
Scalable up genomic analysis with ADAM
27
Scaling up genomic analysis with ADAM Frank Austin Nothaft, UC Berkeley AMPLab [email protected], @fnothaft 10/27/2014
-
Upload
fnothaft -
Category
Engineering
-
view
415 -
download
2
Transcript of Scalable up genomic analysis with ADAM

Scaling up genomic analysis with ADAM
Frank Austin Nothaft, UC Berkeley AMPLab [email protected], @fnothaft
10/27/2014

What is ADAM?• An open source, high performance, distributed
platform for genomic analysis
• ADAM defines a:
1. Data schema and layout on disk*
2. A Scala API
3. A command line interface
* Via Avro and Parquet

What’s the big picture?ADAM:!
Core API + CLIs
bdg-formats:!Data schemas
RNAdam:!RNA analysis on
ADAM
avocado:!Distributed local
assembler
xASSEMBLEx:!GraphX-based de novo assembler
bdg-services:!ADAM clusters
PacMin:!String graph assembler

Implementation Overview
• 34k LOC (96% Scala)
• Apache 2 licensed OSS
• 23 contributors across 10 institutions
• Pushing for production 1.0 release towards end of year

Key Observations• Current genomics pipelines are I/O limited
• Most genomics algorithms can be formulated as a data or graph parallel computation
• These algorithms are heavy on iteration/pipelining
• Data access pattern is write once, read many times
• High coverage, whole genome will become main sequencing target (for human genetics)

Principles for Scalable Design in ADAM
• Parallel FS and data representation (HDFS + Parquet) combined with in-memory computing eliminates disk bandwidth bottleneck
• Spark allows efficient implementation of iterative/pipelined Map-Reduce
• Minimize data movement: send code to data

• An in-memory data parallel computing framework
• Optimized for iterative jobs —> unlike Hadoop
• Data maintained in memory unless inter-node movement needed (e.g., on repartitioning)
• Presents a functional programing API, along with support for iterative programming via REPL
• Used at scale on clusters with >2k nodes, 4TB datasets

Why Spark?• Current leading map-reduce framework:
• First in-memory map-reduce platform
• Used at scale in industry, supported in major distros (Cloudera, HortonWorks, MapR)
• The API:
• Fully functional API
• Main API in Scala, also support Java, Python, R
• Manages node/job failures via lineage, data locality/job assignment
• Downstream tools (GraphX, MLLib)

Data Format• Avro schema encoded by
Parquet
• Schema can be updated without breaking backwards compatibility
• Read schema looks a lot like BAM, but renormalized
• Actively removing tags
• Variant schema is strictly biallelic, a “cell in the matrix”
record AlignmentRecord { union { null, Contig } contig = null; union { null, long } start = null; union { null, long } end = null; union { null, int } mapq = null; union { null, string } readName = null; union { null, string } sequence = null; union { null, string } mateReference = null; union { null, long } mateAlignmentStart = null; union { null, string } cigar = null; union { null, string } qual = null; union { null, string } recordGroupName = null; union { int, null } basesTrimmedFromStart = 0; union { int, null } basesTrimmedFromEnd = 0; union { boolean, null } readPaired = false; union { boolean, null } properPair = false; union { boolean, null } readMapped = false; union { boolean, null } mateMapped = false; union { boolean, null } firstOfPair = false; union { boolean, null } secondOfPair = false; union { boolean, null } failedVendorQualityChecks = false; union { boolean, null } duplicateRead = false; union { boolean, null } readNegativeStrand = false; union { boolean, null } mateNegativeStrand = false; union { boolean, null } primaryAlignment = false; union { boolean, null } secondaryAlignment = false; union { boolean, null } supplementaryAlignment = false; union { null, string } mismatchingPositions = null; union { null, string } origQual = null; union { null, string } attributes = null; union { null, string } recordGroupSequencingCenter = null; union { null, string } recordGroupDescription = null; union { null, long } recordGroupRunDateEpoch = null; union { null, string } recordGroupFlowOrder = null; union { null, string } recordGroupKeySequence = null; union { null, string } recordGroupLibrary = null; union { null, int } recordGroupPredictedMedianInsertSize = null; union { null, string } recordGroupPlatform = null; union { null, string } recordGroupPlatformUnit = null; union { null, string } recordGroupSample = null; union { null, Contig} mateContig = null;}

