
Engineering\CADD Systems Office CADD Manager's Series Site Integration.
Upload
barrie-laneCategory
view
212download
0
R. S. Pearlman1, Y. Wu1, K. M. Smith2, and B. B. Masek2
1 Laboratory for the Development of CADD Software, College of Pharmacy, University of Texas, Austin TX
2 Tripos, Inc., 1699 S. Hanley Rd., St. Louis MO
Cheminformatics for Computational Chemistry and
Computer-Aided Molecular Discovery

R. S. Pearlman1, Y. Wu1, K. M. Smith2, and B. B. Masek2
1 Laboratory for the Development of CADD Software, College of Pharmacy, University of Texas, Austin TX
2 Tripos, Inc., 1699 S. Hanley Rd., St. Louis MO
Dealing with the Concepts of both
“Chemical Compound” and
“Chemical Structure”

A few words about “passion” • over the years, my colleagues and I developed 15 substantial CADD-
related programs which were distributed (by Tripos or Optive Research or through my UT Lab) to industrial & academic users around the world
• several factors contributed to our success but the primary driving force was my “passion” for addressing CADD-related issues I felt needed to be addressed
• I am genuinely passionate about issues discussed in this talk
• but ... passion is a “double-edged sword”
• I am genuinely frustrated about no longer being in a position to advance the concepts and software to which I will allude during this presentation
• I apologize in advance in case my frustration gets the better of me

Outline
• introductory comments
• terminology
• motivating factors
• software / implementation
• summary

Accepted perspectives
• can’t (shouldn’t) take a single step in the wet lab until all “composition of chemical matter” issues have been formally addressed (i.e., are we using the right compound?)– avoid ambiguous, erroneous, misleading experimental results– industry-standard “best practice” approach to doing good science
• after performing wet lab experiments or measurements (e.g. HTS effort or logPerm measurement), discard results?– of course not!– recognize results as valuable intellectual property of potential utility
for current and future research efforts– take appropriate steps to associate resulting data with “cmpd-IDs”
to ensure facile access to data for future applications

Traditional cheminformatics technologies
• identify chemical compounds ... establish cmpd-IDs
• associate and store experimental data/results with corresponding compounds
• developed for traditional chemical wet-lab research purposes performed in “the real world” ... Mother Nature’s world
• not developed for Comp. Chemistry or Computer Assisted Molecular Discovery purposes performed in the “in silico” world serious problem!

Nature’s world vs the in silico world
• we deal with compounds: real entities
• cmpd can exist as different structures
• M. N. determines predominent structure based on chem. pot. (μ) of environment
• measured property is a consequence of structure(s) chosen by Mother Nature
– typically, we don’t know which structure(s)
• measured data should be associated with cmpd on which it was measured
• measurements might be wrong due to experimental error
• we deal with structures: CTs
• cmpd represented by single structure
• DB curator or chemist determines structure based on “drawing rules”
• predicted property is a consequence of structure chosen by a human
• predicted data should be associated with struct on which it was calculated
• predictions (even from “perfect” SW) will be wrong unless we consider the structure chosen by Mother Nature
When trying to understand or predict properties of compounds in silico, we must address the same range of structures that those compounds can adopt in Nature.

Looking at compounds the way Mother Nature sees them:
Nature’s world vs the in silico world
• we deal with compounds: real entities
• cmpd can exist as different structures
• M. N. determines predominent structure based on chem. pot. (μ) of environment
• measured property is a consequence of structure(s) chosen by Mother Nature
– typically, we don’t know which structure(s)
• measured data should be associated with cmpd on which it was measured
• measurements might be wrong due to experimental error
• we deal with structures: CTs
• cmpd represented by single structure
• DB curator or chemist determines structure based on “drawing rules”
• predicted property is a consequence of structure chosen by a human
• predicted data should be associated with struct on which it was calculated
• predictions (even from “perfect” SW) will be wrong unless we consider the structure chosen by Mother Nature
“Nature’s Way”

Accepted perspectives
• can’t (shouldn’t) take a single step in the wet lab until all “composition of chemical matter” issues have been formally addressed (i.e., are we using the right compound?)– avoid ambiguous, erroneous, misleading experimental results– industry-standard “best practice” approach to doing good science
• after performing wet lab experiments or measurements (e.g. HTS effort or logPerm measurement), discard results?– of course not!– recognize results as valuable intellectual property of potential utility
for current and future research efforts– take appropriate steps to associate resulting data with “cmpd-IDs”
to ensure facile access to data for future applications

Evolving perspectives
• can’t (shouldn’t) take a single step on the computer until all “composition of chemical matter” issues have been formally addressed (i.e., are we considering the right structure?)– avoid ambiguous, erroneous, misleading computational results– industry-standard “best practice” approach to doing good science
• after performing in silico experiments or calculations (e.g. vHTS effort or logPerm estimate), discard results?– of course not!– recognize results as valuable intellectual property of potential utility
for current and future research efforts– take appropriate steps to associate resulting data with “struct-IDs” and
“cmpd-IDs” to ensure facile access to data for future applications

Traditional cheminformatics technologies
• identify chemical compounds ... establish cmpd-IDs
• associate and store experimental data/results with corresponding compounds

Required cheminformatics technologies
• identify chemical compounds ... cmpd-IDs
• associate and store experimental data/results with corresponding compounds
• identify the various structures which a given compound can adopt in various chemical environments ... struct-IDs
• associate and store computational (and some experimental) data/results with corresponding structures
• associate all relevant structures with their corresponding compounds ... via canonically determined, unique cmpd-ID

