
Recent progress in understanding chemical shifts
25
SOLID STATE Nuclear Magnetic Resonance ELSEVIER Solid State Nuclear Magnetic Resonance 6 (1996) 101-125 Review Recent progress in understanding chemical shifts 1 Angel C. de Dios, Eric Oldfield * Department of Chemistry, University of Illinois, 600 S. Mathews Avenue, Urbana, IL 61801, USA Received 18 January 1995; accepted 16 August 1995 Abstract In the past three or four years computer hardware and software developments have reached the stage where the nuclear magnetic resonance (NMR) spectra of many molecular systems can now be accurately evaluated. Detailed analysis of chemical shifts may soon become a routine part of solid (and liquid) state NMR structure prediction in chemistry and biology, and this Article covers the development of the topic from its earliest beginnings. Keywords: Chemical shift; Electrostatics; Protein; Shielding; Tensor 1. Introduction In 1949-1950, Knight observed large differ- ences in nuclear magnetic resonance (NMR) fre- quency between a metal and its salt [l], and Dickinson 121, Lindstriim [3], Thomas [4], Gutowsky and Hoffman [5] and Proctor and Yu [6] observed large frequency shifts between differ- ent chemical compounds, the latter “chemical effects” being denoted chemical shifts. For physi- cists, chemical shifts were a source of both fasci- nation and mild annoyance since as Proctor and Yu stated “Until it is clearly understood, the accuracy of magnetic moments determined under certain chemical conditions remain somewhat in doubt”. For chemists, however, a cornucopia had * Corresponding author. ’ This work was supported in part by the U.S. National Institutes of Health (grants HL-19481 and GM-50694). A.C.D. is an American Heart Association Inc. (Illinois Affiliate) Postdoctoral Research Fellow (Grant FW-01). arrived. In this Article, we trace some of the developments which have led to an understand- ing of how “chemical conditions” influence NMR resonance shifts. NMR spectroscopy is now one of the most important tools for characterizing the structures of organic, biological, and to a some- what lesser extent, inorganic chemical systems, but only recently have computational capabilities advanced to the state where the chemical shifts of most molecules can be accurately predicted. If a resonance is detected, its chemical shift can be measured, and at least in principle, useful struc- tural information can be derived. The availability of powerful workstations now greatly facilitates this task, and in what follows we describe theoret- ical developments, and recent applications to small molecules, as well as macromolecular sys- tems, such as proteins. In the next few years, it is likely that “turnkey” (hardware and software) packages will become available for routinely predicting the NMR spec- tra of most organic chemicals and natural prod- 0926-2040/96/$15.00 0 1996 Elsevier Science B.V. All rights reserved SSDI 0926-2040(95)01207-9
Transcript of Recent progress in understanding chemical shifts

SOLID STATE Nuclear Magnetic Resonance
ELSEVIER Solid State Nuclear Magnetic Resonance 6 (1996) 101-125
Review
Recent progress in understanding chemical shifts 1
Angel C. de Dios, Eric Oldfield *
Department of Chemistry, University of Illinois, 600 S. Mathews Avenue, Urbana, IL 61801, USA
Received 18 January 1995; accepted 16 August 1995
Abstract
In the past three or four years computer hardware and software developments have reached the stage where the nuclear magnetic resonance (NMR) spectra of many molecular systems can now be accurately evaluated. Detailed analysis of chemical shifts may soon become a routine part of solid (and liquid) state NMR structure prediction in chemistry and biology, and this Article covers the development of the topic from its earliest beginnings.
Keywords: Chemical shift; Electrostatics; Protein; Shielding; Tensor
1. Introduction
In 1949-1950, Knight observed large differ- ences in nuclear magnetic resonance (NMR) fre- quency between a metal and its salt [l], and Dickinson 121, Lindstriim [3], Thomas [4], Gutowsky and Hoffman [5] and Proctor and Yu [6] observed large frequency shifts between differ- ent chemical compounds, the latter “chemical effects” being denoted chemical shifts. For physi- cists, chemical shifts were a source of both fasci- nation and mild annoyance since as Proctor and Yu stated “Until it is clearly understood, the accuracy of magnetic moments determined under certain chemical conditions remain somewhat in doubt”. For chemists, however, a cornucopia had
* Corresponding author. ’ This work was supported in part by the U.S. National
Institutes of Health (grants HL-19481 and GM-50694). A.C.D. is an American Heart Association Inc. (Illinois Affiliate) Postdoctoral Research Fellow (Grant FW-01).
arrived. In this Article, we trace some of the developments which have led to an understand- ing of how “chemical conditions” influence NMR resonance shifts. NMR spectroscopy is now one of the most important tools for characterizing the structures of organic, biological, and to a some- what lesser extent, inorganic chemical systems, but only recently have computational capabilities advanced to the state where the chemical shifts of most molecules can be accurately predicted. If a resonance is detected, its chemical shift can be measured, and at least in principle, useful struc- tural information can be derived. The availability of powerful workstations now greatly facilitates this task, and in what follows we describe theoret- ical developments, and recent applications to small molecules, as well as macromolecular sys- tems, such as proteins.
In the next few years, it is likely that “turnkey” (hardware and software) packages will become available for routinely predicting the NMR spec- tra of most organic chemicals and natural prod-
0926-2040/96/$15.00 0 1996 Elsevier Science B.V. All rights reserved SSDI 0926-2040(95)01207-9

102 A.C. de Dies, E. Oldfeld /Solid State Nuclear Magnetic Resonance 6 (1996) 101-125
ucts - even systems as large as proteins and nucleic acids. While this might have been a some- what speculative claim two or three years ago, recent developments support the idea that even protein spectra can now be successfully predicted [7-101, and current progress in the direction of deducing structure from experimental spectra by using quantum chemical methods [ll] offer even more promise in utilizing the chemical shift. Af- ter 45 years, “chemical effects” - at least for first and second row elements - can now be reliably evaluated, and given the amazing im- provements in computer hardware price/perfor- mance ratios over the same time period (espe- cially when contrasted with the same ratios for NMR spectrometers themselves), it seems likely that computational quantum chemistry will soon become a routine complement to experimental studies of NMR chemical shifts. In what follows, we trace some of the key developments since the first observations of “chemical effects”, and we begin by briefly reviewing work in days when Mflops were unheard of.
2. The early years
As noted early on [l-6], in the presence of an applied magnetic field the observed nuclear mag- netic resonance frequency for a particular nu- cleus is influenced by its chemical environment. Nuclei are normally observed amidst a back- ground of a sea of electrons which are in continu- ous motion. The applied field induces electrical currents, which in turn induce a secondary local magnetic field at a given nuclear site. Hence, the actual field felt at the nucleus of interest, Z?Oc, becomes slightly different from the applied exter- nal magnetic field, Wxt, giving the familiar rela- tion:
B’G(l-cr)BeX’ (I)
where u is a so-called screening or shielding constant. A positive value of (+ (shielding) thus results in a smaller Bloc, and a lower NMR reso- nance frequency, while a negative (T (deshielding) results in an increase in resonance frequency. Experimentally, one usually measures a chemical
shift, 6, which has the opposite sign (and a differ- ent reference state) to the shielding, u, but the two are simply related.
Now, in a solid, it is easy to see that the screening or shielding - of say a benzene ring or a CO group - will be orientation dependent, and the shielding is in fact more usefully repre- sented as a second rank tensor quantity. Thus, Eq. (1) can be recast as:
BE = (1 - CQ)B;~ (4
where BE is the magnitude of the field felt at the site of the nucleus along the o-direction induced by electronic currents brought about by an applied magnetic field along the P-direction. The shielding is thus composed of nine indepen- dent components, although symmetry generally reduces this number. In the liquid and gas phase, or in solution NMR, the free Brownian motion of molecules usually averages the shielding tensor without preference to any particular orientation with respect to the applied field. Thus, what is observed is referred to as the isotropic shielding, which is simply an average of the principal com- ponents:
uisO = - :( fill + u22 + 933) (3)
Both the applied magnetic field, B, and the nu- clear moment, CL, which acts like a tiny magnet, cause changes in the electronic wavefunctions and energies of the molecule, and these changes in energy due to simultaneous magnetic perturba- tions define shielding from a different, quantum chemical, perspective [12]:
a2E U 4
=- auw B B-O,p=O
(4)
where E is the total molecular eigenenergy. Being electronic in its origin, the history of the
theoretical analysis of the shielding property par- allels advances in evaluating E. Theories of mag- netic shielding begin with Ramsey’s formulation [13], which made use of second-order perturba- tion theory. Upon collecting all terms in energy that are proportional to both p and B obtained after a perturbation treatment, Ramsey catego-

A.C. de Dies, E. Oldfield/Solid State Nuclear Magnetic Resonance 6 (1996) 101-125 103
rized two distinctly different terms which com- prise shielding: a diamagnetic term and paramag- netic term. The diamagnetic contribution, Us, de- rived from the first-order correction to the en- ergy, involves only the ground state wavefunction, IO>:
Its evaluation is straightforward since Eq. (5) requires only knowledge of the positions of the electrons with respect to the nucleus of interest, rn, and with respect to an origin chosen for the magnetic vector potential, rO. The difficulty in calculating shielding lies however in evaluating the paramagnetic part:
up = - c 4
mainly because it requires both the wavefunc- tions, (q I, and energies, E,, of the excited states. Once again, since the phenomenon arises from a simultaneous perturbation, L, refers to the angu- lar momentum with respect to the nucleus of interest, while L, corresponds to the origin of the magnetic vector potential. Explicit in both Eqs. (5) and (6) is a gauge origin chosen for the magnetic vector potential. This origin can be placed anywhere, and in Ramsey’s original for- mulation, it was situated at the nucleus of inter- est (r. = r,). The shielding, being an observable, is independent of the choice of the gauge origin. However, its parts, the diamagnetic and paramag- netic terms shown in Eqs. (5) and (61, are both gauge-dependent, and much of the history of nuclear magnetic shielding calculations has re- volved around solving this so-called gauge prob- lem.
As mentioned above, the primary challenge in shielding calculations lies in evaluation of the paramagnetic term. The first simplification, intro- duced by Ramsey, involved use of an average value of (E, - E,,) [14]. However, choosing an appropriate average value implies a knowledge of all of the excited states, which is a very difficult
problem to solve. Thus after Ramsey’s basic for- mulation, two improved approaches were intro- duced. In the first, it was suggested that since the paramagnetic term is gauge-dependent, it should be possible to choose a gauge origin in which the contribution from the sum over the excited states is at a minimum. Chan and Das [15] showed that by placing the gauge origin at the center of elec- tronic charge, the excited state contribution is greatly reduced, and in the case of proton shield- ing in a variety of compounds, their results were very satisfactory. Kern and Lipscomb [16] also employed a gauge transformation in their proton shielding calculations, in which a cancellation of the AE term was again achieved by having the origin in proximity to the centroid of charge dis- tribution.
A distinctly different approach made use of the variational method. Tillieu and Guy [17] rep- resented the new wavefunction as V = pO(l + g . B), where g is a parameter with which the energy is minimized, then later Kolker and Karplus 1181 used a more elaborate function which took into account the nuclear moment: W = p(,,(l +f. B + g. p). The results obtained by using such varia- tional methods were in fairly good agreement with experiment. However, as in the gauge trans- formation approaches described above, success was limited to proton shieldings only.
Prompted by the limited success of calculating chemical shieldings from the unperturbed wave- functions, Stevens and coworkers formulated the first coupled Hartree-Fock method of shielding computation [19-231 in which both the magnetic field and nuclear moment were taken into ac- count in the Hamiltonian. This generated wave- functions correct to first order, and energies cor- rect to second order (excluding configuration in- teraction and electron correlation arising from more than one determinant function), and the shieldings obtained by their methods for the heavier elements in a variety of diatomics, such as LiH, HF, F2, CO and ClF, were in good agree- ment with experiment. Large basis sets, however, were .essential in order to adequately describe the first-order wavefunctions, making it very difficult to apply the same methods to larger molecular systems.

