
Sesión extrahospitalaria 2010 - Parc de Salut Mar · Sistema Complicaciones Sistema Nervioso...
26
NEUROFIBROMATOSIS-1 Enfermedad de Von Recklinghausen Mª Ángeles López Vílchez Sección de Neonatología Parc de Salut Mar
Transcript of Sesión extrahospitalaria 2010 - Parc de Salut Mar · Sistema Complicaciones Sistema Nervioso...

NEUROFIBROMATOSIS-1Enfermedad de Von Recklinghausen
Mª Ángeles López VílchezSección de Neonatología
Parc de Salut Mar

INTRODUCCIÓN
• Afectación predominante piel y sistema nervioso• Prevalencia: 1/3000• Gen Cromosoma 17 q11.2. Expresividad variable
dentro de la misma familia• Herencia: AD. > 50% de novo• > 500 mutaciones• El Dx definitivo alrededor de los 4 años, 95% a los
11 años.

CRITERIOS CLÍNICOSInstituto Nacional de Salud EEUU (NIH) (1987)
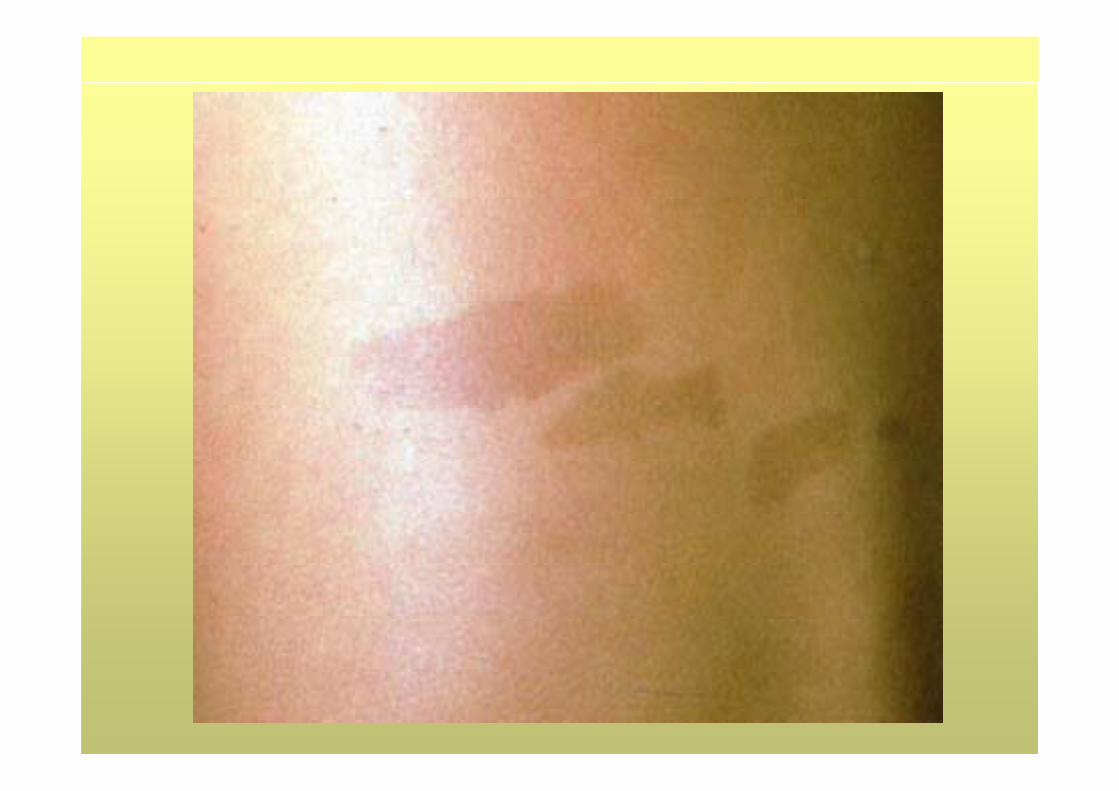
1. ≥6 manchas “café con leche” ≥5 mm prepúber o ≥15 mm resto
2. ≥2 neurofibromas de cualquier tipo o uno plexiforme3. Pecas en axilas o ingles4. Glioma del nervio óptico5. ≥2 nódulos de Lisch6. Lesión ósea: displasia esfenoides o adelgazamiento cortical
huesos largos7. Un familiar de 1º grado afecto

