
Small-Molecule Sigma1 Modulator Induces Autophagic...
14
Metabolism Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1 Christina M. Maher 1 , Jeffrey D. Thomas 1 , Derick A. Haas 1 , Charles G. Longen 1 , Halley M. Oyer 1 , Jane Y.Tong 1 , and Felix J. Kim 1,2 Abstract Emerging evidence suggests that Sigma1 (SIGMAR1, also known as sigma-1 receptor) is a unique ligand-regulated inte- gral membrane scaffolding protein that contributes to cellular protein and lipid homeostasis. Previously, we demonstrated that some small-molecule modulators of Sigma1 alter endo- plasmic reticulum (ER)–associated protein homeostasis path- ways in cancer cells, including the unfolded protein response and autophagy. Programmed death-ligand 1 (PD-L1) is a type I integral membrane glycoprotein that is cotranslationally inserted into the ER and is processed and transported through the secretory pathway. Once at the surface of cancer cells, PD-L1 acts as a T-cell inhibitory checkpoint molecule and suppresses antitumor immunity. Here, we demonstrate that in Sigma1- expressing triple-negative breast and androgen-independent prostate cancer cells, PD-L1 protein levels were suppressed by RNAi knockdown of Sigma1 and by small-molecule inhibition of Sigma1. Sigma1-mediated action was confirmed by phar- macologic competition between Sigma1-selective inhibitor and activator ligands. When administered alone, the Sigma1 inhibitor decreased cell surface PD-L1 expression and sup- pressed functional interaction of PD-1 and PD-L1 in a coculture of T cells and cancer cells. Conversely, the Sigma1 activator increased PD-L1 cell surface expression, demonstrating the ability to positively and negatively modulate Sigma1 associated PD-L1 processing. We discovered that the Sigma1 inhibitor induced degradation of PD-L1 via autophagy, by a mechanism distinct from bulk macroautophagy or general ER stress–asso- ciated autophagy. Finally, the Sigma1 inhibitor suppressed IFNg -induced PD-L1. Our data demonstrate that small-mole- cule Sigma1 modulators can be used to regulate PD-L1 in cancer cells and trigger its degradation by selective autophagy. Implications: Sigma1 modulators sequester and eliminate PD-L1 by autophagy, thus preventing functional PD-L1 expres- sion at the cell surface. This posits Sigma1 modulators as novel therapeutic agents in PD-L1/PD-1 blockade strategies that regulate the tumor immune microenvironment. Visual Overview: http://mcr.aacrjournals.org/content/molcanres/ 16/2/243/F1.large.jpg. Mol Cancer Res; 16(2); 243–55. Ó2017 AACR. Introduction Programmed death-ligand 1 (PD-L1, also known as B7-H1 or CD274) is a type I integral membrane glycoprotein that is cotranslationally inserted into, posttranslationally modified in, and transported through the secretory pathway of a range of cell types, including tumor cells (1–4). In this pathway, the endo- plasmic reticulum (ER) is the primary site of synthesis, folding, and assembly of secreted and integral membrane proteins. Protein homeostasis in the ER relies on the timely convergence of multiple mechanisms that detect protein–concentration thresholds, pro- vide quality control, and regulate the ebb and flow of ER proteins (4–6). Integral membrane proteins are intrinsically dependent on ER protein homeostasis machinery to reach the cell surface. PD-L1 expressed at the surface of tumor cells can act as a T-cell inhibitory checkpoint molecule and can inactivate tumor-infiltrating immune cells that express cell surface pro- grammed death-1 (PD-1; also known as CD279; refs. 1, 3, 7). Immune checkpoint inhibitors that block PD-L1/PD-1 inter- actions are promising therapeutic agents in strategies that exploit antitumor immune responses. However, a relatively small percentage of cancer patients respond to anti–PD-L1/ PD-1 monotherapy, which thus far includes subsets of patients with melanoma, non–small cell lung cancer, metastatic blad- der cancer, and renal cell carcinoma (7–10). Although high PD-L1 expression has been associated with poor prognosis in a range of cancers (11–13), high PD-L1 expression levels do not necessarily correspond with response to anti–PD-L1/PD-1 therapeutic agents (7, 14). For instance, anti–PD-L1/PD-1 agents to treat prostate and triple-negative breast tumors with high levels of PD-L1 have shown minimal efficacy in early clinical trials (7–10, 12–15). Therefore, the search for predic- tive biomarkers of response to PD-L1/PD-1 agents is an active area of investigation (14, 16), as is the search for novel agents and combination therapy approaches that modulate the tumor microenvironment to promote antitumor immunity (7, 14–17). Full-length PD-L1 comprises 229 amino acids leading with an N-terminal signal sequence, IgV- and IgC- extracellular domains, which engage PD-1 on infiltrating immune cells, a single trans- membrane domain, and a relatively short 31 residue cytoplasmic tail, which has no defined functional motifs and whose role in PD-L1 biology has not been determined (1–3, 18). The cellular factors and processes engaged in the synthesis, maturation, and 1 Department of Pharmacology & Physiology, Drexel University College of Medicine, Philadelphia, Pennsylvania. 2 Sidney Kimmel Cancer Center, Philadelphia, Pennsylvania. Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/). Corresponding Author: Felix J. Kim, Department of Pharmacology & Physiol- ogy, Drexel University College of Medicine, 245 N. 15th Street, Philadelphia, PA 19102. Phone: 215-762-2508; Fax: 215-762-2299; E-mail: [email protected] doi: 10.1158/1541-7786.MCR-17-0166 Ó2017 American Association for Cancer Research. Molecular Cancer Research www.aacrjournals.org 243 on August 9, 2019. © 2018 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from Published OnlineFirst November 8, 2017; DOI: 10.1158/1541-7786.MCR-17-0166
Transcript of Small-Molecule Sigma1 Modulator Induces Autophagic...

Metabolism
Small-Molecule Sigma1 Modulator InducesAutophagic Degradation of PD-L1Christina M. Maher1, Jeffrey D. Thomas1, Derick A. Haas1, Charles G. Longen1,Halley M. Oyer1, Jane Y. Tong1, and Felix J. Kim1,2
Abstract
Emerging evidence suggests that Sigma1 (SIGMAR1, alsoknown as sigma-1 receptor) is a unique ligand-regulated inte-gral membrane scaffolding protein that contributes to cellularprotein and lipid homeostasis. Previously, we demonstratedthat some small-molecule modulators of Sigma1 alter endo-plasmic reticulum (ER)–associated protein homeostasis path-ways in cancer cells, including the unfolded protein responseand autophagy. Programmed death-ligand 1 (PD-L1) is a type Iintegral membrane glycoprotein that is cotranslationallyinserted into the ER and is processed and transported throughthe secretory pathway. Once at the surface of cancer cells, PD-L1acts as a T-cell inhibitory checkpoint molecule and suppressesantitumor immunity. Here, we demonstrate that in Sigma1-expressing triple-negative breast and androgen-independentprostate cancer cells, PD-L1 protein levels were suppressed byRNAi knockdown of Sigma1 and by small-molecule inhibitionof Sigma1. Sigma1-mediated action was confirmed by phar-macologic competition between Sigma1-selective inhibitorand activator ligands. When administered alone, the Sigma1inhibitor decreased cell surface PD-L1 expression and sup-
pressed functional interaction of PD-1 and PD-L1 in a cocultureof T cells and cancer cells. Conversely, the Sigma1 activatorincreased PD-L1 cell surface expression, demonstrating theability to positively and negatively modulate Sigma1 associatedPD-L1 processing. We discovered that the Sigma1 inhibitorinduced degradation of PD-L1 via autophagy, by a mechanismdistinct from bulk macroautophagy or general ER stress–asso-ciated autophagy. Finally, the Sigma1 inhibitor suppressedIFNg-induced PD-L1. Our data demonstrate that small-mole-cule Sigma1 modulators can be used to regulate PD-L1 incancer cells and trigger its degradation by selective autophagy.
Implications: Sigma1 modulators sequester and eliminatePD-L1 by autophagy, thus preventing functional PD-L1 expres-sion at the cell surface. This posits Sigma1 modulators asnovel therapeutic agents in PD-L1/PD-1 blockade strategiesthat regulate the tumor immune microenvironment.
Visual Overview: http://mcr.aacrjournals.org/content/molcanres/16/2/243/F1.large.jpg. Mol Cancer Res; 16(2); 243–55. �2017 AACR.
IntroductionProgrammed death-ligand 1 (PD-L1, also known as B7-H1 or
CD274) is a type I integral membrane glycoprotein that iscotranslationally inserted into, posttranslationally modified in,and transported through the secretory pathway of a range of celltypes, including tumor cells (1–4). In this pathway, the endo-plasmic reticulum (ER) is the primary site of synthesis, folding,and assembly of secreted and integralmembraneproteins. Proteinhomeostasis in theER relies on the timely convergence ofmultiplemechanisms that detect protein–concentration thresholds, pro-vide quality control, and regulate the ebb and flow of ER proteins(4–6). Integralmembrane proteins are intrinsically dependent onER protein homeostasis machinery to reach the cell surface.
PD-L1 expressed at the surface of tumor cells can act as aT-cell inhibitory checkpoint molecule and can inactivate
tumor-infiltrating immune cells that express cell surface pro-grammed death-1 (PD-1; also known as CD279; refs. 1, 3, 7).Immune checkpoint inhibitors that block PD-L1/PD-1 inter-actions are promising therapeutic agents in strategies thatexploit antitumor immune responses. However, a relativelysmall percentage of cancer patients respond to anti–PD-L1/PD-1 monotherapy, which thus far includes subsets of patientswith melanoma, non–small cell lung cancer, metastatic blad-der cancer, and renal cell carcinoma (7–10). Although highPD-L1 expression has been associated with poor prognosis in arange of cancers (11–13), high PD-L1 expression levels do notnecessarily correspond with response to anti–PD-L1/PD-1therapeutic agents (7, 14). For instance, anti–PD-L1/PD-1agents to treat prostate and triple-negative breast tumors withhigh levels of PD-L1 have shown minimal efficacy in earlyclinical trials (7–10, 12–15). Therefore, the search for predic-tive biomarkers of response to PD-L1/PD-1 agents is an activearea of investigation (14, 16), as is the search for novel agentsand combination therapy approaches that modulate the tumormicroenvironment to promote antitumor immunity (7, 14–17).