Dealing with compounds in “Nature’s Way”
• it’s not just about ligands and docking !− although that’s still what garners most of the attention
• and it’s not just about “tautomers” !− must also consider protonation state
− must also consider stereochemical issues
− must also consider conformational issues
• it’s about enabling humans and software to be able to automatically use the same structures in silico as Mother Nature uses for a cmpd in the real world

Dealing with compounds in “Nature’s Way”
• it’s about enabling humans and software to be able to automatically use the same structures in silico as Mother Nature uses for a cmpd in the real world
• for example:• when performing a “docking experiment” on a given
cmpd, automatically attempt to dock all reasonable “tautomers” ... as well as all reasonable conformers
• when searching a 2D (or 3D) DB for cmpds satisfying a particular query, automatically consider all reasonable “tautomers” (as well as all reasonable conformers)
• when retrieving cmpds known to dock with given target, return known bound structure(s) ... not just DB default

Dealing with compounds in “Nature’s Way”
• it’s not just about ligands and docking !− although that’s still what garners most of the attention
• and it’s not just about “tautomers” !− must also consider protonation state
− must also consider stereochemical issues
− must also consider conformational issues
• it’s about enabling humans and software to be able to automatically use the same structures in silico as Mother Nature uses for a cmpd in the real world

Outline
• introductory comments
• terminology
• motivating factors
• software / implementation
• summary

Stereochemical Issues: Proto-Invertible Atoms
• protonating I “introduces” chirality in II
• protonating III “introduces” opposite chirality in IV
• A/B equilibria and I III inversion mean that, in reality, II “inverts” to IV
• II and IV are proto-invertible
• neither the II IV proto-inversion nor the I III inversion occurs in silico
• Nature’s Way technology appropriately perceives all 4 pseudo-chiral isomers as different structures of same cmpd
• Nature’s Way technology appropriately identifies and generates all 4 structures to avoid missing potential 3D vHTS hits
NX
YZ
N
X YZ
NX
YZ
N
X YZ
H
H
"S"
"R""R"
"S"
::
(III) (IV)
(I) (II)
• sp3 Nitrogens (I and III) are readily invertible usually not considered chiral

Stereochemical Issues: Proto-Invertible Atoms & Bonds
• assuming Y has greater steric bulk (and higher C-I-P rank) than X, we begin with protomer-I in conformation Ia
• Ia will yield II which could revert to Ia or yield III in conformation IIIa
• assuming that Econf of IIIb is not too high, protomer-IV could also be formed and could yield protomer-I in conformation Ib which, of course, could revert to Ia
X
Y
H
O
Z
X
Y
OH
Z
Y
X
OH
Z
H
Y
X
O
Z
H
Y
X
O
Z
Y
X
H
O
Z
120o
"S" "E" "R"
"R""Z"
(Ia) (II) (IIIa)
(IV)
H
X
Y
O
Z"S"
H
X
Y
O
Z120o
(IIIb)(Ib)
(I)
(III)
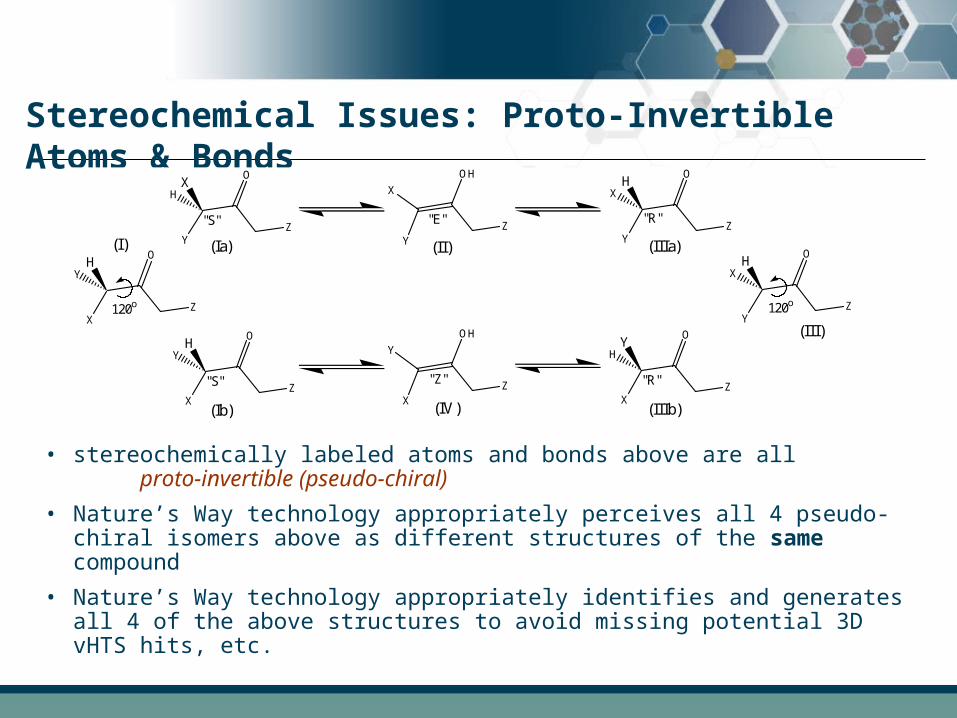
Stereochemical Issues: Proto-Invertible Atoms & Bonds
• stereochemically labeled atoms and bonds above are all proto-invertible (pseudo-chiral)
• Nature’s Way technology appropriately perceives all 4 pseudo-chiral isomers above as different structures of the same compound
• Nature’s Way technology appropriately identifies and generates all 4 of the above structures to avoid missing potential 3D vHTS hits, etc.
X
Y
H
O
Z
X
Y
OH
Z
Y
X
OH
Z
H
Y
X
O
Z
H
Y
X
O
Z
Y
X
H
O
Z
120o
"S" "E" "R"
"R""Z"
(Ia) (II) (IIIa)
(IV)
H
X
Y
O
Z"S"
H
X
Y
O
Z120o
(IIIb)(Ib)
(I)
(III)

