
SolventsasReagentsinOrganicSynthesisdownload.e-bookshelf.de/download/0010/3083/44/L-G-0010308344... ·...
30
Transcript of SolventsasReagentsinOrganicSynthesisdownload.e-bookshelf.de/download/0010/3083/44/L-G-0010308344... ·...



Solvents as Reagents in Organic Synthesis


Solvents as Reagents in Organic Synthesis
Reactions and Applications
Edited by Xiao-Feng Wu

Editor
Dr. Xiao-Feng WuAlbert-Einstein-Str. 29a18059 RostockGermany
Cover credits:Getty Images / d1sk1ss.deviantart
All books published by Wiley-VCH arecarefully produced. Nevertheless, authors,editors, and publisher do not warrant theinformation contained in these books,including this book, to be free of errors.Readers are advised to keep in mind thatstatements, data, illustrations, proceduraldetails or other items may inadvertently beinaccurate.
Library of Congress Card No.:applied for
British Library Cataloguing-in-PublicationDataA catalogue record for this book is availablefrom the British Library.
Bibliographic information published bythe Deutsche NationalbibliothekThe Deutsche Nationalbibliotheklists this publication in the DeutscheNationalbibliografie; detailed bibliographicdata are available on the Internet at<http://dnb.d-nb.de.>
© 2018 Wiley-VCH Verlag GmbH & Co.KGaA, Boschstr. 12, 69469 Weinheim,Germany
All rights reserved (including those of trans-lation into other languages). No part of thisbook may be reproduced in any form – byphotoprinting, microfilm, or any othermeans – nor transmitted or translated intoa machine language without written permis-sion from the publishers. Registered names,trademarks, etc. used in this book, evenwhen not specifically marked as such, arenot to be considered unprotected by law.
Print ISBN: 978-3-527-34196-2ePDF ISBN: 978-3-527-80561-7ePub ISBN: 978-3-527-80563-1Mobi ISBN: 978-3-527-80564-8oBook ISBN: 978-3-527-80562-4
Cover Design Adam-Design, Weinheim,GermanyTypesetting SPi Global, Chennai, IndiaPrinting and Binding
Printed on acid-free paper

v
Contents
List of Contributors xiii
1 The Applications of Water as Reagents in Organic Synthesis 1Zhengkai Chen and Hongjun Ren
1.1 Introduction 11.2 Incorporation of Hydrogen Atom from the Water 21.2.1 1,2,3-Triazoles 41.3 Incorporation of Oxygen Atom from the Water 71.4 Incorporation of Hydroxyl Group from Water 311.5 Traceless Promotion of the Reactions by Water 391.6 Conclusions 44
References 44
2 The Applications of Toluene and Xylenes 49Krishna Nand Singh, Narendra R. Chaubey, and Neetu Singh
2.1 Application of Toluene and Xylenes as Reagents 502.2 Oxidation of Methyl Group into Common Functionalities 502.3 Application of Methyl Group as Acyl Building Block 512.3.1 Synthesis of Carbonyl Compounds 512.3.2 Synthesis of Amides 532.3.3 Synthesis of N-Aroyl Sulfoximines 542.3.4 Synthesis of Esters 552.3.5 Synthesis of Thioesters 562.4 Application as Alkyl Building Block 602.4.1 Synthesis of Nitriles 602.4.2 Synthesis of 2-Phenyl Acetic Acid Derivatives 612.4.3 Alkylation of Sulfonamides 622.4.4 Alkylation of Thiophenols 632.4.5 Synthesis of Trifluoromethyl Sulfides 642.4.6 Synthesis of Benzyl Esters 642.4.7 Synthesis of Phosphate Esters 662.4.8 Synthesis of Carbamates, Thioamides, and Esters 662.4.9 Synthesis of 3-Benzyl Coumarin Derivatives 662.4.10 Decarboxylative Benzylation of Cinnamic Acids 682.4.11 Synthesis of Functionalized Oxindoles 68

vi Contents
2.4.12 Synthesis of Dihydroquinolinones 692.4.13 Benzylation of Enones 692.4.14 Coupling with 1,3-Dicarbonyl Compounds 702.4.15 Benzylation of Pyridine-N-Oxide 702.4.16 Synthesis of Dihydrofurans 702.4.17 Synthesis of Quinoline Derivatives 712.4.18 Reaction with Ethyl Diazoacetate 722.4.19 Synthesis of Benzo[b]phosphole Oxides 722.4.20 Synthesis of β-Aromatic α-Amino Acid Derivatives 732.4.21 Halogenation Reactions 732.4.22 N-Benzylation of Isoquinolines 752.5 Application as Esters Building Block 762.6 Application as Alcohols Building Block 77
References 77
3 The Applications of 1,4-Dioxane, THF, and Ethers as VersatileBuilding Blocks in Organic Synthesis 81Ping Liu, Guanghui Zhang, and Peipei Sun
3.1 Introduction 813.2 Cleavage of C(sp3)–H of Ethers 823.2.1 Cross-Dehydrogenative Coupling Reactions of Ethers 823.2.1.1 C–C Bond Formation 833.2.1.2 C–N Bond Formation 913.2.1.3 C–O Bond Formation 933.2.2 The Formation of C–S Bond 973.2.3 Addition of Ethers to C=C and C≡C Bonds 983.2.4 Decarboxylative Alkenylation or Alkylation Reactions 1043.2.5 Radical Alkenylation and Alkynylation of Ethers 1063.2.6 Radical α-C–H Hydroxyalkylation and Aminoalkylation of
Ethers 1073.2.7 Intermolecular Carbenoid Insertion to α-C–H Bond of Ethers 1093.2.8 C(sp3)–H Arylation with Arylmetal or Arylboron Reagents 1093.3 Cleavage of C–O of Ethers 1123.4 Cleavage of C–C Bonds of Ethers 1173.5 Conclusion 118
References 118
4 The Application of Dichloromethane and Chloroform asReagents in Organic Synthesis 125Anis Tlili and Johannes Schranck
4.1 The Application of Dichloromethane and Chloroform as Reagents inOrganic Synthesis 125
4.1.1 Dichloromethane 1254.1.1.1 Reactions of Dichloromethane with Posttransition Metals 1264.1.1.2 Reactions of Dichloromethane with Transition Metals 1274.1.1.3 Reactions of Dichloromethane with Phosphines 135

