
Regulation of Caspase-9 by Natural and Synthetic Inhibitors
239
University of Massachusetts Amherst University of Massachusetts Amherst ScholarWorks@UMass Amherst ScholarWorks@UMass Amherst Open Access Dissertations 5-2012 Regulation of Caspase-9 by Natural and Synthetic Inhibitors Regulation of Caspase-9 by Natural and Synthetic Inhibitors Kristen L. Huber University of Massachusetts Amherst Follow this and additional works at: https://scholarworks.umass.edu/open_access_dissertations Part of the Chemistry Commons Recommended Citation Recommended Citation Huber, Kristen L., "Regulation of Caspase-9 by Natural and Synthetic Inhibitors" (2012). Open Access Dissertations. 554. https://doi.org/10.7275/jr9n-gz79 https://scholarworks.umass.edu/open_access_dissertations/554 This Open Access Dissertation is brought to you for free and open access by ScholarWorks@UMass Amherst. It has been accepted for inclusion in Open Access Dissertations by an authorized administrator of ScholarWorks@UMass Amherst. For more information, please contact [email protected].
Transcript of Regulation of Caspase-9 by Natural and Synthetic Inhibitors

University of Massachusetts Amherst University of Massachusetts Amherst
ScholarWorks@UMass Amherst ScholarWorks@UMass Amherst
Open Access Dissertations
5-2012
Regulation of Caspase-9 by Natural and Synthetic Inhibitors Regulation of Caspase-9 by Natural and Synthetic Inhibitors
Kristen L. Huber University of Massachusetts Amherst
Follow this and additional works at: https://scholarworks.umass.edu/open_access_dissertations
Part of the Chemistry Commons
Recommended Citation Recommended Citation Huber, Kristen L., "Regulation of Caspase-9 by Natural and Synthetic Inhibitors" (2012). Open Access Dissertations. 554. https://doi.org/10.7275/jr9n-gz79 https://scholarworks.umass.edu/open_access_dissertations/554
This Open Access Dissertation is brought to you for free and open access by ScholarWorks@UMass Amherst. It has been accepted for inclusion in Open Access Dissertations by an authorized administrator of ScholarWorks@UMass Amherst. For more information, please contact [email protected].

REGULATION OF CASPASE-9 BY NATURAL AND SYNTHETIC INHIBITORS
A Dissertation Presented
by
KRISTEN L. HUBER
Submitted to the Graduate School of the
University of Massachusetts Amherst in partial fulfillment
of the requirements for the degree of
DOCTOR OF PHILOSOPHY
MAY 2012
Chemistry

© Copyright by Kristen L. Huber 2012
All Rights Reserved

REGULATION OF CASPASE-9 BY NATURAL AND SYNTHETIC INHIBITORS
A Dissertation Presented
by
KRISTEN L. HUBER
Approved as to style and content by:
_________________________________________
Jeanne A. Hardy, Chair
_________________________________________
Lila M. Gierasch, Member
_________________________________________
Robert M. Weis, Member
_________________________________________
Peter Chien, Member
______________________________________
Craig T. Martin, Department Head
Department of Chemistry

DEDICATION
For my mother, Elizabeth Huber, who taught me perseverance and to always follow my
dreams, no matter how big or small
For my father, Kenneth Huber, who taught me to believe in myself and to remember you
can always find a new beginning in tomorrow
For my sister, Elyse Huber, for being the person who inspires me every single day and
who taught me to be proud of who I am

v
ACKNOWLEDGMENTS
To my mentor, Professor Jeanne Hardy, I thank you for guidance and support over this
long and somewhat bumpy road. We have both grown in many ways since our first year
here at UMass; you as a mentor, me as a budding scientist and both as individuals.
Between the memories of our scientific travels and your enthusiastic high fives, you have
truly made this an experience I will never forget.
To my committee, Dr. Lila Gierasch, Dr. Bob Weis and Dr. Peter Chein, you have
pushed me to be a better scientist by teaching me to think outside the box and appreciate
the meaning of testing a hypothesis. I thank you.
To the Hardy Lab members, my scientific family, both past and present, you will always
be held near and dear to my heart. From the Monday morning sports talk to the mid week
frustrations followed up by the Friday fireside chats and all of the fun times, you filled
my days with laughter and enjoyment. Thank you for being the everyday rock I could
always count on.
To my all my friends, you have been a shoulder to lean on and a breath of fresh air when
I needed it most. To Mike Wilson Jr. and Shannon Coates Flagg, thank you for always
listening, understanding, and making me smile. This road would have been difficult
without all of your support so let the good times roll!
And last but certainly not least, to my family, I would not be where I am today or the
person I am today without your unconditional love, support, and understanding. Thank
you, not only for being my biggest fans but for supporting me through the tough times
and keeping me grounded as a person. Mama, thank you for encouraging me to not only
dance to the beat of a different drummer but to polka whenever I had the chance. Pappy,
thank you for always finding the right words to make me feel at ease and giving me the
encouragement to always stand up for what I believe in. Elyse, my partner in crime, thank
you for teaching me all of the important life skills I needed to accomplish my goals. You
have taught me how to think on my feet, especially when getting framed for taking
cookies out of the kitchen, how to accept constructive criticism by convincing me there is
always room for improvement, particularly when it comes to my touchdown dance
moves, and patience by forgetting to find me when playing hide-n-go-seek. We are truly
two peas in a pod, SBN! I love you all!
“You have brains in your head.
You have feet in your shoes
You can steer yourself
any direction you choose.
You're on your own. And you know what you know.
And YOU are the guy who'll decide where to go.”
Oh, the Places You’ll Go!
-Dr. Seuss

vi
ABSTRACT
REGULATION OF CASPASE-9 BY NATURAL AND SYNTHETIC INHIBITORS
MAY 2012
KRISTEN L. HUBER, B.S., QUINNIPIAC UNIVERSITY
Ph.D., UNIVERSITY OF MASSACHUSETTS AMHERST
Directed by: Professor Jeanne A. Hardy
Tight regulation of caspase-9, a key initiator of apoptosis, is required to uphold
cellular homeostasis. Although it is controlled on a multifactorial level, misregulation of
this process does occur, which is a characteristic of a variety of diseases from ischemic
injury to cancer. Therefore it remains important to gain a detailed understanding of the
mechanisms behind native caspase-9 regulatory pathways and harness these mechanisms
for therapeutic purposes.
Based on known mechanisms, such as the unique inhibitory complex of caspase-
9 and XIAP-BIR3, development of synthetic regulators can be envisioned, while other
mechanisms such as zinc-mediated inhibition and CARD activation of caspse-9 remain
undefined. Intrigued by the multiple ways to control caspase-9’s activity, we sought after
designing synthetic caspase-9 inhibitors in addition to defining the mechanistic details
metal regulation and CARD domain activation.
We report the first stabilized α-helical peptides that harness the native regulatory
mechanism of caspase-9 and the BIR3 domain which lead to the understanding of the
importance of exosites in inhibitory complexes. Our studies also revealed that there are
two distinct zinc binding sites, one at the active site and another at a novel zinc binding
site of yet unknown function in caspase-9 however this site may have the potential to

vii
control caspase-6 based on its regulatory mechanism. Furthermore, an interaction was
discovered between CARD and the catalytic core of caspase-9 in the presence of a
properly formed substrate binding groove, a potential mechanism utilized by the
apoptosome for activation of the enzyme.
All in all, the regulation of caspase-9 occurs on a variety of levels that requires
almost every surface of the enzyme. Through exploring these underlying molecular
details behind the various mechanisms, not only has the field of caspase-9 regulation
mechanisms been extended, essential information was gained for further pursuit in an
advancement towards the design of caspase-9 activators and inhibitors.

viii
TABLE OF CONTENTS
Page
ACKNOWLEDGMENTS ...................................................................................................v
ABSTRACT ....................................................................................................................... vi
LIST OF TABLES ........................................................................................................... xiii
LIST OF FIGURES ......................................................................................................... xiv
CHAPTER I. APOPTOSIS, DISEASE AND THE REGULATION OF CASPASE-9 .........................1
1.1. The Roles of Apoptosis in Disease ...................................................................1
1.2. Caspases: Facilatators of Apoptosis..................................................................4
1.3. Caspase Active Site and Catalytic Mechanism .................................................6
1.4. Apoptotic Pathways ..........................................................................................9
1.5. Natural Regulation of Caspase-9 ....................................................................11
1.6. Synthetic Regulation of Caspase-9 .................................................................15
1.7. Caspase-9 and Its Role in Disease ..................................................................19
1.8. Refrences.........................................................................................................21 II. ROBUST PRODUCTION OF A LIBRARY OF CASPASE-9 INHIBITOR
PEPTIDES USING METHODOLOGICAL SYNCHRONIZATION .........................30
2.1. Introduction .....................................................................................................31
2.2. Results .............................................................................................................34
2.2.1. Construction of Peptide-Fusion Expression Vector and
aPP Variants ....................................................................................34
2.2.2. Expression and Purification of a Recombinant Peptide Library ......36
2.3. Discussion .......................................................................................................41
2.4. Materials and Methods ....................................................................................45

ix
2.4.1. Cloning of Recombinant Peptide-Fusion Variants ..........................45
2.4.2. Expression and Purification of Recombinant
Peptide-Fusion Variants ...................................................................46
2.4.3. Expression and Purification of Human Rhinovirus 3C Protease .....47
2.4.4. Peptide-Fusion Cleavage by Human Rhinovirus 3C Protease and
Separation from Fusion Partners ......................................................48
2.4.5. Mass Spectromerty...........................................................................49
2.5. References .......................................................................................................49
III. INHIBITION OF CASPASE-9 BY STABILIZED PEPTIDES TARGETING
THE DIMERIZATION INTERFACE .........................................................................54
3.1. Introduction .....................................................................................................55
3.2. Results .............................................................................................................61
3.2.1. Native and Aib-Stabilized Peptides .................................................61
3.2.2. aPP-Scaffolded Peptides ..................................................................66
3.2.3. Aliphatic Stapled Peptides ...............................................................71
3.3. Discussion .......................................................................................................73
3.4. Materials and Methods ....................................................................................77
3.4.1. Caspase-9 Expression and Purification ............................................77
3.4.2. Caspase-7 WT and Caspase-7 C186A
Expression and Purification .............................................................79
3.4.3. Peptide Production ...........................................................................79
3.4.3.1. Synthesis of N-Fmoc-S-2-(2'-pentyl) alanine..........................79
3.4.3.2. Synthesis of N-Fmoc-S-2-(2'-octyl) alanine............................82
3.4.4 Activity Assays .................................................................................84
3.4.5. Mass Spectromerty...........................................................................84

x
3.4.6. Secondary Structure Analysis by Circular Dichroism .....................85
3.4.7. Computational Structure Prediction .................................................85
3.5. References .......................................................................................................86
IV. MECHANISM OF ZINC-MEDIATED INHIBITION OF CASPASE-9 ...................94
4.1. Introduction .....................................................................................................94
4.2. Results .............................................................................................................95
4.2.1. Metal Affects on the Properties of Caspase-9 ..................................95
4.2.2. Determining the Location of Zinc Binding Sites .............................99
4.3. Discussion .....................................................................................................102
4.4. Materials and Methods ..................................................................................104
4.4.1. Caspase-9 Expression and Purification ..........................................104
4.4.2. Prediction of Metal Lingands and Construction of
Ligand Substitution Variants .........................................................106
4.4.3. Caspase-7 C186A Expression and Purification .............................107
4.4.4 Activity Assays ...............................................................................107
4.4.5. Oligomeric-State Determination ....................................................109
4.4.6. Secondary Structure Analysis ........................................................110
4.4.7. Zinc Binding Analysis by ICP-OES ..............................................110
4.4.8. Model of Zinc Binding to Caspase-9 .............................................111
4.5. Structure Determination trials of Caspase-9 and Zinc ..................................112
4.5.1. Crystallization of Caspase-9 in the Presence and
Absence of Zinc ............................................................................112
4.5.2. Data Collection on Crystals of Caspase-9 .....................................114
4.5.3. Zinc Soaks of Caspase-9 Crystals ..................................................115
4.6. References .....................................................................................................117

xi
V. CASPASE-9 CARD:CORE DOMAIN INTERACTIONS REQUIRE A
PROPERLY-FORMED ACTIVE SITE .....................................................................121
5.1. Introduction ...................................................................................................121
5.2. Results ...........................................................................................................124
5.2.1. The Influence of CARD on the Oligomeric State and
Stability of Caspase-9 ....................................................................124
5.2.2. Determining an Interaction Site Between Caspase-9
Catalytic Core and CARD Domains .............................................130
5.3. Discussion .....................................................................................................134
5.4. Materials and Methods ..................................................................................137
5.4.1. Caspase-9 Expression and Purification ..........................................137
5.4.2. Oligomeric State Determination ....................................................139
5.4.3. CARD Expression and Purification ...............................................140
5.4.4 Thermal Stability and Secondary Structure Analysis
by Circular Dichroism....................................................................141
5.4.5. Caspase-3 Expression and Purification ..........................................142
5.4.6. Native Gel Analysis and Ni-NTA Pull Down Assay to
Determine in trans Interactions .....................................................142
5.4.7. Activity Assays ..............................................................................143
5.5. References .....................................................................................................145 VI. A SURFACE-WIDE VIEW OF THE NATIVE REGULATORY
MECHANISMS OF CASPASE-9 ..............................................................................147
6.1. Regulation of Caspase-9 Occurs at a Variety of Locations
on its Surface.................................................................................................147
6.2. References .....................................................................................................153

xii
APPENDICES
A. SMALL MOLECULE ACTIVATION OF CASPASE-9 ...........................................157
B. DESIGN OF AN ACTIVATIABLE INITIATOR CASPASE ...................................170
C. EXPRESSION AND PURIFICATION OF YEAST METACASPASE
YCA1 ...........................................................................................................................180
BIBLIOGRAPHY ............................................................................................................188

xiii
LIST OF TABLES
Table Page
2.1. Peptide Expression, Yield, and Chemical Characteristics .................................. 40 3.1. Overall Inhibition of Caspsae-9 Full-Length by Peptides .................................. 65
4.1. Zinc Binding Analysis of Caspase-9 Variants .................................................. 100
5.1. Kinetic Parameters for Full-Length and ΔCARD variants of Caspase-9 ...................................................................................................... 125
5.2. Kinetic Parameters for Full-Length Caspase-9
Charge Swap Variants....................................................................................... 134

xiv
LIST OF FIGURES
Figure Page
1.1. Caspase Structure .................................................................................................. 5
1.2. Caspsae Active Site Structure and Mechanism .................................................... 7
1.3. Intrinsic and Extrinsic Pathways of Apoptosis ..................................................... 9
1.4. Inhibitory Complex of Caspase-9 with XIAP-BIR3 Domain ............................. 14
1.5. Synthetic Inhibitors of Caspase-9 Activity ......................................................... 16
2.1. aPP Structure ....................................................................................................... 33
2.2. Schematic Representation of Expression Vector, pET32-Peptide ...................... 34
2.3. Structure of Peptide Library................................................................................ 35
2.4. Peptide-Fusion Purifications ............................................................................... 37
2.5. Peptide-Thioredoxin Fusion Protein Cleavage ................................................... 38
2.6. HPLC Purification Chromatograms of Cleaved Peptide-Fusion Samples ......... 39
2.7. Mass Spectra of Peptides .................................................................................... 39
2.8. Methodological Synchronization ........................................................................ 44
3.1. Interactions Required for Inhibition of Caspase-9 are Clustered
in the α5 Helix.................................................................................................... 58
3.2. Sequences of the α5 Region of XIAP-BIR3 and Peptides 1-9 ........................... 62
3.3. Circular Dichroism Spectra of Native Peptide 5 and the
Aib-Stabilized Peptide 8 ..................................................................................... 63
3.4. Native and Aib-Stabilized Peptides Show Some Inhibition of
Caspase-9 Activity .............................................................................................. 63
3.5. Peptide 2 Non-Specifically Activates Caspase-9 ................................................ 66
3.6. BIR3 Interactions Grafted onto Stabilized Miniature Protein aPP ..................... 67
3.7. aPP and Peptide Structures Predicted Computationally by Rosetta ................... 69
3.8. Properties of aPP Based Peptides ....................................................................... 69
3.9. Aliphatic Stapled Peptide Design ....................................................................... 71
3.10. Properties of Aliphatically Stapled Peptides....................................................... 72
3.11. Analysis of Aliphatically Stapled Peptides ......................................................... 73
3.12. Full-Length BIR3 is Required for High-Affinity Caspase-9 Inhibition ............. 76
4.1. Zinc Exclusively Inhibits Caspase-9 Activity..................................................... 96
4.2. Kinetics of Full-Length Caspase-9 Wild-Type in the Presence of
0-50 μM ZnCl2 ................................................................................................... 97
4.3. Zinc Does Not Alter the Biophysical Properties of Caspase-9 ........................... 98
4.4. Zinc Binding in Caspase-9 .................................................................................. 99
4.5. Kinetics of Full-Length Caspase-9 C272A Variant in the Presence of
0-50 μM ZnCl2 .................................................................................................. 102
4.6. A Model of Caspase-9 Active-Site Ligand Interactions with
a Modeled Zinc Ion ........................................................................................... 103
4.7. Crystals of Wild-Type Caspase-9 Full-Length Pre and Post Optimization ...... 114
4.8. Diffraction Image of Apo Wild Type Caspase-9 Crystals
Soaked in a 20% PEG 400 Cryoprotectant for One Hour ............................... 115
4.9. Diffraction Image of Apo Wild Type Caspase-9 Crystals Soaked in ZnCl2 .... 117

xv
5.1. Model Depicting the Increase in Enzymatic Activity of Caspase-9 ................. 123
5.2. Size Exclusion Chromatography of Caspase-9 Full-Length and ΔCARD
in the Presence and Absence of Active Site Ligand z-VAD-FMK .................. 125
5.3. Thermal Denaturation Analysis and Circular Dichroism Spectrum of
Monomeric, Cleaved Caspase-9 Full-Length, ΔCARD, and CARD Only ....... 126
5.4. Thermal Denaturation Analysis and Circular Dichroism Spectrum of
Dimeric, Cleaved Caspase-9 Full-Length and ΔCARD ................................... 127
5.5. Overlay of the Circular Dichroism Spectrum for Full-Length Caspase-9
in the Presence and Absence of Active Site Ligand z-VAD-FMK .................. 128
5.6. Thermal Denaturation Analysis and Circular Dichroism
Spectrum of Monomeric, Uncleaved caspase-9 and Monomeric,
Cleaved Caspase-9 Full-Length ........................................................................ 129
5.7. SDS-PAGE Analysis of Full-Length Uncleaved Caspase-9 in the
Presence and Absence of 3% Active Caspase-3 Enzyme ................................. 129
5.8. Analysis of CARD:Caspse-9 Core Domain Interactions in trans .................... 130
5.9. Characteristics of the Ser-Gly Linker Extension Caspase-9 Variant ................ 131
5.10. Top RosettaDock Models of the Potential Interaction Site Between
CARD and the Catalytic Core of Caspase-9 ..................................................... 133
5.11. Model of Caspase-9 Activation States in the Presence of CARD .................... 136
6.1. Structure of Caspase-9 with Mapped Regulatory Surfaces .............................. 151
A.1. Molecular Structure of FlAsH-EDT2 ................................................................ 157
A.2. Models of the Caspase-9 FlAsH Binding Variants ........................................... 159
A.3. Analysis of FlAsH Binding to Caspase-9 ΔCARD .......................................... 163
A.4. Analysis of FlAsH Binding to Full-Length Caspase-9 ..................................... 165
A.5. Western Blot Analysis of FlAsH Binding to Full-Length Caspase-9 ............... 165
B.1. Model of the Designed Caspase-9 Disulfide Cross-Linked Variant ................. 172
B.2. Disulfide Cross-Linking of Wild Type Caspase-9
Full-Length and Disulfide Variant .................................................................... 174
B.3. Chemical Cross-Linking of Wild Type Caspase-9
Full-Length and Disulfide Variant .................................................................... 176
C.1. Schematic Representation of Expression Vector, pYCA1 ............................... 182
C.2. Purification of YCA1 ........................................................................................ 184

1
CHAPTER I
APOPTOSIS, DISEASE, AND THE REGULATION OF CASPASE-9
Apoptosis, otherwise known as programmed cell, has been established as a key
component for a variety of disease. Pivotal roles are played by the facilitators of this
process, the caspases, with caspase-9 (ICE-LAP6, Mch6) being of particular interest due
to its role in initiating the cascade. Its activation mechanism and regulatory checkpoints
are targets for treatment of a variety of apoptosis-related diseases. Thus, an improved
understanding of these processes would improve our ability to create new therapies to
treat diseases in which apoptosis has gone awry.
1.1. The Roles of Apoptosis in Disease
Cells go through a highly regulated process called apoptosis. Apoptosis is critical
for the normal development and stability or homeostasis of all multi-cellular organisms.
Apoptosis plays a role in sculpting and structuring developing tissue and organs
throughout the all stages of an organism‟s development. It is responsible for eliminating
tissue or appendages that were once required in early development but are no longer
necessary for the mature stages of life, for controlling the number of cells during the
developmental process to achieve proper organ function, as well as, for eliminating
defective or damaged cells from the developing system1,2
. Therefore, any disturbance in
the apoptotic process is harmful to the system.
Apoptosis has been found to play a role in a variety of diseases. Upregulation of
apoptosis has been observed in a variety of cardiovascular diseases such as myocardial
infarction, dilated cardiomyopathy and end-stage heart failure3-6
. A build-up of apoptotic
macrophages and smooth muscle cells have been observed in rupturing atherosclerotic

2
plaques resulting in macrophage build-up, clogging and weakening of the arterial cell
wall. Furthermore, increased levels of cardiac-myocyte apoptosis is observed in patients
suffering from chronic heart failure, which reduces the ability of the heart to maintain
contractile function, providing a strong impetus for further investigating the role of
apoptosis in heart disease. In addition, diabetes, a disease characterized by inappropriate
blood glucose levels and insulin imbalance, has also been linked to an increase in
apoptosis7-10
. For both Type 1 and Type 2 diabetic patients, apoptotic beta cells in the
islets of the pancreas have been observed. Although not clearly understood, beta-cell
apoptosis in Type 1 diabetes is thought to be the main cause of the disease whereas the
beta-cell death found in Type 2 diabetes is thought to be triggered by increased toxins in
the beta cells themselves, such as glucose, saturated fatty acids, and islet amyloid
polypeptides, which together induce oxidative stress and thus apoptosis. Given that 25.8
million children and adults in America have diabetes11
, an improved understanding of the
involvement of apoptosis is strongly warranted.
Further evidence of the harmful effects of a apoptotic imbalance can be observed
in chronic airway inflammatory diseases such as bronchial asthma. This condition affects
34 million Americans12
and is caused by damage to the epithelium lining of the airways
also caused by an increase in apoptotic cells. Although apoptosis is a normal process for
clearing damaged cells from the epithelium layer of the nasal passage, trachea, and
bronchia, an excessive amount of apoptosis is undesirable and thought to highly
contribute to the pathogenesis of this class of disease13,14
. Hepatitis C, a viral disease of
the liver has been shown to cause an increase in apoptosis of infected cells, a process
thought to provide a mechanism for viral shedding15
in addition to the host‟s own

3
mechanism of eliminating the virus from liver cells16
. It has also been shown that during
a systematic inflammatory response caused by a bacterial infection, otherwise known as
sepsis, the body tries to regain homeostasis via initiating a compensatory anti-
inflammatory response. Although this process is not highly understood, one area of focus
has been lymphocyte apoptosis, characterized by reduced levels of CD4, B-, T- and
dendritic cells17-19
. Treatment of these cells with apoptotic inhibitors has shown increased
survival rates in sepsis models17,20
. Furthermore, neuronal cell death has been shown as a
characteristic of neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS),
Huntington's disease, Alzheimer's disease, and Parkinson's disease. Upregulation of the
apoptotic machinery is evident in early and late stages of disease progression,
characterized by increased activation of pro-apoptotic proteins. Treatment with apoptotic
inhibitors, however, provides protection from additional cell death in mouse models,
further highlighting apoptosis as an important area of study.
Down regulation of apoptosis is also prevalent in a variety of diseases, such as
rheumatoid arthritis and cancer. Rheumatoid arthritis is a chronic inflammatory disorder
affecting 2.1 million adults and approximately 1 in every 250 children under the age of
18 in America. Rheumatoid arthritis is caused by an imbalance between proliferating and
apoptotic cells resulting in synovial hyperplasia and angiogenesis21
. A defective apoptotic
pathway by means of increase apoptotic inhibitors in synovial tissue has been discovered
and prevention of apoptotic cell death is thought to restore the synovial membrane
tissue22
. In a similar respect, the apoptotic machinery is found to be absent or in low
cellular levels in breast, gastric, colorectal and lung cancers, which also show an
increased level of apoptotic inhibitors23-25
. Not only is there a change in oncogenic and

4
pro-apoptotic-protein expression levels in cancer cells, mutations in other pro-apoptotic
proteins26-31
have also been observed.
Because it is an ongoing area of research interest, irregularities within apoptosis
are continually being discovered in a wide array of diseases. It is thought that
irregularities in apoptosis may account for up to 50% of all diseases in which there are no
suitable therapies32
. Therefore, a further understanding of the apoptotic process and its
regulators is still necessary in order to understand how diseases are able to evade this
important cellular process.
1.2. Caspases: Facilitators of Apoptosis
Facilitators of this lethal cellular mechanism of apoptotic cell death are the
cysteine aspartate proteases, or caspases. Caspases are classified into two subgroups
based on their function in the cascade, structural characteristics, and substrate
specificity33
. The apoptotic caspases are commonly classified as in the initiator or
executioner subgroups. Initiator caspases, caspase-2, -8, -9, and -10 emerge in the initial
stages of the apoptotoic cascade and are known for their ability to be involved in large
oligomeric activation assemblies. The executioner caspases-3, -6, and -7 are located in
the latter half of the cascade and are responsible for the majority of proteolytic events
resulting in the orderly demise of the cell. Caspases-1, -4, -5, -11, -12 and -14 are
involved in the inflammatory response or other cell processes, but play no known role in
apoptosis.
Caspases are synthesized as three-domain polypeptide chains (Fig. 1.1 A). The N-
terminal domain, called the prodomain, is unique to each caspase, ranging from 16 to 220
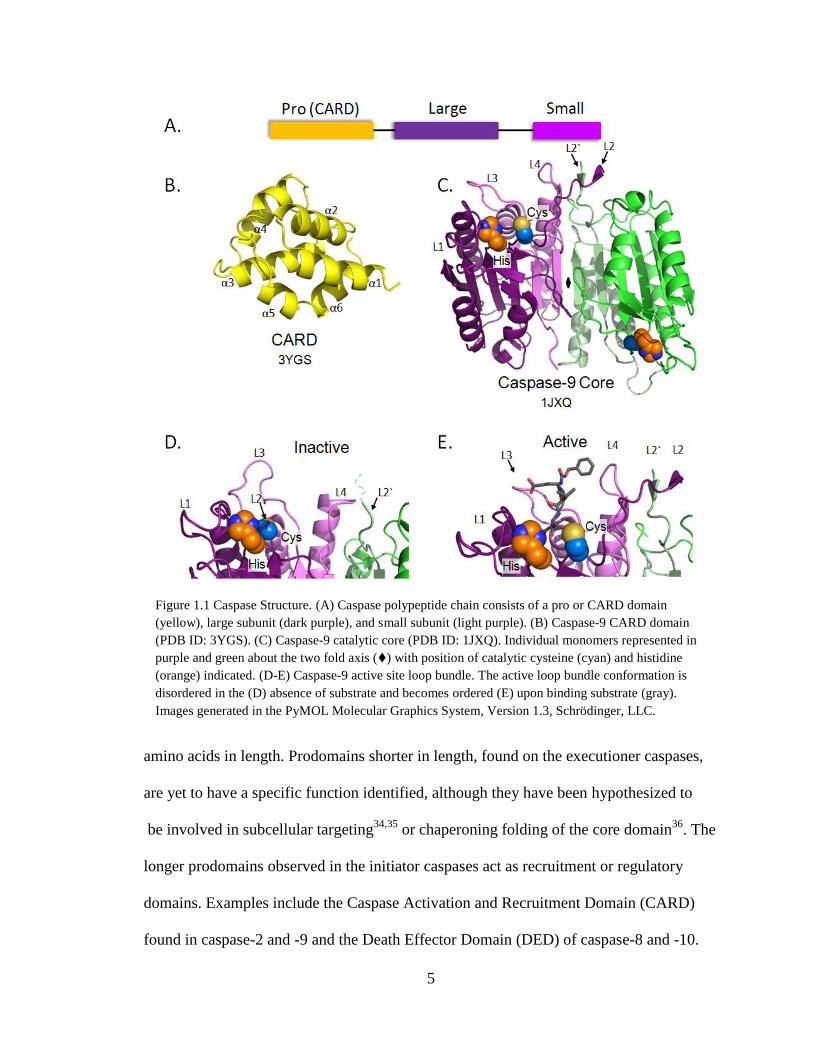
5
amino acids in length. Prodomains shorter in length, found on the executioner caspases,
are yet to have a specific function identified, although they have been hypothesized to
be involved in subcellular targeting34,35
or chaperoning folding of the core domain36
. The
longer prodomains observed in the initiator caspases act as recruitment or regulatory
domains. Examples include the Caspase Activation and Recruitment Domain (CARD)
found in caspase-2 and -9 and the Death Effector Domain (DED) of caspase-8 and -10.
Figure 1.1 Caspase Structure. (A) Caspase polypeptide chain consists of a pro or CARD domain
(yellow), large subunit (dark purple), and small subunit (light purple). (B) Caspase-9 CARD domain
(PDB ID: 3YGS). (C) Caspase-9 catalytic core (PDB ID: 1JXQ). Individual monomers represented in
purple and green about the two fold axis () with position of catalytic cysteine (cyan) and histidine
(orange) indicated. (D-E) Caspase-9 active site loop bundle. The active loop bundle conformation is
disordered in the (D) absence of substrate and becomes ordered (E) upon binding substrate (gray).
Images generated in the PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC.

6
Although CARD and DED domains are highly dissimilar, both fold into an anti-parallel
six-α-helix Greek key structure and facilitate the formation of large protein complexes
(Fig, 1.1 B). The two domains C-terminal to this prodomain region are the large and
small subunits connected via the intersubunit linker. Together, these domains fold into a
heterodimer structure, common to all caspases. Homodimers of the heterodimer large and
small subunits are formed about a two-fold axis which in turn makes up the catalytic core
of the enzyme. The large subunit, averaging 17-20 kDa in size, houses the catalytic Cys-
His dyad. The small subunit, averaging 10-12 kDa, comprises the main portion of the
homodimer interface. This catalytic core region of the caspases folds into a canonical 12-
strand β-sheet core packed between two α-helical layers (Fig 1.1 C). Specific loops which
connect portions of the catalytic core secondary structure known as L1, L2 and L2„(the
product of cleavage of the intersubunit linker), L3 and L4 also form the catalytic loop
bundle. These loops are known to alter their structural position based upon the activation
state of the protease. In the apo unliganded, state, these loops are disordered (Fig. 1.1 D)
however upon binding of substrate, the L2 loop from one of the monomers interacts with
the L2` loop from the opposing monomer (Fig. 1.1 E). Furthermore, L1, L3, and L4 loops
are in an ordered state around the substrate binding groove to complete the formation of
the catalytic loop bundle in the active state.
1.3. Caspase Active Site and Catalytic Mechanism
Caspases have evolved to be highly specific enzymes with the moderate catalytic
properties: Km in the range of 4 to 408 μM and a kcat of 0.2 to 9.1 s-1
37
. Caspase-3 is the
fastest in its class while caspase-9 possesses the slowest catalytic rates overall. The
differences in catalytic properties most likely stem from their active site architecture and

7
target specificity. The active site structure of a caspase consists of a substrate binding
groove, the catalytic Cys-His dyad, and four mobile loops. The substrate-binding groove
(Fig 1.2 A) provides the specificity and binding determinants for a particular substrate
sequence via four interaction interfaces or specificity subsites (S1 – S4). Closest to the
catalytic cysteine lies the S1 pocket. This region provides the highest specificity for
substrate recognition, essentially always accommodating an aspartate residue, although
caspase-9 occasionally recognizes glutamate residues. The S2 substrate-binding site is
thought to be involved in substrate differentiation38-40
. S3 provides an anchoring function
for substrate while S4 provides specificity between the subclasses of caspases.
Figure 1.2 Caspase Active Site Structure and Mechanism. (A) Model of the caspase substrate binding
groove with peptide z-EVD bound (gray). Catalytic Cys (blue) and His (orange) are in close proximity
to the Asp recognition element found in the S1 pocket (white). S2 (yellow), S3 (tan), and S4 (green),
accommodates the remainder of the peptide. (B) Catalytic mechanism of caspases adapted from
Fuentes-Prior and Salvesen43
.

8
Executioner caspases prefer to bind acidic amino acids in their S4 pocket41
while initiator
caspase-9 prefers bulkier hydrophobic residues (reviewed in42
).
The cysteine and histidine catalytic residues responsible for cleavage of the
substrate peptide bond are located on the C-terminal end of the β4 strand and the N-
terminal end of the βI strand respectively. Caspases are thought to have similar catalytic
mechanisms (Fig. 1.2 B) as that of other cysteine proteases (reviewed in43
) where the P1
recognition element is anchored through hydrogen bonding to the oxyanion hole, thus
polarizing the carbonyl carbon of the peptide bond. Deprotonation of the catalytic
cysteine thiol via the catalytic acid/base, a histidine residue, occurs as the initial step of
the cleavage mechanism. The anionic sulfur of the now deprotonated catalytic cysteine
then performs a nucleophilic attack on the substrates carbonyl carbon, C-terminal to the
P1 aspartate recognition element in the case of the caspases. The catalytic histidine then
protonates the α-amino group of the peptide leaving group. After release of this C-
terminal peptide of the cleaved substrate, the deprotonated histidine abstracts a proton
from water to restore its native state. Activated water then hydrolizes the thioester bond
that links the substrates carboxy-terminus to the cysteine thiol and thus restores the
enzyme to its native, unliganded state.
Alternative mechanisms to the classical cysteine protease reaction mechanism
have also been proposed. For example, the catalytic histidine Nγ is proposed to stabilize
the developing charge on the leaving group while a water is utilized as the proton donor
for the peptide leaving group instead of a protonated histidine residue44
. Additionally,
based upon known crystal structures of the caspase family of proteases, the distance of
the caspase catalytic residues are observed to be approximately 5Å apart which would be

9
too far for hydrogen abstraction of the cysteine thiol by the catalytic acid/base, making
the common cysteine protease reaction mechanism difficult to achieve. Therefore, an
alternative mechanism has been proposed by having the active site cysteine nucleophile
develop along the reaction coordinate instead of being polarized by the catalytic
histidine45
. This particular part of the mechanism would be governed by the pH optimum
requirement for these enzymes where the executioner caspases lie more toward the
neutral to slightly basic pH range of 7.0-8.0 while initiator caspases-8 and -9 have a
lower pH optimum of 6.5-7.037
. Further experimental data is required however, in order
to discern which reaction mechanism is proper for caspase catalytic function.
1.4. Apoptotic Pathways
Although apoptosis‟ final phenotype is cell death, the cascade can be initiated via
different mechanisms which would activate either the extrinsic and intrinsic pathways
(Fig 1.3). The decision of which pathway to initiate depends on the type of cellular stress
signal received.
Figure 1.3 Intrinsic and extrinsic pathways of apoptosis. Caspases and other important proteins
involved in apoptosis are shown. Figure by Kristin Paczkowski.

10
Activation of the extrinsic pathway through triggers such as cytokine release
induces signaling of the pro-apoptotic receptors associated with the TNF or TRAIL
family of proteins that reside on the cell surface. These membrane bound receptors bind
ligands, such as FasL, TNF-α, or TRAIL, triggering the recruitment and clustering of
adapter molecules, such as Fas-associated death domain (FADD), to the cytoplasmic
death domain region of the receptor. Additional molecules, caspase-8 or -10 are also
recruited ultimately forming the death inducing signaling complex (DISC)46-49
. Once
bound, the caspases are thought to undergo autoproteolysis and thus activation. As
activated caspases, the initiators -8 and -10 can continue the cascade by cleaving their
downstream targets, caspases-3, -6, and -7.
If a stress signal, such as DNA damage or build up of reactive oxygen species,
occurs from within the boundaries of the cellular membrane, the intrinsic pathway is
initiated. This type of stress induces recruitment of the pro-apoptotic Bcl-2 proteins to
invade the outer mitochondrial membrane. This event, which controls mitochondrial
integrity, induces translocation of other stress dependent proteins known as Bax50
, Bad51
,
or Bid52
to the mitochondrial membrane, resulting in pore formation. As a result, a
release of cytochrome c from the mitochondria into the cytosol occurs. Within the cytosol
an inactive form of apoptotic protease activating factor-1 (Apaf-1) resides. Upon binding
of cytochrome c, Apaf-1 undergoes a conformational change which exposes the Apaf-1
CARD53,54
resulting in the oligomerization of the molecule in an ATP/dATP dependent
manner55-57
. The now heptameric Apaf-1 complex with exposed CARD domains is able
to recruit the zymogen form of caspase-9 through an interaction between the caspase
recruiting domains (CARD) on both Apaf-1 and the caspase-9 full length protein. This

11
allows for multiple procaspase-9 monomers to be within close distance for activation and
for intermolecular self-processing. This large oligomeric complex formed of cytochrome
c, Apaf-1 and pro-caspase-9 is known as the apoptosome. Cryo electron microscopy of
the holo form of the apoptosome58,59
indicate the seven Apaf-1 monomers oligomerize
into a wheel-shape with each monomer representing one spoke. There is a central hub
which houses Apaf-1 CARD which associates with caspase-9. Once bound to the
apoptosome, procaspase-9 can become activated and cleave the executioner caspases-3, -
6, and -7 while still associated to the apoptosome.
1.5. Natural Regulation of Caspase-9
Regulating the caspases is a crucial step in controlling unwanted cell death. For
this reason, a variety of regulatory check points have evolved to make sure caspase
activation does not go awry. These precautions throughout the apoptotic pathway ensure
that caspase activation occurs only upon the appropriate cellular signals. In particular,
multiple levels of regulation exist for the initiator caspase of the intrinsic pathway,
caspase-9. Caspase-9 is a 416 amino acid protein consisting of a 15 kDa CARD domain
which facilitates its binding to the apoptosome, an 18 kDa large subunit which houses the
catalytic Cys287 and His 237 residues and a 10 kDa small subunit. Caspase-9 not only
begins the apoptotic cleavage cascade of the intrinsic pathway, it is a focal point for
regulation.
In a natural non-apoptotic state, caspase-9 exists as an inactive, monomeric
zymogen. In order to become active, the CARD domain of caspase-9 becomes involved
in a protein-protein interaction with the Apaf-1 CARD resulting in a profound increase in
caspase-9 activity55,57,60
. This is further supported by the increase in activity gained by a

12
stepwise addition of the domains involved in this interaction to the caspase-9 catalytic
core, the simplest active unit which possesses the least amount of enzymatic activity.
Addition of caspase-9 CARD increases enzymatic activity by 20 % which is five-fold
further enhanced by the addition of Apaf-1 CARD61
. Maximal activity is only achieved
upon association with the entire apoptosome. Furthermore, caspases are known to be
active as dimers. The dimerization constant, KD, for caspase-9 is in the μM range62,63
,
enforcing a fairly weak complex considering cellular concentrations of ~20 nM54
. In
comparison, KD of the executioner caspases is <50 nM64
. Initial models of the
apoptosome predict a monomeric caspase-9 which dimerizes via increasing the local
concentration of monomeric caspase-9 resulting in recruitment of the dimeric partner65
or
dimerization amongst the apoptosome bound monomers66
or induced conformations
around the active site region of the enzyme67
. However, evidence is emerging from high-
resolution cryo electron microscopy that caspase-9 monomers are active bound to the
apoptosome58
.
In general, for caspases to reach their full activity, they need to be proteolytically
processed within their intersubunit linker, which connects the large and small subunits of
the enzyme. Full activation upon this cleavage serves as a secondary form of regulation.
In the case of caspase-9 this occurs either through autoprocessing or aid from an
additional caspase or other protease such a granzyme B. As a cleaved dimer, the L2 loop
from one half of the dimer is able to interact with the L2` loop from the other half in the
presence of substrate, thus positioning the active site loop bundle62
for catalysis. Caspase-
9, which becomes cleaved at Asp315 has a ten-fold increase in activity upon cleavage in
vitro45
. This value rises to 2000-fold54
when caspase-9 is associated as part of the

13
apoptosome. However, it should be noted that caspase-9 does not require cleavage of its
intersubunit linker to have some minimal activity54
. Furthermore, proteolytic removal of
the CARD domain from the large subunit, which results in decreased activity, has also
been observed in addition to processed caspase-9 being displaced from the apoptosome
by additional molecules of procaspase-968
. Without these early checks and balances of
caspase-9 activation in place, caspase-9 would prematurely activate the caspase cleavage
cascade resulting in unwanted cell death.
As a failsafe, protein-based inhibitors also exist in the cell to control caspase
activity. The most potent inhibitors69
of both the intrinsic and extrinsic pathways of
apoptosis70
are known as the inhibitors of apoptosis or IAP‟s. These multidomain
inhibitors consist of baculoviral IAP zinc-binding repeat (BIR) domains which are
approximately 70 to 80 residues in length and contain a characteristic zinc-binding motif
of -CX2CX16HX6C-. A single IAP consists of one to three BIR domains. Typically a
carboxy-terminal RING domain is also associated, which allows targeting for
ubiquitination and thus proteosomal degredation. Mammalian IAPs including X-
chromosome-linked inhibitors of apoptosis (XIAP), cIAP1, cIAP2, Livin/ML-IAP, and
neuronal apoptosis inhibitor protein also share this same baculoviral zinc motif.
XIAPs have been shown to inhibit both the initiator and executioner caspases in
human cells. They consist of three tandem BIR domains named BIR1, BIR2 and BIR3
followed by a ring domain consisting of one zinc ion chelated to three cysteines and one
histidine and an additional zinc ion bonded to four cysteines. Although similar in
structure and sequence with approximately 30% sequence identity, each BIR domain
possesses its own individual functions. Caspases-3 and -7 can be regulated by blocking of
14
the active site through binding of a small linker region N-terminal to the BIR2 domains of
IAPs (residues 164–235). Binding this linker prevents substrate from entering the active
site71-73
. In addition, a BIR2 domain interaction with the caspase interaction binding motif
distal from the active site of the enzyme74
with a Ki of 0.2-5 nM is present73,75
. In the case
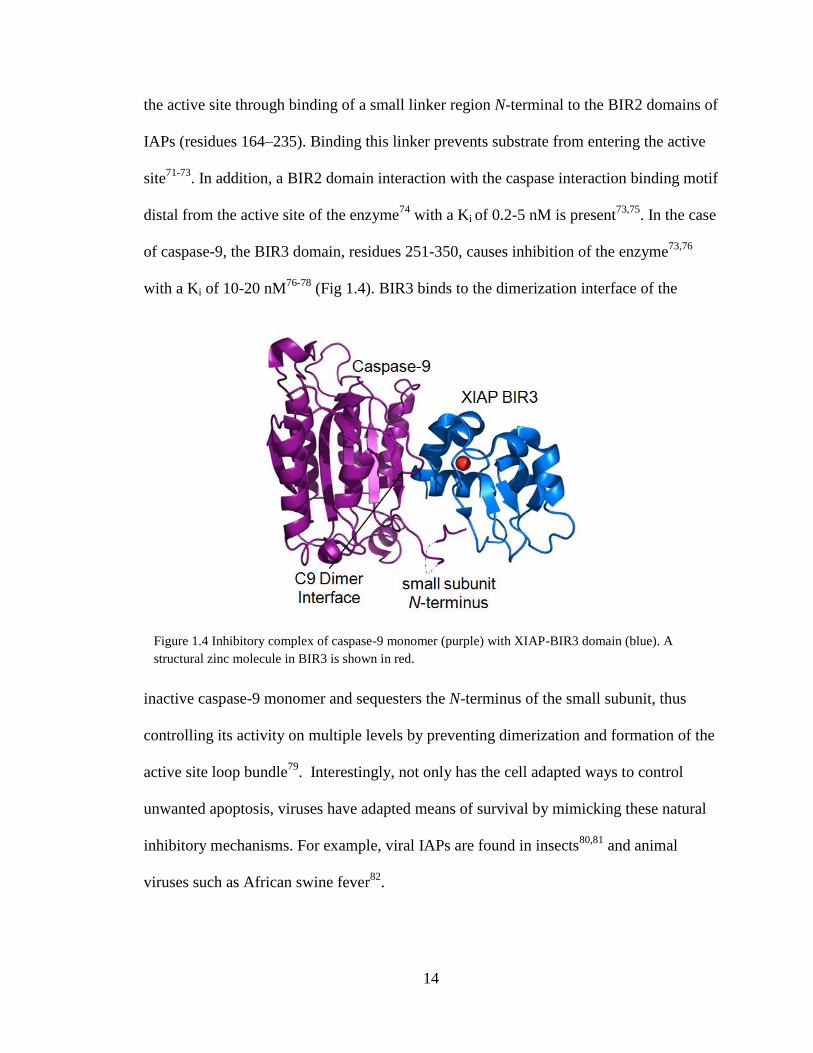
of caspase-9, the BIR3 domain, residues 251-350, causes inhibition of the enzyme73,76
with a Ki of 10-20 nM76-78
(Fig 1.4). BIR3 binds to the dimerization interface of the
inactive caspase-9 monomer and sequesters the N-terminus of the small subunit, thus
controlling its activity on multiple levels by preventing dimerization and formation of the
active site loop bundle79
. Interestingly, not only has the cell adapted ways to control
unwanted apoptosis, viruses have adapted means of survival by mimicking these natural
inhibitory mechanisms. For example, viral IAPs are found in insects80,81
and animal
viruses such as African swine fever82
.
Figure 1.4 Inhibitory complex of caspase-9 monomer (purple) with XIAP-BIR3 domain (blue). A
structural zinc molecule in BIR3 is shown in red.

15
In addition to enzymatic cleavage, binding platforms and protein based inhibitors,
binding of metal ions and post-translational modifications, such as phosphorylation, can
alter the function of the caspase class of enzymes. Metal ions commonly used for both
biological structure and cellular processes, such as zinc and copper, have been observed
as specific regulators of apoptosis by targeting the caspases83
. HeLa cells depleted of zinc
for nine hours resulted in the processing of caspase-984
while the presence of zinc
protected caspase-9 activation from manganese mediated apoptosis85
. Further correlations
linking zinc and caspases have been observed via the cellular localization of zinc and
caspases under apoptotic and zinc deficient conditions13
, however, the cellular processes
which trigger metal based regulation in addition to how metal ions regulate caspase
activity still remain unclear. Kinases, on the other hand, such as Akt, ERK MAPK‟s, and
c-Abl regulate caspase-9 activity, both through activation and inhibition of the enzyme.
This occurs based on the position of the phosphorylation. For example, phosphorylation
at position T153 increases the activity of the enzyme86
whereas T125 phosphorylation
prevents processing of caspase-9 and subsequent executioner caspase activation87
. These
alternate modes of caspase regulation further enhance the idea of a multi-factorial
requirement for controlling caspase activity and unwanted cell death.
1.6. Synthetic Regulation of Caspase-9
Discovery of the caspases and their crucial role in apoptosis opened a new area of
therapeutic interest and thus the discovery and design of small synthetic inhibitors. Initial
inhibitor designs were inspired by the substrate specificity elements of the enzymes
themselves. Designed amino acid analogues (Fig 1.5 A) utilized the caspase substrate
recognition element aspartate to target all caspases, including caspase-9. Additional

16
analogues branched out into di-, tri-, and tetrapeptides that incorporated the sequence
requirements for the remainder of the substrate binding groove. Sequences such as Val-
Asp and Val-Ala-Asp targeted a broad spectrum of caspases and are classified as pan-
caspase inhibitors88
. As sequence specificities for individual caspases were determined,
specific peptide inhibitors were designed such as Leu-Glu-His-Asp (LEHD) in case of
caspase-933
(Fig 1.5 A).
Figure 1.5 Synthetic Inhibitors of Caspase-9 Activity. (A) Protected amino acid monomers to
tetrapeptide substrate mimics are tuned for caspase-9 active site specificity. The recognition element
Asp is utilized for capped amino acids while LEHD is utilized for tetrapeptide sequences which can be
functionalized on the N- and C- terminus. (B) N-terminal protective groups and C-terminal electrophilic
warheads provide variation and increase potency of the peptide-based inhibitors. (C-E) Small molecule
and α-helical peptide inhibitors are designed to disrupt caspase-9 activation via binding to Apaf-1 and
preventing the CARD-CARD interactions. Images generated in ChemBioDraw Ultra 12.0,
CambridgeSoft Corporation and the PyMOL Molecular Graphics System, Version 1.3, Schrödinger,
LLC.

17
Caspase targeting and potency of peptide-based inhibitors can be altered through
addition of protective groups, fluorophores, and reactive electrophilic warheads (Fig. 1.5
B). Functionalization on the N-terminus of the peptide sequence has included protective
groups such as the N-acetyl (ac), benzyloxycarbonyl (Z), or quinolyl (Q) moieties.
Fluorophores such as FITC have also been included on the amino terminal end of this
class of peptides to enable detection. The C-terminal portion of the peptide inhibitors
typically comprise electrophilic warheads that react with the catalytic nucleophile.
Aldehydes, nitriles and ketones are used for reversibility, while halomethylketones (e.g.
FMK, fluoromethyketone; CMK, chloromethylketone), diazomethylketones and
acylomethylketones serve as irreversible warheads33,89,90
. The C-terminal O-phenoxy
group was also incorporated into the warhead class of substrate based inhibitors due to its
improved leaving properties and reactivity compared to fluoromethylketones. Peptide
inhibitor Q-VD-Oph was determined to have an IC50 of 25-430 nM against a broad
spectrum of caspases, with caspase-9 being on the highest end of the spectrum91
. Potency
and specificity vary greatly amongst these inhibitors with dissociation constants ranging
anywhere from pM to μM. Caspase-9-specific, warhead driven inhibitors such as LEHD-
FMK, are potent against enzymatic activity. Compound IDN-6556 ((3 2-[(2-tert-butyl-
phenylaminooxalyl)-amino]-propionylamino 4-oxo-5-(2,3,5,6-tetrafluoro-phenoxy)-
pentanoic acid)) (Fig 1.5 C), a broad-spectrum irreversible inhibitor of the caspases with
an IC50 of 25 nM92
, encompasses a similar premise with incorporation of a peptide
backbone and the aspartate recognition element however, IDN-6566 utilizes an N-
terminal oxamide and C-terminal tetrafluorophenoxy warhead. This class of substrate
mimicking inhibitors have been studied in the treatment of acute liver failure93
,

18
myocardial ischemia-reperfusion injury94
, and traumatic brain injury95
. In this thesis, we
have explored an entirely new class of inhibitors (Chapter III).
Caspase-9‟s mechanism for activation via binding to the apoptosome complex has
also been targeted to control its enzymatic activity. Small molecule and α-helical peptide
inhibitors have been designed to either disrupt oligomerization of Apaf-1, which creates
the bulk of the apoptosome platform, or caspase-9‟s association with the apoptosome. N-
alkylglycine peptoid inhibitors (Fig 1.5 D) and their solubility-enhancing analogues96
bind to Apaf-1 (KD = 57 ± 12 nM) and prevent recruitment of caspase-9 to the
apoptosome. Fusion of cell penetrating peptides and a polymeric carrier to the N-
alkylglycine peptoid inhibitors have also been explored to enhance membrane
permeability and efficacy respectively for their ability to decrease apoptosis in neonatal
rat cardiomyoctyes under hypoxic conditions. Diarylurea based compounds also inhibit
formation of the apoptosome97
however the mechanism is slightly less clear. This class of
inhibitor is determined to either prevent recruitment cytochrome c, dATP, or caspase-9 to
Apaf-1. α-helical polypeptide modulators on the other hand, specifically target the
CARD-CARD mediated interaction of caspase-9 and Apaf-198
. Synthetic peptides were
derived from the acidic patch region of helices 2 and 3 of Apaf-1 CARD domain (Fig 1.5
D) and the basic patch of helices 1 and 3 of the caspase-9 CARD. This class of inhibitors
successfully disrupted the interaction of Apaf-1 and caspase-9, thus preventing caspase-9
activation both in vitro and in cell extracts with an IC50 of 68-102 μM for apoptosome
activity as judged by cleavage of caspase-3 and 63-113 μM in human embryonic kidney
293 (HEK 293) cellular extracts.

