
Standards and Best Practices for Cell, Gene and Tissue ... · Raw and Ancillary materials – USP ...
34
Standards and Best Practices for Cell, Gene and Tissue-based Therapies Rebecca Potts, Ph.D. July 18, 2017
Transcript of Standards and Best Practices for Cell, Gene and Tissue ... · Raw and Ancillary materials – USP ...

Standards and Best Practices for
Cell, Gene and Tissue-based Therapies
Rebecca Potts, Ph.D.
July 18, 2017

Introduction- USP
USP <1046> Cellular and Tissue-based Products
– Quality Systems, Qualification of Materials
– Manufacturing of Cell and Tissue-based Products
– Release Tests for Cell and Tissue-based Products
USP <1047> Gene Therapy Products
– Manufacturing Gene Therapy Products, Safety Concerns
– Analytical Methods for assessing gene therapy product quality
Raw and Ancillary materials
– USP <1043> Ancillary Materials for Cell, Gene and Tissue-
Engineered Products
– Risk-based categories of Ancillary Materials
Reference Standards - Examples
Conclusions2
Outline
3
U.S. Pharmacopeial Convention (USP)
Work with more than 900
scientists, practitioners
and regulators to develop
standards that help
protect public health
Internationally recognized
and globally focused
Headquarters in Rockville,
MD
Laboratory facilities
in U.S., India, China,
Brazil and Ghana
Offices in Switzerland,
Ethiopia, Indonesia, the
Philippines and Nigeria
Founded in 1820,
nonprofit, private,
independent and
self-funded
Values-driven
organization focused
on quality standards to
protect the public’s health
More than 1,000
employees worldwide
4
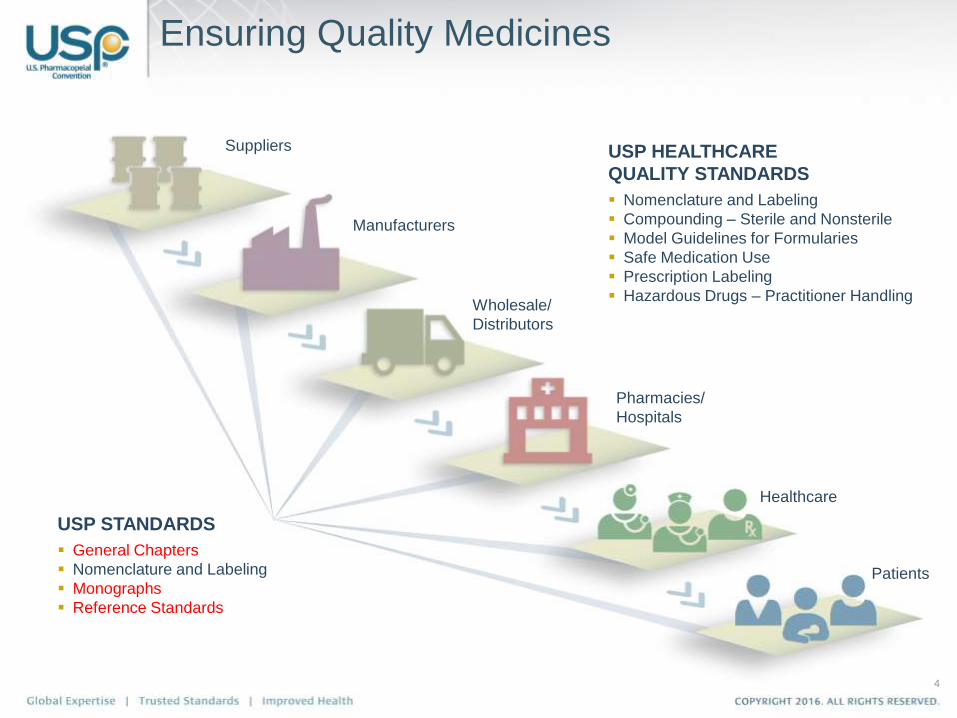
Ensuring Quality Medicines
Suppliers
Manufacturers
Wholesale/
Distributors
Pharmacies/
Hospitals
Healthcare
Patients
USP STANDARDS
General Chapters
Nomenclature and Labeling
Monographs
Reference Standards
USP HEALTHCARE
QUALITY STANDARDS
Nomenclature and Labeling
Compounding – Sterile and Nonsterile
Model Guidelines for Formularies
Safe Medication Use
Prescription Labeling
Hazardous Drugs – Practitioner Handling

