
Review Scanning tunneling microscopy applications in...
13
www.elsevier.nl/locate/jelechem Journal of Electroanalytical Chemistry 492 (2000) 81 – 93 Review Scanning tunneling microscopy applications in electrochemistry — beyond imaging N.J. Tao *, C.Z. Li, H.X. He Department of Physics, Florida International Uni6ersity, Miami, FL 33199, USA Received 15 May 2000; received in revised form 27 July 2000; accepted 28 July 2000 Abstract Scanning tunneling microscopy (STM) has gradually matured into a powerful tool for imaging electrode surfaces in the electrochemical environment with atomic resolution. It has been used to elucidate numerous old puzzling structural issues and to reveal many new interesting phenomena. As an imaging tool, it will continue to contribute to the understanding of various electrochemical processes on electrode surfaces. STM is more than an imaging tool for structural characterizations, other important electrochemical applications, such as probing electron transfer processes, fabricating nanostructures and studying fast electrochemical kinetics, have also been actively pursued. These later unconventional applications are the focus of this discussion. © 2000 Elsevier Science B.V. All rights reserved. Keywords: Scanning tunnel microscopy; Probing electron transfer; Fabricating nanostructures 1. Introduction Since the pioneering applications of scanning tunnel- ing microscopy (STM) in an aqueous environment nearly 15 years ago [1,2], the technique has become a powerful tool for studying various electrochemical phe- nomena with atomic or molecular resolution in real space. Important applications include atomic or molec- ular resolution studies of well-defined electrode surfaces [3 – 7], electrochemical deposition and dissolution [8 – 11] and molecular adsorption [12 – 16]. More complex sys- tems and phenomena, such as biological molecules [17–20] and electrochemical reactions of organic molecules [21,22] have also been studied with molecular resolution. While the progress in the last 15 years has been impressive, the full potential of STM applications in electrochemistry has yet to be reached, and this de- pends critically on several developments. One of these developments is better instrumentation. Today’s STM designs and costs are not that different from those of 10 – 15 years ago (excluding the computers). High qual- ity atomic-resolution images still need frequent ‘mas- sages’, which often challenges the patience of new users. Revolutionary designs of user-friendlier STM are highly desired and should also be possible given the rapid development in micromachining capability and elec- tronics. Another critical development is a more com- plete understanding of the STM imaging mechanism in the electrochemical environment. In principle, the tun- neling current measured by STM contains information about electron transfer through water layers, which is extremely important for nearly all electrochemical pro- cesses. It also contains valuable information about the electronic states of adsorbed molecules, which could be used not only to identify the molecules, but also to study the reactivity of the molecules. Extracting the information requires a good understanding of the STM tunneling/imaging mechanism, which is still incomplete. Increasing theoretical and experimental efforts have been devoted to understanding the STM tunneling/ imaging mechanism, and to extract molecular identity and reactivity information, which are discussed in this paper. Efforts to improve STM temporal resolution for studying fast electrochemical kinetics are also briefly described here. Another unconventional electrochemi- * Corresponding author. Tel.: +1-305-3483954; fax: +1-305- 3486700. E-mail address: taon@fiu.edu (N.J. Tao). 0022-0728/00/$ - see front matter © 2000 Elsevier Science B.V. All rights reserved. PII:S0022-0728(00)00295-3
Transcript of Review Scanning tunneling microscopy applications in...

www.elsevier.nl/locate/jelechem
Journal of Electroanalytical Chemistry 492 (2000) 81–93
Review
Scanning tunneling microscopy applications inelectrochemistry — beyond imaging
N.J. Tao *, C.Z. Li, H.X. HeDepartment of Physics, Florida International Uni6ersity, Miami, FL 33199, USA
Received 15 May 2000; received in revised form 27 July 2000; accepted 28 July 2000
Abstract
Scanning tunneling microscopy (STM) has gradually matured into a powerful tool for imaging electrode surfaces in theelectrochemical environment with atomic resolution. It has been used to elucidate numerous old puzzling structural issues and toreveal many new interesting phenomena. As an imaging tool, it will continue to contribute to the understanding of variouselectrochemical processes on electrode surfaces. STM is more than an imaging tool for structural characterizations, otherimportant electrochemical applications, such as probing electron transfer processes, fabricating nanostructures and studying fastelectrochemical kinetics, have also been actively pursued. These later unconventional applications are the focus of this discussion.© 2000 Elsevier Science B.V. All rights reserved.
Keywords: Scanning tunnel microscopy; Probing electron transfer; Fabricating nanostructures
1. Introduction
Since the pioneering applications of scanning tunnel-ing microscopy (STM) in an aqueous environmentnearly 15 years ago [1,2], the technique has become apowerful tool for studying various electrochemical phe-nomena with atomic or molecular resolution in realspace. Important applications include atomic or molec-ular resolution studies of well-defined electrode surfaces[3–7], electrochemical deposition and dissolution [8–11]and molecular adsorption [12–16]. More complex sys-tems and phenomena, such as biological molecules[17–20] and electrochemical reactions of organicmolecules [21,22] have also been studied with molecularresolution.
While the progress in the last 15 years has beenimpressive, the full potential of STM applications inelectrochemistry has yet to be reached, and this de-pends critically on several developments. One of thesedevelopments is better instrumentation. Today’s STMdesigns and costs are not that different from those of
10–15 years ago (excluding the computers). High qual-ity atomic-resolution images still need frequent ‘mas-sages’, which often challenges the patience of new users.Revolutionary designs of user-friendlier STM are highlydesired and should also be possible given the rapiddevelopment in micromachining capability and elec-tronics. Another critical development is a more com-plete understanding of the STM imaging mechanism inthe electrochemical environment. In principle, the tun-neling current measured by STM contains informationabout electron transfer through water layers, which isextremely important for nearly all electrochemical pro-cesses. It also contains valuable information about theelectronic states of adsorbed molecules, which could beused not only to identify the molecules, but also tostudy the reactivity of the molecules. Extracting theinformation requires a good understanding of the STMtunneling/imaging mechanism, which is still incomplete.
Increasing theoretical and experimental efforts havebeen devoted to understanding the STM tunneling/imaging mechanism, and to extract molecular identityand reactivity information, which are discussed in thispaper. Efforts to improve STM temporal resolution forstudying fast electrochemical kinetics are also brieflydescribed here. Another unconventional electrochemi-
* Corresponding author. Tel.: +1-305-3483954; fax: +1-305-3486700.
E-mail address: [email protected] (N.J. Tao).
0022-0728/00/$ - see front matter © 2000 Elsevier Science B.V. All rights reserved.PII: S 0022 -0728 (00 )00295 -3

N.J. Tao et al. / Journal of Electroanalytical Chemistry 492 (2000) 81–9382
cal application is to use STM as a nanofabrication tool,which is discussed at the end of the paper. As this paperis focused on unconventional applications of STM inelectrochemistry, readers who are interested in struc-tural studies or images may wish to refer to recentreviews [23–29]. Other closely related and equally im-portant techniques, such as atomic force microscopy(AFM) [30] and scanning electrochemical microscopy(SECM) [31], are also excluded here. We note that thisarticle is not intended to be an exhaustive review; itspurpose is to highlight some of the novel unconven-tional applications of STM in electrochemistry anddiscuss new challenges that face the applications.
2. Electron tunneling mechanism in electrochemicalSTM
In vacuum, a simple one-dimensional square-wellbarrier has been used to describe electron tunnelingbetween the STM tip and the substrate (Fig. 1). Thesimple model predicts an exponential decay in thetunneling current, I, as the substrate–tip distance, s,increases, by
I8exp(−1.025fs) (1)
where f is the work function or the tunneling barrierheight in electron volts and s is in angstrom. Theexponential decay was verified experimentally over awide range of tunneling currents despite the fact thatthe simple model ignores factors, such as the imagingforce experienced by the tunneling electrons. The de-pendence of the logarithm of the tunneling current withrespect to distance is a measure of the tunneling barrierheight [32], according to
f='2
8m�d ln I
ds�2
(2)
The tunneling barrier height is typically measured byvarying the tip–substrate distance through the z-piezo,often with an ac modulation. Because the simple modeldoes not include the imaging force, the square barrierbecomes ill defined when the tip–substrate distance isvery small.