Parquet• ASF Incubator project, based on
Google Dremel
• http://www.parquet.io
• High performance columnar store with support for projections and push-down predicates
• 3 layers of parallelism:
• File/row group
• Column chunk
• Page
Image from Parquet format definition: https://github.com/Parquet/parquet-format

Filtering
• Parquet provides pushdown predication
• Evaluate filter on a subset of columns
• Only read full set of projected columns for passing records
• Full primary/secondary indexing support in Parquet 2.0
• Very efficient if reading a small set of columns:
• On disk, contig ID/start/end consume < 2% of space
Image from Parquet format definition: https://github.com/Parquet/parquet-format

Compression• Parquet compresses
at the column level:
• RLE for repetitive columns
• Dictionary encoding for quantized columns
• ADAM uses a fully denormalized schema
• Repetitive columns are RLE’d out
• Delta encoding (Parquet 2.0) will aid with quality scores
• ADAM is 5-25% smaller than compressed BAM

Parquet/Spark Integration• 1 row group in Parquet maps
to 1 partition in Spark
• We interact with Parquet via input/output formats
• These apply projections and predicates, handle (de)compression
• Spark builds and executes a computation DAG, manages data locality, errors/retries, etc.
RG 1 RG 2 RG n…Parquet
RG 1 RG 2 RG n…Parquet
SparkParquet Input Format
Parquet Output Format
Partition 1
Partition 2
Partition n
…

Long-read assembly with PacMin

The State of Analysis• Conventional short-read alignment based pipelines
are really good at calling SNPs
• But, we’re still pretty bad at calling INDELs, and SVs
• And are slow: 2 weeks to sequence, 1 week to analyze. Not fast enough for clinical use.
• If we move away from short reads, do we have other options?

Opportunities
• New read technologies are available
• Provide much longer reads (250bp vs. >10kbp)
• Different error model… (15% INDEL errors, vs. 2% SNP errors)
• Generally, lower sequence specific biasLeft: PacBio homepage, Right: Wired, http://www.wired.com/2012/03/oxford-nanopore-sequencing-usb/

If long reads are available…• We can use conventional methods:
Carneiro et al, Genome Biology 2012

But!• Why not make raw assemblies out of the reads?
=?
Find overlapping reads Find consensus sequencefor all pairs of reads (i,j):
i j
…ACACTGCGACTCATCGACTC…
• Problems:
1. Overlapping is O(n2) and single evaluation is expensive anyways
2. Typical algorithms find a single consensus sequence; what if we’ve got polymorphisms?