Introduction- USP
USP <1046> Cellular and Tissue-based Products
– Quality Systems, Qualification of Materials
– Manufacturing of Cell and Tissue-based Products
– Release Tests for Cell and Tissue-based Products
USP <1047> Gene Therapy Products
– Manufacturing Gene Therapy Products, Safety Concerns
– Analytical Methods for assessing gene therapy product quality
Raw and Ancillary materials
– USP <1043> Ancillary Materials for Cell, Gene and Tissue-
Engineered Products
– Risk-based categories of Ancillary Materials
Reference Standards - Examples
Conclusions
5
Outline

Cellular product: living human or animal cells or tissue
that have been manipulated or are used in ways that result in
their regulation as somatic cellular therapies as defined by the
U.S. Food and Drug Administration (FDA)
Tissue-based product: human tissues that do not require
pre-market approval by FDA and only need to comply with good
tissue practices (GTP regulations)
Combination product: cells combined with medical
devices such as natural or synthetic scaffold
* <1046> is undergoing revision
6
Cell, Gene and Tissue-based Products
Definitions— USP <1046>
Source Material for Cell and Tissue-based products: human
and animal derived cells
– Procurement of HCT/Ps for medical use in adherence with donor eligibility
requirements
Isolation
– Solid organs or tissues are usually dissected to expose desired
region
– Enzymatic digestion of extracellular connective tissue is another
common method for dissecting tissue.
Selection
– Varying force during centrifugation selectively enrich cell
populations
– Monoclonal antibodies directed against specific cell-surface
proteins used for both negative and positive selection
7
Source Material for Cell and Tissue-Based Products

Manufacturers of cellular or tissue-based products:
– Ensure that all components used in manufacturing are
appropriately qualified
– Components used in production: source cells and tissues,
natural or synthetic biomaterials, ancillary material and excipients
used in formulation products
Qualification: process of acquiring and evaluating data to establish
the source, identity, purity, biological safety and overall suitability of a
specific component
Material qualification more comprehensive as the product
progresses in development.
Testing for key quality attributes is critical
Cell-based therapies regulated as biological products must comply
with applicable sections of 21 CFR 211 and 21 CFR 610
8
Quality Systems for Components in Cellular and
Tissue-based Products

Analytical methods are used to establish in-process controls
and product release criteria.
Quality specifications for cell and tissue products confirm the
product’s quality, safety and potency.
Specifications are established through characterization of the
product during development phase.
Terminal sterilization is not possible for a living cell-based
product. Cell-based products are required to meet
acceptance criteria for product tests such as sterility,
mycoplasma and endotoxin.
In-process controls and specifications should be anchored by
the use of an appropriate reference standard.
9
Analytical Methods for Cell and Tissue-based
Products

Identity
– Lot release testing for cell-based products must include an
identity test
– Identity test serves to unequivocally identify the product
• Differential surface markers are frequently used to ascertain
product identity
• Cell morphology may also be used to distinguish specific cell types
Purity
– Purity methods specifically quantify the intended active product
components
– Testing for impurities as part of lot release should reflect the
safety risks associated with the impurity and the ability of the
process to remove the impurity
10
Final Product Release Testing for Cell and
Tissue-base Products

