
Qualification / Validation Qualification / Validation - °KEV
28
GMP-Conference / 10.-11. June 2004 / Istanbul 1 Andreas Brutsche Novartis Switzerland GMP Conference, 11.06.04, Istanbul Qualification / Validation Quality Risk Assessment in the Pharmaceutical Industry - Challenges and Opportunities -
Transcript of Qualification / Validation Qualification / Validation - °KEV

GMP-Conference / 10.-11. June 2004 / Istanbul 1
Andreas BrutscheNovartis Switzerland
GMP Conference, 11.06.04, Istanbul
Qualification / ValidationQuality Risk Assessment in the
Pharmaceutical Industry- Challenges and Opportunities -
Qualification / ValidationQuality Risk Assessment in the
Pharmaceutical Industry- Challenges and Opportunities -

GMP-Conference / 10.-11. June 2004 / Istanbul 2
Qualification, Validation, CalibrationQualification, Validation, Calibration
Visible Process
UnvalidatedProcess
New System
Validation/Qualification Iceberg

GMP-Conference / 10.-11. June 2004 / Istanbul 3
Validation / Qualification Approach
User RequirementSpecifications
Functional Specifications
DesignSpecifications
System Build
InstallationQualification
OperationalQualificaton
PerformanceQualificationrelated to
related to

GMP-Conference / 10.-11. June 2004 / Istanbul 4
Quality Risk Assessment in the Pharmaceutical IndustryQuality Risk Assessment in the Pharmaceutical Industry
Risk Assessment has become a key concept in the decision making process of today’s pharmaceutical industry. Its impact affects many disciplines including:
EngineeringManufacturingProject ManagementQualitySafetyEnvironmental
GMP-Conference / 10.-11. June 2004 / Istanbul 5
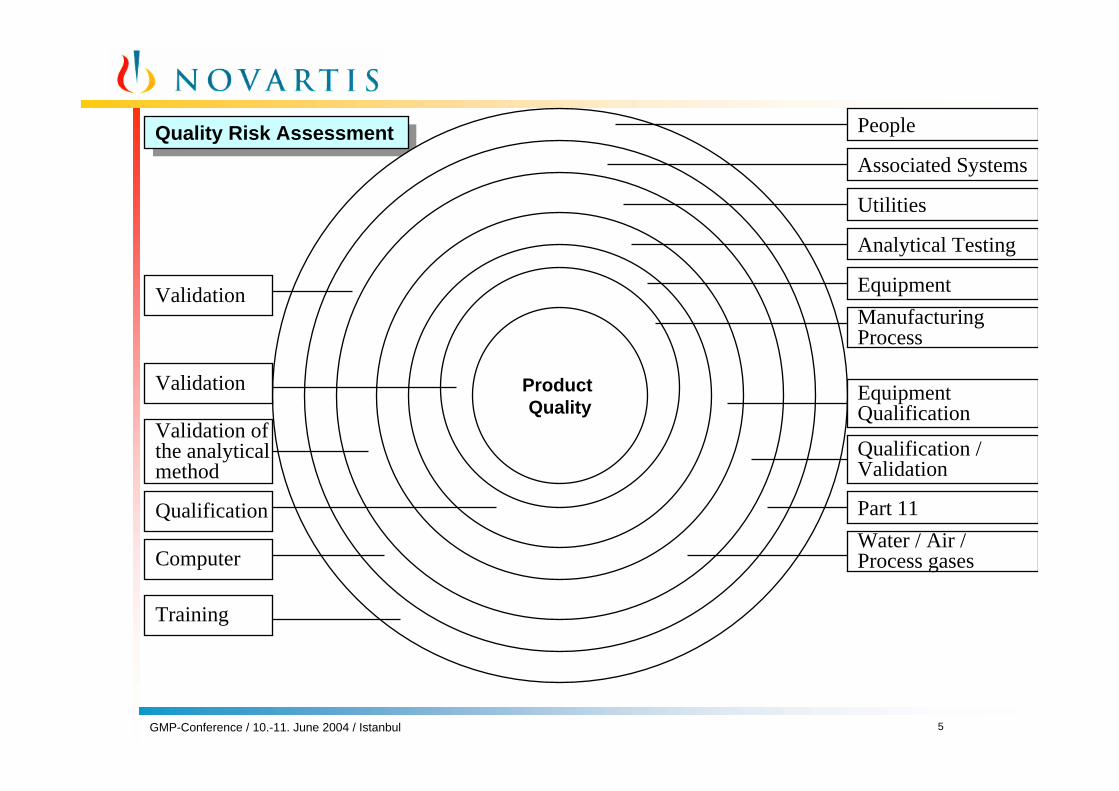
Quality Risk AssessmentQuality Risk Assessment
Product Quality
Associated Systems
Utilities
Analytical Testing
EquipmentManufacturingProcess
EquipmentQualification
Qualification /Validation
Part 11Water / Air /Process gases
Validation
Validation ofthe analytical method
Validation
Qualification
People
Computer
Training

