
QUÁ TRÌNH THOÁT CỦA PROTEIN MỚI SINH TẠI ĐƯỜNG HẦM …
41
BỘ GIÁO DỤC VIỆN HÀN LÂM KHOA HỌC VÀ ĐÀO TẠO VÀ CÔNG NGHỆ VIỆT NAM HỌC VIỆN KHOA HỌC VÀ CÔNG NGHỆ ----------------------------- Phạm Thị Thơm QUÁ TRÌNH THOÁT CỦA PROTEIN MỚI SINH TẠI ĐƯỜNG HẦM THOÁT RIBOSOME LUẬN VĂN THẠC SĨ VẬT LÝ Hà Nội - 2019
Transcript of QUÁ TRÌNH THOÁT CỦA PROTEIN MỚI SINH TẠI ĐƯỜNG HẦM …

BỘ GIÁO DỤC VIỆN HÀN LÂM KHOA HỌC
VÀ ĐÀO TẠO VÀ CÔNG NGHỆ VIỆT NAM
HỌC VIỆN KHOA HỌC VÀ CÔNG NGHỆ
-----------------------------
Phạm Thị Thơm
QUÁ TRÌNH THOÁT CỦA PROTEIN MỚI SINH
TẠI ĐƯỜNG HẦM THOÁT RIBOSOME
LUẬN VĂN THẠC SĨ VẬT LÝ
Hà Nội - 2019

BỘ GIÁO DỤC VIỆN HÀN LÂM KHOA HỌC
VÀ ĐÀO TẠO VÀ CÔNG NGHỆ VIỆT NAM
HỌC VIỆN KHOA HỌC VÀ CÔNG NGHỆ
-----------------------------
Phạm Thị Thơm
QUÁ TRÌNH THOÁT CỦA PROTEIN MỚI SINH
TẠI ĐƯỜNG HẦM THOÁT RIBOSOME
Chuyên ngành: Vật lý lý thuyết và vật lý toán
Mã số: 8440103
LUẬN VĂN THẠC SĨ VẬT LÝ
NGƯỜI HƯỚNG DẪN KHOA HỌC :
Hướng dẫn : PGS. TS. Trịnh Xuân Hoàng
Hà Nội - 2019

Lời cam đoan
Tôi xin cam đoan những gì viết trong luận văn là do sự tìm tòi, học hỏi
của bản thân và sự hướng dẫn tận tình của Thầy - PSG.TS Trịnh Xuân Hoàng.
Mọi kết quả nghiên cứu cũng như ý tưởng của tác giả khác, nếu có đều được
trích dẫn cụ thể. Đề tài luận văn này cho đến nay chưa được bảo vệ tại bất kỳ
một hội đồng bảo vệ luận văn thạc sĩ nào và cũng chưa hề được công bố trên
bất kỳ một phương tiện nào. Tôi xin chịu trách nhiệm về những lời cam đoan
trên
Hà Nội, tháng 04 năm 2019
Tác giả luận văn
Phạm Thị Thơm

Lời cảm ơn
Trước khi trình bày nội dung chính của khóa luận, em xin bày tỏ lòng
biết ơn sâu sắc tới Thầy – PGS.TS. Trịnh Xuân Hoàng, người đã giúp đỡ em
hoàn thành khóa luận này. Thầy đã tận tình chỉ bảo cung cấp cho em nhiều
kiến thức quan trọng cũng như đã truyền đạt cho em những kinh nghiệm quý
báu trong quá trình học tập và nghiên cứu.
Em cũng xin bày tỏ lòng biết ơn chân thành tới toàn thể các thầy cô
giáo Học viện Khoa học Công nghệ đã dạy bảo em tận tình trong suốt quá
trình học tập.
Em thực sự cảm ơn gia đình, bạn bè đã luôn bên em, cổ vũ, động viên,
giúp đỡ em trong suốt quá trình học tập và thực hiện khóa luận tốt nghiệp.
Hà Nội, tháng 04 năm 2019
Học viên
Phạm Thị Thơm

Danh mục các hình vẽ, đồ thị
1.1 (a) Cấu trúc chung của amino acid, (b) Cấu trúc của proline……..
1.2 Chuỗi polypeptide…………………………………………………
1.3 Biểu diễn mạch xương sống của các cấu trúc bậc hai phổ biến của
protein: (a) 2RJX xoắn 𝛼 và (b) 1SHG phiến β…………………..
1.4 Cấu trúc xoắn alpha………………………………………………
3.1 Cấu trúc trạng thái tự nhiên của protein SH3 domain (1SHG) và
protein Villin (2RJX)……………………………………………..
3.2 Sự phụ thuộc của năng lượng (E) và số hạt thoát ra ngoài đường
hầm (Nout)vào thời gian trong một quỹ đạo mô phỏng cho protein
1SHG tại nhiệt độ T = 0.8 /kB……………………………………
3.3 Phân bố thời gian thoát của protein 1SHG tại 3 nhiệt độ khác
nhau. Các biểu đồ thu được từ dữ liệu mô phỏng. Đường liền nét
là hàm phân bố thu được bởi lý thuyết khuyếch tán 1 chiều được
khớp (fit) với biểu đồ…………………………………………….
3.4 Phân bố thời gian thoát của protein 2RJX tại 3 nhiệt độ khác
nhau. Các biểu đồ thu được từ dữ liệu mô phỏng. Đường liền nét
là hàm phân bố thu được bởi lý thuyết khuyếch tán 1 chiều được
khớp (fit) với biểu đồ…………………………………………….
3.5 Sự phụ thuộc của độ lệch chuẩn của thời gian thoát vào thời gian
thoát trung bình cho 2protein 1SHG và 2RJX. Đường đứt nét
tương ứng với mô hình khuyếch tán 1 chiều với k không đổi….
3.6 Sự phụ thuộc của thời gian thoát trung bình vào nhiệt độ cho
protein 1SHG (a) và protein 2RJX (b) trên thang tọa độ log-log.
Đường đứt nét có độ dốc bằng −1 tương ứng với 𝜇𝑇 ∼ 𝑇−1……...

3.7 Sự phụ thuộc của thời gian thoát trung bình (t) vào nhiệt độ (T)
cho 2 protein 1SHG và 2RJX……………………………………


1
MỤC LỤC
Mở đầu
Chương 1: Tổng quan về quá trình cuốn của các protein mới sinh
1.1. Thành phần hóa học và cấu trúc của protein
1.2. Hiện tượng cuốn protein trong ống nghiệm
1.3. Quá trình tổng hợp protein trong tế bào
1.4. Quá trình cuốn đồng dịch mã của protein
1.5. Đường hầm thoát ribosome
1.6. Ảnh hưởng của đường hầm thoát ribosome lên quá trình cuốn của
các protein mới sinh
Chương 2: Các mô hình và phương pháp mô phỏng
2.1. Mô hình Go cho protein
2.2. Mô hình đường hầm thoát ribosome
2.3. Phương pháp động lực học phân tử dựa trên phương trình Langevin
2.4. Mô hình khuyếch tán cho quá trình thoát protein
Chương 3: Một số kết quả nghiên cứu
3.1. Sự phụ thuộc của thời gian thoát của protein vào nhiệt độ.
3.2. Sự phụ thuộc của thời gian thoát vào cấu trúc của protein.
Chương 4: Kết luận và kiến nghị
Tài liệu tham khảo

2
MỞ ĐẦU
Nghiên cứu về vật lý các phân tử sinh học hiện nay đang là một hướng
nghiên cứu nổi bật trong các lĩnh vực khoa học liên ngành do vai trò đặc biệt
quan trọng của các phân tử sinh học đối với cơ thể sống và khả năng mang lại
những ứng dụng trong y sinh học. Vật lý các phân tử sinh học áp dụng các
phương pháp và dựa trên các nền tảng vật lý để nghiên cứu các vấn đề sinh
học nhằm mang lại những hiểu biết căn bản cũng như những tiên đoán mang
tính định lượng về các hệ sinh học. Bên cạnh các phương pháp thực nghiệm
tiên tiến, thì các phương pháp mô hình hóa và mô phỏng máy tính đang được
sử dụng rất rộng rãi trong vật lý sinh học phân tử và là các công cụ rất hiệu
quả, giúp tiết kiệm thời gian và chi phí nghiên cứu.
Trong sinh học phân tử thì việc nghiên cứu về protein rất được quan tâm
bởi lẽ protein là một thành phần cơ bản của vật chất sống, chúng có mặt trong
thành phần nhân và tế bào chất của mọi tế bào [1]. Cấu trúc phức tạp và đa
dạng cho phép các protein thực hiện rất nhiều chức năng. Tế bào xâu chuỗi 20
loại amino acid thành mạch thẳng, tạo nên protein. Protein có thể đóng vai trò
cấu trúc, ví dụ tạo thành khung tế bào. Protein với khả năng biến đổi hình
dạng khi nhiệt độ, nồng độ ion hoặc các tính chất khác của tế bào thay đổi có
thể giữ vai trò cảm biến. Protein có thể hấp thu và bài xuất vật chất qua màng
tế bào. Chúng có thể là enzyme, chất xúc tác giúp phản ứng hóa học xảy ra
nhanh hơn rất nhiều. Chúng có thể là phân tử tín hiệu ngoại bào (do một tế
bào giải phóng ra để giao tiếp với các tế bào khác) hoặc phân tử tín hiệu nội
bào (mang thông tin bên trong tế bào). Chúng có thể là mô-tơ sử dụng năng
lượng hóa học (ATP) để vận động quanh phân tử khác.
Thông tin mỗi loại protein được tạo ra như thế nào, khi nào và ở đâu nằm
trong vật liệu di truyền, axit deoxyribonucleic (DNA). Cấu trúc lập thể của
DNA chứa hai mạch xoắn, cuộn quanh một trục chung, tạo thành cấu trúc
xoắn kép. Chuỗi DNA cấu thành từ các đơn phân gọi là nucleotide (còn được
gọi là bazơ do chứa bazơ hữu cơ mạch vòng). Trong DNA có bốn loại
nucleotide khác nhau bởi các gốc nucleobase là Adenine (viết tắt là A),
Thymine (viết tắt là T), Cytosine (tiếng Việt còn gọi là Xytosine, viết tắt

