
Structure Intro Part1
33
1 Protein structure Predictive methods and experimental methodologies Sources for this lecture Bioinformatics (course text: Baxevanis and Ouellette) Chapter 8: Predictive methods using protein sequences (Ofran and Rost) 198-219 Chapter 9: Protein structure prediction and analysis (Wishart) 224-247 Chapter 12: Creation and analysis of protein multiple sequence alignments (Barton) pgs 333-336; (Required reading during weeks 3-4 on multiple sequence alignment) Proteins: Structures and molecular properties (Creighton) Introduction to Protein Structure (Branden & Tooze) Much of the text in the slides that follow are drawn either verbatim or paraphrased from these texts.
-
Upload
marylaranjo -
Category
Documents
-
view
17 -
download
2
description
Mmoaoa
Transcript of Structure Intro Part1

1
Protein structure
Predictive methods and
experimental methodologies
Sources for this lecture
Bioinformatics (course text: Baxevanis and Ouellette)
Chapter 8: Predictive methods using protein sequences (Ofran and Rost) 198-219
Chapter 9: Protein structure prediction and analysis (Wishart) 224-247
Chapter 12: Creation and analysis of protein multiple sequence alignments (Barton)
pgs 333-336; (Required reading during weeks 3-4 on multiple sequence alignment)
Proteins: Structures and molecular properties (Creighton)
Introduction to Protein Structure (Branden & Tooze)
Much of the text in the slides that follow are drawn either verbatim
or paraphrased from these texts.

2
Topics Covered
• Methods for solving protein structures experimentally
• Overview of protein structure: primary, secondary, tertiary,
and quaternary
• Overview of protein folding
• Protein structure classification resources
– CATH
– SCOP
• The Structural Genomics Initiative
The importance of protein structure
“Bioinformatics is much more than just sequence
analysis…many of the most interesting and exciting
applications in bioinformatics today actually are
concerned with structure analysis.”
“The origins of bioinformatics actually lie in the field of
structural biology.”
“Proteins are perhaps the most complex chemical entities in
nature. No other class of molecule exhibits the variety and
and irregularity in shape, size, texture and mobility that
can be found in proteins.”
Baxevanis & Ouellette (Ch. 9, p.224, Wishart)

