Reactivity of zeolitic bronsted acidic sites · Chem. Rev. 1995, 95, 637-660 637 Reactivity Theory...
25
Reactivity of zeolitic bronsted acidic sites Citation for published version (APA): Santen, van, R. A., & Kramer, G. J. (1995). Reactivity of zeolitic bronsted acidic sites. Chemical Reviews, 95(3), 637-660. https://doi.org/10.1021/cr00035a008 DOI: 10.1021/cr00035a008 Document status and date: Published: 01/01/1995 Document Version: Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers) Please check the document version of this publication: • A submitted manuscript is the version of the article upon submission and before peer-review. There can be important differences between the submitted version and the official published version of record. People interested in the research are advised to contact the author for the final version of the publication, or visit the DOI to the publisher's website. • The final author version and the galley proof are versions of the publication after peer review. • The final published version features the final layout of the paper including the volume, issue and page numbers. Link to publication General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal. If the publication is distributed under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license above, please follow below link for the End User Agreement: www.tue.nl/taverne Take down policy If you believe that this document breaches copyright please contact us at: [email protected] providing details and we will investigate your claim. Download date: 23. Jul. 2021
Transcript of Reactivity of zeolitic bronsted acidic sites · Chem. Rev. 1995, 95, 637-660 637 Reactivity Theory...

Reactivity of zeolitic bronsted acidic sites
Citation for published version (APA):Santen, van, R. A., & Kramer, G. J. (1995). Reactivity of zeolitic bronsted acidic sites. Chemical Reviews, 95(3),637-660. https://doi.org/10.1021/cr00035a008
DOI:10.1021/cr00035a008
Document status and date:Published: 01/01/1995
Document Version:Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers)
Please check the document version of this publication:
• A submitted manuscript is the version of the article upon submission and before peer-review. There can beimportant differences between the submitted version and the official published version of record. Peopleinterested in the research are advised to contact the author for the final version of the publication, or visit theDOI to the publisher's website.• The final author version and the galley proof are versions of the publication after peer review.• The final published version features the final layout of the paper including the volume, issue and pagenumbers.Link to publication
General rightsCopyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright ownersand it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights.
• Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal.
If the publication is distributed under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license above, pleasefollow below link for the End User Agreement:www.tue.nl/taverne
Take down policyIf you believe that this document breaches copyright please contact us at:[email protected] details and we will investigate your claim.
Download date: 23. Jul. 2021

Chem. Rev. 1995, 95, 637-660 637
Reactivity Theory of Zeolitic Brarnsted Acidic Sites
R. A. van Santen*
Schuit lnstitute of Catalysis, Faculty of Chemical Engineering, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands
G. J. Kramer
Shell Research b. v., Koninkl~ke/Shell-Laboratorium, Amsterdam, The Netherlands
Received June 27, 1994 (Revised Manuscript Received December 20, 1994)
Contents I. General Introduction 0. The Br~nsted Acidic Hydroxyl
A. Background B. Quantum-Chemical Cluster Calculations C. Vibrational Spectroscopy of the Zeolitic OH
Group
A. Introduction B. Lattice Stability C. Deprotonation Energies
IV. Hydrogen Bonding versus Zwitterion Formation A. Background B. Zwitterion Formation C. Vibrational Dynamics of the Disturbed Zeolitic
Hydroxyl D. Solid Acid Br~nsted Acidity
V. Ionic Intermediate versus Transition State Formation A. Background B. The CH5+ Carbonium Ion, CH30H2+, and
C. The Activation of Hydrocarbons
Ill. Lattice Stability and Proton Affinity
Bifunctional Acidity
IV. Conclusion
637 639 639 640 642
644 644 645 646 647 647 647 649
652 652
652 653
656 657
1. General Introduction The unique environment of the Bransted acidic
protons in the micropores of zeolites controls the overall catalytic behavior of zeolites to a significant extent. One of the most obvious consequences for reactivity is the resulting stereoselectivity of zeolitic catalysts. Molecules too large to enter the zeolite will not be converted. Also, large molecules that are formed in the microcavities of the zeolite but are unable to pass through the smallest micropore di- ameter on their path outward of a zeolite particle will not appear as a product.
Molecules that move through the micropores of a zeolitic particle can have a significant interaction with the walls of the zeolite micropores. The strength of this interaction can be deduced from heats of adsorption measurements. I t appears to be a sensi- tive function of zeolite micropore diameter or shape.lj2 The interaction energy tends to increase as the zeolite micropores get smaller. The smaller the micropore diameters, the shorter the average distance between the molecular adsorbate atoms and the micropore
0009-2665/95/0795-0637$15.50/0
atoms. This causes the enhanced interaction energy. The interaction energy with the zeolite wall atoms is mainly due to the dispersive van der Waals interaction that is typical for systems without cova- lent interactions. Changes in zeolite structure can affect the heat of adsorption dramatically. The heat of adsorption of a molecule will determine its con- centration in the zeolite. Clearly, for reactions with an overall reaction rate that has a positive order in the reactant, the rate of reaction is enhanced when the reactant concentration is increased in the zeolite micropore.
The rate of diffusion of molecules through zeolites may also differ ~ i d e l y . ~ In reactions with zeolite crystallites of such a size that diffusion effects are important, the overall rate of reaction will change when zeolites are used with widely varying molecular diffusion constants.
It is important to realize the relevance of adsorp- tion and diffusion effects to the overall catalytic performance of zeolitic particles. The physical prop- erties of zeolitic protons and their reactivity with reactant molecules can only be translated to overall catalyst performance when adsorption and diffusion effects are properly included.
It is not a simple matter to define the Bransted acidity of protons at the solid-gas interface. It would be natural to relate the overall acidity to the number of protons per unit accessible micropore surface area or per unit zeolite volume. The intrinsic acidity of a zeolitic proton should relate to the response of the zeolitic proton to an interacting molecule. When the interacting molecule is a strong base, it may become protonated and the heat of adsorption of this mol- ecule may provide a measure of this response. However as we will discuss in section III.B, the protonation energy of a molecule depends to a sig- nificant extent on the interaction of the protonated molecule with the zeolite wall. So it only partially reflects the response of the proton to the adsorbing molecule. We have included a section dealing with the spectroscopy of hydrogen-bonded adsorbates to zeolites in order to illustrate that the response of protons to weakly interacting basic molecules pro- vides important information on the polarizability of the OH bond. This has a direct relation to the activation energy of the proton transfer reaction that occurs in Bronsted-catalyzed reactions.
The spectroscopic properties of the zeolitic protons attached to the lattice oxygen atoms that bridge
0 1995 American Chemical Society

638 Chemical Reviews, 1995, Vol. 95, No. 3 van Santen and Kramer
Theory has contributed significantly to an under- standing of the zeolitic OH bond. A major theoretical chemical question is the covalent versus ionogenic nature of the zeolitic framework bonds as well as that of the zeolitic OH bond. Proper understanding of the chemical nature of zeolitic Brgnsted acidity will depend on a resolution of this issue. A question of relevance to theoretical results is the validity of the cluster approximation and its relation to solid state chemical properties.
We will extensively discuss the use of classical molecular mechanics calculations on extended sys- t e m ~ ~ using force fields deduced from ab initio quantum-chemical cluster ~a lcu la t ions .~~~~’ This en- ables a detailed description of local distortions in zeolites around the protonic site. We will try to show the usefulness of this approach, which is based on the prediction of physical properties and structures of a set of zeolitic Si02 polymorphs. It appears that the zeolitic lattices are quite flexible and that relax- ation of the lattice atoms around the protonated site is responsible to a significant extent for the difference in deprotonation energies! Analysis of lattice vibra- tional spectra has demonstrated the validity of this conclusion?
The reactivity of the zeolitic Brgnsted acidic site will be discussed in the three following sections. An approach will be followed in which theoretical and experimental data are compared. Theory and experi- ment appear to be highly complementary.
As mentioned above, vibrational analysis of the response of the zeolitic OH group to adsorbing mol- ecules appears to be very useful as a probe of the intrinsic acidity of the zeolitic OH group. A discus- sion on the problem of proton transfer follows. The essential issue in proton transfer is the charge sepa- ration between the positively-charged protonated in- termediate and the negatively-charged zeolite lattice.
As will be shown, zeolites are essentially isolators. Protonated molecules have to be considered “zwit- terions”, stabilized by strong electrostatic interactions between the charged zeolite wall and the protonated adsorbate. Especially for polar molecules this raises the question whether such molecules can be consid- ered as hydrogen bonded to the zeolite lattice or whether protonated intermediates such as hydro- nium or hydroxonium ions are formed. Clearly, in order to settle this question theoretically one must understand the nature of the long-range electrostatic interactions in zeolites.
For acid catalysis an understanding of the nature and energetics of carbonium and carbenium ion formation is essential.
Finally, in order to predict overall catalytic per- formance one needs the values of the rate constants of the elementary reaction steps that form the catalytic reaction cycle.
Transition state rate theoryl0 expressions can be used once the transition state of the reaction is known. State of the art ab initio quantum chemical methods can currently be used to compute the minima and saddlepoint of potential energy surfaces of a reaction path between reactants and hence can predict the energetics and geometry of transition states.
1
Rutger Anthony van Santen was bom in 1945 in Langedijk, The Netherlands. He received his Ph.D. in 1971 in theoretical organic chemistry at the University of Leiden, on a thesis titled ”On the theoly of resonant scanering” with promoter Pro1 Dr. L. J. Ooslerhoff. From 1971 to 1972 he continued research as a postdoc at Stanford Research Institute, Menlo Park, CA, in the Molecular Physics department headed by Dr. F. T. Smith. During the period 1972-1988 he had several different lunctions at Shell Research, mainly in Amsterdam. At Shell his main research interest became catalysis. In 1976 he was a visiting Professor in Theoretical Chemistry at the Free University Amsterdam. From 1982 to 1984, he was assigned to Shell Development Co. in Houston, TX. In 1986 he became Professor Exlraordinarius in Sudace Chemistly at Eindhoven, University of Technology. In 1988 the appointment to full Professor 01 Catalysis followed. He is at present director 01 the Schuit Institute of Catalysis at Eindhoven. In 1981 he received the golden medal lor excellence in research of the Royal Dutch Chemical Society. In 1991 he was awarded the Chiapetla Lectureship by the North American Catalysis Society. His present research interests are molecular aspects of heterogeneous catalysis. The two main themes of his research are computational studies of sudace chemical reactivity and mechanism in heterogeneous catalysis.
\ Gert Jan Kramer was born in The Hague, The Netherlands, in 1961. In 1988 he obtained a Ph.D. in experimental solid slate physics lrom Leiden University. He then joined She& Amsterdam Laboratory where he worked for five years on the application of quantum chemistry in the area of zeolite catalysis. Presently. he works in the reactor engineering group of the same laboratoly, maintaining a vivid interest in the theoretical aspects of catalysis.
tetrahedrally coordinated three valent (Al, Ga, etc.) and four valent (Si) atoms have been studied experi- mentally, mainly by infrared and NMR spectros- COPY.
As will be discussed in the next section, the OH vibrational modes are a sensitive function of the zeolite composition. The dependence on zeolite struc- ture is not very large except when additional interac- tions occur by hydrogen bonding with a second lattice oxygen atom.

Reactivity Theory of Zeolitic Brsnsted Acidic Sites Chemical Reviews, 1995, Vol. 95, No. 3 639
Quantum-chemical calculations indicate that the charge on the proton is very low (<0.1 eu).12 Proton NMR datal3 indicate a slight upward chemical shift compared to that of the free silanol. A small weak- ening of the OH bond is demonstrated by a compari- son of the stretching infrared frequencies. Whereas the silanol infrared frequency is typically 3750 cm-l, the zeolitic hydroxyl groups have frequencies between 3550 and 3650 cm-l.
The stretching frequency of the zeolitic OH group is significantly higher than the highest lattice fun- damental modes, which have their maximum values around 1300 cm-l. This does not simply imply that the OH bond is much stronger than the lattice TO bonds, but reflects the low proton mass.
Values for the covalent OH bond strength have been deduced by the group of Kazansky.14 They used the overtone spectrum of OH groups to deduce the potential of OH using the Morse potential approxi- mation. They found a covalent dissociation energy of -500 kJ/mol, with very small differences in covalent bond strength between OH groups on many different solids.
This indicates, as we will confirm later, that the acidic nature of the OH group becomes apparent only when proton transfer itself, or the response of the OH group on an interacting basic molecule, is mea- sured.
Clearly, when the OH bond is broken heterolyti- cally (for a bridging hydroxyl group the energy cost is -1250 kJ/mol15), the overall bond energy will sensitively depend on the degree of stabilization of the negative charge left on the oxygen atom. The silanol group (with a heterolytic bond dissociation energy of 1600 kJ/mol15) is less acidic that the zeolitic hydroxyl, because the negative charge on the oxygen atom is only stabilized by orbital interactions with one Si4+ ion in the case of the silanol, but by interactions with a Si4+ and another T3+ ion on the bridging site.
As will be discussed, the polarizability of the zeolitic OH group is also much higher than that of the free silanol group. This polarizability relates of course to the electronic interactions between the oxygen and neighboring atoms.
A classical interpretation of the high Bransted acidity of the zeolite is the large Pauling valency excess on the three coordinated oxygen atom.16 Whereas this valency excess is zero on the oxygen atom of free silanol, it is +3/4 on the bridging zeolitic oxygen atom. This represents the excess in effective formal positive charge of the nonclassically coordi- nated oxygen atom. As we will show, this simple electrostatic view of
the zeolitic OH bond can be misleading. The OH as well as the lattice OT bonds have to be considered as strong covalent bonds superposed by small (long- range) electrostatic interactions.
Apart from the analysis of the chemical bond of zeolitic materials, as computed from electronic struc- ture calculations and the direct computation of the electrostatic potential in the channels of the zeolite,l' analysis of measured and computed vibrational spec- tra18J9 and changes in bonding geometry upon pro- tonation20vzl provide additional information on the
/H n 1 - o;si-o,
/O
Silanol Zeolitic Brsnsted acidic hydroxyl
Figure 1. (a) Schematic representations of silanol and (b) zeolitic Bransted acidic hydroxyl.
We will discuss such computed transition states for a few proton transfer reactions and show that in zeolites carbonium ions have to be considered as transition states and not intermediates. Again it will be shown that the stabilizing interaction with the negatively-charged zeolite wall is essential.
Understanding of the nature of the transition states of the protonation reaction has led to new insights into the factors that determine differences in acid catalytic performance.'l We will show that the ionicity of the transition state and its geometry determine whether acidity relates to the difference in proton affinity of the two oxygen atoms involved in protonation, or that the value of the strong oxygen-proton bond controls protonation.
We also comment on the importance of differences in the proton bond strength to overall catalytic performance. Some additional factors that cause differences in catalytic performance when different zeolites are compared, will also be discussed.
I/# The Br0nsted Acidic Hydroxy/
A. Background The Bransted acidic proton consists of a hydrogen
atom bonded to the oxygen atom that connects the tetrahedrally-coordinated cations, which form the zeolitic framework (see Figure 1).
Zeolites can be considered to be constructed of tetrahedra, with oxygen atoms as apices and cations in their center. The tetrahedra form a three- dimensional system by sharing of one oxygen atom between each two tetrahedra. With Si4+ cations the zeolitic framework is a polymorph of quartz and has Si02 stoichiometry. The zeolitic framework loses neutrality when lattice Si4+ cations become replaced by lattice A13+ cations. The excess lattice negative charge now has to be compensated for by positively charged cations. Often alkali ions are used, which find a location in the microporous zeolite channel system. The zeolitic acidic site can be generated in several ways. NH4+ ions can be introduced in the zeolitic microcavity by ion exchange and by heating they can subsequently be decomposed into NH3 and H+. The NH3 molecule desorbs, and the proton is left bonded to a bridging lattice oxygen atom, which connects a tetrahedron with a four valent (Si4+) cation and one that contains a three valent (A3+, Ga3+, Fe3+, ... ) cation.
Formally the 3-fold coordination of the oxygen bridge is a nonclassical bonding situation, also known for instance for the hydronium ion H@+. Compared to silanol (Figure la), which is only weakly acidic, the acidity of the proton is enhanced, which is due to a silanol that undergoes Lewis acid promotion by Al3'.