GMP-Conference / 10.-11. June 2004 / Istanbul 6
Risk Assessment:Risk Assessment:
Each step in a process is assessed on its influence on product quality.
• Critical steps have to be validated/qualified
• Uncritical steps no activities are required

GMP-Conference / 10.-11. June 2004 / Istanbul 7
Equipment QualificationEquipment Qualification
• User Requirement Specifications (URS)• Design Qualification
• Conceptual Design• Basic Design• Detail Design
• Installation Qualification

GMP-Conference / 10.-11. June 2004 / Istanbul 8
Equipment QualificationEquipment Qualification
• Operational Qualification
• Performance Qualification
Calibration and Maintenance Programs
Retrospective Qualification

GMP-Conference / 10.-11. June 2004 / Istanbul 9
Equipment QualificationEquipment Qualification
Design Qualification
“Defining the quality parameters required of the equipment and manufacturer”

GMP-Conference / 10.-11. June 2004 / Istanbul 10
Equipment QualificationEquipment Qualification
Installation Qualification
“Assurance that the intended equipment is received as designed and specified.”

GMP-Conference / 10.-11. June 2004 / Istanbul 11
Equipment QualificationEquipment Qualification
Operational Qualification
“Confirmation that the equipment functions as specified and operates correctly.”

GMP-Conference / 10.-11. June 2004 / Istanbul 12
Equipment QualificationEquipment Qualification
Performance Qualification
“Confirmation that the equipment consistently continues to perform as required”.

GMP-Conference / 10.-11. June 2004 / Istanbul 13
Validation
To prove that a process works is, in a nutshell, what we mean by the verb to
validate.
E. Frey, FDA

GMP-Conference / 10.-11. June 2004 / Istanbul 14
Validation
Process ValidationCleaning Validation Computer system ValidationValidation of Analytical MethodsPart 11 Compliance
Risk based approach is key !!!Focus on Product, not only on technology