Full-length PD-L1 comprises 229 amino acids leading with anN-terminal signal sequence, IgV- and IgC- extracellular domains,which engage PD-1 on infiltrating immune cells, a single trans-membrane domain, and a relatively short 31 residue cytoplasmictail, which has no defined functional motifs and whose role inPD-L1 biology has not been determined (1–3, 18). The cellularfactors and processes engaged in the synthesis, maturation, and
1Department of Pharmacology & Physiology, Drexel University College ofMedicine, Philadelphia, Pennsylvania. 2Sidney Kimmel Cancer Center,Philadelphia, Pennsylvania.
Note: Supplementary data for this article are available at Molecular CancerResearch Online (http://mcr.aacrjournals.org/).
Corresponding Author: Felix J. Kim, Department of Pharmacology & Physiol-ogy, Drexel University College of Medicine, 245 N. 15th Street, Philadelphia, PA19102. Phone: 215-762-2508; Fax: 215-762-2299; E-mail: [email protected]
doi: 10.1158/1541-7786.MCR-17-0166
�2017 American Association for Cancer Research.
MolecularCancerResearch
www.aacrjournals.org 243
on August 9, 2019. © 2018 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst November 8, 2017; DOI: 10.1158/1541-7786.MCR-17-0166

transport of PD-L1 are also not well defined, and few PD-L1–regulatory proteins have been identified (1–3, 18).
Sigma1 is a unique ligand-operated integral membrane chap-erone or scaffolding protein that is highly expressed in the ER of arange of cancer cell lines (19). Initially thought to be an opioidreceptor (19), Sigma1 lacks homology with any known mamma-lian protein and is a distinct protein, unrelated to any traditionalreceptor (20, 21). A growing body of evidence demonstrates thatinhibition of Sigma1 can suppress growth, proliferation, andinduce apoptosis in multiple cancer cell lines (19) and demon-strates the regulation of cell-intrinsic properties of cancer cells bySigma1; however, antitumor immunity activity induced by Sig-ma1 inhibition has not been determined (19). Emerging datasuggest Sigma1 is amultifunctional chaperone (22) or scaffoldingprotein (19, 23) involved in maintaining ER protein homeostasisand supporting the increased demand for secretory pathwayprotein synthesis associated with tumor growth (19, 24). In thisregard, we have shown that selective small-molecule Sigma1modulators can be used to regulate protein translation andactivate the unfolded protein response (UPR) and autophagy ina pharmacologically controllable manner (24, 25).
Autophagy describes a set of cellular sequestration and degra-dation mechanisms by which cells can maintain energy levelsunder conditions of metabolic stress as well as a mechanism bywhich large aggregates of misfolded proteins and cellular compo-nents are sequestered into membrane-bound vesicles called auto-phagosomes and subsequently targeted for lysosomal degradation(26, 27). Multiple types of autophagy have been identified cor-responding to distinguishing mechanistic features, autophagiccargo proteins, and organelles involved. These include bulk macro-autophagy, chaperone-mediated autophagy, secretory autophagy,ubiquitin-selective autophagy, lipophagy, mitophagy, and mostrecently ER-phagy (26–32). Emerging reports demonstrate thatcomponents of the secretory pathway membrane remodeling andprotein trafficking machinery contribute to autophagy (28–32).
We previously reported a role for Sigma1 in autophagy incancer cells and the ability to pharmacologically control autop-hagy with small-molecule Sigma1modulators (24). In this study,we asked whether the ER protein homeostasis regulating prop-erties of small-molecule Sigma1modulators could be exploited toregulate the transport and stability of PD-L1 in cancer cells andthus suppress the surface expression of PD-L1 and potentiallyabrogate PD-L1/PD-1–mediated inactivation of immune cells.
Materials and MethodsChemicals and reagents
IPAG (1-(4-Iodophenyl)-3-(2-adamantyl) guanidine)was pur-chased from Tocris. SA4503 was purchased from Axon MedChem. Bafilomycin A1, wortmannin, rapamycin, and thapsigarginwere purchased from Sigma-Aldrich. Cycloheximide was pur-chased from Alfa Aesar, and recombinant human Interferon (IFN)was purchased from Gibco. Endoglycosidase H (Endo H) andpeptide:N-glycosidase F (PNGase F) were purchased from NewEngland Biolabs. Trypan Blue was purchased from Invitrogen.
Cell lines and transfectionsMDA-MB-231 and PC3 cells were purchased from ATCC and
were authenticated by short tandem repeat profiling. ATG5þ/þ
wild-type (WT) and ATG5�/� knockout (KO) mouse embryofibroblasts (MEF) described elsewhere were obtained from
Dr. Noboru Mizushima via RIKEN (33). All cancer cell lines weremaintained in RPMI1640 supplemented with 10% FBS (Corn-ing), and ATG5þ/þ and ATG5�/� MEFs were grown in DMEMsupplemented with 10% FBS (Corning). Cells were seededapproximately 24 hours prior to the start of drug treatment inmost assays. Human Sigma1 siRNA (sc-42250) and control-AsiRNA (sc-37007) were purchased from Santa Cruz Biotechnology.siRNA transfections (100 nmol/35-mm well) were performedwith INTERFERin (PolyPlus) transfection reagent accordingto the manufacturer's procedures. Forty-eight hours later, cellswere reseeded, allowed to attach and recover for 24 hours, andtransfected again. Cells were harvested 48 hours after thesecond transfection. Sigma1 shRNAs (TRCN0000296908 andTRCN0000291305), ATG5 shRNAs (TRCN0000330392 andTRCN0000151474), andATG7 shRNA (TRCN0000007588)werepurchased from Sigma-Aldrich. Infections were done with 48-hour and72-hour lentivirus and�8mg/mLpolybrene (SantaCruzBiotechnology). Cells were treated 5 days postinfection andharvested after indicated time point. Transient transfections ofthe pcDNA3.1, C-terminal–tagged Sigma1-HA (34), and C-ter-minal–tagged PD-L1-FLAG (Sino Biological Inc.) plasmid con-structs were performed using jetPRIME transfection reagent (Poly-Plus) per the manufacturer's protocol. In these experiments, 2 mgof DNAwere transfected per 10-cm plate, and cells were harvested48 hours posttransfection.
Immunoblots and antibodiesCell lysis, protein extraction, SDS-PAGE, and immunoblotting
were performed as described previously (24), with a fewmodifica-tions. The Luminata Western HRP Substrate ChemiluminescenceKit (Millipore) was used to reveal immunoblotted proteins. Theanti-Sigma1 antibody was generated in our laboratory as describedelsewhere (24). The anti–b-actin (C4), anti-GFP (B-2), mouse anti-FLAG (OctA-probe H-5), anti–MHC-I (W6/32), and rabbit andmouse anti-HA (Y-11 and F-7) antibodies were purchased fromSanta Cruz Biotechnology. The rabbit anti–PD-L1 (E1L3N XP),mouse anti–PD-L1 (405.9A11), anti–PD-L1 extracellular domainspecific (D8T4X), anti-LC3B (D11XP), anti-ATG5 (D5F5U), rabbitanti-FLAG epitope (DYKDDDDK tag), anti-GAPDH (D16H11XP), and horseradish peroxidase–conjugated secondary antibodieswere purchased fromCell Signaling Technology. The anti-Vinculinantibody (E00153) was purchased from Sigma-Aldrich.
Confocal microscopyCells were seeded onto #1.5 (0.17 mm) borosilicate glass
coverslips (Electron Microscopy Sciences) coated with 0.1 mg/mL75,000 to 150,000 MW poly-D-lysine substrate (Sigma-Aldrich)36 hours prior to fixation. The cells were washed with roomtemperature Dulbecco's modified PBS solution (DPBS) and fixedin 4% formaldehyde (Pierce) for 15 minutes. After DPBS washes,cells were permeabilized using ice-cold methanol at �20�C for15minutes andwerewashed.Next, cells were subjected to antigenretrieval using a 1% (v/v) citrate-based unmasking solution(Vector) for 10minutes at 95�C. To reduce nonspecific secondarystaining, cells were incubated with Image-iT FX solution (Molec-ular Probes) for 30minutes at room temperature andwashedwithDPBS. Cells were blocked in 5% (v/v) normal goat serum (Invi-trogen), 0.3% (v/v) Triton X-100 in DPBS for 1 hour. Primariesdiluted in 1% (w/v) BSA, 0.3% (v/v) Triton X-100 in DPBS at1:200 were added to the coverslips and incubated overnight at4�C. After the primary stain, cells were washed with DPBS and
Maher et al.
Mol Cancer Res; 16(2) February 2018 Molecular Cancer Research244
on August 9, 2019. © 2018 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst November 8, 2017; DOI: 10.1158/1541-7786.MCR-17-0166

stained with the corresponding secondary Alexa Fluor 488 goatanti-rabbit IgG (HþL) or Alexa Fluor 594 goat anti-mouse(Molecular Probes) in 1% (w/v) BSA, 0.3% (v/v) Triton X-100in DPBS for 2 hours at room temperature at 1:1,000. Cells werewashed with DPBS, nuclear counterstained with 0.1 mg/mL DAPI(Pierce) for 90 seconds, and thenmounted onto glass slides usingProlong Gold (Molecular Probes). Coverslips were sealed usingnail polish 36 hours after mounting. Images were acquired usingthe Olympus FV1000 inverted confocal microscope using a 60 �1.42 NA oil immersion objective at a scanning resolution of0.0294 mm/pixel. In the studies involving PD-L1 colocalizationwith GFP-LC3, five randomly selected microscopic fields weretaken for each condition. For the PD-L1 cell surface localizationstudies, 11 and 12 randomly selected microscopic fields forvehicle and SA4503, respectively, were taken for each condition.Confocal images weremedian filtered (median intensities of 32�3-pixel squares centered at the target pixel were subtracted fromthe target pixel, respectively) as described previously (35). ImageJwas used to generate 3D surface plots.
PD-L1/PD-1 blockade assayThe Promega PD-L1/PD-1 Blockade Assay Kit (J1250) was
utilized to determine functional changes in PD-1/PD-L1 interac-tions with Sigma1 modulation. PC3 or MDA-MB-231 cells (1 �104) were plated per well on Wallac B&W 96-well Isoplates withwhite wells and bottoms and black matrix. Approximately 24hours later, drug treatment of IPAG (0–20 mmol/L) was added for16 hours. The following day, the drug-containing media wereremoved, and1�104 Jurkat cellswere added to eachof the treatedwells. Six hours later, Promega Bio-Glo reagent was added to eachwell. After a 5-minute incubation, the plate was read with aGloMax 96 Microplate Luminometer (Promega).