Contents vii
4.1.1.4 Reactions of Dichloromethane with Amines and Phosphines 1364.1.1.5 Reactions of Dichloromethane with Amines 1364.1.1.6 Reactions of Dichloromethane with Amines and Nucleophilic Carbon
Derivatives 1394.1.1.7 Reaction of Dichloromethane with Nucleophilic Sulfur 1414.1.2 Chloroform 1424.1.2.1 Reaction of Chloroform with Hydrogen Fluoride 1424.1.2.2 Reactions of Chloroform with Post-Transition Metals 1424.1.2.3 Reactions of Chloroform with Transition Metals 1434.1.2.4 Formation and Use of Dichlorocarbene 146
References 154
5 The Applications of Acetone and Ethyl Acetate 161Jie-Ping Wan
5.1 Acetone 1615.1.1 Aldol Reaction 1615.1.2 Claisen–Schmidt Reaction 1675.1.3 Mannich Reaction 1705.1.4 Miscellaneous 1755.2 Ethyl Acetate 1825.2.1 Transesterification 1835.2.2 Amidation 1875.2.3 Miscellaneous 190
References 192
6 N,N-Dimethylformamide and N,N-Dimethylacetamide asCarbon, Hydrogen, Nitrogen, and/or Oxygen Sources 199Jean Le Bras and Jacques Muzart
6.1 Introduction 1996.2 Amination 2006.2.1 Benzylic and (Hetero)aryl Halides 2006.2.2 Benzyl and Allyl Acetates 2056.2.3 Ketones and Aldehydes 2066.2.4 Azoles 2076.2.5 Others 2086.3 Amidation and Thioamidation 2096.3.1 Using the DM Dimethylamine Moiety 2096.3.1.1 Aryl and Alkenyl Halides or Triflates 2096.3.1.2 Acyl Halides 2106.3.1.3 Carboxylic Acids, α-Ketoacids, Esters, Peresters, and Anhydrides 2116.3.1.4 Primary Alcohols and Aldehydes 2166.3.1.5 Methyl Ketones 2176.3.1.6 Nitriles 2186.3.1.7 Dibenzyldisulfanes 2196.3.2 Using the DM Dimethylcarbamoyl Moiety 2196.3.2.1 Aryl Halides 2196.3.2.2 Ketones 221

viii Contents
6.3.2.3 β-Dicarbonyl Compounds 2216.3.2.4 Phenols 2216.3.2.5 Thiophenols 2236.3.2.6 Alkenes 2236.3.2.7 Alkynes 2236.3.2.8 Amines 2246.3.2.9 Amides 2256.3.2.10 Nitriles 2256.3.2.11 Isonitriles 2266.3.2.12 Benzothiazoles 2266.3.2.13 Selenides and Sulfides 2266.3.2.14 Aryl-Tethered Activated Alkenes 2276.3.3 Using the DM Formyl/Acetyl Moiety 2276.3.4 Using the DMF Dimethylamino-Carbon Moiety 2316.3.5 Using CH, CHCONMe2, CH2CONMe2, or H(Me)CONMeCH2 Moiety
of DM 2336.3.5.1 Alcohols 2336.3.5.2 Aldehydes and Ketones 2346.3.5.3 Carboxylic Acids and α-Ketoacids 2356.3.5.4 Amines 2356.3.5.5 Imides and Amides 2366.3.5.6 Alkenes 2376.3.5.7 Sulfides 2376.3.5.8 (Hetero)arenes 2386.3.5.9 Domino Reactions 2406.4 Amidination 2416.4.1 Sulfonamides 2416.4.2 Enamines 2426.5 Formylation and Related Domino Reactions 2426.5.1 Vilsmeier-Mediated Formylations 2436.5.1.1 (Hetero)arenes 2436.5.1.2 Alkenes 2456.5.1.3 O-Silylated Ethers 2466.5.1.4 Alcohols and Phenols 2466.5.1.5 Enamines 2476.5.1.6 Activated Methyl Groups 2476.5.2 Vilsmeier-Mediated Domino Reactions 2476.5.2.1 Formylation and Cyclization 2476.5.2.2 Haloformylation 2496.5.2.3 Haloformylation and Cyclization 2506.5.2.4 Ring Opening, Haloformylation, and Cyclization 2526.5.2.5 Diformylation 2536.5.2.6 Intramolecular Formylation-Intermediate Trapping 2546.5.3 Using Organolithiens or Organomagnesiens 2566.5.4 Hydroformylation 2586.5.5 Formoxylation 2596.6 Carbonylation 260

Contents ix
6.6.1 Carbonyl from DMF 2606.6.2 Carbon from DM Dimethylamine Moiety 2626.7 Cyanation 2646.7.1 Carbon from DMF Carbonyl 2646.7.1.1 Vilsmeier Procedure 2646.7.1.2 n-BuLi (or Mg)/I2/NH3 Procedure 2656.7.2 Carbon from DM Dimethyl 2666.7.2.1 Pd/Cu Procedure 2666.7.2.2 Cu Procedure 2676.7.3 Carbon and Nitrogen from DM Dimethyl 2696.7.3.1 Pd Procedure 2696.7.3.2 Cu Procedure 2706.8 Insertion Reactions 2716.8.1 Alkenes 2716.8.2 Alkynes 2726.8.3 Arynes 2746.8.4 Imines 2766.8.5 Carbenes 2766.8.6 Nitriles 2786.9 Miscellaneous Reactions 2786.9.1 Cycloaddition 2786.9.2 Methylenation 2786.9.2.1 Of Benzylic Carbons 2786.9.2.2 Of Aromatic Carbons 2806.9.2.3 Of Enolic Carbons 2826.9.3 Methylidynation 2836.9.4 Acetylation 2856.9.5 Ether Formation 2856.9.6 Anhydride Formation 2866.9.7 Substitution 2866.9.8 Hydrogen Delivery 2896.9.9 Acetalization of DMF 2946.9.10 Thionation of DM 2946.9.11 Hydrodeoxygenation of DMF 294
Acknowledgments 295References 296
7 The Applications of DMSO 315Jia-Chen Xiang, Qing-He Gao, and An-Xin Wu
7.1 A Brief Introduction of DMSO 3157.2 Name Reactions 3167.2.1 Swern Oxidation 3167.2.2 Parikh–Doering Oxidation 3167.2.3 Pfitzner–Moffatt Oxidation 3177.2.4 Kornblum Oxidation 3177.3 As Reaction Reagents 3187.3.1 Providing –OH (Hydroxylation Reagent) 318