3
là X hoặc C), và Guanine (viết tắt là G). Ba nucleotide liên tiếp trên mạch mã
gốc DNA của gene, sẽ quy định một loại amino acid nhất định. Do đó, mã di
truyền còn được gọi là mã bộ ba, và tổ hợp ba nucleotide được gọi là một bộ
ba mã hoá.. Các bộ ba nucleotide trong mỗi mạch đơn của chuỗi xoắn kép
DNA khi giản phân, là một tổ hợp của 3 trong bốn loại nucleotide này nối liền
nhau trong mạch DNA sao cho phần bazơ nhô ra từ khung mạch xoắn. DNA
xoắn kép có cấu trúc khá đơn giản: A trên một mạch bắt cặp với T trên mạch
kia và G bắt cặp với C. Hai mạch bắt cặp bổ sung mạnh đến mức sau khi bị
tách rời chúng sẽ tự gắn lại với nhau nếu điều kiện nhiệt độ và nồng độ muối
cho phép. DNA mang thông tin di truyền trong trật tự nucleotide dọc theo
mạch thẳng. Phần DNA mang thông tin được chia thành các đơn vị chức năng
nằm phân tán gọi là gene. Độ dài của một gene điển hình nằm trong khoảng
5.000 đến 100.000 nucleotide. Gene mã hóa cho protein thường gồm hai
phần: Vùng mã hóa đặc hiệu cho trình tự amino acid của protein và vùng điều
hòa kiểm soát thời điểm, vị trí tạo protein.
Tế bào sử dụng hai chuỗi quá trình liên tiếp để chuyển thông tin mã hóa
trong DNA thành protein. Chuỗi quá trình thứ nhất gọi là phiên mã: Sao chép
vùng mã hóa của gene thành axit ribonucleic (RNA) mạch đơn. Trong quá
trình này enzyme RNA polymerase sử dụng một mạch khuôn DNA để xúc tác
liên kết các nucleotide thành chuỗi RNA mới tạo ra được gia công thành phân
tử RNA thông tin ( mRNA) nhỏ hơn. mRNA sau đó chuyển tới tế bào chất,
nơi ribosome (bộ máy phân tử rất phức tạp cấu thành từ RNA và protein) thực
hiện quá trình thứ hai gọi là dịch mã. Khi dịch mã, ribosome lắp ráp và liên
kết các amino acid với nhau theo trật tự chính xác, do trình tự mRNA quy
định theo mã di truyền gần như chung cho mọi sinh giới. Ngoài vai trò truyền
thông tin từ nhân tới tế bào chất, RNA còn tham gia vào bộ máy phân tử. Ví
dụ, ribosome chứa bốn chuỗi RNA kết hợp với hơn 50 protein, tạo thành bộ
đọc mRNA và tổng hợp protein rất chính xác và hiệu quả.
Các protein được tổng hợp bởi các ribosome trong tế bào và được chiết
xuất vào trong tương bào (cytosol) hoặc lưới nội chất (endoplasmic reticulum)
trước khi được vận chuyển tới các địa chỉ khác nhau bên trong hoặc ngoài tế

4
bào. Các protein mới sinh chịu ảnh hưởng của đường hầm thoát ribosome
(ribosomal exit tunnel), là nơi protein cần phải di chuyển qua để đi vào tế bào
chất. Đường hầm thoát ribosome là một ống chật hẹp, có cấu trúc phức tạp
[2], có ảnh hưởng rất lớn đến quá trình cuốn của các protein mới sinh. Các
nghiên cứu thực nghiệm cho thấy protein có thể hình thành các cấu trúc bậc
hai (xoắn alpha và phiến beta) bên trong đường hầm thoát [3, 4]. Ngoài ra,
đường hầm thoát có thể đóng vai trò tích cực trong việc nhận dạng trình tự
amino acid cũng như điều khiển quá trình sinh tổng hợp protein [5, 6].
Các nghiên cứu mô phỏng gần đây [7, 8] cho thấy quá trình thoát của
protein tại đường hầm thoát ribosome xảy ra song song với quá trình cuốn và
hai quá trình này có ảnh hưởng qua lại lẫn nhau. Quá trình cuốn làm cho
protein thoát ra nhanh hơn, trong khi quá trình thoát xảy ra đủ chậm giúp
protein cuốn chính xác hơn. Việc protein thoát ra đủ chậm cũng như cuốn
chính xác giúp giảm thiểu khả năng kết tụ [11] của các protein mới sinh bên
trong tế bào. Ngoài ra, các kết quả mô phỏng cho thấy thời gian thoát phù hợp
với mô hình khuyếch tán đơn giản trong trường thế tuyến tính [7, 8]. Điều này
mở ra khả năng áp dụng mô hình khuyếch tán trong việc phân tích các kết quả
thực nghiệm.
Trong luận văn này, trên cơ sở các nghiên cứu trước đây [7, 8], chúng tôi
nghiên cứu quá trình thoát của protein tại đường hầm thoát ribosome nhằm
làm rõ hơn sự phụ thuộc của thời gian thoát vào nhiệt độ và vào cấu trúc trạng
thái tự nhiên của protein. Để thực hiện điều này, chúng tôi chọn hai protein
nhỏ đơn miền có cấu trúc hoàn toàn khác nhau, một protein chỉ bao gồm các
phiến beta và một protein chỉ bao gồm các xoắn alpha, để so sánh.
Nghiên cứu trong luận văn sử dụng phương pháp mô hình hóa và mô
phỏng máy tính. Protein được xét trong mô hình Go là mô hình đơn giản coi
mỗi amino acid là một hạt và sử dụng thế năng Lennard-Jones. Đường hầm
thoát ribosome được mô hình hóa là một ống trụ rỗng có tương tác đẩy với
các amino acid ở khoảng cách gần. Phương pháp mô phỏng động lực học
phân tử với phương trình Langevin được áp dụng để mô phỏng các quá trình

5
mọc protein và quá trình thoát protein tại đường hầm. Các kết quả mô phỏng
được phân tích trên cơ sở lý thuyết của mô hình khuyếch tán một chiều.
Các kết quả chính của luận văn đó là chỉ ra rằng các protein khác nhau có
phân bố thời gian thoát cũng như sự phụ thuộc của thời gian thoát vào nhiệt
độ tương tự nhau. Tuy vậy, protein có cấu trúc phiến beta có thời gian thoát
nhanh hơn so với protein có cấu trúc xoắn alpha.
Bố cục của luận văn được chia thành 4 chương, với các nội dung như
sau:
1. Chương 1: Tổng quan về quá trình cuốn của các protein mới sinh.
2. Chương 2: Các mô hình và phương pháp mô phỏng.
3. Chương 3: Một số kết quả nghiên cứu.
4. Chương 4: Kết luận và kiến nghị.

6
CHƯƠNG 1: TỔNG QUAN VỀ QUÁ TRÌNH CUỐN CỦA CÁC
PROTEIN MỚI SINH
1.1. THÀNH PHẦN HÓA HỌC VÀ CẤU TRÚC CỦA PROTEIN
1.1.1. Thành phần hóa học của protein
Protein(protid hay đạm) là những đại phân tử được cấu tạo theo
nguyên tắc đa phân mà các đơn phân là amino acid. Chúng kết hợp với nhau
thành một mạch dài nhờ các liên kết peptide (gọi là chuỗi polypeptide). Các
chuỗi này có thể xoắn cuộn hoặc gấp theo nhiều cách để tạo thành các bậc cấu
trúc không gian khác nhau của protein. Chuỗi polypeptide là polymer được
cấu thành từ 20 loại amino acid. Trong đó, 19 loại có cấu trúc cơ bản giống
nhau như Hình 1.1 (a). Mỗi amino acid có cấu trúc đặc thù gồm một nguyên
tử carbon alpha (Cα) gắn với bốn nhóm hóa học khác: Nhóm amin (NH2),
axit carboxylic hay nhóm carboxyl (COOH) (bởi đó có tên là amino acid),
nguyên tử hydro (H) và một nhóm biến đổi gọi là chuỗi bên hay nhóm R.
Loại amino acid thứ 20 còn lại là proline có cấu trúc tương tự như các amino
acid khác, nhưng nguyên tử C thuộc chuỗi bên của nó liên kết với nguyên tử
N của nhóm amin tạo thành một mạch vòng (Hình 1.1 (b)).
Hình 1.1: (a) Cấu trúc chung của amino acid, (b) Cấu trúc của proline [ 7]
Đặc tính duy nhất của amino acid quyết định vai trò của amino acid
trong chuỗi polypeptide là do các tính chất vật lý và hóa học của chuỗi bên R
xác định. Các amino acid có thể phân thành các nhóm kị nước (không phân
cực), ưa nước (phân cực), tích điện âm (nếu chuỗi bên mang điện tích âm),
tích điện dương (nếu chuỗi bên mang điện tích dương).

7
Trong protein, các amino acid gắn với nhau thông qua liên kết cộng hóa
trị gọi là liên kết peptide, tạo thành mạch thẳng, không phân nhánh. Trong các
protein có chứa cysteine, một loại amino acid với chuỗi bên có nguyên tử lưu
huỳnh (S), đôi khi các nguyên tử S của hai chuỗi bên khác nhau tạo liên kết
cộng hóa trị với nhau gọi là liên kết disulfide. Liên kết peptide hình thành
giữa nhóm amin của một amino acid và nhóm carboxyl của amino acid khác
và giải phóng một phân tử nước (dehydrat hóa). Kết nối lặp giữa –NH, carbon
α (Cα), và –CO và nguyên tử O trong mỗi amino acid cấu thành bộ khung của
phân tử protein. Các chuỗi bên (nhóm R) nhô ra từ bộ khung này. Do liên kết
peptide, khung có tính định hướng vì tất cả các nhóm –NH nằm cùng một
phía với Cα. Do đó một đầu của protein có nhóm amin tự do (không liên kết)
gọi là đầu N và đầu kia có nhóm cacboxyl tự do gọi là đầu C. Trình tự của
chuỗi protein thường được viết theo thứ tự đầu N nằm bên trái, đầu C nằm
bên phải và các amino acid được đánh số thứ tự bắt đầu từ đầu N (số 1).
Hình 1.2: Chuỗi polypeptide [7]
1.1.2 Cấu trúc của protein
Protein có hình dạng và kích thước rất đa dạng. Cấu trúc lập thể 3 chiều
đa dạng của chúng chủ yếu do những khác biệt trong chiều dài và trình tự
amino acid quyết định. Nhìn chung, mỗi protein với một trình tự amino acid
xác định tại các điều kiện sinh lý bình thường sẽ chỉ gấp nếp thành một hoặc
một số ít hình dạng lập thể giống nhau – gọi là cấu hình native. Những khác
biệt trong cấu hình native và tính chất hóa học của các chuỗi bên của các
amino acid quyết định chức năng của protein. Do đó protein có khả năng thực
hiện rất nhiều chức năng khác nhau bên trong và bên ngoài tế bào. Những

