
Rdr priones
45
UNIVERSIDAD NACIONAL FEDERICO VILLARREAL FACULTAD "HIPÓLITO UNANUE" INFECTOLOGÍA 2015 DOCENTE: DR. RAMÍREZ ERAZO, JULIO GRUPO 1: ALVA HUARAJ, ROSA ALEJANDRA ARTEZANO SÁNCHEZ, LUZ ESTEPHANIE CHÁVEZ ORTIZ, SILVIA LISSET DEPAZ MONTAÑEZ, GISELLA
-
Upload
rosa-alva -
Category
Health & Medicine
-
view
138 -
download
2
Transcript of Rdr priones

UN
IVER
SID
AD
NA
CIO
NA
L FE
DER
ICO
VIL
LAR
REA
LFA
CU
LTA
D "
HIP
ÓLI
TO U
NA
NU
E"IN
FEC
TOLO
GÍA
20
15
DOCENTE: DR. RAMÍREZ ERAZO, JULIO
GRUPO 1:
ALVA HUARAJ, ROSA ALEJANDRA
ARTEZANO SÁNCHEZ, LUZ ESTEPHANIE
CHÁVEZ ORTIZ, SILVIA LISSET
DEPAZ MONTAÑEZ, GISELLA

Dar a conocer la importancia de los priones , ya que
es un agente infeccioso que causa graves
enfermedades neurodegenerativas, con la finalidad de
informar y alertar a la población para poder tomar
algunas medidas preventivas y que este problema de
salud no sea más grande.

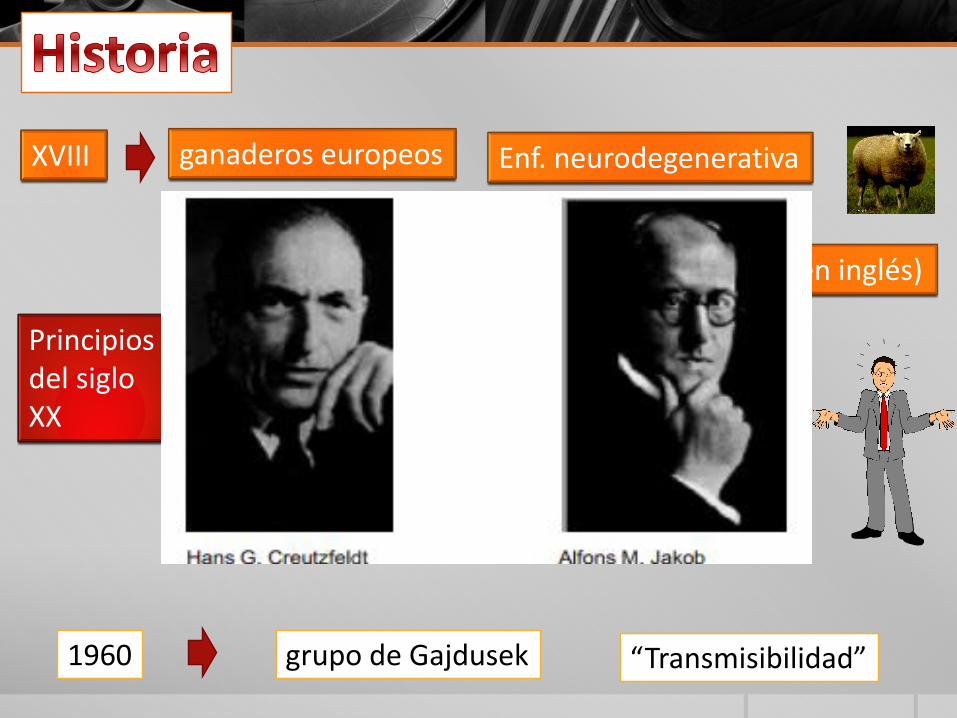
XVIII ganaderos europeos Enf. neurodegenerativa
“tembladera” (scrapie, en inglés)“Espongiforme”
Principios del siglo XX
Creutzfeldt y Jakob Encefalopatía espongiforme en el hombre
“Enfermedad de Creutzfeldt-Jacob”.
1960 grupo de Gajdusek “Transmisibilidad”
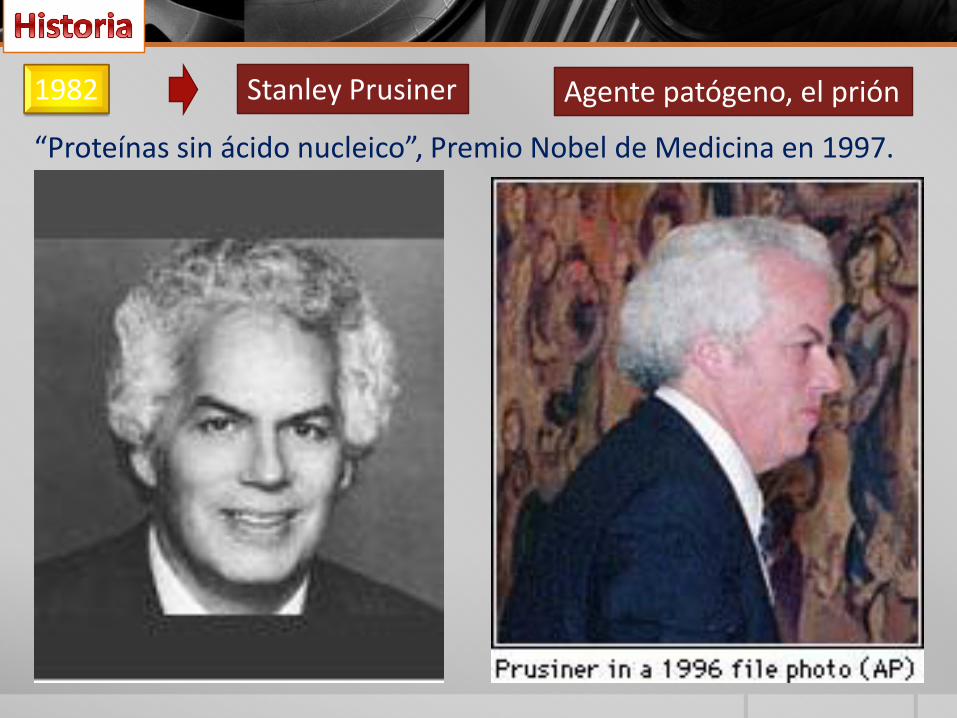
1982 Stanley Prusiner Agente patógeno, el prión
“Proteínas sin ácido nucleico”, Premio Nobel de Medicina en 1997.