Potency
– With dose, potency defines the biological activity of each lot.
– Potency may be assessed by in vitro or in vivo bioassays, or a
combination of the two
• Assays require a well-defined reference material that can serve as
a positive control for the assay. The positive control serves to
qualify the performance of an individual assay
Dose-Defining Assays
– An assay that precisely measures the amount of product
– Cell therapy products may be dosed on the basis of
enumeration of one or more cell populations
11
Final Product Release Testing for Cell and
Tissue-base Products (Continued)
12
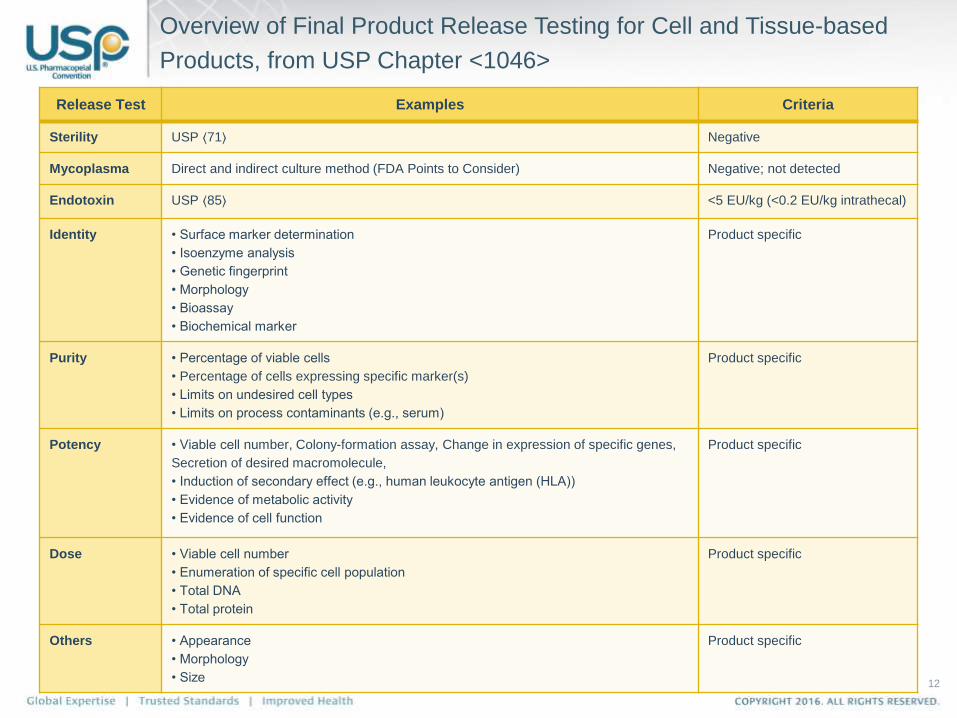
Overview of Final Product Release Testing for Cell and Tissue-based
Products, from USP Chapter <1046>
Release Test Examples Criteria
Sterility USP ⟨71⟩ Negative
Mycoplasma Direct and indirect culture method (FDA Points to Consider) Negative; not detected
Endotoxin USP ⟨85⟩ <5 EU/kg (<0.2 EU/kg intrathecal)
Identity • Surface marker determination
• Isoenzyme analysis
• Genetic fingerprint
• Morphology
• Bioassay
• Biochemical marker
Product specific
Purity • Percentage of viable cells
• Percentage of cells expressing specific marker(s)
• Limits on undesired cell types
• Limits on process contaminants (e.g., serum)
Product specific
Potency • Viable cell number, Colony-formation assay, Change in expression of specific genes,
Secretion of desired macromolecule,
• Induction of secondary effect (e.g., human leukocyte antigen (HLA))
• Evidence of metabolic activity
• Evidence of cell function
Product specific
Dose • Viable cell number
• Enumeration of specific cell population
• Total DNA
• Total protein
Product specific
Others • Appearance
• Morphology
• Size
Product specific

Stability of cell or tissue-based products and the
components used to create them will vary depending on
the nature of the product, the intended clinical use,
specific attributes, storage, packaging and shipping
conditions.
Stability studies must verify that the storage conditions
maintain the quality attributes of the product
Where feasible, stability testing should be carried out in
accordance with the principles described in ICH
guideline Q5C.
13
Stability of Cell or Tissue-based Products