SINTOMATOLOGÍA
• 50% sintomatología leve• 1/3 retraso mental leve• Frecuente manchas café con leche desde el nacimiento, luego
aumentan• Pecas en pliegues (75%) 3-5 años• Neurofibromas plexiformes (25%), congénitos• Neurofibromas dérmicos en período puberal• Displasia tibial y ala esfenoides congénitas• Nódulos de Lisch sólo 30% en < 6 años. Inicio en la adolescencia• Gliomas del nervio óptico. 15% niños <6 años. Sólo el 5% afectación
grave (pérdida visual, proptosis, hidrocefalia).
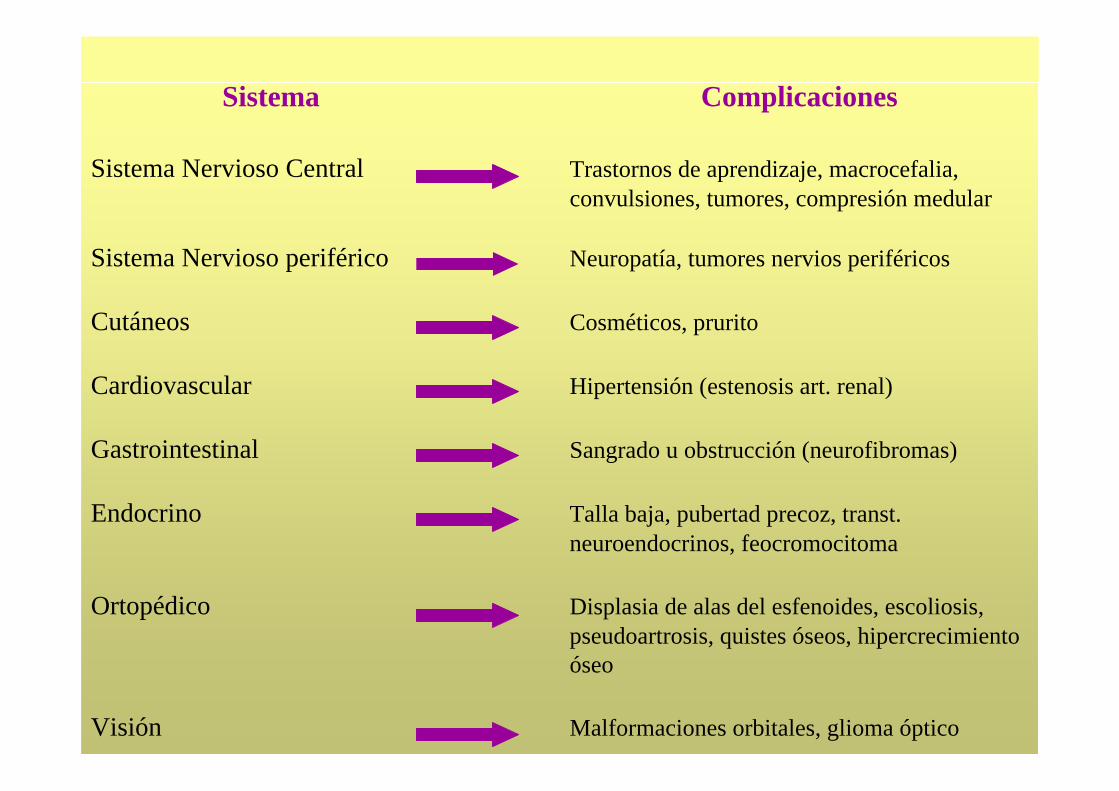
Sistema Complicaciones
Sistema Nervioso Central Trastornos de aprendizaje, macrocefalia, convulsiones, tumores, compresión medular
Sistema Nervioso periférico Neuropatía, tumores nervios periféricos
Cutáneos Cosméticos, prurito
Cardiovascular Hipertensión (estenosis art. renal)
Gastrointestinal Sangrado u obstrucción (neurofibromas)
Endocrino Talla baja, pubertad precoz, transt. neuroendocrinos, feocromocitoma
Ortopédico Displasia de alas del esfenoides, escoliosis,pseudoartrosis, quistes óseos, hipercrecimiento óseo
Visión Malformaciones orbitales, glioma óptico