3
Primary,Secondary,
Tertiary andQuaternaryStructure
Secondary structure

4
! helix first described byLinux Pauling in 1951
Avg length: 10 residues(3 turns)
range: from 4 to over 40residues
good helix formers:A, E, L, M
very poor formers:P, G, Y, S
most common location:on surface, one side
buried, one side exposed
transmembrane heliceshave almost entirely
hydrophobic side chains(Branden & Tooze)
http://www.web-books.com/MoBio/Free/Ch2C4.htm
Amphipathicalpha helix
http://www.web-books.com/MoBio/Free/Ch2C4.htm
5
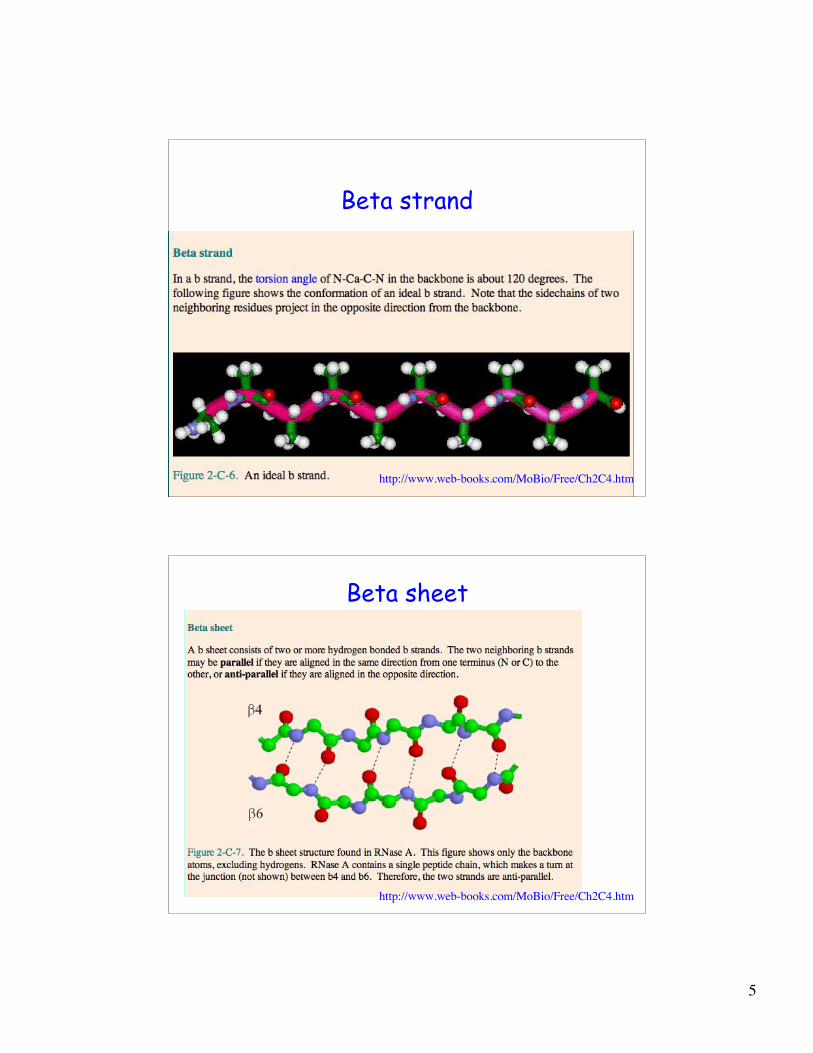
Beta strand
http://www.web-books.com/MoBio/Free/Ch2C4.htm
Beta sheet
http://www.web-books.com/MoBio/Free/Ch2C4.htm
6
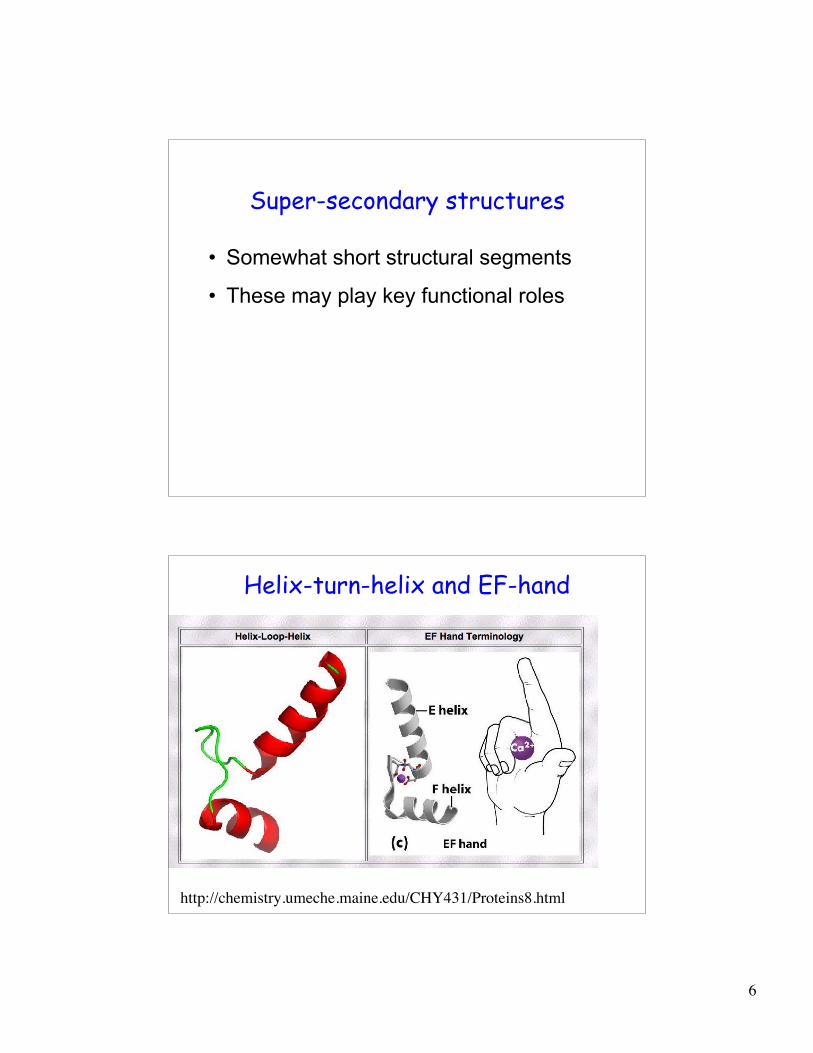
Super-secondary structures
• Somewhat short structural segments
• These may play key functional roles
Helix-turn-helix and EF-hand
http://chemistry.umeche.maine.edu/CHY431/Proteins8.html

