
REVIEW OF LITERATURE -...
44
Chapter - II REVIEW OF LITERATURE II. 1. Human Serum Albumin Human serum albumin (HSA) is the most abundant protein in blood plasma with one liter of plasma containing about 60% HSA. There is about 42 ± 3.5 g of HSA per liter of human blood. HSA is a non glycoprotein, one of the few secreted proteins which lacks carbohydrate (Peters, 1985). Produced in the liver, HSA is largely responsible for maintaining normal osmolarity in the bloodstream but also functions as a carrier for numerous endogenous and exogenous compounds such as fatty acids, hormones, toxic metabolites, bile acids, amino acids, drugs, and metals (Furukawa, 2011). Besides being present in the plasma, HSA is also found in tissues and body secretions, skins and lymph cavities. Under normal physiological conditions HSA contributes 80% of the colloid osmotic pressure that provides the driving force to retain fluid within blood vessels (25–33 mmHg). A reduction in the concentration of HSA in the blood circulation results in edema, ascites, and pleural effusion. In addition to the osmotic function, HSA provides a high-capacity reservoir to stabilize the concentration of free ligands (Kragh-Hansen, 1990). HSA also functions as a major antioxidant in human body (Hiroshi et al., 2001). Clinically, it is being used as a plasma expander. The administration of HSA is generally considered to be the gold standard for treating severe hypoalbuminemia as a result of conditions such as burns, nephritic syndrome, reduction in the synthesis of HSA induced by chronic liver cirrhosis, and hemorrhagic shock (Kazuaki et al., 2012). HSA is the most well studied plasma protein and an attractive macromolecular carrier due to its availability in pure form and its biodegradability, non-toxicity and non immunogenicity (Kratz, 2008).
Transcript of REVIEW OF LITERATURE -...

Chapter - II
REVIEW OF LITERATURE
II. 1. Human Serum Albumin
Human serum albumin (HSA) is the most abundant protein in blood
plasma with one liter of plasma containing about 60% HSA. There is about 42 ±
3.5 g of HSA per liter of human blood. HSA is a non glycoprotein, one of the
few secreted proteins which lacks carbohydrate (Peters, 1985). Produced in the
liver, HSA is largely responsible for maintaining normal osmolarity in the
bloodstream but also functions as a carrier for numerous endogenous and
exogenous compounds such as fatty acids, hormones, toxic metabolites, bile
acids, amino acids, drugs, and metals (Furukawa, 2011). Besides being present in
the plasma, HSA is also found in tissues and body secretions, skins and lymph
cavities. Under normal physiological conditions HSA contributes 80% of the
colloid osmotic pressure that provides the driving force to retain fluid within
blood vessels (25–33 mmHg). A reduction in the concentration of HSA in the
blood circulation results in edema, ascites, and pleural effusion. In addition to the
osmotic function, HSA provides a high-capacity reservoir to stabilize the
concentration of free ligands (Kragh-Hansen, 1990). HSA also functions as a
major antioxidant in human body (Hiroshi et al., 2001). Clinically, it is being
used as a plasma expander. The administration of HSA is generally considered to
be the gold standard for treating severe hypoalbuminemia as a result of
conditions such as burns, nephritic syndrome, reduction in the synthesis of HSA
induced by chronic liver cirrhosis, and hemorrhagic shock (Kazuaki et al., 2012).
HSA is the most well studied plasma protein and an attractive macromolecular
carrier due to its availability in pure form and its biodegradability, non-toxicity
and non immunogenicity (Kratz, 2008).

16 Chapter II
II. 1. 1. Functional and physical properties of HSA
HSA has various in vivo functional and physical properties which have
been exploited in a number of biopharmaceutical applications (Mead et al.,
2007). Its amphiphilic properties makes it suitable as an additive to inhibit
adsorption of the active protein to the container via competitive adsorption
mechanisms. Its surface active characters enable it to fulfill the role of surfactant,
thereby preventing protein aggregation. In some instances, it stabilizes the
conformational structure of active molecule to maintain its bioactivity throughout
the product shelf-life. The presence of 17 disulphide linkage provides the protein
remarkable stability and in vivo half-life of 15-20 days. HSA also has a high glass
transition temperature, which in combination with its amphiphilic nature makes it
an ideal vehicle for cryopreservation. In vitro, the molecule’s stability is
increased and it remains in solution at room temperature to sustain the shelf-life
of biopharmaceutical products. During manufacture, HSA can withstand heating
to 60°C for 10 hrs to facilitate viral inactivation (Burnouf, 2007).
II. 1. 2. Pharmaceutical applications of HSA
HSA is one of the most widely used proteins in the pharmaceutical
industry (Mead et al., 2007). It is clinically in great demand as a replacement
fluid of plasma expander or as a compensator for blood loss. It is also used as a
clinical tool for the delivery of many biological products (Peters, 1985; Peters,
1996; Varshney et al., 2010). HSA is widely used clinically to treat serious burn
injuries, plasmaphoresis, hemorrhagic shock, hypoproteinemia, fetal
erythroblastosis, ascites caused by cirrhosis of the liver diuretic-resistant oedema
and for some groups of surgical patients (Hastings and Wolf, 1992; Goodey,
1993). The protein has been described as the major colloid that retains fluid in the
vascular system, acting as a tramp streamer by dithering. HSA transport a mixed
cargo of metabolites around various organs (Kragh- Hansen, 1990). Among these
metabolites, long chain fatty acids are quantitatively the most abundant, with the
normal loading being approximately two fatty acids per molecule of albumin

Review of Literature 17
(Petitpas et al., 2001). HSA also binds bilirubin (Petersen et al., 2000; Weisiger
et al., 2001), amino acids, numerous drugs (Ozer and Tacal, 2001) and heavy
metals and is implicated in the transfer of many ligands across organ-circulatory
interfaces such as liver, intestine, kidney and brain. The molecule has been
succinctly described as the protein that makes blood thicker than water. In
addition to the use of HSA alone, drugs that can be transported in conjunction
with HSA have been recently approved by many countries and provide beneficial
effects. Products like, Abraxane® (American Bioscience, Los Angeles, CA),
which is an albumin-paclitaxel nanoparticle with a mean particle size of 130 nm,
was approved for the treatment of metastatic breast cancer. Albumin and
paclitaxel, in the form of a complex, have several beneficial effects; Abraxane®
accumulates in tumors through the enhanced permeability and retention (EPR)
effect of solid tumors, but a further albumin transport pathway mediated by Gp60
located on the surface of endothelial cells appears to be responsible for the tumor
uptake and tumor distribution of Abraxane® and the subsequent release of
paclitaxel (Schnitzer et al., 1992; Tiruppathi et al., 1997; Wang et al., 2009).
Several drugs and agents that use an albumin as a drug carrier were also
approved, and there are new drugs based on albumin currently in the
development and in the clinical trial stages (Table II-1) (Morales et al., 2007;
Flisiak et al., 2009; Flisiak et al., 2010; Kim et al., 2011; Kazuaki et al., 2012).
Albuferon® (Human Genome Sciences, Rockville, Maryland), a fusion protein of
albumin and interferon-α-2b for the treatment of hepatitis C infections, is
expected to improve the treatment by allowing a monthly dosing schedule in
clinics (Nelson et al., 2010; Zeuzem et al., 2010). In addition to drug
development in the pharmaceutical industry, there is a growing interest in
exploring various pharmaceutical applications of albumin as a drug carrier in
academic research.

18 Chapter II
Table II-1 - Albumin-based drugs that have received market approval or are in clinical trial
Product Name Formation with Albumin Application Eff ect of Albumin
Levemir® (Novo nordisk, Princeton, NJ)
The albumin-binding derivative of human insulin
Type 1 and 2 diabetes
Increment of half-life (native insulin, 4–6 min; Lavemir® , 5–7 h)
Nanocoll® (Nycomed Amersham Sorin, Milan, Italy)
Tecnetium99m (99mTc) aggregated Albumin
γ-emitting radionuclide imaging agent
Increment of the accumulation in targeting area
Albures® (Sorin Biomedical, Irvine, CA)
,,
,,
,,
Pulmolite® (Du Pont Pharma, North Billerica, NA)
,,
,,
,,
Draximage® (draxlmage Inc., London, UK)
,,
,,
,,
Abraxane® Albumin–paclitaxel nanoparticle
Solid tumors Increment of accumulation and uptake in tumor
Albuferon™ Fusion protein of albumin and interferon-α-2b
Hepatitis C infection
Increased blood retention
Vasovist® (Schering AG, Berlin, Germany
Gadofosveset reversibly binds to human serum albumin.
Magnetic resonance contrast agent
Higher relaxivity and extended intravascular enhancement
MARS™ (Gambro, Lund, Sweden)
Dialysis solution containing human serum albumin across a high-flux permeable membrane.
Liver failure Increment of effectively removing albumin-bound endogenous or exogenous toxin.
Many researchers have used different approaches to develop novel
albumin-based drug carriers and these can be generally categorized into three
main modifications: (a) low-molecular-weight proteins fused with albumin
(Furukawa et al., 2011), (b) polymerization (Bae et al., 2012, Ozgur et al., 2012),
(c) surface modification (Lu et al., 2007; Hirata et al., 2010). The main objectives
of these modifications were to prolong blood retention or to enhance the delivery
of a drug to the target area, and their effectiveness and usefulness for various
reports. Among these modifications, albumin dimerization has great potential and
advantages for clinical applications as both a plasma expander and a drug carrier,

Review of Literature 19
whereas other forms of albumin modifications could achieve only one of these two
clinical applications (Taguchi et al., 2012). Furthermore, several investigators have
clearly shown that increased concentrations of albumin dimers are present in the
circulating blood of patients with chronic renal disease as compared with age-
matched healthy individuals (Scorza and Minetti, 1998; Ogasawara et al., 2006), as
well as the concentration of products resulting from oxidative damage in the blood
(Mimic-Oka et al., 1999; Richard et al., 1991). These results imply that the
albumin dimer also has the potential for its use as a biomarker of oxidative stress
(Kazuaki et al., 2012). Many other novel uses for HSA in biological applications
have recently been explored, such as carrier of oxygen (Tsuchida et al., 2009),
nanodelivery of drugs (Cai et al., 2006), and fusion of peptides (Subramanian et
al., 2007).
Almost all of the HSA used therapeutically or non-therapeutically is
currently purified from donated human plasma or whole blood (pHSA). Although
pHSA is generally considered to be safe, the supply and purity of plasma itself is
threatened by known or other emerging pathogens.
II. 1. 3. Traditional HSA manufacture
HSA is currently used in greater volume than any other pharmaceutical
solution, with worldwide manufacture in the order of hundreds of tons annually
(Matejtschuk et al., 2000). Since 1940, it has been produced by fractionation of
plasma obtained from donors (Cohn, 1941). Although blood plasma represents
the richest source of HSA, there are problems associated with the purity of HSA
obtained from human donor blood, currently the most available source of blood
plasma. Donated blood plasma can carry viruses like Human Immunodeficiency
Virus (HIV), Hepatitis C Virus (HCV), Hepatitis A Virus (HAV), variant
Creutzfeldt-Jakob disease (vCJD) and West Nile virus. Such viruses must be
removed during the process of HSA recovery from donated plasma and, although
historically HSA has been a safe product, it is constantly subject to potential risks