SEGUIMIENTO RECIÉN NACIDOSAmerican Academy of Pediatrics 1995
• Confirmar Dx por las manifestaciones cutáneas (manchas café con leche, pecas axilares)
• Padres exámen oftalmológico: nódulos de Lisch. 90% adultos, infrecuente en < 3 años
• No se recomienda TAC/RMN en pacientes asintomáticos

SEGUIMIENTO 1 MES-AÑOAmerican Academy of Pediatrics 1995
• Crecimiento y desarrollo neurológico• Vigilar manchas café con leche• Controlar proptosis, aumento rápido del PC,
focalidad neurológica• Malformaciones esqueléticas (espalda, piernas)• Remitir a especialistas si indicado

SEGUIMIENTO 1-5 AÑOSAmerican Academy of Pediatrics 1995
• Buscar neurofibromas y nuevas pecas• Fotografías lesiones para seguimiento tamaño• Examinar la visión• Recomendar visita a oftalmología/año• RMN si clínica (cefalea, aumento rápido PC,
problemas visuales, neuroma plexiforme cabeza)• Examinar el habla

SEGUIMIENTO 5-13 AÑOSAmerican Academy of Pediatrics 1995
• Buscar tumores cutáneos que desfiguren• Signos de pubertad. Su pubertad precoz: glioma
nervio óptico o lesión hipotalámica• Descartar problemas de aprendizaje o
comportamiento, hiperactividad• Revisar las posibles modificaciones sociales

SEGUIMIENTO 13-21 AÑOSAmerican Academy of Pediatrics 1995
• Revisión oftalmológica anual• Signos de pubertad o hipogonadismo• Examen neurológico para buscar neurofibromas
plexiformes profundos• Consulta con cirujano si signos de compresión de
estructuras profundas

BIBLIOGRAFÍA• National Institutes of Health (NIH). Consensus
Development Conference Statement. Neurofibromatosis. Betesa Md., July 13-15, 1987. Neurofibromatosis 1988;1(3):172-178
• Committee on Genetics. American Academy of PediatricsHealth supervision for children with neurofibromatosis. Pediatrics 1995;96: 368-372
• E. Galán Gómez. Neurofibromatosis tipo I. Protocolos Genética. Anales Españoles de Pediatría. En www.aeped.es/protocolos/genética
• L. Puig Sanz. Síndromes neurocutáneos. Protocolos de Dermatología. Anales Españoles de Pediatría. En www.aeped.es/protocolos/dermatología

Reticulohistiocitosis congénita autolimitada
Enfermedad de Hashimoto-Pritzker
Mª Ángeles López VílchezSección de Neonatología
Parc de Salut Mar