¿Qué es un prión?
Los priones no son organismos vivos, son solo
proteínas sin ácido nucleico. Aunque no son virus,
tienen también características patógenas e
infecciosas.
Esta proteína ha perdido su función normal y ha
adquirido la capacidad de transformar la normal
en patógena

¿Cómo se descubrieron?• Las primeras referencias a las enfermedades
espongiformes transmisibles se remontan alsiglo XVIII, cuando varios ganaderoseuropeos describieron una enfermedadneurodegenerativa letal que afectaba a lasovejas y a las cabras, mal al que se denominó"tembladera" (en inglés, scrapie). El cerebrode estos animales presentaba un aspecto deesponja, de donde proviene el término"espongiforme". Posteriormente se demostróque estas enfermedades eran transmisibles.El agente patógeno, el prion, fue descubiertoen 1982 por Stanley Prusiner, quien demostróque se trataba de partículas puramenteproteicas sin ácido nucleico.
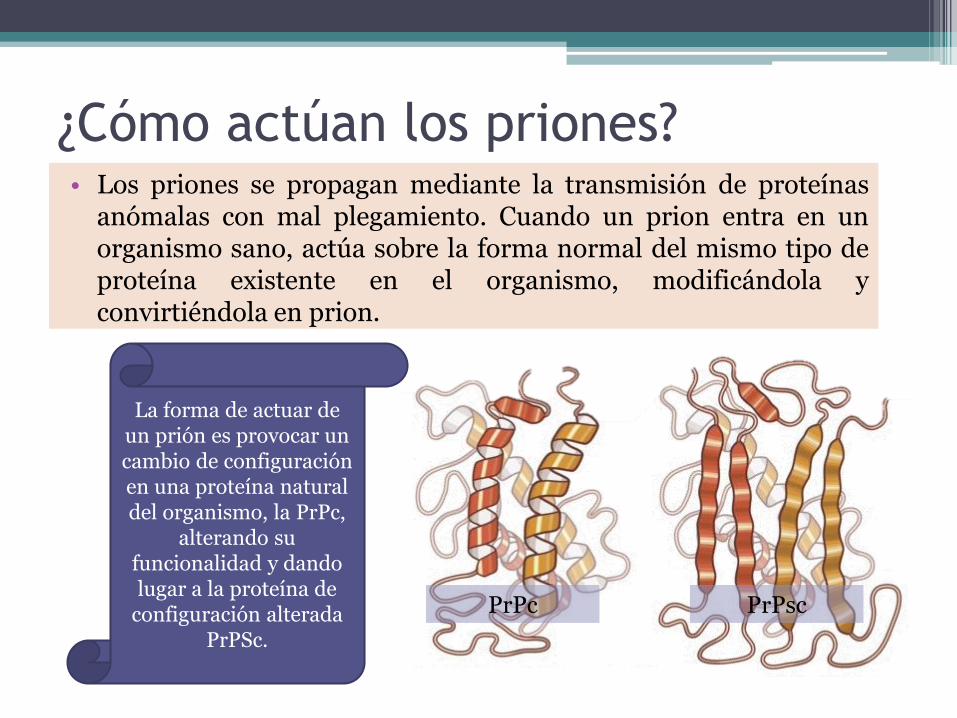
¿Cómo actúan los priones?• Los priones se propagan mediante la transmisión de proteínas
anómalas con mal plegamiento. Cuando un prion entra en unorganismo sano, actúa sobre la forma normal del mismo tipo deproteína existente en el organismo, modificándola yconvirtiéndola en prion.
La forma de actuar de un prión es provocar un cambio de configuración en una proteína natural del organismo, la PrPc,
alterando su funcionalidad y dando lugar a la proteína de
configuración alterada PrPSc.
PrPc PrPsc
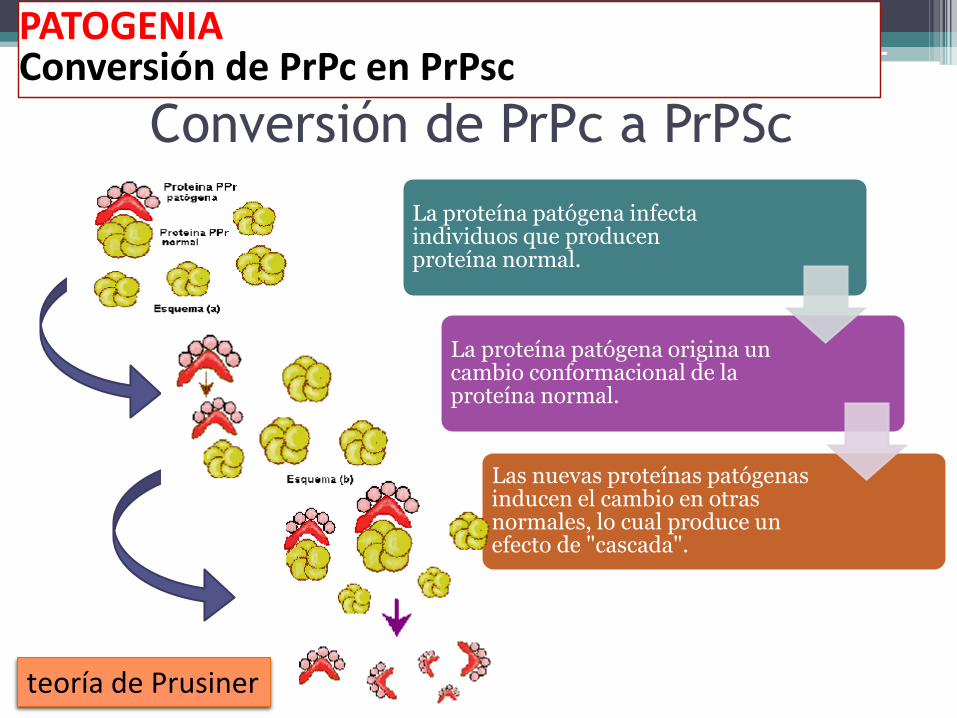
Conversión de PrPc a PrPSc
La proteína patógena infecta individuos que producen proteína normal.
La proteína patógena origina un cambio conformacional de la proteína normal.
Las nuevas proteínas patógenas inducen el cambio en otras normales, lo cual produce un efecto de "cascada".
PATOGENIAConversión de PrPc en PrPsc
teoría de Prusiner
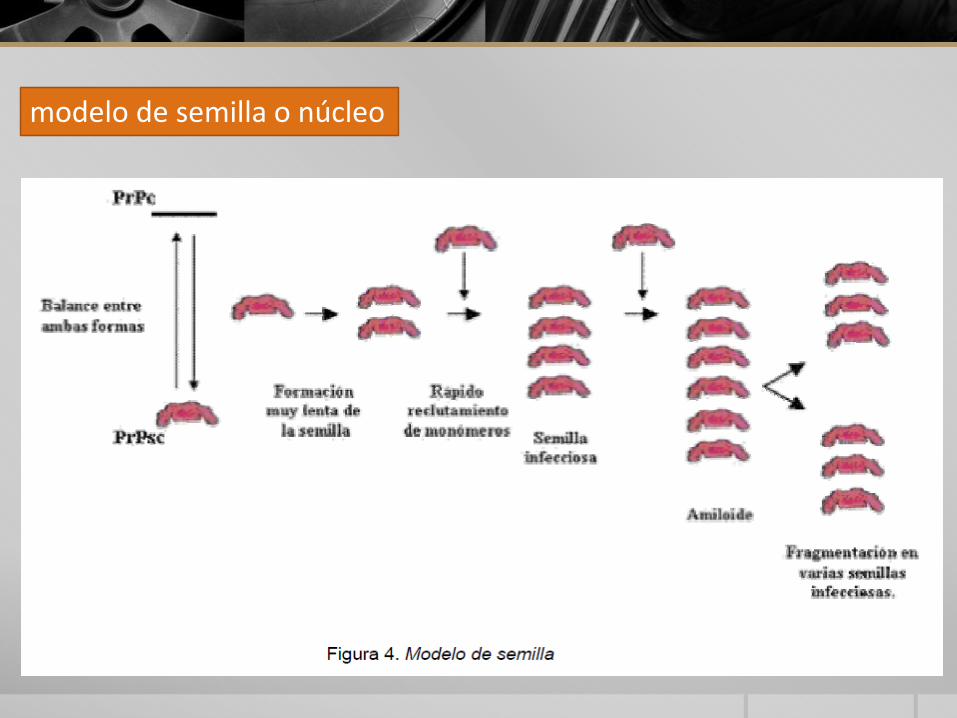
modelo de semilla o núcleo
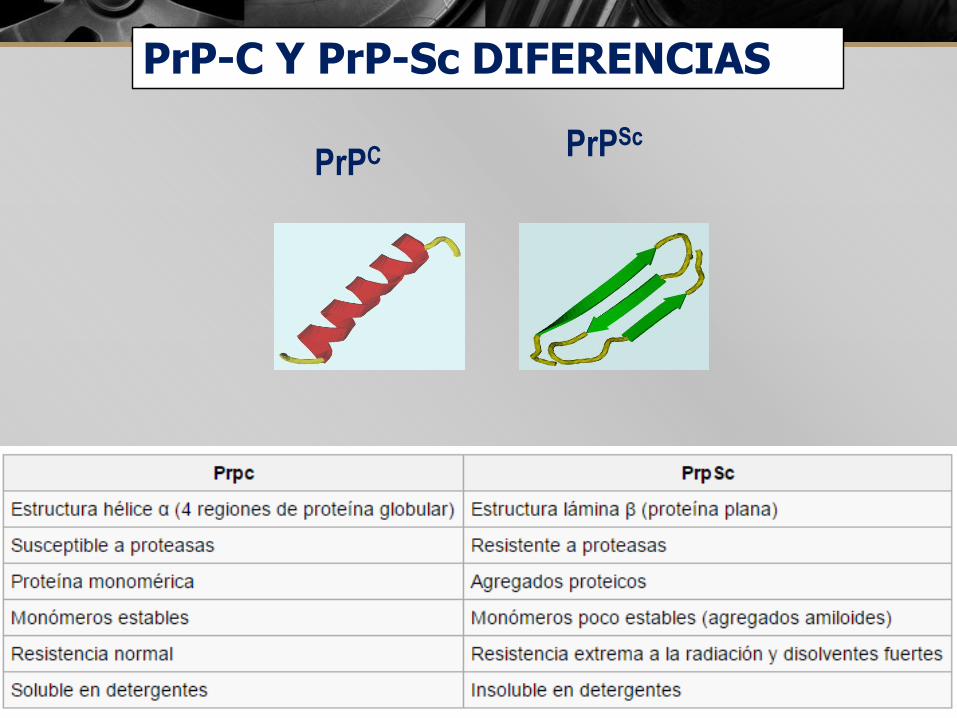
PrPC PrPSc
PrP-C Y PrP-Sc DIFERENCIAS
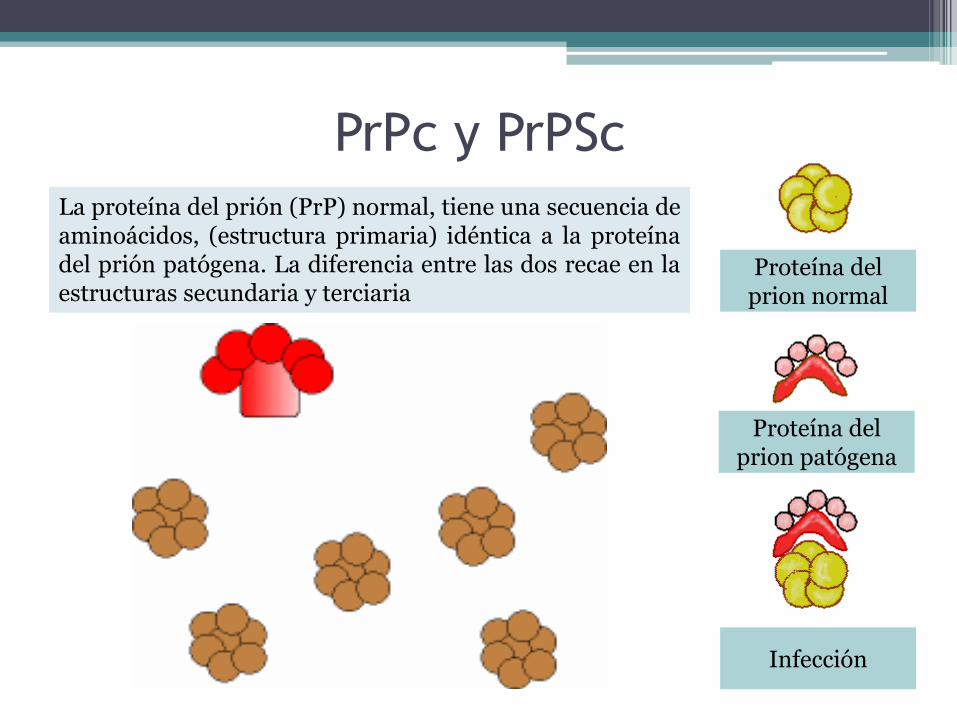
PrPc y PrPSc
La proteína del prión (PrP) normal, tiene una secuencia deaminoácidos, (estructura primaria) idéntica a la proteínadel prión patógena. La diferencia entre las dos recae en laestructuras secundaria y terciaria
Proteína del prion normal
Proteína del prion patógena
Infección

