
RReegguullaattiioonnss aanndd … · 1 RReegguullaattiioonnss aanndd RReeqquuiirreemmeennttss...
19
1 R R e e g g u u l l a a t t i i o o n n s s a a n n d d R R e e q q u u i i r r e e m m e e n n t t s s f f o o r r C C o o n n d d u u c c t t i i n n g g C C l l i i n n i i c c a a l l T T r r i i a a l l s s o o n n D D r r u u g g s s Version 1.1 Date issued 06/07/2015 Date of implementation 06/07/2015
-
Upload
hoangxuyen -
Category
Documents
-
view
217 -
download
0
Transcript of RReegguullaattiioonnss aanndd … · 1 RReegguullaattiioonnss aanndd RReeqquuiirreemmeennttss...

1
RRReeeggguuulllaaatttiiiooonnnsss aaannnddd
RRReeeqqquuuiiirrreeemmmeeennntttsss fffooorrr
CCCooonnnddduuuccctttiiinnnggg CCCllliiinnniiicccaaalll
TTTrrriiiaaalllsss ooonnn DDDrrruuugggsss
Version 1.1
Date issued 06/07/2015
Date of implementation 06/07/2015

2
Regulations and Requirements for Conducting Clinical Trials on Drugs
Version 1.1
Drug Sector
Saudi Food & Drug Authority
Please visit the SFDA’s website at http://www.sfda.gov.sa/En/Drug
for the latest updates.
For inquiries: [email protected]
For comments or suggestions: [email protected]

3
Drug Sector
Vision and Mission
Vision
To be the leading regional drug regulatory authority for pharmaceuticals and cosmetic products, with
professional excellence and services that contribute to the protection and advancement of public health
in the Kingdom of Saudi Arabia.
الرؤية
قلميياً الصحة ية ومس تحرضات التجميل، ويقدم خدماته مبهنية ممتزية تسهم يف حامية وتعزيزيف الرقابة عىل ال دو أ ن يكون قطاع ادلواء رائداً ا
يف اململكة العربية السعودية.
Mission
To protect public health by ensuring the safety, quality, efficacy, and accessibility of human and
veterinary drugs and biological products as well as the safety of cosmetics through administration of a
national regulatory system, which is consistent with international best practices. We also aim to provide
accurate and scientific-based information to both the public and health-care professionals.
الرساةل
حامية الصحة العامة من خالل ضامن أ مان وجودة وفعالية وتوفر ال دوية البرشية والبيطرية واملنتجات احليوية وسالمة مواد التجميل عرب
.املهنيني الصحينيو تطبيق نظام وطين للرقابة متوافق مع أ فضل املامرسات ادلولية وتقدمي املعلومات ادلوائية املبنية عىل أ سس علمية للعامة
4
Document Control
Version Date Author(s) Comments
1.0 06/07/2015 Clinical Trials Department Final
1.1 27/10/2016 Clinical Trials Department Update*
Note: For a list of the most recent updates, please refer to appendix.

5
Regulations and Requirements for Conducting Clinical Trials on Drugs
1. According to memo no. 1794, issued on 20/1/1432 H, all clinical trials involving drugs must
be registered with the SFDA Drug Sector’s Clinical Trials Department by the researcher, the
sponsor, or the Contract Research Organization (CRO) through the Saudi Clinical Trials
Registry (SCTR), knowing that the registration of a clinical trial does not indicate
approval. The registration processes should accord with the SCTR’s guidance, which is
available at https://sctr.sfda.gov.sa/Guidance.aspx.
2. The researcher, sponsor, and CRO must adhere to the regulations of Research Ethics Code on
Living Creatures issued by Royal decree no. M/59 on 14/9/1431 H.
3. It is mandatory to adhere to SFDA memos E/15481 and E/15482 (13/5/1434 H) in regard to
the registration of local institutional review boards (IRBs).
4. The researcher, sponsor, and CRO must adhere to good clinical practice (GCP) in accordance
with the ICH-E6 guideline.
5. According to the memo E/9699 (23/4/1432 H), the sponsor or CRO must pay 15,000 Saudi
Riyals for each submitted trial as an evaluation fee for the clinical trial, excluding phase IV
from such fees.
6. Phase IV Trials:
Clinical trials that are conducted on registered drugs at the SFDA to gather more data about
said drugs.
A. The researcher, sponsor, and CRO can start the clinical trial after obtaining local IRB
approval. They should notify the SFDA by registering the trial at the SCTR and sending
the requirements in Table 1 to the Clinical Trials Department ([email protected])
within 20 working days after obtaining local IRB approval.
B. When changing or adding a clinical trial site, the researcher, sponsor, or CRO should
notify the SFDA by sending the IRB approval form via e-mail to the Clinical Trials
Department.
C. It is mandatory to obtain SFDA approval before conducting clinical trials on registered
drugs), which are not phase IV trials, such as trials for
new indication or off-label use;
change in dosage regimen or route of administration; or
change in dosage form.
D. Concerning phase IV trials on unregistered direct purchased drugs, the researcher must
adhere to memo E/1811 (16/1/1436 H).