8
chức năng này rất quan trọng cho sự sống. Các protein phối hợp hoạt động để
duy trì sự sống và đảm bảo chức năng chính xác của tế bào.
1.1.2.1. Cấu trúc bậc một của protein
Cấu trúc bậc một của protein là trình tự amino acid trong protein. Có
nhiều thuật ngữ dùng để chỉ chuỗi polymer tạo bởi các amino acid. Chuỗi
amino acid ngắn liên kết với nhau bằng liên kết peptide và có trình tự xác
định gọi oligopeptide hay chỉ đơn giản là peptide; chuỗi dài gọi là
polypeptide. Peptide thường chứa ít hơn 30 amino acid, còn polypepide
thường dài từ vài chục tới vài trăm amino acid.
1.1.2.2. Cấu trúc bậc hai của protein
Cấu trúc bậc hai là thành tố đặc trưng của cấu trúc protein. Cấu trúc bậc
hai là sự sắp xếp không gian địa phương của các vùng trong chuỗi
polypeptide. Những phân đoạn này gắn với nhau thông qua liên kết hydro
giữa các nhóm –NH và –CO của mạch khung, thường tạo thành các mô hình
cấu trúc có tính lặp. Các cấu trúc bậc hai cơ sở là xoắn α, phiến β và đoạn
ngoặt β ngắn hình chữ U.
Hình 1.3: Biểu diễn mạch xương sống của các cấu trúc bậc hai phổ biến của
protein: (a) 2RJX xoắn 𝛼 và (b) 1SHG phiến β [7]
Cấu trúc xoắn α: Trong phân đoạn polypeptide có kiểu gấp nếp xoắn
α, mạch khung xoắn lại. Ở cấu trúc dạng này O thuộc nhóm –CO của mỗi liên
kết peptide tạo liên kết hydro với H thuộc nhóm –NH của amino acid thứ tư

9
trong chuỗi. Trong xoắn α, mọi nhóm –NH và –CO của mạch khung liên kết
hydro với nhau, trừ các nhóm tại hai đầu tận cùng của chuỗi polypeptide.
Kiểu sắp xếp liên kết theo chu kì này tạo thành cấu trúc xoắn từ đầu amin đến
đầu carboxyl với chiều cao mỗi xoắn là 5,4 Å. Mỗi vòng xoắn chứa 3,6 amino
acid, do đó khoảng cách giữa hai nguyên tử Cα liền nhau theo phương trục
xoắn là 1,5 Å.
Trong xoắn α, liên kết hydro ổn định của các amino acid làm mạch
khung có dạng trụ dài, thẳng, từ đó nhóm R của amino acid quay ra ngoài xác
định tính chất ưa nước hay kỵ nước của xoắn. Trong dung dịch xoắn kỵ nước
thường vùi trong lõi của protein đã cuốn, trong khi xoắn ưa nước thường nằm
tại bề mặt của protein, nơi chúng có thể tương tác với môi trường nước.
Hình 1.4: Cấu trúc xoắn alpha
Cấu trúc phiến β: Chứa các mạch β là một phân đoạn ngắn (5 đến 8
amino acid) và hầu như trải ra hết cỡ. Không như xoắn α, liên kết hydro trong
phiến β hình thành giữa các nguyên tử nằm trên khung của các mạch β riêng
biệt nhưng liền kề nhau trong không gian ( Hình 1.3 (b)). Liên kết hydro trong
mạch phẳng phiến giữ các mạch β với nhau và các nhóm R gắn phía trên hoặc
phía dưới mặt phẳng này. Phiến β có tính định hướng do chiều của liên kết
peptide. Do đó, trong phiến xếp nếp, các mạch β sát nhau có thể nằm cùng

10
chiều (song song) hoặc ngược chiều (đối song song). Kẹp tóc 𝛽 ( 𝛽 – harpin)
là một cấu trúc đơn giản của phiến 𝛽 chỉ gồm hai mạch 𝛽 phản song song
được nối với nhau bởi một đoạn uốn cong chứa từ 2 đến 5 amino acid.
Cấu trúc đoạn ngoặt β: Gồm 4 amino acid tạo thành một đường cong
hẹp, uốn mạch khung của chuỗi polypeptide quay ngược lại. Cấu trúc bậc hai
hình chữ U ngắn này được ổn định nhờ liên kết hydro giữa hai đầu. Đoạn
ngoặt β giúp các protein lớn gấp lại rất gọn. Có 6 loại đoạn ngoặt khác nhau
được xác định, cấu trúc chi tiết của chúng phụ thuộc vào sự sắp xếp không
gian và liên kết hydro.
1.1.2.3. Cấu trúc bậc ba của protein
Cấu trúc bậc ba của protein là cấu hình tổng thể của chuỗi polypeptide
hay sắp xếp ba chiều của toàn bộ các amino acid. Cấu trúc bậc ba chủ yếu ổn
định nhờ tương tác kỵ nước giữa các nhóm R không phân cực, liên kết hydro
giữa các nhóm R phân cực và tương tác van der Waals. Các tương tác này yếu
nên cấu trúc bậc ba của protein không cứng nhắc mà luôn dao động nhỏ và
liên tục. Thậm chí một số phân đoạn cấu trúc bậc ba của protein linh động đến
mức chúng được coi là bị mất trật tự (không có cấu trúc lập thể bền vững,
tường minh). Tuy vậy, sự biến thiên cấu trúc này lại quan trọng đối với chức
năng và sự điều hòa của protein.
Tính chất hóa học của nhóm R của amino acid quyết định cấu trúc bậc
ba của protein. Amino acid có chuỗi bên phân cực, ưa nước hay tích điện
thường nằm trên bề mặt của protein. Những amino acid này tương tác với
nước, giúp protein hòa tan trong dung dịch và tạo tương tác không cộng hóa
trị với các phân tử không hòa tan trong nước, bao gồm các protein khác.
Ngược lại, amino acid với chuỗi bên không phân cực, kỵ nước thường bị cô
lập cách xa bề mặt tiếp xúc nước của protein. Trong nhiều trường hợp chúng
tạo thành lõi trung tâm không hòa tan trong nước (gọi là mô hình giọt dầu của
protein cầu vì lõi tương đối kỵ nước hay “có tính dầu”). Nhóm R phân cực,
ưa nước, không tích điện có thể nằm trong bề mặt cũng như trong lõi của
protein.

11
Dựa trên cấu trúc bậc ba có thể chia protein thành ba loại: protein sợi,
protein cầu và protein xuyên màng. Protein sợi là phần tử lớn, dài, cứng và
thường cấu thành từ nhiều trình tự ngắn lặp liên tiếp, tạo thành cấu trúc bậc
hai lặp đơn. Protein sợi thường kết tụ thành sợi lớn gồm nhiều protein và
không hòa tan trong nước. Chúng thường có vai trò tạo nên cấu trúc hoặc
tham gia vào sự vận động của tế bào. Protein cầu thường chứa tập hợp các
cấu trúc bậc hai, hòa tan trong nước, gấp nếp chặt và không có hình cầu hoàn
hảo. Protein xuyên màng nhúng trong lớp phospholipid kép của màng. Màng
này đóng vai trò bức tường của tế bào và cơ quan tử. Ba loại protein này
không hoàn toàn độc lập, một số protein cấu thành từ tổ hợp của hai hoặc
thậm chí cả ba loại này.
1.1.2.4. Cấu trúc bậc bốn của protein
Một số protein gồm các chuỗi polypeptide tập hợp thành một đại phân
tử chức năng. Cấu trúc bậc bốn là cấu trúc của protein do sự tập hợp các tiểu
đơn vị polypeptide đó. Cấu trúc bậc bốn là sự sắp xếp không gian giữa các
cấu trúc bậc ba của các protein. Ví dụ về các tổ hợp đại phân tử với chức năng
cấu trúc bao gồm capsid bao bọc bộ gen của virus và các bó sợi khung tế bào
giúp chống đỡ và tạo hình tế bào chất. Các tổ hợp đại phân tử khác hoạt động
như các bộ máy phân tử, thực hiện hầu hết các quá trình phức tạp của tế bào
bằng cách tích hợp những chức năng riêng lẻ thành một chỉnh thể thống nhất.
1.2. HIỆN TƯỢNG CUỐN PROTEIN TRONG ỐNG NGHIỆM
Tế bào phải thực hiện các quá trình nghiêm ngặt để tạo ra những
protein với đầy đủ các chức năng. Sau khi các ribosome liên kết các amino
acid thành các chuỗi polypeptide không phân nhánh (là cấu trúc bậc một của
protein) thì mỗi chuỗi polypeptide này được cuốn nếp, biến đổi thành cấu
hình lập thể mang cấu trúc bậc hai, bậc ba và có thể bậc bốn. Cấu hình này
được gọi là trạng thái native (trạng thái tự nhiên của protein). Với hầu hết
protein, trạng thái tự nhiên là cấu hình bền vững nhất và theo quan điểm nhiệt
động học thì trạng thái này có năng lượng tự do thấp nhất. Quá trình cuốn
protein là quá trình biến đổi động lực học của chuỗi polypeptide từ trạng thái
duỗi với cấu trúc bậc một tới trạng thái tự nhiên của protein. Tùy thuộc vào

12
điều kiện bên ngoài mỗi chuỗi polypeptide có thể cuốn thành rất nhiều các
hình dạng khác nhau. Vì vậy quá trình cuốn có thể cho sản phẩm là một cấu
hình khác với cấu hình tự nhiên của protein, gọi là quá trình cuốn lỗi. Protein
sẽ mất đi các chức năng sinh học vốn có khi nằm ở trạng thái cuốn lỗi.
Như vậy, yếu tố nào quyết định cấu hình tự nhiên của protein? Cơ chế
nào giúp cho protein cuốn về trạng thái tự nhiên nhanh và chính xác? Trước
hết, dễ thấy mặt phẳng liên kết peptide hạn chế phương thức cuốn nếp của
chuỗi polypeptide. Liên kết peptide có tính chất tương đối giống liên kết đôi
nên nguyên tử C của nhóm –CO và N của nhóm –NH và các nguyên tử gắn
trực tiếp với chúng phải nằm trên cùng một mặt phẳng cố định. Liên kết
peptide không thể tự quay quanh nó. Do vậy biến thiên cấu hình của chuỗi
polypeptide chỉ do độ linh động của khung quyết định. Độ linh động này lại
do góc quay của các mặt phẳng cố định tạp thành từ liên kết giữa Cα với N
của nhóm –NH (góc quay gọi là ) và liên kết giữa Cα với C của nhóm –CO
(góc quay gọi là ) quyết định ( Hình 1.3). Các góc xoay và bị hạn chế
bởi các ràng buộc địa phương về không gian của các nguyên tử trong chuỗi
polypeptide.
Theo nghịch lý Levinthal [9], nếu thời gian protein nằm ở mỗi cấu hình
là 10–12 s thì để trải qua tất cả các cấu hình protein sẽ cần một khoảng thời
gian lớn hơn tuổi vũ trụ. Thực nghiệm quan sát được thời gian cuốn của
protein cỡ từ vài phần nghìn giây tới vài giây. Nghĩa là protein sẽ không trải
qua tất cả các cấu hình trước khi đạt tới trạng thái tự nhiên. Các ràng buộc về
không gian và các ràng buộc trong liên kết peptide đã hạn chế được đáng kể
các cấu hình trung gian. Tuy nhiên, số lượng các cấu hình khả dĩ vẫn tăng
theo hàm số mũ của N là số amino acid trong protein vì vậy không loại bỏ
được nghịch lý Levinthal.
Các thí nghiệm của Christian Anfinsen [10] về quá trình cuốn của
protein biến tính (denatured) trong ống nghiệm cho thấy trình tự bậc một là
thông tin cần cho cấu trúc lập thể chính xác. Một số yếu tố, ví dụ như năng
lượng nhiệt động từ nhiệt độ, pH khắc nghiệt với khả năng thay đổi điện tích
chuỗi bên của amino acid và các hóa chất làm biến tính (denaturants), có thể