640 Chemical Reviews, 1995, Vol. 95, No. 3
Table 1. Computed Dielectric Constants E (m) and Plasmon Vibrational Frequencies m,,~.amon18*a
van Santen and Kramer
parameters used in the rigid ion and shell model have been obtained empirically from the physical proper- ties of quartz, the potential parameters of the partial charge model have been obtained from the potential energy surfaces obtained from ab initio quantum- chemical calculation^.^>^^ The charge on Si, according to partial charge models, is approximately half of the formal ~ a l e n c y . ~ Inspection of Table 1 shows that both the rigid ion and the shell models give too large long-range electrostatic interactions. Polarization effects clearly reduce the contribution of the long- range electrostatic interactions. The long-range elec- trostatic interactions have been also reduced in the partial charge calculations, due to the lower ionic charges, but are still too high because of the absence of polarization effects. Recently we developed shell potential parameters completely based on a fit to the potential energy surface as well as the electrostatics from ab initio calculations on small clusters.6 We now have satisfactory agreement between computed and predicted dielectric constants.
Table 1 also shows the large decrease in long-range electrostatic effects with a density decrease of the system. For zeolites such as faujasite, long-range electrostatic interactions contribute only -5% to the calculated vibrational frequency difference^.^^
We conclude that siliceous zeolites are isolators with a very low dielectric constant. The interaction of a weak base with the zeolitic hydroxyl can then be considered as a local bond that is very similar to the hydrogen bonding that occurs between gas-phase acidic molecules, e.g. between HC1 and NH3. Only when cations such as Na+, Kf or Mg2+, and Ca2+ are located in the zeolite micropore next to tetrahedra that contain trivalent cations such as A13+ instead of Si4+, are large electrostatic fields generated. They are short ranged, and the positive cation charges are compensated for by corresponding negative lattice charges.
B. Quantum-Chemical Cluster Calculations To study the zeolitic proton in more detail, it is
useful to analyze chemical bonding of the OH bond in clusters that represent part of the zeolite lattice. As we will argue in the next section, when chosen adequately clusters can often be fruitfully used to study chemical bonding questions in zeolites.
We will study the changes in geometry of a ring of four tetrahedra (Figure 2) terminated by hydrox- y l ~ . ~ ~ ! ~ ~ , ~ ~ Figure 2a shows the predicted geometry of such a cluster consisting of tetrahedra that contain only Si. Figure 2b shows the change in geometry of the cluster when one of the Si4+ ions is substituted by an A13+ ion. The Al-0 distances are longer in alumina tetrahedra compared to that in the silicon tetrahedron, because of the weaker Al-0 bond strength. A signature of covalent bonding in the clusters is the shortening of the Si-0 bond lengths that neighbor the Al-containing tetrahedron. Note also that the next neighbor bond lengths increase.
The alternating extensions and contractions of bond length with distance from the substituting center are typical for systems dominated by covalent bonding. It can be best understood on the basis of the principle of Bond Order C o n ~ e r v a t i o n . ~ ~ J ~ , ~ ~ ~ ~ ~
shell model rigid ion model Wplasmon, Wplasmon,
species u a , A 3 c ( m ) cm-1 ua,A3 c (-1 cm-1 a-quartzC 36.107 2.1163 366.80 42.367 l.OOOOb 557.80 a-quartzd 36.107 2.1397 254.24 42.367 l.OOOOb 394.43 sodalite 56.168 1.6992 166.73 59.110 l.OOOOb 248.50 faujasite 74.066 1.5030 107.53 62.51 l.OOOOb 149.98 a-quartzcae 37.66 2.383 139.57 a-quartzdSe 37.66 2.356 98.55
a In the calculations the length of the wave vectors 2 is about 0.001 reciprocal lattice units. * In the rigid ion model the high- frequency dielectric constants are exactly one. The zz com- ponent of the dielectric tensor and the shift of the A2 modes are taken. The xx component of the dielectric tensor and the splitting of the E modes are taken. e Experimental value.
relative importance of electrostatic versus covalent interactions.
The difference in frequencies of the transversal optical and longitudinal optical modes in solids is a direct measure of the long-range electrostatic inter- action in a solid.22
Whereas in high-symmetry crystals infrared radia- tion only excites the transversal optical modes, the longitudinal modes are accessible by Raman or neutron excitation. The longitudinal modes, which correspond to atomic motions along a symmetry axis of a crystal, usually have a slightly higher frequency than transversal modes, which correspond to atomic motions perpendicular to a symmetry axis of a crystal. Long-range electrostatic interactions enforce themselves for longitudinal modes, but cancel each other for transversal modes. The average difference of longitudinal and transversal optical mode differ- ences relates to the vibrational plasmon frequency:
The vibrational plasmon is a collective charge oscillation. It is related to the elementary charge of the charge carriers and the dielectric constant of a solid:
ua is the molar volume, ~ ( m ) the dielectric constant contribution due to electronic polarization, mi the atomic masses, and &i the effective ionic charge.
In Table 1, computed vibrational plasmon frequen- cies and measured values are compared for siliceous zeolitic polymorphs.18 The calculated values have been obtained from extended lattice calculations using rigid ion, partial charge, or shell model poten- tial parameters.
In the rigid ion model the charges on the ions are chosen to be equal to their formal valencies (e.g. the silicon charge is 4+). The shell model potential parameters are also based on formal valencies. Whereas polarization effects cannot be accounted for in the rigid ion approximation, in the shell model the oxygen atoms are considered to be p~larizable.~ The shell model enables a prediction of E ( = ) . Whereas the

Reactivity Theory of Zeolitic Brernsted Acidic Sites
a
Chemical Reviews, 1995, Vol. 95, No. 3 641
Ho, H O
b
A OH OH
H 0.965: bond length (-0.97). Mulliken charge
Figure 2. (a) Si0 and SiOH bond lengths in geometry-optimized [SiO(OH)234 cluster (STO-3G level),16 (b) bond lengths in geometry-optimized [SiO(OH)~l~[A1O(OH)~l- cluster (STO-3G level),16 (c) a four ring cluster containing two silicon and two aluminum atoms and one proton @TO-3G (d) a four ring cluster containing two silicon and two aluminum atoms and two protons [bond strengthened (s) or weakened (w) by second proton (STO-3G (a, b: Reprinted from ref 16. Copyright 1991 World Scientific. c, d: Reprinted from ref 25. Copyright 1994 American Chemical Society.)
According to this hypothesis, each atom has a con- stant valency to be distributed over the bonds to the atom. As a consequence, when one of the bond strengths decreases, e.g. by substitution with a weaker bonding atom, the others increase. In this way the total valency, that is the sum of the bond orders, remains constant. The alternating weaken- ing (corresponding to a stretching of a bond) and strengthening (corresponding to a shortening of a bond) of bond lengths is a consequence of this bond order conservation principle.
Changes in the geometry at the acidic site upon protonation and deprotonation illustrate also the highly covalent character of an acidic OH bond. In Figure 2c and Figure 2d the geometries of a four- membered tetrahedral ring are given for the situation where 2 Al ions have been substituted by 2 Si atoms and one ring is partially deprotonated.
Comparison of calculations with ring systems where Al- and Si-containing tetrahedra do not alter- nate shows the alternating system to be the most favorable.25 This agrees with the empirical Lowen- stein rule that forbids the presence of Al cations in neighboring tetrahedra. The geometries of the ring systems have been completely relaxed, the only constraint being that the ring cations are located in the same plane.
The computed geometry of the Si-O(H)-Al group is very close to that deduced for protonated sites using Al NMR and H NMR spectroscopy.28
Compared to the nonprotonated [Si-0-All bridge the bond lengths of the Si-0 and Al-0 bonds have
become lengthened upon protonation. This again agrees with the bond order conservation principle. The requirement that the 0 electrons bind to hydro- gen weakens the Si-0 and Al-0 bonds. The next neighboring 4-0 and Si-0 bonds are found to be shortened, as was expected because the alternation of bond length increases and decreases in covalently- bonded systems. There is a small decrease in the Si- 0-Al angle due to protonation, which is due to the change of approximate sp hybridization from nearly linear [Si-0-All to sp2 hybridization in the bent protonated system.
The results discussed so far have been obtained for a small cluster. The question is whether the struc- tural changes as computed for the cluster will remain when it is embedded in the zeolite crystal.
This will be discussed extensively in the next section, where it will be pointed out that the zeolite structure is quite flexible. Therefore, relaxation of the structure as found for clusters will, at least partially, also occur in the extended solid.
Quantum-chemical calculations have also been done for clusters of two tetrahedra as sketched in Figure la, in which the angle Q, of the T-0-T' bridge was kept constant. The OH deprotonation energy decreases with increasing angle qZ9 This again relates to the covalent nature of the OH bond. When the angle Q, equals M O O , hybridization is sp and the proton is bonded to the weakly-bonding 2p, or 2py oxygen orbitals. When Q, decreases (in zeolites ip has an average value of approximately 150°), hybridiza- tion around oxygen changes from its initial sp

642 Chemical Reviews, 1995, Vol. 95, No. 3
Table 2. Total Energies (in au) and Proton Affinities (in ev) of Four Ring Clusters"
van Santen and Kramer
geometry (A) STO3G (optimized) (B) 3-21G (ST03G) (C) 3-21G (optimized)
ring E (au) PA (eV) E (au) PA (eV) E (au) PA (eV) 1 SiSiSiSi -203.318 558 -2048.522 833 2 SiHSiSiSi -203.779 993 12.56 -2048.839 850 8.63 3 AlSiSiSi -201.331 399 -2001.777 284 -2001.850 643 4 AIHSiSiSi -202.002 739 18.27 -2002.279 709 13.67 -2002.374 317 14.25 5 AISiHSiSi -201.925 292 16.16 -2002.208 117 11.72 -2002.309 532 12.49
7 AlSiAlSi 2002.988 098 8 AlAlSiSi -199.161 940 -1954.885 995 9 AIHSiAISi -200.024 318 21.98 -1955.542 784 16.77 -1955.626 118 17.36
10 AIHSiHAISi -200.633 693 16.58* -1955.994 844 12.29* (-1956.120 927 13.46*) 11 AIHSiAIHSi -200.726 481 19.10* -1956.067 643 14.27* -1956.163 893 14.63* 12 AISiHAIHSi -200.683 936 17.94* -1956.024 463 13.11* -1956.125 6167 13.58* a Complete geometry optimizations were performed at the STO-3G level; the 3-21G results refer to the ST03G-optimized
geometry with an additional optimization of the proton position. Results marked by an asterisk refer to the abstraction of one proton only; the result in parentheses is influenced by a different configuration of the terminal hydrogen atoms.25
6 AIHSiSiSiH -2002.749 718 10.22*
hybridized state to spz hybridization when ~1 reaches 120"; the increased s character of the OH bond enhances its bond strength. Hence we predict that the intrinsic OH acidity increases with increasing 9. Again the amount of relaxation in the embedded system will determine to what extent the angle dependence of the deprotonation energy will be relevant in the extended system.
Quantum-chemical calculations on dimer systems have also been done to compare the effect of different trivalent ions by substituting Al by Fe or Ga.30 The changes in the OH stretching frequencies, as ob- served in low T' containing ZSM-5, have been studied e~perimental ly .~~ Whereas a value of 3610 cm-l is typical for the hydroxyl next to A13+, for Ga3+ and Fe3+ higher values are measured, implying a stronger OH covalent bond. The strengthening of the OH bond (weaker acidity) relates to the larger radii of the Ga3+ and Fe3+ tetrahedra. According to the bond order conservation principle the weaker OT' bonds result in stronger OH bonds.
Similar changes in the OH frequency have been observed when the concentration of T' atoms is changed. I t has been experimentally demonstrated that the strength of the OH bond is only sensitive to substitutional changes in the next-nearest neighbor tetrahedra with respect to the OH bond con~idered .~~ Changes in copposition in tetrahedra further re- moved from the OH bond have hardly any effect.
For the four ring systems computed changes of the deprotonation energy are given in Table 2 as a function of Al concentration as well as degree of protonation.
Note the stronger proton bond for the system with an N S i = 1 and the weakening of this bond when a second proton is coordinated to the ring system. The direction of the proton bond energy changes can again be predicted using the bond order conservation rule. The protons always prefer the oxygen position next to Al.
In the one ring system two SiO(0H)Z units were replaced by an (HO)AOP(OH)zO unit. The hydroxyl bond energy is found to increase. This agrees with the weaker intrinsic acidity of HSAPO's compared to that of the corresponding siliceous systems.33
1
- ? - W
c 2 0.5 a 2
0 I
I I I
3500 3600 3700 Wavenumber ( cm-1)
Figure 3. F'roton infrared absorption spectra of W Y , HY, and DY. (Reprinted from ref 35. Copyright 1994 Butter- worths.)
Of course the weakening and strengthening of the OH bond as a function of nextneighbor tetrahedra composition are also reflected in changes of electronic structure in the system. Considerations employing the HOMO-LUMO interaction concept have been applied and changes in reactivity have been cor- related with the corresponding parameter^.^^ A stronger bond is reflected in bonding orbitals of lower energy or a depletion of antibonding orbitals. These are the electronic features that are responsible for the chemical bonding rules, as are for instance expressed by bond order c ~ n s e r v a t i o n . ~ ~ ~ ~ ~
C. Vibrational Spectroscopy of the Zeolitic OH Group
In Figure 3 the infrared spectra of a NaHY zeolite with different WNa ratios35 can be compared. Three spectral features are observed. The very weak absorption around 3750 cm-l is due to surface silanol groups. A high-frequency (HF) feature is seen at approximately 3650 cm-l and a low-frequency (LF) feature at around 3550 cm-1.36 With decreasing proton concentration a small shift to higher frequen- cies is found, which is in agreement with the theo- retical results discussed in the previous subsection.
Calorimetric measurements of the adsorption of NH337 have shown that a decrease in proton concen-