104 A.C. de Dies, E. Oldfield/Soiid State Nuclear Magnetic Resonance 6 (1996) 101-125
3. Modem approaches
The coupled Hartree-Fock calculations de- scribed above follow exactly the formalisms of Eqs. (5) and (6), that is, contributions from all orbitals in the molecule are evaluated with re- spect to a single gauge origin, which is usually placed at the site of the nucleus in question. Both diamagnetic and paramagnetic terms are gauge- origin dependent, and a change in the choice of gauge origin leads to additional terms. In princi- ple these additional gauge-dependent terms are exactly equal in magnitude but of opposite sign, rendering the total shielding gauge-invariant. In practice, the desired cancellation does not occur exactly due to the practical difficulties encoun- tered in calculating the paramagnetic term. The paramagnetic term involves electronic excited states, which can only be described adequately with (very large) basis sets which provide an am- ple set of virtual orbitals. In addition, molecular orbitals obtained from self-consistent field (SCF) calculations are normally optimized with respect to an origin within the region of interest, and assigning a common gauge origin is clearly unde- sirable for orbitals far from this origin. For these reasons, common origin calculations only yield accurate values for shielding in the limit of ex- tremely large atomic basis sets. Thus, only the smallest molecules are accessible using these methods.
In order to overcome the unrealistic depen- dence of the calculated shielding on the choice of origin, several methods have been proposed. The first one can be traced back to the early seminal work of London 1241, and introduces field depen- dence into the basis functions. These gauge-in- cluding atomic orbitals (GIAOs) differ from the field-independent basis by an exponential gauge factor:
&Jr, B) =&Jr) exp [ -i(B XR,) *r/2c]
(7)
which takes into account the value of the vector potential (B X R,) at the position of the centroid R,, of the atomic orbital &Jr). The incorpora- tion of field dependence into basis functions for
shielding calculations was first demonstrated by Ditchfield 1251. The reduction in the gauge de- pendence of the calculated shielding values re- sults from the fact that GIAOs closely resemble the first-order wavefunctions in the presence of a magnetic field (see e.g. Ref. [261X GIAOs no longer allow for an arbitrary choice of origin, and more importantly, having each orbital with its own gauge origin leads to a much more accurate computation of what one might refer to as a local paramagnetic term. Although GIAOs were ideal for achieving the desired gauge independence, the gauge factors also led to perturbation of the two-electron part of the Hamiltonian, necessitat- ing additional (time-consuming) two-electron in- tegral calculations. This again restricted utility to small molecules, or big computers [27].
In the 198Os, Kutzelnigg [28] and Schindler and Kutzelnigg [291 proposed an alternative method, individual gauge for localized orbitals (IGLO), in which localized molecular orbitals are made field-dependent. The gauge origin for each localized molecular orbital is placed at its cen- troid of charge-analogous to previous ideas [15,16]. Immediately after its introduction, shield- ings for more than a hundred compounds were calculated [30]. The results were very satisfactory, and since the shieldings were evaluated with lo- calized ?rbitals, individual contributions from var- ious parts of the molecules could be obtained. As in GIAO, however, there was a price to pay for having more than one gauge origin, and in this case, so-called additional “resonance and ex- change” correction terms needed to be evaluated. Moreover, an integral transformation was also necessary to convert from an atomic orbital to a molecular orbital basis. Hansen and Bouman [31] thus proposed another method, which simply left out calculation of the large terms of opposite sign in the diamagnetic and paramagnetic contribu- tions, which should ideally cancel in the limit of an infinite basis. By invoking commutation and closure relations in a localized orbital-local ori- gin (LORG) formalism, the large errors associ- ated with calculating the paramagnetic term in a finite basis set were avoided.
The IGLO and LORG methods are thus very similar, since they both involve localized molecu-

A. C. de Dios, E. Oldfield / Solid State Nuclear Magnetic Resonance 6 (19!?6) 101-125 105
lar orbitals, invoke closure relations, and require integral transformations. GIAO on the other hand, has to evaluate additional two-electron in- tegrals, and does not provide insight regarding bond contributions to shielding. The massive amount of integral calculations in the GIAO method thus made it less attractive. However, by taking advantage of gradient theory [32], Wolinski et al. [33] formulated a much more efficient im- plementation of the old GIAO, making it about three times faster. In addition, semi-direct ver- sions of both GIAO and IGLO, in which two- electron integrals are computed on an “as- needed” basis, have recently been introduced [34,35], and promise to alleviate basis size con- cerns - albeit at the expense of increased com- putational time (loss of a factor of = 3).
Going beyond Coupled Hartree-Fock (CHF), while keeping the advantages gained from local origin methods, has also been of recent interest. The first successful results were reported in 1990 by Bouman and Hansen [36], who applied the second-order polarization propagator approxima- tion of Geertsen and Oddershede [37]. The new method, referred to as SOLO (second-order LORG) was shown to satisfactorily reproduce experimental shifts for 31P in small phosphorus- containing compounds [36], as well as 15N in azines, for example [38]. Implementation of Moller-Plesset theory with GIAOs has been made by Gauss [39], and very recently, van Wullen and Kutzelnigg incorporated multi-configuration SCF wavefunctions into the IGLO method [40]. Thus, all three local origin methods now have the option of going beyond Hartree-Fock-again al- beit at the expense of an increase in computa- tional time on the order of N, the number of basis functions.
Fortunately, in most cases it is not essential to describe all atoms in a molecule with large basis sets, since this adds tremendously to computa- tional time, which varies between = N4 and (N log Nj2 for CHF calculations [41]. Thus, for ac- curate shielding calculations, larger basis sets can usually be restricted to the atom or small group of atoms which carry the nucleus of interest, while a significantly smaller set is provided for the other atoms. The amount of computational
time required, and disk storage space for inte- grals, are thus greatly reduced with such a “lo- cally dense” basis [42], while the results remain satisfactory when compared with experiment, and calculations with a uniform basis. Use of either modern GIAO or IGLO methods on high perfor- mance workstations means that shielding in most molecular systems can now be confidently ad- dressed, and we present below numerous illustra- tions on systems of biomolecular interest.
4. Gas phase NMR and absolute shielding scales
Before we move to consider large molecular systems, we need to consider the accuracy of shielding calculations. Here, the proper evalua- tion of shielding calculations would not have been possible without reference to an extensive gas phase NMR literature ([43], and references cited therein). The reason for this is that shielding calculations are usually performed on an isolated molecule at its equilibrium geometry, and it is therefore essential to have the same conditions when determining the experimental values, if a valid comparison is to be made. With measure- ments of the spin-rotation coupling constants in molecular beams, and the demonstration of an intimate relation between the spin-rotation con- stant and the paramagnetic component of shield- ing [44], absolute shielding values (referenced to the bare nucleus) can be obtained, an essential prerequisite for testing the theoretical methods. Moreover, as soon as the absolute shielding of a particular nucleus in one molecule is known, an absolute shielding scale can be established for the same nucleus in other molecules by determining their chemical shifts with respect to the molecule whose absolute shielding value was obtained from the molecular beam study. There is one caveat, however, which is that the chemical shifts be free of intermolecular effects. Fortunately, gas phase NMR offers the ability to vary the density of a sample, which leads to a direct way of eliminating most intermolecular contributions to shielding.
The dependence of any molecular electronic property, such as shielding, on density in a pure

106 A.C. de Dies, E. Oldfield /Solid State Nuclear Magnetic Resonance 6 (1996) 101-125
0 100 200 300 400 500 600 700 800 900 1000
- bpmi
Fig. 1. Absolute shielding scale for phosphorus-31 (from Ref. 1461).
gas was first formulated by Buckingham and Pople [45] as:
(8)
where the shielding, (T, is expressed as a virial expansion in terms of the density, p. v~(-O(T> is the shielding in the isolated molecule at a given tem- perature, T, and is derived by an extrapolation to zero density using values obtained from samples of low to intermediate density.
Measurements of a,(T) in a variety of molecules have provided a means of establishing absolute shielding scales. These numbers, al- though they still contain intramolecular effects, are free of medium effects, and have enabled the establishment of absolute shielding scales for 13C, 15N, 19F, 29Si and 31P (see e.g. Ref. 1461, and references cited therein). An example of an abso- lute shielding scale (for 31P) is shown in Fig. 1. Shielding calculations for these nuclei have there- fore been properly checked by comparison with experimental data obtained under conditions close to what the calculations have assumed. Of course, it is also highly desirable for ab initio methods to reproduce not only the measured absolute shieldings of an isolated molecule frozen in an equilibrium configuration, but also to evalu- ate the effects that come from dynamics, confor- mational changes, as well as intermolecular inter- actions, and we now discuss these topics in detail,
since these basic ideas are all of importance for understanding chemical shifts in macromolecules, both in solution and in the solid-state.
5. Intramolecular effects on shielding
The effects of geometrical parameters, such as bond lengths, bond angles and dihedral angles, on nuclear shielding are most readily studied in small molecules [47,48]. The simplest systems to study are diatomics, in which the only parameter that requires consideration is the internuclear separation. Initial theoretical work aimed at in- vestigating how shielding changes with internu- clear separation near the equilibrium geometry was carried out by Stevens and coworkers on LiH [20], HF [21], CO [49] and N, [50], using common-origin CHF methods. It was noted that, except for the 7Li in LiH, shielding decreases with increasing internuclear separation, and em- ploying the GIAO method, Ditchfield verified these early results [51]. Chesnut and Foley then observed a systematic variation of the shielding derivative, au/at-, in the hydrides across the Peri- odic Table U.521, and references cited therein). For both first and second row hydrides, the shielding derivative starts out being small and positive for the alkali metals. There is a small increase for the alkaline earth hydrides, then a decrease for boron and aluminum, which contin- ues all the way across the Periodic Table to the halogen hydrides, where the shielding derivatives are (generally) large and negative. The first gen- eral explanation for these trends was made by Jameson and de Dios 1531 who proposed a univer- sal shape for the dependence of shielding on internuclear separation (shown in Fig. 21, based on work with 23Na in NaH [48] and 39Ar in Ar, [53]. The explanation for this behavior, which is in accord with the first exact and complete shield- ing trace for Hz+ by Hegstrom [54], is that from the united atom value, the shielding decreases monotonically until it reaches a minimum, then it continues to rise and approach the value for infinitely separated atoms. The alkali hydrides have equilibrium separations that are longer than the separation where the shielding minimum oc-

A.C. de Dies, E. OldfEld/Sokd State Nuclear Magnetic Resonance 6 (1996) 101-125 107
curs, hence, positive shielding derivatives are ob- served. On the other hand, halides have bond lengths inside the minimum, where the shielding decreases with increasing internuclear distance.
In systems involving more than two atoms, the number of geometrical factors which can influ- ence shielding increases, and more complicated shielding surfaces, not just traces, may be neces- sary [55]. Lazzeretti et al. made use of the sym- metry coordinates of the molecule in their theo- retical study of CH, [56], and the effects on shielding of changing each symmetry coordinate were obtained separately. When the same theo- retical strategies were applied to the shielding of i5N in NH, [57] and 31P in PH, [58], the follow- ing trends became apparent: (1) the first and second derivatives with respect to stretching coor- dinates are negative; (2) the experimentally ob- served secondary deuterium-induced isotope shifts are dominated by stretching contributions, and (3) the experimentally observed temperature dependencies of the shielding come mainly from centrifugal stretching. Representative shielding traces for these molecules are shown in Fig. 3.
In more complex molecules, the addition of heavy atoms often leads to the introduction of a new geometrical factor which requires considera- tion, the torsion angle. In ethane, Chesnut et al.
h c ‘is
“5 Fig. 2. Universal shape proposed for the dependence of shielding on internuclear separation (from Ref. [53]).
.70
35
0
b
-50 IL -70 -0.2 -0.1 0.0 0.1 0.2 Symmetry Coordinate. Angstroms
Fig. 3. Shielding traces for “‘P in PH, (filled symbols) and “N in NH, (open symbols) as functions of the symmetry displace- ment coordinates: S, (symmetric stretch 0, l ), S, (asymmet- ric stretch A, A ), S4 (asymmetric bend 0, n ) (from Ref. Dgl).
[59] and Kutzelnigg et al. [60] have concluded that = 90% of the change in 13C shielding ob- served as a function of the torsion angle change is directly due to the torsion, rather than a change in other geometric factors (i.e. bond length or bond angle changes). For ethane, experimental results are unfortunately not available, however, in more complex hydrocarbons, Barfield [61] and Kurosu et al. [62] have reported work aimed at reproducing the “y-gauche” effect first observed by Paul and Grant 1631, and agreement with ex- periment has been quite satisfactory. As we dis- cuss below in more detail, torsional effects on shielding are exceptionally important for under- standing experimental chemical shift patterns in macromolecules such as proteins, where back- bone (4, $1 and sidechain (~1 torsion angles appear to be major contributors to 13C shielding. For example, for alanine residues, C” and Ca shifts are dominated by backbone ~,IJ torsions, while valine C*, Cp shifts are influenced by 4,# and x [8,11], and once the 4,$,x dependencies on shielding are determined, then experimental