13
phá hủy các tương tác không cộng hóa trị yếu, làm entropy tăng nhanh và làm
duỗi protein. Tập hợp protein biến tính này chứa rất nhiều loại cấu hình không
có hoạt tính sinh học và không trong trạng thái tự nhiên. Các thí nghiệm của
Anfinsen cho thấy khi đưa mẫu chứa một loại protein tinh sạch, ở trạng thái
biến tính về điều kiện thường (nhiệt độ sinh lý, pH bình thường, pha loãng
hoặc loại bỏ chất biến tính) thì hầu hết các chuỗi polypeptide đã biến tính có
thể tự phục hồi cấu hình tự nhiên mang hoạt tính sinh học. Điều này khiến
Anfinsen đi đến kết luận rằng trình tự chuỗi amino acid là đủ để xác định cấu
trúc ba chiều của protein. Quá trình cuốn lại của protein biến tính là tự phát và
không cần sự hỗ trợ của các yếu tố liên quan.
1.3. QUÁ TRÌNH TỔNG HỢP PROTEIN TRONG TẾ BÀO
Quá trình sinh tổng hợp protein là một quá trình trung tâm của sinh học
phân tử, là sự truyền thông tin di truyền vào các protein mang chức năng. Quá
trình sinh tổng hợp protein là một quá trình phức tạp và bao gồm hai giai
đoạn: phiên mã và dịch mã.
1.3.1. Quá trình phiên mã
Phiên mã (sao mã) là quá trình tổng hợp mRNA (messenger RNA –
RNA thông tin). Trình tự nucleotide (trong ngôn ngữ bốn bazơ A, G, C và T)
trên phân tử DNA được sao chép thành trình tự nucleotide (trong ngôn ngữ
bốn bazơ A, G, C và U) trên phân tử mRNA. Quá trình này gồm 3 bước sau:
Khởi đầu phiên mã: Enzyme polymerase liên kết vào đoạn trình tự
DNA khởi đầu phiên mã hình thành phức hệ đóng với DNA. Trong giai đoạn
này, DNA vẫn duy trì ở dạng sợi kép, trong khi enzyme liên kết vào bề mặt
của chuỗi xoắn kép. Sau đó polymerase làm biến đổi vùng DNA xoắn kép gần
vị trí khởi đầu hình thành phức hệ mở, DNA tách thành hai mạch đơn.
Kéo dài chuỗi mRNA: Enzyme polymerase di chuyển dọc theo mạch
khuôn DNA theo chiều từ 3’ đến 5’ và thay đổi cấu hình để liên kết ổn định
vào mạch khuôn đồng thời thực hiện một loạt các chức năng khác: Giãn xoắn
mạch DNA ở phía trước, tổng hợp chuỗi RNA theo nguyên tắc bổ sung, tách
chuỗi RNA khỏi mạch khuôn DNA và đóng xoắn mạch DNA ở phía sau.

14
Kết thúc phiên mã: Khi RNA polymerase đã phiên mã hết chiều dài
gene nó dừng lại giải phóng RNA đã tổng hợp xong và tách khỏi DNA. Sau
khi được tổng hợp, mRNA di chuyển tới các ribosome để trực tiếp tổng hợp
protein cho tế bào.
1.3.2 Quá trình dịch mã
Là quá trình tổng hợp protein từ tế bào chất, ngôn ngữ bốn bazơ của
mRNA được dịch mã thành ngôn ngữ 20 amino acid của protein. Quá trình
giải mã trình tự nucleotide trên mRNA thành trình tự amino acid cần sự tham
gia của các phân tử tRNA (transfer RNA hay RNA vận chuyển). Ở tế bào vi
khuẩn, hiện đã xác định được 30 – 40 loại tRNA và ở tế bào động vật và thực
vật có khoảng 50 – 100 loại tRNA. Như vậy, mỗi amino acid (20 loại) có thể
gắn với nhiều tRNA. Ngoài ra một loại tRNA có thể bắt cặp với nhiều codon
trong mã di truyền (61 loại). Để tổng hợp protein, tRNA mang amino acid
được hoạt hóa tới khớp với bộ ba nucleotide (còn gọi là bộ ba hay codon) trên
mRNA để amino acid đã hoạt hóa gắn vào chuỗi polypeptide đang được tổng
hợp. Quá trình dịch mã bao gồm các bước cơ bản sau:
Hoạt hóa amino acid tự do trong tế bào chất: Amino acid được hoạt
hóa nhờ gắn với hợp chất giàu năng lượng adenosinetriphosphate (ATP). Sau
đó, các amino acid đã được hoạt hóa liên kết với tRNA tương ứng để tạo nên
phức hợp amino acid – tRNA.
Mở đầu chuỗi polypeptide: Các tiểu phần của ribosome lắp ráp gần vị
trí khởi đầu dịch mã trên mRNA cùng với tRNA mang methionine có gắn đầu
amin sẽ liên kết bazơ với codon mở đầu. Phức hợp amino acid mở đầu –
tRNA (aa0 – tRNA) tiến vào ribosome đối mã của nó khớp với mã mở đầu
trên mRNA theo nguyên tắc bổ sung.
Kéo dài chuỗi polypeptide: tRNA vận chuyển amino acid thứ nhất tiến
vào ribosome đối mã với nó (theo nguyên tắc bổ sung). Enzyme xúc tác tạo
thành liên kết peptide giữa amino acid mở đầu và amino acid thứ nhất.
Ribosome dịch chuyển đi một bộ ba trên mRNA làm cho tRNA mở đầu rời
khỏi ribosome. Tiếp đó aa2 – tRNA tiến vào ribosome, đối mã của nó khớp

15
với mã thứ hai trên mRNA. Liên kết peptide giữa aa1 và aa2 được tạo thành.
Sự dịch chuyển lại xảy ra, và cứ tiếp tục như vậy cho đến khi ribosome tiếp
giáp với bộ ba kết thúc. Trong mỗi chu trình kéo dài chuỗi, ribosome trải qua
hai lần biến đổi hình dạng. Lần đầu tiên cho phép tRNA đang tới gắn chặt vào
mRNA và tRNA đã hoàn thành nhiệm vụ giải phóng khỏi mRNA. Lần biến
đổi hình dạng thứ hai dẫn tới sự dịch chuyển.
Kết thúc chuỗi polypeptide: Ribosome chuyển dịch sang bộ ba kết thúc
và ngừng quá trình dịch mã. Hai tiểu phần của ribosome tách nhau ra. Một
enzyme đặc hiệu loại bỏ amino acid mở đầu giải phóng chuỗi polypeptide.
1.4. QUÁ TRÌNH CUỐN ĐỒNG DỊCH MÃ CỦA PROTEIN
Quá trình cuốn của protein xảy ra đồng thời với quá trình tổng hợp
protein (hay dịch mã mRNA) được gọi là cuốn đồng dịch mã (cotranslational
folding). Protein chỉ có thể cuốn hoàn chỉnh khi được tổng hợp xong và chui
ra ngoài đường hầm thoát.
Để có thể tổng hợp một lượng protein đủ nhanh, mỗi phân tử mRNA
thường được dịch mã cùng một lúc bởi nhiều ribosome. Tương tác kỵ nước dễ
dẫn đến sự kết tụ của các chuỗi polypeptide mới sinh, đặc biệt đối với các
chuỗi mới sinh được tổng hợp gần nhau từ cùng một mRNA. Sau khi
ribosome đầu tiên kết hợp với mRNA thực hiện dịch mã được một đoạn ngắn,
thì ở phía sau, nhiều ribosome khác cũng bám vào mRNA để dịch mã. Cùng
một lúc, tại vị trí rất gần nhau trong tế bào sẽ xuất hiện rất nhiều các protein
mới sinh có cấu trúc bậc một giống hệt nhau, lại chưa đạt đến cấu trúc cuốn
bền vững và chỉ được cuốn một phần. Dịch mã tương đối chậm nên cấu trúc
một phần và nhạy cảm với kết tụ sẽ tồn tại trong một khoảng thời gian tương
đối dài. Các chuỗi mới sinh đã cuốn một phần rất dễ bám chặt vào nhau, kết
tụ thành một khối lớn, không thể tiếp tục cuốn bình thường và cũng không thể
thực hiện được các chức năng sinh học nữa. Do hiệu ứng kỵ nước những
nhánh kỵ nước lộ ra trên các phân tử khác nhau sẽ bám vào nhau khi chưa kịp
vùi vào lõi của protein, thúc đẩy sự kết tụ. Sự kết tụ là nguyên nhân chính gây
ra bệnh Alheimer do các protein kết tụ thành các sợi amyloid không hòa tan
và hủy các tế bào thần kinh.