GMP-Conference / 10.-11. June 2004 / Istanbul 15
Link between Validation/ Qualification
Risk Analysis Equipment Qualification
Risk Analysis Process Validation
Product
GMP-Conference / 10.-11. June 2004 / Istanbul 16
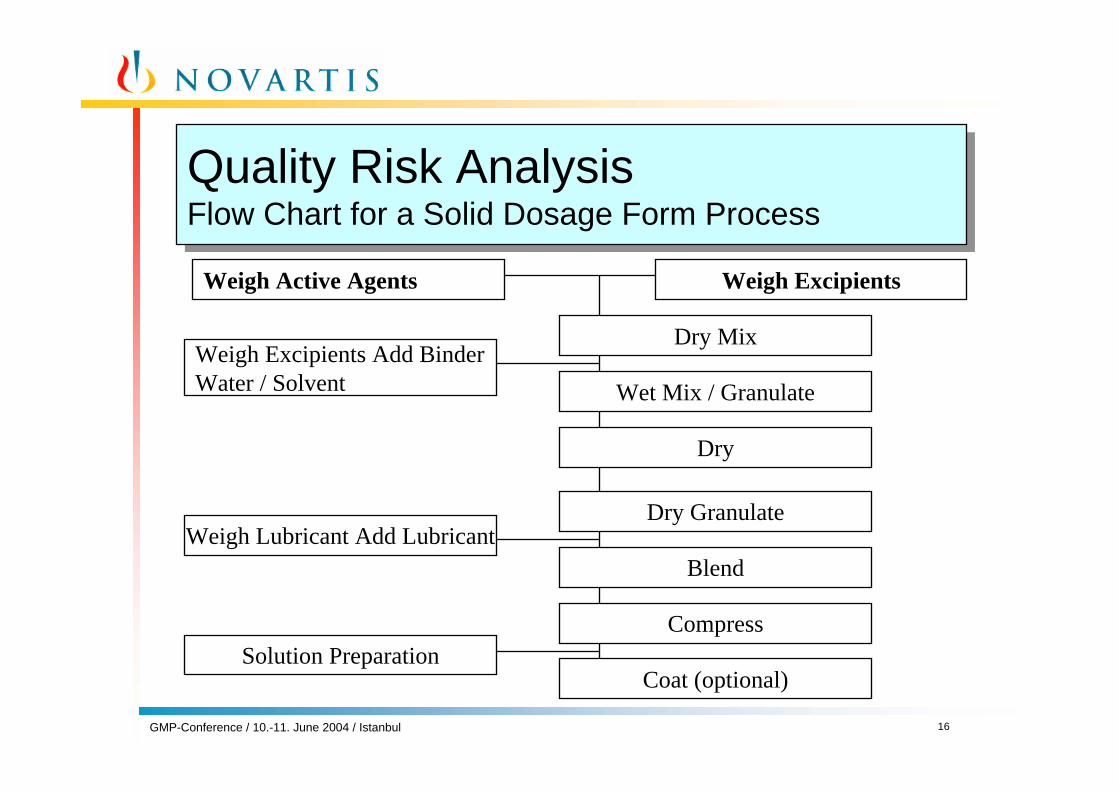
Quality Risk AnalysisFlow Chart for a Solid Dosage Form ProcessQuality Risk AnalysisFlow Chart for a Solid Dosage Form Process
Weigh Active Agents
Weigh Excipients Add Binder Water / Solvent
Weigh Lubricant Add Lubricant
Solution Preparation
Weigh Excipients
Dry Mix
Wet Mix / Granulate
Dry
Dry Granulate
Blend
Compress
Coat (optional)

GMP-Conference / 10.-11. June 2004 / Istanbul 17
Quality Risk AnalysisKey ParametersQuality Risk AnalysisKey ParametersProcess Stage Parameter Worst Case Values___________________________________________________________________________________________________________________________________________
__________________
Raw materials Particle size Min-Max valuesActive agent Surface areaWet mixing Binder temperature Target +/- x°C
Binder volume Min-Max used in Pilot ScaleMixing time Normal time +/- x minutes
Drying Granule moisture Upper-lower in process specBlending Granule particle size Upper-lower in process specCompression Compressor speed Normal production range
(e.g. 100000/hr-150000/hr)Pre-compressionpressure Min-Max
Coating Inlet air temperature Target + / - xSpray rate target rate + / - x

GMP-Conference / 10.-11. June 2004 / Istanbul 18
Quality Risk AnalysisCritical Process ParameterQuality Risk AnalysisCritical Process Parameter
Parameters: Colette gral mixerAgitator - Speed 2Chopper - offMixing time - 5 minutes
Sampling After mixing take 12 samples from the mixing bowl using a Regimen: sample thief. Samples to be taken 4 top, 4 middle, 4
bottom. Sample to be 300 mg approx.
Testing: Analyse each sample for active agent content
Acceptance All individual values to be within + 7 - 10 % of the group Critemean RSD to be less than 6 %. The mean to be within + 7 - 5 % of the theoretical value
______________________________________________________________________
Solid dosage form