x Contents
7.3.2 Providing –CO (Carbonylation Reagent) 3207.3.3 Providing –SO2Me (Sulfonylation Reagent) 3217.3.4 DMSO Serves as a Source of Sulfur 3247.3.4.1 Providing –MeSMe Group 3247.3.4.2 Providing –SMe Group 3267.3.4.3 Providing –SOMe Group 3317.3.5 As One-Carbon Synthon 3327.3.5.1 Methylation Reagent 3327.3.5.2 Formylation Reagent 3327.3.5.3 Cyanation Reagent 3357.3.5.4 One-Carbon Unit to Participate in the Ring or Bridge Formation 3367.3.6 Dimsyl Anion Activation Reagent 3397.4 As Multifunctional Catalyst/Reagent in Self-Sorting Reaction
System 3427.5 Summary and Perspectives 349
Acknowledgments 349References 349
8 Acetonitrile as Reagents in Organic Synthesis: Reactions andApplications 355Shun-Yi Wang, Xue-Qiang Chu, Yi Fang, and Shun-Jun Ji
8.1 Introduction 3558.2 Transition-Metal-Catalyzed Cross-Coupling of Acetonitrile and
Nitriles 3558.3 Free-Radical-Initiated C–H Functionalization of Acetonitrile and
Nitriles 3668.4 Summary and Outlook 374
Acknowledgments 374References 374
9 The Applications of Nitromethane as Reagent and Solvent inOrganic Synthesis 377Xinxin Qi, Jin-Bao Peng, and Xiao-Feng Wu
9.1 Introduction 3779.2 Reactions with Aldehydes 3779.3 Reactions with Imines 3859.4 Reactions with Ketones 3879.5 Michael Reaction 3889.6 Other Reactions 391
References 395
10 Alcohol as a Reagent in Homogeneous Catalysis 403Feng Han, Wei Sun, Chungu Xia, and Chao Liu
10.1 Introduction 40310.2 Alcohol as O-nucleophile 40310.2.1 Esterification Reaction 403

Contents xi
10.2.2 Oxa-Michael Addition 41110.2.3 Etherification of Alcohol 41410.2.3.1 Etherification of Alcohol with Halide 41410.2.3.2 Etherification of Alcohol with C–H 41810.3 Alcohol Oxidation or α-C–H Functionalization (Alcohol as
C-nucleophile) 42110.3.1 Oxidation 42110.3.2 α-C–H Functionalization 42210.4 Alcohol as Electrophile 42410.4.1 Amination (Amine as Nucleophilic Reagent) 42410.4.2 Alkylation Reaction with Alcohol (Alcohol as Electrophiles) 42610.4.2.1 Alcohol and Alkene 42610.4.2.2 Alcohol and Alkyne 42810.4.2.3 Alcohol and Indole 43010.4.2.4 Alcohol with Other Aromatic Systems 43210.4.3 Ritter Reaction of Alcohol and Nitrile 43510.4.3.1 Brønsted Acid Catalyst 43510.4.3.2 Lewis Acid and Metal Catalysis 43610.5 Conclusion 436
References 437
11 Synchronous Application of Hydrocarbons as Solvents andReagents in Transition-Metal Catalysis 449Jian Cao and Li-Wen Xu
11.1 Introduction 44911.2 Aromatic Hydrocarbons 44911.2.1 C–C Bond Formation 44911.2.1.1 Arene–Arene Coupling 44911.2.1.2 Arene–Alkene Coupling 45811.2.1.3 Arene–Alkyne Coupling 46811.2.1.4 Arene–Haloarenes Coupling 47111.2.1.5 Arene–Arylboronic Acid Coupling 47611.2.1.6 Arene–CO–Alcohol/Amine Coupling 47711.2.2 C–N Bond Formation 47811.2.3 C–O Bond Formation 48211.2.4 C–B Bond Formation 48411.2.5 C–Si Bond Formation 48711.3 Aliphatic Hydrocarbons 49111.3.1 C–C Bond Formation 49111.3.1.1 Alkane–Arene Coupling 49111.3.1.2 Alkane–Alkene Coupling 49311.3.1.3 Alkane-Alkyne Coupling 49711.3.1.4 Alkane–Ketone Coupling 49711.3.1.5 Alkane–Aldehyde Coupling 49811.3.1.6 Alkane–Isocyanide Coupling 49911.3.1.7 Alkane–CO–Amine Coupling 49911.3.1.8 Alkane–Carbene Coupling 500

xii Contents
11.3.1.9 Alkane–Arylboronic Acid Coupling 50111.3.2 C–N Bond Formation 50211.3.3 C–O Bond Formation 50411.3.4 C–S Bond Formation 50411.3.5 C–B Bond Formation 50611.4 Conclusions 507
Acknowledgments 507References 508
Index 515

xiii
List of Contributors
Jian CaoHangzhou Normal UniversityKey Laboratory of OrganosiliconChemistry and Material Technologyof Ministry of EducationScience Park of HZNUHangzhou 311121PR China
Narendra R. ChaubeyBanaras Hindu UniversityDepartment of Chemistry(Centre of Advanced Study)Institute of ScienceVaranasi 221005India
Zhengkai ChenZhejiang Sci-Tech UniversityDepartment of ChemistryHangzhou 310018PR China
Xue-Qiang ChuSoochow UniversityKey Laboratory of Organic Synthesisof Jiangsu ProvinceCollege of Chemistry, ChemicalEngineering and Materials Science &Collaborative Innovation Center ofSuzhou Nano Science and TechnologySuzhou 215123PR China
Yi FangSoochow UniversityKey Laboratory of Organic Synthesisof Jiangsu ProvinceCollege of Chemistry, ChemicalEngineering and Materials Science &Collaborative Innovation Center ofSuzhou Nano Science and TechnologySuzhou 215123PR China
Qing-He GaoXinxiang Medical UniversitySchool of PharmacyDistrict of HongqiHenanXinxiang 453003PR China
Feng HanState Key Laboratory for OxoSynthesis and Selective OxidationSuzhou Research Institute of LICPLanzhou Institute of ChemicalPhysics (LICP)Chinese Academy of SciencesLanzhou 730000PR China

xiv List of Contributors
Shun-Jun JiSoochow UniversityKey Laboratory of Organic Synthesisof Jiangsu ProvinceCollege of Chemistry, ChemicalEngineering and Materials Science &Collaborative Innovation Center ofSuzhou Nano Science and TechnologySuzhou 215123PR China
Jean Le BrasCNRS – Université de ReimsChampagne-ArdenneInstitut de Chimie Moléculaire deReimsUMR 7312, B.P. 103951687 Reims Cedex 2France
Ping LiuNanjing Normal UniversityCollege of Chemistry and MaterialsScienceJiangsu Provincial Key Laboratory ofMaterial Cycle Processes andPollution ControlJiangsu Collaborative InnovationCenter of Biomedical FunctionalMaterialsNanjing 210023PR China
Chao LiuState Key Laboratory for OxoSynthesis and Selective OxidationSuzhou Research Institute of LICPLanzhou Institute of ChemicalPhysics (LICP)Chinese Academy of SciencesLanzhou 730000PR China
Jacques MuzartCNRS – Université de ReimsChampagne-ArdenneInstitut de Chimie Moléculaire deReimsUMR 7312, B.P. 103951687 Reims Cedex 2France
Krishna Nand SinghBanaras Hindu UniversityDepartment of Chemistry(Centre of Advanced Study)Institute of ScienceVaranasi 221005India
Jin-Bao PengZhejiang Sci-Tech UniversityDepartment of ChemistryHangzhou 310018PR China
Xinxin QiZhejiang Sci-Tech UniversityDepartment of ChemistryXiasha CampusHangzhou 310018PR China
Hongjun RenZhejiang Sci-Tech UniversityDepartment of ChemistryHangzhou 310018PR China
Johannes SchranckSolvias AGRömerpark 24303 KaiseraugstSwitzerland