Reactivity Theory of Zeolitic Brsnsted Acidic Sites Chemical Reviews, 1995, Vol. 95, No. 3 643
88 Figure 4. Schematic representation of the faujasite structure of zeolite Y. The corners denote the atoms (T = Si or Al) and the lines represent the bridging oxygen atoms. The small pictures show the 4 of the protons that are covalently bonded to the oxygen atoms around an aluminum atom.
positions different
of the T positions
tration gives a concomitant decrease in the protona- tion energy of NH3. This agrees with the increased OH bond energy as deduced for higher OH frequency.
In apparent disagreement with this observation, the HF proton has a higher reactivity than the LF proton. For instance, CO adsorption has been shown to disturb the HF frequency bond, but not the LF frequency bond.38
Crystallographic data show the oxygen atoms to which the protons are coordinated.20,21 In the fauja- site structure of zeolite Y, four distinct 0 atoms can be distinguished, labeled 0(1), 0 ( 2 ) , 0 ( 3 ) , and o(4) (Figure 4). The HF protons have been identified with the O(1) site, which is the oxygen bridge that connects the six rings in the double six ring unit formed by 12 tetrahedra. O(2) has been found to be only partially occupied.
Whereas the O(1) site has been identified with the HF bond, the o(3) site has been identified with the LF bond.36 Experimental support for this is given by ion-exchange experiments. When Cs+ ions ex- change with protons, the LF band intensity remains constant but the HF band intensity decreases.39 The proposed assignment follows as Cs+ cannot exchange with positions in the sodalite cage and the double six ring because of its size. An O(3) proton (Figure 4) is shown directed inwards toward the center of the six ring structure, inaccessible to Cs+.
The O(3) proton can form a weak hydrogen bond to another oxygen atom of the tetrahedral six ring in which it participates. This additional hydrogen bond will weaken the OH bond (bond order conservation), which explains its low OH frequency and also its lower reactivity. Additional evidence for this hydro- gen bonding derives from spectroscopic analysis of the corresponding bending mode frequencies and from vibrational dynamics studies.
We will first discuss the OH bending mode fre- quencies.
The frequencies of the in-plane OH bonding (plane defined as the triangle Si-0-Al) modes and out- plane OH bonding mode (perpendicular to the tri- angle Si-0-Al) are respectively -1100 and 400
cm-l. The zeolitic lattice modes cause these frequen- cies to overlap with the intense modes. They are therefore not directly observable by infrared spec- troscopy. However, these modes have been identified using neutron s p e c t r o s ~ o p y . ~ ~ ~ ~ ~ These values are in agreement with the computed values of the bending modes for a tetrahedral dimer cluster.41 This is in accordance with the local nature of the bonding interactions. When one exchanges the protons by deuterium, the mode frequencies are lowered. The in-plane bending mode shifts to -870 cm-', which happens to be located in the absorption window between the asymmetric and symmetric lattice TO modes.42 The in-plane bending modes of deuterated hydroxyls are therefore directly observable by infra- red spectroscopy.
The spectrum observed for zeolite Y is shown in Figure 5a. Two maxima are observed. The lowest intensity has a higher frequency than the low- frequency peak. The low-frequency peak at -870 cm-l is due to the proton at the O(1) site, the high- frequency peak at -890 cm-l is due to the proton coordinated to O(3). The upward shift is due to its hydrogen bridging with another six ring oxygen atom. This restricts the motion of the proton, with a resulting upward-shifted frequency. The bending- mode frequencies can also be indirectly observed as overtones of the stretching frequencies, as has been d e m o n ~ t r a t e d . ~ ~ , ~ ~
Picosecond infrared laser spectroscopy can be used to probe the decay times of vibrationally-excited protons. Such measurements show large differences in lifetime of the excited vibrational OH stretching mode^.^^,^ For instance the HF OH mode has a decay time of approximately 320 ps, whereas the LF OH has a decay time of 60 f 30 P S . ~
The shorter decay time of the LF mode is due to the changed lattice coupling of the 0(3) site due to interaction with the bridging oxygen atom.
The proton bonded to the O(2) site has also a weaker OH bond, resulting in stronger Si0 and AlO bonds. The vibrational coupling is thus stronger and a shorter decay time can also be expected. As shown

644 Chemical Reviews, 1995, Vol. 95, No. 3
Y ‘5 2.00 -
g a, 0
6
2 1.50 - a
van Santen and Kramer
a 2.00 1 a72
v) c ‘5 1.40 a, 0
m g 2 0.80 a
I r
0.20 I I I I I
840 880 920 960 lo00 Wavenumber (cm-1)
b
2.50 1
1.00 I I I I r 840 a80 920 960 lo00
Wavenumber (cm-1) Figure 5. (a) Comparison of in-plane OD bending modes of DY (SUN = 5 ) in the absence and presence of interacting CCl&N and CD3CN and (b) in-plane OD bending modes of deuterated zeolite Y (SUN = 18) in the absence42 and presence of CD3CN. The spectrum of nondeuterated HY is also shown. (b: Reprinted from ref 42. Copyright 1993 Elsevier.)
X NaHY/0.07
0 (Na)HY/O22
T I
50
0 ‘ I I I -20 0 20
detuning 6v, (cm-1)
Figure 6. Frequency dependence of TI when the laser is scanned over the LF absorption bands for the three zeolites. The x axis denotes the detuning of the top of the laser band from the maximum of the LF absorption band. (Reprinted from ref 43. Copyright 1994 Elsevier.)
in Figure 6, the decay time of the LF bond is found to change with frequency. This is in contrast with the frequency independence of the HF bond (not shown). The frequency dependence of the LF bond agrees with the proposition that it is partially due
.E 3 ““1 1.20
NH,, Y I -7
I 1 1 1 I ( ( [ I I I I I I xx) 400 600 800 1000 1200 1400 1600
wavenumbers (cm-1)
Figure 7. Infrared-active lattice modes of NHkY, HY, and DY. (Reprinted from ref 9. Copyright 1993 Buttenvorths.)
to the overlap of O(2) and 0(3) vibrational stretching frequencies.
The dependence of the relaxation time on proton concentration has been shown to be a Forster proton- proton virtual radiative relaxation mechanism43 that is superposed on the zeolitic bond relaxation chan- nels. Infrared spectroscopy can also provide infor- mation on the degree of lattice deformation that occurs when a proton binds to a lattice oxygeng
Figure 7 compares the lattice frequencies of zeolite Y in the NH4+ form and the protonated zeolite. A dramatic change in the intensity occurs around 600 cm-l. The intensity loss in this regime corresponds to a change in lattice modes, which are mainly totally symmetric breathing modes of the double six ring.45 It is a consequence of bond distance and angle relaxation. The upward shift of the modes at ap- proximately 1100 cm-l is due to mode coupling of the OH in-plane frequencies with the TO stretching modes.lg Deprotonation of the zeolite lattice by N&+ formation due to adsorption of NH3 has also been shown to lead to very large local geometry changes using diffraction methods.46 This confirms the pres- ence of large lattice relaxation effects that accompany protonation or deprotonation of the zeolite lattice.
The differences in proton-bond energies for zeolite Y as well as the lattice relaxation effects have also been predicted theoretically and will be discussed in the next section.
Ill. Lattice Stabilify and Proton Affinity
A. Introduction Quantum-mechanical calculations of the electronic
structure of extended systems is currently a field of extensive research. Such methods have also been applied to zeolitic system^.^^,^' Whereas most of the methods are very appropriate for a calculation of the electronic structure, to compute the stability or reactivity of a solid is still a formidable task. First, an accurate description of the electronic structure on the reacting atoms has to be given; second, we need to predict the details of the local interaction geom- etry. Whereas the “crystal” approach is especially promising,48 there is a need for alternative develop- ments that focus on the embedding of an accurately described local deformation into the extended lat- tice.49r50

Reactivity Theory of Zeolitic Brernsted Acidic Sites
Table 3 a. Absolute Differences IE, - EnIQ
m n 3 4 5 6
Chemical Reviews, 1995, Vol. 95, No. 3 645
3 2.14 2.33 2.34 4 8.94 0.20 0.21 5 9.77 0.82 0.01 6 9.81 0.86 0.04
b. ST03G Equilibrium Geometry of Ring Frameb 3 4 5 6
Si-0 1.622 1.602 1.595 1.592 Si-Si 2.942 3.042 3.063 3.067 Si-0-Si 130.2 143.5 147.5 148.8 0-Si-0 105.7 108.3 108.4 108.1
a Values in the upper triangle are in kcal/mol; lower triangle, kJ/m01.~~ Distances in A; angles in deg.54
We will return to the question of cluster size and embedding in the next section.
In the following subsections we will discuss results obtained using approaches that predict properties of extended lattices by molecular mechanics methods based on potentials from ab initio cluster calculations. We first discuss calculations on the relative stability of siliceous zeolitic structures. Then we will focus on the deprotonation energy as a function of lattice structure. The local relaxation effects due to substi- tution by Al and proton attachment will also be discussed. For a more general discussion of zeolite stability as a function of composition we refer to ref 51.
B. Lattice Stability With only a few exceptions, zeolitic structures are
constructed of four, five, or six rings of tetrahedral units. Three ring tetrahedral units contain unusual cations such as Zn in their f r a m e w ~ r k . ~ ~ , ~ ~ Rings larger than six are often present. They can however be considered as a combination of bonds in smaller ring systems.
As an introduction to our discussion it is useful to compare the relative stability of the SiO(0H)Z unit in three, four, five, or six ring clusters.54 The energies of formation of the ring system per SiO(0H)Z unit is given in Table 3a. With the exception of the three ring, the energies of formation of the four, five, or six rings are essentially the same. To understand the reason for this one has to inspect the angles of the Si-0-Si and 0-Si-0 units as well as the Si-0 bond lengths. The 0-T-0 angle is very similar to the tetrahedral values for the larger ring systems, which show a widening of the Si-0-Si angle and a small decrease of the Si-0 bond length with increas- ing ring size. The lesser stability of the three ring stems from ring strain that deforms the tetrahedral units. The four, five, and six rings appear to be nonstrained and of essentially the same energy. A small change of the Si-0-Si angle is used to release strain that is built up with change in ring size. The shortened Si-0 bond length is due to the changed oxygen hybridization state.
As long as deformation energies are not too large, they can be accommodated by a change in the Si- 0-Si angle. The energy cost is very small (-10 kJ/ 10 degrees per mole of SiOz), due to the very shallow
BO -
60 - - R 3 4
stishovite
0
.. @ a n n
O i 1 - 2 0 4 , , , , ,
0 10 20 30 40 50
Density (W T/1000A*3)
Figure 8. Lattice-energy differences of zeolitic Si02 poly- morphs; also included are stishovite (six-coordinated Si) and three ring structures. 34 contains tetrahedra con- structed from three rings. (Reprinted from ref 51. Copy- right 1989 American Chemical Society.)
Si-0-Si potential energy ~ u r f a c e . ~ ~ ~ ~ ~ The low de- formation energy of the Si-0-Si angle appears to be the key to lattice relaxation.
As already outlined in section I1 several potential parameter sets are available to compute lattice energies and to predict the structure of systems using molecular mechanics appro ache^.^>^^ The potentials often have the form of a Lennard-Jones two-body potential that describes the covalent attraction and repulsion effects, as well as an electrostatic potential. A Born potential is sometimes used to describe the repulsive part of the potential. In the shell model an additional term for the atomic polarization has been added.
A proper balance of electrostatic and covalent interactions is necessary to describe the zeolitic bond adequately. Semiempirical partial charge rigid ion potential calculations on quartz, employing param- eters derived from potential energy surfaces of ab initio calculations on small clusters, have provided evidence that the maximum charge of Si has to be less than 2 eu.5,54 For larger values of the silicon charge, ,&quartz is more stable than a-quartz, which is contrary to experimental results. In Figure 8 the calculated lattice energies of geometry-optimized Si02 polymorphs, based on the partial charge model, are shown plotted as a function of density.51 Note the small density dependence. The partial charge model predicts differences. Less than 10 kJ/mol for zeolitic structures that contain only four, five, or six ring tetrahedral systems, a result very similar to that found for the isolated rings (Table 3). Experimental measurements of heats of formation for siliceous zeolitic systems have been reported that are in complete agreement with this result.58
For the zeolites with four, five, or six rings, energy differences can be readily accommodated by lattice relaxation involving small changes of the Si-0-Si angle. Zeolites with the smaller three rings are less stable due to lattice strain effects59 that deform the tetrahedra. Clearly, long-range electrostatic interac- tions do not dominate differences in stability of the Si02 zeolitic polymorphs.
Partial charge Alp04 parameters5 have been used for a comparable study of the stabilities of the Alp04