Fast Overlapping with MinHashing
• Wonderful realization by Berlin et al1: overlapping is similar to document similarity problem
• Use MinHashing to approximate similarity:
1: Berlin et al, bioRxiv 2014
Per document/read, compute signature:!!
1. Cut into shingles 2. Apply random
hashes to shingles 3. Take min over all
random hashes
Hash into buckets:!!Signatures of length l can be hashed into b buckets, so we expect
to compare all elements with similarity ≥ (1/b)^(b/l)
Compare:!!For two documents with signatures of length l, Jaccard similarity is
estimated by (# equal hashes) / l
!
• Easy to implement in Spark: map, groupBy, map, filter
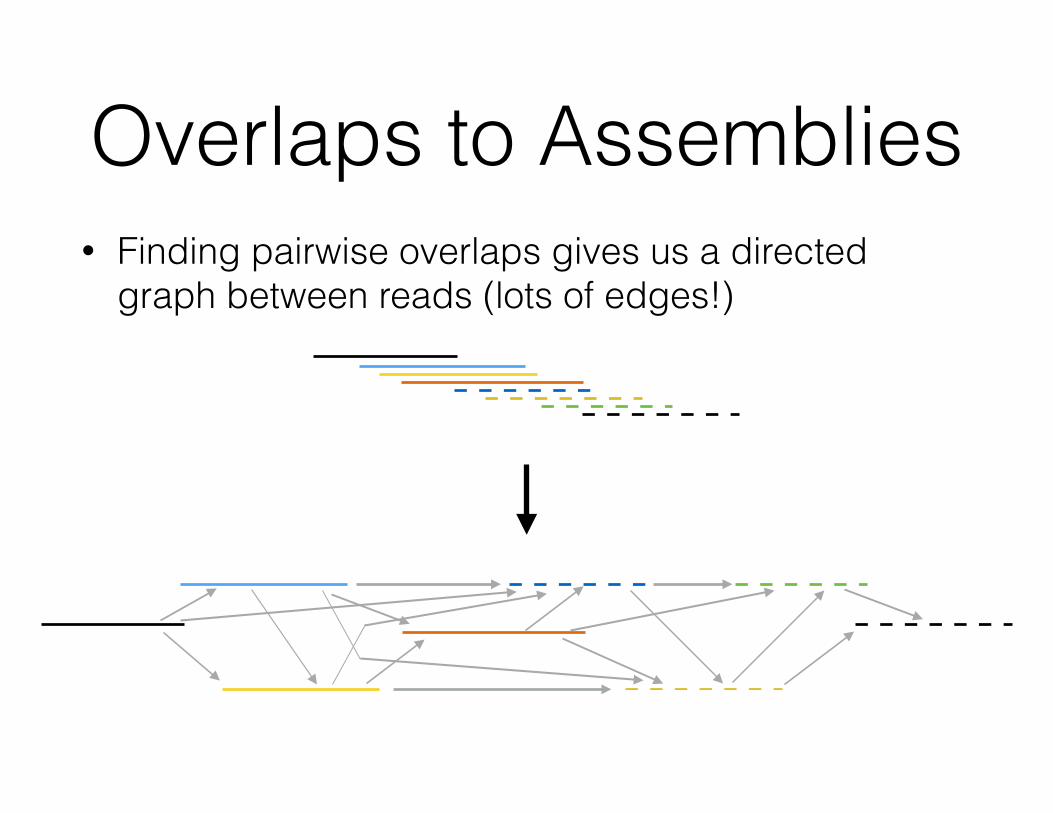
Overlaps to Assemblies• Finding pairwise overlaps gives us a directed
graph between reads (lots of edges!)
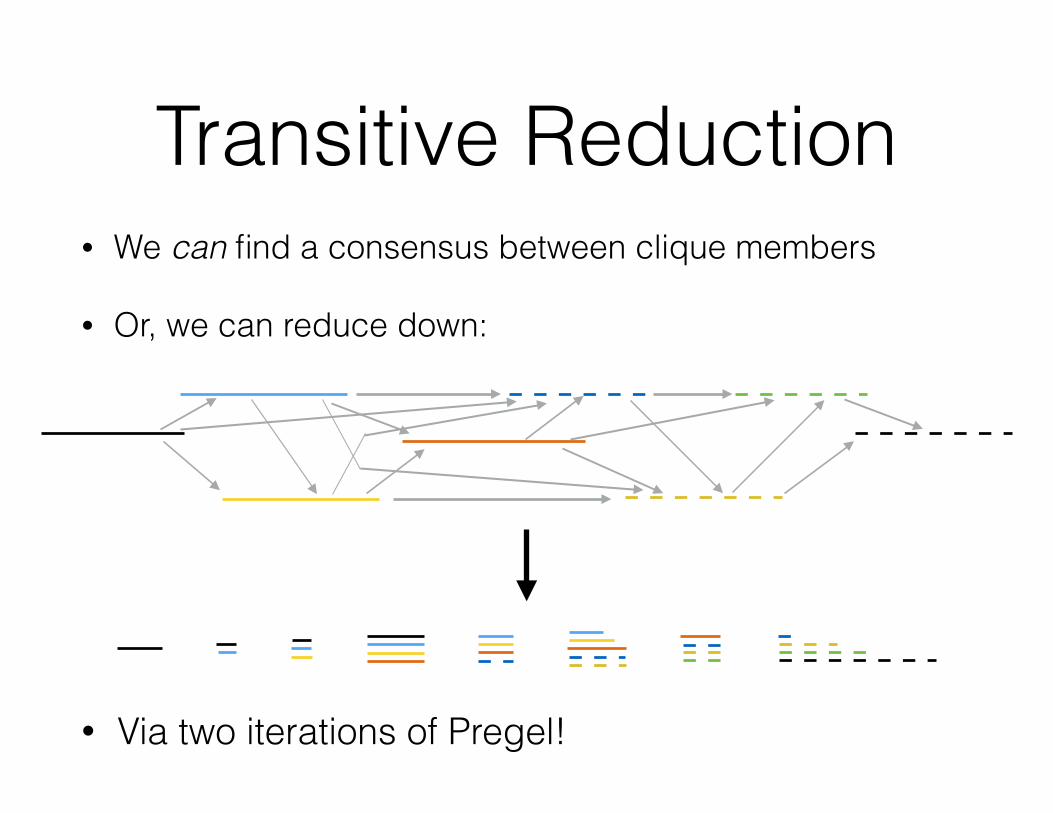
Transitive Reduction• We can find a consensus between clique members
• Or, we can reduce down:
• Via two iterations of Pregel!

