
Sonchus Yellow Net Rhabdovirus Nuclear Viroplasms Contain...
11
JOURNAL OF VIROLOGY, 0022-538X/98/$04.0010 July 1998, p. 5669–5679 Vol. 72, No. 7 Copyright © 1998, American Society for Microbiology. All Rights Reserved. Sonchus Yellow Net Rhabdovirus Nuclear Viroplasms Contain Polymerase-Associated Proteins CLAUDIA R. F. MARTINS,² JENNIFER A. JOHNSON, DIANE M. LAWRENCE, TAE-JIN CHOI,‡ ANNA-MARIA PISI,§ SARA L. TOBIN,\ DENISE LAPIDUS, JOHN D. O. WAGNER,# STEVEN RUZIN, KENT MCDONALD,²² AND ANDREW O. JACKSON* Department of Plant and Microbial Biology, University of California, Berkeley, California 94720 Received 16 December 1997/Accepted 23 March 1998 We have initiated a study of the cytopathology of nucleorhabdoviruses by analyzing the subcellular local- ization of sonchus yellow net virus (SYNV) genomic and antigenomic RNAs and the encoded polymerase proteins. In situ hybridizations demonstrated that the minus-strand genomic RNA sequences are restricted to the nuclei of infected cells, while the complementary plus-strand antigenomic RNA sequences are present in both the nuclei and the cytoplasm. Immunofluorescence and immunogold labeling experiments also revealed that the nucleocapsid (N) protein and phosphoprotein (M2) are primarily localized to discrete regions within the nuclei and in virus particles that accumulate in perinuclear spaces. The N protein antiserum specifically labeled the nuclear viroplasms, whereas the M2 antiserum was more generally distributed throughout the nuclei. Antibody detection also indicated that the polymerase (L) protein is present in small amounts in the viroplasm. When the N and M2 proteins were expressed individually from the heterologous potato virus X (PVX) vector, both proteins preferentially accumulated in the nuclei. In addition, viroplasm-like inclusions formed in the nuclei of cells infected with the PVX vector containing the N gene. Fusions of the carboxy terminus of b-glucuronidase to N and M2 resulted in staining of the nuclei of infected cells following expression from the PVX vector. Deletion analyses suggested that multiple regions of the N protein contain signals that are important for nuclear localization. Sonchus yellow net virus (SYNV) belongs to the Rhabdoviri- dae family of nonsegmented, negative-strand RNA viruses. This family is one of the most widely distributed virus families in nature, and it contains members that infect animals, plants, and insects (47). With this diversity, it is not surprising that individual members of the family differ with respect to require- ments for infection and the sites of viral replication and mor- phogenesis. All of the well-characterized animal rhabdoviruses replicate in the cytoplasm; however, the plant rhabdoviruses are divided into two groups depending on whether their mor- phogenesis occurs in association with cytoplasmic or nuclear membranes (16). The cytorhabdoviruses, which replicate in the cytoplasm and bud in association with the endoplasmic retic- ulum, include lettuce necrotic yellows virus, strawberry crinkle virus, and barley yellow striate mosaic virus, among others. The nucleorhabdoviruses, of which SYNV, sowthistle yellow vein virus, potato yellow dwarf virus, and maize mosaic virus are members, appear to replicate in the nucleus and bud from the inner nuclear envelope into perinuclear spaces. Furthermore, over 50 plant rhabdoviruses have yet to be assigned to specific groups because of insufficient information about their replica- tion and morphogenesis (50). SYNV is the most extensively characterized plant rhabdovi- rus, and its genome has been completely sequenced (5, 6, 10, 12, 14, 42, 51, 52). The 13,720-nucleotide (nt) SYNV RNA encodes six proteins in a negative-sense orientation (see Fig. 1), and all of these are found in association with virus particles (42). A ribonucleoprotein core that can be released from viri- ons (17), and from the nuclei of infected plants (44), consists of the nucleocapsid (N) protein, the phosphoprotein (M2), and the polymerase (L) protein complexed with SYNV RNA. The glycoprotein (G) is thought to traverse the viral lipid envelope and associate with the matrix (M1) protein, which probably also has a role in coiling of the ribonucleoprotein core (16). A sixth protein, sc4, has no known function but can be released from virions by treatment with mild nonionic detergents (42). Two putative regulatory regions, located at the 39 and 59 ends of the SYNV genome, flank the six minus-sense genes. The 144-nt leader sequence is located at the 39 end of the genome (44, 51), and the 160-nt untranslated trailer sequence is present at the 59 end of the genomic RNA. The transcribed plus-sense leader RNA is encapsidated by the N protein, and the extent of encapsidation is thought to regulate transcription versus rep- lication (49). Sixteen of the eighteen 59-terminal trailer nucle- otides are complementary to those at the 39 terminus of the genome and potentially could form a circular structure by base pairing (6). Several studies have provided evidence that the nucleus is the site of SYNV replication. Nuclear inclusions that can be visualized by light microscopy are a prominent feature of leaf cells infected with SYNV (7, 19). Electron microscopy studies have also shown that many enveloped bacilliform particles are present in the perinuclear spaces surrounding the nuclei, and viral cores at various stages of morphogenesis can be seen * Corresponding author. Mailing address: Department of Plant and Microbial Biology, University of California, 111 Koshland Hall, Berkeley, CA 94720-3102. Phone: (510) 642-3906. Fax: (510) 642-9017. E-mail: [email protected]. ² Present address: Departamento de Biologia Celular, Universidade de Brasilia, Brasilia, Brazil. ‡ Present address: Department of Microbiology, Pukyong National University, Pusan, Korea. § Present address: Istituto di Patologia Vegetale, 40126 Bologna, Italy. \ Present address: Program for Genomics, Ethics & Society, Center for Biomedical Ethics, Stanford University, Palo Alto, Calif. # Present address: Division of Biology, California Institute of Tech- nology, Pasadena, Calif. ²² Present address: Electron Microscope Laboratory, University of California, Berkeley, Calif. 5669
Transcript of Sonchus Yellow Net Rhabdovirus Nuclear Viroplasms Contain...

JOURNAL OF VIROLOGY,0022-538X/98/$04.0010
July 1998, p. 5669–5679 Vol. 72, No. 7
Copyright © 1998, American Society for Microbiology. All Rights Reserved.
Sonchus Yellow Net Rhabdovirus Nuclear Viroplasms ContainPolymerase-Associated Proteins
CLAUDIA R. F. MARTINS,† JENNIFER A. JOHNSON, DIANE M. LAWRENCE, TAE-JIN CHOI,‡ANNA-MARIA PISI,§ SARA L. TOBIN,\ DENISE LAPIDUS, JOHN D. O. WAGNER,# STEVEN RUZIN,
KENT MCDONALD,†† AND ANDREW O. JACKSON*
Department of Plant and Microbial Biology, University of California, Berkeley, California 94720
Received 16 December 1997/Accepted 23 March 1998
We have initiated a study of the cytopathology of nucleorhabdoviruses by analyzing the subcellular local-ization of sonchus yellow net virus (SYNV) genomic and antigenomic RNAs and the encoded polymeraseproteins. In situ hybridizations demonstrated that the minus-strand genomic RNA sequences are restricted tothe nuclei of infected cells, while the complementary plus-strand antigenomic RNA sequences are present inboth the nuclei and the cytoplasm. Immunofluorescence and immunogold labeling experiments also revealedthat the nucleocapsid (N) protein and phosphoprotein (M2) are primarily localized to discrete regions withinthe nuclei and in virus particles that accumulate in perinuclear spaces. The N protein antiserum specificallylabeled the nuclear viroplasms, whereas the M2 antiserum was more generally distributed throughout thenuclei. Antibody detection also indicated that the polymerase (L) protein is present in small amounts in theviroplasm. When the N and M2 proteins were expressed individually from the heterologous potato virus X(PVX) vector, both proteins preferentially accumulated in the nuclei. In addition, viroplasm-like inclusionsformed in the nuclei of cells infected with the PVX vector containing the N gene. Fusions of the carboxyterminus of b-glucuronidase to N and M2 resulted in staining of the nuclei of infected cells followingexpression from the PVX vector. Deletion analyses suggested that multiple regions of the N protein containsignals that are important for nuclear localization.