16
1.5. ĐƯỜNG HẦM THOÁT RIBOSOME
Trong bán cầu lớn tại PTC (peptidyl transferase center) của ribosome
chuỗi polypeptide được sinh tổng hợp. Trong bán cầu lớn này có một đường
hầm thoát hẹp dẫn protein mới sinh từ nơi được tổng hợp tới môi trường tế
bào. Đường kính của đường của đường hầm thoát có thể thay đổi từ 10 Å – 20
Å và không hoàn toàn thẳng [2]. Đường hầm thoát chật hẹp nhất và hơi uốn
cong ở chỗ thắt cách PTC khoảng 20 Å. Đường hầm ribosome được xây dựng
bởi các đoạn của phân tử 23S rRNA và một số protein ribosome (L4, L22,
L39). Đường hầm có chiều dài trong khoảng 80 Å – 100 Å tùy thuộc vào cách
định nghĩa cổng ra. Đường hầm có thể chứa trọng vẹn một chuỗi peptide
thẳng gồm 30 amino acid hoặc một chuỗi có cấu trúc xoắn α gồm 60 amino
acid. Mặt trong của đường hầm thoát có bản chất ưa nước cho phép chuỗi mới
sinh chui ra dễ dàng không bị cản trở.
1.6. ẢNH HƯỞNG CỦA ĐƯỜNG HẦM THOÁT RIBOSOME LÊN QUÁ
TRÌNH CUỐN CÁC PROTEIN MỚI SINH
Đường hầm thoát ribosome chật hẹp đã tạo ra một số rào cản không
gian cho sự hình thành các cấu trúc của protein mới sinh [2]. Các đơn vị cấu
trúc đơn giản như xoắn α và phiến có thể hình thành bên tại đường hầm
thoát [3]. Và một số vùng cấu trúc nhỏ bậc ba khác có thể hình thành ở cổng
ra của đường hầm thoát [4]. Vì vậy, quá trình protein cuốn hoàn thiện chỉ xảy
ra bên ngoài đường hầm thoát. Thực nghiệm cho thấy ngay cả khi protein nằm
bên ngoài đường hầm ribosome, sự có mặt của ribosome vẫn giúp cho protein
cuốn chính xác hơn [12].
Thí nghiệm của Ito và cộng sự [5, 6] cho thấy ribosome ngừng dịch mã
khi trong đường hầm xuất hiện trình tự SecM trong chuỗi polypeptide. Điều
này gợi ý rằng đường hầm thoát có vai trò nhận biết trình tự amino acid và
tham gia tích cực vào quá trình tổng hợp protein. Nghiên cứu mô phỏng trong
mô hình đầy đủ các nguyên tử của Pande và các cộng sự cho thấy đường hầm
ribosome tạo nên hàng rào năng lượng tự do đối với các amino acid và có cơ
chế then cửa (gate-latch mechanism) kiểm soát sự thoát ra của các amino acid
[13].

17
Nghiên cứu mô phỏng gần đây của Hoàng và cộng sự [7] cho thấy quá
trình cuốn protein tại đường hầm thoát ribosome xảy ra đồng thời với quá
trình thoát protein ra khỏi đường hầm, và điều này giúp cho protein cuốn
chính xác hơn. Nghiên cứu ảnh hưởng của chiều dài đường hầm lên quá trình
thoát protein [8] cũng gợi ý rằng chiều dài của đường hầm ribosome đã được
tự nhiên lựa chọn sao cho quá trình khuyếch tán protein ra khỏi đường hầm
được hiệu quả đồng thời không làm cho protein thoát ra quá nhanh để tránh
các nguy cơ cuốn nhầm và kết tụ.
CHƯƠNG 2: CÁC MÔ HÌNH VÀ PHƯƠNG PHÁP MÔ PHỎNG
2.1 MÔ HÌNH GO CHO PROTEIN
Mô hình Go [14] là một loại mô hình đơn giản hóa cho protein cho phép
nghiên cứu động lực học của quá trình cuốn protein. Các thế năng tương tác
trong mô hình Go được xây dựng trên cấu trúc trạng thái native của protein
sao cho trạng thái này có thế năng cực tiểu. Mô hình Go áp dụng thế năng hút
cho các tương tác giữa các amino acid có tiếp xúc trong trạng thái native và
thế năng đẩy cho các tương tác giữa các amino acid không tiếp xúc với nhau
trong trạng thái native. Ngoài ra, giữa các amino acid lân cận nhau dọc theo
chuỗi polypeptide người ta có thể áp dụng các thế năng cho các góc liên kết
và góc nhị diện. Các nghiên cứu mô phỏng cho thấy mô hình Go cho cơ chế
cuốn phù hợp với các kết quả thực nghiệm [15, 16].
Trong luận văn này, chúng tôi sử dụng một phiên bản mô hình Go [15]
với thế năng Lennard-Jones 10-12 và các thế năng góc. Trong mô hình này,
mỗi amino acid được coi là một hạt có khối lượng m và có vị trí trùng với vị
trí của nguyên tử Cα. Gọi N là số amino acid trong chuỗi protein. Cấu hình
phân tử protein được xác định bởi hệ vectơ {𝑟𝑖⃗⃗ }i=1,2,…,N là tọa độ của tất cả các
hạt. Năng lượng của protein tại một cấu hình cho trước được xác định bởi:
E = VNative + VRepulsive + VBond + VAngles (2.1)
Số hạng thứ nhất trong phương trình (2.1) là tổng các thế năng Lennard-Jones
cho tương tác giữa các amino acid có tiếp xúc trong trạng thái native:

18
𝑉Native = ∑ 휀 [5(𝜎𝑖𝑗
𝑟𝑖𝑗)
12
− 6(𝜎𝑖𝑗
𝑟𝑖𝑗)
10
]
native
𝑖,𝑗>𝑖+3
(2.2)
trong đó rij là khoảng cách giữa hai hạt thứ i và j, ε là tham số năng lượng ứng
với độ sâu của thế năng, 𝜎𝑖𝑗 = 𝑟𝑖𝑗𝑛𝑎𝑡𝑖𝑣𝑒
là tham số được chọn bằng khoảng
cách giữa hạt i và j trong trạng thái native. Để xác định xem hai amino acid
trong trạng thái native có tiếp xúc nhau hay không, chúng tôi sử dụng tiêu chí
đề xuất bởi Best và Hummer [17] bằng cách xét tất cả các nguyên tử nặng
trong hai amino acid (trừ các nguyên tử hydro). Nếu giữa hai amino acid thứ i
và thứ j với i + 3 < j tồn tại một cặp nguyên tử nặng có khoảng cách nhỏ hơn
4,5 Å thì hai amino acid được cho là có tiếp xúc với nhau.
Số hạng thứ hai trong phương trình (2.1) là tổng thế năng đẩy được cho
bởi:
𝑉𝑅𝑒𝑝𝑢𝑙𝑠𝑖𝑣𝑒 = ∑ 4
non-native
𝑖,𝑗>𝑖+3
𝜖 [(𝜎
𝑟𝑖𝑗)
12
− (𝜎
𝑟𝑖𝑗)
6
] 𝛩(21 6⁄ 𝜎 − 𝑟𝑖𝑗) (2.3)
trong đó tổng theo i và j được lấy theo tất cả các cặp amino acid không có tiếp
xúc trong trạng thái native, (x) là hàm bước Heavyside, σ = 5 Å.
Các hạt liên tiếp nhau trong chuỗi protein tương tác thông qua thế năng
điều hòa. Số hạng thứ ba trong phương trình (2.1) là tổng thế năng điều hòa:
𝑉Bond = 100휀 ∑ (𝑟𝑖,𝑖+1 − 𝑑0)2𝑁−1
𝑖=1 , (2.4)
trong đó d0 = 3.8 Å là khoảng cách giữa hai nguyên tử carbon alpha liên tiếp
trong chuỗi protein.
Những hạn chế về không gian liên quan tới mặt phẳng liên kết peptide
trong protein được đặc trưng bởi số hạng thứ tư trong phương trình (2.1)
𝑉𝐴𝑛𝑔𝑙𝑒𝑠 = 𝑉𝐵𝐴 + 𝑉𝐷𝐴, (2.5)

19
trong đó các số hạng vế phải của phương trình (2.5) lần lượt là thế năng liên
quan tới các góc liên kết (bond angle) và các góc nhị diện (dihedral angle)
trong chuỗi.
𝑉𝐵𝐴 = ∑𝑘0
2(𝜃𝑖 − 𝜃0,𝑖)
2,
𝑁−1
𝑖=2
(2.6)
với góc liên kết 𝜃𝑖là góc tạo bởi ba nguyên tử carbon alpha liên tiếp, 𝜃0,𝑖 là
góc liên kết ở trạng thái native, k0= 20 /rad2 là tham số.
𝑉𝐷𝐴 = ∑{𝐴[1 + 𝑐𝑜𝑠(𝜙𝑖 − 𝜙0,𝑖)] + 𝐵[1 + 𝑐𝑜𝑠3(𝜙𝑖 − 𝜙0,𝑖)]}
𝑁−3
𝑖=2
(2.7)
với góc nhị diện 𝜙𝑖 là góc tạo bởi bốn nguyên tử carbon alpha liên tiếp nhau
trong chuỗi, 𝜙0,𝑖 là góc nhị diện ở trạng thái gốc, A = − và B = − 0.5 là các
tham số.
2.2. MÔ HÌNH ĐƯỜNG HẦM THOÁT RIBOSOME
Chúng tôi sử dụng mô hình đơn giản hóa cho đường hầm thoát của
ribosome [7, 8] để khảo sát ảnh hưởng của đường hầm thoát lên quá trình
thoát của các protein mới sinh. Đường hầm thoát được mô hình hóa như một
ống hình trụ rỗng có chiều dài L = 80 Å, đường kính d = 15 Å và tâm chạy
dọc theo trục x, với một đáy kín và một đáy mở gắn vào một bức tường phẳng
mô phỏng mặt ngoài của ribosome. Giả sử tâm của đáy ống trụ (x = 0) chính
là vị trí mọc của protein mới sinh hay PTC (nơi amino acid được gắn vào
chuỗi polypeptide đang được tổng hợp). Protein từ đường hầm thoát chui ra
ngoài qua đáy còn lại của đường hầm thoát. Tương tác giữa amino acid và
tường đường hầm thoát là thế năng đẩy cho bởi thế năng Lennard-Jones bị cắt
tại cực tiểu của thế năng:
𝑉𝑤𝑎𝑙𝑙(𝑟) = {4휀 [(
𝜎
𝑟)12
− (𝜎
𝑟)6
] + 휀, 𝑟 ≤ 21 6⁄ 𝜎
0, 𝑟 > 21 6⁄ 𝜎
(2.8)