For electrochemical STM, tunneling through waterlayers has to be taken into account. The tunnelingbarrier in aqueous solutions has been measured by anumber of groups [33–39] and found to be close to orsomewhat smaller than the vacuum value. Lindsay andco-workers [37,38] carried out a systematic study of thetunneling barrier in STM as a function of bias voltageand electrochemical potential in the presence and ab-sence of organic adsorbates. They found a strikingnon-exponential dependence of the tunneling resistanceon the tip–substrate distance (Fig. 2). In vacuum tun-neling, non-exponential behavior occurs only at an
extremely small tip–substrate distance (a fewangstroms) where the tunneling barrier vanishes. Thenon-exponential behavior observed by Lindsay and co-workers, occurs at a much greater distance and isrelated to the existence of a water layer between the tipand substrate electrodes [38]. For example, if the struc-ture of the water layers changes as the gap changes dueto either a variation in the influence of the structure bythe electrodes or the intrinsic size-dependent structureof the water clusters, then a varying tunneling willoccur, which can lead to the non-exponential behavior.By extrapolating the resistance to the value of thequantum-point contact, they estimated the tunnellinggap between the tip and substrate at 109 V (e.g. 0.1 nAset point at 0.1 V of bias voltage) to be ca. 20 A, , which
Fig. 1. One–dimensional substrate–vacuum–STM tip tunneling junc-tion.
Fig. 2. Log (base 10) of the tunneling resistance plotted against thedistance the tip moved from the position at which R=109 V.Increasing negative displacement is towards the surface. From Ref.[38] with permission.

N.J. Tao et al. / Journal of Electroanalytical Chemistry 492 (2000) 81–93 83
is considerably larger than the theoretical gap ofSchmickler [40]. With such a large gap, it is quitesurprising that atomic resolution images can still beobtained under the imaging conditions (tunneling resis-tance ca. 109 V).
Another striking observation of electron tunnelingthrough water layers is the dependence of the tunnelingcurrent on the polarity of the bias voltage. Hong et al.[39] reported that the tunneling barrier for the electrontunneling from tip to substrate was remarkably differ-ent from the reverse direction, and the asymmetrybecame more pronounced when the tunneling gap wasdecreased below 4 A, . Such large asymmetry was at-tributed to the change in the orientation of watermolecules on the electrodes upon change of the biaspolarity. An X-ray study by Toney et al. has shown thereorientation of water molecules as the electrode poten-tial changes [41]. The change in the water orientationcan lead to a huge asymmetric tunneling according to anumerical simulation by Benjamin et al. [42]. However,a change in water orientation alone is not adequate toexplain the asymmetry. Because STM involves twoelectrodes (tip and substrate), if the water molecules onthe two electrodes behave in the same way, the tunnel-ing should be always symmetric. So the asymmetrymust also be related to the sharp geometry of the STMtip near which water molecules may arrange into adifferent structure.
Both the non-exponential decay and asymmetric tun-neling demand a better theoretical description of elec-tron tunneling in the electrochemical STM. Schmicklerand Henderson presented the first model of electrontunneling through water layers in the STM junction[43]. They treated the water molecules in the tunnelinggap as a dielectric medium and predicted a smallertunneling barrier than in vacuum. Vigorovich et al.have considered the effects of solvent fluctuations onthe tunneling barrier height and tunneling rate [44].They found that the solvent fluctuations could lead toan enhancement in the tunneling rate, and therefore aneffective lower barrier height, which provides an alter-native explanation of the observed barrier height inelectrochemical STM.
More elaborate models involve electronic states ofthe water molecules. Sass and Gimzewski proposedelectron tunneling through water via two intermediatestates: a delocalized dry state and a localized wet state[45]. While tunneling via the dry state is the same as themodel by Schmickler and Henderson, tunneling via thewet state is a different mechanism. Repphun andHalbritter considered resonant tunneling via dipole-in-duced states at the Fermi energy [46]. Lindsay andco-workers proposed tunneling via virtual resonanceswith localized states on the water molecules [37].Schmickler carried out the first ab initio calculation inwhich jellium slabs were used for the metal electrodes
and molecular dynamics for water molecules with aone-electron potential [40]. These studies demonstratedthe importance of treating the water structure explicitlyin three dimensions. What is still missing in the abinitio simulations is the ability to replace the planargeometry for the tip electrode with a more realisticgeometry, which seems to be necessary to explain STMimages and the asymmetric tunneling barrier.
In the presence of molecular adsorbates, the STMimaging mechanism is even less well understood. Manymodels have been proposed for adsorbates in vacuum[47–53] and in aqueous solutions [54–61]. Kuznetsovand coworkers pointed out that a strong couplingbetween the electronic states of a molecule and theenvironmental fluctuations could affect the tunnelingcurrent by lowering the effective tunneling barrier andby momentarily bringing a molecular state to align withthe substrate/tip Fermi levels [54,55]. The lowering inthe effective barrier should lead to an enhancement inthe tunneling current through molecular adsorbates.The momentary alignment of the molecular state withthe substrate/tip Fermi levels could result in a two-stepsequential tunneling in which electrons tunnel from thetip (substrate) to the molecule then to the substrateelectrode (tip) after the molecule is fully relaxed. Inaddition to the adsorbate–solvent coupling, Schmicklerand Widrig took into account the adsorbate–substratecoupling [57]. They considered explicitly a resonanttunneling process when the molecular state was broughtto align with the substrate/tip Fermi levels by thecouplings.
Lindsay and co-workers considered that a tip forceapplied on the imaged molecules could significantlyshift the molecular states thus inducing resonant tun-neling [48]. Wang et al. extracted the interaction of theSTM tip with a guanine monolayer on graphite bydetermining the tip-induced deformation in the organicmonolayer [61]. They found that even at the smallesttunneling current, 3 pA, the force was still in the orderof a few nanonewtons per molecule, which confirmedthe importance of tip–sample interaction in STM asproposed by Lindsay and co-workers
3. Comparative STM and AFM
Given the importance of tip–sample interaction inthe STM imaging mechanism, it is desirable to measurelocal tip–sample interactions in STM. Using a conduc-tive AFM tip, the tunneling current and tip–sampleinteraction have been simultaneously measured in airfor organic adsorbates [62,63]. A recent example is themeasurement of conductance of single thiolatedcarotene molecules attached to a gold electrode byLindsay’s group [64]. The thiolated carotene moleculeswere embedded in insulating n-alkanethiol self-assem-

N.J. Tao et al. / Journal of Electroanalytical Chemistry 492 (2000) 81–9384
bled monolayers. They found that at a contact force ofa few nanonewtons, a carotenoid molecule behavedohmically with a resistance of approximately 4.290.7G V, which is over a million times more conductivethan an alkane chain of similar length. Unfortunately,the approach cannot be easily extended to measuringthe tip–sample interactions in the electrochemical envi-ronment because of the difficulty in coating the AFMtip, which is necessary to reduce the ionic conductioncurrent. This technical difficulty is likely to be over-come in the near future.
An indirect method to obtain the force and tunnelinginformation is to image a sample with STM and AFMseparately. One example is 2,2%-bipyridine on Au(111)as shown in Fig. 3. The molecules stand vertically whenadsorbed on the Au surface and stack into polymer-likechains via p–p interaction [65]. The AFM image (Fig.3A) shows that the polymer-like chains line up inparallel (along the white line in the figure). Along theparallel chains, each molecule is revealed as two elon-gated blobs that are tilted away from the chain direc-tion. The two blobs correspond to the positions of thetwo rings of 2,2%-bipyridine. The STM image (Fig. 3B)shows similar parallel chains, but each molecule isrevealed as two round blobs that are much closer toeach other than the elongated blobs in the AFM image.The distance between the two round blobs in the STMimage is ca. 0.3 nm which matches the distance betweenthe two nitrogen atoms in 2,2%-bipyridine and is muchsmaller than the separation between the two rings ob-served in the AFM image. This observation confirmsthat the nitrogen atoms bind directly to the substrateand provide electronic coupling between the molecularand the substrate states that is probed by STM. Thisexample demonstrates that STM may provide chemicalbonding information between adsorbed molecules andthe underlying electrode surface.