Introduction- Background
USP <1046> Cellular and Tissue-based Products
– Quality Systems, Qualification of Materials
– Manufacturing of Cell and Tissue-based Products
– Release Tests for Cell and Tissue-based Products
USP <1047> Gene Therapy Products
– Manufacturing Gene Therapy Products, Safety Concerns
– Analytical Methods for assessing gene therapy product quality
Raw and Ancillary materials
– USP <1043> Ancillary Materials for Cell, Gene and Tissue-
Engineered Products
– Risk-based categories of Ancillary Materials
Reference Standards - Examples
Conclusions14
Outline

Gene therapy product: the administration of nucleic acids to
modify the genetic material of cells
– Delivery of nucleic acids can be through
• viral vectors, nucleic acid in simple formulation (naked DNA) or
nucleic acids formulated with agents that enhance the ability to
penetrate the cell
– Genetic material introduced can either cause expression of new genes
or inhibit expression of already expressed genes and products
Cell Therapy products can be modified by treatment with
integrating or non-integrating genetic materials (DNA, RNA,
siRNA), so that the pattern of gene expression is changed.
– Typically cells are taken from the patient and modified outside the body
before they are returned to the patient
– Regulatory bodies consider ex-vivo gene modified cellular product to be
a gene therapy product.
15
Gene Therapy Products

Manufacturing of gene therapy products requires:
Choosing a vector:
– The vector will include: the vector backbone, a promoter, the
therapeutic gene and a polyadenylation signal
Targeting Transduction
– Vector must find and transduce the target cell
– Viruses have a natural host range that is influenced by the
expression of cell-surface receptors, the cell cycle and the route
of administration
– Barrier to transduction is humoral immune response to the vector
16
Manufacturing Gene Therapy Products

Safety Testing focuses on 3 issues:
– Detecting contamination from adventitious sources during product
processing
– Preventing the use of packaging cell lines and plasmids that permit
genetic recombination between packaging cell lines or plasmids
– Testing the final product to ensure a safe level of undesired genetic
and/or structural variants or other viruses used in processing
Viral Gene Therapy Products
– Occurrence of undesired genetic variants
Non-Viral Gene Therapy Products
– Safety testing should be performed on non-viral formulated material
– Focuses on methods similar to those described in USP <88>
Biological Reactivity Tests, In Vivo and <71> Sterility
17
Safety Concerns with Gene Therapy Products

Approved gene therapy products must comply with applicable
sections of 21 CFR 211 and 610 to ensure identity, dose, potency,
purity and safety
Samples must be retained after product release testing is completed.
In-Process Controls
– Manufacturing processes should have well-defined decision criteria that
are established for key in-process manufacturing steps
Specifications
– Specifications for gene therapy product should be chosen to ensure the
safety and efficacy of the product before use
– Development and setting of specifications for cell and gene therapies
should follow the principles outlined in ICH Q6B
– Procedures in a specification for a product should be anchored by
appropriate reference standards
18
Analytical Methods for Assessing Product Quality
of Gene Therapy Products

Dose Defining Assays
– Precisely measures the amount of product
– Example: particle concentration is a commonly used measure
for viral vector dose
Potency
– Defined as the therapeutic activity of the drug product. Dose
and potency define the biological activity of each lot
– Bioassays to measure the potency of viral and non-viral gene
therapies involve infection or transfection of susceptible cell line
in vitro, followed by a measurement of the expressed gene of
interest
19
Tests for Gene Therapy Products