20
trong đó σ = 5 Å và r là khoảng cách ngắn nhất từ tâm các amino acid đến
tường của đường hầm cộng thêm 2.5 Å (tương ứng với bán kính Van der
Waals của các hạt ảo được giả định là cấu trúc thành mặt trong của đường
hầm thoát). Thế năng đẩy cho bởi (2.8) cũng được sử dụng cho tương tác đẩy
của cổng ra đường hầm thoát và tường phẳng bao ngoài đáy đường hầm thoát
đối với các amino acid.
2.3. PHƯƠNG PHÁP ĐỘNG LỰC HỌC PHÂN TỬ DỰA TRÊN PHƯƠNG
TRÌNH LANGEVIN
Động lực học phân tử là kĩ thuật mô phỏng sử dụng để nghiên cứu sự
biến đổi theo thời gian trạng thái của một hệ các hạt cổ điển tương tác với
nhau qua việc tích phân phương trình chuyển động của chúng. Phương pháp
động lực học phân tử cho phép xác định trạng thái của hệ ở một thời điểm t
bất kì khi biết trạng thái ban đầu của hệ. Phương pháp động lực học phân tử
có thể dùng để nghiên cứu các quá trình động lực học của protein. Trong luận
văn này, chúng tôi sử dụng phương pháp động lực học phân tử xét tới các tác
động ngẫu nhiên của dung môi lên hạt chuyển động dựa trên phương trình
Langevin.
2.3.1. Phương trình Langevin
Chuyển động của các hạt cổ điển tuân theo các phương trình Newton.
Trong trường hợp chuyển động của các hạt chịu tác động của các yếu tố ngẫu
nhiên do môi trường bên ngoài người ta sử dụng phương pháp động học phân
tử dựa trên phương trình Langevin.
Phương trình Langevin có thể được sử dụng để mô tả chuyển động của
các hạt lớn như các amino acid trong protein trong dung môi là nước. Ở nhiệt
độ T, phương trình Langevin một chiều cho một hạt có dạng:
𝑚�̈�(𝑡) = 𝑓(𝑡) − 휁�̇�(𝑡) + 𝛤(𝑡) (2.9)
trong đó m là khối lượng của hạt, f là lực tác dụng lên hạt gây bởi các thế
năng tương tác 𝑓 = −𝜕𝑉(𝑥)
𝜕𝑥; 휁 là hệ số ma sát của dung môi tác động lên hạt;
là lực ngẫu nhiên của dung môi tác động lên hạt, có phân bố Gauss với

21
trung bình bằng không và hàm tương quan theo thời gian thỏa mãn định luật
biến thiên tiêu tán:
⟨𝛤(𝑡)𝛤(𝑡 + 𝑡′)⟩ = 2휁𝑚𝑘𝐵𝑇𝛿(𝑡′), (2.10)
với 𝛿(𝑡′) là hàm delta Dirac. Khi = 0 thì phương trình Langevin trở thành
phương trình Newton.
2.3.2. Thuật toán Verlet cho phương trình Langevin
Chúng tôi sử dụng thuật toán Verlet cho phương trình Langevin được
đề xuất trong tài liệu [7]. Từ phương trình Langevin ta có:
�̈�(𝑡) =1
𝑚[𝑓(𝑡) − 휁�̇�(𝑡) + 𝛤(𝑡)]. (2.11)
Khai triển tọa độ x theo chuỗi Taylor đến bậc hai ta có:
𝑥(𝑡 + ∆𝑡) ≈ 𝑥(𝑡) + �̇�(𝑡)∆𝑡 +1
2�̈�(𝑡)∆𝑡2, (2.12)
𝑥(𝑡 − ∆𝑡) ≈ 𝑥(𝑡) − �̇�(𝑡)∆𝑡 +1
2�̈�(𝑡)∆𝑡2, (2.13)
trong đó ∆𝑡 là bước thời gian.
Từ (2.12) và (2.13) ta có:
�̇�(𝑡) =𝑥(𝑡+∆𝑡)−𝑥(𝑡−∆𝑡)
2∆𝑡, (2.14)
𝑥(𝑡 + ∆𝑡) ≈ 2𝑥(𝑡) − 𝑥(𝑡 − ∆𝑡) + �̈�(𝑡)∆𝑡2. (2.15)
Thay (2.17) và (2.14) và (2.18) ta thu được:
𝑥(𝑡 + ∆𝑡) = (1 +∆𝑡
2𝑚휁)
−1
{2𝑥(𝑡) − (1 −∆𝑡
2𝑚휁)𝑥(𝑡 − ∆𝑡)
+∆𝑡2
𝑚[𝑓(𝑡) + 𝛤(𝑡)]}.
(2.16)

22
Thuật toán Verlet cho phép ta tính tọa độ tại thời điểm t + t khi biết tọa độ tại
các thời điểm t và t + t.
Trong các mô phỏng, nhiệt độ được biểu thị bằng đơn vị ε/kB, thời gian
được đo bằng đơng vị 𝜏 = √𝑚𝜎2 휀⁄ . Bước thời gian được chọn là ∆t =
0.002τ, và các mô phỏng được thực hiện với hệ số ma sát ζ = 2.5 mτ−1.
2.4. MÔ HÌNH KHUYẾCH TÁN CHO QUÁ TRÌNH THOÁT PROTEIN
Các nghiên cứu trước đây [7, 8] đã đưa ra mô hình khuyếch tán đơn
giản cho quá trình thoát của protein ra khỏi đường hầm ribosome, trong đó
quá trình thoát được coi là quá trình khuyếch tán một chiều của một hạt trong
một trường thế. Giả sử ta có trường thế U(x), sự khuyếch tán của hạt trong
trường thế được mô tả bởi phương trình Smoluchowski một chiều:
𝜕
𝜕𝑡𝑝(𝑥, 𝑡; 𝑥0, 𝑡0) =
𝜕
𝜕𝑥𝐷 (𝛽
𝜕𝑈(𝑥)
𝜕𝑥+
𝜕
𝜕𝑥) 𝑝(𝑥, 𝑡; 𝑥0, 𝑡0), (2.17)
trong đó 𝑝(𝑥, 𝑡; 𝑥0, 𝑡0) là mật độ xác suất tìm thấy hạt ở tọa độ x vào thời
điểm t, nếu tại thời điểm t0 hạt được tìm thấy tại tọa độ x0; 𝜕 𝜕⁄ 𝑡 và 𝜕 𝜕⁄ 𝑥 là
các đạo hàm riêng phần theo thời gian và tọa độ; D là hằng số khuyếch tán
được giả thiết rằng không phụ thuộc vào tọa độ; 𝛽 = (𝑘𝐵𝑇)−1 là nghịch đảo
của nhiệt độ. Đối với khuyếch tán thông thường trong chất lỏng, hằng số
khuyếch tán D thỏa mãn hệ thức Einstein-Smoluchowski:
𝐷 =𝑘𝐵𝑇
𝜁, (2.18)
trong đó là hệ số ma sát nhớt. Nếu U(x) là thế năng tuyến tính theo tọa độ:
𝑈(𝑥) = −𝑘𝑥, (2.19)
với k là một hằng số thì phương trình (2.17) có nghiệm chính xác cho bởi
𝑝(𝑥, 𝑡) ≡ 𝑝(𝑥, 𝑡; 0,0) =𝐷𝛽𝑘
√4𝜋𝐷𝑡𝑒𝑥𝑝 [
−(𝑥 − 𝐷𝛽𝑘𝑡)2
4𝐷𝑡], (2.20)
với điều kiện ban đầu là p(x,0) = (x). Hàm phân bố (2.20) cho ta tọa độ trung
bình của hạt:

23
⟨𝑥⟩ = (𝐷𝛽𝑘)𝑡. (2.21)
Phương trình trên cho thấy 𝐷𝛽𝑘 là tốc độ khuyếch tán.
Thời gian thoát của protein tại đường hầm ribosome có chiều dài L trong
mô hình khuyếch tán chính là thời gian tới đầu tiên (first passage time) của
hạt khuyếch tán có điều kiện ban đầu p(x,0) = (x) tại thời điểm t = 0 và điều
kiện biên hấp thụ p(L,t) = 0 tại x = L. Lời giải phân bố thời gian tới đầu tiên
cho phương trình Smoluchowski với các điều kiện biên như trên có thể tìm
thấy trong tài liệu [18]. Phân bố thời gian tới đầu tiên được cho bởi
𝑔(𝑡) =𝐿
√4𝜋𝐷𝑡3𝑒𝑥𝑝 [
−(𝐿−𝐷𝛽𝑘)2
4𝐷𝑡]. (2.22)
Hàm phân bố này cho ta thời gian thoát trung bình:
𝜇𝑡 ≡ ⟨𝑡⟩ = ∫ 𝑡 𝑔(𝑡)𝑑𝑡 =𝐿
𝐷𝛽𝑘, (2.23)
và độ lệch chuẩn của thời gian thoát:
𝜎𝑡 ≡ (⟨𝑡2⟩ − ⟨𝑡⟩2)1 2⁄ =√2𝛽𝑘𝐿
𝐷(𝛽𝑘)2 (2.24)
Ta thấy rằng tỷ lệ 𝜎𝑡 𝜇𝑡⁄ không phụ thuộc vào hằng số khuyếch tán D mà chỉ
phụ thuộc vào k:
𝜎𝑡
𝜇𝑡= √
2
𝛽𝑘𝐿. (2.25)
Chú ý rằng cả 𝜎𝑡và 𝜇𝑡 đều phân kỳ (bằng vô cùng) khi k = 0, khi đó hàm phân
bố g(t) trở thành hàm phân bố Lévy. Mô hình khuyếch tán cho thấy quá trình
thoát chỉ hiệu quả với thời gian thoát trung bình hữu hạn khi k > 0.

