
Refined Non Parametric Methods for Genomic inference Refined Non Parametric Methods for Genomic...
47
Refined Non Parametric Refined Non Parametric Methods for Genomic Methods for Genomic inference inference Peter J. Bickel Peter J. Bickel Department of Statistics Department of Statistics University of California at University of California at Berkeley, USA Berkeley, USA work with Nancy R. Zhang (Stanford), James B. Brown and Haiyan Huang (UCB)
-
Upload
byron-garrett -
Category
Documents
-
view
235 -
download
1
Transcript of Refined Non Parametric Methods for Genomic inference Refined Non Parametric Methods for Genomic...

Refined Non Parametric Refined Non Parametric Methods for Genomic Methods for Genomic
inferenceinference Peter J. BickelPeter J. Bickel
Department of StatisticsDepartment of StatisticsUniversity of California at Berkeley, USAUniversity of California at Berkeley, USA
Joint work with Nancy R. Zhang (Stanford), James B. Brown (UCB) and Haiyan Huang (UCB)

Motivating QuestionsMotivating Questions

Association of functional annoAssociation of functional annotations in Human Genometations in Human Genome
5' 3'
→ Transcription Start Sites (TSSs)
→ GENCODE Exons
3' 5'
The ENCODE Consortium found that many Transcription The ENCODE Consortium found that many Transcription Start Sites are anti-sense to GENCODE exonsStart Sites are anti-sense to GENCODE exons
They also found vastly more TSSs than previously They also found vastly more TSSs than previously supposedsupposed
Is the association between TSSs and exons in the anti-Is the association between TSSs and exons in the anti-sense direction real, or experimental noise in TSS sense direction real, or experimental noise in TSS identification? identification?

Association of experimental annotatioAssociation of experimental annotations across whole chromosomesns across whole chromosomes
Do two factors tend to bind together more closely or more often than other pairs of factors? Does a factor’s binding site relative to TSSs tend to change across genomic regions?

The statistical relation of TranscriptioThe statistical relation of Transcription Start Sites and protein binding sitesn Start Sites and protein binding sites
Normalized Chip-chIP signals around GENCODE Normalized Chip-chIP signals around GENCODE TSSs in ENCODE regionsTSSs in ENCODE regions
Most peak over the TSS and are clearly significantMost peak over the TSS and are clearly significant Does the upstream bump in CTCF constitute good Does the upstream bump in CTCF constitute good
evidence of enchancer binding activity?evidence of enchancer binding activity?
Normalized signal intensity
Figure from ENCODE Consortium Paper: Nature, June 14th, 2007
Enchancer activity?

What is a non-parametric What is a non-parametric model for the Genome and model for the Genome and
why is it needed?why is it needed?

Feature Overlap: the Feature Overlap: the questionquestion
A mathematical question arises:A mathematical question arises:
Do these features overlap more, or Do these features overlap more, or less than “expected at random”? less than “expected at random”?
5' 3'
→Transcription Fragments
→ Conserved sequence

Our formulationOur formulation
Defining “expectation” and “at Defining “expectation” and “at random”:random”: The genome is highly structuredThe genome is highly structured Analysis of feature inter-dependence Analysis of feature inter-dependence
must account for superficial structuremust account for superficial structure ““Expected at random” becomes:Expected at random” becomes:
Overlap between two feature sets Overlap between two feature sets bearing structure, under no biological bearing structure, under no biological constraintsconstraints

Naïve MethodNaïve Method Treating bases as being independent with same distTreating bases as being independent with same dist
ribution (ordinary bootstrap)ribution (ordinary bootstrap) Hypothesis: Feature markings are independent Hypothesis: Feature markings are independent Specific Object Test based on Specific Object Test based on % Feature Overlap – (% Feature1)(% Feature2) % Feature Overlap – (% Feature1)(% Feature2) and standard statistics and standard statistics
Why naïve ? Bases are NOT independentWhy naïve ? Bases are NOT independent Better method: keeping one type of feature fixed anBetter method: keeping one type of feature fixed an
d simulating moving start site of another feature und simulating moving start site of another feature uniformly (feature bootstrap)iformly (feature bootstrap)
Why still a problem?Why still a problem? Even if feature occurrences are independent functionally, tEven if feature occurrences are independent functionally, t
here can be clumping caused by the complex underlying ghere can be clumping caused by the complex underlying genome sequence structure enome sequence structure
(i.e. inhomogeneity, local sequence dependence) (i.e. inhomogeneity, local sequence dependence)