6
E. Concerning post-authorization safety studies (PASS), the researcher, sponsor, or CRO
should adhere to the Guideline on Good Pharmacovigilance Practices (GVP) which
published on SFDA’s website (Guideline’s page) .
7. Phase II and III Trials:
A. It is mandatory to obtain SFDA approval before conducting phase II and III clinical
trials, in accordance with the requirements in Table 1.
B. The researcher, sponsor, or CRO must annually submit a progress report on the ongoing
trials by completing the Annual Progress Report (Form No. 1).
C. Clinical Trials Amendments:
In case of non-substantial amendments, the SFDA should be notified via the
annual progress report.
In case of substantial amendments, it is mandatory to obtain SFDA approval
before implementing any amendments, in accordance with the requirements in
Table 2.
The SFDA allows urgent safety measures to be taken, without prior approval,
to protect trial subjects from immediate hazards. However, the SFDA must be
notified about such measures as soon as possible.
If the amendment contains changes to or the addition of a clinical trial site, the
researcher, sponsor, or CRO must adhere to the requirements in Table 2.
8. Importing Drugs/Study Materials Related to Clinical Trials:
It is mandatory to obtain an importation license for investigational drugs or study materials
from the SFDA Drug Sector’s Importation Department in accordance with “Regulations and
Requirements for Importing and Clearance of Medications and Medical Supplies for Clinical
Trials,” which can be found on the SFDA website.
9. Exporting Clinical Trial Bio-samples:
A. The researcher, sponsor, and CRO must adhere to the regulations of the Research Ethics
Code on Living Creatures, issued by Royal decree no. M/59 on 14/9/1431 H, which
regulates bio-sample exportation.
B. The researcher, sponsor, or CRO must provide the SFDA with a copy of the local IRB
exportation permission.
10. Clinical Trials Adverse Drug Reactions Reporting:
A. It is mandatory to inform the SFDA immediately about any suspected unexpected
serious adverse reactions (SUSAR) on “Form No. 2” as soon as possible, no later than
15 days followed by the follow-up report as soon as possible. If the SUSAR is fatal or
life threating, SFDA must be informed as soon as possible, no later than seven days in

7
accordance with the ICH-E2A guideline, with a follow-up report succeeding it within
15 days.
B. It is mandatory to inform the SFDA of any SUSAR that occurs internationally to an
investigational drug involved in ongoing clinical trials in Saudi Arabia as soon as
possible within the same time limits specified above.
C. SUSARs should be reported through the National Pharmacovigilance Center via e-mail
([email protected]). The e-mail subject must be “SUSAR Case.”
D. The sponsor or CRO must send SUSARs in XML format in addition to completing the
CIOMS-1 Form (Form No. 2). Researchers, however, may be exempted from reporting
in XML format.
E. The researcher, sponsor, or CRO must annually send a development safety update
report (DSUR) to the Clinical Trials Department in accordance with the ICH-E2F
guideline.
11. Completion, Termination, or Suspension of Clinical Trials:
The researcher, sponsor, or CRO must inform the SFDA within 60 days with the need to
attach proof of IRB notification. In addition, it is mandatory to submit the final clinical trial
report within one year of the end of the trial in accordance with ICH-E3 guidelines.
12. The Qualifications of the Clinical Trial Research Team:
To ensure the safety of clinical trial subjects, the research team must provide proof of
adequate training in GCP. It is mandatory that the latest training occurred within the last
two years.
13. The period (timeline) needed to respond to the researcher, sponsor, or CRO requests after
completing all the required documents are, at maximum,
10 working days for phase IV trials;
30 working days for phase III trials; and
60 working days for phase II trials.