Stereochemical issues: Summary
• tautomeric transforms can change stereochemistry• protonation/deprotonation can change stereochemistry• protomeric transforms can change stereochemistry• ProtoPlexing MUST be followed by StereoPlexing• the concepts are inextricably linked !!• cheminformatics and all Computer-Assisted Drug Discovery
(CADD) applications should recognize and address the important difference between truly chiral atoms and bonds and protomerically-invertible, pseudo-chiral atoms and bonds
• failure to do so will guarantee that computer-based models of chemical behaviors do not accurately reflect the behaviors due the MetaStructures observed in Nature

New terminology for some “new” concepts
• two types of stereo-centers: truly chiral atoms and bonds• stereomers: different stereochemical isomers (hence,
different chemical compounds)• two types of proto-centers: acid/base & tautomeric D/A pairs• protomers: different protonation states and/or tautomeric
states of a single given compound• protomeric state: refers to both protonation state and
tautomeric state of a given protomer
• protomeric transform: protomeric-statei → protomeric-statej
• proto-stereomers: different stereomers of protomers of a given compound which differ ONLY with respect to chiralities of invertible or proto-invertible (pseudo-chiral) centers
• proto-stereo-conformers: different 3D conformations of the proto-stereomers of a given compound

New terminology for some “new” concepts
• proto-stereomers: different stereomers of protomers of a given compound which differ ONLY with respect to chiralities of invertible or proto-invertible (pseudo-chiral) centers
• proto-stereo-conformers: different 3D conformations of the proto-stereomers of a given compound
• 2D-MetaStructure of a compound: the set of all proto-stereomers of a given compound; i.e., set of all 2.5D connection tables which could be achieved by and which should be associated with a given compound
• 3D-MetaStructure of a compound: the set of all proto-stereo-conformers of a given compound; i.e., set of all 3D conformations of all 2.5D connection tables which could be achieved by and which should be associated with a given compound

Outline
• introductory comments
• terminology
• motivating factors
• software / implementation
• summary

Dealing with compounds in “Nature’s Way”
motivated by:
• CADD/QSPR-related reasons (in addition to docking)
• cheminformatic reasons
• business-related reasons
• IP-related reasons

Dealing with compounds in “Nature’s Way”
• it’s not just about ligands and docking !− although that’s still what garners most of the attention
• and it’s not just about “tautomers” !− must also consider protonation state
− must also consider stereochemical issues
− must also consider conformational issues
• it’s about enabling humans and software to be able to automatically use the same structures in silico as Mother Nature uses for a cmpd in the real world

Dealing with compounds in “Nature’s Way”
• it’s not just about ligands and docking !− subject of ACS Symposium I organized in S-2002
− subject of ACS Symposium Paul Labute (CCG) was organizing for F-2007 but post-poned to Sp-2008
− primary focus of paper CINF-89 being presented on Thursday at 4:25pm on by Hongyao Zhu (Plexxikon, Inc.)
− a couple of “obligatory” examples …

ProtoPlex generates 4 neutral tautomeric forms
(plus additional charged protomers)
Example: Ricin Inhibitors - Pterins
Pterin(1) Pterin(2) Pterin(4)
Ionized Protomers not shown
N
NH
N N
O
H2N
N
N
N N
OH
H2N
N
N
HN N
O
H2N
Pterin(3)
HN
N
N N
O
H2N
receptor-bound tautomer (protomer) may not be the protomer most prevalent in solution

Example: Ricin Inhibitors - Pterins
“A tautomer of pterin that is not in the low energy form in either the gas phase or in aqueous solution has the best interaction with the enzyme.”
S. Wang, et. al., Proteins, 31, 33-41 (1998)
Pterin(1) protomer is preferred in both gas and aqueous soln
Pterin(3) protomer is preferred in receptor binding site
HN
N
N N
O
H2N
N H
OGly121
Tyr123
NH2+
H2N NHArg 180
HO
O
N
H
Val81
Ser176
Redrawn from Wang, et. al, Proteins, 31, 33-41(1998)
Pterin(1) Pterin(2) Pterin(4)
Ionized Protomers not shown
N
NH
N N
O
H2N
N
N
N N
OH
H2N
N
N
HN N
O
H2N
Pterin(3)
HN
N
N N
O
H2N

Example: Barbiturate Matrix Metalloproteinase Inhibitors
ProtoPlex generates 5 neutral tautomeric forms
(plus additional charged protomers)
N
HN OHO
O
N
Ph
OH
N
HN OO
OH
N
Ph
OH
HN
HN OO
O
N
Ph
OH
Enol Form (A) Enol Form (B) Keto Form
Ionized Protomers not shown
N
N OHO
OH
N
Ph
OH
Di-Enol Form (D)
N
N OHO
OH
N
Ph
OH
Di-Enol Form (E)
• the receptor-bound tautomer (protomer) might not be the keto protomer which is most prevalent in aqueous solution
• which protomer does the receptor prefer?
• which protomer(s) will be used for vHTS???