A non parametric modelA non parametric model
Requirements:Requirements:a)a) It should roughly reflect known It should roughly reflect known
statistics of the genomestatistics of the genome
b)b) It should encompass methods listedIt should encompass methods listed
c)c) It should be possible to do inference, It should be possible to do inference, tests, set confidence bounds tests, set confidence bounds meaningfullymeaningfully

Segmented Stationary Segmented Stationary ModelModel
Let Let XXi i = = base at position base at position i, i=1,…,ni, i=1,…,n
such that for each such that for each k=1,…,rk=1,…,r, is: , is: Stationary (homogeneity within blocks) Stationary (homogeneity within blocks) Mixing (bases at distant positions are nearly independent)Mixing (bases at distant positions are nearly independent) rr << << n n
1 111 1 1( ,..., ) ( ,..., ,..., ,..., ),
n n rn r rX X X X X X 1 ... rn n n
{ :1 }jk kX j n
…
1n 2n 1rn rn

Empirical InterpretationsEmpirical Interpretations Within a segment:Within a segment:
For For kk small compared to minimum segment small compared to minimum segment length, statistics of random kmers do not length, statistics of random kmers do not differ between large subsegments of segmentdiffer between large subsegments of segment
Knowledge of the first kmer does not help in Knowledge of the first kmer does not help in predicting a distant kmerpredicting a distant kmer
Remark: Remark: If this model holds it also applies to derived If this model holds it also applies to derived
local features, e.g. {local features, e.g. {II11,…,,…,IInn} where } where IIkk = 1 if = 1 if position position kk belongs to binding site for given belongs to binding site for given factorfactor

Mentioned other models Mentioned other models are special cases for are special cases for rr = 1 = 1
Independent identically distributed Independent identically distributed (bootstrap)(bootstrap)
Stationary MarkovStationary Markov Uniform displacement of start sites Uniform displacement of start sites
(Homogeneous Poisson Process) (Homogeneous Poisson Process)

Is the Effect Serious?Is the Effect Serious?
Ordinary Ordinary bootstrapbootstrap Base-by-base Base-by-base
sampling sampling randomly from randomly from observed observed sequence for sequence for two features two features separatelyseparately
Feature Feature randomization:randomization: Keep one type of Keep one type of
feature fixed and feature fixed and randomizing the randomizing the start positions start positions of the other of the other
Example Statistic: Overlap between two features in a binary sequence of 10K bases (region statistic in the ENCODE studies) Feature 1: occurrence of motif 111000; Feature 2: more than six 1’s in 10 consecutive bases
True distribution: Mean=5.23 SD=0.53
Ordinary Bootstrap: Mean=4.83 SD=0.26
Feature Randomization: Mean=6.19 SD=0.81
Block Bootstrap: Mean=4.81 SD=0.55

Evidence for Segmented Evidence for Segmented StationarityStationarity
DNA sequence is known to bDNA sequence is known to be inhomogeneouse inhomogeneous
However, it has been segmenHowever, it has been segmented into homogeneous domaited into homogeneous domains based on:ns based on: Base composition (e.g. finding IBase composition (e.g. finding I
sochores) sochores) CpG densityCpG density Density of higher order features Density of higher order features
(e.g. ORFS, palindromes, TFBS)(e.g. ORFS, palindromes, TFBS) Our model aims to capture thOur model aims to capture th
ese “domain-specific” effeese “domain-specific” effects, while avoiding parametricts, while avoiding parametric assumptions within domaic assumptions within domainsns
Figure from Li, 2001:
References: Elton (1974, J. Theoretical Bio.), Braun and Müller (1998, Statistical Science), Li et al. (1998, Genome Res.), Liu and Lawrence (1999, Bioinformatics)

Inference with our modelInference with our model
Use Use XX11,…,,…,XXnn for basic data, but for basic data, but XXkk could be base identity, feature could be base identity, feature identity, a vector of feature identity, a vector of feature identities obeying segmented identities obeying segmented stationarity assumption. stationarity assumption.