20 Chapter II
of contamination. In addition, large–scale production of HSA aggravates the
limited source of human blood (EMEA,CHMP 2004).
These concerns have impelled the regulatory authorities worldwide for
creating a myriad of regulations to limit the use of plasma-derived materials with
the aim of minimizing transmission risks and necessitating a dedicated drive from
within the industry to develop substitute products and ever-more sophisticated
tools for the detection, clearance and removal of adventitious agents from serum-
derived products. Beyond purity, safety and other issues, consistence of supply is
another major issue in HSA production. The fractionation industry has not always
been capable of providing an adequate HSA supply and shortages have been
encountered, particularly when there are shortages of donated human blood
plasma. To address this issue, pharmaceutical companies have turned towards the
development of recombinant versions of HSA.
II. 2. Recombinant HSA (rHSA)
The shortcomings of therapeutic protein production from blood plasma
have inspired the production of recombinant human serum albumin (rHSA). In
1981, Lawn and co-workers (Genentch Company, America) for the first time
expressed rHSA in E. coli successfully and achieved the patent; however, the
expression level is milligram level without the capability of industrialization.
Recombumin® is a recombinant albumin produced and patented by Aventis (a US
pharmaceutical company) (Chuang et al., 2002).
Equivalent blood plasma products using DNA technologies with
recombinant therapeutic proteins offer several potential advantages over human
donor plasma. Most notably, because they are expressed in bacteria or animal
cells, recombinant proteins are (theoretically) 100 percent risk-free from the viral
contaminants that human plasma derived products may contain. It would be a
great advantage to be able to use genetic engineering to obtain rHSA in good
yield and at lower cost, with no danger of contamination by human pathogens.
For this reason, great efforts have been dedicated to the production of this protein

Review of Literature 21
on a large scale by transgenic organisms. To date, expression of rHSA has been
reported in a number of expression systems from bacteria to mammalian cells as
listed in table II-2.
Table II-2: Recombinant HSA expression in different heterologous organisms
Organism Expression level
(Highest reported yet)
Reference
Escherichia coli 7 % of TBP** (Laltta et al., 1990)
Saccharomyces cervisiae 85 mg/l (Okabayashi et al., 1991; Sleep et al., 1990)
Pichia pastoris 270 mg/l (Ohtani et al., 1998)
Kluyvermyces lactis 1g/l (Fleer et al., 1991; Saliola et al., 1995)
Bacillus subtilis 0.135% of TSP (Saunders et al., 1987; Mao et al., 2012)
Solanum tuberosum 0.2% of TSP (Sijmons et al., 1990; Farran et al., 2002)
Niccotiana tobaccum 0.7% of TSP (Millan et al., 2003; Sun et al., 2011)
Hansenula polymorpha 24.6 g/l (Kang et al., 2001; Youn et al., 2010)
Hevea brasiliensis 33 µg/ml (Arokiaraj et al., 2002)
Transgenic mice 11.9 g/l (Barash et al., 1996; Shani et al., 1992, Wu et al., 2012)
Oryza sativa 10.58% of TSP (Huang et al., 2005; He et al., 2011)
* TSP-Total soluble protein, **TBP- Total bacterial protein
The rHSA has to be priced competitively to make any impact on the HSA
market. So the production costs must be kept to an absolute minimum. Therefore,
any production process involving the use of expensive media of the type used in
mammalian cell culture is effectively excluded from consideration. Another
constraint is the high number of disulfide bridges and the size of the protein (66.5
kDa), which in terms of heterologous expression considered to be large. Attempts
to produce large quantities of such proteins in an intracellular form in
recombinant organisms often result in the accumulation of the protein in an
insoluble denatured form in inclusion bodies. In such cases the disulfide bonds
are usually formed incorrectly or not at all.

22 Chapter II
II. 2. 1. rHSA producton - Current strategies
The global annual demand for HSA has increased significantly from
about 300 tons in 2006 to about 500 tons in 2011(He et al., 2011). The ever-
growing emergence of new therapies and applications based on HSA protein has
been a major driving force in the development of new, safe and large-scale,
platforms for rHSA production. To meet growing consumer demands, bioreactors
are becoming increasingly more important aspect for the production of large
amounts of recombinant proteins, particularly in cases where traditional sources
are limited due to safety, cost and/or availability. With the aim of establishing an
rHSA-producing organism for large scale industrial use, studies have been
performed on number of expression systems. The advances made in this aspect
have seen the development rHSA production in a number of expression system
based on a wide variety of living cells and organisms.
II. 2. 1a. Bacterial expression
The first expression system to produce rHSA was E. coli (Lawn et al.,
1981). The expression of heterologous genes in bacteria is the simplest and most
inexpensive way available for obtaining large amounts of desired proteins for
research, industrial and clinical research purposes. The E. coli expression system
has a proven track record since the first expression of somatostatin by Itakura and
co workers (1977), and is considered to be a milestone in genetic engineering.
Bacterial expression is the first expression system introduced in the biotech
industry (recombinant insulin) and gram negative bacteria is still used for
expression of a wide range of biopharmaceutical products.
Generally, there are three forms of foreign proteins expressed in E. coli
bacteria: (i) Fusion proteins are formed by fusing the gene of interest to other
sequences encoding a FLAG epitope tag, a poly-histidine tag, a fluorescent
protein, etc. This can facilitate further protein enrichment (e.g., by
immunoprecipitation), purification, or monitoring, and often affords protection
against degradation by intracellular proteases (Kim et al., 2002; Magliery et al.,

Review of Literature 23
2005; Rosenfeld et al., 2005). (ii) Secreted proteins can accumulate in either the
periplasm or extracellular medium. This process can reduce potential degradation
by intracellular proteases, simplify the purification procedure, and contribute to
the correct formation of spatial structures in proteins secreted into the medium
(Stader and Silhavy, 1990; Makrides, 1996). Although some recombinant
proteins expressed in E. coli can be secreted into the periplasm, they cannot be
secreted into the extracellular medium in most circumstances. (iii) Inclusion
bodies are largely protected from proteolytic degradation by host cell enzymes.
However, the protein product may typically occur as an insoluble, misfolded
inclusion body, so subsequent solubilization and refolding steps arerequired
(Clark, 1998b; Marston, 1986).
rHSA has been expressed as insoluble aggregates in the cytoplasm of
E.coli (Lawn et al., 1981). However, there are distinct disadvantages in
producing HSA in E.coli. For example, conversion of the insoluble product to the
soluble form, which is frequently inefficient in vitro, is both time-consuming and
expensive. Besides, HSA derived from E.coli may be contaminated by endotoxin,
which causes an adverse reaction in patients (Okabayashi et al., 1991).
Saunders and his co-workers have studied the secretion of rHSA from
Bacillus subtilis using bacterial signal sequences (Saunders et al., 1987). The key
advantages of such a route are that the secreted protein is, by definition, soluble
and will not require disruption of the host organism prior to protein purification
(Goodey, 1993). One problem with rHSA accumulation that has been observed is
that a proportion of the secreted rHSA molecules are shorter than the full-length
protein, the nature and amount of these truncated molecules vary according to the
producer organism, growth regime leader sequence adopted (Sleep et al., 1990).
These molecules may be the result of proteolytic degradation of the full-length
protein or incomplete translation of the mRNA. In case of Bacillus subtilis,
however, incorrect processing of rHSA was reported and improvement by gene
manipulation resulted in only minimal secretion. It was found that the efficiency

24 Chapter II
of processing of the leader peptides is inversely correlated with the productivity.
For commercial production of rHSA at an economically feasible level, several
grams of rHSA secretion into the culture broth seemed to be the minimum
requirement for the starting material to be purified (Saunders et al., 1987).
The disadvantages of bacterial expression system include intra-cellular
expression in gram-negative bacteria (although some systems have been designed
for periplasmatic expression of the target protein). Lack of post-translational
modifications and protein folding mechanism limits hetrologus protein
expression.
II. 2. 1b. Yeast expression of rHSA
Currently, Saccharomyces cerevisiae, Kluyveromyces lactis, Hansenula
polymorph and Pichia pastoris (Sleep et al., 1990; Fleer et al., 1991; Kang et al.,
2001; Kobayashi et al., 2000) are the common yeast systems for heterologous
expression of rHSA. The yeast expression system offer high efficiency in terms
of short doubling times, high cell densities, high yields and low fermentation
costs. The production levels ranges from milligrams product per liter of culture to
several grams, depending on choice of yeast strain, type of product and culture
conditions. Yeast is easy to grow in large scale with simple nutritional demands
that lower the media cost. Yeast is given generally regarded as safe (GRAS)
status. In contrast to E. coli, yeast can express correctly folded proteins directly to
the medium, which greatly facilitates purification (low level of host cell proteins,
non-viscous solution, low level of DNA). The rigid cell wall renders the use of all
sorts of bioreactors possible regardless of stirring and shaking mechanisms.
S. cerevisiae has long been known to secrete few or no extracellular
proteases, however investigations carried out in recent years have begun to reveal
that extracellular proteolysis represents one of the most significant barriers to the
secretory production of heterologous proteins in this host organism. The main
problem in the production of rHSA in S. cerevisiae was found to be the
extracellular proteolytic cleavage of the recombinant protein during prolonged

Review of Literature 25
culture (Kang et al., 2000). In another study, rHSA expressed in the yeast S.
cerevisiae accumulated in the insoluble fraction in a denatured form (Quirk et al.,
1989). Extraction of the product requires cell breakage, solubilization, reduction
and renaturation prior to purification (Goodey, 1993). Such downstream
processing would not be practical at the scale envisaged for rHSA and would
certainly not be cost effective. It was reported that the electrophoretic karyotype
of S. cerevisiae may not necessarily be stable. For example, during serial
cultivation of S. cerevisiae wild strain, modifications of electrophoretic karyotype
were observed at 55 generations (Longo and Vezinhet, 1993).
The methylotrophic yeast Pichia pastoris seemed an attractive candidate
as its system usually possesses a high production level. The high stability of
chromosomal DNA in large-scale fermentation is required for stable production,
especially for pharmaceutical products. From this point of view, the gene stability
of a P. pastoris recombinant strain expressing rHSA after long-term serial
cultivation under the conditions of vegetative and non- selective growth was
investigated (Ohi et al., 1998). Isolates from 83 generations showed HSA
production, suggesting that elimination of HSA expression constructs hardly
occurred during serial cultivation. In addition, the cells of each generation
produced the almost same amount of HSA, approximately 110 µg/ml. Thus, P.
pastoris was found to have superior gene stability; therefore, it is regarded as a
suitable host for rHSA gene expression even on an industrial scale.
Kluyveromyces lactis is another promising organism that can be used as
an alternative host due to its good secretory properties. K. lactis is aerobic yeast
which is able to grow on lactose as the sole carbon source. The conditions used
for cultivation of this organism on the large scale have been well established. K.
lactis has been successfully used as an alternative host to S. cerevisiae for
heterologous gene expression, and good transformation systems and stable
multicopy vectors are now available for the genetic manipulation of this yeast
(Fleer, 1992; Romanos et al., 1992). Under the control of the KlADH4 promoter

