
Regulation of -catenin phosphorylation and nuclear...
42
1 © 2014. Published by The Company of Biologists Ltd. Journal of Cell Science Accepted manuscript JCS Advance Online Article. Posted on 4 February 2014
Transcript of Regulation of -catenin phosphorylation and nuclear...

1
Regulation of β-catenin phosphorylation and nuclear/cytoplasmic transport by
APC and its cancer-related truncated form.
Authors,
Lili Wang1,2,*, Xiaoyong Liu2, Ekaterina Gusev2, Chuanxin Wang1#, and François Fagotto2, *
Affiliations:
1Department of Clinical Laboratory, Qilu Hospital, Shandong University, Jinan, China, and
2Department of Biology, McGill University, Montreal, Canada
*To whom correspondence should be addressed, François Fagotto, Department of Biology, McGill University 1205 Dr. Penfield Ave., room W5/15 Montreal, QC H3A 1B1, Canada Email, [email protected]
#, Equal last author, Chuanxin Wang, Department of Clinical Laboratory, Qilu Hospital, Shandong University 107 Wenhua Xi Road Jinan, 250012, P.R. China Email, [email protected]
© 2014. Published by The Company of Biologists Ltd.Jo
urna
l of C
ell S
cien
ceA
ccep
ted
man
uscr
ipt
JCS Advance Online Article. Posted on 4 February 2014

2
Abstract
We report the first direct analysis of the endogenous β-catenin phosphorylation activity in colon
cancer SW480 cells. By comparing parental SW480 cells that harbor a typical truncated APC form,
cells expressing full length APC and APC-depleted cells, we provide the formal demonstration that
APC is necessary for β-catenin phosphorylation, both for priming at residue serine 45 and for the
subsequent phosphorylation of residues 33, 37 and 41. Truncated APC still sustains a surprisingly high
phosphorylation activity, which requires β-catenin binding to 20AA repeats for APC, thus providing
biochemical explanation for the precise truncations found in cancer cells. We also discovered that
most of the β-catenin phosphorylation activity is associated with a dense insoluble fraction. We finally
examined the impact of full length and truncated APC on β-catenin nuclear transport. We observed
that β-catenin was transported much faster than previously thought. While this fast translocation was
largely insensitive to the presence of wild type or truncated APC, the two forms appeared to limit the
pool of β-catenin available for transport, which could have an impact on β-catenin nuclear activities in
normal and cancer cells.
Introduction
The Wnt-β-catenin pathway is a major signaling route that controls embryonic patterning and tissue
homeostasis. Its deregulation is involved in many cancers. The pathway is in particular over-activated
in virtually all colon cancer, due to mutations of the adenomatous polyposis coli (APC) tumor
suppressor gene, which could actually represent the initiating event for this type of cancer (Polakis,
2007). The pathway revolves around β-catenin, which, among many other functions, is responsible for
the transduction of Wnt signals into gene regulation through its ability to act as transcriptional
coactivator (Valenta et al, 2012). β-catenin appears to be controlled in the cytoplasm by a complex
built on the scaffold protein Axin. In the absence of Wnt signal, β-catenin is inactivated by the Axin
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

3
complex, also called “β-catenin destruction complex”, Soluble β-catenin is captured by Axin, and
sequentially phosphorylated, first by casein kinase 1 (CK1) of serine residue 45, which serves as
priming for the subsequent action of glycogen synthase 3 (GSK3) on three consecutive residues,
threonine 41, serine 37 and serine 33. N-terminally phosphorylated β-catenin is then ubiquitinated and
rapidly degraded. Upon activation of the pathway by Wnt ligand binding to Frizzled and LRP5/6
receptors, the complex is inhibited by a still poorly understood mechanism (Li et al, 2012; Roberts et
al, 2012; Taelman et al, 2010). This results in the accumulation of soluble β-catenin that can enter the
nucleus, where it interacts with transcription factors of the TCF/LEF1 family to regulate a series of
target genes (Valenta et al, 2012).
Behind this seemingly simple picture of the Wnt pathway, the actual mechanisms that regulate β-
catenin remain highly controversial (Hernandez et al, 2012; Li et al, 2012; Roberts et al, 2012; Taelman
et al, 2010). The role of APC is in particular quite unclear, and the consequences of the mutations
found in cancer cells still ill defined. What appears well established is the fact that the recruitment of
the two kinases, CK1 and GSK3, and their substrate, β-catenin, within a single complex strongly
increases the efficiency of the reaction. It is thus commonly accepted that Axin, CK1 and GSK3
constitute the minimally required “core complex”. Many studies have shown that APC is also essential,
since β-catenin accumulates when APC is mutated or depleted (Munemitsu et al, 1995). That the APC
function is quite proximal to the activity of the Axin complex is strongly suggested by APC’s ability to
associate with both β-catenin and Axin (Fagotto et al, 1999; Hart et al, 1998; Hinoi et al, 2000; Kishida
et al, 1998; Rubinfeld et al, 1993; Su et al, 1993). APC binds directly β-catenin via two different types
of short repeats, called 15 and 20 amino acid repeats. Affinity to the 20AA repeats is strongly increases
by phosphorylation (Ha et al, 2004; Rubinfeld et al, 1996). APC also binds directly to Axin, via short
“SAMP” motifs (Behrens et al, 1998) and indirectly via the Arm repeats (Roberts et al, 2011). APC has
therefore been considered to be a bona fide constituent of the destruction complex (Ha et al, 2004;
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

4
Hinoi et al, 2000; Xing et al, 2003). In one model, Axin and APC are thought to act as coordinate
scaffolds that ensure the specificity of β-catenin phosphorylation and of its regulation by the Wnt
pathway. In vitro experiments using pure recombinant proteins have indeed demonstrated that APC
further increases the efficiency of the Axin-GSK3 complex to phosphorylate β-catenin (Hinoi et al,
2000). The presence of low and high affinity binding sites led to a refined version of this model, which
states that different sites are used depending on the β-catenin levels (Ha et al, 2004). The fact that
phosphorylated 20AA repeats compete with Axin for β-catenin (Ha et al, 2004) suggested a different
model, where APC helped dissociating phosphorylated β-catenin from Axin, creating a catalytic cycle
of binding and release of the substrate (Kimelman & Xu, 2006). Others have suggested that APC acts
either upstream, by gathering or even transporting cytosolic β-catenin to the complex (Bienz, 2002), or
on the contrary downstream of the phosphorylation reactions, recruiting the ubiquitin ligase βTrCP to
the complex (Li et al, 2012; Su et al, 2008). A last interesting variant presented APC functioning both
in β-catenin phosphorylation and its subsequent release from the complex (Roberts et al, 2011).
This uncertainty partly stems from the fact that the central process in this pathway, regulation of β-
catenin phosphorylation, has only been studied in vitro, using purified proteins, or inferred from
observation of steady state levels, rather than by direct measurement of the endogenous kinase
activity. The in vitro data, while demonstrating the role of Axin and APC in making β-catenin
phosphorylation more efficient, also left open a relative wide range of possible reactions. For instance,
GSK3 could phosphorylate β-catenin even in the absence of APC or CK1 (Hinoi et al, 2000), and
even in the absence of Axin, as a matter of fact (Yost et al, 1996), while APC showed some enhancing
effect even in the absence of Axin (Hinoi et al, 2000). Whether APC could also act on the priming
reaction was not tested. A direct transposition of these in vitro data to the in vivo situation is far from
straightforward, since we still know very little about the actual concentrations, activities, associations
and localization of the endogenous components. This raises the question of whether all components
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

5
are required in vivo for the entire process, or whether different partial complexes may be in charge of
partial tasks. Different complexes may also be active under different conditions, for instance low basal
β-catenin levels or high levels during Wnt stimulation (Ha et al, 2004), or even in different cellular
compartments.
Relating the in vitro and in vivo situations would necessarily require investigating specific reactions under
endogenous conditions. Measurement of levels of endogenous phosphorylated β-catenin and
detection of phosphorylated β-catenin at particular cellular locations is clearly not sufficient, and can
be interpreted in opposite ways (e.g. local enrichments could be considered on the contrary as sites of
stabilized β-catenin (Faux et al, 2010). A few studies measured GSK3 activity in the context of the
Wnt pathway, but used substrates irrelevant to the pathway (Stambolic et al, 1996; Taelman et al,
2010). Such measurements almost certainly included activities independent of the Axin-APC complex.
It is even likely that the complex may exclude or at least be poorly accessible to substrates other than
β-catenin. Thus, none of these available data provides adequate information about the actual function
of the pathway. Note also that one still needs to solve the simple question of whether APC is an
intrinsic component of the machinery or merely a modulator. Axin has been indeed considered to be
the limiting factor in the pathway, based on measurements of the relative concentrations in Xenopus
egg extracts (Lee et al, 2003), but see (Tan et al, 2012), and Axin overexpression was found to rescue
lower β-catenin signaling in APC-mutated cancer cells, suggesting that APC may be dispensable when
Axin levels are sufficiently high (Behrens et al, 1998; Cliffe et al, 2003; Faux et al, 2008; Hart et al,
1998); Nakamura et al, 1998).
Considering the many unknowns about APC biology, it comes to no surprise that the precise effect of
the mutations found in colon cancers is similarly unclear and matter of lively debates. The
overwhelming majority of the mutations so far identified in both sporadic (Kinzler & Vogelstein,
1996) and familial colon cancers lead to early termination and thus production of a truncated protein.
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

6
Although recessive, these mutations are not random, since many are located in a small region in the
middle of APC coding sequence, indicating that there is a selection in cancer for maintaining the
expression of an intact N-terminus half of the protein (Furuuchi et al, 2000). APC is a very large
(>300kDa) and complex protein, constituted of multiple domains interacting with a variety of cellular
components, and it has been implicated is several different cellular processes, from transcriptional
regulation to mitotic spindle positioning and cell migration (Nathke, 2004). However, the loss-of-
function produced by the deletion of its C-terminus in cancer cells has been definitively linked to the
Wnt pathway (Polakis, 2000). Most truncations remove all but the first of the seven 20AA repeats, as
well as the Axin-binding SAMP repeats (Fig.1A, Kohler et al, 2008). The loss of most of the high
affinity β-catenin binding sites or the inability to bind Axin were thus prime suspects for the abnormal
accumulation of soluble non-phosphorylated β-catenin and for the activation of its transcriptional
targets observed in colon cancer cells. These models have been challenged, however, and different
alternative hypothesis have been proposed, including downstream effects on nuclear localization,
retention or transcription (Bienz, 2002; Krieghoff et al, 2006; Sierra et al, 2006).
As a first step to attempt to clarify some of these issues, we have set a kinase assay to monitor
endogenous activities for β-catenin S45 priming and for S33/S37/T41 phosphorylation. This study
proposes to address the following basic questions, is APC required for β-catenin phosphorylation,
and, if so, for which of the two kinase reactions? What is the effect of APC truncation on β-catenin
phosphorylation?
Another important and still poorly explored question is the subcellular location of the active
complexes responsible for β-catenin phosphorylation. Besides the absence of direct information on
the enzymatic activity, even localization of the components of the complex has not been solidly
established. Detection of endogenous proteins by immunofluorescence has suffered from the lack of
specificity of the antibodies (see e.g. Brocardo et al, 2005), while exogenously expressed constructs
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