GMP-Conference / 10.-11. June 2004 / Istanbul 19
Tool boxRisk AssessmentTool boxRisk Assessment
• FMEA (= Failure Mode and Effect Analysis)
• HACCP (= Hazard Analysis Critical Control Points)
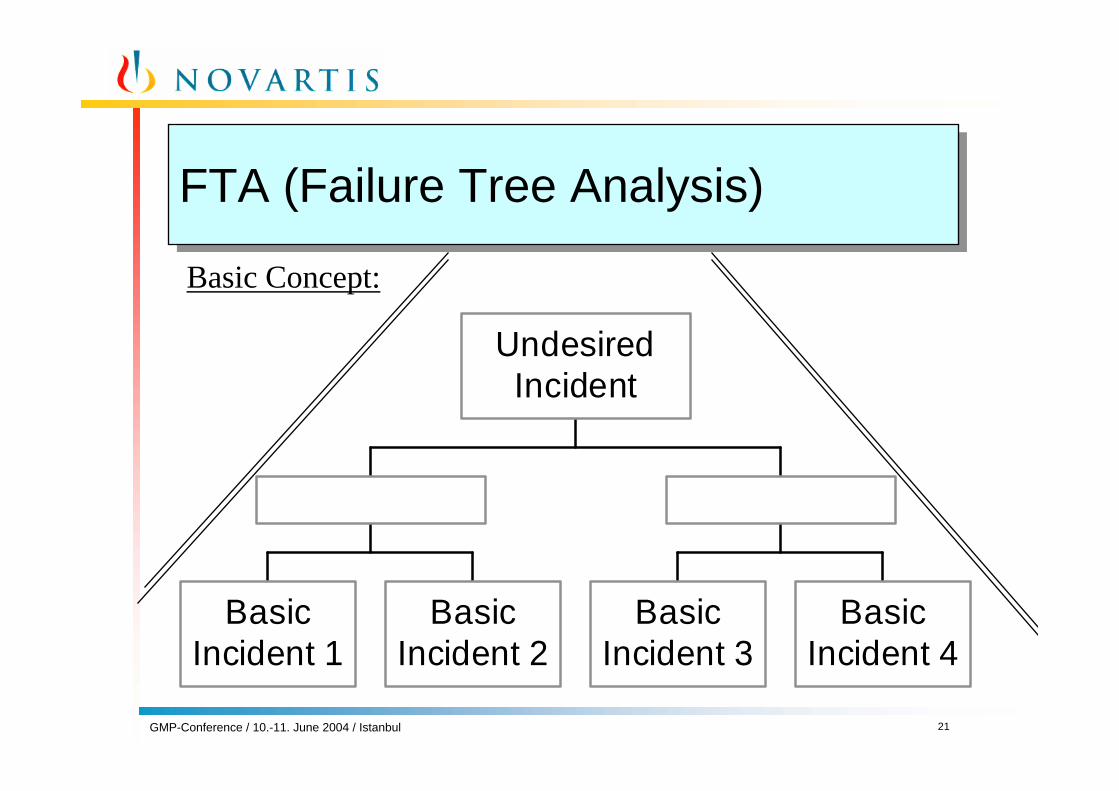
• FTA (= Failure Tree Analysis)
• System Kepner-Tregoe• Analysis of the situation• Analysis of the problem • Analysis of the decisions• Analysis of potential problems

GMP-Conference / 10.-11. June 2004 / Istanbul 20
Tool boxRisk AssessmentTool boxRisk Assessment
• FMEA (Failure Mode and Effect Analysis)
→ Define the “Risk Priority number” ( - RPN)Example: Steam sterilisation Process
- Steampressure / Temperature in the autoclave- Sterilisation Time- Measures to avoid air in the autoclave- Treatment of the product before and after the
process
Risk
decreasing
GMP-Conference / 10.-11. June 2004 / Istanbul 21
FTA (Failure Tree Analysis)FTA (Failure Tree Analysis)
BasicIncident 1
BasicIncident 2
BasicIncident 3
BasicIncident 4
UndesiredIncident
Basic Concept:

GMP-Conference / 10.-11. June 2004 / Istanbul 22
Key Success Factor:
Training and Skills of people
• Scientific skills
• Broad - Experience in pharmaceutical Industry
• Leadership, Responsibilities
Risk AssessmentRisk Assessment

GMP-Conference / 10.-11. June 2004 / Istanbul 23
Change / DeviationChange / Deviation
“Change” usually refers to a planned alteration which is documented and considered before implementation. Normally, changes are either permanent or have a fixed period of validity.
“Deviation” represents a change which may occur for a variety of reasons during operations, or may be observed to have happened after the operation.
Normally, deviations are not permanent and represent single events“Planned Deviations” ??