Upon coculture of Jurkat and PC3 or MDA-MB-231 cells, theJurkat cells are activated through the T-cell receptor complex. Thisactivation results in the expression of genes that are downstreamof the nuclear factor of activated T-cells (NFAT) response element.Here, the luciferase gene is downstream of the NFAT responseelement, resulting in only activated T cells showing luminescentsignal. This activation of the NFAT response element can beinhibited via interaction between PD-1 and PD-L1. When PD-1on Jurkat cells binds PD-L1 on cancer cells, the NFAT responseelement will become inactive, and the luminescence will abate. Ifan agent prevents the interaction of PD-1 and PD-L1, Jurkat cellswill remain active, and the luminescent signal will increase inintensity when compared with the control.
Isopycnic density-gradient centrifugationMDA-MB-231 cells were harvested 24 hours after seeding by
scraping with a rubber policeman and washed with DPBS. Thecells were pelleted by centrifugation at 200� gmax for 7minutes at4�C in aGH-3.8 rotor (BeckmanCoulter),washedwithDPBS, andpelleted once more. The cells were resuspended in ice-cold iso-tonic homogenization buffer [HB; 250 mmol/L D-sucrose, 150mmol/L sodium chloride, 10 mmol/L HEPES pH 7.4, 1 mmol/LEDTA, supplemented with Halt Protease and Phosphatase Inhi-bitors (Pierce)] with the volume normalized against the pelletsize. Cells weremechanically lysed using a syringe-driven stainlesssteel–encased Balch ball-bearing homogenizer (Isobiotec). Thecells were passed though the tungsten carbide ball bore 14 timeswith a high tolerance clearance of 12 mm. The postnuclear super-natant (PNS) was generated by separation of the homogenate
using centrifugation at 1,000 � gmax for 15 minutes at 4�C in anF45-30-11 rotor (Eppendorf). The iodinated density gradientmedia OptiPrep (Axis-Shield) was used to create a discontinuousdensity gradient. Stock (60% w/v) Optiprep was diluted in HB toobtain a 0.9 mL bottom layer of 50% (1.272 g/mL) Optiprep, a0.8 mL middle layer of 30% (1.175 g/mL) Optiprep, and a 2 mLtop layer of 10%w/v (1.079 g/mL)Optiprep loaded intoOptiSealpolypropylene tubes (Beckman Coulter). A 1 mL aliquot of PNSwas loaded on top of the OptiPrep and separated by ultracentri-fugation at 48,000� gmax for 19 hours at 4�C in a TLA 100.4 fixedangle rotor (Beckman Coulter). Fourteen 300 mL fractions werecollected by tube puncture and gravity flow.
Coimmunoprecipitation assayMDA-MB-231 cells were transiently transfectedwith pcDNA3.1
(empty vector plasmid), Sigma1-HA, and/or PD-L1-FLAG plas-mid constructs using jetPRIME Transfection Reagent (PolyPlus).Forty-eight hours posttransfection, cells were harvested and lysedin HB as described above (see isopycnic density-gradient centri-fugation). Subsequently, lysates were solubilized in an equalvolume of 2�NP-40 wash buffer (300mmol/L sodium chloride,25 mmol/L Tris pH 7.6, 10% glycerol, 2% NP-40 detergent)supplemented with both Halt Protease and Phosphatase Inhibi-tors (Pierce). Detergent solubilized lysates were precleared onProtein G Magnetic Beads (Bio-Rad) coupled with mouse serum(Sigma-Aldrich). The precleared lysates were then layered onProtein G Magnetic Beads (Bio-Rad) coupled with mouse mono-clonal anti-FLAG (H-5) antibody or mouse monoclonal anti-HA(F-7) antibody and incubated for 1 hour at 4�C with constantrotation. TheProteinGMagnetic Beadswere thenwashedwith 1�NP-40 wash buffer (300 mmol/L sodium chloride, 25 mmol/LTris pH 7.6, 10% glycerol, 1% NP-40 detergent). The immuno-precipitated protein complexes were eluted by heating at 95�C inLaemmli sample loading buffer supplementedwith dithiothreitol(DTT) and b-mercaptoethanol.
Plasma and intracellular membrane fractionationFollowing 16-hour drug treatment, cells were harvested and
lysed in HB as described above (see isopycnic density-gradientcentrifugation). The PNSwas separated into a cytosol supernatantandmembrane pellet by ultracentrifugation at 100,000� gmax for1 hour at 4�C in a TLS-55 swinging bucket rotor (BeckmanCoulter). To isolate plasmamembrane from the total membrane,the membrane pellet was suspended in Upper Phase solutionprovided by theMembrane Protein Extraction Kit (BioVision Inc).The sample was mixed with an equal volume of Lower Phasesolution from the same kit, vortexed and incubated on wet ice for10minutes. Centrifugation at 1,000� gmax for 5minutes at 4�C inan F45-30-11 rotor (Eppendorf) produced two phases separatedby a visible interface. The upper phase was collected and saved onice, while the lower phase was again mixed and incubated withupper phase solution before a repeat centrifugation. The collectedupper phases were combined and mixed with fresh Lower Phasesolution, incubated, and centrifuged. The upper phase was col-lected and contained the plasma membrane. The lower phaseswere combined and contained the intramembrane (ER/Golgi).Both phases were diluted in 5 volumes of ultrapure water andincubated at 4�C overnight, followed by centrifugation at 20,000� gmax for 15 minutes at 4�C in an F45-30-11 rotor (Eppendorf).After aspiration, the resultant dry pellet was lysed in RIPA bufferand run on SDS-PAGE.
Autophagic Degradation of PD-L1 by a Sigma1 Modulator
www.aacrjournals.org Mol Cancer Res; 16(2) February 2018 245
on August 9, 2019. © 2018 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst November 8, 2017; DOI: 10.1158/1541-7786.MCR-17-0166

Flow cytometryMDA-MB-231 cells were seeded, and 24 hours later, IPAG
treatment was applied for 16 hours. At the time of harvest, cellswere washed with PBS and then scraped into PBS. The cellswere fixed in 4% formaldehyde for 10 minutes at 37�C andplaced on ice for one minute. Cells were washed twice inincubation buffer (0.5% BSA in PBS) and aliquoted such thateach tube contained 7.5� 104 cells. Cells were stained with thePD-L1 extracellular domain–specific antibody at 1:100 for onehour at room temperature. Cells were washed in incubationbuffer then stained with anti-rabbit Alexa Fluor 488 secondaryantibody (Cell Signaling Technology cat. no. 4412) at 1:500for 30 minutes at room temperature. Cells were washed inincubation buffer and resuspended in 500 mL incubationbuffer.
Cell fluorescence was analyzed using a Millipore Guava Easy-Cyte instrument, and data were analyzed using FlowJo V10. Cellswere gated such that only single cells were analyzed. Meanfluorescence intensity (MFI) was measured for each population.Average and SEM values were calculated for the MFI of biologicaltriplicates. Unpaired, two-tailed t test was performed to comparethe treatment groups.
Evaluation of IFNg induced PD-L1 transcripts by qRT-PCRMDA-MB-231 and PC3 cells were treated 24hours after seeding
with 100 ng/mL IFNg for 24 hours in the absence or presence ofthe Sigma1 inhibitor, IPAG (10 mmol/L) for the final 16 hours.Total cellular RNA was extracted from the treated cells using theRNeasy Kit (Qiagen) per the manufacturer's protocol. IFNg-induced transcription of PD-L1 was confirmed by qRT-PCR. Taq-Man primer probe sets were purchased from Life Technologies,and the genes and catalog numbers used for the qRT-PCR experi-ments are the following: PD-L1 (Hs00204257_m1) and GAPDH(Hs99999905-m1). qRT-PCR was performed using the 7900HT-Fast Real Time PCR System (Applied Biosystems), and the 9reactions were performed in triplicate using the Brilliant II qRT-PCR Master Mix 1-Step Kit (Agilent Technologies) following themanufacturer's instructions. Data were normalized to GAPDHtranscript levels and presented as fold increase or percent changeof PD-L1 transcripts relative to DMSO-treated controls.
Trypan blue exclusion assayCells were harvested and gently mixed at a 1:1 ratio of cell
suspension:trypan blue. A hemocytometer was used to count thepercentage of cells that excluded the trypan blue, which representsthe percentage of live cells in the suspension. Values were gener-ated from at least three independent determinations.
Statistical analysisTests of statistical significance of single comparisons were
performed using Student t test. Statistical significance of multiplecomparisons was determined by one-way ANOVA followed byBonferroni posttest using Prism software (GraphPad).
ResultsSigma1 physically associates with PD-L1
PD-L1 is a type I integral membrane glycoprotein that issynthesized in, posttranslationally modified in, and transportedthrough the cellular secretory pathway to reach the plasma mem-brane of tumor cells. The MDA-MB-231 triple-negative breast
cancer cell line has been reported to express high levels of PD-L1under standard cell culture conditions (11). By confocal micros-copy, we confirmed that PD-L1 is enriched in the cytoplasm ofMDA-MB-231 cells as well as present on the surface of these cells(Fig. 1A). Sigma1 is a unique integral membrane chaperone orscaffolding protein in the secretory pathway and is enriched in theER of cancer cells. We performed isopycnic centrifugation todemonstrate that Sigma1 cofractionates with PD-L1 in MDA-MB-231 cells. We show that 50% of PD-L1 and 78% of Sigma1coisolated in density gradient fractions 5, 6, and 7 (Supplemen-tary Fig. S1). This suggests that Sigma1 and PD-L1 occupy mem-brane domains with similar density and thus similar cellularcompartments. Therefore, we asked whether Sigma1 could phys-ically interact with PD-L1.