7
tend to aggregate (e.g. Fagotto et al, 1999; Faux et al, 2008). In addition, soluble cytosolic components
are known to leak during fixation (Liu & Fagotto, 2011). Studies by cell fractionation, on the other
hand, have been plagued by systematic co-purification of nuclear and plasma membrane insoluble
fractions and by the omission of adequate markers to validate the identity of the fractions (Liu &
Fagotto, 2011). We have recently established a fractionation protocol that cleanly separates the major
cellular components that may be involved in β-catenin regulation (Liu & Fagotto, 2011). Here we have
used this protocol combined to our kinase assay to compare the activity of the various compartments.
The second key process tackled in this study is β-catenin nuclear transport. There have been
conflicting views about a possible role of APC in β-catenin nuclear localization. APC was proposed to
mediate β-catenin export, carrying it via a piggy-back mechanism and that β-catenin nuclear
accumulation in colon cancer cells was due to failure of the truncated APC to fulfill this function
(Bienz, 2002), although the nuclear localization of truncated APC was later contested (Henderson &
Fagotto, 2002) and may be an artifact due to unspecific antibody staining (Brocardo et al, 2005). A
diametrically different mechanism proposed that β-catenin freely shuttles through the nuclear pore
(Fagotto et al, 1998; Koike et al, 2004; Sharma et al, 2012). Kinetic analysis of transport showed that
overexpression of APC or other binding partners such as Axin rather decreased nuclear import
(Krieghoff et al, 2006), supporting the hypothesis that these proteins influenced β-catenin distribution
by sequestration in particular compartments (Krieghoff et al, 2006; Roberts et al, 2011). It remained
however possible that the observed retention was due to the artificial elevated levels of APC and that
endogenous APC may act differently. The effect of truncated APC on β-catenin nuclear translocation
also remained unexplored. We thus included in this study the analysis of β-catenin transport in cells
expressing physiological levels of wild type APC, truncated APC, or cells depleted of APC.
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

8
Results
Characterization of components of the Wnt pathway in SW480 and SW480APC cells
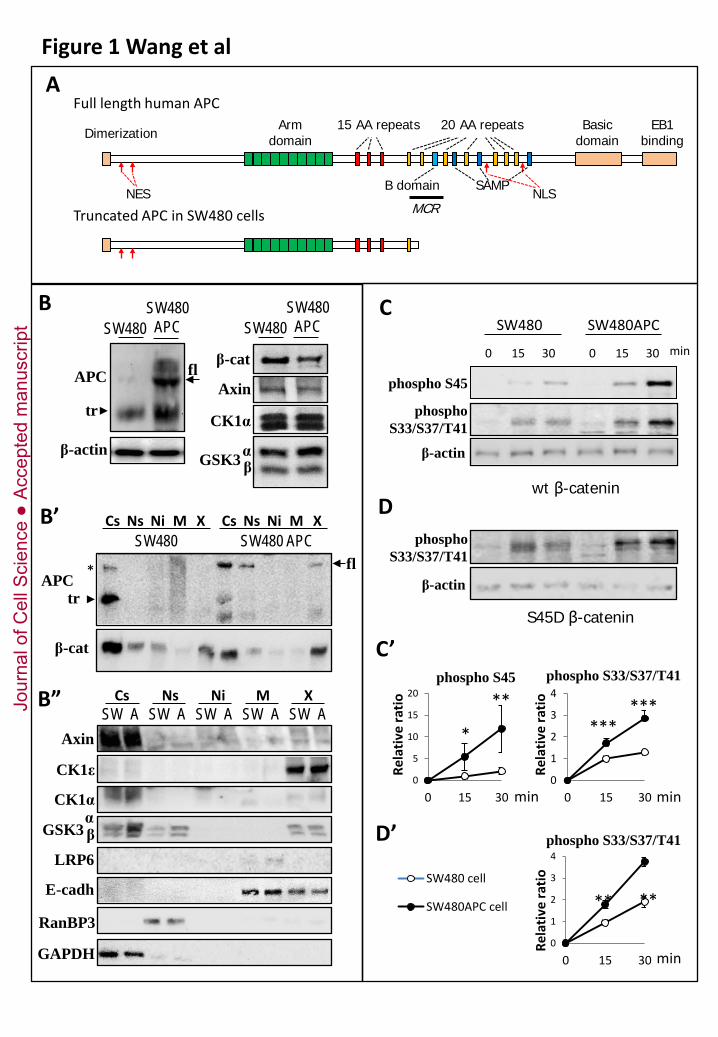
In this study, we compared parental SW480 cells with a SW480 cell line stably expressing full length
APC (Faux et al, 2004). We verified that SW480APC cells expressed relatively low levels of full length
APC (Fig. 1B, arrow). Several lower fragments were also detected, including a major band, which,
according to its migration, probably represented the endogenous truncated APC (Fig. 1B, arrowhead).
Note that APC levels are controlled by proteasomal degradation, both wild type and truncated forms
being target for ubiquitination (Choi et al, 2004). Thus one may not necessarily expect identical levels
of the truncated form in the absence of presence of wild type APC. Note also that contribution from a
cleavage product of full the full length protein is also possible, since similar fragments are commonly
observed various cell lines wild type for APC (e.g. Kishida et al, 1998, Liu and Fagotto, unpublished).
The signal was globally decreased in siRNA-transfected cells, demonstrating that all bands were related
to APC (Fig.2C). We also compared the levels of the major components of the Axin complex
(examples in Fig.1B, quantification in supplementary Fig.S1). Axin, GSK3 and CK1 were expressed
roughly at similar levels, with the exception of GSKα, slightly higher in SW480APC. On the contrary,
steady-state levels of β-catenin were lower, consistent with the original report (Faux et al, 2004).
We used our cell fractionation protocol to examine the subcellular distribution of these components.
This protocol yields five fractions (Fig.1B’,B”), cytosol (Cs), nucleosol (Ns), nuclear insoluble fraction
(Ni), membranes (M) and dense insoluble material (X). This latter fraction is mainly composed of
cytoskeleton components, with a minor contribution of nuclear material (Liu & Fagotto, 2011). Most
components of the destruction complex were distributed in similar patterns in parental and APC-
rescued cells (Fig.1B’, quantification in supplementary Fig.1SB). The bulk of Axin, GSK3 and CK1α
were found in the cytosol. On the contrary, CK!ε was strongly enriched in fraction X. Full length APC
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

9
was mostly cytosolic, with a second significant pool in fraction X and low levels in the nucleosol. In
both cell lines, truncated APC was enriched in the cytosol, with smaller pools in nuclear and dense
insoluble fractions (Fig.1B”). As for β-catenin (Fig.1B’), cytosolic levels were high in parental SW80
cells, consistent with previous reports (Munemitsu et al, 1995), but was significantly lower in
SW480APC cells. The dense insoluble fraction contained the second largest pool in both cells lines.
Nucleosolic levels were also slightly lower in SW480APC cells. This characterization whoed that
fraction X constituted the second major subcellular pool for all the components of the destruction
complex. This fraction is generally discarded in cell fractionation experiments, since it has been long
considered as “cell debris”, and has never been analyzed in the context of the Wnt pathway. Note that
Triton X100 was present in the last step of the fractionation, thus fraction X constituted a bona fide
“detergent-insoluble” fraction. We performed APC immunoprecipitation for the Triton-soluble
fraction of the cells (thus fraction X not included). It showed robust co-precipitation of all
components of the destruction complex in both cell lines (supplementary Fig.S2).
β-catenin phosphorylation in SW480 and SW480APC cells
We established a specific in vitro kinase assay to monitor the endogenous activity responsible for β-
catenin N-terminal phosphorylation. Recombinant β-catenin was used as substrate, at the
concentration of 100nM, which corresponds to the estimated cytosolic levels in non-stimulated cells
(Lee et al, 2003). At the dilutions used in this assay, contribution from endogenous β-catenin present
in the cell extracts was negligible (supplementary Fig.S3A). Priming at residue S45 and subsequent
phosphorylation at sites S33/S37/T41 were detected with specific antibodies (pS45 and
pS33/S37/T41) by quantitative immunoblotting (see Materials and Methods). Note that pre-
absorption of the anti-phospho-S33/S37/S41 antibody was essential, because this polyclonal antibody
showed strong cross-reactivity toward non-phosphorylated β-catenin (see Material and Methods).
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

10
Antibody concentrations were optimized to obtain a linear response over the relevant range of signal
intensities.
Comparison of kinase activities in SW480 and SW480APC crude extracts readily yielded an
unambiguous result: the activity was significantly higher in SW480APC cells, both for S45 and for
S33/S37/T41 (Fig.1C). The differences (~ five folds for S45, ~ two folds for S33/S37/T41) were
very reproducible (Fig.1C’), highlighting the robustness of the assay and the consistency of
endogenous activities. Similar difference between the two cell lines were also observed when β-catenin
concentration was raised to 1μM (Supplementary Fig.S3B).
Although not absolutely required for phosphorylation by GSK3 in in vitro experiments, S45 priming is
nevertheless considered essential in vivo. Since full length APC rescue enhanced priming significantly
more than S33/S37/T41 phosphorylation, we wanted to determine whether APC was directly
required for the latter reaction, or if increased S33/S37/T41 phosphorylation was simply a
consequence of accelerated priming. We isolated the S33/S37/T41 phosphorylation step by using a
“constitutively primed” phospho-mimetic β-catenin variant, S45D. We found that the kinase activity
toward S45D β-catenin was also higher in SW480APC cell extracts (Fig.1D). We conclude that full
length APC is required for full activity of both phosphorylation steps. The fact that S33/S37/T41
phosphorylation of wild type and S45D-β-catenin was enhanced to a similar degree indicated that
priming was not limiting in SW480 cells.
The observed two fold increase in overall β-catenin phosphorylation appeared surprisingly modest if
one assumed that APC was absolutely required. Various explanations could account for this rather
mild enhancement: a) Consistent with in vitro experiments, Axin could be sufficient for β-catenin
phosphorylation. APC would then only improve the efficiency of the reaction. b) Axin could be
limiting in these cells, and APC expression could enhance phosphorylation only up to the maximal
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