108 A. C. de Dios, E. OIdfeM / Solid State Nuclear Magnetic Resonance 6 (I 996) 101-l 25
measurements of say C*, Cp chemical shifts can give important information on what &$, and in some case x values, must be present in order to reproduce the observed shifts.
6. Weak intermolecular interactions and short- range repulsion
For polar molecules, Raynes et al. [64] first suggested a potentially useful categorization of the intermolecular effects on shielding into bulk susceptibility, magnetic anisotropy, electric field and van der Waals contributions:
~1 = apUlk + alanisotropy + a;lectric field
+ @.;an der Waals (9)
Such a categorization is useful since it allows one to formulate computational methods specific for a particular kind of contribution ([65] and refer- ences cited therein), although it is a somewhat arbitrary distribution of effects.
The bulk susceptibility correction takes into account the sample shape, orientation and sus- ceptibility factors often of importance when com- paring chemical shift results obtained in electro- magnet vs. superconducting magnet geometries, a]anisotropy takes into account the magnetizabilities of adjacent groups, u1 ‘lectric fie’d is an electrostatic field contribution to shielding, discussed in more detail below, and ~“,a” der Was’s is the somewhat enigmatic dispersion contribution, due to the fluctuating instantaneous moments of polarizable molecules [66]. Such a categorization has been of limited use, however, since the electrostatic and van der Waals dispersion contributions are par- ticularly difficult to evaluate, and indeed, for non-polar gases, short-range contributions arising from orbital overlap - not considered in Eq. (9) - appear to be very significant [53]. While shielding in rare gases may appear to be rather removed from our primary interests in biomolecu- lar and solid-state NMR chemical shifts, they are in fact a paradigm for the van der Waals shift, a question of interest for so many years, in pro- teins.
For example, in recent work a scaling scheme based on the dependence of the chemical shift range of a particular rare gas on the characteris- tic (ao3/r3> of the free atom in its ground state [67], and the scaling of intermolecular potential functions given by Maitland et al. [68], has been proposed to explain observed gas-to-solution shifts of 21Ne, s3Kr and ‘29Xe ([69,70], and refer- ences cited therein), as well as the measured second virial coefficient (a,) and its temperature dependence, for ‘29Xe in rare gas mixtures [71,72]. Moreover, the sensitivity of the ‘29Xe chemical shift to its environment has also been utilized to characterize zeolites, polymers and other microp- orous solids [73,74]. For example in zeolites, the ‘29Xe chemical shift is found to be a function of loading, cavity size and temperature, and an un- derstanding of how the chemical shift is influ- enced by these factors has led to a better under- standing of the structure and dynamics of Xe clusters trapped in these cavities, as well as help- ing to clarify the importance of ~\;a” der Was’s on shielding.
Based on earlier ideas, van der Waals disper- sion effects might have been thought to be very important in determining rare gas shielding. However, it has been shown that scaling ab initio shielding functions for Ar in Ar, [53], in Ar-NaH, and in Ar-OH, [47,48] (which mimic the main rare gas interactions in the zeolite cage) to those of ‘29Xe has recently permitted the successful interpretation of the trends in 129Xe chemical shifts observed in Xe-zeolite adsorption studies [75,76]. The so-called alpha cage in which the xenon atoms are trapped is illustrated in Fig. 4, which also shows a typical ‘29Xe NMR spectrum. Clearly, there are a large number of resonances due to different cage occupancies. Nevertheless, the experimentally observed chemical shifts and intensity distributions have been successfully computed by using Monte Carlo methods [75] combined with ‘29Xe shielding functions obtained by scaling of Ar functions, and excellent agree- ment between theory and experiment can be seen in Fig. 5, results which were obtained simply by assuming an additivity of the intermolecular ef- fects. This work also indicated that no specific structures for the Xe, clusters were responsible
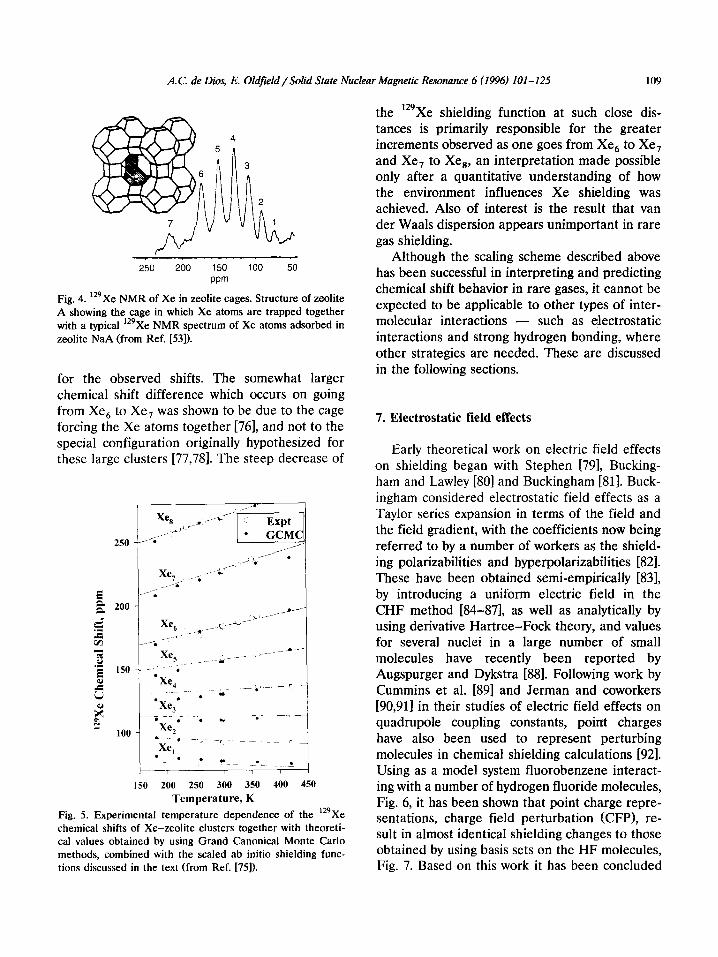
A.C. de Dies, E. Oldfield/Solid State Nuclear Magnetic Resonance 6 (1996) 101-12.5 109
/” 250 200 150 100 50
wm
Fig. 4. 129Xe NMR of Xe in zeolite cages. Structure of zeolite A showing the cage in which Xe atoms are trapped together with a typical ‘29Xe NMR spectrum of Xe atoms adsorbed in zeolite NaA (from Ref. [53]).
for the observed shifts. The somewhat larger chemical shift difference which occurs on going from Xe, to Xe, was shown to be due to the cage forcing the Xe atoms together [76], and not to the special configuration originally hypothesized for these large clusters [77,78]. The steep decrease of
250
f & 200
& ‘2 CA Z .g IS0 4J
G
k? R
too
i
c-
150 200 250 300 350 400 450
Temperature, K
Fig. 5. Experimental temperature dependence of the ‘%Xe chemical shifts of Xe-zeolite clusters together with theoreti- cal values obtained by using Grand Canonical Monte Carlo methods, combined with the scaled ab initio shielding func- tions discussed in the text (from Ref. [75]).
the ‘29Xe shielding function at such close dis- tances is primarily responsible for the greater increments observed as one goes from Xe, to Xe, and Xe, to Xe,, an interpretation made possible only after a quantitative understanding of how the environment influences Xe shielding was achieved. Also of interest is the result that van der Waals dispersion appears unimportant in rare gas shielding.
Although the scaling scheme described above has been successful in interpreting and predicting chemical shift behavior in rare gases, it cannot be expected to be applicable to other types of inter- molecular interactions - such as electrostatic interactions and strong hydrogen bonding, where other strategies are needed. These are discussed in the following sections.
7. Electrostatic field effects
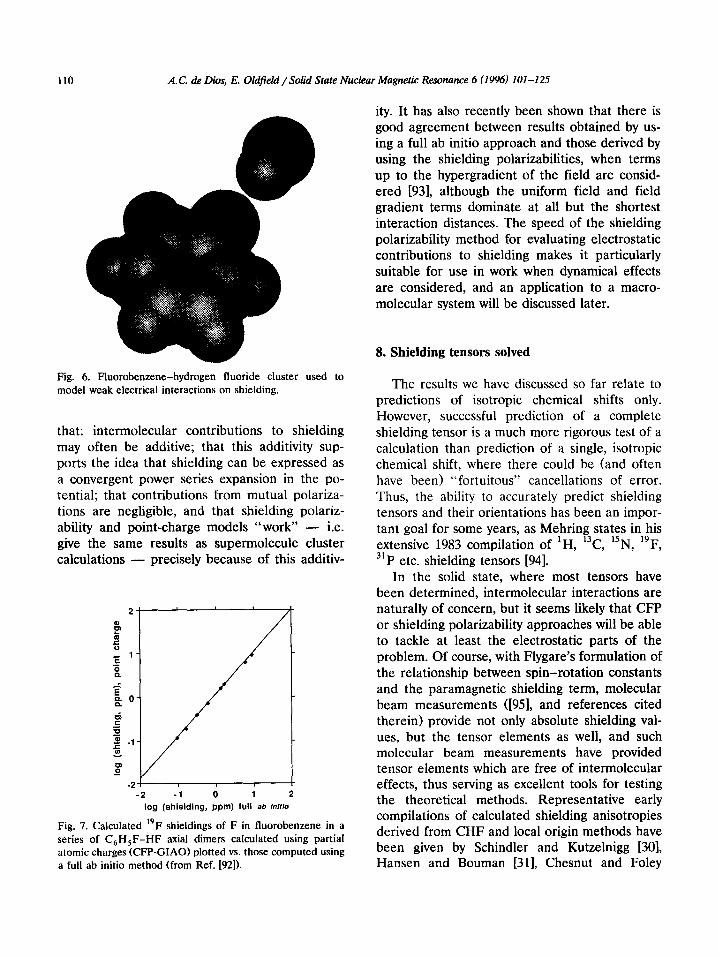
Early theoretical work on electric field effects on shielding began with Stephen 1791, Bucking- ham and Lawley [80] and Buckingham [81]. Buck- ingham considered electrostatic field effects as a Taylor series expansion in terms of the field and the field gradient, with the coefficients now being referred to by a number of workers as the shield- ing polarizabilities and hyperpolarizabilities [82]. These have been obtained semi-empirically [83], by introducing a uniform electric field in the CHF method [84-871, as well as analytically by using derivative Hartree-Fock theory, and values for several nuclei in a large number of small molecules have recently been reported by Augspurger and Dykstra 1881. Following work by Cummins et al. [89] and Jerman and coworkers [90,911 in their studies of electric field effects on quadrupole coupling constants, point charges have also been used to represent perturbing molecules in chemical shielding calculations [92]. Using as a model system fluorobenzene interact- ing with a number of hydrogen fluoride molecules, Fig. 6, it has been shown that point charge repre- sentations, charge field perturbation (CFP), re- sult in almost identical shielding changes to those obtained by using basis sets on the HF molecules, Fig. 7. Based on this work it has been concluded
110 A.C. de Dios, E. Oldfield /Solid State Nuclear Magnetic Resonance 6 (1996) 101-125
Fig. 6. Fluorobenzene-hydrogen fluoride cluster used to model weak electrical interactions on shielding.
that: intermolecular contributions to shielding may often be additive; that this additivity sup- ports the idea that shielding can be expressed as a convergent power series expansion in the po- tential; that contributions from mutual polariza- tions are negligible, and that shielding polariz- ability and point-charge models “work” - i.e. give the same results as supermolecule cluster calculations - precisely because of this additiv-
I -2 -1 0 1 i
log (shielding, ppm) full ab lnfflo
Fig. 7. Calculated 19F shieldings of F in fluorobenzene in a series of C,H,F-HF axial dimers calculated using partial atomic charges (CFP-GIAO) plotted vs. those computed using a full ab initio method (from Ref. 1921).
ity. It has also recently been shown that there is good agreement between results obtained by us- ing a full ab initio approach and those derived by using the shielding polarizabilities, when terms up to the hypergradient of the field are consid- ered 1931, although the uniform field and field gradient terms dominate at all but the shortest interaction distances. The speed of the shielding polarizability method for evaluating electrostatic contributions to shielding makes it particularly suitable for use in work when dynamical effects are considered, and an application to a macro- molecular system will be discussed later.
8. Shielding tensors solved
The results we have discussed so far relate to predictions of isotropic chemical shifts only. However, successful prediction of a complete shielding tensor is a much more rigorous test of a calculation than prediction of a single, isotropic chemical shift, where there could be (and often have been) “fortuitous” cancellations of error. Thus, the ability to accurately predict shielding tensors and their orientations has been an impor- tant goal for some years, as Mehring states in his extensive 1983 compilation of iH, 13C, i5N, i9F, 31P etc. shielding tensors [94].
In the solid state, where most tensors have been determined, intermolecular interactions are naturally of concern, but it seems likely that CFP or shielding polarizability approaches will be able to tackle at least the electrostatic parts of the problem. Of course, with Flygare’s formulation of the relationship between spin-rotation constants and the paramagnetic shielding term, molecular beam measurements ([951, and references cited therein) provide not only absolute shielding val- ues, but the tensor elements as well, and such molecular beam measurements have provided tensor elements which are free of intermolecular effects, thus serving as excellent tools for testing the theoretical methods. Representative early compilations of calculated shielding anisotropies derived from CHF and local origin methods have been given by Schindler and Kutzelnigg [30], Hansen and Bouman [31], Chesnut and Foley