Fig. 4 compares the STM and AFM images ofxanthine on graphite. The molecule is known to lie flaton the surface and form a hydrogen bonded network[21]. In the AFM image (Fig. 4A), each xanthinemolecule appears as a bright blob. In sharp contrast,the STM image (Fig. 4B) reveals much more complexblob-like features. Since the dimensions of the blobs inthe STM image are much smaller than the dimensionsof a xanthine molecule, these blobs must be due to theinternal structure of the molecule. However, the blobsdo not simply coincide with the positions of the individ-ual atoms in the molecule, instead they reflect thespatial distribution of the electronic density of stateswithin the molecule.
4. Molecular identification and reactivity
Because STM probes the electronic properties of the
Fig. 3. AFM (A) and STM (B) images of 2,2%-bipyridine monolayeron Au(111) in 0.1 NaClO4. The molecules stand vertically on thesurface (C) with the two N atoms facing the Au(111), and thevertically standing molecules stack into a chain-like structure onsurface.
Fig. 4. Comparison of AFM (A) and STM (B) for xanthine monolay-ers grown on graphite basal plane from 0.1 M NaCl.
molecules, it has the potential to identify structurallysimilar molecules based on the difference in the elec-tronic states of the molecules. Knowing the electronicstates will also allow one to determine the reactivity ofthe molecules. Precisely extracting molecular electronicinformation ultimately depends on the understandingof STM imaging mechanism, which, as discussed above,is still a subject of ongoing research. Nevertheless, evenan incomplete understanding of the STM imagingmechanism is useful for us to obtain important elec-tronic information in some cases. For example,Schmickler and Widrig [57] suggested that the LUMO(lowest unoccupied molecular orbital) and HOMO(highest occupied molecular orbital) of molecules ad-sorbed on an electrode could be probed by STM. TheLUMO (HOMO) is broadened and shifted due to thecoupling of the molecule to the environment (solventand electrodes), which is described as the density ofoxidized (reduced) states. In principle, the density ofstates can be determined by performing scanning tun-

N.J. Tao et al. / Journal of Electroanalytical Chemistry 492 (2000) 81–93 85
neling spectroscopy (STS) in which the tunneling cur-rent, I, is measured as a function of the tip–substratebias voltage, Vbias. A more useful quantity in STS is(dI/dV)(V/I) because it is directly related to the densityof electronic states.
In vacuum, STS is usually obtained by measuring thetunneling current with a lock-in amplifier as a smallmodulation which is superimposed on a linearly sweep-ing bias voltage. The method is, however, difficult toapply to STM in the electrochemical environment be-cause the modulation and sweeping of the bias voltageinduce a large polarization current that can overwhelmthe tunneling current. Furthermore, the faradic currentat large bias voltage limits the spectroscopy to a rela-tively small range of bias voltage. Despite thedifficulties, useful electronic information aboutmolecules adsorbed onto electrodes has been obtainedby performing tunneling spectroscopy outside of elec-trochemical cells or by indirect methods.
An early attempt to extract electron density of statesof redox molecules from tunneling current was madeby Morisaki et al. [66]. They used thin SiO2 tunnelbarriers on Pt silicide substrates for tunneling spectro-scopy of complex ions, such as Fe(CN)6
3−/4− andFe(EDTA)1−/2−. The energy dependent electronic den-sities of states extracted from the tunneling spectra werein good agreement with the classical Marcus–Gerischermodel [67]. The experiment was macroscopic because itmeasured a large number of molecules. In contrast,STM based tunneling spectroscopy has the capability ofprobing the electronic states of a single molecule oreven local structure within a molecule.
Snyder and White measured electron transferthrough Fe-protoporphyrin IX multilayer films ad-sorbed on a graphite electrode using STM spectroscopyin air [68]. They observed an enhancement that wasidentical for both signs of the tip–substrate biasvoltage. The symmetric current versus bias voltagecurves were explained in terms of discrete sequentialelectron transfer steps through the films between theSTM tip and substrate. They, as well as Schmickler[57], pointed out, that STM could be used to probe fastelectron transfer reactions.
Tao studied the redox active molecules, Fe-protopor-phyrins IX, coadsorbed with redox in-active protopor-phyrins IX on a graphite substrate in aqueous solution[69]. By adjusting the substrate electrochemical poten-tial, the Fermi levels of the substrate and tip wereshifted relative to the energy levels of Fe–protopor-phyrins IX. When the Fermi levels were aligned to amolecular energy level of Fe-protoporphyrins IX, alarge increase in the tunneling current occurred (Fig. 5).This observation was explained tentatively as the reso-nant tunneling via the molecular state [69,70]. Thetunneling current peaks near the formal potential, E0,while the oxidized state should be located at E0+l
where l is the reorganization energy. This may be dueto the fact that l is small for a molecule sitting betweenthe substrate and STM tip. An alternative explanationis a two-step sequential tunneling process that leads toa maximum tunneling current at E0 [55]. The study
Fig. 5. STM image of Fe–protoporphyrin IX molecule embedded inan ordered array of protoporphyrin IX when the substrate was heldat −0.15 (A), −0.30 (B), −0.42 (C), −0.55 (D) and −0.65 Vversus Ag � AgCl (E), respectively. From Ref. [69] with permission.
N.J. Tao et al. / Journal of Electroanalytical Chemistry 492 (2000) 81–9386
Fig. 6. Derivatives (dI/dV) of the tunneling current-voltage curves ofFe, Mn, Zn and H embedded in tetraphenol porphyrin (TPP). FromRef. [71] with permission.
bias-dependent features were not observed in the lesselectroactive molecules, so the STM was shown to becapable of distinguishing electroactive molecules fromnon-electroactive molecules. The I–V curves are asym-metric which are in contrast to the work of Snyder andWhite [68]. Because the porphyrin molecules are mobilein the multilayer films that adsorbed on both the tipand substrate electrodes, electron transfer on both elec-trodes can occur and results in the symmetric I–Vcurves, observed by Snyder and White [68].
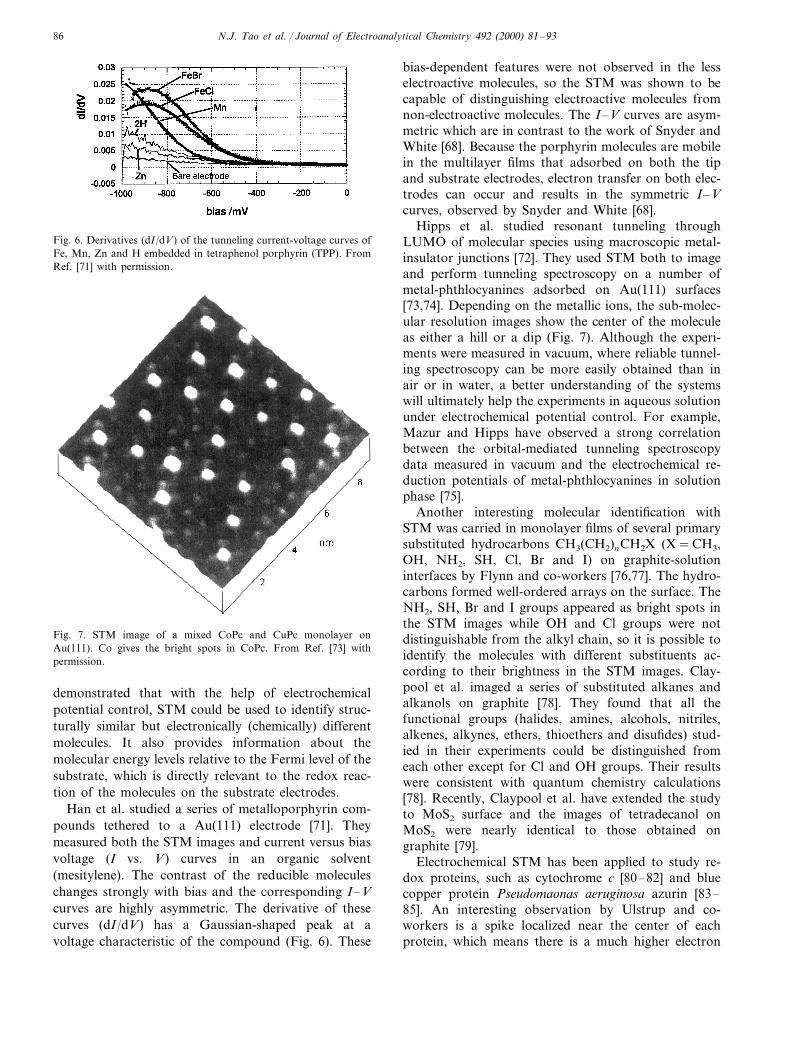
Hipps et al. studied resonant tunneling throughLUMO of molecular species using macroscopic metal-insulator junctions [72]. They used STM both to imageand perform tunneling spectroscopy on a number ofmetal-phthlocyanines adsorbed on Au(111) surfaces[73,74]. Depending on the metallic ions, the sub-molec-ular resolution images show the center of the moleculeas either a hill or a dip (Fig. 7). Although the experi-ments were measured in vacuum, where reliable tunnel-ing spectroscopy can be more easily obtained than inair or in water, a better understanding of the systemswill ultimately help the experiments in aqueous solutionunder electrochemical potential control. For example,Mazur and Hipps have observed a strong correlationbetween the orbital-mediated tunneling spectroscopydata measured in vacuum and the electrochemical re-duction potentials of metal-phthlocyanines in solutionphase [75].