List of Contributors xv
Neetu SinghBanaras Hindu UniversityDepartment of ChemistryCentre of Advanced StudyInstitute of ScienceVaranasi 221005India
Peipei SunNanjing Normal UniversityCollege of Chemistry and MaterialsScienceJiangsu Provincial Key Laboratory ofMaterial Cycle Processes andPollution ControlJiangsu Collaborative InnovationCenter of Biomedical FunctionalMaterialsNanjing 210023PR China
Wei SunState Key Laboratory for OxoSynthesis and Selective OxidationSuzhou Research Institute of LICPLanzhou Institute of ChemicalPhysics (LICP)Chinese Academy of SciencesLanzhou 730000PR China
Anis TliliUniversité Lyon 1Institute of Chemistry andBiochemistry (ICBMS-UMR CNRS5246), CNRSVilleurbanneFrance
Jie-Ping WanJiangxi Normal UniversityCollege of Chemistry and ChemicalEngineeringNanchang 330022PR China
Shun-Yi WangSoochow UniversityKey Laboratory of Organic Synthesisof Jiangsu ProvinceCollege of Chemistry, ChemicalEngineering and Materials Science &Collaborative Innovation Center ofSuzhou Nano Science and TechnologySuzhou 215123PR China
An-Xin WuKey Laboratory of Pesticide &Chemical BiologyMinistry of EducationCollege of ChemistryCentral China Normal UniversityHubeiWuhan 430079PR China
Xiao-Feng WuZhejiang Sci-Tech UniversityDepartment of ChemistryXiasha CampusHangzhou 310018PR China
and
Universität RostockLeibniz-Institut für Katalysee.V. an derAlbert-Einstein-Straße 29a18059 RostockGermany
Chungu XiaState Key Laboratory for OxoSynthesis and Selective OxidationSuzhou Research Institute of LICPLanzhou Institute of ChemicalPhysics (LICP)Chinese Academy of SciencesLanzhou 730000PR China

xvi List of Contributors
Jia-Chen XiangKey Laboratory of Pesticide &Chemical BiologyMinistry of EducationCollege of ChemistryCentral China Normal UniversityHubeiWuhan 430079PR China
Li-Wen XuHangzhou Normal UniversityKey Laboratory of OrganosiliconChemistry and Material Technologyof Ministry of EducationScience Park of HZNUHangzhou 311121PR China
Guanghui ZhangPurdue UniversitySchool of Chemical EngineeringWest LafayetteIN 47907USA

1
1
The Applications of Water as Reagents in Organic SynthesisZhengkai Chen and Hongjun Ren
Zhejiang Sci-Tech University, Department of Chemistry, Hangzhou 310018, PR China
1.1 Introduction
Water due to its low cost, easy availability, nontoxic and nonflammableproperties has been considered one of the most ideal and promising solvents inorganic synthesis from the green and sustainable point of view. Furthermore,with regard to enormous enzyme-catalyzed biosynthesis in nature, water servesas a favorable medium for the versatile synthesis of a variety of complicatedmolecules and compounds. Over the past decades, considerable efforts had beendevoted into the organic reactions by using water as solvent from economy andenvironment perspectives [1]. Even more, the proposed concept of “in-water”and “on-water” further stimulated the booming development of the utilizationof water as solvent for organic synthesis [1e, 2]. Therefore, in recent years, moreand more general organic reactions were successfully exploited to perform inwater instead of organic solvents to achieve sustainable and environmentalbenefits.
From a different perspective, the water itself could also be applied as a usefulreagent to participate in the reaction through incorporating a hydrogen oroxygen atom or hydroxyl group into the target product. Generally, water isindispensable for various hydrolysis reactions. As a hydrogen source, wateris used to quench numerous susceptible reaction systems by providing activehydrogen. Meanwhile, as a versatile nucleophile, the hydroxyl group could bereadily introduced into the specific reaction sites by the employment of wateras hydroxyl precursor. The hydroxyl group could also be readily oxidized tocarbonyl group during the reaction. It is worth mentioning that, in some cases,the presence of water could obviously improve the efficiency of the reaction,albeit the exact reason is elusive for some special reactions.
This chapter is divided into the following four parts for further discussion: (i)incorporation of hydrogen atom from water; (ii) incorporation of oxygen atomfrom water; (iii) incorporation of hydroxyl group from water; and (iv) tracelesspromotion of the reactions by water.
Solvents as Reagents in Organic Synthesis: Reactions and Applications,First Edition. Edited by Xiao-Feng Wu.© 2018 Wiley-VCH Verlag GmbH & Co. KGaA. Published 2018 by Wiley-VCH Verlag GmbH & Co. KGaA.

2 1 The Applications of Water as Reagents in Organic Synthesis
1.2 Incorporation of Hydrogen Atom from the Water
Aggarwal and coworkers demonstrated a versatile strategy through lithia-tion/borylation/protodeboronation of a homoallyl carbamate for the highlyenantioselective synthesis of (+)-sertraline and (+)-indatraline, which servedas potent inhibitors (Scheme 1.1) [3]. It was observed that the presence of thealkene could hamper the lithiation/borylation process, so the modifications ofthe reaction conditions were necessary by the use of 12-crown-4, TMSCl, H2Oor a solvent switch to achieve 1,2-metalate rearrangement in order to ensurehigh yields and enantioselectivity. As for the protodeboronation step of tertiaryboronic ester, the amount of water played a crucial role in the reaction. In 2010,the same group had disclosed a simple approach for the protodeboronationof tertiary boronic esters employing CsF-H2O or TBAF⋅3H2O with completestereoselectivity to access to diverse enantioenriched tertiary alkanes [4].
A catalyst-free sulfonylation of activated alkenes with sulfonyl hydrazidesin water for highly efficient construction of monosubstituted ethyl sulfoneswas demonstrated by Wang and coworkers (Scheme 1.2) [5]. Remarkably, thereaction proceeded through without any catalyst, additive, ligand, or organicsolvent, with the release of N2 as single by-product. The results of controlexperiments indicated that an anion pathway was involved in the reaction andthe α-hydrogen atom of β-sulfone esters originated from water.
The sulfinyl anion I was first formed assisted by water with the release of onemolecule of N2, which could transform into sulfur-centered anion II throughresonance process in the presence of water. The sulfur-centered anion readilyadded to the activated alkene to give the oxygen-centered anion III, followed byanother resonance interaction leading to the carbon-centered anion IV. Finally,the proton transfer of intermediate IV from hydronium ions to deliver the desiredβ-sulfone ester product.
OCb (1) sBuLi (1.3 equiv.), 1 h, –78 °C
(2) ArBpin (1.5 equiv.), 2 h, –78 °C
(3) 12-crown-4 (1.2 equiv.),
H2O (0.1 equiv.), 1 h, –78 °C
(4) TMSCl (1.2 equiv.), 16 h, rt
Bpin
ClCl
CsF (1.5 equiv.)H2O (2.5 equiv.)
CH2Cl2, rt, overnight
Cl
Cl
H
(+)-Sertraline
(+)-Indatraline
98% ee
98% ee
95% ee
sBuLi, ArBpin, CHCl3
Scheme 1.1 Enantioselective synthesis of (+)-sertraline and (+)-indatraline.