A. C. de Dies, E. Oldfield / Solid State Nuclear Magnetic Resonance 6 (I 996) 101-125 111
[52], Chesnut 1961, and Holler and Lischka [97]. All calculations compare favorably with experi- ment.
More recently, much larger molecular species have been investigated, e.g. i3C and 15N shielding tensors in the retinal Schiff base of interest in vision 1981, as well as solid saccharides such as methyl+D-glucopyranoside and sucrose [99,100], and Grant et al. have shown how peak assign- ments can be made, based on ab initio calcula- tion. For example, in the methyl glycoside, per- mutation of C, and C, tensors gives a much better agreement between prediction and experi- ment for each atom, supporting a reassignment. Clearly, some care needs to be taken when reas- signing an experimental shift based on theory - but the case can be made quite convincing when full tensors are used.
In comparing calculated and observed tensors, one should also give considerable attention to the orientation of the principal components. Direc- tion cosines with respect to a molecular reference frame are normally used, however, direct compar- ison between calculated and experimental direc- tion cosines is difficult to visualize. In addition to not being able to translate directly errors in di-
3
Fig. 8. Icosahedral axis system used for shielding tensors. Top:
relationship between the six icosahedral axes and the Carte-
sian coordinate system. Bottom: icosahedron showing rela-
tionships between X, y, z and the icosahedral vertices (from
Ref. 1991).
rection cosines to errors in degrees or radians, a fair evaluation involving tensors that are small or almost spherical is difficult. Efforts have been made by Grant and coworkers [99,100] at solving this problem, and they have proposed a novel way of expressing shielding tensors called the icosahe- dral representation. Here, the shift or shielding tensor is represented by six quantities associated with the shifts measured when the applied field is along the six unique directions of an icosahedron, as shown in Fig. 8. The six icosahedral compo- nents are related to the Cartesian components or the principal components via the following rela- tionships:
6, = a2Sxx + b2S,, - 2abS,, = (al, - bm,)2S,,
+ (a& - bm2)2S,2 + (a4 - bm3)2S33
S, = a2Sxx + b2S,, + 2abS,, = (a!, + bm,)2S, 1
+ (al2 + bm2j2b2 + (4 + bmJ2h
6, = a2S,, + b2S,, - 2abS,,=(am, -bn,)2S,,
+ (am2 -bn2)2S,2 + (am, - bnJ2S3,
6, = a2S,, + b2S,, + 2abS,, = (am, + bnl)‘S,,
+ ( am2 + bn2)2S22 + ( am3 + bn,)2S33
S, = a2S,, + b2S,, - 2abS,,= (an, - bf,)2S,,
+ ( an2 - b12)2S22 + ( an3 - bZ3)2S,3
S, = a2SLZ + b2S,, + 2abS,,= (an, + bl,)2S,,
+ (an, + b12)2S22 + ( an3 + bQ2S3,
where S,,, SyY, S,,, SXy, S,, and S,, are the Cartesian components in the laboratory frame. The constants a and b are cos 4 and sin 4 (4 = 31.72”), respectively, Sll, S,, and S,, are the principal components, and li, mi and ni are the direction cosines of the ith principal axis. In this representation, all six components can be com- pared visually and carry the same weight, thereby enabling a fair comparison between two tensors. Moreover, this representation is equally applica- ble in any coordinate frame. Thus, as long as the orientation of the shielding tensor is known ex- perimentally, additional evaluations of the calcu-

112 A.C. de Dies, E. Oldfield/Solid State Nuclear Magnetic Resonance 6 (1996) 101-125
lated tensor, primarily the direction of its princi- pal components, can be made.
The shielding tensors for several other large molecular systems have also now been success- fully predicted using ab initio methods. For exam- ple, there is an excellent correlation between theory and experiment for the fluorobenzenes first studied in the 1950s by Gutowsky and coworkers [.5,1011 whose tensors were represented more than a decade ago as a major challenge for theory [94]. However, in recent work by others [102,103] it has again been suggested that van $r Waals (vdW) dispersion forces influence F shielding. This approach uses a modified Buck- ingham-Stephen description for the electrical contributions to shielding:
S,=AE,+B(P+ (P)) -B(P) (10)
in which the fluctuating field (E2> is derived from the London dispersion formula, such that the overall vdW shift is:
6 vdw = B C (3P,Zi/2r,‘:) (11)
with B taken as 67.7 X low6 A3 eV-’ [103,104]. Unfortunately, this approach is seriously flawed. The principal reason for this is that the entire theory is based on very early semi-empirical work in which numerous parameters (AE, P, I, r, A, E, B) were used in an attempt to fit i9F shifts in fluorobenzenes. However, over the past 30 years
there have been many theoretical developments which have led to a complete revision of these early vdW ideas. In particular, in the rare gases, where vdW dispersion effects were always as- sumed to dominate, recent work using SOLO and LORG [53] has clearly indicated that dispersion does not affect shielding perceptibly, with ‘29Xe shifts being well accounted for in the gas phase and in zeolite lattices by SCF-level calculations - which do not include vdW dispersion [75]. Also, the values of the A parameter, computed by using derivative Hartree-Fock theory [93,105], are now known to be two orders of magnitude larger than assumed by previous workers. For the fluorobenzenes, GIAO-SCF level calculations show very good correlations between isotropic liquid state chemical shifts and theoretical shield- ing (23 points, slope = 0.94, R2 = 0.975, 3 ppm r.m.s. deviation over a 63 ppm range) as well as between experimental solid state shifts/shielding tensor elements and computed values (21 points, slope = 0.95, R2 = 0.989, 6.5 ppm r.m.s. deviation over a 237 ppm shift range), as shown in Fig. 9 [9]. These results do not contain any second-order or vdW dispersion contributions, yet accurately predict all shift and shielding tensor elements with no adjustable parameters (the r.m.s. devia- tions are already close to the experimental uncer- tainties in the solid). Of particular importance is the accurate prediction of all pentafluorobenzene
320 - -180 -150 -120 -90
experimental shift (ppm)
-130 -65 0 65 130
experimental shielding (ppm)
Fig. 9. Experimental vs. theoretical 19F chemical shift/shielding for fluorobenzenes. (A) Liquid state isotropic chemical shifts
plotted vs. computed shielding values. (B) Solid-state shielding principal tensor components (in ppm from CLF6) plotted vs
computed shielding tensor elements (from Ref. [9]).
A. C. de Dios, E. Olafield / Solid State Nuclear Magnetic Resonance 6 (1996) IO1 -125 113
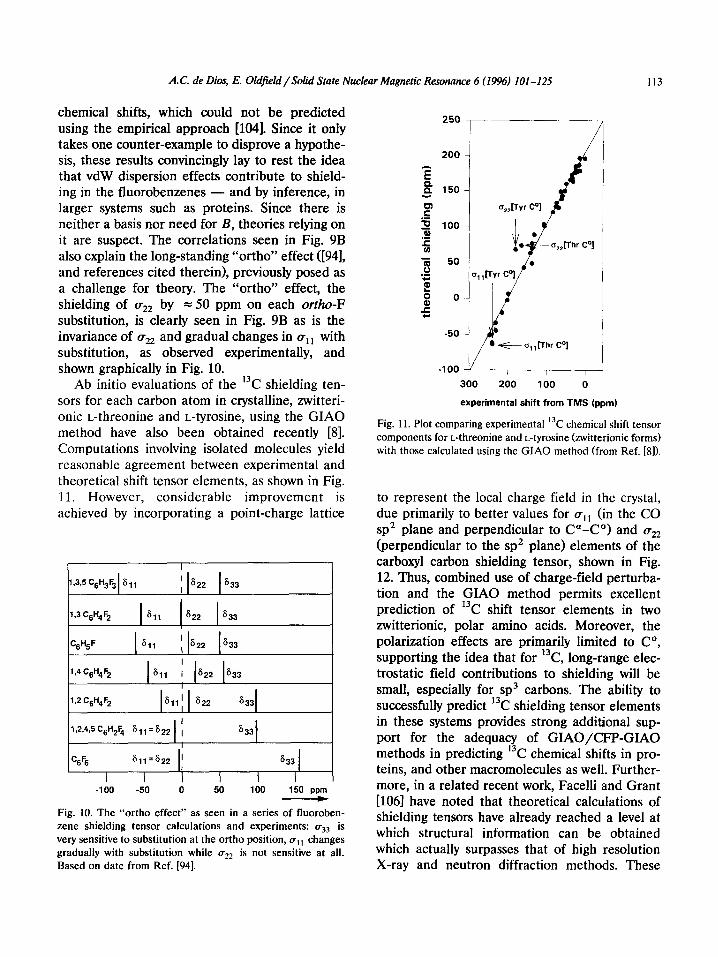
chemical shifts, which could not be predicted using the empirical approach [NM]. Since it only takes one counter-example to disprove a hypothe- sis, these results convincingly lay to rest the idea that vdW dispersion effects contribute to shield- ing in the fluorobenzenes - and by inference, in larger systems such as proteins. Since there is neither a basis nor need for B, theories relying on it are suspect. The correlations seen in Fig. 9B also explain the long-standing “ortho” effect ([94], and references cited therein), previously posed as a challenge for theory. The “ortho” effect, the shielding of a,, by = 50 ppm on each ortho-F substitution, is clearly seen in Fig. 9B as is the invariance of a,, and gradual changes in gll with substitution, as observed experimentally, and shown graphically in Fig. 10.
Ab initio evaluations of the 13C shielding ten- sors for each carbon atom in crystalline, zwitteri- onic L-threonine and L-tyrosine, using the GIAO method have also been obtained recently [8]. Computations involving isolated molecules yield reasonable agreement between experimental and theoretical shift tensor elements, as shown in Fig. 11. However, considerable improvement is achieved by incorporating a point-charge lattice
I
l,3,5C6H3F3 611 ' 622 533
7.3C6H4F2 bl 622 633
%%F 611 [ 622 633
I
',4c6H4F2 611 I 622 633 I 1
12 C6H4F2 611' 622 633
1,2,4,6C6H2Fq 6,,=622 1 ; 633) I I I I
C6% 611=S22 ' 633 I
I I I I I
-100 -50 0 50 100 150 ppm )
Fig. 10. The “ortho effect” as seen in a series of fluoroben-
zene shielding tensor calculations and experiments: a,, is
very sensitive to substitution at the ortho position, (+, , changes
gradually with substitution while flZ2 is not sensitive at all.
Based on date from Ref. [94].
250
‘I
J
experimental shift from TMS (ppm)
Fig. 11. Plot comparing experimental ‘k chemical shift tensor components for L-threonine and L-tyrosine bitterionic forms) with those calculated using the GIAO method (from Ref. [8]).
to represent the local charge field in the crystal, due primarily to better values for (+11 (in the CO sp2 plane and perpendicular to Ca-Co> and uZ2 (perpendicular to the sp2 plane) elements of the carboxyl carbon shielding tensor, shown in Fig. 12. Thus, combined use of charge-field perturba- tion and the GIAO method permits excellent prediction of 13C shift tensor elements in two zwitterionic, polar amino acids. Moreover, the polarization effects are primarily limited to Co, supporting the idea that for 13C, long-range elec- trostatic field contributions to shielding will be small, especially for sp3 carbons. The ability to successfully predict 13C shielding tensor elements in these systems provides strong additional sup- port for the adequacy of GIAO/CFP-GIAO methods in predicting 13C chemical shifts in pro- teins, and other macromolecules as well. Further- more, in a related recent work, Facelli and Grant [106] have noted that theoretical calculations of shielding tensors have already reached a level at which structural information can be obtained which actually surpasses that of high resolution X-ray and neutron diffraction methods. These