CASO CLÍNICO
• Recién nacido de sexo masculino que ingresa en la Unidad Neonatal por taquipnea y riesgo de infección por FIP
• Antecedentes: Gestación controlada, sin incidencias. 383
semanas. Amniorrexis 5 horas, liquido teñido. Fiebre materna de 38,5ºC. Parto eutócico. Apgar 9/10. pH vasos: 7,27/7,26. PN: 3000 gr, talla: 48 cm, PC: 35 cm
• EF: taquipnea (FR 80 x’), Saturación 95% (FiO2 21%), FC 160 x’. Pápulas contenido parduzco diseminadas, alguna en fase de costra. Resto, normal.
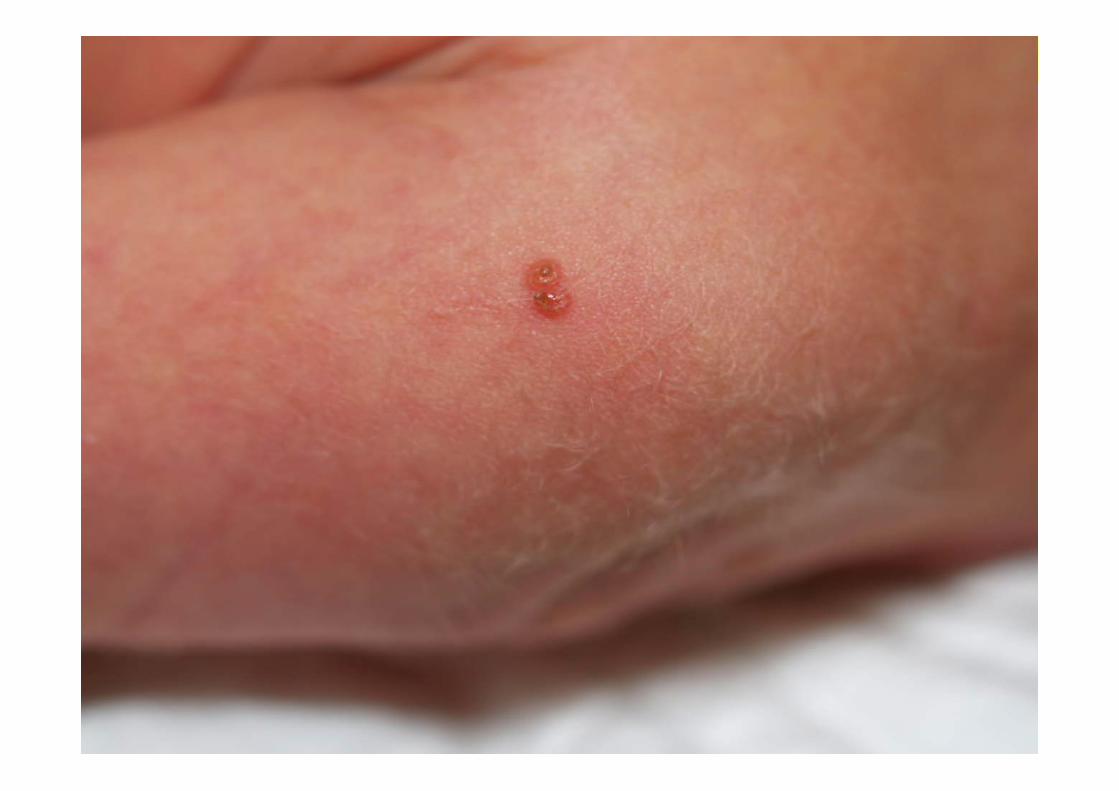