11
rate allowed by Axin. c) Alternatively, the APC mutation of SW480 cells may not constitute a
complete loss-of-function and the resulting truncated APC may still supply part of APC function in β-
catenin phosphorylation.
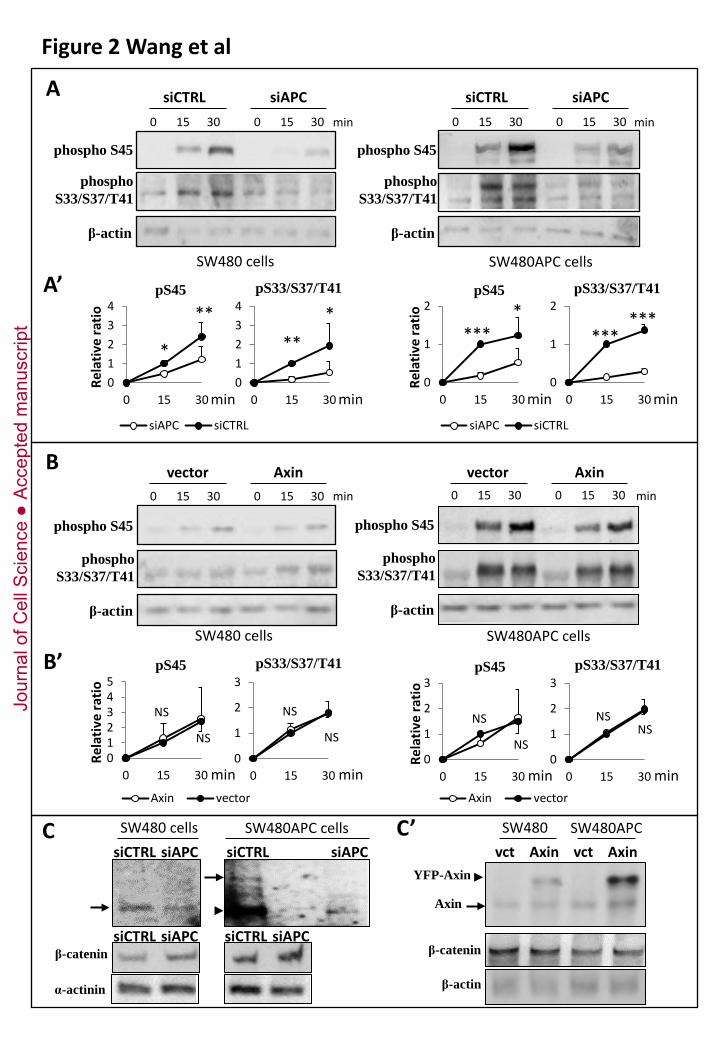
To discriminate between these possibilities, we depleted both wild type and truncated APC by RNA
interference (Fig.2A). If truncated APC is not a complete loss-of-function, one would expect that its
depletion would further decrease β-catenin phosphorylation. Otherwise, depletion would have no
effect, and might even increase activity, as the truncated fragment could potentially have an inhibitory
effect on this process. Transfection of APC siRNA led to a ~ 50% depletion of truncated APC in
SW480 cells and ~ 80-90% depletion of full length APC SW480APC cells (Fig. 2C). Both activities
toward S45 and S33/S37/T41 were further reduced, both in SW480 cells and in SW480APC cells. The
decrease was roughly proportional to the reduction in the levels of full length, respectively truncated
APC. Steady-state levels of endogenous β-catenin were also consistently increased (Fig.2C). We
inferred that the presence of APC (or at least of its N-terminal half) was absolutely required for β-
catenin phosphorylation.
We then asked whether Axin was limiting, in which case one would expect that higher levels may
boost β-catenin phosphorylation in SW480APC cells and perhaps even compensate for the absence of
full length APC in SW480 cells. However, mild Axin overexpression (2.3+/-0.4 folds in parental
SW480 cells and 2.5+/-0.9 in SW480APC cells, Fig.2C’) failed to stimulate of β-catenin
phosphorylation (Fig. 2B). On the contrary S45 phosphorylation in SW480 cells was slightly but
reproducibly decreased. We conclude that, contrary to common assumptions, Axin is not limiting, at
least not in SW480 cells and for the specific reaction of β-catenin phosphorylation. These results
further support the notion that APC is crucial and cannot be substituted by increasing Axin levels.
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

12
Altogether, these experiments led to two important conclusions: They showed that APC is required
for both phosphorylation steps, and they also demonstrated that truncated APC has still a significant
activity.
Direct binding to APC is required for β-catenin phosphorylation
We verified that the role of APC in β-catenin phosphorylation required direct APC-β-catenin binding.
For this purpose, we used recombinant β-catenin variants with point mutations that specifically
impaired binding to APC. To distinguish between binding to 15 AA and 20 AA repeats (Fig.1A), we
tested three separate mutations (Fig.3), APCΔ15(R386A), defective in binding to 15AA repeats, and
APCΔ20(K345A) and APCΔ20(W383A), both defective in binding to the 20AA repeats (von Kries et
al, 2000). Loss of binding to a specific type of repeat was confirmed by in vitro pull down
(supplementary Fig.S4). These mutant substrates were tested for both S45 and S33/S37/T41
phosphorylation, in SW480 and in APC-rescued cells. In all cases, the activity was lower than for wild
type β-catenin. The difference was relatively mild for the mutant lacking binding to the 15AA repeats,
but quite strong for the two other mutants. Double 345/383 mutation led to a slight but not
statistically significant decrease in the S33/S37/T41 phosphorylation activity compared to the single
mutants. We conclude that both types of interactions are required for full activity, with a stronger
requirement for the 20 AA repeats. These results also highlight the importance of the only 20AA
repeat left in the truncated APC of SW480 cells (see discussion).
β-catenin phosphorylation occurs mainly in an insoluble fraction
We determined the subcellular distribution of β-catenin phosphorylation activity using the above-
mentioned cell fractionation protocol. The distribution of the kinase activity was largely similar in
SW480 and SW480APC cells, and for all three measured activities, i.e. pS45 priming and S33/S37/T41
phosphorylation on wild type β-catenin as well as S33/S37/T41 phosphorylation on constitutively
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

13
primed S45D β-catenin (Fig.4). Quite surprisingly, the dense insoluble fraction “X” was by far the
most active pool accounting for ~ 60-70% of the total cell activity. Comparatively, the cytosol showed
only a modest activity (10-20%), despite the fact that it contained the largest pools of APC, Axin,
CK1α and GSK3 (Fig.1B’, B”). The other significant pool was the nuclear insoluble fraction, which
matched and even surpassed the cytosol in the case of S45D phosphorylation (Fig.4B,D). Nucleosol
and membrane fractions showed low to negligible activity.
Effect of APC and APC truncation on β-catenin nuclear transport
To directly investigate the effect of truncated and wild type APC on β-catenin nuclear transport, we
performed FRAP experiments on SW480 cells transfected with YFP-β-catenin (Fig.5). The import and
export kinetics in parental SW480 cells were roughly similar to those measured in HEK293 and NIH
3T3 cells (Krieghoff et al, 2006; Sharma et al, 2012). However, the use of the spinning confocal
microscope allowed us to obtain information about the initial phase of recovery, which had not been
studied so far. We measured surprisingly fast transport kinetics, both for import and for export (Fig.5
and Table 1). The resulting recovery kinetics clearly fitted a two phase association model, with kinetics
sensibly similar in both directions (Table 1). The first phase of translocation was extremely rapid (K ~
0.1/sec, half-life <10 sec), in fact almost as fast as for GFP, used to monitor free diffusion of a small
protein (Fig.5D,E, supplementary Fig.S5, and Table 1). The second phase was an order of magnitude
slower (K ~ 0.01/sec, half-life > 1 min). Neither import nor export seemed to reach full recovery
after 5 minutes, but approached a plateau around 60-80%. These observations generally suggested the
existence of at least three potential β-catenin pools, one free to diffuse through the nucleopores, the
second subjected to partial retention, and a third pool apparently unavailable for nuclear translocation
on this time scale. Note that β-catenin was transported more efficiently than Cherry-NLS, used as
reporter for classical importin-mediated import (supplementary Fig.S4G,H).
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

14
We also compared transport kinetics of APC-rescued and APC-depleted cells (Fig. 5B-E and Table 1).
For import, the kinetics of the fast phase was sensibly the same under all conditions. We observed
however differences in the contribution of the slow phase (% recovery - % fast phase), which was
larger in APC-rescued cells and smaller in APC-depleted cells. The kinetics of this phase was also
significantly slower in the presence of full length APC, with a half live shifted from ~ 1 min to ~ 5
min. These results indicated that APC acted as a reversible retention component, with the full length
form retaining β-catenin more strongly than the truncated form. As for export, expression of full
length APC had no effect, but depletion again stimulated the process by increasing the fast moving
fraction.
We included in the analysis the effect of inhibition of classical CRM1-mediated export by the drug
Leptomycin B (LMB) (Supplementary Fig.S4A-D). Because nucleocytoplasmic shuttling is a very fast
process, nuclear accumulation of shuttling proteins is expected to be observed within less than one
hours of LMB treatment. In the case of β-catenin, however, a 4-hours treatment had no detectable
effect, neither on import nor on export. In both parental and APC-rescued cells, the recovery curves
perfectly superimposed with those from control cells. In fact, APC distribution under these conditions
was not significantly affected (supplementary Fig.S6). Longer treatments (8hrs) did show however an
effect on import (but not export): Import was altogether increased, with the initial recovery phase
becoming even faster than for APC-depleted cells, approaching the kinetics of free GFP
(supplementary Fig.S5, insert E’).
We also compared the relative nuclear and cytoplasmic steady-state distribution of β-catenin by
measuring the relative fluorescence of YFP-β-catenin in transiently transfected cells. We found that
the nuclear signal was generally close to the cytoplasmic signal (median ~1.2, supplementary Fig. 7A),
although it varied from cell to cell. The ratio was largely similar for parental, APC-rescued and APC-
depleted cells and was not affected by LMB treatment. We also verified that cell to cell variations were
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

15
not related to levels of expression (supplementary Fig.S7B). Note that a very similar ratio was
measured for free GFP, which is considered to freely equilibrate between both compartments. Note
also that even GFP appeared to have an immobile nuclear fraction (suppl. Fig.S5), which probably
account for its nuclear/cytoplasmic ratio being slightly higher than 1.
Discussion
In this study, we have explored systematically three crucial aspects of APC biology. We have
demonstrated the requirement for APC in β-catenin phosphorylation, we have identified that the
activity is largely restricted to an insoluble compartment, and we have formally verified that β-catenin
nuclear transport is independent of APC, which rather acts as its main retention factor. This study has
also confirmed that a typical cancer-related APC truncated form is not a null mutant in terms of Wnt
regulation, but can still promote a significant β-catenin phosphorylation activity. In addition, it still
plays a significant role in β-catenin retention in the context of nuclear transport.
Direct comparison of the function of truncated and full length APC
The APC-rescued SW480 cell line produced by Burgess’ group has provided a powerful tool to
examine APC function in Wnt signaling. Note Faux et al (2004) observed that re-introduction of full
length APC had effects on cell behavior, and in particular on cell adhesion. We have performed a
series of verifications, which did not reveal any overt differences in levels or subcellular distribution of
the components of the Axin complex. We only detected a slight increase in soluble GSK3α, which
does not impact on the interpretation of our results, since the kinase activities are concentrated in the
dense insoluble fraction. Even assuming the existence of other small differences, they would not
account for the dramatic increase in β-catenin phosphorylation measured in APC-rescued cells. A
second issue inherent to cancer cells, and as matter of fact to all immortalized cell lines, is the likely
occurrence of additional unknown mutations. This caveat was circumvented by the comparison with
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