26 Chapter II
K. lactis cells grow in complex and defined media resulting in a high biomass
production (70 to 80 g dry-cell weight/l), and a significant amount of rHSA
production was observed in fed-batch cultures (1 g/l) when compared to batch
cultures (0.2 to 0.3 g/l) (Saliola et al., 1999).
Yeast cell cultures may be infected with bacteria and fungi. Filtration,
centrifugation and expanded bed technologies are used to harvest the cell culture.
Expanded bed technology combines separation of cells and product and capture
as the target protein binds to the suspended resin, while cells and culture medium
are passing through the column. If filtration or centrifugation is used, the
supernatant is applied directly to the capture column or after ultra-filtration with
the purpose of sample concentration. Hydrophobic interaction chromatography
should be avoided in the capture operation, as anti-foam agents added during
fermentation will bind to the column thus reducing the binding capacity. The
following purification and polishing steps are straight forward and yeast is
regarded as one of the most safe and simple expressions in use. As with E. coli,
this expression system is not able to produce post-translational modified proteins.
Viruses are of no concern with this expression system and relative low levels of
host cell proteins, nucleic acids and endotoxins (e.g. microbial contamination
during downstream operations) should be expected. But, problems are with
correct post-translational modifications, extensive proteolysis of target proteins
can occur. These restrictions limit the use of this expression system from
different heterologous proteins.
II. 2. 1c. Mammalian cells
Mammalian cell expression systems have several advantages over the
systems discussed above in that they promote signal synthesis, process, and can
secrete and glycosylate proteins, particularly eukaryotic proteins. Glycoproteins
are generally synthesized in mammalian cells because common microbial hosts
like E. coli lack the requisite machinery to synthesize appropriate glycoforms of
proteins. Several rodent or human derived cells like 3T3, CHO, BHK, HeLa and

Review of Literature 27
HEpG2 are frequently used in biomedical research for hetrologus protein
expression. Despite the availability of a plenitude of cell lines, nearly 70% of all
recombinant protein therapeutics produced today is made in Chinese Hamster
Ovary (CHO) cells. The current annual sales for biological produced using CHO
cells alone exceed US $30 billion worldwide.
Mammalian cell expression systems can be used for either transient or
stable expression, depending upon the purpose of the expressed product. COS-
cell-based transient expression systems (COS TES) are commonly used for the
former and Chinese hamster ovary (CHO) cell stable expression systems (CHO
SES) for the latter (Edwards and Aruffo, 1993). CHO cells are used for rHSA
fusion protein production to improved half-life of precious proteins. Recombinant
human albumin-linker-erythropoietin (HSA-EPO) fusion protein was expressed
in CHO cells with EPO immunogenicity, which serve as foundation for the
development of long-lasting recombinant HSA-EPO protein (Joung et al., 2009;
Huang et al., 2011).
Since the introduction of continuous mammalian cell lines for the
production of recombinant biopharmaceuticals (e.g. interferon), this expression
system has been the second most used despite relatively high fermentation costs,
slow growth, relatively low yields and potential risk for viral infections. This
major achievement was made possible by a combination of technical progress
(bioreactors) and strict demands to cell line safety and characterization, use of
tested raw materials, good manufacturing practices, in process control and drug
substance/product quality control programs.
The relative low expression levels combined with high prices on culture
media and expensive quality control programs makes it generally more expensive
to produce recombinant proteins in animal cells than in microbial systems. This
system is capable of expression and secretion of complex posttranslational
modified proteins. The availability of good regulatory track record and
commercially available vectors are plus points of this expression system.

28 Chapter II
Mammalian cell-based bioreactors are, however, very expensive to develop and
maintain, and are plagued by complex nutrient requirements, poor oxygen and
nutrient distribution, waste accumulation, contamination by pathogens, and high
sensitivity of cells to shear stress (Wurm, 2004; Zhang et al., 2010).
Proteins produced in cell culture systems are generally purified from the
culture supernatant of lysed cells, an expensive process. Although the relatively
high cost, complicated technology, and potential contamination with animal
viruses of mammalian cell expression have been bottlenecks for its use in large-
scale industrial production, this system is often utilized to express many
heterologous proteins including viral structural protein and bioactive peptide for
specific functional analysis (Zimmer et al., 2003; Nagpal et al., 2004; Ren et al.,
2006).
II. 2. 1d. Transgenic animals
Since Gordon and team (1987) first reported that plasminogen could be
produced in mammary glands of the lactating mouse by transgenic technology,
there has almost been no arguement for the advantages and possibilities when
using transgenic animal bioreactors to generate the valuable pharmaceuticals.
Even complex post-translational modified proteins are successfully expressed in
their native biologically active form, thus making it possible to produce plasma
proteins, human antibodies and other proteins not easily derived from other
sources at industrial scale. It takes 18-33 months from introduction of gene to
production at usable levels. The animal husbandry and milking procedures are
known technologies upgraded to Good Agricultural Practices (GAP). The target
protein is usually expressed in the mammary gland, often at high protein
concentrations (5-50 g/l milk) far exceeding other expression systems.
Human serum albumin was first expressed in mammary glands of
transgenic mice (Huang et al., 2001). Though earlier study by Barsh and team in
1996 gave a suppression in hALB expression (Barsh et al., 1996), the results of
Ying and co-workers (2001) were positive and high level of hALB expression

Review of Literature 29
was reported. The harvest procedure used for cell-based cultures is replaced by
fat and casein removal operations known from the dairy industry. Recently a high
level expression of HSA in milk from transgenic mice was reported (Wu et al.,
2012)
In comparison, the cost of producing 100 kg of protein from transgenic
goats’ milk has been estimated at $105/gram and manufacture using mammalian
cell culture costs $300-3000/gram depending on the product yield and other
operating factors (Young et al., 1997). Two adventitious agents are of concern:
viruses and prions; the latter have never been observed in milk and for that reason
the safety risk is expected low. Never the less, the safety of transgenic animal
derived products has been continuously challenged and is perhaps the main
reason why this expression system has not found widespread usage. The
downstream process very much resembles that of mammalian cell expression
systems including virus inactivation and filtration steps. Relative long period
from introduction of gene to production at usable levels and co-expression of
animal protein is also a major concern with this expression system.
II. 2. 1e. Plant-based bioreactors
The use of plants and plant cell suspension cultures is a relatively new
strategy for the production of recombinant proteins and has been adopted to
express many important pharmaceutical proteins (Brandsma et al., 2010; Shih
and Doran, 2009). Protein production in plants provides a number of advantages
not found in other production platforms. First, a major advantage that all plant
protein production systems have over cell culture systems (including bacteria,
yeast, and mammalian cell culture) is the potential for significant reduction in
cost. It is estimated that protein production in transgenic plants can be as much as
four orders of magnitude less expensive than production in mammalian cell
culture, on a per gram of unpurified protein basis (Dove, 2002). Secondly, plant-
produced proteins are not susceptible to viral or prion contamination that can
harm humans, as is always a concern with animal cell culture (Chebolu and

30 Chapter II
Daniell, 2009). Third, as eukaryotes plants possess the chaperones and cellular
machinery required to fold complex human proteins that bacteria and yeast may
not be able to process properly (Franklin and Mayfield, 2004). Finally, most
species of plants are considered GRAS (generally regarded as safe) (Rosenberg et
al., 2008), meaning that if the protein can be expressed in a bioavailable form;
purification steps could potentially be eliminated altogether.
Studies have shown that rHSA can be produced in leaves of transgenic
tobacco and potato and tobacco suspension cells (Sijmons et al., 1990). The
transgenic constructs contained either the authentic HSA signal peptide or a
preseqeunce from the extracellular PR-S protein from tobacco fused to the coding
region of HSA. The secreted protein was either isolated from leaf tissue or from
plant cell suspension cultures. Chromatographically purified rHSA was analysed
by N-terminal amino acid sequencing. The recombinant protein containing the
human signal peptide was not correctly processed, while the construct with the
plant specific prepropeptide allowed the production of correctly cleaved rHSA in
transgenic plants, and transgenic tobacco suspension cell lines. Plant specific
signal sequence facilitates secretion of 0.25µg rHSA/mg protein into the culture
medium. However, expression levels were, too low for most commercial
applications being 0.02% of the total soluble protein in shoot and young leaves,
and five times lower in roots and tubers.
Farran and his co-workers studied expression of rHSA in tubers of
transgenic potato plants (Farran et al., 2002). HSA gene was cloned under the
B33 promoter of patatin (a specific tuber promoter). The rHSA amino terminal
was fused to a tuber signal peptide (protease inhibitor II) and Agrobacterium
mediated gene transfer to potato leaves was performed. rHSA was detected in the
tubers of transgenic potato plants, the highest accumulation level reaching to
0.2% of the tuber soluble protein. This higher level than the previous report
(Sijmons et al., 1990) was attributed to the differences in the expression cassettes
used for transformation.

Review of Literature 31
Ideally, in order for the bioreactor system to be commercially viable, the
expression levels should be in excess of 5% TSP. The expression levels of
recombinant proteins in transgenic plants are highly variable and range from
levels in the order of 0.01% (Mason et al., 1992) to 46.1% (De Cosa et al., 2001)
of total soluble protein (TSP). Studies have shown that the cost of producing
recombinant proteins in plants could be 10-50-fold lower than producing the
same protein by E. coli fermentation, depending on the crop, and that this gap
will widen as production reaches agricultural scale (Kusnadi et al., 1997). A
recent study in Oryza sativa has come up with high level of rHSA production,
10.58% of TSP (He et al., 2011) which is found to be a promising result for
commercial production.
Generally plants are not hosts to major pathogens that can contaminate
bacterial and mammalian systems (Herbers and Sonnewald, 1999). There is
however at least one pathogen, Pseudomonas aeruginosa, that can cause disease
in both plants and animals (Rhame et al., 1997), and some toxin-producing
bacteria and fungi occur in plants. To date, plant viruses are not known to infect
humans or animals. Potentially pathogenic human and animal viruses are not
capable of replicating in plant cells (Miele, 1997). There are, however, potential
concerns that must be addressed. These include: containment of genetically-
modified plants in the environment (gene flow); allergic reactions to plant protein
glycans and other plant antigens; product contamination by mycotoxins,
pesticides, herbicides and endogenous plant secondary metabolites and regulatory
uncertainty for proteins requiring approval for human drug use.
Although several systems for the expression of rHSA have been tested
and found to be successful, none is yet commercially viable (Desai et al., 2010).
This knowledge has lead research community to search for newer, more efficient
and economical expression systems. Microalgae are expected to be ideal
candidates for this requirement.