114 A. C. de Dies, E. Oldfeld /Solid State Nuclear Magnetic Resonance 6 (I 996) IO1 -125
300 200 100 0
experimental shift from TMS (ppm)
Fig. 12. Experimental and theoretical 13C NMR chemical shift/shielding tensor elements for L-threonine and L-tyro- sine, with charge field perturbation (from Ref. 181).
authors note that (especially) solid-state NMR methods should be useful for refining structural data of biomolecules with molecular weights in the lo-20 X lo3 Da range, where diffraction data have large structural errors, since chemical shift data are not sensitive to many of the structural imperfections which degrade diffraction data. While such single crystal studies have not yet been reported, their ideas nevertheless lead us naturally to the question of predicting chemical shifts in macromolecules, such as proteins, where at least in solution, there is already a consider- able (= 10’) body of uninterpreted chemical shift data [107], and solid-state data are now beginning to become available. Once chemical shifts in pro- teins (and DNA, polysaccharides, etc.) become interpretable, then structure prediction, refine- ment and validation follow naturally.
9. Predicting protein chemical shifts
Protons are the most abundant nuclei in macromolecules such as proteins (or nucleic
acids), and hydrogen is the most sensitive natu- rally occurring nucleus, so early NMR studies of macromolecules were of course overwhelmingly dominated by ‘H NMR investigations. For most proton resonances, it was found early on that ring currents from aromatic residues dominated ‘H chemical shifts - at least for side-chain groups [108], where r.m.s. differences between ring cur- rent prediction and experiment, in, e.g. hen egg white lysozyme and the bovine pancreatic trypsin inhibitor, were found to be small. However, for backbone (Ha and HN) protons, worse agree- ment was found, and this was suggested to be due to the effects of the anisotropy in the diamagnetic susceptibility of the peptide groups, as well as electrostatic field effects. Such influences were proposed quite early on by Sternlicht and Wilson [1091, who showed an upfield or shielding effect on helix formation from a random coil. However, it was only much later that a large number of groups began to report the general observation that H” are shifted = 0.39 ppm upfield in helices but = 0.37 ppm downfield in sheets (see e.g. Refs. [llO-1141). This soon led to detailed analy- ses of H” shifts in terms of magnetic susceptibil- ity (ring current and peptide group), electrostatic field and random coil shifts [115-1171, with the result that for H”, experimental chemical shifts can now be quite accurately predicted (at least in populous $,I) regions) by using what are basically empirical methods.
For the heavier elements frequently studied in proteins, 13C, r5N and r9F, the situation is more complex. For r3C, good empirical correlations between secondary structure and “secondary” chemical shifts - the shifts from the random coil position - have been reported [118,119], but for 15N, the correlations are much weaker [114,120], and for r9F, only a general picture that buried residues are often deshielded has developed [121]. The reason for the lack of any significant devel- opments until recently in the analysis of shielding of the heavier elements is due primarily to the fact that the problem looks so daunting. The paramagnetic term dominates shielding, and this needs to be evaluated quantum mechanically - in a protein! Moreover, both X-ray and solution NMR structures of proteins have accuracies much

A. C. de Dios, E, Oldfield / Soki State Nuclear Magnetic Resonance 6 (1996) 101-l 25 115
less than typically expected for chemical shift calculations. Fortunately, however, the task can be reduced to manageable proportions by first separating the total shielding, at, into three parts, basically akin to Eq. (9):
a, = 0, + ue + u (12) and using unif:rm bond lengths [122]. a, is the short-range or “electronic” contribution to shielding, and has to be evaluated by quantum chemical means; a, is the electrostatic field con- tribution, and can be determined either by using the shielding polarizabilities, or by representing atoms outside of a “core fragment” by partial atomic charges - the CFP approach; and a,.,, represents the other, primarily magnetic contri- butions, such as the long-range peptide and aro- matic “ring current” effects noted. If necessary these can be evaluated using the semi-classical approaches used for ‘H NMR.
As with ‘H NMR, there are well defined ex- perimental correlations between secondary struc- ture and chemical shift in 13C NMR, and we show in Fig. 13A the secondary chemical shifts for C* and Cp residues in the data base of Spera and Bax [118]. As can be seen from Fig. 13A, C” resonances are shielded in P-sheet regions but deshielded in cw-helices, while CB shows the op- posite trend, both in solution and in the solid-state 11231. How can these trends be investigated in more detail? Clearly, it is not at present feasible
a-heli
k
expt p-sheet
a-hel
A a-helix
? m
theory -sheet
I wm SCp,
Fig. 13. 13C NMR shielding in proteins. (A) Histograms show- ing separation of a-helical and P-sheet chemical shifts for C” and CD sites in proteins (based on global database of Spera and Bax, Ref. [1181X (B) Histogram showing C” and C@ shifts for model o-helical and p-sheet (alanine only) fragments, based on ab initio shielding surface calculations (from Refs. [118,1221).
to carry out ab initio calculations on a complete protein. However, if it is reasonable to suppose that if 4,$ torsion angle effects dominate shield- ing - as suggested by the empirical observations on helical and sheet residues - then the shield- ing changes due to these geometric parameters would propagate through the bonding framework, and would therefore only operate over very short ranges. Thus, only the local geometry might need be considered, i.e. u, in Eq. (lo), which would require only a small number of atoms requiring basis functions. Of course, if more indirect effects were important - such as differential hydrogen bonding patterns - or if longer-range (e.g. helix dipole) electrostatic field effects dominate, then simple 4,3/ correlations might not be readily de- tected.
The effects of 4, 4 alone on shielding can be determined by using, e.g. the GIAO method, and we first consider as a general example C” and C” shielding trends of alanine residues in proteins. Construction of a simple molecular fragment, N- formyl-L-alanine amide:
permits incorporation of two peptide planes, so that +,I) effects can be studied. Use of a rela- tively small (6-311G * * ) basis enables a relatively rapid determination of C” and C@ shielding as a function of &$, and results obtained using this fragment are shown in Fig. 13B. There is clearly good general agreement with the experimental trends reported by Spera and Bax [118,122]. Of course, Figs. 13A and 13B show only general trends, and not predictions of specific experimen- tal chemical shifts. This can be achieved by use of a larger basis, and good agreement between the- ory and experiment has been achieved for specifi- cally assigned C* and Cp resonances of the twelve Ala residues in Staphylococcal nuclease [122] (SNase). The effects of hydrogen bonding can be incorporated by locating formamide molecules on the coordinates of hydrogen bond partners, and long-range electrostatic field effects are included

116 A.C. de Dies, E. Oldfield /Solid State Nuclear Magnetic Resonance 6 (19%) 101-125
by using CFP - although both effects are quite minor. There is obviously good agreement with experiment, as shown previously [122], even though the protein itself has a molecular weight of over 13000 Da.
10. Carbon-13 shielding surfaces
The +,1+5 influences on shielding are not seen in a particularly clear way from the results shown in Fig. 13, and it is desirable to have a more direct way of converting 4, + information into experimental chemical shifts. This would enable spectral assignments to be rapidly verified, and could lead to ways of deducing structure directly from chemical shifts - if 4, I++ effects dominate.
-180 -120 -60 0 60 120 180
Such a nexus between structure and shielding is indeed implicit in the work of Wishart et al. [114] and Spera and Bax [118], and we show in Figs. 14A and 14B results for C”, C? as a function of 4, I,+ [118,124]. The work of Spera and Bax in- volved use of a “global” data base containing all types of residue, but it nevertheless clearly sug- gests that C*, C? shifts vary within the helical and sheet regions. This behavior is seen much more readily when individual shielding surfaces are computed for particular amino acids, and Figs. 14C and 14D show computed C”, CB shield- ing surfaces for alanine, again using a locally dense approach [124]. The general trend seen in the empirical surface of the sheet region contain- ing shielded C* but deshielded CB, with the opposite trend in the helical region, is apparent,
ccx
120
60
-60
-180 -120 -60 0 60 120 180
Fig. 14. Global ‘“C 4, $ shielding surfaces for (A) C”, (B) Ca from Spera and Bax [118], together with theoretical alanine ‘“C 4, JI
shielding surfaces for (C) C”: (II) CB, from Ref. 11241.
A. C. & Dies, E. Oldfield /Solid State Nuclear Magnetic Resonance 6 (1994) 101 -I 25 117
130 - 70 60 50 40
experimental shift (ppm from TSP)
Fig. 15. Computed 13Cu shielding for Gly, Ala and Val
residues in SNase and calmodulin; 57 data points, r.m.s. deviation from fitted curve, 1.5 ppm (from Ref. [lo]).
with the added advantage over the empirical sur-
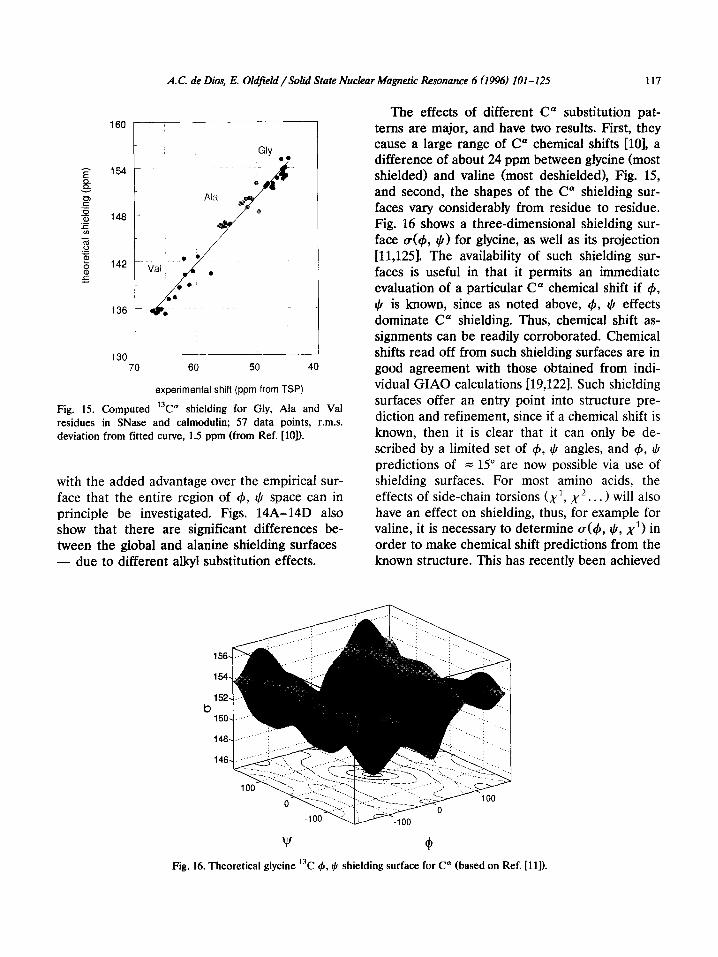
face that the entire region of 4, $ space can in principle be investigated. Figs. 14A-14D also show that there are significant differences be- tween the global and alanine shielding surfaces - due to different alkyl substitution effects.
The effects of different C” substitution pat- terns are major, and have two results. First, they cause a large range of C” chemical shifts [lo], a difference of about 24 ppm between glycine (most shielded) and valine (most deshielded), Fig. 15, and second, the shapes of the C” shielding sur- faces vary considerably from residue to residue. Fig. 16 shows a three-dimensional shielding sur- face ~(4, $) for glycine, as well as its projection [11,125]. The availability of such shielding sur- faces is useful in that it permits an immediate evaluation of a particular C* chemical shift if 4, $ is known, since as noted above, 4, 4 effects dominate C” shielding. Thus, chemical shift as- signments can be readily corroborated. Chemical shifts read off from such shielding surfaces are in good agreement with those obtained from indi- vidual GIAO calculations [19,122]. Such shielding surfaces offer an entry point into structure pre- diction and refinement, since if a chemical shift is known, then it is clear that it can only be de- scribed by a limited set of 4, $ angles, and 4, I) predictions of = 15” are now possible via use of shielding surfaces. For most amino acids, the effects of side-chain torsions (x1, x2.. . > will also have an effect on shielding, thus, for example for valine, it is necessary to determine (~(4, I), x1> in order to make chemical shift predictions from the known structure. This has recently been achieved
b
w (4 Fig. 16. Theoretical glycine “C 4, I) shielding surface for C” (based on Ref. [l I]).
118 A. C. de Dim, E. Oldfield /Solid State Nuclear Magnetic Resonance 6 (1996) 101-125
[126], and the principal conclusion, based on both shielding surface predictions and spin-spin cou- pling constant information [127,128], is that x1 torsions for valine can vary considerably between crystal and solution states, at least in the calmod- ulin system investigated.
11. Electrostatics and dynamics in proteins
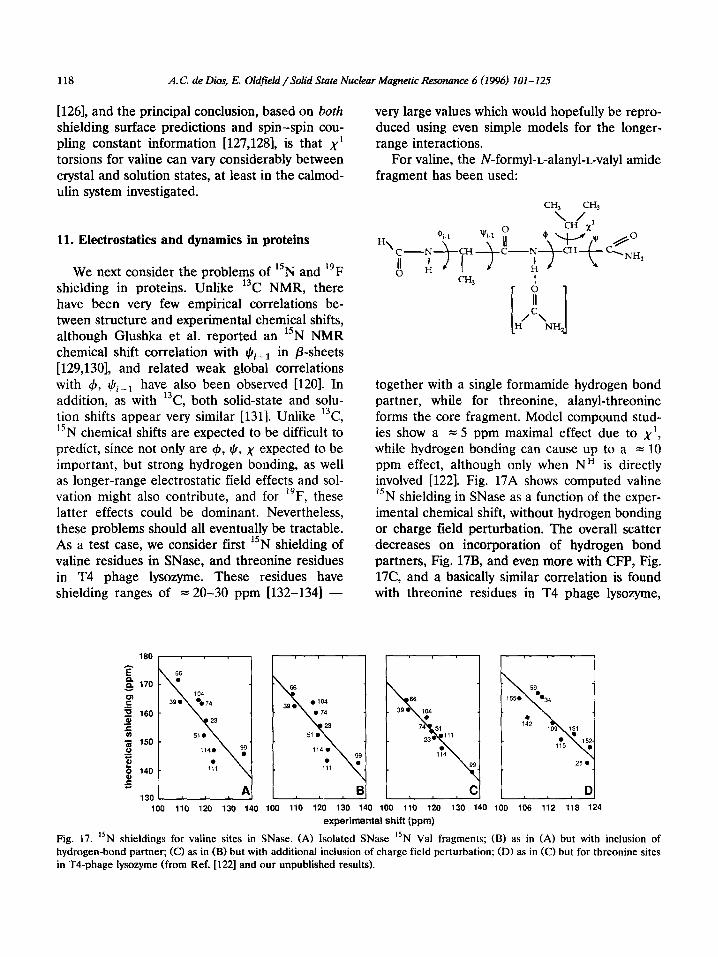
We next consider the problems of “N and 19F shielding in proteins. Unlike 13C NMR, there have been very few empirical correlations be- tween structure and experimental chemical shifts, although Glushka et al. reported an “N NMR chemical shift correlation with qQi__r in P-sheets [129,130], and related weak global correlations with 4, rcli_r have also been observed [120]. In addition, as with 13C, both solid-state and solu- tion shifts appear very similar [1311. Unlike 13C, 15N chemical shifts are expected to be difficult to predict, since not only are (b, $, x expected to be important, but strong hydrogen bonding, as well as longer-range electrostatic field effects and sol- vation might also contribute, and for l”F, these latter effects could be dominant. Nevertheless, these problems should all eventually be tractable. As a test case, we consider first “N shielding of valine residues in SNase, and threonine residues in T4 phage lysozyme. These residues have shielding ranges of = 20-30 ppm 1132-1341 -
160
very large values which would hopefully be repro- duced using even simple models for the longer- range interactions.
For valine, the N-formyl-r_-alanyl-L-valyl amide fragment has been used:
;I [ 1 H/c\
NH,
together with a single formamide hydrogen bond partner, while for threonine, alanyl-threonine forms the core fragment. Model compound stud- ies show a = 5 ppm maximal effect due to x’, while hydrogen bonding can cause up to a = 10 ppm effect, although only when NH is directly involved [122]. Fig. 17A shows computed valine 15N shielding in SNase as a function of the exper- imental chemical shift, without hydrogen bonding or charge field perturbation. The overall scatter decreases on incorporation of hydrogen bond partners, Fig. 17B, and even more with CFP, Fig. 17C, and a basically similar correlation is found with threonine residues in T4 phage lysozyme,
100 110 120 130 140 100 110 120 130 140 100 110 120 130 140 100 106 112 118 124
experimental shift (ppm)
Fig. 17. “N shieldings for valine sites in SNase. (A) Isolated SNase 15N Val fragments; (B) as in (A) but with inclusion of
hydrogen-bond partner; (C) as in (B) but with additional inclusion of charge field perturbation; (D) as in (C) but for threonine sites
in T4-phage lysozyme (from Ref. [122] and our unpublished results).