19
1.7. Caspase-9 and its Role in Disease
Caspase-9 is of high interest due to its entry point into the apoptotic pathway and
its involvement in disease. As an initiator caspase in the intrinsic pathway of apoptosis,
targeting caspase-9 provides flexibility between the upstream apoptotic signals and the
cleavage cascade, making this a critical target point for cell survival or cell death. This
role is reflected by its specific involvement in a variety of diseases.
Down regulation of caspase-9 has been observed in cancers as well as viral
infections. In colonic carcinoma cells, expression levels are found to be low or even
absent when compared to normal cells99
suggesting caspase-9 as the critical apoptotic
checkpoint within this cell line. In the case of testicular cancer, caspase-9 fails to
activate100
. Through the targeting caspase-9 in this manner, the testicular cancer cells are
also able to resist treatment with cisplatin, a treatment in which cell death is a priority,
further confirming the importance of this cell death protease in cancer. The viral infection
caused by the vaccinia virus, has also adopted means of controlling caspase-9. Viral
protein F1L has been found not only to mimic apoptosis-like proteins upstream of the
caspase cascade101,102
but to directly affect caspase-9 function103
. F1L inhibits caspase-9
activity by preventing zymogen caspase-9‟s association with the apoptosome in addition
to blocking the enzymes protease function, driving home the significance of caspase-9 in
controlling apoptosis.
Up regulation of caspase-9 has also been observed in a variety of neurological
diseases and immune disorders. Amyotrophic lateral sclerosis (ALS) exhibits increased
caspase-9 activity in the spinal motor neurons of ALS mouse models104
and has been
found to be a key player in the progression of the disease. Increased caspase-9 expression

20
levels and activity are also observed in the severe neuropathological cases of
Huntington‟s disease patients and in a Huntington mouse model, implicating caspase-9‟s
involvement in neuronal death at the end stage of the disease105
. The same trend is also
observed in the endothelial cells of patients with a rare multisystemic immune disorder
known as Behçet's disease106
, further confirming caspase-9‟s pivotal role in the
progression of a disease.
In addition to direct action of caspase-9, diseases also co-opt the regulators of
caspase-9 in order to ensure activation of the apoptotic pathway. A characteristic of renal
cell carcinoma107,108
, lung cancer109
, testicular germ cell tumors110
and hepatocellular
cancer111
is the release of caspase inhibitor antagonist, Smac/Diablo, which is a direct
antagonists of BIR3 resulting in the release its inhibition over caspase-9. This allows
caspase-9 to activate and initiate the caspase cascade. In a similar fashion, while
experiencing ischemia-reperfusion injury of the heart, the BIR3 antagonist, Omi/HtrA2,
is released thus causing activation of caspase-9 as well112
.
Given these unique roles of caspase-9 in a wide variety of diseases, fully
understanding the process of apoptosis is vital to understanding the normal homeostasis
of a cell and to controlling a variety of diseases in which the cellular death process has
been altered. By focusing on the caspases, the ultimate “hit men” of the cell, control can
be achieved, specifically when targeting the initiator caspase-9. By controlling caspase-9
activity, the transmission of the cell death signal can be controlled as well. For this
reason, the focus of this thesis is to harness and further understand the natural regulatory
events inhibition of caspase-9 by BIR3, CARD domain activation and metal inhibition
via biochemical and biophysical means.

21
1.8. References
1. Jacobson, M. D., Weil, M. & Raff, M. C. Programmed cell death in animal
development. Cell 88, 347-354, (1997).
2. Meier, P., Finch, A. & Evan, G. Apoptosis in development. Nature 407, 796-801,
(2000).
3. Aharinejad, S. et al. Programmed cell death in idiopathic dilated cardiomyopathy
is mediated by suppression of the apoptosis inhibitor Apollon. The Annals of
thoracic surgery 86, 109-114, (2008).
4. Narula, J. et al. Apoptosis in myocytes in end-stage heart failure. The New
England journal of medicine 335, 1182-1189, (1996).
5. Olivetti, G. et al. Acute myocardial infarction in humans is associated with
activation of programmed myocyte cell death in the surviving portion of the heart.
Journal of molecular and cellular cardiology 28, 2005-2016, (1996).
6. Saraste, A. et al. Apoptosis in human acute myocardial infarction. Circulation 95,
320-323, (1997).
7. Augstein, P., Elefanty, A. G., Allison, J. & Harrison, L. C. Apoptosis and beta-
cell destruction in pancreatic islets of NOD mice with spontaneous and
cyclophosphamide-accelerated diabetes. Diabetologia 41, 1381-1388, (1998).
8. Butler, A. E. et al. Beta-cell deficit and increased beta-cell apoptosis in humans
with type 2 diabetes. Diabetes 52, 102-110, (2003).
9. Kurrer, M. O., Pakala, S. V., Hanson, H. L. & Katz, J. D. Beta cell apoptosis in T
cell-mediated autoimmune diabetes. Proceedings of the National Academy of
Sciences of the United States of America 94, 213-218, (1997).
10. O'Brien, B. A., Harmon, B. V., Cameron, D. P. & Allan, D. J. Apoptosis is the
mode of beta-cell death responsible for the development of IDDM in the
nonobese diabetic (NOD) mouse. Diabetes 46, 750-757, (1997).
11. Diabetes Association. (2011).
12. American Lung Association. Epidemiology & Statistics Unit, Research and
Program Services, (2007).
13. Carter, J. E. et al. Involvement of redox events in caspase activation in zinc-
depleted airway epithelial cells. Biochemical and biophysical research
communications 297, 1062-1070, (2002).

22
14. Truong-Tran, A. Q., Grosser, D., Ruffin, R. E., Murgia, C. & Zalewski, P. D.
Apoptosis in the normal and inflamed airway epithelium: role of zinc in epithelial
protection and procaspase-3 regulation. Biochemical pharmacology 66, 1459-
1468, (2003).
15. Kountouras, J., Zavos, C. & Chatzopoulos, D. Apoptosis in hepatitis C. Journal of
viral hepatitis 10, 335-342, (2003).
16. Calabrese, F. et al. Liver cell apoptosis in chronic hepatitis C correlates with
histological but not biochemical activity or serum HCV-RNA levels. Hepatology
31, 1153-1159, (2000).
17. Hotchkiss, R. S. et al. Prevention of lymphocyte cell death in sepsis improves
survival in mice. Proceedings of the National Academy of Sciences of the United
States of America 96, 14541-14546, (1999).
18. Hotchkiss, R. S. et al. Sepsis-induced apoptosis causes progressive profound
depletion of B and CD4+ T lymphocytes in humans. J Immunol 166, 6952-6963,
(2001).
19. Keel, M. et al. Interleukin-10 counterregulates proinflammatory cytokine-induced
inhibition of neutrophil apoptosis during severe sepsis. Blood 90, 3356-3363,
(1997).
20. Hotchkiss, R. S. et al. Caspase inhibitors improve survival in sepsis: a critical role
of the lymphocyte. Nature immunology 1, 496-501, (2000).
21. Tak, P. P. & Bresnihan, B. The pathogenesis and prevention of joint damage in
rheumatoid arthritis: advances from synovial biopsy and tissue analysis. Arthritis
and rheumatism 43, 2619-2633, (2000).
22. Smith, M. D. et al. Apoptosis in the rheumatoid arthritis synovial membrane:
modulation by disease-modifying anti-rheumatic drug treatment. Rheumatology
(Oxford) 49, 862-875, (2010).
23. Ambrosini, G., Adida, C. & Altieri, D. C. A novel anti-apoptosis gene, survivin,
expressed in cancer and lymphoma. Nature medicine 3, 917-921, (1997).
24. Tamm, I. et al. Expression and prognostic significance of IAP-family genes in
human cancers and myeloid leukemias. Clinical cancer research : an official
journal of the American Association for Cancer Research 6, 1796-1803, (2000).
25. Vucic, D., Stennicke, H. R., Pisabarro, M. T., Salvesen, G. S. & Dixit, V. M. ML-
IAP, a novel inhibitor of apoptosis that is preferentially expressed in human
melanomas. Current biology : CB 10, 1359-1366, (2000).
26. Miyashita, T. et al. Tumor suppressor p53 is a regulator of bcl-2 and bax gene
expression in vitro and in vivo. Oncogene 9, 1799-1805, (1994).

23
27. Miyashita, T. & Reed, J. C. Tumor suppressor p53 is a direct transcriptional
activator of the human bax gene. Cell 80, 293-299, (1995).
28. Nakano, K. & Vousden, K. H. PUMA, a novel proapoptotic gene, is induced by
p53. Molecular cell 7, 683-694, (2001).
29. Oda, E. et al. Noxa, a BH3-only member of the Bcl-2 family and candidate
mediator of p53-induced apoptosis. Science 288, 1053-1058, (2000).
30. Sax, J. K. et al. BID regulation by p53 contributes to chemosensitivity. Nature
cell biology 4, 842-849, (2002).
31. Yu, J., Zhang, L., Hwang, P. M., Kinzler, K. W. & Vogelstein, B. PUMA induces
the rapid apoptosis of colorectal cancer cells. Molecular cell 7, 673-682, (2001).
32. Reed, J. C. & Tomaselli, K. J. Drug discovery opportunities from apoptosis
research. Current opinion in biotechnology 11, 586-592, (2000).
33. Thornberry, N. A. et al. A combinatorial approach defines specificities of
members of the caspase family and granzyme B. Functional relationships
established for key mediators of apoptosis. The Journal of biological chemistry
272, 17907-17911, (1997).
34. Denault, J. B. & Salvesen, G. S. Human caspase-7 activity and regulation by its
N-terminal peptide. The Journal of biological chemistry 278, 34042-34050,
(2003).
35. Meergans, T., Hildebrandt, A. K., Horak, D., Haenisch, C. & Wendel, A. The
short prodomain influences caspase-3 activation in HeLa cells. The Biochemical
journal 349, 135-140, (2000).
36. Feeney, B., Pop, C., Swartz, P., Mattos, C. & Clark, A. C. Role of loop bundle
hydrogen bonds in the maturation and activity of (Pro)caspase-3. Biochemistry 45,
13249-13263, (2006).
37. Garcia-Calvo, M. et al. Purification and catalytic properties of human caspase
family members. Cell death and differentiation 6, 362-369, (1999).
38. Blanchard, H. et al. Caspase-8 specificity probed at subsite S(4): crystal structure
of the caspase-8-Z-DEVD-cho complex. Journal of molecular biology 302, 9-16,
(2000).
39. Chereau, D., Kodandapani, L., Tomaselli, K. J., Spada, A. P. & Wu, J. C.
Structural and functional analysis of caspase active sites. Biochemistry 42, 4151-
4160, (2003).
40. Wei, Y. et al. The structures of caspases-1, -3, -7 and -8 reveal the basis for
substrate and inhibitor selectivity. Chemistry & biology 7, 423-432, (2000).

24
41. Stennicke, H. R., Renatus, M., Meldal, M. & Salvesen, G. S. Internally quenched
fluorescent peptide substrates disclose the subsite preferences of human caspases
1, 3, 6, 7 and 8. The Biochemical journal 350 Pt 2, 563-568, (2000).
42. Shi, Y. Mechanisms of caspase activation and inhibition during apoptosis.
Molecular cell 9, 459-470, (2002).
43. Fuentes-Prior, P. & Salvesen, G. S. The protein structures that shape caspase
activity, specificity, activation and inhibition. The Biochemical journal 384, 201-
232, (2004).
44. Brady, K. D. et al. A catalytic mechanism for caspase-1 and for bimodal
inhibition of caspase-1 by activated aspartic ketones. Bioorganic & medicinal
chemistry 7, 621-631, (1999).
45. Stennicke, H. R. & Salvesen, G. S. Catalytic properties of the caspases. Cell death
and differentiation 6, 1054-1059, (1999).
46. Boldin, M. P. et al. A novel protein that interacts with the death domain of
Fas/APO1 contains a sequence motif related to the death domain. The Journal of
biological chemistry 270, 7795-7798, (1995).
47. Chinnaiyan, A. M., O'Rourke, K., Tewari, M. & Dixit, V. M. FADD, a novel
death domain-containing protein, interacts with the death domain of Fas and
initiates apoptosis. Cell 81, 505-512, (1995).
48. Kischkel, F. C. et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated
proteins form a death-inducing signaling complex (DISC) with the receptor. The
EMBO journal 14, 5579-5588, (1995).
49. Wang, J., Chun, H. J., Wong, W., Spencer, D. M. & Lenardo, M. J. Caspase-10 is
an initiator caspase in death receptor signaling. Proceedings of the National
Academy of Sciences of the United States of America 98, 13884-13888, (2001).
50. Gross, A., Jockel, J., Wei, M. C. & Korsmeyer, S. J. Enforced dimerization of
BAX results in its translocation, mitochondrial dysfunction and apoptosis. The
EMBO journal 17, 3878-3885, (1998).
51. Yang, E. et al. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax
and promotes cell death. Cell 80, 285-291, (1995).
52. Ge, X., Fu, Y. M., Li, Y. Q. & Meadows, G. G. Activation of caspases and
cleavage of Bid are required for tyrosine and phenylalanine deficiency-induced
apoptosis of human A375 melanoma cells. Archives of biochemistry and
biophysics 403, 50-58, (2002).

25
53. Hu, Y., Benedict, M. A., Ding, L. & Nunez, G. Role of cytochrome c and
dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis.
The EMBO journal 18, 3586-3595, (1999).
54. Stennicke, H. R. et al. Caspase-9 can be activated without proteolytic processing.
The Journal of biological chemistry 274, 8359-8362, (1999).
55. Rodriguez, J. & Lazebnik, Y. Caspase-9 and APAF-1 form an active holoenzyme.
Genes & development 13, 3179-3184, (1999).
56. Saleh, A., Srinivasula, S. M., Acharya, S., Fishel, R. & Alnemri, E. S.
Cytochrome c and dATP-mediated oligomerization of Apaf-1 is a prerequisite for
procaspase-9 activation. The Journal of biological chemistry 274, 17941-17945,
(1999).
57. Zou, H., Li, Y., Liu, X. & Wang, X. An APAF-1.cytochrome c multimeric
complex is a functional apoptosome that activates procaspase-9. The Journal of
biological chemistry 274, 11549-11556, (1999).
58. Yuan, S. et al. The holo-apoptosome: activation of procaspase-9 and interactions
with caspase-3. Structure 19, 1084-1096, (2011).
59. Yu, X., Wang, L., Acehan, D., Wang, X. & Akey, C. W. Three-dimensional
structure of a double apoptosome formed by the Drosophila Apaf-1 related killer.
Journal of molecular biology 355, 577-589, (2006).
60. Pop, C., Timmer, J., Sperandio, S. & Salvesen, G. S. The apoptosome activates
caspase-9 by dimerization. Molecular cell 22, 269-275, (2006).
61. Shiozaki, E. N., Chai, J. & Shi, Y. Oligomerization and activation of caspase-9,
induced by Apaf-1 CARD. Proceedings of the National Academy of Sciences of
the United States of America 99, 4197-4202, (2002).
62. Renatus, M., Stennicke, H. R., Scott, F. L., Liddington, R. C. & Salvesen, G. S.
Dimer formation drives the activation of the cell death protease caspase 9.
Proceedings of the National Academy of Sciences of the United States of America
98, 14250-14255, (2001).
63. Donepudi, M., Mac Sweeney, A., Briand, C. & Grutter, M. G. Insights into the
regulatory mechanism for caspase-8 activation. Molecular cell 11, 543-549,
(2003).
64. Bose, K. & Clark, A. C. Dimeric procaspase-3 unfolds via a four-state
equilibrium process. Biochemistry 40, 14236-14242, (2001).
65. Acehan, D. et al. Three-dimensional structure of the apoptosome: implications for
assembly, procaspase-9 binding, and activation. Molecular cell 9, 423-432,
(2002).

26
66. Salvesen, G. S. & Dixit, V. M. Caspase activation: the induced-proximity model.
Proceedings of the National Academy of Sciences of the United States of America
96, 10964-10967, (1999).
67. Shi, Y. Caspase activation: revisiting the induced proximity model. Cell 117, 855-
858, (2004).
68. Malladi, S., Challa-Malladi, M., Fearnhead, H. O. & Bratton, S. B. The Apaf-
1*procaspase-9 apoptosome complex functions as a proteolytic-based molecular
timer. The EMBO journal 28, 1916-1925, (2009).
69. Deveraux, Q. L. & Reed, J. C. IAP family proteins--suppressors of apoptosis.
Genes & development 13, 239-252, (1999).
70. Fesik, S. W. Insights into programmed cell death through structural biology. Cell
103, 273-282, (2000).
71. Chai, J. et al. Structural basis of caspase-7 inhibition by XIAP. Cell 104, 769-780,
(2001).
72. Riedl, S. J. et al. Structural basis for the inhibition of caspase-3 by XIAP. Cell
104, 791-800, (2001).
73. Takahashi, R. et al. A single BIR domain of XIAP sufficient for inhibiting
caspases. The Journal of biological chemistry 273, 7787-7790, (1998).
74. Scott, F. L. et al. XIAP inhibits caspase-3 and -7 using two binding sites:
evolutionarily conserved mechanism of IAPs. The EMBO journal 24, 645-655,
(2005).
75. Deveraux, Q. L., Takahashi, R., Salvesen, G. S. & Reed, J. C. X-linked IAP is a
direct inhibitor of cell-death proteases. Nature 388, 300-304, (1997).
76. Sun, C. et al. NMR structure and mutagenesis of the third Bir domain of the
inhibitor of apoptosis protein XIAP. The Journal of biological chemistry 275,
33777-33781, (2000).
77. Liu, Z. et al. Structural basis for binding of Smac/DIABLO to the XIAP BIR3
domain. Nature 408, 1004-1008, (2000).
78. Vucic, D. et al. Engineering ML-IAP to produce an extraordinarily potent caspase
9 inhibitor: implications for Smac-dependent anti-apoptotic activity of ML-IAP.
The Biochemical journal 385, 11-20, (2005).
79. Shiozaki, E. N. et al. Mechanism of XIAP-mediated inhibition of caspase-9.
Molecular cell 11, 519-527, (2003).

27
80. Birnbaum, M. J., Clem, R. J. & Miller, L. K. An apoptosis-inhibiting gene from a
nuclear polyhedrosis virus encoding a polypeptide with Cys/His sequence motifs.
Journal of virology 68, 2521-2528, (1994).
81. Crook, N. E., Clem, R. J. & Miller, L. K. An apoptosis-inhibiting baculovirus
gene with a zinc finger-like motif. Journal of virology 67, 2168-2174, (1993).
82. Nogal, M. L. et al. African swine fever virus IAP homologue inhibits caspase
activation and promotes cell survival in mammalian cells. Journal of virology 75,
2535-2543, (2001).
83. Perry, D. K. et al. Zinc is a potent inhibitor of the apoptotic protease, caspase-3. A
novel target for zinc in the inhibition of apoptosis. The Journal of biological
chemistry 272, 18530-18533, (1997).
84. Chimienti, F., Seve, M., Richard, S., Mathieu, J. & Favier, A. Role of cellular
zinc in programmed cell death: temporal relationship between zinc depletion,
activation of caspases, and cleavage of Sp family transcription factors.
Biochemical pharmacology 62, 51-62, (2001).
85. Schrantz, N. et al. Zinc-mediated regulation of caspases activity: dose-dependent
inhibition or activation of caspase-3 in the human Burkitt lymphoma B cells
(Ramos). Cell death and differentiation 8, 152-161, (2001).
86. Raina, D. et al. c-Abl tyrosine kinase regulates caspase-9 autocleavage in the
apoptotic response to DNA damage. The Journal of biological chemistry 280,
11147-11151, (2005).
87. Allan, L. A. et al. Inhibition of caspase-9 through phosphorylation at Thr 125 by
ERK MAPK. Nature cell biology 5, 647-654, (2003).
88. Garcia-Calvo, M. et al. Inhibition of human caspases by peptide-based and
macromolecular inhibitors. The Journal of biological chemistry 273, 32608-
32613, (1998).
89. Thornberry, N. A. et al. Inactivation of interleukin-1 beta converting enzyme by
peptide (acyloxy)methyl ketones. Biochemistry 33, 3934-3940, (1994).
90. Nicholson, D. W. et al. Identification and inhibition of the ICE/CED-3 protease
necessary for mammalian apoptosis. Nature 376, 37-43, (1995).
91. Caserta, T. M., Smith, A. N., Gultice, A. D., Reedy, M. A. & Brown, T. L. Q-VD-
OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties.
Apoptosis : an international journal on programmed cell death 8, 345-352,
(2003).
92. Linton, S. D. et al. First-in-class pan caspase inhibitor developed for the treatment
of liver disease. Journal of medicinal chemistry 48, 6779-6782, (2005).

28
93. Yoshida, N. et al. Improvement of the survival rate after rat massive hepatectomy
due to the reduction of apoptosis by caspase inhibitor. Journal of
gastroenterology and hepatology 22, 2015-2021, (2007).
94. Mocanu, M. M., Baxter, G. F. & Yellon, D. M. Caspase inhibition and limitation
of myocardial infarct size: protection against lethal reperfusion injury. British
journal of pharmacology 130, 197-200, (2000).
95. Abrahamson, E. E. et al. Caspase inhibition therapy abolishes brain trauma-
induced increases in Abeta peptide: implications for clinical outcome.
Experimental neurology 197, 437-450, (2006).
96. Malet, G. et al. Small molecule inhibitors of Apaf-1-related caspase- 3/-9
activation that control mitochondrial-dependent apoptosis. Cell death and
differentiation 13, 1523-1532, (2006).
97. Lademann, U. et al. Diarylurea compounds inhibit caspase activation by
preventing the formation of the active 700-kilodalton apoptosome complex.
Molecular and cellular biology 23, 7829-7837, (2003).
98. Palacios-Rodriguez, Y. et al. Polypeptide modulators of card-card-mediated
protein-protein interactions. The Journal of biological chemistry, (2011).
99. Palmerini, F., Devilard, E., Jarry, A., Birg, F. & Xerri, L. Caspase 7
downregulation as an immunohistochemical marker of colonic carcinoma. Human
pathology 32, 461-467, (2001).
100. Mueller, T. et al. Failure of activation of caspase-9 induces a higher threshold for
apoptosis and cisplatin resistance in testicular cancer. Cancer research 63, 513-
521, (2003).
101. Postigo, A., Cross, J. R., Downward, J. & Way, M. Interaction of F1L with the
BH3 domain of Bak is responsible for inhibiting vaccinia-induced apoptosis. Cell
death and differentiation 13, 1651-1662, (2006).
102. Wasilenko, S. T., Banadyga, L., Bond, D. & Barry, M. The vaccinia virus F1L
protein interacts with the proapoptotic protein Bak and inhibits Bak activation.
Journal of virology 79, 14031-14043, (2005).
103. Zhai, D. et al. Vaccinia virus protein F1L is a caspase-9 inhibitor. The Journal of
biological chemistry 285, 5569-5580, (2010).
104. Inoue, H. et al. The crucial role of caspase-9 in the disease progression of a
transgenic ALS mouse model. The EMBO journal 22, 6665-6674, (2003).
105. Kiechle, T. et al. Cytochrome C and caspase-9 expression in Huntington's disease.
Neuromolecular medicine 1, 183-195, (2002).

29
106. Oztas, P. et al. Caspase-9 expression is increased in endothelial cells of active
Behcet's disease patients. International journal of dermatology 46, 172-176,
(2007).
107. Kempkensteffen, C. et al. Expression levels of the mitochondrial IAP antagonists
Smac/DIABLO and Omi/HtrA2 in clear-cell renal cell carcinomas and their
prognostic value. Journal of cancer research and clinical oncology 134, 543-550,
(2008).
108. Mizutani, Y. et al. Downregulation of Smac/DIABLO expression in renal cell
carcinoma and its prognostic significance. Journal of clinical oncology : official
journal of the American Society of Clinical Oncology 23, 448-454, (2005).
109. Sekimura, A. et al. Expression of Smac/DIABLO is a novel prognostic marker in
lung cancer. Oncology reports 11, 797-802, (2004).
110. Kempkensteffen, C. et al. The equilibrium of XIAP and Smac/DIABLO
expression is gradually deranged during the development and progression of
testicular germ cell tumours. International journal of andrology 30, 476-483,
(2007).
111. Bao, S. T., Gui, S. Q. & Lin, M. S. Relationship between expression of Smac and
Survivin and apoptosis of primary hepatocellular carcinoma. Hepatobiliary &
pancreatic diseases international : HBPD INT 5, 580-583, (2006).
112. Liu, H. R. et al. Role of Omi/HtrA2 in apoptotic cell death after myocardial
ischemia and reperfusion. Circulation 111, 90-96, (2005).

30
CHAPTER II
ROBUST PRODUCTION OF A LIBRARY OF CASPASE-9-INHIBITOR
PEPTIDES USING METHODOLOGICAL SYNCHRONIZATION
This chapter was published as: Huber, K. L., Olsen, K. D.* and Hardy, J. A., 2009. “Robust
Production of a Peptide Library using Methodological Synchronization.” Protein Expression
and Purification, 67,139-147. KLH designed initial parent construct and mutational sites, in
addition to performing metal ion affinity and reverse HPLC purifications, ESI-MS, and peptide
expression analysis. KDO performed site directed mutagenesis and affinity purification steps.
Abstract
Peptide libraries have proven to be useful in applications such as substrate profiling, drug
candidate screening and identifying protein-protein interaction partners. However, issues
of fidelity, peptide length and purity have been encountered when peptide libraries are
chemically synthesized. Biochemically produced libraries, on the other hand, circumvent
many of these issues due to the fidelity of the protein synthesis machinery. Using
thioredoxin as an expression partner, a stably folded peptide scaffold (avian pancreatic
polypeptide) and a compatible cleavage site for human rhinovirus 3C protease, we report
a method that allows robust expression of a genetically-encoded XIAP-Bir3 inspired
peptide library, which yields peptides of high purity. In addition, we report the use of
methodological synchronization, an experimental design created for the production of a
library, from initial cloning to peptide characterization, within a five-week period of time.
Total peptide yields ranged from 0.8 to 16%, which corresponds to 2-70 milligrams of
pure peptide per one liter of cell culture. Additionally, no correlation was observed
between the ability to be expressed or overall yield of peptide-fusions and the intrinsic
chemical characteristics of the peptides, indicating that this system can be used for a wide
variety of peptide sequences with a range of chemical characteristics.

31
2.1. Introduction
Peptide libraries are commonly used in a variety of endeavors including identifying
peptide-DNA interactions 1, as surrogates for screening protein-protein interactions
2-5,
and as a basis for finding potential peptidic drug molecules. Peptide libraries have also
been used for profiling substrate specificities of proteases6-11
, phosphatases12-14
, kinases15-
18 and other drug targets
19-22. Thus, the production of high quality peptide libraries is of
wide interest for many applications.
Typically, peptides for libraries have been produced via solid-phase synthesis, which
involves sequential coupling of amine-protected amino acids to resin-bound amino acids.
The resulting libraries generally contain peptides no longer than 15-20 amino acids,
which can prove limiting in applications requiring longer peptides. The length constraints
for chemically synthesized peptides are the result of coupling and deprotection
efficiencies at each step, such that an exponential decrease in sequence fidelity is
observed as a function of length. In addition, enantiomerization of nineteen of the twenty
naturally-occurring amino acids and the associated difficulties in purification of the D-
isomer-containing peptides from the L-isomers set practical limits to peptide lengths in
library synthesis23,24
. Sometimes peptide sequences are restricted due to steric clash of
adjacent amino acids with bulky chemical catalysts or intermolecular aggregation which
results in a low efficiency of the chemical coupling reaction25-27
. Overall, chemically
synthesized peptide libraries of longer lengths tend to have decreased sample quality, be
costly and have increased production time.
Genetically-encoded peptide libraries offer several advantages in library design. The

32
resulting libraries can contain longer peptide lengths and significantly increased yields
while avoiding the more common limitations of chemical synthesis. Biological synthesis
of peptides of longer lengths can be particularly important for production of isotopically-
labeled peptides for use in heteronuclear Nuclear Magnetic Resonance (NMR)
spectroscopy. The principle advantages of biological production result from the fidelity
of protein machinery, particularly because the ribosome is not limited in sequence or
length of synthesized peptides and has an error rate of only 0.01% error per amino acid
added28
. Genetically-encoded peptide libraries have not been widely used due to
difficulties in the expression and purification of small peptides. This problem has been
overcome by the use of protein-expression fusion partners such as glutathione-s-
transferase29
, maltose binding protein30
and thioredoxin31
being the three most widely-
used fusions. While fusion proteins often promote production, this method can be time
consuming and requires post expression protease processing and purification of the
peptide from the protease. However, the addition of fusion partners has resulted in the
improvement of the overall yields by enhancing solubility of the peptide of choice.
We report a method that allows robust expression of a genetically-encoded XIAP-
Bir3 inspired peptide library that addresses the issues of purity, yield, length of the
purified peptide, batch-to-batch variability which we have observed with chemically
synthesized peptides, in addition to cost and time of production. This genetically-encoded
system features thioredoxin as a fusion tag for the stably-folding peptide scaffold avian
pancreatic polypeptide (aPP). aPP is a 36 amino acid peptide that contains a hydrophobic
core and hydrogen bond network between the α-helix and polyproline helix of the

33
Figure 2.1 aPP structure. (A) aPP backbone (lightest
gray) has a hydrophobic core created by three
prolines located on the poly-proline helix (medium
gray) and various residues on the α-helix (dark gray).
(B) aPP’s internal hydrogen bond network,
highlighted in dashed lines, is responsible for the
impressive structural stabilization of a peptide that is
just 36 amino acids long.
peptide32,33
(Fig. 2.1 A and B). These
properties are responsible for its fold
and stability. Previous work has
demonstrated that the presence of the
hydrophobic core and hydrogen bond
network render aPP insensitive to
mutations over much of the amino acid
sequence34,35
or to a c-terminal
truncation33,36
.
A common difficulty with peptide
expression is residual amino acid overhangs following the protease cleavage event. The
native sequence of aPP allows inclusion of an N-terminal cleavage site for human
rhinovirus 3C protease, a highly specific protease, without any unwanted amino acid
additions to the peptides. Human rhinovirus 3C protease cleaves the sequence LEVLFQ-
GP, generating a gly-pro overhang upon cleavage. In aPP the first two amino acids are
gly-pro, so no residual amino acids are left upon cleavage. Combining this optimized
expression system with methodological synchronization of site-directed mutagenesis,
protein expression and purification, a twenty-member aPP variant library of peptides 27
amino acids in length was generated. This method allowed production of the desired
library with overall improved yields, purity, and cost, all on a time frame that is
comparable to synthetic peptide library methods on the same production scale.

34
Figure 2.2 Schematic representation of expression vector,
pET32-Peptide. The T7 promoter, a fusion partner
thioredoxin (TrxA), 6xHis tag, human rhinovirus 3C
cleavable linker, gene for peptide, and ampicillin resistance
are shown.
2.2. Results
2.2.1. Construction of Peptide-Fusion Expression Vector and aPP Variants
A peptide-expression system for the library of aPP-based peptides was generated
that expressed thioredoxin fusion proteins. A goal of this project was to streamline all
steps in the process of cloning,
expression and purification. In
order to limit subsequent
subcloning events, the parent
vector (Fig. 2.2) was created
through a single PCR reaction
that resulted in the gene for a
human rhinovirus 3C protease
cleavage site (LEVLFQ) and
the truncated aPP sequence
(GPSQPTYPGDDAPVE-DLIRFYNDLQQY). This gene product was then ligated into
the thioredoxin fusion gene and 6xHis purification tag containing vector, pET32b via
restriction sites NcoI and XhoI. The resultant thioredoxin-peptide fusion construct then
served as the first base sequence for further mutagenesis.
aPP variant sequences were created by mutating codons for six to twelve of the
amino acids in the truncated 27 amino acid peptide scaffold (Fig. 2.3 A) via a
QuikChange (Stratagene) mutagenesis strategy. This limited library was designed as aPP
mimics of the natural caspase inhibitor, XIAP-BIR3. Mutations included the desired
variations in the original peptide sequences and took into consideration amino acids that

35
are known to be structurally important for scaffold stability. Sites of mutagenesis are
throughout the truncated aPP sequence and are spread across the aPP structural elements
(Fig. 2.3 B). Mutational sites include Q4, T6, D10 and D11 on the polyproline helix and
D16, D17, I18, Y21, D23, L24 Q25, and Y26 on the α-helix. In addition D11 was
selected as a site to be extensively interrogated. By focusing our investigation in one
region, we were able to produce many variants in a limited number of mutagenic steps.
Moreover, all mutational oligonucleotides were designed to limit cost and allow rapid
production of the new peptide encoding DNA constructs. These designs focused on
producing an array of peptide sequences using the minimum number of rounds of
Figure 2.3 Structure of peptide library. (A) Amino acid sequences of the twenty-member peptide
library. Mutated residues are bold. (B) Cartoon representation of truncated aPP peptide scaffold
with sites of mutations shown in stick representation and highly interrogated site D11 represented
in spheres.

36
mutagenesis. If mutagenesis was performed not by sequential means, 123 rounds of
mutagenesis would have been performed to complete the library. Mutations were quickly
assessed by growing the transformed E. coli cells for eight hours and plasmid prepping
the DNA in time for same day sequencing by an outsourced company (Genewiz Inc).
Upon sequence analysis, one out of two or more clones tested obtained the desired
mutations. The success rate was directly related to the thoroughness of the DpnI digestion
step during the QuikChange protocol (data not shown).
2.2.2. Expression and Purification of a Recombinant Peptide Library
Once the desired genetic mutations were verified, the vectors were transformed
into the BL21(DE3) strain of E. coli, expressed and harvested. The resulting cell pellets
were lysed and prepared for purification. Various avenues were tested to optimize the
purification of the peptide-fusion from other bacterial proteins. This was done in order to
minimize the potential additive loss of protein during each purification step. Therefore, a
nickel affinity step gradient, alone or in combination with ion exchange methods as well
as nickel affinity linear gradients were explored to obtain a peptide-fusion sample of
adequate purity without sacrificing yield. For example, peptide B3NK was purified via a
Ni-affinity step gradient and ion exchange methods while peptide B3NR was purified by
a Ni-affinity linear gradient only (Fig. 2.4). It is clear that a two-step purification yields
protein of higher purity compared to Ni-gradient alone. However, since only a few
contaminants are removed by the ion-exchange chromatography step in
spite of a significant loss of sample, the single-step linear Ni-gradient became the
preferred method of purification.

37
Once the peptide-fusions were purified to a satisfactory degree, cleavage of the
peptide from its fusion partner and linkers was accomplished by the highly specific
human rhinovirus 3C protease (Fig. 2.5). Various proteases to peptide fusion ratios were
examined for optimal cleavage in minimal time. As a 5-hour digestion with a 1:25
protease to protein ratio was sufficient for full cleavage of peptide from the fusion tag,
this method emerged as the time-optimized protocol. To separate the liberated peptide
from the other components in solution, reverse phase HPLC was employed. By
employing a three phase gradient which changes in organic phase from steep for buffer
component elution, to shallow for peptide elution, to a second steep phase to regenerate
Figure 2.4 Peptide-fusion purifications. (A) SDS-PAGE gels of peptide-fusion B3NK purified by
a Ni-Affinity Step gradient (Load-L, Flow through 1-F1, Flow through 2-F2, Flow through 3-F3,
Wash 1-W1, Wash 2-W2, Wash3 W3, and Elution-E) and ion exchange (fractions A-G) are
indicated. (B) SDS-PAGE gel of peptide-fusion B3NR purified by Ni-affinity linear gradient
(Load-L, Wash1-W1, Wash 2-W2 and elution fractions A-C 250 mM Imidazole. Fraction D is the
elution from 1M imidazole and shows tightly bound B3NK and contaminants.) Uncleaved
peptide-fusion MW = 24 kDa.
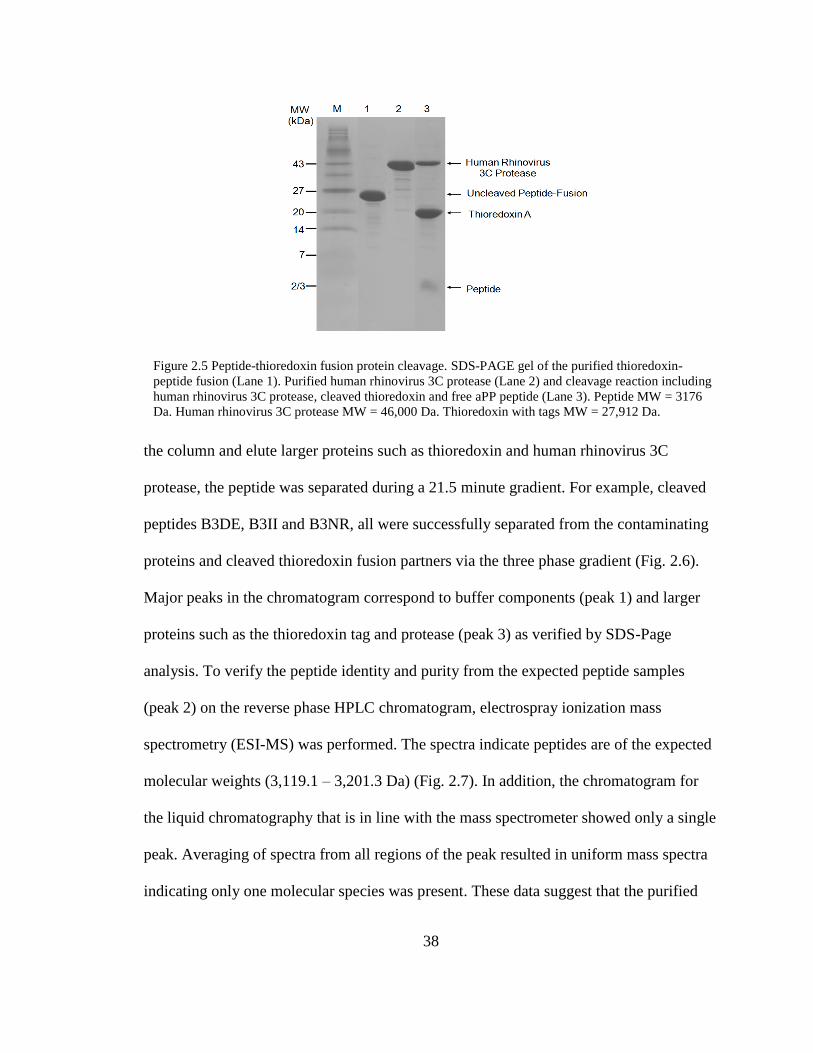
38
the column and elute larger proteins such as thioredoxin and human rhinovirus 3C
protease, the peptide was separated during a 21.5 minute gradient. For example, cleaved
peptides B3DE, B3II and B3NR, all were successfully separated from the contaminating
proteins and cleaved thioredoxin fusion partners via the three phase gradient (Fig. 2.6).
Major peaks in the chromatogram correspond to buffer components (peak 1) and larger
proteins such as the thioredoxin tag and protease (peak 3) as verified by SDS-Page
analysis. To verify the peptide identity and purity from the expected peptide samples
(peak 2) on the reverse phase HPLC chromatogram, electrospray ionization mass
spectrometry (ESI-MS) was performed. The spectra indicate peptides are of the expected
molecular weights (3,119.1 – 3,201.3 Da) (Fig. 2.7). In addition, the chromatogram for
the liquid chromatography that is in line with the mass spectrometer showed only a single
peak. Averaging of spectra from all regions of the peak resulted in uniform mass spectra
indicating only one molecular species was present. These data suggest that the purified
Figure 2.5 Peptide-thioredoxin fusion protein cleavage. SDS-PAGE gel of the purified thioredoxin-
peptide fusion (Lane 1). Purified human rhinovirus 3C protease (Lane 2) and cleavage reaction including
human rhinovirus 3C protease, cleaved thioredoxin and free aPP peptide (Lane 3). Peptide MW = 3176
Da. Human rhinovirus 3C protease MW = 46,000 Da. Thioredoxin with tags MW = 27,912 Da.
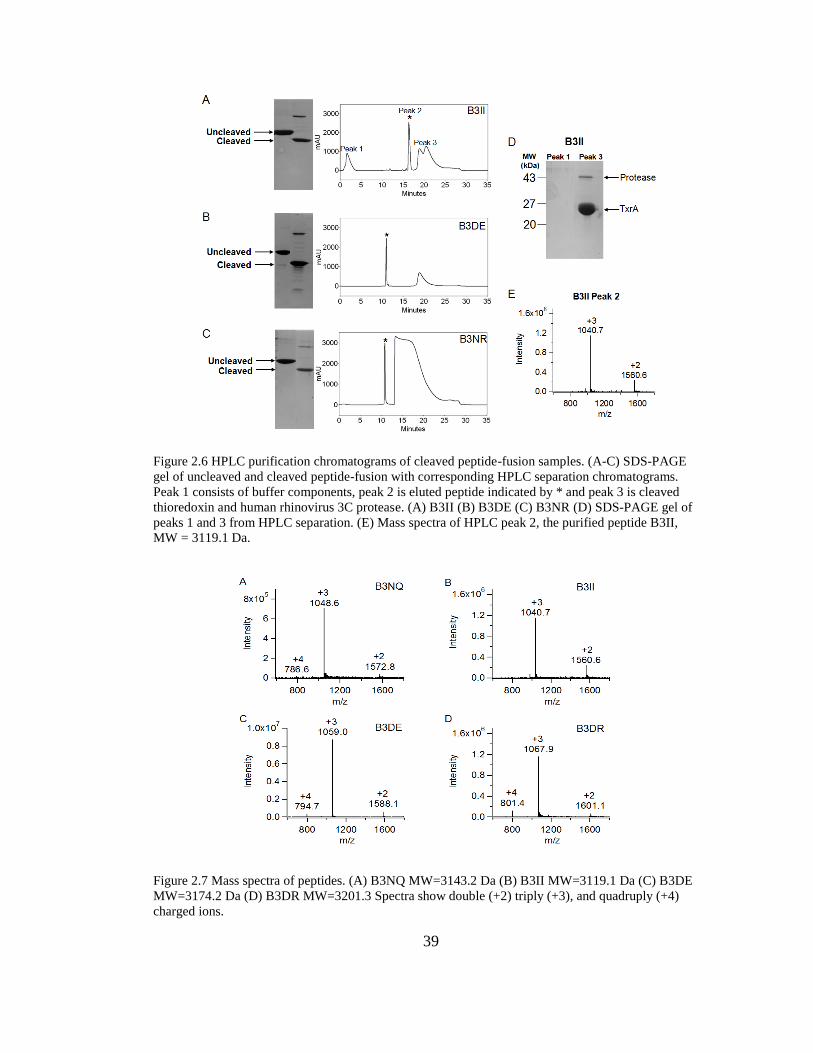
39
Figure 2.7 Mass spectra of peptides. (A) B3NQ MW=3143.2 Da (B) B3II MW=3119.1 Da (C) B3DE
MW=3174.2 Da (D) B3DR MW=3201.3 Spectra show double (+2) triply (+3), and quadruply (+4)
charged ions.
Figure 2.6 HPLC purification chromatograms of cleaved peptide-fusion samples. (A-C) SDS-PAGE
gel of uncleaved and cleaved peptide-fusion with corresponding HPLC separation chromatograms.
Peak 1 consists of buffer components, peak 2 is eluted peptide indicated by * and peak 3 is cleaved
thioredoxin and human rhinovirus 3C protease. (A) B3II (B) B3DE (C) B3NR (D) SDS-PAGE gel of
peaks 1 and 3 from HPLC separation. (E) Mass spectra of HPLC peak 2, the purified peptide B3II,
MW = 3119.1 Da.

40
peptides are of extremely high purity.
To assess the overall utility of the peptide library construction system, the total
amount of peptide fusion that was expressed in E. coli lysates is compared to the overall
yield of each of the purification methods (Table 2.1). Total peptide yield ranged from 0.8
to 16% from the overall expression. A major contributor to low peptide yield is
likely loss of protein at the step of column loading in the flow through during initial
purification due to overloaded resin. In addition, other contributing losses are observed at
the ionic exchange and reverse phase HPLC portions of the purification. For each
Table 2.1 Peptide Expression, Yield and Chemical Characteristics. Total peptide fusion expressed is
compared to the overall yield of the peptide as a function of purification method used. Peptide
characteristics such as charge, isoelectric point (IEP), the number of acidic, basic, polar, and non-
polar amino acid residues are also tabulated.
Percent peptide in raw E. coli lystates was calculated using GeneTools (SynGene). * Indicates %
peptide calculated was estimated by inspection of the stained SDS-PAGE gels. Ni: Ni-Affinity
chromatography. IE: ionic exchange chromatography. Acidic, Basic, Polar and Non-polar represent
the number of amino acids in the 27-residue peptide that have the particular chemical characteristic.

41
peptide, the ability to be expressed and the overall yield after purification was compared
to different peptide features including charge, isoelectric point, and amino acid
characteristics. Peptide charge ranged from -0.06 to -4.06 and calculated isoelectric point
from 3.77 to 6.92. The number of acidic residues in the peptide sequences ranged from
two to five while one to three basic residues, six to twelve polar residues and seven to
nine hydrophobic residues were present in the various sequences. Favorably, no
correlation between the expressability of the peptide-fusion and intrinsic chemical
characteristics of the peptides was found. Likewise, no correlation between peptidic
character and overall yield of pure peptide was observed. This suggests that our system
can be used for a wide variety of peptide sequences with a range of chemical
characteristics.
2.3.Discussion
During chemical synthesis of peptides, sequence errors are a likely result due to
the imperfect coupling efficiency at each step. For chemically synthesized peptides, the
contaminants are extremely similar to the desired product in size and chemical
characteristics and are therefore difficult to separate based on chromatography. Peptides
produced by biochemical synthesis by the E. coli ribosome are expected to be very
homogeneous due to the ribosome’s low error rate. In the system described here, any
contaminants that remain after affinity purification and protease cleavage of the
biochemically synthesized peptide fusions are unlikely to be chemically similar to the
peptides. Our method for production of peptides uses reverse-phase HPLC purification as
the final step. Because the non-peptide components are not chemically related to the

42
peptides themselves, HPLC purification allows for simultaneous removal of the fusion
partner, the protease, and any residual components of the buffer solution. This makes a
simple, two-step purification possible. In addition, because of the high fidelity of
biochemical synthesis, the homogeneity of our peptides is very impressive. This results in
a library of extremely high purity.
All of the peptide-fusion proteins described here were purified using a six-
histidine affinity tag. Following a Ni-affinity step gradient purification the peptide
fusions were approximately 50% pure. We probed the effect of more extensively
purifying the peptide fusions to >95% purity by subsequent anion exchange
chromatography or to 80% purity by using a linear gradient of imidazole on the Ni-
affinity column. Employing reverse-phase HPLC, we were able to robustly purify the
cleaved peptide to homogeneity after either of these protocols. This finding significantly
shortened our production time by eliminating the ion-exchange purification step.
An additional advantage of the method described is that it is unnecessary for the
peptide and the protease to be tagged with the same affinity tag, as is the case in some
commercial protein-fusion purification systems. The method described here is compatible
with any fusion partner and any protease. Since both the fusion partner and the protease
are likely to be much larger than peptide products, it will be possible to separate the
product from the contaminants in the final reverse-phase HPLC purification step. This
fact is of particular importance in production of peptide libraries where additional amino
acids at either the N- or C-termini can have significant effects on the overall properties of
the peptide. Since any protease can be used, the overhanging amino acids from the

43
protease cleavage site can be matched with the desired peptide sequence, or a protease
that does not leave any overhang can be selected37
. The facts that this method is not
dependent on use of any particular affinity tags or on utilizing matching affinity tags also
limit sub-cloning steps that are necessary to use the library production method.
This method of peptide production using genetically encoded peptides offers clear
advantages in the length and fidelity of peptides that can be produced and the resultant
purity of the final samples. In the field of chemical peptide synthesis, the concept of
“difficult” sequences (e.g. hydrophobic sequences) exists. Conversely, the genetically-
encoded production method described here was insensitive to peptide sequence such that
every sequence attempted worked the first time with no optimization of expression or
purification procedure required. To make this method truly competitive with chemical
synthesis, it is essential that libraries can be produced on the same general time scale as
chemical synthesis. The scheduling strategy for the twenty-member library we present
here lays the foundation for production of much larger libraries using the same processes.
To this end, we have time-optimized our expression, purification and characterization
protocols. Using the protocols we report and have routinely executed, we have
constructed a map for methodological synchronization (Fig. 2.8). By coordinating
mutagenesis cycles, sequencing, transformation, expression, purification and
characterization, this peptide library can be produced rapidly. We generated this map
reflecting the methods that were actually used; however, we have also performed
sequencing reactions after an eight-hour growth of cultures followed by mini-prepping of
DNA for sequencing. If this were applied to each peptide construct, it would save one

44
Figure 2.8 Methodological Synchronization. An experimental map showing how synchronization of
QuikChange-based mutagenesis and protein preparatory procedures allows the production of 20
peptides in a five-week time frame. Mutagenesis daily cycle (top) outlines the number of days in
which cloning steps such as QuikChange mutagenesis, transformation, DNA purification and
sequencing proceeds. 1:3, 2:3, and 3a:3 represents mutagenesis steps to create first peptide construct
B3. Protein daily cycle (bottom) outlines the number of days in which expression, purification, and
characterization for each peptide proceeds. The resultant DNA constructs are represented in light gray
and purified peptides are represented in dark gray.
day from each segment of the mutagenesis portion of the plan. In addition, we have
developed methodology so that QuikChange-mutagenized plasmids can be directly
transformed into an expression strain of bacteria (such as BL21(DE3)) rather than into a
cloning strain, which can save one step in the protocol. With these developments, the
entire library could be generated even more rapidly. As presented, this methodological
synchronization scheme allows construction of twenty peptide-expressing plasmids and
purification of the resultant peptides to homogeneity in a five-week period using one

45
chromatography system, one HPLC and one LC-MS.
In this work, we have used no more than two liters of E. coli culture per peptide and
have accepted losses at the affinity purification step. Nevertheless, the protocols
discussed here could facilely be scaled up to produce even greater yields of pure peptide.
For some of the peptide fusions we have constructed, the yield of the expressed fusion
protein as a fraction of total E. coli proteins is high enough (45% of total E. coli protein)
that we can nearly envision skipping purification of the fusion protein altogether. By co-
expressing the protease and the fusion protein it may be possible to perform a one-step
purification by HPLC to yield large quantities of pure peptide.
2.4. Materials and Methods
2.4.1. Cloning of Recombinant Peptide-Fusion Variants
In order to create the parent vector, pET32-Peptide, for the peptide-fusion DNA
variants, the aPP gene was amplified by PCR from the pJC2035
vector (from Alana
Schepartz and Doug Daniels). Using the forward primer 5’GTACAACCATGGCTGGA-
AGTGCTGTTTCAGGGTCCGTCCCAGCCGACCTACCC3’ that included the human
rhinovirus 3C cleavage sequence (bold) and NcoI endonuclease restriction site (italics)
and the reverse primer 5’TCGAGCCTCGAGCTAGTAACGGTGACGGGTAACAACG-
TTCAGG3’ which encodes the XhoI restriction site (italics) and stop codon (bold), the
desired gene was produced. This gene product was then ligated into the thioredoxin
fusion tag, 6xHis purification tag containing vector, pET32b (Novagen) via restriction
sites NcoI and XhoI. Insertion was confirmed by sequence analysis (Genewiz, Inc.).
The newly constructed vector, pET32-Peptide, was used for PCR-based site directed

46
mutagenesis via QuikChange (Stratagene) to create the variable peptide sequences. Initial
base constructs, B3 and B3II were created using two sets of primers. Mutational sites
include Q4, T6, L17, I18, Y21, D23, Q25, L24 and Y27. Secondary base constructs
B3DQ and B3NQ were produced from the B3 parent sequence by mutating sites D10,
D11, and D16 while B3IQ and 3INQ were produced from the B3II sequence using two
sets of primers. Remaining peptide sequences were produced from single step
mutagenesis at position D11 utilizing one set of primers. The nucleotide sequences were
optimized for expression in E. coli and all constructs were verified via sequence analysis.
2.4.2. Expression and Purification of Recombinant Peptide-Fusion Variants
DNA sequences encoding desired peptide sequences were transformed into E. coli
strain BL21(DE3) competent cells (Novagen) for expression. These cells were inoculated
into 2xYT media containing 100 μg/mL ampicillin (Sigma). Cells were grown at 37°C
until an OD600 of 0.6 was reached. Isopropyl β-D-1-thiogalactopyranoside (IPTG),
(Anatrace) was then added to a final concentration of 1mM. Cells were allowed to grow
at 37°C for an additional 3 hrs and then harvested by centrifugation at 5000 rpm (Sorvall
SLC-4000 rotor) for 10 minutes.
Cells were resuspended in lysis buffer (50 mM NaH2PO4 pH 8.0, 300 mM NaCl,
2mM imidazole) and lysed using a microfluidizer system (Microfludics). The resulting
solution was then centrifuged at 15,000 rpm (Sorvall SS-34 rotor) for one hour at 4°C to
remove cellular debris. Lysates were passed over HiTrapTM
Chelating HP columns (GE
Healthcare). aPP, B3, B3DQ, B3DE, B3DK, B3DR, B3NE, B3NQ, B3NK, and B3II
bound proteins were washed and eluted using a step gradient that consisted of a wash step

47
using 50 mM NaH2PO4 pH 8.0, 300 mM NaCl, 10 mM imidazole and an elution step
using 50 mM NaH2PO4 pH 8.0, 300 mM NaCl, 250 mM imidazole. Peptide fusions B3,
B3DK, B3DR, B3NQ, B3NE, B3NK, B3II eluted proteins were subjected to ion
exchange chromatography to obtain additional purity. The previously eluted proteins
were diluted 1:5 using Buffer A (20 mM Tris pH 8.0, 2mM dithiothreitol (DTT)) and
bound to Macro-Prep HighQ cartridge (Biorad) at 5% Buffer B (Buffer B: 20 mM Tris
pH 8.0, 2 mM DTT, 1M NaCl). Peptide-fusion proteins were eluted via a linear gradient
that ranged from 50 mM to 350 mM NaCl over 240 minutes. Peptide fusions B3NR,
B3IQ, B3IK, B3IE, B3IR, B3IL, 3INQ, 3INE, 3INR and 3INK were purified by a Ni-
affinity gradient going from 0 mM imidazole to 50 mM imidazole over 300 minutes. All
peptide-fusions were analyzed by 16% SDS-PAGE gels that were stained with comassie
brilliant blue (Sigma). Peptide fusions were stored at -20°C until required for cleavage
and separation.
2.4.3. Expression and purification of Human Rhinovirus 3C Protease
The human rhinovirus 3C gene in the pGEX vector (GE Healthcare) was
transformed into E. coli strain BL21(DE3) competent cells (Novagen) for expression.
These cells were inoculated into 2xYT media containing 100 μg/mL ampicillin (Sigma).
Cells were grown at 37°C until an OD600 of 0.6 was reached. IPTG was then added to a
final concentration of 1mM. Cells were allowed to grow at 20°C for 18 hours and then
harvested by centrifugation at 5000 rpm (Sorvall SLC-4000 rotor) for 10 minutes.
Cells were resuspended in binding buffer (20 mM NaH2PO4 pH 7.4, 150 mM NaCl, 2mM
DTT) and lysed using a microfluidizer system (Microfludics). The resulting solution was

48
then centrifuged at 15,000 rpm (Sorvall SS-34 rotor) for one hour at 4°C to remove
cellular debris. Lysates were passed over GSTrapTM
FF column (GE Healthcare) and
washed with 50 mM Tris HCl, 5mM reduced glutathione, 2mM DTT to remove loosely
binding contaminants. The majority of the protease was eluted using 50 mM Tris HCl,
15mM reduced glutathione, 5mM DTT followed by a second elution step of 50 mM Tris
HCl, 35mM reduced glutathione, 5mM DTT to release remaining protease and recharge
the column.
The elution was diluted 1:5 using Buffer A (20 mM Tris pH 8.0, 2mM DTT) and
bound to Biorad Macro-Prep HighQ cartridge at 2% Buffer B (Buffer B: 20 mM Tris pH
8.0, 2mM DTT, 1M NaCl). Protease was eluted via a linear gradient from 20 mM to 100
mM NaCl over 50 minutes.
2.4.4. Peptide-Fusion Cleavage by Human Rhinovirus 3C and Separation from
Fusion Partners
Peptide-fusion and protease were incubated at 4°C for a 5-hour digestion period
in a 1:25 protease to protein ratio. Reverse phase HPLC was carried out on the cleaved
peptide-fusion samples to separate the peptide from the protein components. Cleaved
peptide-fusion samples were filtered through 0.22 μm syringe filter and loaded onto a
Waters Sunfire Prep C18 column (10x50 mm, 5μm, and 300Å). Separation of the peptide
from fusion protein and contaminants was carried out by reverse phase HPLC on a
Shimadzu HPLC system equipped with a SPD-20AV Prominence UV/Vis detector and
LC-20AT Prominence liquid chromatograph. The mobile phase was 0.1% trifluoroacetic
acid in water (Buffer A), with an elutant of 0.1% trifluoroacetic acid in acetonitrile
(Buffer B). The column was developed with a three phase gradient consisting of a steep

49
change in organic (5-30% Buffer B over 2.5 minutes) for buffer component elution, a
shallow step (30-45% Buffer B over 15 minutes) for peptide elution, followed by a steep
gradient (45-100% Buffer B over 4 minutes) to regenerate the column and elute larger
proteins such as thioredoxin and human rhinovirus 3C. Absorbance was monitored at
214nm and 280 nm.
2.4.5. Mass spectrometry
To determine peptide molecular weight and degree of purity, peptide samples were
analyzed using electrospray mass spectrometry. Lyophilized peptide samples were
resuspended in water to a peptide concentration of 50 μM. 5 μl of sample was analyzed
using an Esquire-LC electrospray ion trap mass spectrometer (Bruker Daltonics, Inc) set
up with an ESI source and positive ion polarity. This system was equipped with an
HP1100 HPLC system (Hewlett Packard). Scanning was carried out between 200 m/z and
2000 m/z, and the final spectra obtained were an average of 10 individual spectra.
Equipment used was located at the Mass Spectrometry Center of the University of
Massachusetts Amherst.
2.5. References
1. Winston, R. L. & Gottesfeld, J. M. Rapid identification of key amino-acid–DNA
contacts through combinatorial peptide synthesis. Chemistry & Biology 7, 245-
251, (2000).
2. Rodriguez, M., Li, S. S. C., Harper, J. W. & Songyang, Z. An Oriented Peptide
Array Library (OPAL) Strategy to Study Protein-Protein Interactions. Journal of
Biological Chemistry 279, 8802, (2004).
3. Sweeney, M. C. et al. Decoding protein-protein interactions through
combinatorial chemistry: sequence specificity of SHP-1, SHP-2, and SHIP SH2
domains. Biochemistry 44, 14932–14947, (2005).