32 Chapter II
II. 3. Microalgae
Unicellular green microalgae are of interest in biological research as
simple models for higher plants and from a biotechnological perspective as a
natural source of high value compounds for use in health and medical
applications. The current world production of raw microalgal biomass exceeds
5000 tons, generating an estimated $1–1.25 billion in revenue every year (Pulz
and Gross, 2004; Walker et al., 2005a; Spolaore et al., 2006). Fundamental
research has focused on the alga Chlamydomonas reinhardtii as a model plant
system for the study of photosynthetic and other metabolic pathways, the
biogenesis of subcellular components and cell-cycle control. On the other hand,
species of Dunaliella, Haematococcus, Chlorella and Euglena produce
carotenoids such as β-carotene, astaxanthin and canthaxanthin used as pigments
in food products and cosmetics, vitamin A antioxidant supplements, health food
products, and feed additives for poultry, livestock, fish and crustaceans
(Borowitzka, 1988; Lorenz and Cysewski, 2000). For example, over 80% of the
world’s supply of natural β-carotene comes from the halophilic green alga
Dunaliella salina harvested from large saline ponds (Curtain, 2000).
Microalgae are still mainly used as nutritional supplements for human and
animal consumption due to their high protein and vitamin contents. They have,
however, also elicited interest as production systems for a host of valuable
naturally-produced compounds (Harun et al., 2010). The most notable of these
applications is the cultivation of the green algae Haematococcus pluvialis for its
high-level production scaling up to 50 mg/g dry cell weight (DCW) of
astaxanthin, a carotenoid pigment widely used as a feed additive in fish
aquaculture, and prized by the pharmaceutical and cosmetic industries for its
antioxidant properties (Hyunsuk et al., 2005; Kathiresan and Sarada, 2009).
Microalgae could potentially be a future commercial source of a number
of other vitamins including vitamin C, E and B12 (Borowitzka, 1988).
Pharmacologically active compounds have also been reported in microalgae,

Review of Literature 33
including compounds with anticancer, antimicrobial, antiviral, and various
neurological activities (Schwartz et al., 1990; Cannell, 1993; Codd, 1995; Moore,
1996) as well as lipids that are rich in long-chain polyunsaturated acids of interest
in the treatment of heart disease (Simons et al., 1985) and as anti-inflammatory
agents (Lee et al., 1985).
Given their relatively high oil content, microalgae are of considerable
interest for the biodiesel industry (Williams and Laurens, 2010). Although
microalgal oil contents as high as 75% DCW have been reported, such contents
are highly unusual and obtained only under of specific cultivation conditions, and
therefore do not apply to current industrial systems. The microalgal systems used
in practical applications typically have oil contents closer to 20– 40% DCW. It
has been argued that microalgae may present the only economically and
environmentally viable feedstock for biofuel production. Estimates based on oil
contents of 70% DCW, which some believe to eventually be reachable as
research progresses, place the cost of microalgae-derived biodiesel at 0.72 USD/l,
which currently exceeds the viability of alternate biofuel feedstocks (Chisti,
2007). Considerable progress has been achieved in regards to increasing the oil
yield of microalgae (Courchesne et al., 2009), and several reviews on
microalgae-based biodiesel production have recently been published (Sialve et
al., 2009; Brennan and Owende, 2010; Greenwell et al., 2010; Mata et al., 2010;
Smith et al., 2010; Williams and Laurens, 2010). Several reviews are available on
microalgae based cultivation methods (Carvalho et al., 2006; Eriksen, 2008 and
Xu et al., 2009).
Microalgae have been used in health food and cosmetic industry for a
long time as they represent a natural source of lipids, vitamins, pigments and
antioxidants (Potvin and Zhang, 2010). The harvesting of products from
microalgal culture has a number of advantages from an industry perspective,
which has led to an increased interest in the biotechnology of this group of
organisms. Microalgae are photoautotrophic obviating the need for an exogenous

34 Chapter II
carbon source for energy and making their large-scale culture comparatively
cheap. Many microalgae grow in saline to hyper-saline waters and thus their
large-scale culture does not compete with conventional agriculture for the limited
resources of arable land and fresh water. Furthermore, microalgae such as
Dunaliella, Haematococcus and Chlorella which do not produce toxins and are
classified as food sources. Naturally, the potential for large-scale culture in
conditions unsuitable for conventional crops makes microalgae a desirable target
for both increased production of natural compounds by metabolic engineering
and for exploitation as biological factories for the synthesis of novel high-value
compounds. While the utility of Chlamydomonas as a model organism has been
greatly facilitated by the development of genetic transformation procedures
(Stevens and Purton, 1997), the lack of efficient genetic transformation systems
for other microalgal species has been a major limitation in the manipulation of
these organisms, and current strategies for increasing product yield rely on
standard techniques such as mutagenesis and selection, or trial and error testing
of growth conditions.
The development of systems for algal transformation system has
advanced significantly in recent years; success with Chlamydomonas has led to
the development of transformation systems for related algae, most notably Volvox
carteri, and several diatom species. Success in this area has required novel
approaches as most algal groups have shown significant natural resistance to
many of the antibiotics and herbicides commonly used for selection (Apt et al.,
1996) also the high salt conditions required for the growth of many marine algae
may reduce the activities of antibiotics (Allnutt et al., 2000).
II. 3. 1. Microalgae: as an expression system
In addition to their value as platforms for the production of naturally-
produced compounds, microalgae have been eliciting considerable interest over
the last decade as recombinant protein expression systems, as they combine the
rapid growth and ease of cultivation inherent to many microorganisms with the

Review of Literature 35
ability of plant cells to perform post-transcriptional and post-translational
modifications. Research into transgenic microalgae is fuelled by the worldwide
demand for recombinant proteins and other bio products, the market for which is
growing exponentially and was expected to be $70 billion in 2010 (Pavlou and
Reichert, 2004).
Algae possess a number of advantages over transgenic plant systems for
the production of recombinant proteins. They can be grown in contained
bioreactors, reducing the risk of contamination of the production system by
airborne contaminants, and also protecting the environment from any potential
flow of transgenes into the surrounding ecosystem. Growth in containment also
greatly reduces the potential for loss of the crop due to predation or pathogen
attack. Algae progress from initial transformation to large-scale protein
production in a matter of weeks, compared to timescales on the order of months
or years in higher plants such as corn or tobacco (Franklin and Mayfield, 2004).
As micro-algae are all a single cell type, there should also be less variation in
recombinant protein accumulation, making downstream processing more
uniform.
II. 3. 2. Advantages of therapeutic protein production in microalgae
Although transgenic microalgal technology is still in its infancy,
microalgae may represent the ‘best of both worlds’ by combining simple and
inexpensive growth requirements and capabilities for post-transcriptional and
post-translational processing of plants, with the rapid growth rate and potential
for high-density culture of microorganisms (Walker et al., 2005a). Unicellular
photosynthetic green algae are most commonly used for protein production as
they only require inexpensive salt-based media, carbon dioxide and light for
growth. Most green algae are also classified as generally regarded as safe
(GRAS), making purification and processing of expressed products much less
onerous for many targeted applications. Contrary to transgenic plants which must
be strictly contained to avoid the transfer of transgenic material to surrounding
36 Chapter II
wild-type flora by airborne vectors, microalgae can be cultivated in open
facilities as no such transfer can occurs. On the economics side, based on
recombinant antibody production studies, the cost of production per gram of
functional antibody is $150, $0.05 and $0.002 in mammalian, plant and
microalgal bioreactor systems respectively, making the latter system very
economically attractive (Mayfield et al., 2003). A comparison of different
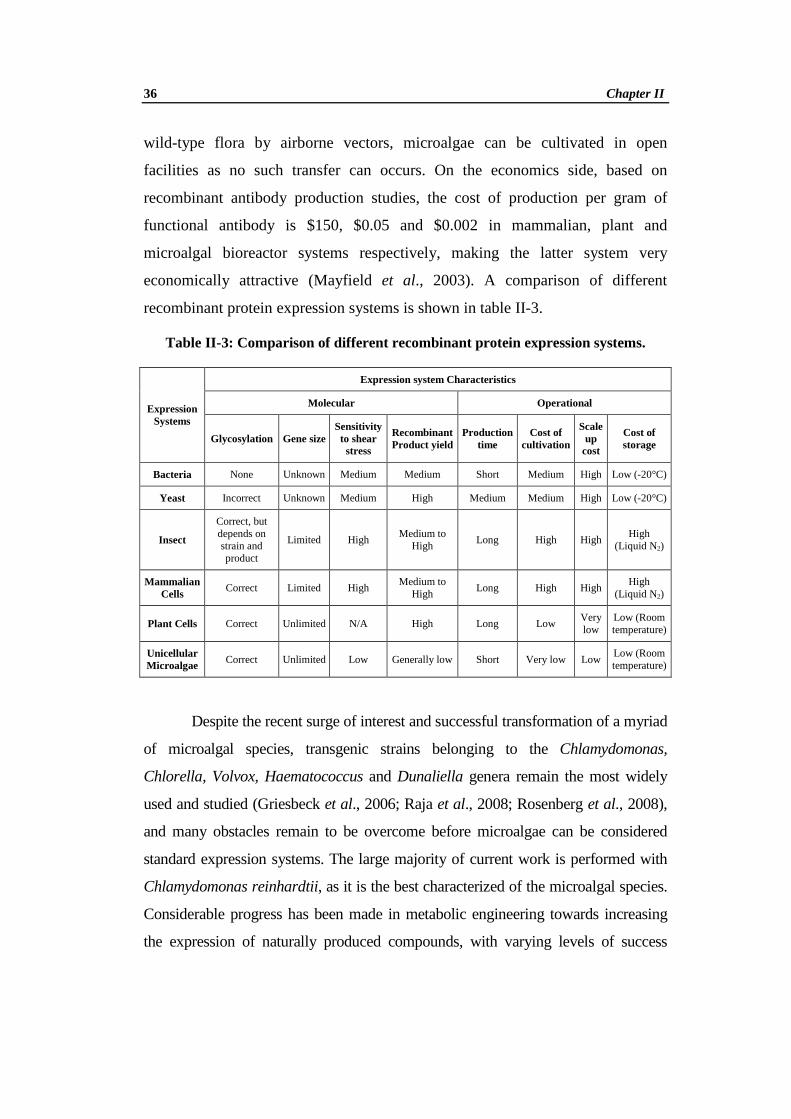
recombinant protein expression systems is shown in table II-3.
Table II-3: Comparison of different recombinant protein expression systems.
Expression Systems
Expression system Characteristics
Molecular Operational
Glycosylation Gene size Sensitivity to shear stress
Recombinant Product yield
Production time
Cost of cultivation
Scale up
cost
Cost of storage
Bacteria None Unknown Medium Medium Short Medium High Low (-20°C)
Yeast Incorrect Unknown Medium High Medium Medium High Low (-20°C)
Insect
Correct, but depends on strain and product
Limited High Medium to
High Long High High
High (Liquid N2)
Mammalian Cells
Correct Limited High Medium to
High Long High High
High (Liquid N2)
Plant Cells Correct Unlimited N/A High Long Low Very low
Low (Room temperature)
Unicellular Microalgae Correct Unlimited Low Generally low Short Very low Low
Low (Room temperature)
Despite the recent surge of interest and successful transformation of a myriad
of microalgal species, transgenic strains belonging to the Chlamydomonas,
Chlorella, Volvox, Haematococcus and Dunaliella genera remain the most widely
used and studied (Griesbeck et al., 2006; Raja et al., 2008; Rosenberg et al., 2008),
and many obstacles remain to be overcome before microalgae can be considered
standard expression systems. The large majority of current work is performed with
Chlamydomonas reinhardtii, as it is the best characterized of the microalgal species.
Considerable progress has been made in metabolic engineering towards increasing
the expression of naturally produced compounds, with varying levels of success