A.C. de Dios, E. Oldfield/Solid State Nuclear Magnetic Resonance 6 (1996) 101-125 119
Fig. 17D. Since these results are the first predic- tions of 15N shielding in a protein, they can be regarded as quite promising. In crystalline SNase, Va151 is not observed, possibly due to disorder, but the shifts for the other residues appear very similar in crystals and in solution, and are well predicted from X-ray structures.
The final nucleus we consider is 19F. Fluorine is a non-native probe of structure in proteins, but has been used for many years in NMR studies of protein structure in solution [121,135-1391, and more recently systems as large as the nicotinic acetylcholine receptor have begun to be probed using solid-state techniques 11401. Conventional wisdom has suggested that van der Waals disper- sion interactions dominate 19F chemical shifts in proteins, but this now seems unlikely, since the basis for the B term has disappeared [9]. In particular, we now know that 19F chemical shifts and shift tensors can be well described at the CHF level, without vdW dispersion, and we also know that the shielding polarizabilities of fluoro- aromatics are extraordinarily large-about 2000 ppm a.u.-’ [93,105]. Also there is evidence to suggest that electrostatic fields of up to = 0.008 a.u. exist in proteins [141-1431, so electrostatic field induced shifts of = 2000 X 0.008 = 16 ppm are predicted for 19F - about the values found experimentally. It thus seems plausible that a, dominates i9F shielding in proteins, and in order to test this hypothesis, it is necessary to investi- gate a protein having a rigorously assigned i9F
10 0 127 133 103 195 284
NMR spectrum, whose structure is also known. The galactose binding protein (GBP) from Es- chekhiu coli fits the bill since it has five trypto- phan residues, all of which have been labeled with [5F]Trp, and the chemical shifts cover a = 10 ppm range [137].
In an SCF calculation, it is clearly a difficult matter to decide on the correct atomic charges in a system as complex as a protein, and also to decide on whether or not to consider dielectric effects. Since it is known that surface charge field modification does not appreciably perturb the r9F NMR spectrum of hen egg white lysozyme [144], surface charges need not be considered, with only whole electroneutral residues being used to eval- uate the electrostatic field contributions to shield- ing in GBP [122]. Fig. 18A shows the electrostatic field contributions to shielding for four of the five [5F]Trp residues in GBP - results in good accord with experiment 11371. Trpzs4 is exposed to solvent, and here the alternate route of using the shielding polarizabilities is required in order to evaluate solvent effects. Based on the model compound studies discussed above [93], the field and field gradient contributions (dipole and quadrupole shielding polarizabilities) are ex- pected to dominate the electrostatic contributions to shielding, and these have been evaluated by using a molecular dynamics approach. When the field and field gradients are multiplied by the corresponding shielding polarizabilities, the elec- trostatic contributions to shielding are obtained
-4 -2 0 2 4 6 experiment (Ppm) shielding (ppm)
.4 -2 i i h i experiment (ppm)
Fig. 18. “F shielding results for the five [S-r”F]-Trp residues in the Escherichia coli galactose binding protein. (A) GIAO shielding
results for Trp residues 127, 133, 183 and 195; (B) individual 20 ps shielding trajectories for each of the five [S-F] Trp residues; (C)
graph showing experimental vs. theoretical shielding results from the trajectories in (B) (from Refs. [7,122]).

120 A. C. de Dies, E. Oldfield /Solid State Nuclear Magneiic Resonance 6 (1996) 101-l 25
over, in this case 5 X 20 psec MD trajectories, Fig. 18B. The time-average shielding values ob- tained from the shielding trajectories are in good accord with the experimental chemical shifts, and the results of both CFP and shielding polarizabil- ity approaches are in good accord with experi- ment (Fig. 18). For the CFP method, the shield- ing range is slightly overestimated - possibly because of the lack of dielectric contributions. The shielding trajectory method can also of course be combined with, e.g. C”, C? shielding surfaces in order to incorporate the effects of molecular motion, and should be of use in making even more accurate predictions of experimental pro- tein NMR spectra in the future.
The basic physics of the CFP/MSP weak elec- trical interaction model is quite different from the vdW dispersion approach, also recently pro- posed as a means of predicting 19F chemical shifts. On the one hand, the SCF-related meth- ods give good accord with experimental shifts as well as shift tensors for all fluorobenzenes, with- out exception, and with no adjustable parameters, while the vdW dispersion method fails in several cases, and can best be thought of as an empirical fitting procedure, with no physical basis for A,B, etc.
12. Protein structure prediction and refinement
In essentially all cases examined so far, known (generally X-ray) structures have been used in order to predict experimental NMR spectra. In many cases this is an important step in itself, but clearly one of the main goals of our quantum chemical work is also to be able to deduce struc- ture from experimental chemical shifts. Chemists routinely employ empirical methods, essentially look up tables, to deduce structure from shielding in small molecules, and more recently this ap- proach is beginning to be applied to macro- molecules such as proteins. We thus consider first the empirical approach to structure prediction, and then show how the method can be extended to incorporation of shielding surfaces obtained by using ab initio methods.
For proteins, there are = 130000 reported
chemical shifts [107], although perhaps only 1 or 2% have been used directly to facilitate structure analysis. The simplest empirical approach is the so-called chemical shift index (CSI) reported by Wishart and coworkers [119,145]. The idea here is simple and is that for CO, H*, C’, and to a lesser extent Cp, there are clear positive or nega- tive secondary chemical shifts from the random coil chemical shift position which are structure (helix, sheet) related - albeit the nature of the relation may be quite different for say C” and C’. The CSI method uses a tri-state method in which, e.g. for C”, a “1” is assigned to a helical shift, a “0” to a random coil shift, or a “ - 1” to a P-sheet shift. When a certain number of consecu- tive residues exhibit, e.g. helical behavior, then the presence of helical secondary structure is inferred. The method is particularly powerful when C*, Ha and C’ indices are combined into a consensus CSI pattern, and the method gives a generally excellent accord with experiment 11191.
The success of the empirical chemical shift index method immediately leads one to ask - why is it successful, and can it be improved? The method is successful because chemical shifts are structure dependent. Since many such chemical shifts can now be predicted using ab initio meth- ods, it is thus logical to see if additional, more detailed, structural information can be obtained, since as shown in Fig. 14, there are considerable chemical shift differences within a particular structure type region. That is, helical and sheet residues do not have just two chemical shifts, rather, a more or less continuous distribution. How then can more detailed structural informa- tion be derived from these distribution functions?
One approach is essentially an analog version of the digital CSI method, and makes use of e.g. C”, Cp and H” shift surfaces. The idea is basi- cally to estimate the probability, Z, that a given chemical shift, Pexpt corresponds to a particular 4,J, torsion angle pair. For P =fCc$, $), the prob- ability 2
Z = exp
is given by:
P expt - P(47 $1 * - W (13)
A. C. de Dies, E. Okffiehi /Solid State Nuclear Magnetic Resonance 6 (I 996) 101-125 121
A C 190
90
0
-90
8
4 -‘*O - 180 3
90
0
-90
-iso -W d 9b tad -ho -9b
’
90 180
B 4 (deal D
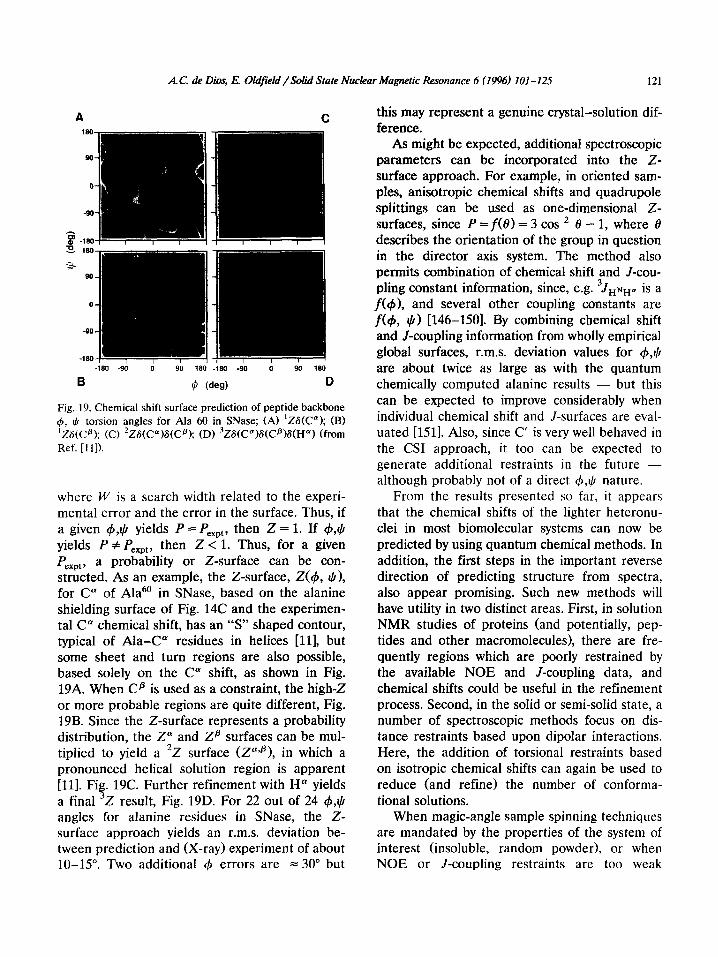
Fig. 19. Chemical shift surface prediction of peptide backbone 4, I) torsion angles for Ala 60 in SNase; (A) ‘Z6(C”); (B) ‘ZG(Cfi); (C) ‘ZS(C”)S(Cp); (D) 3ZS(C”)S(Cs)S(Hu) (from Ref. [I 11).
where W is a search width related to the experi- mental error and the error in the surface. Thus, if a given 4,1) yields P = Pexpt, then Z = 1. If 4,$ yields P # Pexpt, then Z < 1. Thus, for a given P a probability or Z-surface can be con- siE:ted. As an example, the Z-surface, Z(r), I)), for C” of Ala6’ in SNase, based on the alanine shielding surface of Fig. 14C and the experimen- tal C” chemical shift, has an “S” shaped contour, typical of Ala-C” residues in helices [ll], but some sheet and turn regions are also possible, based solely on the C” shift, as shown in Fig. 19A. When Cp is used as a constraint, the high-Z or more probable regions are quite different, Fig. 19B. Since the Z-surface represents a probability distribution, the Z” and Za surfaces can be mul- tiplied to yield a ‘2 surface (ZaTp), in which a pronounced helical solution region is apparent [ll]. Fig. 19C. Further refinement with H” yields a final 3Z result, Fig. 19D. For 22 out of 24 $,tj angles for alanine residues in SNase, the Z- surface approach yields an r.m.s. deviation be- tween prediction and (X-ray) experiment of about 10-15”. Two additional 41 errors are = 30” but
this may represent a genuine crystal-solution dif- ference.
As might be expected, additional spectroscopic parameters can be incorporated into the Z- surface approach. For example, in oriented sam- ples, anisotropic chemical shifts and quadrupole splittings can be used as one-dimensional Z- surfaces, since P = f(e) = 3 cos * 8 - 1, where 8 describes the orientation of the group in question in the director axis system. The method also permits combination of chemical shift and J-cou- pling constant information, since, e.g. 3JH~Ha is a f(4), and several other coupling constants are fC$, $1 [146-1501. By combining chemical shift and J-coupling information from wholly empirical global surfaces, r.m.s. deviation values for 4,$ are about twice as large as with the quantum chemically computed alanine results - but this can be expected to improve considerably when individual chemical shift and J-surfaces are eval- uated [151]. Also, since C’ is very well behaved in the CSI approach, it too can be expected to generate additional restraints in the future - although probably not of a direct 4,rl, nature.
From the results presented so far, it appears that the chemical shifts of the lighter heteronu- clei in most biomolecular systems can now be predicted by using quantum chemical methods. In addition, the first steps in the important reverse direction of predicting structure from spectra, also appear promising. Such new methods will have utility in two distinct areas. First, in solution NMR studies of proteins (and potentially, pep- tides and other macromolecules), there are fre- quently regions which are poorly restrained by the available NOE and J-coupling data, and chemical shifts could be useful in the refinement process. Second, in the solid or semi-solid state, a number of spectroscopic methods focus on dis- tance restraints based upon dipolar interactions. Here, the addition of torsional restraints based on isotropic chemical shifts can again be used to reduce (and refine) the number of conforma- tional solutions.
When magic-angle sample spinning techniques are mandated by the properties of the system of interest (insoluble, random powder), or when NOE or J-coupling restraints are too weak