7
Supersecondary structure:Helix-turn-helix
4-helical bundles

8
Moreabout 4-helicalbundles
Coiled Coil

9
A small selection of common folds
TM proteins: Porin & Rhodopsin
http://www.mhl.soton.ac.uk/public/research/projects/current/rhodopsin/index.html
10
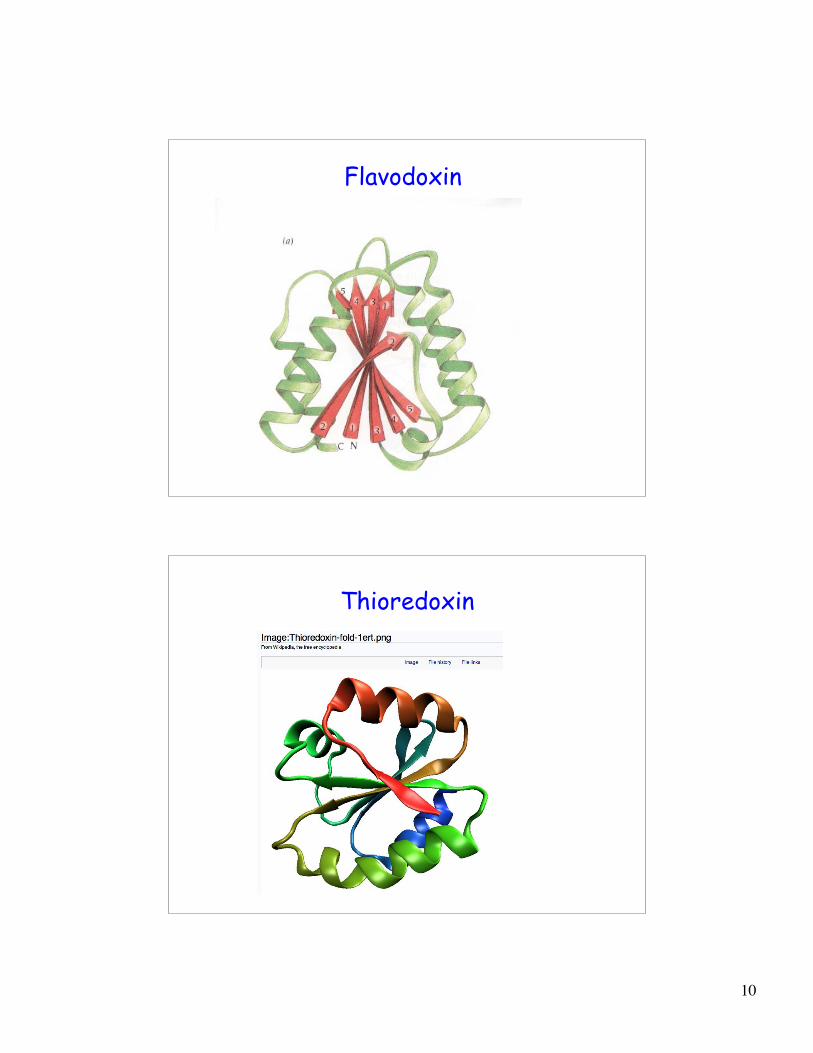
Flavodoxin
Thioredoxin