Sonchus yellow net virus (SYNV) belongs to the Rhabdoviri-dae family of nonsegmented, negative-strand RNA viruses.This family is one of the most widely distributed virus familiesin nature, and it contains members that infect animals, plants,and insects (47). With this diversity, it is not surprising thatindividual members of the family differ with respect to require-ments for infection and the sites of viral replication and mor-phogenesis. All of the well-characterized animal rhabdovirusesreplicate in the cytoplasm; however, the plant rhabdovirusesare divided into two groups depending on whether their mor-phogenesis occurs in association with cytoplasmic or nuclearmembranes (16). The cytorhabdoviruses, which replicate in thecytoplasm and bud in association with the endoplasmic retic-ulum, include lettuce necrotic yellows virus, strawberry crinklevirus, and barley yellow striate mosaic virus, among others. Thenucleorhabdoviruses, of which SYNV, sowthistle yellow veinvirus, potato yellow dwarf virus, and maize mosaic virus aremembers, appear to replicate in the nucleus and bud from theinner nuclear envelope into perinuclear spaces. Furthermore,
over 50 plant rhabdoviruses have yet to be assigned to specificgroups because of insufficient information about their replica-tion and morphogenesis (50).
SYNV is the most extensively characterized plant rhabdovi-rus, and its genome has been completely sequenced (5, 6, 10,12, 14, 42, 51, 52). The 13,720-nucleotide (nt) SYNV RNAencodes six proteins in a negative-sense orientation (see Fig.1), and all of these are found in association with virus particles(42). A ribonucleoprotein core that can be released from viri-ons (17), and from the nuclei of infected plants (44), consists ofthe nucleocapsid (N) protein, the phosphoprotein (M2), andthe polymerase (L) protein complexed with SYNV RNA. Theglycoprotein (G) is thought to traverse the viral lipid envelopeand associate with the matrix (M1) protein, which probablyalso has a role in coiling of the ribonucleoprotein core (16). Asixth protein, sc4, has no known function but can be releasedfrom virions by treatment with mild nonionic detergents (42).Two putative regulatory regions, located at the 39 and 59 endsof the SYNV genome, flank the six minus-sense genes. The144-nt leader sequence is located at the 39 end of the genome(44, 51), and the 160-nt untranslated trailer sequence is presentat the 59 end of the genomic RNA. The transcribed plus-senseleader RNA is encapsidated by the N protein, and the extent ofencapsidation is thought to regulate transcription versus rep-lication (49). Sixteen of the eighteen 59-terminal trailer nucle-otides are complementary to those at the 39 terminus of thegenome and potentially could form a circular structure by basepairing (6).
Several studies have provided evidence that the nucleus isthe site of SYNV replication. Nuclear inclusions that can bevisualized by light microscopy are a prominent feature of leafcells infected with SYNV (7, 19). Electron microscopy studieshave also shown that many enveloped bacilliform particles arepresent in the perinuclear spaces surrounding the nuclei, andviral cores at various stages of morphogenesis can be seen
* Corresponding author. Mailing address: Department of Plantand Microbial Biology, University of California, 111 Koshland Hall,Berkeley, CA 94720-3102. Phone: (510) 642-3906. Fax: (510) 642-9017.E-mail: [email protected].
† Present address: Departamento de Biologia Celular, Universidadede Brasilia, Brasilia, Brazil.
‡ Present address: Department of Microbiology, Pukyong NationalUniversity, Pusan, Korea.
§ Present address: Istituto di Patologia Vegetale, 40126 Bologna,Italy.
\ Present address: Program for Genomics, Ethics & Society, Centerfor Biomedical Ethics, Stanford University, Palo Alto, Calif.
# Present address: Division of Biology, California Institute of Tech-nology, Pasadena, Calif.
†† Present address: Electron Microscope Laboratory, University ofCalifornia, Berkeley, Calif.
5669

budding through the inner nuclear envelope (7, 15, 19, 43).Incubation with tunicamycin, a glycosylation inhibitor, inter-rupts morphogenesis and leads to accumulation of a strikingarray of nucleocapsid cores at the periphery of the nuclei andat the outer edges of greatly enlarged viroplasms (43). Anactive polymerase complex contained within the nucleoproteincore has also been isolated from purified tobacco nuclei (20,44, 45). This complex is involved in transcription because it iscapable of synthesizing the polyadenylated leader RNA andeach of the six mRNAs in a polar fashion to yield decreasingamounts as follows: N . M2 . sc4 . M1 . G . L (44, 45).A temporal switch from transcription to replication is thoughtto occur at the intermediate stages of the infection processbecause SYNV genomic RNAs accumulate later than themRNAs in infected protoplasts (22). This strategy of transcrip-tion and replication is similar to that of vesicular stomatitisvirus (VSV) and other monopartite negative-strand viruses (2).
In SYNV-infected tissue, a dynamic series of events occurthat suggest intimate interactions of host and viral compo-nents. The nuclei swell, distinct nuclear inclusions appear, andlarge numbers of virus particles accumulate in the perinuclearspaces (7, 16). Observations of SYNV in protoplasts indicatethat following entry into the cell, one of the early events in-volves fusion of the viral envelope with the endoplasmic retic-ulum and subsequent release of the nucleocapsid core into thecytoplasm (43). These cores are then thought to enter thenucleus and establish a discrete viroplasm that functions intranscription and replication. To mediate these integral eventsin the viral life cycle, the host macromolecular transport ma-chinery must carry out several essential steps requiring nucle-ocytoplasmic transport of viral components. For example, un-spliced SYNV mRNAs are thought to be transcribed in thenucleus (44, 45) and exported to the cytoplasm, where they arepartitioned into free and membrane-associated polysomes dur-ing translation (30, 31). After protein synthesis, some of thevirus-encoded proteins are postulated to enter the nucleus andparticipate in formation of the viroplasm complex. However,the interactions culminating in formation of the nuclear viro-plasm are not yet understood. To begin to investigate theseprocesses, we examined the subcellular localization of SYNVRNAs and the polymerase-associated proteins in infected cells.Additional experiments were performed to determine the fateof exogenously expressed N and M2 proteins and to identifyregions of the N protein that are required for nuclear local-ization.
MATERIALS AND METHODS
General. SYNV (ATCC PV-263) was propagated by serial passages in Nico-tiana benthamiana under ambient greenhouse conditions (18). SYNV and potatovirus X (PVX) systemically infected tissue was harvested 11 to 14 days postin-oculation (dpi) for various experiments.
Construction and expression of PVX expression vectors. A wild-type versionof the N gene (p3ZN2, nt 203 to 1627 in the 39-to-59 orientation of the SYNVgenome; GenBank accession no. L32603) was constructed from plasmids p3ZN1(44) and pAS41 (52). The XbaI/PstI fragment of p3ZN1 which contains a pre-mature termination codon in the N coding sequence was removed and replacedwith the wild-type N sequence by insertion of the analogous XbaI/PstI fragmentfrom pAS41 to generate plasmid pNWT1. The N gene of pNWT1 was thendigested with BamHI and EcoRI, blunted with Klenow enzyme, and cloned intothe EcoRV site of the PVX vector pPC2S (4) to construct the vector PVX-N. TheM2 gene (nt 1750 to 2837 of the SYNV genome) was cloned into the EcoRV siteof pPC2S by digestion of the cDNA clone pJ3T1 (44) with EcoRI and PstI andsubsequently blunted with Klenow enzyme to produce PVX-M2. The b-glucu-ronidase (GUS) gene was excised from plasmid pRTL2-GUS/NIaDBam (36) bydigestion with NcoI and BglII, blunted with Klenow enzyme, and cloned into theEcoRV site of pPC2S to form PVX-GUS. The N gene was cloned as an in-framefusion with the 39 end of the GUS gene (GUS:N fusion) in pRTL2-GUS/NIaDBam to generate plasmid pRTL2-GUS:N. This was accomplished by di-gestion of plasmid pRTL2-GUS/NIaDBam with BglII, followed by a partial fill-in(A, G, and T) reaction using Klenow enzyme prior to digestion with BamHI. The
N gene was digested with NheI, partially filled in (A, C, and T) with Klenowenzyme, and subsequently digested with BamHI. The resulting plasmid, pRTL2-GUS:N, was digested with NcoI and BamHI, blunted with Klenow enzyme, andcloned into the EcoRV site of pPC2S, to produce PVX-GUS:N. The putativenuclear localization signal (NLS) of N (amino acids 446 to 461) was fused to thecarboxy terminus of GUS. Four phosphorylated primers (59GATCTAGGAAAAGAAGGAGTGACGCT39, 59TGTAAGAGCGTCACTCCTTCTTTTCCTA39, 59CTTACAACTGAGAAGCCTAAGAAGGCTTAGG39, and 59TCGACCTAAGCCTTCTTAGGCTTCTCAGT 39) encoding the NLS sequence with BglIIand SalI sites located at the 59 and 39 termini, respectively, were annealed priorto ligation into pRTL2-GUS/NIaDBam that had been digested with BglII andSalI to generate plasmid pRTL2-GUS:NLS. PVX-GUS:NLS was then made bydigesting pRTL2-GUS:NLS with NcoI and BamHI, and the resulting fragmentwas blunted with Klenow enzyme and cloned into the EcoRV site of pPC2S. Tofacilitate cloning of GUS:N gene mutations, the ApaI-to-SacI fragment of PVX-GUS:N, which contains nt 4945 to 6545 of the PVX genome and the GUS:Nfusion, was cloned into pGem5Zf(1) (Promega, Madison, Wis.) which had beencut with ApaI and SacI. This clone is referred to as pAS-GUS:N. Followingsite-directed mutagenesis or deletions of the N gene, the ApaI/SacI fragment wassubcloned back into PVX-GUS:N. The glutamate (GAG) at position 435 of theN protein was altered to a stop codon (TAG) by site-directed mutagenesis (23),using the primer 59CAACCACCTAATCCTCCTTG39 (bold indicates stopcodon) to generate PVX-GUS:Nstop. To construct PVX-GUS:Nstu, pAS-GUS:N was digested with StuI and SalI, blunted with Klenow enzyme, andreligated.