646 Chemical Reviews, 1995, Vol. 95, No. 3 van Santen and Kramer
Table 4. Energy Changes in Four Ring Cluster (kJ/m01)~~
ab initio force field system 6E system SQ 6E
4R-AI 114.3 FAU-AI 0.96 107.0 0.48 61.1
4R-AlNa 129.2 FAU-AlNa 1.00 119.3 4R-Al0~ 211.4 FAU-A~OH 1.00 3 0 4 i ~ 9
0.50 2 6 2 i 8
polymorphs as a function of density, with very similar results. Now the difference in effective charges controls the relative stability of the structures. The Alp04 lattices are also flexible and can easily deform by bending of the T-0-T' angles. It also appears that physical properties such as the infrared spectra are much more sensitive to parameter values than the structure.60
C. Deprotonation Energies As we have argued in the previous sections, the
zeolitic lattice is a covalently bonded flexible network. This implies that substitution of lattice Si ions by ions of different size, for instance Al, will cause a local rearrangement of bond lengths and angles. These changes in geometry can be accommodated due to the very small energy changes involved with variation of the T-0-T angles.
For this reason, quantum-chemical calculations on substitutional effects that do not allow for changed geometries may have some value in estimating trends in electronic structure, but are of little value in elucidating energetic effects.
We will compare the energetics of substitution of an Al ion and protonation of a four ring cluster as computed for a geometry-optimized system according to a quantum-chemical calculation with the results found for an extended system based on molecular mechanics calculations involving full lattice relax- ation.61 The partial charge potentials used have been derived by fitting them to the energy surfaces as computed for small clusters using ab initio quantum- chemical calculations. The extended lattice calcula- tions have been done for the faujasite lattice. In Table 4, a comparison is given of the quantum- chemical and molecular mechanics results. There is agreement between the energy changes according to the cluster calculation and those of the embedded system. The relaxation effect of H+ addition and AI substitution is approximately twice that of the Naf addition case, because in the first case two bond lengths are significantly affected, whereas in the latter case only one. (The Naf ion has only a weak covalent interaction.)
It can be seen in Figure 9, which shows results for fully geometry-optimized extended lattices, that the geometry changes are quite local.61 Substitution causes local lattice deformations that are accom- modated mainly by changes in the T-0-T bond angles. The positions of tetrahedral sites that are next nearest neighbors of the [Si-O(H)-All unit are hardly affected.
Table 5 shows a comparison of the predicted deprotonation energies based on Kramers potential parameter^^,^^ and these of Schroder et a1.62 The O(3)
Figure 9. Lattice relaxation due to protonation of the double-six ring in the faujasite lattice. (Reprinted from ref 56. Copyright 1991 American Physical Society.)
Table 5. Comparison of Deprotonation Energy Differences Computed by Kramer et al.sl and by Schrtjder et a1.82 with Experimental Resultsao on OH Site Occupancies OH expt Kramer et al. Schroder et al. site (fractional occupancy) (eV) (eV) 01 0.23 & 0.06 0.36 0.05 02 0 0.74 0.21 0 3 0.34 i= 0.08 0 0 0 4 0 0.92 0.25
site is most stable, followed by the O ( I ~ and O(2, sites. The computational models do not incorporate the hydrogen bonding with opposing lattice oxygen at- oms.
The stronger bond of the 0(3)-H hydroxyl would imply that the corresponding stretching frequency is high. In contrast, as discussed in section II.C, the LF band in H-Y has a lowered stretching frequency due to hydrogen bonding of the O(31-H group with an additional 0 atom. The O(11-H bond corresponds to the HF vibrational absorption band. The O(2l-H hydroxyl bond has a low occupancy due to its low bond energy and the absence of hydrogen bonding should result in a lower vibrational frequency than for the 0(1)-H bond.
We conclude that the molecular mechanics simula- tions predict a ranking of proton-stability sites that is consistent with experimental observations.
Much less is known about the distribution of deprotonation energies in other zeolitic systems. By the same molecular mechanics approaches as dis- cussed above, the deprotonation energies of protons adsorbed to aluminum-containing silicalite (MFI) have also been s t ~ d i e d . ~ ~ , ~ ~ Two modifications exist with 12 different T-sites. Now the differences in deprotonations energy of protons on different oxygen atoms are less than in the faujasite case. According to Kramer et al.25 they can be categorized into two groups. Strongly bonded protons coordinated to oxygen atoms in ten rings and more weakly bonded protons coordinated to oxygen atoms of the twelve rings forming the zeolite microchannels. Interest- ingly Datka et al.63 identified two different kinds of

Reactivity Theory of Zeolitic Brmsted Acidic Sites Chemical Reviews, 1995, Vol. 95, No. 3 647
3 1 A 9
1.59 1.60 1.61 1.62 average TO distance
Figure 10. Relation between average T-0 distance, 0-0 distance, and T-0-T angle in the relaxed all-silica struc- ture of MFI, and the (averaged) proton energy of the corresponding oxygen site. (Reprinted from ref 25. Copy- right 1993 American Chemical Society.) protons in ZSM-5 (Al-containing silicalite) using probe molecule experiments which will be discussed in the next section. Catalytic experiments have suggested a large heterogeneity of the proton reactiv- ity in zeolites with a large number of crystallographi- cally different T site^.^^^^^ Because of the many different deprotonation energies computed for ZSM-5 a statistical analysis could be made of lattice param- eters of the completely siliceous system that would correlate with sites of different deprotonation energy. In systems that are dominated by differences in electrostatic potential one would expect a correlation with the local electrostatic potential (local Madelung potential). When the zeolitic atoms are supposed to be nonrelaxable one would expect a correlation with the T-0-T' angle. It appears that the best param- eters that correlate with proton affinity are the lengths of the T-0 bonds involved with the acidic hydroxyl. The larger the T-T' distances between the T atoms concerned, the stronger the corresponding hydroxyl bond. This result implies that the mis- match between the effective size of the [Si-O(H)- All system and the original [Si-0-Si] volume is the main parameter controlling the deprotonation energy (Figure 10). We thus conclude that the deprotonation energy is determined by local differences in lattice strain. For four, five, or six rings, ring-strain effects are minimal. However, the three-dimensional struc- ture imposes local strain. This can be considered a long-range effect,66 which is not of an electrostatic nature but of a geometric nature. For the three rings of the tetrahedra strain is released when the ring is protonated, because the effective ring diameter then increases and deformation of the tetrahedra is de- creased. Protons attached to three rings are expected to be strongly bonded and hence to be weakly acidic.
The need to fully geometry optimize the clusters in computations of acidity is now widely recognized. Highly accurate calculations are available that study the details of the proton bond as a function of quantum-chemical accuracy and cluster composi- t i ~ n . ~ '
IV. Hydrogen Bonding versus Zwitterion Formation A. Background
When a proton is transferred between zeolite lattice and substrate, separation of charge occurs
C
Y Figure 11. Clusters showing the interaction of NH3 with the Bronsted acidic site: (a) hydrogen-bonded NH3, (b) bidentate-coordinated m+, and (c) tridentate-coordination of N H d + . (Reprinted from ref 68. Copyright 1992 American Chemical Society.)
between the now positive charge of the protonated substrate and the negative charge of the deproto- nated lattice site.
In section 1I.A we mentioned the low dielectric constant of zeolites. A consequence of this is that screening interactions are absent and that a large attractive interaction develops between charged spe- cies in a zeolite.
Understanding of the factors that determine the energy cost of charge separation is essential to a proper theory of zeolite acidity. In particular, ac- curate quantum-chemical calculations with small zeolite structure like clusters that interact with basic molecules such as ammonia have significantly en- hanced the analysis of the acidity problem.
In the next subsection we will discuss the main conclusions from these studies. It appears that, instead of protonation, hydrogen bonding between zeolitic proton and adsorbate is often found in such calculations. Also, in situations where proton trans- fer occurs, there are large local rearrangements of the zeolite atom positions near the protonating site.
In the final subsection we will provide an analysis of the infrared spectra of hydrogen-bonded adsorbates with respect to the question of intrinsic acidity of the proton.
B. Zwitterion Formation Studies of ammonia protonation using neutral
small [Si-O(H)-All dimer clusters that are termi- nated by hydrogen or hydroxyls, using an adsorption geometry as sketched in Figure l l a , show that hydrogen-bonded NH3 is the stable groundstate.68 A quantum-chemical method that properly describes a

648 Chemical Reviews, 1995, Vol. 95, No. 3
weak bond, like the hydrogen bond, is required. It implies the use of large basis sets and adequate configuration interaction methods. Clearly, when a negatively charged cluster was used, as is sometimes done based on the intuitive, but incorrect notion that the zeolite is an ionic solid,69 the energy cost of proton transfer will be enhanced compared to deprotonation from a neutral cluster and hydrogen bonding would be even more preferable.
There is no doubt that ammonia ions are present in zeolites. This follows for instance from infrared spectros~opy~~ or NMR spectroscopic data.39 The question is whether the predicted hydrogen bonding is due to the cluster approximation or has other
A rather obvious question relates to the absence of the long-range lattice Madelung potential contri- bution, an interaction that may arise when the cluster is embedded into the lattice.
M ~ r t i e r ~ ~ has developed an attractive scheme to compute the Madelung energy based on charges close to their quantum-chemical values. Indeed, some results have been presented that indicate that such Madelung effects may be c o n ~ i d e r a b l e . ~ ~ , ~ ~ Several reservations must be made however. First, when one starts with a nonneutral, negatively-charged cluster, the electrostatic embedding effect will be very large, because the positive lattice potential has to compen- sate for the negative charge of the cluster. Second, when a neutral cluster is used, embedding of the cluster into a lattice considered to exist of ions requires a careful treatment of the potential overlap effects that arise between cluster edge atoms and lattice ions. We have recently proposed a solution to this problem50 and have used this to address the question at what cluster size electrostatic embedding effects can be ignored. We conclude that for neutral clusters the electron distribution and protonation is significantly affected by changes in tetrahedra that are next neighboring the protonated bond. This conclusion agrees with the experimental observation that substitution effects are only effective when they occur within the length scale (see section 1I.B). Of course these effects can be also partially electrostatic. Substitution of Si4+ by A13+ necessarily implies the introduction of compensating cations. In section 1I.B we also discussed the effect of replacing the cation by a proton and the preference of relative proton positioning. Finally, for a proper evaluation of the embedding effect, cluster calculations have to be of high quality and geometry optimization has to be included.
We will argue that ion formation will only occur, when a careful study is made of the adsorption state of the ion against the negatively-charged lattice. This is illustrated in Figure 11, parts B and C. The geometries are the ground states of NH4+ adsorbed to a negatively-charged HA(OH)z- or HAl(OH)3 cluster.
NH4+ is adsorbed bidentate or tridentate to the two or three negatively-charged oxygen atoms around aluminum. Whereas in the geometry of Figure 11A which corresponds to hydrogen bonding, the interac- tion energy between ammonia and the proton is -60 kJ/mol, the reaction energy of ammonium to am-
van Santen and Kramer
monia for situations parts B and C is approximately 120 kJ/mol. Compare this with 150 kJ/mol, which is a typical value of NH3 adsorption to acidic zeo- l i t e~ .3~,~3
The result confirms earlier suggestions based on semiempirical consideration^^^ that proper compen- sation of the separation of charges by optimal coor- dination of the positively-charged adsorbate to the negatively-charged oxygen atoms left around the reaction site makes protonation possible. The result- ing state can be best described as a “zwitterionic” state.
As we will see in section V, which deals with the activation of hydrocarbons, the zwitterion state is key to the kinetics of such reactions.
The difference in computed and experimental am- monia interaction energy probably arises from the absence of neighboring tetrahedra. Of great impor- tance is the observation that significant local geom- etry changes occur around the Al ion when the proton is transferred. The AlO- as well as Si0 distances contract next to the oxygen atom involved with the proton bond. The aluminum tetrahedra becomes more symmetrical and the 0-4-0 angle decreases. Clearly, embedding of the solid will not only change electronic structural details but may also influence the degree of relaxation of the reacting acidic site. Calculations with large clusters indeed indicate that relaxability of the protonation site may have very large effect^.^^^^^ In the previous section we con- cluded that at least those lattice atoms should be allowed to relax that are the direct neighbors of the lattice atoms that change coordination during pro- tonation.
It is of interest to note that the picture of proto- nation as essentially occurring in a vacuum, and the need to stabilize the protonated adsorbate, agrees with the experimental observation that heat of adsorption and base proton a f f i n i t ~ ~ ~ , ~ ~ show a uni- form relationship in zeolites. For instance, pyridine adsorbs more strongly than NH3 to an acidic zeolite. It interacts by formation of pyridinium.
On the other hand NH3 dissolved in water is a stronger base than pyridine. The gas-phase proto- nation energy of pyridine is 90 kJ/mol larger than that of NH3.75 The difference in behavior is due to the larger solvation energy of NH4+ by water mol- ecules than pyridinium, but the dominance of differ- ence in protonation energies in the zeolite.
Inelastic neutron scattering data77 are consistent with the proposal of bidentate or tridentate NH4+ bonding. Also, neutron diffraction studies on zeolite 0 are to be interpreted by such NH4+ adsorption states.46
The finding that several of the protons of the NH4+ ion have a short distance to negatively-charged lattice atoms has important consequences for the analysis of protonation reaction paths. We will discuss this extensively in the next section, but will illustrate this here by indicating its consequences for models of water or methanol adsorption.
The question of interest is the state of adsorbed water or methanol. Is water or methanol only hydrogen bonded or adsorbed to the zeolitic proton as a hydronium or methoxonium ion? We will

Reactivity Theory of Zeolitic Bronsted Acidic Sites Chemical Reviews, 1995, Vol. 95, No. 3 649
Table 6. NOD) Band and Transmission Window Minimum Wavenumbers (cm-1)8s
Br~nsted CD3CN adsorption CC13CN adsorption site &OD) &OD) shift window d(OD) shift
Hydrogen bonding versus protonation
H
I H
I
Hydrogen bonding Hydronium
rH3 0
,*.*-,’’ \ gH3
H / O T H
Hydrogen bonding Methoxonium Figure 12. Hydrogen bonding versus protonation: (-1 short OH bond and (- - -) long OH bond.
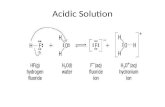
provide a conclusive answer at the end of the next subsection, after an extensive discussion of the infrared spectra of these molecules adsorbed to zeolitic protons. Figure 12 illustrates the chemical bonding.
As we learned from the ammonia interaction case, protonation requires not only interaction of a lattice proton and a basic adsorbate atom, but also the back- bonding of “acidic” adsorbate protons with the basic oxygen atoms around the three-valent cation-con- taining site.79
For methanol the hydrogen-bonded state would result in a short lattice proton-lattice oxygen bond as well as short molecular proton-molecular oxygen bonds, but two hydrogen bond lengths. In the meth- oxonium ion, the two proton-molecular oxygen bonds would be shorter than their corresponding proton- lattice oxygen bonds. Protonation of methanol can be considered a Lewis base-BrGnsted acid interac- tion. This concept appears to be general and key to understanding the mechanism of protonation.
A consequence of the requirement that the proto- nated ions have an optimum with the negatively- charged zeolitic sites is that their position does not necessarily follow directly from that of the protons. For zeolite Y, NH4+ has three nonequivalent adsorp- tion sites, two within the sodalite cage and one in the s ~ p e r c a g e . ~ ~
Before discussing this in the next subsection, an analysis of the infrared spectra of hydrogen-bonded adsorbates will be given.
C. Vibrational Dynamics of the Disturbed Zeolitic Hydroxyl
The interaction of a basic molecule with a zeolitic proton changes the stretching or in-plane bending
SiODAL 894 988 94 1977 970 76 SiODFe 865 950 85 1902 930 65
modes of a zeolitic proton in different ways. The OH stretching mode frequency will weaken, but the motion of the OH bending mode will become frus- trated and its frequency will increase.
Because the change of the stretching frequencies may be quite complex, we will start the discussion with experimental observations of changes in the bending mode frequencies and then return to the shifted stretching mode.
Figure 5 parts a and b, shows the upward shifts of the in-plane bending modes of deuterated zeolite Y. Compared to the upward shift of the zeolite Y, LF deuteron, the interaction with the basic adsorbates results in a significantly larger shift. The shift relates to the basicity of the interacting molecule as well as the acidity of the proton (see Table 6). The stronger the interaction is, the larger the restriction of the bending motion and the more the frequency shifts upward.
In contrast with the upward shift of the bending mode, the frequency of the OH stretching modes moves downward when the hydroxyl interacts with a basic molecule. The OH stretching frequency is very sensitive to such interactions. The interaction with molecules such as CO results already in sub- stantial shifts.80 As long as the downward shifts of the hydroxyl frequencies are not too large, each type of hydroxyl gives a broad absorption bond with a single maximum. The corresponding infrared ab- sorption intensity increases substantially.
This spectral feature is a characteristic of hydrogen bonding.81 In Figure 13 this is illustrated for the interaction of CH&N with the weakly acidic POH hydroxyl of a VAP011.82 The lowering in frequency stems from the weakening of the OH bond, which is due to sharing of the proton valency between the zeolite oxygen and the adsorbing molecule. The increase in intensity is due to polarization of the OH bond. The repulsive interaction due to electron density overlap of occupied adsorbate orbitals and the OH 0 bond electrons is reduced by polarization of the OH bond such that electron density is reduced on the proton. Hydrogen bonding between OH and the Lewis basic adsorbate induces a positive charge on the proton. The larger the intrinsic acidity is of the proton concerned, the larger the polarization of the OH bond and the resulting OH dipole moment. The increased OH dipole moment gives the enhanced infrared absorption intensity. The increased positive charge on the H in contact with basic molecules has been confirmed by proton NMR measurement^.^^
The broadening of the adsorption band is due to the considerable anharmonic coupling with the OH-B modes of the basic molecule with respect to the proton. Whereas the OH frequency is typically -3000 cm-l, the OH-B fundamental frequency is typically -50 cm-l. This stems partially from the high molecular adsorbate mass and partially from the