50
4. Baltrusch, S., Lenzen, S., Okar, D. A., Lange, A. J. & Tiedge, M. Characterization
of Glucokinase-binding Protein Epitopes by a Phage-displayed Peptide Library
Identification of 6-Phosphofructo-2-kinase/fructose-2, 6-Bisphosphatase as a
Novel Interaction Partner. Journal of Biological Chemistry 276, 43915-43923,
(2001).
5. Newman, J. R. S. & Keating, A. E. Vol. 300 2097-2101 (American Association
for the Advancement of Science, 2003).
6. Choe, Y. et al. Substrate Profiling of Cysteine Proteases Using a Combinatorial
Peptide Library Identifies Functionally Unique Specificities. Journal of
Biological Chemistry 281, 12824, (2006).
7. Gosalia, D. N., Salisbury, C. M., Ellman, J. A. & Diamond, S. L. High
Throughput Substrate Specificity Profiling of Serine and Cysteine Proteases
Using Solution-phase Fluorogenic Peptide Microarrays*. Molecular & Cellular
Proteomics 4, 626-636, (2005).
8. Gosalia, D. N., Salisbury, C. M., Maly, D. J., Ellman, J. A. & Diamond, S. L.
Profiling serine protease substrate specificity with solution phase fluorogenic
peptide microarrays. PROTEOMICS 5, 1292-1298, (2005).
9. Harris, J. L. et al. Vol. 97 7754-7759 (National Acad Sciences, 2000).
10. Turk, B. E., Huang, L. L., Piro, E. T. & Cantley, L. C. Determination of protease
cleavage site motifs using mixture-based oriented peptide libraries. Nature
Biotechnology 19, 661-667, (2001).
11. Cuerrier, D., Moldoveanu, T. & Davies, P. L. Determination of Peptide Substrate
Specificity for {micro}-Calpain by a Peptide Library-based Approach: The
Importance of Primed Side Interactions. Journal of Biological Chemistry 280,
40632, (2005).
12. Garaud, M. & Pei, D. Substrate profiling of protein tyrosine phosphatase PTP1B
by screening a combinatorial peptide library. J. Am. Chem. Soc 129, 5366-5367,
(2007).

51
13. Huyer, G. et al. Affinity Selection from Peptide Libraries to Determine Substrate
Specificity of Protein Tyrosine Phosphatases. Analytical Biochemistry 258, 19-30,
(1998).
14. Vetter, S. W. & Zhang, Z. Y. Probing the Phosphopeptide Specificities of Protein
Tyrosine Phospha-tases, SH2 and PTB Domains with Combinatorial Library
Methods. Current Protein and Peptide Science 3, 365-397, (2002).
15. Hutti, J. E. et al. A rapid method for determining protein kinase phosphorylation
specificity. Nature Methods 1, 27-29, (2004).
16. Mah, A. S. et al. Substrate specificity analysis of protein kinase complex Dbf2-
Mob1 by peptide library and proteome array screening. BMC Biochemistry 6, 22,
(2005).
17. Yaffe, M. B. Study of Substrate Specificity of MAPKs Using Oriented Peptide
Libraries. Methods in Molecular Biology 250, 237-250, (2004).
18. Turk, B. E., Hutti, J. E. & Cantley, L. C. Determining protein kinase substrate
specificity by parallel solution-phase assay of large numbers of peptide substrates.
Nat Protoc 1, 375-379, (2006).
19. Bartsevich, V. V. & Juliano, R. L. Regulation of the MDR1 Gene by
Transcriptional Repressors Selected Using Peptide Combinatorial Libraries.
Molecular Pharmacology 58, 1-10, (2000).
20. El-Mousawi, M. et al. A Vascular Endothelial Growth Factor High Affinity
Receptor 1-specific Peptide with Antiangiogenic Activity Identified Using a
Phage Display Peptide Library*. Journal of Biological Chemistry 278, 46681-
46691, (2003).
21. Jahnke, A. et al. Leukemia targeting ligands isolated from phage display peptide
libraries. Leukemia 21, 411, (2007).
22. Obata, T. et al. Peptide and Protein Library Screening Defines Optimal Substrate
Motifs for AKT/PKB. Journal of Biological Chemistry 275, 36108-36115,
(2000).

52
23. Benoiton, N. L. Chemistry Of Peptide Synthesis. (Taylor & Francis, 2006).
24. Scriba, G. K. E. in Encyclopedia of Analytical Chemistry (2006).
25. Tam, J. P. & Lu, Y. A. Coupling Difficulty Associated with Interchain Clustering
and Phase Transition in Solid Phase Peptide Synthesis. Journal of the American
Chemical Society 117, 12058-12063, (1995).
26. Kent, S. B. H. in Proceedings of the 9th Annual American Peptide Symposium.
(ed VJ Hruby and KD Kopple CM Deber) 407-414.
27. Tam, J. P. in Proceedings of the 9th Annual American Peptide Symposium. (ed VJ
Hruby and KD Kopple CM Deber) 423-425.
28. Weaver, R. F. Molecular Biology. (McGraw-Hill College, 2007).
29. Smith, D. B. & Johnson, K. S. Single-step purification of polypeptides expressed
in Escherichia coli as fusions with glutathione S-transferase. Gene 67, 31-40,
(1988).
30. Guan, C., Li, P., Riggs, P. D. & Inouye, H. Vectors that facilitate the expression
and purification of foreign peptides in Escherichia coli by fusion to maltose-
binding protein. Gene 67, 21-30, (1988).
31. LaVallie, E. R. et al. A Thioredoxin Gene Fusion Expression System That
Circumvents Inclusion Body Formation in the E. coli Cytoplasm. Bio/Technology
11, 187-193, (1993).
32. Blundell, T. L., Pitts, J. E., Tickle, I. J., Wood, S. P. & Wu, C. W. X-Ray
Analysis (1. 4-angstrom Resolution) of Avian Pancreatic Polypeptide: Small
Globular Protein Hormone. Proceedings of the National Academy of Sciences 78,
4175-4179, (1981).
33. Tonan, K., Kawata, Y. & Hamaguchi, K. Conformations of isolated fragments of
pancreatic polypeptide. Biochemistry 29, 4424-4429, (1990).

53
34. Zondlo, N. J. & Schepartz, A. Highly Specific DNA Recognition by a Designed
Miniature Protein. Nature (London) 363, 38, (1993).
35. Chin, J. W., Grotzfeld, R. M., Fabian, M. A. & Schepartz, A. Methodology for
optimizing functional miniature proteins based on avian pancreatic polypeptide
using phage display. Bioorganic & Medicinal Chemistry Letters 11, 1501-1505,
(2001).
36. Shimba, N., Nomura, A. M., Marnett, A. B. & Craik, C. S. Herpesvirus Protease
Inhibition by Dimer Disruption. Journal of Virology 78, 6657-6665, (2004).
37. Feeney, B., Soderblom, E. J., Goshe, M. B. & Clark, A. C. Novel protein
purification system utilizing an N-terminal fusion protein and a caspase-3
cleavable linker. Protein Expression and Purification 47, 311-318, (2006).

54
CHAPTER III
INHIBITION OF CASPASE-9 BY STABILIZED PEPTIDES TARGETING
THE DIMERIZATION INTERFACE
This chapter has been in collaboration with Sumana Ghosh. Sumana perfomed all of
the solid phase peptide synthesis and structural characterization of the resultant
peptides by circular dichroism spectroscopy. The author performed all other aspects
of the work described here.
Abstract
Caspases comprise a family of dimeric cysteine proteases that control apoptotic
programmed cell death and are therefore critical in both organismal development and
disease. Specific inhibition of individual caspases has been repeatedly attempted, but has
not yet been attained. Caspase-9 is an upstream or initiator caspase that is regulated
differently from all other caspases, as interaction with natural inhibitor XIAP-BIR3
occurs at the dimer interface maintaining capsase-9 in an inactive monomeric state. One
route to caspase-9-specific inhibition is to mimic this interaction, which has been
localized to the α5 helix of XIAP-BIR3. We have developed three types of stabilized
peptides derived from the α5 helix, using incorporation of amino-isobutyric acid, the
avian pancreatic polypeptide-scaffold or aliphatic staples. The stabilized peptides are
helical in solution and achieve up to 32 μM inhibition, indicating that this allosteric site at
the caspase-9 dimerization interface is regulatable with low-molecular weight synthetic
ligands and is thus a druggable site. The most potent peptides are the avian pancreatic
polypeptide-scaffolded peptides. Given that all of the peptides attain helical structures but
cannot recapitulate the high-affinity inhibition of the intact BIR3 domain, it has become
clear that interactions of caspase-9 with the BIR3 exosite are essential for high-affinity
binding. These results explain why the full XIAP-BIR3 domain is required for maximal

55
inhibition and suggest a path forward for achieving allosteric inhibition at the
dimerization interface using peptides or small molecules.
3.1. Introduction
The apoptotic process of programmed cell death has been well studied for its
involvement in development in all multi-cellular organisms as well as for its roles in
conditions including cancer, heart attack, stroke, Alzheimer's and Huntington disease.
Irregularities in apoptosis may contribute up to 50% of all diseases in which there are no
suitable therapies1, underscoring the need to both understand and control apoptosis.
One promising approach to controlling apoptosis is through caspase regulation. The
caspases are cysteine aspartate proteases known to propagate apoptosis through a cascade
of cleavage reactions ultimately leading to the demise of the cell. Apoptotic caspases are
typically classified into two groups, the initiator caspases, caspase-8 and -9, and the
executioners, caspase-3, -6 and -7. Caspase-8 is activated in response to upstream signals,
which are transmitted to caspase-8 by the DISC complex2-4
while caspase-9 is activated
by association with the apoptosome5-7
. Once activated, caspase-8 and -9 further propagate
the apoptotic cascade by cleaving and thus activating the executioner caspases, caspase-3,
-6, and -78-11
. The executioner caspases are responsible for cleaving specific intracellular
targets, ultimately resulting in cell death. Due to its central role in initiating and
propagating apoptosis and it’s unique regulatory mechanism12
, we have focused on
controlling the apoptosis initiator, caspase-9.
Caspase-9 is synthesized as this three-domain polypeptide, which is able to form
homodimers. Structurally, caspase-9 consists of three domains. A prodomain, categorized
as a Caspase Activation and Recruitment Domain (CARD), a large subunit, which houses

56
the catalytic Cys-His dyad, and the small subunit which comprises the main portion of
the dimer interface 13
. The zymogen caspase-9 has very low activity as a monomer.
Activity increases upon dimerization13
and increases even more profoundly upon
interaction with the apoptosome5-7
. The apoptosome is a heptameric complex formed by
Apaf-1 and cytochrome c in an ATP dependent process10,14,15
. The uncleaved zymogen
form of caspase-9 is recruited to the apoptosome by binding of its CARD domain to the
Apaf-1 CARD domain. For many years the oligomeric state of caspase-9 bound to the
apoptosome has been debated. Evidence is emerging from high-resolution cryo electron
microscopy that caspase-9 monomers are activated while bound to the apoptosome16
.
Caspase-9 does not require intersubunit cleavage for activation. However, when bound to
the apoptosome, caspase-9 is processed at Asp315, resulting in a CARD-large subunit
portion of the enzyme and a small subunit beginning with the N-terminal sequence of
ATPF. Proteolytic removal of the CARD domain from the large subunit, which results in
decreased activity, has also been observed. Processed caspase-9 can be displaced from
the apoptosome by additional molecules of procaspase-917
, resulting in release of cleaved
caspase-9, which can form active dimers. As a cleaved dimer, the L2 loop from one half
of the dimer is able to interact with the L2` loop from the other half in the presence of
substrate similar to other caspases13
. In this state, the protein is catalytically competent to
process substrate. Like other caspases, caspase-9 recognizes its substrates and cleaves
after specific aspartic acid residues.
Once the caspase cascade is activated, apoptosis rapidly ensues. Treating diseases
in which apoptosis is activated requires specific apoptotic caspase inhibitors. A naturally
occurring family of caspase inhibitors, the inhibitor of apoptosis (IAP) proteins, show

57
promise as models for caspase-specific inhibition. Caspase-9 is inhibited by the x-linked
inhibitor of apoptosis protein (XIAP) in a manner that is distinct from the inhibition of
other apoptotic caspases, including caspase-3 and -7. XIAP comprises three baculovirus
inhibitory repeats (BIR). The linker between BIR1 and BIR2 binds to the active sites of
caspase-3 and -7 dimers, blocking access to substrate and thereby inhibiting these
caspases. The third domain of XIAP, BIR3, docks at the dimer interface of caspase-9
holding capase-9 in an inactive monomeric state. The most critical interactions occur
between the α5 helix in BIR3 and the 8 strand of caspase-9 (Fig 3.1 A). In addition, the
N-terminus of the caspase-9 small subunit binds to an exosite on the BIR3 domain.
Critical interactions within these two regions of the BIR3 domain have been highlighted
as essential for BIR3’s interactions with caspase-9 (Fig 3.1 B) and its inhibitory
properties12
. Many of these interactions are hydrogen bonds and salt bridges between the
α5 helix of BIR3 and caspase-9, often BIR3 side chains interacting with caspase-9
backbone atoms. In addition, some hydrophobic interactions are critical. For example,
mutations of both polar H343 and hydrophobic L344 in the α5 helix of BIR3 result in a
complete loss of its inhibitory properties12,18
. Substitution of these two residues, within
similar but non-functional IAP’s, such as cIAP1 and ML-IAP, restored caspase-9
inhibition enforcing the idea of the α5 helix as a main component in the BIR3 inhibitory
mechanism19,20
. Another region of BIR, comprised of the amino acid sequence WYPG
(residues 323 to 326), also improved the inhibitory properties when substituted in other
IAP’s, suggesting that this region is also important for the interaction. Although these
key regions are localized to the first three helices of BIR3, truncation of BIR3’s N-
terminal residues 245-261 resulted in an extremely unstable protein with low potency for

58
caspase-9 inhibition21
. Additionally, the C-terminal truncation, BIR3 residues 348-356
had no affect on protein stability. Potency for caspsae-9 was increased only in the
presence of the N-terminal region indicating that stabilization of the C-terminal region of
BIR3 is required for its inhibitory properties against caspase-921
. We hypothesize that this
unique mechanism of BIR3 inhibition of caspase-9 may enable development of caspase-9
specific inhibitors if the α5 helix can be sufficiently stabilized.
Figure 3.1 Interactions required for inhibition of caspase-9 are clustered in the 5 helix. (A) Structure
of a caspase-9 monomer (purple) bound to XIAP-BIR3 (blue) observed in 1NW9 (upper panel). A
structural zinc (red sphere) is observed in XIAP-BIR3. The exosite is marked with a circle. The boxed
region is highlighted in the lower panel. Hydrogen bonds (black dotted lines) between caspase-9
monomer and the 5 helix from XIAP-BIR3 provide recognition specificity to the complex. These
interactions are recapitulated in the native peptides. (B) Specific interactions observed in the structure
of caspase-9 bound to XIAP-BIR3 and those present in the three classes of designs are listed.

59
To date, the vast majority of work towards caspase-specific inhibition has focused
on creating small-molecule and peptide-based active-site inhibitors. Achieving
specificity at the active site has been difficult because all caspases have a stringent
requirement for binding aspartic acids in the S1 pocket. This means that both apoptotic
and inflammatory caspases are subject to inhibition by very similar inhibitors. Most
caspase inhibitors have consisted of a peptide or peptidomimetic with an acid
functionality to bind in the S1 pocket of the active site and some kind of cysteine-reactive
warhead. Utilizing the BIR3 mechanism of inhibition is promising in that it is unique to
caspase-9 and should avoid inhibition of other caspases. In this work we have developed
peptide-based inhibitors that mimic BIR3 inhibition of caspase-9.
Peptide mimics as surrogates for large protein-protein interactions have been
extensively pursued. In one early example, an entire interface of the vascular endothelial
growth factor was reduced to a 20-amino acid peptide which could then be optimized via
phage display to achieve the needed binding affinity22
. The atrial natriuretic peptide was
similarly minimized to half the original size using rational design and phage display and
still bound23
. Some proteins, like the β-adrenergic receptor, can be controlled with small,
unstructured peptides in which the principal requirement for binding is an available
amine. On the other hand, successful mimics of the vast majority of protein-protein
interactions attempted to date have required a structured peptide to mimic the native
protein interactions. Thus a number of approaches have been developed to stabilize
fragments of proteins in their native structure. For helices the prominent methods are to
initiate or stabilize helix formation or use small scaffolding proteins. Some approaches
have targeted naturally occurring capping motifs or side-chain cross-linking methods

60
such as disulfide bonds, lactam bridges and metal mediated bridges24-33
. Main chain
conformational restrictions using unnatural α-methylated amino acids, such as
aminoisobutyric acid (Aib), have also been explored. The placement of Aib in peptides
restricts the conformations of the peptide bonds, promoting proper torsion angles for α-
helical formation34
. More recently, all hydrocarbon cross-linking side chains, sometimes
called peptide staples have been introduced to maintain helical conformations of small
peptides35
. These hydrocarbon staples have been shown to improve helicity as well as cell
permeability of peptides derived from a BID BH3 helix36
, the NOTCH transcription
factor complex37
, and a p53 helix38
. In this ‘stapling’ method, unnatural α-methylated
amino acids containing olefinic side-chains of the appropriate length, are placed at the i
and i+4 or i and i+7 positions of the helix to be stabilized. These positions are selected
because one turn of an α-helix is 3.6 residues in length thus the staples would span one or
two turns of the helix. Cyclization of these amino acids is performed through an olefin
metathesis reaction, facilitated by Grubbs catalyst, thus locking the amino acids into a
staple formation reinforcing nucleation of the α-helix39
. Others have enforced main-chain
interactions through hydrogen bond surrogates which places a covalent link between the
backbone of the i and i+4 positions utilizing a similar olefin metathesis reaction40
. An
orthogonal approach utilizing stabilized scaffolds called miniature proteins has also
proven useful in helix stabilization41,42
. The critical amino acids for the protein-protein
interface are grafted onto a stable helical scaffold in the appropriate register to obtain the
proper amino acid orientation for the interaction. For example, the scaffold of avian
pancreatic polypeptide (aPP) has been used to design Bcl-2 based inhibitors43
, p53-hDM2
interaction inhibitors44
, herpes virus protease dimer disrupters45
, as well as mimicking

61
protein-DNA interactions46-48
. More recently, Peptide YY has also been used as a
miniature protein scaffold42
.
Upon analysis of the caspase-9/BIR3 complex and by understanding the successes
of stabilized helices, our approach to caspase-9 inhibition is two-fold. First, we aim to
investigate which regions of the α5 helix of BIR3 are essential to achieve BIR3-like
inhibition. Secondly, we aim to utilize the α5 helix region of BIR3 in order to achieve
caspase-9 inhibition via small α-helical peptides42
.
3.2. Results
To achieve inhibition of caspase-9 utilizing the interactions observed between the
BIR3 α5 helix and the caspase-9 8 strand (Fig 3.1A), we predicted that some type of
helix stabilization or helical scaffold would be required. We have designed and
synthesized three different classes of α5-stabilized peptides and tested them for their
ability to inhibit caspase-9. These three classes: native and Aib-stabilized, aPP-
scaffolded, and aliphatic stapled peptides take three entirely different approaches to helix
stabilization, but each of them recapitulates the most important interactions from the α5
helix (Fig 3.1 B). The stabilized peptides are markedly more helical and are better
inhibitors than the analogous native, non-stabilized peptides.
3.2.1. Native and Aib-Stabilized Peptides
Our first approach was to isolate native sequences from the BIR3 α5 helix (Fig
3.1 A, 3.2). In some sequences, aminoisobutyric acid (Aib) was inserted at various
positions to stabilize the helical conformation of the peptide. Aib is an α-branched amino
acid analogous to the naturally-occurring amino acid alanine, but derivatized at the α-
carbon with two equivalent methyl groups. The presence of two methyl groups on the α-

62
carbon locks adjacent
peptide bonds into the α-
helical confirmation which
supports the desired
dihedral angles, and thus
has been widely used to
stabilize helical
peptides34,49-51
. We
designed peptides ranging from 11 to 35 amino acids in length. The longest,
Peptide 1, composed of BIR3 residues 315-350, was designed to contain the α3-α5
helices. The α3 helix forms part of the exosite; the 4 helix orients the WYPG region and
the α5 helix contains the major elements for recognition of caspase-9. Peptides 2-4 are
21-25 amino acids long and contain all of the core interactions from α3, α4 and α5
helices. Peptide 9 is similar to Peptide 2 but is stabilized in four non-interacting positions
by Aib. Peptide 5 is 11 residues long and derived directly from the α5 helix. In Peptide 6
we added helix capping residues at the N- and C-termini. Peptides 7 and 8 are stabilized
at non-interacting position by Aib. The circular dichroism (CD) spectra suggested that the
native peptides were less helical than the Aib-stabilized peptides (Fig 3.3), predicting that
the Aib-containing peptides were closer to their target structures than the peptides
constructed solely from native amino acids derived directly from BIR3.
Caspase-9 is most active prior to catalytic removal of the 16 kD caspase
activation and recruitment domain (CARD). The CARD domain is responsible for
associating caspase-9 with the apoptosome. However, in the absence of the apoptosome,
Figure 3.2 Sequences of the α5 region of XIAP-BIR3 (BIR3) and
peptides 1-9. * represents aminoisobutyric acid (Aib)

63
the location of the CARD
domain remains
uncharacterized.
Due to the size of
the CARD and its ability to
enhance caspase-9’s basal
catalytic activity through an as yet unknown mechanism, we reasoned that the peptide
inhibitors may show a preference for interaction with either full-length caspase-9 (C9FL:
containing the CARD, large and small subunits) due to positive interactions with the
CARD or a preference for
ΔN caspase-9 (C9ΔN:
containing only the large
and small subunits) due to
negative interactions with
the CARD. Finding no
difference in the ability of
the inhibitors to influence
full-length or ΔN caspase-9
would suggest that the
peptides do not interact in
any way with CARD. We
tested both C9FL (Fig 3.4
A) and C9ΔN (Fig 3.4 B)
Figure 3.3 Circular dichroism spectra of the native Peptide 5 and
the Aib-stabilized Peptide 8. These spectra demonstrate that the
presence of Aib increases the helicity of the peptides.
Figure 3.4 Native and Aib-stabilized peptides show some
inhibition of caspase-9 activity. Peptides (1-9) exhibit some
inhibition of (A) full-length caspase-9 (C9 FL) or (B) the N-
terminal CARD-domain deleted caspase-9 (C9 N) activity to
cleave a natural caspase-9 substrate, the caspase-7 zymogen (C7
C186A) to the caspase-7 large (C7 Lg) and small (C7 Sm)
subunits in an in vitro cleavage assay monitored by gel mobility.

64
in an in vitro gel-based caspase-9 assay, which monitors cleavage of caspase-7 (substrate)
(Fig 3.4, Table 3.1) and in a fluorescent peptide cleavage assay in which we observed no
difference between the two enzymes (Table 3.1). Peptide concentrations were calculated
by absorbance at 280 nm or by using densitometry measurements on the purified peptides
analyzed by SDS-PAGE. Although it is difficult to quantify peptides in this manner, we
have confirmed that the error in the determination of peptide concentrations is no more
than 2-fold.
For peptides in this study an apparent IC50 (IC50 app) was fit from an inhibitor
titration at a fixed substrate concentration. This was compared to the amount of inhibition
observed in a gel-based assay where the peptide concentration was fixed at 66.7 μM, a
concentration in excess of the best IC50 app observed (Table 3.1). The full BIR3 domain,
with a reported Ki of 10-20 nM (18, 20, 52) is an effective inhibitor in both the
fluorescence assay, which uses a small fluorogenic peptide substrate mimic and in the gel
based assay, which uses a natural substrate. In both activity assay formats, we observed
moderate inhibition by many of the peptides. Other investigators have traditionally relied
on gel-based assays to assess caspase-9 inhibition and activation. This is likely due to the
greater reproducibly and lowered requirements for peptide consumption in the gel-based
assay, which we have also observed. The inhibition that we observe in the two assays
formats, agrees qualitatively for the peptides we have tested. All of the native and Aib-
stabilized peptides showed an IC50 app of >100 μM.
Peptide 2 routinely activated caspase-9 in samples of aged peptide (incubated for
three weeks at 4°C). Initially we were intrigued by the ability of aged Peptide 2 to
activate caspase-9. We confirmed that samples of aged Peptide 2 were of the same
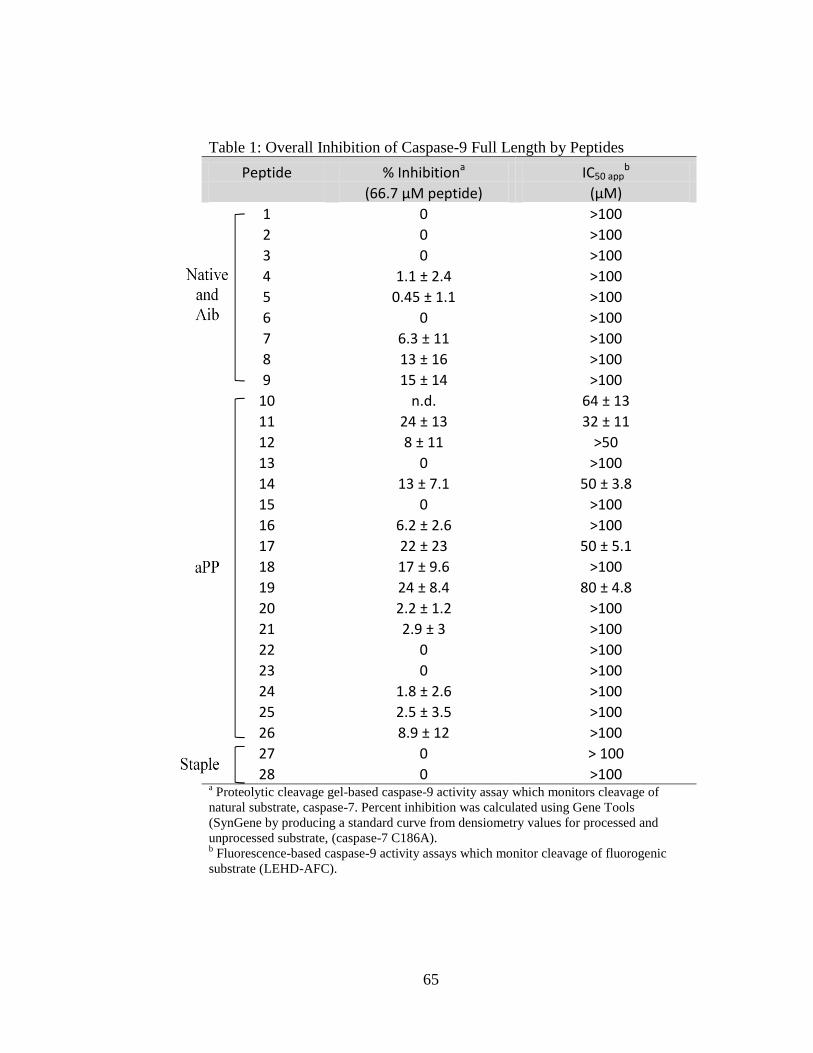
65
Table 1: Overall Inhibition of Caspase-9 Full Length by Peptides
Peptide % Inhibitiona IC50 appb
(66.7 μM peptide) (μM)
1 0 >100
2 0 >100
3 0 >100
4 1.1 ± 2.4 >100
5 0.45 ± 1.1 >100
6 0 >100
7 6.3 ± 11 >100
8 13 ± 16 >100
9 15 ± 14 >100
10 n.d. 64 ± 13
11 24 ± 13 32 ± 11
12 8 ± 11 >50
13 0 >100
14 13 ± 7.1 50 ± 3.8
15 0 >100
16 6.2 ± 2.6 >100
17 22 ± 23 50 ± 5.1
18 17 ± 9.6 >100
19 24 ± 8.4 80 ± 4.8
20 2.2 ± 1.2 >100
21 2.9 ± 3 >100
22 0 >100
23 0 >100
24 1.8 ± 2.6 >100
25 2.5 ± 3.5 >100
26 8.9 ± 12 >100
27 0 > 100
28 0 >100 a Proteolytic cleavage gel-based caspase-9 activity assay which monitors cleavage of
natural substrate, caspase-7. Percent inhibition was calculated using Gene Tools
(SynGene by producing a standard curve from densiometry values for processed and
unprocessed substrate, (caspase-7 C186A). b Fluorescence-based caspase-9 activity assays which monitor cleavage of fluorogenic
substrate (LEHD-AFC).

66
molecular weight as the
freshly diluted Peptide 2
immediately following
synthesis, confirming that
there were no chemical
modifications upon aging
(Fig 3.5 A). Freshly
diluted Peptide 2 samples
were incapable of
activation, whereas the
incubated peptides were
(Fig 3.5 B). Although
aged Peptide 2 samples were capable of activating caspase-9 four-fold over basal rates of
hydrolysis of the fluorogenic LEHD-AFC peptide, they were similarly capable of
activating other caspases, including caspase-7 (Fig 3.5 C). Caspase active sites are highly
mobile and are thus responsive to molecular crowding agents, including polymers like
polyethylene glycol (PEG)52
. The type of activation from aged Peptide 2 is similar to that
observed by PEG 8000 (Fig 3.5 C), suggesting that aggregation occurring over a three-
week aging process at 4°C was responsible for this level of activation.
3.2.2. aPP-Scaffolded Peptides
Our second approach for developing stabilized α5 helices used the avian
pancreatic polypeptide (aPP) scaffold (Fig 3.6). aPP is a 36-amino acid peptide derived
from the pancreas of turkey. Its native role is in the feedback inhibition loop halting
Figure 3.5 Peptide 2 non-specifically activates caspase-9. (A) Peptide
2 (MW 3118) is chemically stable even after a 3-week, 4˚C
incubation. (B) Aged peptide 2 activates caspase-9 activity (C) 3-week
aged peptide non-specifically activates both caspase-9 and caspase-7
in a manner similar to the polymeric crowding agent PEG 8,000

67
pancreatic secretion after a meal (reviewed in53
). In the crystal structure, aPP is composed
of a standard alpha helix flanked by a polyproline helix (Fig 3.6 A)54,55
. The interaction
between these two helices provides stability for the folded conformation.
aPP is amenable to substitution on many faces41,48
and can withstand a C-terminal
truncation55
without negatively effecting the structure or function. α5-mimicing peptides
were designed by structurally aligning the α-helical region of aPP with the α5 helix in the
BIR3-Caspase-9 complex. aPP was aligned principally by superposition of the Cα
positions and tested for maximum overlap in all possible helical registers. We ultimately
selected one register (Fig 3.2, 3.6 A) because it maximized overlap of productive
interactions between naturally-occurring aPP amino acids and caspase-9, such as the
WYPG amino acid region which was found to be critical in converting other IAP’s to
inhibit caspase-919,20,45
. In addition this orientation predicts no steric clashes between the
Figure 3.6 BIR3 interactions grafted onto stabilized miniature protein aPP. (A) Modeled interactions of
designed peptides based on an aPP scaffold (yellow) were designed to mimic the interactions between
caspase-9 (purple) and the 5 helix. The polyproline helix labeled in the upper panel, has been removed
from the lower panel for clarity. (B) Sequences of native BIR3, derivative Peptides 10-26 and native aPP,
in which critical residues from the 5 region of XIAP-BIR3 have been inserted into the aPP scaffold.
Highly varied positions are shaded.

68
aPP N-terminus, the polyproline helix, and caspase-9. Ultimately we chose to truncate
aPP to prevent steric clash between the aPP C-terminus and caspase-9. To make Peptide
10, we grafted the critical BIR3 residues 323-6, 336-7 and 340-4 onto aPP at positions
17-18 and 21-25 (Fig 3.6 B). We also grafted residues 323-326, which compose the
sequence WYPG between the α3 and α4 helices of BIR3, at positions 6-9 of aPP. aPP
positions 6-9 are in the polyproline helix and are designed to support the interactions of
the WYPG region of BIR3 with caspase-9. In Peptides 11-14, we varied the identity of
position 11 to enable novel interactions with the loop region in caspase-9 and changed all
of the aspartates to glutamates. Caspases cleave exclusively following aspartate residues,
so substitution of these residues was designed to limit their caspase substrate
characteristic. In Peptides 15-18, the aspartates were substituted by asparagines to
prevent caspase cleavage and position 11 was varied to introduce novel interactions. In
Peptides 19-26, we systematically returned positions that had previously been shown to
be important for the fold and stability of aPP to the identity in the native aPP sequence.
To produce this series of caspase-9 inhibitors, Peptides 10-26, we developed a novel
expression system that allowed robust production and purification of aPP-based
peptides56
. The mass spectra of peptides 10-26 indicated that each of these peptides are
produced accurately in bacteria and that the peptides could be purified to a very
homogenous state using our methodology56
. The conformation of these truncated
versions of aPP were predicted by Rosetta57,58
to fold into a helical structure (Fig 3.7). In
all of our aPP scaffolded designs, additional interactions are created between Y7, D10
and D11 of aPP’s polyproline helix and the caspase-9 interface. Neither truncation of aPP
nor the insertion of the α5-derived residues appears to have a substantial effect on the

69
structure of aPP-derived peptides as the CD spectra of the designed peptides show the
same conformation as the aPP parent (Fig 3.8 A). All spectra indicate that these peptides
are mainly composed of polyproline helix character with a negative peak at 198-206
nm59,60
and a predicted 5% of α-helical content61
. Although these data suggest that the
aPP peptides fold into a less helical conformation that the target helical conformation of
the scaffold, the aPP-based peptides were still tested for their ability to inhibit full-length
Figure 3.8 Properties of aPP based peptides. (A) The CD spectra of the aPP and Peptides 11 and 25
indicate that the structure of the designed peptides is very similar to that of native aPP in solution. (B)
aPP-based peptides show some inhibition of full-length caspase-9 (C9 FL) or the N-terminal CARD-
domain deleted caspase-9 (C9 N) to cleave a natural caspase-9 substrate, the caspase-7 zymogen (C7
C186A) to the caspase-7 large (C7 Lg) and small (C7 Sm) subunits in an in vitro cleavage assay
monitored by gel mobility.
Figure 3.7 aPP and peptide structures predicted computationally (black) by Rosetta. Predicted structures
are superimposed on the known structure of aPP (gray). RMSD for backbone atoms suggests the
majority of the designed aPP-based peptides should adopt a helical conformation in aqueous solution.

70
and N caspase-9 in the gel based (Fig 3.8 B) and fluorescent peptide cleavage assays
(Table 3.1). Of this class, the best inhibitors were peptides 10, 11, 14, 17 and 19 with
IC50 app of 64, 32, 50, 50 and 80 M respectively. The majority of the aPP-based peptides
had IC50 app greater than 100 M. Our four best peptides 10, 11, 14, and 17 each contain
all of the interacting residues that make up the BIR3 interface while Peptide 19
incorporates those residues important for the stability of the aPP scaffold. Exclusion of
any of the critical BIR3 residues appears to be unfavorable for the peptides inhibitory
properties based on our findings considering Peptide 19 could potentially pose as an
active site competitor with three aspartate groups at positions ten, eleven and sixteen.
Peptides 11, 14 and 17 are devoid of aspartate residues, which decreases the propensity of
the peptides to interact with the caspase-9 substrate binding groove and thus increases the
probability of interaction with the caspase-9 dimer interface. Interestingly, the only
difference between peptides 10 and 11 is the substitution of three aspartates present in
peptide 10 for three glutamates. Thus the improved potency of Peptide 11 over Peptide
10 could be related to differences in caspase-9-mediated cleavage. In addition, position
eleven on the scaffold in Peptide 11, 14, and 17 contains a glutamate, arginine or lysine,
respectively, at residue 11. These residues were designed to form an additional salt bridge
with the caspase-9 interface (not present in the native BIR3 domain) thus enhancing their
ability to inhibit caspase-9 activity. This appears to have been successful as these
peptides have the best inhibition properties. Interestingly, the addition of a glutamine at
position 11 did not show any favorable effects on the inhibitory properties of the
peptides, suggesting that glutamate, arginine, and lysine can form specific interactions
that are not possible for glutamine.

71
3.2.3. Aliphatic Stapled Peptides
The third
approach to achieve
caspase-9 allosteric
inhibition at the
dimer interface was
to incorporate
aliphatic staples to
stabilize short
helices derived
directly from the α5
helix in BIR3 (Fig
3.9A). Aliphatic
staples have been
used successfully to stabilize helices and increase cell permeability of the peptides in
which they are incorporated36,62
. To synthesize the stapled peptides, we adapted the
synthetic route reported by Seebach et.al63-65
to synthesize three unnatural Fmoc-
protected amino acids used in the stapling reactions (Fig 3.9B). In this synthetic approach
we used a mild organic base, potassium trimethyl-silanolate that could perform both the
CBz deprotection and five-membered ring opening reactions in a single step to obtain the
final α-α-disubstituted amino acids. This method is less hazardous than traditional two-
step methods35,66
, which require the use of sodium in liquid ammonia for hydrogenolysis
of the five-membered ring, followed by a reaction with TFA to deprotect the Boc group
Figure 3.9 Aliphatic stapled peptide design. (A) Modeled interactions of α5-
derived peptides stabilized by aliphatic staples (green) with caspase-9
(purple). (B) Synthetic route for production of the pentyl alanine amino acid
used to generate the aliphatic peptide staples
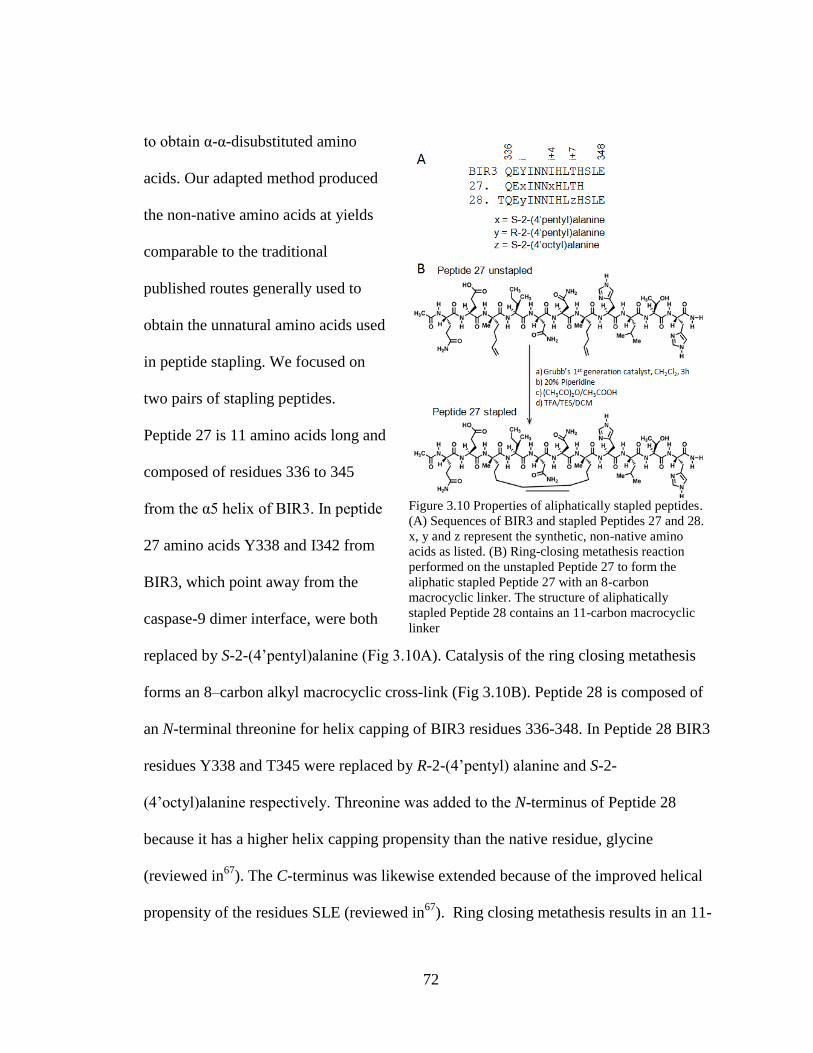
72
to obtain α-α-disubstituted amino
acids. Our adapted method produced
the non-native amino acids at yields
comparable to the traditional
published routes generally used to
obtain the unnatural amino acids used
in peptide stapling. We focused on
two pairs of stapling peptides.
Peptide 27 is 11 amino acids long and
composed of residues 336 to 345
from the α5 helix of BIR3. In peptide
27 amino acids Y338 and I342 from
BIR3, which point away from the
caspase-9 dimer interface, were both
replaced by S-2-(4’pentyl)alanine (Fig 3.10A). Catalysis of the ring closing metathesis
forms an 8–carbon alkyl macrocyclic cross-link (Fig 3.10B). Peptide 28 is composed of
an N-terminal threonine for helix capping of BIR3 residues 336-348. In Peptide 28 BIR3
residues Y338 and T345 were replaced by R-2-(4’pentyl) alanine and S-2-
(4’octyl)alanine respectively. Threonine was added to the N-terminus of Peptide 28
because it has a higher helix capping propensity than the native residue, glycine
(reviewed in67
). The C-terminus was likewise extended because of the improved helical
propensity of the residues SLE (reviewed in67
). Ring closing metathesis results in an 11-
Figure 3.10 Properties of aliphatically stapled peptides.
(A) Sequences of BIR3 and stapled Peptides 27 and 28.
x, y and z represent the synthetic, non-native amino
acids as listed. (B) Ring-closing metathesis reaction
performed on the unstapled Peptide 27 to form the
aliphatic stapled Peptide 27 with an 8-carbon
macrocyclic linker. The structure of aliphatically
stapled Peptide 28 contains an 11-carbon macrocyclic
linker

73
carbon aliphatic linker (Fig 3.10B) which has been shown to be the optimal staple for i to
i+7 stapling35
. As predicted, both peptides appeared to be substantially more helical in
aqueous solution following the ring closing metathesis reaction to generate the aliphatic
staple than prior to closure of the staple (Fig 3.11A). Thus in both cases we can conclude
that the peptides attain the designed helical structure. The stapled peptides were both
tested against caspase-9 full-length or ΔN caspase-9 (Fig 3.11B), but did not show strong
inhibition at concentrations below 100 μM in either assay format on either caspase-9
construct.
3.3. Discussion
All three classes of peptides are capable of attaining helical structures to present
the appropriate amino acids to the caspase-9 dimer interface. The length and the type of
scaffold did appear to have a tremendous influence on the potency of the peptides. The
aPP-scaffolded peptides were more successful than the native, Aib-stabilized or stapled
peptides. Our data suggest peptides composed of 15 amino acids or less in length such as
Figure 3.11 Analysis of aliphatic stapled peptides. (A) The CD spectra of Peptides 27 and 28 indicate a
significant increase in the helicity following the formation of the aliphatic staple. (B) Stapled Peptides
27 and 28 show some inhibition of full-length caspase-9 (C9 FL) or the N-terminal CARD-domain
deleted caspase-9 (C9 ΔN) to cleave a natural caspase-9 substrate, the caspase-7 zymogen (C7 C186A)
to the caspase-7 large (C7 Lg) and small (C7 Sm) subunits in an in vitro cleavage assay monitored by
gel mobility.

74
Peptides 5-8 of the native and Aib stabilized class or Peptides 27-28 of the stapled
peptides may be too short to have the requisite interaction for high-affinity binding to
caspase-9. Models of these small peptides bound to caspase-9 bury 900Å2 of surface area
in contrast to the 1700Å2 for models of the aPP-scaffolded peptides and the 2200Å
2
interface found in the native BIR3-caspase-9 complex. It is possible that other non-
critical residues found in the large interface potentially provide a hydrophobic interaction,
which may be lacking in the short peptide sequences, thus decreasing their ability to
efficiently inhibit caspase-9.
Peptides ranging in length from 11 to 39 amino acids have achieved high affinity
binding utilizing the various stabilizing methods we have employed here37,38,44-47,68-70
. An
efficient peptide inhibitor of the hdm2–p53 interaction was shown to improve potency
based on additional helical constraints imposed by Aib where incorporation of four Aib
residues into a 12 amino acid peptide was required68
. Our peptides utilized two Aib
residues for an 11 amino acid stretch, so perhaps a greater number of Aib residues could
further improve binding properties. The aPP-scaffolded Kaposi’s sarcoma-associated
herpesvirus protease (KSHV Pr) inhibiting peptide, which was 30 amino acids long, was
also designed with a five amino acid C-terminal truncation and altered ten amino acids
within the α-helical region of aPP. This peptide was successful at disruption of
dimerization, however it required 200-fold molar excess of peptide in order to obtain
50% inhibition of protease activity45
. Our best peptides were at least as effective. We
observe 50% inhibition with an average of ~30-fold molar excess peptide. Additionally, a
13 amino acid stapled peptide co-activator mimic of the estrogen receptor was able to
achieve high affinity binding. The authors noted however, that in these short peptides, the

75
hydrocarbon staple can alter the binding geometry via its ability to interact with
hydrophobic interfaces thus perturbing the desired interactions required for inhibition70
.
Thus, although our peptides were not as potent as the parent BIR3 domain, they are of
similar efficacy as other stabilized peptides. Most importantly, these peptides
demonstrate that the dimerization interface of caspase-9 can be targeted by small-
molecular weight inhibitors, indicating that this is indeed a druggable site.
Based on spectroscopic data, it is clear that all three classes of peptides we
designed and synthesized have been stabilized in a helical conformation by incorporation
of Aib, aliphatic staples or by the aPP scaffold. Despite the measured helicity, we have
not been able to recapitulate the 10-20 nM interaction of BIR3 with caspase-918,20,71
.
Consideration of critical residues, additional interface interactions, scaffold requirements,
and fold led to production of some peptide inhibitors of caspase-9 in the micromolar
range. The most potent inhibitors were all members of the aPP scaffolded class of
peptides. This class provided the secondary structural requirements needed to mimic the
α5 helix interactions, provided the additional interactions of the WYPG region of BIR3
as well as providing a larger surface area coverage, which the other peptide classes lack.
Although native and Aib-stabilized Peptides 1-9 and the stapled Peptides 27-28 contain
all of the critical contacts for interacting with caspase-9 through the α5 helix and were
helical in solution, they were still not highly potent inhibitors. This suggests that the α5
helix alone is not sufficient for full inhibition of the intact BIR3 domain even when it is
presented in a stable, helical structure. This observation further suggests that the exosite
of BIR3 (composed of residues 306-314) which binds the N-termini of the small subunit
of caspase-9 provides additional, mainly hydrophobic interactions, required for high
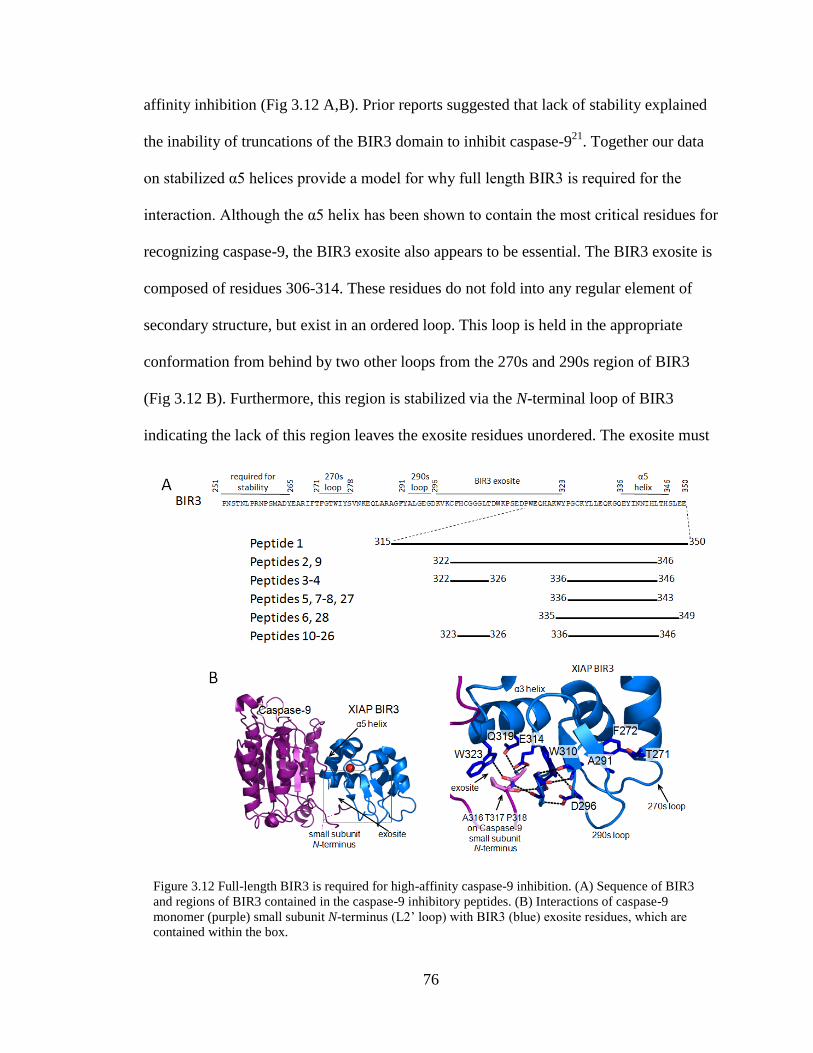
76
affinity inhibition (Fig 3.12 A,B). Prior reports suggested that lack of stability explained
the inability of truncations of the BIR3 domain to inhibit caspase-921
. Together our data
on stabilized α5 helices provide a model for why full length BIR3 is required for the
interaction. Although the α5 helix has been shown to contain the most critical residues for
recognizing caspase-9, the BIR3 exosite also appears to be essential. The BIR3 exosite is
composed of residues 306-314. These residues do not fold into any regular element of
secondary structure, but exist in an ordered loop. This loop is held in the appropriate
conformation from behind by two other loops from the 270s and 290s region of BIR3
(Fig 3.12 B). Furthermore, this region is stabilized via the N-terminal loop of BIR3
indicating the lack of this region leaves the exosite residues unordered. The exosite must
Figure 3.12 Full-length BIR3 is required for high-affinity caspase-9 inhibition. (A) Sequence of BIR3
and regions of BIR3 contained in the caspase-9 inhibitory peptides. (B) Interactions of caspase-9
monomer (purple) small subunit N-terminus (L2’ loop) with BIR3 (blue) exosite residues, which are
contained within the box.