Example: Barbiturate Matrix Metalloproteinase Inhibitors
“The enol form (A) of the barbiturate is thus favored by the protein matrix over the tautomeric keto form, which dominates in solution.”
H. Brandstetter, et. al., J. Biol. Chem., 276(20), 17405-17412 (2001)
N
N OO
O
P1'
P2'
H
Zn+2
N
O
NO
N
O
Pro217 Asn218
Tyr219
-O O
O
N
O
N
Ala160
Ala161
Glu198
Redrawn from Branstetter, J. Biol. Chem

What if ???
did not know certain barbiturate is a good MMP inhibitor and ...• vHTS effort only considered the expected keto-form of the cmpd?
– missed vHTS hit missed lead missed drug? missed $billions??
did know certain barbiturate is a good MMP inhibitor but ...• vHTS-method assessment only considered the expected keto-form?
– “false negative” “unreliable method” wasted CADD-scientist time
– missed hits from not using what is actually a reliable CADD method
• pharmacophore determination only considered the “default” keto-form?– failure to identify pharmacophore model wasted CADD-scientist time
– missed hits from not having what could have been good 3D-search query, alignment rule, CoMFA model, etc.

CADD motives for adopting “Nature’s Way”
• better results from docking-based vHTS– compounds can and will adopt different structures under different
conditions or in different Natural environments
• μreceptor ≠ μwater, etc. we simply must adjust our preconceptions
– what’s the cost of not screening a potential lead cmpd predicted to dock poorly due to failure to consider the structure preferred by the receptor?
– problems re: dock-scoring-functions or problems due to wrong structure?
– need to enumerate the set of structures which a compound can adopt

CADD motives for adopting “Nature’s Way”
• better results from docking-based vHTS
– better vHTS docking scores and docking poses
– better understanding better structure-based design
• better CoMFA models (better alignments)
• better field-matching, shape-matching
• better pharmacophore perception
• better 3D searching
• better clustering, etc., etc.

QSPR motives for adopting “Nature’s Way”
• better ADME and other SPR and QSPR models– protomeric state of a “solute” depends on the chemical
potential presented by the surrounding “solvent” or molecular environment (often different than aqueous soln)
– partition coefficients (two solvent environments to consider!)

S
O
ON
OH
O
HN
N
N
O
SO
O
OH
N
N
Piroxicam
CLogP = 1.90 CLogP = 0.60
Improve property prediction by considering Protomeric State
• which structure is most probable in water? in lipoidal membrane?
• even the best logP software doesn’t treat even the unionized species correctly with respect to an easy physical property
• how many structure-based models of BBB permeability, CaCo-2 permeability, oral BA, etc. currently address tautomerism?
• what should we expect (or plan for) in the not too distant future?

QSPR motives for adopting “Nature’s Way”
• better ADME and other SPR and QSPR models– protomeric state of a “solute” depends on the chemical
potential presented by the surrounding “solvent” or molecular environment (often different than aqueous soln)
– partition coefficients (two solvent environments to consider)– permeability coefficients (depend on donor-phase and membrane)– solubilities (depend on crystalline and solvent environments)– melting points (crystal packing can favor unusual protomeric forms)

Example: effect of crystal environment
Two different protomers observed in the SAME unit cell!
“Coexistence of both histidine tautomers in the solid state and stabilisation of the unfavoured N-H form by intramolecular hydrogen bonding: crystalline L-His-Gly hemihydrate” T. Steiner and G. Koellner, Chem. Commun., 1997, 1207.
Protomeric transform was induced by intramolecular interaction which was induced by a conformational change which was induced by intermolecular interactions.

QSPR motives for adopting “Nature’s Way”
• better ADME and other SPR and QSPR models– protomeric state of a “solute” depends on the chemical potential
presented by the surrounding “solvent” or molecular environment (often different than aqueous soln)
– partition coefficients (two solvent environments to consider)– permeability coefficients (depend on donor-phase and membrane)– solubilities (depend on crystalline and solvent environments)– melting points (crystal packing can favor unusual protomeric forms)– need to “select” protomeric forms according to user-specs
• better models better decisions – about what to screen– about which “hits” to promote to “leads”– about route of administration and/or formulation– about which leads to promote to candidacy

Cheminformatic motives for adopting “Nature’s Way”
• better storage of data– measured properties of compound should be associated with
the compound (with notations re: experimental conditions)– predicted properties “of a compound” should be associated with
(stored under) the particular structure used for the prediction– that structure, in turn, should be associated with the compound– need a unique identifier that can tie any proto-stereomeric
structure to the compound to which it corresponds
• better use of data– enable “data-mining” of both measured and computed data
• discard wet HTS data? save for future “data-mining?” • discard virtual HTS data? save for future “data-mining?”
• better (more robust) results when searching for compounds, data, structures, and substructures

Business & IP motives for adopting “Nature’s Way”
companies must be able to recognize when
two different structures correspond to the same compound!
need a canonically unique identifier that can tie any proto-stereomeric structure
to the compound to which it corresponds

Business & IP motives for adopting “Nature’s Way”
• companies allocate resources for cmpds, not structures– resource-related decisions (what should we purchase, synthesize,
screen?) should be based on compounds, not structures• to properly manage corporate inventories• to avoid costly, unintended duplications (acquisitions and screening)• to avoid far more costly failure to screen active compounds for which
the representative (DB) structures were predicted to be inactive
• companies own & intend to patent cmpds, not structures– offensive and defensive “Freedom To Operate” strategies are far
stronger when all structures of patented cmpds are considered– failure to realize that a competitor’s “novel compound” is merely a
different structure of your patented compound can cost $billions• at least one acknowledged example already exists!!
• patent offices need to revise their procedures

Outline
• introductory comments
• motivating factors
• terminology
• software / implementation
• summary

