-2-Benzothiazolylazo]-4-Hydroxy ...](https://static.fdocuments.net/doc/165x107/5895c0161a28ab73208bf1aa/synthesis-of-the-new-2-6-nitro-2-benzothiazolylazo-4-hydroxy-.jpg)


![Applications of Boronic Acids in Organic Synthesis · 2015-07-20 · 2.2.3.1 Synthesis of (1-Hydroxy-1H-benzo[d][1,2,3]triazol-7-yl)boronic Acid 57 2.2.3.2 Synthesis of “Sulfur-Armed”](https://static.fdocuments.net/doc/165x107/5f48fd34aac6fc6584477de7/applications-of-boronic-acids-in-organic-synthesis-2015-07-20-2231-synthesis.jpg)