77
interact with the very flexible N-terminus of the caspase-9 small subunit also called the
L2’ loop. Because the exosite region does not adopt any regular secondary structure it is
difficult to envision how to recapitulate the high-affinity BIR3:caspase-9 interaction with
any smaller, contiguous region of BIR3. Thus, we now understand why full-length BIR3
is required for full-potency inhibition of caspase-9 via the dimer interface. Furthermore,
these studies provide a roadmap for design of future BIR3-like small-molecule or peptide
inhibitors. High potency inhibitors that block caspase-9 inhibition by a BIR3-type
mechanism must necessarily encompass all the critical interactions in the exosite as well
those in the α5 helix.
3.4. Materials and Methods
3.4.1. Caspase-9 Expression and Purification
The caspase-9 full-length gene (human sequence) construct in pET23b (Addgene
plasmid 1182972
) was transformed into the BL21 (DE3) T7 Express strain of E. coli
(NEB). The cultures were grown in 2xYT media with ampicillin (100 mg/L, Sigma-
Aldrich) at 37°C until they reached an optical density of 1.2. The temperature was
reduced to 15°C and cells were induced with 1 mM IPTG (Anatrace) to express soluble
6xHis-tagged protein. Cells were harvested after 3 hrs to obtain single site processing.
Cell pellets stored at -20°C were freeze-thawed and lysed in a microfluidizer
(Microfluidics, Inc.) in 50 mM sodium phosphate pH 8.0, 300 mM NaCl, 2 mM
imidazole. Lysed cells were centrifuged at 17K rpm to remove cellular debris. The
filtered supernatant was loaded onto a 5 ml HiTrap Ni-affinity column (GE Healthcare).
The column was washed with 50mM sodium phosphate pH 8.0, 300 mM NaCl, 2 mM
imidazole until base lined. The protein was eluted using a 2-100mM imidazole gradient

78
over the course of 270 mL. The eluted fractions containing protein of the expected
molecular weight and composition were diluted by 10-fold into 20 mM Tris pH 8.5, 10
mM DTT buffer to reduce the salt concentration. This protein sample was loaded onto a 5
ml Macro-Prep High Q column (Bio-Rad Laboratories, Inc.). The column was developed
with a linear NaCl gradient and eluted in 20 mM Tris pH 8.5, 100 mM NaCl, and 10 mM
DTT buffer. The eluted protein was stored in -80°C in the above buffer conditions. The
identity of the purified caspase-9 was analyzed by SDS-PAGE and ESI-MS to confirm
mass and purity.
Caspase-9ΔCARD was expressed from a two plasmid expression system52,73,74
.
Two separate constructs, one encoding the large subunit, residues 140-305 and the other
encoding the small subunit, residues 331-416, both in the pRSET plasmid, were
separately transformed into the BL21 (DE3) T7 Express strain of E. coli (NEB). The
recombinant large and small subunits were individually expressed as inclusion bodies.
Cultures were grown in 2xYT media with ampicillin (100 mg/L, Sigma-Aldrich) at 37°C
until they reached an optical density of 0.6. Protein expression was induced with 0.2mM
IPTG. Cells were harvested after 3 hrs at 37°C. Cell pellets stored at -20°C were freeze-
thawed and lysed in a microfluidizer (Microfluidics, Inc.) in 10mM Tris pH 8.0 and 1mM
EDTA. Inclusion body pellets were washed twice in 100mM Tris pH 8.0, 1mM EDTA,
0.5M NaCl, 2% Triton, and 1M urea, twice in 100mM Tris pH 8.0, 1mM EDTA and
finally resuspended in 6M guanidine hydrochloride. Caspase-9 large and small subunit
proteins in guanidine hydrochloride were combined in a ratio of 1:2, large:small subunits,
and rapidly diluted dropwise into refolding buffer composed of 100mM Tris pH 8.0, 10%
sucrose,0.1% CHAPS, 0.15M NaCl, and 10mM DTT, allowed to stir for one hour at

79
room temperature and then dialyzed four times against 10mM Tris pH 8.5, 10mM DTT,
and 0.1mM EDTA buffer at 4°C. The dialyzed protein was spun for 15 minutes at
10,000rpm to remove precipitate and then purified using a HiTrap Q HP ion exchange
column (GE Healthcare) with a linear gradient from 0 to 250mM NaCl in 20 mM Tris
buffer pH 8.5, with 10 mM DTT. Protein eluted in 20 mM Tris pH 8.5, 100 mM NaCl,
and 10 mM DTT buffer was stored in -80°C. The identity of the purified caspase-
9ΔCARD was analyzed by SDS-PAGE and ESI-MS to confirm mass and purity.
3.4.2. Caspase-7 WT and Caspase-7 C186A Expression and Purification
The caspase-7 full-length human gene in the pET23b vector or the caspase-7
C186A variant (zymogen), made by Quikchange mutagenesis (Stratagene) of the human
caspase-7 gene in pET23b (gift of Guy Salvesen72
) were transformed into BL21 (DE3)
T7 Express strain of E. Coli. Induction of caspase-7 was at 18°C for 18 hrs. These
proteins were purified as described previously for caspase-775
. The eluted proteins were
stored in -80°C in the buffer in which they were eluted. The identity of purified caspase-7
was assessed by SDS-PAGE and ESI-MS to confirm mass and purity.
3.4.3. Peptide Production
Peptides 1-9 were synthetically produced by New England Peptide (Gardner,
MA). The aPP scaffolded peptides 10-26 were designed and purified using methods
previously described56
. Unnatural amino acids for Peptides 27-28 were synthesized by the
following reactions:
3.4.3.1. Synthesis of N-Fmoc-S-2-(2’-pentyl)alanine (5)
(2S,4S)-2-Phenyl-3-(carbobenzyloxy)-4-methyloxazolidin-5-one(2S,4S), (1)

80
To a stirred solution of Z-D-Alanine (0.4 g, 1.8 mmol) and benzaldehyde dimethyl acetal
(0.4 ml, 2.61 mmol) in Et2O (5 mL) was added 2 ml (15.7 mmol) of BF3• Et2O slowly by
maintaining the reaction temperature at -78 °C. The mixture was allowed to reach -20°C
and then stirring was continued at -20 °C for 4 days. At the end of this time period, the
reaction mixture was slowly added to ice-cooled, saturated aqueous NaHCO3 (10 ml),
and the mixture was stirred for additional 30 min at 0°C. After the aqueous work-up, the
separated organic layer was washed thrice with saturated NaHCO3 and H2O and then
dried over Na2SO4. The solvent was removed in vacuo. The residue was dissolved in 11
ml of Et2O/hexane (4/7) for recrystallization and afforded white crystals of (2S,4S)-1
(0.37 g, 70%). 1H NMR (CDCl3, 400 MHz): δ ppm 1.59 (d, J = 6.8 Hz, 3H), 4.49 (q, J =
6.8 Hz, 1H), 5.13-5.16 (m, 2H), 6.64 (s, 1H), 7.26-7.42 (m, 10H).
(2S,4R)-Benzyl-4-methyl-5-oxo-4-(pent-4-enyl)-2-phenyloxazolidine-3carboxylate,
(2S,4R), (3)
(2S,4S)-1 (0.22 g, 0.71 mmol) was dissolved in anhydrous THF/HMPA (4:1, 2
ml) under an argon atmosphere. The resultant solution was cooled to -78o
C. Into that 1M
LiHMDS solution (1.1 ml, 1.065 mmol) in THF was added slowly under nitrogen at -
78°C. After the addition of LiHMDS, the slightly yellow solution was stirred at this
temperature for 1 h. Next 5-iodo-1-pentene (0.21 g, 1.06 mmol, synthesized from 5-
Bromo-1-pentene) was added dropwise into the reaction mixture under an argon
atmosphere and the resultant mixture was slowly allowed to reach room temperature. The
resultant reaction mixture was stirred at room temperature overnight. At end, saturated
NH4Cl solution was added, and the reaction mixture was extracted in ether. The organic
layer was washed subsequently with saturated NaHCO3 and saturated aqueous NaCl
solutions, respectively. The organic phase was dried over Na2SO4 and evaporated. The

81
product was purified by flash column chromatography and elution with 30% DCM in
hexane gives the gummy product (0.08 gm) with 30 % isolated yield. 1H NMR (CDCl3,
400 MHz): δ ppm 1.26-1.33 (m, 2H), 1.69 (s, 1H), 1.79 (s, 3H), 2.06-2.08 (m, 2H), 2.14-
2.196 and 2.49-2.56 (m, 1H), 4.91-5.035 (m, 4H), 5.56-5.77 (m, 1H), 6.4 (d, 1H), 6.88 (d,
J = 7.0 Hz, 1H), 7.20-7.416 (m, 10 H).
S-2-Amino-2-methylhept-6-enoic-acid, (4)
A solution of 3 (1.1 gm, 2.9 mmol) in tetrahydrofuran (52 ml) was treated with
potassium trimethylsilanolate (90% pure; 1.1 gm, 8.6 mmol) and the mixture was heated
at 75°C for 2 h 30 min by which time all starting material was consumed. The mixture
was diluted with methanol and the solvents were removed under reduced pressure. The
resultant mixture was diluted with dichloromethane and evaporated under vacuum. The
crude solid was used directly for the next step without further purification.
Fmoc-S-2-(2’-pentyl)alanine, (5)
To a solution of 4 (0.1 gm, 0.64 mmol) in water (8 ml), DIEA (0.167 ml, 0.96
mmol) was added followed by the addition of Fmoc-OSu (0.237gm, 0.7 mmol) in
acetonitrile (8 ml). The resultant mixture was stirred at room temperature for 5-6 h. The
solvent was evaporated and the resultant solution was dissolved in water and
subsequently acidified with 2N HCl solution. The aqueous layer was extracted with
ethylacetate for 3 times and purified by silica column chromatography using
hexane/DCM and later MeOH/DCM as eluent. The product elutes with 3% MeOH/DCM
mixture. This gives rise to 0.12 gm of the product with 50% isolated yield. 1H NMR
(CDCl3, 400 MHz): δ ppm 1.24-1.43 (m, 2H), 1.63 (s, 3H), 1.87-1.89 (m, 1H), 2.06-2.16
(m, 3H), 4.22 (t, J = 6.48 Hz, 1H), 4.41(bs, 2H), 4.96-5.03 (m, 2H), 5.52 (bs, 1H), 5.76

82
(m, 1H), 7.315 (t, J1= J2= 7.4 Hz, 2H), 7.40 (t, J = 7.4 Hz, 2H), 7.6 (d, J = 7.4 Hz, 2H),
7.76 (d, J = 7.48 Hz, 2H), 10.66 (bs, 1H).
5-iodo-1-pentene, (2)
5-Bromo-1-pentene (0.8 ml, 6.71 mmol) was added to a solution of sodium iodide
(2.0 gm, 13.3 mmol) in acetone (22 ml). The reaction mixture was heated at 60 oC for 2
h. The mixture was cooled to room temperature and diluted with water (100 ml) and
extracted with pentane 3-times. The pentane layers were combined, washed with brine,
dried over sodium sulfate and concentrated to give 5-iodo-1-pentene. The product was
obtained as 90% isolated yield (1.2 gm). 1H NMR (CDCl3, 400 MHz): δ ppm 1.91 (p, J1
= 7.12 Hz, J2 = 7.0 Hz, 2H), 2.16 (q, J1 = 6.7 Hz, J2 = 6.82 Hz, 2H), 3.19 (t, J1 = 6.88 Hz,
J2 = 6.92 Hz, 2H), 5.00-5.10 (dd, J1 = 17.12 Hz, J2 = 10.12 Hz, 2H), 5.69-5.80 (m, 1H).
Similarly Fmoc-S-2-(2’-octyl alanine)alanine, was obtained starting from Z-D-alanine
using 8-iodo-1octene. Fmoc-R-2-(2’-pentyl)alanine was obtained starting from Z-L-
alanine using similar reaction conditions as described above.
3.4.3.2. Synthesis of N-Fmoc-S-2-(2’-octyl)alanine
(2S,4R)-Benzyl-4-methyl-5-oxo-4-(oct-4-enyl)-2-phenyloxazolidine-3carboxylate,
(2S,4R)
1H NMR (CDCl3, 400 MHz): δ ppm 1.05-1.31 (m, 8H), 1.69 (s, 1H), 1.8 (s, 3H), 2.03-2.2
(m, 3H), 4.94-5.35 (m, 4H), 5.78-5.82 (m, 1H), 6.41 (d, 1H), 6.89 (m, 1H), 7.21-7.42 (m,
10H).
N-Fmoc-S-2-(2’-octyl)alanine
1H NMR (CDCl3, 400 MHz): δ ppm 1.09-1.37 (m, 8H), 1.66 (s, 3H), 1.88 (bs, 1H), 2.03-
2.18 (m, 3H), 4.25 (s, 1H), 4.43-4.72 (m, 2H), 4.96-5.05 (m, 2H), 5.71 (bs, 1H), 5.78-

83
5.88 (m, 1H), 7.34 (t, J1 = 7.24, J2 = 7.32 Hz, Hz, 2H), 7.42 (t, J1 = 7.24, J2 = 7.0, 2H),
7.63 (bs, 2H), 7.79 (d, J = 7.44 Hz, 2H), 11.6 (bs, 1H).
8-iodo-1-octene
1H NMR (CDCl3, 400 MHz): δ ppm 1.25-1.43 (m, 6H), 1.82 (p, J1 = 7.12 Hz, J2 = 7.4
Hz, 2H), 2.04 (q, J1 = 6.76 Hz, J2 = 6.84 Hz, 2H), 3.18 (t, J1 = J2 = 7.04 Hz, 2H), 4.92-
5.02 (m, 2H), 5.74-5.81 (m, 1H).
Peptides 27-28 were synthesized on solid phase by sequentially adding
appropriate amino acids along with the peptide coupling reagents (HATU, DIEA) onto
the Fmoc-Rink Amide resin76,77
. Each amino acid coupling was done for 1.5 h to 2 h.
Both peptides were obtained in 60-80% isolated yields after purification by RP-HPLC
and characterized by ESI-MS. All solvents and reagents used were of highest purity
available. Fmoc-Asp(tBu)-OH, 1 was purchased from Novabiochem and N,O-dimethyl
hydroxylamine hydrochloride was bought from Acros. 1H-NMR spectra were recorded
on Bruker 400 spectrometer. Chemical shifts () are reported in ppm downfield from the
internal standard (TMS).
Peptide concentrations were tested by absorbance at 280 nm or densitometry
measurements for the gel-based activity measurements. A consistent error of the peptide
concentration was determined to be no more than two-fold more concentrated than the
expected amount. The inhibitory constant, Ki, was adjusted accordingly. However, the
ranking of our best peptides were not changed, therefore our overall conclusions remain
the same.

84
3.4.4. Activity Assays
For measurements of caspase activity, 700 nM freshly purified protein was
assayed over the course of 10 minutes in a caspase-9 activity assay buffer containing 100
mM MES pH 6.5, 10% PEG 8,000 and 10 mM DTT78
. 300 μM fluorogenic substrate,
LEHD-AFC, (N-acetyl-Leu-Glu-His-Asp-AFC (7-amino-4-fluorocoumarin), Enzo
Lifesciences) Ex395/Em505, was added to initiate the reaction. Assays were performed
in duplicate at 37°C in 100 μL volumes in 96-well microplate format using a Molecular
Devices Spectramax M5 spectrophotometer. Initial velocities versus inhibitor
concentration were fit to a rectangular hyperbola using GraphPad Prism (Graphpad
Software) to calculate Ki app.
Proteolytic cleavage gel-based caspase-9 activity assays were performed for
testing the ability of each peptide to inhibit caspase-9 against a natural substrate.
Cleavage of the full-length procaspase-7 variant C186A, which is catalytically inactive
and incapable of self-cleavage, was used to report activity. 1 μM full-length procaspase-
7 C186A was incubated with 1 μM active caspase-9 in a minimal assay buffer containing
100 mM MES pH 6.5 and 10 mM DTT in a 37°C water bath for 1 hr. Samples were
analyzed by 16% SDS-PAGE to confirm the exact lengths of the cleavage products.
Percent inhibition was calculated using Gene Tools (SynGene) by producing a standard
curve from densiometry values for process and unprocessed substrate (caspase-7 C186A).
3.4.5. Mass Spectrometry
Peptide 2 in water was diluted in α-cyano-4-hydroxycinnamic acid matrix to a
final concentration of 5 µM. 1µl was spotted onto a target for analysis by Omniflex
MALDI-TOF mass spectrometer (Bruker Daltonics, Inc, Billerica, MA) using linear

85
detection mode. Peptides 27 and 28 in water were diluted in 0.1% formic acid to a final
concentration of 5 µM for direct injection onto Esquire-LC electrospray ion trap mass
spectrometer (Bruker Daltonics, Inc.) set up with an ESI source and positive ion polarity.
The MS instrument system was equipped with an HP1100 HPLC system (Hewlett-
Packard). Scanning was carried out between 600-1400 m/z and the final spectra obtained
were an average of 10 individual spectra. All mass spectral data were obtained at the
University of Massachusetts Mass Spectrometry Facility, which is supported, in part, by
the National Science Foundation.
3.4.6. Secondary Structure Analysis by Circular Dichroism
The secondary structure of the peptide variants was monitored via CD spectra
(250-190 nm) measured on a J-720 circular dichroism spectrometer (Jasco) with a peltier
controller. Peptides 2, 3, 5, 8, 27, and 28 in water and Peptides 11, 25 and aPP in 20mM
Tris pH 8.0 were diluted to 15 μM and analyzed at room temperature. Peptides 27 and 28
were monitored for secondary structure formation before and after the metathesis
reaction.
3.4.7. Computational Structure Prediction
The structures of aPP variants were predicted computationally using Rosetta57,58
.
Folding simulations were performed on the designed peptide sequences 10-26 using the
scaffold aPP as a control. The structure of aPP was reasonably well simulated as assessed
by backbone alignment and rotomer confirmation of the top 15 lowest scored predicted
conformations. Structural visualization and model interrogation was performed in The
PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC79
.

86
3.5. References
1. Reed, J. C. & Tomaselli, K. J. Drug discovery opportunities from apoptosis
research. Curr Opin Biotechnol 11, 586-592, (2000).
2. Scaffidi, C., Medema, J. P., Krammer, P. H. & Peter, M. E. FLICE is
predominantly expressed as two functionally active isoforms, caspase-8/a and
caspase-8/b. J Biol Chem 272, 26953-26958, (1997).
3. Medema, J. P. et al. FLICE is activated by association with the CD95 death-
inducing signaling complex (DISC). EMBO J 16, 2794-2804, (1997).
4. Lavrik, I. et al. The active caspase-8 heterotetramer is formed at the CD95 DISC.
Cell Death Differ 10, 144-145, (2003).
5. Zou, H., Li, Y., Liu, X. & Wang, X. An APAF-1· cytochrome c multimeric
complex is a functional apoptosome that activates procaspase-9. Journal of
Biological Chemistry 274, 11549, (1999).
6. Pop, C., Timmer, J., Sperandio, S. & Salvesen, G. S. The apoptosome activates
caspase-9 by dimerization. Mol Cell 22, 269-275, (2006).
7. Rodriguez, J. & Lazebnik, Y. Caspase-9 and APAF-1 form an active holoenzyme.
Genes Dev 13, 3179-3184, (1999).
8. Stennicke, H. R. et al. Pro-caspase-3 is a major physiologic target of caspase-8. J
Biol Chem 273, 27084-27090, (1998).
9. Muzio, M. et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is
recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 85,
817-827, (1996).
10. Li, P. et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9
complex initiates an apoptotic protease cascade. Cell 91, 479-489, (1997).
11. Slee, E. A. et al. Ordering the cytochrome c–initiated caspase cascade:
hierarchical activation of caspases-2,-3,-6,-7,-8, and-10 in a caspase-9–dependent
manner. The Journal of cell biology 144, 281, (1999).

87
12. Shiozaki, E. N. et al. Mechanism of XIAP-mediated inhibition of caspase-9.
Molecular cell 11, 519-527, (2003).
13. Renatus, M., Stennicke, H. R., Scott, F. L., Liddington, R. C. & Salvesen, G. S.
Dimer formation drives the activation of the cell death protease caspase 9.
Proceedings of the National Academy of Sciences 98, 14250, (2001).
14. Liu, X., Kim, C. N., Yang, J., Jemmerson, R. & Wang, X. Induction of apoptotic
program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86,
147-157, (1996).
15. Cain, K., Brown, D. G., Langlais, C. & Cohen, G. M. Caspase activation involves
the formation of the aposome, a large ( 700 kDa) caspase-activating complex.
Journal of Biological Chemistry 274, 22686, (1999).
16. Yuan, S. et al. The Holo-Apoptosome: Activation of Procaspase-9 and
Interactions with Caspase-3. Structure 19, 1084-1096, (2011).
17. Malladi, S., Challa-Malladi, M., Fearnhead, H. O. & Bratton, S. B. The Apaf-
1*procaspase-9 apoptosome complex functions as a proteolytic-based molecular
timer. EMBO J 28, 1916-1925, (2009).
18. Sun, C. et al. NMR structure and mutagenesis of the third Bir domain of the
inhibitor of apoptosis protein XIAP. Journal of Biological Chemistry 275, 33777,
(2000).
19. Eckelman, B. P. & Salvesen, G. S. The human anti-apoptotic proteins cIAP1 and
cIAP2 bind but do not inhibit caspases. J Biol Chem 281, 3254-3260, (2006).
20. Vucic, D. et al. Engineering ML-IAP to produce an extraordinarily potent caspase
9 inhibitor: implications for Smac-dependent anti-apoptotic activity of ML-IAP.
Biochemical Journal 385, 11, (2005).
21. Shin, H. et al. The BIR domain of IAP-like protein 2 is conformationally
unstable: implications for caspase inhibition. Biochemical Journal 385, 1, (2005).
22. Fairbrother, W. J. et al. Novel peptides selected to bind vascular endothelial
growth factor target the receptor-binding site. Biochemistry 37, 17754-17764,
(1998).

88
23. Li, B. et al. Minimization of a polypeptide hormone. Science 270, 1657-1660,
(1995).
24. Felix, A. M. et al. Synthesis, biological activity and conformational analysis of
cyclic GRF analogs. Int J Pept Protein Res 32, 441-454, (1988).
25. Osapay, G. & Taylor, J. W. Multicyclic polypeptide model compounds. 2.
Synthesis and conformational properties of a highly. alpha.-helical uncosapeptide
constrained by three side-chain to side-chain lactam bridges. Journal of the
American Chemical Society 114, 6966-6973, (1992).
26. Phelan, J. C., Skelton, N. J., Braisted, A. C. & McDowell, R. S. A general method
for constraining short peptides to an -helical conformation. Journal of the
American Chemical Society 119, 455-460, (1997).
27. Taylor, J. W. The synthesis and study of side chain lactam bridged peptides.
Peptide Science 66, 49-75, (2002).
28. Jackson, D. Y., King, D. S., Chmielewski, J., Singh, S. & Schultz, P. G. General
approach to the synthesis of short. alpha.-helical peptides. Journal of the
American Chemical Society 113, 9391-9392, (1991).
29. Leduc, A. M. et al. Helix-stabilized cyclic peptides as selective inhibitors of
steroid receptor–coactivator interactions. Proceedings of the National Academy of
Sciences 100, 11273, (2003).
30. Ghadiri, M. R. & Choi, C. Secondary structure nucleation in peptides. Transition
metal ion stabilized. alpha.-helices. Journal of the American Chemical Society
112, 1630-1632, (1990).
31. Kelso, M. J. et al. Alpha-Turn Mimetics: Short Peptide-Helices Composed of
Cyclic Metallopentapeptide Modules. Journal of the American Chemical Society
126, (2004).
32. Ruan, F., Chen, Y. & Hopkins, P. B. Metal ion-enhanced helicity in synthetic
peptides containing unnatural, metal-ligating residues. Journal of the American
Chemical Society 112, 9403-9404, (1990).

89
33. Brunel, F. M. & Dawson, P. E. Synthesis of constrained helical peptides by
thioether ligation: application to analogs of gp41. Chem. Commun., 2552-2554,
(2005).
34. Karle, I. L. & Balaram, P. Structural characteristics of. alpha.-helical peptide
molecules containing Aib residues. Biochemistry 29, 6747-6756, (1990).
35. Schafmeister, C. E., Po, J. & Verdine, G. L. An all-hydrocarbon cross-linking
system for enhancing the helicity and metabolic stability of peptides. Journal of
the American Chemical Society 122, 5891-5892, (2000).
36. Walensky, L. D. et al. A stapled BID BH3 helix directly binds and activates
BAX. Molecular cell 24, 199-210, (2006).
37. Moellering, R. E. et al. Direct inhibition of the NOTCH transcription factor
complex. Nature 462, 182-188, (2009).
38. Bernal, F. et al. A stapled p53 helix overcomes HDMX-mediated suppression of
p53. Cancer Cell 18, 411-422, (2010).
39. Blackwell, H. E. & Grubbs, R. H. Highly efficient synthesis of covalently cross
linked peptide helices by ring closing metathesis. Angewandte Chemie
International Edition 37, 3281-3284, (1998).
40. Patgiri, A., Jochim, A. L. & Arora, P. S. A hydrogen bond surrogate approach for
stabilization of short peptide sequences in -helical conformation. Accounts of
chemical research 41, 1289-1300, (2008).
41. Chin, J. W., Grotzfeld, R. M., Fabian, M. A. & Schepartz, A. Methodology for
optimizing functional miniature proteins based on avian pancreatic polypeptide
using phage display. Bioorganic & medicinal chemistry letters 11, 1501-1505,
(2001).
42. Hodges, A. M. & Schepartz, A. Engineering a monomeric miniature protein.
Journal of the American Chemical Society 129, 11024-11025, (2007).
43. Chin, J. W. & Schepartz, A. Design and evolution of a miniature Bcl 2 binding
protein. Angewandte Chemie 113, 3922-3925, (2001).

90
44. Kritzer, J. A. et al. Miniature protein inhibitors of the p53-hDM2 interaction.
ChemBioChem 7, 29-31, (2006).
45. Shimba, N., Nomura, A. M., Marnett, A. B. & Craik, C. S. Herpesvirus protease
inhibition by dimer disruption. Journal of virology 78, 6657, (2004).
46. Chin, J. W. & Schepartz, A. Concerted evolution of structure and function in a
miniature protein. Journal of the American Chemical Society 123, 2929-2930,
(2001).
47. Montclare, J. K. & Schepartz, A. Miniature homeodomains: high specificity
without an N-terminal arm. Journal of the American Chemical Society 125, 3416-
3417, (2003).
48. Zondlo, N. J. & Schepartz, A. Highly specific DNA recognition by a designed
miniature protein. Journal of the American Chemical Society 121, 6938-6939,
(1999).
49. Marshall, G. R. et al. Factors governing helical preference of peptides containing
multiple alpha, alpha-dialkyl amino acids. Proceedings of the National Academy
of Sciences 87, 487, (1990).
50. Prasad, B. & Balaram, P. The stereochemistry of peptides containing alpha-
aminoisobutyric acid. CRC critical reviews in biochemistry 16, 307, (1984).
51. Toniolo, C. et al. Preferred conformations of peptides containing , disubstituted
amino acids. Biopolymers 22, 205-215, (1983).
52. Garcia-Calvo, M. et al. Purification and catalytic properties of human caspase
family members. Cell Death Differ 6, 362-369, (1999).
53. Lonovics, J., Devitt, P., Watson, L. C., Rayford, P. L. & Thompson, J. C.
Pancreatic polypeptide: A review. Archives of Surgery 116, 1256, (1981).
54. Blundell, T., Pitts, J., Tickle, I., Wood, S. & Wu, C. W. X-ray analysis (1. 4-Å
resolution) of avian pancreatic polypeptide: Small globular protein hormone.
Proceedings of the National Academy of Sciences 78, 4175, (1981).

91
55. Tonan, K., Kawata, Y. & Hamaguchi, K. Conformations of isolated fragments of
pancreatic polypeptide. Biochemistry 29, 4424-4429, (1990).
56. Huber, K. L., Olson, K. D. & Hardy, J. A. Robust production of a peptide library
using methodological synchronization. Protein expression and purification 67,
139-147, (2009).
57. Simons, K. T., Strauss, C. & Baker, D. Prospects for ab initio protein structural
genomics1. Journal of molecular biology 306, 1191-1199, (2001).
58. Rohl, C. A., Strauss, C. E. M., Misura, K. & Baker, D. Protein structure
prediction using Rosetta. Methods in enzymology 383, 66-93, (2004).
59. Naganagowda, G. A., Gururaja, T. L. & Levine, M. J. Delineation of
conformational preferences in human salivary statherin by 1H, 31P NMR and CD
studies: sequential assignment and structure-function correlations. J Biomol Struct
Dyn 16, 91-107, (1998).
60. Arnott, S. & Dover, S. D. The structure of poly-L-proline II. Acta Crystallogr B
24, 599-601, (1968).
61. Andrade, M. A., Chacon, P., Merelo, J. J. & Moran, F. Evaluation of secondary
structure of proteins from UV circular dichroism spectra using an unsupervised
learning neural network. Protein Eng 6, 383-390, (1993).
62. Walensky, L. D. et al. Activation of apoptosis in vivo by a hydrocarbon-stapled
BH3 helix. Science 305, 1466, (2004).
63. Coe, D. M., Perciaccante, R. & Procopiou, P. A. Potassium trimethylsilanolate
induced cleavage of 1, 3-oxazolidin-2-and 5-ones, and application to the synthesis
of (< i> R</i>)-salmeterol. Org. Biomol. Chem. 1, 1106-1111, (2003).
64. Procopiou, P. A., Ahmed, M., Jeulin, S. & Perciaccante, R. Synthesis of (R)- -
benzylmethionine: a novel rearrangement during alkylation of the Seebach (R)-
methionine oxazolidinone. Organic & biomolecular chemistry 1, 2853-2858,
(2003).

92
65. Seebach, D. & Fadel, A. N, O Acetals from Pivalaldehyde and Amino Acids for
the Alkylation with Self Reproduction of the Center of Chirality. Enolates of 3
Benzoyl 2 (tert butyl) 1, 3 oxazolidin 5 ones. Helvetica chimica acta 68, 1243-
1250, (1985).
66. Williams, R. M. & Im, M. N. Asymmetric synthesis of monosubstituted and.
alpha.,. alpha.-disubstituted. alpha.-amino acids via diastereoselective glycine
enolate alkylations. Journal of the American Chemical Society 113, 9276-9286,
(1991).
67. Aurora, R. & Rose, G. Helix capping. Protein science: a publication of the
Protein Society 7, 21, (1998).
68. Banerjee, R., Basu, G., Roy, S. & Chène, P. Aib based peptide backbone as
scaffolds for helical peptide mimics. The Journal of peptide research 60, 88-94,
(2002).
69. Bird, G. H. et al. Hydrocarbon double-stapling remedies the proteolytic instability
of a lengthy peptide therapeutic. Proceedings of the National Academy of
Sciences 107, 14093, (2010).
70. Phillips, C. et al. Design and structure of stapled peptides binding to estrogen
receptors. Journal of the American Chemical Society, (2011).
71. Liu, Z. et al. Structural basis for binding of Smac/DIABLO to the XIAP BIR3
domain. Nature 408, 1004-1008, (2000).
72. Stennicke, H. R. & Salvesen, G. S. Caspases: preparation and characterization.
Methods 17, 313-319, (1999).
73. Rotonda, J. et al. The three-dimensional structure of apopain/CPP32, a key
mediator of apoptosis. Nat Struct Biol 3, 619-625, (1996).
74. Garcia-Calvo, M. et al. Inhibition of human caspases by peptide-based and
macromolecular inhibitors. J Biol Chem 273, 32608-32613, (1998).
75. Witkowski, W. A. & Hardy, J. A. L2' loop is critical for caspase-7 active site
formation. Protein Sci 18, 1459-1468, (2009).

93
76. Barany, G., Kneib Cordonier, N. & Mullen, D. G. Solid phase peptide synthesis: a
silver anniversary report*. International Journal of Peptide and Protein Research
30, 705-739, (1987).
77. Fields, G. B. & Noble, R. L. Solid phase peptide synthesis utilizing 9
fluorenylmethoxycarbonyl amino acids. International Journal of Peptide and
Protein Research 35, 161-214, (1990).
78. Stennicke, H. R. & Salvesen, G. S. Biochemical characteristics of caspases-3,-6,-
7, and-8. Journal of Biological Chemistry 272, 25719, (1997).
79. Schrödinger, L. The PyMOL Molecular Graphics System, Version 1.3, (2010).

94
CHAPTER IV
MECHANISM OF ZINC-MEDIATED INHIBITION OF CASPASE-9
Abstract
Zinc-mediated inhibition is implicated in global caspase regulation, with relief of
zinc-mediated inhibition central to both small-molecule and natively induced caspase
activation. As an initiator, caspase-9 regulates the upstream stages of the apoptotic
caspase cascade, making it a critical control point. Here we identify two distinct zinc-
binding sites on caspase-9. The first site, composed of H237, C239, C287 includes the
active site dyad and is primarily responsible for zinc-mediated inhibition. The second
binding site at C272 plays is distal from the active site. Given the amino-acid
conservation in both regions, these sites appear to be conserved across the caspase family.
4.1. Introduction
Apoptotic cell death is carried out by caspases, a family of cysteine proteases.
Caspases function in a cascade of cleavage events leading to orderly destruction of cells,
culminating in cell death. Initiator caspase-8 and -9 activate the executioner caspases-3,
-6 and -7, which cleave select targets enabling cell death. Apoptosis is important for both
organismal development and homeostasis and is implicated in diseases ranging from
ischemic injury to cancer. Understanding the mechanisms of the native regulatory
pathways of caspases provides new insights for controlling and harnessing apoptosis.
Exposure of the apoptotic machinery to metal ions has been reported to influence
apoptosis. Zinc, a metal commonly used for both biological structure and function, has
been highlighted as a specific regulator of apoptosis, where even the smallest fluctuations

95
in cellular zinc concentrations can tip a cell towards survival or apoptotic cell death1.
Zinc-based regulation of apoptosis had previously been thought to act upstream of the
caspases2. However, in vitro and in vivo studies implicate caspases as the targets for zinc-
mediated inhibition of apoptosis3,4
.
The basis of the necessity for zinc regulation of caspases is not well understood,
but may relate to protection of the essential active-site sulfhydryls during oxidative stress
(reviewed in5). Misregulation of zinc and changes in caspase activity have been linked to
a number of diseases. Patients with asthma and chronic bronchitis show a correlation
between zinc deficiency and increased levels of apoptosis in airway epithelial cells6,7
,
suggesting a protective role for zinc-mediated caspase inhibition. Heliobacter Pylori
based infections have also been linked to zinc-mediated regulation of the caspases8,9
,
where release of zinc from the bacterium inhibits caspase activity to avoid cell death.
PAC-1, a serendipitous small-molecule caspase activator, relieves zinc-mediated caspase-
3 inhibition10
further suggesting that regulation of zinc-inhibition of caspases may be
therapeutically exploited. In this work, we aim to understand the molecular mechanism of
zinc-mediated caspase inhibition.
4.2. Results
4.2.1. Metal Affects on the Properties of Caspase-9
Caspase inhibition by zinc has been explored in the executioner caspases -3, -6,
and -7, however, little is known about zinc’s function on initiator caspases, including
caspase-9. Inhibition of the initiator caspases is particularly critical as the initiators

96
control the upstream steps of the proteolytic cascade. Previous studies have suggested
that zinc disrupts caspase-9 activation and thus activity11,12
; however a full analysis of
zinc’s affect on caspase-9 or the mechanism of inhibition has not been investigated. We
interrogated the effects of a panel of metal cations on the ability of caspase-9 to cleave its
natural substrate, caspase-7. Both full-length caspase-9 (Fig 4.1 A) and caspase-9 lacking
its pro domain (Caspase Activation and Recruitment Domain, CARD) (Fig 4.1 B) were
inhibited by zinc but not by any other cations, including cobalt (Fig 4.1 C). Full inhibition
of both caspase-9 variants by zinc indicates that the CARD domain is not involved in
zinc’s mechanism of inhibition. Other caspases including caspase-3, -6, -7 and -8 have
Figure 4.1 Zinc exclusively inhibits caspase-9 activity. (A, B) Zinc is the predominant metal cation to
inhibit full-length caspase-9 (C9 FL) (A) or the N-terminal CARD-domain deleted caspase-9 (C9 N)
(B) when monitored by cleavage of a natural caspase-9 substrate, the caspase-7 zymogen (C7 C186A)
to the caspase-7 large (C7 Lg) and small (C7 Sm) subunits in an in vitro cleavage assay monitored by
gel mobility. (C) Caspase-9 full-length activity was not inhibited by cobalt or cadmium as judged by
cleavage of fluorogenic peptide substrate LEHD-AFC. (D) Competition with metal chelator, EDTA,
indicates zinc inhibition is reversible, as monitored by the cleavage of fluorogenic peptide substrate
LEHD-AFC.

97
previously been shown to be inhibited in the presence of zinc3; however other metal
cations, including copper also inhibit caspase-3 function4. In contrast, caspase-9 appears
to be inhibited specifically by zinc but not by other metals. Zinc-mediated inhibition was
fully reversible by the metal chelator EDTA (Fig 4.1 D) suggesting a regulatory role for
zinc rather than a non-specific mechanism such as promotion of an irreversible protein
aggregate. Inhibition by zinc lead to Michaelis-Menten like curves that fit best to a mixed
model of inhibition (Fig 4.2). The observed error within the fit of the individual curves
was not due to quenching of the fluorophore, AFC, by zinc (data not shown). This
suggests zinc inhibition functions allosterically or binds near the active site thus
influencing the binding of substrate. The inhibitory constant, Ki, of 1.5 ± 0.3 μM is
similar that reported for zinc inhibition of caspase-3, -6, -7 and -8 (IC50 = 8.8, 0.3, 1.7,
Figure 4.2 Kinetics of full-length caspase-9 wild type in the presence of 0-50 μM ZnCl2. Measured data
were fit to competitive, noncompetitive, and mixed models of inhibition. R2 values are presented for
individual zinc concentrations and for the overall global fit of the curves fit by each model in which
mixed model of inhibition represented the best fit model.
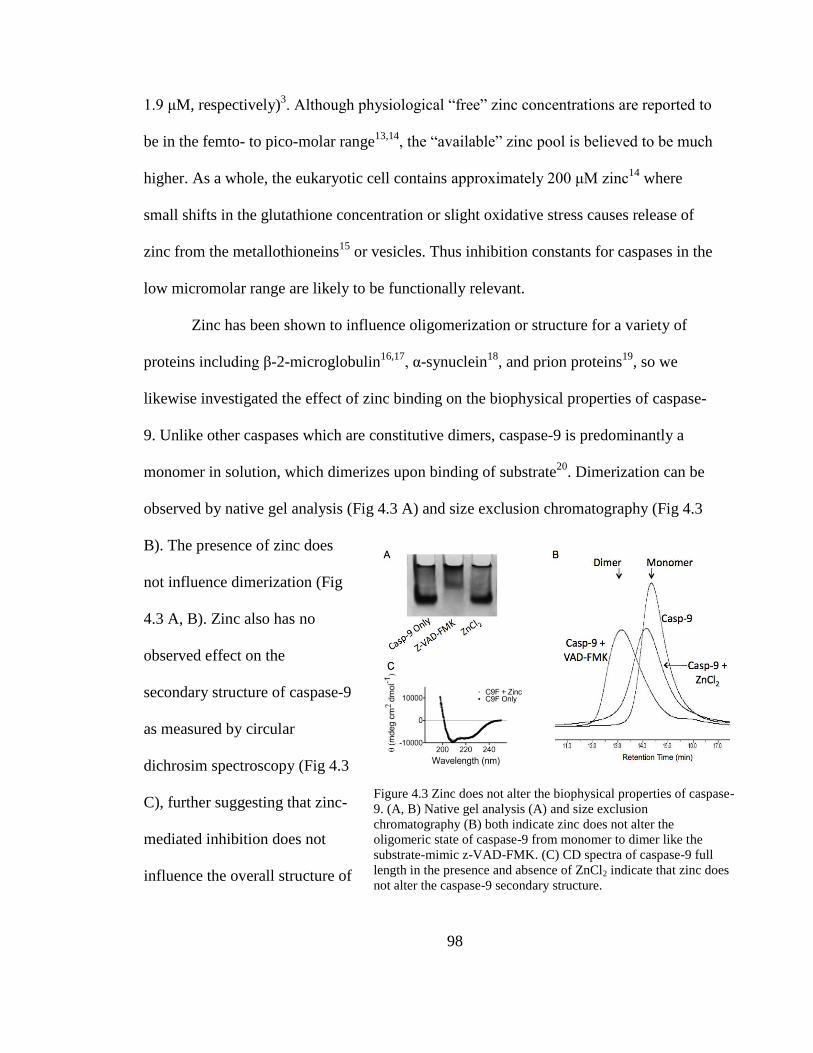
98
1.9 μM, respectively)3. Although physiological “free” zinc concentrations are reported to
be in the femto- to pico-molar range13,14
, the “available” zinc pool is believed to be much
higher. As a whole, the eukaryotic cell contains approximately 200 μM zinc14
where
small shifts in the glutathione concentration or slight oxidative stress causes release of
zinc from the metallothioneins15
or vesicles. Thus inhibition constants for caspases in the
low micromolar range are likely to be functionally relevant.
Zinc has been shown to influence oligomerization or structure for a variety of
proteins including β-2-microglobulin16,17
, α-synuclein18
, and prion proteins19
, so we
likewise investigated the effect of zinc binding on the biophysical properties of caspase-
9. Unlike other caspases which are constitutive dimers, caspase-9 is predominantly a
monomer in solution, which dimerizes upon binding of substrate20
. Dimerization can be
observed by native gel analysis (Fig 4.3 A) and size exclusion chromatography (Fig 4.3
B). The presence of zinc does
not influence dimerization (Fig
4.3 A, B). Zinc also has no
observed effect on the
secondary structure of caspase-9
as measured by circular
dichrosim spectroscopy (Fig 4.3
C), further suggesting that zinc-
mediated inhibition does not
influence the overall structure of
Figure 4.3 Zinc does not alter the biophysical properties of caspase-
9. (A, B) Native gel analysis (A) and size exclusion
chromatography (B) both indicate zinc does not alter the
oligomeric state of caspase-9 from monomer to dimer like the
substrate-mimic z-VAD-FMK. (C) CD spectra of caspase-9 full
length in the presence and absence of ZnCl2 indicate that zinc does not alter the caspase-9 secondary structure.

99
caspase-9 monomers.
4.2.2. Determining the Location of Zinc Binding
Zinc binding to caspase-9 was assessed by inductively coupled plasma-optical
emissions spectroscopy (ICP-OES). Each wild-type caspase-9 monomer was observed to
bind two zinc molecules. Caspase-9 monomers contain thirteen cysteine and eight
histidine residues (Fig 4.4 A, B), all potential ligands for zinc. Most of these residues are
located in the large subunit, either clustered near the active site or at the bottom of the
210’s helix (Fig 4.4 C, D) suggesting the locations of two potential zinc-binding sites.
Figure 4.4 Zinc binding in caspase-9. (A) Caspase-9, monomer, highlights clusters of cys and his metal-
binding residues. (B) Cys and his residues are mainly located in the large subunit of caspase-9. (C) Cys-
His clusters are conserved throughout the caspase family. (D) Location of the conserved active-site and
210’s helix exosite-ligand clusters on caspase-9 (pdb ID 1JXQ).

100
The active site cluster comprises the conserved catalytic residues C287 and H237, in
proximity to two cysteines that are unique to caspase-9, C172 and C239. Interestingly, the
210’s helix region contains a conserved cysteine-histidine-cysteine triad of unknown
function, comprising residues 272, 224, 230 respectively. Furthermore, both the active
site region and the triad are highly conserved in the apoptotic class of caspases (Fig 4.4
C).
We interrogated five positions within
these two cys-his clusters in caspase-9 for
involvement in zinc binding (Table 4.1).
Whereas two zinc molecules bind to one
monomer of wild-type caspase-9, caspase-9
variants which substitute the active-site residues
C287A or H237A bind just one molecule of
zinc. This suggests that the active-site region is one of the zinc binding sites. The two
most likely proximal zinc-binding ligands were also substituted. C172A had little effect
on zinc binding, but C239S lead to a more substantial loss of zinc binding. The
C287A/C239S variant also lost binding of one molecule of zinc, suggesting that the
active site zinc binding cluster comprises residues C239, H237 and C287. Although the
identity at residue C239 is not conserved across caspases, it is Glu in all the executioner
caspases, suggesting that this residue might be a conserved zinc ligand position across the
apoptotic caspase family.
Substitution of the residue C272A to alanine in the conserved 210’s-helix triad,
Table 4.1 Zinc bincing as monitored by
ICP-OES.

101
also resulted in loss of one zinc molecule (Table 4.1), suggesting that C272 is at the core
of the second zinc-binding site. The loss of zinc binding at C272A was not due to
unfolding of the protein as the substitution C272A has no effect on basal activity of the
enzyme. The catalytic efficiency of C272A is nearly the same as wild-type caspase-9, 2.0
± 0.16 and 2.7 ± 0.25 respectively. Given the proximity, we suggest that the second
binding site comprises residues H224, C230 and C272. Together these data suggest that
there are two distinct zinc-binding sites in caspase-9 (Fig 4.4 D). The presence of two
independent binding sites was confirmed by caspase-9 C287A/C272A variant in which
zinc binding is ablated.
Binding of zinc to both the catalytic residues of caspase-9 is expected to block
nucleophilic attack of the peptide bond by C287 rendering the enzyme inactive. The
potential contribution of the second binding site to inhibition is less obvious. If the C272
site contributes directly to zinc-mediated caspase-9 inhibition, then the caspase-9 C272A
variant C272A might be expected to show a competitive mechanism of inhibition.
Surprisingly, the C272A variant also shows a mixed model of inhibition (Fig 4.5), like
wild-type caspase-9 and exhibited a comparable Ki value of 5.0 ± 2.8 μM. The Ki for zinc
with the C272A variant is statically indistinguishable from that observed with wild type
enzyme (Student’s t-test), suggesting this site does not contribute appreciably to the
mechanism of caspase-9 inhibition by zinc.

102
4.3. Discussion
Although no regulatory function was observed in the case of the caspase-9 second
zinc binding site, this 210’s helix region (the 90’s helix in caspase-6) has been implicated
in the mechanism of caspase-6 activation21
. A 21° pivot about the bottom of this helix has
been observed. The open conformation is adopted when caspase-6 is in an inactive state
whereas the closed conformation is characteristic of the active enzyme. In light of the fact
that zinc inhibition is most potent in caspase-6 (IC50 = 0.3 μM )3,22
, and that this structural
region is sensitive and critical for caspase-6 activity, it is tempting to hypothesize that
zinc could control caspase-6 allosterically, via binding to this caspase conserved exosite
triad.
Our findings indicate that the reversible binding of zinc to the caspase-9 active
Figure 4.5 Kinetics of full-length caspase-9 C272A variant in the presence of 0-50 μM ZnCl2. Measured
data were fit to competitive, noncompetitive, and mixed models of inhibition. R2 values are presented
for individual zinc concentrations and for the overall global fit of the curves fit by each model in which
mixed model of inhibition represented the best fit model.

103
site residues H237, C239, and
C287 is the predominant
mechanism for caspase-9
inhibition. A plausible model
with reasonable metal-to-ligand
bond distances between active-
site residues and zinc was
obtained simply by changing
the rotomeric conformations of
H237, C239, and C287 (Fig
4.6). Like the majority of zinc-
binding sites in proteins, the binding site is likely to be four-coordinate, tetrahedral. The
fourth ligand could be water, solvent, or potentially a nearby glutamate residue, E290
(Fig 4.6). E290 resides on a flexible loop just 3.8Å from the proposed position of the zinc
ion. Optimal E290 ligand-binding distances may be achieved by loop movement or by a
water-mediated interaction.
Taken together, these results suggest a model for how zinc binds to the active site
and inhibits caspase-9 and caspases generally. The function of the secondary zinc-binding
exosite remains an open question; however the conservation of this site suggests that
perhaps zinc does play a physiological role at this site as well. The fact that zinc does not
function in a strictly competitive manner with substrate at the active site, may suggest
some flexibility in the zinc-binding ligands. Given that the zinc-mediated inhibition
Figure 4.6 A model of caspase-9 active-site ligand interactions
with a modeled zinc ion. Model was obtained by altering the
H237, C239 and C287 rotomers.

104
constants for all the caspases are similar and that ligands are conserved in the active site
region, suggests that the mechanism of active-site inhibition of the apoptotic caspases is
conserved. Thus, utilization of zinc-based inhibition of caspases, like that used by PAC-
110
, remains a promising avenue for controlling caspases and apoptosis.
4.4. Materials and Methods
4.4.1. Caspase-9 Expression and Purification
The caspase-9 full-length gene (human sequence) construct, encoding amino acids
1-416, in pET23b (Addgene plasmid 1182923
) was transformed into the BL21 (DE3) T7
Express strain of E. coli (NEB). The cultures were grown in 2xYT media with ampicillin
(100 mg/L, Sigma-Aldrich) at 37°C until they reached an optical density at 600 nm of
1.2. The temperature was reduced to 15°C and cells were induced with 1 mM IPTG
(Anatrace) to express soluble 6xHis-tagged full-length protein. Cells were harvested
after 3 hrs to obtain single site processing at Asp315. Cell pellets stored at -20°C were
freeze-thawed and lysed in a microfluidizer (Microfluidics, Inc.) in 50 mM sodium
phosphate pH 8.0, 300 mM NaCl, and 2 mM imidazole. Lysed cells were centrifuged at
17,000 rpm to remove cellular debris. The filtered supernatant was loaded onto a 5-ml
HiTrap Ni-affinity column (GE Healthcare). The column was washed with a buffer
containing 50 mM sodium phosphate pH 8.0, 300 mM NaCl, 2 mM imidazole until 280
nm absorbance returned to base line. The protein was eluted using a linear imidazole
gradient of 2 to100 mM over the course of 270 mL. The eluted fractions containing
protein of the expected molecular weight and composition were diluted by 10-fold into a
buffer composed of 20 mM Tris pH 8.5, 10 mM DTT to reduce the salt concentration.