NW requirements revealed by motives for adoption
1. need to enumerate the set of proto-stereomeric structures which a compound can adopt (2D-MetaStructure) and the set of proto-stereo-conformers (3D-MetaStructure) subject to user specifications and limitations
2. need to “normalize” protomeric form according to user-specs
3. need a unique identifier that can tie any proto-stereomeric structure to the compound to which it corresponds
4. need a chemical database system designed to handle 2D and 3D MetaStructures and computational data associated therewith – i.e., a MetaStructure-oriented DB, not just a compound-oriented DB

Current and future state of “Nature’s Way” technology
• currently in use as three separate Discovery Engines:– ProtoPlex, StereoPlex, and Confort (for 3D conformer generation)– Engines communicate via unix/linux pipes or file I/O– OK, but … each program sees input as “new” structures
• software data-structures must be reloaded• some information must be recomputed• proto-stereomer and proto-stereo-conformer naming is non-optimal

Example Nature’s Way Protocol
Database
Raw, 2D Input
CompoundFilter
Filtered, 2D Input
ProtoPlex StereoPlex Confort
Multiple, 2D Protomers
Multiple, 2.5D Proto-Stereomers
2D App.
vHTS
Multiple, 3D Proto-Stereo-Conformers
For each compound …– many Proto-Stereomers
– One 2D-MetaStructure
– Many Proto-Stereo-Conformers
– One 3D-MetaStructure • associate structure-based data with corresponding structure of each compound pulled from DB

Current and future state of “Nature’s Way” technology
• currently in use as three separate Discovery Engines:– ProtoPlex, StereoPlex, and Confort– Engines communicate via unix/linux pipes or file I/O– OK, but … each program sees input as “new” structures
• software data-structures must be reloaded• some information must be recomputed• proto-stereomer and proto-stereo-conformer naming is non-optimal
• coming soon ... a single integrated Discovery Component:– Nature’s Way Web Service
– comprised of ProtoPlex, StereoPlex, and Confort Web Services– communicate via SOAP or embeddable within 3rd-party code
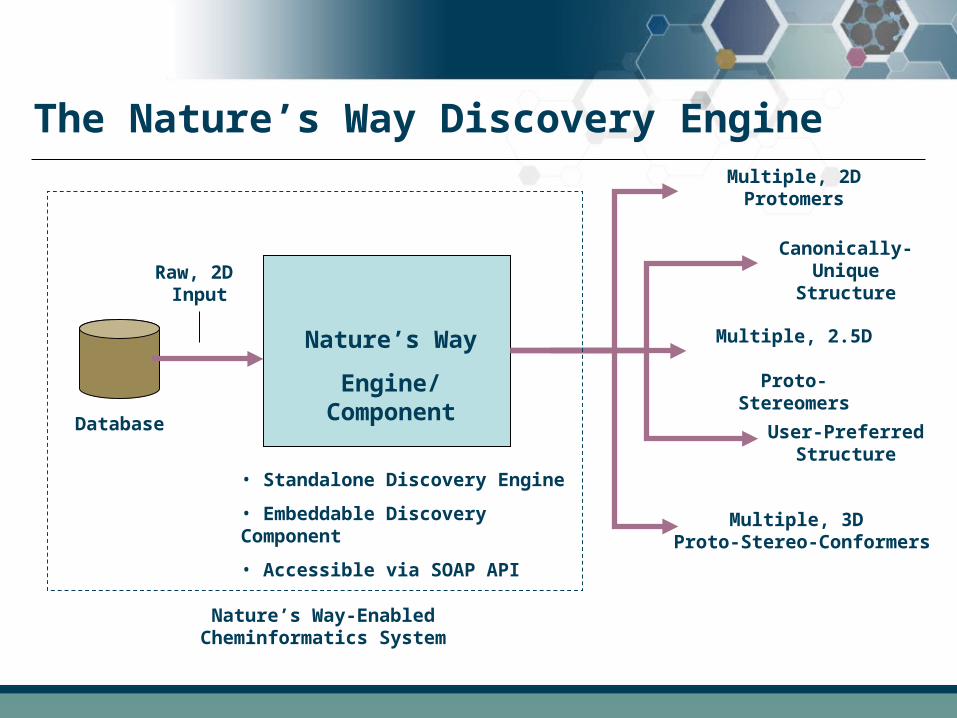
The Nature’s Way Discovery Engine
Database
Raw, 2D Input
Multiple, 2D Protomers
Multiple, 2.5D Proto-Stereomers
Multiple, 3D Proto-Stereo-Conformers
Nature’s Way
Engine/Component
• Standalone Discovery Engine
• Embeddable Discovery Component
• Accessible via SOAP API
Nature’s Way-Enabled Cheminformatics System
Canonically-Unique Structure
User-Preferred Structure

StereoPlex
• for general purposes, provides user-controlled “multiplexing” of all truly chiral, invertible, and proto-invertible stereocenters
– addresses atom-centered (R/S) and bond-centered (E/Z) chirality– automatically excludes “stereochemical junk” (e.g., 254 out of 256
combinations of R’s and S’s for chiral, substituted cubane)– outputs a user-specified number of stereomers selected according
to a user-specified priority rule• multiplexing unspecified stereocenters ensures that CADD results
don’t suffer due to (necessarily) “random” stereochemistry introduced when converting from 2D to 3D -- -- a concept we introduced in 1986
• multiplexing specified stereocenters provides “stereochemical diversity” for vHTS applications – just as important as “structural diversity”
• for “Nature’s Way” purposes, provides user-controlled “multiplexing” of all invertible & proto-invertible stereocenters
– yields proto-stereomers