Purity
– Process related impurities for gene therapy products include
residual production medium components (ie FBS, antibiotics,
cytokines, E.coli chromosomal DNA in a plasmid product)
– Product related impurities are specific for each type of gene
therapy product (viral or nonviral)
– Viral gene therapy product-related impurities include aggregates and
defective immature particles that may be produced during the
manufacture or purification of the recombinant vector
– Nonviral gene therapy product-related impurities include nicked
plasmid forms
Identity
– Lot release testing for gene therapy products must include an identity
test
– Identity test clearly identifies the product and confirms that
inadvertent substitution with another product has not occurred.
20
Tests for Gene Therapy Products (continued)
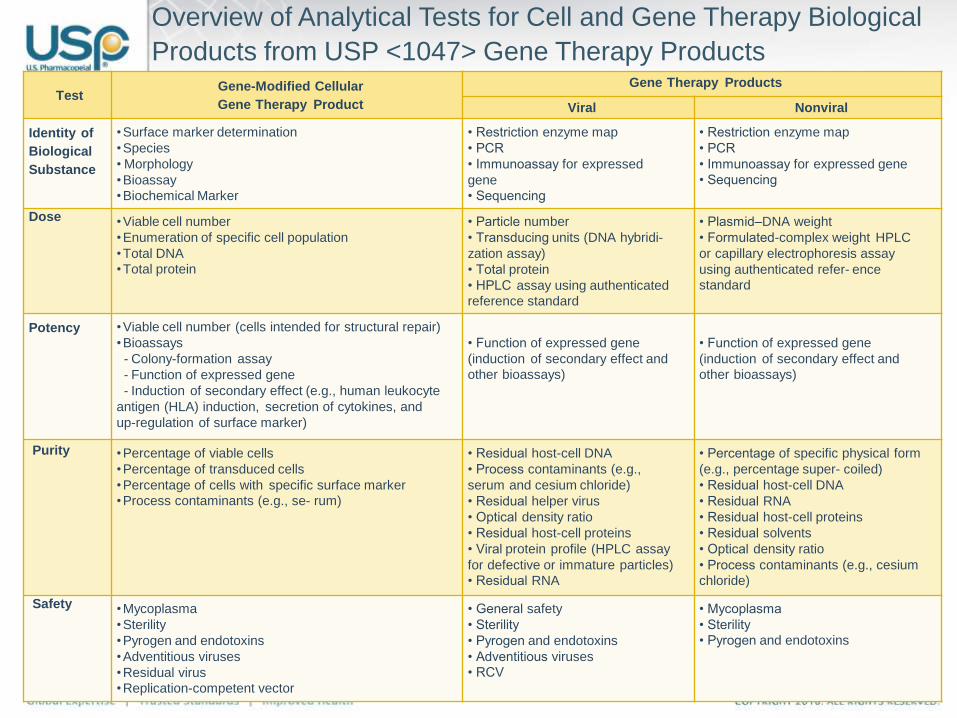
21
Overview of Analytical Tests for Cell and Gene Therapy Biological
Products from USP <1047> Gene Therapy Products
TestGene-Modified Cellular
Gene Therapy Product
Gene Therapy Products
Viral Nonviral
Identity of
Biological
Substance
•Surface marker determination
•Species
• Morphology
•Bioassay
•Biochemical Marker
• Restriction enzyme map
• PCR
• Immunoassay for expressed
gene
• Sequencing
• Restriction enzyme map
• PCR
• Immunoassay for expressed gene
• Sequencing
Dose •Viable cell number
•Enumeration of specific cell population
•Total DNA
•Total protein
• Particle number
• Transducing units (DNA hybridi-
zation assay)
• Total protein
• HPLC assay using authenticated
reference standard
• Plasmid–DNA weight
• Formulated-complex weight HPLC
or capillary electrophoresis assay
using authenticated refer- ence
standard
Potency •Viable cell number (cells intended for structural repair)
•Bioassays
- Colony-formation assay
- Function of expressed gene
- Induction of secondary effect (e.g., human leukocyte
antigen (HLA) induction, secretion of cytokines, and
up-regulation of surface marker)
• Function of expressed gene
(induction of secondary effect and
other bioassays)
• Function of expressed gene
(induction of secondary effect and
other bioassays)
Purity •Percentage of viable cells
•Percentage of transduced cells
•Percentage of cells with specific surface marker
•Process contaminants (e.g., se- rum)
• Residual host-cell DNA
• Process contaminants (e.g.,
serum and cesium chloride)
• Residual helper virus
• Optical density ratio
• Residual host-cell proteins
• Viral protein profile (HPLC assay
for defective or immature particles)
• Residual RNA
• Percentage of specific physical form
(e.g., percentage super- coiled)
• Residual host-cell DNA
• Residual RNA
• Residual host-cell proteins
• Residual solvents
• Optical density ratio
• Process contaminants (e.g., cesium
chloride)
Safety •Mycoplasma
•Sterility
•Pyrogen and endotoxins
•Adventitious viruses
•Residual virus
•Replication-competent vector
• General safety
• Sterility
• Pyrogen and endotoxins
• Adventitious viruses
• RCV
• Mycoplasma
• Sterility
• Pyrogen and endotoxins