105
This protein sample was loaded onto a 5-ml Macro-Prep High Q column (Bio-Rad
Laboratories, Inc.). The column was developed with a linear NaCl gradient and eluted in
20 mM Tris pH 8.5, 100 mM NaCl, and 10 mM DTT buffer. The eluted protein was
stored in -80°C in the above buffer conditions. The identity of the purified caspase-9 was
analyzed by SDS-PAGE and ESI-MS to confirm mass and purity. Caspase-9 variants in
the full-length expression construct (C287A, H237A, C172A, C239S, C287A/C239S,
C272A, C272A/C287A) were purified by the same method as described here for the
wild-type protein.
Caspase-9 ΔCARD was expressed from a two-plasmid expression system. Two
separate constructs, one encoding the large subunit, residues 140-305, and the other
encoding the small subunit, residues 331-416, each in the pRSET plasmid, were
separately transformed into the BL21 (DE3) T7 Express strain of E. coli (NEB). The
recombinant large and small subunits were individually expressed as inclusion bodies for
subsequent reconstitution. Cultures were grown in 2xYT media with ampicillin (100
mg/L, Sigma-Aldrich) at 37°C until they reached an optical density at 600 nm of 0.6.
Protein expression was induced with 0.2 mM IPTG. Cells were harvested after 3 hrs at
37°C. Cell pellets stored at -20°C were freeze-thawed and lysed in a microfluidizer
(Microfluidics, Inc.) in 10 mM Tris pH 8.0 and 1 mM EDTA. Inclusion body pellets were
washed twice in 100 mM Tris pH 8.0, 1 mM EDTA, 0.5 M NaCl, 2% Triton, and 1M
urea, twice in 100 mM Tris pH 8.0, 1 mM EDTA and finally resuspended in 6 M
guanidine hydrochloride. Caspase-9 large and small subunit proteins in guanidine
hydrochloride were combined in a ratio of 1:2, large:small subunits, and rapidly diluted

106
dropwise into refolding buffer composed of 100 mM Tris pH 8.0, 10% sucrose, 0.1%
CHAPS, 0.15 M NaCl, and 10 mM DTT, allowed to stir for one hour at room
temperature and then dialyzed four times against 10 mM Tris pH 8.5, 10 mM DTT, and
0.1mM EDTA buffer at 4°C. The dialyzed protein was centrifuged for 15 minutes at
10,000 rpm to remove precipitate and then purified using a HiTrap Q HP ion exchange
column (GE Healthcare) with a linear gradient from 0 to 250 mM NaCl in 20 mM Tris
buffer pH 8.5, with 10 mM DTT. Protein eluted in 20 mM Tris pH 8.5, 100 mM NaCl,
and 10 mM DTT buffer was stored in -80°C. The identity of the purified caspase-
9ΔCARD was analyzed by SDS-PAGE and ESI-MS to confirm mass and purity.
4.4.2. Prediction of Metal Ligands and Construction of Ligand Substitution
Variants
Potential zinc-binding ligands were predicted via the computational server which
predicts metal ion-binding sites, HotPatch version 4.0 , by a computational program
specific for zinc binding sites, PREDZINC version 1.1 , and by visual inspection of zinc
ligand clusters and distances based on the caspase-9ΔCARD crystal structure (pdb ID:
1JXQ) via the PyMOL Molecular Graphics System, Version 1.3 (Schrödinger, LLC).
Sites predicted as potential zinc ligands were C172, C239, C272, H237, and C287.
Individual active-site knockout variants, H237A and C287A, and cysteine knockout
variants, C172A, C239S, and C272A, in the caspase-9 full-length gene were made by a
single round of QuikChange site directed mutagenesis (Stratagene). Double knockout
variants, C239S/C287A and C272A/C287A, were made by an additional round of
QuikChange site directed mutagenesis in the background of the caspase-9 full-length

107
C287A variant. Formation of the proper variant was confirmed by sequence analysis
(Genewiz, Inc.).
4.4.3. Caspase-7 C186A Expression and Purification
The active site knockout variant, C186A, was made by QuikChange mutagenesis
(Stratagene) of the human caspase-7 gene in pET23b (gift of Guy Salvesen ) The
construct was transformed into BL21 (DE3) T7 Express strain of E. coli and protein
expression was induced with 1 mM IPTG at 18°C for 18 hrs. The protein was purified as
described previously for caspase-7. The eluted protein was stored in -80°C in the buffer
in which they eluted. The identity of purified caspase-7 C186A was assessed by SDS-
PAGE and ESI-MS to confirm mass and purity.
4.4.4. Activity Assays
For measurements of caspase activity, proteolytic cleavage gel-based caspase-9
activity assays were performed for testing the ability of a panel of metal cations to inhibit
caspase-9 activity against a natural substrate. Cleavage of the full-length procaspase-7
variant C186A, which is catalytically inactive and incapable of self-cleavage, was used to
report caspase-9 activity. 1μM caspase-9 full-length in a minimal assay buffer containing
100 mM MES pH 6.5 and 10 mM DTT was incubated in the presence of 1mM metal
cation for 1hr at room temperature. 1 μM full-length procaspase-7 C186A was added to
the reaction mix and incubated in a 37°C water bath for 1 hr. Samples were analyzed by
16% SDS-PAGE to confirm the exact lengths of the cleavage products.
To test the reversibility of zinc binding, 700 nM freshly purified caspase-9 full-
length was incubated in the presence and absence of 5 equivalents excess ZnCl2 for 1hr at

108
room temperature. Half of each protein sample was treated with 25-fold excess EDTA
just prior to fluorescence activity assay measurements. Samples were assayed for activity
over the course of 10 minutes in a caspase-9 activity assay buffer containing 100 mM
MES pH 6.5, 10% PEG 8,000 and 10 mM DTT. 300 μM fluorogenic substrate, LEHD-
AFC, (N-acetyl-Leu-Glu-His-Asp-7-amino-4-fluorocoumarin), (Enzo Lifesciences)
Ex395/Em505, was added to initiate the reaction. Assays were performed in duplicate at
37°C in 100 μL volumes in 96-well microplate format using a Molecular Devices
Spectramax M5 spectrophotometer.
The inhibitory constant for zinc, Ki, was determined by incubating 700 nM
caspase-9 full-length protein, diluted in 100 mM MES pH 6.5, 10% PEG 8,000, and 5
mM DTT, in the presence of 0-50 μM ZnCl2 for 1.5 hours at room temperature.
Samples from each ZnCl2 concentration were subjected to a substrate titration, performed
in the range of 0-300 μM fluorogenic substrate, LEHD-AFC (Ex365/Em495) which was
added to initiate the reaction. Assays were performed in duplicates at 37°C in 100 μL
volumes
in 96-well microplate format using a Molecular Devices Spectramax M5 spectro-
photometer. Initial velocities versus substrate concentration were globally fit to
competitive, noncompetitive and mixed models of inhibition using GraphPad Prism
(Graphpad Software) to determine inhibitory constant, Ki. Determination for the type of
inhibition was based on four criteria. R2 values for the individual curve fits, criterion for
the α-value reported by the mixed model of inhibition, overall visual inspection of the
curve fits with the data and knowledge of the system were all taken into consideration.

109
The R2 values and visual inspection of the curve fits with the data ruled out competitive
inhibition whereas these criteria were less clear between the noncompetitive and mixed
models of inhibition. However, a slight improvement in the R2 values for the individual
curve fits for the mixed model of inhibition was observed, suggesting that this is the best
fit for the data. The α-value obtained from the mixed model of inhibition fits also reports
on the inhibition mechanism. If the α-value is equal to one, the mixed model of inhibition
is identical to the non-competitive model whereas if this value is very large, the model
becomes identical to the competitive model of inhibition. The reported α-value for the
fits of both full-length caspase-9 wild-type and variant C272A match neither of these
criteria, therefore the mechanism of inhibition is appears to follow a mixed model.
Furthermore, based on our enzymatic system and the determined location of metal
binding, a mixed model of inhibition appears to be the most likely.
4.4.5. Oligomeric-State Determination
Caspase-9 wild-type, full-length protein samples in 20 mM Tris pH 8.5, 110 mM
NaCl, and 5 mM DTT were incubated alone (monomer), with covalent inhibitor z-VAD-
FMK (carbobenzoxy-Val-Ala-Asp-fluoromethylkeytone) (dimer) or with 5 equivalents
excess of ZnCl2 for 2 hours at room temperature. The oligomeric state of the caspase-9
samples were determined via gel filtration and native gel analysis. 100 μL of 0.75 mg/ml
protein sample was loaded onto a Superdex 200 10/300 GL (GE Healthcare) gel-filtration
column. Apo and z-VAD-FMK incubated protein samples were eluted with 20 mM Tris
pH 8.0, 100 mM NaCl, and 2 mM DTT while ZnCl2-incubated protein was eluted in the
same buffer and one containing 50 μM ZnCl2 to ensure zinc was not be lost during the gel

110
filtration process. Eluted peaks were analyzed by SDS-PAGE to ensure protein identity.
Four different molecular weight standards from the gel-filtration calibration kit LMW
(GE Healthcare) were run in the same conditions and a standard plot was generated to
determine whether the peaks were caspase-9 monomer or dimer. For native gel analysis,
samples were mixed with glycerol loading dye and fractionated on a 10% Tris/Glycine
pH 8.3 polyacrylamide gel. The oligomeric state of the ZnCl2 treated caspase-9 was
identified by comparison to the Apo (monomer) and z-VAD-FMK (dimer) caspase-9 full-
length samples.
4.4.6. Secondary Structure Analysis by Circular Dichroism
Caspase-9 full-length protein was buffer exchanged via dialysis against 100 mM
sodium phosphate pH 7.0, 110 mM NaCl, and 2 mM DTT, and diluted to 7 μM. The
sample was split in half and incubated in the presence or absence of five equivalents of
ZnCl2 for 1hr at room temperature. The circular dichroism spectra of caspase-9 full-
length in the presence and absence of zinc was monitored from 250-190 nm, measured on
a J-720 circular dichroism spectrometer (Jasco) with a peltier controller.
4.4.7. Zinc Binding Analysis by ICP-OES
Purified caspase-9 full-length, wild-type and variants (15 μM) in 20 mM Tris pH
8.5, 110 mM NaCl, and 5 mM DTT were incubated with 5 equivalents excess of ZnCl2
for 1 hour at room temperature. Unbound zinc was removed by incubating samples of 1
mL volume in the presence of approximately 125 mg of Chelex® 100 for 1 hour, mixing
every 20 minutes. A control sample of 1 mL of 20 mM Tris pH 8.5, 110 mM NaCl, and 5
mM DTT buffer only was treated in the exact same manner to judge the completeness of

111
unbound zinc chelation. Wild-type caspase-9 full-length samples either treated with zinc
or untreated were also used as controls for metal content pre and post zinc incubation.
Zinc content of the sample supernatant was quantified using a PerkinElmer Optima 4300
DV inductively coupled plasma-optical emission spectrometer (ICP-OES) equipped with
a 40 MHz free-running generator and a segmented-array charge-coupled device (SCD)
detector and a sample introduction system consisted of a concentric nebulizer with a
cyclonic spray chamber. The concentration of zinc in each sample was determined at
206.2 nm and converted from ppm to μM to obtain the zinc to monomer of caspase-9
full-length ratio. Post ICP-OES analysis, remaining samples were tested via absorbance
at 280 nm determine protein concentration. A 16% SDS-PAGE gel visualized with the
Pierce Silver Stain Kit (Thermo Scientific) was used to determine the percentage of low
concentration contaminants so the total caspase-9 concentration could be adjusted
accordingly. This adjustment did not alter the overall trend observed.
4.4.8. Model of Zinc Binding to Caspase-9
A proposed model for zinc binding at the active site of caspase-9 ΔCARD was
developed in PyMOL Molecular Graphics System, Version 1.3 (Schrödinger, LLC). The
zinc ionic radius was contoured to the reported values for a tetrahedral geometry of
ligand binding. The rotomeric conformations of the ligands which coordinate the zinc
ion, H237, C239, and C287, as well as a putative ligand, E290, were sampled to obtain
the best metal to ligand geometry and allowed bond distances for ligand character.

112
4.5. Structure Determination Trials of Caspase-9 and Zinc
4.5.1. Crystallization of Caspase-9 in the Presence and Absence of Zinc
To further understand the molecular details of zinc-mediated inhibition at the
active site as well as zinc binding at the secondary binding site we aimed to obtain the
crystal structure of caspase-9 full-length, wild-type protein in the presence of zinc.
Previous caspase-9 structures have shown a dimeric caspase-9 enzyme without the N-
terminal CARD domain (ΔCARD) in complex with covalent peptide inhibitor z-VAD-
FMK (PDB ID: 1JXQ)20
, a variant which shifts the enzymes oligomeric state from
monomer to dimer by re-engineering the dimerization interface to mimic caspase-3 (PDB
ID: 2AR9)24
, and monomeric caspase-9 ΔCARD in complex with the protein inhibitor
XIAP-Bir3 (PDB ID: 1NW9)25
. Crystals from dimeric caspase-9 enzyme lacking the N-
terminal CARD domain (ΔCARD) in complex with a covalent peptide inhibitor grew in a
0.1 M MES pH 6.0 and 12% PEG 20,000 solution when incubated at 20 °C. The sitting-
drop vapor diffusion method was utilized for crystal growth. On the other hand, the
dimeric caspase-9 variant, (residues 140–416) made by altering the dimeric interface to
mimic that of caspase-3 and knocking out the catalytic Cys (C287S), crystallized in
grown in 5% (w/v) PEG 5,000 monomethylether, 0.1M MES pH 6.0 and 3% tacsimate
when incubated at 17 °C utilizing the hanging drop vapor diffusion method. Crystals of
the caspase-9/XIAP-Bir3 inhibitory complex were grown also grown using this method
where drops were set up in 0.1 M Tris pH 8.0, 1.0 M potassium monohydrogen
phosphate, and 0.2 M sodium chloride. These three conditions served as a starting point
for initial screening for caspase-9 crystals in the presence of zinc.

113
Purified wild type caspase-9 full-length protein (residues 1-416) in 20 mM Tris
pH 8.5, 100 mM NaCl, and 5 mM DTT was concentrated to 11 mg/mL using an Amicon
Ultracell 3K concentrator (Millipore). The concentrated protein was then incubated with
five equivalents excess of ZnCl2 for one hour at room temperature and filtered using a
durapore-PVDF 0.22 μM filter (Millipore) to remove any dust, debris or protein
precipitate prior to crystallization trials. Initial crystallization trials were set up using the
hanging drop, vapor diffusion method in all known conditions previously described.
However upon drop set-up, immediate protein precipitate was observed and no crystal
formation was seen over time. Therefore, naïve crystallization screens were tested with 8
to 20 mg/mL of the full-length form of caspase-9 wild-type protein pre-incubated with
zinc. No crystal formation was observed in the naïve screens when using Crystallization
Screen I & II, Index Screen, and JCSG+ Suite from Hampton Research. Crystallization
trials of the apo wild-type caspase-9 in the full-length form were then pursued using the
known crystallization conditions using 11 mg/mL protein. Initial drops result in a white
cloudy precipitate in the drop, however, crystal growth was observed after 24 hours in the
condition 0.1 M MES pH 6.0 and 12% PEG 20,000 in a 1:1 protein-to-mother-liquor
ratio incubated at 20 °C. The crystals obtained formed clusters of thin plates measuring
730 x 260 x 50 μM in size, which were unusable for data collection (Fig 4.7 A).
Further optimization was performed to obtain single crystals of caspase-9.
Optimizations included varying the pH between 5.2 to 6.0, the identity of the PEG
precipitant from PEG 200 to 20,000 average molecular weight and the concentration of
PEG from 10 to 20%. Final conditions were 0.1 M MES pH 5.8 and 14% PEG 6,000,

114
resulting in thin single crystals of approximately 100 x 40 x 10 μM in size and form in
the shape of a half of a hexagon or a full hexagon in some instances (Fig 4.7 B).
Microseeding, macroseeding, and additive screen experiments in addition to further
purifying the protein via a HiLoad 26/600 Superdex gel filtration column in 20 mM Tris
pH 8.5, 100 mM NaCl, and 2 mM DTT were used to improve consistency of the crystal
form within a drop and overall thickness and size of crystals. Unfortunately these
approaches did not result in a significant improvement in crystal morphology or growth.
4.5.2. Data Collection on Crystals of Caspase-9
Crystals of full length caspase-9 wild-type apo protein in final mother liquor
conditions of 0.1 M MES pH 6.0 and 15% PEG 6,000 were incubated in 20% glycerol,
ethylene glycol or PEG 400 as a cryoprotectant for one hour and flash frozen in liquid
nitrogen for storage. During crystal screening at a variety of different angles at the
Brookhaven National Laboratory synchrotron x-ray light source, the caspase-9 wild-type
Figure 4.7 Crystals of wild type caspase-9 full-length (A) pre and (B) post optimization.

115
crystals diffracted to approximately 3.0 Å, with nice rounded spot shape and minimal
mosaicity, but a relatively weak intensity (Fig 4.8). This indicates that optimal
crystallization and freezing conditions were obtained. Indexing the diffraction images
was performed with ease, and indicated that the crystals showed C2 symmetry, similar to
that of the previously reported N-terminal CARD domain (ΔCARD) in complex with
covalent peptide inhibitor z-VAD-FMK (PDB ID: 1JXQ)20
.
4.5.3. Zinc Soaks of caspase-9 Crystals
Therefore, these conditions were used for further experimentation. In order to
obtain a structure of caspase-9 in the presence of zinc, full-length caspase-9 wild-type
crystals needed to be soaked in mother liquor containing the zinc ion. The 0.1 M MES pH
5.8 and 15% PEG 6,000 mother liquor was prepared to include 10 mM ZnCl2. Crystals
Figure 4.8 Diffraction image of apo wild type caspase-9 crystals soaked in a 20% PEG 400
cryoprotectant for one hour.

116
were transferred from their growth drops to a 3 μl drop of zinc containing mother liquor.
Soaking of the crystals lasted for the duration of 15 minutes, 30 minutes, 1 hour, 2 hours,
3 hours, 5 hours, or 18 hours. Crystals appeared to remain intact during the entire soaking
process as no microfractures were observed microscopically. The zinc-soaked crystals
were then either preserved in cryoprotectant containing zinc to ensure the bound zinc ions
are not lost during the cryoprotectant incubation time due to the on/off rate of zinc to the
binding sites or backsoaked in cryoprotectant to ensure excess zinc binding to random
free thiols did not occur. Cryoprotectant incubation times lasted anywhere from 15
minutes to overnight. Crystals were preserved by flash freezing in liquid nitrogen. Data
collection was carried out at Brookhaven National Labs Synchrotron x-ray Light Source
beamline X6A on the caspase-9 wild type crystals in the presence of zinc. These crystals
did diffract, however, the diffraction was limited to approximately 7Å (Fig 4.9). In
addition, within the diffraction image, a smeary/mosaic spot shape was observed as well
as an anisotropic diffraction pattern. These characteristics provide clues as to the changes
in conformation that may have occurred within the caspase-9 crystal upon binding zinc.
The observed smeary spot shape, suggests increased mosaicity in the crystal form, which
could not be corrected by annealing. In addition the diffraction images themselves
indicate that crystals are directionally dependent or anisotropic in nature.
Based on the quality of the diffraction images for the apo caspase-9
crystals, conditions and methods for soaking of the caspase-9 crystal in zinc soak should
be reevaluated. Alternate metal salts such as zinc sulfate and zinc acetate which also
inhibit enzymatic function could be explored. Furthermore, soaking methodology such as

117
the zinc concentration and time could also improve diffraction of zinc bound caspase-9
crystal.
References:
1. Zalewski, P. D., Forbes, I. J. & Betts, W. H. Correlation of apoptosis with change
in intracellular labile Zn(II) using zinquin [(2-methyl-8-p-toluenesulphonamido-6-
quinolyloxy)acetic acid], a new specific fluorescent probe for Zn(II). Biochem J
296 ( Pt 2), 403-408, (1993).
2. Wolf, C. M., Morana, S. J. & Eastman, A. Zinc inhibits apoptosis upstream of
ICE/CED-3 proteases rather than at the level of an endonuclease. Cell Death
Differ 4, 125-129, (1997).
3. Stennicke, H. R. & Salvesen, G. S. Biochemical characteristics of caspases-3, -6, -
7, and -8. J Biol Chem 272, 25719-25723, (1997).
Figure 4.9 Diffraction image of wild type caspase-9 soaked in ZnCl2. Crystals were soaked for 2
hours and a 20% ethylene glycol for ten minutes.

118
4. Perry, D. K. et al. Zinc is a potent inhibitor of the apoptotic protease, caspase-3. A
novel target for zinc in the inhibition of apoptosis. J Biol Chem 272, 18530-
18533, (1997).
5. Truong-Tran, A. Q., Carter, J., Ruffin, R. E. & Zalewski, P. D. The role of zinc in
caspase activation and apoptotic cell death. Biometals 14, 315-330, (2001).
6. Carter, J. E. et al. Involvement of redox events in caspase activation in zinc-
depleted airway epithelial cells. Biochem Biophys Res Commun 297, 1062-1070,
(2002).
7. Truong-Tran, A. Q., Grosser, D., Ruffin, R. E., Murgia, C. & Zalewski, P. D.
Apoptosis in the normal and inflamed airway epithelium: role of zinc in epithelial
protection and procaspase-3 regulation. Biochem Pharmacol 66, 1459-1468,
(2003).
8. Kohler, J. E. et al. Monochloramine-induced toxicity and dysregulation of
intracellular Zn2+ in parietal cells of rabbit gastric glands. Am J Physiol
Gastrointest Liver Physiol 299, G170-178, (2010).
9. Kohler, J. E. et al. Monochloramine impairs caspase-3 through thiol oxidation and
Zn2+ release. J Surg Res 153, 121-127, (2009).
10. Peterson, Q. P. et al. PAC-1 activates procaspase-3 in vitro through relief of zinc-
mediated inhibition. J Mol Biol 388, 144-158, (2009).
11. Chimienti, F., Seve, M., Richard, S., Mathieu, J. & Favier, A. Role of cellular
zinc in programmed cell death: temporal relationship between zinc depletion,
activation of caspases, and cleavage of Sp family transcription factors. Biochem
Pharmacol 62, 51-62, (2001).
12. Schrantz, N. et al. Zinc-mediated regulation of caspases activity: dose-dependent
inhibition or activation of caspase-3 in the human Burkitt lymphoma B cells
(Ramos). Cell Death Differ 8, 152-161, (2001).
13. Bozym, R. A., Thompson, R. B., Stoddard, A. K. & Fierke, C. A. Measuring
picomolar intracellular exchangeable zinc in PC-12 cells using a ratiometric
fluorescence biosensor. ACS Chem Biol 1, 103-111, (2006).

119
14. Krezel, A. & Maret, W. Zinc-buffering capacity of a eukaryotic cell at
physiological pZn. J Biol Inorg Chem 11, 1049-1062, (2006).
15. Krezel, A., Hao, Q. & Maret, W. The zinc/thiolate redox biochemistry of
metallothionein and the control of zinc ion fluctuations in cell signaling. Arch
Biochem Biophys 463, 188-200, (2007).
16. Eakin, C. M., Knight, J. D., Morgan, C. J., Gelfand, M. A. & Miranker, A. D.
Formation of a copper specific binding site in non-native states of beta-2-
microglobulin. Biochemistry 41, 10646-10656, (2002).
17. Morgan, C. J., Gelfand, M., Atreya, C. & Miranker, A. D. Kidney dialysis-
associated amyloidosis: a molecular role for copper in fiber formation. J Mol Biol
309, 339-345, (2001).
18. Goers, J., Uversky, V. N. & Fink, A. L. Polycation-induced oligomerization and
accelerated fibrillation of human alpha-synuclein in vitro. Protein Sci 12, 702-
707, (2003).
19. Millhauser, G. L. Copper binding in the prion protein. Acc Chem Res 37, 79-85,
(2004).
20. Renatus, M., Stennicke, H. R., Scott, F. L., Liddington, R. C. & Salvesen, G. S.
Dimer formation drives the activation of the cell death protease caspase 9. Proc
Natl Acad Sci U S A 98, 14250-14255, (2001).
21. Vaidya, S., Velazquez-Delgado, E. M., Abbruzzese, G. & Hardy, J. A. Substrate-
induced conformational changes occur in all cleaved forms of caspase-6. J Mol
Biol 406, 75-91, (2011).
22. Takahashi, A. et al. Cleavage of lamin A by Mch2 alpha but not CPP32: multiple
interleukin 1 beta-converting enzyme-related proteases with distinct substrate
recognition properties are active in apoptosis. Proc Natl Acad Sci U S A 93, 8395-
8400, (1996).
23. Stennicke, H. R. et al. Caspase-9 can be activated without proteolytic processing.
J Biol Chem 274, 8359-8362, (1999).

120
24. Chao, Y. et al. Engineering a dimeric caspase-9: a re-evaluation of the induced
proximity model for caspase activation. PLoS Biol 3, e183, (2005).
25. Shiozaki, E. N. et al. Mechanism of XIAP-mediated inhibition of caspase-9. Mol
Cell 11, 519-527, (2003).

121
CHAPTER V
CASPASE-9 CARD:CORE DOMAIN INTERACTIONS REQUIRE A
PROPERLY-FORMED ACTIVE SITE
Abstract
The activation mechanism of caspase-9 is unique in that the addition of individual
domains or when bound to the large protein complex, the apoptosome, has a great affect
on enzymatic activity. The N-terminal prodomain, CARD, of caspase-9 is the smallest
unit capable of increasing caspase-9 activity. We investigated CARD with respect to the
activation, oligomerization and stability of caspase-9. Our data suggest an increase in
dimerization due to the presence of CARD is not the cause of the increased activity
observed. We determined that caspase-9 undergoes conformational acrobatics by
adopting one conformation in the uncleaved, monomeric form and cleaved active dimeric
state where the active site is in an ordered state, while adopting another in the cleaved
monomeric form in the presence of a disordered active site.
5.1. Introduction
Caspase-9 is as a critical initiator of the intrinsic pathway of apoptosis or
programmed cell death. It is responsible for activating downstream executioner caspases-
3, -6 and -7 which ultimately results in the breakdown of the cell. Defects in this pathway
are a characteristic of diseases ranging from autoimmune disorders to cancer. In these
diseases, activation of caspase-9 is particularly critical1-3
, however, a full mechanistic
understanding of caspase-9 activation is lacking.
In general, caspases, or cysteine aspartate proteases, are functional as homodimers
of monomeric units that comprise an N-terminal prodomain and a catalytic large and

122
small subunit connected by an intersubunit linker. Upon dimerization and cleaveage at a
specific aspartate residue within the intersubuint linker, the enzyme becomes active.
Unlike the executioner caspases, caspase-9 is active even in the absence of cleavage of
the intersubunit linker4. Furthermore, the executioner caspases have short N-terminal
prodomains whereas caspase-9’s catalytic core is preceded by a long regulatory domain
called the Caspase Activation and Recruitment Domain (CARD). The caspase-9 CARD
facilitates a protein-protein interaction with the CARD of the apoptotic protease
activation factor 1(Apaf-1), which anchors caspase-9 to the activation platform known as
the apoptosome5-7
. This platform is created upon release of cytochrome c from the
mitochondria following an internal cellular stress signal. Cytochrome c is then able to
bind to Apaf-1. This binding event, initiates a conformational change in Apaf-1, resulting
in the heptameric oligomerization of the protein, driven in an ATP-dependent manner.
This Apaf-1 platform is then responsible for the recruitment and activation of caspase-9.
The molecular details of caspase-9 activation by the apoptosome have not been
elucidated.
The interaction of caspase-9 with the apoptosome increases caspase-9’s activity
by approximately 2000-fold4. However, even in the absence of this platform, the presence
of individual domains influence caspase-9 activity. Caspase-9 is 20% more active when
caspase-9’s catalytic core remains covalently linked to the CARD domain8 (Fig 5.1). The
authors hypothesized that increased tangling of CARD domains lead to an increase in the
dimeric fraction of full-length caspase-9 over ΔCARD-caspase-9. Addition of the Apaf-1
CARD in isolation further enhances caspase-9 activity by five-fold, in vitro. The greatest
increase in activity is when full-length caspase-9, including both the CARD and core

123
domains, bind to the apoptosome through Caspase-9 CARD: Apoptosome CARD
interactions.
Early models of the apoptosome activation of caspase-9 argued that the increased
activity is due to a change in the oligomeric state of the enzyme via increasing the local
concentration of monomeric caspase-9. Recruitment of additional molecules were
hypothesized to participate as partners in dimerization9 or facilitate dimerization amongst
the apoptosome-bound monomers10
. This model is supported by the evidence that
enhanced activity is associated with dimerized capase-9 molecules11
. Alternative,
competeting views of the activation mechanism invoke induced conformational changes
around the active site region of the enzyme12
. This conformational change model is
potentially supported by the new evidence emerging from high-resolution cryo electron
microscopy that shows that caspase-9 is monomeric when bound to the apoptosome in
the highly active state and that caspase-9 activation can be achieved without apoptosome
formation13
.
Figure 5.1 Model depicting the increase in enzymatic activity of caspase-9. Activity is lowest
for the monomeric unit of the catalytic core (C9ΔCARD) and increases to the highest when
full-length caspase-9 is bound to the apoptosome.

124
Taking inspiration from the proposed mechanisms of capase-9 activation by the
apoptosome we want to further investigate the affect of CARD with respect to the
activation, oligomerization and stability of caspase-9. A goal in this work is to understand
whether CARD influences the oligomeric state of caspase-9, specifically the ratio of
monomer to dimer in vitro, thus causing the observed increase in activity. We probe
interactions between the caspase-9 CARD and core domains. Finally we investigate
whether the caspase-9 CARD induces or takes advantage of conformational changes in
the loops that form the active site region as has been observed for caspase-6 and -714,15
. In
caspase-6 and -7 conformational changes in the active-site loops can be observed by both
thermal denaturation and circular dichroism spectral measurements. Defining CARD’s
role in the activation of caspase-9 will provide much needed insight for exploiting
caspase-9 for therapeutic means.
5.2. Results
5.2.1. The Influence of CARD on the Oligomeric State and Stability of Caspase-9
Activity measurements of caspase-9’s catalytic core (caspase-9 ΔCARD), in the
absence of the apoptosome platform, have been previously observed to have a lower
catalytic efficiently when compared to the full-length version of the enzyme. We
interrogated the affect the presence of the CARD has on these specific biophysical
properties of the capase-9 enzyme. To ensure that the caspase-9 ΔCARD and full-length
reagents used for this study were comparable to the studies that show CARD’s ability to
enhance caspase-9 activity, measurements of the enzymes catalytic properties were
performed (Table 5.1). As previously reported, the ΔCARD variant of caspase-9 has a
decreased catalytic efficiency when compared to the full-length version. Therefore we

125
reasoned that any differences observed between the two caspase-9 variants would be due
to the presence or absence of the CARD domain.
One potential reason for the increased in activity of full-length caspase-9 would
be due to an altered oligomeric state of the enzyme in the presence of CARD as predicted
previously8. Full-length and ΔCARD versions of the caspase-9 enzyme were subjected to
size exclusion chromatography to determine the oligomeric state of both enzymes (Fig
5.2). Both enzymes are predominantly monomeric in solution to such a great extent that
no dimeric fraction could be observed. Both the full-length and ΔCARD versions of
caspase-9 are both capable of completely converting to the dimeric state as observed
when the enzyme is in the presence of an active-site ligand such as the covalent active-
site inhibitor z-VAD-FMK. Thus the CARD does not have a great influence on the
Table 5.1 Measured kinetic parameters of full-length caspase-9 (C9FL) and caspase-9 in the absence of
the N-terminal CARD domain (C9ΔCARD).
Figure 5.2 Size exclusion chromatography of full-length caspase-9 (C9F)
and caspase-9 ΔCARD (C9 ΔCARD) in the presence and absence of active
site ligand z-VAD-FMK.

126
oligomeric state of the enzyme and cannot be the cause of the increased activity observed
in the presence of CARD.
Another potential reason for the increased activity of caspase-9 full-length is an
increased in stability in the presence of the CARD. Thermal denaturation studies were
then pursued to determine if the presence of CARD changes the thermal stability of the
enzyme and thus the reason for increased activity. Both the full-length and ΔCARD
caspase-9 enzymes in their monomeric and dimeric forms were analyzed for changes in
their thermal stability measurements. Full-length capase-9, which has been cleaved at the
intersubunit linker between the large
and small subunits, and includes the
CARD domain, shows a three-state
unfolding curve (Fig 5.3 A). The first
melting transition occurs at 49 ± 2 °C
while the second occurs at 62 ± 2 °C.
To determine which component of the
full-length caspase-9 enzyme was
contributing to which melting
transition, the catalytic core and CARD
domains were expressed independently
and interrogated in a similar fashion.
The catalytic core of the enzyme
(caspase-9ΔCARD), which is also
cleaved at the intersubunit linker, is
Figure 5.3 Thermal denaturation analysis and circular
dichroism spectrum of monomeric, cleaved, caspase-
9 (A) full-length, (B) ΔCARD, and (C) CARD only.

127
responsible for the first melting transition at 49 ± 1 °C (Fig 5.3 B) while the second
transition corresponds to CARD only with a melting temperature of 61 ± 2 °C (Fig 5.3
C). These results indicate that when caspase-9 is in its cleaved monomeric state, the
presence of CARD does not affect the overall thermal stability of the caspase-9 catalytic
core and the two portions of the enzyme unfold independently. Caspase-9 can be forced
to a dimeric state by binding of active-site ligand. Thermal denaturation studies were
performed on the cleaved full-length and ΔCARD versions of caspase-9 when the
enzyme is in a dimeric state with active site ligand bound (Fig 5.4). Both versions of
caspase-9 show an increase in thermal stability. Caspase-9ΔCARD (Fig 5.4 A) has a
14°C increase in thermal stability which is similar to the increase in stability observed
when caspase-7 binds active site ligand15
, while full-length caspase-9 (Fig 5.4 B) shows
only a 6 °C increase in thermal stability, more similar to the 3 °C increase in stability
observed for caspase-6 upon ligand bidning14
. Strikingly, the three-state unfolding of the
full-length caspase-9 is no longer observed in the presence of substrate. It appears that
binding ligand to the active site of caspase-9, which induces dimerization and ordering of
the active site loop bundle, transitions caspase-9 to a two-state unfolding mechanism.
Figure 5.4 Thermal denaturation analysis and circular dichroism spectrum of dimeric, cleaved,
caspase-9 (A) full-length, (B) ΔCARD.

128
Full-length caspase-9 is completely unfolded at 90 °C as observed in the circular
dichroism spectrum. The single transition observed when cleaved full-length caspase-9 in
the dimeric state indicates that binding substrate and dimerization either results in the
complete unfolding of CARD or causes the catalytic core and CARD to unfold as one
cooperative unit. An overlay of the circular dichorism spectra from the full-length
cleaved caspase-9 in the monomeric and dimeric states (Fig 5.5) indicates that there is not
a change in the secondary structure content.
Therefore, CARD does not appear to unfold in
the presence of substrate so the CARD and the
caspase-9 catalytic core must be unfolding as
a single cooperative unit. These results
suggest that a physical interaction between the
CARD and core domains occurs, which
causes the two domains to unfold as a single
unit.
It has been reported that uncleaved caspase-9 acts as an active zymogen 4 possibly
due to its increased intersubunit linker length which enables the enzyme to support a
properly formed active site. Therefore, this form of caspase-9 can be utilized to
interrogate whether the interaction observed between CARD and the catalytic core of
caspase-9 is due to changes in the active site conformation. Analysis of the full-length
monomeric caspase-9 in the uncleaved state, where the complete polypeptide chain is still
intact, resulted in the similar two-state unfolding mechanism as observed for the cleaved
active dimeric state (Fig 5.6). Therefore, the monomeric, uncleaved caspase-9 enzyme
Figure 5.5 Overlay of the circular dichroism
spectrum for full-length caspase-9 (C9F) in
the presence and absence of active site ligand
z-VAD-FMK
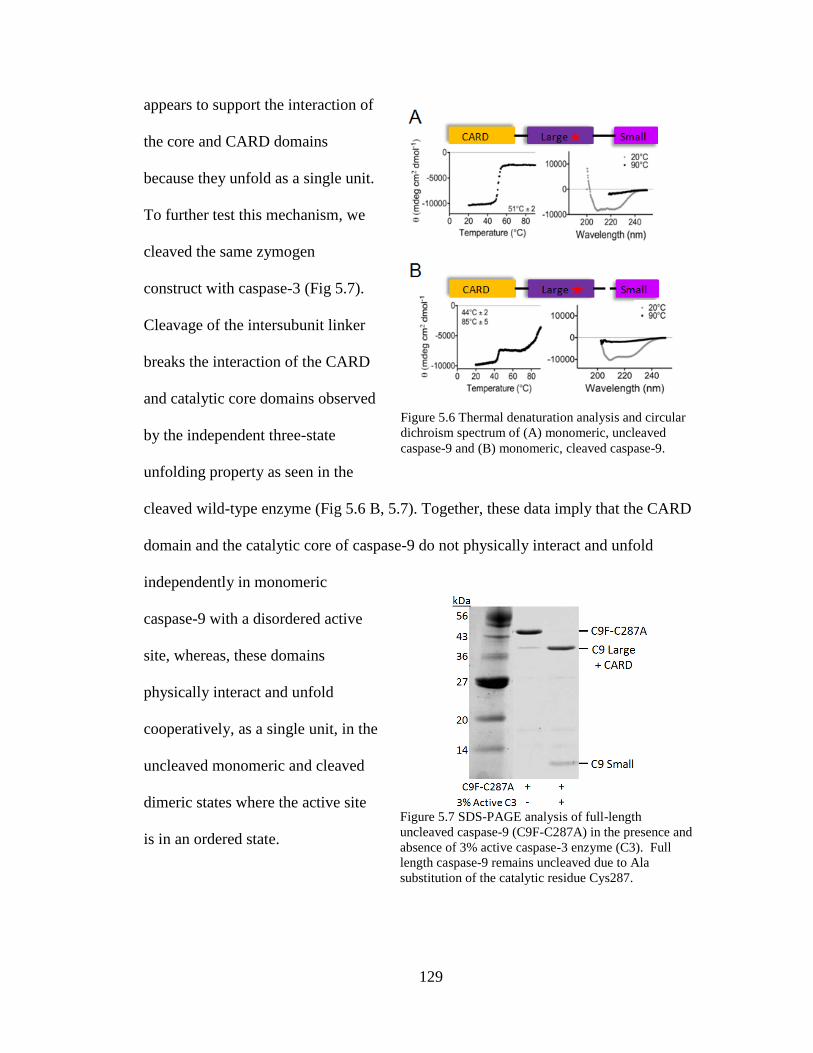
129
appears to support the interaction of
the core and CARD domains
because they unfold as a single unit.
To further test this mechanism, we
cleaved the same zymogen
construct with caspase-3 (Fig 5.7).
Cleavage of the intersubunit linker
breaks the interaction of the CARD
and catalytic core domains observed
by the independent three-state
unfolding property as seen in the
cleaved wild-type enzyme (Fig 5.6 B, 5.7). Together, these data imply that the CARD
domain and the catalytic core of caspase-9 do not physically interact and unfold
independently in monomeric
caspase-9 with a disordered active
site, whereas, these domains
physically interact and unfold
cooperatively, as a single unit, in the
uncleaved monomeric and cleaved
dimeric states where the active site
is in an ordered state.
Figure 5.6 Thermal denaturation analysis and circular
dichroism spectrum of (A) monomeric, uncleaved
caspase-9 and (B) monomeric, cleaved caspase-9.
Figure 5.7 SDS-PAGE analysis of full-length
uncleaved caspase-9 (C9F-C287A) in the presence and
absence of 3% active caspase-3 enzyme (C3). Full
length caspase-9 remains uncleaved due to Ala
substitution of the catalytic residue Cys287.
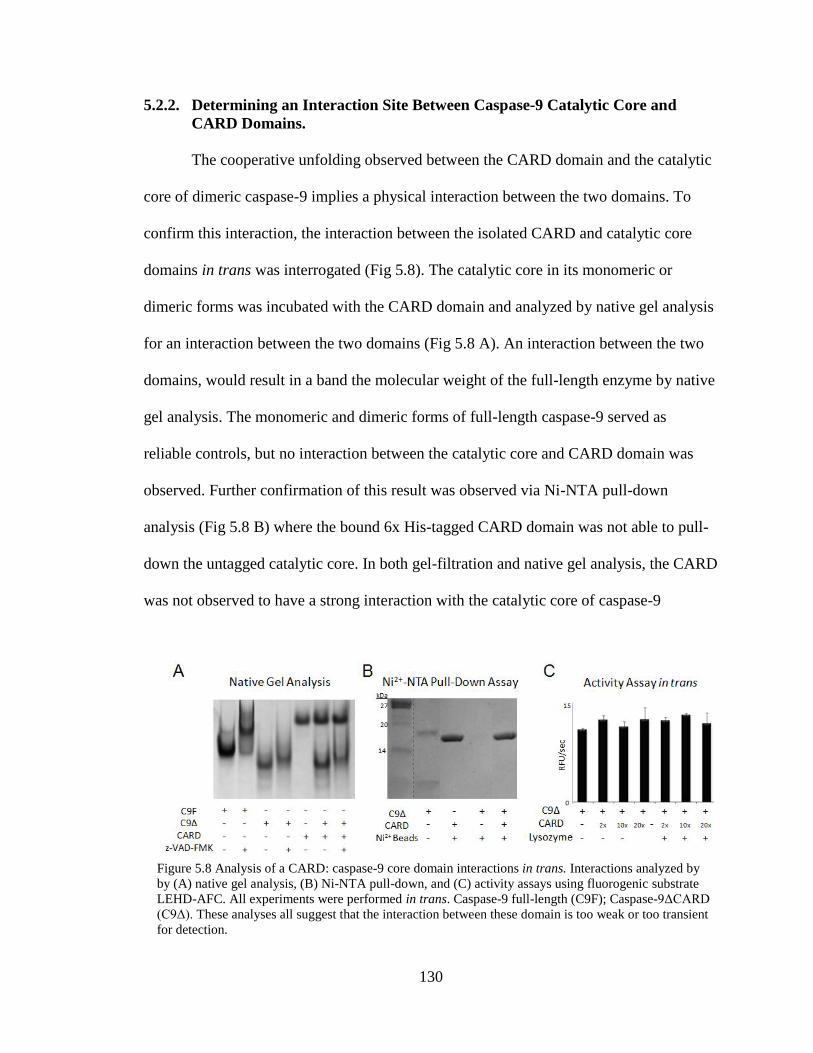
130
5.2.2. Determining an Interaction Site Between Caspase-9 Catalytic Core and
CARD Domains.
The cooperative unfolding observed between the CARD domain and the catalytic
core of dimeric caspase-9 implies a physical interaction between the two domains. To
confirm this interaction, the interaction between the isolated CARD and catalytic core
domains in trans was interrogated (Fig 5.8). The catalytic core in its monomeric or
dimeric forms was incubated with the CARD domain and analyzed by native gel analysis
for an interaction between the two domains (Fig 5.8 A). An interaction between the two
domains, would result in a band the molecular weight of the full-length enzyme by native
gel analysis. The monomeric and dimeric forms of full-length caspase-9 served as
reliable controls, but no interaction between the catalytic core and CARD domain was
observed. Further confirmation of this result was observed via Ni-NTA pull-down
analysis (Fig 5.8 B) where the bound 6x His-tagged CARD domain was not able to pull-
down the untagged catalytic core. In both gel-filtration and native gel analysis, the CARD
was not observed to have a strong interaction with the catalytic core of caspase-9
Figure 5.8 Analysis of a CARD: caspase-9 core domain interactions in trans. Interactions analyzed by
by (A) native gel analysis, (B) Ni-NTA pull-down, and (C) activity assays using fluorogenic substrate
LEHD-AFC. All experiments were performed in trans. Caspase-9 full-length (C9F); Caspase-9ΔCARD
(C9Δ). These analyses all suggest that the interaction between these domain is too weak or too transient
for detection.

131
suggesting that the interaction between the CARD and core domains is either too weak or
too transient for detection by these methods or is exquisitely sensitive to buffer
conditions.
It is possible that the conditions and experimental requirements for the native gel
and pull-down analysis could disrupt the CARD and core interaction, so CARD’s ability
to increase caspase-9 activity in trans was tested under conditions in which activity can
be monitored (Fig 5.8 C). This equilibrium-based experiment should report on the
strength of the interaction between the CARD and catalytic core domains and define the
importance of the linker that attaches the CARD and core domains together. No change
in caspase-9 ΔCARD activity was observed in the presence of excess CARD alone or in
the presence of excess lysozyme as a crowding agent. This indicates that the tether
between CARD and the catalytic core of caspase-9 is necessary for CARD’s impact on
enzymatic activity.
Since the linker between the CARD and core domains is essential to increase the
activity of caspase-9, we reasoned that
perhaps there were either specific
interactions with the tether and the
adjacent domains or a length-dependence
to the interaction. To test this, a five
amino acid Ser-Gly extension was
inserted within the linker between CARD
and the large subunit of caspase-9’s
catalytic core (Fig 5.9). This variant
Figure 5.9 Characteristics of the Ser-Gly linker
extension caspase-9 variant. Kinetic parameters,
thermal denaturation analysis, and circular
dichroism spectra of the Ser-Gly linker extension
caspase-9 variant.

132
behaves like the native full-length-form of caspase-9 in both its catalytic parameters and
thermal stability suggesting that longer potentially more flexible linker does not
negatively impact the function of caspase-9. Further investigation into the linker length
would be required to determine if a certain distance or flexibility can disrupt the
activation property of CARD or if a tether alone is sufficient to obtain the desired effect
no matter the length. Moreover, it is intriguing to consider, shortening the linker length
between the CARD and core domains to determine if restricting the flexibility of CARD
will reduce the activation effect of CARD. This will provide information on the shortest
length required for the interaction to occur.
The previous studies suggest that the interaction between the CARD and core
domains of caspase-9 are present, but are weak and potentially transient. Docking studies
between reported crystal structures of the CARD and the dimeric form of the caspase-9
catalytic core were performed using the RosettaDock server16
to predict potential
interactions between the CARD and core domain to guide mutational studies. Protein
structure files for CARD (PDB ID: 3YGS17
) and the catalytic core of caspase-9 (PDB ID:
1JXQ11
) were submitted for docking studies. The top docking models were analyzed
using two criteria i) avoiding interactions with residues involved in the caspase-9
CARD/Apaf-1 CARD complex and ii) not allowing negative interactions to occur in the
presence of Apaf-1 CARD. Two models, Model 2 and Model 10, fit these criteria (Fig
5.10). In both, the CARD is positioned behind the substrate-binding groove where it
could potentially affect active site loops L3 and L4 and there are no negative interactions
observed. Charge swap variations were made in both Model 2, which places CARD in a
helix-to-helix interaction with the catalytic core of caspase-9 (Fig 5.10 A), and Model 10,

133
which places CARD in a loop-to-helix interaction with the catalytic core of caspase-9
(Fig 5.10 B), in an attempt to disrupt the activating effect of CARD. Single variations of
Arg7 and Arg11 and in combination, both changed to glutamate, would test Model 2
while Asp23 and Arg51 charge swapped to glutamate would test Model 10. These
variations were selected to be in the CARD domain only so the observed effect would
report on CARD and not on change the catalytic activity, which is solely due to the
catalytic portion of the enzyme. One variant, Glu365Arg, was selected in the catalytic
core which had potential to disrupt either model of CARD binding to the catalytic core of
caspase-9.
We expected that mutations that disrupted the interaction between the CARD and
core domain would decrease the catalytic efficiency of the variant versions of caspase-9.
Upon analysis of the charge swap affects on the catalytic efficiency of caspase-9, a two-
fold decrease in catalytic efficiency was observed for variant R7E (Table 5.2) which
would support Model 2, the helix-to-helix binding mode of CARD to caspase-9. A
similar affect was observed by the single site variant R11E which further supports a
helix-to-helix binding model of CARD and the catalytic core of caspase-9. However the
Figure 5.10 Top RosettaDock models of the potential interaction site between CARD and the catalytic
core of caspase-9. (A) Model 2 provided a helix-to-helix mode of binding while (B) Model 10 provides
a loop-to-helix mode of binding.

134
double site variant of R7E/R11E did not show an enhancement of this affect (Table 5.2).
This could indicate that R7 and R11E are included in the activating affect of CARD;
however, the combination of both these variants is not strong enough to completely
eliminate the activation property of CARD, as the catalytic efficiency of the ΔCARD
version of the enzyme was not recapitulated. This suggests that this region of the CARD
domain is responsible for its activating effects however additional interactions are
required.
5.3. Discussion
Caspase activation mechanisms are relevant for treatment of diseases in which
apoptosis has gone awry. Caspase-9 in particular has a unique activation mechanism
including changes in its conformational and oligomeric state and its association with an
activation platform called the apoptosome. Furthermore, individual domains which are
linked to the enzyme such as CARD or which are associated with the caspase-9 activation
mechanism, such as the Apaf-1 CARD also have the ability to increase enzymatic
Table 5.2 Measured kinetic parameters of caspase-9 variants designed to disrupt the activation affect
of the CARD domain.

135
activity. The property of changing enzymatic activity by presence of additional domains
such as the one observed with CARD and caspase-9 is also relevant in other proteins such
as PAS Kinase18
, Dnmt1 DNA methyltransferase19
, and ADAMTS-420
. Therefore
studying the individual activation effects of a particular domain provides further insights
towards how caspase-9 becomes activated on the apoptosome.
Here we have investigated the effect of the caspase-9 CARD domain with respect
to its ability to increase enzymatic activity. We have determined that the oligomeric state
of both caspase-9 full-length and ΔCARD were comparable with respect to the presence
of the monomeric and dimeric caspase-9 states, indicating the presence of CARD does
not shift the oligomeric state equilibrium from monomer to dimer. Thus, the presence of
CARD is not the cause for the increased enzymatic activity observed. Furthermore, we
observed that the CARD and catalytic core domains of caspase-9 unfold independently
and do not physically interact in the monomeric state of the cleaved enzyme where the
active site is not able to form a properly ordered substrate-binding groove. However,
these domains unfold cooperatively, or as a single unit, in a dimeric state indicating a
physical interaction when the cleaved enzyme dimerizes and has a properly ordered
active site. Moreover, cooperative unfolding of CARD and the catalytic core of caspase-9
is also observed in the uncleaved form of full-length caspase-9 zymogen. Intriguingly this
CARD-core domain interaction is alleviated by cleavage of the intersubunit linker by
caspase-3. Thus, CARD appears to be interacting with any version of caspase-9
presenting a properly formed substrate-binding groove. The substrate-binding groove is
ordered in dimeric cleaved caspases (-1, -3, -6, -7 and -9) and can also be formed in
uncleaved zymogen of caspase-9, due to linkage effects which allow the intersubunit

136
linker to buttress the L3 and L4 loops in an ordered conformation (Fig 5.11). Shortening
of the intersubunit linker would show the length requirement necessary this stabilizing
effect.
This alteration in conformational state of CARD observed between the cleaved
inactive monomer and the uncleaved monomer or cleaved active dimer states is
reminiscent of the conformational acrobatics observed in caspase-6. In this case, a helical
conformation is observed in the mature enzyme which converts to a β-strand
conformation in the active state. Furthermore, the uncleaved zymogen of caspase-6
adopts a similar β-strand as observed in the active state of the enzyme. Our data suggest
caspase-9 follows a similar trend to caspase-6 by adopting one conformation in the
uncleaved, monomeric form and cleaved active dimeric state while adopting another in
the cleaved monomeric state (Fig. 5.11).Therefore, it appears that the most unique
regions within the caspase class of enzymes, the pro domain and intersubunit linker may
combine efforts to regulate the activity of caspase-9, providing evidence for the active
site stabilizing model of apoptosome activation.
Figure 5.11 Model of caspase-9 activation states in the presence of CARD.