An in-frame fusion of GUS and M2 (GUS:M2 fusion) was generated byligation of an EcoRI/BglII fragment from plasmid pRTL2-GUS/NIaDBam, con-taining the tobacco etch virus untranslated leader followed by the GUS gene,into the EcoRI and NcoI sites in the plasmid p3ZT1 (44) to produce p3ZGT1.Prior to ligation, the BglII site at the 39 end of the GUS gene and the NcoI siteat the 59 end of the M2 gene were made compatible by filling in with Klenowenzyme. The resulting plasmid, p3ZGT1, was digested with EcoRI and HindIII,and the fragment containing the tobacco etch virus untranslated leader followedby the GUS:M2 fusion was blunted with Klenow enzyme and cloned into theEcoRV site of pPC2S to generate PVX-GUS:M2.
All the PVX expression vectors were linearized with SpeI prior to in vitrotranscription reactions. Capped transcripts were synthesized in vitro by usingbacteriophage T7 polymerase and used to inoculate N. benthamiana plants asdescribed elsewhere (41).
In situ hybridization. The SYNV plasmids pJ3N1 (nt 380 to 1633), pJ3T1 (nt2011 to 2837), and pJ3L1 (nt 12788 to 13630) were used as templates to generateprobes for recognition of the N, M2, and L regions, respectively, of the genomicand antigenomic RNAs (44). A probe designed to recognize the genomic orantigenomic leader RNAs was constructed by PCR amplification of pJW2 (44)with the following primers: 59GGGGTACCAGAGACAGAAACTCAGAAAATAC39 and 59CGCGAGCTCCTTTAATCTACTAATCAG39. The PCR fragmentwas digested with KpnI and SacI sites (indicated in bold in the primer sequences)and cloned into the KpnI and SacI sites of pBSKS1 II (Stratagene, La Jolla,Calif.). This cDNA clone is referred to as pJ3ld1.
Digoxigenin-labeled probes were synthesized by using either bacteriophage T3or T7 polymerase. Probes recognizing regions of the negative-strand (genomic)leader, N, M2, and L genes were generated with T7 polymerase, using thefollowing templates linearized with the designated restriction enzymes: pJ3N1-BamHI, pJ3TI-HindIII, pJ3L1-SacI, and pJ3ld1-SacI. The pJ3N1-HindIII,pJ3T1-XbaI, pJ3L1-KpnI, and pJ3ld1-KpnI linearized plasmids provided tem-plates for T3 polymerase transcription of probes recognizing the leader, N, M2,and L positive-strand (antigenomic) RNAs, respectively. Uncapped transcriptswere generated as described previously (41) except that the reactions included amodified digoxigenin nucleoside triphosphate labeling mix (250 mM digoxigenin-11-UTP [Boehringer Mannheim, Indianapolis, Ind.]; 0.5 mM ATP, GTP, andCTP; 0.25 mM UTP). Following transcription, DNase (5 U) was added andreaction mixtures were incubated for 15 min at 37°C before ethanol precipitationof the probes. The N, M2, and L probes were hydrolyzed for 45 min before usein an equal volume of 23 carbonate buffer (80 mM NaHCO3, 120 mM Na2CO3[pH 10.2]), neutralized with 0.5% (vol/vol) acetic acid, precipitated, and resus-pended in 50% formamide. Probe concentrations were estimated by analyzingaliquots on 1% agarose gels.
SYNV-infected and uninfected N. benthamiana leaf tissue was vacuum infil-trated with FAA (3.7% formalin, 5% acetic acid, 50% ethanol) and fixed over-night at room temperature. Samples were embedded in paraffin, sectioned,prehybridized, and hybridized with individual probes (Fig. 1) as previously de-scribed (21). Posthybridization washes, digoxigenin detection, and slide mount-ing were performed as described elsewhere (28). The slides were viewed with aZeiss Axiophot microscope using differential interference contrast.
Electron microscopy. Virus-infected and uninfected N. benthamiana leaf tissuewas processed by using a modified 800-W safety-exhausted microwave designedfor microscopy (model 345 and accessories; Ted Pella Inc., Redding, Calif.) asdescribed previously (9). Samples were fixed twice in 2% paraformaldehyde–0.25% glutaraldehyde in phosphate-buffered saline (PBS; 10 mM phosphatebuffer [pH 7.4], 150 mM NaCl) by treatment for 40 s in the microwave. The tissuesamples were then rinsed in PBS and dehydrated through a graded (50 to 90%)acetone series. Tissue was infiltrated in the microwave for 15 min in 1:1 ace-
5670 MARTINS ET AL. J. VIROL.

tone-LR white (Polysciences, Inc., Warrington, Pa.) followed by two additional15-min microwave treatments in 100% LR white. All of the steps above tookplace at 45°C. Samples in LR white were polymerized at 80°C for 45 min inBEEM capsules (Ted Pella Inc.) held under water in the microwave.
Immunogold labeling was performed at room temperature as described pre-viously (29). Silver sections (;60 nm thick) were incubated on Formvar-coatednickel grids in blocking buffer (0.1% [vol/vol] cold water fish gelatin, 0.8%[wt/vol] bovine serum albumin [BSA], 0.02% [vol/vol] Tween 80), rinsed in PBS,and incubated for 1 h in primary antibody. All antibodies were preabsorbedovernight with an N. benthamiana extract obtained by grinding ;1 g of leaf tissuein 10 ml of PBS. Antisera specific for SYNV (18) and the N (52), M2 (12), andL (5) proteins were diluted in blocking buffer (anti-N, 1:450; anti-M2, 1:450;anti-L, 1:1,100). Grids were rinsed in PBS and incubated for 1 h in secondarygold-conjugated (10-nm gold particles) goat anti-rabbit immunoglobulin G at a1:20 dilution in blocking buffer. The grids were then rinsed in PBS, fixed in 0.5%glutaraldehyde in PBS, rinsed in water, and stained with 2% aqueous uranylacetate and Reynold’s lead citrate (37). Sections were viewed at 80 kV in a JEOL100 CX transmission electron microscope.
GUS fusion and immunofluorescence analyses. Protoplasts were isolated frominfected and uninfected N. benthamiana leaves as described previously (22).Localizations of GUS:N and GUS:M2 fusions were determined in PVX-infectedprotoplasts isolated from 11- to 14-dpi systemically infected leaves. The proto-plasts were incubated in GUS substrate buffer (50 mM Na2PO4 [pH 7.0], 10 mMEDTA [pH 8.0], 0.5 mM K3Fe[CN]6, 0.5 mM K4Fe[CN]6 z 3H2O, and 0.25 mg of5-bromo-4-chloro-3-indolyl-b-D-glucuronic acid per ml) for 1 h at 37°C beforevisualization under bright-field illumination with a Zeiss Axiophot microscope(40). Computer images were acquired with an Optronics 450 Color charge-coupled device camera and captured at high resolution by using a Scion CG-7RGB framegrapper board. Figures were assembled on a Power Macintosh com-puter using Photoshop and Canvas software.