1.2 Incorporation of Hydrogen Atom from the Water 3
+R1
O
R2 S
O
O
NHNH265 °C, air, 24 h
H2OR1
O
S
O
OR2
R2S
O
O
NHNH2
H2O H3O+
N2
R2S
O–
OR2 S
O
O
-
R1
O
O–
R1 S
O
O
R2
O
R1 S
O
O
R2-R1
O
S
O
OR2 H3O+H2O
H
I II
IIIIV
Scheme 1.2 Catalyst-free sulfonylation of activated alkenes in water.
HO
R1
R2
+ TMS-N3
N3
OHR1
R2
Ag2CO3 (10 mol%)
DMSO (trace amount of H2O)
80 °C, N2, 1–2 h
[Ag]
HO
R1
R2Ag
[Ag]
HN3OH
R1
R2H
Ag
H2OH2O
I IIN3
Scheme 1.3 Silver(I)-catalyzed hydroazidation of ethynyl carbinols.
A silver(I)-catalyzed chemo- and regioselective hydroazidation of ethynylcarbinols for the construction of 2-azidoallyl alcohols was developed by Biand coworkers (Scheme 1.3) [6]. In this transformation, trimethylsilyl azide(TMS-N3) was chosen as the optimal azide source and the pendent hydroxylgroup directed the chemo- and regioselectivity of hydroazidation by stabilizingthe vinyl azide products. Catalyzed by 10 mol% Ag2CO3, a wide range ofsecondary and tertiary ethynyl carbinols bearing different substituents could betransformed into the corresponding products in good to excellent yields.
The observation data of control experiments implied that the residual water inthe DMSO played the critical role in the reaction. The initial step of the plausiblepathway involved the generation of silver acetylide intermediate I. Meanwhile,the hydrazoic acid (HN3) was in situ formed by silver-catalyzed hydrolysis ofTMS-N3, which added to the intermediate I to lead to vinyl silver intermediate II.Under the promotion of a trace amount of H2O in the DMSO solvent, the proto-nation of intermediate II released the final product. As a class of functionalized

4 1 The Applications of Water as Reagents in Organic Synthesis
synthetic intermediates, 2-azidoallyl alcohols could be readily transformed intoNH aziridines under the mild reaction conditions.
1.2.1 1,2,3-Triazoles
Inspired by the previous work of silver(I)-catalyzed hydroazidation of ethynylcarbinols, Bi and coworkers extended their study to the general hydroazidationof unactivated alkynes (Scheme 1.4a) [7]. The key point of the progress lay inthe necessity of a trace amount of water in DMSO with regard to hydroazidationof ethynyl carbinols. Therefore, it was speculated that a stoichiometric amountof H2O could enhance the reactivity of the reaction and the relevant experimentsconfirmed the hypothesis. The condition screening of the amount of H2O demon-strated 2.0 equiv. of H2O was appropriate for high efficiency. Furthermore, it wasessential to control the reaction time to circumvent the further conversion ofvinyl azides to nitriles. The protocol featured readily accessible starting materials,mild reaction conditions, broad substrate scope, and good scalability.
The significance of the aforementioned synthetic method was embodiedin the application for the assembly of several valuable heterocyclic frame-works. In 2015, the strategy of hydroazidation of unactivated alkynes wascombined with alkyne-azide 1,3-dipolar cycloaddition reaction to access to avariety of piperidine-fused 1,2,3-triazoles by Bi and coworkers (Scheme 1.4b)[8]. Under silver-catalyzed conditions, the treatment of diyne with TMS-N3in the presence of H2O gave rise to pharmaceutically relevant 1,5-fused1,2,3-triazoles in excellent yields. The reaction was assumed to undergo thetandem hydroazidation/alkyne-azide 1,3-dipolar cycloaddition sequence, whichpresented a concise method for the synthesis of structurally complicated fusedheterocyclic compounds in one pot.
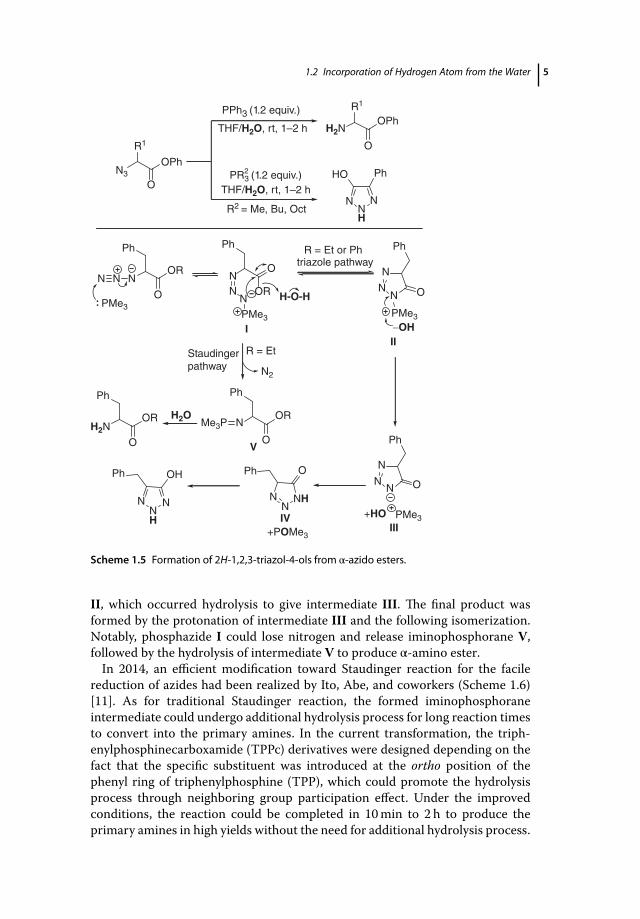
Taylor and coworkers developed a variant of Staudinger reaction on α-azidoesters with trialkyl phosphines for the formation of 2H-1,2,3-triazol-4-ols(Scheme 1.5) [9]. In this reaction, phosphazides were generated from the reac-tion of trialkyl phosphines with α-azido esters in THF/H2O, which underwentintramolecular cyclization to afford the desired products. Upon using PPh3, themajor product was the reduced amine from the classic Staudinger pathway [10].As shown in Scheme 1.5, phosphazide I could cyclize to deliver intermediate
+ TMS-N3
Ag2CO3 (10 mol%)
H2O (2.0 equiv.)
DMSO, 80 °C, 20–60 min
R1
R2
R3
N3
R1
R2
R3
X
R1
R4
R5
R2
R3
+ TMS-N3
Ag2CO3 (10 mol%)
H2O (2.0 equiv.)
DMSO, 80 °C, 3–5 h
X R4
R5
R2
R3
N
N N
R1
(a)
(b)
X = C, N, O
Scheme 1.4 Silver-catalyzed hydroazidation of alkynes and the application to access to1,5-fused 1,2,3-triazoles.
1.2 Incorporation of Hydrogen Atom from the Water 5
N3
OPh
O
R1
PPh3 (1.2 equiv.)
THF/H2O, rt, 1–2 h
PR23 (1.2 equiv.)
THF/H2O, rt, 1–2 h
R2 = Me, Bu, Oct
NNH
N
PhHO
H2NOPh
O
R1
NOR
O
Ph
NN
PMe3
NO
OR
Ph
NN
PMe3
H-O-H
R = Et or Phtriazole pathway
N
NN
Ph
O
PMe3
−OHI
IIR = Et
N2
Staudinger
pathway
NOR
O
Ph
Me3PH2O
H2NOR
O
Ph
V
N
NN
Ph
O
+HO PMe3
III
NN
NH
Ph O
IV
+POMe3
NNH
N
Ph OH
Scheme 1.5 Formation of 2H-1,2,3-triazol-4-ols from α-azido esters.
II, which occurred hydrolysis to give intermediate III. The final product wasformed by the protonation of intermediate III and the following isomerization.Notably, phosphazide I could lose nitrogen and release iminophosphorane V,followed by the hydrolysis of intermediate V to produce α-amino ester.
In 2014, an efficient modification toward Staudinger reaction for the facilereduction of azides had been realized by Ito, Abe, and coworkers (Scheme 1.6)[11]. As for traditional Staudinger reaction, the formed iminophosphoraneintermediate could undergo additional hydrolysis process for long reaction timesto convert into the primary amines. In the current transformation, the triph-enylphosphinecarboxamide (TPPc) derivatives were designed depending on thefact that the specific substituent was introduced at the ortho position of thephenyl ring of triphenylphosphine (TPP), which could promote the hydrolysisprocess through neighboring group participation effect. Under the improvedconditions, the reaction could be completed in 10 min to 2 h to produce theprimary amines in high yields without the need for additional hydrolysis process.