122 A.C. de Dies, E. Oldfield/Solid State Nuclear Magnetic Resonance 6 (1996) 101-125
(surface loops, large proteins, small peptides), then chemical shifts offer an important route for structure refinement and prediction, and in re- cent work, Case and coworkers [152,1531 have begun to demonstrate the utility of ‘H chemical shifts in the refinement of a relatively low resolu- tion solution NMR structure of the heme protein, carbonmonoxymyoglobin. The general approach can be extended to more than a two-dimensional 4,1(1 surface, and recently we have evaluated a three-dimensional shielding hypersurface for va- line, a(+, I), x), for use in refinement and pre- diction [126]. An interesting result of these stud- ies is that chemical shift refined structures have less scatter (in 4, 4, x> than unrestrained struc- tures, and appear very close to X-ray values.
13. Conclusions and prospects
Recent advances in computer performance to- gether with faster algorithms have begun to en- able the widespread application of computational chemistry to permit the accurate prediction of both solid-state and liquid-state NMR chemical shifts in most molecular systems - from small organic molecules to macromolecules such as proteins. However, of even more interest in the long term are the development of new methods of directly predicting structure from experimental spectra. In proteins, the empirical chemical shift index and its quantum chemical analog, the chemical shift or Z-surface approach, can facili- tate secondary structure prediction and assign- ment verification, and represent the first steps toward structure prediction, refinement and vali- dation in proteins, using the chemical shift pa- rameter. The chemical shifts (and indeed the shielding tensors) of most light (and many heavy) nuclei in most molecular systems can now be accurately predicted via quantum chemistry, us- ing small workstations or workstation clusters. Moreover, the sensitivity of NMR chemical shifts to structural parameters means that chemical shifts and quantum chemistry can be used to- gether to refine structures obtained by using other (e.g. diffraction) methods. For example, typical X-ray bond lengths found in proteins are quite
inconsistent with experimental chemical shifts [lo]. After over forty years of continuous develop- ment, most of the “chemical effects” observed in the 1950s can now be predicted. Given the gener- ally “sparse” nature of distance restraints in solid-state systems, the results we have outlined above suggest that chemical shift analysis - both isotropic and anisotropic - will be a welcome addition to solid-state structure determination. At present, chemical shift calculations are still unpleasantly slow. However, recent progress with density functional theory (DFT) [154] rather than Hartree-Fock methods suggest significant speed improvements, together with the incorporation of electron correlation, and even at the Hartree- Fock level, considerable time savings can be ex- pected in favorable cases, e.g. where accurate shielding surface shapes can be obtained using very small basis sets [126]. The evaluation of metal shifts is also now possible using especially DFT methods [154], and it seems likely that use of periodic Hartree-Fock methods [155] may per- mit analysis of shielding in inorganic crystal lat- tices in the near future. Given predictable in- creases in CPU speed over the next few years, the use of ab initio shielding calculations for macro- molecular structure prediction, refinement and validation in chemistry and biology is sure to flourish.
Acknowledgments
We are grateful to C.J. Jameson, P. Pulay and C.E. Dykstra for their continuous flow of advice, and P. Pulay, J.F. Hinton, K. Wolinski and W. Kutzelnigg for providing their computer pro- grams.
References
[l] W.D. Knight, Phys. Rev., 76 (1949) 1259. [2] W.C. Dickinson, Phys. Rev., 77 (1950) 736. [3] G. Lindstriim, Phys. Rev., 78 (1950) 817.
[4] H.A. Thomas, Phys. Rev., 80 (1950) 901. [5] H.S. Gutowsky and C.J. Hoffman, Phys. Rev., 80 (1950)
110-111. [6] W.G. Proctor and F.C. Yu, Phys. Rev., 81 (1951) 20.

A. C. de Dios, E. Oldfield / Solid State Nuclear Magnetic Resonance 6 (1996) IO1 -125 123
[7] J.G. Pearson, E. Oldfield, F.S. Lee and A. Warshel, J. Am. Chem. Sot., 115 (1993) 6851.
[8] AC. de Dios, D.D. Laws and E. Oldfield, J. Am. Chem. Sot., 116 (1994) 7784.
[9] A.C. de Dios and E. Oldfield, J. Am. Chem. Sot., 116 (1994) 7453.
[lo] D.D. Laws, A.C. de Dios and E. Oldfield, J. Biomol. NMR, 3 (1993) 607.
[l l] H. Le, J.G. Pearson, A.C. de Dios and E. Oldfield, J. Am. Chem. Sot., 117 (1995) 3800.
[12] C.E. Dykstra, Ab Initio Calculation of the Structures and Properties of Molecules, Elsevier, Amsterdam, 1988.
[13] N.F. Ramsey, Phys. Rev., 78 (1950) 699. [14] N.F. Ramsey, Phys. Rev., 86 (1952) 243. [15] S.I. Chan and T.P. Das, J. Chem. Phys., 37 (1962) 1527. [16] C.J. Kern and W.N. Lipscomb, J. Chem. Phys., 37 (1962)
260. [17] J. Tillieu and G.J. Guy, Chem. Phys., 24 (1956) 1117. [18] H.J. Kolker and M.J. Karplus, Chem. Phys., 41 (1964)
1259. [19] R.M. Stevens, R.M. Pitzer and W.N. Lipscomb, J. Chem.
Phys., 38 (1963) 550.
[Zl] R.M. Stevens and W.N. Lipscomb, J. Chem. Phys., 41 (1964) 184.
[20] R.M. Stevens and W.N. Lipscomb, J. Chem. Phys., 40 (1964) 2238.
[22] R.M. Stevens and W.N. Lipscomb, J. Chem. Phys., 41 (1964) 3710.
[23] R.M. Stevens and W.N. Lipscomb, J. Chem. Phys., 42 (1964) 4302.
[24] F. London, J. Phys. Radium, 8 (1937) 397. [2.5] R. Ditchfield, Mol. Phys., 27 (1974) 789. 1261 L. Salem, The Molecular Orbital Theory of Conjugated
Systems, W.A. Benjamin, New York, 1966. [27] R. Ditchfield and P.D. Ellis, Top. Carbon-13 NMR
Spectrosc., 1 (1974) 1-51. [28] W. Kutzelnigg, Ix. J. Chem., 19 (1980) 193. [29] M. Schindler and W. Kutzelnigg, J. Chem. Phys., 76
(1982) 1919. [30] M. Schindler and W. Kutzelnigg, J. Am. Chem. Sot.,
105 (1983) 1360. [31] A.E. Hansen and T.D. Bouman, J. Chem. Phys., 82
(1985) 5035. [32] P. Pulay, Adv. Chem. Phys., 69 (1987) 241. [33] K. Wolinski, J.F. Hinton and P. Pulay, J. Am. Chem.
Sot., 112 (199018251. [34] P. Pulay, personal communication, 1994. [35] M. H%er, R. Ahlrichs, H.P. Baron, P. Weis and H.
Horn, Theor. Chim. Acta, 83 (1992) 455. [36] T.D. Bouman and A.E. Hansen, Chem. Phys. Lett., 175
(19901 292. (371 J. Geertsen and J.J. Oddershede, Chem. Phys., 90 (19841
301. [38] A.E. Hansen and T.D. Bouman, in J.A. Tossell (Ed.),
Nuclear Magnetic Shieldings and Molecular Structure, NATO ASI Series C, Vol. 386, Kluwer, Dordrecht, 1993, pp. 117-140.
[39] J. Gauss, Chem. Phys. Lett., 191 (1992) 614.
1401 C. van Wiillen and W. Kutzelnigg, Chem. Phys. Lett., 205 (1993) 563.
[41] V. Dyczmons, Theor. Chim. Acta, 28 (1973) 307. [42] D.B. Chesnut and K.D. Moore, J. Comput. Chem., 10
(1989) 648. 1431 C.J. Jameson, Chem. Rev., 91 (1991) 1375. [44] W.H. Flygare, J. Chem. Phys., 41 (1964) 793. [45] A.D. Buckingham and J.A. Pople, Discuss. Faraday
Sot., 22 (19561 17. [46] C.J. Jameson, A.C. de Dios and A.K. Jameson, Chem.
Phys. L&t., 167 (1990) 575. [47] C.J. Jameson and A.C. de Dios, in J.A. Tossell (Ed.),
Nuclear Magnetic Shieldings and Molecular Structure, NATO AS1 Series C, Vol. 386, Kluwer, Dordrecht, 1993, pp. 95-l 16.
[48] C.J. Jameson and A.C. de Dios, J. Chem. Phys., 98 (1993) 2208.
[49] R.M. Stevens and M.J. Karplus, Chem. Phys., 49 (1968) 1094.
[SO] E.A. Laws, R.M. Stevens and W.N. Lipscomb, J. Chem. Phys., 54 (1971) 4269.
[51] R. Ditchfield, Chem. Phys., 63 (1981) 185. [52] D.B. Chesnut and C.K. Foley, J. Chem. Phys., 85 (1986)
2814. [53] C.J. Jameson and A.C. de Dios, J. Chem. Phys., 97
(1992) 417. (541 R.A. Hegstrom, Phys. Rev., 19 (1979) 17. [55] P.W. Fowler, G. Riley and W.T. Raynes, Mol. Phys., 42
(1981) 1463. [56] P. Lazzeretti, R. Zanasi, A. Sadlej and W.T. Raynes,
Mol. Phys., 62 (1987) 605. [57] C.J. Jameson, A.C. de Dios and A.K. Jameson, J. Chem.
Phys., 95 (19911 1069. [58] C.J. Jameson, A.C. de Dios and A.K. Jameson, J. Chem.
Phys., 95 (1991) 9042. [59] D.B. Chesnut, D.W. Wright and R.A. Macphail, Chem.
Phys. Lett., 151 (1988) 415. (601 W. Kutzelnigg, C. van Wullen, U. Fleischer, R. Franke
and T. van Mourik, in J.A. Tossell (Ed.), Nuclear Mag- netic Shieldings and Molecular Structure, NATO AS1 Series C, Vol. 386, KIuwer, Dordrecht, 1993, pp. 141- 162.
(611 M. Barfield, in J.A. Tossell (Ed.), Nuclear Magnetic Shieldings and Molecular Structure, NATO AS1 Series C, Vol. 386, Kluwer, Dordrecht, 1993, pp. 523-538.
[62] H. Kurosu, I. Ando and G. Webb, Magn. Reson. Chem., 31 (1993) 399.
[63] E.G. Paul and D.M. Grant, J. Am. Chem. Sot., 85 (1963) 1701.
[64] W.T. Raynes, A.D. Buckingham and H.J. Bernstein, J. Chem. Phys., 36 (1961) 3481.
[65] W.T. Raynes, in J.A. Tossell (Ed.), Nuclear Magnetic Shieldings and Molecular Structure, NATO AS1 Series C, Vol. 386; Kluwer, Dordrecht, 1993, pp. 401-420.
[66] F. London, 2. Phys. Chem. B, 11 (1930) 222. [67] C.J. Jameson and J. Mason, in J. Mason (Ed.), Multinu-
clear NMR, Plenum, New York, 1987, pp. 51-88. [68] G.C. Maitland, M. Rigby, E.B. Smith and W.A. Wake-