Many genomic statistics are function of one or more sums of the form:
e.g. is 1 or 0 depending on the presence or absence of a feature or features
Using our model for inferenceUsing our model for inference
When the summands are small compared to When the summands are small compared to SS::
Gaussian case Gaussian case
Example: Region overlap for common Example: Region overlap for common features, or rare features over large regions features, or rare features over large regions
n
iiUgS
1
Under segmented stationarity, these distributions can be estimated from the data
kXg

Distributions of feature Distributions of feature overlapsoverlaps
The Block BootstrapThe Block Bootstrap Can’t observe independent Can’t observe independent
occurrences of ENCODE regions, but occurrences of ENCODE regions, but if our hypothesis of segmented if our hypothesis of segmented stationarity holds then the distribution stationarity holds then the distribution of sum statistics and their functions of sum statistics and their functions can be approximated as followscan be approximated as follows

Block Bootstrap for r = 1Block Bootstrap for r = 1Algorithm 4.1: a) Given L << n choose a number N uniformly at random from
b) Given the statistics Tn(X1,…,Xn) , under the assumption that X1,…,Xn is stationary, compute
c) Repeat B times to obtain d) Estimate the distribution of by the empirical
distribution:
By Theorem 4.2.1 of Politis, Romano and Wolf (1999)
Ln ,...,1
*L1L TT LN1N ,..., XX
*LB
*L1 TT ,...,
nn T
BjXXL
nnnLjB 1,,...,1
** TT
,0* NB

Block Bootstrap AnimationBlock Bootstrap Animationr = 1r = 1
*1X )( *
1*
1 XfS
*BX
Observed Sequence (X): Statistic:
S=f(X)
…… …
)( **BXfS B
Draw a block of length L from original sequence, this is the block-bootstrapped sequence.
Calculate statistic on the block bootstrapped sequence.Repeat this procedure identically B times.
*2X )( *
2*
2 XfS

Observing the distributionsObserving the distributions
Block bootstrap distribution of the Region Overlap Statistic
Shown here with the PDF of the normal distribution with the same mean and variance
QQplot of BB distribution vs. standard normal
The histogram of
Is approximately the same as density of

What if What if r r > 1> 1
The estimated distribution is always The estimated distribution is always heavier tailed leading to heavier tailed leading to conservative conservative p p valuesvalues
But it can be enormously so if the But it can be enormously so if the segment means of the statistic differ segment means of the statistic differ substantiallysubstantially
Less so but still meaningful if the Less so but still meaningful if the means agree but variances differ means agree but variances differ

Simulation StudySimulation Study
For simplicity, we concatenate 2 For simplicity, we concatenate 2 homogeneous regions generated as homogeneous regions generated as aboveabove

Simulation Results and Simulation Results and comparison to a naïve comparison to a naïve
methodmethodTrue distribution
Uniform Start Site Shuffling
Block Bootstrap without Segmentation
Block Bootstrap with True Segmentation

SolutionsSolutions
1)1) Segment using biological Segment using biological knowledgeknowledge
Essentially done in ENCODE: poor Essentially done in ENCODE: poor segmentation occasionally led to non-segmentation occasionally led to non-Gaussian distributions (excessively Gaussian distributions (excessively conservative)conservative)
2)2) Segment using a particular linear Segment using a particular linear statistic which we expect to statistic which we expect to identify homogeneous segments identify homogeneous segments

Block Bootstrap with Block Bootstrap with SegmentationSegmentation
Draw a block from each sub-segment Draw a block from each sub-segment and concatenate to form a block and concatenate to form a block bootstrap sample bootstrap sample

Block Bootstrap given Block Bootstrap given SegmentationSegmentation
1. Draw Subsample of length L:
f1L f2L f3L
2. Compute statistic on subsample:
T(X*)
3. Do this B times: T(X1*),…T(XB
*)

Simulation Results, with Simulation Results, with segmentationsegmentation
True distribution
Uniform Start Site Shuffling
Block Bootstrap without Segmentation
Block Bootstrap with True Segmentation
Block Bootstrap with Estimated Segmentation

Dyadic SegmentationDyadic Segmentation
Define,Define,
Find Find jjmaxmax maximizing maximizing MM((jj) creating intervals I) creating intervals Ileft left and Iand Irightright
If length of both intervals falls below a stopping If length of both intervals falls below a stopping criterion, stopcriterion, stop
Else, repeat process for IElse, repeat process for Ileft left and/or I and/or Irightright, whichever , whichever are longer than stopping criterion, with redefined are longer than stopping criterion, with redefined MM((jj) )
nijXAvejiXAve
n
j
n
jjM
iij
j
1:1:
1 2