Another interesting molecular identification withSTM was carried in monolayer films of several primarysubstituted hydrocarbons CH3(CH2)nCH2X (X=CH3,OH, NH2, SH, Cl, Br and I) on graphite-solutioninterfaces by Flynn and co-workers [76,77]. The hydro-carbons formed well-ordered arrays on the surface. TheNH2, SH, Br and I groups appeared as bright spots inthe STM images while OH and Cl groups were notdistinguishable from the alkyl chain, so it is possible toidentify the molecules with different substituents ac-cording to their brightness in the STM images. Clay-pool et al. imaged a series of substituted alkanes andalkanols on graphite [78]. They found that all thefunctional groups (halides, amines, alcohols, nitriles,alkenes, alkynes, ethers, thioethers and disufides) stud-ied in their experiments could be distinguished fromeach other except for Cl and OH groups. Their resultswere consistent with quantum chemistry calculations[78]. Recently, Claypool et al. have extended the studyto MoS2 surface and the images of tetradecanol onMoS2 were nearly identical to those obtained ongraphite [79].
Electrochemical STM has been applied to study re-dox proteins, such as cytochrome c [80–82] and bluecopper protein Pseudomaonas aeruginosa azurin [83–85]. An interesting observation by Ulstrup and co-workers is a spike localized near the center of eachprotein, which means there is a much higher electron
Fig. 7. STM image of a mixed CoPc and CuPc monolayer onAu(111). Co gives the bright spots in CoPc. From Ref. [73] withpermission.
demonstrated that with the help of electrochemicalpotential control, STM could be used to identify struc-turally similar but electronically (chemically) differentmolecules. It also provides information about themolecular energy levels relative to the Fermi level of thesubstrate, which is directly relevant to the redox reac-tion of the molecules on the substrate electrodes.
Han et al. studied a series of metalloporphyrin com-pounds tethered to a Au(111) electrode [71]. Theymeasured both the STM images and current versus biasvoltage (I vs. V) curves in an organic solvent(mesitylene). The contrast of the reducible moleculeschanges strongly with bias and the corresponding I–Vcurves are highly asymmetric. The derivative of thesecurves (dI/dV) has a Gaussian-shaped peak at avoltage characteristic of the compound (Fig. 6). These

N.J. Tao et al. / Journal of Electroanalytical Chemistry 492 (2000) 81–93 87
tunneling through the region of the spike [83,84]. Thespike could be due to the redox center, copper, in theazurin molecule, which enhances the tunneling current.This study provides the first evidence of local spectro-scopic information of redox proteins with STM.
5. Fast kinetics
The temporal resolution of STM is typically on theorder of seconds due to the limited scan rate of theinstruments, which prohibits STM from directly imag-ing fast kinetics on the surfaces. Magnussen et al. haverecently achieved video rate electrochemical STM [86].A number of indirect methods have also been devel-oped to study fast kinetics with the current STM setup.One is to monitor and analyze the fluctuations in thetunneling current. Binning et al. showed that the diffu-sion of adsorbed species gave rise to fluctuations in theSTM current that could be used to evaluate diffusiveproperties of the adsorbates [87]. Lozano and Tringidesstudied the surface diffusion of oxygen adsorbed onstepped Si(111) from the time dependence of the tunnel-ing current [88]. The power spectrum (the Fouriertransform of the current correlation function) was de-
termined and fitted to the expected theoretical form[89–91] for a single diffusion process, from which thediffusion coefficient and activation energy were ex-tracted. In the case of electrochemical STM, thermalfluctuations of solvent molecules between the STM tipand substrate electrode are expected to affect the tun-neling current [44]. So, in principle, a time-resolvedtunneling current provides important informationabout the thermal fluctuations of solvent moleculesnear the electrode surface. An experimental challenge isto build a much faster STM preamplifier than thecurrent ones in order to probe water molecules at roomtemperature.
Another method is to scan repeatedly along a singleline that is perpendicular to a surface step. The growthor dissolution of the step can be rapidly recorded whichprovides fast kinetic information about the depositionor dissolution at the step. This method was used tostudy the kinetics of Pb underpotential deposition onAu(111) and Ag(111) [8,92,93] in electrolyte and thedynamics of kinks along surface steps of Au and Agcrystals in vacuum [94]. In a recent application of themethod, Giesen et al. studied step dynamics on Cu(100)and Ag(111) electrodes in an aqueous electrolyte [95].From the time dependence of the step fluctuations, theydetermined the mass transport involved in the fluctua-tions as a function of the electrode potential and ex-tracted the activation energy of the mass transport.
Magnussen and Vogt developed a novel STM time-of-walk (TOW) method to measure kink motions alonga surface step during Cu(100) dissolution in HCl solu-tion [96]. Direct imaging of the kink motions with STMis difficult because the rate of the motions exceeds thetypical STM rate even at the very onset of Cu dissolu-tion. In the TOW method, they recorded STM scanlines across a surface step alternately at two locations,y1 and y2, separated by a distance of Dy (Fig. 8). Ateach location, a passing kink was shown as a distinctchange in the step position along x. The positionalchanges at y1 and y2 could be correlated with the timedifference Dt corresponding to the propagation time ofthe kink from y1 to y2. The method allowed them todetermine the average velocity of a single kink andreaction rate. They found that at the onset of Cudissolution, the average kink velocity and the reactionrates were in the range of 103 and 105 atoms s−1,respectively. Using the microscopic data, Magnussenand Vogt estimated the macroscopic dissolution currentdensity which was in good agreement with the densitydetermined from the quasi-static electrochemical polar-ization curves.
The above methods studied fast kinetics of electro-chemical deposition or dissolution at surface steps inthe lateral directions. For fast kinetics perpendicular tothe electrode surface, Li and Tao used a STM tunnelingjunction method to study fast kinetics of electrochemi-cal deposition and dissolution on the atomic scale [97].
Fig. 8. Measurements of kink propagation velocities during Cu(100)dissolution in 0.01 M HCl by the TOW method. (a) An in situ STMimage (170×450 A, 2) recorded at −0.25 V prior to the experiment,solid (recorded scan lines) and dotted (backscan) lines illustrate thepath of the scanning tip during one TOW cycle. (b) Small part of theTOW data, recorded after a potential step to −0.14 V at positionsy1 and y2 (Dy=306 A, ; time per TOW cycle =100 ms). At time t1
a kink in the leftmost step with a width of Dx=3.6 A, , i.e. one(2×2)R45°unit, passes through y1 and y2, whereas no posi-tional changes occur at the other steps. (c) Step positions x(t) at y1and y2, extracted from similar TOW experiments and showing thepassing of a kink with a width of single (t=20.5 s) and double (t=16s) one and two (2×2)R45°units, respectively. From Ref. [96]with permission.