Review of Literature 37
(Rosenberg et al., 2008). Although recombinant protein production is notably
hindered by low expression levels, the continuing development of genetic
engineering tools for microalgae has allowed the expression of fully functional
antibodies (Franklin and Mayfield, 2005; Tran et al., 2009), therapeutics (Boehm,
2007; Weathers et al., 2010), and bactericides (Li and Tsai, 2009) at economically
viable levels. Despite this progress, however, success essentially remains anecdotal
and no wide-ranging system or protocol leading to high-level expression has been
established till date. The recent advances made in C. reinhardtii makes this
microalga an ideal candidate for future studies.
II. 4. Chlamydomonas reinhardtii
Chlamydomonas is a unicellular, freshwater dwelling green microalga.
Highly adaptable, these green algae live in many different environments throughout
the world. Chlamydomonas can grow on a simple medium of inorganic salts in the
light, using photosynthesis to provide energy. They can also grow in total darkness if
acetate is provided as a carbon source for chemosynthesis. Chlamydomonas is the
model organism for physiological, biochemical, and molecular studies of
photosynthesis and flagella function (Harris, 2001), Chlamydomonas reinhardtii
over the years emerged as the predominant laboratory species of Chlamydomonas,
primarily owing to its ability to grow non-photosynthetically with acetate as its sole
carbon source.
The wild-type C. reinhardtii cell averages about 10 mm in diameter (with
significant variation through the cell cycle) and is enclosed within a wall consisting
primarily of hydroxyproline-rich glycoproteins that resemble plant extensins.
Contrary to a few erroneous early reports that have unfortunately been perpetuated in
some textbooks, the C. reinhardtii cell wall does not contain cellulose. The wild-type
wall comprises seven principal layers. Genes for some wall components have been
cloned and sequenced, and many mutants with defects in cell wall biogenesis have
been isolated. Cell wall mutants have found widespread use as recipients for
transformation with exogenous DNA, a process that is much more efficient with

38 Chapter II
wall-less cells. Vegetative cells are normally haploid, but stable diploids can be
selected using auxotrophic markers.
The 15,758 bp mitochondrial genome is linear and contains only a few
genes: cob, cox1, five subunits of mitochondrial NADH dehydrogenase, the
mitochondrial rRNAs (which are fragmented in the DNA sequence); three tRNAs,
and an opening reading frame that resembles a reverse transcriptase (GenBank #
NC001638) (Vahrenholz et al., 1993). In 2002, the complete sequence of the
Chlamydomonas chloroplast genome was released (GenBank #BK000554) which
consists of 203,395 bp (Maul et al., 2002). DOE Joint Genome Institute in 2007
completed the whole genome sequencing of Chlamydomonas. The nuclear genome
size is estimated to be 121-megabase (Mb), generated by whole-genome, shotgun
end sequencing of plasmid and fosmid libraries, followed by assembly into ~1500
scaffolds. The Chlamydomonas nuclear genome comprises 17 linkage groups
presumably corresponding to 17 chromosomes (Merchant et al., 2007).
Chlamydomonas is also highly amenable to genetic analysis with
transformation systems for all three genomes (nuclear, chloroplast and
mitochondrial). The complete characterization of the genomes of the mitochondrion
and the chloroplast along with the recent completion of the nuclear genome sequence
provides unique opportunities in functional genomics and will further add to the
value of this organism as a model system. For the selection of an ideal expression
system, major requirements are the availability of various selectable markers,
reporter genes and promoters. In C. reinhardtii a lot of studies have been carried out
in this regard successfully. In addition to that, a vast number of methods for genetic
manipulation have been developed for C. reinhardtii over the last few years making
it a top candidate for biotechnological applications (Fuhrmann et al., 2002).
II. 4. 1. Therapeutic protein production in Chlamydomonas reinhardtii
Expressing therapeutic proteins in micro-alga C. reinhardtii has several
advantages over other systems employed today. The length of time required from the
initial transformation event to having usable (milligram to gram) quantities of a

Review of Literature 39
protein can be relatively quick in algae. Stable transgenic lines can be generated in as
little at 10 days, and scale-up to production volumes can potentially be achieved a
few weeks after this. In addition, algae can easily be grown in full containment
reducing any concern about environmental contamination of the therapeutic protein.
Moreover, because algae do not have pollen, there is no potential for the introduction
of transgenes into food crops, as potentially could occur in higher plants by gene
flow (via pollen) to surrounding plants (Mayfield et al., 2007). Finally, many green
algae fall into the category of Generally Recognized as Safe (GRAS), meaning they
are safe to eat, and are therefore potentially a source for the oral delivery of
therapeutic proteins, perhaps with little or no purification.
There is a growing interest in exploiting Chlamydomonas for
biotechnological purposes, for the contained and cost-effective expression of
biopharmaceuticals, an area commonly referred to as molecular farming (Franklin
and Mayfield, 2004; Walker et al., 2005a). Several transgenes with a pharmaceutical
or biotechnological background have been successfully expressed in C. reinhardtii
(Table II-4) (modified from Specht et al., 2010). They can be grouped into proteins
involved in metal metabolism, antigenic proteins, and antibodies. Whole
Chlamydomonas cells could be used as edible vaccines in industrial farming
processes superseding the purification of the immunogen. It has been suggested that
an oral application of such modified algal strains in food or drinking water can also
stimulate antibody production in mammals.

40 Chapter II
Table II-4: Recent successes in therapeutic protein production in Chlamydomonas reinhardtii
Gene expressed
Function Expression
level Source
HSV8-lsc First mammalian protein expressed, antibody
Detectable (Mayfield et al., 2003)
CTB-VP1 Cholera toxin B subunit fused to foot and mouth disease VP1
3% TSP (Sun et al., 2003)
HSV8-scFv Classic single-chain antibody 0.5% TSP (Mayfield et al., 2005)
hMT-2 Human metallothionine-2 Detectable (Zhang et al., 2006)
hTRAIL Human tumor necrosis factor-related apoptosis-inducing ligand
~0.67% TSP (Yang et al., 2006)
M-SAA Bovine mammary-associated serum amyloid
~5% TSP Manuell et al., (2007)
CSFV-E2 Swine fever virus E2 viral protein ~2% TSP (He et al., 2007)
hGAD65 Diabetes-associated anutoantigen human glutamic acid decarboxylase 65
~0.3% TSP (Wang et al., 2008)
ARS2-crEpo Human erythropoietin fused to ARS2 export sequence
100 lg/l culture (Eichler-Stahlberg et
al., 2009)
83K7C Full-length IgG1 human monoclonal antibody against anthrax protective antigen 83
0.01% dry algal biomass
(Tran et al., 2009)
IgG1 Murine and human antibodies (LC and HC) Detectable (Tran et al., 2009)
VP28 White spot syndrome virus protein 28 ~10.5% TSP (Surzycki et al., 2009)
CTB-D2 D2 fibronectin-binding domain of Staphylococcus aureus fused with the cholera toxin B subunit
0.7% TSP (Dreesen et al., 2010)
10NF3, 14FN3
Domains 10 and 14 of human fibronectin, potential antibody mimics
14FN3: 3% TSP 10FN3:
detectable (Rasala et al., 2010)
M-SAA-Interferon β1
Multiple sclerosis treatment fused to M-SAA
Detectable (Rasala et al., 2010)
Proinsulin Blood sugar level-regulating hormone, type I diabetes treatment
Detectable (Rasala et al., 2010)
VEGF Human vascular endothelial growth factor isoform 121
2% TSP (Rasala et al., 2010)
HMGB1 High mobility group protein B1 2.5% TSP (Rasala et al., 2010)
MSP1 Vaccine against Malaria Detectable (Deauville et al., 2010)
Pfs25, Pfs28 Elicits Antibody that inhibit Malarial infection
Detectable (Gregory et al., 2012)

Review of Literature 41
II. 4. 2. Strategies for foreign gene expression in C. reinhardtii
Three basic strategies are commonly used for the expression of foreign
protein in C. reinhardtii; i) Chloroplast transformation ii) Mitochondrial
transformation and iii) Stable nuclear transformation
II. 4. 2a. Chloroplast Transformation
Stable chloroplast transformation was first accomplished in 1988 when
Boynton and co-workers restored the photosynthetic capacity of a C. reinhardtii
mutant by cell bombardment with high-velocity micro-projectiles coated with the
wild-type gene. Once inside the organelle, the DNA integrates into the genome by
homologous recombination (Boynton et al., 1988). While the Chlamydomonas
chloroplast contains around 80 copies of its genome, homoplasmic transformants, in
which all genome copies contain the change, can be obtained after several rounds of
culturing on selective medium (Rochaix, 1997). Several approaches have been used
to select cells following chloroplast transformation and a detailed list of selectable
markers is presented in section. Early selection methods were based on cloned
chloroplast genes used to rescue photosynthesis mutants (Boynton et al., 1988).
Chloroplast genes that confer resistance to antibiotics or herbicides have also been
widely used (Newman et al., 1992). However, a major breakthrough in chloroplast
transformation came with the development of dominant selectable markers based on
bacterial genes for antibiotic resistance (Goldschmidt- Clermont, 1991).
Foreign protein expression in the chloroplast of C. reinhardtii for the first
time involved the bacterial neomycin phosphotransferase (Blowers et al., 1989) and
β-glucuronidase genes (Blowers et al., 1990), both driven by C. reinhardtii
chloroplast promoters. To date, only a handful of therapeutic proteins have been
expressed in algal chloroplasts. Production of recombinant proteins in chloroplasts
also possesses several unique attributes. At present transgenic proteins can
accumulate to much higher levels in the chloroplast than when expressed from the
nuclear genome, mainly because plastids lack gene silencing mechanisms and other
mechanisms that reduce recombinant protein production from nuclear encoded genes

42 Chapter II
(Bock, 2007). Chloroplasts can be transformed with multiple genes in a single event,
due to the availability of multiple insertion sites, as well as an ability to process
polycistronic transcripts, allowing an entire gene cassette to be regulated by a single
promoter (Rymarquis et al., 2006; Bock, 2007). Additionally, proteins produced
within the chloroplast are not glycosylated (Franklin and Mayfield, 2005), which can
prove useful in many applications such as producing antibodies that are similar to
native antibodies in their ability to recognize their antigen, but whose lack of
glycosylation prevents them from recruiting killer cells (Tran et al., 2009).
II. 4. 2b. Mitochondrial Transformation
The mitochondrial genome of C. reinhardtii has also been transformed using
the biolistic method. Function was restored to a respiratory-deficient C. reinhardtii
mutant (dum-1) with a 1.5 kb deletion in the apocytochrome b gene (cob) by
bombardment with partially purified total mitochondrial DNA (Randolph-Anderson
et al., 1993) and selection for growth in the dark on acetate-containing media. Since
the mitochondrial genome of C. reinhardtii is small and contains very few genes, it is
unlikely to be developed further as a system for genetic engineering.
II. 4. 2c. Nuclear Transformation
The nuclear genome of C. reinhardtii was first transformed using the biolistic
method over 23 years ago, making it the first photosynthetic organism in which both
the nuclear and chloroplast genomes were transformed (Debuchy et al., 1989;
Boynton et al., 1988). However, using this method, transformants were recovered at
consistently low levels and the foreign DNA was usually introduced into the genome
in multiple copies (Kindle et al., 1989). Today, glass beads have superseded
biolistics as the most commonly used method for the nuclear transformation C.
reinhardtii. This method is simple and yields high numbers of transformants, but it
requires cells lacking a cell wall. The nuclear genome of wild-type C. reinhardtii
cells has also been transformed using silicon carbide (SiC) whiskers (Dunahay,
1993) and electroporation (Brown et al., 1991).