11
TIM Barrel
Rossman fold

12
Repeat structures: LRR
LRR structuresacross the Tree
of Life

13
Repeat structures: TPR
Structure Classification Databases
SCOP and CATH

14
CATH
CATH classification

15
Structural Classification ofProteins (SCOP)
and the Astral datasets
16
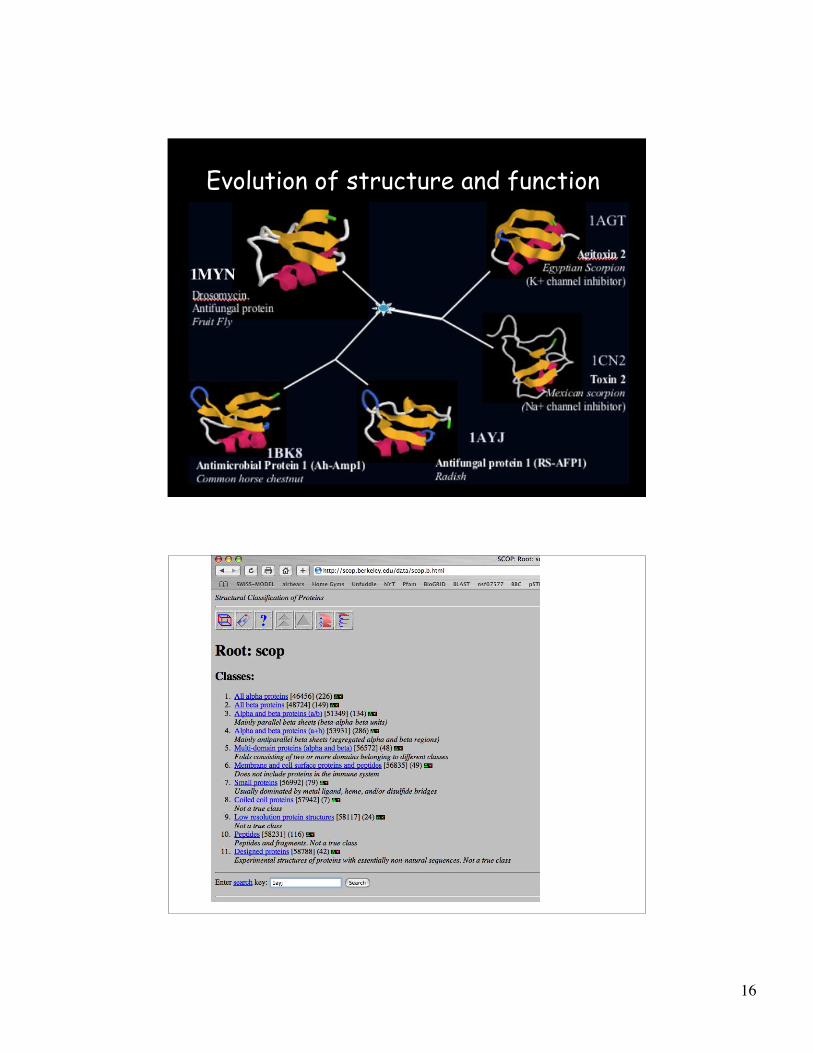
Evolution of structure and function
Anti-fungal defensin
(Radish) Scorpion toxin
Extend function prediction through inclusion of
structure prediction and analysis
Drosomycin
(Drosophila)
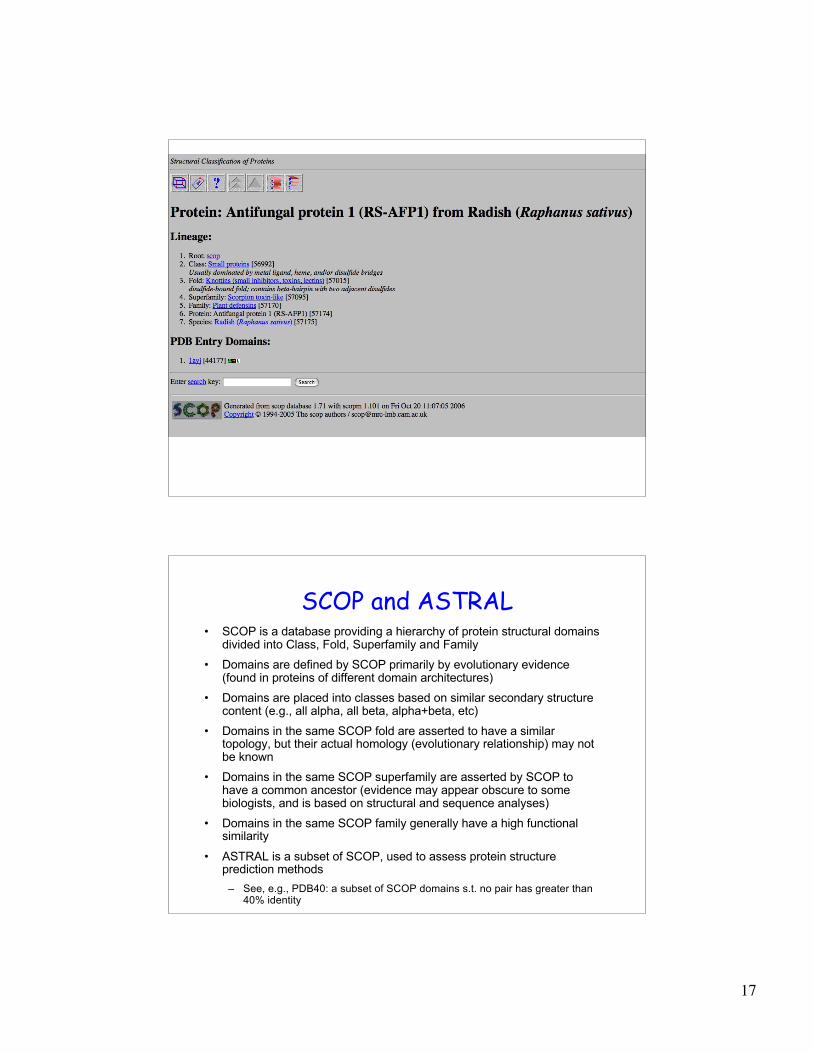
17
SCOP and ASTRAL• SCOP is a database providing a hierarchy of protein structural domains
divided into Class, Fold, Superfamily and Family
• Domains are defined by SCOP primarily by evolutionary evidence(found in proteins of different domain architectures)
• Domains are placed into classes based on similar secondary structurecontent (e.g., all alpha, all beta, alpha+beta, etc)
• Domains in the same SCOP fold are asserted to have a similartopology, but their actual homology (evolutionary relationship) may notbe known
• Domains in the same SCOP superfamily are asserted by SCOP tohave a common ancestor (evidence may appear obscure to somebiologists, and is based on structural and sequence analyses)
• Domains in the same SCOP family generally have a high functionalsimilarity
• ASTRAL is a subset of SCOP, used to assess protein structureprediction methods
– See, e.g., PDB40: a subset of SCOP domains s.t. no pair has greater than40% identity
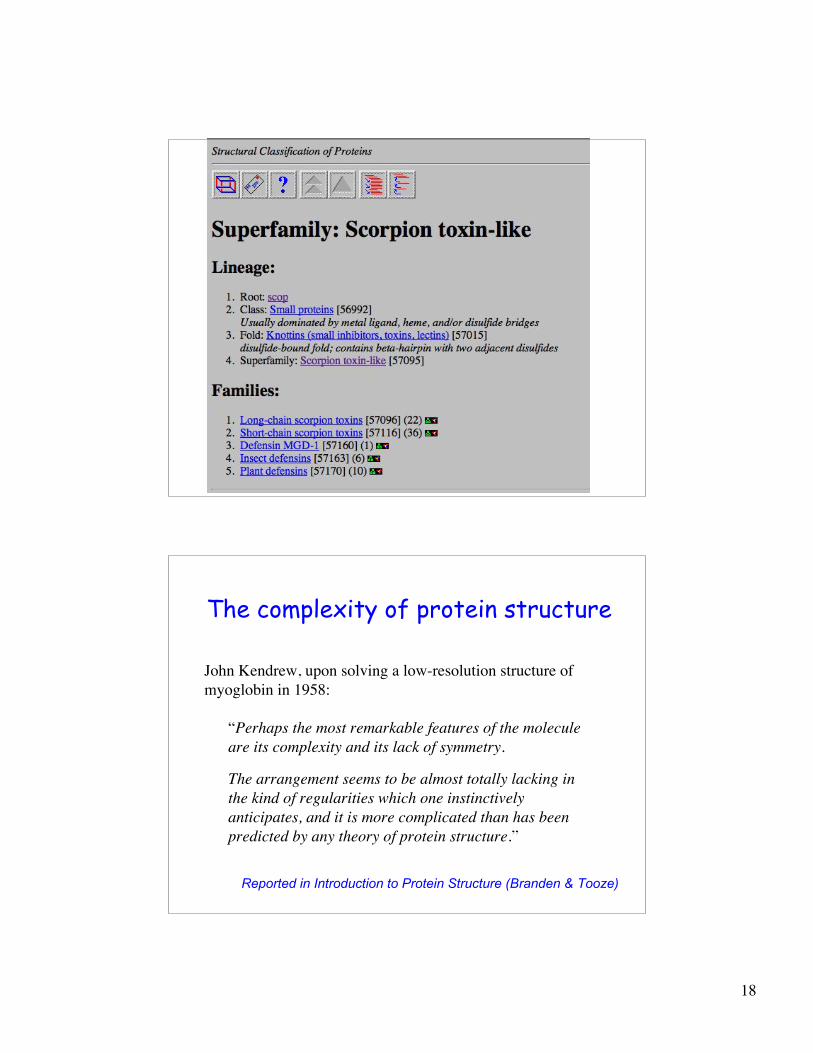
18
The complexity of protein structure
Reported in Introduction to Protein Structure (Branden & Tooze)
John Kendrew, upon solving a low-resolution structure of
myoglobin in 1958:
“Perhaps the most remarkable features of the molecule
are its complexity and its lack of symmetry.
The arrangement seems to be almost totally lacking in
the kind of regularities which one instinctively
anticipates, and it is more complicated than has been
predicted by any theory of protein structure.”