16
siRNA-mediated depleted cells: all our results turned out to be extremely coherent. They can all be
fully explained by the sole contribution of APC.
APC is required for β-catenin phosphorylation
While there is abundant evidence in the literature for a requirement of APC in β-catenin
phosphorylation, this has never been proven in vivo. We provide here the first formal demonstration of
this key point. Our results support those models in which APC is a core component of the Axin
complex (Ha et al, 2004; Hinoi et al, 2000; Roberts et al, 2012; Roberts et al, 2011; Xing et al, 2003).
The intimate involvement of APC in β-catenin phosphorylation is further confirmed by the findings
that APC is required for both S45 priming and subsequent S33/S37/T41 phosphorylation, and that it
requires β-catenin binding directly to APC. While Axin seems to compensate for APC truncations and
rescue “normal” β-catenin in SW480 cells when highly overexpressed (Behrens et al, 1998; Hart et al,
1998), APC seems to be absolutely necessary under more physiological conditions. Note that while
measurement in Xenopus egg extracts suggested that Axin is limiting (Salic et al, 2000; Lee et al, 2003),
Axin and APC are expressed at similar levels in SW480 and SW480APC cells, and Axin is even more
abundant in other mammalian cells (Tan et al, 2012). In Drosophila embryo, β-catenin regulation is
equally sensitive to APC and Axin levels (Roberts et al, 2012; Roberts et al, 2011).
β-catenin phosphorylation occurs in an insoluble fraction
The weak phosphorylation activity of the cytosol was another surprise of our study. The cytosol
seemed indeed to contain an excess of all components required to build active complexes. CK1ε was
the only component absent from this fraction, but CK1α is generally considered to be at least as
effective for β-catenin priming. Dilution is unlikely to explain this low activity, because in our assays
cytosolic fractions were in fact more concentrated than the crude extracts. Furthermore, according to
our immunoprecipitation data (supplementary Fig.S2), all interactions seemed to resist well dilution.
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

17
Note also that the conditions for cytosol extraction were milder than those used for
immunoprecipitation (very low digitonin as opposed to high Triton X100). We conclude to the
existence of large cytosolic Axin and APC pools that are poorly or not active at all in β-catenin
phosphorylation. We suggest that cytosol complexes may either participate to a dynamic, perhaps
regulated, equilibrium with fully active insoluble complexes, and/or fulfill other functions, such as
JNK signaling for Axin or cytoskeleton regulation for APC.
Several studies have investigated the possible nature of APC complexes (Mahadevaiyer et al, 2007;
Maher et al, 2009; Penman et al, 2005; Reinacher-Schick & Gumbiner, 2001). A systematic study by
cell fractionation showed that a significant fraction of APC was sedimentable and detergent insoluble
(Reinacher-Schick & Gumbiner, 2001). Other evidence for the association of APC with dense
structures came from the images of APC positive granules in cell protrusions (Mili et al, 2008) or with
the plasma membrane (Reinacher-Schick & Gumbiner, 2001). Junctional localization of APC, Axin
and GSK3 was also reported in SW480 cells (Maher et al, 2009).
Unfortunately, it is difficult to compare our data with any of the previous biochemical investigations,
because they all used so-called “post-nuclear” supernatants of an initial centrifugation. This standard
step of all classical fractionation protocols aims at getting rid of nuclei and unbroken cells. The
content of the discarded pellet, however, contains a large fraction of the cytoskeleton and of the dense
plasma membranes, and significantly overlaps with our fraction X (Liu and Fagotto, 2011). We now
demonstrate that this dense insoluble fraction contains most of the β-catenin phosphorylation activity.
Since this activity fraction was likely missing from all previous analyses (including Bilic et al, 2007; Li
et al, 2012; Taelman et al, 2010), the nature and regulation of the Axin-based β-catenin destruction
complex(es) need to be revisited. Note that a fraction of E-cadherin is also recovered in fraction X,
which is most likely to corresponds to the a detergent-insoluble, cytoskeleton-associated junctional
pool. This probably explains also the relatively high β-catenin levels in this fraction. However, this
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

18
pool is bound to be independent of the Axin-APC complex, since β-catenin interactions with APC
and cadherin are mutually exclusive (Huelsken et al, 1994).
The effect of C-terminal truncations and the relative role of 15 and 20AA repeats
The reason for the strong selection of the cancer-related APC truncations has been abundantly
discussed (e.g. Kohler et al, 2008). The initial theory proposed that loss of the 20AA repeats was
causal to β-catenin stabilization. This view was later challenged and was largely substituted by a model
where the loss of the Axin-binding SAMP motifs had the central role. Recent evidence has re-
questioned this view, showing in particular that APC can interact with Axin independently of SAMP
motifs. It has also become clearer that both the type and number of β-catenin binding repeats are
important in regulation of the Wnt pathway (Roberts et al, 2011). These results have brought back the
original model in the front scene.
In any case, the fact that most truncations left more than half of the APC protein intact indicated that
the distal region of this portion still bore an important function and that there was a strong selection
in cancer cells to preserve it. However, whether this residual function was directly related to β-catenin
phosphorylation has remained an open question. It has been even suggested that the truncated forms
may have acquired some dominant activity. Our data confirm the model of Peifer and colleagues
(Roberts et al, 2011) and unequivocally establish that in terms of β-catenin phosphorylation, APC
truncation behaves as a bona fide loss-of-function, yet not a null but rather a relatively weak allele that
still retains a surprisingly high activity.
Thank to our specific assay, we have been able to further dissect the requirements and confirm/infirm
several previous assumptions: We demonstrate that both types of APC repeats are needed for full
activity. The 20AA repeats are, as predicted, particularly important. This is not only true for cells
expressing full length APC, but even for parental SW480 cells, thus providing a clear explanation for
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

19
the maintenance of one 20AA repeat in truncated APC. On the other hand, we found that binding to
the 15AA, which was thought to have little influence on β-catenin degradation (Roberts et al, 2011;
Roberts et al, 2012), was in fact quite important (Fig.3). The relative contribution of the two types of
repeats correlated quite well with their relative number in full length and truncated APC: the 15AA
repeats played a particularly important role for pS33/S37/T41 phosphorylation in parental SW480
cells, while the 20AA repeats seemed to take care of most of the function for full length APC. These
results do not exclude other specific defects due to APC truncation (we did observe differences in
retention), but in principle the observed decreased rate of β-catenin phosphorylation appears sufficient
to account for its stabilization and over-activation of the pathway.
APC and β-catenin nuclear transport
Although APC (and Axin) was proposed to mediate nuclear export of β-catenin (Bienz, 2002), there is
strong evidence that β-catenin can freely diffuse in and out of the nucleus (Fagotto et al, 1998; Kose et
al, 1997; Wiechens & Fagotto, 2001; Henderson & Fagotto, 2002; Sharma et al, 2012) and that APC
and other partners of β-catenin, all negatively affect β-catenin translocation (Krieghoff et al, 2006).
Our observation of very fast transport kinetics further corroborate the notion of selective diffusion,
while the analysis of transport in APC-depleted cells confirms that APC is not required for
translocation. Note also that the speed of recovery observed in APC-depleted cells, where apparent
retention is very low, approaches the one of freely diffusible GFP, which is quite remarkable, also
considering the differences in size and shape (28kDa and globular for GFP versus rodlike and
>90kDa for β-catenin).
Another definitive argument against a role of APC, or of any other potentially shuttling protein such
as Axin, in β-catenin export is brought by the lack of measurable changes after 4 hrs of LMB-
treatment. In fact, APC does not appear to be a freely shuttling protein, since this 4 hrs treatment is
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

20
not sufficient to cause any significant nuclear accumulation. We had previously obtained similar results
with Axin (Wiechens et al, 2004). It thus seems clear that the ability of Axin and APC to be re-
exported has no short term implications on β-catenin. This property probably serves to maintain on
the long term a proper distribution of these important scaffold proteins in the various cellular
compartments. Obviously complete block of export for longer hours will impact on this distribution,
and eventually also on the pool of β-catenin retained on each side of the nucleus.
In terms of the impact of APC on β-catenin retention, our results perfectly confirm previous results
by Behrens and coworkers (Krieghoff et al, 2006). Our specific contribution is to demonstrate that
APC has measurable effects on β-catenin transport even when expressed at physiological levels. The
fact that retention is significant in APC-expressing cells, and almost nil in the absence of APC suggests
that APC is a major, possibly the main, factor controlling the pool of β-catenin available for transport
in these cells. Consistent with the data of Peifer and colleagues (Roberts et al, 2011; Roberts et al,
2012), truncated APC was still able to retain β-catenin in the cytoplasm, although as argued above, its
high residual activity in β-catenin phosphorylation may have at least as much impact on regulation of
β-catenin signaling in cancer cells. Note that cadherins constitute another component that sequesters
β-catenin at the plasma membrane. Because the association if very strong, we expect that it would
contribute to the immobile fraction in the time scale of our experiments. We do not believe that
cadherins had any significant impact in our FRAP measurements, for the simple reason that we used
single spread cells grown at low density. Under these conditions, cadherin levels are extremely low in
SW480 cells (data not shown). This is consistent with the live images, where GFP-β-catenin was only
weakly and generally not detectable at the membrane, even in cells expressing very low levels of this
construct.
In conclusion, APC truncations are certainly not “null mutants” in term of β-catenin regulation and
still fulfill an unexpectedly large part of APC function, fully consistent with the necessity even for
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