N.J. Tao et al. / Journal of Electroanalytical Chemistry 492 (2000) 81–9388
They placed a STM tip at various fixed distances froman electrode surface, and monitored the tunneling cur-rent as Cu was deposited (dissolved) onto (from) theAu electrode or the STM tip. The deposition or dissolu-tion changes the tunneling gap between the tip and theAu substrate, which is reflected in the measured tunnel-ing current. They observed stepwise changes in thetunneling current, which may be attributed to the dis-crete nature of atoms. The deposition rate estimatedfrom the slope of the logarithm of the current versustime is about 10 A, s−1. For a 10 kHz STM pream-plifier, a deposition/dissolution rate faster than 104 A,s−1 can be measured.
6. Nanofabrication based on electrochemical depositionand dissolution
The quest toward further miniaturization of deviceshas driven a growing activity in the interdisciplinaryarea of nanometer scale science and technology. Acritical task is to develop methods to fabricate nanome-ter-scale structures. Shortly after the invention of STMand AFM, the techniques were used to fabricate nanos-tructures on surfaces, and a large body of work hasbeen published. Here we discuss only some of thenanofabrication methods that are based on STM andelectrochemical processes, more comprehensive reviewsof STM or related AFM lithography can be found inthe literature [98,99]. Electrochemical STM-based nano-fabrication techniques allow one not only to fabricatenanostructures for applications, but also to study vari-ous electrochemical phenomena on the nanometerscale.
Electrochemical STM nanofabrication techniquesusually involve localized etching (dissolution) and plat-ing (deposition) on the surface via the sharp STM tip.The first demonstration of nanofabrication on Si(100)and GaAs(100) surfaces via localized etching was car-ried out by Nagahara et al. [100]. By maintaining atip–substrate bias of 1.4 V (tip is positive) and acurrent of 1 nA in 0.05% HF, they observed that thearea scanned by the STM was etched. Using the tech-nique, they fabricated nanostructures of 20 nm wideand 1–5 nm deep. Xie et al. have reported anotherlocal etching of H-terminated Si(111). They proposedthat the tunneling of electrons from the valence band ofthe Si electrode into the empty states at the STM tipleads to local oxidation or etching of the Si electrode[101].
Recently Xie and Kolb presented a systematic studyof STM tip-induced local etching of a metal electrode,Cu [102]. They found that Cu can be dissolved from anelectrode at potentials below the Cu/Cu2+ equilibriumpotential. The dissolution rate depends sensitively onboth the tip and electrode potentials. Similar to the
local etching of H-terminated Si(111) [101], they pro-posed a mechanism that involves an electron transferfrom Cu directly into the empty states of the STM tip.
Crooks’ group used STM to locally etch Au(111)electrode coated with a self-assembled thiol monolayer(SAM), and they were able to fabricate features from afew tens of nanometers to several micrometers [103–108]. The STM etched features may work as ultrami-croelectrodes because the rest of the electrode iscovered with the SAM [103]. They studied local etchingof the Au(111) electrode in F- and Cu-electrolytes. Atthe open circuit potential, they found no etching in 0.1M KF with a 0.1 V bias and 0.5 nA tunneling current[105,108]. However, at −0.15 V (vs. Ag � AgCl), theyobserved that pits with a monoatomic depth wereformed underneath the scanning STM tip. The etchingdepends on the electrolytes and tip–substrate bias po-larity. For example, in 0.001 M KCN, they observedthe etching at potentials above −0.52 V. Based on theexperimental data, they provided a detailed model forthe STM nanofabrication mechanism that involves pen-etration of the STM tip into the SAM, concerteddisruption of the SAM, and subsequent removal of theSAM due to electrochemical etching [106].
In addition to etching, electrochemical deposition hasbeen applied to STM-based nanofabrications. Penner’sgroup used the bias pulse-induced pits on graphite asnucleation sites for the deposition of metal nanostruc-tures [109,110]. The deposition was carried out in dilutemetal-plating solutions by applying bias pulses betweenthe STM tip and the substrate. The method allowedthem to fabricate silver and copper nanoclusters of20–50 nm wide and 1–7 nm high. They further demon-strated that underpotential deposition of silver mono-layers on the STM tip provided the precursor silverrequired for the formation of silver nanoclusters on thegraphite substrate. Breuer et al. also used STM as atool for nanomodifications on graphite electrode togenerate holes and copper clusters of nanometer size[111]. In addition to metallic clusters, the method wasused to fabricate nanometer-sized polyaniline on agraphite electrode [112]. The experiment was performedwith a Pt STM in an aqueous electrolyte that containedaniline monomers. The electropolymerization of anilinewas initiated by two bias voltage pulses: the first onecreated a pit on a graphite electrode and the secondpulse induced polymerization of aniline at the pit.Using the procedure, polyaniline particles of 10–60 nmin diameter and 1–20 nm in height were fabricated.
Kolb’s group developed a method to deposit coppernanoclusters locally onto gold surfaces from copperplating solutions with an electrochemical STM [113–116]. In the method, they first deposited copper ontothe STM tip, and then transferred the copper from thetip onto the surface via a mechanical contact betweenthe tip and substrate. Using a fully automated proce-

N.J. Tao et al. / Journal of Electroanalytical Chemistry 492 (2000) 81–93 89
Fig. 9. (A) STM image of 12 Cu clusters on an Au(111) electrode in0.05 M H2SO4+1 mM CuSO4. Esample= +10 mV; Etip= −50 mV;IT=2 nA. The clusters were generated by 12 programmed ap-proaches of the tip to the surface caused by external voltage pulses tothe z-piezo. Tip displacement Dz=1.6 nm; pulse duration Dt=10ms. (B) Cross section through the center of the circle. From Ref. [113]with permission.
energy gaps between the electronic states of the clusters[116]. The high stability of the STM fabricated nan-oclusters appears to be in accordance with the observa-tion that subnanometer silver clusters exhibitunexpected electrochemical metastability on graphite atpositive potentials reported by Ng et al. [120]. Theyconsidered two possible explanations: the first one isthat silver particles undergo a metal to nonmetal transi-tion during dissolution, and that interfacial electrontransfer to the resulting nonmetallic silver clusters isslow for a variety of reasons. The second possibilityinvolves the existence on these clusters of a stronglycoordinated anion layer that impedes their dissolution.Further studies will be needed for a complete under-standing of these interesting observations.
Hofmann et al. showed that the mechanical contactbetween tip and substrate in the method by Kolb’sgroup could be avoided [121]. They first deposited ametal onto the STM tip and quickly stripped it off,which caused local transient oversaturation of the metalion near the STM tip. The oversaturation shifted theNernst potential and resulted in the local deposition ofthe metal onto the substrate. Lorenz and co-workersdiscovered a similar procedure and successfully de-posited lead nanoclusters onto atomically flat terracesof an n-type Si(111) using STM [118]. Schuster et al.argued that the electrochemical reaction was governedby the potential drop in the double layer and chemicalsnear the electrode surface [122]. An applied voltagecaused rapid redistribution of ions within the doublelayer and the potential varied essentially across theHelmoltz layers of the working and counter electrodes.So the electrochemical reaction was not localized nearthe apex of the STM tip. To overcome the problem,they applied voltages shorter than 100 ns to the STMtip, which induced electrochemical deposition and dis-solution far from equilibrium.
The metal nanoclusters fabricated by the abovemethods provide nice systems for studying electrochem-istry on the nanometer scale. For many electronic ap-plications, nanoclusters have to be, weakly coupled tothe substrate electrode. For example, in order to ob-serve the single electron charging or Coulomb blockadeeffects, the tunneling resistance between the substrateand the nanoclusters has to be much greater than h/e2
(ca. 25.5 kV) [123]. So directly depositing nanoclustersonto a conductive substrate will not meet the require-ment and an insulating buffer layer between the nan-oclusters and substrate has to be provided. By locallyoxidizing a titanium line prefabricated on an oxidecovered Si substrate with a STM tip, Matsumoto et al.fabricated a nanostructure that exhibits a single elec-tron charging effect [124,125].