650 Chemical Reviews, 1995, Vol. 95, No. 3
-0.40-
2309 1
3550 3740 CD3 CN
1
0.4
0.3
v)
c 3 P)
m
L .-
2 0.2 g 9 In
0.1
0
van Santen and Kramer
CoAPOll(1.0)
I I I I I I I I I I
1500 2000 2500 3000 3500 4000 Wavenumber (cm-1)
Figure 13. Difference FT-IR spectra of CoAPO-11 for adsorbed CD3CN. (Reprinted from ref 82. Copyright 1993 Eindhoven, University of Technology.)
1 I I I I
1500 2000 2500 3000 3500
Wavenumber (cm-1)
Figure 14. Difference FT-IR spectra of adsorbed CD3CN.
relatively weak OH-B interaction. Because of the differences in energy of the modes concerned, high OH-B vibrational states can become further excited. This results in the broadening of the absorption band. There exists a linear relationship between Aus, the shift of the maximum of the OH stretching peak, and W,, the width of the shifted absorption band.84
The large anharmonicity of the OH bond itself gives rise to interesting additional features when molecules of larger Lewis basicity or proton affinity are ad- sorbed. Figure 14 shows infrared absorption differ- ence spectra of CDsC=N on H-ZSM-5 as well as HY zeolites.85
Instead of a single shifted adsorption band, now two broad doubly-peaked features are observed in the 2300-3000 cm-l spectral range. Around 1800 cm-I
new spectral features appear that are also due to the infrared absorption of the shifted and broadened OH bond. These peaks will be denoted as the A, B, and C bands. Overlapped over these features are sharp absorption bands due to intramolecular infrared absorption bands. For instance, for CD3C=N the modes around 2200 cm-l are due to shifted C=N modes. When one uses CD3CN instead of CHsCN, these shifts can be easily interpreted. Protonation of the C=N bond depopulates antibonding CN elec- tron orbitals and the CN stretching frequency in- creases.
This is more difficult in the case of CH3CN because of Fermi resonance coupling between CH3 and CN modes. The disturbance of the CN frequencies is different when the CN molecular fragment interacts

Reactivity Theory of Zeolitic Brensted Acidic Sites
with a proton or a Lewis acid. Lewis acidic sites may arise from the small alumina oligomers that are occluded in the microcavities of the zeolite. Also signals can be observed of CD3CN physically ab- sorbed in the zeolite. In the experiments of Figure 14, the Bransted acidic interaction results in a CEN frequency at 2300 cm-l, the Lewis acidic interaction at 2322 cm-l and physically-adsorbed CD3CN has a CN mode at 2265 cm-l.
As we will also see later, when we discuss the interaction of a proton with HzO and CHsOH, anal- ysis of the disturbed intramolecular modes can as- sist interpretation of the adsorption bands in terms of the adsorption geometry of the adsorbed mole- cule.
The A, B, and C peaks that are part of the broadened and shifted OH stretching frequencies have their analogues in liquid-phase hydrogen bond- ing.86 The interaction with CH3CN is so large that the OH band is shifted nearly 1000 cm-l downward and as a consequence has a width of -700 cm-l. As in molecules, Fermi resonance coupling can also occur between localized modes in a solid. In this particular case the broadened and shifted OH band overlaps with the 26 overtone of the in-plane upward-shifted OH bending mode. This is approximately 2 x 1300 cm-l. In Table 6 (86) a comparison is given of the measured frequencies of the fundamental 6 modes and corresponding Fermi resonances for the deuter- ated systems. Note the agreement between the values of the 28 overtones and Fermi resonance values. The spectral features in the C peak area arise from Fermi resonance with the combination band of the in-plane 6 and out-of-plane y OH bands. The 26 overtone as well as the 6, y combination bands have very low direct infrared absorption intensities. Anharmonic coupling of these bending mode features with the infrared active OH stretching frequency leads to intensity stealing in the broad OH stretching adsorption band. Intensity stealing leads to reso- nance minima at the 26 (-2600 cm-l) and 6 + y (-1800 cm-l) positions. In the spectral theory of hydrogen bonding, these minima are called Evans
Theoretical treatments of these reso- nances can be found in refs 87 and 88. The A, B, and C peaked absorption bands can be considered a signature of hydrogen bonding of adsorption mol- ecules. For instance, infrared absorption features of adsorbed N H 3 do not show such bands, notwithstand- ing extensive adsorption in the same spectral re- gime.'O This proves that in that case ionic NH4+ species have formed A, B, and C peaked adsorption bands have been observed for many adsorbing mol- ecules but have been (incorrectly) given very different interpretations by some authors. For instance, for the adsorption of acetone,89 they have been inter- preted as an equilibrium between hydrogen-bonded and protonated acetone. In the case of water, the A and B spectra have also been interpreted as the symmetric and asymmetric vibrational modes of protonated water.g0
Figure 15 shows the spectrum of adsorbed HzO on the Bransted acidic hydroxyl of SAP0-34.90 The A, B, and C peaked adsorption features are very clear. In addition, the shifted frequencies of the two H20
Chemical Reviews, 1995, Vol. 95, No. 3 651
v1
3
L .-
3
I I I I I 3500 3000 2500 2000 1500
Wavenumber (cm-1)
Figure 15. Difference spectrum for adsorption of HzO to dehydrated H-SAPO-34. (Reprinted from ref 90. Copy- right 1992 American Chemical Society.)
protons are observable. One of the protons is directed to a basic surface oxygen as shown in Figure 12. Calculationsg1 indicate that it weakly interacts and is shifted downward by -250 cm-'. The other proton interacts only very weakly with a third oxygen atom in the tetrahedron around AI. It is shifted downward by 50 cm-'. The interpretation of these spectral features is consistent with presence of hydrogen- bonded water, instead of hydroxonium.
When HzO is adsorbed with higher molar ratios than those which correspond to the zeolitic proton, the spectra change dramatically and the A-C spec- tral features tend to disappear.92 This is a strong indication of hydronium formation. The hydronium ion becomes solvated with H20 molecules. Complete solvation is expected when four HzO molecules are adsorbed per one H+ (see Figure 16Lg3
Broad line 'H NMRg4 leads to similar conclusions. For water concentrations about equal to the concen- tration of bridging hydroxyl groups two configura- tions are deduced: watermolecules hydrogen bonded to the bridging OH group and the hydroxonium ion. The hydrogen-bonded species dominates.
A similar interpretation can be made of the spectra of adsorbed methanol. These spectra sometimes become complicated by the presence of additional features due to methoxylation of strained SiOSi bands.95 This may especially occur in H-ZSM-5.
Figure 17 shows the spectrum of CH30H adsorbed at low concentration to a zeolitic proton of HNaY. The A, B, and C features of the zeolitic hydrogen bond are present. The methanol OH band is shifted downward by -200 cm-l on ZSM-Eig6 A low intensity of the A, B, and C features is found. This implies a very large shift of the zeolitic OH band and the bonding situation may be close to that of protonated
We will discuss the chemisorption of CH3OH more CH30H.
fully in the next section.

652 Chemical Reviews, 1995, Vol. 95, No. 3 van Santen and Kramer
H
HI'LH Figure 16. The solvated hydronium (schematic): proton complex of four water molecules.
3400 3000 2600 2200 1800 1400 Wavenumbers (cm-1)
Figure 17. Difference IR spectrum of methanol mbar) adsorbed at HNaY. (Reprinted from ref 92. Copy- right 1989 American Chemical Society.)
The small disturbance of the CH3 modes agrees with this interpretation. This interpretation is very different from the theoretical predictions found in ref 69. As we argued before, the cluster approximation used in these calculations, namely the choice of a negatively-charged silicate cluster embedded in a lattice Madelung field, may result in a chemical- bonding situation that tends to reflect model cluster choice rather than the actual chemical structure.
D. Solid Acid Brarnsted Acidity In case of equilibrium between a protonated BH+
and nonprotonated basic molecule B, the van't Hoff relation defines the corresponding proton transfer energy Qp:
[BH+],, is the maximum amount of protonated Base that can be adsorbed to the solid.
The proton transfer energy Qp equals
(3)
In eq 3, the symbols PA stand for the proton affini- ties. PA::: is the protonation energy of a gas-phase molecule in the vacuum; PA:', is equal to the deprotonation energy of a surface hydroxyl. E,i is the
zwitterion stabilization energy between protonated base and negatively-charged zeolite wall. In eq 3 it should be used with a negative sign.
We have extensively discussed in the previous section that the downward shift of the OH stretch frequency by contact with a weak base relates to proton affinity of acid as well as base. As long as the interaction remains weak, it has been shown by Paukshtis and Y u r ~ h e n k o ~ ~ that the relation between shift and protonaffinity difference is linear. They propose eq 4 as a relation to use to obtain the PA:::: (OH):
with AVOH the stretch frequency shift of the zeolitic proton concerned and AvESH that of a surface sil- anol group with the same base. The empirical relationship 4 has been deduced based on proton affinity data of organic acids in the gas phase and their measured hydroxyl stretch frequency shifts upon contact with a weakly adsorbing basic molecule. The unknown quantity in eq 3 then is the zwitterion energy E,i. This can be deduced directly from experi- ment by measuring [Bl and [BH+l as a function of temperature or by direct calculation. The earlier quantum-chemical studies of the protonation of NH3 are an example. A characteristic value of E,i - -450 kJ/mol. Clearly when a linear re la t ion~hip~~ is derived for the relation between Qp and APA, E,i can be considered a constant.
V. Ionic Intermediate versus Transition State Formation A. Background
In this section the mechanism of proton activation of some prototype molecules will be discussed. The molecules have been chosen such that they enable study of a few key elementary reaction steps in acid catalysis. One of the most important types of ele- mentary reaction steps is the activation of CH and C-C bonds. Another reaction of importance is the dehydration reaction.
In particular, mechanistic studies of superacid- catalyzed organic transformation reactions (carried out approximately 25 years ago) have provided the basic concepts of acid c a t a l y s i ~ . ~ ~ ~ ~ ~
Activation of alkanes in homogeneous superacids proceeds via carbonium ions, with pentacoordinated carbon atoms and three-center two-electron bonds. It has been proposed that these carbonium ions are the intermediates for dehydrogenation or cracking reactions that produce another ion, the carbenium ion (see Figure 18). The carbenium ion has a positive charge and a three-coordinated carbon atom. Car- benium ions can also be formed directly by protona- tion of an alkene.
Whereas the intermediates formed in acid-cata- lyzed reactions in zeolites are very similar to those formed in superacids, the energetics of their forma- tion is very different. In zeolites the proton is initially strongly bonded to the zeolite and its bond energy has to be overcome in order for the reaction to take place.

Reactivity Theory of Zeolitic Bronsted Acidic Sites Chemical Reviews, 1995, Vol. 95, No. 3 653
a) Elementary reaction step via carbonium ion
H Y7 I H3C-C-CH2- CH2- CH, -> R- Cj-R' + H2
IO H
H
R"- -k -H + R"
carbenium ion
H3C-CH2 - C H ~ - C H ~ - C C H S
H+
Carbonium ion
b) Hydride transfer
H H I I I
H3C-C-CH2-CH2- CH3 H3C- C-CH2- CH2- CH3 + H + I
I I
+ +
H3C- C-CH2- CH2- CH3 H,
H3C-C-CH2 -CH3
CH3 CH3 Figure 18. (a) Elementary reaction steps via carbonium ion and (b) hydride transfer.
Secondly, in contrast to the homogeneous me- d i ~ m , ~ ~ solvation effects are absent in the zeolite. Instead carbonium ion and carbenium ion formation requires stabilization by negatively-charged sites of the zeolite wall. As we will see, the resulting interactions are very similar to those discussed in section N.B.
Another reaction of importance is the hydride transfer reaction. It requires a lower activation energy than carbonium ion formation. It is il- lustrated in Figure 18b. From the reactant alkane a carbenium ion is generated and another carbenium ion is converted into alkane product. Most of the chemistry involving rearrangement of intra- or in- termolecular bonds are reaction steps of the carbe- nium ions which are well known from classical organic chemistry. Examples are alkylation, methyl transfer, or isomerization reactions. In superacids these reactions occur at low temperatures, whereas zeolites usually require higher reaction temperatures. In some cases this is caused by the temperature requirement for regeneration of protons by desorption of the products. An example is dehydration chem- istry that requires desorption of water to liberate the protons. The overall reaction rate will also depend on the activation energies of the acid-catalyzed elementary reaction steps and the concentration of reactants in the zeolite micropore, which is deter- mined by the heats of their adsorption.
The atomistic detail of the protonation event provides the microscopic basis to these reaction steps. As we will illustrate for carbonium ion formation, the nature of the transition state has immediate conse- quences for the relation between catalytic activity and deprotonation energy.
It will also appear that our classical ideas on the formation of protonated intermediates are not always valid. Application of ab initio quantum-chemical calculations has demonstrated that carbonium ions
in zeolites are not always intermediates but are sometimes transition states.99 Information on the transition states can be used to compute reaction rate constants of the elementary reaction steps occurring in acid catalysis reactions.
The passage through a transition state or forma- tion of intermediates appears to depend on the relative stability of the carbenium ions. This varies with the primary, secondary, or tertiary nature of the carbon atom that carries the positive charge. Again the ionicity versus covalency of the interaction with the zeolite atoms becomes an important issue. Fig- ure 19 illustrates the difference in potential energy dependence for a reaction that proceeds via a transi- tion state or an activated intermediate.
B. The CHs+ Carbonium Ion, CH30H2+, and Bifunctional Acidity
Methane has a very low reactivity in a zeolite. Only recently has acid-catalyzed activation of CHI been demonstrated by zeolitic protons.99 Using CD4 the rate of the hydrogen-deuterium exchange reac- tion was followed by infrared spectroscopy measure- ment of the rate of disappearance of the zeolitic proton signal. On H-ZSM-5 the reaction was found to proceed with an activation energy of 130 kJ/mol. By using ab initio calculations its reaction path has been studied by a computation of the ground and activated states on small three tetrahedral clusters.
No stable carbonium intermediate was found as intermediate. The interaction energy of CH4 with a zeolitic proton, in the adsorbed state in which CH4 can be considered hydrogen bonded, is only of the order of 10 kJ/mol.
In order to become protonated the CH4 molecule has to deform. The protonated CH5+ is not a stable intermediate, but has to be considered a transition state. If CH5+ were an intermediate, it would have a local minimum on the potential energy surface