For immunofluorescence studies, protoplasts isolated from SYNV-infectedand uninfected tissue were mounted on poly-L-lysine-coated slides, fixed in 3.7%formaldehyde–10% mannitol for 30 min at 20°C, washed twice in 10% mannitolfor 5 min, permeabilized in PBS containing 3% BSA and 0.05% Tween 20 for 5min, washed twice in buffer A (3% BSA in PBS) for 5 min, and incubated inprimary antibody (anti-N, 1:500 in buffer A; anti-M2, 1:400; anti-SYNV, 1:500)overnight. Then, the protoplasts were washed twice in buffer A for 5 min,incubated in fluorescein isothiocyanate (FITC)-conjugated secondary antibody(1:250 in buffer A) for 1 h, washed three times for 5 min in buffer A, andincubated in 49,6-diamidino-2-phenylindole (DAPI; 0.1 mg/ml in PBS) for 30min. The samples were subsequently examined in a Zeiss Axiophot microscopewith epifluorescence. Two sets of filters were used: DAPI was visualized byexcitation at 365 nm and emission at 397 nm, and FITC was viewed by excitationat 450 to 490 nm and emission at 520 nm.
RESULTS
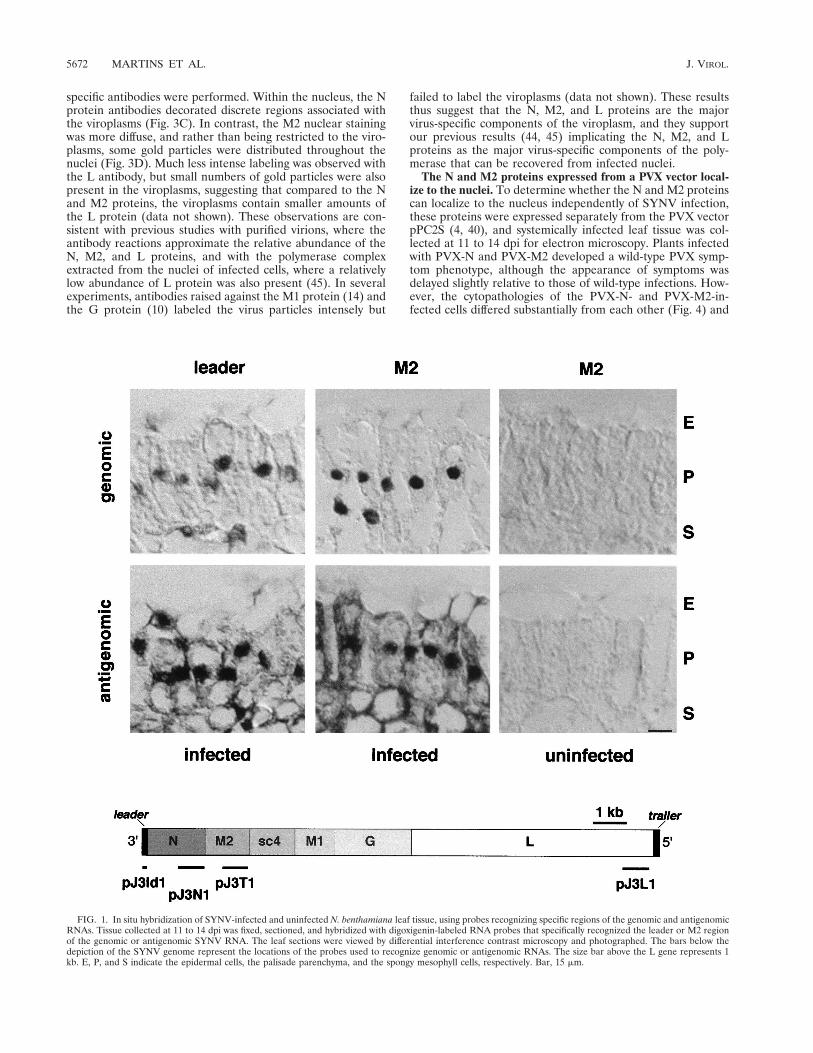
Genomic and antigenomic RNAs are abundant in nuclei ofSYNV-infected cells. Electron microscopy has previously indi-cated the presence of viroplasms in the nuclei of SYNV-in-fected cells and accumulation of virions in the perinuclearspaces (16), but the subcellular localization of SYNV RNAshas not been visualized directly in infected cells. To obtain thisinformation, in situ hybridization experiments were performedwith single-stranded riboprobes designed to recognize specificregions of the genomic or antigenomic RNAs (Fig. 1). Thespecificity of the probes used in these experiments was verifiedby Northern blot analysis of poly(A)1 RNA extracted fromSYNV-infected and uninfected leaves (not shown). Probes de-signed to recognize the plus-strand leader RNA, and the N andM2 mRNAs hybridized to the appropriately sized poly(A)1
RNAs (38). Corresponding complementary probes hybridizedto RNA blots containing the minus-sense genomic sequences(44). In sections of fixed tissue, probes specific for the leaderand M2 regions of the genomic RNA hybridized within nucleithroughout infected mesophyll tissue but failed to hybridize touninfected tissue (Fig. 1). Similar results were obtained withthe N and L probes (data not shown). These results thussuggest that the overwhelming proportion of the genomicRNA is confined to the nucleus, a finding that is consistentwith previous experiments suggesting that SYNV replicationoccurs in the nucleus (16, 45). The antigenomic RNA alsoappeared to be concentrated predominantly in the nuclei, asexpected from its putative role as the template for genomicRNA synthesis. However, in contrast to the strict nuclear hy-
bridization of the genome specific probes, the antigenomicleader, N, M2, and L RNA probes produced a diffuse cyto-plasmic labeling pattern (Fig. 1 and data not shown). Theseobservations provide additional verification of previous hybrid-ization results indicating cytoplasmic partitioning of polyade-nylated SYNV mRNAs (30, 31). In addition, our results showunexpectedly that a portion of the polyadenylated leader RNAis present in the cytoplasm (Fig. 1), where it possibly influenceshost metabolism and/or has a role in regulating viral replica-tion.
The N, M2, and L proteins are components of the nuclearinclusion bodies. No direct experiments have previously beenconducted to determine which SYNV proteins are associatedwith the nuclear viroplasms observed in SYNV-infected cells(16) or to identify the subcellular location of the N, M2, and Lpolymerase-associated proteins (45). Therefore, we conductedimmunofluorescence experiments to determine the subcellularlocalization of the SYNV proteins in combination with DAPIstaining to identify nuclei. As shown in Fig. 2, SYNV-specificantibodies raised against disrupted virus particles resulted inintense fluorescence around the nuclear envelope, with fainterfluorescence within the nuclei of protoplasts isolated frominfected N. benthamiana. The nuclear envelope is the site ofSYNV morphogenesis (16, 43), and hence the bright signalaround the periphery of the nuclei correlates with the presenceof large numbers of virus particles that accumulate in theperinuclear spaces and react with antibodies that recognize theG protein on the surface of the virions. In contrast, antibodiesraised against the N and M2 proteins were not as readilydetected in virus particles because N and M2 within the virionsare less accessible to the antibodies. Therefore, the N and M2antibodies exhibited less intense fluorescence at peripheralregions and more intense reactions at discrete sites within thenuclei of SYNV-infected protoplasts (Fig. 2). Moreover, areasshowing the most intense signal following labeling by the N andM2 antibodies resided within regions of the nucleus which hadminimal DAPI staining, suggesting that chromatin is excludedfrom sites containing the N and M2 proteins. Staining was notobserved in protoplasts isolated from uninfected tissue withany of the antibodies described above (Fig. 2 and data notshown).