Dyadic SegmentationDyadic Segmentation
change in mean of the statistic
Statistic as a function of position
First cut maximizes the difference between the means in the new segments
All subsequent cuts are greedy, making maximal splits
The mean is recomputed in each segment, so long as the segment is longer than a set threshold
No new cuts exist, the segmentation is complete

True distribution
Uniform Start Site Shuffling
Block Bootstrap without Segmentation
Block Bootstrap with True Segmentation
Block Bootstrap with Estimated Segmentation

Confidence Bounds: Confidence Bounds: rr > 1 > 1
Given a statistic, e.g. basepair % Given a statistic, e.g. basepair % overlap:overlap:
Find such that:
as small as possible
“Average basepair overlap over all potential genomes for the region considered”

Use Algorithm 4.1Use Algorithm 4.1
For each segment pick random block For each segment pick random block of length proportional to segment of length proportional to segment lengthlength
Concatenate to get block of length LConcatenate to get block of length L Compute % bp overlap for blockCompute % bp overlap for block Repeat many timesRepeat many times Use 100(1-Use 100(1-αα) percentiles of this for ) percentiles of this for

Testing AssociationTesting Association
Question: How do we estimate Question: How do we estimate null distribution given only data null distribution given only data for which we believe the null is for which we believe the null is false?false?

Testing Association (bp Testing Association (bp overlap)overlap)
1X 2X
Observed Sequence (Feature 1 = , Feature 2 = ):
Sample two blocks of equal length.
1Y
2Y
2X 1X
1Y
2Y
Align Feature 1 of first block with Feature 2 of second block,And vice versa.
Calculate overlap in the blocks after swapping = (X2)(Y1)+(X1)(Y2)Statistic is: (X2)(Y1)+(X1)(Y2), properly normalized and set to mean 0. Under the null hypothesis of independence, this should be Gaussian.

Test StatisticTest Statistic
H H : Features : Features not not associated in each segment (so-called associated in each segment (so-called “dummy overlap”)“dummy overlap”)
Then has a Gaussian distribution. Then has a Gaussian distribution.
We form the test statistic:We form the test statistic:
where: where:
Length of segment i/n
% of basepairs in segment i identified as Feature 1
% of basepairs in segment i identified as Feature 2

Null DistributionNull Distribution
Choose pairs of blocks at randomChoose pairs of blocks at random Compute false (“dummy”) overlap Compute false (“dummy”) overlap HH Compute Compute II = % Feature 1 and = % Feature 1 and JJ = % = %
Feature 2Feature 2 Block bootstrapped Null: Block bootstrapped Null: H – IJH – IJ
If r > 1, pairs of blocks are chosen in each If r > 1, pairs of blocks are chosen in each region, region, HH and and IJ IJ are weighted sums across are weighted sums across regions.regions.
The Null is mean zero, and has the The Null is mean zero, and has the correct variancecorrect variance