Using carboxy-terminal (C-terminal) FLAG-tagged PD-L1 (PD-L1-FLAG) and C-terminal HA-tagged Sigma1 (Sigma1-HA), wefound that Sigma1 coimmunoprecipitates with PD-L1 (Fig. 1B).The PD-L1-FLAG protein was detected as multiple bands, a pre-dicted approximately 33-kDa unmodified species as well as anumber of higher molecular weight (MW), slower migratingbands. PD-L1 is glycosylated as it is processed and maturesthrough the secretory pathway, which is reflected in the hetero-geneous, smeared migration pattern of PD-L1 bands on theimmunoblot. We performed a peptide:N-glycosidase F (PNGaseF) digestion of immunoprecipitated PD-L1-FLAG eluate andfound that the PD-L1 smear was reduced to a single band ofapproximately 33 kDa, confirming that the higher MW, slowermigrating bands are glycosyl–PD-L1 (Fig. 1C).
Interestingly, in the reciprocal co-IP, only the approximately33-kDa nonglycosylated species and an approximately 45 kDaPD-L1-FLAG species coimmunoprecipitated with Sigma1-HA(Fig. 1D). PNGase F digestion of the immunoprecipitated sam-ples reduced the higher MW PD-L1 band to approximately 33kDa, confirming that the approximately 45 kDa band is a glyco-sylated form of the protein (Fig. 1E). Digestion of co-IP eluateswith EndoH also reduced the approximately 45 kDa species to 33kDa (Fig. 1F). Whereas PNGase F cleaves nearly all types ofN-linked (Asn-linked) glycosylation, including complex glycans,Endo H cleaves only high mannose and some hybrid types ofN-linked carbohydrates but not complex glycans. Thus, oligosac-charide structures beyond enzymatic modification by Golgialpha-mannosidase II in the secretory pathway are resistant toEndo H cleavage. This differential sensitivity to PNGase F andEndo H is used to track the maturation of glycoproteins. Thepattern observed here suggests that Sigma1 physically associatesprimarily with glycosyl-PD-L1 formed early in the proximalsecretory pathway, ER, and early Golgi compartments (Fig. 1F).
PD-L1 protein levels are suppressed by RNAi-mediatedknockdown and pharmacologic inhibition of Sigma1
We propose that Sigma1 is a ligand-operated scaffolding pro-tein that promotes the stability, processing, assembly, and traf-ficking of specific proteins in the secretory pathway of cancer cells.In support of this hypothesis, we found that siRNA-mediatedknockdown of Sigma1 resulted in a significant decrease in PD-L1protein levels in triple-negative MDA-MB-231 breast cancer andandrogen-independent PC3 prostate cancer cells (Fig. 2A). Wealso confirmed this effectwith twodistinct Sigma1 shRNAs,whichproduced similar trends in both of these cell lines (Fig. 2B). Thesedata demonstrate that Sigma1 is required to sustain PD-L1proteinlevels in these cancer cells. It is important to note that Sigma1
Maher et al.
Mol Cancer Res; 16(2) February 2018 Molecular Cancer Research246
on August 9, 2019. © 2018 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst November 8, 2017; DOI: 10.1158/1541-7786.MCR-17-0166

knockdown affects only a specific subset of proteins; shRNA-mediated knockdown of Sigma1 did not result in a significantdecrease in HSP90 or Akt protein levels (Supplementary Fig. S2).
Subsequently, we asked whether a small-molecule inhibitor ofSigma1 could mimic the effects of Sigma1 knockdown. Here, weused IPAG, a prototypic selective small-molecule inhibitor ofSigma1 that we previously have shown engages the UPR, theubiquitin proteasome system, and autophagy (23, 24). We foundthat IPAG induced a significant dose-responsive decrease of totalPD-L1 protein in PC3 and MDA-MB-231 cells (Fig. 3A). Insupport of the selectivity of proteins affected/targeted by Sigma1modulator treatment, we determined that MHC-I, a related secre-tory pathway associated membrane protein, was not eliminatedunder standard cell culture conditions in MDA-MB-231 cells andwas only slightly decreased in PC3 cells (Fig. 3B). Thus, both RNAiknockdown and pharmacologic inhibition of Sigma1 selectivelysuppressed PD-L1 protein levels in cancer cells.
To determine the operative mechanism by which the Sigma1modulator eliminated PD-L1, we evaluated the potential andrelative contribution of protein synthesis and degradation bytreating with IPAG in the presence of the translation inhibitor,cycloheximide. Whereas translation arrest by cycloheximidealone did not result in a decrease in PD-L1 protein levels over
8 hours, the combination of cycloheximide and IPAG resulted insignificant decreases in PD-L1 protein levels after 4 and 8 hours oftreatment (Supplementary Fig. S3). These data suggest thatalthough IPAG-mediated translation arrest may contribute todecreased PD-L1 levels in response to long-term IPAG treatment,protein degradation plays a significant role in the elimination ofPD-L1.
Pharmacologic inhibition of Sigma1 decreases PD-L1 cellsurface expression and immune checkpoint activity in vitro
Mature PD-L1 expressed at the surface of tumor cells binds itscognate receptor PD-1 on the surface of infiltrating T cells and thuscontributes to suppression of antitumor immunity. Blockade ofPD-L1 binding to PD-1, that is, immune checkpoint inhibition,prevents inactivation of T cells and promotes antitumor immu-nity. Here, we utilized an NFAT transcriptional response elementluciferase reporter assay (NFAT-luciferase) to evaluate the effectsof Sigma1 inhibition on PD-L1/PD-1 interaction blockade (Fig.3C). This assay involved coculture of PD-L1–expressing MDA-MB-231 or PC3 cancer cells with PD-1–expressing NFAT-lucifer-ase transfected Jurkat T-cells. PD-L1/PD-1 binding results in T-cellinactivation and the abrogation of luminescent signal, whereasblockade of PD-L1/PD-1 binding prevents inactivation of genes
Figure 1.
PD-L1 association with Sigma1. A, Confocal micrograph showing cytoplasmic and cell surface staining of PD-L1. MDA-MB-231 cells in standard culture medium,cells fixed and immunostained 72 hours after seeding onto poly-D-lysine–coated coverslip. Scale bar, 10 mm. B, Coimmunoprecipitation of Sigma1-HA withPD-L1-FLAG from transiently transfected MDA-MB-231 cells. Glycosylated (�) and nonglycosylated (o) PD-L1-FLAG indicated. Immunoglobulin (IgG) andGAPDH loading controls for immunoprecipitated and input fractions, respectively. C, Anti-FLAG immunoprecipitated samples treated with PNGase F.Nonglycosylated (o) PD-L1-FLAG. D, Coimmunoprecipitation of PD-L1-FLAG with Sigma1-HA from transiently transfected MDA-MB-231 cells. Glycosylated (�)and nonglycosylated (o) PD-L1-FLAG. E and F, Anti-HA immunoprecipitated samples treated with PNGase F (E) and endoglycosidase H (Endo H; F).Nonglycosylated (o) PD-L1-FLAG.
Autophagic Degradation of PD-L1 by a Sigma1 Modulator
www.aacrjournals.org Mol Cancer Res; 16(2) February 2018 247
on August 9, 2019. © 2018 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst November 8, 2017; DOI: 10.1158/1541-7786.MCR-17-0166

regulated by the NFAT response element and results in lumines-cent signal (Fig. 3C).
MDA-MB-231 and PC3 cells were treated with multiple con-centrations of IPAG (3, 10, and 20 mmol/L) for 16 hours, removeddrug-containing media, and then cocultured with Jurkat NFAT-luciferase reporter T cells in the absence of drug. The transcrip-tionally induced bioluminescent signal increased in a concentra-tion related manner in both MDA-MB-231 and PC3 cell cultures,indicating that IPAG disrupted PD-L10s checkpoint activity (Fig.3D). Importantly, we confirmed that the concentrations of IPAGand 16-hour treatment time used in these experiments did notinduce cell death (Fig. 3E). On the basis of these data, weperformed all subsequent mechanism-focused experiments well
within the 16-hour time point to minimize potential confoundsassociated with cell death.
We next assessed how Sigma1 inhibition affects the expressionof PD-L1 at the cell surface. Using flow cytometry, we demon-strated that treatment with IPAG (20 mmol/L) for 16 hourssignificantly decreased cell surface expression of PD-L1 onMDA-MB-231 cells (Fig. 3F and G). These data suggest thatSigma1 inhibition effectively disrupts PD-L1 function at least inpart by decreasing its functional expression on the plasmamembrane.
We also performed a biochemical membrane fractionationassay, which separates and isolates the cell surface plasma mem-brane from intracellularmembranes, such as the ER and theGolgi,based on the affinity of membranes to distinct polymer phases(36). We used this technique to determine the extent to whichmature PD-L1 at the plasmamembrane and intracellular PD-L1 issuppressed by IPAG. IPAG treatment for 16 hours at a concen-tration of 10 mmol/L resulted in a salient decrease in the intra-cellular membrane PD-L1 and a more modest decrease in plasmamembrane PD-L1 (Supplementary Fig. S4). These data suggestthat IPAG treatment decreases nascent PD-L1 protein stability,thus preventing the trafficking of functional PD-L1 complexes tothe cell surface.
A selective positive modulator of Sigma1 blocks the IPAG-mediated decrease of PD-L1 and promotes its cell surfaceexpression
SA4503 is a Sigma1-selective putative agonist or activator(positive modulator) (37). Cotreatment of SA4503 with IPAGblocked the decrease in PD-L1 protein levels, demonstratingSigma1-specific pharmacologic activity (Fig. 4A). Interestingly,by confocal microscopy, treatment with SA4503 alone resulted inincreased cell surface expression of PD-L1 (Fig. 4B and C),suggesting that PD-L1 stability and trafficking can be differentiallypromoted and inhibited by positive and negative modulators ofSigma1, respectively.
In some cells, SA4503 treatment was associated with intenselocalized intracellular clusters of PD-L1 staining. The nature ofthis clustered staining pattern is unclear.
Treatment with a small-molecule Sigma1 inhibitor inducesselective autophagic degradation of PD-L1
We previously demonstrated that IPAG could induce autop-hagy in cancer cells (24). Therefore, we asked whether autophagycontributes to the decrease in PD-L1 protein levels in this study.An IPAG treatment time course (0, 4, 8, and 16 hours) experimentrevealed increased LC3B corresponding with decreased PD-L1levels (Fig. 5A). Thus, autophagosome formation coincides withdecreasing PD-L1, suggesting a role for autophagy.