Electron microscopy in combination with immunogold label-ing was used to investigate the subcellular localization of theSYNV proteins at higher resolution. Our observations re-vealed large numbers of bacilliform particles present in theperinuclear spaces and occasional particles that appeared to bebudding from the inner nuclear envelope (Fig. 3A, inset). Vi-roplasms of different shapes were also present in the swollennuclei of infected tissue that were reminiscent of the granularstructures previously observed in SYNV-infected cowpea pro-toplasts (43). The nuclei of SYNV-infected tissue were en-larged considerably and, from observations of a large numberof sections, were estimated to be two- to threefold greater involume than those of uninfected tissue. The sizes of the viro-plasms varied considerably, and their shapes ranged from ovalto spherical, possibly as a reflection of varied orientationsrelative to the plane of sectioning. Neither virus particles nornuclear inclusion bodies were detected in control experimentswith uninfected tissue (data not shown).
Immunogold labeling with the SYNV antiserum revealedthe presence of large numbers of gold particles located overthe virions and in the viroplasms (Fig. 3B). The remaining goldparticles were restricted to the nuclei and nuclear membranes,but no appreciable label was found in the cytoplasm (data notshown). To localize each of the polymerase-associated proteins(N, M2, and L) in the infected cells, labeling experiments using
VOL. 72, 1998 NUCLEORHABDOVIRUS VIROPLASM 5671
specific antibodies were performed. Within the nucleus, the Nprotein antibodies decorated discrete regions associated withthe viroplasms (Fig. 3C). In contrast, the M2 nuclear stainingwas more diffuse, and rather than being restricted to the viro-plasms, some gold particles were distributed throughout thenuclei (Fig. 3D). Much less intense labeling was observed withthe L antibody, but small numbers of gold particles were alsopresent in the viroplasms, suggesting that compared to the Nand M2 proteins, the viroplasms contain smaller amounts ofthe L protein (data not shown). These observations are con-sistent with previous studies with purified virions, where theantibody reactions approximate the relative abundance of theN, M2, and L proteins, and with the polymerase complexextracted from the nuclei of infected cells, where a relativelylow abundance of L protein was also present (45). In severalexperiments, antibodies raised against the M1 protein (14) andthe G protein (10) labeled the virus particles intensely but
failed to label the viroplasms (data not shown). These resultsthus suggest that the N, M2, and L proteins are the majorvirus-specific components of the viroplasm, and they supportour previous results (44, 45) implicating the N, M2, and Lproteins as the major virus-specific components of the poly-merase that can be recovered from infected nuclei.
The N and M2 proteins expressed from a PVX vector local-ize to the nuclei. To determine whether the N and M2 proteinscan localize to the nucleus independently of SYNV infection,these proteins were expressed separately from the PVX vectorpPC2S (4, 40), and systemically infected leaf tissue was col-lected at 11 to 14 dpi for electron microscopy. Plants infectedwith PVX-N and PVX-M2 developed a wild-type PVX symp-tom phenotype, although the appearance of symptoms wasdelayed slightly relative to those of wild-type infections. How-ever, the cytopathologies of the PVX-N- and PVX-M2-in-fected cells differed substantially from each other (Fig. 4) and
FIG. 1. In situ hybridization of SYNV-infected and uninfected N. benthamiana leaf tissue, using probes recognizing specific regions of the genomic and antigenomicRNAs. Tissue collected at 11 to 14 dpi was fixed, sectioned, and hybridized with digoxigenin-labeled RNA probes that specifically recognized the leader or M2 regionof the genomic or antigenomic SYNV RNA. The leaf sections were viewed by differential interference contrast microscopy and photographed. The bars below thedepiction of the SYNV genome represent the locations of the probes used to recognize genomic or antigenomic RNAs. The size bar above the L gene represents 1kb. E, P, and S indicate the epidermal cells, the palisade parenchyma, and the spongy mesophyll cells, respectively. Bar, 15 mm.
5672 MARTINS ET AL. J. VIROL.

from that of the PVX-infected cells (data not shown). PVX-infected cells contained large paracrystalline inclusion bodiesin the cytoplasm, but the nuclei were similar in size to those ofuninfected cells and contained no obvious inclusions. The cy-toplasmic paracrystalline inclusion bodies consisted of rod-shaped PVX particles whose morphology was typical of thebanded inclusions observed frequently in potexvirus infections(34). In sharp contrast, the PVX-N-infected cells had greatly
enlarged nuclei containing pronounced viroplasm-like inclu-sions (Fig. 4A). The N antibody label in the nuclei was re-stricted primarily to the viroplasms (Fig. 4A, inset), althoughsmall amounts of label were present in the cytoplasm associ-ated with the PVX banded inclusions.
The nuclei of PVX-M2-infected cells were also larger thanthose of PVX infected cells (data not shown), but unlike thePVX-N infection, the PVX-M2 infection did not induce viro-plasm-like inclusions (Fig. 4B). Instead, the immunogold la-beling of PVX-expressed M2 was typical of that observed inSYNV-infected cells, since label was distributed more uni-formly throughout the nucleus (Fig. 4B, inset). However, largenumbers of amorphous electron-dense aggregates werepresent in the nuclei of PVX-M2-infected cells. These aggre-gates did not appear to contain M2 protein because gold la-beling was not concentrated within the aggregates (Fig. 4B,inset). Although the vast majority of the M2 label was associ-ated with the nuclei, some signal was also present in the cyto-plasm in association with the paracrystalline PVX inclusions(Fig. 4B). These minor amounts of cytoplasmic M2 may resultfrom a combination of high levels of expression of M2 from thePVX vector coupled with inefficient transport of M2 to thenucleus. Antibodies raised against PVX virions labeled thecytoplasmic PVX inclusions very heavily, whereas the nucleiwere devoid of label (data not shown). These results demon-strate that the N and M2 proteins are able to accumulate in theplant cell nucleus independently of the presence of otherSYNV proteins.
GUS:N and GUS:M2 protein fusions are transported to thenuclei. Examination of the N protein sequence revealed thepresence of a putative nucleoplasmin-like bipartite NLS fromamino acids 446 to 461 located near the carboxy terminus butthat the M2 protein did not have regions with obvious homol-ogy to previously described NLSs (11, 33). To investigate nu-clear import of the N and M2 proteins further, GUS:N andGUS:M2 fusions were expressed from PVX vectors, and theprotoplasts isolated from systemically infected leaves were an-alyzed for GUS expression (Fig. 5). Protoplasts recovered fromPVX-infected leaves expressing GUS alone had uniform stain-ing throughout the cytoplasm (Fig. 5C). In contrast, proto-plasts infected with PVX-GUS:N exhibited intense nuclearstaining (Fig. 5A). The PVX-GUS:M2 derivatives also elicitedGUS activity in the nuclei of infected protoplasts, but somecytoplasmic staining was also observed (Fig. 5B). These resultsindicate that the 122-kDa GUS:N and 106-kDa GUS:M2 fu-sion proteins can localize to the nucleus independently, al-though the GUS:M2 localization was less pronounced thanthat of the GUS:N. The PVX-GUS:N fusion results also rein-forced the pronounced nuclear localization obtained in theimmunogold labeling experiments. This strict nuclear localiza-tion permitted us to examine the putative nucleoplasmin-likeNLS and to conduct mutagenesis experiments to define thedistribution of the NLSs residing within the N protein.
To identify the karyophilic signals in the N protein and todelineate their functional activity in PVX-GUS:N, three mu-tations were constructed in the N gene sequence. These mu-tations resulted in (i) removal of about 50% of the carboxy-terminal portion of the N protein (GUS:Nstu), (ii)introduction of a stop codon prior to the putative bipartiteNLS sequence from the N protein (GUS:Nstop), and (iii)fusion of the putative NLS to the carboxy terminus of GUS(GUS:NLS). Each of these alterations compromised GUS nu-clear staining to some extent. The GUS:Nstu fusion, contain-ing amino acids 1 to 210, reduced nuclear transit considerablybecause GUS staining was detected in the cytoplasm of theinfected protoplasts, although some nuclear staining was also
FIG. 2. Immunofluorescence localization of SYNV proteins, the N protein,or the M2 protein in SYNV-infected N. benthamiana protoplasts, isolated fromsystemically infected leaves at 11 to 14 dpi, or uninfected protoplasts. Followingfixing and permeabilization, the protoplasts were incubated with rabbit antiseraraised against disrupted SYNV particles, purified N protein, or M2 protein,followed by immunofluorescence (IF) using a secondary antiserum conjugated toFITC. The protoplasts were also incubated with DAPI before epifluorescenceexamination. Note that the most intense areas of immunofluorescence and DAPIexclusion coincide. Bar, 5 mm.