Li and Tao reported that a metallic quantum wirecould be fabricated with an electrochemical STM [126].In the method, they electrochemically deposited Cu
dure, they were able to fabricate arrays of coppernanoclusters within minutes (Fig. 9). They also showedthat the height of the copper clusters can be controlledbetween two and five atomic layers (i.e. 0.4–1 nm) bychanging the tip approach [114]. The width of theclusters measured from the STM images was about 3–4nm. The actual width is probably even somewhatsmaller because of the finite size of the STM tip.Recently, they fabricated arrays of Pd clusters on aAu(111) electrode, which could be used to study sizeeffects in electrocatalysis [117].
An interesting observation is that the STM-fabri-cated clusters are surprisingly stable against anodicdissolution [114,116], and are referred to as zero dimen-sional phase materials by Lorenz and co-workers [118].The high stability of the STM-fabricated clusters wasinitially attributed to tip shielding [113,119], but thiswas rejected later because the clusters were found to behighly stable even when the tip was withdrawn from theelectrode surface. Another explanation of the high sta-bility was suggested to be due to the lack of defects forsmall clusters [114]. More recently, Kolb et al. pointedout that the high stability might be related to quantumconfinement of the electrons in the clusters that leads to

N.J. Tao et al. / Journal of Electroanalytical Chemistry 492 (2000) 81–9390
onto a sharp STM tip that was held at a fixed distance(10–150 nm) from the electrode (Fig. 10). The selectivedeposition onto the tip was achieved by keeping thesubstrate potential slightly more positive than the bulkdeposition potential. When the growing Cu reached thesubstrate, a wire between the substrate and tip wasformed which was reflected by a sharp increase in thecurrent that flows between the tip and substrate (Fig.11, top). They found the conductance of the wireincreases in a quantized fashion with a step height ofca. G0=2e2/h. The phenomenon was completely re-versible, when dissolving Cu from the nanowires (Fig.11, bottom).
Conductance quantization in metallic nanowires oc-curs when the diameter of the wire is comparable to the
wavelength of the conduction electrons [127–132]. Asimplified picture is that the electron waves in thetransverse direction of the nanowires form a well-defined quantum mode (standing waves). Because eachmode contributes equally to the total conductance, thetotal conductance is proportional to the number ofmodes, and thus quantized. The wavelength of conduc-tion electrons in a typical metal is on the order of a fewangstroms, so the diameter of the wire must be on theorder of a few atoms when the conductance is at thelast quantum step. This was directly verified by trans-mission electron microscopy [133]. The phenomenon ofconductance quantization was first observed in thenanowires formed by mechanically separating twometallic electrodes from contact. An important advan-tage of the electrochemical method is because no me-chanical movement is involved, nanowires withlong-term stability can be fabricated. Recently, Li et al.have demonstrated that the nanowires can last formany hours [134]. Another advantage is that polymernanowires can also be fabricated using the method.
The fabrication of nanowires in the electrochemicalenvironment allows one to study various electrochemi-cal phenomena that take place on the nanowires. Forexample, upon changing the electrochemical potential,an interesting fractional conductance quantization wasobserved [135]. At positive potentials, the conductanceis quantized near integer multiples of G0 (2e2/h) asshown by well-defined peaks in the conductance his-togram. Below a certain potential, however, additionalpeaks near 0.5G0 and 1.5G0 appear in the histogram.The fractional conductance steps are as stable and welldefined as the integer steps. The experimental data maybe interpreted in terms of electrochemical potential-in-duced defect scattering and Fermi energy shift but acomplete theory of the phenomenon is yet to bedeveloped.
7. Conclusions
Fifteen years after the first application of STM inelectrochemistry, STM has become a powerful imagingtool. The spatial resolution of STM allows one toresolve individual atoms, but the temporal resolution istypically on the order of seconds, which prevents STMfrom imaging fast kinetics of electrochemical processes.Encouraging progress has been made to speed up STMimaging acquisition in recent years, and various indirectnovel STM-based methods have also been developed toprobe fast kinetics. In addition to structural studies,STM has proven to be useful in extracting electrontransfer properties and electronic states of the probedmolecules. The electronic information allows one toidentify structurally similar molecules and obtain chem-ical reactivity of the molecules. To extract reliably the
Fig. 10. Formation of atomically thin wire between a STM tip andsubstrate by depositing Cu onto the STM tip that is held at a fixeddistance from a substrate.
Fig. 11. Conductance of a Cu nanowire as its width is being increasedby electrochemical deposition (top) and decreased by etching (bot-tom). The stepwise change is due to conductance quantization.

N.J. Tao et al. / Journal of Electroanalytical Chemistry 492 (2000) 81–93 91
information, however, a better understanding of STMimaging mechanism in the electrochemical environmentis needed. Electrochemical STM has been used to fabri-cate various nanostructures. Although it is not clear ifSTM could fabricate practical devices because of itsslow speed, it is certainly useful to fabricate desirablenanostructures for studying electrochemistry on thenanometer scale. So far, the focus has been largely onhow to fabricate nanostructures, it is desirable to seemore studies on the fundamental electrochemical andelectronic properties of the STM-fabricated nanostruc-tures in relation to applications.
Acknowledgements
Financial support is acknowledged through grantsfrom NSF (CHE-9818073), PRF (33516-AC5, adminis-tered by the ACS) and AFOSR (F49620-99-1-0112),and NIH (GM-08205).
References
[1] R. Sonnenfeld, P.K. Hansma, Science 232 (1986) 211.[2] H.Y. Liu, F.R.F. Fan, C.W. Lin, A. Bard, J. Am. Chem. Soc.
108 (1986) 3838.[3] K. Itaya, E. Tomita, Surf. Sci. 201 (1988) L507.[4] D.J. Trevor, C.E.D. Chidsey, D.N. Loiacono, Phys. Rev. Lett.
62 (1989) 929.[5] J. Wiechers, T. Twomey, D.M. Kolb, R.J. Behm, J. Elec-
troanal. Chem. 248 (1988) 451.[6] X. Gao, A. Hamelin, M.J. Weaver, Phys. Rev. B 44 (1991)
10983.[7] N.J. Tao, S.M. Lindsay, J. Appl. Phys. 70 (1991) 5141.[8] M.P. Green, K.J. Hanson, D.A. Scherson, X. Xing, M.
Rochter, P.N. Ross, R. Carr, I. Lindau, J. Phys. Chem. 93(1989) 2181.
[9] O.M. Magnussen, J. Hotlos, R.J. Nichols, D.M. Kolb, R.J.Behm, Phys. Rev. Lett. 64 (1990) 2929.
[10] T. Hachiya, H. Honbo, K. Itaya, J. Electroanal. Chem. 315(1991) 275.
[11] N.J. Tao, J. Pan, Y. Li, P.I. Oden, J.A. DeRose, S.M. Lindsay,Surf. Sci. Lett. 271 (1992) L344.
[12] S.L. Yau, C.M. Vitus, B.C. Schardt, J. Am. Chem. Soc. 112(1990) 3677.
[13] R.L. MaCarley, A.J. Bard, J. Phys. Chem. 95 (1991) 9618.[14] N.J. Tao, S.M. Lindsay, J. Phys. Chem. 96 (1992) 5213.[15] X. Gao, Y. Zhang, M.J. Weaver, J. Phys. Chem. 96 (1992)
4156.[16] J. Schott, C.R. Arana, H.D. Abruna, H.H. Petoch, G.M. Elliot,
H.S. White, J. Phys. Chem. 96 (1992) 5222.[17] S.M. Lindsay, B. Barris, J. Vac. Sci. Technol. A6 (1988) 544.[18] R. Srinivasan, J.C. Murphy, R. Fainchtein, J. Electroanal.
Chem. 312 (1991) 293.[19] N.J. Tao, J.A. DeRose, S.M. Lindsay, J. Phys. Chem. 97 (1993)
910.[20] M. Holzle, T. Wandlowski, D.M. Kolb, Surf. Sci. 335 (1995)
281.[21] (a) N.J. Tao, Z. Shi, Surf. Sci. 321 (1994) L149. (b) N.J. Tao,
Z. Shi, J. Phy. Chem. 98 (1994) 7422.[22] K.M. Richard, A.A. Gewirth, J. Phys. Chem. 99 (1995) 12288.
[23] J. Lipkowski, P.N. Ross (Eds.), Imaging of Surfaces and Inter-faces, Wiley–VCH, New York, 1999.