We confirmed that IPAG induced autophagy by treating WTand ATG5 KOMEFs with IPAG and demonstrating that LC3B wasinduced in the WT but not in the ATG5 KO MEFs (Fig. 5B).Subsequently, we confirmed that autophagy contributes to Sig-ma1 modulator induced PD-L1 degradation by blocking theprocess in MDA-MB-231 cells by RNAi knockdown of ATG5 (Fig.5C; Supplementary Fig. S5A and S5B), of ATG7 (SupplementaryFig. S5C), and by pharmacologically inhibiting autophagywith wortmannin and bafilomycin A1 (Baf A1; Fig. 5D and E).First, we performed shRNA-mediated knockdown (KD) of ATG5,the product of an essential autophagy gene required for theformation of autophagosomes (26, 27, 31). Knockdown of ATG5
Figure 2.
Sigma1 knockdown results in decreased PD-L1. A and B, Knockdown of Sigma1by siRNA (A) and by two distinct shRNAs (291305, 296908) compared with thecontrol shRNA (PKLO.1; B) in PC3 and MDA-MB-231 cells and correspondingdecrease in PD-L1 protein levels. For all blots, bands were quantified bydensitometry, and data are presented as levels of PD-L1 relative to control siRNAor shRNA (pKLO.1). Bars represent mean and SEM from at least threeindependent determinations. �� , P < 0.01; ��� , P < 0.001.
Maher et al.
Mol Cancer Res; 16(2) February 2018 Molecular Cancer Research248
on August 9, 2019. © 2018 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst November 8, 2017; DOI: 10.1158/1541-7786.MCR-17-0166

inMDA-MB-231 cells blocked IPAG-mediated degradationof PD-L1 (Fig. 5C; Supplementary Fig. S5A and S5B). This experimentshowed that autophagosome formation is required for IPAG-induced degradation of PD-L1. We replicated the results of thisstudy with two distinct ATG5 shRNAs (data shown forTRCN0000330392 in Fig. 5C and in Supplementary Fig. S5A,and similar results were obtainedwith TRCN0000151474, shownin Supplementary Fig. S5B). Furthermore,we confirmed this effectwith another essential autophagy gene, ATG7. ATG7 shRNAknockdown also mitigated PD-L1 degradation by IPAG whencompared with the control (Supplementary Fig. S5C), confirmingthe fundamental role of autophagy in IPAG-mediated degrada-tion of PD-L1.
Furthermore, we showed that pharmacologic inhibition ofearly and late stages of autophagy prevented PD-L1 decreaseduring IPAG treatment. Wortmannin is a Vps34 inhibitor thatblocks autophagosome formation (26, 27, 38). Consistent with
ATG5 knockdown, cotreatment with IPAG and wortmanninabrogated the PD-L1–decreasing effect of IPAG (Fig. 5D).Bafilomycin A1 (Baf A1) inhibits vacuolar Hþ ATPase, whichblocks fusion of autophagosomes with lysosomes, and thusblocks autolysosomal degradation or autophagic flux (26, 27).We previously showed that IPAG induces autophagic flux in anumber of cancer cell lines (24) and confirmed that IPAGinduces autophagic flux here as well (Supplementary Fig. S6).Baf A1 blocked the decrease in PD-L1 in IPAG-treated MDA-MB-231 cells (Fig. 5E).
Next, we tested whether PD-L1 protein levels would be sup-pressed by other inducers of autophagy. Interestingly, inductionof bulk macroautophagy by cell starvation in Hank's bufferedsaline solution (HBSS) or treatment with rapamycin (26, 27) didnot alter PD-L1 protein levels (Fig. 5F andG). As IPAG induces ERstress–associated autophagy (24), we asked whether this was ageneral mechanism by which PD-L1 could be degraded. In
Figure 3.
Suppression of PD-L1 protein levels in cancer cells and corresponding activation of cocultured T cells in response to small-molecule inhibition of Sigma1. A,Immunoblot of PD-L1 from whole-cell protein extracts from MDA-MB-231 and PC3 cells treated for 16 hours with DMSO (0), 3, 10, and 20 mmol/L IPAG (Sigma1inhibitor). Immunoblots were quantified by densitometry for each cell line. Data represent mean values from at least three independent determinations, anderror bars represent SEM. �, P < 0.05; �� , P < 0.01; ��� , P < 0.001. B, Immunoblot of MHC-I protein levels with DMSO (0), 3, 10, and 20 mmol/L IPAG treatment for16 hours in PC3 and MDA-MB-231 cells, highlighting Sigma1 modulator specificity. C, Schematic illustration of PD-L1/PD-1 blockade assay. D, PD-L1/PD-1blockade assay performed with PC3 and MDA-MB-231 cells treated with 3, 10, and 20 mmol/L IPAG for 16 hours. Jurkat NFAT-luciferase reporter cells (10,000cells/well) were added, and cells were cocultured for 6 hours. Data are presented as fold induction over nontreated control cocultures. �� , P < 0.01; ��� , P < 0.001.E, Evaluation of cell death by Trypan blue exclusion following treatment with DMSO (0), 3, 10, and 20 mmol/L IPAG for 16 hours. No significant (NS) celldeath was observed under these conditions. F, Cell surface expression of PD-L1 in response to treatment with small-molecule Sigma1 inhibitor. Flow cytometryof PD-L1 on the surface of MDA-MB-231 cells treated with IPAG (20 mmol/L) for 16 hours. G, Quantification of MFI of PD-L1 cell surface expression from threeindependent determinations. �� , P < 0.01.
Autophagic Degradation of PD-L1 by a Sigma1 Modulator
www.aacrjournals.org Mol Cancer Res; 16(2) February 2018 249
on August 9, 2019. © 2018 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst November 8, 2017; DOI: 10.1158/1541-7786.MCR-17-0166

contrast to IPAG, we found that a widely used chemical inducer ofER stress–associated autophagy, thapsigargin (TG), which blocksthe sarco/endoplasmic reticulum Ca2þ ATPase (SERCA; refs. 26,27), did not induce PD-L1 degradation under the experimentalconditions evaluated herein (Fig. 5H). Altogether, these datasuggest that Sigma1 modulators induce a form of selectiveautophagy.
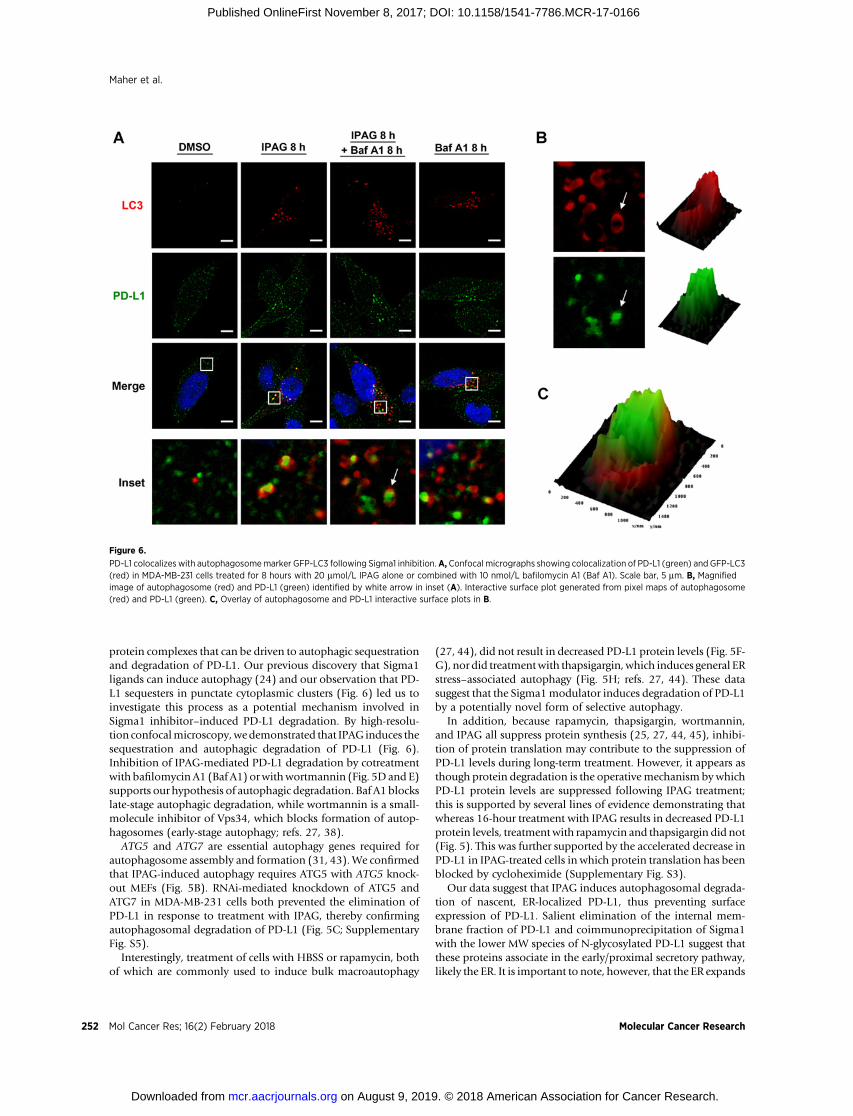
Finally, we performed confocal microscopy to visualizeautophagic sequestration and degradation of PD-L1 duringIPAG treatment (Fig. 6). We treated MDA-MB-231 cells stablyexpressing a GFP-LC3 construct with IPAG alone or in thepresence of Baf A1 (Fig. 6A). The formation of GFP-LC3punctae indicates autophagosome formation. Baf A1 blocksautophagic flux and causes accumulation of autophagosomesand their cargo. This is detected as increased numbers and insome cases size of GFP-LC3 punctae. Consistent with the resultsdescribed above, IPAG treatment produced GFP-LC3 labeledautophagosomes and induced their colocalization with PD-L1(Fig. 6A). In contrast, treatment with Baf A1 alone resulted inincreased GFP-LC3 autophagosomes but did not induce thesame colocalization (Fig. 6A). Cotreatment with IPAG and BafA1 resulted in increased numbers of GFP-LC3–positive punctaethat colocalized with PD-L1, indicating an accumulation ofPD-L1–containing autophagosomes (Fig. 6A–C). Under con-ditions of treatment with IPAG alone and cotreatment of IPAGand Baf A1, PD-L1 appears to be engulfed in autophagosomes(Fig. 6B and C). Altogether, these data suggest that autophagy isthe operative mechanism by which IPAG induces intracellulardegradation of PD-L1.