6 1 The Applications of Water as Reagents in Organic Synthesis
R1 N3
P
R′
R′
Ph
Ph
N2
PPh
Ph N
R1
SlowH2O addition
TPP: R′ = H
–PPh3=OR1 NH2
Aza-phosphine ylide
FastH2O addition
TPPc: R′ = CONR2
R3
PPh
Ph N
R1
N
O
R2
R3
PPh
Ph
N
O
R2
R3
N
R1H
OH
Staudinger reaction
PPh
Ph OO
N
R3
R2
Scheme 1.6 Triphenylphosphinecarboxamide: An effective reagent for the reduction ofazides.
+R1
R2
[Ph3PCF2Br]+Br−
Visible lightfac-[Ir(ppy)3] (3 mol%)
PPh3 (5.0 equiv.)NaI (5.0 equiv.)
KHCO3 (5.0 equiv.)
H2O (10 equiv.)
THF, rt, N2, 10 h
R1
R2 H
CF2H
[Ph3PCF2Br]Br−H2O
[ CF2] + 2HBr + Ph3P=O
NaI, H2O
HCF2I
PPh3
+
+[Ph3PCF2H]X−
HCF2Br
PPh3
Ph3P-CF2
+ –
H2O or HBr
PPh3
SET
CF2H + PPh3 + X−
Scheme 1.7 Visible-light-induced hydrodifluoromethylation of alkenes.
A visible-light-induced hydrodifluoromethylation of alkenes by the use of bro-modifluoromethylphosphonium bromide as the precursor of difluorocarbene forthe direct synthesis of the difluoromethylated alkanes was achieved by Qing andcoworkers (Scheme 1.7) [12]. For the first time, the CF2H radical was generatedfrom fluorinated phosphonium salts, which could be readily synthesized fromthe reaction of PPh3 and CF2Br2 in quantitative yield. The results of mechanistic

1.3 Incorporation of Oxygen Atom from the Water 7
investigations implied that the formation process of CF2H radical (Scheme 1.7).The reaction of bromodifluoromethylphosphonium bromide with H2O gen-erated difluorocarbene, from which the HCF2I or HCF2Br was formed andreacted with PPh3 to give difluoromethylphosphonium salts. Another pathwayinvolved the capture of difluorocarbene by PPh3 and subsequent treatment ofH2O or HBr to deliver difluoromethylphosphonium salts, which could undergoa single-electron transfer (SET) process mediated by fac-[IrIII(ppy)3]* to lead tothe CF2H radical.
1.3 Incorporation of Oxygen Atom from the Water
Two facile one-pot copper-catalyzed reactions for the generation of furo[3,2-c]coumarins and chlorofuro[3,2-c]coumarins using 2-(1-alkynyl)-2-alken-1-onederivatives as starting materials was disclosed by Hu and Cheng (Scheme 1.8)[13]. One reaction involved CuCl-catalyzed cascade addition/cyclization/oxidation sequence of 2-(1-alkynyl)-2-alken-1-one in the presence of water.Another protocol utilized CuBr as catalyst and excess CuCl2 as chlorinatedreagent. The proposed mechanism indicated that in the two processes, Lewisacid Cu(I) salt activated the carbonyl group to facilitate the Michael addition ofH2O to the C=C bond, which was the key step of the reaction.
An unexpected example about the hydroxyphosphinylation reaction of3-cyclopropylideneprop-2-en-1-ones for the introduction of hydroxyl group andphosphorus group via C—P bond cleavage was developed by Wu and coworkers(Scheme 1.9) [14]. The transformation utilized a highly activated analogue of
R1
O
O R2
CuCl (20 mol%)
H2O (10 equiv.)
DMF, 90 °C, 10 h
R1
O
O
O
R2
CuBr (10 mol%)CuCl2 (4.2 equiv.)H
2O (10 equiv.)DMF, 75 °C, 10 h R1
O
O
O
R2
Cl
Scheme 1.8 Two cascade reactions to synthesize substituted furocoumarins.
+R2
O
R1
P R″
R″
R″′
R″′R′
H2O (7.5 equiv.)
Acetone, 0 °C, air, 10 h
R2 R1
O
OH
P O
Scheme 1.9 Hydroxyphosphinylation reaction of 3-cyclopropylideneprop-2-en-1-ones.