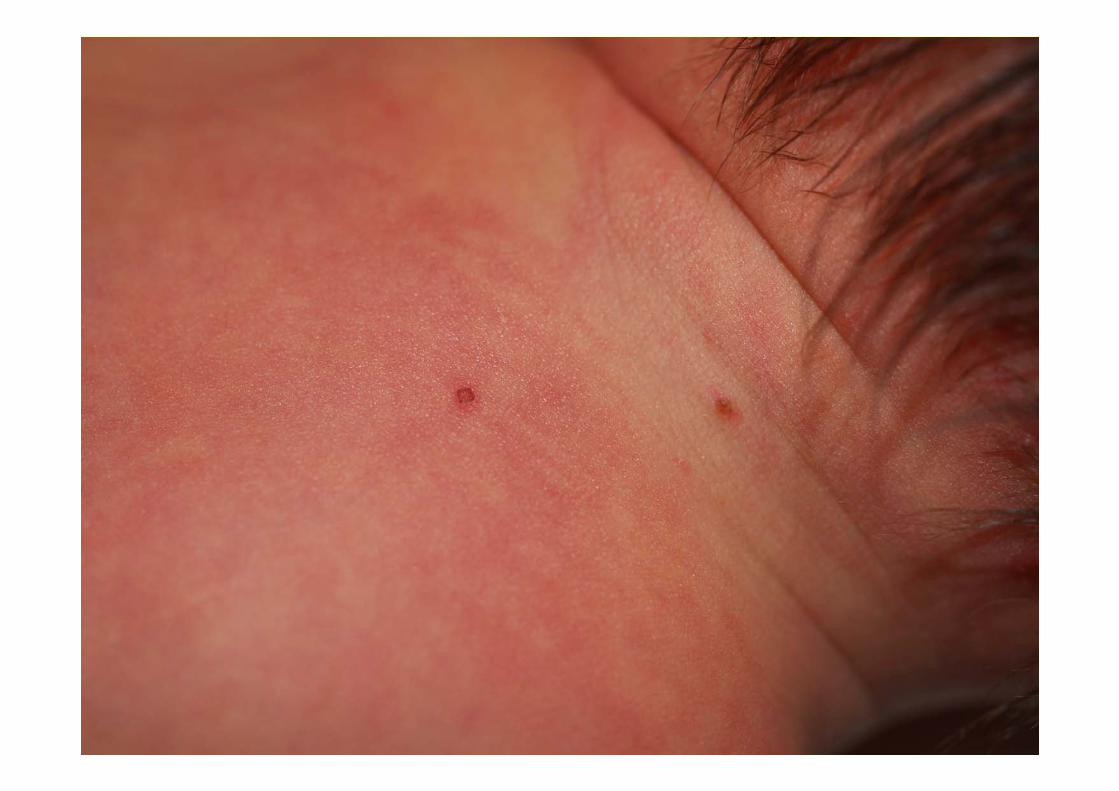

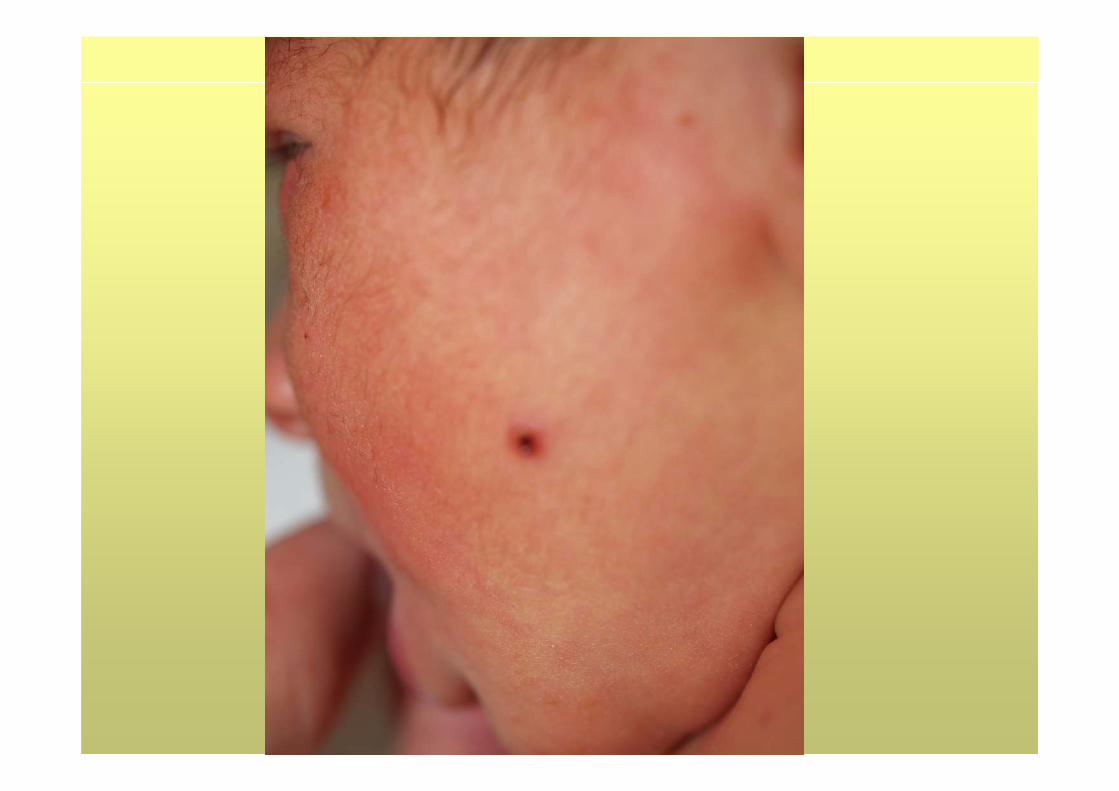

CASO CLÍNICO• Evolución: desaparición progresiva de la taquipnea.
Controles analíticos normales.• Consulta dermatología. OD: reticulohistiocitosis
congénita autoinvolutiva (RCA)– Cultivo lesiones (bacterias, hongos, virus) negativos– Test Tzank negativo. Visualizan histiocitos– Bx piel (AP y ME) Inmunohistoquímica: marcada
expresión de CD1a, langherina, CD68 y proteina S-100. ME: abundantes células de Langerhans con cuerpos de Birbeck.

CASO CLÍNICO
• Seguimiento en CCEE dermatología y neonatología:– EECC: analítica, ecografía abdominal, serie ósea y Rx tórax,
normales– Lesiones nuevas hasta el mes y medio de vida– Desaparición de lesiones a los 2 meses y medio– A los 3 meses: cicatrices residuales atróficas en calota y EE– Último control a los 6 meses: asintomático

RETÍCULOHISTIOCITOSIS CONGÉNITA AUTOINVOLUTIVA
• Paul Langerhans en 1868: células dendríticas epidermis células de Langerhans: positividad S100, expresión CD-1a y gránulos de Birbeck (ME)
• 1973 Hashimoto y Pritzker describen la RCA• Clínica:
– Inicio al nacimiento o pocos días de vida, hasta los 2 meses– Lo más frecuente: pápulas y nódulos marronáceos– Formas atípicas: vesículas, ampollas, aspecto angiomatoso– Diseminadas (palmas y plantas)– Puede afectar mucosas– Involución espontánea 3-4 meses/6-8 meses– Carácter imprevisible

RETÍCULOHISTIOCITOSIS CONGÉNITA AUTOINVOLUTIVA
• Dx: biopsia + autoinvolución lesiones• Dx diferencial:
– Lesiones solitarias: xantogranuloma infantil, nevus de Spitz, mastocitoma