137
Further studies on the interconversion properties of the caspase-9 catalytic core
and CARD domains, utilizing a preformed caspase-9 dimer21
, could provide insight into
the dependence of the oligomeric state on the cooperative unfolding property of the full-
length enzyme. Single charge swap mutations on the surface of the protein distal from the
active site were not strong enough or properly positioned to disrupt the activating affect
of CARD. A more extensive alanine scanning mutagenesis study or charge repulsion
analysis around the substrate-binding groove could further define the region of
interaction between the CARD and core domains mediated by the active-site region of
the enzyme.
5.4. Materials and Methods
5.4.1. Caspase-9 Expression and Purification
The caspase-9 full-length gene (human sequence) construct, encoding amino acids
1-416, in pET23b (Addgene plasmid 118294) was transformed into the BL21 (DE3) T7
Express strain of E. coli (NEB). The cultures were grown in 2xYT media supplemented
with ampicillin (100 mg/L, Sigma-Aldrich) at 37°C until they reached an optical density
at 600 nm of 1.2. The temperature was reduced to 15°C and cells were induced with 1
mM IPTG (Anatrace) to express soluble 6xHis-tagged full-length protein. Cells were
harvested after 3 hrs to obtain single-site processing at Asp315. Cell pellets stored at -
20°C were freeze-thawed and lysed in a microfluidizer (Microfluidics, Inc.) in 50 mM
sodium phosphate pH 8.0, 300 mM NaCl, and 2 mM imidazole. Lysed cells were
centrifuged at 17,000 rpm to remove cellular debris. The filtered supernatant was loaded
onto a 5-ml HiTrap Ni-affinity column (GE Healthcare). The column was washed with a
buffer containing 50 mM sodium phosphate pH 8.0, 300 mM NaCl, and 2 mM imidazole

138
until 280 nm absorbance returned to base line. The protein was eluted using a linear
imidazole gradient of 2 to100 mM over the course of 270 mL. The eluted fractions
containing protein of the expected molecular weight and composition were diluted by 10-
fold into a buffer composed of 20 mM Tris pH 8.5, 10 mM DTT to reduce the salt
concentration. This protein sample was loaded onto a 5-ml Macro-Prep High Q column
(Bio-Rad Laboratories, Inc.). The column was developed with a linear NaCl gradient and
eluted in 20 mM Tris pH 8.5, 100 mM NaCl, and 10 mM DTT buffer. The eluted protein
was stored in -80°C in the above buffer conditions. Purified caspase-9 was analyzed by
SDS-PAGE and ESI-MS to confirm mass and purity. Caspase-9 variants, C287A, R7E,
R11E, D23E, R51E E365R, R7E/R11E and the Ser-Gly linker extension, were produced
by the Quikchange mutagenesis method (Stratagene), in the full-length expression
construct, and were purified by the same method as described here for the wild-type
protein.
Caspase-9ΔCARD was expressed from a two-plasmid expression system. Two
separate constructs, one encoding the large subunit, residues 140-305, and the other
encoding the small subunit, residues 331-416, each in the pRSET plasmid, were
separately transformed into the BL21 (DE3) T7 Express strain of E. coli (NEB). The
recombinant large and small subunits were individually expressed as inclusion bodies for
subsequent reconstitution. Cultures were grown in 2xYT media supplemented with
ampicillin (100 mg/L, Sigma-Aldrich) at 37°C until they reached an optical density at
600 nm of 0.6. Protein expression was induced with 0.2 mM IPTG. Cells were harvested
after 3 hrs at 37°C. Cell pellets stored at -20°C were freeze-thawed and lysed in a
microfluidizer (Microfluidics, Inc.) in 10 mM Tris pH 8.0 and 1 mM EDTA. Inclusion

139
body pellets were washed twice in 100 mM Tris pH 8.0, 1 mM EDTA, 0.5 M NaCl, 2%
Triton, and 1M urea, twice in 100 mM Tris pH 8.0, 1 mM EDTA and finally resuspended
in 6 M guanidine hydrochloride. Caspase-9 large and small subunit proteins in guanidine
hydrochloride were combined in a ratio of 1:2, large:small subunits, and rapidly diluted
dropwise into refolding buffer composed of 100 mM Tris pH 8.0, 10% sucrose, 0.1%
CHAPS, 0.15 M NaCl, and 10 mM DTT, allowed to stir for one hour at room
temperature and then dialyzed four times against 10 mM Tris pH 8.5, 10 mM DTT, and
0.1mM EDTA buffer at 4°C. Typically 5 mL of of mixed caspase large and small
subunits was diluted into 80 mL in refolding buffer and dialyzed against 5 L of dialysis
buffer. The first and last dialysis steps were allowed to proceed for 4 hours at 4 °C while
the second dialysis proceeded overnight at 4 °C. The dialyzed protein was centrifuged for
15 minutes at 10,000 rpm to remove precipitate and then purified using a HiTrap Q HP
ion exchange column (GE Healthcare) with a linear gradient from 0 to 250 mM NaCl in
20 mM Tris buffer pH 8.5, with 10 mM DTT. Protein eluted in 20 mM Tris pH 8.5, 100
mM NaCl, and 10 mM DTT buffer was stored in -80°C. The identity of the purified
caspase-9ΔCARD was analyzed by SDS-PAGE and ESI-MS to confirm mass and purity.
5.4.2. Oligomeric-State Determination
Caspase-9 wild-type, full-length and ΔCARD variant protein samples in 20 mM
Tris pH 8.5, 110 mM NaCl, and 5 mM DTT were incubated alone (monomer) or with
covalent inhibitor z-VAD-FMK (carbobenzoxy-Val-Ala-Asp-fluoromethylketone, Enzo
Lifesciences) (dimer) for 2 hours at room temperature. The oligomeric state of the
caspase-9 samples were determined via gel filtration. 100 μL of 0.5 mg/ml protein sample
was loaded onto a Superdex 200 10/300 GL (GE Healthcare) gel-filtration column. Apo

140
and z-VAD-FMK-incubated protein samples were eluted with 20 mM Tris pH 8.0, 100
mM NaCl, and 2 mM DTT. Eluted peaks were analyzed by SDS-PAGE to identify the
eluted protein. Four different molecular weight standards from the gel-filtration
calibration kit LMW (GE Healthcare) were run in the same conditions and a standard plot
was generated to determine whether the peaks were caspase-9 monomer or dimer.
5.4.3. CARD Expression and Purification
The CARD only construct (amino acids 1-138) in pET23b was made by
QuikChange mutagenesis (Stratagene) using the oligo-nucleotide primer 5`-CCCAGA-
CCAGTGGACATTGGTTCTGGAGGATTCGGTGATCACCACCACCACCACCAC
TAAGTCGGTGCTCTTGAGAGTTTGAGGGGAAATGCAGATTTGG-3`and its
reverse compliment on the caspase-9 full-length gene (Addgene plasmid 11829). These
oligo-nucleotide primers insert a 6xHis-tag and a stop codon after the last amino acid of
the CARD domain (D138), leaving the remaining portion of the caspase-9 gene in the
plasmid. The construct was transformed into BL21 (DE3) T7 Express strain of E. coli.
The cultures were grown in 2xYT media supplemented with ampicillin (100 mg/L,
Sigma-Aldrich) at 37°C until they reached an optical density at 600 nm of 0.6. The
temperature was reduced to 15°C and cells were induced with 1 mM IPTG (Anatrace) to
express soluble 6xHis-tagged full-length protein. Cells were harvested after 18 hrs. Cell
pellets stored at -20°C were freeze-thawed and lysed in a microfluidizer (Microfluidics,
Inc.) in 50 mM sodium phosphate pH 8.0, 300 mM NaCl, and 2 mM imidazole. Lysed
cells were centrifuged at 17,000 rpm to remove cellular debris. The filtered supernatant
was loaded onto a 5-ml HiTrap Ni-affinity column (GE Healthcare). The column was
washed with a buffer containing 50 mM sodium phosphate pH 8.0, 300 mM NaCl, 2 mM

141
imidazole until 280 nm absorbance returned to base line. The column was washed with
50 mM phosphate pH 8.0, 300 mM NaCl, 50 mM imidazole and the protein was eluted
with 50 mM phosphate pH 8.0, 300 mM NaCl, 250 mM imidazole. The eluted fraction
was diluted by 10-fold into a buffer containing 20 mM Tris pH 8.0 and 2 mM DTT to
reduce the salt concentration. This protein sample was loaded onto a 5 ml Macro-Prep
High Q column (Bio-Rad Laboratories, Inc.). The column was developed with a linear
NaCl gradient. Protein eluted in 20 mM Tris pH 8.0, 2 mM DTT, and 130 mM NaCl.
Eluted protein was analyzed by SDS-PAGE to assess purity and stored in -80°C.
5.4.4. Thermal Stability and Secondary Structure Analysis by Circular Dichroism
Caspase-9 variants and CARD proteins were buffer exchanged via dialysis against
100 mM sodium phosphate pH 7.0, 110 mM NaCl, and 5 mM TCEP and diluted to 7 μM.
The samples were split in half and incubated in the presence or absence of four molar
equivalents of active site ligand VAD-FMK for 3 hours at room temperature. To ensure
complete binding of the active site ligand to the protein, remaining enzymatic activity
was assayed using 300 μM substrate, LEHD-AFC (N-acetyl-Leu-Glu-His-Asp-7-amino-
4-fluorocoumarin), (Enzo Lifesciences). Once full inhibition was achieved, samples were
buffer-exchanged six times with 100 mM phosphate buffer pH 7.0, 100 mM NaCl and 5
mM TCEP using an Amicon Ultracell 3K concentrator (Millipore) to remove unbound
inhibitor. For cleavage of the unprocessed caspase-9 C287A variant, the 7μM protein
sample was incubated with 3% active caspase-3 protein for two hours at room
temperature. Full processing of caspase-9 C287A by caspase-3 was determined by SDS-
PAGE analysis. Thermal denaturation of caspase-9 variants and CARD was monitored by
loss of CD signal at 222 nm over a range of 20–90 °C. The circular dichroism spectra

142
were monitored from 250-190 nm. Both were performed on a J-720 CD spectrometer
(Jasco) with a Peltier controller. Data were collected four separate times on different days
from different batches of purified proteins. Curves were fit with Origin Software
(OriginLab) using sigmoid fit to determine the melting temperature.
5.4.5. Caspase-3 Expression and Purification
Caspase-3 full-length gene (human sequence) in pET23b (Addgene plasmid
1182122
) was transformed into BL21 (DE3) T7 Express strain of E. coli and protein
expression was induced with 1 mM IPTG at 30°C for 3 23
. The protein was purified as
described previously for caspase-3 24
. The eluted protein was stored in -80°C in the buffer
in which they eluted. The identity of purified caspase-3 was assessed by SDS-PAGE and
ESI-MS to confirm mass and purity.
5.4.6. Native Gel Analysis and Ni-NTA Pull Down Assay to Determine in trans
Interactions
For native gel analysis to diagnose an interaction between caspase-9ΔCARD and
caspase-9 CARD in trans, full-length caspase-9, caspase-9 ΔCARD and the CARD
domain only were dialyzed twice against 100 mM phosphate pH 7.0 and 2 mM DTT for
90 minutes to rid of excess salt. Samples were incubated either alone or combined with
CARD to achieve a 1:1 ratio of caspase-9ΔCARD plus CARD. Each protein sample was
diluted to a final concentration 10 μM in the dialysis buffer. To induce dimerization
samples were incubated with 5-fold excess z-VAD-FMK. All samples were allowed to
incubate at room temperature for one hour. All samples were mixed with glycerol loading
dye and fractionated on a 10% Tris/Glycine pH 8.3 polyacrylamide gel. The oligomeric
state of the mixed caspase-9ΔCARD and CARD samples were identified by comparison

143
to the Apo (monomer) and z-VAD-FMK (dimer) of both the caspase-9 full-length and
caspase-9ΔCARD protein in addition to the CARD only sample.
For Ni-NTA pull-down analysis of caspase-9ΔCARD plus CARD in trans,
samples were diluted to 10 μM in 100 mM phosphate pH 8.0, 5 mM TCEP and 100 mM
NaCl, to avoid nonspecific interactions of the caspase-9 enzyme with the Ni-NTA beads.
Samples were incubated either alone or combined with CARD to achieve a 1:1 ratio of
caspase-9ΔCARD (no tag) plus CARD (6x His-tag). Each protein sample was diluted to a
final concentration of 10 μM in the dialysis buffer. To induce dimerization samples were
incubated with 5-fold excess z-VAD-FMK for 15 minutes. 200 μL of protein sample was
added to a tube containing 50 μL of Ni-NTA beads (Qiagen) that were washed three
times in water and twice in buffer. Ni-NTA plus caspase-9ΔCARD and CARD samples
were incubated for 3 hours at 4°C while rocking. Samples were centrifuged for 5 minutes
at 13,000 rpm to pellet the Ni-NTA beads. Supernatant was aspirated and the beads were
washed five times with 100 mM phosphate pH 8.0, 5 mM TCEP and 100 mM NaCl to
remove any unbound or weakly bound protein. Protein elution was then carried out by
incubating the Ni-NTA beads with phosphate buffer containing 300 mM imidazole for 10
minutes at room temperature. Samples were centrifuged for 5 minutes at 13,000 rpm to
pellet the Ni-NTA beads and the supernatant was used for SDS-PAGE analysis. The Ni-
NTA beads were also tested to ensure there were no remaining proteins bound to the
resin.
5.4.7. Activity Assays
For kinetic measurements of caspase activity, 700 nM caspase-9 full-length
protein was diluted in 100 mM MES pH 6.5, 10% PEG 8,000, and 5 mM DTT. Each

144
sample was subjected to a substrate titration, performed in the range of 0-300 μM
fluorogenic substrate, LEHD-AFC, (Ex365/Em495) which was added to initiate the
reaction. Assays were performed in duplicates at 37°C in 100 μL volumes in 96-well
microplate format using a Molecular Devices Spectramax M5 spectrophotometer. Initial
velocities versus substrate concentration were fit to a rectangular hyperbola using
GraphPad Prism (Graphpad Software) to determine kinetic parameters Km and kcat.
Enzyme concentrations were determined by active site titration with quantitative,
inhibitor z-VAD-FMK. Active site titrations were incubated over a period of 3 h in 100
mM MES pH 6.5, 10% PEG 8,000, and 5 mM DTT. Optimal labeling was observed
when protein was added to VAD-FMK solvated in dimethylsulfoxide in 96-well V-
bottom plates, sealed with tape, and incubated at room temperature in a final volume of
200 μL. 90 μL aliquots were transferred to black-well plates in duplicate and assayed
with 300 μM substrate. The protein concentration was determined to be the lowest
concentration at which full inhibition was observed.
To test the ability of CARD to activate caspase-9ΔCARD in trans, 700 nM
freshly purified caspase-9ΔCARD was incubated in the presence and absence of 2-, 10-,
or 20-fold excess of CARD protein for 15 minutes. Half of each protein sample was
treated with excess lysozyme as a crowding agent. Samples were assayed for activity
over the course of 10 minutes in a caspase-9 activity assay buffer containing 100 mM
MES pH 6.5, 10% PEG 8,000 and 10 mM DTT. 300 μM of the fluorogenic substrate,
LEHD-AFC (Ex365/Em495), was added to initiate the reaction. Assays were performed
in duplicate at 37°C in 100 μL volumes in 96-well microplate format using a Molecular
Devices Spectramax M5 spectrophotometer.

145
5.5. References
1. Zhai, D. et al. Vaccinia virus protein F1L is a caspase-9 inhibitor. J Biol Chem
285, 5569-5580, (2010).
2. Oztas, P. et al. Caspase-9 expression is increased in endothelial cells of active
Behcet's disease patients. Int J Dermatol 46, 172-176, (2007).
3. Sekimura, A. et al. Expression of Smac/DIABLO is a novel prognostic marker in
lung cancer. Oncol Rep 11, 797-802, (2004).
4. Stennicke, H. R. et al. Caspase-9 can be activated without proteolytic processing.
J Biol Chem 274, 8359-8362, (1999).
5. Rodriguez, J. & Lazebnik, Y. Caspase-9 and APAF-1 form an active holoenzyme.
Genes Dev 13, 3179-3184, (1999).
6. Zou, H., Li, Y., Liu, X. & Wang, X. An APAF-1.cytochrome c multimeric
complex is a functional apoptosome that activates procaspase-9. J Biol Chem 274,
11549-11556, (1999).
7. Pop, C., Timmer, J., Sperandio, S. & Salvesen, G. S. The apoptosome activates
caspase-9 by dimerization. Mol Cell 22, 269-275, (2006).
8. Shiozaki, E. N., Chai, J. & Shi, Y. Oligomerization and activation of caspase-9,
induced by Apaf-1 CARD. Proc Natl Acad Sci U S A 99, 4197-4202, (2002).
9. Acehan, D. et al. Three-dimensional structure of the apoptosome: implications for
assembly, procaspase-9 binding, and activation. Mol Cell 9, 423-432, (2002).
10. Salvesen, G. S. & Dixit, V. M. Caspase activation: the induced-proximity model.
Proc Natl Acad Sci U S A 96, 10964-10967, (1999).
11. Renatus, M., Stennicke, H. R., Scott, F. L., Liddington, R. C. & Salvesen, G. S.
Dimer formation drives the activation of the cell death protease caspase 9. Proc
Natl Acad Sci U S A 98, 14250-14255, (2001).
12. Shi, Y. Caspase activation: revisiting the induced proximity model. Cell 117, 855-
858, (2004).
13. Manns, J. et al. Triggering of a novel intrinsic apoptosis pathway by the kinase
inhibitor staurosporine: activation of caspase-9 in the absence of Apaf-1. FASEB J
25, 3250-3261, (2011).
14. Vaidya, S. & Hardy, J. A. Caspase-6 latent state stability relies on helical
propensity. Biochemistry 50, 3282-3287, (2011).

146
15. Witkowski, W. A. & Hardy, J. A. L2' loop is critical for caspase-7 active site
formation. Protein Sci 18, 1459-1468, (2009).
16. Lyskov, S. & Gray, J. J. The RosettaDock server for local protein-protein
docking. Nucleic Acids Res 36, W233-238, (2008).
17. Qin, H. et al. Structural basis of procaspase-9 recruitment by the apoptotic
protease-activating factor 1. Nature 399, 549-557, (1999).
18. Rutter, J., Michnoff, C. H., Harper, S. M., Gardner, K. H. & McKnight, S. L. PAS
kinase: an evolutionarily conserved PAS domain-regulated serine/threonine
kinase. Proc Natl Acad Sci U S A 98, 8991-8996, (2001).
19. Fatemi, M., Hermann, A., Pradhan, S. & Jeltsch, A. The activity of the murine
DNA methyltransferase Dnmt1 is controlled by interaction of the catalytic domain
with the N-terminal part of the enzyme leading to an allosteric activation of the
enzyme after binding to methylated DNA. J Mol Biol 309, 1189-1199, (2001).
20. Kashiwagi, M. et al. Altered proteolytic activities of ADAMTS-4 expressed by C-
terminal processing. J Biol Chem 279, 10109-10119, (2004).
21. Chao, Y. et al. Engineering a dimeric caspase-9: a re-evaluation of the induced
proximity model for caspase activation. PLoS Biol 3, e183, (2005).
22. Zhou, Q. et al. Target protease specificity of the viral serpin CrmA. Analysis of
five caspases. J Biol Chem 272, 7797-7800, (1997).
23. Stennicke, H. R. & Salvesen, G. S. Caspases: preparation and characterization.
Methods 17, 313-319, (1999).
24. Stennicke, H. R. & Salvesen, G. S. Biochemical characteristics of caspases-3, -6, -
7, and -8. J Biol Chem 272, 25719-25723, (1997).

147
CHAPTER VI
A SURFACE-WIDE VIEW OF THE
NATIVE REGULATORY MECHANSIMS OF CASPASE-9
6.1. Regulation of Caspase-9 Occurs at a Variety of Locations on its Surface
Control of cellular processes plays an essential role in maintaining the
homeostasis of a cell. This control is maintained by a complex network composed of
specific regulatory mechanisms for each individual protein within the cell. These
checkpoints monitor a variety of different protein characteristics ranging from their initial
synthesis to their final activation states and even their degradation pathways. In order to
fully understand how to externally regulate a particular protein, a fundamental
understanding of the native regulatory pathways and their interactions within those native
pathways is required.
Protein regulatory mechanisms can be categorized into a variety of different
classes. Regulation can occur by altering the genetic expression levels and timing of
activation of a particular protein or enzyme. This allows the cell to control not only when
a protein will be produced but the quantity of protein production, resulting in an
increased versatility during the lifespan of the cell or organism. Post protein production,
sequestration or compartmentalization of the protein from its functional partner(s) is
utilized to control a protein function. In the case of proteases, processing of the zymogen
form of an enzyme is frequently required to initiate its activation pathway. Alternate
means of regulation also include post-translational modifications or even allosteric
regulation. Through the utilization of all or a variety of these regulatory checkpoints, a
tight control of a protein’s function can be obtained. Ideally, each point within these

148
native regulatory pathways can be targeted to create tools to further study enzymatic
function or for therapies in cases where there has been disruption of the regulation of a
particular protein.
This dissertation focused on discovering new regulatory pathways for caspase-9
as well as exploiting a known regulatory pathway of the enzyme due to its pivotal role in
apoptosis and disease. Our work aimed to mimic the native inhibitory complex of
caspase-9 with XIAP-BIR3 by stabilized peptides to create a tool to control enzymatic
activity. Instead, this work underscored the importance of bi-functional binding sites.
Critical interactions at the protein-protein interface as well as at a distal exosite region
provide two distinct avenues for protein control. In the case of caspase-9 and XIAP-
BIR3, both sites are necessary for potent inhibition. In other protein:inhibitor complexes
this bi-functionality determines specificity or can individually be used as independent
regulatory sites. The viral inhibitor, human adenovirus type 5, of human granzyme B, a
protein that is essential for targeting cell death of natural killer cells and cytotoxic CD8+
T-cells, utilizes exosite interactions as a specificity factor1. Common homologues of
human granzyme B are capable of recognizing human adenovirus type 5 in the substrate
binding pocket, however, the exosite was critical for specific inhibition of the human
enzyme. A similar exosite requirement has also been observed with plasminogen
activator inhibitor-1 which prevents conversion of plasminogen into plasmin through its
interactions with both tissue-type plasminogen activator and urokinase-type
plasminogen2. Furthermore, when designing blockades to prevent heparin cofactor II
binding to α-thrombin, targeting of the exosite position was an essential requirement for
specificity and affinity3,4
. These examples not only suggest that the presence of an exosite

149
is a common feature within protein complexes; they also provide an alternate avenue for
designing regulatory molecules.
In addition to underscoring the importance of targeting the multiple regulatory
regions within a native protein complex, our work has also provided insights towards
alternate regulatory sites within caspase-9. When determining the regulatory mechanism
of zinc-mediated inhibition of caspase-9, not only did we discover that one zinc ion binds
to the catalytic region of the protein, an alternate zinc binding site also exists. The main
regulatory site, at the active site, sequesters the active site residues which would prevent
catalysis of the peptide bond of a substrate. Besides controlling function, a possible role
for active-site zinc binding is to protect the active-site ligands from oxidation via a
similar mechanism to that observed for protein tyrosine phosphatase5. The other binding
site identified for zinc is distal from the active site and lies within a conserved Cys-His
region of the enzyme. Although the functional details of this site remain unidentified, this
region has been previously shown to be conformationally sensitive in caspase-66.
Therefore, zinc binding to this site could potentially have a site-specific impact on
caspase-6 activity via an allosteric mechanism. Furthermore, the distal zinc binding site
could potentially be utilized as a stabilization factor for a particular conformation in
caspase-6 or other caspases, similar to that observed in voltage-gated potassium channel7.
The non-native control of protein activity by a metal also occurs in a variety of enzymes
which natively do not utilize zinc. Glycerol 3-phosphate dehydrogenase8, tyrosine
phosphatase9, aldehyde dehydrogenase
10, and the caspases
11, are all capable of binding
zinc which results in a loss of enzymatic function. This suggests that a wide variety of
non-metalloproteins have evolved to be influenced by metals as an underlying mode of

150
regulation and that these sites also exist in locations distal from the active site of these
enzymes.
Furthermore, our work in elucidating the mechanism by which CARD activates
the enzymatic core of caspase-9 revealed yet another alternate regulatory site on the
surface of caspase-9. The mechanism reveals that CARD not only mediates interactions
with Apaf-1 of the apoptosome, it potentially facilitates proper organization of the active-
site loop bundle and strengthens these interactions resulting in a fully active enzyme. An
alternate role for CARD has also been observed where free caspase-9 CARD has the
ability to cause activation of NF-κB12
. Furthermore, identification of an interaction
between the CARD domain of a caspase-9 and the substrate-binding groove provides a
unique regulatory mechanism instilled on this cell-death protease that could be targeted
for further study. Although this interaction is unique to caspase-9 due to the secondary
structural elements involved, globally similar observations have been made with the
prodomains of the executioner caspases -3, -6, and -7 and their individual catalytic cores.
The prodomain of caspase-3 has been shown to play a role in determining the proper
active-site conformation13
, in which the presence of the prodomain enhances the
efficiency of forming the native active-site conformation as if it was an intermolecular
chaperone. In caspase-6, a similar structural conformation is observed in the zymogen
and active states of the enzyme6 where an alternate form is adopted for the cleaved
mature form as seen in caspase-9. Unlike caspase-9, caspase-7 has an observed increase
in catalytic activity upon removal of the prodomain14
, suggesting that the prodomain
inhibits the function of the enzyme and possibly the proper formation of the active site
loop bundle. Nonetheless, in all cases, the prodomain affects the catalytic function of the

151
caspase class of enzymes. It appears that the prodomains play more active role in
regulating caspase activity than previously thought and affect function through an
allosteric binding event. Therefore, investing time into understanding this mode of
regulation and the location of these particular interactions sites would provide valuable
information not only to the field of caspase function but also to the field of allosteric
regulation.
By gaining an
understanding of the additional
modes of regulation imposed on
caspase-9 function, it is clear that
this enzyme uses the majority of
its surface for regulatory control
(Fig 6.1). This characteristic can
also be found for a variety of other
proteins15-17
which exemplifies the
dynamic nature of the caspase
class of enzymes and opens up the possibility of the caspases involvement in alternate
roles within the cell. Furthermore, the discovery of these new regulatory sites native to
the enzyme and possibly specific to caspase-9, increases the potential for specific drug
targeting of an individual caspases and even makes the search for additional sites within
the caspase class of enzymes an alluring concept.
Determination of these novel sites would be critical to distinguish one caspase
from the next in a regulatory or drug targeting standpoint. Although their general
Figure 6.1 Structure of caspase-9 with mapped regulatory
surfaces.

152
structural properties may be similar, it is suggested by Ranganathan and coworkers, that a
statistical coupling between amino acids exists within a protein’s architecture and this
coupling has the potential to impose functional constraints on the enzyme18-22
. These
networks provide a means of communication from a surface site on the protein to its
functional active site and provide the underlying scheme that drives the co-evolution of
new “hot spots” for protein regulation23,24
. Investigation into these networks allowed for
design of non-native allosteric control of dihydrofolate reductase24
and the PDZ family of
proteins23
. Therefore it is plausible to think that all proteins have these sensitive
regulatory sites over the entire surface and at some point these sites will be exploited by
nature to increase the ultimate fitness of the cell.
A study into the co-evolution of these “hot spot” networks via and computational
simulations of correlated motions within the family of caspases could help distinguish
unique patterns within the caspase architecture. An alternate yet parallel investigation
utilizing NMR dynamics analysis or even thermal fluctuation analysis of crystal
structures could provide insight into the dynamic properties of these enzymes and provide
hints into the location of these networks as observed in the case of PDZ domains 20,25-27
.
Hints towards these mechanisms within the caspase class of enzymes may already exist
in the literature. Because thermodynamic values relating to protein stability under
various conditions has been linked to functional cooperativity (reviewed in28
), the
presence of these networks in caspases seems likely. These studies would provide the
information necessary for unique regulation of the caspases via a diverse set of novel
allosteric sites.

153
A potential venue to observe such networks may already exist by understanding
the phosphorylation mechanisms of the caspase class of enzymes. Phosphorylation of a
protein has been shown to increases the possibilities for protein-protein interactions as
well as conformational regulation in a phosphorylation pathway. Therefore, it is
intriguing to speculate how the co-evolving networks might change as a result of the
phosphorylation patterns imposed on the enzyme during cellular signaling. Within the
caspase class of enzymes, the function of some phosphorylation sites have been
characterized to disrupt active site formation such as phosphorylation of S257 in caspase-
6 (unpublished data). However, the molecular details of most phosphorylation sites are
unknown and determining those mechanisms could potentially link or even expose
alternate “hot spot” networks to diversify the caspase function at a selected point in
cellular signaling.
In summary, the emergence of allosteric mechanisms that occurs during the
evolutionary process has the ability to diversify members of a single protein family.
Therefore, these mechanisms provide a means of specifically controlling a single protein
from that of an otherwise undistinguishable class of enzymes. For this reason it is
important to further explore native regulatory mechanisms of not only the caspases but
other proteins as well to aid in understanding the protein function as well as be a guide
for future design.
6.2. References
1. Andrade, F., Casciola-Rosen, L. A. & Rosen, A. A novel domain in adenovirus
L4-100K is required for stable binding and efficient inhibition of human
granzyme B: possible interaction with a species-specific exosite. Mol Cell Biol
23, 6315-6326, (2003).

154
2. Ibarra, C. A., Blouse, G. E., Christian, T. D. & Shore, J. D. The contribution of
the exosite residues of plasminogen activator inhibitor-1 to proteinase inhibition.
J Biol Chem 279, 3643-3650, (2004).
3. Rogers, S. J., Pratt, C. W., Whinna, H. C. & Church, F. C. Role of thrombin
exosites in inhibition by heparin cofactor II. J Biol Chem 267, 3613-3617, (1992).
4. Becker, D. L., Fredenburgh, J. C., Stafford, A. R. & Weitz, J. I. Exosites 1 and 2
are essential for protection of fibrin-bound thrombin from heparin-catalyzed
inhibition by antithrombin and heparin cofactor II. J Biol Chem 274, 6226-6233,
(1999).
5. Haase, H. & Maret, W. Protein tyrosine phosphatases as targets of the combined
insulinomimetic effects of zinc and oxidants. Biometals 18, 333-338, (2005).
6. Vaidya, S., Velazquez-Delgado, E. M., Abbruzzese, G. & Hardy, J. A. Substrate-
induced conformational changes occur in all cleaved forms of caspase-6. J Mol
Biol 406, 75-91, (2011).
7. Bixby, K. A. et al. Zn2+-binding and molecular determinants of tetramerization in
voltage-gated K+ channels. Nat Struct Biol 6, 38-43, (1999).
8. Maret, W., Yetman, C. A. & Jiang, L. Enzyme regulation by reversible zinc
inhibition: glycerol phosphate dehydrogenase as an example. Chem Biol Interact
130-132, 891-901, (2001).
9. Zander, N. F. et al. Purification and characterization of a human recombinant T-
cell protein-tyrosine-phosphatase from a baculovirus expression system.
Biochemistry 30, 6964-6970, (1991).
10. Maret, W., Jacob, C., Vallee, B. L. & Fischer, E. H. Inhibitory sites in enzymes:
zinc removal and reactivation by thionein. Proc Natl Acad Sci U S A 96, 1936-
1940, (1999).
11. Stennicke, H. R. & Salvesen, G. S. Biochemical characteristics of caspases-3, -6, -
7, and -8. J Biol Chem 272, 25719-25723, (1997).

155
12. Stephanou, A., Scarabelli, T. M., Knight, R. A. & Latchman, D. S. Antiapoptotic
activity of the free caspase recruitment domain of procaspase-9: a novel
endogenous rescue pathway in cell death. J Biol Chem 277, 13693-13699, (2002).
13. Feeney, B. & Clark, A. C. Reassembly of active caspase-3 is facilitated by the
propeptide. J Biol Chem 280, 39772-39785, (2005).
14. Denault, J. B. & Salvesen, G. S. Human caspase-7 activity and regulation by its
N-terminal peptide. J Biol Chem 278, 34042-34050, (2003).
15. Gopal, V. K., Francis, S. H. & Corbin, J. D. Allosteric sites of phosphodiesterase-
5 (PDE5). A potential role in negative feedback regulation of cGMP signaling in
corpus cavernosum. Eur J Biochem 268, 3304-3312, (2001).
16. Birdsall, N. J., Lazareno, S., Popham, A. & Saldanha, J. Multiple allosteric sites
on muscarinic receptors. Life Sci 68, 2517-2524, (2001).
17. Hardy, J. A., Lam, J., Nguyen, J. T., O'Brien, T. & Wells, J. A. Discovery of an
allosteric site in the caspases. Proc Natl Acad Sci U S A 101, 12461-12466,
(2004).
18. Ferguson, A. D. et al. Signal transduction pathway of TonB-dependent
transporters. Proc Natl Acad Sci U S A 104, 513-518, (2007).
19. Hatley, M. E., Lockless, S. W., Gibson, S. K., Gilman, A. G. & Ranganathan, R.
Allosteric determinants in guanine nucleotide-binding proteins. Proc Natl Acad
Sci U S A 100, 14445-14450, (2003).
20. Lockless, S. W. & Ranganathan, R. Evolutionarily conserved pathways of
energetic connectivity in protein families. Science 286, 295-299, (1999).
21. Shulman, A. I., Larson, C., Mangelsdorf, D. J. & Ranganathan, R. Structural
determinants of allosteric ligand activation in RXR heterodimers. Cell 116, 417-
429, (2004).
22. Suel, G. M., Lockless, S. W., Wall, M. A. & Ranganathan, R. Evolutionarily
conserved networks of residues mediate allosteric communication in proteins. Nat
Struct Biol 10, 59-69, (2003).

156
23. Reynolds, K. A., McLaughlin, R. N. & Ranganathan, R. Hot spots for allosteric
regulation on protein surfaces. Cell 147, 1564-1575, (2011).
24. Lee, J. et al. Surface sites for engineering allosteric control in proteins. Science
322, 438-442, (2008).
25. Ota, N. & Agard, D. A. Intramolecular signaling pathways revealed by modeling
anisotropic thermal diffusion. J Mol Biol 351, 345-354, (2005).
26. Fuentes, E. J., Der, C. J. & Lee, A. L. Ligand-dependent dynamics and
intramolecular signaling in a PDZ domain. J Mol Biol 335, 1105-1115, (2004).
27. Kong, Y. & Karplus, M. Signaling pathways of PDZ2 domain: a molecular
dynamics interaction correlation analysis. Proteins 74, 145-154, (2009).
28. Luque, I., Leavitt, S. A. & Freire, E. The linkage between protein folding and
functional cooperativity: two sides of the same coin? Annu Rev Biophys Biomol
Struct 31, 235-256, (2002).

157
APPENDIX A
SMALL MOLECULE ACTIVATION OF CASPASE-9
A.1. Introduction
A major factor in the activation of caspase-9 is regulation of its oligomeric state
where the enzyme transitions from an inactive monomer to an active dimer1. Disruption
of this mechanism leads to a down regulation of apoptosis, which is a characteristic of a
variety of many cancer cells2-6
. For this reason, molecules to control caspase activation
are highly desired. To date, the molecule PAC-1 has been developed to activate the
caspases through relieving their zinc mediated inhibition7,8
, however, a molecule which
directly acts on the initiator of the intrinsic pathway of the apoptotic cascade (caspase-9)
has not been developed. An external trigger of caspase-9 dimerization would be a useful
tool for understanding the detailed mechanism of caspase-9 activation and provide a
proof of principal for small molecule development of caspase-9 activators.
The small fluorescent molecule, 4`, 5`-bis(1, 3, 2-dithioarsolan-2-yl)fluorescein-
(1, 2-ethanedithiol), otherwise known as Fluorescein Arsenical Hairpin or FlAsH9
recognizes a genetically encoded peptide tag making it
a useful tool for studying protein localization, folding,
stability and conformational changes10-14
and thus a
tool for controlling caspase-9 dimerization. This
fluorescein based molecule contains As(III)
substituents on the 4`- and 5`- positions (Fig A.1)
which form a covalent bond with a reduced sulfhydryl
found on a cysteine residue. The As(III) substituents are protected by 1,2-dithiol such as
Figure A.1 Molecular structure
of FlAsH-EDT2.

158
ethandithiol (EDT) to prevent non-specific binding to cysteine pairs in the protein, in
addition to limiting toxicity and background fluorescence. The FlAsH recognition
sequence includes two cysteine pairs separated by a two amino acid spacer (C-C-X-X-C-
C) where X could be any amino acid, although tightest binding is observed using Pro-Gly
in those positions15
. FlAsH is non-fluorescent in its unbound state. Upon binding to the
FlAsH recognition motif the small molecule becomes fluorescent with an excitation
maximum of 508 nm and emission of 528 nm.
We aimed to engineer a FlAsH binding caspase-9 variant in which the FlAsH
specific binding motif was incorporated across the dimer interface, similar to the split
motif designed as a structural sensor of CRABP I10
. FlAsH binding at this engineered
site would lock the caspase-9 in the dimeric active form of the enzyme. Furthermore, the
FlAsH fluorescence will be used to confirm binding and the global location of this
interaction within the enzyme. Ultimately, this study would provide a proof of concept
that a small molecule can facilitate activation of caspase-9 through inducing dimerization
of the enzyme and thus activation.
A.2. Results
A.2.1. Design of a FlAsH-Activatable Caspase-9
Harnessing the natural activation mechanism of caspase-9 will allow development
of tools to further study this form of caspase regulation and therapeutics for diseases such
as testicular cancer3 and infection of the vaccinia virus
16 in which the caspase-9
activation pathway is targeted. Therefore, our goal is to lock caspase-9 into a dimeric
state via the small fluorescent molecule FlAsH. This would shift the enzymes equilibrium

159
from inactive monomer to active dimer which would result in an overall increase in
enzymatic activity.
Suitable sites for positioning the FlAsH binding motif across the dimer interface
were selected based on four criteria; (1) the site would be able to accommodate not only
the aresnic binding end of the molecule but also the bulky fluorescent portion of the
probe, (2) the location of binding must be able to facilitate proper FlAsH binding
geometry, (3) the designs must consider positions that would not disrupt the active site
loop conformations and (4) if available, place half of the FlAsH motif on a homogeneous
caspase-9 monomer where upon dimerization, the full binding motif could be formed.
Based on these outlined criteria, inspection of the wild-type caspase-9 structure
(PDB ID: 1JXQ) resulted in two FlAsH-binding models (Fig A.2 A). The first model,
C9-X, would place FlAsH on the periphery of the protein (Fig A.2 B). This site would
link the C-terminal end of the α5 helix in caspase-9 to its symmetry partner on the other
half of the dimer. This model is called C9-helix or C9-X. Amino acid variations at
Figure A.2 Models of the caspase-9 FlAsH binding variants. (A) An overall view of the caspase-9
dimer (blue/cyan) representing FlAsH binding sites for C9-X and C9-β within the dimer interface.
(B) C9-X model of FlAsH binding to the α5 helix of caspase-9 at positions Q381C and S382C across
the dimer interface. (C) C9-β model of FlAsH binding to the β6 strands of caspase-9 at positions
C403 and N405C across the dimer interface.

160
Gln381 and Ser382 to cysteine would facilitate the formation of the FlAsH binding motif
upon dimerization of caspase-9 and create a covalent cross-link between the small
subunit of one monomer to the small-subunit of the opposite monomer. These surface
exposed residues would allow free movement of the FlAsH molecule while the
positioning of this site is distant from the active site loop bundle which allows the C9-X
model of FlAsH binding to fit all the desired criteria.
The second designed FlAsH binding motif focuses on the allosteric cavity
observed within the dimer interface of the caspases17
(Fig A.2 C). The base of this site is
lined with two β6 strands which are critical for the dimerization of caspase-918
. This
strand contains a native cysteine residue, Cys403, which became a focus for the design
due to its location in the allosteric pocket and its close proximity to its symmetry related
partner about the two-fold axis. The nearby residue, Asn405, sits directly across the
dimer interface from the native cysteine, Cys403, and is in proper positioning for FlAsH
binding. A cross-link at this position would link the small subunit from one monomer to
the small-subunit of the opposite monomer. Therefore, Asn405 would complete the
FlAsH binding motif with native Cys403 for the C9-β model of FlAsH activation. Further
inspection also suggests this pocket would be able to accommodate the entire FlAsH
molecule. However, the C9-β model could potentially disrupt the inactive down
conformation of the active site loops. Although avoiding disruption of loop conformation
was a criterion for the design, at this position, binding of FlAsH could potentially shift
the active site loop equilibrium towards the up or active conformation. Therefore, this
model would not affect the overall goal of designing an active caspase-9 enzyme. Both

161
the C9-X and C9-β FlAsH binding motifs fit the design criteria for activation of caspase-
9 via FlAsH binding.
A.2.2. Production of Caspse-9 FlAsH Variants
The caspase-9 gene construct encoding amino acids for the small subunit only,
residues 331-416, in the pRSET was used for converting the wild type gene sequence into
a version of the caspase-9 ΔCARD FlAsH variants. The oligo-nucleotide pimer 5`-
GCTCACTCTGAAGACCTGTGTTGCCTCCTCTTAGGGTCGC-3`was used to convert
caspase-9 wild type sequence into the C9small-X variant for FlAsH binding studies via
converting Gln381 and Ser382 to cysteine. Due to an error within the original primer
sequence, a second oligo-nucleotide primer, 5`-CCTGCAGTCCCTCCTGCTTAGGG-
TCGCTAATGC-3`, was designed for correction of a base pair deletion at Leu384 which
can be found in position 54 of the Hardy Lab DNA Archive Box 1. To make C9small-β,
the oligo-nucleotide primer 5`-CAGATGCCTGGTTGTTTTTGCTTCCTCCGGAAA-
AAACTTTTC-3` was used for converting N405 to cysteine, found in position 51 of the
Hardy Lab DNA Archive Box 1. The previously described primers were also used for
conversion of the caspase-9 full-length gene (human sequence), encoding amino acids 1-
416, in the pET23b vector (Addgene plasmid 11829) into a full-length version of the
caspase-9 FlAsH variants, C9F-X and C9F-β, located in position 33 and 35 of the Hardy
Lab DNA Archive Box 2 Respectively. Furthermore, a FlAsH binding control variant
was produced in the caspase-9 full-length construct (C9F-FC). The oligo-nucleotide
primer, 5`- CCGGAAAAAACTTTTCTTTAAAACATCATGCTGCCCGGGCTGCT-
GCCACCACCACCACCACCACTAATCTAGCG-3`, was designed to incorporate the
FlAsH binding motif on the C-terminal end of the protein sequence just prior to the 6xHis

162
tag. All caspase-9 FlAsH variants were produced via the Quikchange mutagenesis
method (Stratagene).
The C9small-X and -β FlAsH constructs were separately transformed into the BL21
(DE3) T7 Express strain of E. coli (NEB). The recombinant large and small subunits
were individually expressed as inclusion bodies for subsequent reconstitution. Cultures
were grown in 2xYT media with ampicillin (100 mg/L, Sigma-Aldrich) at 37°C until
they reached an optical density at 600 nm of 0.6. Protein expression was induced with 0.2
mM IPTG. Cells were harvested after 3 hrs at 37°C. Cell pellets stored at -20°C for future
use. The resultant inclusion bodies were refolded and purified ion exchange column
purification step only which is outlined in Chapter III.
C9F-X and -β FlAsH variants were also transformed into the BL21 (DE3) T7
Express strain of E. coli (NEB). The cultures were grown in 2xYT media with ampicillin
(100 mg/L, Sigma-Aldrich) at 37°C until they reached an optical density at 600 nm of
0.8. The temperature was reduced to 20°C and cells were induced with 1 mM IPTG
(Anatrace) to express soluble 6xHis-tagged full-length protein. Cells were harvested
after 18 hrs and stored at -20°C. The resultant protein was purified using the standard
caspase-9 imidazole gradient purification followed by an ion-exchange column
purification step outlined in Chapter IV. The purified C9F-X and -β FlAsH variants
proteins are estimated to be approximately 95% pure with a two-site cleavage (at residues
D315 and E306 (Large) or D330 (small)) within its intersubunit linker was used for
further analysis.
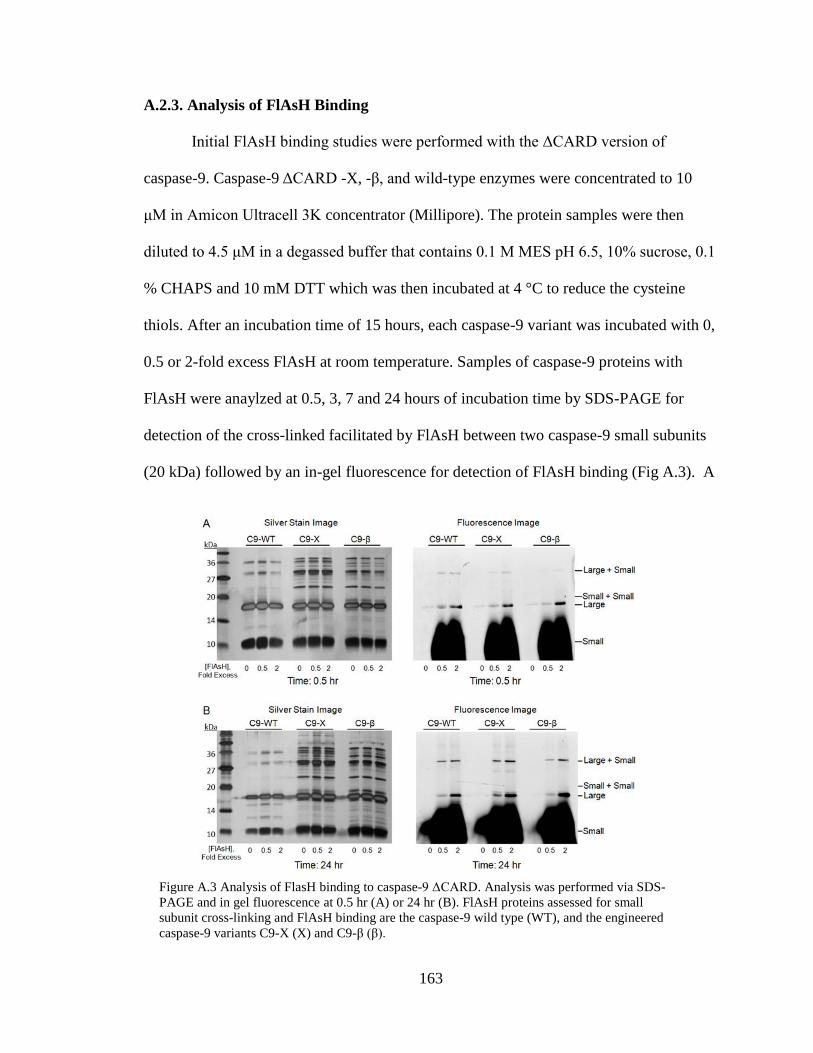
163
A.2.3. Analysis of FlAsH Binding
Initial FlAsH binding studies were performed with the ΔCARD version of
caspase-9. Caspase-9 ΔCARD -X, -β, and wild-type enzymes were concentrated to 10
μM in Amicon Ultracell 3K concentrator (Millipore). The protein samples were then
diluted to 4.5 μM in a degassed buffer that contains 0.1 M MES pH 6.5, 10% sucrose, 0.1
% CHAPS and 10 mM DTT which was then incubated at 4 °C to reduce the cysteine
thiols. After an incubation time of 15 hours, each caspase-9 variant was incubated with 0,
0.5 or 2-fold excess FlAsH at room temperature. Samples of caspase-9 proteins with
FlAsH were anaylzed at 0.5, 3, 7 and 24 hours of incubation time by SDS-PAGE for
detection of the cross-linked facilitated by FlAsH between two caspase-9 small subunits
(20 kDa) followed by an in-gel fluorescence for detection of FlAsH binding (Fig A.3). A
Figure A.3 Analysis of FlasH binding to caspase-9 ΔCARD. Analysis was performed via SDS-
PAGE and in gel fluorescence at 0.5 hr (A) or 24 hr (B). FlAsH proteins assessed for small
subunit cross-linking and FlAsH binding are the caspase-9 wild type (WT), and the engineered
caspase-9 variants C9-X (X) and C9-β (β).

164
fluorescent signature was observed just below the 20 kDa molecular weight marker as
expected of a cross-link between two small subunits within the dimer of caspase-9. Upon
further inspection of the wild type control, it was discovered that FlAsH was non-
specifically binding to the large subunit of the enzyme (18 kDa). This phenomenon has
been reported prior for other cysteine rich proteins19
. To alleviate fluorescence within the
expected 20 kDa region, the caspase-9 -X and -β variants were produced in the full length
version of the enzyme in which the large subunit is connected to the CARD domain
resulting in a 36.5 kDa fragment while the small subunit still remains at approximate 10
kDa. By using this form of the enzyme, a clear image of FlAsH fluorescence can be
observed around the 20 kDa marker. A full-length caspase-9 variant which incorporates
the FlAsH binding motif -CCPGCC- on the C-terminus was also created for a FlAsH
binding control, labeled as C9F-FC (position 38 in the Hardy Lab DNA Archive Box 2).
10 μM of the C9F-WT, C9F-X, C9F-β and C9F-FC FlAsH variants were diluted to 4.5
μM in a reducing buffer containing 0.1 M MES pH 6.5, 10 % sucrose, 50 mM TCEP,
which was used as a non-thiol based reductant. This mixture was allowed to incubate for
one hour at room temperature. Samples were then subjected to FlAsH binding by addition
of 0.5 mM 1,2-ethandithiol and a 2-fold excess of the FlAsH fluorescent compound.
Samples were then anayzed by SDS-PAGE (Fig A.4 A) and in-gel fluorescence methods
(Fig A.4 B). The small subunit of C9F-FC variant appears as a fluorescent band
indicating these conditions result in a functional assay for detecting FlAsH fluorescence.
Furthermore, fluorescent bands are also observed for a molecular weight band
corresponding to large + CARD (36 kDa) and large + small (28 kDa) as expected from
the non-specific binding of the FlAsH molecule observed in the caspase-9ΔCARD
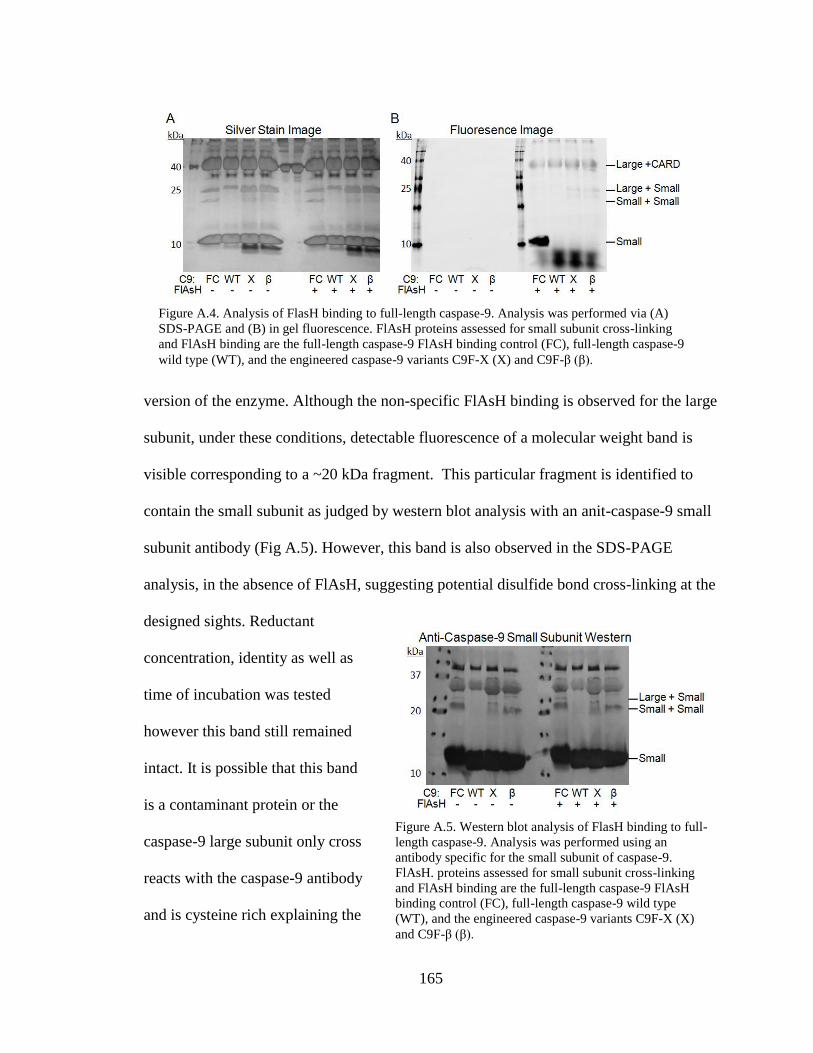
165
version of the enzyme. Although the non-specific FlAsH binding is observed for the large
subunit, under these conditions, detectable fluorescence of a molecular weight band is
visible corresponding to a ~20 kDa fragment. This particular fragment is identified to
contain the small subunit as judged by western blot analysis with an anit-caspase-9 small
subunit antibody (Fig A.5). However, this band is also observed in the SDS-PAGE
analysis, in the absence of FlAsH, suggesting potential disulfide bond cross-linking at the
designed sights. Reductant
concentration, identity as well as
time of incubation was tested
however this band still remained
intact. It is possible that this band
is a contaminant protein or the
caspase-9 large subunit only cross
reacts with the caspase-9 antibody
and is cysteine rich explaining the
Figure A.5. Western blot analysis of FlasH binding to full-
length caspase-9. Analysis was performed using an
antibody specific for the small subunit of caspase-9.
FlAsH. proteins assessed for small subunit cross-linking
and FlAsH binding are the full-length caspase-9 FlAsH
binding control (FC), full-length caspase-9 wild type
(WT), and the engineered caspase-9 variants C9F-X (X)
and C9F-β (β).
Figure A.4. Analysis of FlasH binding to full-length caspase-9. Analysis was performed via (A)
SDS-PAGE and (B) in gel fluorescence. FlAsH proteins assessed for small subunit cross-linking
and FlAsH binding are the full-length caspase-9 FlAsH binding control (FC), full-length caspase-9
wild type (WT), and the engineered caspase-9 variants C9F-X (X) and C9F-β (β).

166
minimal FlAsH fluorescence detected. Further studies are needed to determine whether
this 20 kDa band is a pre-formed small-small dimer, caspase-9 large subunit only, or a
protein contaminate.
Considering there was not a robust appearance of FlasH induced caspase-9
dimerization or fluorescence, efforts turned towards identifying the location of the non-
specific FlAsH binding for further characterization and potential future design. The full-
length caspase-9 contains thirteen cysteines in its amino acid sequence. Therefore, the
cysteine residues that are solvent exposed were targeted as potential FlAsH binding sites.
Selected positions, C162, C172, C229, and C239 were interrogated for their role in the
non-specific FlAsH binding observed due to their proximity to other cysteine residues.
Each position was converted to either a serine or an alanine by Quikchange mutagenesis
(Stratagene) and tested for a reduction in FlAsH binding. Single caspase-9 variants,
C162A, C172A, C229A, C239S, and the double variant C229A/C230A did not show a
significant decrease in FlAsH fluorescence. This suggests that an alternate positioning of
the FlAsH binding site exists or there is a need for construction of a caspase-9 variant
which incorporates multiple cysteine locations other than C229/C230 due to the fact that
fluorescence by FlAsH has been reported to occur when bound to less than four
cysteines19
.
A.3. Discussion
The initiator caspase of the intrinsic pathway of apoptosis, caspase-9, has the
potential to be a good target for regulation by a small molecule. Caspase-9 plays a pivotal
role in the activation of the cleavage cascade of apoptosis and its activation mechanism is
targeted in a variety of diseases anywhere from viral infections to cancer. Taking

167
inspiration from natural activation pathways of caspase-9, such as dimer formation, we
designed two variants of caspase-9, C9-X and C9-β that could potentially dimerize upon
binding of a small fluorescent molecule known as FlAsH. This would provide a proof-of-
concept that caspase activation can be controlled by a small molecule regulator.
Upon analysis, distinct induction of the caspase-9 dimer was not observed, in
addition to the lack of a robust fluorescence signal expected from binding of the FlAsH
molecule, which was observed for the control protein. Although precautions were taken
when designing FlAsH binding sites across the dimer interface, C9-X and C9-β were
unsuccessful. Unforeseen pitfalls such as improper geometric binding angles for FlAsH
binding within the designed cysteine motif or limited access to the designed site could
have prevented FlAsH from binding the engineered caspases. In addition, it is possible
that FlAsH could only bind to a pre-formed dimer of caspase-9 which is limited in vitro1.
This hypothesis can be tested by addition of a covalent active site inhibitor which shifts
the enzyme’s equilibrium from monomer to dimer. Further analysis of the geometric
restrictions imposed by these sites as well as the discovery of alternative FlAsH binding
sites or alternative molecules could result in a caspase-9 variant that can be activated
through a small molecule trigger.
Although the desired outcome was not obtained, new information was discovered
about caspase-9. A natural FlAsH binding site exists within the large subunit of caspase-
9. Once the location of the non-specific FlAsH site is determine, this site can be
optimized to obtain a more robust FlAsH signal and potentially exploit the site for
allosteric control as in the case of protein tyrosine phosphate 1B20
.
A.4. References

168
1. Renatus, M., Stennicke, H. R., Scott, F. L., Liddington, R. C. & Salvesen, G. S.
Dimer formation drives the activation of the cell death protease caspase 9. Proc
Natl Acad Sci U S A 98, 14250-14255, (2001).
2. Palmerini, F., Devilard, E., Jarry, A., Birg, F. & Xerri, L. Caspase 7
downregulation as an immunohistochemical marker of colonic carcinoma. Hum
Pathol 32, 461-467, (2001).
3. Mueller, T. et al. Failure of activation of caspase-9 induces a higher threshold for
apoptosis and cisplatin resistance in testicular cancer. Cancer Res 63, 513-521,
(2003).
4. Mizutani, Y. et al. Downregulation of Smac/DIABLO expression in renal cell
carcinoma and its prognostic significance. J Clin Oncol 23, 448-454, (2005).
5. Sekimura, A. et al. Expression of Smac/DIABLO is a novel prognostic marker in
lung cancer. Oncol Rep 11, 797-802, (2004).
6. Kempkensteffen, C. et al. The equilibrium of XIAP and Smac/DIABLO
expression is gradually deranged during the development and progression of
testicular germ cell tumours. Int J Androl 30, 476-483, (2007).
7. Peterson, Q. P. et al. PAC-1 activates procaspase-3 in vitro through relief of zinc-
mediated inhibition. J Mol Biol 388, 144-158, (2009).
8. Wolan, D. W., Zorn, J. A., Gray, D. C. & Wells, J. A. Small-molecule activators
of a proenzyme. Science 326, 853-858, (2009).
9. Griffin, B. A., Adams, S. R. & Tsien, R. Y. Specific covalent labeling of
recombinant protein molecules inside live cells. Science 281, 269-272, (1998).
10. Krishnan, B. & Gierasch, L. M. Cross-strand split tetra-Cys motifs as structure
sensors in a beta-sheet protein. Chem Biol 15, 1104-1115, (2008).
11. Luedtke, N. W., Dexter, R. J., Fried, D. B. & Schepartz, A. Surveying polypeptide
and protein domain conformation and association with FlAsH and ReAsH. Nat
Chem Biol 3, 779-784, (2007).