21
cancer cells to maintain β-catenin activity at bay, as proposed in the “just-right” hypothesis (Furuuchi
et al, 2000).
Materials and methods
Cell culture
SW480 cells and SW480APC cells are kind gifts from Antony Burgess (Ludwig Institute, Melbourne,
Australia). Cells were cultured in RPMI 1640 medium, supplemented with 10% fetal bovine serum,
genetecin (1.5 mg/ml) and 1% penicillin/streptomycin.
Antibodies
The antibodies employed in the study were, mouse anti-APC (ALi 12-28, Santa Cruz Biotechnology),
rabbit anti-APC (C-20, Santa Cruz Biotechnology), rabbit anti-APC (M-APC, generous gift of Dr.
Inke Näthke, University of Dundee ( Nathke et al, 1996), affinity purified rabbit anti-Axin (Wiechens
et al, 2004), anti-β-catenin (H102, Santa Cruz Biotechnology), mouse anti-β-catenin (6F9Sigma), rabbit
anti-phospho-β-catenin (Ser33/Ser37/Thr41, Cell Signaling), rabbit anti-phospho-β-catenin (Ser45,
Cell Signaling), mouse anti-GSK3α/β (05-412, Millipore), rabbit anti-casein kinase 1α (sc-28886, Santa
Cruz Biotechnology), goat anti-casein kinase 1ε (sc-6471, Santa Cruz Biotechnology), mouse anti-
casein kinase 1ε (sc-365259, Santa Cruz Biotechnology), mouse anti-GAPDH (6C5, Applied
Biosystems), mouse anti-RanBP3 (BD Biosciences), rabbit anti-LRP6 (C-10, Santa Cruz
Biotechnology), goat anti-γ-Tubulin (sc-7396, Santa Cruz Biotechnology), mouse anti-γ-tubulin
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

22
(ab11316, Abcam), rabbit anti-pericentrin (ab4448, Abcam), rabbit anti-β-actin (ab25894, Abcam), rat
anti-α-actinin (BT-GB-276S, Babraham tech).
Plasmids and recombinant β-catenin construction
eGFP (Clontech), myc-tagged-full length mouse Axin (Zeng et al, 1997), pET-His-β-catenin
(Wiechens & Fagotto, 2001). YFP-βcatenin was constructed by adding the eYFP sequence followed
by a five glycine linker upstream of Xenopus β-catenin in pCS2+MT. CherryNLS was constructed by
adding a classical nuclear localization sequence (KKKRK) to the C terminus of Cherry fluorescent
protein subcloned into the pCS2-vector. Various His-tag-β-catenin and YFP-β-catenin mutants (S45D,
APCΔ15(W386A), APCΔ20(K345A), APCΔ15(W383A) APCΔ20(K345A/W383A) were produced by
site directed mutagenesis based on pCS2-YFP-β-catenin and pET-His-β-catenin using the
QuikChange II XL Site-Directed Mutagenesis Kit, according to the manufacturer’s protocol. All
constructs were confirmed by sequencing. GSTP-APCr15 and r20 were constructed using
oligonucleotides coding for the sequences LDTPINYSLKYSDEQ (1st 15AA repeat of human APC)
and EDTPICFSRCSSLSSLSSAED (1st 20AA repeat). APC-siRNA (sc-29702) and Control siRNA (sc-
37007) were purchased from Santa Cruz Biotechnology. Cells were transfected with plasmids or
siRNAs using Lipofectamine 2000 (Invitrogen) according to the manufacture’s protocol.
Cell homogenization and cell fractionation
For preparation of total homogenates, cells were cultured in 6 cm plastic dishes and harvested by
scraping in 300 μl osmolysis buffer (20mM Hepes-NaOH pH 7.4, 0.2mM EDTA), homogenized with
40 strokes in a tight fitted Douce homogenizer, and equal volume of high Na Buffer (400mM Sucrose,
300mM NaCl, 20mM Hepes-NaOH pH 7.4, 0.2mM EDTA) was added followed by 40 additional
strokes. Our cell fractionation protocol was previously described (Liu & Fagotto, 2011). The
separation yielded five fractions (of the following volumes), cytosol (3 ml), nucleosol (0.5 ml), nuclear
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

23
insoluble (1.35 ml), membranes (1.35 ml) and dense insoluble material (fraction “X”), recovered at the
bottom of the Percoll Gradient and resuspended in 450 μl low Na buffer (150mM NaCl, 10mM
Hepes-NaOH , pH 7.4, 0.1mM EDTA). Note that the Percoll Gradient contained 0.6% Triton-X100
(Liu & Fagotto, 2011).
Western blot
Samples were separated on SDS-PAGE according to the regular protocol, expect for the detection of
APC and Pericentrin which were resolved in a 4% gel without stacking gel. The blots were developed
using a chemiluminescence detection reagent (WBKLS0500, Millipore), and images acquired with a 12
bits digital camera (Alpha Innotech MultiImage system). Data were quantified using the Gene Tools
software (Syngene). Dilution series of were used to verify linearity of the signal. Note that in several
cases a large number of samples had to be blotted simultaneously, which required to run several gels in
parallel. To insure perfectly equal conditions of transfer, incubation with antibodies and development,
two to three gels were transferred on one single nitrocellulose membrane. In all cases presented where
collages are presented, they presented conditions from a single membrane, with identical exposure
time and contrast.
In vitro kinase assay
Substrates were recombinant His-tagged-β-catenin proteins. Proteins were expressed in E. coli BL21
(DE3), purified on Ni-NTA agarose column and exchanged into kinase buffer (150mM NaCl, 20mM
Hepes-NaOH). Reactions were carried out in a total volume of 50μl, containing 100nM recombinant
β-catenin substrates, 1mM ATP, 1mM MgCl2, 10mM Creatine Phosphate and 10U Creatine Kinase,
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

24
and the following amounts of sample, total cell lysates, 20μl; cell fractions, 40μl (undiluted for Cs, Ns,
Ni and Mem fractions, diluted 1:3 in the case of fraction “X”). Volumes were adjusted using kinase
buffer. The reaction was started by adding the samples, was carried on at 37oC, and stopped by
addition of 4x Laemmli sample buffer with 20mM EDTA and boiled immediately for 3 min at 98°C.
Relative levels of phosphorylated S45 and S33/S37/T41 were determined by quantitative immunoblot
using corresponding phospho-specific antibodies. Several commercial anti-phospho-β-catenin
antibodies were tested. Except for the anti-phospho-S45 (Cell Signaling), all other antibodies showed
strong reactivity toward non-phosphorylated β-catenin. This reactivity was eliminated by preabsorbing
anti-phospho-S33/S37/T41 with ~ 50 μg/ml recombinant β-catenin for 60 min before incubation on
the membrane. Band intensities were quantified as above, and relatively activities were calculated after
background subtraction. For samples from crude extracts, as results were expressed as the ratio
between the signal intensity for phosphorylated β-catenin and β-actin used as loading control.
Confocal microscopy and fluorescence recovery after photobleaching
Cells were grown on Fluorodish and transfected with YFP-β catenin, eGFP or CherryNLS. Cells were
maintained in a FCS2 live cell chamber at 37oC, 5% CO2 chamber. Images were acquired using a
Quorum WaveFX spinning disk confocal system (QuorumTechnologies Inc.), with a 40x HCX PL
APO CS, NA= 1.25 oil objective. For photobleaching experiments, samples were photobleached with
a solid state 405nm laser (475mW) using a mosaic digital diaphragm (Andor Technology PLC., Belfast
UK). Either the nucleus or the cytoplasm was bleached for 1 sec at 100% laser power. The samples
were images continuously with a separate 488nm laser line. 5-20 frames of a single z plane were
collected every 200ms before and immediately following bleaching, followed by frames taken at 2-
second intervals. Average nuclear and cytoplasmic intensities were measured using Metamorph or
ImageJ softwares. After background subtraction, the nucleus to cytoplasm (or cytoplasm to nucleus
for export) ratios were calculated. The pre-bleach ratio was set to 100% and ratio in the first post-
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

25
bleach image was set to 0. The recovery curves shown are the averages of at least 8-15 cells from at
least three independent experiments. Curve fitting and statistical calculations were computed using
GraphPad Prism 6.0 and Excel softwares.
Acknowledgements
We thank Drs. Faux and Burgess for generous gift of SW480APC cells, and Laura Canty for providing
the Cherry-NLS construct. We acknowledge the support of the McGill University Biology department
Cell Imaging and Analysis Network (CIAN) for confocal microscopy. L. Wang was recipient of a
Shandong University Joint-Ph.D. training program studentship. This work was supported by a grant
from the Canadian Cancer Research Society to F.F.
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

26
References
Behrens, J., Jerchow, B.A., Wurtele, M., Grimm, J., Asbrand, C., Wirtz, R., Kuhl, M., Wedlich,
D., Birchmeier, W. (1998). Functional interaction of an axin homolog, conductin, with beta- catenin,
APC, and GSK3beta. Science 280, 596-599.
Bienz, M. (2002). The subcellular distribution of APC proteins. Nature Rev. Mol. Cell Biol. 3, 328-338
Bilic, J., Huang, Y.L., Davidson, G., Zimmermann, T., Cruciat, C.M., Bienz, M., Niehrs, C.
(2007). Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation.
Science 316, 1619-1622.
Brocardo, M., Naethke, I.S., Henderson, B.R. (2005) Redefining the subcellular location and
transport of APC, new insights using a panel of antibodies. EMBO Rep 6, 184-190.
Choi , J. Park, S.-Y., Costantini, F., Jho, E.-h., Joo, C.-K. (2004) Adenomatous Polyposis Coli Is
Down-regulated by the Ubiquitin-Proteasome Pathway in a Process Facilitated by Axin. J. Biol. Chem.
279, 49188–49198.
Cliffe, A., Hamada, F., Bienz, M. (2003). A Role of Dishevelled in Relocating Axin to the Plasma
Membrane during Wingless Signaling. Curr. Biol. 13, 960-966.
Fagotto, F., Gluck, U., Gumbiner, B.M. (1998). Nuclear localization signal-independent and
importin/karyopherin- independent nuclear import of beta-catenin. Curr. Biol. 8, 181-190.
Fagotto, F., Jho, E., Zeng, L., Kurth, T., Joos, T., Kaufmann, C., Costantini, F. (1999).
Domains of axin involved in protein-protein interactions, Wnt pathway inhibition, and intracellular
localization. J. Cell Biol. 145, 741-756.
Faux, M.C., Coates, J.L., Catimel, B., Cody, S., Clayton, A.H., Layton, M.J., Burgess, A.W.
(2008). Recruitment of adenomatous polyposis coli and beta-catenin to axin-puncta. Oncogene 27, 5808-
5820.
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