8 1 The Applications of Water as Reagents in Organic Synthesis
allene and tertiary phosphine as starting material to construct highly func-tionalized 1-dialkylphinyl-3-oxo-(1Z)-alkenyl cyclopropanols in a regio- andstereoselective manner under the metal-free conditions. Mechanistic study indi-cated that the high strain in the C=C bond of allene substrate played a significantrole in the hydroxyphosphinylation reaction. Intriguingly, based on the proposedmechanism, the oxygen atom of hydroxyl group in the product originated fromO2 and the oxygen atom of P=O bond came from additional H2O.
A metal-free TfOH-catalyzed domino cycloisomerization/hydrolytic defluori-nation reaction of n-perfluoroalkyl allenones for the assembly of furanyl perflu-oroalkyl ketones was developed by Ma and coworkers (Scheme 1.10) [15]. Theadditional water was necessary for the reaction since the oxygen atom of the car-bonyl group in the product came from H2O based on 18O-labelling experiments.The reaction proceeded through nucleophilic attack from the carbonyl oxygen,1,2-phenyl shift, aromatization, nucleophilic attack of water, and elimination ofHF sequence. The whole domino reaction process was solely catalyzed by H+
from TfOH to deliver a wide range of furan-2-yl perfluoroalkyl ketones in goodyields.
In 2013, Sun and coworkers described an I2–H2O mediated highly chemoselec-tive synthesis of benzyl derivatives through oxidation of stilbenes without the useof any acid and metal (Scheme 1.11) [16]. A wide variety of substituted stilbeneswere viable substrates for the protocol and the corresponding benzyl productscould be constructed in high yields. Isotopic labeling experiments verified thatthe oxygen atom of the benzils derived from water and molecular oxygen partic-ipated in the reaction. The reaction pathway presumably involved the generationof an iodonium ion, the attack of water to the iodonium ion and sequential oxi-dation with iodine in water under air.
A Cu(0)/Selectfluor mediated oxidative cyclization of 1,5-enynes in the pres-ence of water with C—C bond cleavage for the synthesis of 3-formyl-1-indenonederivatives was described by Liu and coworkers (Scheme 1.12) [17]. The reac-tion involved water-participated oxygen-insertion β-carbon elimination and thecleavage of C—C bond sequence. The 18O-labeling experiment unambiguouslysuggested that both of the carbonyl oxygen atoms in the product came from water.On the basis of preliminary mechanistic studies, the o-alkynyl epoxide served
TfOH (10 mol%)
H2O (1.8 equiv.)R3
R2
R1
O
CF2Rf Toluene, 100 °C, 2–21 hO
R3R1
R2Rf
O
Scheme 1.10 TfOH-catalyzed domino cycloisomerization/hydrolytic defluorination of2,3-allenyl perfluoroalkyl ketones.
H2O, 140 °C, 20 hR1
R2
I2 (2.0 equiv.)
R1
R2O
O
Scheme 1.11 Synthesis of benzyl derivatives via oxidation of stilbenes in an I2–H2O system.

1.3 Incorporation of Oxygen Atom from the Water 9
+R1 Ph
O
R2
Cu(0) (5 mol%)Selectfluor (2.0 equiv.)NaHCO3 (2.0 equiv.)
MeCN/H2O (150 : 1)
80 °C, 4 h
R1
CHO
O
R2
Ph F
O
Scheme 1.12 Copper-catalyzed oxidative cyclization of 1,5-enynes to access to3-formyl-1-indenones.
as an intermediate for the reaction and oxycupration of the triple bond and thefollowing ring opening of the epoxide moiety constituted the key steps of thereaction. The C—C bond was cleaved assisted by Selectfluor to deliver benzoylfluoride.
A Rh(II)-catalyzed denitrogenative hydration reaction of N-sulfonyl-1,2,3-triazoles with water for the synthesis of a series of biologically active α-aminoketones was demonstrated by Murakami and coworkers (Scheme 1.13) [18].N-Sulfonyl-1,2,3-triazoles could be readily available from copper-catalyzed1,3-dipolar cycloaddition reaction (CuAAC) of N-sulfonyl azides and terminalalkynes. In the transformation, the key intermediate α-imino rhodium-(II) car-benoid II was in situ generated assisted by rhodium catalyst, which underwentinsertion into the O—H bond of water to produce α-imino alcohol III. Finally, theimine–enamine tautomerization and the following keto–enol tautomerizationsequence led to the desired α-amino ketone products. The protocol achievedregioselective 1,2-aminohydroxylation of terminal alkynes, which served as acomplement to the example reported by Chang and Fokin employing a copper(I)catalyst to realize 1,1-aminohydroxylation of terminal alkynes [19].
The Wacker oxidation reaction was a typical and powerful tool for the oxidationof olefins to synthesize ketones in the presence of palladium(II) catalyst, water,and co-oxidant [20]. Usually, the reaction occurred in Markovnikov selectivityfrom the majority of terminal olefins. In 1959, researchers at Wacker Chemieutilized catalytic PdCl2 and a stoichiometric amount of CuCl2 with bubbling O2to realize the hydration of olefins (Scheme 1.14). Afterward, the Wacker process
N NN
R1
SO2R2
+ H2O
(10 equiv)
Rh2(Oct)4 (0.5 mol%)
CHCl3, 15 min
140 °C/MWR1
O HN
SO2R2
SO2R2
SO2R2SO2R2
SO2R2SO2R2N N
N
R1
N2 N
R1H H
Rh(II)
–N2
[Rh] N
R1 HI II
H2O
–Rh(II)
N
R1 HIII
HOH HN
R1 HIV
HOTM
Scheme 1.13 Rh(II)-catalyzed denitrogenative hydration reaction of N-sulfonyl-1,2,3-triazoles.