169
12. Ignatova, Z. & Gierasch, L. M. Monitoring protein stability and aggregation in
vivo by real-time fluorescent labeling. Proc Natl Acad Sci U S A 101, 523-528,
(2004).
13. Ignatova, Z. & Gierasch, L. M. A method for direct measurement of protein
stability in vivo. Methods Mol Biol 490, 165-178, (2009).
14. Ignatova, Z. et al. From the test tube to the cell: exploring the folding and
aggregation of a beta-clam protein. Biopolymers 88, 157-163, (2007).
15. Adams, S. R. et al. New biarsenical ligands and tetracysteine motifs for protein
labeling in vitro and in vivo: synthesis and biological applications. J Am Chem
Soc 124, 6063-6076, (2002).
16. Zhai, D. et al. Vaccinia virus protein F1L is a caspase-9 inhibitor. J Biol Chem
285, 5569-5580, (2010).
17. Hardy, J. A., Lam, J., Nguyen, J. T., O'Brien, T. & Wells, J. A. Discovery of an
allosteric site in the caspases. Proc Natl Acad Sci U S A 101, 12461-12466,
(2004).
18. Chao, Y. et al. Engineering a dimeric caspase-9: a re-evaluation of the induced
proximity model for caspase activation. PLoS Biol 3, e183, (2005).
19. Stroffekova, K., Proenza, C. & Beam, K. G. The protein-labeling reagent
FLASH-EDT2 binds not only to CCXXCC motifs but also non-specifically to
endogenous cysteine-rich proteins. Pflugers Arch 442, 859-866, (2001).
20. Zhang, X. Y. & Bishop, A. C. Site-specific incorporation of allosteric-inhibition
sites in a protein tyrosine phosphatase. J Am Chem Soc 129, 3812-3813, (2007).

170
APPENDIX B
DESIGN OF AN ACTIVATABLE INITIATOR CASPASE
B.1. Introduction
Caspases, proteases that control apoptosis, have been a target in cancer research
for many years. In particular, caspase-9, an initiator of the caspase cleavage cascade, has
been shown to be down regulated in tumor cells1,2
. Therefore, activating the caspase
cleavage cascade, via the intrinsic pathway of apoptosis by utilizing caspase-9 within
these tumors, would be a useful tool to study apoptosis in tumors and to halt tumor
progression. Critical steps within the caspase-9 activation pathway, such as dimerization
and active site loop conformation, can be used as inspiration for designing such a tool.
Caspase-9’s equilibrium in solution lies toward the monomeric state3. Therefore
designing a way to shift the equilibrium of caspase-9 from monomer to dimer would aid
in increasing the active population of caspase-9 molecules. Additionally, if creating a
dimeric variant of caspase-9 could also be facilitated via control of the active site loop
bundle, further enhancement of caspase-9 activity would be expected.
Nature provides naturally occurring amino acids, such as cysteines, that form
covalent, redox-controllable cross-links, which became a useful protein-engineering tool
for controlling a proteins conformation. The disulfide cross-link has been used for a
variety of applications including stabilization of protein conformational states and
analyzing functional mechanisms4-10
on anywhere from viral proteins11
to critical cellular
transcription factors12
and even for release from a delivery system13-17
. Wide applicability
and controlability of this technique, based upon redox environment18,19
, makes disulfide
cross-linking an intriguing tool for activating caspase-9. Therefore we aim to rationally

171
design a caspase-9 variant to become activated by inducing dimerization and formation of
the active site loop bundle via a disulfide cross-link.
B.2. Results
B.2.1. Rational Redox Controllable Caspase-9
Like other caspases, caspase-9 undergoes distinct structural conformation changes
based upon the state of the enzyme. Caspase-9 exists as an inactive monomer within the
cytosol and becomes active upon dimerization3. Crystallographic evidence shows an
active site loop bundle that adopts a disordered conformation in the absence of substrate
and changes conformation to a more ordered state upon binding substrate3. The active
state of the enzyme is a dimer in which the active site loop, L2, from one half of the
dimer interacts with the L2` loop from the other half. Our goal is to lock caspase-9 into
the active state, overcoming the slow step of the caspase-9 activation process and thus
preparing it to cleave substrate.
The natural residue, cysteine, was utilized to create a redox controllable link
between both halves of the dimer. Wild-type caspase-9 protein contains thirteen native
cysteine residues, where a natural redox regulation event or a native disulfide bond
linkage has not been observed within the enzyme to date. These thirteen native cysteines
within the caspase-9 amino acid sequence were initially investigated for potentially
forming a natural disulfide link. The only native cysteine capable of this would be C403,
which resides within the dimer interface. Based on conformation with the β-sheet
secondary structure and distance from the corresponding C403 across the two fold axis of
the dimer interface, disulfide cross-linking at this site would not be feasible. Therefore a
computational tool to predict the availability of residues for disulfide engineering, known

172
as MODIP, was utilized to predict which amino acid positions would facilitate proper
amino acid pairing for potential disulfide linkage. The best prediction across the dimer
interface comprised the residues Lys399, found at the top of the L4 loop, and Pro339 of
the L2` loop from the opposite monomer. However, cross-linking two active site loops
which have different functions would most likely have a negative effect on activity of the
enzyme. Therefore, the caspase-9 crystal structure, in the active state, was inspected for
alternate designs. Inspired by MODIP’s prediction of locking active site loop, interaction
between L2 and L2`of caspase-9 in the substrate bound state (PDB ID: 1NW9) was
investigated. The L2 and L2` loops naturally interact upon binding of substrate therefore
the addition of a cross-link at this position would enhance the probability of the
conformational state. Upon inspection of the caspase-9 active conformation, Glu297 on
the L2 loop and Ser334C on L2` appeared to be within optimal distance and
conformation as they reside just past the intersection point of the L2 and L2` (Fig B.1).
Optimal distance for disulfides was defined as 4.4-6.8 Å from Cα-Cα20
. A disulfide at
this site would link the large and
small subunits of the enzyme
together. Therefore these two
sites were chosen as the link site
for the redox controllable
caspase-9 variant.
B.2.2 Production of a Redox
Controllable Caspase-9
The caspase-9 full-length
Figure B.1 Model of the designed caspase-9 disulfide
cross linked variant.

173
gene (human sequence) construct, encoding amino acids 1-416, in pET23b (Addgene
plasmid 11829 ) was used to design a genetic construct for expression and purification
from Bl21(DE3) E.coli strain of bacteria of the disulfide activatiable caspase-9 variant.
The oligo-nucleotide pimer 5’ GCAGAAAGACCATGGGTTTTGCGTGGCCTCCA-
CTTCCCC 3’converted position 297 from a Glu to a Cys while and a second primer was
designed, 5’ GCTGGACGCCATATCTTGTTTGCCC-ACACCC 3’, to convert position
344 from a Ser to a Cys utilizing the Quikchange mutagenesis method. First, C9F-E297
was changed to cysteine, found in position 37 of the Hardy Lab DNA Archive Box 2. The
final construct of C9F-E297C/S334C, alternately named C9F-DS was made by
performing Quikchange mutagenesis using the Ser334 oligo-primer in the background of
the C9F-E298 construct stored in the Hardy Lab DNA Archive Box 2 position 38.
The C9F-DS construct was transformed into the BL21 (DE3) T7 Express strain of
E. coli (NEB) for expression. The cultures were grown in 2xYT media with ampicillin
(100 mg/L, Sigma-Aldrich) at 37°C until they reached an optical density at 600 nm of
0.8. The temperature was reduced to 20°C and cells were induced with 1 mM IPTG
(Anatrace) to express soluble 6xHis-tagged full-length protein. Cells were harvested
after 18 hrs and stored at -20°C. The resultant protein was purified using the standard
caspase-9 imidazole gradient purification followed by an ion exchange column
purification step, in which a detailed protocol can be found in Appendix D: Protocol D1.
The purified C9F-DS protein, estimated to be approximately 95% pure with a two site
cleavage within its intersubunit linker was used for further analysis.
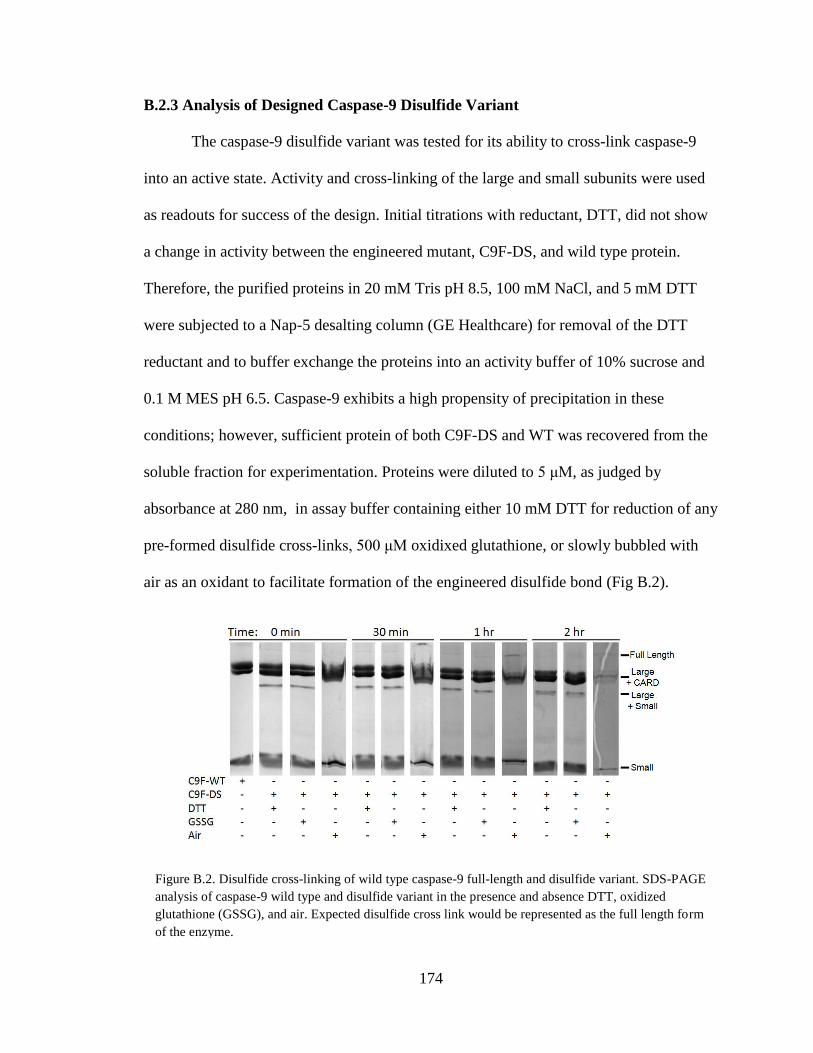
174
B.2.3 Analysis of Designed Caspase-9 Disulfide Variant
The caspase-9 disulfide variant was tested for its ability to cross-link caspase-9
into an active state. Activity and cross-linking of the large and small subunits were used
as readouts for success of the design. Initial titrations with reductant, DTT, did not show
a change in activity between the engineered mutant, C9F-DS, and wild type protein.
Therefore, the purified proteins in 20 mM Tris pH 8.5, 100 mM NaCl, and 5 mM DTT
were subjected to a Nap-5 desalting column (GE Healthcare) for removal of the DTT
reductant and to buffer exchange the proteins into an activity buffer of 10% sucrose and
0.1 M MES pH 6.5. Caspase-9 exhibits a high propensity of precipitation in these
conditions; however, sufficient protein of both C9F-DS and WT was recovered from the
soluble fraction for experimentation. Proteins were diluted to 5 μM, as judged by
absorbance at 280 nm, in assay buffer containing either 10 mM DTT for reduction of any
pre-formed disulfide cross-links, 500 μM oxidixed glutathione, or slowly bubbled with
air as an oxidant to facilitate formation of the engineered disulfide bond (Fig B.2).
Figure B.2. Disulfide cross-linking of wild type caspase-9 full-length and disulfide variant. SDS-PAGE
analysis of caspase-9 wild type and disulfide variant in the presence and absence DTT, oxidized
glutathione (GSSG), and air. Expected disulfide cross link would be represented as the full length form
of the enzyme.

175
Samples of the reaction were pulled at time points 0, 0.5, 1, and 2 hours to monitor the
potential cross-link. A cross-link between the large and small subunits, forming the
expected intact monomer, was not observed pre or post treatment with oxidized
glutathione. However, a minimal amount of cross-linked monomer was detected upon
oxidation with air at the one and two hour time points. Reduction in protein concentration
was also observed due to precipitation of protein during the experimental timeframe,
indicating a need for further optimization of experimental conditions. Protein precipitate
was also observed for the wild type version of the enzyme (data not shown), therefore
alternate ways to obtain the cross-linked form of C9F-DS need further exploration.
The designed redox controllable variant, C9F-DS, was successful to some degree
under air oxidizing conditions only. Due to protein precipitation issues, studies of
enzymatic activity posed to be troublesome, therefore investigating whether this variant
can induced dimerization of the small and large subunits via a cross-link between the L2
and L2` loops was perused. This would verify this site alone can enforce the dimer form
of the enzyme, which is required for activity. Small maleimide-based cross-linkers,
BMOE (bis-maleimidoethane) and BMDB (1,4 bismaleimidyl-2,3-dihydroxybutane)
(Pierce), of arm spacer lengths of 8.0 Å and 10.2 Å respectively, were used as cross-
linking agents (Fig B.3 A-B). 300 μM of BOME or BMDB was added to 10 μM C9F-DS
and WT protein that was reduced for 30 minutes in 5mM TCEP and equilibrated in 0.1 M
phosphate buffer pH 7.0, 0.15 M NaCl and 5 mM EDTA. Samples were incubated for
two hours at room temperature and immediately analyzed via SDS-PAGE (Fig B.3 C-D).
For both BMOE and BMDB cross-linking compounds, the expected large plus small
molecular weight band is present; however, it is only slightly more concentrated than

176
samples that do not include the maleimide compounds. This result indicates that the
E297C/S334C site is capable of associating the large and small subunits via sulfhydryl
cross-linking compounds in addition to formation of this complex under different buffer
conditions.
B.3. Discussion
The initiator of the caspase cleavage cascade for the intrinsic pathway of
apoptosis, caspase-9, is a promising target for activating apoptosis in tumor cells. Taking
inspiration from natural activation pathways of caspase-9, such as dimer formation and
active site loop orientation, we designed a disulfide cross-linked version of the caspase-9
enzyme. A cross-link between the large and small subunits of caspase-9, due to the
altered amino acids E297C and S334C within the intersubunit linker, was observed under
air oxidation as well as the presence of sulfhydryl cross-linking agents. However,
precipitation of both the control wild type protein, as well as the disulfide variant posed
Figure B.3. Chemical cross-linking of wild type caspase-9 full-length and disulfide variant. (A-B)
Small spacer arm maliemide based cross linking compounds. (A) BMOE (B) BMDB. (C-D) SDS-
PAGE analysis of caspase-9 wild type and disulfide variant in the presence and absence of BMOE
(C) and BMDB (D). Expected disulfide cross link would be represented as the full length form of the
enzyme.

177
as a large issue within these experiments potentially due to intramolecular disulfides
being formed between the thirteen native cysteines within the enzyme. The incidence of
cross-linking between the large and small subunits of caspase-9 was observed to a greater
degree (Fig B.3.D) than in the control experiments, suggesting induced dimerization of
the enzyme. However protein precipitation and the intrinsic requirement of reductant
prevented activation analysis.
Verification of caspase-9 increased activation is critical for making this disulfide
mediated caspase-9 variant a useful tool. Therefore, optimization of the assay conditions
is required to enforce the disulfide cross-link, while still being able to assay for activity
and avoid precipitation of the enzyme. Identification of the lowest amount of reductant
necessary for caspase-9 activity and solubility would also be useful for solving these
issues. Potentially a redox buffer that includes both reduced and oxidized glutathione is
more closely related to that of the chemical cross-linking studies, would be more
appropriate. In addition, removal of any surface exposed cysteines would aid in this
endeavor.
B.4. References
1. Mueller, T. et al. Failure of activation of caspase-9 induces a higher threshold for
apoptosis and cisplatin resistance in testicular cancer. Cancer Res 63, 513-521,
(2003).
2. Palmerini, F., Devilard, E., Jarry, A., Birg, F. & Xerri, L. Caspase 7
downregulation as an immunohistochemical marker of colonic carcinoma. Hum
Pathol 32, 461-467, (2001).
3. Renatus, M., Stennicke, H. R., Scott, F. L., Liddington, R. C. & Salvesen, G. S.
Dimer formation drives the activation of the cell death protease caspase 9. Proc
Natl Acad Sci U S A 98, 14250-14255, (2001).

178
4. Jeong, M. Y., Kim, S., Yun, C. W., Choi, Y. J. & Cho, S. G. Engineering a de
novo internal disulfide bridge to improve the thermal stability of xylanase from
Bacillus stearothermophilus No. 236. J Biotechnol 127, 300-309, (2007).
5. Ma, K., Temiakov, D., Anikin, M. & McAllister, W. T. Probing conformational
changes in T7 RNA polymerase during initiation and termination by using
engineered disulfide linkages. Proc Natl Acad Sci U S A 102, 17612-17617,
(2005).
6. Mitchinson, C. & Wells, J. A. Protein engineering of disulfide bonds in subtilisin
BPN'. Biochemistry 28, 4807-4815, (1989).
7. Qureshi, S. H., Yang, L., Manithody, C., Iakhiaev, A. V. & Rezaie, A. R.
Mutagenesis studies toward understanding allostery in thrombin. Biochemistry 48,
8261-8270, (2009).
8. Seeger, M. A. et al. Engineered disulfide bonds support the functional rotation
mechanism of multidrug efflux pump AcrB. Nat Struct Mol Biol 15, 199-205,
(2008).
9. Shandiz, A. T., Capraro, B. R. & Sosnick, T. R. Intramolecular cross-linking
evaluated as a structural probe of the protein folding transition state. Biochemistry
46, 13711-13719, (2007).
10. Witkowski, W. A. & Hardy, J. A. A designed redox-controlled caspase. Protein
Sci, (2011).
11. Lee, J. K., Prussia, A., Snyder, J. P. & Plemper, R. K. Reversible inhibition of the
fusion activity of measles virus F protein by an engineered intersubunit disulfide
bridge. J Virol 81, 8821-8826, (2007).
12. Zheng, M., Aslund, F. & Storz, G. Activation of the OxyR transcription factor by
reversible disulfide bond formation. Science 279, 1718-1721, (1998).
13. Bauhuber, S., Hozsa, C., Breunig, M. & Gopferich, A. Delivery of nucleic acids
via disulfide-based carrier systems. Adv Mater 21, 3286-3306, (2009).
14. Hong, R. et al. Glutathione-mediated delivery and release using monolayer
protected nanoparticle carriers. J Am Chem Soc 128, 1078-1079, (2006).

179
15. Navath, R. S. et al. Dendrimer-drug conjugates for tailored intracellular drug
release based on glutathione levels. Bioconjug Chem 19, 2446-2455, (2008).
16. Ouyang, D., Shah, N., Zhang, H., Smith, S. C. & Parekh, H. S. Reducible
disulfide-based non-viral gene delivery systems. Mini Rev Med Chem 9, 1242-
1250, (2009).
17. Singh, R. & Lillard, J. W., Jr. Nanoparticle-based targeted drug delivery. Exp Mol
Pathol 86, 215-223, (2009).
18. Russo, A., DeGraff, W., Friedman, N. & Mitchell, J. B. Selective modulation of
glutathione levels in human normal versus tumor cells and subsequent differential
response to chemotherapy drugs. Cancer Res 46, 2845-2848, (1986).
19. Jones, D. P., Brown, L. A. & Sternberg, P. Variability in glutathione-dependent
detoxication in vivo and its relevance to detoxication of chemical mixtures.
Toxicology 105, 267-274, (1995).
20. Richardson, J. S. The anatomy and taxonomy of protein structure. Adv Protein
Chem 34, 167-339, (1981).

180
APPENDIX C
EXPRESSION AND PURIFICATION OF A YEAST METACASPASE YCA1
C.1. Introduction
Saccharomyces cerevisiea yeast has served as a model organism for a huge
number of genetic and regulatory pathways due to its ease of genetic manipulation and
strikingly conserved pathways in comparison to other eukaryotic organisms.
Furthermore, this model system has demonstrated apoptotic-like cell death with such
markers as DNA fragmentation and chromatin condensation1. Since the discovery of
apoptosis in yeast, a number of yeast orthologs to key players in the mammalian
apoptotic cycle have been identified, indicating conserved cell death pathways for
proteasomal and mitochondrial-type cell death2.
An apoptotic-like phenotype in yeast has even been observed upon a variety of
triggers including exposure to reactive oxygen species 3 and aging
4. Characteristic
homologues for apoptotic regulators such as the Bcl-2 family of proteins have not yet
been discovered in the yeast model system; however, a caspase-like protein has been
identified in the S. cerevisiea genome. Yeast caspase-1 (YCA1 also known as
YOR197W) has been classified as an evolutionary ancestor to human caspases5, or a
metacaspase, due to primary sequence identification of a catalytic cysteine-histidine diad
as well as a predicted caspase like fold. YCA1 has been highlighted as the protein
responsible for apoptotic like cell death in yeast6. For example, knockout of the YCA1
gene in S. cervisiea has been shown to abolish an H2O2 induced form of apoptosis7. In
addition to YCA1’s ability to control the apoptotic properties of yeast, this protease
shows caspase like character in both its cleavage properties and substrate recognition

181
sequences, VEID and IETD. YCA1 studies have mainly been performed in whole yeast
cells to understand how overexpression of the enzyme affects cell survival rates as well
as how different apoptosis inducing factors, such as H2O2, affect survival when in the
presence and absence of the YCA1 gene4,6,8,9
. Additional studies involved removal of the
soluble protein fractions from the yeast cells for measurement of YCA1 expression levels
under normal and stress conditions7 and utilizing cellular extracts to test the enzymatic
activity of YCA110
. These caspase-like characteristics therefore make this metacaspase
an intriguing target for protein engineering and structural studies. We aimed to express
and purify YCA1 to perform biochemical characterizations for further elucidation of the
proteases similarities to its human homologues. This ultimately would allow solution of
the crystal structure of YCA1 to observe its structural similarities and to design alternate
ways to regulate the enzyme though protein engineering which can be easily tested in
yeast as the model system.
C.2. Results
C.2.1. Gene Construction
The S. cervisiea YCA1 gene was used to design and prepare a genetic construct
for expression and purification from Bl21(DE3) E.coli strain of bacteria. The gene was
amplified from a yeast plasmid by PCR for insertion into the pET21(+)b expression
vector. Oligo-nucleotide pimers, 5`-AGTCGTGCTAGCATGTATCCAGGTAGTGGA-
CGTTACACC-3` and 5`-AGTATACTCGAGCTACATAATAAATTGCAGATTTAC-
GTCAATAGGG-3`, encode the NheI and Xho1 endonuclease restriction sites for the 5`
and 3` primers, respectively, were utilized for the amplification. The PCR product was
digested to expose complementary DNA overhangs and gel purified, resulting in a 64

182
ng/μl digested YCA1 DNA gene product. Ligation of the YCA1 gene to the digested the
pET21(+)b expression vector was performed through incubating one molar ratio of vector
with three molar ratios of YCA1 gene insert with T4 DNA ligase at 16°C overnight. The
ligation reactions were transformed into the TAM1 cloning strain of E.coli cells and
plated onto 100 mg/ml ampicillin plates. Transformants were tested for successful
ligations through enzymatic digest with NheI and XhoI to reveal the insertion of the
YCA1 gene which was subsequently confirmed through DNA sequencing. The resulting
plasmid encodes the YCA1 gene with a C-terminal 6xHis tag driven by the T7 promoter
with apmicillin resistance (Fig C.1). This plasmid is housed in the Hardy Lab DNA
archive box 1 position 9.
C.2.2. Protein Sequence
10 20 30 40 50 60
MYPGSGRYTY NNAGGNNGYQ RPMAPPPNQQ YGQQYGQQYE QQYGQQYGQQ NDQQFSQQYA
70 80 90 100 110 120
PPPGPPPMAY NRPVYPPPQF QQEQAKAQLS NGYNNPNVNA SNMYGPPQNM SLPPPQTQTI
130 140 150 160 170 180
QGTDQPYQYS QCTGRRKALI IGINYIGSKN QLRGCINDAH NIFNFLTNGY GYSSDDIVIL
Figure C.1. Schematic representation of expression vector, pYCA1. The T7 promoter, YCA1 gene,
6xHis tag, and ampicillin resistance are shown.

183
190 200 210 220 230 240
TDDQNDLVRV PTRANMIRAM QWLVKDAQPN DSLFLHYSGH GGQTEDLDGD EEDGMDDVIY
250 260 270 280 290 300
PVDFETQGPI IDDEMHDIMV KPLQQGVRLT ALFDSCHSGT VLDLPYTYST KGIIKEPNIW
310 320 330 340 350 360
KDVGQDGLQA AISYATGNRA ALIGSLGSIF KTVKGGMGNN VDRERVRQIK FSAADVVMLS
370 380 390 400 410 420
GSKDNQTSAD AVEDGQNTGA MSHAFIKVMT LQPQQSYLSL LQNMRKELAG KYSQKPQLSS
430
SHPIDVNLQF IM
C.2.3. YCA1 Expression
The YCA1 gene is expected to produce ~50 kDa 6xHis-tagged protein of 438
amino acids in length. YCA1 is reported to undergo autoprocessing upon induction of
apoptosis or overexpression of the enzyme in yeast. Therefore, two cleavage products
would be observed, a ~12 kDa portion of the enzyme which is noted as the small subunit
and ~38 kD molecular weight piece of the remaining enzyme that would comprise the
large and pro-domains.
In order to obtain a homogenous sample of the active form of the YCA1 protein,
expression and purification of the YCA1 was performed. Cell growth of the BL21(DE3)
strain of E.coli transformed with the YCA1-pET21(+)b plasmid in 2xYT media was
performed. When OD600 reached 0.6, cells were induced for protein expression with
1mM IPTG at 14, 30, or 37 °C. A standard 6x-His tag purification protocol was utilized
in order to obtain the desired protein from cellular lysates as outlined by the Current
Protocols in Protein Science11
. Under these standard expression and purification
conditions, protein induced at OD600= 0.6 with 1 mM IPTG 14°C for 18 hrs produced an
elution fraction that contained minimal amount of protein of the expected molecular
weight of full length YCA1 as judged by Western blot analysis with an antibody specific
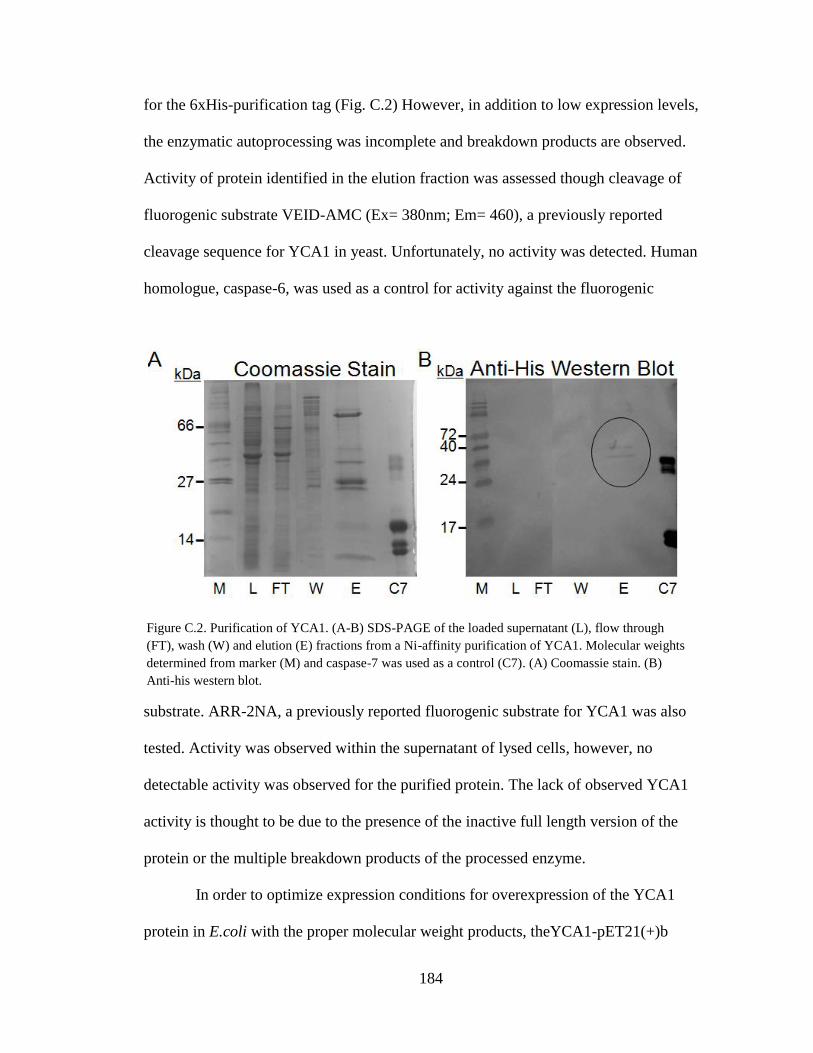
184
for the 6xHis-purification tag (Fig. C.2) However, in addition to low expression levels,
the enzymatic autoprocessing was incomplete and breakdown products are observed.
Activity of protein identified in the elution fraction was assessed though cleavage of
fluorogenic substrate VEID-AMC (Ex= 380nm; Em= 460), a previously reported
cleavage sequence for YCA1 in yeast. Unfortunately, no activity was detected. Human
homologue, caspase-6, was used as a control for activity against the fluorogenic
substrate. ARR-2NA, a previously reported fluorogenic substrate for YCA1 was also
tested. Activity was observed within the supernatant of lysed cells, however, no
detectable activity was observed for the purified protein. The lack of observed YCA1
activity is thought to be due to the presence of the inactive full length version of the
protein or the multiple breakdown products of the processed enzyme.
In order to optimize expression conditions for overexpression of the YCA1
protein in E.coli with the proper molecular weight products, theYCA1-pET21(+)b
Figure C.2. Purification of YCA1. (A-B) SDS-PAGE of the loaded supernatant (L), flow through
(FT), wash (W) and elution (E) fractions from a Ni-affinity purification of YCA1. Molecular weights
determined from marker (M) and caspase-7 was used as a control (C7). (A) Coomassie stain. (B)
Anti-his western blot.

185
expression vector was transformed into various forms of the BL21(DE3) E.coli cell line
including the original BL21(DE3) strain as well as the PLysS, Codon +, and Rosetta
strains. Induction of protein expression was performed at an OD600 = 0.4 and 0.6 in a 5
mL 2xYT media culture format by addition of IPTG in a concentration intervals of 0.05,
0.2, 0.5, 0.75, and 1 mM of protein growth or through utilizing auto-induction media.
Protein expression was allowed to be induced for a time course of 3, 6 or 18 hours at
temperatures of 15, 28 or 37 °C. Cells cultures were stored in a 96-well format at -80°C
for future analysis. Stored cells were collected by centrifugation and resuspended in
phosphate buffer pH 8.0 containing lysozyme. The samples were subjected to five freeze-
thaw cycles to lyse cells, spun at 3700 rpm to remove cellular debris. Supernatants
diluted in 0.1M HEPES pH 7.5, 0.1% CHAPS, 10% sucrose, and 5mM β-Me. buffer
were then analyzed for activity using the fluorogenic substrate VEID-AMC. Activity was
only observed for control protein caspase-6, indicating that changing the bacterial strain
induction conditions such as cell density, temperature, and [IPTG] did not improve
YCA1 expression levels or cleavage patterns to result in an active enzyme judged by
protease activity.
C.3. Discussion
Yeast caspase 1, a homologue of human caspases, would be a good surrogate for
studying regulation of YCA1 in the yeast apoptotic cascade. A genetic construct
containing the YCA1 gene was created for expression of YCA1 in E.coli. Upon analysis
of expression, induction, and purification conditions, minimal amounts of the inactive full
length form of protein was obtained. In addition, a non-homogeneous mixture of cleavage
products was observed, which did not possess any enzyme activity. Due to this lack of

186
activity, the observed cleaved forms of the enzyme are thought to be breakdown products
which occurred during expression or purification by an endogenous E.coli protease. This
would also reflect on the low yield that was obtained. Due to these issues, further
progression of this project was halted.
Future studies could include purifications in which protease inhibitors such as
PMSF are utilized. These inhibitor cocktails, however, will also target the enzyme of
interest, limiting analysis to only structural based methods in which a peptide is bound to
the active site. Additionally, avenues to express YCA1 in its native system, yeast, could
also be explored. Although over-expression of YCA1 in yeast has been shown to induce
apoptotic cell death, potential purification from natural abundance or leaky expression
would most likely lead to the proper cleavage of the enzyme resulting in the active
enzyme.
C.4. References
1. Madeo, F., Frohlich, E. & Frohlich, K. U. A yeast mutant showing diagnostic
markers of early and late apoptosis. J Cell Biol 139, 729-734, (1997).
2. Carmona-Gutierrez, D. et al. Apoptosis in yeast: triggers, pathways, subroutines.
Cell Death Differ 17, 763-773, (2010).
3. Madeo, F. et al. Oxygen stress: a regulator of apoptosis in yeast. J Cell Biol 145,
757-767, (1999).
4. Herker, E. et al. Chronological aging leads to apoptosis in yeast. J Cell Biol 164,
501-507, (2004).
5. Uren, A. G. et al. Identification of paracaspases and metacaspases: two ancient
families of caspase-like proteins, one of which plays a key role in MALT
lymphoma. Mol Cell 6, 961-967, (2000).

187
6. Madeo, F. et al. A caspase-related protease regulates apoptosis in yeast. Mol Cell
9, 911-917, (2002).
7. Khan, M. A., Chock, P. B. & Stadtman, E. R. Knockout of caspase-like gene,
YCA1, abrogates apoptosis and elevates oxidized proteins in Saccharomyces
cerevisiae. Proc Natl Acad Sci U S A 102, 17326-17331, (2005).
8. Bettiga, M., Calzari, L., Orlandi, I., Alberghina, L. & Vai, M. Involvement of the
yeast metacaspase Yca1 in ubp10Delta-programmed cell death. FEMS Yeast Res
5, 141-147, (2004).
9. Reiter, J., Herker, E., Madeo, F. & Schmitt, M. J. Viral killer toxins induce
caspase-mediated apoptosis in yeast. J Cell Biol 168, 353-358, (2005).
10. Watanabe, N. & Lam, E. Two Arabidopsis metacaspases AtMCP1b and
AtMCP2b are arginine/lysine-specific cysteine proteases and activate apoptosis-
like cell death in yeast. J Biol Chem 280, 14691-14699, (2005).
11. Petty, K. J. Metal-Chelate Affinity Chromatography. Vol. 2 (John Wiley and
Sons, Inc., 2000).

188
BIBILOGRAPHY
1-45,47,46,48-84,86,85,87-203,205,204,206-250 251-282,284,283,285-347
Abrahamson EE, Ikonomovic MD, Ciallella JR, Hope CE, Paljug WR, Isanski BA, Flood
DG, Clark RS, DeKosky ST (2006) Caspase inhibition therapy abolishes brain
trauma-induced increases in Abeta peptide: implications for clinical outcome.
Experimental neurology 197:437-450.
Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW (2002) Three-
dimensional structure of the apoptosome: implications for assembly, procaspase-9
binding, and activation. Molecular cell 9:423-432.
Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, Llopis J, Tsien RY
(2002) New biarsenical ligands and tetracysteine motifs for protein labeling in
vitro and in vivo: synthesis and biological applications. J Am Chem Soc
124:6063-6076.
Aharinejad S, Andrukhova O, Lucas T, Zuckermann A, Wieselthaler G, Wolner E,
Grimm M (2008) Programmed cell death in idiopathic dilated cardiomyopathy is
mediated by suppression of the apoptosis inhibitor Apollon. The Annals of
thoracic surgery 86:109-114.
Aiuchi T, Mihara S, Nakaya M, Masuda Y, Nakajo S, Nakaya K (1998) Zinc ions prevent
processing of caspase-3 during apoptosis induced by geranylgeraniol in HL-60
cells. Journal of biochemistry 124:300-303.
Alberts IL, Nadassy K, Wodak SJ (1998) Analysis of zinc binding sites in protein crystal
structures. Protein science : a publication of the Protein Society 7:1700-1716.
Allan LA, Morrice N, Brady S, Magee G, Pathak S, Clarke PR (2003) Inhibition of
caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nature cell
biology 5:647-654.
Ambrosini G, Adida C, Altieri DC (1997) A novel anti-apoptosis gene, survivin,
expressed in cancer and lymphoma. Nature medicine 3:917-921.
Andrade F, Casciola-Rosen LA, Rosen A (2003) A novel domain in adenovirus L4-100K
is required for stable binding and efficient inhibition of human granzyme B:
possible interaction with a species-specific exosite. Molecular and cellular
biology 23:6315-6326.

189
Andrade MA, Chacon P, Merelo JJ, Moran F (1993) Evaluation of secondary structure of
proteins from UV circular dichroism spectra using an unsupervised learning
neural network. Protein engineering 6:383-390.
Andreini C, Bertini I, Rosato A (2009) Metalloproteomes: a bioinformatic approach. Acc
Chem Res 42:1471-1479.
Aoyagi M, Zhai D, Jin C, Aleshin AE, Stec B, Reed JC, Liddington RC (2007) Vaccinia
virus N1L protein resembles a B cell lymphoma-2 (Bcl-2) family protein. Protein
science : a publication of the Protein Society 16:118-124.
Arnott S, Dover SD (1968) The structure of poly-L-proline II. Acta crystallographica
Section B: Structural crystallography and crystal chemistry 24:599-601.
American Lung Association. Trends in Asthma Morbidity and Mortality. (2007).
Research and Program Services.
Diabetes Association. Diabetes Stastics. (2011).
Augstein P, Elefanty AG, Allison J, Harrison LC (1998) Apoptosis and beta-cell
destruction in pancreatic islets of NOD mice with spontaneous and
cyclophosphamide-accelerated diabetes. Diabetologia 41:1381-1388.
Aurora R, Rose G (1998) Helix capping. Protein science: a publication of the Protein
Society 7:21.
Baltrusch S, Lenzen S, Okar DA, Lange AJ, Tiedge M (2001) Characterization of
Glucokinase-binding Protein Epitopes by a Phage-displayed Peptide Library
Identification of 6-Phosphofructo-2-kinase/fructose-2, 6-Bisphosphatase as a
Novel Interaction Partner. Journal of Biological Chemistry 276:43915-43923.
Banerjee R, Basu G, Roy S, Chène P (2002) Aib based peptide backbone as scaffolds for
helical peptide mimics. The Journal of peptide research 60:88-94.
Bao ST, Gui SQ, Lin MS (2006) Relationship between expression of Smac and Survivin
and apoptosis of primary hepatocellular carcinoma. Hepatobiliary & pancreatic
diseases international : HBPD INT 5:580-583.

190
Barany G, Kneib Cordonier N, Mullen DG (1987) Solid phase peptide synthesis: a silver
anniversary report*. Int J Pept Protein Res 30:705-739.
Bartsevich VV, Juliano RL (2000) Regulation of the MDR1 Gene by Transcriptional
Repressors Selected Using Peptide Combinatorial Libraries. Molecular
Pharmacology 58:1-10.
Bauhuber S, Hozsa C, Breunig M, Gopferich A (2009) Delivery of nucleic acids via
disulfide-based carrier systems. Adv Mater 21:3286-3306.
Becker DL, Fredenburgh JC, Stafford AR, Weitz JI (1999) Exosites 1 and 2 are essential
for protection of fibrin-bound thrombin from heparin-catalyzed inhibition by
antithrombin and heparin cofactor II. The Journal of biological chemistry
274:6226-6233.
Benoiton NL. 2006. Chemistry Of Peptide Synthesis, Taylor & Francis.
Benyamini H, Friedler A (2010) Using peptides to study protein–protein interactions.
Future 2:989-1003.
Bernal F, Wade M, Godes M, Davis TN, Whitehead DG, Kung AL, Wahl GM, Walensky
LD (2010) A stapled p53 helix overcomes HDMX-mediated suppression of p53.
Cancer cell 18:411-422.
Bettiga M, Calzari L, Orlandi I, Alberghina L, Vai M (2004) Involvement of the yeast
metacaspase Yca1 in ubp10Delta-programmed cell death. FEMS yeast research
5:141-147.
Bird GH, Madani N, Perry AF, Princiotto AM, Supko JG, He X, Gavathiotis E, Sodroski
JG, Walensky LD (2010) Hydrocarbon double-stapling remedies the proteolytic
instability of a lengthy peptide therapeutic. Proceedings of the National Academy
of Sciences 107:14093.
Birdsall NJ, Lazareno S, Popham A, Saldanha J (2001) Multiple allosteric sites on
muscarinic receptors. Life sciences 68:2517-2524.
Birnbaum MJ, Clem RJ, Miller LK (1994) An apoptosis-inhibiting gene from a nuclear
polyhedrosis virus encoding a polypeptide with Cys/His sequence motifs. Journal
of virology 68:2521-2528.

191
Bixby KA, Nanao MH, Shen NV, Kreusch A, Bellamy H, Pfaffinger PJ, Choe S (1999)
Zn2+-binding and molecular determinants of tetramerization in voltage-gated K+
channels. Nature structural biology 6:38-43.
Blackwell HE, Grubbs RH (1998) Highly efficient synthesis of covalently cross linked
peptide helices by ring closing metathesis. Angewandte Chemie International
Edition 37:3281-3284.
Blanchard H, Donepudi M, Tschopp M, Kodandapani L, Wu JC, Grutter MG (2000)
Caspase-8 specificity probed at subsite S(4): crystal structure of the caspase-8-Z-
DEVD-cho complex. Journal of molecular biology 302:9-16.
Blundell T, Pitts J, Tickle I, Wood S, Wu CW (1981) X-ray analysis (1. 4-Å resolution)
of avian pancreatic polypeptide: Small globular protein hormone. Proceedings of
the National Academy of Sciences 78:4175.
Blundell TL, Pitts JE, Tickle IJ, Wood SP, Wu CW (1981) X-Ray Analysis (1. 4-
angstrom Resolution) of Avian Pancreatic Polypeptide: Small Globular Protein
Hormone. Proceedings of the National Academy of Sciences 78:4175-4179.
Boldin MP, Varfolomeev EE, Pancer Z, Mett IL, Camonis JH, Wallach D (1995) A novel
protein that interacts with the death domain of Fas/APO1 contains a sequence
motif related to the death domain. The Journal of biological chemistry 270:7795-
7798.
Bose K, Clark AC (2001) Dimeric procaspase-3 unfolds via a four-state equilibrium
process. Biochemistry 40:14236-14242.
Bozym RA, Thompson RB, Stoddard AK, Fierke CA (2006) Measuring picomolar
intracellular exchangeable zinc in PC-12 cells using a ratiometric fluorescence
biosensor. ACS chemical biology 1:103-111.
Brady KD, Giegel DA, Grinnell C, Lunney E, Talanian RV, Wong W, Walker N (1999)
A catalytic mechanism for caspase-1 and for bimodal inhibition of caspase-1 by
activated aspartic ketones. Bioorganic & medicinal chemistry 7:621-631.
Brand IA, Kleineke J (1996) Intracellular zinc movement and its effect on the
carbohydrate metabolism of isolated rat hepatocytes. The Journal of biological
chemistry 271:1941-1949.

192
Brunel FM, Dawson PE (2005) Synthesis of constrained helical peptides by thioether
ligation: application to analogs of gp41. Chem Commun:2552-2554.
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta-cell
deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes
52:102-110.
Cain K, Bratton SB, Langlais C, Walker G, Brown DG, Sun XM, Cohen GM (2000)
Apaf-1 oligomerizes into biologically active approximately 700-kDa and inactive
approximately 1.4-MDa apoptosome complexes. The Journal of biological
chemistry 275:6067-6070.
Cain K, Brown DG, Langlais C, Cohen GM (1999) Caspase activation involves the
formation of the aposome, a large ( 700 kDa) caspase-activating complex. Journal
of Biological Chemistry 274:22686.
Cain K, Brown DG, Langlais C, Cohen GM (1999) Caspase activation involves the
formation of the aposome, a large (approximately 700 kDa) caspase-activating
complex. The Journal of biological chemistry 274:22686-22692.
Calabrese F, Pontisso P, Pettenazzo E, Benvegnu L, Vario A, Chemello L, Alberti A,
Valente M (2000) Liver cell apoptosis in chronic hepatitis C correlates with
histological but not biochemical activity or serum HCV-RNA levels. Hepatology
31:1153-1159.
Carmona-Gutierrez D, Eisenberg T, Buttner S, Meisinger C, Kroemer G, Madeo F (2010)
Apoptosis in yeast: triggers, pathways, subroutines. Cell death and differentiation
17:763-773.
Carter JE, Truong-Tran AQ, Grosser D, Ho L, Ruffin RE, Zalewski PD (2002)
Involvement of redox events in caspase activation in zinc-depleted airway
epithelial cells. Biochemical and biophysical research communications 297:1062-
1070.
Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL (2003) Q-VD-OPh, a broad
spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis : an
international journal on programmed cell death 8:345-352.

193
Chai F, Truong-Tran AQ, Ho LH, Zalewski PD (1999) Regulation of caspase activation
and apoptosis by cellular zinc fluxes and zinc deprivation: A review. Immunology
and cell biology 77:272-278.
Chai J, Shiozaki E, Srinivasula SM, Wu Q, Datta P, Alnemri ES, Shi Y (2001) Structural
basis of caspase-7 inhibition by XIAP. Cell 104:769-780.
Chao Y, Shiozaki EN, Srinivasula SM, Rigotti DJ, Fairman R, Shi Y (2005) Engineering
a dimeric caspase-9: a re-evaluation of the induced proximity model for caspase
activation. PLoS biology 3:e183.
Chereau D, Kodandapani L, Tomaselli KJ, Spada AP, Wu JC (2003) Structural and
functional analysis of caspase active sites. Biochemistry 42:4151-4160.
Chimienti F, Seve M, Richard S, Mathieu J, Favier A (2001) Role of cellular zinc in
programmed cell death: temporal relationship between zinc depletion, activation
of caspases, and cleavage of Sp family transcription factors. Biochemical
pharmacology 62:51-62.
Chin JW, Grotzfeld RM, Fabian MA, Schepartz A (2001) Methodology for optimizing
functional miniature proteins based on avian pancreatic polypeptide using phage
display. Bioorganic & medicinal chemistry letters 11:1501-1505.
Chin JW, Schepartz A (2001) Concerted evolution of structure and function in a
miniature protein. Journal of the American Chemical Society 123:2929-2930.
Chin JW, Schepartz A (2001) Design and evolution of a miniature Bcl 2 binding protein.
Angewandte Chemie 113:3922-3925.
Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM (1995) FADD, a novel death domain-
containing protein, interacts with the death domain of Fas and initiates apoptosis.
Cell 81:505-512.
Choe Y, Leonetti F, Greenbaum DC, Lecaille F, Bogyo M, Bromme D, Ellman JA, Craik
CS (2006) Substrate Profiling of Cysteine Proteases Using a Combinatorial
Peptide Library Identifies Functionally Unique Specificities. Journal of Biological
Chemistry 281:12824.

194
Coe DM, Perciaccante R, Procopiou PA (2003) Potassium trimethylsilanolate induced
cleavage of 1, 3-oxazolidin-2-and 5-ones, and application to the synthesis of (< i>
R</i>)-salmeterol. Org Biomol Chem 1:1106-1111.
Coleman JE (1992) Zinc proteins: enzymes, storage proteins, transcription factors, and
replication proteins. Annual review of biochemistry 61:897-946.
Crook NE, Clem RJ, Miller LK (1993) An apoptosis-inhibiting baculovirus gene with a
zinc finger-like motif. Journal of virology 67:2168-2174.
Cuerrier D, Moldoveanu T, Davies PL (2005) Determination of Peptide Substrate
Specificity for {micro}-Calpain by a Peptide Library-based Approach: The
Importance of Primed Side Interactions. Journal of Biological Chemistry
280:40632.
Cvetkovic A, Menon AL, Thorgersen MP, Scott JW, Poole FL, 2nd, Jenney FE, Jr.,
Lancaster WA, Praissman JL, Shanmukh S, Vaccaro BJ, Trauger SA, Kalisiak E,
Apon JV, Siuzdak G, Yannone SM, Tainer JA, Adams MW (2010) Microbial
metalloproteomes are largely uncharacterized. Nature 466:779-782.
Davoodi J, Mohammad-Gholi A, Es-Haghi A, MacKenzie A (2007) W323S variant of
Xiap-Bir3 binds to SMAC but not caspase-9. Journal of biochemistry 141:293-
299.
Denault JB, Salvesen GS (2003) Human caspase-7 activity and regulation by its N-
terminal peptide. The Journal of biological chemistry 278:34042-34050.
Deveraux QL, Reed JC (1999) IAP family proteins--suppressors of apoptosis. Genes &
development 13:239-252.
Deveraux QL, Takahashi R, Salvesen GS, Reed JC (1997) X-linked IAP is a direct
inhibitor of cell-death proteases. Nature 388:300-304.
Donepudi M, Mac Sweeney A, Briand C, Grutter MG (2003) Insights into the regulatory
mechanism for caspase-8 activation. Molecular cell 11:543-549.
Eakin CM, Knight JD, Morgan CJ, Gelfand MA, Miranker AD (2002) Formation of a
copper specific binding site in non-native states of beta-2-microglobulin.
Biochemistry 41:10646-10656.

195
Eckelman BP, Salvesen GS (2006) The human anti-apoptotic proteins cIAP1 and cIAP2
bind but do not inhibit caspases. The Journal of biological chemistry 281:3254-
3260.
El-Mousawi M, Tchistiakova L, Yurchenko L, Pietrzynski G, Moreno M, Stanimirovic
D, Ahmad D, Alakhov V (2003) A Vascular Endothelial Growth Factor High
Affinity Receptor 1-specific Peptide with Antiangiogenic Activity Identified
Using a Phage Display Peptide Library*. Journal of Biological Chemistry
278:46681-46691.
Enninga J, Mounier J, Sansonetti P, Tran Van Nhieu G (2005) Secretion of type III
effectors into host cells in real time. Nat Methods 2:959-965.
Fairbrother WJ, Christinger HW, Cochran AG, Fuh G, Keenan CJ, Quan C, Shriver SK,
Tom JYK, Wells JA, Cunningham BC (1998) Novel peptides selected to bind
vascular endothelial growth factor target the receptor-binding site. Biochemistry
37:17754-17764.
Fatemi M, Hermann A, Pradhan S, Jeltsch A (2001) The activity of the murine DNA
methyltransferase Dnmt1 is controlled by interaction of the catalytic domain with
the N-terminal part of the enzyme leading to an allosteric activation of the
enzyme after binding to methylated DNA. Journal of molecular biology
309:1189-1199.
Feeney B, Clark AC (2005) Reassembly of active caspase-3 is facilitated by the
propeptide. The Journal of biological chemistry 280:39772-39785.
Feeney B, Pop C, Swartz P, Mattos C, Clark AC (2006) Role of loop bundle hydrogen
bonds in the maturation and activity of (Pro)caspase-3. Biochemistry 45:13249-
13263.
Feeney B, Soderblom EJ, Goshe MB, Clark AC (2006) Novel protein purification system
utilizing an N-terminal fusion protein and a caspase-3 cleavable linker. Protein
Expression and Purification 47:311-318.
Felix AM, Heimer EP, Wang CT, Lambros TJ, Fournier A, Mowles TF, Maines S,
Campbell RM, Wegrzynski BB, Toome V, et al. (1988) Synthesis, biological
activity and conformational analysis of cyclic GRF analogs. Int J Pept Protein Res
32:441-454.

196
Ferguson AD, Amezcua CA, Halabi NM, Chelliah Y, Rosen MK, Ranganathan R,
Deisenhofer J (2007) Signal transduction pathway of TonB-dependent
transporters. Proceedings of the National Academy of Sciences of the United
States of America 104:513-518.
Fesik SW (2000) Insights into programmed cell death through structural biology. Cell
103:273-282.
Fields GB, Noble RL (1990) Solid phase peptide synthesis utilizing 9
fluorenylmethoxycarbonyl amino acids. Int J Pept Protein Res 35:161-214.
Fuentes-Prior P, Salvesen GS (2004) The protein structures that shape caspase activity,
specificity, activation and inhibition. The Biochemical journal 384:201-232.
Fuentes EJ, Der CJ, Lee AL (2004) Ligand-dependent dynamics and intramolecular
signaling in a PDZ domain. Journal of molecular biology 335:1105-1115.
Fukuda H, Paredes SR, Batlle AM (1988) Active site histidine in pig liver aminolevulic
acid dehydratase modified by diethylpyrocarbonate and protected by Zn2+ ions.
Comparative biochemistry and physiology B, Comparative biochemistry 91:285-
291.
Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien
RY, Ellisman MH (2002) Multicolor and electron microscopic imaging of
connexin trafficking. Science 296:503-507.
Garaud M, Pei D (2007) Substrate profiling of protein tyrosine phosphatase PTP1B by
screening a combinatorial peptide library. J Am Chem Soc 129:5366-5367.
Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA (1998)
Inhibition of human caspases by peptide-based and macromolecular inhibitors.
The Journal of biological chemistry 273:32608-32613.
Garcia-Calvo M, Peterson EP, Rasper DM, Vaillancourt JP, Zamboni R, Nicholson DW,
Thornberry NA (1999) Purification and catalytic properties of human caspase
family members. Cell death and differentiation 6:362-369.