ProtoPlex
• identifies and ensures that invertible and proto-invertible (pseudo-chiral) atoms and bonds are not labeled as chiral– essential for canonically unique compound identification
• can output a “normalized” protomer based on a user-specified selection rule – useful for generating input for certain CADD or QSPR applications– useful for implementing corporate “drawing rules” for preferred
representation at registration time
• can output a user-specified number of protomers selected according to a user-specified priority rule– useful for limiting the types as well as the numbers of protomers
considered and used for various CADD purposes
• offers rational protomer-naming options

ProtoPlex • under development since 1999
– achieving chemical and cheminformatic robustness is not easy!
– benefited from feedback received from large pharma Collaborators
• can generate all plausible protomers by exhaustively “multiplexing” the corresponding protomeric transforms– simultaneously addresses all acid/base and tautomeric transforms
• simultaneity is critically important for cheminformatic robustness
– automatically excludes implausible “protochemical junk”
• generates output in a canonically unique protomer-order and each protomer is expressed in a canonically unique atom-order
• can output canonically unique protomer selected/based on an Optive Standard canonical Normalization rule– resulting OSN protomer yields canonically unique compound ID

Protomer enumeration is a non-trivial task!
• don’t want to enumerate “implausible” protomers
• don’t want to miss any “plausible” protomers
• we must adjust our preconceptions regarding “plausible” but … we must still consider the energy required for the protomeric transforms; i.e., we must not consider energetically implausible protomers
• we need to consider protomers within a user-specified E-window, analogous to the E-window concept used when considering conformers
• meanwhile, use heuristics (rules)
– most programs use relatively simple heuristics
– ProtoPlex uses very detailed heuristics
{{coming soon!}} {{was coming soon }}
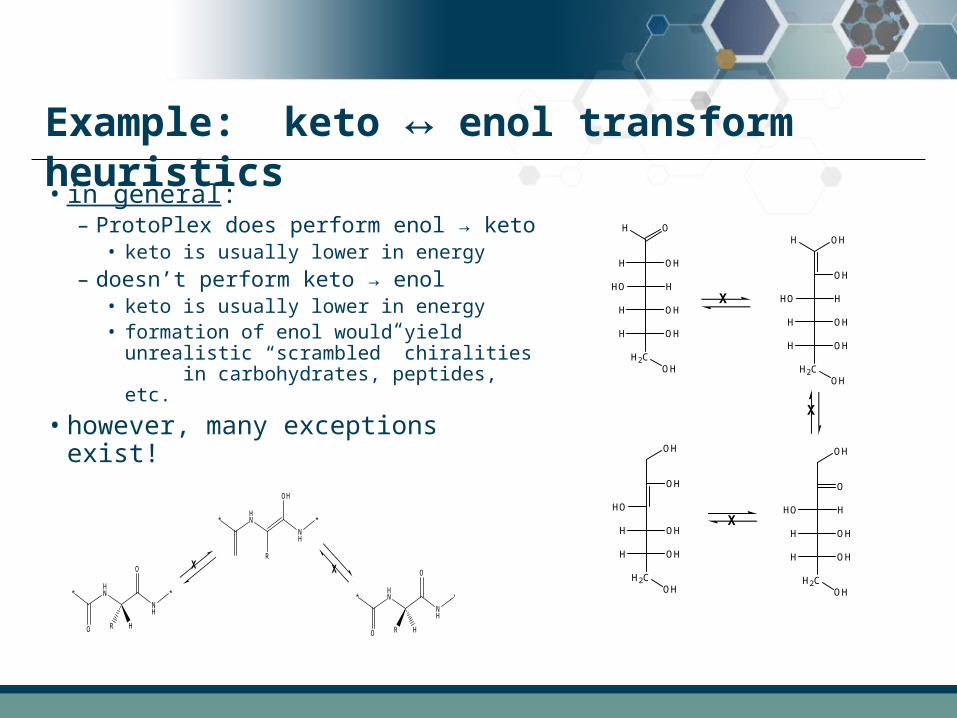
Example: keto ↔ enol transform heuristics
• in general:– ProtoPlex does perform enol → keto
• keto is usually lower in energy
– doesn’t perform keto → enol• keto is usually lower in energy• formation of enol would yield
unrealistic “scrambled” chiralities in carbohydrates, peptides, etc.
• however, many exceptions exist!
H2C
OH
H OH
HO H
H OH
H OH
OH
X
H2C
OHH
OH
HO H
H OH
H OH
OH
H2C
OH
O
HO H
H OH
H OH
OH
H2C
OH
OH
HO
H OH
H OH
OH
X
X
NH
*
O X
HN
O
*
HR
NH
*
OH
HN*
R
NH
*
O
HN
O
*
HR
X
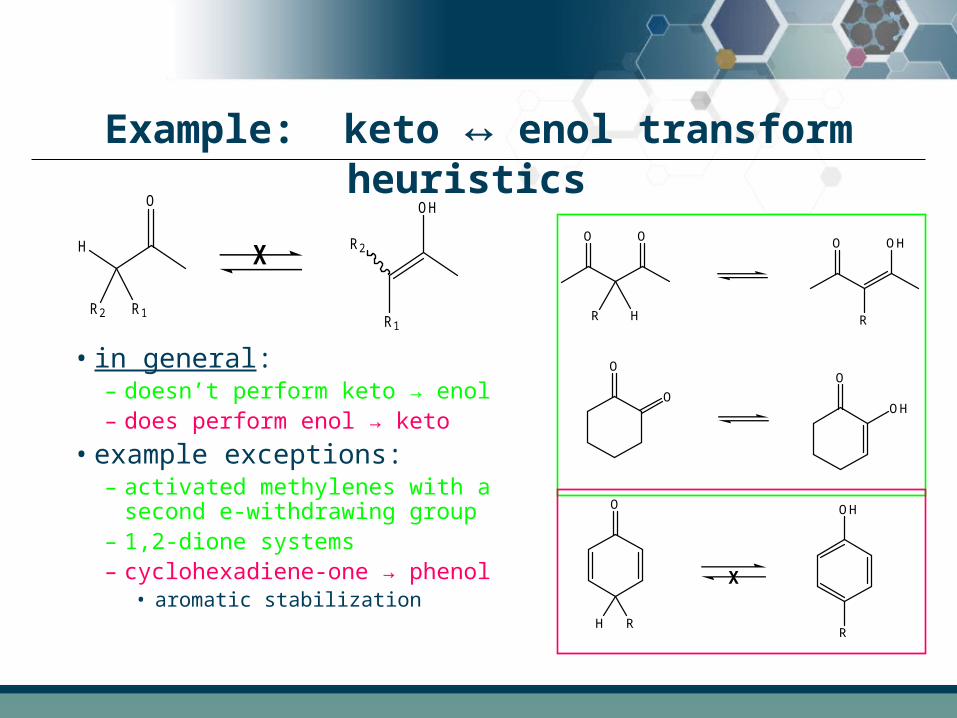
Example: keto ↔ enol transform heuristics
O
R2
OH
R1
H X
R1
R2O O O OH
R
O
O
O
OH
O
H R
OH
R
X
HR
• in general:– doesn’t perform keto → enol– does perform enol → keto
• example exceptions: – activated methylenes with a
second e-withdrawing group– 1,2-dione systems– cyclohexadiene-one → phenol
• aromatic stabilization