27
Faux, M.C., Coates, J.L., Kershaw, N.J., Layton, M.J., Burgess, A.W. (2010). Independent
interactions of phosphorylated beta-catenin with E-cadherin at cell-cell contacts and APC at cell
protrusions. PloS One 5, e14127.
Faux, M.C., Ross, J.L., Meeker, C., Johns, T., Ji, H., Simpson, R.J., Layton, M.J., Burgess,
A.W. (2004). Restoration of full-length adenomatous polyposis coli (APC) protein in a colon cancer
cell line enhances cell adhesion. J. Cell Sci. 117, 427-439.
Furuuchi, K., Tada, M., Yamada, H., Kataoka, A., Furuuchi, N., Hamada, J., Takahashi, M.,
Todo, S., Moriuchi, T. (2000). Somatic mutations of the APC gene in primary breast cancers. Am. J.
Pathol. 156, 1997-2005.
Ha, N.C., Tonozuka, T., Stamos, J.L., Choi, H.J., Weis, W.I. (2004). Mechanism of
phosphorylation-dependent binding of APC to β-catenin and its role in β-catenin degradation. Mol.
Cell 15, 511-521.
Hart, M.J., de los Santos, R., Albert, I.N., Rubinfeld, B., Polakis, P. (1998). Downregulation of
β-catenin by human Axin and its association with the APC tumor suppressor, β-catenin and GSK3β.
Curr. Biol. 8, 573-581.
Henderson, B., Fagotto, F. (2002). The ins and outs of APC and β-catenin nuclear transport.
EMBO Rep. 3, 834-839.
Hernandez, A.R., Klein, A.M., Kirschner, M.W. (2012). Kinetic responses of β-catenin specify the
sites of Wnt control. Science 338, 1337-1340.
Hülsken, J., Birchmeier, W., Behrens, J. (1994) E-cadherin and APC compete for the interaction
with beta-catenin and the cytoskeleton. J. Cell Biol. 127, 2061-2069.
Hinoi, T., Yamamoto, H., Kishida, M., Takada, S., Kishida, S., Kikuchi, A.K. (2000). Complex
formation of adenomatous polyposis coli gene product and axin facilitates glycogen synthase kinase-3
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

28
beta-dependent phosphorylation of β-catenin and down-regulates β-catenin. J Biol Chem 275, 34399-
34406.
Kimelman, D., Xu, W. (2006). β-catenin destruction complex, insights and questions from a
structural perspective. Oncogene 25, 7482-7491.
Kinzler, K.W., Vogelstein, B. (1996). Lessons from hereditary colorectal cancer. Cell 87, 159-170
Kishida, S., Yamamoto, H., Ikeda, S., Kishida, M., Sakamoto, I., Koyama, S., Kikuchi, A.
(1998). Axin, a negative regulator of the Wnt signaling pathway, directly interacts with adenomatous
polyposis coli and regulates the stabilization of β-catenin. J. Biol. Chem. 273, 10823-10826.
Kohler, E.M., Derungs, A., Daum, G., Behrens, J., Schneikert, J. (2008). Functional definition of
the mutation cluster region of adenomatous polyposis coli in colorectal tumours. Hum Mol Genet 17,
1978-1987.
Koike, M., Kose, S., Furuta, M., Taniguchi, N., Yokoya, F., Yoneda, Y., Imamoto, N. (2004).
β-Catenin shows an overlapping sequence requirement but distinct molecular interactions for its
bidirectional passage through nuclear pores. J. Biol. Chem. 279, 34038-34047.
Kose, S., Imamoto, N., Tachibana, T., Shimamoto, T., Yoneda, Y. (1997). Ran-unassisted
nuclear migration of a 97-kD component of nuclear pore-targeting complex. J. Cell Biol. 139, 841-849.
Krieghoff, E., Behrens, J., Mayr, B. (2006). Nucleo-cytoplasmic distribution of Beta-catenin is
regulated by retention. J. Cell Sci. 119, 1453-1463.
Lee, E., Salic, A., Kruger, R., Heinrich, R., Kirschner, M.W. (2003). The roles of APC and Axin
derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biology 1, E10.
Li, V.S., Ng, S.S., Boersema, P.J., Low, T.Y., Karthaus, W.R., Gerlach, J.P., Mohammed, S.,
Heck, A.J., Maurice, M.M., Mahmoudi, T., Clevers, H. (2012). Wnt signaling through inhibition
of β-catenin degradation in an intact Axin1 complex. Cell 149, 1245-1256.
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

29
Liu, X., Fagotto, F. (2011). A method to separate nuclear, cytosolic, and membrane-associated
signaling molecules in cultured cells. Sci. Signal. 4, pl2.
Mahadevaiyer, S., Xu, C., Gumbiner, B.M. (2007). Characterization of a 60S complex of the
adenomatous polyposis coli tumor suppressor protein. Biochim. Biophys. Acta 1773, 120-130.
Maher, M.T., Flozak, A.S., Stocker, A.M., Chenn, A., Gottardi, C.J. (2009). Activity of the β-
catenin phosphodestruction complex at cell-cell contacts is enhanced by cadherin-based adhesion.
J.Cell Biol. 186, 219-228.
Mili, S., Moissoglu, K., Macara, I.G. (2008). Genome-wide screen reveals APC-associated RNAs
enriched in cell protrusions. Nature 453, 115-119.
Munemitsu, S., Albert, I., Souza, B., Rubinefeld, B., Polakis, P. (1995). Regulation of
intracellular β-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc.
Natl. Acad. Sci. USA 92, 3046-3050.
Nakamura, T., Hamada, F., Ishidate, T., Anai, K., Kawahara, K., Toyoshima, K., Akiyama, T.
(1998). Axin, an inhibitor of the Wnt signalling pathway, interacts with β-catenin, GSK-3β and APC
and reduces the β-catenin level. Genes Cells 3, 395-403.
Nathke, I.S. (2004). The adenomatous polyposis coli protein, the Achilles heel of the gut epithelium.
Ann. Rev. Cell Dev. Biol. 20, 337-366.
Nathke, I.S., Adams, C.L., Polakis, P., Sellin, J.H., Nelson, W.J. (1996). The adenomatous
polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell
migration. J. Cell Biol. 134, 165-179.
Penman, G.A., Leung, L., Nathke, I.S. (2005). The adenomatous polyposis coli protein (APC)
exists in two distinct soluble complexes with different functions. J. Cell Sci. 118, 4741-4750.
Polakis, P. (2000). Wnt signaling and cancer. Genes Dev. 14, 1837-1851.
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

30
Polakis, P. (2007). The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 17, 45-51.
Reinacher-Schick, A., Gumbiner, B.M. (2001). Apical membrane localization of the Adenomatous
polyposis coli tumor sppressor protein and subcellular distribution of the beta-catenin destruction
complex in polarized epithelial cells. J. Cell Biol. 152, 491-502.
Roberts, D.M., Pronobis, M.I., Alexandre, K.M., Rogers, G.C., Poulton, J.S., Schneider, D.E.,
Jung, K.C., McKay, D.J., Peifer, M. (2012). Defining components of the ss-catenin destruction
complex and exploring its regulation and mechanisms of action during development. PLoS One 7,
e31284.
Roberts, D.M., Pronobis, M.I., Poulton, J.S., Waldmann, J.D., Stephenson, E.M., Hanna, S.,
Peifer, M. (2011). Deconstructing the β-catenin destruction complex, mechanistic roles for the tumor
suppressor APC in regulating Wnt signaling. Mol. Biol. Cell 22, 1845-1863.
Rubinfeld, B., Albert, I., Porfiri, E., Fiol, C., Munemitsu, S., Polakis, P. (1996). Binding of
GSK3β to the APC-β-catenin complex and regulation of complex assembly. Science 272, 1023-1026.
Rubinfeld, B., Souza, B., Albert, I., Müller, O., Chamberlain, S.H., Masiarz, F.R., Munemitsu,
S., Polakis, P. (1993). Association of the APC gene product with β-catenin. Science 262, 1731-1734.
Sharma, M., Jamieson, C., Johnson, M., Molloy, M.P., Henderson, B.R. (2012). Specific
armadillo repeat sequences facilitate β-catenin nuclear transport in live cells via direct binding to
nucleoporins Nup62, Nup153, and RanBP2/Nup358. J. Biol. Chem. 287, 819-831.
Sierra, J., Yoshida, T., Joazeiro, C.A., Jones, K.A. (2006). The APC tumor suppressor counteracts
β-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev. 20, 586–600.
Stambolic, V., Ruel, L., Woodgett, J.R. (1996). Lithium inhibits glycogen synthase kinase-3 activity
and mimics wingless signalling in intact cells. Curr. Biol. 6, 1664-1668.
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

31
Su, L.K., Vogelstein, B., Kinzler, K.W. (1993). Association of the APC tumor suppressor protein
with catenins. Science 262, 1734-1737.
Su, Y., Fu, C., Ishikawa, S., Stella, A., Kojima, M., Shitoh, K., Schreiber, E.M., Day, B.W., Liu,
B. (2008). APC is essential for targeting phosphorylated beta-catenin to the SCFbeta-TrCP ubiquitin
ligase. Mol. Cell 32, 652-661.
Taelman, V.F., Dobrowolski, R., Plouhinec, J.L., Fuentealba, L.C., Vorwald, P.P., Gumper, I.,
Sabatini, D.D., De Robertis, E.M. (2010). Wnt signaling requires sequestration of glycogen synthase
kinase 3 inside multivesicular endosomes. Cell 143, 1136-1148.
Tan, C.W., Gardiner, B.S., Hirokawa, Y., Layton, M.J., Smith, D.W., Burgess, A.W. (2012).
Wnt signalling pathway parameters for mammalian cells. PLoS One 7, e31882.
Valenta, T., Hausmann, G., Basler, K. (2012). The many faces and functions of β-catenin. EMBO
J. 31, 2714-2736.
von Kries, J.P., Winbeck, G., Asbrand, C., Schwarz-Romond, T., Sochnikova, N., Dell'Oro, A.,
Behrens, J., Birchmeier, W. (2000). Hot spots in β-catenin for interactions with LEF-1, conductin
and APC. Nat. Struct. Biol. 7, 800-807.
Wiechens, N., Fagotto, F. (2001). Crm1- and Ran-independent nuclear export of β-catenin. Curr.
Biol. 11, 18-27.
Wiechens, N., Heinle, K., Englmeier, L., Schohl, A., Fagotto, F. (2004). Nucleo-cytoplasmic
shuttling of Axin, a negative regulator of the Wnt-beta-catenin Pathway. J. Biol. Chem. 279, 5263-5267.
Xing, Y., Clements, W.K., Kimelman, D., Xu, W. (2003). Crystal structure of a β-catenin/Axin
complex suggests a mechanism for the β-catenin destruction complex. Genes Dev. 17, 2753-2764.
Yost, C., Torres, M., Miller, J.R., Huang, E., Kimelman, D., Moon, R.T. (1996). The axis-
inducing activity, stability, and subcellular distribution of β-catenin is regulated in Xenopus embryos
by glycogen synthase kinase 3. Genes Dev. 10, 1443-1454.
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

32
Zeng, L., Fagotto, F., Zhang, T., Hsu, W., Vasicek, T.J., Perry, W.L.3rd, Lee, J.J., Tilghman,
S.M., Gumbiner, B.M., Costantini, F. (1997). The mouse Fused locus encodes Axin, an inhibitor of
the Wnt signaling pathway that regulates embryonic axis formation. Cell 90, 181-192.
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