197
Ge X, Fu YM, Li YQ, Meadows GG (2002) Activation of caspases and cleavage of Bid
are required for tyrosine and phenylalanine deficiency-induced apoptosis of
human A375 melanoma cells. Archives of biochemistry and biophysics 403:50-
58.
Ghadiri MR, Choi C (1990) Secondary structure nucleation in peptides. Transition metal
ion stabilized. alpha.-helices. Journal of the American Chemical Society
112:1630-1632.
Glover I, Haneef I, Pitts J, Wood S, Moss D, Tickle I, Blundell T (1983) Conformational
flexibility in a small globular hormone: X ray analysis of avian pancreatic
polypeptide at 0.98 Å resolution. Biopolymers 22:293-304.
Goers J, Uversky VN, Fink AL (2003) Polycation-induced oligomerization and
accelerated fibrillation of human alpha-synuclein in vitro. Protein science : a
publication of the Protein Society 12:702-707.
Gopal VK, Francis SH, Corbin JD (2001) Allosteric sites of phosphodiesterase-5 (PDE5).
A potential role in negative feedback regulation of cGMP signaling in corpus
cavernosum. European journal of biochemistry / FEBS 268:3304-3312.
Gosalia DN, Salisbury CM, Ellman JA, Diamond SL (2005) High Throughput Substrate
Specificity Profiling of Serine and Cysteine Proteases Using Solution-phase
Fluorogenic Peptide Microarrays*. Molecular & Cellular Proteomics 4:626-636.
Gosalia DN, Salisbury CM, Maly DJ, Ellman JA, Diamond SL (2005) Profiling serine
protease substrate specificity with solution phase fluorogenic peptide microarrays.
PROTEOMICS 5:1292-1298.
Griffin BA, Adams SR, Tsien RY (1998) Specific covalent labeling of recombinant
protein molecules inside live cells. Science 281:269-272.
Gross A, Jockel J, Wei MC, Korsmeyer SJ (1998) Enforced dimerization of BAX results
in its translocation, mitochondrial dysfunction and apoptosis. The EMBO journal
17:3878-3885.
Guan C, Li P, Riggs PD, Inouye H (1988) Vectors that facilitate the expression and
purification of foreign peptides in Escherichia coli by fusion to maltose-binding
protein. Gene 67:21-30.

198
Haase H, Maret W (2005) Protein tyrosine phosphatases as targets of the combined
insulinomimetic effects of zinc and oxidants. Biometals 18:333-338.
Hardy JA, Lam J, Nguyen JT, O'Brien T, Wells JA (2004) Discovery of an allosteric site
in the caspases. Proceedings of the National Academy of Sciences of the United
States of America 101:12461-12466.
Harris JL, Backes BJ, Leonetti F, Mahrus S, Ellman JA, Craik CS. Rapid and general
profiling of protease specificity by using combinatorial fluorogenic substrate
libraries. (2000). National Acad Sciences, pp. 7754-7759.
Hatley ME, Lockless SW, Gibson SK, Gilman AG, Ranganathan R (2003) Allosteric
determinants in guanine nucleotide-binding proteins. Proceedings of the National
Academy of Sciences of the United States of America 100:14445-14450.
Helmersson A, von Arnold S, Bozhkov PV (2008) The level of free intracellular zinc
mediates programmed cell death/cell survival decisions in plant embryos. Plant
physiology 147:1158-1167.
Henchey LK, Jochim AL, Arora PS (2008) Contemporary strategies for the stabilization
of peptides in the [alpha]-helical conformation. Current opinion in chemical
biology 12:692-697.
Herker E, Jungwirth H, Lehmann KA, Maldener C, Frohlich KU, Wissing S, Buttner S,
Fehr M, Sigrist S, Madeo F (2004) Chronological aging leads to apoptosis in
yeast. J Cell Biol 164:501-507.
Hodges AM, Schepartz A (2007) Engineering a monomeric miniature protein. Journal of
the American Chemical Society 129:11024-11025.
Holm RH, Kennepohl P, Solomon EI (1996) Structural and Functional Aspects of Metal
Sites in Biology. Chemical reviews 96:2239-2314.
Hong R, Han G, Fernandez JM, Kim BJ, Forbes NS, Rotello VM (2006) Glutathione-
mediated delivery and release using monolayer protected nanoparticle carriers. J
Am Chem Soc 128:1078-1079.

199
Hotchkiss RS, Chang KC, Swanson PE, Tinsley KW, Hui JJ, Klender P, Xanthoudakis S,
Roy S, Black C, Grimm E, Aspiotis R, Han Y, Nicholson DW, Karl IE (2000)
Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte.
Nature immunology 1:496-501.
Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, Korsmeyer
SJ, Karl IE (1999) Prevention of lymphocyte cell death in sepsis improves
survival in mice. Proceedings of the National Academy of Sciences of the United
States of America 96:14541-14546.
Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE, Jr., Hui JJ, Chang KC, Osborne
DF, Freeman BD, Cobb JP, Buchman TG, Karl IE (2001) Sepsis-induced
apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes
in humans. J Immunol 166:6952-6963.
Hu Y, Benedict MA, Ding L, Nunez G (1999) Role of cytochrome c and dATP/ATP
hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. The EMBO
journal 18:3586-3595.
Huber KL, Olson KD, Hardy JA (2009) Robust production of a peptide library using
methodological synchronization. Protein expression and purification 67:139-147.
Hutti JE, Jarrell ET, Chang JD, Abbott DW, Storz P, Toker A, Cantley LC, Turk BE
(2004) A rapid method for determining protein kinase phosphorylation specificity.
Nature Methods 1:27-29.
Huyer G, Kelly J, Moffat J, Zamboni R, Jia Z, Gresser MJ, Ramachandran C (1998)
Affinity Selection from Peptide Libraries to Determine Substrate Specificity of
Protein Tyrosine Phosphatases. Analytical Biochemistry 258:19-30.
Ibarra CA, Blouse GE, Christian TD, Shore JD (2004) The contribution of the exosite
residues of plasminogen activator inhibitor-1 to proteinase inhibition. The Journal
of biological chemistry 279:3643-3650.
Ignatova Z, Gierasch LM (2004) Monitoring protein stability and aggregation in vivo by
real-time fluorescent labeling. Proceedings of the National Academy of Sciences
of the United States of America 101:523-528.
Ignatova Z, Gierasch LM (2009) A method for direct measurement of protein stability in
vivo. Methods Mol Biol 490:165-178.

200
Ignatova Z, Krishnan B, Bombardier JP, Marcelino AM, Hong J, Gierasch LM (2007)
From the test tube to the cell: exploring the folding and aggregation of a beta-
clam protein. Biopolymers 88:157-163.
Inoue H, Tsukita K, Iwasato T, Suzuki Y, Tomioka M, Tateno M, Nagao M, Kawata A,
Saido TC, Miura M, Misawa H, Itohara S, Takahashi R (2003) The crucial role of
caspase-9 in the disease progression of a transgenic ALS mouse model. The
EMBO journal 22:6665-6674.
Jabbour AM, Ekert PG, Coulson EJ, Knight MJ, Ashley DM, Hawkins CJ (2002) The
p35 relative, p49, inhibits mammalian and Drosophila caspases including
DRONC and protects against apoptosis. Cell death and differentiation 9:1311-
1320.
Jackson DY, King DS, Chmielewski J, Singh S, Schultz PG (1991) General approach to
the synthesis of short. alpha.-helical peptides. Journal of the American Chemical
Society 113:9391-9392.
Jacobson MD, Weil M, Raff MC (1997) Programmed cell death in animal development.
Cell 88:347-354.
Jahnke A, Wilmes T, Adebahr S, Pfeifer D, Berg T, Trepel M (2007) Leukemia targeting
ligands isolated from phage display peptide libraries. Leukemia 21:411.
Jeong MY, Kim S, Yun CW, Choi YJ, Cho SG (2007) Engineering a de novo internal
disulfide bridge to improve the thermal stability of xylanase from Bacillus
stearothermophilus No. 236. Journal of biotechnology 127:300-309.
Jones DP, Brown LA, Sternberg P (1995) Variability in glutathione-dependent
detoxication in vivo and its relevance to detoxication of chemical mixtures.
Toxicology 105:267-274.
Karle IL, Balaram P (1990) Structural characteristics of. alpha.-helical peptide molecules
containing Aib residues. Biochemistry 29:6747-6756.
Kashiwagi M, Enghild JJ, Gendron C, Hughes C, Caterson B, Itoh Y, Nagase H (2004)
Altered proteolytic activities of ADAMTS-4 expressed by C-terminal processing.
The Journal of biological chemistry 279:10109-10119.

201
Keel M, Ungethum U, Steckholzer U, Niederer E, Hartung T, Trentz O, Ertel W (1997)
Interleukin-10 counterregulates proinflammatory cytokine-induced inhibition of
neutrophil apoptosis during severe sepsis. Blood 90:3356-3363.
Kelso MJ, Beyer RL, Hoang HN, Lakdawala AS, Snyder JP, Oliver WV, Robertson TA,
Appleton TG, Fairlie DP (2004) Alpha-Turn Mimetics: Short Peptide-Helices
Composed of Cyclic Metallopentapeptide Modules. Journal of the American
Chemical Society 126.
Kempkensteffen C, Hinz S, Christoph F, Krause H, Magheli A, Schrader M, Schostak M,
Miller K, Weikert S (2008) Expression levels of the mitochondrial IAP
antagonists Smac/DIABLO and Omi/HtrA2 in clear-cell renal cell carcinomas
and their prognostic value. Journal of cancer research and clinical oncology
134:543-550.
Kempkensteffen C, Jager T, Bub J, Weikert S, Hinz S, Christoph F, Krause H, Schostak
M, Miller K, Schrader M (2007) The equilibrium of XIAP and Smac/DIABLO
expression is gradually deranged during the development and progression of
testicular germ cell tumours. International journal of andrology 30:476-483.
Kenji Tonan YK, and Kozo Hamaguchi (1990) Conformations of isolated fragments of
pancreatic polypeptide. Biochemistry 29:4424-4429.
Kent SBH. Peptides:Structure and Function. In: CM Deber VHaKK, Ed. (1985)
Proceedings of the 9th Annual American Peptide Symposium. Rockford, pp. 407-
414.
Khan MA, Chock PB, Stadtman ER (2005) Knockout of caspase-like gene, YCA1,
abrogates apoptosis and elevates oxidized proteins in Saccharomyces cerevisiae.
Proceedings of the National Academy of Sciences of the United States of America
102:17326-17331.
Kiechle T, Dedeoglu A, Kubilus J, Kowall NW, Beal MF, Friedlander RM, Hersch SM,
Ferrante RJ (2002) Cytochrome C and caspase-9 expression in Huntington's
disease. Neuromolecular medicine 1:183-195.
Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME
(1995) Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a
death-inducing signaling complex (DISC) with the receptor. The EMBO journal
14:5579-5588.

202
Kohler JE, Dubach JM, Naik HB, Tai K, Blass AL, Soybel DI (2010) Monochloramine-
induced toxicity and dysregulation of intracellular Zn2+ in parietal cells of rabbit
gastric glands. American journal of physiology Gastrointestinal and liver
physiology 299:G170-178.
Kohler JE, Mathew J, Tai K, Blass AL, Kelly E, Soybel DI (2009) Monochloramine
impairs caspase-3 through thiol oxidation and Zn2+ release. The Journal of
surgical research 153:121-127.
Kong Y, Karplus M (2009) Signaling pathways of PDZ2 domain: a molecular dynamics
interaction correlation analysis. Proteins 74:145-154.
Korichneva I, Hoyos B, Chua R, Levi E, Hammerling U (2002) Zinc release from protein
kinase C as the common event during activation by lipid second messenger or
reactive oxygen. Journal of Biological Chemistry 277:44327.
Kountouras J, Zavos C, Chatzopoulos D (2003) Apoptosis in hepatitis C. Journal of viral
hepatitis 10:335-342.
Krezel A, Hao Q, Maret W (2007) The zinc/thiolate redox biochemistry of
metallothionein and the control of zinc ion fluctuations in cell signaling. Archives
of biochemistry and biophysics 463:188-200.
Krezel A, Maret W (2006) Zinc-buffering capacity of a eukaryotic cell at physiological
pZn. Journal of biological inorganic chemistry : JBIC : a publication of the
Society of Biological Inorganic Chemistry 11:1049-1062.
Krishnan B, Gierasch LM (2008) Cross-strand split tetra-Cys motifs as structure sensors
in a beta-sheet protein. Chemistry & biology 15:1104-1115.
Kritzer JA, Zutshi R, Cheah M, Ran FA, Webman R, Wongjirad TM, Schepartz A (2006)
Miniature protein inhibitors of the p53-hDM2 interaction. ChemBioChem 7:29-
31.
Kurrer MO, Pakala SV, Hanson HL, Katz JD (1997) Beta cell apoptosis in T cell-
mediated autoimmune diabetes. Proceedings of the National Academy of Sciences
of the United States of America 94:213-218.

203
Lademann U, Cain K, Gyrd-Hansen M, Brown D, Peters D, Jaattela M (2003) Diarylurea
compounds inhibit caspase activation by preventing the formation of the active
700-kilodalton apoptosome complex. Molecular and cellular biology 23:7829-
7837.
LaVallie ER, DiBlasio EA, Kovacic S, Grant KL, Schendel PF, McCoy JM (1993) A
Thioredoxin Gene Fusion Expression System That Circumvents Inclusion Body
Formation in the E. coli Cytoplasm. Bio/Technology 11:187-193.
Lavrik I, Krueger A, Schmitz I, Baumann S, Weyd H, Krammer PH, Kirchhoff S (2003)
The active caspase-8 heterotetramer is formed at the CD95 DISC. Cell death and
differentiation 10:144-145.
Leduc AM, Trent JO, Wittliff JL, Bramlett KS, Briggs SL, Chirgadze NY, Wang Y,
Burris TP, Spatola AF (2003) Helix-stabilized cyclic peptides as selective
inhibitors of steroid receptor–coactivator interactions. Proceedings of the National
Academy of Sciences 100:11273.
Lee J, Natarajan M, Nashine VC, Socolich M, Vo T, Russ WP, Benkovic SJ,
Ranganathan R (2008) Surface sites for engineering allosteric control in proteins.
Science 322:438-442.
Lee JK, Prussia A, Snyder JP, Plemper RK (2007) Reversible inhibition of the fusion
activity of measles virus F protein by an engineered intersubunit disulfide bridge.
Journal of virology 81:8821-8826.
Li B, Tom JY, Oare D, Yen R, Fairbrother WJ, Wells JA, Cunningham BC (1995)
Minimization of a polypeptide hormone. Science 270:1657-1660.
Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG (2004) A small
molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death.
Science 305:1471-1474.
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997)
Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex
initiates an apoptotic protease cascade. Cell 91:479-489.

204
Linton SD, Aja T, Armstrong RA, Bai X, Chen LS, Chen N, Ching B, Contreras P, Diaz
JL, Fisher CD, Fritz LC, Gladstone P, Groessl T, Gu X, Herrmann J, Hirakawa
BP, Hoglen NC, Jahangiri KG, Kalish VJ, Karanewsky DS, Kodandapani L,
Krebs J, McQuiston J, Meduna SP, Nalley K, Robinson ED, Sayers RO, Sebring
K, Spada AP, Ternansky RJ, Tomaselli KJ, Ullman BR, Valentino KL, Weeks S,
Winn D, Wu JC, Yeo P, Zhang CZ (2005) First-in-class pan caspase inhibitor
developed for the treatment of liver disease. Journal of medicinal chemistry
48:6779-6782.
Liu HR, Gao E, Hu A, Tao L, Qu Y, Most P, Koch WJ, Christopher TA, Lopez BL,
Alnemri ES, Zervos AS, Ma XL (2005) Role of Omi/HtrA2 in apoptotic cell
death after myocardial ischemia and reperfusion. Circulation 111:90-96.
Liu X, Kim CN, Yang J, Jemmerson R, Wang X (1996) Induction of apoptotic program
in cell-free extracts: requirement for dATP and cytochrome c. Cell 86:147-157.
Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, Herrmann J, Wu JC, Fesik
SW (2000) Structural basis for binding of Smac/DIABLO to the XIAP BIR3
domain. Nature 408:1004-1008.
Lockless SW, Ranganathan R (1999) Evolutionarily conserved pathways of energetic
connectivity in protein families. Science 286:295-299.
Lonovics J, Devitt P, Watson LC, Rayford PL, Thompson JC (1981) Pancreatic
polypeptide: A review. Archives of Surgery 116:1256.
Ludewig R, Dong J, Zou H, Scriba GK (2010) Separation of peptide diastereomers using
CEC and a hydrophobic monolithic column. Journal of separation science
33:1085-1089.
Luedtke NW, Dexter RJ, Fried DB, Schepartz A (2007) Surveying polypeptide and
protein domain conformation and association with FlAsH and ReAsH. Nature
chemical biology 3:779-784.
Luque I, Leavitt SA, Freire E (2002) The linkage between protein folding and functional
cooperativity: two sides of the same coin? Annual review of biophysics and
biomolecular structure 31:235-256.
Lyskov S, Gray JJ (2008) The RosettaDock server for local protein-protein docking.
Nucleic acids research 36:W233-238.

205
Ma K, Temiakov D, Anikin M, McAllister WT (2005) Probing conformational changes
in T7 RNA polymerase during initiation and termination by using engineered
disulfide linkages. Proceedings of the National Academy of Sciences of the
United States of America 102:17612-17617.
Madeo F, Frohlich E, Frohlich KU (1997) A yeast mutant showing diagnostic markers of
early and late apoptosis. J Cell Biol 139:729-734.
Madeo F, Frohlich E, Ligr M, Grey M, Sigrist SJ, Wolf DH, Frohlich KU (1999) Oxygen
stress: a regulator of apoptosis in yeast. J Cell Biol 145:757-767.
Madeo F, Herker E, Maldener C, Wissing S, Lachelt S, Herlan M, Fehr M, Lauber K,
Sigrist SJ, Wesselborg S, Frohlich KU (2002) A caspase-related protease
regulates apoptosis in yeast. Molecular cell 9:911-917.
Mah AS, Elia AEH, Devgan G, Ptacek J, Schutkowski M, Snyder M, Yaffe MB,
Deshaies RJ (2005) Substrate specificity analysis of protein kinase complex Dbf2-
Mob1 by peptide library and proteome array screening. BMC Biochemistry 6:22.
Malet G, Martin AG, Orzaez M, Vicent MJ, Masip I, Sanclimens G, Ferrer-Montiel A,
Mingarro I, Messeguer A, Fearnhead HO, Perez-Paya E (2006) Small molecule
inhibitors of Apaf-1-related caspase- 3/-9 activation that control mitochondrial-
dependent apoptosis. Cell death and differentiation 13:1523-1532.
Malladi S, Challa-Malladi M, Fearnhead HO, Bratton SB (2009) The Apaf-1*procaspase-
9 apoptosome complex functions as a proteolytic-based molecular timer. The
EMBO journal 28:1916-1925.
Manns J, Daubrawa M, Driessen S, Paasch F, Hoffmann N, Loffler A, Lauber K, Dieterle
A, Alers S, Iftner T, Schulze-Osthoff K, Stork B, Wesselborg S (2011) Triggering
of a novel intrinsic apoptosis pathway by the kinase inhibitor staurosporine:
activation of caspase-9 in the absence of Apaf-1. The FASEB journal : official
publication of the Federation of American Societies for Experimental Biology
25:3250-3261.
Maret W, Jacob C, Vallee BL, Fischer EH (1999) Inhibitory sites in enzymes: zinc
removal and reactivation by thionein. Proceedings of the National Academy of
Sciences of the United States of America 96:1936-1940.

206
Maret W, Yetman CA, Jiang L (2001) Enzyme regulation by reversible zinc inhibition:
glycerol phosphate dehydrogenase as an example. Chemico-biological
interactions 130-132:891-901.
Marshall GR, Hodgkin EE, Langs DA, Smith GD, Zabrocki J, Leplawy MT (1990)
Factors governing helical preference of peptides containing multiple alpha, alpha-
dialkyl amino acids. Proceedings of the National Academy of Sciences 87:487.
Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME
(1997) FLICE is activated by association with the CD95 death-inducing signaling
complex (DISC). The EMBO journal 16:2794-2804.
Meergans T, Hildebrandt AK, Horak D, Haenisch C, Wendel A (2000) The short
prodomain influences caspase-3 activation in HeLa cells. The Biochemical
journal 349:135-140.
Meier P, Finch A, Evan G (2000) Apoptosis in development. Nature 407:796-801.
Mesner PW, Bible KC, Martins LM, Kottke TJ, Srinivasula SM, Svingen PA, Chilcote
TJ, Basi GS, Tung JS, Krajewski S (1999) Characterization of caspase processing
and activation in HL-60 cell cytosol under cell-free conditions. Journal of
Biological Chemistry 274:22635.
Millhauser GL (2004) Copper binding in the prion protein. Acc Chem Res 37:79-85.
Mitchinson C, Wells JA (1989) Protein engineering of disulfide bonds in subtilisin BPN'.
Biochemistry 28:4807-4815.
Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, Hoffman
B, Reed JC (1994) Tumor suppressor p53 is a regulator of bcl-2 and bax gene
expression in vitro and in vivo. Oncogene 9:1799-1805.
Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of
the human bax gene. Cell 80:293-299.

207
Mizutani Y, Nakanishi H, Yamamoto K, Li YN, Matsubara H, Mikami K, Okihara K,
Kawauchi A, Bonavida B, Miki T (2005) Downregulation of Smac/DIABLO
expression in renal cell carcinoma and its prognostic significance. Journal of
clinical oncology : official journal of the American Society of Clinical Oncology
23:448-454.
Mocanu MM, Baxter GF, Yellon DM (2000) Caspase inhibition and limitation of
myocardial infarct size: protection against lethal reperfusion injury. British
journal of pharmacology 130:197-200.
Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, Kung AL,
Gilliland DG, Verdine GL, Bradner JE (2009) Direct inhibition of the NOTCH
transcription factor complex. Nature 462:182-188.
Montclare JK, Schepartz A (2003) Miniature homeodomains: high specificity without an
N-terminal arm. Journal of the American Chemical Society 125:3416-3417.
Morgan CJ, Gelfand M, Atreya C, Miranker AD (2001) Kidney dialysis-associated
amyloidosis: a molecular role for copper in fiber formation. Journal of molecular
biology 309:339-345.
Mueller T, Voigt W, Simon H, Fruehauf A, Bulankin A, Grothey A, Schmoll HJ (2003)
Failure of activation of caspase-9 induces a higher threshold for apoptosis and
cisplatin resistance in testicular cancer. Cancer research 63:513-521.
Muslin EH, Li D, Stevens FJ, Donnelly M, Schiffer M, Anderson LE (1995) Engineering
a domain-locking disulfide into a bacterial malate dehydrogenase produces a
redox-sensitive enzyme. Biophysical journal 68:2218-2223.
Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C,
Bretz JD, Zhang M, Gentz R (1996) FLICE, a novel FADD-homologous
ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing
signaling complex. Cell 85:817-827.
Naganagowda GA, Gururaja TL, Levine MJ (1998) Delineation of conformational
preferences in human salivary statherin by 1H, 31P NMR and CD studies:
sequential assignment and structure-function correlations. Journal of biomolecular
structure & dynamics 16:91-107.

208
Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53.
Molecular cell 7:683-694.
Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, Schmidt U,
Semigran MJ, Dec GW, Khaw BA (1996) Apoptosis in myocytes in end-stage
heart failure. The New England journal of medicine 335:1182-1189.
Navath RS, Kurtoglu YE, Wang B, Kannan S, Romero R, Kannan RM (2008)
Dendrimer-drug conjugates for tailored intracellular drug release based on
glutathione levels. Bioconjugate chemistry 19:2446-2455.
Newman JRS, Keating AE. Comprehensive Identification of Human bZIP Interactions
with Coiled-Coil Arrays. (2003). American Association for the Advancement of
Science, pp. 2097-2101.
Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y,
Griffin PR, Labelle M, Lazebnik YA, et al. (1995) Identification and inhibition of
the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376:37-43.
Nogal ML, Gonzalez de Buitrago G, Rodriguez C, Cubelos B, Carrascosa AL, Salas ML,
Revilla Y (2001) African swine fever virus IAP homologue inhibits caspase
activation and promotes cell survival in mammalian cells. Journal of virology
75:2535-2543.
O'Brien BA, Harmon BV, Cameron DP, Allan DJ (1997) Apoptosis is the mode of beta-
cell death responsible for the development of IDDM in the nonobese diabetic
(NOD) mouse. Diabetes 46:750-757.
Obata T, Yaffe MB, Leparc GG, Piro ET, Maegawa H, Kashiwagi A, Kikkawa R,
Cantley LC (2000) Peptide and Protein Library Screening Defines Optimal
Substrate Motifs for AKT/PKB. Journal of Biological Chemistry 275:36108-
36115.
Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T,
Tanaka N (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate
mediator of p53-induced apoptosis. Science 288:1053-1058.

209
Olivetti G, Quaini F, Sala R, Lagrasta C, Corradi D, Bonacina E, Gambert SR, Cigola E,
Anversa P (1996) Acute myocardial infarction in humans is associated with
activation of programmed myocyte cell death in the surviving portion of the heart.
Journal of molecular and cellular cardiology 28:2005-2016.
Osapay G, Taylor JW (1992) Multicyclic polypeptide model compounds. 2. Synthesis
and conformational properties of a highly. alpha.-helical uncosapeptide
constrained by three side-chain to side-chain lactam bridges. Journal of the
American Chemical Society 114:6966-6973.
Ota N, Agard DA (2005) Intramolecular signaling pathways revealed by modeling
anisotropic thermal diffusion. Journal of molecular biology 351:345-354.
Ouyang D, Shah N, Zhang H, Smith SC, Parekh HS (2009) Reducible disulfide-based
non-viral gene delivery systems. Mini reviews in medicinal chemistry 9:1242-
1250.
Oztas P, Lortlar N, Polat M, Alli N, Omeroglu S, Basman A (2007) Caspase-9 expression
is increased in endothelial cells of active Behcet's disease patients. International
journal of dermatology 46:172-176.
Palacios-Rodriguez Y, Garcia-Lainez G, Sancho M, Gortat A, Orzaez M, Perez-Paya E
(2011) Polypeptide modulators of card-card-mediated protein-protein interactions.
The Journal of biological chemistry.
Palmerini F, Devilard E, Jarry A, Birg F, Xerri L (2001) Caspase 7 downregulation as an
immunohistochemical marker of colonic carcinoma. Human pathology 32:461-
467.
Park CM, Sun C, Olejniczak ET, Wilson AE, Meadows RP, Betz SF, Elmore SW, Fesik
SW (2005) Non-peptidic small molecule inhibitors of XIAP. Bioorg Med Chem
Lett 15:771-775.
Patgiri A, Jochim AL, Arora PS (2008) A hydrogen bond surrogate approach for
stabilization of short peptide sequences in -helical conformation. Accounts of
chemical research 41:1289-1300.

210
Perry DK, Smyth MJ, Stennicke HR, Salvesen GS, Duriez P, Poirier GG, Hannun YA
(1997) Zinc is a potent inhibitor of the apoptotic protease, caspase-3. A novel
target for zinc in the inhibition of apoptosis. The Journal of biological chemistry
272:18530-18533.
Peterson QP, Goode DR, West DC, Ramsey KN, Lee JJ, Hergenrother PJ (2009) PAC-1
activates procaspase-3 in vitro through relief of zinc-mediated inhibition. Journal
of molecular biology 388:144-158.
Pettit FK, Bare E, Tsai A, Bowie JU (2007) HotPatch: a statistical a pproach to finding
biologically relevant features on protein surfaces. Journal of molecular biology
369:863-879.
Petty KJ. 2000. Metal-Chelate Affinity Chromatography, John Wiley and Sons, Inc.
Phelan JC, Skelton NJ, Braisted AC, McDowell RS (1997) A general method for
constraining short peptides to an -helical conformation. Journal of the American
Chemical Society 119:455-460.
Phillips C, Bazin R, Bent A, Davies NL, Moore R, Pannifer AD, Pickford AR, Prior SH,
Read CM, Roberts LR (2011) Design and structure of stapled peptides binding to
estrogen receptors. Journal of the American Chemical Society.
Pop C, Timmer J, Sperandio S, Salvesen GS (2006) The apoptosome activates caspase-9
by dimerization. Molecular cell 22:269-275.
Postigo A, Cross JR, Downward J, Way M (2006) Interaction of F1L with the BH3
domain of Bak is responsible for inhibiting vaccinia-induced apoptosis. Cell death
and differentiation 13:1651-1662.
Prasad B, Balaram P (1984) The stereochemistry of peptides containing alpha-
aminoisobutyric acid. CRC critical reviews in biochemistry 16:307.
Procopiou PA, Ahmed M, Jeulin S, Perciaccante R (2003) Synthesis of (R)- -
benzylmethionine: a novel rearrangement during alkylation of the Seebach (R)-
methionine oxazolidinone. Organic & biomolecular chemistry 1:2853-2858.

211
Qin H, Srinivasula SM, Wu G, Fernandes-Alnemri T, Alnemri ES, Shi Y (1999)
Structural basis of procaspase-9 recruitment by the apoptotic protease-activating
factor 1. Nature 399:549-557.
Qureshi SH, Yang L, Manithody C, Iakhiaev AV, Rezaie AR (2009) Mutagenesis studies
toward understanding allostery in thrombin. Biochemistry 48:8261-8270.
Raina D, Pandey P, Ahmad R, Bharti A, Ren J, Kharbanda S, Weichselbaum R, Kufe D
(2005) c-Abl tyrosine kinase regulates caspase-9 autocleavage in the apoptotic
response to DNA damage. The Journal of biological chemistry 280:11147-11151.
Ray CA, Black RA, Kronheim SR, Greenstreet TA, Sleath PR, Salvesen GS, Pickup DJ
(1992) Viral inhibition of inflammation: cowpox virus encodes an inhibitor of the
interleukin-1 beta converting enzyme. Cell 69:597-604.
Reed JC, Tomaselli KJ (2000) Drug discovery opportunities from apoptosis research.
Current opinion in biotechnology 11:586-592.
Reiter J, Herker E, Madeo F, Schmitt MJ (2005) Viral killer toxins induce caspase-
mediated apoptosis in yeast. J Cell Biol 168:353-358.
Renatus M, Stennicke HR, Scott FL, Liddington RC, Salvesen GS (2001) Dimer
formation drives the activation of the cell death protease caspase 9. Proceedings
of the National Academy of Sciences of the United States of America 98:14250-
14255.
Reynolds KA, McLaughlin RN, Ranganathan R (2011) Hot spots for allosteric regulation
on protein surfaces. Cell 147:1564-1575.
Richardson JS (1981) The anatomy and taxonomy of protein structure. Advances in
protein chemistry 34:167-339.
Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC,
Salvesen GS (2001) Structural basis for the inhibition of caspase-3 by XIAP. Cell
104:791-800.
Rodriguez J, Lazebnik Y (1999) Caspase-9 and APAF-1 form an active holoenzyme.
Genes & development 13:3179-3184.

212
Rodriguez M, Li SSC, Harper JW, Songyang Z (2004) An Oriented Peptide Array
Library (OPAL) Strategy to Study Protein-Protein Interactions. Journal of
Biological Chemistry 279:8802.
Rogers SJ, Pratt CW, Whinna HC, Church FC (1992) Role of thrombin exosites in
inhibition by heparin cofactor II. The Journal of biological chemistry 267:3613-
3617.
Rohl CA, Strauss CEM, Misura K, Baker D (2004) Protein structure prediction using
Rosetta. Methods in enzymology 383:66-93.
Rotonda J, Nicholson DW, Fazil KM, Gallant M, Gareau Y, Labelle M, Peterson EP,
Rasper DM, Ruel R, Vaillancourt JP, Thornberry NA, Becker JW (1996) The
three-dimensional structure of apopain/CPP32, a key mediator of apoptosis.
Nature structural biology 3:619-625.
Ruan F, Chen Y, Hopkins PB (1990) Metal ion-enhanced helicity in synthetic peptides
containing unnatural, metal-ligating residues. Journal of the American Chemical
Society 112:9403-9404.
Russo A, DeGraff W, Friedman N, Mitchell JB (1986) Selective modulation of
glutathione levels in human normal versus tumor cells and subsequent differential
response to chemotherapy drugs. Cancer research 46:2845-2848.
Rutter J, Michnoff CH, Harper SM, Gardner KH, McKnight SL (2001) PAS kinase: an
evolutionarily conserved PAS domain-regulated serine/threonine kinase.
Proceedings of the National Academy of Sciences of the United States of America
98:8991-8996.
Ryan CA, Stennicke HR, Nava VE, Burch JB, Hardwick JM, Salvesen GS (2002)
Inhibitor specificity of recombinant and endogenous caspase-9. The Biochemical
journal 366:595-601.
Saleh A, Srinivasula SM, Acharya S, Fishel R, Alnemri ES (1999) Cytochrome c and
dATP-mediated oligomerization of Apaf-1 is a prerequisite for procaspase-9
activation. The Journal of biological chemistry 274:17941-17945.
Salvesen GS, Dixit VM (1999) Caspase activation: the induced-proximity model.
Proceedings of the National Academy of Sciences of the United States of America
96:10964-10967.

213
Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki LM (1997)
Apoptosis in human acute myocardial infarction. Circulation 95:320-323.
Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El-Deiry WS (2002) BID
regulation by p53 contributes to chemosensitivity. Nature cell biology 4:842-849.
Scaffidi C, Medema JP, Krammer PH, Peter ME (1997) FLICE is predominantly
expressed as two functionally active isoforms, caspase-8/a and caspase-8/b. The
Journal of biological chemistry 272:26953-26958.
Schafmeister CE, Po J, Verdine GL (2000) An all-hydrocarbon cross-linking system for
enhancing the helicity and metabolic stability of peptides. Journal of the
American Chemical Society 122:5891-5892.
Schimmer AD, Welsh K, Pinilla C, Wang Z, Krajewska M, Bonneau MJ, Pedersen IM,
Kitada S, Scott FL, Bailly-Maitre B, Glinsky G, Scudiero D, Sausville E,
Salvesen G, Nefzi A, Ostresh JM, Houghten RA, Reed JC (2004) Small-molecule
antagonists of apoptosis suppressor XIAP exhibit broad antitumor activity. Cancer
cell 5:25-35.
Schrantz N, Auffredou MT, Bourgeade MF, Besnault L, Leca G, Vazquez A (2001) Zinc-
mediated regulation of caspases activity: dose-dependent inhibition or activation
of caspase-3 in the human Burkitt lymphoma B cells (Ramos). Cell death and
differentiation 8:152-161.
Schrödinger L. The PyMOL Molecular Graphics System, Version 1.3, . (2010).
Scott FL, Denault JB, Riedl SJ, Shin H, Renatus M, Salvesen GS (2005) XIAP inhibits
caspase-3 and -7 using two binding sites: evolutionarily conserved mechanism of
IAPs. The EMBO journal 24:645-655.
Scriba GKE. Peptide Diastereomers, Separation of. (2006) Encyclopedia of Analytical
Chemistry.
Seebach D, Fadel A (1985) N, O Acetals from Pivalaldehyde and Amino Acids for the
Alkylation with Self Reproduction of the Center of Chirality. Enolates of 3
Benzoyl 2 (tert butyl) 1, 3 oxazolidin 5 ones. Helvetica chimica acta 68:1243-
1250.

214
Seeger MA, von Ballmoos C, Eicher T, Brandstatter L, Verrey F, Diederichs K, Pos KM
(2008) Engineered disulfide bonds support the functional rotation mechanism of
multidrug efflux pump AcrB. Nature structural & molecular biology 15:199-205.
Sekimura A, Konishi A, Mizuno K, Kobayashi Y, Sasaki H, Yano M, Fukai I, Fujii Y
(2004) Expression of Smac/DIABLO is a novel prognostic marker in lung cancer.
Oncology reports 11:797-802.
Shandiz AT, Capraro BR, Sosnick TR (2007) Intramolecular cross-linking evaluated as a
structural probe of the protein folding transition state. Biochemistry 46:13711-
13719.
Shi Y (2002) Mechanisms of caspase activation and inhibition during apoptosis.
Molecular cell 9:459-470.
Shi Y (2004) Caspase activation: revisiting the induced proximity model. Cell 117:855-
858.
Shimaoka M, Lu C, Palframan RT, von Andrian UH, McCormack A, Takagi J, Springer
TA (2001) Reversibly locking a protein fold in an active conformation with a
disulfide bond: integrin alphaL I domains with high affinity and antagonist
activity in vivo. Proceedings of the National Academy of Sciences of the United
States of America 98:6009-6014.
Shimba N, Nomura AM, Marnett AB, Craik CS (2004) Herpesvirus protease inhibition
by dimer disruption. Journal of virology 78:6657.
Shin H, Renatus M, Eckelman BP, Nunes VA, Sampaio CA, Salvesen GS (2005) The
BIR domain of IAP-like protein 2 is conformationally unstable: implications for
caspase inhibition. The Biochemical journal 385:1-10.
Shin H, Renatus M, Eckelman BP, Nunes VA, Sampaio CAM, Salvesen GS (2005) The
BIR domain of IAP-like protein 2 is conformationally unstable: implications for
caspase inhibition. Biochemical Journal 385:1.
Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, Alnemri ES, Fairman R,
Shi Y (2003) Mechanism of XIAP-mediated inhibition of caspase-9. Molecular
cell 11:519-527.

215
Shiozaki EN, Chai J, Shi Y (2002) Oligomerization and activation of caspase-9, induced
by Apaf-1 CARD. Proceedings of the National Academy of Sciences of the
United States of America 99:4197-4202.
Shu N, Zhou T, Hovmoller S (2008) Prediction of zinc-binding sites in proteins from
sequence. Bioinformatics 24:775-782.
Shulman AI, Larson C, Mangelsdorf DJ, Ranganathan R (2004) Structural determinants
of allosteric ligand activation in RXR heterodimers. Cell 116:417-429.
Simons KT, Strauss C, Baker D (2001) Prospects for ab initio protein structural
genomics1. Journal of molecular biology 306:1191-1199.
Singh R, Lillard JW, Jr. (2009) Nanoparticle-based targeted drug delivery. Experimental
and molecular pathology 86:215-223.
Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed
JC, Nicholson DW, Alnemri ES (1999) Ordering the cytochrome c–initiated
caspase cascade: hierarchical activation of caspases-2,-3,-6,-7,-8, and-10 in a
caspase-9–dependent manner. The Journal of cell biology 144:281.
Smith DB, Johnson KS (1988) Single-step purification of polypeptides expressed in
Escherichia coli as fusions with glutathione S-transferase. Gene 67:31-40.
Smith MD, Weedon H, Papangelis V, Walker J, Roberts-Thomson PJ, Ahern MJ (2010)
Apoptosis in the rheumatoid arthritis synovial membrane: modulation by disease-
modifying anti-rheumatic drug treatment. Rheumatology (Oxford) 49:862-875.
Songyang Z, Cantley LC (1998) The Use of Peptide Library for the Determination of
Kinase Peptide Substrates. Methods in Molecular Biology 87:87-98.
Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES (1998) Autoactivation of
procaspase-9 by Apaf-1-mediated oligomerization. Molecular cell 1:949-957.
Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee RA, Robbins PD,
Fernandes-Alnemri T, Shi Y, Alnemri ES (2001) A conserved XIAP-interaction
motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis.
Nature 410:112-116.

216
Srivastava V, Rawall S, Vijayan VK, Khanna M (2009) Influenza a virus induced
apoptosis: inhibition of DNA laddering & caspase-3 activity by zinc
supplementation in cultured HeLa cells. The Indian journal of medical research
129:579-586.
Stennicke HR, Deveraux QL, Humke EW, Reed JC, Dixit VM, Salvesen GS (1999)
Caspase-9 can be activated without proteolytic processing. The Journal of
biological chemistry 274:8359-8362.
Stennicke HR, Jurgensmeier JM, Shin H, Deveraux Q, Wolf BB, Yang X, Zhou Q,
Ellerby HM, Ellerby LM, Bredesen D, Green DR, Reed JC, Froelich CJ, Salvesen
GS (1998) Pro-caspase-3 is a major physiologic target of caspase-8. The Journal
of biological chemistry 273:27084-27090.
Stennicke HR, Renatus M, Meldal M, Salvesen GS (2000) Internally quenched
fluorescent peptide substrates disclose the subsite preferences of human caspases
1, 3, 6, 7 and 8. The Biochemical journal 350 Pt 2:563-568.
Stennicke HR, Salvesen GS (1997) Biochemical characteristics of caspases-3, -6, -7, and
-8. The Journal of biological chemistry 272:25719-25723.
Stennicke HR, Salvesen GS (1999) Caspases: preparation and characterization. Methods
17:313-319.
Stennicke HR, Salvesen GS (1999) Catalytic properties of the caspases. Cell death and
differentiation 6:1054-1059.
Stephanou A, Scarabelli TM, Knight RA, Latchman DS (2002) Antiapoptotic activity of
the free caspase recruitment domain of procaspase-9: a novel endogenous rescue
pathway in cell death. The Journal of biological chemistry 277:13693-13699.
Stroffekova K, Proenza C, Beam KG (2001) The protein-labeling reagent FLASH-EDT2
binds not only to CCXXCC motifs but also non-specifically to endogenous
cysteine-rich proteins. Pflugers Archiv : European journal of physiology 442:859-
866.
Suel GM, Lockless SW, Wall MA, Ranganathan R (2003) Evolutionarily conserved
networks of residues mediate allosteric communication in proteins. Nature
structural biology 10:59-69.

217
Sun C, Cai M, Meadows RP, Xu N, Gunasekera AH, Herrmann J, Wu JC, Fesik SW
(2000) NMR structure and mutagenesis of the third Bir domain of the inhibitor of
apoptosis protein XIAP. The Journal of biological chemistry 275:33777-33781.
Sweeney MC, Wavreille AS, Park J, Butchar JP, Tridandapani S, Pei D (2005) Decoding
protein-protein interactions through combinatorial chemistry: sequence specificity
of SHP-1, SHP-2, and SHIP SH2 domains. Biochemistry 44:14932–14947.
Tak PP, Bresnihan B (2000) The pathogenesis and prevention of joint damage in
rheumatoid arthritis: advances from synovial biopsy and tissue analysis. Arthritis
and rheumatism 43:2619-2633.
Takahashi A, Alnemri ES, Lazebnik YA, Fernandes-Alnemri T, Litwack G, Moir RD,
Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC (1996) Cleavage of
lamin A by Mch2 alpha but not CPP32: multiple interleukin 1 beta-converting
enzyme-related proteases with distinct substrate recognition properties are active
in apoptosis. Proceedings of the National Academy of Sciences of the United
States of America 93:8395-8400.
Takahashi R, Deveraux Q, Tamm I, Welsh K, Assa-Munt N, Salvesen GS, Reed JC
(1998) A single BIR domain of XIAP sufficient for inhibiting caspases. The
Journal of biological chemistry 273:7787-7790.
Tam JP. Peptides:Structure and Function. In: CM Deber VHaKK, Ed. (1985) Proceedings
of the 9th Annual American Peptide Symposium. Rockford, pp. 423-425.
Tam JP, Lu YA (1995) Coupling Difficulty Associated with Interchain Clustering and
Phase Transition in Solid Phase Peptide Synthesis. Journal of the American
Chemical Society 117:12058-12063.
Tamm I, Kornblau SM, Segall H, Krajewski S, Welsh K, Kitada S, Scudiero DA, Tudor
G, Qui YH, Monks A, Andreeff M, Reed JC (2000) Expression and prognostic
significance of IAP-family genes in human cancers and myeloid leukemias.
Clinical cancer research : an official journal of the American Association for
Cancer Research 6:1796-1803.
Taylor JW (2002) The synthesis and study of side chain lactam bridged peptides. Peptide
Science 66:49-75.

218
Tejwani GA, Pedrosa FO, Pontremoli S, Horecker BL (1976) Dual role of Zn2+ as
inhibitor and activator of fructose 1,6-bisphosphatase of rat liver. Proceedings of
the National Academy of Sciences of the United States of America 73:2692-2695.
Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C,
Burns K, Bodmer JL, Schroter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J
(1997) Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by
death receptors. Nature 386:517-521.
Thornberry NA (1998) Caspases: key mediators of apoptosis. Chemistry & biology
5:R97-103.
Thornberry NA, Peterson EP, Zhao JJ, Howard AD, Griffin PR, Chapman KT (1994)
Inactivation of interleukin-1 beta converting enzyme by peptide (acyloxy)methyl
ketones. Biochemistry 33:3934-3940.
Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M,
Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson
DW (1997) A combinatorial approach defines specificities of members of the
caspase family and granzyme B. Functional relationships established for key
mediators of apoptosis. The Journal of biological chemistry 272:17907-17911.
Tomishige M, Vale RD (2000) Controlling kinesin by reversible disulfide cross-linking.
Identifying the motility-producing conformational change. J Cell Biol 151:1081-
1092.
Tonan K, Kawata Y, Hamaguchi K (1990) Conformations of isolated fragments of
pancreatic polypeptide. Biochemistry 29:4424-4429.
Toniolo C, Bonora GM, Bavoso A, Benedetti E, di Blasio B, Pavone V, Pedone C (1983)
Preferred conformations of peptides containing , disubstituted amino acids.
Biopolymers 22:205-215.
Truong-Tran AQ, Carter J, Ruffin RE, Zalewski PD (2001) The role of zinc in caspase
activation and apoptotic cell death. Biometals 14:315-330.
Truong-Tran AQ, Grosser D, Ruffin RE, Murgia C, Zalewski PD (2003) Apoptosis in the
normal and inflamed airway epithelium: role of zinc in epithelial protection and
procaspase-3 regulation. Biochemical pharmacology 66:1459-1468.

219
Turk BE, Huang LL, Piro ET, Cantley LC (2001) Determination of protease cleavage site
motifs using mixture-based oriented peptide libraries. Nature Biotechnology
19:661-667.
Turk BE, Hutti JE, Cantley LC (2006) Determining protein kinase substrate specificity by
parallel solution-phase assay of large numbers of peptide substrates. Nature
protocols 1:375-379.
Uren AG, O'Rourke K, Aravind LA, Pisabarro MT, Seshagiri S, Koonin EV, Dixit VM
(2000) Identification of paracaspases and metacaspases: two ancient families of
caspase-like proteins, one of which plays a key role in MALT lymphoma.
Molecular cell 6:961-967.
Vaidya S, Hardy JA (2011) Caspase-6 latent state stability relies on helical propensity.
Biochemistry 50:3282-3287.
Vaidya S, Velazquez-Delgado EM, Abbruzzese G, Hardy JA (2011) Substrate-induced
conformational changes occur in all cleaved forms of caspase-6. Journal of
molecular biology 406:75-91.
Vallee BL, Auld DS (1993) Zinc: biological functions and coordination motifs. Acc
Chem Res 26:543-551.
Vetter SW, Zhang ZY (2002) Probing the Phosphopeptide Specificities of Protein
Tyrosine Phospha-tases, SH2 and PTB Domains with Combinatorial Library
Methods. Current Protein and Peptide Science 3:365-397.
Vucic D, Franklin MC, Wallweber HJ, Das K, Eckelman BP, Shin H, Elliott LO,
Kadkhodayan S, Deshayes K, Salvesen GS, Fairbrother WJ (2005) Engineering
ML-IAP to produce an extraordinarily potent caspase 9 inhibitor: implications for
Smac-dependent anti-apoptotic activity of ML-IAP. The Biochemical journal
385:11-20.
Vucic D, Franklin MC, Wallweber HJA, Das K, Eckelman BP, Shin H, Elliott LO,
Kadkhodayan S, Deshayes K, Salvesen GS (2005) Engineering ML-IAP to
produce an extraordinarily potent caspase 9 inhibitor: implications for Smac-
dependent anti-apoptotic activity of ML-IAP. Biochemical Journal 385:11.

220
Vucic D, Stennicke HR, Pisabarro MT, Salvesen GS, Dixit VM (2000) ML-IAP, a novel
inhibitor of apoptosis that is preferentially expressed in human melanomas.
Current biology : CB 10:1359-1366.
Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine
GL, Korsmeyer SJ (2004) Activation of apoptosis in vivo by a hydrocarbon-
stapled BH3 helix. Science 305:1466.
Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL,
Korsmeyer SJ (2006) A stapled BID BH3 helix directly binds and activates BAX.
Molecular cell 24:199-210.
Wang J, Chun HJ, Wong W, Spencer DM, Lenardo MJ (2001) Caspase-10 is an initiator
caspase in death receptor signaling. Proceedings of the National Academy of
Sciences of the United States of America 98:13884-13888.
Wasilenko ST, Banadyga L, Bond D, Barry M (2005) The vaccinia virus F1L protein
interacts with the proapoptotic protein Bak and inhibits Bak activation. Journal of
virology 79:14031-14043.
Watanabe N, Lam E (2005) Two Arabidopsis metacaspases AtMCP1b and AtMCP2b are
arginine/lysine-specific cysteine proteases and activate apoptosis-like cell death in
yeast. The Journal of biological chemistry 280:14691-14699.
Weaver RF. 2007. Molecular Biology, McGraw-Hill College.
Wei Y, Fox T, Chambers SP, Sintchak J, Coll JT, Golec JM, Swenson L, Wilson KP,
Charifson PS (2000) The structures of caspases-1, -3, -7 and -8 reveal the basis
for substrate and inhibitor selectivity. Chemistry & biology 7:423-432.
Williams RM, Im MN (1991) Asymmetric synthesis of monosubstituted and. alpha.,.
alpha.-disubstituted. alpha.-amino acids via diastereoselective glycine enolate
alkylations. Journal of the American Chemical Society 113:9276-9286.
Winston RL, Gottesfeld JM (2000) Rapid identification of key amino-acid–DNA contacts
through combinatorial peptide synthesis. Chemistry & biology 7:245-251.
Witkowski WA, Hardy JA (2009) L2' loop is critical for caspase-7 active site formation.
Protein science : a publication of the Protein Society 18:1459-1468.

221
Witkowski WA, Hardy JA (2011) A designed redox-controlled caspase. Protein science :
a publication of the Protein Society.
Wolan DW, Zorn JA, Gray DC, Wells JA (2009) Small-molecule activators of a
proenzyme. Science 326:853-858.
Wolf CM, Eastman A (1999) The Temporal Relationship between Protein Phosphatase,
Mitochondrial CytochromecRelease, and Caspase Activation in Apoptosis.
Experimental cell research 247:505-513.
Wolf CM, Morana SJ, Eastman A (1997) Zinc inhibits apoptosis upstream of ICE/CED-3
proteases rather than at the level of an endonuclease. Cell death and
differentiation 4:125-129.
Yaffe MB (2004) Study of Substrate Specificity of MAPKs Using Oriented Peptide
Libraries. Methods in Molecular Biology 250:237-250.
Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ (1995) Bad, a
heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell
death. Cell 80:285-291.
Yoshida N, Iwata H, Yamada T, Sekino T, Matsuo H, Shirahashi K, Miyahara T, Kiyama
S, Takemura H (2007) Improvement of the survival rate after rat massive
hepatectomy due to the reduction of apoptosis by caspase inhibitor. Journal of
gastroenterology and hepatology 22:2015-2021.
Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B (2001) PUMA induces the rapid
apoptosis of colorectal cancer cells. Molecular cell 7:673-682.
Yu X, Wang L, Acehan D, Wang X, Akey CW (2006) Three-dimensional structure of a
double apoptosome formed by the Drosophila Apaf-1 related killer. Journal of
molecular biology 355:577-589.
Yuan S, Yu X, Asara JM, Heuser JE, Ludtke SJ, Akey CW (2011) The holo-apoptosome:
activation of procaspase-9 and interactions with caspase-3. Structure 19:1084-
1096.

222
Zalewski PD, Forbes IJ, Betts WH (1993) Correlation of apoptosis with change in
intracellular labile Zn(II) using zinquin [(2-methyl-8-p-toluenesulphonamido-6-
quinolyloxy)acetic acid], a new specific fluorescent probe for Zn(II). The
Biochemical journal 296 ( Pt 2):403-408.
Zander NF, Lorenzen JA, Cool DE, Tonks NK, Daum G, Krebs EG, Fischer EH (1991)
Purification and characterization of a human recombinant T-cell protein-tyrosine-
phosphatase from a baculovirus expression system. Biochemistry 30:6964-6970.
Zhai D, Yu E, Jin C, Welsh K, Shiau CW, Chen L, Salvesen GS, Liddington R, Reed JC
(2010) Vaccinia virus protein F1L is a caspase-9 inhibitor. The Journal of
biological chemistry 285:5569-5580.
Zhang XY, Bishop AC (2007) Site-specific incorporation of allosteric-inhibition sites in a
protein tyrosine phosphatase. J Am Chem Soc 129:3812-3813.
Zheng M, Aslund F, Storz G (1998) Activation of the OxyR transcription factor by
reversible disulfide bond formation. Science 279:1718-1721.
Zhou Q, Snipas S, Orth K, Muzio M, Dixit VM, Salvesen GS (1997) Target protease
specificity of the viral serpin CrmA. Analysis of five caspases. The Journal of
biological chemistry 272:7797-7800.
Zondlo NJ, Schepartz A (1993) Highly Specific DNA Recognition by a Designed
Miniature Protein. Nature (London) 363:38.
Zondlo NJ, Schepartz A (1999) Highly specific DNA recognition by a designed
miniature protein. Journal of the American Chemical Society 121:6938-6939.
Zoog SJ, Schiller JJ, Wetter JA, Chejanovsky N, Friesen PD (2002) Baculovirus
apoptotic suppressor P49 is a substrate inhibitor of initiator caspases resistant to
P35 in vivo. The EMBO journal 21:5130-5140.
Zou H, Li Y, Liu X, Wang X (1999) An APAF-1.cytochrome c multimeric complex is a
functional apoptosome that activates procaspase-9. The Journal of biological
chemistry 274:11549-11556.

223
Zou H, Li Y, Liu X, Wang X (1999) An APAF-1· cytochrome c multimeric complex is a
functional apoptosome that activates procaspase-9. Journal of Biological
Chemistry 274:11549.


