8
Table 1: Clinical Trial Requirements
Documents Phase II / III Phase IV 1. Arabic-Headed Letter to SFDA Executive Vice President for
Drug Affairs, Including SCTR Registration No. √
2. IRB Approval √ √
3. Informed Consent Form (Arabic and English) √ √
4. Trial Protocol √ √
5. Investigator Brochure √
6. Case Report Form √
7. Labeling of the Study Drug √
8. Clinical Trial Agreement √
9. Financial Disclosure of Principal Investigator (Form No. 3) √
10. Confidentiality Agreement √
11. Certificate of Analysis for the Study Drug √
12. GMP Certificate √
13. Subject’s Insurance √
14. Delegation/Authorization Letter for CRO (if applicable) √ √
15. CVs of Principal Investigator & Coordinator. √
16. Principal Investigator GCP Certificate √
17. Statement of Investigator (Form No. 4) √ √
18. Documents must be submitted as hard and soft copies √
9
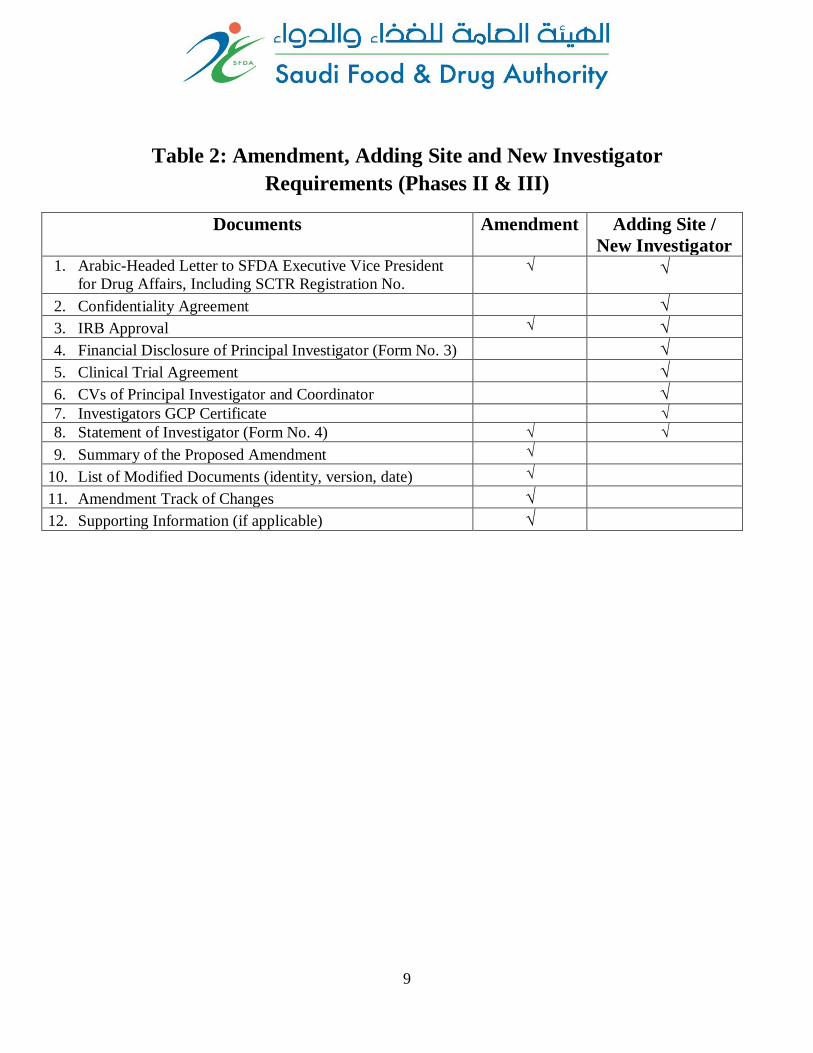
Table 2: Amendment, Adding Site and New Investigator
Requirements (Phases II & III)
Documents Amendment Adding Site /
New Investigator 1. Arabic-Headed Letter to SFDA Executive Vice President
for Drug Affairs, Including SCTR Registration No.
√ √
2. Confidentiality Agreement √
3. IRB Approval √ √
4. Financial Disclosure of Principal Investigator (Form No. 3) √
5. Clinical Trial Agreement √
6. CVs of Principal Investigator and Coordinator √ 7. Investigators GCP Certificate √ 8. Statement of Investigator (Form No. 4) √ √
9. Summary of the Proposed Amendment √
10. List of Modified Documents (identity, version, date) √
11. Amendment Track of Changes √
12. Supporting Information (if applicable) √