10 1 The Applications of Water as Reagents in Organic Synthesis
PdCl2 (10 mol%)
CuCl2 (10 mol%)
DMF/H2O, 60–70 °C
bubbling O2
RR Me
O
Scheme 1.14 The Wacker oxidation reaction for the synthesis of methyl ketones.
+
PdCl2(CH3CN)2 (2.5 mol%)
BQ (1.15 equiv.)
t-BuOH, 85 °C, 1 hAr H2O Ar
O
Scheme 1.15 Efficient and highly aldehyde selective Wacker oxidation.
was extensively investigated by synthetic chemists to overcome several key lim-itations. In recent years, the aldehyde-selective Wacker oxidation had been welldeveloped in the field of anti-Markovnikov functionalization of unbiased alkenes,which could produce a range of synthetically versatile aldehydes.
A Pd(II)-catalyzed aldehyde-selective Wacker oxidation of aryl-substitutedolefins in the presence of 1,4-benzoquinone was described by Grubbs andcoworkers (Scheme 1.15) [21], which was greatly different from the classicalWacker oxidation of providing methyl ketones. As demonstrated by the previousworks, t-BuOH was used in Wacker oxidation to improve the aldehyde selectivityand BQ was widely applied as a hydrogen acceptor and two-electron oxidant inPd(II)-catalyzed reactions. Noteworthy was that high yield of aldehyde productswas obtained with respect to more electron-deficient aromatic substrates. Thereaction mechanism was similar to the previous work of the same author, inwhich the Pd-catalyzed oxidation and acid-catalyzed hydrolysis process wereinvolved in the reaction.
The direct oxygenation of an allylic C—H bond catalyzed by palladium usingH2O as oxygen source for the production of (E)-alkenyl aldehydes was developedby Jiang and coworkers (Scheme 1.16) [22]. During the process, allylic C—H bondcleavage occurred and an allyl-palladium species was formed as the key interme-diate. The choice of DDQ as oxidant could greatly promote the reaction efficiency.The substrate scope was broad and diverse (E)-alkenyl aldehyde products wereafforded in high yields with good stereoselectivity. The kinetic isotopic experi-ments implied that the activation of the allyl C(sp3)—H bond was involved in therate-determining step.
A plausible reaction mechanism is shown in Scheme 1.16. π-Allylpalladiumspecies I was generated from the reaction of alkene and Pd(II) through the allylicC—H bond activation. Subsequently, the nucleophilic attack of H2O into inter-mediate I afforded the oxidative allylic oxygenation products III and III′ , whichexisted as an equilibrating mixture. Finally, the desired aldehyde product wasformed by DDQ promoted oxidation of III and III′ .
The direct anti-Markovnikov dehydrogenative oxygenation of β-alkyl styrenesunder external-oxidant-free conditions by the use of the synergistic effectof photocatalysis and proton-reduction catalysis was presented by Lei andcoworkers (Scheme 1.17) [23]. In this transformation, water was applied as singleterminal oxidant for the construction of a series of carbonyl compounds. The

1.3 Incorporation of Oxygen Atom from the Water 11
+
PdCl2 (10 mol%)
DDQ (2.0 equiv.)R H2O
DCE, 50 °C, 2 h RCHO
Pd(II)R
H
HX
R
PdX
I
R
PdX
II
H2OH2O
Pd(0)
NC
NC
Cl
Cl
OH
OH
H2O
HX
NC
NC
Cl
Cl
O
ODDQ
R
OH
R OH
III′ III
NC
NC
Cl
Cl
O
ODDQ
NC
NC
Cl
Cl
OH
OH
RCHO
Scheme 1.16 Pd(II)-catalyzed direct oxygenation of allylic C—H bond with H2O.
Ar
R1
R2
+ H2O
Acr+-Mes ClO4–(5 mol%)
Co(dmgH)2pyCl (3 mol%)
CH3CN, rt, blue LEDsAr
R1
R2
O
NClO4
–
+
Co
N
NN
N
Me
Me
NMe
Me ClO
O
H
OO
H
Acr+-Mes ClO4– Co(dmgH)2pyCl
Scheme 1.17 Anti-Markovnikov oxidation of β-alkyl styrenes with H2O as the terminal oxidant.

12 1 The Applications of Water as Reagents in Organic Synthesis
reaction proceeded through an alkene radical cation intermediate, which wasin situ formed by the photoinduced system, followed by the nucleophilic attackof water to give distonic radical cation. Subsequently, the deprotonation of thedistonic radical cation produced the anti-Markovnikov intermediate, whichunderwent single-electron oxidation, elimination, and keto–enol tautomerismsequence to lead to the final carbonyl products. The synergistic effect of this dualcatalytic system was vital for the single anti-Markovnikov selectivity. The resultsof control experiments indicated that the oxygen atom of carbonyl group wasderived from water.
A transition-metal-free carboxyamidation reaction by the use of aryl diazo-nium tetrafluoroborates and isocyanides as coupling partners for the synthesisof arylcarboxyamides under mild conditions was achieved by Zhu and Xia(Scheme 1.18) [24]. It is worth mentioning that arylcarboxyamides could notbe directly assembled by the aminocarbonylation of aryl diazonium salts withamines in the presence of CO. The reaction was realized in the absence oftransition-metal catalysts in aqueous media at low temperature and exhibitedbroad substrate scope with moderate to high efficiency.
A radical mechanism involving hydroxide- or polar-solvent-induced dediazo-niation is proposed in Scheme 1.18. Aryl radical I was first formed via homolyticdediazoniation process, which included SET from hydroxide or acetone to aryldiazonium salt. The reaction of aryl radical I with isocyanide delivered a keyimidoyl radical intermediate II, followed by oxidation by aryl diazonium cationto lead to a nitrilium intermediate III. Finally, the hydration and tautomeriza-tion of intermediate III in the presence of H2O could afford the desired arylcar-boxyamide product.
One year later, the transition-metal-free multicomponent reaction involvingarynes and isocyanides with H2O for the preparation of benzamide derivativeswas reported by Biju and coworkers (Scheme 1.19) [25]. The 1,3-zwitterionicintermediate generated from isocyanide and aryne could be intercepted bydifferent electrophiles for the synthesis of diverse benzannulated heterocycles.The treatment of isocyanide and 2-(trimethylsilyl)aryl triflate mediated by KF
+N2
+BF4–
R1CN R2
Cs2CO3 (1.1 equiv.)acetone/H2O (5 : 2)
0 °C, Ar, 20 minR1
NH
O
R2
Ar N2+
Solvent/OH–
–N2
Ar
C N R
Ar C
N R
I
II
–N2
Ar N2+Ar N R
+
III
H2O
–H+Ar
O
NH
R
Scheme 1.18 Synthesis of arylcarboxyamides from aryl diazonium salts and isocyanides.


