19
Experimental methods for solvingprotein 3D structure
Experimental determination ofprotein structure
• X-ray crystallography
• NMR spectroscopy
Many experimental issues:
Some proteins are hard to solve (e.g., membrane proteins)
Long proteins normally hard to solve, and are often divided intoindividual domains
Getting the domain boundaries correct is hard
Numerous other problems (which is why predicting proteinstructure is useful)

20
X-ray crystallography
• Most accurate; can be applied to larger proteins
• Oldest method; first structure (myoglobin) determined in late 1950s(Kendrew et al 1958). More than 20K structures solved to date
• Method:
– Small protein crystals (measuring <1mm) exposed to X-ray beams
– X-rays (which have a wavelength of 1-2Angstroms) are scattered ordiffracted by the protein atoms in the crystal
– Diffraction pattern appears as tens of thousands of tiny spots arrayed incomplex circular patterns
– XYZ coordinates determined from these diffraction patterns, along withintensity and phase information
– Molecular replacement (comparative models based on homologousstructures) can facilitate that process.
Baxevanis & Ouellette (Ch. 9, Wishart)
Limitations of X-ray crystallography
• Structures are solved in an artificial solid-state (crystalline) environmentunlike the natural (liquid) environment of cell.
– Structures can be altered by crystal packing and solvent exclusion effects.
• Some regions can not be resolved properly (especially very mobileregions)
– these can be left as gaps in the 3D structure, or simply be “fuzzy”
• R factor: measure of agreement (actually divergence: smaller is better)between calculated structure and experimental data
– 0.25 (for good protein structures of normal size)
– 0.05 (for small molecules)
– Completely wrong structures can have an R factor of 0.59
• It is not unusual for many protein structures to have some errors,ambiguities or inaccuracies in atomic positions (+/- 0.5 Angstroms), orto be missing atoms and residues (e.g., TrwB, PDB 1E9RA)
Baxevanis & Ouellette (Ch. 9, Wishart)