van Santen and Kramer
Table 7. The Activation Energies for Hydrogen-Deuterium Exchange (not Corrected for Zero Point Vibrational Energies) as Well as the Coordinates of the Transition State Carbonium Ion C&+ According to Density Functional Theory"
CHs+ Symmetric Activation Energies
Eb = 125 kJ/mol (NLSD) E,& = 164 kJ/mol (LSD)
distances in (.&I
654 Chemical Reviews, 1995, Vol. 95, No. 3
- r a
Reaction proceeds via intermediate
- b r Reaction proceeds via transition state
Figure 19. Schematic illustration of difference between intermediate I and transition states # (a) reaction pro- ceeds via intermediate and (b) reaction proceeds via transi- tion state.
Figure 20. Calculated transition-state geometry for the isotope exchange process between methane and an acid zeolite cluster. All small circles represent hydrogen or deuterium atoms. The arrows indicate the main compo- nents of the displacement vectors along the reaction coordinate. (Reprinted from ref 99. Copyright 1993 Mac Millan.)
defined by its degrees of freedom. This is position I as denoted in Figure 19. In a transition state denoted by the symbol "#", the state is also a stationary point on the potential energy surface. However, it is only a minimum with respect to N-7 coordinates (6 are the translational and rotational degrees of freedom of methane). It is a maximum in one degree of freedom, which according to transition state theory is the reaction coordinate. The com- puted atomic displacements of this reaction coordi- nate are indicated in Figure 20. The motion of one proton with respect to the carbon atom of C& is shown and that of one of the other C& protons to another basic oxygen around Al.
r(OH*) 1.32 r(Si0) 1.67 r(CH*) 1.33 r(Al0) 1.82 r(CH) 1.11
The transition state geometry of CH5+ is analogous to that of adsorbed N&+ in that it is coordinated with several, in this case two, of its protons to lattice oxygen atoms. Table 7loo presents the relatively short OH distances and lengthened C-H bonds of the corresponding CHs+ bonds. From an analysis of the distribution of charge in the complex, we conclude that the interaction of this CHb+ transition state with the oxygen atoms is more covalent than ionic.
The overall effect of the exchange reaction of the protonated zeolite with CD4 is that the deuterium atom left-bonded to the zeolite lattice after exchange is coordinated to a different oxygen atom than that onto which the proton was bonded before the reac- tion.
This has an important consequence for the concept of acidity as applied to this reaction. This can be analyzed by studying the activation energy of the proton exchange as a function of the proton affinity of the two lattice oxygen atoms involved in the re- action complex. According to the Brgnsted-Polanyi relation (see lo), the activation energy can be con- sidered a continuous function of the initial and final state energies as long as the reaction mechanism does not change.
On the clusters studied to produce Figure 20, cluster termination is accommodated by saturation of terminating bonds with hydrogen atoms. The proton affinity of the oxygen atoms around Al can be vaned in such a cluster by changes in the hydride bond lengths. This is the action of the bond order conservation principle as discussed extensively in section 1I.B. The chemical bonds between atoms will alternatively strengthen and weaken neigh- boring and next-neighboring bonds. By applying this technique it could be established that hE,,t x 0.7hEd,protonation.101 AE~eprotonation is the difference in proton affinities of the two oxygen atoms around Al before proton or deuteron exchange occurs.
The strong dependence of the activation energy on AE deprotonation implies that for the exchange reaction it is not the absolute deprotonation energy value, but differences in the deprotonation energies of the oxygen atoms accessible as bonding sites in the reaction, control the effective acidity of the ~ystem.9~
This bifunctional concept, Lewis base-assisted Brgn- sted acidity, could be used to predict the difference in the rate of the exchange reaction for H-ZSM-5 and dealuminated HY.
In section III.C, we discussed computational results on the deprotonation energies of these systems. We

Reactivity Theory of Zeolitic Brmsted Acidic Sites Chemical Reviews, 1995, Vol. 95, No. 3 655
Figure 21. Transition state for dehydrogenation; the CH bond stretches; Hz and u-bonded carbenium ion are gener- ated. (Reprinted from ref 100. Copyright 1994 American Chemical Society.)
"9
Figure 22. The computed transition state for the dehy- drogenation of CH, according to a density functional calculation. Esd = 318 kJ/mol (LSD). Distances (in A): r(OzHd, 1.512; r(OaCls), 1.992; r(ClsHd, 1.104; r(SilOz), 1.670; r(Al304). 1.861; r(HBHIG), 0.901; r(CI5Hlfi), 1.665; r(ClsHl~,ld, 1.087; r(OzA13), 1.880; r(04Sis), 1.660. (Re- printed from ref 100. Copyright 1994 American Chemical Society.)
concluded that the maximum difference in deproto- nation energies on the zeolitic oxygen atoms in H-ZSM-5 is similar to that in the HY system. However the energy differentials are much smaller in H-ZSM-5 than H Y because of the much larger number of crystallographically different 0-atom posi- tions in the former case. The overall rate of the reaction has to be considered as an averaged rate of reaction of the different proton-occupied oxygen sites. The experimental and theoretical results are in complete agreement and confirm that the bifunction- al acidity model can be used to predict the reaction rate. Because of the smaller average AEE~.~,,~,.~~, values H-ZSM-5 is more active than dealuminated HY.
Whereas hydrogen-deuterium exchange is an in- teresting probe reaction, it does not represent a model for activation of a hydrocarbon molecule. In Figure 21, dehydrogenation of an alkane is also proposed to proceed via a carbonium ion.
The same concept as generated for the hydrogen- deuterium reaction should essentially be valid for the transition state that leads to dehydrogenation.loO Using density functional theorylOO as well as Har- tree-Fock ab initio theory,lol this transition state has recently been found (Figure 22). Both methods give very similar results. Whereas in the hydrogen- deuterium exchange reaction the Lewis basic oxygen stabilized one of the CD4 atoms, now the Lewis basic
" I O
Figure 23. Geometry of the zeolitic methoxy group ac- cording to density functional theory. Distances (in r(ClsOa), 1.44; r(SilOz), 1.64; r(04Sis), 1.71; ~(CIOHI~, I~ , I~) , 1.10; r(O&), 1.73; r(&04), 1.90. (Reprinted from ref 100. Copyright 1994 American Chemical Society.)
oxygen compensates for the positive charge that develops on the CH3+ part of the reaction complex. The proton now attacks the C-H bond. The car- bonium ion is again a transition state, but different from the transition state shown in Figure 20. The activation energy for dehydrogenation of CHI is approximately 80 kJ/mol larger than that for the hydrogen-deuterium exchange reaction.
Experimentally the activation energy for butane has been measured and a value of the order of 110 kJ/mol has been found.lo2 In the case of butane a secondary carbenium ion is formed, with a significant stabilization of charge. The difference in energies between a primary and a secondary carbenium ion is of the order 100 kJ/mol?? We will return to activation of larger molecules in the next subsection.
In the case of methane dehydrogenation it is of interest to consider also the final state. This is a CH3+ ion, a carbenium ion, strongly covalently- bonded to the cluster oxygen atom. It can be con- sidered a methoxy group. The strong covalent nature of this bond is apparent from the long T-0 bond distances next to the 0-C bond (Figure 23). Changes in proton affinity will affect the strength of this bond and, hence, according to the Brgnsted-Polanyi prin- ciple, the activation energy for the dehydrogenation reaction. The bifunctional acidity concept will thus also apply to dehydrogenation of CH4.
In the next subsection we will discuss the limita- tion of the bifunctional acidity concept. We will show that the degree of ionicity of the transition state- lattice interaction is an important parameter. The more ionic the transition state, the smaller the correlation between activation energy and differences in proton affinity between the oxygen atom that is deprotonated and the oxygen atom that becomes populated after reaction. A correlation is now ex- pected to be found with the deprotonation energy itself. Inspection of the transition state for CH4 dehydrogenation (Figure 22) reveals a large differ- ence between carbon coordination. Whereas the carbon in the CH5+ carbonium transition state for hydrogen-deuterium exchange is covalently bonded to all of the five hydrogen atoms, the CH5+ carbonium ion transition state shows very different carbon

656 Chemical Reviews, 1995, Vol. 95, No. 3
2.22;
H
00
'*.. 2.94
0.98/
0
van Santen and Kramer
H
4 b) c) Figure 24. The structures of the quantum chemically calculated intermediates in ethoxylation of the zeolite surface: (a) n-complex of ethylene, (b) the transition state of the ethoxylation reaction, and (c) the final ethoxide structure. (Reprinted from ref 107. Copyright 1994 J. C. Baltzer AG.)
coordination. It is to be considered as a planar CH3+ ion, weakly interacting with negatively-charged zeolitic oxygen anion and leaving hydride ion. One expects that correlation of activation energy with the proton affinity of the second oxygen atom now will be weak.
Whereas there is no evidence for direct methoxy formation from CH4, NMR data strongly indicate the formation of methoxy intermediates from metha- nol.lo3 Instead of a dehydrogenation reaction, dehy- dration of CH30H will produce H20 and a surface methoxy group. Due to the presence of the oxygen atom in methanol, the activation energy for its formation will be considerably lower than from CH4. The geometry of the transition state will be analogous to that for methane dehydrogenation. The surface methoxy group has been postulatedlo4 as an inter- mediate for C-C bond formation in the conversion reaction of methanol to higher hydrocarbons. Ad- sorption of methanol is also of fundamental interest because it undergoes very rapid hydrogen-deuterium exchange with zeolitic protons.
Careful ab initio studies have shown that the ground state of methanol is the hydrogen-bonded state (Figure 12). However, the energy difference between the CH30HD+ transition state for proton exchange with a geometry close to that shown for the methoxonium state in Figure 12b is less than 10 kJ/ mol.lo5 Therefore the adsorbed state may be dynamic in the sense that, on a time scale of the order of the inverse of the proton exchange rate, there is an alternation of OH bond lengths to the two lattice oxygen atoms in contact with the hydrogen atoms. It also implies that the average state of the molecule is partially hydrogen bonded and partially of the methoxonium type. Only at very low temperature can the complex be frozen in one hydrogen-bonded state. These conclusions agree with the infrared experimental evidence that indicates a hydrogen- bonded state close to methoxonium, and NMR proton chemical shiftslo6 that also indicate a state in be- tween hydrogen bonding and methoxonium.
C. The Activation of Hydrocarbons The work by Kazansky and Senchenyalo7 and A.
G. Pelmenschikov et a1.1°8 on the protonation of ethylene initially demonstrated the importance of the
interaction of the resulting carbenium ion with the zeolite lattice. When ethylene interacts with a proton it can adsorb in a n adsorption state or a a-bonded state (see Figure 24).
Protonation proceeds via a transition state, where the zeolitic proton attaches to one of the ethylene carbon atoms and the positive charge on the other ethylene carbon atom becomes stabilized by the negative charge that develops on one of the other oxygen atoms around the catalytically active center that initially was not bonded to a zeolitic proton. This ionic interaction is converted to a covalent final state, the a state, where the carbenium ion becomes strongly and covalently bonded to lattice oxygen atoms to form an ethoxy species, analogous to the methoxy species discussed above. Such alkoxy species have indeed been observed by 13C NMR spectroscopy.103 Only at low temperatures will the heat of adsorption of ethylene correspond to n adsorption.
The energy differences of primary versus secondary or tertiary carbenium ion formation are very large. The difference between a primary and secondary carbenium ion is typically -100 kJ/mol, that between secondary and tertiary -40 kJ/m01.~~ Calculations indicate that the covalent-bond energies of the u-bond- ed states only differ by a few kilojoules/mol. How- ever, the activation energies for their formation show large differences because of the highly ionic nature of the zwitterion transition states. Because of the differences in stability of secondary and primary carbenium ions, the R+ 0- interaction in the transi- tion state becomes increasingly more ionic. For instance for butane the protonation reaction of lowest activation energy is likely to be secondary carbenium ion formation with an activation energy of -100 kJ1 mol. This protonation mode would initiate butane dehydrogenation. This competes with butane crack- ing which would generate a primary carbenium ion.lo2 In a consecutive step the a-bonded butoxylate is converted to n-bonded butene. Dehydrogenation and cracking via carbonium ion transition states are dominant reaction routes in narrow pore zeolites that suppress the bimolecular recombination reactions that lead to hydride transfer. At high temperatures, acid-catalyzed alkane conversion by H-ZSM-5 is also monomolecular and hence the reaction proceeds via carbonium ion transition states.log