[24] A.A. Gewirth, B.K. Niece, Chem. Rev. 97 (1997) 1129.[25] D.M. Cyr, B. Venhataraman, G.W. Flynn, Chem. Mater. 8
(1996) 1600.[26] A.J. Bard, H.D. Abruna, C.E. Chidsey, L.R. Faulkner, S.W.
Feldberg, K. Itaya, M. Majda, O. Melroy, R.W. Murray, M.D.Porter, M.P. Soriaga, H.S. White, J. Phys. Chem. 97 (1993)7147.
[27] T.P. Moffat, Electroanal. Chem. 21 (1999) 211.[28] D.M. Kolb, Electrochim. Acta 45 (2000) 2387.[29] J.E.T. Andersen, J.-D. Zhang, Q. Chi, A.G. Hansen, J.U.
Nielsen, E.P. Friis, J. Ulstrup, A. Boisen, H. Jensnius, TrendsAnal. Chem. 18 (1999) 665.
[30] S. Manne, P.K. Hansma, J. Massie, A.A. Gewirth, Science 251(1991) 183.
[31] A.J. Bard, F-R. Fan, J. Kwak, O. Lev, Anal. Chem. 61 (1989)132.
[32] C.J. Chen, Introduction to Scanning Tunneling Microscopy,Oxford University Press, New York, 1993.
[33] J.H. Coombs, J.B. Pethica, IBM J. Res. Dev. 20 (1986) 455.[34] R. Christoph, H. Siegenthaler, H. Rohrer, W. Wiese, Elec-
trochim. Acta 34 (1989) 1011.[35] J. Wiechers, T. Twomey, D.M. Kolb, R.J. Behm, J. Elec-
troanal. Chem. 248 (1988) 451.[36] M. Bingelli, D. Carnal, R. Nyffenegger, H. Siegenthaler, R.
Christoph, H. Rohrer, J. Vac. Sci. Technol. B9 (1991) 1985.[37] J. Pan, T.W. Jing, S.M. Lindsay, J. Phys. Chem. 98 (1993)
4205.[38] A. Vaught, T.W. Jing, S.M. Lindsay, Chem. Phys. Lett. 236
(1995) 306.[39] Y.A. Hong, J.R. Hahn, H. Kang, J. Chem. Phys. 108 (1998)
4367.[40] W. Schmickler, Surf. Sci. 335 (1995) 416.[41] M.F. Toney, J.N. Howard, J. Richer, G.L. Borges, J.G. Gor-
don, O.R. Melroy, D.G. Wiesler, D. Yee, L.B. Sorensen, Na-ture 368 (1994) 444.
[42] I. Benjamin, D. Evans, A Nitzan, J. Chem. Phys. 106 (1997)1291.
[43] W. Schmickler, D. Henderson, J. Electroanal. Chem. 290 (1990)283.
[44] M.V. Vigdorovich, A.M. Kuznetsov, J. Ulstrup, Chem. Phys.176 (1993) 539.
[45] J.K. Saas, J.K. Gimzewski, J. Electroanal. Chem. 308 (1991)333.
[46] G. Repphun, J. Halbritter, J. Vac. Sci. Technol. A 13 (1995)1693.
[47] J. Tersoff, D.R. Hamann, Phys. Rev. B 31 (1985) 805.[48] S.M. Lindsay, O.F. Sankey, Y. Li, C. Herbst, A. Rupprecht, J.
Phys. Chem. 94 (1990) 4655.[49] N.D. Lang, IBM J. Res. Dev. 30 (1986) 374.[50] C. Joachim, P. Sautet, in: R.J. Behm, N. Garcia, H. Rohner
(Eds.), Scanning Tunneling Microscopy and Related Methods,Kluwer, Netherlands, 1990.
[51] V. Mujica, M. Kemp, M.A. Ratner, J. Chem. Phys. 101 (1994)6849.
[52] H. Ou-Yang, B. Kallebring, R.A. Marcus, J. Chem. Phys. 98(1993) 7565.
[53] A.J. Fisher, P.E. Blochl, Phys. Rev. Lett. 70 (1993) 3263.[54] A.M. Kuznetsov, P. Sommer-Larsen, J. Ulstrup, Surf. Sci. 275
(1992) 52.[55] A.M. Kuznetsov, J. Ulstrup, Mol. Phys. 87 (1992) 1189.[56] J.E.T. Andersen, A.A. Kornyshev, A.M. Kuzenetsov, L.L.
Madsen, P. Moller, J. Ulstrup, Electrochim. Acta 42 (1997)819.
[57] (a) W. Schimickler, J. Electroanal. Chem. 296, (1990) 283. (b)W. Schmickler, C. Widrig, J. Electroanal. Chem. 336 (1992)213.

N.J. Tao et al. / Journal of Electroanalytical Chemistry 492 (2000) 81–9392
[58] H. Sumi, J. Phys. Chem. B 102 (1998) 1833.[59] A.K. Mishra, S.K. Rangarajan, J. Mole. Struc. 361 (1996) 101.[60] (a) D.N. Futaba, S. Chiang, J. Vac. Sci. Technol. A 15 (1997)
1295. (b) V.M. Hallmark, S. Chiang, Surf. Sci. 329 (1995) 255.[61] X.W. Wang, N.J. Tao, F. Cunha, J. Chem. Phys. 105 (1996)
3747.[62] R. Durig, O. Zuger, B. Michel, L. Haussling, H. Ringsdorf, J.
Chem. Phys. 48 (1993) 1711.[63] M. Specht, F. Ohnesorge, W.M. Heckl, Surf. Sci. 257 (1991)
L653.[64] G. Leatherman, E.N. Durantini, D. Gust, T.A. Moore, A.L.
Moore, S. Stone, Z. Zhou, P. Rez, Y.Z. Liu, S.M. Lindsay, J.Phys. Chem. 103 (1999) 4006.
[65] (a) F. Cunha, N.J. Tao, X.W. Wang, Q. Jin, B. Duong, J.D’Agnese, Langmuir 12 (1996) 6410. (b) F. Cunha, N.J. Tao,Phys. Rev. Lett. 75 (1995) 2376.
[66] H. Morisaki, H. Nishkawa, H. Ono, K. Yazawa, J. Elec-trochem. Soc. 137 (1990) 2759.
[67] H. Gerischer, Z. Phys. Chem. N.F. 27 (1961) 48.[68] S.R. Snyder, H.S. White, J. Electroanal. Chem. 394 (1995) 177.[69] N.J. Tao, Phys. Rev. Lett. 76 (1996) 4066.[70] W. Schmickler, N.J. Tao, Electrochim. Acta 42 (1997) 2809.[71] W. Han, E.N. Durantini, T.A. Moore, A.L. Moore, D. Gust, P.
Rez, G. Leatherman, G.R. Seely, N.J. Tao, S.M. Lindsay, J.Phys. Chem. 101 (1997) 10719.
[72] K.W. Hipps, X. Lu, X.D. Wang, U. Mazur, J. Phys. Chem. 100(1996) 11207.
[73] X. Lu, K.W. Hipps, X.D. Wang, U. Mazur, J. Am. Chem. Soc.118 (1996) 7197.
[74] X. Lu, K.W. Hipps, J. Phys. Chem. B 101 (1997) 5397.[75] U. Mazur, K.W. Hipps, J. Phs. Chem. 99 (1995) 6684.[76] D.M. Cyr, B. Venkataraman, G.W. Flynn, A. Black, G.W.
Whitesides, J. Phys. Chem. 100 (1996) 13748.[77] H.B. Fang, L.C. Giancarlo, G.W. Flynn, J. Phys. Chem. B 103
(1999) 5712.[78] C.L. Claypool, F. Faglioni, W.A. Goddard, H.B. Gray, N.S.
Lewis, R.A. Marcus, J. Phys. Chem. B 101 (1997) 5978.[79] C.L. Claypool, F. Faglioni, W.A. Goddard, N.S. Lewis, J.