Review of Literature 43
Chloroplasts are generally preferred for foreign protein expression in
microalgae due to high expression levels. But nuclear expression is necessary for the
expression of complex proteins which require post-transcriptional and post-
translational processing and targeting despite low yields. Transgene expression from
the nuclear genome offers several advantages over chloroplast expression, such as
glycosylation and other post-translational modifications and heterologous protein-
targeting to sub-cellular locations or for secretion (Leon-Banares, 2004).
Nuclear expression of foreign proteins remains very low, for reasons that are
as yet not well understood. Positional effects, RNA silencing, a prohibitively
compact chromatin structure and non-conventional epigenetic effects have been
proposed as possible causes. Several advancements have been developed for
improved nuclear transgene expression. For nuclear-expressed proteins, degradation
can be minimized by targeting protein synthesis to the ER rather than to the cytosol
(Conrad and Fiedler, 1998), a strategy that lead to a 104-fold increase in recombinant
growth factor expression in tobacco (Wirth et al., 2004). A recent study describes the
selection of highly expressed nuclear transgenes following UV-induced mutations of
transformed strains. Using this protocol, yields of foreign proteins accounting for
0.2% total soluble protein (TSP) were achieved, which is relatively high for nuclear
expression (Neupert et al., 2009). A possible strategy to minimize proteolytic
degradation of proteins requiring modification would therefore be to target the
nuclear-expressed proteins to the chloroplast for storage (Potvin and Zhang, 2010).
Inserting introns from native genes in heterologous sequences under the control of
that gene's promoter has been shown to increase protein yields. Expression of
recombinant genes in the nuclear genome of C. reinhardtii also improved following
the insertion of the first RBCS2 intron, which has been shown to contain an enhancer
element (Lumbreras et al., 1998; Berthold et al., 2002).
Agrobacterium mediated genetic transformation of C. reinhardtii was
reported in 2004 by Kumar and co-workers (Kumar et al., 2004). The
Agrobacterium-mediated transformation method offers several advantages over

44 Chapter II
direct gene transfer methods including its feasibility to transfer large DNA
fragments, low copy number of transgene integration with little rearrangement,
preferential integration into transcriptionally active regions and its simplicity (Cha et
al., 2011). Recent advances made in the Agrobacterium mediated genetic
transformation technology offer great opportunities for higher and more consistent
transgene expression in C. reinhardtii.
II. 4. 3. Molecular Factors Influencing Transgene Expression in C. reinhardtii
Choice of suitable selectable marker genes, reporter genes and efficient
promoters are key factors governing transformation. These molecular tools can
greatly enhance, reduce or even silence transgene expression. The studies conducted
so far in this regard facilitated the improvement of Chlamydomonas transformation
studies in recent years.
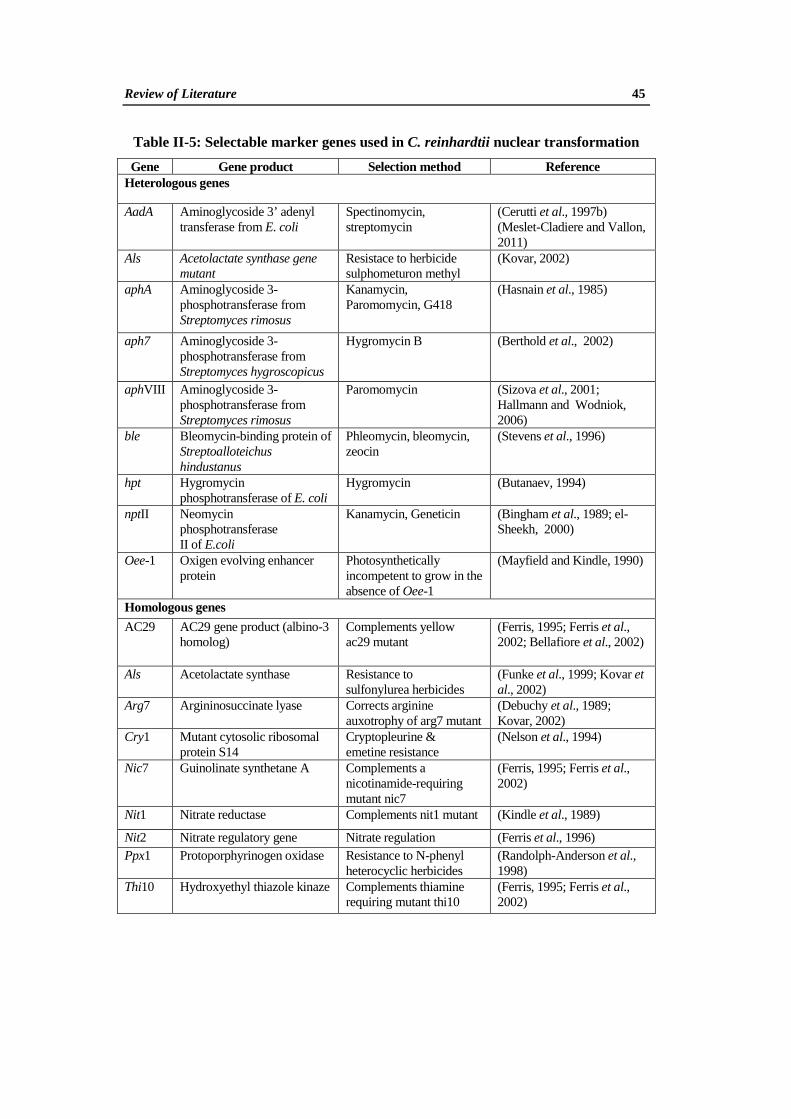
II. 4. 3a. Selectable Marker Genes
An important aspect of transformation is the choice of an appropriate
selective agent and its corresponding resistance gene. Selectable marker gene may
encode an enzyme capable of either detoxifying a phytotoxic compound (negative
selection) or metabolising a substrate (e.g. a carbon source) that wild-type cells
cannot utilize (positive selection). In Chlamydomonas transformation studies a
number of homologous and heterologous selectable marker genes have been used for
both nuclear (Table II-5) and chloroplast (Table II-6) transformation. The
heterologous genes introduced into the nuclear genome of C. reinhardtii are often
poorly expressed. Possible reasons for this failure include: epigenetic suppression of
gene expression, inefficient transcription due to lack of the appropriate promoter
and/or enhancer elements, the lack of introns required for efficient RNA processing,
and poor translation of foreign mRNA due to biased codon usage (Kindle and
Sodeinde, 1994; Stevens et al., 1996; Cerutti et al., 1997a). The nptII gene was only
weakly expressed and direct selection for kanamycin resistance proved unsuccessful
(Hall et al., 1993).
Review of Literature 45
Table II-5: Selectable marker genes used in C. reinhardtii nuclear transformation
Gene Gene product Selection method Reference Heterologous genes
AadA
Aminoglycoside 3’ adenyl transferase from E. coli
Spectinomycin, streptomycin
(Cerutti et al., 1997b) (Meslet-Cladiere and Vallon, 2011)
Als
Acetolactate synthase gene mutant
Resistace to herbicide sulphometuron methyl
(Kovar, 2002)
aphA
Aminoglycoside 3- phosphotransferase from Streptomyces rimosus
Kanamycin, Paromomycin, G418
(Hasnain et al., 1985)
aph7
Aminoglycoside 3- phosphotransferase from Streptomyces hygroscopicus
Hygromycin B
(Berthold et al., 2002)
aphVIII
Aminoglycoside 3- phosphotransferase from Streptomyces rimosus
Paromomycin
(Sizova et al., 2001; Hallmann and Wodniok, 2006)
ble
Bleomycin-binding protein of Streptoalloteichus hindustanus
Phleomycin, bleomycin, zeocin
(Stevens et al., 1996)
hpt
Hygromycin phosphotransferase of E. coli
Hygromycin
(Butanaev, 1994)
nptII
Neomycin phosphotransferase II of E.coli
Kanamycin, Geneticin
(Bingham et al., 1989; el- Sheekh, 2000)
Oee-1
Oxigen evolving enhancer protein
Photosynthetically incompetent to grow in the absence of Oee-1
(Mayfield and Kindle, 1990)
Homologous genes AC29
AC29 gene product (albino-3 homolog)
Complements yellow ac29 mutant
(Ferris, 1995; Ferris et al., 2002; Bellafiore et al., 2002)
Als
Acetolactate synthase
Resistance to sulfonylurea herbicides
(Funke et al., 1999; Kovar et al., 2002)
Arg7
Argininosuccinate lyase
Corrects arginine auxotrophy of arg7 mutant
(Debuchy et al., 1989; Kovar, 2002)
Cry1
Mutant cytosolic ribosomal protein S14
Cryptopleurine & emetine resistance
(Nelson et al., 1994)
Nic7 Guinolinate synthetane A
Complements a nicotinamide-requiring mutant nic7
(Ferris, 1995; Ferris et al., 2002)
Nit1 Nitrate reductase Complements nit1 mutant (Kindle et al., 1989)
Nit2 Nitrate regulatory gene Nitrate regulation (Ferris et al., 1996) Ppx1
Protoporphyrinogen oxidase Resistance to N-phenyl heterocyclic herbicides
(Randolph-Anderson et al., 1998)
Thi10
Hydroxyethyl thiazole kinaze Complements thiamine requiring mutant thi10
(Ferris, 1995; Ferris et al., 2002)

46 Chapter II
Table II-6: Selectable marker genes used in C. reinhardtii chloroplast transformation
Gene Gene product Selection method Reference Heterologous genes aadA
Aminoglycoside 3’ adenyl transferase from E. coli
Spectinomycin, streptomycin
(Goldschmidt-Clermont, 1991)
aphA6
Aminoglycoside 3 phosphotransferase from Streptomyces rimosus
Kanamycin, Amikacin
(Bateman and Purton, 2000)
Cat
Chloramphenicol acetyltransferace
Resistace to chloramphenicol
(Brown et al., (1991)
nptII
Neomycin phoshotransferace gene from E. coli
Resistance to G418 (Butanaev, 1994)
nitl Nitrate reductase Nitrate reduction (Kindle et al., 1989) Homologous genes atpA,B,E
α-, β-, & ε-subunits of the CF1 ATP synthase complex
Restores Photosynthesis
(Lemaire and Wollman, 1989; Kindle et al. 1991; Leu et al., 1992; Boynton et al., 1988)
chlL/chlN
Protochlorophyllide reductase Yellow in dark
(Choquet et al. 1992; Suzuki and Bauer, 1992)
petA & D
Subunits I and IV of cytochrome b6/f complex
Restores photosynthesis
(Buschlen et al., 1991; Baymann et al., 1999)
psaA Photosystem I reaction centre protein
Restore photosynthesis
(Choquet et al., 1988)
psaB Photosystem I reaction centre protein
Restores photosynthesis
(Bingham et al., 1991)
psaC Iron sulphur protein of photosystem I
Restores Photosynthesis
(Takahashi et al., 1991)
psbA
Photosystem II reaction centre protein D1
Restores photosynthesis & confers DCMU resistance on wild type
(Przibilla et al., 1991; Hatano- Iwasaki et al., 2000)
psbC P6 photosystem II core protein
Restores photosynthesis
(Rochaix et al., 1989)
psbD D2 protein of photosystem II Restores photosynthesis
(Erickson et al., 1986)
rbcL Rubisco large subunit Restores photosynthesis
(Newman et al., 1991)
rps4 &rps12
Ribosomal protein S4 & S12
Streptomycin dependence to independence
(Liu et al. 1989; Randolph- Anderson et al. 1995)
tscA Photosystem 1 compex Restores photosynthesis
(Goldschmidt-Clermont, 1991)
16S rDNA
16S rRNA
Streptomycin, Spectinomycin, Kanamycin resistance
(Newman et al., 1990)
23S rDNA 23S rRNA Erythromycin (Newman et al., 1992)