Reactivity Theory of Zeolitic Brcansted Acidic Sites
A prototype of the cracking reaction is methane formation from ethane. The transition state for eth- ane cracking has been found using a similar zeolitic cluster as for the methane activation reaction.'1°
The nature of the transition state follows the same principle as discussed above. Proton transfer to the adsorbate is accompanied by stabilization of the developing positive charge by neighboring basic oxygen atoms.
In the case of the cracking reaction, the proton moves to the approximate middle of the C-C bond. In the transition state one part of the molecule develops to the CH3+, intermediate with a planar geometry as a free carbenium ion that in the final state becomes o-bonded to one of the basic oxygen atoms bonded to Al. The other part of the molecule is converted to methane.
The computed activation energy for' dehydrogena- tion is lower for ethane than for the dehydrogenation of CH4. This is mainly due to the difference in stability of a primary ethyl ion versus CH3 ion.
Reactions that proceed via carbonium ion interme- diates have high activation energies and hence will be suppressed at lower temperatures in favour of the hydride transfer reactionl'l (see Figure 18b). The transition states for the hydride transfer reaction can be deduced from that for the dehydrogenation of CH4. We will illustrate this for the termination of the isomerization reaction of hexane.
Hexane can be initially activated to a secondary carbenium ion and hydrogen via a carbonium ion transition state. It isomerizes via an n-hexyl cation intermediate to produce an isohexylcarbenium ion. In a bimolecular reaction mechanism the reaction is terminated by hydride transfer between n-hexane to give a n-hexyl cation, which initiates the reaction, and isohexane, which terminates the reaction (see Figure 18b).
The hydride transfer reaction starts with the isohexyl o complex. The reaction with hexane is the analogue of the dehydrogenation reaction in which a zeolitic proton reacts with a CH bond to give Ha and a carbenium ion. In the hydride transfer reac- tion the intermediate transition state consists of an (isohexy1)C-(hexane)H bond. The now positively- charged carbon atom of hexane starts to interact with a basic lattice oxygen. The low activation energy of hydride transfer compared to that of the dehydroge- nation reaction relates to the difference in dissocia- tion energy of the zeolitic OH bond versus that of the tertiary carbenium ion o-bond. This is at least 140 kJ/mol. Clearly a low activation energy requires a small difference between the ground state alkoxy 0-C bond and the (zwitterionic) transition state. This will be found in strongly acidic materials. Hydride transfer competes with conversion of the a-bonded alkoxy state to x-bonded alkene, which would result in proton back donation to the lattice.
Finally, we comment on the possibility of the formation of carbocationic intermediates versus tran- sition states. Because of their existence in the free state,l12 it has been investigated whether cracking of ethane proceeds by the asymmetrical transition state, shown in Figure 25a, to also form a sym- metrical intermediate as illustrated in Figure 25b.
Chemical Reviews, 1995, Vol. 95, No. 3 657
a
/O\ /O\ / Si- \
1 si ' A b
C"
1.1291 H I H
j1.589
AI 1 . 7 1 i \ m W
=P P 0 0
Figure 25. (a) Transition state for cracking (schematic); C-C bond stretches; CH4 and a-bonded carbenium are generated (arrows indicate direction of atomic motion)lo2 and (b) a symmetrical intermediate for cracking. (Re- printed from ref 110. Copyright 1994 J. C. Baltzer AG.)
Calculations show that compound 25b is indeed an intermediate, but of an energy approximately 100 kJ/ mol higher than the transition state in Figure 25a.110 The high value stems from the indirect stabilization of the positive charge by the 0-H interactions instead of 0-C stabilization in the transition state.
This example shows that carbocationic intermedi- ate formation cannot be excluded in zeolites. We conclude that for primary carbon atoms the corre- sponding energies will be too high. This may be different when secondary or tertiary carbenium ion intermediates become involved.
In zeolites the activation energy for reactions that proceed via primary carbocations can be lowered be- cause the primary carbocation state can become sta- bilized by strong 0 0-C bonds with the zeolite lattice. It is interesting to note that recently zeolitic catalysts have been proposed for the isomerization of butene that may proceed via such a transition state.l13
IV. Conclusion The detailed molecular understanding being gained
on the mechanism of Br~nsted acid-catalyzed reac- tions has provided a new understanding of the reactivity of the carbocations which are the proposed

658 Chemical Reviews, 1995, Vol. 95, No. 3
intermediates. The most essential need is to include explicitly in the mechanism the interaction with negatively-charged basic oxygen ions. This leads to the concept of n- and a-bonded intermediates. The interaction with the zeolite lattice directly affects the geometry of transition states, as we have shown for the hydrogen-deuterium reaction of CH4 versus its dehydrogenation.
With regard to secondary or tertiary carbenium ions, the ionic nature of the carbocationic zeolite interaction in the transition state implies that the chemistry of most reactions will be analogous to that in the homogeneous superacidic phase.
Differences stem from the altered proton transfer energies between solid and reactant as well as the weak complexation energies of the carbenium ions with the solid. In addition, they are steric constraints due to microcavity shape and size.
Most important is the presence of strongly Lewis basic oxygen atoms in Brprnsted acidic zeolites. This resolves a long-standing paradox involving the con- version of methanol to higher hydrocarbons. The mechanisms proposed in this reaction proceed via intermediateslo4 that form a C-C bond driven by the back-donation of a proton to the zeolite lattice.
The presence of the Lewis basic oxygen also assists proton transfer in reactions that proceed via covalent transition states. Such transition states usually involve primary carbon atoms. The activation of methanol proceeds via such a transition state as well as “classically” forbidden reactions such as butene isomerization. Also, acid-catalyzed reactions that produce light hydrocarbons via carbonium ion-medi- ated reaction paths require zeolite stabilization of primary carbenium ions.l13
The ionic transition states or intermediates involv- ing secondary or tertiary carbenium ions are less bifunctional in nature because now the stabilization of the transition state or intermediate is electrostatic in nature, and does not directly relate to the covalent part of the deprotonation energy. There the depen- dence on hEdeprotonation will be small and proton activation will directly relate to the heterolytic OH bond strength.
In conclusion, we will summarize the intrinsic kinetic factors that determine the difference in activ- ity of acidic zeolites. We will support our argumen- tation with the example of a monomolecular reaction.
The overall rate of the reaction will be given by a (highly simplified) expression:
r = k6R (5)
k is a reaction rate constant and OR the site concen- tration of reactant.
In reactions with several reactants, expression 5 will of course be more complex. Expression 6 es- sentially involves a concentration, expressed as a site occupation density, OR, and a reaction rate constant for the adsorbed molecule. In its most simplified form OR is a Langmuir adsorption isotherm:
van Santen and Kramer
At low OR, it is a first-order reaction in p~ and the
effective activation energy equals:
When however the occupancy of the zeolite is high, the effective activation energy equals ICact, the activa- tion energy of the elementary rate constant, k, and is independent of the heat of adsorption.
In zeolites the heat of adsorption may vary signifi- cantly with chain length or size of the hydrocarbon. The heat of adsorption also varies with size of the micropore cavities.1,2 Reactions that have a positive order in their reactants therefore may be expected to have different overall rates, because the difference in heat of adsorption varies the reactant concentra- tion in the zeolite. This has been very elegantly shown by Lercher and Vinek114 for the cracking of alkanes as a function of chain length in ZSM-5. Differences in overall reaction rate between zeolites can also be controlled by differences in heat of adsorption of the reactants.
The composition of a zeolite may in addition affect the overall rate by an altered microcavity concentra- tion of reactant as well as the proton affinity of the Br~nsted acidic sites.
The details of the elementary steps that control zeolite-catalyzed reactions depend of course on the type of reaction. In complex reactions competitive adsorption effects may also play a role. The reaction temperature then also depends on the conditions being suitable for reactant desorption or product formation. For example, the thermodynamics of dehydrogenation and aromatization reactions re- quires a high temperature.
When adsorption effects inhibit the reaction the application of wide porous zeolitic systems or even amorphous silica-aluminas may be beneficial. Such materials have significantly lower heats of adsorption than the narrow pore materials. Amorphous silica- aluminas usually contain a few sites where tetrahe- drally coordinated silica and alumina tetrahedra share oxygen bridges.66 The corresponding Br~nsted acidic site can be considered zeolitic. These sites are responsible for strongly acidic catalytic behavior. The difference in acidity between zeolites and amorphous silica-aluminas stems partially from the high alu- mina concentration close to the zeolitic Brprnsted acidic site, affecting the corresponding deprotonation energies as well as from the altered local concentra- tions of reactants compared to the situation in zeolites. The latter is mainly due to differences in heat of adsorption on the silica-aluminas compared to that on zeolites.
References Stach, H.; Lohse, U.; Thamm, H.; Schirmer, W. Zeolites 1986, 6,74. Stach, H.; Fiedler, K.; Jhchen, J. Pure Appl. Chem. 1993, 65,2193. Jhchen, J.; Stach, H.Adsorpt. Sei. Technol. 1986,3, 3. Derouane, E. G.; Andre, J. M.; Lucas, A. A. J. Catal. 1988, 11 0 , 58. Derouane, E. G. In Guidelines for Mastering the properties of molecular sieves; Bartomeuf, D., Derouane, E. G., Holderich, W., Eds.; NATO AS1 221; Plenum: New York, 1990; p 225. Eic, M.; Ruthven, D. M. Zeolites 1988,8,472; 1988,8,40. Karger, J.; Ruthven, D. M. J. Chem. Soc., Faraday Trans. 1 1981, 77, 1485. Karger, J.; Pfeifer, H. Zeolites 1987, 7, 90. Post, M. F. M.

Reactivity Theory of Zeolitic Br~nsted Acidic Sites
In Introduction to zeolite science and practice; van Bekkum, H., Flanigan, E. M., Jansen, J. C., Eds. Stud. Surf Sci. Catal. 1991, 58, 392. Catlow, C. R. A.; Mackrodt, W. C. Computer simulation of solids. Lecture notes on Physics; Springer: Berlin, 1982. van Beest, B. W. H.; Kramer, G. J.; van Santen, R. A. Phys. Rev. Lett. 1990, 64, 1955. de Boer, K.; Jansen, A. P. J.; van Santen R. A. Chem. Phys. Lett. 1994, 223, 46. Hill, J. R.; Sauer, J. Z. Phys. Chem. (Leipzig) 1989,270, 203. van Santen, R. A.; de Man, A. J. M.; Jacobs, W. P. J. H.; Teunissen, E. M.; Kramer, G. J. Catal. Lett. 1991, 9, 273. Jacobs, W. P. J. H.; van Wolput, J. H. M. C.; van Santen, R. A. Zeolites 1993, 13, 170. Laidler, K J. Theories of Chemical Reaction Rates; McGraw- Hill: New York, 1969. Glasstone, S.; Laidler, K. J.; Eyring, H. The theory of rate process; McGraw-Hill: New York, 1941. Kramer, G. J.; van Santen, R. A,; Emeis, C. A.; Nowak, K. A. Nature 1993,363, 529. Sauer, J . Chem. Rev. 1989,89, 199. Pfeifer, H.; Freude, D.; Hunger, M. Zeolites 1985, 5, 274. See: Kazansky, V. B. Acc. Chem. Res. 1991,24,379. See: Sauer, J. In Modelling of structure and reactivity in zeolites; Catlow, C. R. A., Eds.; Academic Press: New York, 1992; Chapter 8. Brand, H. V.; Curtiss, L. A.; Iton, L. E. J. Phys. Chem. 1993,97, 12773. See: van Santen, R. A. Theoretical heterogeneous catalysis. World Scientific lecture and course notes in Chemistry 5; World Scientific: Singapore, 1991; p 291; p 204. Pisani, C. J. Mol. Catal. 1993,82,229. Saunders, V. R.; Freyeria- Fava, C.; Dovesi, R.; Salasco, L.; Roetti, C. Mol. Phys. 1992, 77, 629. Apri, E.; Dovesi, R.; Freyeria-Fava, C.; Pisani, C.; Roetti, C.; Saunders, V. R. Mod. S i n . Mater. Sci. Eng. 1993,1, 297. van Santen, R. A,; van Beest, B. W. H.; de Man A. J. M. In Guidelines for Mastering the properties of molecular sieves; Bartomeuf, D., Derouane, E. G., Holderich, W., Eds.; NATO AS1 221; Plenum: New York, 1990; p 493 a.f. Jacobs, W. P. J. H.; van Wolput, J . H. M. C.; van Santen, R. A,; Jobic, H. Zeolites 1994, 14, 117. Jirak, Z.; Vratislav, V.; Bosacek, V. Phys. Chem. Solids 1980, 41, 1089. Czjzek, M.; Jobic, H.; Fitch, A.; Vogt, T. J. Phys. Chem. 1992, 96, 1535. Bok , M.; Huang, K Llynumic theory of crystal lattices; Clarendon Press: Oxford, 1954. Tsuneyuki, S.; Tsukada, M.; Aoki, H.; Matsui, Y. Phys. Rev. Lett. 1988, 61, 869. de Man, A. J. M.; van Beest, B. W. H.; Leslie, M.; van Santen, R. A. J. Phvs. Chem. 1990.94, 2524. Kramer, G.- J.; van Santen; R. A. J . Am. Chem. SOC. 1993,115, 2887. van Santen R. A,; de Man A. J . M.; Kramer, G. J . In Zeolite microporous solids: synthesis structure and reactivity; Derouane, E. G., Lemos, F., Naccache, C., Ramoa Ribeiro, F., Eds.; NATO AS1 352; Kluwer: Dordrecht, 1992; p 493 a.f. Shustorovich, E. M. S u ~ . Sci. Rep. 1986, 6, 1. van Santen, R. A. Recueil des Trau. Chim. Pays-Bas 1990,109, 59. Stevenson, R. L. J. Catal. 1971,21, 113. Freude, D.; Klinnoski, J.; Hamdan, H. Chem. Phys. Lett. 1988,149, 355. Hunger, M.; Freude, D.; Fenzke, D.; Pfeiffer, H. Chem. Phys. Lett. 1992,191, 391. Hunger, M.; Anderson, M. W.; Ojo, A.; Pfeifer, H. Mi- croporous Mater. 1993, l , 17. OMalley, P. J.; Dwyer, J . J . Phys. Chem. 1988, 92, 3005. O'Malley, P. J.; Dwyer, J. Chem. Phys. Lett. 1988, 143, 97. Chu, C. T. W.; Chang, C. D. J. Phys. Chem. 1985,89, 1569. Post, M. F. M.; Huizinga, T.; Emeis, C. A.; Nanne, J. M.; Stork, W. H. J. In Zeolites as catalysts sorbents and detergent builders. Applications and Innovations; Karge, H., Weitkamp, H. G. J., Eds.; Elsevier: Amsterdam, 1989; p 365. Wachter, W. A. Proc. 6th International Zeolite Conference, 1984; p 141. Bartomeuf, D. Mater. Chem. Phys. 1987,17, 49. Briend, M.; Bartomeuf, D. In Proc. 9th International Zeolite Conference; Higgins, J. B., von Ballmoos, R., Tracy, M. M. J., Eds.; Buttenvorth-Heinemann: New York, 1993. Su, B. L.; Barthomeuf, D. Zeolites 1993, 628. Corma, A.; Sastre, G.; Viruela, R.; Zicovich-Wilson, C. J . Catal. 1992,136, 521. Jacobs, W. P. J . H.; Jobic, H.; van Wolput, J . H. M. C.; van Santen, R. A. Zeolites 1992, 12, 315. Jacobs, P. A,; Uytterhoeven, J . B. J . Chem. SOC., Faraday Trans. 1973,169, 359. Lohse, U.; Janchen, J. Unpublished results. See also: van Santen, R. A,; Kramer, G. J.; Jacobs, W. P. J . H. In Elementary reaction steps in heterogeneous catalysis; Joyner, R. W., van Santen, R. A,, Eds.; Kluwer: Dordrecht, 1993; p 113 e.v. Kustov, L. M.; Borovkov, V. Y.; Kazansky, V. B. J . Catal. 1981, 72, 149. White, J. L.; Beck, L. W.; Haw, J. F. J . Am. Chem. SOC. 1992, 114,6182. Jacobs, W. P. J. H.; de Haan, J . W.; van de Ven, L. J . M.; van Santen, R. A. J. Phys. Chem. 1993,97, 10394.
Chemical Reviews, 1995, Vol. 95, No. 3 659
Jobic, H. J. Catal. 1991,131, 289. Sauer. J. J. Mol. Catal. 1989.54. 312. Jacobs, W. P. J. H.; van Wolput, J. H. M. C.; van Santen R. A. Chem. Phys. Lett. 1993,210 32. Brugmans, M. J. P.; Kleyn, A. W.; Lagendijk, A.; Jacobs, W. P. J. H.: van Santen. R. A. Chem. Phvs. Lett. 1994.217. 117. Kubota, J.; F&, M.; Goto, Y.; Kondo, J.; Wada, A:; Domen, D.; Hirose, C. Chem. Phys. Lett. 1993,204, 273. de Man, A. J. M.; van Santen, R. A. Zeolites 1992,12, 269. Wax, M. J.; Cavanagh, R. R.; Rush, J. J.; Stucky, G. D.; Abrahams, L.; Corbin, D. R. J. Phys. Chem. 1986,90, 532. Teunissen, E. H.; Roetti, C.; Pisani, C.; de Man, A. J. M.; Jansen, A. P. J.; Orlando, R.; van Santen, R. A.; Dovesi, R. Mod. Sim. Mater. Sci. Eng. 1994,2, 921. Pisani, C.; Dovesi, R.; Roetti, C. Hartree-Fock ab initio treat- ment of crystalline systems. Lecture Notes in Chemistry 48; Springer-Verlag: Berlin, 1988. Pisani, C. Phys. Reu. 1978, B17, 1314. van Santen, R. A.; Toneman, L. H. Int. J . Quantum Chem. 1977,12, suppl. 2, 83. Pisani, C.; Orlando, R.; Cori, F. J. Chem. Phys. 1992,97,4195. Pisani, C.; Dovesi, R.; Nada, R.; Kantaroich, L. N. J. Chem. Phys. 1990,92, 7448. Teunissen, E. H.; van Santen, R. A.; Jansen, A. P. J.; Orlando, R.; Dovesi, R.; Pisani, C. J. Chem. Phys. 1994, 101 (7), 5865. van Santen, R. A.; Ooms, G.; den Ouden, C. J. J.; van Beest, B. W.; Post, M. F. M. In Zeolite synthesis; Occelli, M. L., Robson, H. E., Eds.; ACS symposium series 398; American Chemical Society: Washington, DC, 1989. Merlino, S. Acta Cryst. 1981, A37, C189. Annen, M. J.; Davis, M. E.; Higgins, J. B.; Schlenker, J. L., J . Chem. SOC., Chem. Comm. 1991, 1175. Lawton, S. L.; Rohr- baugh, W. J. Science 1990,247, 1319. van Beest, B. W.; Verbeek, J.; van Santen, R. A. Catal. Lett. 1988, 1, 147. van Santen, R. A.; de Bruyn, D. P.; den Ouden, C. J . J.; Smit, B. In Introduction to Zeolite Science and Practice; van Bekkum, C. H., Flanigan, E. M., Jansen, J. C., Eds. Stud. Surf. Sci. Catal. 1991. 58. 317. K r k e r , G. J.; Farragher, N. P.; van Beest, B. W. H.; van Santen, R. A. Phys. Rev. 1991, B43, 5068. Kramer, G. J.; van Beest, B. W. H.; van Santen, R. A. Nature 1991,351, 636. Patarin. J.: Kessler. H.: Soulard. M.: Guth. J. L. In Zeolite synthesis; American Chemical Society:' Washkgton, DC, 1989; ACS symposium series 398; p 221. Petrovic, I.; Navrotsky, A.; Davis, M. E.; Zones, S. I. Chem. Mater. 1993,5, 1805. de Man, A. J. M.; Ueda, S.; Annen, M. J.; Davis, M. E.; van Santen, R. A. Zeolites 1992,12, 789. de Man, A. J . M.; Vogt, E. T. L.; van Santen, R. A. J. Phys. Chem. 1992.96. 10460. Kramer, G. J.; de Man, A. J . M.; van Santen, R. A. J. Am. Chem. SOC. 1991,113 (7), 6435. Schroder, K. P.; Sauer, J.; Leslie, M.; Catlow, C. R. A.; Thomas, J. M. Chem. Phys. Lett. 1990,173, 26. Datka. J.: Bocren, M.: Rvmarowicz. P. J. Catal. 1988,114, 368. Lombardo, E. A.;'Siel, A. A.; Hall,' W. R. J. Catal. 1989,' 119, 426. Fritz, P. 0.; Lunsford, J . H. J. Catal. 1989, 118, 85. Gajda, G. J.; Rabo, J . Catal. Rev. Sci. Tech. 1989-1990, 31, 385. Alvarado-Swaisgood, A. E.; Barr, M. K.; Hag, P. J.; Redondo, A. J . Phys. Chem. 1991,95, 10031. Kassab, E.; Seiti, K.; Aleavena, M. J. Phys. Chem. 1991,95,9425. Brand, H. V.; Curtiss, L. A.; Iton, L. E. J . Phys. Chem. 1992, 96, 7725. Cook, S. J. ; Chakraborty, A. K.; Bell, A. T.; Theodorou, P. N. J. Phys. Chem. 1993,97,6679. Sauer, J.; Kolmel, C.; Mill, R.; Allrichs, R. Chem. Phys. Lett. 1989, 164, 193. (a) Teunissen, E. H.; van Duyneveldt, F. B.; van Santen, R. A. J . Phys. Chem. 1992,96,366. (b) Teunissen, E. H.; van Santen, R. A.; Jansen, A. P. J.; van Duyneveldt, F. B. J. Phys. Chem. 1993,97, 203. Vetrivel, R.; Catlow, C. R. A,; Colbourn, E. A. J. Phys. Chem. 1989,93, 4594. Vetrivel, R.; Catlow, C. R. A.; Colbourn, E. A. Proc. R. SOC. London 1988, A417, 81. Teunissen, E. H.; Jacobs, W. P. J. M.; Jansen, A. P. J.; van Santen, R. A. In Proc. 9th International Zeolite Conference; van Ballmoos, R., Ed.; Montreal, 1992; Butterworth: London, 1993; Vnl I D 629
- 7 r
1 hiavena, M.; Seiti, K.; Kassab, E.; Ferenccy, Gy; Angyan, J . G. Chem. Phys. Lett. 1990, 168, 461. Kassab, E.; Fouquet, J.; Allavena, M.; Evleth, E. M. J. Phys. Chem. 1993, 97, 9034.
(72) Mortier, W. J. Proc. 6th International Zeolite Conference, 1984; pp 734-746. Baekelandt, B. G.; Mortier, W. J.; Schoonheydt, R. A. In Modelling of Structure and Reactivity in Zeolites; Catlow, C. R. A,, Ed.; Academic Press: New York, 1992.
(73) Paukshtis, E. A,; Yurchenko, E. N. Uspekki Khim. 1983,52,426. Cardona Martinez, N.; Dumesic, J . A. Adu. Catal. 1992,38,149.
(74) Paukshtis, E. A.; Pankrat'ev, Yu. D.; Pelmenschikov, A. G.; Burgina, E. B.; Turkov, V. M.; Yurchenko, E. N.; Zhdinirov, G. M. Kinet. Katal. 1986,27, 1440.