Phys. Chem. B 103 (1999) 7077.[80] J.E.T. Andersen, P.P. Moller, M.V. Pedersen, J. Ulstrup, Surf.
Sci. 325 (1995) 193.[81] B. Zhang, E. Wang, Probe Micros. 1 (1997) 57.[82] J. Zhang, Q. Chi, S. Dong, E. Wang, Bioelectrochem. Bioenerg.
39 (1996) 267.[83] E.P. Friis, J.E.T. Andersen, Yu I. Kharkats, A.M. Kuznetsov,
R.J. Nichols, J.D. Zhang, J. Ulstrup, Proc. Natl. Acad. Sci.USA 96 (1999) 1379.
[84] E.P. Friis, J.E.T. Andersen, P. Moller, J. Ulstrup, J. Elec-troanal. Chem. 431 (1997) 35.
[85] J.J. Davis, C.M. Halliswell, H.O.A. Hill, G.W. Canters, M.C.van Amsterdam, M.P. Verbeer, New J. Chem. 22 (1998) 1119.
[86] O.M. Magnussen, L. Zitzler, B. Gleich, R.J. Behm, Presenta-tion for the Electrochemical Society, Honolulu, October, 1999.
[87] G. Binning, H. Fuchs, E. Stoll, Surf. Sci. 169 (1986) L295.[88] M.L. Lozano, M.C. Tringides, Europhys. Lett. 30 (1995) 537.[89] M. Sumetskii, A.A. Kornyshev, Phys. Rev. B 48 (1993) 17493.[90] A.S. Prostnev, V.I. Chernysh, B.R. Shub, Chem. Phys. Rep. 18
(1999) 13.[91] M. Sumetskij, A.A. Kornyshev, U. Stimming, Surf. Sci. 307
(1994) 23.[92] T. Will, M. Dietterle, D.M. Kolb, in: A.A. Gewirth, H. Siegen-
thaler (Eds.), Nanoscale Probes of the Solid-Liquid Interface,Nato ASI, vol. E288, Kluwer, Dordrecht, 1995, p. 137.
[93] M. Giesen-Seibert, R. Jentjens, M. Poensgen, H. Ibach, Phys.Rev. Lett. 71 (1993) 3521.
[94] M.S. Hoogeman, L. Kuipers, D.C. Schloßer, J.W.M. Frenken,Surf. Sci. 447 (2000) 25.
[95] M. Giesen, R. Randler, S. Baier, H. Ibach, D.M. Kolb, Elec-trochim. Acta 45 (1999) 527.
[96] O.M. Magnussen, M.R. Vogt, Phys. Rev. Lett. 85 (2000) 357.[97] C.Z. Li, N.J. Tao, unpublished.[98] R.N. Nyffenegger, R.M. Penner, Chem. Rev. 97 (1997) 1195.[99] W. Wlieth, W.J. Lorenz (Eds.), Electrochemical Nanotechnol-
ogy—in situ Local Probe Techniques at Electrochemical Inter-faces, Wiley–VCH, Weinheim 1998.
[100] L.A. Nagahara, T. Thundat, S.M. Lindsay, Appl. Phys. Lett.57 (1990) 270.
[101] Z.X. Xie, X.W. Cai, J. Tang, Y.A. Chen, B.W. Mao, Chem.Phys. Lett. 322 (2000) 219.
[102] Z.X. Xie, D.M. Kolb, J. Electroanal. Chem. 481 (2000) 177.[103] C.B. Ross, L. Sun, R.M. Crooks, Langmuir 9 (1993) 632.[104] J.K. Schoer, C.B. Ross, R.M. Crooks, T.S. Corbitt, M.J.
Hampdensmith, Langmuir 10 (1994) 615.[105] J.K. Schoer, F.P. Zamborini, R.M. Crooks, J. Phys. Chem. 100
(1996) 11086.[106] J.K. Schoer, R.M. Crooks, Langmuir 13 (1997) 2323.[107] F.P. Zamborini, R.M. Crooks, J. Am. Chem. Soc. 120 (1998)
9700.[108] Y.Q. Li, O. Chailapakul, R.M. Crooks, J. Vac. Sci. Technol. B
13 (1995) 1300.[109] W.J. Li, J.A. Nirtanen, R.M. Penner, Appl. Phys. Lett. 60
(1992) 1181.[110] W. Li, G.S. Hsiao, D. Harris, R.M. Nyffenegger, J.A. Virtanen,
R.M. Penner, J. Phys. Chem. 100 (1996) 20103.[111] N. Breuer, U. Stimming, R. Vogel, Surf. Coatings Technol. 67
(1994) 145.[112] R.M. Nyffenegger, R.M. Penner, J. Phys. Chem. 100 (1996)
17041.[113] D.M. Kolb, R. Ullmann, T. Will, Science 275 (1997) 1097.[114] D.M. Kolb, R. Ullmann, J.C. Ziegler, Electrochim. Acta 43
(1998) 2751.[115] R. Ullmann, T. Will, D.M. Kolb, Ber. Bunsenges. Phys. Chem.
99 (1995) 1414.[116] D.M. Kolb, G.E. Engelmann, J.C. Ziegler, Angew. Chem. Int.
Ed. Engl. 39 (2000) 1123.[117] G.E. Engelmann, J.C. Ziegler, D.M. Kolb, J. Electrochem. Soc.
145 (1998) L33.[118] R.T. Potzshke, G. Staikov, W.J. Lorentz, W. Wisebeck, J.
Electrochem. Soc. 146 (1999) 141.[119] U. Stimming, R. Vogel, D.M. Kolb, T. Will, J. Power Sources
43 (1993) 169.[120] K.H. Ng, H. Liu, R.M. Penner, Langmuir 16 (2000) 4016.[121] D. Hofmann, W. Schindler, J. Kirchner, Appl. Phys. Lett. 73
(1998) 3279.[122] R. Schuster, V. Kirchner, X.H. Xia, A.M. Bittner, G. Ertl,
Phys. Rev. Lett. 80 (1998) 5599.[123] M.H. Devoret, H. Grabert, in: H. Grabert, M.H. Devoret
(Eds.), Single Charge Tunneling, Plenum Press, New York,1992.
[124] K. Matsumoto, M. Ishii, K. Segawa, Y. Oka, B.J. Vartanian, S.Harris, Appl. Phys. Lett. 68 (1996) 34.
[125] K. Matsumoto, Y. Gotoh, T. Maeda, J.A. Dagata, J.S. Harris,Appl. Phys. Lett. 76 (2000) 239.
[126] C.Z. Li, N.J. Tao, Appl. Phys. Lett. 72 (1998) 894.[127] J.M. Krans, J.M. van Ruitenbeek, V.V. Fisun, I.K. Yanson,
L.J. de Jongh, Nature 375 (1995) 767.[128] K. Gimzewski, R. Moller, Phy. Rev. B36 (1987) 1284.[129] U. Landman, W.D. Luedtke, N.A. Burman, R.J. Colton, Sci-
ence 248 (1990) 454.[130] J.I. Pascual, J. Mendez, J. Gomez-Herrero, A.M. Baro, N.
Garcia, Binh V.Thien, Phys. Rev. Lett. 71 (1993) 1852.[131] J.I. Pascual, J. Mendez, J. Gomez-Herrero, A.M. Baro, N.
Garcia, U. Landman, W.D. Luedtke, E.N. Bogachek, H-PCheng, Science 267 (1995) 1793..

N.J. Tao et al. / Journal of Electroanalytical Chemistry 492 (2000) 81–93 93
[132] L. Olesen, E. Lægsgaard, I. Stensgaad, F. Besenbacher, J.Schiotz, P. Stoltze, K.W. Jacobsen, J.K. Norskov, Phys. Rev.Lett. 72 (1994) 2251.
[133] H. Ohnishi, Y. Kondo, K. Takayanagi, Nature 395 (1998) 780.
[134] C.Z. Li, A. Bogozi, W. Huang, N.J. Tao, Nanotechnology 10(1999) 221.
[135] C. Shu, C.Z. Li, X. He, A. Bogozi, J.S. Bunch, N.J. Tao, Phys.Rev. Lett. 84 (2000) 5196.
.


