Review of Literature 47
The most commonly used selectable markers for Chlamydomonas nuclear
transformation are Chlamydomonas genes that complement mutations in the
corresponding structural genes. The most widely used markers have been Nit1,
which encodes nitrate reductase (Fernandez et al., 1989; Kindle et al., 1989), and
Arg7, which encodes argininosuccinate lyase (Debuchy et al., 1989). A mutated
version (Cry1) of the RPS14 gene encoding ribosomal protein S14, was the first
dominant selectable marker allowing direct transformation of wild-type
Chlamydomonas and confers resistance to the translation inhibitors emetine and
cryptopleurine (Nelson et al., 1994). Recently, transformation of C. reinhardtii
with a mutated acetolactate synthase gene (ALS) under the control of the native
RbcS2 promoter allowed recovery of colonies resistant to the herbicide
sulfometuron methyl (Kovar et al., 2002). Drug resistance markers that have also
been used in Chlamydomonas including bacterial genes that confer resistance to
bleomycin (ble gene encodes a bleomycin-binding protein) (Stevens et al., 1996)
and spectinomycin/streptomycin (aadA gene, encodes aminoglycoside adenine
transferase; Cerutti et al., 1997b). The bacterial aadA gene is also the most
commonly used dominant marker for C. reinhardtii chloroplast transformation
(Goldschmidt-Clermont, 1991).When fused to the 5` and 3` untranslated regions
of endogenous chloroplast genes it was shown to confer resistance to
spectinomycin and streptomycin in both Chlamydomonas (Goldschmidt-
Clermont, 1991) and higher plants (Svab and Maliga, 1993). Two strategies for
removal of the aadA cassette from the chloroplast genome have also been
developed (Fischer et al., 1996). The first relies on homologous recombination
between direct repeats flanking the aadA cassette to produce direct excision of
the cassette when selection is removed. A second is to co-transform with a
plasmid containing an essential chloroplast gene disrupted by the aadA
cassetteand with a plasmid containing a modified non-essential chloroplast gene.
Under selective conditions the second mutation is transferred to all chloroplast
copies while the aadA insertion remains heteroplasmic and is subsequently lost
when selection is removed. Another selectable marker gene is the bacterial

48 Chapter II
aminoglycoside 3-phosphotransferase gene (aphA6), which allows for the direct
selection of chloroplast transformants on selective media containing kanamycin
or amikacin and can also be used to modify or inactivate specific chloroplast
genes by insertional inactivation (Bateman and Purton, 2000).
II. 4. 3b. Promoters
One important aspect in the development of a transformation system has
been the choice of promoter to drive expression of the transgene. One of the most
widely used promoters in plant molecular biology is the cauliflower mosaic virus
35S (CaMV 35S) promoter. Although the CaMV 35S promoter drives strong and
constitutive expression in most dicotyledonous and some monocotyledonous
plants (Benfey et al., 1990), it has not shown to be a useful promoter in most
algal species. Algal transformation has been most successful using promoters
derived from highly expressed algal genes (Table II-7). A widely used promoter
for Chlamydomonas transformation is derived from the 5’ untranslated region of
the C. reinhardtii ribulose bisphosphate carboxylase/oxygenase small subunit
(RbcS2) (Stevens et al., 1996). It was also shown that transformation frequency
was significantly increased when Chlamydomonas introns (particularly the first
intron of RbcS2) were introduced into the coding region of the ble selectable
marker gene (Lumbreras et al., 1998). This intron appears to contain a
transcriptional enhancer element as it can act in an orientation-independent
manner and is effective when placed either upstream or downstream of the
promoter. Synthetic promoters have also been developed by fusing the promoter
from the Chlamydomonas Hsp70A (heat shock protein 70A) gene to other
Chlamydomonas promoters. The Hsp70A promoter serves as a transcriptional
enhancer of promoters RbcS2, β2-tubulin and Hsp70B leading to high-level
expression under inducing conditions (Schroda et al., 2000).

Review of Literature 49
Table II-7: Promoters shown to drive transgene expression in C. reinhardtii
Promoter Derivation Regulation Reference
Nuclear
Amt
Chlorella virus adenine methyltransferase
Constitutive
(Kang et al., 2000)
Ca1/ Ca2 Carbonic anhydrase CO2 and light (Villand et al., 1997)
CabII-1
Chlorophyll a/b-binding protein of photosystem I
Light
(Blankenship and Kindle 1992)
CaMV 35S
Cauliflower mosaic virus 35S promoter
Constitutive
(Kumar et al., 2004; Ruecker et al., 2008)
Hsp70A Heat shock protein 70A Heat shock, light
(Schroda et al., 2000)
Hsp70B Heat shock protein 70B Heat shock, light
(Schroda et al., 2000)
Nia1 (Nit1)
NAD(P)H nitrate reductase
Light, nitrate
(Ohresser et al., 1997; Loppes et al., 1999)
Nos
Agrobacterium tumefaciens nopaline synthase
Constitutive
(Hall et al., 1993)
PsaD Photosystem II subunit D Light (Fischer and Rochaix, 2001)
RbcS2
Small subunit of ribulose-1,5 bisphosphate carboxylase/
Oxygenase
Light
(Stevens et al., 1996)
TubB2 β2-tubulin Deflagellation (Davies et al., 1992)
Hsp70A-RbcS2
Heat shock protein 70A- RbcS2
Heat shock, light
(Wu et al., 2008)
SV40 Animal adenovirus SV40 Constitutive (Butanaev, 1994)
Chloroplast
atpA
α-subunit of the ATP
synthase CF1 complex
Light
(Blowers et al., 1990)
atpB
β-subunit of the ATP
synthase CF1 complex
Light
(Blowers et al., 1990)
chlL UV resistance Light (Zang et al., 2006)
petD
Subunit IV of
cytochrome b6/f complex
Light
(Sakamoto et al., 1993)
rbcL
Large subunit of ribulose
bisphosphate carboxylase
Light
(Klein et al., 1994)
rrn16 16S rRNA Light (Klein et al., 1992)

50 Chapter II
II. 4. 3c. Reporter Genes
Transformation systems are generally developed using an efficient
reporter gene, which encodes a protein that can be easily detected and quantified
in transgenic lines. The most commonly used reporter genes in higher plants are
uidA, which encodes β- glucuronidase (GUS) and the green fluorescent protein
gene (gfp) from the bioluminescent jellyfish Aequorea victoria (Chalfie et al.,
1994). There has been considerable difficulty to date in the expression of
heterologous reporter genes in the nuclear genome of Chlamydomonas. The uidA
gene has not been a successful reporter gene for nuclear transformation of C.
reinhardtii even when under the control of a native promoter (Blankenship and
Kindle, 1992).
The use of GFP as a reporter gene in both the nuclear and chloroplast
genome of Chlamydomonas has been demonstrated (Fuhrmann et al., 1999;
Franklin et al., 2002). Unfortunately, the unmodified GFP gene under the control
of heterologous promoters gave poor expression when used as a reporter for
nuclear transformation of Chlamydomonas (Fuhrmann et al., 1999). Reasons for
poor expression may include the predominance of A/T rich codons in the native
gene and the possible presence or absence of sequence motifs that regulate
transcription or the processing and targeting of the transcript. To overcome these
limitations a modified gfp gene has been synthesised utilising the codon
preference of C. reinhardtii nuclear genes (Fuhrmann et al., 1999). Additional
modifications known to modify the spectral properties of GFP-protein for
fluorescence imaging were also introduced. This modified gene was fused in-
frame to the ble gene and expressed under the control of the RbcS2 promoter.
GFP fluorescence was observed in the nucleus demonstrating nuclear
accumulation of the GFP-BLE fusion protein (Fuhrmann et al., 1999). GFP-
fusions have proven to be a useful tool for the study in vivo of dynamic processes
such as flagellar and centriole assembly and cell cycle events in Chlamydomonas
(Ruiz-Binder et al., 2002).

Review of Literature 51
A modified gfp gene for Chlamydomonas chloroplast transformation has
also been synthesized by optimizing its codon usage to reflect that of the major
chloroplast encoded proteins (Franklin et al., 2002). Chloroplasts transformed
with this modified gene under the control of the C. reinhardtii rbcL 5’- and 3’-
UTRs were shown to accumulate ~80-fold more GFP than those transformed
with the native gene. The GFP accumulated to ~0.5% of the total soluble protein
(TSP), ~50-fold higher than reports of uidA (GUS) expression under the control
of the same promoter (Ishikura et al., 1999). In contrast to the high-levels of GFP
visible in the nucleus GFP fluorescence in the chloroplast was not easily
visualized using fluorescence microscopy (Franklin et al., 2002). This is most
likely due to the chlorophyll and other pigments within the chloroplast absorbing
much of the incident light targeted to GFP, or some of the light being emitted by
the GFP being reabsorbed by the chloroplast pigments. GFP has been visualized
in the chloroplasts of higher plants (Reed et al., 2001), but only when the levels
of GFP accumulated to over 5% of the TSP, approximately 10-fold higher than
levels presently obtained in C. reinhardtii. The most encouraging development in
chloroplast transformation is the recent report of successful GFP visualization in
the Chlamydomonas chloroplast using a gfp gene that had been modified for
tobacco chloroplast expression (Komine et al., 2002).
The most commonly used reporter genes for Chlamydomonas
transformation are homologous genes (Table. II-8). Two Chlamydomonas genes
confer easily discernible phenotypes. The ARS, gene encodes the periplasmic
enzyme arylsulfatase, which is normally expressed only under conditions of
sulphur starvation. Arylsulfatase activity can be detected in colonies by spraying
plates with the chromogenic substrate 5- bromo-4-chloro-3-indolyl sulphate
(XSO4) or assayed in solution with p-nitrophenyl sulphate or α-naphthylsulphate
(Davies et al., 1992). The second commonly used reporter gene PC1, encodes
NADPH: protochlorophyllide oxidoreductase (POR) and is responsible for light-
dependent protochlorophyll reduction (Li and Timko, 1996). Wild-type strains of
Chlamydomonas make chlorophyll and are green when grown in both the light
52 Chapter II
and the dark. Strains containing mutations in both PC1 and in genes of the light-
independent protochlorophyllide-reduction pathways are unable to produce
chlorophyll under any light conditions. After transformation with pc1 they form
green colonies in the light only.
Table II-8: Commonly used reporter genes for transformation studies in C. reinhardtii
Gene Gene Product Assay Method Reference
Nuclear
Ars
Arylsulfatase
Chromogenic detection using X-SO4 as a substrate
(Davies et al., 1992)
GFP
Green fluorescent protein of Aequora victoria
Luminescence or antibody based
(Fuhrmann et al., 1999)
PC1
NADPH: protochlorophyllide
Oxidoreductase
Assay protochlophyllide
reductase mRNA or enzyme activity
(Li and Timko., 1996)
Cgluc luciferase of Gaussia princeps Luminescence (Ruecker et al., 2008)
Rluc
luciferase of Renilla reniformis
Luminescence (Fuhrmann et al., 2004)
Chloroplast
aadA
Aminoglycoside 3’ adenyl transferase from E. coli
AAD assay
(Zerges et al., 1997)
GFP
Green fluorescent protein of Aequora victoria
Fluorescence or antibody based
(Franklin et al., 2002; Komine et al., 2002)
LUC
Luciferase from Renilla reniformis
Luminescence or antibody based
(Minko et al., 1999)
uidA
β-glucuronidase of E. coli
Histochemical or flurometric
(Sakamoto et al., 1993; Ishikura et al., 1999; el-Sheekh 2000)
II. 4. 4. Methods used for the transformation of C. reinhardtii
Appropriate transformation method used for the delivery of foreign DNA
to target microalgal genome greatly influences transformation efficiency and
expression. Research works over the years have developed in a number of