10
(Form No. 1)
ANNUAL PROGRESS REPORT TO SFDA
(This report should be completed by authorized personnel.) A soft copy of this form can be found in the drug sector portal under Forms Section
1. Sponsor Details
Name of sponsor/CRO:
Address:
City:
Contact person:
Contact number:
2. Study Details
Study title:
Protocol number:
Current study status: Completed Terminated Ongoing Other (please
specify):
SCTR number (if applicable) :
3. Start and Completion Dates
Did the study begin in Saudi Arabia?
Yes / No
If yes, what was the actual start date in Saudi Arabia?
If no, what are the reasons for not beginning the study in
Saudi Arabia?
What is the expected start date?

11
Has the study concluded?
Yes / No
If no, what is the expected completion date?
If you do not expect the study to be completed, give
reason(s)
4. Investigational Site Information
4.1
Total number of participants globally (if applicable):
Total number of participants in Saudi Arabia:
Number of sites proposed in original application:
Number of sites recruited to date:
Do you plan to increase the total number of sites proposed
for the study?
Yes / No
4.2
Name of site:
Name of principal investigator:
Number of participants on this site:
Number of withdrawals, to date, from trial due to:

12
(a) withdrawal of consent: ______
(b) loss to follow-up: ______
(c) death (where not the primary outcome): ______
Total study withdrawals: ______
Number of treatment failures to date (prior to reaching primary outcome) due to:
(a) adverse events: ______
(b) lack of efficacy: ______
Total treatment failures: ______
*(For 4.2, fill out each site of the study separately)
4.3
Have there been any serious difficulties in recruiting
participants?
Yes / No
If yes, provide details:
Do you plan to increase the planned recruitment of study
participants?
Yes / No

13
5. Safety Reports
Have there been any suspected unexpected serious adverse
reactions (SUSARs) in this trial in Saudi Arabia?
Yes / No
Have these SUSARs been reported to the SFDA within 7
or 15 days in accordance with the SFDA’s Regulations
and Requirements for Conducting Clinical Drug Trials?
If no, please arrange urgently and provide give reasons for
late notification.
Yes / No
Has a DSUR been submitted?
Yes / No / Not yet due
When is the next DSUR due?
6. Amendments
Have any substantial amendments been made to the trial?
Yes / No
If yes, please give the date and amendment number for
each substantial amendment made.
7. Serious Deviations in Protocol or Good Clinical Practice
Have any serious protocol or GCP deviations occurred in
relation to this trial? Yes / No
If yes, please give the date of each notification to the
SFDA.