IFNg induction of PD-L1IFNg is an immunostimulatory cytokine that has long been
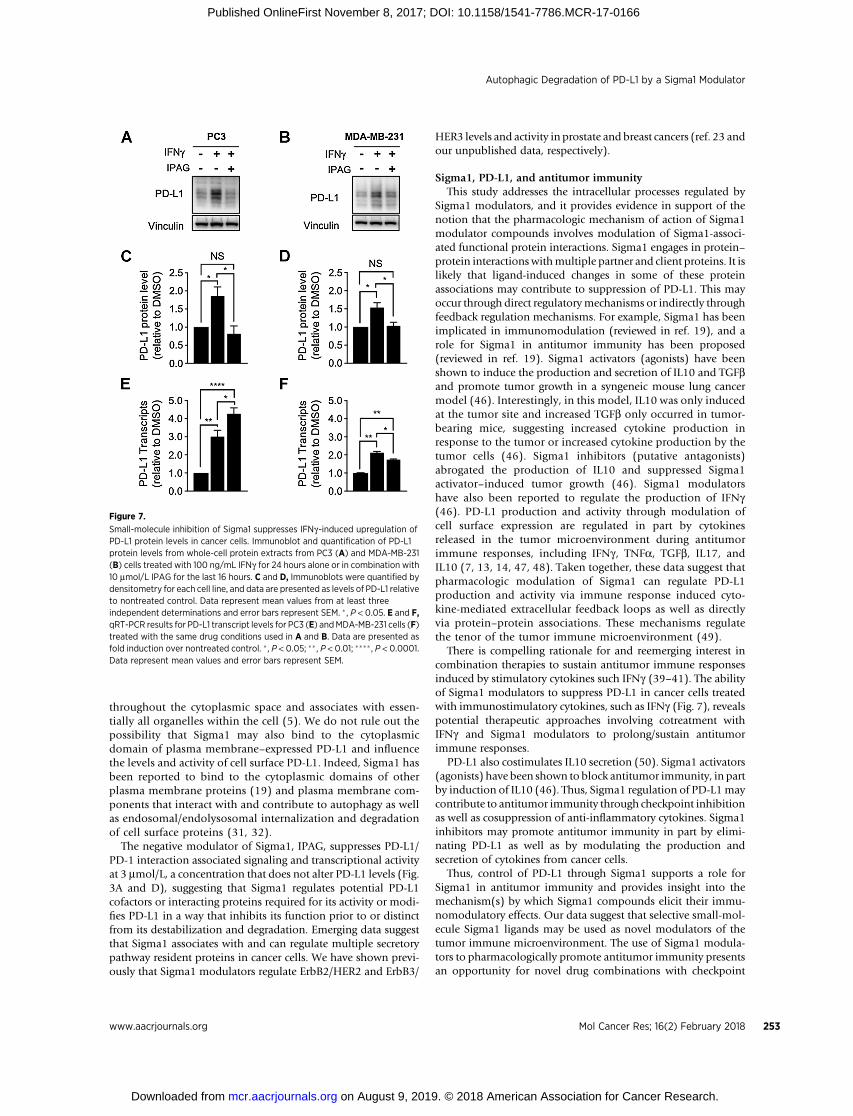
of interest for its potential in promoting antitumor immunity(39, 40). However, exposure to IFNg also induces PD-L1expression in cancer cells, thereby suppressing the antitumorimmune response through adaptive immune resistance (39–41). As IFNg is present in the tumor microenvironment, weasked whether small-molecule Sigma1 inhibitors could sup-press/eliminate PD-L1 induction by IFNg treatment. In ourmodel, we found that IFNg (100 ng/mL for 24 hours) inducedPD-L1 protein levels (Fig. 7A–D) and transcripts (Fig. 7E and F)in MDA-MB-231 and PC3 cells. Cotreatment with IPAG (10mmol/L for the final 16 hours of the 24-hour IFNg treatment)decreased PD-L1 protein levels back to levels similar to or lessthan DMSO-treated conditions (Fig. 7A–D), with a slightdecrease and a slight increase in PD-L1 transcripts in MDA-MB-231 and PC3 cells, respectively (Fig. 7E and F). It isimportant to note that, under IFNg combined with IPAGconditions, PD-L1 transcript levels remain statistically signifi-cantly higher than DMSO control, whereas the protein leveldecreases to similar levels as the control. Therefore, under theexperimental conditions used here, that is, 16-hour treatmentwith IPAG, the decrease in PD-L1 protein can be attributed toposttranslational effects, likely protein degradation.
DiscussionTo our knowledge, this is the first demonstration of a direct
physical and functional interaction between Sigma1 and PD-L1,the first demonstration that Sigma1 modulators can be used tosequester and degrade secretory pathway proteins by a potentiallynovel form of selective autophagy, and the first demonstration ofautophagic degradation of PD-L1. These data suggest that Sigma1modulators can be used to regulate PD-L1 transport and stabilitythrough regulation of pharmacologically responsive proteincomplexes.
Sigma1 modulator concept: positive and negative modulators,inhibitors, and activators
Traditionally, small molecules with affinity for Sigma1 havebeen classified as agonist or antagonist based primarily on datafrom rodent behavior assays (19). Differences between putativeSigma1 antagonists and agonists, however, have remainedunresolved at the molecular and cellular level. Sigma1 doesnot have the properties of a traditional receptor, and therefore,designation of Sigma1 selective compounds as classicallydefined receptor antagonists or agonists may be inaccurate.Emerging evidence suggests that Sigma1 may function either asan allosteric cofactor protein associated with bona fide receptorsystems or as a novel chaperone (19) or scaffolding protein.Therefore, it may be more appropriate to use the term Sigma1modulator, and not agonist or antagonist, for compounds thatpharmacologically alter Sigma1 activity. Here, we provide evi-dence in support of this concept with opposing actions of theputative Sigma1 antagonist IPAG (negative modulator orinhibitor) and the putative agonist SA4503 (positive modula-tor or activator). The Sigma1-selective actions of these com-pounds have been demonstrated elsewhere (24, 37, 42). Inthese studies, we also show that SA4503 blocked IPAG-medi-ated decreases in PD-L1 protein levels, pharmacologicallydemonstrating Sigma1-specific actions (Fig. 4A).
Figure 4.
Opposing effects of small-molecule Sigma1 activator and inhibitor on PD-L1protein levels and cell surface expression. A, Immunoblot of whole-cell lysatesfrom MDA-MB-231 cells treated with IPAG (10 mmol/L), SA4503 (20 mmol/L), orcotreated with IPAG (10 mmol/L) and SA4503 (20 mmol/L) for 16 hours.Immunoblot bands were quantified by densitometry, and data are presentedas levels of PD-L1 relative to nontreated control. B, Confocal micrographsshowing PD-L1 and DAPI in MDA-MB-231 cells treated with SA4503 (10 mmol/L)for 48 hours. C, Quantification of the proportion of PD-L1 expressed onthe cell surface compared with total cellular PD-L1. Data represent mean andSEM from at least three independent determinations. ��� , P < 0.001.
Maher et al.
Mol Cancer Res; 16(2) February 2018 Molecular Cancer Research250
on August 9, 2019. © 2018 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst November 8, 2017; DOI: 10.1158/1541-7786.MCR-17-0166

Insight into the role of Sigma1 in cancerA number of publications report the cancer cell growth and
survival inhibiting effects of compounds with affinity for Sigma1(19); however, the specific mechanisms underlying these actionsand the physiologic role of Sigma1 in cancer cells have remainedunclear. Our data suggest that Sigma1 may play a role in regu-lating the components or compartments of the secretory pathwayof cancer cells. Modulation of PD-L1 stability, transport, andactivity by Sigma1 is consistent with such a physiologic role.Although the precise biochemical and molecular mechanismsgoverning Sigma1 actions in the secretory pathway of cancer cellsremain to be determined, we posit that Sigma1modulators cooptcomponents of the ER protein homeostasis machinery to regulatethe transport of PD-L1 transiting the secretory pathway to the
plasmamembrane. Demonstration that Sigma1 inhibitors can beused to sequester secretory pathway proteins into autophago-somes supports this notion. These results raise questions regard-ing what other secretory pathway-dependent proteins are regu-lated by Sigma1.
Sequestration and degradation of PD-L1 by pharmacologicallyinduced selective autophagy
The biochemical and molecular mechanisms that govern PD-L1 transcription, translation, processing, assembly, transport, andfunctional binding partners (the intracellular proteins and cellu-lar factors with which PD-L1 associates throughout its life cycle)remain poorly defined. Our data show that Sigma1 can physicallyinteract with intracellular PD-L1 in pharmacologically responsive
Figure 5.
Inhibition of Sigma1 results in the degradation of PD-L1 via selective autophagy.A, Immunoblot of whole-cell protein extracts fromMDA-MB-231 cells treatedwith 20mmol/L IPAG for 4, 8, and 16 hours. B, Immunoblot of proteins from WT and ATG5 knockout (KO) MEFs treated with 10 and 20 mmol/L IPAG for 16 hours. C,Immunoblot showing PD-L1 protein levels in IPAG-treated cells following shRNA knockdown of ATG5 (initial stages of autophagy, autophagosome formation).��� , P < 0.001. D and E, IPAG combined with 50 nmol/L wortmannin (D; early stages of autophagy) or IPAG with 10 nmol/L bafilomycin A1 (E; Baf A1; late stages ofautophagy, autolysosomal degradation) for 8 hours. � , P < 0.05; �� , P < 0.01; ��� , P < 0.001. Data represent mean and SEM from at least three independentdeterminations. Bulk macroautophagy induced by culturing cells in HBSS (F) for 8 hours or treatment with rapamycin (G; 100 nmol/L) for 16 hours. H, ER-stressassociated autophagy induced by thapsigargin (0.3 mmol/L) for 16 hours. Immunoblot bands were quantified by densitometry, and data are presented aslevels of PD-L1 relative to nontreated control. Data represent mean and SD from at least two independent determinations.