Stability studies verify that the storage conditions
maintain the purity and potency of the product for a
defined period so the product administered to the
patient is still capable of meeting the stability
specifications
Stability assessment should include evaluation of
product functionality (potency)
Stability studies analogous to accelerated aging
studies typically used in pharmaceutical stability
monitoring programs
22
Stability of Gene Therapy Products

23
Outline
Introduction- Background
USP <1046> Cellular and Tissue-based Products
– Quality Systems, Qualification of Materials
– Manufacturing of Cell and Tissue-based Products
– Release Tests for Cell and Tissue-based Products
USP <1047> Gene Therapy Products
– Manufacturing Gene Therapy Products, Safety Concerns
– Analytical Methods for assessing gene therapy product quality
Raw and Ancillary materials
– USP <1043> Ancillary Materials for Cell, Gene and Tissue-
Engineered Products
– Risk-based categories of Ancillary Materials
Reference Standards - Examples
Conclusions

Raw and Ancillary Materials are used in Manufacturing
Cell, Gene Therapy and Tissue-based Products
Raw materials may or may not remain in the final
therapeutic product as active substances or as excipients
Ancillary materials are a subset of raw materials.
– Ancillary products may exert an effect on a therapeutic
substance (e.g. a cytokine may activate a population of
cells), but they are not intended to be in final formulation
Some components are more critical than others. Risk
assessment strategies are required to ensure quality
– A critical material will come in contact with cells with a
potential to alter either the growth characteristics of the cells
or the ability of the cell culture to meet lot release
specifications.

Ancillary materials for Cell Therapy/Tissue-based products
– Culture media, buffers, growth factors, cytokines, cultivation and
processing components, monoclonal antibodies and cell-
separation devices
– Residual amounts of ancillary materials used in the manufacturing
process may be antigenic, so the removal must be assessed and
limits on the amount remaining must be set in some cases.
Ancillary materials for Gene Therapy products
– Raw materials used in manufacturing: cells, tissues, biological
fluids, growth factors and monoclonal antibodies
– Quality of the ancillary materials can affect the safety, potency and
purity of the product.
– Qualification of raw and ancillary materials used in manufacturing
Gene Therapy products is necessary
25
Ancillary Materials Used in Cell and Tissue-based
Products and Gene Therapy Products

Raw Materials: Quality Approaches
Reliance on Suppliers
– Specifications: lot to lot consistency and materials for multiple use
– Test methods: are these validated?
– Certificate of Analysis (CoA)
– Pharmacopeial procedures and which pharmacopeia?
– Testing beyond the CoA
Risk-based approaches and critical raw materials
– Use of multi-suppliers materials
– Impact of materials on final product (quality attributes, residuals)
– Define failures mode -use of quantitative tools- and mitigate risk
– Example: USP <1043>, Ancillary Materials (AMs) used in cell
therapy manufacturing