Existe la hipótesis de queesta última estransmitida al hombre porla ingestión de animalesinfectados (vacas y ovejas).
LA BARRERA ENTRE ESPECIES
La secuencia de aminoácidos de la PrP codifica el tipo de prion, yéste obtiene su secuencia PrPS c del último mamífero por el que ha pasado
Existe homología entre la secuencia de aminoácidos de la P r P c del hospedadorinoculado y la de la PrPS c del prion que se inocula.
En la actualidad se acepta que laenfermedad de Creutzfeldt – Jakob esel homólogo humanode la encefalopatía espongiformebovina (EEB).
Procesos neurodegenerativos producidos por el metabolismo aberrante de una proteína priónica.
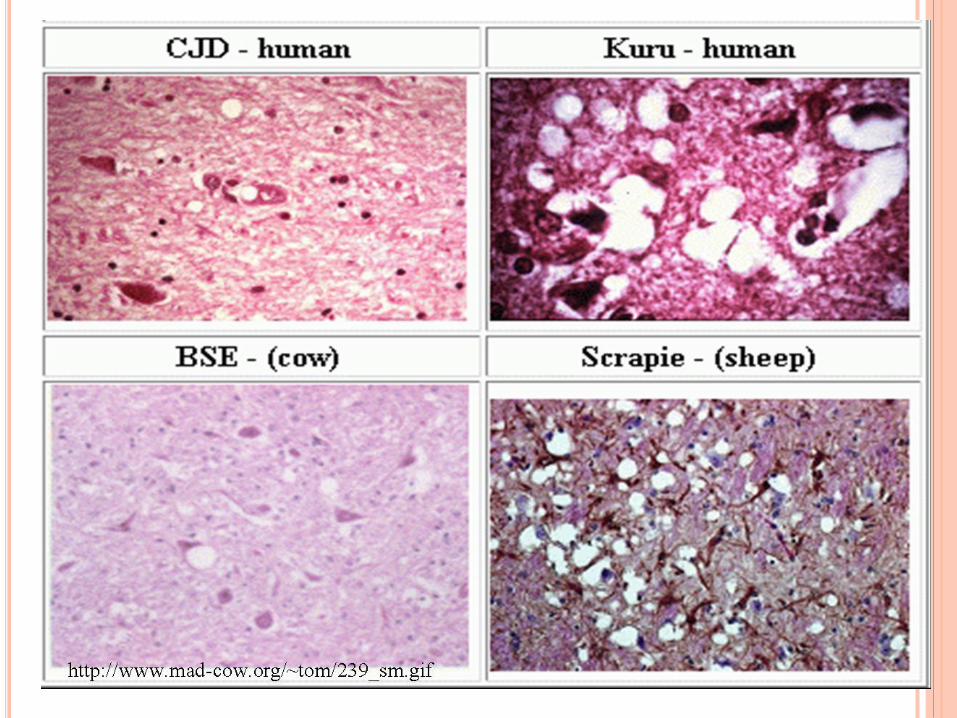
ENFERMEDADES POR PRIONES
Afecta Aspecto espongiforme
de incubación prolongado
carácter transmisible