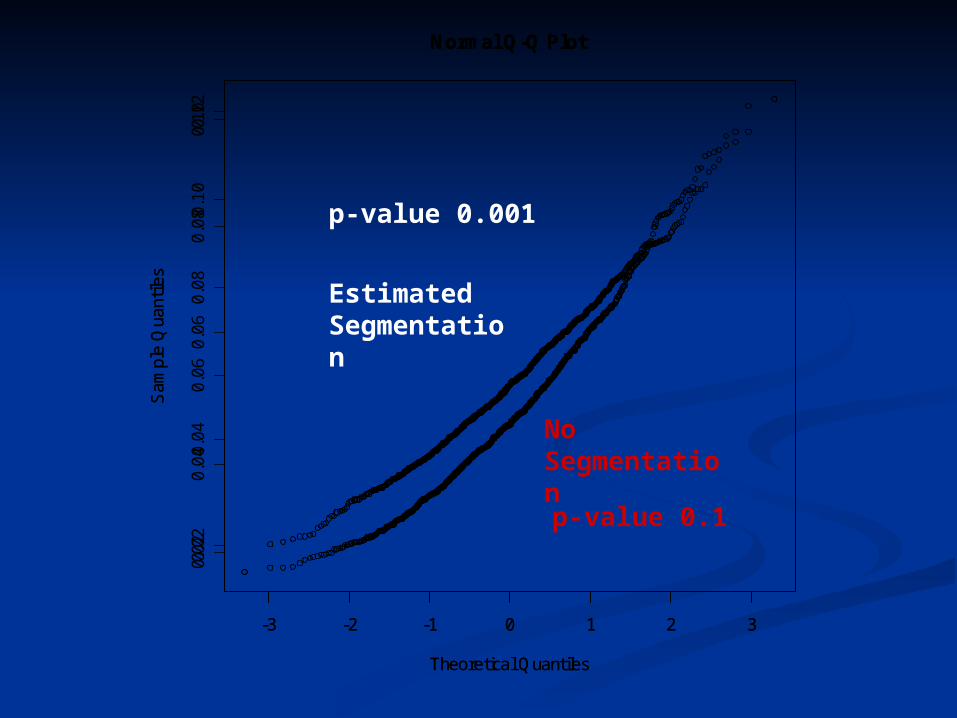
Example from ENCODE Example from ENCODE datadata
ENm001: ENCODE Consortium annotated over ENm001: ENCODE Consortium annotated over 2500 feature-instances exclusive of UTRs and 2500 feature-instances exclusive of UTRs and CDSs CDSs
Question: “Do these (largely) non-coding Question: “Do these (largely) non-coding features exhibit more overlap with constrained features exhibit more overlap with constrained sequences than expected at random?”sequences than expected at random?”
To answer, we used the block bootstrap to To answer, we used the block bootstrap to obtain null distribution obtain null distribution
When null is Gaussian, it has the correct When null is Gaussian, it has the correct variancevariance
When not, it is overly conservativeWhen not, it is overly conservative Segmentation can reduce conservativeness, and Segmentation can reduce conservativeness, and
detect significance that would otherwise be detect significance that would otherwise be missedmissed
-3 -2 -1 0 1 2 3
0.02
0.04
0.06
0.08
0.10
0.12
Normal Q-Q Plot
Theoretical Quantiles
Sam
ple
Qua
ntile
s
No Segmentation
-3 -2 -1 0 1 2 3
0.02
0.04
0.06
0.08
0.10
Normal Q-Q Plot
Theoretical Quantiles
Sam
ple
Qua
ntile
s
Estimated Segmentation
p-value 0.001
p-value 0.1

There are two L’sThere are two L’s
LLss : the minimum segment length : the minimum segment length during segmentationduring segmentation To be discussedTo be discussed
L L : the length of blocks during : the length of blocks during subsamlingsubsamling Chosen on grounds of stabilityChosen on grounds of stability

A philosophical question:A philosophical question:The Issue of ScaleThe Issue of Scale
Relevant probability assessments Relevant probability assessments depend on segmentationdepend on segmentation
Segmentation depends on scaleSegmentation depends on scale Things which seem surprising on Things which seem surprising on
small scales, may not be at larger small scales, may not be at larger onesones
E.g. differences in GC contentE.g. differences in GC contentMy view: It’s only My view: It’s only
determinable determinable biologicallybiologically

Some Future DirectionsSome Future Directions KS type tests KS type tests
Beyond overlap, KS-type tests can compare the distributions of Beyond overlap, KS-type tests can compare the distributions of features, e.g. “Does the pattern of constrained sequence in coding features, e.g. “Does the pattern of constrained sequence in coding regions differ from that in non-coding regions?” regions differ from that in non-coding regions?”
MaximaMaxima Aggregative plots can summarize one feature in the neighborhood of Aggregative plots can summarize one feature in the neighborhood of
another, e.g. “Does binding data (such as Chip-chIP) show that a another, e.g. “Does binding data (such as Chip-chIP) show that a given regulatory factor tends to bind near TSSs?”given regulatory factor tends to bind near TSSs?”
Other types of associationOther types of association Does wavelet analysis offer significant support for the large scale Does wavelet analysis offer significant support for the large scale
association of replication timing and conservation?association of replication timing and conservation? Many others arising from ENCODE, modENCODE, and elsewhereMany others arising from ENCODE, modENCODE, and elsewhere
Other types of segmentationOther types of segmentation Dyadic segmentation is analytically convenient, but other Dyadic segmentation is analytically convenient, but other
segmentations may be usefulsegmentations may be useful