27
Risk-based Approach USP Chapter <1043>Revision available online July 3, 2017
Criteria that define the level of risk
Tier 1:
Low risk
Intended for use as licensed drugs, biologics or medical devices▪
• Suitability for use as a manufacturing component is required
• Quality aspects of these materials may change after use in the manufacturing
process.
Tier 2:
Low risk
Intended to be used as ancillary materials.
Well-characterized
Produced under quality systems well-suited for biological manufacturing,
Material is not a licensed medical product.
Many are produced specifically for the manufacture of biological products.
Tier 3:
Moderate risk
These are research-grade materials not intended for use in biological
manufacturing
• Sometimes approved by regulatory agencies as part of an in vitro diagnostic
device.
More qualification than Tier 1 or Tier 2 materials.
Tier 4:
High risk
Produced as industrial or research-grade materials and may contain harmful
impurities.
May also contain animal- or human-derived components with potential
contaminants.
Requires extensive qualification before use as component in biological product
manufacturing.
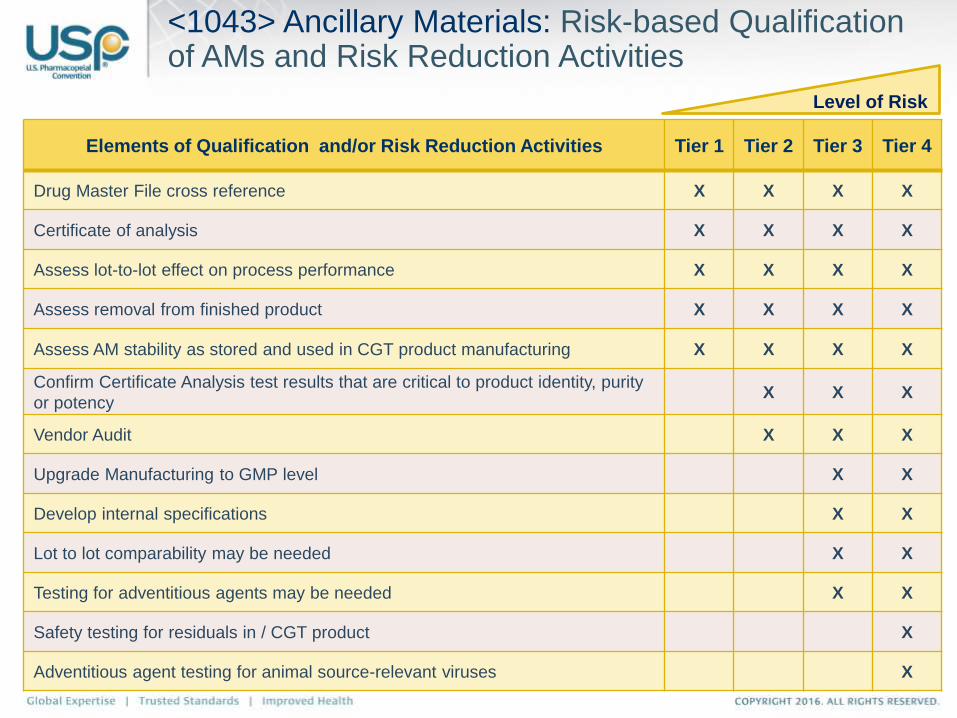
<1043> Ancillary Materials: Risk-based Qualification of AMs and Risk Reduction Activities
Elements of Qualification and/or Risk Reduction Activities Tier 1 Tier 2 Tier 3 Tier 4
Drug Master File cross reference X X X X
Certificate of analysis X X X X
Assess lot-to-lot effect on process performance X X X X
Assess removal from finished product X X X X
Assess AM stability as stored and used in CGT product manufacturing X X X X
Confirm Certificate Analysis test results that are critical to product identity, purity
or potencyX X X
Vendor Audit X X X
Upgrade Manufacturing to GMP level X X
Develop internal specifications X X
Lot to lot comparability may be needed X X
Testing for adventitious agents may be needed X X
Safety testing for residuals in / CGT product X
Adventitious agent testing for animal source-relevant viruses X
Level of Risk

USP’s Ancillary Materials Standards
<1043> Ancillary Materials (AMs) for Cell, Gene, and Tissue-based Products
Specific AM Chapters
<90> FBS Quality Attributes
<130> Protein A
<92> Cytokines and Growth Factors<89> Enzymes
Reference Standards:
- FBS- Protein A- Interleukin-4- Trypsin- Collagenase I and II
General
Information
Chapter
Ancillary Material
Requirements for
Specific AMs
Ancillary Materials
Reference
Standards

Introduction- USP
USP <1046> Cellular and Tissue-based Products
– Quality Systems, Qualification of Materials
– Manufacturing of Cell and Tissue-based Products
– Release Tests for Cell and Tissue-based Products
USP <1047> Gene Therapy Products
– Manufacturing Gene Therapy Products, Safety Concerns
– Analytical Methods for assessing gene therapy product quality
Raw and Ancillary materials
– USP <1043> Ancillary Materials for Cell, Gene and Tissue-
Engineered Products
– Risk-based categories of Ancillary Materials
Reference Standards - Examples
Conclusions30
Outline