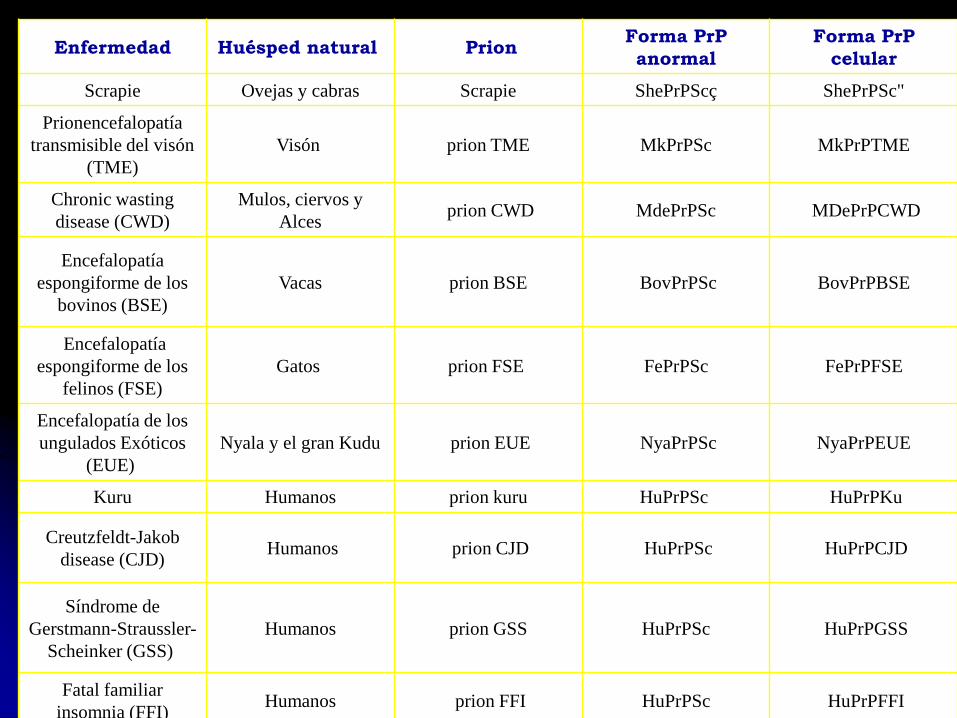
Enfermedad Huésped natural PrionForma PrP
anormal
Forma PrP
celular
Scrapie Ovejas y cabras Scrapie ShePrPScç ShePrPSc"
Prionencefalopatía
transmisible del visón
(TME)
Visón prion TME MkPrPSc MkPrPTME
Chronic wasting
disease (CWD)
Mulos, ciervos y
Alcesprion CWD MdePrPSc MDePrPCWD
Encefalopatía
espongiforme de los
bovinos (BSE)
Vacas prion BSE BovPrPSc BovPrPBSE
Encefalopatía
espongiforme de los
felinos (FSE)
Gatos prion FSE FePrPSc FePrPFSE
Encefalopatía de los
ungulados Exóticos
(EUE)
Nyala y el gran Kudu prion EUE NyaPrPSc NyaPrPEUE
Kuru Humanos prion kuru HuPrPSc HuPrPKu
Creutzfeldt-Jakob
disease (CJD)Humanos prion CJD HuPrPSc HuPrPCJD
Síndrome de
Gerstmann-Straussler-
Scheinker (GSS)
Humanos prion GSS HuPrPSc HuPrPGSS
Fatal familiar
insomnia (FFI)Humanos prion FFI HuPrPSc HuPrPFFI

KURU
1955:Un explorador australiano en Nueva Guinea describió a una niña temblando y riendo.
1956: Carelton Gadusek fue a Nueva Guinea a estudiar el “Kuru”: temblor y sacudidas.
Etnia Fore en Papua
Nueva Guinea.
Kuru en idioma fore: temblores de frío y miedo.
Kuru
. Progresa usualmente hasta la muerte en aproximadamente 1 año.
Aparecía después de la ingestiónde tejidos cerebrales de personasfallecidas con la finalidad deadquirir su sabiduría.
- Predomina en niños y mujeres adultas
Período de incubación puede prolongarse hasta 30 años
- De desarrollo lento