24
CHƯƠNG 3: MỘT SỐ KẾT QUẢ NGHIÊN CỨU
Sử dụng các mô hình và phương pháp mô phỏng, chúng tôi thực hiện
nghiên cứu quá trình thoát của protein mới sinh tại đường hầm thoát
ribosome. Nghiên cứu được thực hiện cho hai protein nhỏ đơn miền là protein
SH3 domain (Mã PDB: 1SHG) và protein Villin (Mã PDB: 2RJX). Hai
protein này được chọn có kích thước tương tự nhau (SH3 domain có chiều dài
57 amino acids, Villin có chiều dài 67 amino acids) nhưng hình dạng của
trạng thái tự nhiên khác nhau để so sánh. Để thuận tiện chúng tôi gọi hai
protein này là 1SHG và 2RJX. Trạng thái tự nhiên của 1SHG và 2RJX được
thể hiện trên Hình 3.1. Cấu trúc của 1SHG gồm hai phiến beta đối song song
bó vào nhau, trong khi 2RJX gồm 5 xoắn alpha xếp gần nhau.
1SHG 2RJX
Hình 3.1 Cấu trúc trạng thái tự nhiên của protein SH3 domain (1SHG) và protein Villin
(2RJX). Các cấu trúc phiến beta được tô màu vàng, xoắn alpha được tô màu đỏ.
Các mô phỏng được thực hiện cho cả 2 protein tại nhiều nhiệt độ khác
nhau trong khoảng từ T = 0.2 /kB tới T = 2.4 /kB. Tại mỗi một nhiệt độ, 500
quỹ đạo mô phỏng độc lập được thực hiện cho quá trình cuốn đồng dịch mã
và quá trình thoát của protein. Quá trình cuốn đồng dịch mã được mô phỏng
với protein được mọc dài dần từ đầu N tới đầu C và thời gian để chuỗi
polypeptide mọc dài thêm một hạt là tg = 100 . Sau khi được dịch mã hoàn
25
chỉnh, protein tiếp tục được mô phỏng cho tới khi thoát ra hoàn toàn khỏi
đường hầm ribosome. Thời gian thoát được tính từ khi protein được dịch mã
hoàn chỉnh cho tới khi protein thoát ra hoàn toàn.
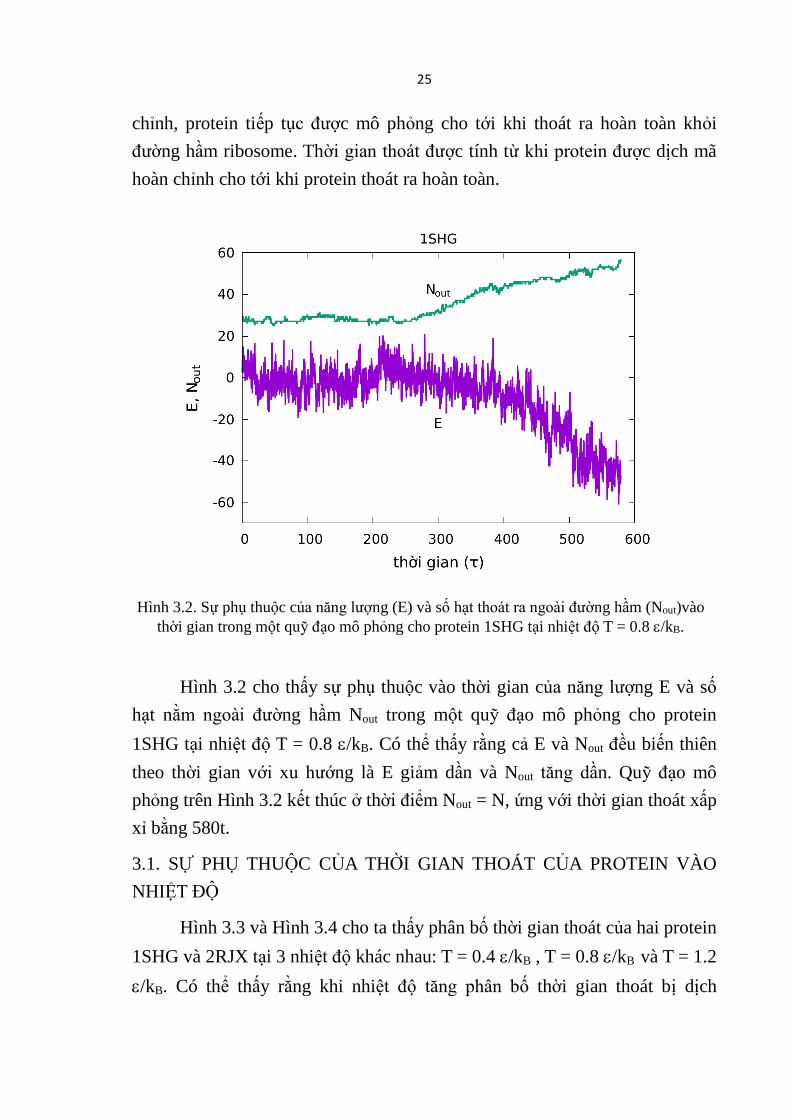
Hình 3.2. Sự phụ thuộc của năng lượng (E) và số hạt thoát ra ngoài đường hầm (Nout)vào
thời gian trong một quỹ đạo mô phỏng cho protein 1SHG tại nhiệt độ T = 0.8 /kB.
Hình 3.2 cho thấy sự phụ thuộc vào thời gian của năng lượng E và số
hạt nằm ngoài đường hầm Nout trong một quỹ đạo mô phỏng cho protein
1SHG tại nhiệt độ T = 0.8 /kB. Có thể thấy rằng cả E và Nout đều biến thiên
theo thời gian với xu hướng là E giảm dần và Nout tăng dần. Quỹ đạo mô
phỏng trên Hình 3.2 kết thúc ở thời điểm Nout = N, ứng với thời gian thoát xấp
xỉ bằng 580t.
3.1. SỰ PHỤ THUỘC CỦA THỜI GIAN THOÁT CỦA PROTEIN VÀO
NHIỆT ĐỘ
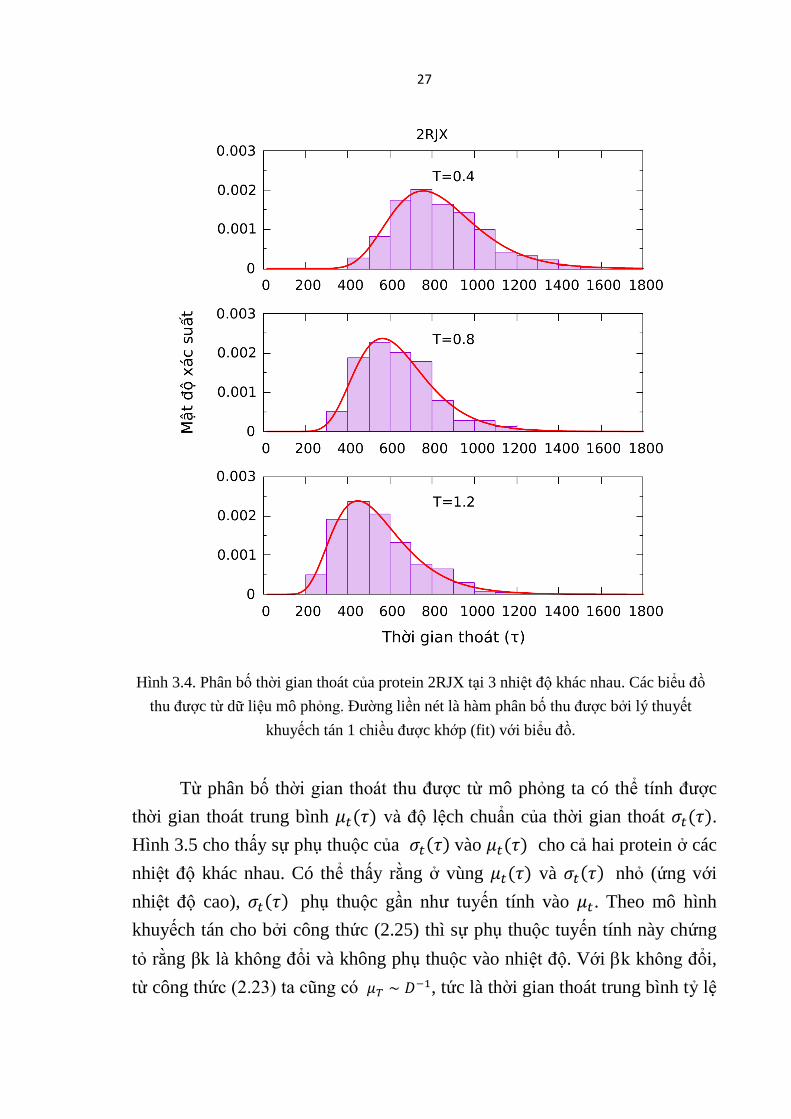
Hình 3.3 và Hình 3.4 cho ta thấy phân bố thời gian thoát của hai protein
1SHG và 2RJX tại 3 nhiệt độ khác nhau: T = 0.4 /kB , T = 0.8 /kB và T = 1.2
/kB. Có thể thấy rằng khi nhiệt độ tăng phân bố thời gian thoát bị dịch
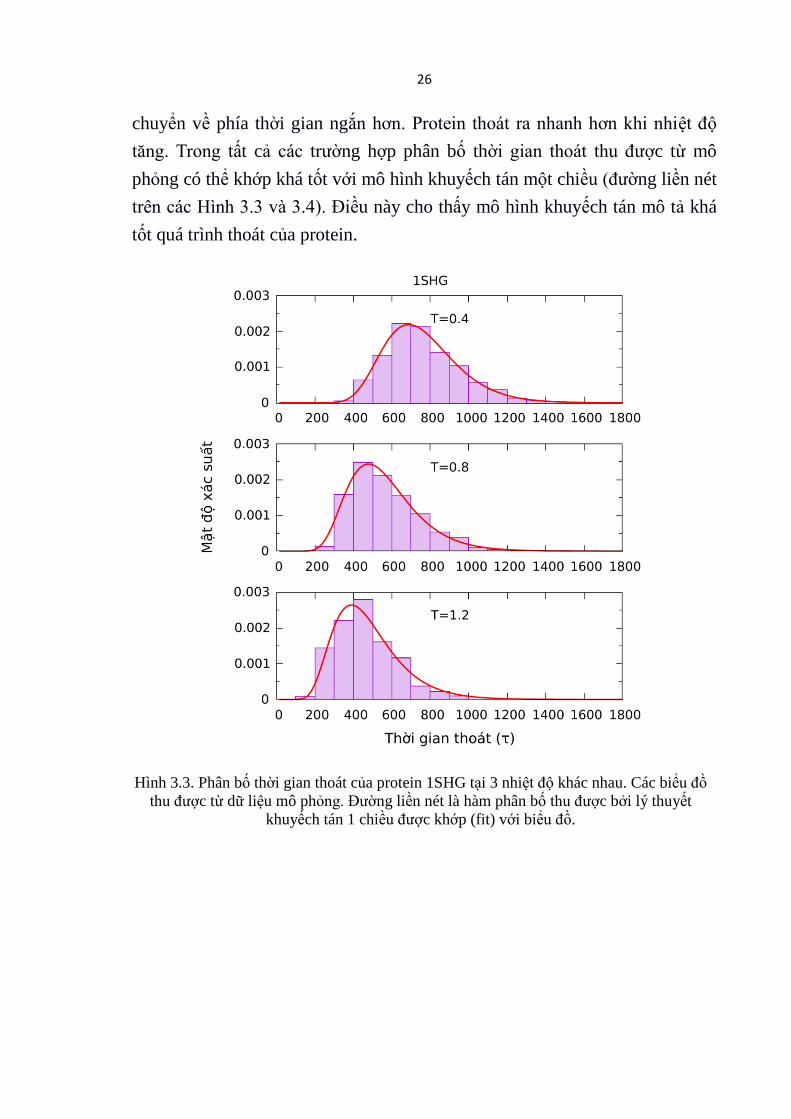
26
chuyển về phía thời gian ngắn hơn. Protein thoát ra nhanh hơn khi nhiệt độ
tăng. Trong tất cả các trường hợp phân bố thời gian thoát thu được từ mô
phỏng có thể khớp khá tốt với mô hình khuyếch tán một chiều (đường liền nét
trên các Hình 3.3 và 3.4). Điều này cho thấy mô hình khuyếch tán mô tả khá
tốt quá trình thoát của protein.
Hình 3.3. Phân bố thời gian thoát của protein 1SHG tại 3 nhiệt độ khác nhau. Các biểu đồ
thu được từ dữ liệu mô phỏng. Đường liền nét là hàm phân bố thu được bởi lý thuyết
khuyếch tán 1 chiều được khớp (fit) với biểu đồ.
27
Hình 3.4. Phân bố thời gian thoát của protein 2RJX tại 3 nhiệt độ khác nhau. Các biểu đồ
thu được từ dữ liệu mô phỏng. Đường liền nét là hàm phân bố thu được bởi lý thuyết
khuyếch tán 1 chiều được khớp (fit) với biểu đồ.
Từ phân bố thời gian thoát thu được từ mô phỏng ta có thể tính được
thời gian thoát trung bình 𝜇𝑡(𝜏) và độ lệch chuẩn của thời gian thoát 𝜎𝑡(𝜏).
Hình 3.5 cho thấy sự phụ thuộc của 𝜎𝑡(𝜏) vào 𝜇𝑡(𝜏) cho cả hai protein ở các
nhiệt độ khác nhau. Có thể thấy rằng ở vùng 𝜇𝑡(𝜏) và 𝜎𝑡(𝜏) nhỏ (ứng với
nhiệt độ cao), 𝜎𝑡(𝜏) phụ thuộc gần như tuyến tính vào 𝜇𝑡. Theo mô hình
khuyếch tán cho bởi công thức (2.25) thì sự phụ thuộc tuyến tính này chứng
tỏ rằng βk là không đổi và không phụ thuộc vào nhiệt độ. Với k không đổi,
từ công thức (2.23) ta cũng có 𝜇𝑇 ∼ 𝐷−1, tức là thời gian thoát trung bình tỷ lệ