124 A.C. de Dios, E. Oldfield /Solid State Nuclear Magnetic Resonance 6 (1996) IO1 -125
ham, Intermolecular Forces, Their Origin and Determi- nation, Clarendon Press, Oxford, 1981.
[69] K.W. Miller, N.V. Reo, A.J.M.S. Uiterkamp, D.P. Sten- gle, T.R. Stengle and K.L. Williamson, Proc. Nat. Acad. Sci. USA, 78 (1981) 4946.
1701 P. Diehl, 0. Muenster and J. Jokisaari, Chem. Phys. L&t., 178 (1991) 147.
[71] C.J. Jameson, AK. Jameson and S.M. Cohen, J. Chem. Phys., 59 (1973) 4540.
[72] C.J. Jameson, A.K. Jameson and SM. Cohen, J. Chem. Phys., 62 (1975) 4224.
[73] J. Fraissard and T. Ito, Zeolites, 8 (1988) 350. [74] P.J. Barrie and J. Klinowski, Prog. NMR Spectrosc., 24
(1992) 91. [75] C.J. Jameson, AK Jameson, B.I. Baello and H. Lim, J.
Chem. Phys., 100 (1994) 5965. [76] C.J. Jameson, A.K. Jameson, H. Lim and B.I. Baello, J.
Chem. Phys., 100 (1994) 5977. [77] P.R. van Tassel, H.T. Davis and A.V. McCormick, Mol.
Phys., 73 0991) 1107. [78] A.V. Vernov, W.A. Steele and L. Abrams, J. Phys.
Chem., 97 (1993) 7660. [79] M.J. Stephen, Mol. Phys., 1 (1958) 223. [80] A.D. Buckingham and K.P. Lawley, Mol. Phys., 3 (1960)
219. 1811 A.D. Buckingham, Can. J. Chem., 38 (1960) 300. [82] J.D. Augspurger, C.E. Dykstra and E. Oldfield, J. Am.
Chem. Sot., 113 (1991) 2447. [83] J.G. Batchelor, J. Am. Chem. Sot., 97 (197.5) 3410. [84] J.P. Riley and W.T. Raynes, Mol. Phys., 32 (1976) 569. [85] B. Day and A.D. Buckingham, Mol. Phys., 32 (1976)
343. [86] A.J. Sadlej and W.T. Raynes, Mol. Phys., 35 (1978) 101. [87] M.J. Packer and W.T. Raynes, Mol. Phys., 69 (1990)
391. [88] J.D. Augspurger and C.E. Dykstra, J. Phys. Chem., 95
(1991) 9230. [89] P.L. Cummins, G.B. Backsay and N.S. Hush, Mol. Phys.,
61 (1987) 795. [90] A.I. Jaman, T.C. Germann, J.D. Augspurger and C.E.
Dykstra, Chem. Phys., 154 (1991) 281. [91] H. Gutowsky, T.C. Germann, J.D. Augspurger and C.E.
Dykstra, J. Chem. Phys., 96 (1992) 5808. [92] A.C. de Dios and E. Oldfield, Chem. Phys. Lett., 205
(1993) 108. [93] J.D. Augspurger, A.C. de Dios, E. Oldfield and C.E.
Dykstra, Chem. Phys. Lett., 213 (1993) 211. [94] M. Mehring, Principles of High Resolution NMR in
Solids, Springer-Verlag, New York, 1983. [95] W.H. Flygare, Chem. Rev., 74 (1974) 653. [96] D.B. Chesnut, in G.A. Webb (Ed.), Annual Reports on
NMR Spectroscopy, Vol. 21, Academic Press, London, 1989, pp. 51-97.
1971 R. Hiiller and H. Lischka, Mol. Phys., 41 (1980) 1017. 1981 J.F. Hinton, P.L. Guthrie, P. Pulay, K. Wolinski and G.
Fogarasi, J. Magn. Reson., 96 (1992) 154. [99] D.M. Grant, J.C. Facelli, D.W. Alderman and M.H.
Sherwood, in J.A. Tossell (Ed.), Nuclear Magnetic
Shieldings and Molecular Structure, NATO AS1 Series C, Vol. 386, Kluwer, Dordrecht, 1993, pp. 367-384.
[lo01 D.W. Alderman, M.H. Sherwood and D.M. Grant, J. Magn. Reson., 101 (1993) 188.
[loll H.S. Gutowsky, D.W. McCall, B.R. McGarvey and L.H. Meyer, J. Am. Chem. Sot., 74 (1952) 4809.
[102] D.H. Gregory and J.T. Gerig, Biopolymers, 31 (1991) 845.
[103] S.E. Chambers, E.Y. Lau and J.T. Gerig, J. Am. Chem. Sot., 116 (1994) 3603.
[104] N. Boden, J.W. Emsley, J. Feeney and L.H. Sutcliffe, Mol. Phys., 8 (1964) 133.
11051 3. Augspurger, J.G. Pearson, E. Oldfield, C.E. Dykstra, K.D. Park and D. Schwartz, J. Magn. Reson., 100 (1992) 342.
[106] J.C. Facelli and D.M. Grant, Nature, 365 (1993) 325. 11071 B.R. Seavey, E.A. Farr, W.M. Westler and J.L. Markley,
J. Biomol. NMR, 1 (1991) 217. [108] S.J. Perkins, Biol. Magn. Reson., 4 (1982) 193. [109] H. Sternlicht and D. Wilson, Biochemistry, 6 (1967)
2881. ill01 A. Pardi, G. Wagner and K. Wiithrich, Eur. J. Biochem.,
137 (1983) 445. [ill] N.J. Clayden and R.J.P. Williams, J. Magn. Reson., 49
(1982) 383. [112] D.C. Dalgarno, B.A. Levine and R.J.P. Williams, Biosci.
Rep., 3 (1983) 443. [113] L. Szil&yi and 0. Jardetzky, J. Magn. Reson., 83 (1989)
441. [114] D.S. Wishart, B.D. Sykes and F.M. Richards, J. Mol.
Biol,, 222 (1991) 311. [115] K. Osapay and D.A. Case, J. Am. Chem. Sot., 113
(1991) 9436. [116] M.P. Williamson, T. Asakura, E. Nakamura and M.
Demura, J. Biomol. NMR, 2 (1992) 83. [117] J. Herranz, C. GonzLlez, M. Rico, J.L. Nieto, J. San-
toro, M.A. JimBnez, M. Bruix, J.L. Niera and F.J. Blanco, Magn. Reson. Chem., 30 (1992) 1012,
11181 S. Spera and A. Bax, J. Am. Chem. Sot., 113 (1991) 5490.
[119] D.S. Wishart and B.D. Sykes, J. Biomol. NMR 4 (1994) 171.
[120] H. Le and E. Oldfield, J. Biomol. NMR, 4 (1994) 341. [121] W.E. Hull and B.D. Sykes, Biochemistry, 15 (1976)
1535. [122] A.C. de Dios, J.G. Pearson and E. Oldfield, Science,
260 (1993) 1491. [123] S.W. Sparks, H.B.R. Cole, D.A. Torchia and P.E.
Young, Chem. Scripta, 29A (1989) 31. [124] A.C. de Dios, J.G. Pearson and E. Oldfield, J. Am.
Chem. Sot., 115 (1993) 9768. 11251 D. Jiao, M. Barfield and V.J. Hruby, J. Am. Chem. Sot.,
115 (1993) 10883. [I261 D.D. Laws, A. de Dios, R.H. Havlin and E. Oldfield, J.
Am. Chem. Sot., 117 (1995) 9542. [127] G.W. Vuister, A.C. Wang and A. Bax, J. Am. Chem.
sot., 115 (1993) 5334. 11281 M. Ikura, personal communication, 1993.

A.C. de Dios, E. Oldfield/Solid State Nuclear Magnetic Resonance 6 (19%) 101-125 125
[129] J. Glushka, M. Lee, S. Coffin and D. Cowbum, J. Am. Chem. Sot., 111 (1989) 7716.
[130] J. Glushka, M. Lee, S. Coffin and D. Cowbum, J. Am. Chem. Sot., 112 (1989) 2843.
[131] H.B.R. Cole and D.A. Torchia, Chem. Phys., 158 (1991) 271.
11321 D.A. Torchia, SW. Sparks and A. Bax, Biochemistry, 28 (1989) 5509.
(1331 J. Wang, A.P. Hinck, S.N. Loh, D.M. LeMaster and J.L. Markley, Biochemistry, 31 (1992) 921.
[134] L.P. McIntosh, A.J. Wand, D.F. Lowry, A.G. Redfield and F.W. Dahlquist, Biochemistry, 29 (1990) 6341.
[135] J.T. Gerig, Methods Enzymol., 177 (1989) 3. [136] L.A. Luck and J.J. Falke, Biochemistry, 30 (1991) 6484. [137] L.A. Luck and J.J. Falke, Biochemistry, 30 (1991) 4248. [138] C. Ho, E.A. Pratt and G.S. Rule, Biochim. Biophys.
Acta, 988 (1989) 173. [139] M.P. Gamcsik, J.T. Gerig and R.B. Swenson, Biochim.
Biophys. Acta, 874 (1986) 372. [140] A. McDermott, personal communication, 1995. [141] W.G. HOI, Prog. Biophys. Mol. Biol., 45 (1985) 149. [142] S. Dao-Ping, D.I. Liao and S.J. Remington, Proc. Nat.
Acad. Sci. USA, 86 (1989) 5361. [143] E. Oldfield, K. Guo, J.D. Augspurger and C.E. Dykstra,
J. Am. Chem. Sot., 113 (1991) 7537.
[144] C. Lian, H. Le, B. Montez, J. Patterson, S. Harrell, D. Laws, I. Matsumura, J. Pearson and E. Oldfield, Bio- chemistry, 33 (1994) 5238.
[145] D. Wishart, F.M. Richards and B.D. Sykes, Biochem- istry, 31 (1992) 1647.
[146] M. Karplus, J. Am. Chem. Sot., 85 (1963) 2870. [1471 V.F. Bystrov, Prog. NMR Spectrosc., 10 (1976) 41. [148] G.W. Vuister, F. Delaglio and A. Bax, J. Am. Chem.
Sot., 114 (1992) 9674. [149] F. Delagho, D. Torchia and A. Bax, J. Biomol. NMR, 1
(1991) 439. [ 1501 G.W. Vuister and A. Bax, J. Am. Chem. Sot., 115 (1993)
7772. 11511 AS. Edison, J.L. Markley and F. Weinhold, J. Phys.
Chem., 97 (1993) 11657. [152] K. Gsapay and D.A. Case, in J.A. Tossell (Ed.) Nuclear
Magnetic Shieldings and Molecular Structure, Khrwer Academic Publishers, Boston, 1993, p. 572.
(1531 D.A. Case and P.E. Wright, in G.M. Clore and A.M. Gronenbom (Eds.), NMR of Proteins, MacMillan, 1993, pp. 53-91.
[154] V.G. Malkin, O.L. Malkina, M.E. Casida and D.R. Salahub, J. Am. Chem. Sot., 116 (1994) 5898.
[155] S. Hammesschiffer and H.C. Andersen, J. Chem. Phys., 101 (1994) 375.












![Patents, Paradigm Shifts, and Progress in Biomedical Science · 2004] Patents, Paradigm Shifts, and Progress 663 framework—what Kuhn calls a scientific paradigm—comprises “normal](https://static.fdocuments.net/doc/165x107/5eb7ffac2f5b8957b72caa8d/patents-paradigm-shifts-and-progress-in-biomedical-science-2004-patents-paradigm.jpg)