Autophagic Degradation of PD-L1 by a Sigma1 Modulator
www.aacrjournals.org Mol Cancer Res; 16(2) February 2018 251
on August 9, 2019. © 2018 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst November 8, 2017; DOI: 10.1158/1541-7786.MCR-17-0166
protein complexes that can be driven to autophagic sequestrationand degradation of PD-L1. Our previous discovery that Sigma1ligands can induce autophagy (24) and our observation that PD-L1 sequesters in punctate cytoplasmic clusters (Fig. 6) led us toinvestigate this process as a potential mechanism involved inSigma1 inhibitor–induced PD-L1 degradation. By high-resolu-tion confocalmicroscopy,wedemonstrated that IPAG induces thesequestration and autophagic degradation of PD-L1 (Fig. 6).Inhibition of IPAG-mediated PD-L1 degradation by cotreatmentwith bafilomycin A1 (Baf A1) orwithwortmannin (Fig. 5D and E)supports our hypothesis of autophagic degradation. Baf A1 blockslate-stage autophagic degradation, while wortmannin is a small-molecule inhibitor of Vps34, which blocks formation of autop-hagosomes (early-stage autophagy; refs. 27, 38).
ATG5 and ATG7 are essential autophagy genes required forautophagosome assembly and formation (31, 43). We confirmedthat IPAG-induced autophagy requires ATG5 with ATG5 knock-out MEFs (Fig. 5B). RNAi-mediated knockdown of ATG5 andATG7 in MDA-MB-231 cells both prevented the elimination ofPD-L1 in response to treatment with IPAG, thereby confirmingautophagosomal degradation of PD-L1 (Fig. 5C; SupplementaryFig. S5).
Interestingly, treatment of cells with HBSS or rapamycin, bothof which are commonly used to induce bulk macroautophagy
(27, 44), did not result in decreased PD-L1 protein levels (Fig. 5F-G), nor did treatmentwith thapsigargin,which induces general ERstress–associated autophagy (Fig. 5H; refs. 27, 44). These datasuggest that the Sigma1modulator induces degradation of PD-L1by a potentially novel form of selective autophagy.
In addition, because rapamycin, thapsigargin, wortmannin,and IPAG all suppress protein synthesis (25, 27, 44, 45), inhibi-tion of protein translation may contribute to the suppression ofPD-L1 levels during long-term treatment. However, it appears asthough protein degradation is the operativemechanism bywhichPD-L1 protein levels are suppressed following IPAG treatment;this is supported by several lines of evidence demonstrating thatwhereas 16-hour treatment with IPAG results in decreased PD-L1protein levels, treatmentwith rapamycin and thapsigargin did not(Fig. 5). This was further supported by the accelerated decrease inPD-L1 in IPAG-treated cells in which protein translation has beenblocked by cycloheximide (Supplementary Fig. S3).
Our data suggest that IPAG induces autophagosomal degrada-tion of nascent, ER-localized PD-L1, thus preventing surfaceexpression of PD-L1. Salient elimination of the internal mem-brane fraction of PD-L1 and coimmunoprecipitation of Sigma1with the lower MW species of N-glycosylated PD-L1 suggest thatthese proteins associate in the early/proximal secretory pathway,likely the ER. It is important to note, however, that the ER expands
Figure 6.
PD-L1 colocalizes with autophagosomemarker GFP-LC3 following Sigma1 inhibition.A, Confocal micrographs showing colocalization of PD-L1 (green) and GFP-LC3(red) in MDA-MB-231 cells treated for 8 hours with 20 mmol/L IPAG alone or combined with 10 nmol/L bafilomycin A1 (Baf A1). Scale bar, 5 mm. B, Magnifiedimage of autophagosome (red) and PD-L1 (green) identified by white arrow in inset (A). Interactive surface plot generated from pixel maps of autophagosome(red) and PD-L1 (green). C, Overlay of autophagosome and PD-L1 interactive surface plots in B.
Maher et al.
Mol Cancer Res; 16(2) February 2018 Molecular Cancer Research252
on August 9, 2019. © 2018 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst November 8, 2017; DOI: 10.1158/1541-7786.MCR-17-0166
throughout the cytoplasmic space and associates with essen-tially all organelles within the cell (5). We do not rule out thepossibility that Sigma1 may also bind to the cytoplasmicdomain of plasma membrane–expressed PD-L1 and influencethe levels and activity of cell surface PD-L1. Indeed, Sigma1 hasbeen reported to bind to the cytoplasmic domains of otherplasma membrane proteins (19) and plasma membrane com-ponents that interact with and contribute to autophagy as wellas endosomal/endolysosomal internalization and degradationof cell surface proteins (31, 32).
The negative modulator of Sigma1, IPAG, suppresses PD-L1/PD-1 interaction associated signaling and transcriptional activityat 3 mmol/L, a concentration that does not alter PD-L1 levels (Fig.3A and D), suggesting that Sigma1 regulates potential PD-L1cofactors or interacting proteins required for its activity or modi-fies PD-L1 in a way that inhibits its function prior to or distinctfrom its destabilization and degradation. Emerging data suggestthat Sigma1 associates with and can regulate multiple secretorypathway resident proteins in cancer cells. We have shown previ-ously that Sigma1 modulators regulate ErbB2/HER2 and ErbB3/
HER3 levels and activity in prostate and breast cancers (ref. 23 andour unpublished data, respectively).
Sigma1, PD-L1, and antitumor immunityThis study addresses the intracellular processes regulated by
Sigma1 modulators, and it provides evidence in support of thenotion that the pharmacologic mechanism of action of Sigma1modulator compounds involves modulation of Sigma1-associ-ated functional protein interactions. Sigma1 engages in protein–protein interactions withmultiple partner and client proteins. It islikely that ligand-induced changes in some of these proteinassociations may contribute to suppression of PD-L1. This mayoccur through direct regulatorymechanisms or indirectly throughfeedback regulation mechanisms. For example, Sigma1 has beenimplicated in immunomodulation (reviewed in ref. 19), and arole for Sigma1 in antitumor immunity has been proposed(reviewed in ref. 19). Sigma1 activators (agonists) have beenshown to induce the production and secretion of IL10 and TGFband promote tumor growth in a syngeneic mouse lung cancermodel (46). Interestingly, in this model, IL10 was only inducedat the tumor site and increased TGFb only occurred in tumor-bearing mice, suggesting increased cytokine production inresponse to the tumor or increased cytokine production by thetumor cells (46). Sigma1 inhibitors (putative antagonists)abrogated the production of IL10 and suppressed Sigma1activator–induced tumor growth (46). Sigma1 modulatorshave also been reported to regulate the production of IFNg(46). PD-L1 production and activity through modulation ofcell surface expression are regulated in part by cytokinesreleased in the tumor microenvironment during antitumorimmune responses, including IFNg , TNFa, TGFb, IL17, andIL10 (7, 13, 14, 47, 48). Taken together, these data suggest thatpharmacologic modulation of Sigma1 can regulate PD-L1production and activity via immune response induced cyto-kine-mediated extracellular feedback loops as well as directlyvia protein–protein associations. These mechanisms regulatethe tenor of the tumor immune microenvironment (49).
There is compelling rationale for and reemerging interest incombination therapies to sustain antitumor immune responsesinduced by stimulatory cytokines such IFNg (39–41). The abilityof Sigma1 modulators to suppress PD-L1 in cancer cells treatedwith immunostimulatory cytokines, such as IFNg (Fig. 7), revealspotential therapeutic approaches involving cotreatment withIFNg and Sigma1 modulators to prolong/sustain antitumorimmune responses.
PD-L1 also costimulates IL10 secretion (50). Sigma1 activators(agonists) have been shown to block antitumor immunity, in partby induction of IL10 (46). Thus, Sigma1 regulation of PD-L1maycontribute to antitumor immunity through checkpoint inhibitionas well as cosuppression of anti-inflammatory cytokines. Sigma1inhibitors may promote antitumor immunity in part by elimi-nating PD-L1 as well as by modulating the production andsecretion of cytokines from cancer cells.
Thus, control of PD-L1 through Sigma1 supports a role forSigma1 in antitumor immunity and provides insight into themechanism(s) by which Sigma1 compounds elicit their immu-nomodulatory effects. Our data suggest that selective small-mol-ecule Sigma1 ligands may be used as novel modulators of thetumor immune microenvironment. The use of Sigma1 modula-tors to pharmacologically promote antitumor immunity presentsan opportunity for novel drug combinations with checkpoint
Figure 7.
Small-molecule inhibition of Sigma1 suppresses IFNg-induced upregulation ofPD-L1 protein levels in cancer cells. Immunoblot and quantification of PD-L1protein levels from whole-cell protein extracts from PC3 (A) and MDA-MB-231(B) cells treated with 100 ng/mL IFNg for 24 hours alone or in combination with10 mmol/L IPAG for the last 16 hours. C and D, Immunoblots were quantified bydensitometry for each cell line, and data are presented as levels of PD-L1 relativeto nontreated control. Data represent mean values from at least threeindependent determinations and error bars represent SEM. � , P < 0.05. E and F,qRT-PCR results for PD-L1 transcript levels for PC3 (E) andMDA-MB-231 cells (F)treated with the same drug conditions used in A and B. Data are presented asfold induction over nontreated control. � , P < 0.05; �� , P < 0.01; ���� , P < 0.0001.Data represent mean values and error bars represent SEM.
Autophagic Degradation of PD-L1 by a Sigma1 Modulator
www.aacrjournals.org Mol Cancer Res; 16(2) February 2018 253
on August 9, 2019. © 2018 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst November 8, 2017; DOI: 10.1158/1541-7786.MCR-17-0166

inhibitor agents to expand the population of patients thatrespond to PD-L1/PD-1–targeted therapies.
Disclosure of Potential Conflicts of InterestF.J. Kim is the scientific founder at, reports receiving commercial research
support from, has ownership interest (including patents) in, and is a consultant/advisory board member for Context Therapeutics. No potential conflicts ofinterest were disclosed by the other authors.
Authors' ContributionsConception and design: C.M. Maher, F.J. KimDevelopment of methodology: C.M. Maher, J.D. Thomas, D.A. Haas,C.G. Longen, H.M. Oyer, F.J. KimAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): C.M. Maher, J.D. Thomas, D.A. Haas, C.G. Longen,H.M. Oyer, J.Y. Tong, F.J. KimAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): C.M. Maher, J.D. Thomas, D.A. Haas, C.G. Longen,H.M. Oyer, J.Y. Tong, F.J. KimWriting, review, and/or revision of the manuscript: C.M. Maher, F.J. Kim
Administrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): C.M. Maher, H.M. Oyer, F.J. KimStudy supervision: F.J. Kim
AcknowledgmentsF.J. Kimwas supported by an American Cancer Society Institutional Research
Grant, Drexel University Clinical and Translational Research Institute Grant,Sidney Kimmel Cancer Center Pilot Study Award, and a Coulter-Drexel Trans-lational Research Partnership Program Award.