Gajdusek descubrió tres etapas en su curso clínico:
Fase ambulatoria
• Aparece temblor generalizado con pérdida de la capacidad para coordinar los movimientos y disartria (Afectación cerebelosa incipiente)
Fase sedentaria:
• Incapacidad de deambulación independiente, temblores más fuertes, ataxia, labilidad emocional, depresión y bradipsiquia.
• Reflejos tendinosos conservados y aún no es perceptible la degeneración muscular.
Fase terminal
• Incapacidad para la sedestación independiente, ataxia severa, temblores, disartria, incontinencia urinaria y fecal, disfagia y ulceraciones cutáneas.
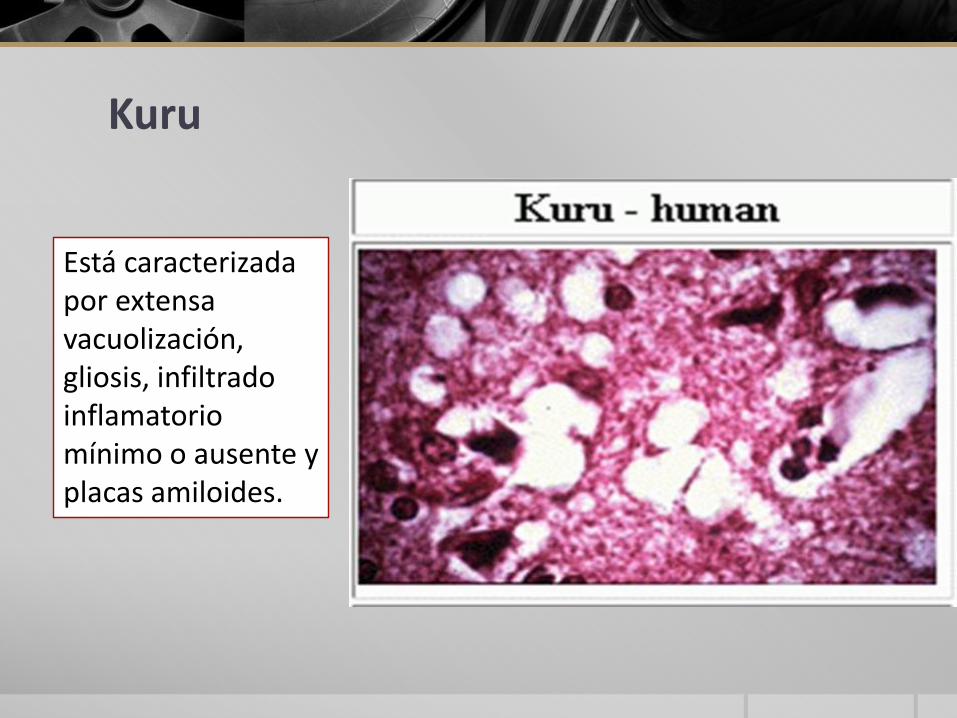
Está caracterizada por extensa vacuolización, gliosis, infiltrado inflamatorio mínimo o ausente yplacas amiloides.
Kuru

Artezano Sanchez Luz E.

Las enfermedades priónicas son procesos
neurodegenerativos producidos por el
metabolismo aberrante de una proteína priónica,
que afectan a seres humanos y animales durante
un período de incubación prolongado, con carácter
transmisible y evolución clínica fatal.
Entre sus manifestaciones clínicas sobresalen:
demencia, ataxia, insomnio, paraplejías,
parestesias y conductas anormales.

1. Resistencia a agentes físicos y químicos.
2. Resistencia a diferentes proteasas.
3. Producen cambios conformacionales en las proteínas priónicas vecinas
y favorecer el progreso de la enfermedad.
4. El daño producido por la proteína priónica anormal puede transmitirse
de un animal a otro.
5. El desarrollo de la enfermedad requiere un período de incubación largo
antes de que se produzcan los síntomas de deterioro neurológico.
6. Penetrancia muy variable depende de: De la cepa de proteína PrPc, del
receptor, de la vía de transmisión y de la carga priónica.
7. Pueden presentarse de forma familiar, esporádica o adquirida.

Según el grado de infecciosidad la OMS diferencia:
Alto riesgo: Cerebro, hipófisis, médula espinal, bazo,
duramadre, timo, amígdalas, placenta, ojos, ganglios
linfáticos e intestino.
Riesgo moderado o bajo: Nervios periféricos, LCR,
páncreas, hígado, glándula suprarrenal, pulmón y médula
ósea.
No relacionado con infectividad en ninguna especie:
Leche, saliva, piel, semen, orina, músculo, sangre, heces,
riñón y hueso.
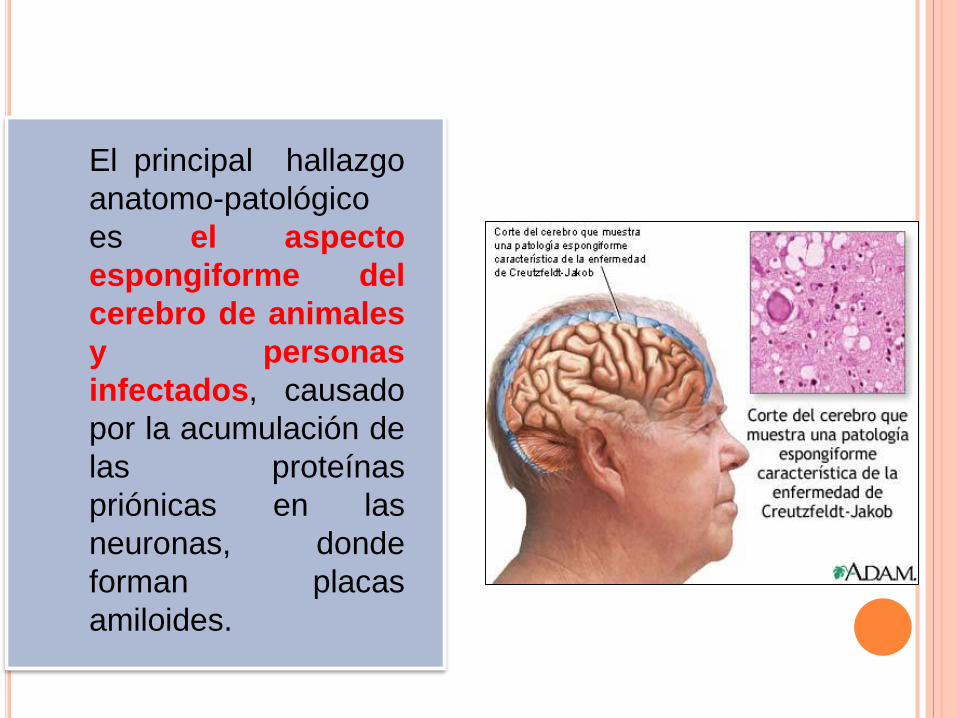
El principal hallazgo
anatomo-patológico
es el aspecto
espongiforme del
cerebro de animales
y personas
infectados, causado
por la acumulación de
las proteínas
priónicas en las
neuronas, donde
forman placas
amiloides.