VOL. 72, 1998 NUCLEORHABDOVIRUS VIROPLASM 5673

FIG. 3. Cytopathology of SYNV-infected N. benthamiana leaf tissue. Tissue isolated 14 dpi was embedded in LR white by using a microwave. (A) Enlarged nucleusof an infected cell that contains virus particles in the perinuclear spaces. The arrow indicates a region containing three incompletely enveloped virions, which aremagnified in the inset. Bar, 750 nm. Panels B to D show reactions with rabbit antisera raised against disrupted virus particles (B), the N protein (C), and the M2 protein(D). Following incubation, sections were washed in PBS and subsequently incubated with gold-conjugated anti-rabbit immunoglobulin G. The sections were stainedwith 2% aqueous uranyl acetate and Reynold’s lead citrate and viewed in a JEOL 100 CX transmission electron microscope operating at 80 kV. Bars, 150 nm. V, virusparticle; VP, viroplasm.
5674 MARTINS ET AL. J. VIROL.

FIG. 4. Subcellular distribution of the N and M2 proteins expressed separately from PVX in infected N. benthamiana. Tissue was prepared 11 dpi, and immunogoldlabeling was performed as described in the legend to Fig. 3. (A) PVX-N-infected tissue incubated with antiserum raised against the N protein; (B) PVX-M2-infectedtissue treated with antiserum specific for the M2 protein. The insets represent higher magnifications of each of the cells to aid in visualization of the gold particles. E,electron-dense aggregates; I, viroplasm-like inclusion; P, PVX particles; VC, vacuole; arrows indicate nuclear envelope. (A) Bar, 750 nm; (B) bar, 500 nm.
VOL. 72, 1998 NUCLEORHABDOVIRUS VIROPLASM 5675

evident (Fig. 5D). The GUS:Nstop derivative, with a stopcodon engineered at amino acid 435, just prior to the putativeNLS, retained appreciable nuclear staining, but substantial dif-fuse staining was also observed in the cytoplasm (Fig. 5E).However, the GUS:NLS construct (NLS amino acids 446 to461) was severely compromised in localization of the fusionprotein to the nucleus, but some nuclear staining was stillpresent (Fig. 5F). Although there are some complications inthe interpretation of these experiments due to possible aber-rant protein folding and instability of PVX constructs, theresults of the carboxy-terminal and NLS deletions suggest thatefficient nuclear import of GUS:N is complex. Although dele-tion of the putative NLS had a substantial impact on localiza-tion, this motif alone appears not to be sufficient to facilitateefficient import of the GUS fusion protein, and so it is evidentthat several other unidentified regions of the N protein havesynergistic effects on the nuclear transit process.
DISCUSSION
Our studies support a model predicting that early in SYNVinfection, virions or viral cores move into the nucleus to estab-
lish a viroplasm that functions to mediate transcription ofmRNAs as well as the antigenomic and genomic replicativeRNAs. According to this model, polyadenylated mRNA tran-scripts are exported to the cytoplasm, where the mRNAs aretranslated differentially on free or membrane-bound poly-somes (30, 38). The present in situ experiments show that boththe genomic and the antigenomic RNAs are associated withthe nuclei, and they most likely are present as replicatingcomplexes within the viroplasms. The results also complementprevious studies showing that the mRNAs accumulate to highabundance in the cytoplasm (30, 31). Interestingly, sequencescorresponding to the positive strand (antigenomic) leaderRNA transcript appear in the cytoplasm as well as in thenucleus. Detection of a cytoplasmic pool of the polyadenylatedSYNV leader RNA is of particular interest because the con-verse situation exists with the animal rhabdovirus VSV, inwhich the majority of the positive-sense leader RNA is tran-siently associated with the nuclei of infected cells very early ininfection (24, 25). These transient associations may have im-portant roles in regulating the rhabdovirus infection cycle be-cause the extent of encapsidation of the leader RNA is thoughtto facilitate the switch between transcription and replication
FIG. 5. Localization of GUS:N and GUS:M2 fusion proteins in N. benthamiana protoplasts recovered from PVX-infected leaves. (A) GUS:N fusions; (B) GUS:M2fusions; (C) GUS alone; (D) GUS:Nstu (GUS fused to amino acids 1 to 210 of the N protein); (E) GUS:Nstop (GUS fused to the N protein prematurely terminatedat amino acid 435); (F) GUS:NLS (GUS fused to the putative NLS of the N protein [amino acids 446 to 461]). Protoplasts were isolated at 11 to 14 dpi from systemicallyinfected leaves, incubated in GUS substrate, and viewed under bright-field illumination with a Zeiss Axiophot microscope. The boxes represent the proteins expressedfrom the PVX vector: blue, GUS protein; red, N protein; yellow, putative NLS; purple, M2 protein.
5676 MARTINS ET AL. J. VIROL.

(1–3, 49). Evidence obtained with VSV suggests that duringthe initial stages of infection, the concentration of the newlysynthesized N protein available to encapsidate the nascentleader RNAs is low and that mRNA transcription predomi-nates. However, as the concentration of the N protein in-creases to levels sufficient to facilitate replication, transcribedleader sequences become increasingly encapsidated, and syn-thesis of full-length antigenomic nucleocapsids commences(49). Although the transient nuclear association of VSV leaderRNAs is still an enigma, it is intriguing that the highest pro-portion of nuclear association occurs during the primary tran-scription phase of replication and that a decrease in abundanceof the leader RNA found in the nuclei coincides with theappearance of antigenomic replicating molecules (24, 25).Thus, it is conceivable that during the early phases of infection,rhabdoviruses such as VSV that use a cytoplasmic mode ofreplication sequester transcribed leader RNAs in the nuclei tofacilitate rapid amplification of replication-competent antige-nomic nucleocapsids. In the case of the nucleorhabdovirusSYNV, transport of the leader RNAs to the cytoplasm couldprovide an analogous mechanism for reducing localized nu-clear concentrations of transcribed leader RNA.
We have also demonstrated that the polymerase-associatedproteins N, M2, and L accumulate in the nuclei of SYNV-infected cells, and the results of DAPI staining suggest that theviroplasms form distinct structures within the nuclei that ex-clude chromatin. Christie et al. (7) first observed that ovalinclusions occur in the nuclei of SYNV-infected plants in as-sociation with bacilliform particles surrounding the nuclei, andthis observation has been verified subsequently in both wholeplant and protoplast infections (16, 19, 43). The present studyprovides the first direct evidence that the N, M2, and L pro-teins are components of the nuclear viroplasms whereas theM1 and G proteins are not. In addition, our immunogoldlabeling experiments suggest that considerably lower amountsof the L protein than the N and M2 proteins colocalize with theviroplasms, a finding that correlates with the relative levels ofthese proteins in the polymerase complex isolated from thenuclei of infected plants (45). Furthermore, these findings pro-vide additional evidence that the polymerase complex recov-ered from nuclei is derived from viroplasms.
Our serological analyses also show that the M2 protein inSYNV infections is present throughout the nucleus. The ob-servation that appreciable amounts of nucleus-associated M2reside outside the viroplasms may be linked to our previousfinding that M2-L protein complexes can be coimmunoprecipi-tated from extracts of SYNV-infected protoplasts (45). Theseexperiments show clearly that the M2-L complex consists ofnonstoichiometric amounts of the two proteins, with a substan-tial excess of M2 predominating (45). We postulate that thenon-viroplasm-associated M2 protein observed in the immu-nogold labeling experiments is a component of an M2-L com-plex that provides a precursor to the viral polymerase. The Nprotein is also localized to the nuclei in SYNV infections but,in contrast to the M2 protein, is concentrated in the viro-plasms. This result suggests that in addition to nuclear target-ing functions, the N protein has signals that mediate its local-ization to particular regions within nuclei.
During the course of SYNV infection, the N, M2, and Lproteins must be transported from their cytoplasmic sites ofsynthesis to the nucleus, where they are required for transcrip-tion, replication, and formation of virus particles. The exoge-nous expression of the N protein from the PVX vector revealsthat the N protein alone is able to localize to the nucleus andconcentrate at localized sites in amounts sufficient to formpseudoviroplasms similar to those found in SYNV-infected
cells. This finding suggests that the karyophilic N protein sig-nals facilitate specific transport associations with host compo-nents that target macromolecules to particular compartmentswithin the nuclei. In addition, it appears that the N proteinalone is sufficient for induction of nuclear swelling and forma-tion of inclusions similar to those found in SYNV-infectedcells. The M2 protein expressed from PVX also moves to thenucleus and results in enlargement of the nuclei. However, M2is distributed throughout the nuclei, and it fails to elicit viro-plasm-like inclusions, although unusual electron-dense aggre-gates of unknown composition are present. Thus, the SYNV-encoded N and M2 proteins differ in the ability to accumulateat specific sites in the nuclei and to alter the nuclear morphol-ogy.