The authors thankDrs. Paul McGonigle and James Barrett for critical readingof the manuscript.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received March 27, 2017; revised September 22, 2017; accepted October 30,2017; published OnlineFirst November 8, 2017.
References1. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in
tolerance and immunity. Annu Rev Immunol 2008;26:677–704.2. Li CW, Lim SO, XiaW, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and
stabilization of programmed death ligand-1 suppresses T-cell activity. NatCommun 2016;7:12632.
3. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpointpathway. N Engl J Med 2016;375:1767–78.
4. Shao S, Hegde RS. Membrane protein insertion at the endoplasmic retic-ulum. Ann Rev Cell Dev Biol 2011;27:25–56.
5. Voeltz GK, Rolls MM, Rapoport TA. Structural organization of the endo-plasmic reticulum. EMBO Rep 2002;3:944–50.
6. Mu TW, Ong DS, Wang YJ, Balch WE, Yates JR 3rd, Segatori L, et al.Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell 2008;134:769–81.
7. SharmaP,Allison JP. Immune checkpoint targeting in cancer therapy: towardcombination strategies with curative potential. Cell 2015;161:205–14.
8. Brahmer JR, Tykodi SS, ChowLQ,HwuWJ, Topalian SL,HwuP, et al. Safetyand activity of anti-PD-L1 antibody in patients with advanced cancer.N Engl J Med 2012;366:2455–65.
9. Topalian SL,Hodi FS, Brahmer JR,Gettinger SN, SmithDC,McDermottDF,et al. Safety, activity, and immune correlates of anti-PD-1 antibody incancer. N Engl J Med 2012;366:2443–54.
10. Hodi FS,O'Day SJ,McDermottDF,Weber RW, Sosman JA,Haanen JB, et al.Improved survival with ipilimumab inpatientswithmetastaticmelanoma.N Engl J Med 2010;363:711–23.
11. Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S,et al. PD-L1 expression in triple-negative breast cancer. Cancer ImmunolRes 2014;2:361–70.
12. Gevensleben H, Dietrich D, Golletz C, Steiner S, Jung M, Thiesler T, et al.The immune checkpoint regulator PD-L1 is highly expressed in aggressiveprimary prostate cancer. Clin Cancer Res 2016;22:1969–77.
13. Bishop JL, Sio A, Angeles A, Roberts ME, Azad AA, Chi KN, et al. PD-L1 ishighly expressed in Enzalutamide resistant prostate cancer. Oncotarget2015;6:234–42.
14. Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancerimmunotherapy. Mol Cancer Ther 2015;14:847–56.
15. Goswami S, Aparicio A, Subudhi SK. Immune checkpoint therapies inprostate cancer. Cancer J 2016;22:117–20.
16. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockadefor cancer therapy: mechanisms, response biomarkers, and combinations.Sci Transl Med 2016;8:328rv4.
17. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade incancer therapy. J Clin Oncol 2015;33:1974–82.
18. Nirschl CJ, Drake CG. Molecular pathways: coexpression of immunecheckpoint molecules: signaling pathways and implications for cancerimmunotherapy. Clin Cancer Res 2013;19:4917–24.
19. Kim FJ, Maher CM. Sigma1 pharmacology in the context of cancer. HandbExp Pharmacol 2017;244:237–308.
20. Hanner M, Moebius FF, Flandorfer A, Knaus HG, Striessnig J, Kempner E,et al. Purification, molecular cloning, and expression of the mammaliansigma1-binding site. Proc Natl Acad Sci U S A 1996;93:8072–7.
21. Schmidt HR, Zheng S, Gurpinar E, Koehl A, Manglik A, Kruse AC. Crystalstructure of the human sigma receptor. Nature 2016;532:527–30.
22. Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrioninterface regulate Ca(2þ) signaling and cell survival. Cell 2007;131:596–610.
23. Thomas JD, Longen CG, Oyer HM, Chen N, Maher CM, Salvino JM, et al.Sigma1 targeting to suppress aberrant androgen receptor signaling inprostate cancer. Cancer Res 2017;77:2439–52.
24. Schrock JM, Spino CM, Longen CG, Stabler SM, Marino JC, Pasternak GW,et al. Sequential cytoprotective responses to Sigma1 ligand-induced endo-plasmic reticulum stress. Mol Pharmacol 2013;84:751–62.
25. Kim FJ, Schrock JM, Spino CM, Marino JC, Pasternak GW. Inhibition oftumor cell growth by Sigma1 ligand mediated translational repression.Biochem Biophys Res Commun 2012;426:177–82.
26. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagyresearch. Cell 2010;140:313–26.
27. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, AcevedoArozena A, et al. Guidelines for the use and interpretation of assays formonitoring autophagy (3rd edition). Autophagy 2016;12:1–222.
28. Molino D, Zemirli N, Codogno P, Morel E. The Journey of the Autophago-some through Mammalian Cell Organelles and Membranes. J Mol Biol2017;429:497–514.
29. Pimentel-Muinos FX, Boada-Romero E. Selective autophagy against mem-branous compartments: Canonical and unconventional purposes andmechanisms. Autophagy 2014;10:397–407.
30. Sica V, Galluzzi L, Bravo-San Pedro JM, Izzo V, Maiuri MC, Kroemer G.Organelle-specific initiation of autophagy. Mol Cell 2015;59:522–39.
31. Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, AkutsuM, et al.Regulation of endoplasmic reticulum turnover by selective autophagy.Nature 2015;522:354–8.
32. Pavel M, Rubinsztein DC. Mammalian autophagy and the plasma mem-brane. FEBS J 2017;284:672–9.
33. Kuma A, HatanoM,Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al.The role of autophagy during the early neonatal starvation period. Nature2004;432:1032–6.
34. Pan YX, Mei J, Xu J, Wan BL, Zuckerman A, Pasternak GW. Cloning andcharacterization of a mouse sigma1 receptor. J Neurochem 1998;70:2279–85.
35. Zinchuk V,Wu Y, Grossenbacher-ZinchukO, Stefani E.Quantifying spatialcorrelations of fluorescent markers using enhanced background reductionwith protein proximity index and correlation coefficient estimations. NatProtoc 2011;6:1554–67.
Maher et al.
Mol Cancer Res; 16(2) February 2018 Molecular Cancer Research254
on August 9, 2019. © 2018 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst November 8, 2017; DOI: 10.1158/1541-7786.MCR-17-0166

36. Schindler J, Nothwang HG. Aqueous polymer two-phase systems: effectivetools for plasma membrane proteomics. Proteomics 2006;6:5409–17.
37. Matsuno K, Nakazawa M, Okamoto K, Kawashima Y, Mita S. Bindingproperties of SA4503, a novel and selective sigma 1 receptor agonist. Eur JPharmacol 1996;306:271–9.
38. Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ.The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002inhibit autophagy in isolated rat hepatocytes. Eur J Biochem 1997;243:240–6.
39. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: impli-cations for cancer therapy. Nat Rev Cancer 2016;16:131–44.
40. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, andacquired resistance to cancer immunotherapy. Cell 2017;168:707–23.
41. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, RodriguezGA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep 2017;19:1189–201.
42. Spruce BA, Campbell LA, McTavish N, Cooper MA, Appleyard MV,O'Neill M, et al. Small molecule antagonists of the sigma-1 receptorcause selective release of the death program in tumor and self-reliantcells and inhibit tumor growth in vitro and in vivo. Cancer Res 2004;64:4875–86.
43. MizushimaN, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights diseasethrough cellular self-digestion. Nature 2008;451:1069–75.
44. Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, CecconiF, et al. Molecular definitions of autophagy and related processes. EMBO J2017;36:1811–36.
45. Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat RevCancer 2017;17:528–42.
46. Zhu LX, Sharma S, Gardner B, Escuadro B, Atianzar K, Tashkin DP, et al. IL-10 mediates sigma1 receptor-dependent suppression of antitumor immu-nity. J Immunol 2003;170:3585–91.
47. ZhaoQ, Xiao X,Wu Y,Wei Y, Zhu LY, Zhou J, et al. Interleukin-17-educatedmonocytes suppress cytotoxic T-cell function through B7-H1 in hepato-cellular carcinoma patients. Eur J Immunol 2011;41:2314–22.
48. Schweizer MT, Drake CG. Immunotherapy for prostate cancer: recent deve-lopments and future challenges. Cancer Metastasis Rev 2014;33:641–55.
49. He J, Hu Y, Hu M, Li B. Development of PD-1/PD-L1 pathway in tumorimmune microenvironment and treatment for non-small cell lung cancer.Sci Rep 2015;5:13110.
50. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7family, co-stimulates T-cell proliferation and interleukin-10 secretion. NatMed 1999;5:1365–9.
www.aacrjournals.org Mol Cancer Res; 16(2) February 2018 255
Autophagic Degradation of PD-L1 by a Sigma1 Modulator
on August 9, 2019. © 2018 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst November 8, 2017; DOI: 10.1158/1541-7786.MCR-17-0166

2018;16:243-255. Published OnlineFirst November 8, 2017.Mol Cancer Res Christina M. Maher, Jeffrey D. Thomas, Derick A. Haas, et al. of PD-L1Small-Molecule Sigma1 Modulator Induces Autophagic Degradation
Updated version
10.1158/1541-7786.MCR-17-0166doi:
Access the most recent version of this article at:
Material
Supplementary
http://mcr.aacrjournals.org/content/suppl/2017/11/08/1541-7786.MCR-17-0166.DC1
Access the most recent supplemental material at:
Overview
Visual
http://mcr.aacrjournals.org/content/16/2/243/F1.large.jpgA diagrammatic summary of the major findings and biological implications:
Cited articles
http://mcr.aacrjournals.org/content/16/2/243.full#ref-list-1
This article cites 50 articles, 13 of which you can access for free at:
Citing articles
http://mcr.aacrjournals.org/content/16/2/243.full#related-urls
This article has been cited by 3 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mcr.aacrjournals.org/content/16/2/243To request permission to re-use all or part of this article, use this link
on August 9, 2019. © 2018 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst November 8, 2017; DOI: 10.1158/1541-7786.MCR-17-0166


