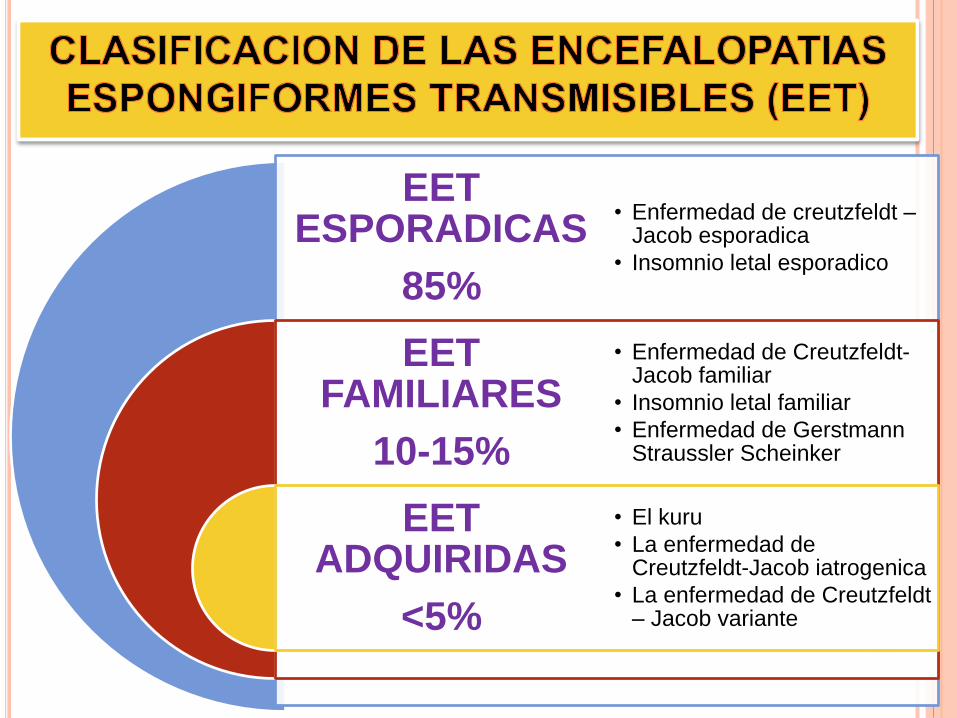
EET ESPORADICAS
85%
EET FAMILIARES
10-15%
EET ADQUIRIDAS
<5%
• Enfermedad de creutzfeldt –Jacob esporadica
• Insomnio letal esporadico
• Enfermedad de Creutzfeldt-Jacob familiar
• Insomnio letal familiar
• Enfermedad de GerstmannStraussler Scheinker
• El kuru
• La enfermedad de Creutzfeldt-Jacob iatrogenica
• La enfermedad de Creutzfeldt– Jacob variante

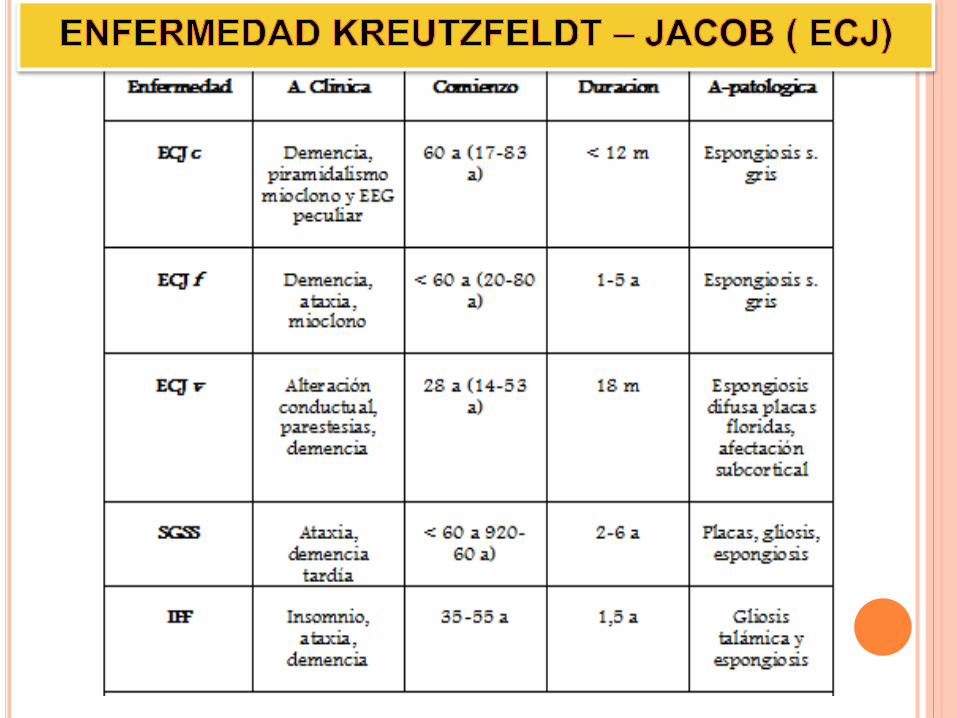
Enfermedad priónica más común.
Son esporádicos (80- 90 %).
Casos en todo el mundo
Afecta a pacientes entre 16 y más de 80 años.
Demencia presenil subaguda
Mioclonías
otros déficit neurológicos:
piramidalismo, ataxia y otros.
La muerte
sobreviene
entre 1 mes -
10 años

Desde 1995 y hasta la mitad del año 2000
casos en Gran Bretaña, Irlanda y Francia
La edad no llega a 30 años con un rango de 14-53 años.
La enfermedad suele superar el año de duración.
Los pacientes presentan:
• Pródromos de alteraciones
psiquiátricas.
• Parestesias en extremidades
durante unos 6 meses