21
NMR spectroscopy• Much newer: first NMR structure in 1983
• Allows biologists to study structure and dynamics of molecules in liquid state(or near-physiological environment)
• Structures solved by measuring how radio waves are absorbed by atomicnuclei
• Absorption measurement allows the determination of how much nuclearmagnetism is transferred from one atom (or nucleus) to another
– Magnetization transfer measured through chemical shifts, J-couplings and nuclearOverhauser effects
– Measured parameters define a set of approximate structural constraints that are fedinto a constraint minimization calculation (distance geometry or simulated annealing)
– Result is an ensemble of (15-50) of structures that satisfy the experimentalconstraints
– These multiple structures are overlaid/superimposed on each other to produce“blurrograms”
• NMR result is potentially more reflective of true solution behavior of proteins;most proteins seem to exist in an ensemble of slightly different configurations
Baxevanis & Ouellette (Ch. 9, Wishart)
Limitations of NMR spectroscopy
• Size limitations: maximum of 30kD (~250aa)
• Solubility of molecule
– cannot be applied to membrane proteins
• Expensive: requires special isotopically labeledmolecules
• Inherently less precise
Baxevanis & Ouellette (Ch. 9, Wishart)

22
Structural superposition
• Superposing protein structures can reveal startlingstructural similarities not apparent from primarysequence comparison
• Structural superposition is the basis for evaluatingmany alignment methods
• Structural superposition has a great degree ofvariability across methods
• Also: proteins can be solved in a bound or unboundconformation
– The two conformations can be significantly different
– Flexible superposition is important
• Popular methods include: DALI, CE, VAST, Structal
Storing and retrieving protein structures
• The Protein Data Bank (PDB)
– First electronic database in bioinformatics
– Set up at Brookhaven National Laboratory by Walter Hamilton in 1971
• 7 protein structures at database initiation
• Coordinates stored and distributed on punch cards and computer tape
– Currently
• 22K structures (as of October 23, 2005) (~46K as of October 2007)
• Coordinate distribution and deposition is electronic (via the world wide web)
• Moved to the Research Collaboratory for Structural Bioinformatics (RSCB) in1998
• Primary archival center for experimentally determined 3D structures of proteins,nucleic acids, carbohydrates and complexes
• Separate repository for theoretical models
Baxevanis & Ouellette (Ch. 9, Wishart)
23
http://www.usm.maine.edu/~rhodes/ModQual/index.html
http://www.usm.maine.edu/~rhodes/ModQual/index.html