CH2
OOH
X
O
OH
OH
O
O
O
NR
OH
OH
NR
O
O
CH2
O
OR
X
X
Example: keto ↔ enol transform heuristics
• assessing influence of aromatic stabilization requires attention to numerous details; e.g., …
− ProtoPlex doesn’t output “keto” tautomers of phenols but …
− ProtoPlex does output “keto” tautomers of most (but not all!) hydroxy furans and pyrroles
• aromatic stabilization is less than for phenyl compounds
• amide or ester resonance stabilizes “keto” form
• assessing aromaticity itself is also quite complex
− goes far beyond “4n+2 rule”

Protomer enumeration is a non-trivial task!
• Collaborator mentioned that the “tautomer enumerator” they were using found “all 55,251 tautomers” of
H2N
N
N
NH2
N
N
NH
N
N
NH2
N
N
NH
N
N
NH2
N
N
NH2NN
N
NN
N
NN
N
Nc1nc2nc(N)nc3nc(Nc4nc5nc(N)nc6nc(Nc7nc8nc(N)nc9nc(N)nc(n7)n98)nc(n4)n56)nc(n1)n23

Protomer enumeration is a non-trivial task!
• Collaborator mentioned that the “tautomer enumerator” they were using found “all 55,251 tautomers” of
H2N
N
N
NH2
N
N
NH
N
N
NH2
N
N
NH
N
N
NH2
N
N
NH2NN
N
NN
N
NN
N
Nc1nc2nc(N)nc3nc(Nc4nc5nc(N)nc6nc(Nc7nc8nc(N)nc9nc(N)nc(n7)n98)nc(n4)n56)nc(n1)n23
H2N
N
N
NH2
N
N
NH
N
N
NH2
N
N
NH
N
N
NH2
N
N
NH2NN
N
NN
N
NN
N

H2N
N
N
NH2
N
N
NH
N
N
NH2
N
N
NH
N
N
NH2
N
N
NH2NN
N
NN
N
NN
N
Nc1nc2nc(N)nc3nc(Nc4nc5nc(N)nc6nc(Nc7nc8nc(N)nc9nc(N)nc(n7)n98)nc(n4)n56)nc(n1)n23
Protomer enumeration is a non-trivial task!
• Collaborator mentioned that the “tautomer enumerator” they were using found “all 55,251 tautomers” of
H2N
N
HN
NH
N
N
NH
N
N
NH2
N
N
NH
HN
N
NH
NH
N
NHNN
N
NN
N
NN
N

Protomer enumeration is a non-trivial task!
• Collaborator mentioned that the “tautomer enumerator” they were using found “all 55,251 tautomers”
• ProtoPlex correctly found only 1 tautomer (!)
• high-level ab initio MO, X-ray, and NMR results all indicate aromatic structure (D3h symmetry) and only 1 tautomer– B. Jurgens et al., J. Am. Chem. Soc., 125, 10288-10300, 2003
• tautomeric transforms which would disrupt aromaticity are highly unlikely due to loss of “aromatic stabilization” (delocalization) energy
• however, definition of “aromatic” is non-trivial and must go far beyond simply checking for “4n+2” compliance

(Outdated) Performance Benchmarks
• performed in September, 2004, on Intel / P4-1.3GHz– distributed processing could have been used but was not
• used MDDR-2000 106,592 entries (“compounds”)
• generate canonically unique representation of compound– 3.53 min, 0.002 sec/cmpd
• found 4,680 pairs of “structural duplicates”– different salt forms (e.g., Cl- vs Br-) of same charged “primary fragment”
• found 233 pairs of protomeric duplicates– 43 due to different tautomer structures used for the same compound

Example duplicates found via OSN representation
NNH
N
S
OCH3O
NN
HN
S
OCH3O
vs.
• tautomeric duplicates:
N
NH
S
O
N
N
HS
O
vs.
N
O
N
N
ONH2
O
Cl
N
HO
N
N
ONH2
O
Cl
vs.