CRITERIOS DE LA OMS PARA LA ENFERMEDAD DE CREUTZFELDT-
JAKOB
I
A Alteración neuropsiquiátrica progresiva
B Duración de la enfermedad mayor de seis meses
C Ningún otro diagnóstico alternativo
D Sin historia de exposición iatrogénica posible
E Sin antecedente familiar de enfermedad por priones
II
A Síntomas psiquiátricos tempranos
B Síntomas sensitivos dolorosos persistentes
C Ataxia
D Mioclonías, corea o distonía
E Demencia
III
A Electroencefalograma sin patrón típico de ECJ o no realizado
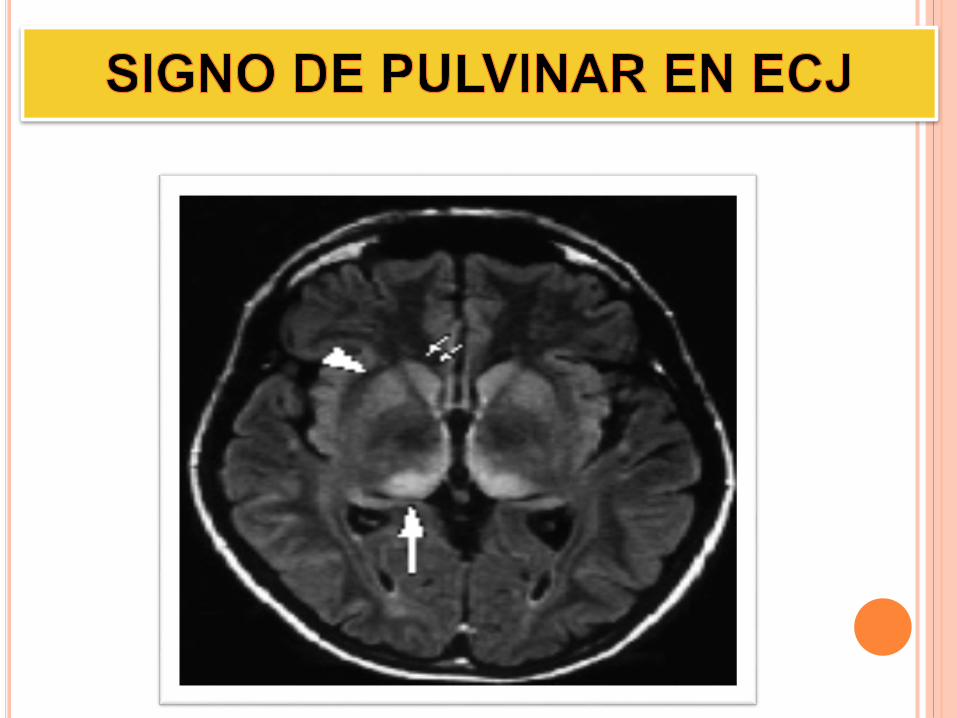
B Signo del pulvinar bilateral en la resonancia magnética
IV
A Biopsia positiva
Definitivo: I A y confirmación neuropatológica.
Probable: I y 4/5 de II y III A y III B o I y IV A
Posible: I y 4/5 o II y III A

Vacuolización de la sustancia gris
Las vacuolas observadas en el microscopio óptico son debidas a hinchazón focal de axones y terminaciones neuronales dendríticas asociado a la pérdida de organelas sinápticas y la acumulación de membranas anormales.
Sustancia blanca subcortical o profunda.
Espongiosis en sustancia gris del
cerebro en ECJ esporádica
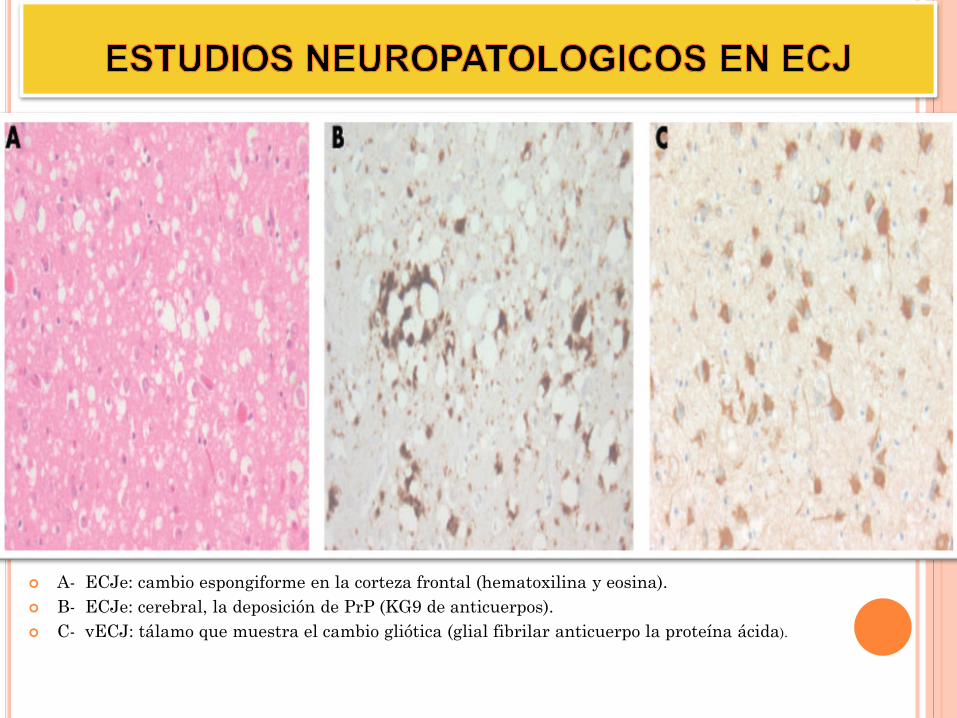
A- ECJe: cambio espongiforme en la corteza frontal (hematoxilina y eosina).
B- ECJe: cerebral, la deposición de PrP (KG9 de anticuerpos).
C- vECJ: tálamo que muestra el cambio gliótica (glial fibrilar anticuerpo la proteína ácida).
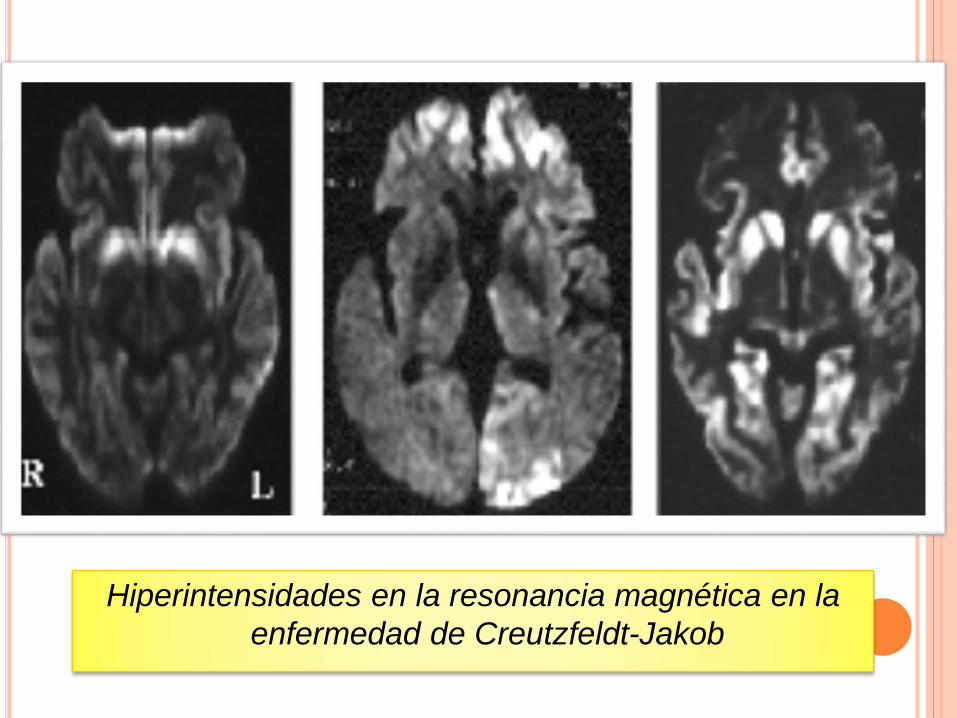
Hiperintensidades en la resonancia magnética en la
enfermedad de Creutzfeldt-Jakob
• Trastorno autosómico dominante
• Causado por una mutación en la
proteína del prión.
• Aparece entre los 23 y los 73 años.
Insomnio
Pérdida del ritmo
circadiano
Alteraciones cognitivas
Disminución de la atención,
la concentración y la
memoria