USP Standards for Fetal Bovine Serum (FBS)
FBS Standard Tests
– Identification: Radial Immunodiffusion (RID): species ID, IgG levels
– Functionality Assays (Growth Curve and Clonal Assay)
– Osmolality: 280-360 mOsm/Kg
– Total Protein: 30-45 mg/mL
– pH: 7.00 - 8.00
– Endotoxin: Less Than 10 units/mL
– Hemoglobin level: Less than 30 mg/dL
– Sterility <71>, meets requirements
Associated Reference Standard (RS)
32
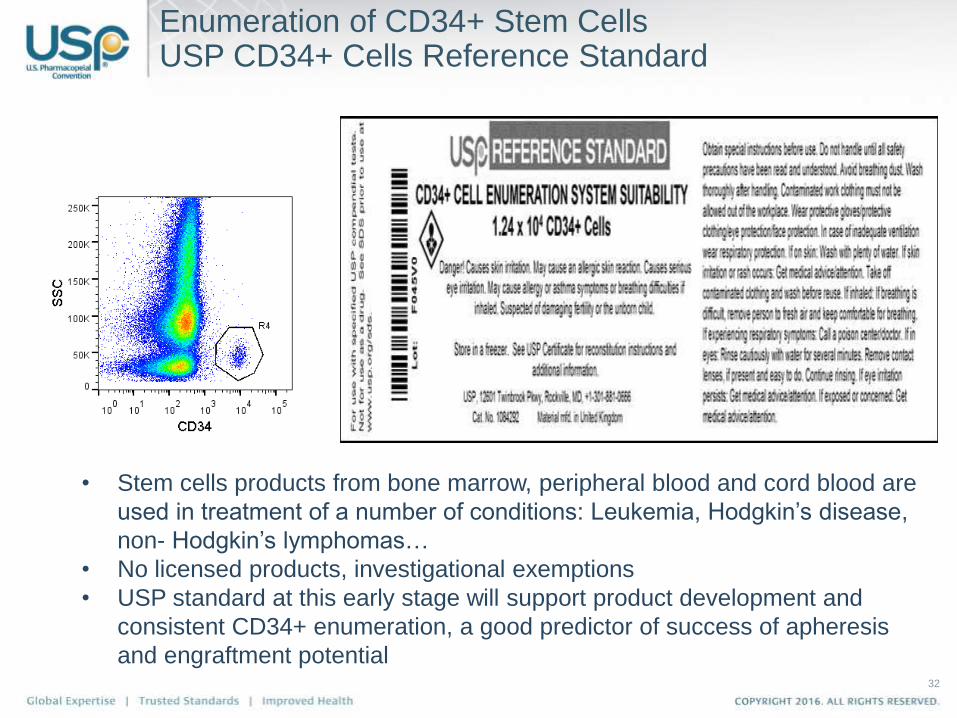
Enumeration of CD34+ Stem Cells USP CD34+ Cells Reference Standard
• Stem cells products from bone marrow, peripheral blood and cord blood are
used in treatment of a number of conditions: Leukemia, Hodgkin’s disease,
non- Hodgkin’s lymphomas…
• No licensed products, investigational exemptions
• USP standard at this early stage will support product development and
consistent CD34+ enumeration, a good predictor of success of apheresis
and engraftment potential

Manufacturers of cell, gene and tissue-based products must
ensure that all components used in manufacturing are
appropriately qualified
Quality specifications for cell, gene and tissue products should
confirm the product’s quality, safety and potency
Standardization of ancillary materials used in manufacturing
allows to control consistency in manufacturing of finished
products
Pharmacopeial standards provide tools for the control of
products development and testing strategies, the standards
can also be used to demonstrate compliance with regulatory
requirements
USP is working on a strategies to include additional standards
for ancillary materials in USP-NF
33
Conclusions

34


