(Outdated) Performance Benchmarks
• performed in September, 2004, on Intel / P4-1.3GHz– distributed processing could have been used but was not
• used MDDR/2000 106,592 entries (“compounds”)
• generate canonically unique representation of compound– 3.53 min, 0.002 sec/cmpd
• found 4,680 pairs of “structural duplicates”– different salt forms (e.g., Cl- vs Br-) of same charged “primary fragment”
• found 233 pairs of protomeric duplicates– 43 due to different tautomer structures used for the same compound
– 190 due to different protonation-state and, possibly, tautomeric-state
• generate all “plausible” proto-stereomers of 101,679 “pri frags”– 30.5 min, 0.018 sec/cmpd (including file-output in smiles format)

Thoughts about chemical DB hierarchy
• performed in September, 2004, on Intel / P4-1.3GHz– distributed processing could have been used but was not
• used MDDR/2000 106,592 entries (“compounds”)
• generate canonically unique representation of compound– 3.53 min, 0.002 sec/cmpd
• found 4,680 pairs of “structural duplicates”– different salt forms (e.g., Cl- vs Br-) of same charged “primary fragment”
• found 233 pairs of protomeric duplicates– 43 due to different tautomer structures used for the same compound
– 190 due to different protonation-state and, possibly, tautomeric-state
• generate all “plausible” proto-stereomers of 101,679 “pri frags”– 30.5 min, 0.018 sec/cmpd (including file-output in smiles format)

• compounds exhibit various “bulk” properties which depend on their “bulk composition” -- i.e., crystal structure and crystal size -- for example ...
– melting point, dissolution rate, solubility, powder flow ...
• once crystals “dissociate” into “molecular” (or ionic) species, cmpds can exhibit “molecular” properties which don’t depend on their “secondary fragment(s)”
– receptor binding, logP from buffered donor phase, ...
• a given “primary fragment” can exist as different protomeric states in different compounds -- for example ...
– dextromethorphan (DXM), DXM.HCl, DXM.HBr, ...
Thoughts about chemical DB hierarchy

• a given “primary fragment” can exist as different protomeric states in different compounds -- for example ...
– dextromethorphan (DXM), DXM.HCl, DXM.HBr, ...
• therefore … improved cheminformatic hierarchy might be:– primary fragment -- which? -- unionized form (e.g. DXM) seems best
– compound(s) -- DXM, DXM.HCl, DXM.HBr, ...
– structures(s) (2D) -- DXM, DXMH+, proto-stereomers thereof
– conformations (3D) -- proto-stereo-conformers
• already supported in ProtoPlex, StereoPlex, Confort …
Thoughts about chemical DB hierarchy

• therefore … improved cheminformatic hierarchy might be:– primary fragment, pf-ID -- (e.g. DXM)
• cmpd1-ID -- (e.g. DXM)– data-type1 re: cmpd-1 (e.g. MW)
– data-type2 re: cmpd-1 (e.g. Solubility @ 25° C)
• cmpd2-ID -- (e.g. DXM.HBr)– data-type1 re: cmpd-2
– data-type2 re: cmpd-2
• struct1-ID (2D proto-stereomer) (e.g. DXMH+, “S”-chirality)– data-type3 re: struct1-ID (e.g. coordinate on axis 4 of corporate std. chem. space)
– conf1,struct1-ID (3D proto-stereo-conformer) (e.g. Concord geometry)
» data-type15 re: conf1,struct1-ID (e.g. binding energy from method abc)
• struct2-ID (2D proto-stereomer) (e.g. DXMH+, “R”-chirality)– data-type7 re: struct2-ID (e.g. polar surface area from method xyz)
– conf1,struct2-ID (3D proto-stereo-conformer) (e.g. MMF geometry)
» data-type15 re: conf1,struct2-ID (e.g. binding energy from method abc)
Thoughts about chemical DB hierarchy

Outline
• introductory comments
• motivating factors
• terminology
• software / implementation
• summary

• it seems so obvious ...– if CAMD doesn’t use same structures as used by Mother Nature,
we greatly reduce the chance of making reliable predictions
– if we go to the trouble of performing calculations and predictions based on structures, it seems silly not to store the results in an easily retrievable manner
• the fundamental technology required already exists
• pharmaceutical industry is already moving in this direction– increasing emphasis and reliance on vHTS and QSAR methods– increasing concern regarding IP issues and competitive strategies
• former Optive collaborators already using NW components • some barriers to broad adoption/implementation but those
barriers are certainly not insurmountable
Summary
... let’s do it!

Thanks for your attention!

Thank you very much!
• nominators(??) and Award Committee
• sponsors (reception, etc.)
• Graham Douglas & Finance Committee
• Osman Guner and Andy Rusinko for organizing Symposium
• Gunther Grethe for very nice press release published in CINF Bulletin (but, regrettably, “chopped” by some junior editor at C&E News)
• MANY Lab colleagues and industrial collaborators without whose contributions I would not be receiving this reward

Thank you very much!• Karl Smith, Brian Masek, Yubin Wu
– former colleagues and coauthors on “my” presentation
• Andrew Rusinko, Jeffrey Skell, Eugene Stewart, Brian Masek
– former colleagues making other presentations in this symposium
• Felix Deanda, Renzo Balducci, Hongyao Zhu, Raed Khashan
– other former colleagues of particular significance
• Uta Lessel, Dora Schnur, Jim Damewood
– former industrial collaborators and presenters in this symposium
• this Award recognizes their contributions as well as my own
• also ... thanks to Martin Grigorov and Stan Young for “rounding out” the Symposium program


