24
Structural Genomics Initiative
From Andras Fiser
Albert Einstein College of Medicine
Why is it useful to know the structure of a
protein not only its sequence?
The 3D structure is more informative than sequence because patterns in space arefrequently more recognizable than patterns in sequence
Evolution tends to conserve function and
function depends more directly on structure
than on sequence, structure is more
conserved in evolution than sequence.
Andras Fiser, Albert Einstein College of Medicine
25
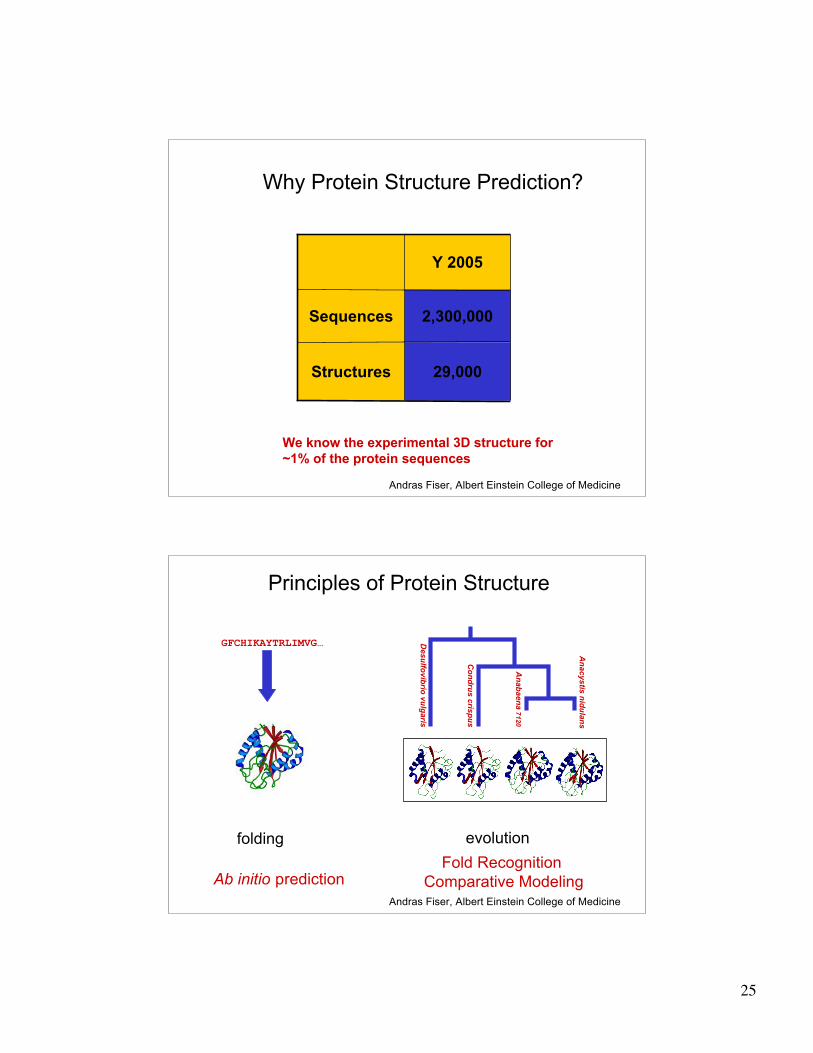
Why Protein Structure Prediction?
Y 2005
29,000Structures
2,300,000Sequences
We know the experimental 3D structure for
~1% of the protein sequences
Andras Fiser, Albert Einstein College of Medicine
Principles of Protein Structure
GFCHIKAYTRLIMVG…
An
ab
ae
na
7120
An
acystis
nid
ula
ns
Co
nd
rus c
risp
us
Desu
lfovib
rio v
ulg
aris
Ab initio predictionFold Recognition
Comparative Modeling
folding evolution
Andras Fiser, Albert Einstein College of Medicine

26
Protein structure modeling
Ab initio prediction Comparative Modeling
Applicable to any sequence
Not very accurate (>4 Ang RMSD),
Attempted for proteins of <100 residues
Accuracy and applicability are limited
by our understanding of the protein
folding problem
Applicable to those sequences only that
share recognizable similarity to a template
structure
Fairly accurate ( <3 Ang RMSD), typically
comparable to a low resolution X-ray
experiment.
Not limited by size
Accuracy and applicability are rather
limited by the number of known folds
Andras Fiser, Albert Einstein College of Medicine
I A small difference in the sequence makes a small
difference in the structure
II Protein structures are clustered into fold families
What makes comparative modeling possible
Andras Fiser, Albert Einstein College of Medicine

27
Structural Genomics
The number of “families” is
much smaller than the number
of proteins
Characterize most protein sequences (red) based on related
known structures (green).
Andras Fiser, Albert Einstein College of Medicine
Structural Genomics
Definition: The aim of structural genomics is to put everyprotein sequence within a “modeling distance” of aknown protein structure.
Size of the problem:
There are a few thousand domain fold families.
There are ~20,000 sequence families (30% sequence id).
Solution:
Determine protein structures for as many differentfamilies as possible.
Model the rest of the family members using comparativemodeling
Andras Fiser, Albert Einstein College of Medicine

28
Protein folding
Information required for folding is (mostly)contained in the primary sequence
• Early on, proteins were shown to fold into their native structures
in isolation
• This led to the belief that structure is determined by sequence
alone (Anfinsen, 1973)
• Over the last decade, a significant number of proteins have
been shown to not fold properly in the test tube (e.g., requiring
the assistance of chaperonins)
• Nevertheless, the native 3D structure is assumed to be in some
energetic minimum
• This led to the development of ab initio folding methods
Baxevanis & Ouellette (Ch. 9, Wishart)

29
Folding pathways
• Evidence that local structure segments form first, and
then pack against each other to form 3D fold
– Exploited in protein fold prediction, Rosetta method
• Simons, Bonneau, Ruczinski & Baker (1999). Ab initio Protein
Structure Prediction of CASP III Targets Using ROSETTA. Proteins
• Semi-stable structural intermediates on folding pathway
to lowest-energy conformation
– Prof. Susan Marqusee, Berkeley
Baxevanis & Ouellette (Ch. 9, Wishart)
Structural studies have providedinsights into protein folding
• When high-resolution studies of myoglobin became available, Kendrewnoticed that amino acids in the interior had almost exclusivelyhydrophobic side chains
• Over time, structural studies have shown the following:
– The main driving force for folding water-soluble globular proteins is topack hydrophobic side chains into the interior, thus creating ahydrophobic core and a hydrophilic surface
– Bringing hydrophobic side chains into the interior requires that the highly polarmain chain (with one hydrogen bond donor, NH, and one hydrogen bondacceptor, C’=0) is brought along
– In a hydrophobic environment, these main-chain polar groups must beneutralized by the formation of hydrogen bonds
– This problem is solved elegantly by the formation of regular secondarystructure in the interior of the molecule
– Internal cavities are usually occupied by water molecules that hydrogen-bondto internal polar groups
From Introduction to Protein Structure (Branden & Tooze)