33
Figure legends
Figure 1
(A) Diagram of human APC and of the truncated form expressed in SW480 cells. The main
domains are represented, including the 15 and 20 amino acid repeats that are responsible for β-catenin
binding, the SAMP motifs that bind Axin, and the B domain (also called β-catenin inhibitory domain),
reported to be required for activity of the destruction complex (Kohler et al, 2008; Roberts et al,
2012). Also included are the confirmed nuclear localization sequences (NLS) and nuclear export
sequences (NES) (Henderson & Fagotto, 2002). MCR, mutation cluster region.
(B-B’’) Expression of the components of the β-catenin destruction complex in parental SW480
cells and in full length APC-rescued cells (SW480APC). (B) Whole extracts. Equal total protein
amounts were loaded. Arrow and arrowhead point respectively to full length (fl) and truncated (tr)
APC. Quantification of relative amounts of full length and truncated APC is presented in
supplementary Figure S1A, and quantification of levels of all the other components in supplementary
Figure S1B. (B’,B”) Subcellular distribution. Cytosolic (Cs), nucleosolic (Ns), nuclear insoluble (Ni),
membrane (M) and dense insoluble (X) fractions were compared between parental (S) and APC-rescue
(A) cells. (B’) APC and β-catenin. Asterisk: non-specific band in parental cells. . (B”) Axin, CK1α and
ε, and GSK. LRP6 and E-cadherin were used as membrane markers, GAPDH and RanBP3 as markers
for cytosol and nucleosol, respectively.See supplementary Fig.S1A for quantification. (B”)
(C,D) Phosphorylation activity toward β-catenin in SW480 and SW480APC cells.
(C,C’) β-catenin phosphorylation was determined in whole extracts using 100nM recombinant β-
catenin as substrate. Levels of phospho-S45 (priming) and phospho-S33/S37/T41 β-catenin were
compared by quantitative immunoblotting using specific antibodies for the corresponding
phosphorylated sites. SW480APC cells showed significantly higher kinase activity than parental cells,
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

34
for both reactions. (C) Western blots from a representative experiment. (C’). Quantification. Relative
intensities were standardized to β-actin input levels. The ratio was arbitrarily set at 1.0 for 15 min
activity in parental SW480 cells. Data are shown as mean intensities ±SD of five independent
experiments. *, **, ***, p<0.05, <0.01, <0.001, respectively, pairwise Student’s t-test.
(D,D’) Recombinant β-catenin with serine 45 mutated to an aspartate (S45D) was used to mimic
constitutive phosphorylation and thus monitor phosphorylation of S33/S37/T41 independently of the
priming step. Phosphorylation was significantly higher in SW480APC cells. (D) Western blots from
representative experiment. (D’) Quantification as for (C).
Figure 2. Effect of APC depletion and Axin overexpression on β-catenin phosphorylation.
(A-A’) APC depletion. Full length and truncated APC forms were depleted in SW480 and
SW480APC cells, respectively, by transfection of siRNA targeting the N-terminal half of the
transcripts. S45 priming and S33/S37/T41 phosphorylation were significantly decreased in both cell
lines. (A) Western blots from representative experiments. (A’) Quantification.
(B-B’) Axin overexpression. SW480 and SW480APC cells transfected with a construct coding for
full length Axin were assayed for β-catenin phosphorylation. Axin overexpression did not increase
phosphorylation. It rather slightly impaired S33/S37/T41 in SW480APC cells. (B) Western blots from
representative experiments. (B’) Quantification. NS, not significant.
(C) Comparison of APC levels in control siRNA and APC siRNA cells. Levels of wild type APC and
truncated APC were decreased respectively to 10-20% and ~50% of normal levels (average of three
experiments). Endogenous β-catenin levels were slightly increased. α-actinin was used as loading
control. (C’) Axin levels in control and YFP-Axin-overexpressing cells. Total Axin levels were
increased 2 to 2.5 folds (average of three experiments). α-actin was used as loading control.
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

35
Figure 3. Phosphorylation of β-catenin variants defective in binding to 15AA or 20AA APC
repeats.
Recombinant β-catenin variants were tested that had single amino acid substitutions that impaired
binding to APC, either via 15AA repeats (ΔAPC15(386)) or via the 20AA repeats (ΔAPC20(345),
ΔAPC20(383), and double mutant ΔAPC20(345/383) (von Kries et al, 2000). Compared to wild type
β-catenin, all mutated proteins were significantly less phosphorylated on residues S45 and
S33/S37/T41. β-catenin mutants lacking 20AA binding were the poorest substrates. *, **, ***, p<
0.05, 0.01, and 0.001, respectively, either compared to wild type substrate (just above columns), or to
ΔAPC15(386) (bars).
Figure 4. Subcellular distribution of β-catenin phosphorylation activity.
SW480 (S) and SW480APC (A) cell extracts were fractionated to separate cytosol (Cs), nucleosol (Ns),
nuclear insoluble fraction (Ni), membranes (M), and dense insoluble material (X). The five fractions
were analyzed for S45 and S33/S37/T41 phosphorylation for wild type β-catenin (A) or S45D β-
catenin (B). The results were similar for both cell lines: phosphorylation activity was largely confined
to fraction X. Smaller contributions were observed in Cs and Ni fractions. (C,D) Quantification of
the relative activities. The total kinase activities in SW480 and in SW480APC cells were calculated as
the sum of the activities in the five cell compartments, taking into account the volume of each
fraction. These values were then used to calculate the % contribution from each fraction.
Quantification was performed using the 7.5 min time points, which were most consistent. Although
the kinase activities in fractionated samples tended to be unstable after longer incubations, and thus
less reliable, the general pattern at 15 min was similar.
Figure 5. β-catenin nuclear transport in parental, APC-rescued, and APC-depleted SW480
cells.
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

36
SW480 cells, SW480APC rescued cells, and SW80 cells transfected with siAPC were transiently
transfected with YFP-β-catenin. YFP-β-catenin nuclear transport was analyzed by fluorescence
recovery after photobleaching (FRAP). Fluorescence recovery was monitored for 300 sec, and
quantified by plotting the nuclear to cytoplasmic ratio for import, and cytoplasmic to nuclear for
export, setting the pre-bleach fluorescent intensity values to 100% and the post-bleach value to 0%.
(A) Examples of nuclear and cytoplasm FRAP. (B-D) Graph showing compiled data (at least 10 cells
from 3-5 independent experiments).
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t
SW480 Cs Ns Ni M X
SW480 APC Cs Ns Ni M X
Figure 1 Wang et al
Dimerization Arm
domain 15 AA repeats 20 AA repeats
SAMP B domain
Basic domain
EB1 binding
NLS NES MCR Truncated APC in SW480 cells
Full length human APC
B
APC β-cat
CK1α
β-actin
Axin
GSK3 β α
SW480 SW480
APC
D’
0
1
2
3
4
0 15 300
5
10
15
20
0 15 30
0
1
2
3
4
0 15 30
phospho S33/S37/T41
phospho S45
β-actin
SW480 SW480APC
0 15 30 0 15 30
wt β-catenin
S45D β-catenin
phospho S33/S37/T41
β-actin
min
SW480 cell
SW480APC cell
Rela
tive
ratio
Rela
tive
ratio
Re
lativ
e ra
tio
phospho S33/S37/T41 phospho S45
phospho S33/S37/T41
min min
min
A
B’
C
C’
D
*
** ***
***
** **
GSK3 β α
CK1ε
LRP6
Axin
CK1α
RanBP3
GAPDH
E-cadh
SW480 SW480
APC
SW A SW A SW A SW A SW A Cs Ns Ni M X B”
tr
fl
*
β-cat
APC tr
fl
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t
SW480 cells
A siCTRL siAPC
0 15 30 0 15 30
phospho S33/S37/T41
β-actin
phospho S45
phospho S33/S37/T41
β-actin
phospho S45
Figure 2 Wang et al
siCTRL siAPC 0 15 30 0 15 30
SW480APC cells
phospho S33/S37/T41
β-actin
phospho S45
vector Axin
0 15 30 0 15 30
SW480 cells
B
phospho S33/S37/T41
β-actin
phospho S45
vector Axin
0 15 30 0 15 30
SW480APC cells
Axin
β-actin
C’ C
A’
01234
0 15 3001234
0 15 300
1
2
0 15 300
1
2
0 15 30
siAPC siCTRL
Rela
tive
ratio
pS33/S37/T41 pS45
min min Re
lativ
e ra
tio
pS33/S37/T41 pS45
min min
B’
012345
0 15 300
1
2
3
0 15 300
1
2
3
0 15 300
1
2
3
0 15 30
Rela
tive
ratio
pS33/S37/T41 pS45
min min
Rela
tive
ratio
pS33/S37/T41 pS45
min min Axin vector
siAPC siCTRL
Axin vector
SW480 cells SW480APC cells siCTRL siAPC siCTRL siAPC
SW480 SW480APC
YFP-Axin
vct Axin vct Axin
min min
min min
**
* ** * *
*** *** ***
NS
NS
NS
NS NS
NS
NS NS
β-catenin β-catenin
α-actinin
siCTRL siAPC siCTRL siAPC
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t
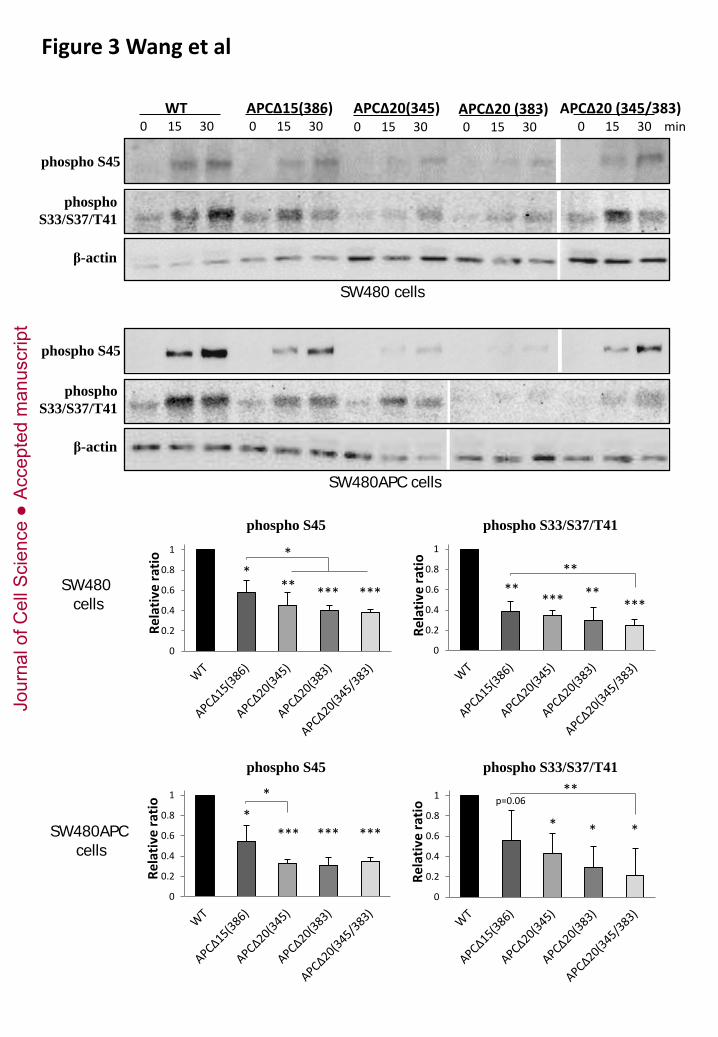
0
0.2
0.4
0.6
0.8
1
Rela
tive
ratio
0
0.2
0.4
0.6
0.8
1
Rela
tive
ratio
SW480 cells
WT APCΔ15(386) 0 15 30 0 15 30
APCΔ20 (383) 0 15 30 0 15 30
APCΔ20(345) APCΔ20 (345/383) 0 15 30
phospho S45
β-actin
phospho S33/S37/T41
phospho S45
β-actin
phospho S33/S37/T41
SW480APC cells
SW480 cells
phospho S45 phospho S33/S37/T41
0
0.2
0.4
0.6
0.8
1
Rela
tive
ratio
0
0.2
0.4
0.6
0.8
1
Rela
tive
ratio
phospho S45 phospho S33/S37/T41
* *** *** ***
p=0.06
* * *
* ** *** *** **
*** ** ***
* **
** *
Figure 3 Wang et al
min
SW480APC cells
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