– Pápulas: histiocitosis de cels. Langerhans multisistémica– Nódulos: hemangiomatosis, eritropoyesis dérmica, sífilis
congénita– Vesiculopustulas: enfermedades infecciosas (varicela, herpes
simple, listeria, sífilis), enf. inflamatorias (eritema tóxico, melanosis pustulosas, incontinentia pigmenti), enf. ampollosas ( epidermiolisis ampollosa, penfingoideestacional)

RETÍCULOHISTIOCITOSIS CONGÉNITA AUTOINVOLUTIVA
Conducta a seguir• Serologías TORCH, cultivo lesiones, frotis de Tzanck,
biopsia• Si Bx diagnóstica de RCA hemograma, frotis sangre
periférica, bioquímica, serie ósea esquelética, Rx tórax, ecografía abdominal, aspirado MO??
• Pronóstico-seguimiento: cautela (recaídas, progresión), seguimiento largo plazo (afectación órganos internos), exploración física minuciosa

BIBLIOGRAFÍA• Chunharas A, Pabunruang W, Hongeng S. Congenital self-
helaing Langerhans cell histiocytosis with pulmonay involvement: spontaneous regression. J Med Assoc Thai 2002;85:1309-1313
• Pastor L, Jiménez A. Reticulohistiocitosis congénita autoinvolutiva. Revisión. Piel 2006;21(9):421-429
• Ersoy-Evans, S, Gursoy T, Yigit S, Akcoren Z, Sahin S. Solitary congenital self-healing reticulohistiocytosis in monozygotic twins. Pediatric dermatology 2006;23(3):273-275
• Pastor L, Tomás L, Clement A, Fuertes A, Villanueva A, Marquina A. Reticulohistiocitosis congénita autoinvolutiva. Med Cutan Iber Lat Am 2007;35(4):193-196








![Convulsiones [autoguardado]](https://static.fdocuments.net/doc/165x107/55b19208bb61eb64198b4608/convulsiones-autoguardado.jpg)