The carboxy terminus of the N protein contains a putativeNLS composed of a bipartite basic sequence that is character-istic of a major class of nuclear import signals (11, 33). How-ever, although fusion of the putative NLS to the reporter genedirects some GUS nuclear import, additional, undefined sig-nals within multiple regions of the N protein are required forefficient localization. This heterogeneous distribution of mul-tiple localization signals has some features in common with thekaryophilic signals present in the proteins of other viruses thatreplicate in the nuclei of their hosts. For example, the influenzavirus N protein has an NLS in the central region that does notresemble any known NLS (8). However, a second recentlyidentified nonconventional NLS (TKRSxxxM) present in theamino terminus of the influenza virus N protein (48) has somesimilarity to a sequence (TSDKxxxM) present in the amino-terminal region (amino acids 69 to 76) of the SYNV N protein.This signal may be involved in nuclear localization because theamino-terminal portion of the SYNV N protein, which con-tains this signal, directs some nuclear import of the fusionprotein. Interestingly, when the two identified NLSs in theinfluenza virus N protein are deleted, nuclear localization iscompromised but some nuclear localization still is evident,suggesting that multiple NLSs are also present in the influenzavirus protein (32). Multiple NLSs also appear to be present inthe N protein of the negative-strand nonsegmented Bornadisease virus (BDV). Of some significance is the presence oftwo forms of the N protein that are essentially identical exceptfor the presence of an amino-terminal 13-amino-acid extensioncontaining an NLS. However, the BDV N protein derivativelacking the amino-terminal extension sequence is still able tolocalize to the nucleus, albeit less efficiently (35). Therefore, itappears that multiple NLSs are present in nucleocapsid pro-teins of several negative-strand viruses that replicate in thenucleus. Discrete signals required for nuclear localization ofthe SYNV M2 protein are not evident, and other than a cen-trally located sequence (AKKKSKA) reminiscent of a simianvirus 40-like NLS (PKKKRKV), no obvious bipartite or M9signals are present (11, 33).
The stringent localization of the N protein in the viroplasmsduring infection, coupled with the more uniform nuclear dis-tribution of M2, raises the possibility that these two compo-nents interact in complexes. Moreover, other viral (for in-stance, M2-L complexes) and/or host nuclear import proteinsprobably facilitate targeting of the polymerase complex pro-teins into specific sites within the nucleus as the viroplasmsform early in infection. Some evidence also exists for the pres-ence of subnuclear localization signals in other viral and non-viral proteins. Such signals are present in a DNA methyltrans-ferase (27) and in the splicing factor Tra (13) proteins. In DNAmethyltransferase, the NLS is distinct from the subnuclearlocalization signals, whereas in Tra the two signals overlap.Neither of these signals is present in the N protein, so the
VOL. 72, 1998 NUCLEORHABDOVIRUS VIROPLASM 5677

determinants that facilitate its localization to specific sites inthe nucleus are presently obscure. Other experiments showingthat M2 is a phosphoprotein (46) also raise the possibility thatthe phosphorylation state of the protein can affect subcellularlocalization as has been suggested for the nucleoprotein ofinfluenza virus (32).
In summary, the results presented in this study provide di-rect evidence that SYNV undergoes a nuclear phase during itsreplication. Despite similarities in transcription and replica-tion, the plant nucleorhabdoviruses differ markedly in severalimportant respects from other characterized members of theRhabdoviridae (47) or Paramyxoviridae (26) that replicate inthe cytoplasm. In this regard, SYNV and the monopartitenegative-strand BDV (39) vary considerably in genome orga-nization and gene expression strategy but are similar in thatboth undergo replication in the nucleus. Therefore, SYNVprovides an excellent model system to compare and contrastthe processes that are involved in establishing sites of replica-tion in plant and animal host cells. These interactions couldhave relevance to the pathology of several different negative-strand viruses.
ACKNOWLEDGMENTS
We thank David Baulcombe and Jim Carrington for generouslyproviding plasmids pPC2S and pRTL2-GUS/NIaDBam, respectively,and Simon Santa Cruz for supplying antisera raised against PVXvirions used in these experiments. We also thank Paula Sicurello at theElectron Microscope Facility at UC-Berkeley, Denise Schichnes at theCNR Center for Biological Imaging at UC-Berkeley, and David Jack-son at the USDA Plant Gene Expression Center for assistance andtechnical advice about various aspects of these experiments. MichaelGoodin, Ignacio Moreno, Teresa Rubio, and Angelika Fath providedsuggestions and editorial comments on the manuscript.
A Conselho Nacional de Desenvolvimento Cientificio e TecnologicoFellowship was awarded to C.R.F.M., and a National Science Foun-dation Visiting Professorship for Women was awarded to S.L.T. Thisresearch was supported by National Science Foundation grant DMB94-18086 awarded to A.O.J.
REFERENCES
1. Arnheiter, H., N. L. Davis, G. Wertz, M. Schubert, and R. A. Lazzarini. 1985.Role of the nucleocapsid protein in regulating vesicular stomatitis virus RNAsynthesis. Cell 41:259–267.
2. Banerjee, A. K., and S. Barik. 1992. Gene expression of vesicular stomatitisvirus genome RNA. Virology 188:417–428.
3. Blumberg, B. M., M. Leppert, and D. Kolakofsky. 1981. Interaction of VSVleader RNA and nucleocapsid protein may control VSV genome replication.Cell 23:837–845.
4. Chapman, S., T. Kavanagh, and D. Baulcombe. 1992. Potato virus X as avector for gene expression in plants. Plant J. 2:549–557.
5. Choi, T.-J., S. Kuwata, E. V. Koonin, L. A. Heaton, and A. O. Jackson. 1992.Structure of the L (polymerase) protein gene of sonchus yellow net virus.Virology 189:31–39.
6. Choi, T.-J., J. D. Wagner, and A. O. Jackson. 1994. Sequence analysis of thetrailer region of sonchus yellow net virus genomic RNA. Virology 202:33–40.
7. Christie, S. R., R. G. Christie, and J. R. Edwardson. 1974. Transmission ofa bacilliform virus of sowthistle and Bidens pilosa. Phytopathology 64:840–845.
8. Davey, J., N. J. Dimmock, and A. Colman. 1985. Identification of the se-quence responsible for the accumulation of the influenza virus nucleoproteinin Xenopus oocytes. Cell 40:667–675.
9. Giberson, R. T., R. S. Demaree, Jr., and R. W. Nordhausen. 1997. Four-hourprocessing of clinical/diagnostic specimens for electron microscopy usingmicrowave technique. J. Vet. Diagn. Invest. 9:61–67.
10. Goldberg, K. B., B. Modrell, B. I. Hillman, L. A. Heaton, T.-J. Choi, andA. O. Jackson. 1991. Structure of the glycoprotein gene of sonchus yellow netvirus, a plant rhabdovirus. Virology 185:32–38.
11. Gorlich, D., and I. W. Mattaj. 1996. Nucleocytoplasmic transport. Science271:1513–1518.
12. Heaton, L. A., D. Zuidema, and A. O. Jackson. 1987. Structure of the M2protein gene of sonchus yellow net virus. Virology 161:234–241.
13. Hedley, M. L., H. Amrein, and T. Maniatis. 1995. An amino acid sequencemotif sufficient for subnuclear localization of an arginine/serine-rich splicing
motif. Proc. Natl. Acad. Sci. USA 92:11524–11528.14. Hillman, B. I., L. A. Heaton, B. G. Hunter, B. Modrell, and A. O. Jackson.
1990. Structure of the gene encoding the M1 protein of sonchus yellow netvirus. Virology 179:201–207.
15. Ismail, I. D., I. A. Hamilton, E. Robertson, and J. J. Milner. 1987. Movementand intracellular location of sonchus yellow net virus within infected Nico-tiana edwardsonii. J. Gen. Virol. 68:2429–2438.