Figure 4 Wang et al
pSer33/Ser37/Thr41
A
B
0.0
0.2
0.4
0.6
0.8
1.0
SW APC SW APC SW APC SW APC SW APC
Cs Ns Ni Mem X
0.0
0.2
0.4
0.6
0.8
1.0
SW APC SW APC SW APC SW APC SW APC
Ns Cs Ni Mem X
S A S A S A S A S A Cs Ns Ni M X
0 7.5 15 7.5 15 7.5 15 7.5 15 7.5 15 7.5 15 7.5 15 7.5 15 7.5 15 7.5 15 0
½
pSer45
C
pSer33/Ser37/Thr41
S A S A S A S A S A Cs Ns Ni M X
0 7.5 15 7.5 15 7.5 15 7.5 15 7.5 15 7.5 15 7.5 15 7.5 15 7.5 15 7.5 15 0
wt β-cat
wt β-cat
S45D β-cat
substrate: wt β-cat
pSer45
substrate: S45D β-cat pSer33/Ser37/Thr41
D
Frac
tion
of to
tal c
ell a
ctiv
ity
0.0
0.2
0.4
0.6
0.8
1.0
SW APC SW APC SW APC SW APC SW APC
Cs Ns Ni Mem X
pSer33/Ser37/Thr41
Frac
tion
of to
tal c
ell a
ctiv
ity
Frac
tion
of to
tal c
ell a
ctiv
ity
min
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t
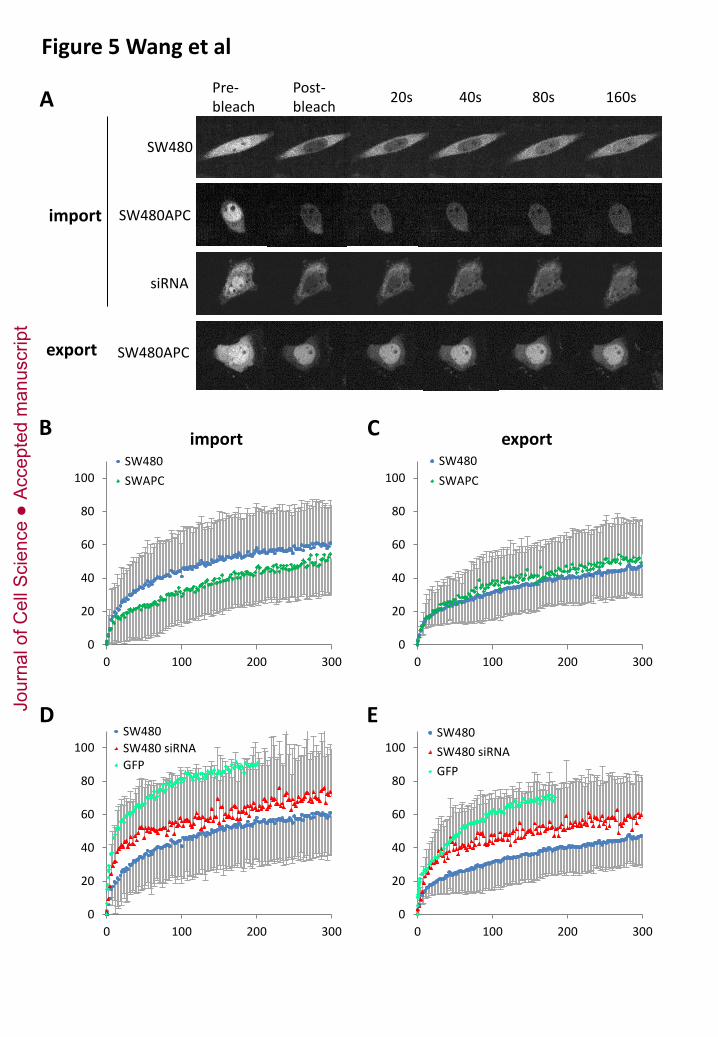
Figure 5 Wang et al
0
20
40
60
80
100
0 100 200 300
SW480SW480 siRNAGFP
0
20
40
60
80
100
0 100 200 300
SW480SWAPC
import
0
20
40
60
80
100
0 100 200 300
SW480SWAPC
export
0
20
40
60
80
100
0 100 200 300
SW480SW480 siRNAGFP
Pre-bleach
Post-bleach 20s 40s 80s 160s
SW480
siRNA
SW480APC
SW480APC
import
export
A
B C
D E Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t
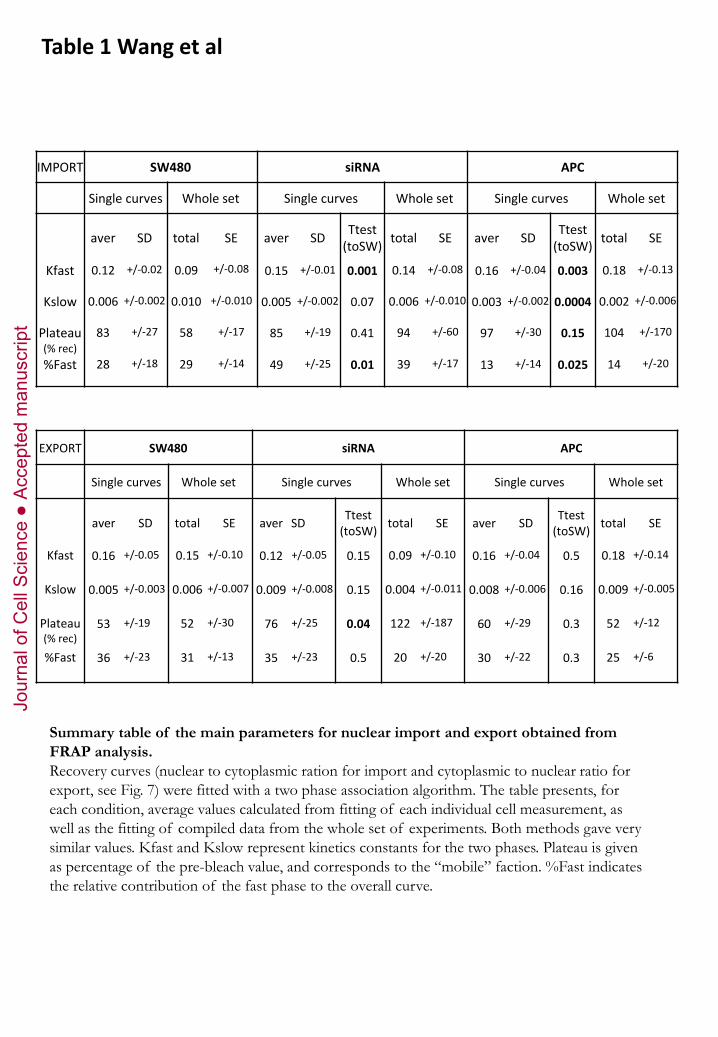
Table 1 Wang et al
IMPORT SW480 siRNA APC
Single curves Whole set Single curves Whole set Single curves Whole set
aver SD total SE aver SD Ttest (toSW) total SE aver SD Ttest
(toSW) total SE
Kfast 0.12 +/-0.02 0.09 +/-0.08 0.15 +/-0.01 0.001 0.14 +/-0.08 0.16 +/-0.04 0.003 0.18 +/-0.13
Kslow 0.006 +/-0.002 0.010 +/-0.010 0.005 +/-0.002 0.07 0.006 +/-0.010 0.003 +/-0.002 0.0004 0.002 +/-0.006
Plateau (% rec)
83 +/-27 58 +/-17 85 +/-19 0.41 94 +/-60 97 +/-30 0.15 104 +/-170
%Fast 28 +/-18 29 +/-14 49 +/-25 0.01 39 +/-17 13 +/-14 0.025 14 +/-20
EXPORT SW480 siRNA APC
Single curves Whole set Single curves Whole set Single curves Whole set
aver SD total SE aver SD Ttest (toSW) total SE aver SD Ttest
(toSW) total SE
Kfast 0.16 +/-0.05 0.15 +/-0.10 0.12 +/-0.05 0.15 0.09 +/-0.10 0.16 +/-0.04 0.5 0.18 +/-0.14
Kslow 0.005 +/-0.003 0.006 +/-0.007 0.009 +/-0.008 0.15 0.004 +/-0.011 0.008 +/-0.006 0.16 0.009 +/-0.005
Plateau (% rec)
53 +/-19 52 +/-30 76 +/-25 0.04 122 +/-187 60 +/-29 0.3 52 +/-12
%Fast 36 +/-23 31 +/-13 35 +/-23 0.5 20 +/-20 30 +/-22 0.3 25 +/-6
Summary table of the main parameters for nuclear import and export obtained from FRAP analysis. Recovery curves (nuclear to cytoplasmic ration for import and cytoplasmic to nuclear ratio for export, see Fig. 7) were fitted with a two phase association algorithm. The table presents, for each condition, average values calculated from fitting of each individual cell measurement, as well as the fitting of compiled data from the whole set of experiments. Both methods gave very similar values. Kfast and Kslow represent kinetics constants for the two phases. Plateau is given as percentage of the pre-bleach value, and corresponds to the “mobile” faction. %Fast indicates the relative contribution of the fast phase to the overall curve.
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t


