Review of Literature 53
methods for genetic manipulation of C. reinhardtii making it a top candidate for
biotechnological applications (Harris, 1989; Fuhrmann, 2002). Although some of
these methods may not have significantly changed since their initial development,
they are still being applied and studied. Recently a number of studies have
presented methods that seem to offer advantages over earlier techniques.
II. 4. 4a. Cell wall-deficient strains
The use of cell wall-deficient strains, or the removal of the cell walls from
wild-type strains, greatly increases the number of transformants recovered
following transformation. Protocols for cell wall removal have been developed
which facilitate the study of microalgae. These protocols involve the mating of
mating type plus (mt+) and mating type minus (mt−) gametes of C. reinhardtii.
The specific cell–cell recognition resulting from flagellar interaction leads to the
release of enzymes, autolysin or lysin, that cause cell wall degradation. These
enzymes can be purified and used as a pre-treatment to transformation. A detailed
protocol for production and purification of these enzymes is given by Buchanan
and Snell (1988) and a detailed study of the mating process was more recently
reported by Hoffmann and Beck (2005).
II. 4. 4b. Particle bombardment
Bombardment of target cells with DNA-coated metallic particles is a
widespread, simple, effective and highly reproducible transformation method.
This method has been successfully employed for the transformation of most
standard cellular expression systems, and it is therefore not surprising that it is
also useful for the study of microalgae. The main drawback of the particle
bombardment method is the cost of the required specialized equipment. Although
the number of transformants recovered following particle bombardment can be
low, it remains the most effective method for the transformation of chloroplasts,
as it allows for the delivery of multiple copies of recombinant DNA through both
the cellular and chloroplast membranes, increasing the chance for a successful
integration event to occur (Boynton and Gillham, 1993). This method has been

54 Chapter II
shown to be effective for the stable nuclear (Mayfield and Kindle, 1990) and
chloroplast (Boynton et al., 1988; El Sheekh, 2000; Ramesh et al., 2011)
transformation of C. reinhardtii, the transformation of Volvox carteri
(Schiedlmeier et al., 1994), Chlorella sorokiana (Dawson et al., 1997), C.
ellipsoidea (Chen et al., 1998) and C. kessleri (El-Sheekh, 1999) species,
transient transformation of H. pluvialis (Teng et al., 2002) and the stable nuclear
transformation of the diatom Phaeodactylum tricornutum (Apt et al., 1996).
Recent work has shown that the particle bombardment method is also effective
for the transformation of more complex algal species, such as the multicellular
Gonium pectorale (Lerche and Hallmann, 2009).
II. 4. 4c. Glass beads method
A simple and effective transformation method consists of agitating cell
wall-deficient microalgal cells with recombinant DNA, polyethylene glycol
(PEG) and glass beads, which greatly increases transformation efficiency.
Despite the drop in cell viability to 25% following agitation with the beads, a
nuclear transformation efficiency of about 103 transformants/µg DNA was
achieved using this method (Kindle, 1990) and an efficiency of 50
transformants/µg DNA was achieved for the transformation of C. reinhardtii
chloroplasts (Kindle, 1990). Compared to the particle bombardment method, the
glass beads method is simpler, more efficient for nuclear transformations, and
much less expensive as it does not require specialized equipment. A recent study
showed that the glass beads method is also more efficient than particle
bombardment for the transformation of Dunaliella salina (Feng et al., 2009).
II. 4. 4d. Silicon carbide whisker method
A similar protocol, using silicon carbon whiskers instead of glass beads to
pierce cells, has also been used successfully (Dunahay, 1993; Wang et al., 1995).
Cells are transformed by mixing with the SiC whiskers and DNA, and vortexing
briefly. Although the exact mechanism for whisker-mediated transformation is
unknown, it is known that fractured SiC crystals readily form sharp cutting edges.

Review of Literature 55
The surface of SiC whiskers is negatively charged which probably results in there
being little affinity between whiskers and DNA in a neutral pH medium. It has
been suggested therefore that the whiskers do not carry the DNA into the treated
cells but function as needles that facilitate DNA delivery by cell perforation and
abrasion during mixing (Wang et al., 1995). The cell viability following agitation
is much improved, but due to low transformation efficiencies, high cost of
materials, and health concerns associated with the handling of the whiskers, the
glass beads are generally preferred. However, SiC whiskers have since been
reported to be extremely hazardous to humans and therefore are rarely used.
II. 4. 4e. Electroporation
Electroporation or electropermeabilization is a transformation technique
that use induction of macromolecular uptake by exposing cell walls to high
intensity electrical field pulses,. The effectiveness of microalgal electroporation,
was first reported by Brown et al., (1991). Electroporation specifically disrupts
lipid bilayers, leading to efficient molecular transport across the plasma
membrane (Azencott et al., 2007). Efficient electroporation mediated
transformation was achieved in both wild-type and cell wall-deficient strains
(Brown et al., 1991). The transformation efficiency of electroporation is two
orders of magnitude higher than the glass beads method, and only requires
relatively simple equipment (Shimogawara et al., 1998). Important parameters
affecting the effectiveness of electroporation include field strength, pulse length,
medium composition, temperature and membrane characteristics (Brown et al.,
1991) as well as the concentration of DNA (Wang et al., 2007a). Electroporation
was successfully used for the transformation microalga D. salina (Geng et al.,
2004; Sun et al., 2005; Wang et al., 2007b; Sun et al., 2008; Feng et al., 2009),
D. viridis (Sun et al., 2006) and D. tertiolecta (Walker et al., 2005b), C.
reinhardtii (Tang et al., 1995; Shimogawara et al., 1998; Kovar et al., 2002;
Ladygin, 2003, 2004), Chlorella sp. (Chow and Tung, 1999; Wang et al., 2007a),
and Nannochloropsis oculata (Chen et al., 2008; Li and Tsai, 2009).

56 Chapter II
II. 4. 4f. Agrobacterium tumefaciens-mediated transformation
Agrobacterium tumefaciens is widely used for genetic transformation of
plants because of its natural ability to transfer foreign DNA into the host plant
genome. It is also a significant plant pathogen that causes crown gall disease on
several agronomically important species, including grape vines, stone fruit, and
nut trees (De Cleene and De Ley, 1976). Transformation results from the
stimulation of cell division by products encoded by T-DNA transferred from
Agrobacterium to the target cell. The T-DNA and virulence (vir) regions are
located on the tumour inducing plasmid (pTi). The vir system processes and
transfers any DNA between the short flanking repeats that delimit the T-DNA
(R&L T-DNA borders), making Agrobacterium an efficient DNA delivery
system (Akhond and Machray, 2009).
The process of T-DNA transfer is initiated by the induction of bacterial
virulence (vir) genes by the phenolic compounds produced and released by the
wounded plant cells through the VirA–VirG two-component signal transduction
system (Zupan et al., 2000). It has been shown that majority of the
dicotyledonous plants produce such phenolic compounds and they have been
routinely transformed by Agrobacterium (Gelvin, 2000). Monocotyledons, which
generally do not produce vir-inducing compounds, could be transformed by the
exogenous addition of such molecules like acetosyringone (AS) (Hiei et al.,
1997). Agrobacterium gene transfer has also been extended to fungi (De Groot et
al., 1998) and human (HeLa) cells (Kunik et al., 2001).
The prospects of A. tumefaciens mediated transformation were first
reported in microalgae by Kumar and co-workers (Kumar et al., 2004). The
microalga C. reinhardtii was successfully transformed with uidA (β-
glucuronidase), gfp (Green Fluorescent Protein) and hpt (hygromycin
phosphotransferase) genes in presence of acetosyringone (AS). Later Kumar and
Rajam reported successful transformation of C. reinhardtii even in the absence of
AS (Kumar and Rajam, 2007), with about fifty-fold increase in transformants

Review of Literature 57
compared to the commonly used glass beads method for genetic transformation in
C. reinhardtii.
The major difficulty associated with the nuclear transformation of C.
reinhardtii was gene silencing as in many other organisms (Manuell and
Mayfield, 2006). Agrobacterium mediated transformation has been reported to
overcome the major limitations of other transformation procedures, providing
stable integration at lower copy number, potentially leading to fewer problems
with transgene co-suppresion, instability (Hansen et al., 1997). Integration of
transgenes into the nuclear genome of C. reinhardtii and their expression without
any silencing was obtained in the studies conducted by Kumar and co-workers
(2004, 2007). This can be attributed to the possible advantages of Agrobacterium
T-DNA to be targeted to and integrated at potentially transcribable regions of the
genome (Hiei et al., 1994). Similar promising results were observed in the
Agrobacterium mediated genetic transformation of other microalgal species,
Haematococcus pluvialis (Kathiresan et al., 2009), Dunaliella bardawil (Anila et
al., 2011), Nannochloropsis sp (Cha et al., 2011), Schizochytrium (Cheng et al.,
2012) and Chlorella vulgaris (Cha et al., 2012).
Though different methods are available for the genetic transformation of
C. reinhardtii, like particle bombardment, glass beads, silicon carbide etc,
transformation frequency and stability of gene in subsequent generations have
been a limiting factor. But in case of Agrobacterium mediated transformation,
cell lines showed high stability in gene integration for up to 18 months in C.
reinhardtii (Kumar et al., 2004).
The Agrobacterium mediated genetic transformation technique is a
simple, stable and efficient method for transgene experiment studies.
Undoubtedly, the recent development of this transformation procedure is
economically valuable for microalgal species and the results obtained in recent
years evidence a promising future. Taking these facts into consideration the
present work was taken up to develop C. reinhardtii as an expression system for

58 Chapter II
the production of recombinant human serum albumin (rHSA) through
Agrobacterium mediated genetic transformation.


