• Duración promedio de la enfermedad
alrededor de 7 a 36 meses.
• Se han identificado 28 familias en el
mundo que portan el gen que provoca la
enfermedad
En la actualidad, el diagnóstico se
realiza mediante la detección de la
proteína 14-3-3 en LCR
Para obtener un diagnóstico de
"definitivo", es necesario que se cumplan
al menos dos de los siguientes criterios:
-Transmisión a animales
-presencia de PrPc o
-Mutaciones en el gen que codifica para la
proteína priónica .
• Herencia autosómico dominanteen un 40% de los casos.
• La aparición clínica: 30 – 60 añosde vida
• La evolución de la enfermedad esrelativamente lenta.
• Progresa hasta la muerte aprox. 5años.
• La mutación más frecuente es en elcodón 102, que determina uncambio de prolina a leucina.
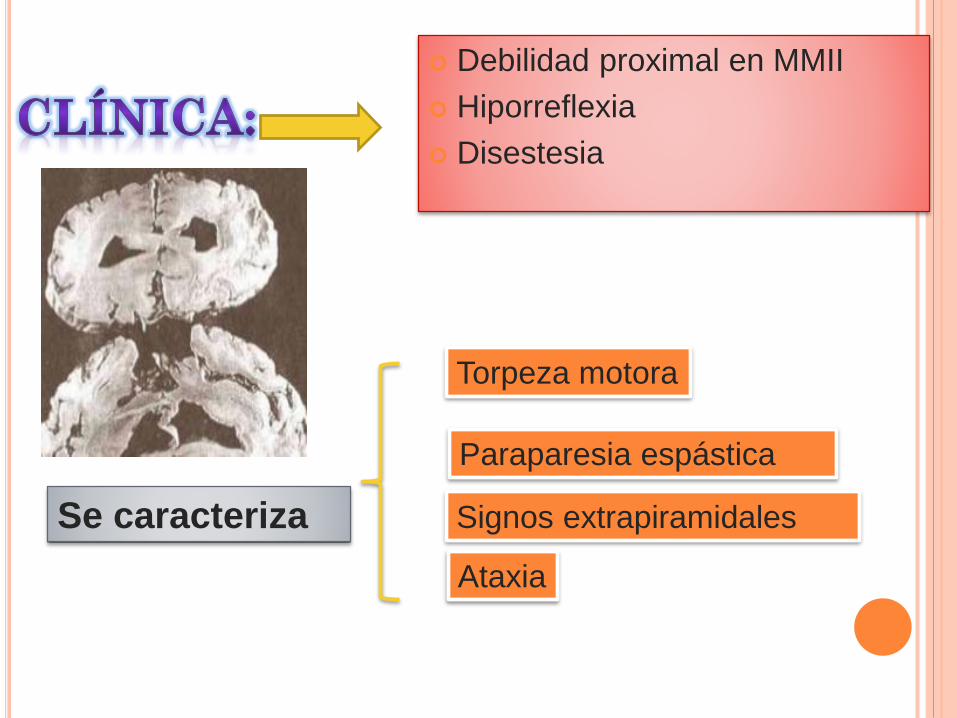
Debilidad proximal en MMII
Hiporreflexia
Disestesia
Se caracteriza
Torpeza motora
Paraparesia espástica
Signos extrapiramidales
Ataxia

Importante diferenciar las enfermedades por
priones genéticas de otras patologías
neurodegenerativas (Alzheimer).
La OMS recomienda hacer un estudio
genético a todos los pacientes con una
historia familiar de enfermedad por priones.
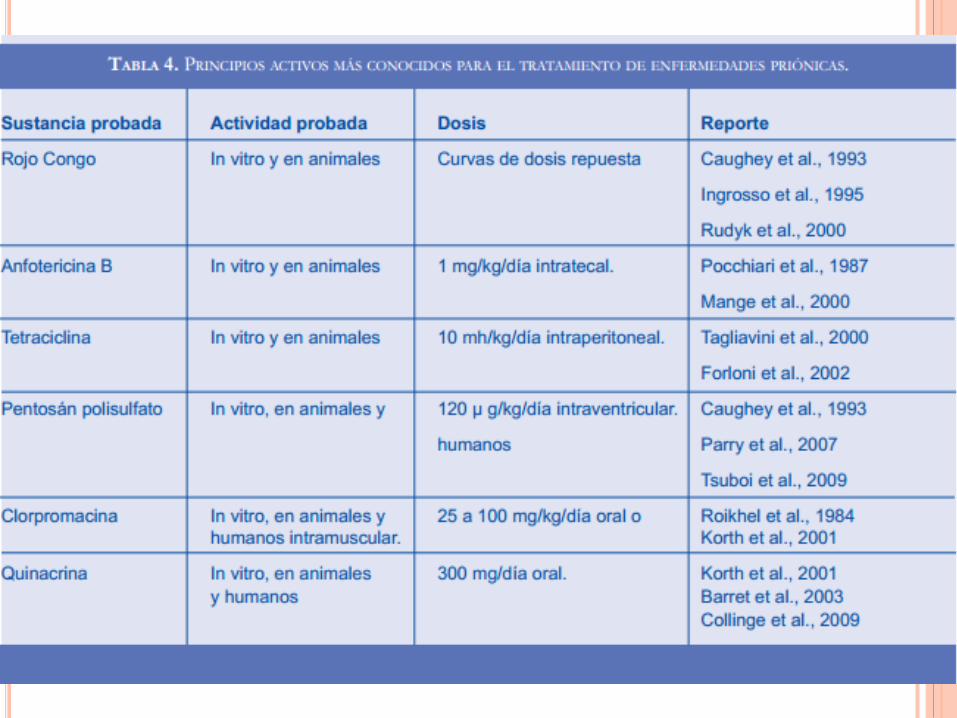
No hay tratamiento
curativo en la
actualidad.
El único tratamiento es
paliativo.

• Lavarse las manos
• Cubrir sus heridas con materiales a prueba de agua.
• Usar guantes de látex.
• Protegerse con ropa resistente a heridas corto-punzantes.
• Usar elementos de protección para la cara.
• Los equipos utilizados, al igual que la camilla y el cabezal,
deben ser cubiertos por laminas de plástico.
• El instrumental quirúrgico será de acero inoxidable, y no se
puede dejar secar tras la intervención.
• El material debe descontaminarse durante una hora en
hipoclorito de sodio, y luego ser sometido a otra hora a 134°C y
3 atm de presión.



