28
nghịch với hằng số khuyếch tán. Ở vùng 𝜇𝑡(𝜏) và 𝜎𝑡(𝜏) lớn (ứng với nhiệt độ
thấp) thì sự phụ thuộc của 𝜎𝑡(𝜏) vào 𝜇𝑡(𝜏) bị lệch khỏi hàm tuyến tính. Lúc
này, cả k và D đều phụ thuộc vào nhiệt độ.
Hình 3.5. Sự phụ thuộc của độ lệch chuẩn của thời gian thoát vào thời gian thoát trung
bình cho 2protein 1SHG và 2RJX. Đường đứt nét tương ứng với mô hình khuyếch tán 1
chiều với k không đổi.
Hình 3.6 mô tả sự phụ thuộc của thời gian thoát trung bình 𝜇𝑡 vào nhiệt
độ T trên thang tọa độ log-log cho hai protein 1SHG và 2RJX. Đối với cả hai
protein thời gian thoát trung bình giảm khi nhiệt độ tăng. Ở vùng nhiệt độ cao
thời gian thoát trung bình phụ thuộc vào nhiệt độ theo quy luật 𝜇𝑇 ∼
𝑇−1(đường tuyến tính đứt nét trên hình vẽ). Điều này chứng tỏ ở nhiệt độ cao
thì hằng số khuyếch tán phụ thuộc tuyến tính vào nhiệt độ, 𝐷 ∼ 𝑇, giống như
trong hệ thức Einstein (phương trình 2.18) cho hạt chuyển động Brown. Như
vậy ở nhiệt độ cao quá trình thoát của protein là tương tự như quá trình
khuếch tán của hạt Brown trong trường thế tuyến tính. Hình 3.6 cho thấy ở

29
vùng nhiệt độ thấp, thời gian thoát trung bình của protein thấp hơn nhiều so
với thời gian thoát của hạt Brown. Điều này khẳng định quá trình cuốn của
protein tại đường hầm ribosome làm cho protein thoát ra nhanh hơn [7, 8].

30
(a)
(b)
Hình 3.6. Sự phụ thuộc của thời gian thoát trung bình vào nhiệt độ cho protein 1SHG (a)
và protein 2RJX (b) trên thang tọa độ log-log. Đường đứt nét có độ dốc bằng −1 tương
ứng với 𝜇𝑇 ∼ 𝑇−1.

31
3.2. SỰ PHỤ THUỘC CỦA THỜI GIAN THOÁT VÀO CẤU TRÚC CỦA
PROTEIN
Các kết quả ở Mục 3.1 cho thấy mặc dù 2 protein 1SHG và 2RJX có
cấu trúc trạng thái tự nhiên rất khác biệt, phân bố thời gian thoát, sự phụ thuộc
của thời gian thoát vào nhiệt độ cũng như các tính chất khuyếch tán của cả hai
protein đều khá tương đồng. Điều này cho thấy quá trình thoát của protein
không phụ thuộc mạnh vào cấu trúc trạng thái tự nhiên.
Hình 3.7. Sự phụ thuộc của thời gian thoát trung bình (t) vào nhiệt độ (T) cho 2 protein
1SHG và 2RJX.
Hình 3.7 so sánh thời gian thoát trung bình của 1SHG và 2RJX tại các
nhiệt độ khác nhau. Có thể thấy rằng 2RJX có thời gian thoát trung bình lớn
hơn so với 1SHG ở hầu hết các nhiệt độ, ngoại trừ ở 2 giá trị nhiệt độ rất thấp
T = 0.2 /kB và T = 0.25 /kB . Tuy vậy, sự chênh lệch thời gian thoát trung
bình giữa hai protein là không nhiều. Ở các vùng nhiệt độ trung bình và cao,
protein 2RJX là protein có cấu trúc xoắn alpha có thời gian thoát trung bình
cao hơn khoảng 10% - 20% so với 1SHG là protein có cấu trúc phiến beta.

32
CHƯƠNG 4. KẾT LUẬN VÀ KIẾN NGHỊ
Trong luận văn này, chúng tôi đã nghiên cứu quá trình thoát của protein
tại đường hầm thoát ribosome sử dụng phương pháp mô phỏng động lực học
phân tử với các mô hình đơn giản hóa cho protein và cho đường hầm thoát
ribosome. Nghiên cứu được thực hiện cho hai protein nhỏ đơn miền với cấu
trúc trạng thái tự nhiên khác nhau. Các kết quả mô phỏng được phân tích và
so sánh với mô hình khuyếch tán trong trường thế tuyến tính một chiều, và
cho các kết luận như sau:
1. Khi nhiệt độ tăng thì thời gian thoát của protein giảm. Phân bố thời
gian thoát phù hợp với mô hình khuyếch tán. Ở vùng nhiệt độ cao thời gian
thoát trung bình tỷ lệ nghịch với nhiệt độ theo hàm mũ 𝑇−1, giống như đối với
hạt chuyển động Brown. Ở vùng nhiệt độ thấp, quá trình cuốn làm protein
thoát ra nhanh hơn so với hạt chuyển động Brown.
2. Thời gian thoát của protein không phụ thuộc mạnh vào cấu trúc trạng
thái tự nhiên của protein. Protein có cấu trúc xoắn alpha thoát ra chậm hơn
10% - 20% so với protein có cấu trúc phiến beta.
Trên đây là các kết quả thu được từ các mô hình đơn giản hóa. Các kết
quả này cần được so sánh với các kết quả của các mô hình chi tiết hơn cho
protein và cho đường hầm thoát ribosome, cũng như được kiểm chứng bởi
thực nghiệm.

33
TÀI LIỆU THAM KHẢO
[1] C. A. Kaiser, M. Krieger, H. Lodish, A. Berk. Molecular Cell Biology, “
Molecular Cell Biology”, WH Freeman (2007).
[2] N. R. Voss, M. Gerstein, T. A. Steitz, and P. B. Moore, “ The geometry
of the ribosomal polypeptide exit tunnel”, J. Mol. Biol. 360, 893 (2006).
[3] J. L. Lu and C. Deutsch, Nat. Struct. Mol. Biol. 12, 1123-1129 (2005).
[4] A. Kosolapov and C. Deutsch, “Tertiary Interactions within the
Ribosomal Exit Tunnel”, Nat. Struct. Mol. Biol. 16, 405-411 (2009).
[5] H. Nakatogawa and K. Ito, “ The Ribosomal Exit Tunnel Functions as a
Discriminating Gate”, Cell 108, 629- 636 (2002).
[6] H. Nakatogawa and K. Ito, Chem. Bio. Chem. 5, 81 (2004).
[7] P. T. Bui and T. X. Hoang, “ Folding anh escape of nascent proteins at
ribosomal exit tunnel” , J. Chem. Phys. 144, 095102 (2016).
[8] P. T. Bui and T. X. Hoang, “ Protein escape at the ribosomal exit tunnel:
Effects of native interactins, tunnel length, and macromolecular crowding”, J.
Chem. Phys. 149, 045102 (2018).
[9] C. Levinthal C, “ Are there pathways for protein floding ?”, Journal de
Chimie Physique. 65, 44-45 (1968).
[10] C. Anfinsen, Biochem,“Principles that govern the folding of protein
chains” , J. 128, 737 (1972).
[11] C. M. Dobson, “ Protein folding and misfolding”, Nature 426, 884 - 890
(2003).
[12] C. M. Kaiser, D. H. Goldman, J. D. Chodera, I. Tinoco, Jr., and C.
Bustamante, “ The ribosome modulates nascent protein folding”, Science 334,
1723 (2011).

34
[13] P. M. Petrone, C. D. Snow, D. Lucent, and V. S. Pande, “ Side – chain
recognition and gating in the ribosome exit tunnel”, Proc. Natl. Acad. Sci. U.
S. A. 105, 16549-16554 (2008).
[14] N. Go, “ Theoretical studies of protein folding”, Ann. Rev. Biophys.
Bioeng. 12, 183-210 (1983).
[15] C. Clementi, H. Nymeyer, and J. N. Onuchic, J. Mol. Biol. 298, 937
(2000).
[16] T. X. Hoang and M. Cieplak, “ Sequencing of folding events in Go-like
proteins”, J. Chem. Phys. 113, 8319 (2000).
[17] R. B. Best and G. Hummer, “Molecular crowding enhances native state
stability and refolding rates of globular proteins” Proc. Natl. Acad. Sci. USA
102, 6732–6737 (2005).
[18] D. R. Cox and H. D. Miller, The Theory of Stochastic Processes
(Chapman and Hall, 1965), pp. 219–223.


