660 Chemical Reviews, 1995, Vol. 95, No. 3 van Santen and Kramer
(75) Teunissen, E. H.; Jansen, A. P. J.; van Santen, R. A,; Orlando, R.; Dovesi, R.; J. Chem. Phys. 1994,101, 5865.
(76) Brand, H. V.; Curtiss, L. A.; Iton, L. E. J. Phys. Chem. 1993, 97, 12773.
(77) Parillo, D. J.; Gorte, R. J.; Farneth, W. E. J. Am. Chem. SOC. 1993,115, 12441.
(78) Jacobs, W. J. P. H.; de Haan, V. 0.; van Santen, R. A,; de Graaf, C. A. J . Phys. Chem., in press. Udovic, T. J.; Cavanagh, R. R.; Rush, J . J.; Wax, M. J.; Stucky, G. D.; Jones, G. A.; Corbin, D. R. J . Phys. Chem. 1987,91, 5968.
(79) Knozinger, H.; Biihl, H.; Kochloefl, K. J. Catal. 1972,29, 57. (80) Kustov, L. M.; Kazansky, V. B.; Beran, S.; Kubelkova, L.; Jiruy,
P. J . Phys. Chem. 1987, 91, 5247. Kubelkova, L.; Beran, S.; Lercher, J. A. Zeolites 1989, 9, 539. Zecchina, A.; Bordiga, S.; Spoto, G.; Marchese, L.; Petrini, G.; Leofanti, G.; Padovan, M. J . Phys. Chem. 1992,96,4991.
(81) Pimentel, G. C.; McClellan, A. L. The hydrogen-bond; Reinold Publ. Co.: New York, 1960.
(82) Peeters, M. Ph.D. Thesis, Eindhoven University of Technology, 1993.
(83) White, J. L.; Beck, L. W.; Haw, J. F. J. Am. Chem. SOC. 1992, 114,6182.
(84) Glazunov, V. P.; Mashkovsky, A. A,; Odinokov, S. E. Zh. Prikl. Spektrosk. 1975,22, 696. Odinokov, S. E.; Mashkovsky, A. A,; Dzizenko, A. Dokl. Akad. Nauk SSSR 1975,220, 1130.
(85) Pelmenschikov, A. G.; van Santen, R. A.; Janchen, J.; Meijer, E. L. J . Phys. Chem. 1993,97, 11071.
(86) Clydon, M. F.; Sheppard, N. Chem. Commun. 1969, 1431. (87) Bratos, S. J. J . Chem. Phys. 1976,63,3499. Bratos, S.; Ratajczak,
H. J . Chem. Phys. 1982, 76, 77. Ratajczak, H.; Yaremko, A,; Baran, J. J . Mol. Struct. 1992,275, 235.
(88) van Santen, R. A. Recl. Trau. Chim. Pays-Bas 1994, 113, 423. (89) Kubelkova, L.; Novakova, J.; Nedomova, K. J. Catal. 1990,124,
441. Kubelkova, L.; Cejka, J.; Novakova, J. Zeolites 1991, 11, 48.
(90) Marchese, L.; Wright, P. A.; Thomas, J. M. J. Phys. Chem. 1993, 97, 8109.
(91) Pelmenschikov, A. G.; van Santen, R. A. J . Phys. Chem. 1993, 97, 10678.
(92) Jenkys, A.;. Warescka, G Dereminsky, M.; Lercher, J. A. J. Phys. Chem. 1989,93, 4837. Mirth, G.; Lercher, J . A. In Natural gas conuersion; Holmen, A,, Ed.; Elsevier: Amsterdam, 1991; p 437.
(93) Librovich, N. B.; Sakum, V. P.; Soholov, N. D. Chem. Phys. 1979, 39,351. Kazansky, V. B.; Senchenya, I. N.; Pankov, A. A. J . Mol. Catal. 1991, 70, 189.
(94) Batamack, P.; Doremieux-Morin, C.; Vincent, R.; Fraissard, J. Chem. Phys. Lett. 1991, 95, 3790. Batamack, P.; Doremieux- Morin, C.; Vincent, R.; Fraissard, J. J . Phys. Chem. 1993, 97, 9779.
(95) Pelmenschikov, A. G.; Morosi, G.; Gamba, A,; Zecchina, A,; Bordiga, S.; Paukshtis, E. A. J . Phys. Chem., in press.
(96) Mirth, G.; Kogelbauer, A.; Lercher, J. A. Proc. 9th International Zeolite Conference, Montreal, 1992; van Ballmoos, R. et al., Eds.; Butterworth-Heineman: Oxford, 1993; p 251. Mirth, G.; Lerch- er, J. A.; Anderson, M. W.; Klinowsky, J. J. Chem. SOC., Faraday Trans. 1990,86,3039. Jentys, A.; Warecka, G.; Derewinsky, M.; Lercher, J. A. J . Phys. Chem. 1989,93, 4837.
(97) Brouwer, D. M.; Hogeveen, H. Prog. Phys. Org. Chem. 1972,9, 179. Olah, G. A,; Prahash, G. K. S.; Sommer, J. Superacids; Wiley: New York, 1985. Kramer, G. J.; van Santen, R. A.; Emeis, C. A.; Novak, A. K. Nature 1993,363, 529. Blaszkowski; S. R.; Nascimento, M. A. C.; Jansen, A. P. J.; van Santen. R. A. J . Phvs. Chem. 1994.98. 11332. Kazansky, V. B.; FGash, M. V.; van Santen,R. A. Catal. Lett. 1994, 28, 211. Lercher, J. A,; van Santen, R. A,; Vinek, H. Catal. Lett. 1994, o w nr ‘ 6 1 , 31.
(103) Gregory Oliver, F.; Munson, E. J . ; Haw, J. F. J . Am. Chem. SOC. 1992, 96, 8106. Munson, E. J . ; Haw, J . F. J. Am. Chem. SOC. 1991,113, 6303. Munson, E. J.; Kheir, A. A.; Lazo, N. D.; Haw, J. F. J . Phys. Chem. 1992,96, 7740.
(104) van den Berg, J. P.; Wolthuizen, J. P.; van Hooff, J . H. C. Proc. 5th Int. Conf. Zeolites; Rees, L. V., Ed.; Heyden: London, 1980. Olah, G. A.; Doggweiler, H.; Feldberg, J . D.; Frohlich, S.; Grilina, M. J.; Karpeles, R.; Heumi, T.; Inabe, S.; Ip, W. M.; Lammetsm, K. A.; Salem, G.; Tabor, D. J. Am. Chem. SOC. 1984,206,2143.
(105) Haase, F.; Sauer, J. Manuscript in preparation. Blaszkowski, S. R.; van Santen, R. A. Manuscript in preparation.
(106) Haase, F.; Sauer, J. J. Phys. Chem. 1994, 98, 3083. (107) Kazansky, V. B.; Senchenya, I. N. J. Catal. 1989, 119, 108.
Kazansky, V. B.; Senchenya, I. N. Catal. Lett. 1991, 8, 317. (108) Pelmenschikov, A. G.; Zhanpeisov, N. U.; Paukshtis, E. A,;
Malyshava, L. v.; Zhdimirov, G. M.; Zamaraev, K. I. Dokl. Akad. Nauk SSSR 1987,393, 915. 1
(109) Haag, W. 0.; Dessau, R. M. Proc. 6th Int. Congress on Catalysis, Berlin, 1984, VZ; p 305. Stefanadis, C.; Gates, B. C.; Haag, W. 0. J . Mol. Catal. 1991,67,363. Krannila, H.; Haag, W. 0.; Gates, B. C. J. Catal. 1992, 135, 115.
(110) Kazansky, V. B.; Senchenya, I. N.; Frash, M.; van Santen, R. A. Catal. Lett. 1994, 27, 345.
(111) Greenfelder, D. S.; Voge, H. H. Znd. Eng. Chem. 1945,37, 514. (112) Hehre, W. J.; Radow, L.; Schleyer, P. v. R.; Pople, J. A. In Ab
initio molecular orbital theory; Wiley: New York, 1986. (113) Mooiweer, H. H.; de Jong, K. P.; Kraushaar-Czarnetzki, B.;
Stork, W. H. J.; Krutzen, B. C. H. In Zeolites and related Microporous Materials; Weitkamp, J., Karge, M. G., Pfeifer, M., Molderich, W., Eds. Stud. Surf. Sci. Catal. 1995,84, 2321.
CR9400301
(114) Lercher, J. A.; Vinek, H. Manuscript in preparation.