30
Hierarchical descriptions of proteins(follows the folding process)
• Primary structure: the amino acid sequence
• Secondary structure: “regular local structure of linear segments of polypeptide chains”(Creighton)
– Helix (~35% of residues): subtypes: !, " and 310
– Beta sheet (~25% of residues)
– Both types predicted by Linus Pauling (Corey and Pauling, 1953; ! helix first described by Pauling in 1951)
– Other less common structures:
• Beta turns
• 3/10 helices
• ! loops
– Remaining unclassifiable regions termed “random coil” or “unstructured regions”
– http://www.chembio.uoguelph.ca/educmat/phy456/456lec01.htm
• Tertiary structure: “Overall topology of the folded polypeptide chain” (Creighton)
– Mediated by hydrophobic interactions between distant parts of protein
• Quaternary structure: “Aggregation of the separate polypeptide chains of a protein”(Creighton)
Baxevanis & Ouellette (Ch. 9, p.224, Wishart)
Folded conformations of globularproteins
• Most proteins are globular: natural proteins in solution are muchsmaller in their dimensions than comparable polypeptides withrandom or repetitive conformations and have roughly sphericalshapes
• Denaturation: Most proteins are robust to changes in theirenvironment, until they (somewhat literally) fall apart:
– Most proteins are robust to changes in temperature, pH andpressure, exhibiting little or no change until a point is reached atwhich there is a sudden change and loss of biological function
– Denaturing proteins has been used to explore folding pathways
e.g.,Understanding how proteins fold: the lysozyme story so far.Dobson CM, EvansPA, Radford SE.Trends Biochem Sci. 1994
Creighton, Proteins Ch. 6

31
Structural domains
• Folded structures of most small proteins are roughly spherical andremarkably compact
• Proteins with >200aa tend to consist of >2 structural units, calleddomains
• Domains interact to varying extents, but less extensively than dostructural elements within domains
– Some domain detection tools make use of this pattern, looking forcovariation between positions as evidence of interaction, and lack ofcovariation as evidence of domain boundaries
• Nagarajan and Yona, Automatic prediction of protein domains from sequenceinformation using a hybrid learning system. Bioinformatics 2004
• Domains may not always be well segregated; some proteins havemultiple domains with 2 or three polypeptide connections betweendomains
– See, for example, the SCOP “interleaved” domains
• Domains may also be connected by flexible “linker” regions
Creighton, Proteins Ch. 6
Structural domains (cont’d)
• Definition of domain is a subjective process done in different waysby different people
• Domains are most evident by their compactness
– Expressed quantitatively as the ratio of the surface area of a domain tothe surface area of a sphere with the same volume
• Observed values are 1.65+/- 0.08
• Course of polypeptide backbone through domain is irregular, butgenerally follows moderately straight course through the domainand then makes a U-turn to re-cross the domain
• Overall impression: segments of somewhat stiff polypeptide chaininterspersed with relatively tight turns or bends (almost always onthe molecule’s surface)
– Compared to behavior of a fire hose dropped in one spot
Creighton, Proteins Ch. 6

32
Driving forces in protein folding
• Complex combination of local and globalforces
– Local forces drive secondary structure formation
• Repulsion between hydrophobic side chains of someamino acids and hydrophilic backbone of protein chain(intra-molecular)
• Interaction between side chains and surrounding solvent
– Subcellular environment (e.g., membrane, secreted, etc.)
– Pauling et al 1951
Baxevanis & Ouellette (Ch. 9, Wishart)
Summary of driving forces in proteinfolding
• Hydrophobicity
– Hydrophobic residues need to be shielded from solvent
• Polar residues to the outside, hydrophobic to the inside
• Stronger interactions
– Hydrogen bonds, disulfide bridges
• Weak interactions
– Van der Waals, electrostatic, etc
Recommended reading: Proteins (Thomas Creighton).

33
Global effects on protein fold
• Long-range interactions (repulsive or
attractive) between distant parts of structure
– These can override local effects
– E.g., chameleon protein:
• 11 amino acids adopt helical structure in one region, and
the same 11 amino acids adopt beta strand in another.
» Minor & Kim, 1996
Baxevanis & Ouellette (Ch. 9, Wishart)


