14
Please provide the IRB/EC with a copy of each
notification of information (unless previously done).
8. Other Issues
Are there any other developments in the trial that you wish
to report to the SFDA?
Yes / No
Are there any ethical issues regarding which further advice
is required?
If yes to either, please attach a separate statement with
details.
Yes / No
9. Declaration
Name and title of authorized person:
Signature:
Date of submission:

15
(Form No. 2)
CIOMS FORM (SUSAR REPORT) A soft copy of this form can be found in the drug sector portal under Forms Section
SUSPECT ADVERSE REACTION REPORT
I. REACTION INFORMATION
1. PATIENT INITIALS (first, last)
1a. COUNTRY
2. DATE OF BIRTH 2a. AGE Years
3. SEX 4–6 REACTION ONSET 8–12 CHECK ALL APPROPRIATE TO ADVERSE REACTION
Day
Month
Year
Day
Month
Year
7 + 13 DESCRIBE REACTION(S) (including relevant tests/lab data)
PATIENT DEATH
INVOLVED OR
PROLONGED
INPATIENT HOSPITALIATION
INVOLVED
PERSISTENT OR SIGNIFICANT DISABILITY OR INCAPACITY
THREAT TO LIFE
II. SUSPECT DRUG(S) INFORMATION
III. CONCOMITANT DRUG(S) AND HISTORY
22. CONCOMITANT DRUG(S) AND DATES OF ADMINISTRATION (exclude drugs used to treat reaction)
23. OTHER RELEVANT HISTORY (e.g., diagnostics, allergies, or pregnancy with last month of period, etc.)
IV. MANUFACTURER INFORMATION
14. SUSPECT DRUG(S) (include generic name)
20. DID REACTION ABATE AFTER STOPPING DRUG?
YES NO NA
15. DAILY DOSE(S)
16. ROUTE(S) OF ADMINISTRATION
21. DID REACTION
REAPPEAR AFTER REINTRO- DUCTION?
17. INDICATION(S) FOR USE
YES NO NA
18. THERAPY DATES (from/to)
19. THERAPY DURATION
24a. NAME AND ADDRESS OF MANUFACTURER
24b. MFR CONTROL NO.
24c. DATE RECEIVED
BY MANUFACTURER
24d. REPORT SOURCE
STUDY LITERATURE HEALTH PROFESSIONAL
DATE OF THIS REPORT 25a. REPORT TYPE INITIAL FOLLOW-UP

16
(Form No. 3) A soft copy of this form can be found in the drug sector portal under Forms Section
Disclosure: Clinical Investigators’ Financial Interests and
Arrangements Form
To Be Completed by Applicant
Study title:
Protocol number:
Study sponsor:
Investigator/sub investigator name:
Study site:
Please indicate by marking YES or NO below whether any of the financial interests or arrangements applies to you, your spouse, dependent children, or any combination thereof.
YES / NO
Are you, your spouse, or any dependent children employed by the study sponsor?
Any financial arrangement entered into between the covered study’s sponsor and the clinical
investigator involved in the covered study’s convict, whereby the study’s outcome might influence
the value of clinical investigator’s compensation for conducting the study.
Any significant payments of other sorts of compensation made by the covered study’s sponsor, such
as a grant to fund ongoing research, compensation in the form of equipment, a retainer for ongoing
consultation, or honoraria.
Any proprietary interest the clinical investigator, his spouse, or any of his dependent children has in
the product tested during the covered study.
Any significant equity interest the clinical investigator, his spouse, or any of his dependent children
has in the covered study’s sponsor.
For each YES response above, please provide detailed information disclosing the nature of the financial arrangement, including total value amount:
ــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــ
ــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــ
ــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــــ
By signing this form, I confirm that all information provided is, to the best of my knowledge and belief, true,
correct, and complete. Furthermore, I will notify SFDA with any updates or changes to the information
provided on this form during the course of the study.
Name:
Signature: Date:

17
(Form No. 4) A soft copy of this form can be found in the drug sector portal under Forms Section
STATEMENT OF INVESTIGATOR
NAME AND ADDRESS OF INVESTIGATOR
Name of principal investigator
Address Saudi Commission for Health Specialties no.
City Qualified area(s) of specialty Telephone no. E-mail
EDUCATION, TRAINING, AND EXPERIENCE THAT QUALIFY THE INVESTIGATOR AS AN EXPERT IN THE CLINICAL INVESTIGATION OF THE DRUG FOR THE USE UNDER INVESTIGATION. ONE OF THE FOLLOWING IS PROVIDED (select one):
Curriculum vitae Other statement of qualifications
Does the investigator have GCP certification?
Yes No If yes, attach your certification.
NAME OF TRIAL SITE
Name of hospital or other research facility
Address City
Telephone no.
NAME AND ADDRESS OF ANY CLINICAL LABORATORY FACILITIES TO BE USED IN THE STUDY (in case of central lab)
Name of clinical laboratory facility
Address
City Province/region Country Postal code
NAME AND ADDRESS OF THE INSTITUTIONAL REVIEW BOARD (IRB) THAT IS RESPONSIBLE FOR REVIEW AND APPROVAL OF THE STUDY/STUDIES
Name of IRB
Address Registration no. at NCBE

18
Details of Study
Study title Protocol no.
Version no. SCTR no.
COMMITMENTS
I agree to conduct the study or studies in accordance with the relevant, current protocol(s) and will make
changes in a protocol only after notifying the sponsor, except when necessary to protect the safety, rights, or
welfare of subjects.
I agree to personally conduct or supervise the described investigation(s).
I agree to inform any patients or any persons used as controls that the drugs are being used for
investigational purposes, and I will ensure that the requirements related to obtaining informed consent and
institutional review board (IRB) review and SFDA regulations are met.
I agree to report to the sponsor adverse experiences that occur in the course of the investigation(s) in
accordance with regulatory requirements. I have read and understand the information in the investigator’s
brochure, including the drug’s potential risks and side effects.
I agree to ensure that all associates, colleagues, and employees assisting in the conduct of the study or
studies are informed about their obligations in meeting the above commitments.
I agree to maintain adequate and accurate records in accordance with GCP E6 and to make those records
available for inspection in accordance with GCP E6.
I will ensure that an IRB that complies with the requirements of the National Committee of Bioethics (NCBE)
is responsible for the initial and continuing review and approval of the clinical investigation. I also agree to
promptly report to the IRB all changes in the research activity and all unanticipated problems involving risks
to human subjects or others. Additionally, I will not make any changes in the research without IRB approval
except where necessary to eliminate apparent and immediate hazards to human subjects.
I agree to comply with all other requirements regarding clinical investigators’ obligations and all other pertinent requirements in the Regulations and Requirements for Conducting Clinical Drug Trials.
NOTE:
INVESTIGATORS SHOULD NOT SEND THIS FORM DIRECTLY TO THE SFDA.
DATE (mm/dd/yyyy) SIGNATURE OF INVESTIGATOR
NAMES OF SUBINVESTIGATORS (if not applicable, enter “none”)

19
ppendixA
What’s New in Regulations and Requirements for Conducting Clinical Trials on
Drugs (Version 1.1)?
The following table shows changes that were added to update version 1.0:
Section Type of Update
6. Phase IV Trials
Statement added:
E. Concerning post-authorization safety studies (PASS),
the researcher, sponsor, or CRO should adhere to the
Guideline on Good Pharmacovigilance Practices (GVP).
7. Phase II and III Trials
Statement updated to:
B. The researcher, sponsor, or CRO must annually submit
a progress report on the ongoing trials by completing the
Annual Progress Report (Form No. 1).
10. Clinical Trials Adverse Drug Reactions
Reporting
Statement updated to:
A. It is mandatory to inform SFDA immediately about
any Suspected Unexpected Serious Adverse Reactions
(SUSAR) “Form No. 2” as soon as possible, no later than
15 days followed by the follow up report as soon as
possible. If the SUSAR is fatal or life threating, SFDA
must be informed as soon as possible, no later than seven
days in accordance with (ICH-E2A) guideline followed
by the follow up report no later than 15 days.
Table 1: Clinical Trial Requirements
Table 2: Amendment/Site-Adding
Requirements (Phases II and III)
Table updated.
Annual Progress Report to SFDA (Form No.
1)
Disclosure: Clinical Investigators’ Financial
Interests and Arrangements Form (Form No.
3)
Statement of Investigator (Form Mo. 4)
Forms added.


