GMP-Conference / 10.-11. June 2004 / Istanbul 24
Documentation
Validation Master PlanQualification Master PlanGMP Risk AnalysisValidation ProtocolTest protocol (including specification)Validation ReportSummary of Deviations / Issues

GMP-Conference / 10.-11. June 2004 / Istanbul 25
Change ManagementChange Management
Who made the changeWhy was the change necessary
When was the change implementedWhat international consequences arise:- Risk / benefit assessments- Cost / benefit assessments- Regulatory impact
Who gave approvals__________________________________________________________________________________________________________________________________________________
Changes are necessary - Uncontrolled changes are dangerous

GMP-Conference / 10.-11. June 2004 / Istanbul 26
Computer Simulations in Conventional Clean Rooms (Sterile Manuf.)
To be in compliance with all microbiological and particulate requirements, the design of the sterile facilities plays the most important role.Computer simulation studies of air flows should be used to analyse the most critical areas.Detailed analysis was made with regard to conflicting areas of class 10`000 and critical class 100 areas in order to prevent any possible contamination of the product.Detailed qualification of the balance is a prerequisite for a successful implementation of these design concepts.
5
GMP-Conference / 10.-11. June 2004 / Istanbul 27
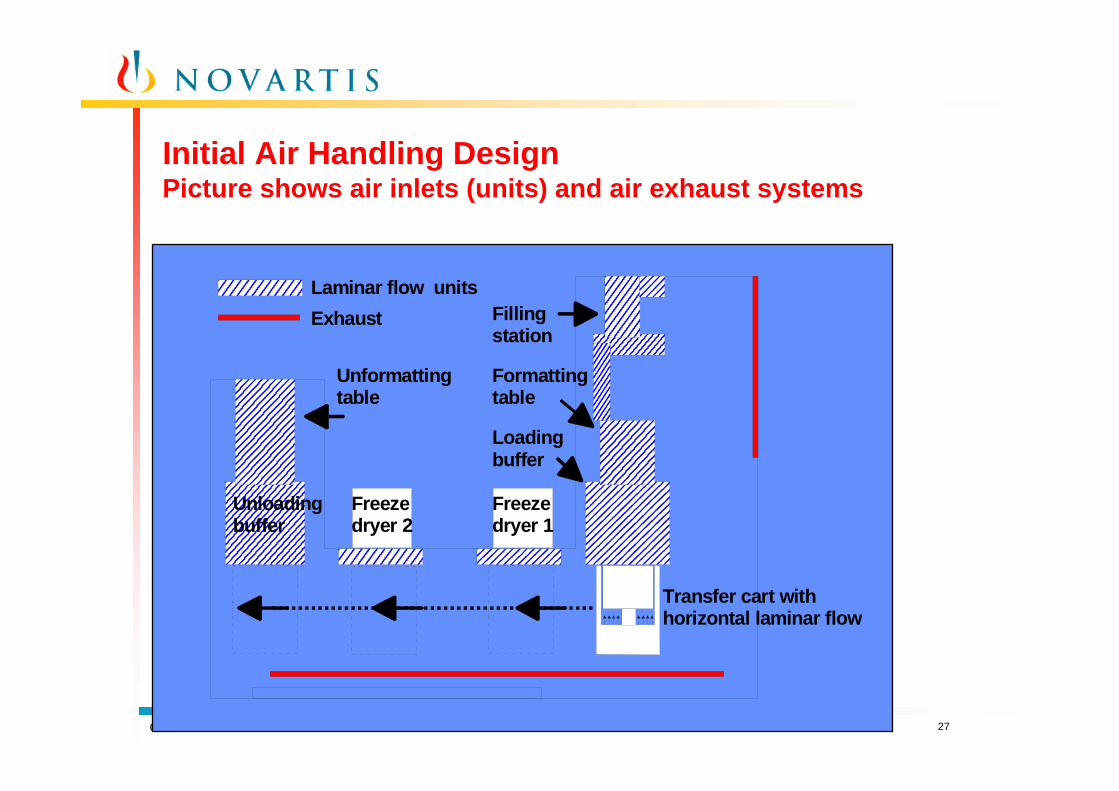
Initial Air Handling DesignPicture shows air inlets (units) and air exhaust systems
Loading buffer
Transfer cart withhorizontal laminar flow
Freezedryer 1
Freezedryer 2
Unloading buffer
Formatting table
Filling station
Unformatting table
Laminar flow unitsExhaust

GMP-Conference / 10.-11. June 2004 / Istanbul 28
Improved Air Handling Design Picture shows new and modified air inlets (LF units) and air exhaust systems
New exhaust
New exhaustson cart
New exhaust
Modified exhaust
New exhaust
New exhaust
New exhaust
New exhaust
Modified inlet on cart
Additional laminar flow units
Modified exhaust
New inlets (background area)


