AcknowledgementsAcknowledgements
The ENCODE ConsortiumThe ENCODE Consortium The MSA and Transcription and The MSA and Transcription and
Regulation GroupsRegulation Groups Especially: Elliot Margulies, Tom Especially: Elliot Margulies, Tom
Gingeras and Ewan BirneyGingeras and Ewan Birney Supported by NIGMS and NHGRISupported by NIGMS and NHGRI

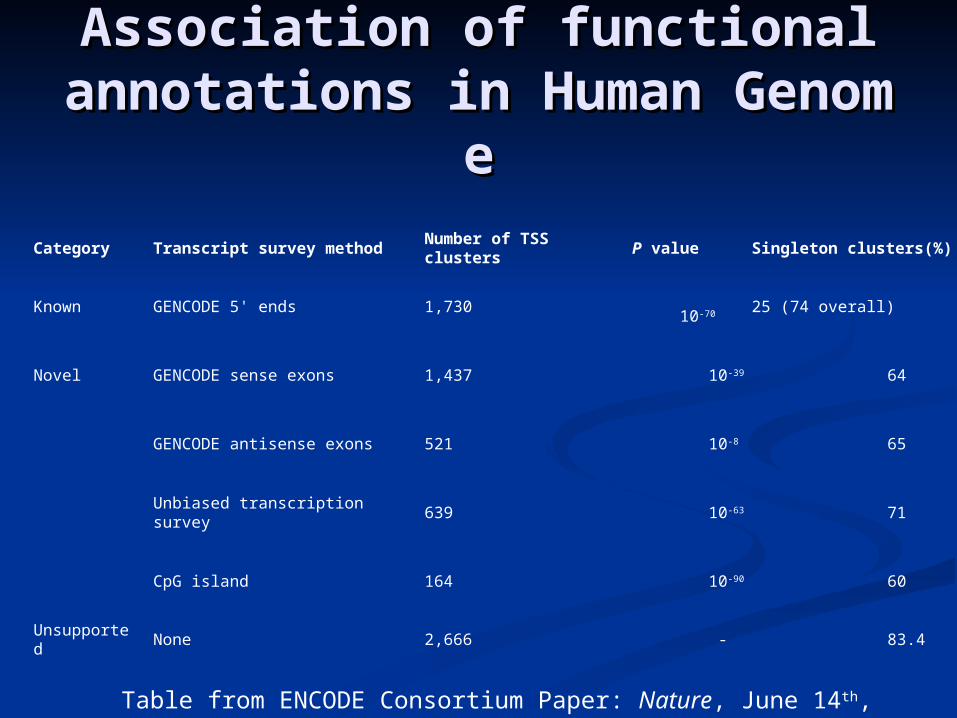
Association of functional annoAssociation of functional annotations in Human Genometations in Human Genome
Category Transcript survey method Number of TSS clusters P value Singleton clusters(%)
Known GENCODE 5' ends 1,730 10-70 25 (74 overall)
Novel GENCODE sense exons 1,437 10-39 64
GENCODE antisense exons 521 10-8 65
Unbiased transcription survey 639 10-63 71
CpG island 164 10-90 60
Unsupported None 2,666 - 83.4
Table from ENCODE Consortium Paper: Nature, June 14th, 2007

Dyadic SegmentationDyadic Segmentation
For a minimum region length Ls and threshold b initialize:
Algorithm 4.8
ntt 10 ,0t
1. For i = 1,…,|t|-1, let M(i)(j) and V(i)(j) be respectively the processes (4.7) and (4.8) computed on the subsequence Xti-1+1,
…,Xti. Let t’i = argmaxjM(i)(k), and mi = min(t’i – ti-1,ti - t’i). Let:
2. Let Vi = V(i)(t’i). Let: If stop, return t.
3. Let i* = argmaxi Bi , and tnew = t’i*
4. Let t = t ∪ tnew reordered so that ti is monotonically increasing in i.
.,0
;,)(
otherwise
LmtMB sii
i
i
Algorithm 4.8
ntt 10 ,0t
b
V
BttIJ
ii
iiii
1 0
iiJ



