16. Jackson, A. O. 1987. Biology, structure, and replication of plant rhabdovi-ruses, p. 427–508. In R. R. Wagner (ed.), The rhabdoviruses. Plenum Press,New York, N.Y.
17. Jackson, A. O. 1978. Partial characterization of the structural proteins ofsonchus yellow net virus. Virology 87:172–181.
18. Jackson, A. O., and S. R. Christie. 1977. Purification and some physiochemi-cal properties of sonchus yellow net virus. Virology 77:344–355.
19. Jackson, A. O., and S. R. Christie. 1979. Sonchus yellow net virus. CMI/AABdescription of plant viruses no. 205. Cambrian News, LTD., Aberystwyth,Wales.
20. Jackson, A. O., and J. D. O. Wagner. 1998. Procedures for plant rhabdoviruspurification, polyribosome isolation, and replicase extraction, p. 77–97. InG. D. Foster and S. C. Taylor (ed.), Plant virology protocols: from virusisolation to transgenic resistance, vol. 81. Humana Press Inc., Totowa, N.J.
21. Jackson, D. 1991. In situ hybridization in plants, p. 163–174. In D. J. Bowles,S. J. Gurr, and M. McPherson (ed.), Molecular plant pathology: a practicalapproach. Oxford University Press, Oxford, England.
22. Jones, R. W., and A. O. Jackson. 1990. Replication of sonchus yellow netvirus in infected protoplasts. Virology 179:815–820.
23. Kunkel, T. A. 1985. Rapid and efficient site-specific mutagenesis withoutphenotypic selection. Proc. Natl. Acad. Sci. USA 82:488–492.
24. Kurilla, M. G., and J. D. Keene. 1983. The leader RNA of vesicular stoma-titis virus is bound by a cellular protein reactive with anti-la lupus antibodies.Cell 34:837–845.
25. Kurilla, M. G., H. Piwnica-Worms, and J. D. Keene. 1982. Rapid and tran-sient localization of the leader RNA of vesicular stomatitis virus in the nucleiof infected cells. Proc. Natl. Acad. Sci. USA 79:5240–5244.
26. Lamb, R. A., and D. Kolakofsky. 1996. Paramyxoviridae: the viruses and theirreplication, p. 577–604. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.),Fundamental virology, 3rd ed. Lippincott-Raven, Philadelphia, Pa.
27. Leonhardt, H., A. W. Page, H. Weier, and T. H. Bestor. 1992. A targetingsequence directs DNA methyltransferase to sites of DNA replication inmammalian nuclei. Cell 71:865–873.
28. Lincoln, C., J. Long, J. Yamaguchi, K. Serikawa, and S. Hake. 1994. Aknotted1-like homeobox gene in Arabidopsis is expressed in the vegetativemeristem and dramatically alters leaf morphology when overexpressed intransgenic plants. Plant Cell 6:1859–1876.
29. McDonald, K. L. 1994. Electron microscopy and EM immunocytochemistry.Methods Cell Biol. 44:411–444.
30. Milner, J. J., M. J. J. Hakkaart, and A. O. Jackson. 1979. Subcellulardistribution of RNA sequences complementary to sonchus yellow net virusRNA. Virology 98:497–501.
31. Milner, J. J., and A. O. Jackson. 1979. Sequence complementarity of sonchusyellow net virus RNA with RNA isolated from the polysomes of infectedtobacco. Virology 97:90–99.
32. Neumann, G., M. R. Castrucci, and Y. Kawaoka. 1997. Nuclear import andexport of influenza virus nucleoprotein. J. Virol. 71:9690–9700.
33. Nigg, E. A. 1997. Nucleocytoplasmic transport: signals, mechanisms andregulation. Nature 386:779–787.
34. Purcifull, D. E., and J. R. Edwardson. 1981. Potexviruses, p. 628–693. In E.Kurstak (ed.), Handbook of plant virus infections and comparative diagnosis.Elsevier/North-Holland Biomedical Press, Amsterdam, The Netherlands.
35. Pyper, J. M., and A. E. Gartner. 1997. Molecular basis for the differentialsubcellular localization of the 38- and 39-kilodalton structural proteins ofborna disease virus. J. Virol. 71:5133–5139.
36. Restrepo, M. A., D. D. Freed, and J. C. Carrington. 1990. Nuclear transportof plant potyviral proteins. Plant Cell 2:987–998.
37. Reynolds, E. S. 1963. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 17:208.
38. Rezaian, M. A., L. A. Heaton, K. Pederson, J. J. Milner, and A. O. Jackson.1983. Size and complexity of polyadenylated RNAs induced in tobaccoinfected with sonchus yellow net virus. Virology 131:221–229.
39. Schneemann, A., P. A. Schneider, R. A. Lamb, and W. I. Lipkin. 1995. Theremarkable coding strategy of borna disease virus: a new member of thenonsegmented negative strand RNA viruses. Virology 210:1–8.
40. Scholthof, H. B., T. M. Morris, and A. O. Jackson. 1993. The capsid proteingene of tomato bushy stunt virus is dispensable for systemic movement andcan be replaced for localized expression of foreign genes. Mol. Plant-Mi-crobe Interact. 6:309–322.
41. Scholthof, H. B., K.-B. G. Scholthof, and A. O. Jackson. 1995. Identificationof tomato bushy stunt virus host-specific symptom determinants by expres-sion of individual genes from a potato virus X vector. Plant Cell 7:1157–1172.
42. Scholthof, K. B., B. I. Hillman, B. Modrell, L. A. Heaton, and A. O. Jackson.1994. Characterization and detection of sc4: a sixth gene encoded by sonchusyellow net virus. Virology 204:279–288.
5678 MARTINS ET AL. J. VIROL.

43. van Beek, N. A. M., D. Lohuis, J. Dijkstra, and D. Peters. 1985. Morpho-genesis of sonchus yellow net virus in cowpea protoplasts. J. Ultrastruct. Res.90:294–303.
44. Wagner, J. D. O., T.-J. Choi, and A. O. Jackson. 1996. Extraction of nucleifrom sonchus yellow net rhabdovirus-infected plants yields a polymerase thatsynthesizes viral mRNAs and polyadenylated plus-strand leader RNA. J.Virol. 70:468–477.
45. Wagner, J. D. O., and A. O. Jackson. 1997. Characterization of the compo-nents and activity of sonchus yellow net rhabdovirus polymerase. J. Virol.71:2371–2382.
46. Wagner, J. D. O., and A. O. Jackson. Unpublished results.47. Wagner, R. R., and J. K. Rose. 1996. Rhabdoviridae: the viruses and their
replication, p. 561–575. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.),Fundamental virology, 3rd ed. Lippincott-Raven, Philadelphia, Pa.
48. Wang, P., P. Palese, and R. E. O’Neill. 1997. The NPI-1/NPI-3 (karyopherina) binding site on the influenza A virus nucleoprotein NP is a nonconven-
tional nuclear localization signal. J. Virol. 71:1850–1856.49. Wertz, G. W., N. L. Davis, and J. Patton. 1987. The role of proteins in
vesicular stomatitis virus RNA replication, p. 271–296. In R. R. Wagner(ed.), The rhabdoviruses. Plenum Press, New York, N.Y.
50. Wunner, W. H., R. G. Calisher, R. G. Dietzgen, A. O. Jackson, E. W.Kitajima, M. Lafron, J. M. Leong, S. Nichols, J. S. Smith, and P. J. Walker.1995. Family Rhabdoviridae, p. 275–288. In F. A. Murphy, C. M. Fauquet,D. H. L. Bishop, S. A. Ghabriel, A. W. Jarvis, G. P. Martelli, M. A. Mayo,and M. D. Summers (ed.), Virus taxonomy: classification and nomenclatureof viruses: sixth report of the International Committee of Taxonomy ofViruses. Springer-Verlag, Vienna, Austria.
51. Zuidema, D., L. A. Heaton, R. Hanau, and A. O. Jackson. 1986. Detectionand sequence of plus-strand leader RNA of sonchus yellow net virus, a plantrhabdovirus. Proc. Natl. Acad. Sci. USA 83:5019–5023.
52. Zuidema, D., L. A. Heaton, and A. O. Jackson. 1987. Structure of thenucleocapsid protein gene of sonchus yellow net virus. Virology 159:373–380.
VOL. 72, 1998 NUCLEORHABDOVIRUS VIROPLASM 5679


