Actually Making Calls• From here, we need to call copy number per edge
• Probably via Newton-Raphson based on coverage; we’re not sure yet.
• Then, per position in each edge, call alleles:
Notes:!Equation is from Li, Bioinformatics 2011
g = genotype state m = ploidy
𝜖 = probability allele was erroneously observed k = number of reads observed
l = number of reads observed matching “reference” allele TBD: equation assumes biallelic observations at site and reference allele; we won’t have either of those conveniences…

An aside: Monoallelic Genotyping
• Traditional probabilistic models for variant calling assume independence at each site
• However, this throws away a lot of information
• Can consider a different formulation of the problem:
• Build a graph of the alleles
• Find the allelic copy numbers that maximize likelihood

Allelic Graph

Allelic Graph
• Edges of graph define conditional probabilities
• E.g., if ACACTCG is covered by 30 reads, and C is covered by 1 read, P(C | ACACTCG) is low
• Can efficiently marginalize probabilities over graph using Eliminate algorithm1, exactly solve for argmax
ACACTCGC
ATCTCA
G
CTCCACACT
1. Jordan, “Probabilistic Graphical Models.”

Output• Current assemblers emit FASTA contigs
• In layperson’s speak: long strings
• We’ll emit “multigs”, which we’ll map back to reference graph
• Multig = multi-allelic (polymorphic) contig
• Working with UCSC, who’ve done some really neat work1 deriving formalisms & building software for mapping between sequence graphs, and GA4GH ref. variation team
1. Paten et al, “Mapping to a Reference Genome Structure”, arXiv 2014.

Acknowledgements• UC Berkeley: Matt Massie, André Schumacher,
Jey Kottalam, Christos Kozanitis, Adam Bloniarz!
• Mt. Sinai: Arun Ahuja, Neal Sidhwaney, Michael Linderman, Jeff Hammerbacher!
• GenomeBridge: Timothy Danford, Carl Yeksigian!
• Cloudera: Uri Laserson!
• Microsoft Research: Jeremy Elson, Ravi Pandya!
• And many other open source contributors: 23 contributors to ADAM/BDG from >10 institutions


















